

Abstract
Mature T- and NK-cell neoplasms (MTNKN) collectively represent a rare disorder, representing less than 15% of all non-Hodgkin lymphoma (NHL) cases and qualifying for orphan disease designation by the U.S. Food and Drug Administration (FDA). These consist of 9 families in the 5th revised World Health Organization (WHO) classification of lymphoid neoplasms, which are made up of over 30 disease subtypes, underscoring the heterogeneity of clinical features, molecular biology, and genetics across this disease group. Moreover, the 5 most common subtypes (peripheral T-cell lymphoma, not otherwise specified; nodal TFH cell lymphoma, angioimmunoblastic type; extranodal NK-cell/T-cell lymphoma; adult T-cell leukemia/lymphoma; and ALK-positive or -negative anaplastic large cell lymphoma) comprise over 75% of MTNKN cases, so other subtypes are exceedingly rare in the context of all NHL diagnoses and consequently often lack consensus on best practices in diagnosis and management. In this review, we discuss the following entities – enteropathy-associated T-cell lymphoma (EATL), monomorphic epitheliotropic intestinal T-cell lymphoma (MEITL), hepatosplenic T-cell lymphoma (HSTCL), subcutaneous panniculitis-like T-cell lymphoma (SPTCL), and primary cutaneous ɣδ T-cell lymphoma (PCGD-TCL) – with an emphasis on clinical and diagnostic features and options for management.
Keywords: EATL, MEITL, HSTCL, SPTCL, PCG-DTCL
Introduction
Although non-Hodgkin lymphoma (NHL) is considered relatively common, representing approximately 4% of all new cancer diagnoses in the United States(1), the majority of these arise from malignant B-cells. Conversely, mature T- and NK-cell neoplasms (MTNKN) comprise less than 15% of all NHL in Western countries and generally have a worse prognosis than B-cell NHL with 5-year overall survival (OS) under 50% for most subtypes(2). Unfortunately, most patients will experience relapsed or refractory disease, after which the median OS is approximately 6 months(3). One of the major challenges in addressing these suboptimal outcomes is the substantial disease heterogeneity across subtypes of MTNKN. The list of MTNKN has been consistently expanding for several decades, informed by an evolving understanding of cells of origin, clinical features of disease, and genomics, and in 2022, over 30 distinct disease entities have been identified in the MTNKN family based on updated classifications of lymphoid neoplasms developed by the World Health Organization (WHO)(4) and the International Consensus Classification (ICC) Clinical Advisory Committee(5).
This raises a unique dilemma in the clinical management of patients with MTNKN because five subtypes – peripheral T-cell lymphoma, not otherwise specified (PTCL, NOS); nodal TFH cell lymphoma, angioimmunoblastic type (NTFH-AITL); extranodal NK-cell/T-cell lymphoma (ENKTL); adult T-cell leukemia/lymphoma (ATLL); and ALK-positive or -negative anaplastic large cell lymphoma (ALCL) – comprise over 75% of all cases (Fig. 1)(2). As such, the vast majority of MTNKN subtypes are underrepresented or excluded in large clinical trials and registry studies impacting changes in standard of care(6, 7, 8).
Figure 1. MTNKN display geographic and phenotypic heterogeneity.
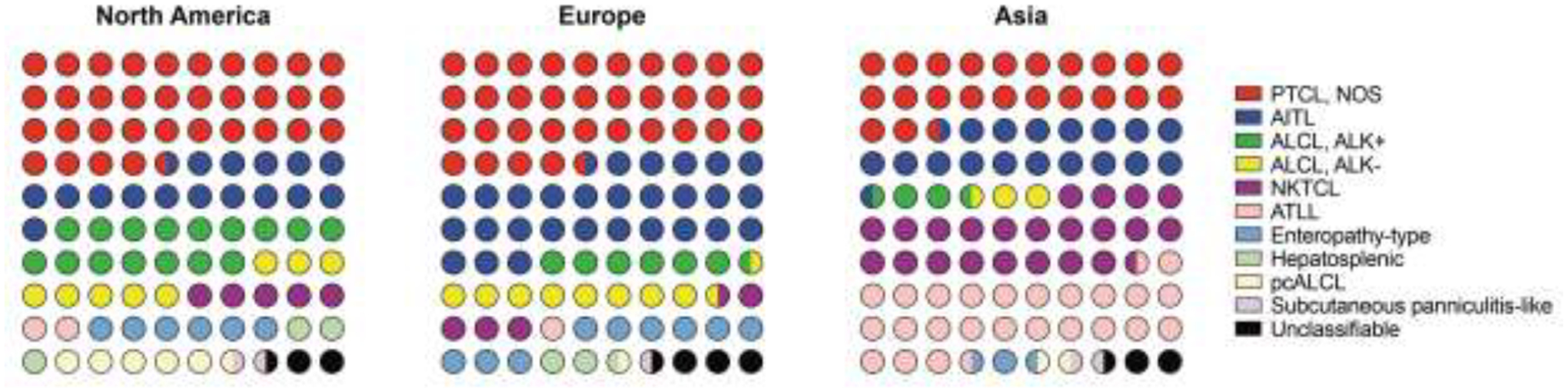
Dot plots (10×10) show relative proportions of MTNKN stratified by geographic population; data adapted from the International T-Cell Lymphoma Project(2). Note that nomenclature/abbreviations are based on the previous WHO classification of lymphoid tumors(93). Abbreviations: Peripheral T-cell lymphoma, not otherwise specified, PTCL, NOS; Angioimmunoblastic T-cell lymphoma, AITL, Anaplastic large cell lymphoma, ALCL; NK/T-cell lymphoma, NKTCL; ATLL, adult T-cell leukemia/lymphoma; pcALCL, primary cutaneous ALCL.
In this review, we will discuss clinical and diagnostic features, outcomes, and treatment considerations of several of the more common “rare” MTNKN, accounting for 6–7% of all diagnoses(2). These include enteropathy-associated T-cell lymphoma (EATL), monomorphic epitheliotropic intestinal T-cell lymphoma (MEITL), hepatosplenic T-cell lymphoma (HSTCL), subcutaneous panniculitis-like T-cell lymphoma (SPTCL), and primary cutaneous ɣδ T-cell lymphoma (PCGD-TCL).
Enteropathy-associated T-cell lymphoma and monomorphic epitheliotropic intestinal T-cell lymphoma
The association of celiac disease with intestinal lymphoma was first described in 1962(9), though subsequent studies described a wide range of histologies and clinical features of what were termed enteropathy-type T-cell lymphomas (ETL)(10, 11, 12, 13). The pathophysiology of this connection is rooted in the progressive accumulation of intraepithelial lymphocytes (IELs) in the setting of chronic intestinal inflammation(14). Secretion of interleukin 15 (IL-15) promotes an anti-apoptotic effect, ultimately selecting for malignant transformation of clonally expanded IELs(15, 16, 17). More recently, DeLeeuw and colleagues performed whole-genome analysis and HLA-genotyping on 30 patients with ETL and observed that there are two distinct morphologic and genetic subtypes, which they termed EATL type I or type II(18). We will henceforth refer to these as EATL or MEITL, respectively, based on updated classification of nomenclature(4, 5). EATL is the most common type of intestinal T-cell lymphoma, accounting for 60–80% of cases, while MEITL represents about 30%(13, 19). EATL classically arises in the setting of refractory celiac disease (RCD) II, though de novo transformation in patients with newly diagnosed or uncomplicated celiac disease has also been described, albeit much less frequently(20, 21). In contrast, MEITL is not associated with celiac disease and sporadically arises from aberrant IELs.
Given this association with celiac disease, it is unsurprising that EATL is largely found in patients of Northern European descent in whom celiac disease is more prevalent(11, 19, 21, 22). MEITL, however, is the most common intestinal T-cell lymphoma in Asian populations, in whom EATL is exceedingly rare(19). These studies have generally observed similar frequency across sexes, though a slight male predominance is seen in MEITL. The incidence of both EATL and MEITL increases with age with a median age of diagnosis of 60–65 years(18, 19, 21, 22, 23). Clinically, patients often may present similarly to an exacerbation of celiac disease with symptoms of abdominal pain/discomfort (80–100%), diarrhea (40–70%), and weight loss (50–80%)(11, 24, 25). Unlike other types of aggressive lymphomas, B symptoms are relatively infrequent, reported in approximately 30% of patients(11, 19, 24, 25, 26), though this should be considered suspicious for clinical progression or higher-risk disease(27). Moreover, signs of bowel perforation or obstruction are frequently seen and are thought, in part, to contribute to the historically poor prognosis for these patients. Laboratory workup for EATL and MEITL is often non-specific and more so may reflect RCD (in the case of EATL), such as hypoalbuminemia or anemia related to nutritional deficiencies; lactate dehydrogenase (LDH) is normal or only mildly elevated in most cases(11, 19, 26). As such, the diagnosis of EATL or MEITL is often found incidentally during endoscopy for gastrointestinal symptoms or during laparotomy for intestinal perforation or obstruction. Tumor involvement presents multifocally in nearly 25% of cases; EATL and MEITL most commonly involve the proximal small bowel, especially the jejunum(28). Although positron emission tomography/computed tomography (PET/CT) or CT imaging is recommended in staging, extra-intestinal involvement at presentation is fairly uncommon(19); furthermore, EATL metabolic activity is usually restricted to sites of active celiac disease, whereas MEITL is more likely to be eumetabolic(29).
Histologically, EATL is characterized by pleomorphic medium to large lymphocytes with increased mitotic index (Ki67 >50%) and an inflammatory milieu of eosinophils, histiocytes, and small lymphocytes. Given its association with celiac disease, surrounding bowel tends to demonstrate crypt hyperplasia and villous atrophy. Angiocentricity and angioinvasion with necrotic tissue is also frequently seen(30). MEITL conversely exhibits transmural infiltration of monomorphic small to medium sized lymphocytes; inflammatory background, as in EATL, is uncommon(4). Immunohistochemistry (IHC), cytogenetics, and genetics furthermore may be helpful in distinguishing these two entities (Table 1). IHC in EATL is typically positive for TIA-1, CD2, and CD3; variably positive for CD8 and CD30; and negative for CD4, CD5, CD7 and CD56, while IHC in MEITL is typically positive for TIA-1, CD2, CD3, CD7, CD8, and CD56, and negative for CD4, CD5, and CD30(13, 18, 19, 30). Although IELs can normally express TCRαβ or TCRɣδ, aberrantly proliferating cytotoxic IELs are often TCRαβ negative though harbor clonal TCRɣ or TCRβ rearrangements(15, 16, 17, 24); accordingly, most cases of EATL lack TCRαβ or TCRɣδ expression, whereas MEITL typically expresses TCRɣδ, or less commonly TCRαβ(30). Of note, a subset of patients with EATL can also have an anaplastic large cell phenotype(18), which is more frequently associated with CD8 negativity and CD30 positivity.
Table 1.
Histopathologic and genetic features of rare MTNKN.
| MTNKN | Immunophenotype | TCR | Cytogenetics | Recurrent genetic mutations |
|---|---|---|---|---|
| EATL | TIA-1+, CD2+, CD3+, CD8+/−, CD30+/−, granzyme B+, perforin+ | TCR-silent > TCRαβ > TCRɣδ | +1q, +5q, +9q, −16q | SET2D, TET2, STAT3, STAT5B, JAK1, JAK3, NRAS, BRAF, BCL11B, TP53, USP10, TERT, DAPK3, SOCS1 |
| MEITL | TIA-1+, CD2+, CD3+, CD8+, CD56+, granzyme B+, perforin+ | TCRɣδ > TCRαβ > TCR-silent | +8q24, +9q, −16q | CREBBP, STAT3, STAT5B, SET2D, GNAI2, JAK1, JAK3, DNAH9, PRR16, ASXL3, MEGF6, EZH2, EP300, TET2, USP10, TERT, KRAS, BRAF, NRAS, BCL11B, TP53 |
| HSTCL | TIA-1+, CD2+, CD3+, CD4−, CD8+/−, FasL+, CD56+, granzyme M+ | TCRɣδ > TCRαβ | i(7q), trisomy 8 | SETD2, INO80, TET3, SMARCA2, PIK3CD, STAT5B, STAT3, IDH2, EZH2, ARID1, DNMT3A |
| SPLTCL | TIA-1+, CD2+, CD3+, CD4−, CD8+, CD56−, granzyme B+, perforin+ | TCRαβ | Nonspecific | HAVCR2, ASXL1, JAK3, PIAS3, PLCG2, KMT2D, KMT2C, BAZ2A, NUP98, DDX11, IDH1, BRD2 |
| PCGD-TCL | TIA-1+, CD2+, CD3+, CD4−, CD8+/−, CD30+/−, CD56+/−, granzyme B+, perforin+ | TCRɣδ | Translocations involving breakpoints at 9p21, 14q11, 14q32, or 16q23 | KRAS, NRAS, MAPK1, MYC, MYCN, FBXW7, STAT3, STAT5B, JAK3, SOCS1, ARID1A, TRRAP, TET2, KMT2D, CDKN2A, IDH2, TP53 |
The most frequent chromosomal abnormalities identified in all types of ETL are 9q gains and 16q losses, which are typically mutually exclusive. EATL specifically is characterized by frequent gains of 1q and 5q while MEITL often harbors 8q24 gains at the MYC locus(18). Additionally, nearly all patients with celiac disease have HLA-DQ2 or HLA-DQ8 positivity(31), which are both also associated with increased risk of EATL(18, 32, 33). While HLA-DQ2/8 positivity should not be considered diagnostic of EATL, negativity of both should bring into question the diagnosis and may favor MEITL. Few studies have specifically compared the somatic mutational landscape between these disease entities (Table 1), though a report by Nicolae et al. found that JAK/STAT pathway and RAS pathway alterations are common in both diseases(23); however, EATL more commonly has STAT3/JAK1 mutations while MEITL more frequently harbors STAT5B/JAK3 mutations. Other groups have also noted that epigenetic regulators (TET2) and DNA damage/apoptotic mediators (TP53, BCL11B) are mutated in approximately 10–15% of patients with either type of lymphoma, though EATL is enriched for SOCS1 and DAPK3 mutations, while MEITL is enriched for alterations in epigenetic modifiers like CREBBP, EP300, EZH2, and SETD2 and MAPK pathway members(33, 34). Moreover, MEITL tends to overexpress MATK, which may aid in appropriate diagnosis(34).
Historically, outcomes for ETL have been poor, owing at least in large part to the frequent presentation of surgical emergencies like bowel perforation or obstruction(11). Many of these patients have poor performance statuses at diagnosis and nearly one-third die prior to any treatment or shortly after completion of a single cycle of chemotherapy. Historical 5-year OS is below 20% with a median OS of 4–11 months(2, 11, 13, 19, 24, 26). Frontline treatment of EATL usually includes multiagent combination chemotherapy. However, in a retrospective analysis of rare T-cell lymphomas, only 30% of patients with ETL achieved a complete response/unconfirmed complete response (CR/CRu) with frontline therapy, which almost universally was anthracycline-based(26). As such, the question arises of whether there would be benefit to intensification of therapy and consolidative hematopoietic cell transplant (HCT) in first remission. At times, this may require resection of involved bowel prior to chemotherapy for debulking, and treatment can be complicated by a high frequency of malnourishment and a significant risk of bowel perforation.
A study from Scotland and Newcastle Lymphoma Group retrospectively analyzed records of patients with EATL (prior to the distinction of EATL vs MEITL) who were treated with a novel regimen consisting of cyclophosphamide, doxorubicin, vincristine, prednisone (CHOP) followed by ifosfamide, epirubicin, etoposide, intermediate-dose methotrexate (IVE/MTX), ultimately culminating in consolidative HCT(35). Most patients had extensive disease and required prior laparotomy for acute abdominal symptoms. When compared to historical CHOP-like regimens, patients who were able to complete the IVE/MTX protocol had better response rates (93% vs 41%) and notably had a nearly 50% reduction in both mortality and lymphoma-related mortality. However, this regimen was associated with higher rates of febrile neutropenia and sepsis compared to CHOP-like therapy alone. A follow up single-arm phase II trial from the same group in 21 patients found that 86% of patients were able to complete the full CHOP plus IVE/MTX regimen with an overall response rate of 71% though with a 1-year OS of 45%(36). A retrospective study from the EBMT Lymphoma Working Party analyzed records of 44 ETL patients who received HCT between 2000–2010, noting that 57% had celiac disease(37). CHOP-like frontline therapy was used in 43% of patients, and 70% of patients underwent HCT while in first CR or partial response (PR). Relapse occurred in 39% of patients at up to 4 years after transplant with only one relapse after 18 months. Moreover, progression free survival (PFS) and OS were 54% and 59% at 4 years, respectively, with a trend toward better OS in those who underwent HCT in first CR/PR. Given the improved OS and possibility for durable remission, consolidative HCT in first remission should strongly be considered in eligible patients.
As previously mentioned, EATL tends to express CD30, so CD30-targeted therapy may offer a novel opportunity in the upfront management of this disease. The EATL-001 study is evaluating the efficacy of brentuximab vedotin, a CD30-directed antibody-drug conjugate, plus cyclophosphamide, doxorubicin, prednisone (BV-CHP) in patients with newly diagnosed CD30 positive EATL(38). In this study, responding patients received consolidative etoposide/high-dose MTX followed by HCT. Preliminary data found an overall response rate of 79% with 64% of patients achieving CR. Although two patients died of septic shock during HCT, at 2-years, no patients had yet relapsed, possibly suggesting a new standard of care for this population when compared to historical cohorts(35, 36, 37, 39).
Given that it has only recently been established as a diagnostic entity, there is no current standard of care for MEITL, though it has been included (as Type II EATL) in several of the aforementioned studies(35, 37, 39) and therefore is often treated similarly to EATL. One retrospective study found that although CHOP was the most frequently used frontline regimen for these patients, CR rates were significantly higher for those who received more intensive therapy than CHOP (71% vs 37%)(40); similarly, one report observed that intensive L-asparaginase-containing regimens are associated with a higher response rate than CHOP(41). Moreover, HCT has been associated with significant improvement in survival and, consistent with the literature in EATL, upfront HCT may portend better outcomes than salvage HCT(40, 41). Ultimately, both EATL and MEITL are associated with poor outcomes and significant disease-related morbidity and typically require intensified chemotherapy regimens with consolidative HCT, as CHOP alone is usually not considered sufficient for most patients (Table 2). Our understanding of these distinct entities is continually evolving, and future prospective studies will be critical to optimizing care for these patients.
Table 2.
Preferred frontline treatment for rare MTNKN.
| MTNKN | Preferred treatment modality for fit patients | Role of transplant | Other considerations | Evidence |
|---|---|---|---|---|
| EATL | Intensified chemotherapy (i.e., CHOP-IVE/MTX, Hyper-CVAD) preferred. Consider BV-CHP if CD30+. | Consolidative HCT preferred in first remission. | Consider prophylactic bowel resection in select patients at high risk for perforation. | (35, 36, 37, 38) |
| MEITL | CHOEP/EPOCH preferred. Consider L-asparaginase-based therapy (i.e., SMILE) or intensified chemotherapy (i.e., CHOP-IVE/MTX, Hyper-CVAD), though this is extrapolated from EATL population. | Consolidative HCT preferred in first remission. | Consider prophylactic bowel resection in select patients at high risk for perforation. | (40, 41) |
| HSTCL | ICE or IVAC preferred. Other platinum-containing high dose regimens, such as GDP may be appropriate. Consider CHOEP/EPOCH if unable to tolerate ifosfamide- or platinum-based therapy. | Consolidative HCT preferred in first remission. Consider allogeneic HCT in eligible patients. | Consider splenectomy if severe thrombocytopenia precludes optimal treatment. | (26, 46, 48, 49, 53, 58, 61, 62) |
| SPTCL | Immunomodulatory therapy (i.e., CsA, MTX/Prednisone) is preferred unless severe symptoms and/or HLH. In those patients, consider multiagent chemotherapy, such as CHO(E)P or ICE. | Consolidative HCT to be considered for patients with severe symptoms and/or HLH. | Relapse after initial immunomodulatory therapy has not been consistently shown to worsen outcomes, so conservative therapy in mildly symptomatic patients is preferred. | (70, 74) |
| PCGD-TCL | CHO(E)P preferred. | Consolidative HCT recommended with preference for allogeneic HCT in eligible patients. | (70, 88, 89) |
Chemotherapy regimen abbreviations: CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone), IVE/MTX (ifosfamide, epirubicin, etoposide, intermediate-dose methotrexate), BV-CHP (brentuximab vedotin, cyclophosphamide, doxorubicin, prednisone), Hyper-CVAD (hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone alternating with methotrexate and cytarabine, CHOEP/EPOCH (cyclophosphamide, doxorubicin, vincristine, prednisone, etoposide), SMILE (dexamethasone, methotrexate, ifosfamide, L-asparaginase, etoposide), ICE (ifosfamide, cisplatin, etoposide), IVAC (ifosfamide, etoposide, high-dose cytarabine), GDP (gemcitabine, dexamethasone, cisplatin), CsA (cyclosporine).
Hepatosplenic T-cell lymphoma
HSTCL is another rare MTNKN, originally reported to arise from ɣδ-T-cells(42, 43), although cases with TCRαβ expression were later described as well. ɣδ-T-cells are the first T cells to develop in the embryonic thymus and are infrequently found in secondary lymphoid tissues. They represent less than 5% of peripheral blood T-cells. After leaving the thymus as CD4/CD8 negative (double negative) T-cells, they circulate and reside in peripheral tissues like skin, adipose tissue, intestine, liver, lungs, and most abundantly in the spleen. They have multiple functions ranging from immune surveillance through excess cytokine production to mucosal barrier maintenance (reviewed in (44)). This immunoregulatory role may play a critical role in both the pathophysiology of HSTCL as well as its clinical presentation, as approximately 20% of cases can arise in patients on immunosuppressive therapy or with immune-dysregulatory disorders(45, 46, 47, 48, 49). Inflammatory bowel disease, particularly Crohn’s disease, has been associated with HSTCL, though this often is thought to occur in the setting of immunosuppressive agents, including anti-tumor necrosis factor (TNF) agents, mercaptopurine, or azathioprine(50, 51, 52). Concomitant use of TNF inhibitors with other immunosuppressive agents is the strongest risk factor for HSTCL(47), suggesting that the pathobiology of disease may be multifactorial stemming from chronic inflammatory antigen exposure with persistence of clonally expanded cytotoxic ɣδ-T-cells of the splenic pool in an immunosuppressed environment, ultimately leading to accumulation of driving genetic alterations. However, as previously mentioned, cases of αβ-T-cell HSTCL have been found, which may suggest a distinct mechanism of disease.
HSTCL occurs in younger patients, with a median age of diagnosis reported between 29–38 years; it is also 2–3-fold more common in males than females(48, 49, 53, 54). B symptoms are seen in 70–80% of patients at presentation. Splenomegaly is almost universal in patients, though hepatomegaly is also seen in 40–88% of patients. Most commonly, this is found in the setting of abdominal discomfort, and signs of hepatic dysfunction, such as jaundice, may occur. Laboratory abnormalities include cytopenias (most commonly thrombocytopenia), elevated LDH, and elevated transaminases. This may be multifactorial due to hypersplenism, bone marrow involvement, or inflammatory myelosuppression. Of note, dysplastic changes in other lineages(55) as well as hemophagocytic syndrome (HPS)(48, 56) have also been reported in patients with HSTCL, though these are unlikely to be common drivers of cytopenias. Given that HSTCL can manifest with cytopenias, elevated LDH, B symptoms, and features of HPS, bone marrow biopsy is an important part of baseline evaluation, as it is involved in 60–70% of patients. While lymphocytosis is uncommon, peripheral blood flow cytometry can detect a neoplastic population of lymphocytes in approximately 50% of patients(46), though overt leukemic phenotypes are seen in less than 2% of patients(48). PET/CT may be non-specific with avidity diffusely in the liver, spleen, and bone marrow, and diagnosis is most commonly made with bone marrow and/or liver biopsy.
Neoplastic cells typically appear as atypical, small to intermediate sized lymphocytes densely infiltrating the sinusoids of the liver and spleen, although blastoid changes have been reported as well(57). However, liver involvement can also manifest as periportal infiltration with spillage into the sinusoids or nodular parenchymal infiltration. Hemophagocytosis can be seen with or without overt HPS. HSTCL cells lack granules, which can help distinguish them from T-cell large granular lymphocytic leukemia. IHC is typically positive for CD2, CD3, CD7, and CD56, while negative for CD4, CD5, CD8, and CD57. Perforin and granzyme B are negative, and TIA-1 and granzyme M are positive, suggestive of a nonactivated cytotoxic phenotype. As previously discussed, approximately 80% of HSTCL are derived from ɣδ-T-cells, typically of the Vδ1 subset.
Notably, αβ variants appear more frequently in females and older patients and are associated with a worse prognosis(48, 58). The most frequent chromosomal abnormalities found in HSTCL are isochromosome 7q and trisomy 8, which are seen in 63% and 50% of cases, respectively, and are not mutually exclusive(2, 54). More recently, next generation sequencing has allowed for the identification of other recurrent genetic abnormalities (Table 1), which may aid in the diagnosis of HSTCL(59, 60). McKinney and colleagues(60) performed whole exome sequencing on 68 HSTCL cases and found recurrent alterations in epigenetic modifiers, JAK/STAT pathway, PI3K pathway, and consensus cancer genes (TP53, UBR5, and IDH2). Mutations seen in other MTNKN such as RHOA and CCR4 are uncommon in HSTCL, while isochromosome 7q and trisomy 8 are rare in other types of MTNKN.
Outcomes for patients with HSTCL are traditionally very poor, with a median OS of 10–12 months and 5-year OS under 10%(2, 26, 46, 48, 60). Certain prognostic factors, like elevated bilirubin, presence of isochromosome 7q and trisomy 8, or αβ variants are associated with worse outcomes. Although CHOP-like regimens can achieve CR in patients with HSTCL, median OS remains under 1 year due to frequent relapses(26, 46, 48, 49, 53, 58, 61). Voss and colleagues reported that 7 patients in a 14-patient series were alive at a median follow up of 65.6 months, 6 of whom received ifosfamide-based induction(61). All surviving patients proceeded to undergo HCT as well, so it is unclear whether the benefit in this subset of patients was driven by transplant-related outcomes or alternative induction strategies. Nevertheless, HCT is an important consideration for patients with HSTCL, as accumulating evidence from case series and retrospective analyses demonstrates that patients undergoing HCT have a higher chance of long-term responses with improvements in OS, though this may not always be durable(26, 46, 48, 49, 53, 58, 61). A retrospective study from the EBMT suggested that patients undergoing allogeneic HCT were less likely to experience relapse than those undergoing autologous HCT, though these data are limited by small sample size and relatively short follow up so should be interpreted cautiously(62).
Cytopenias, in particular, present a clinical challenge due to prolonged treatment-related myelosuppression, infectious complications, and difficulty mobilizing stem cells in those who are eligible for HCT, so splenectomy can be considered if alleviation of thrombocytopenia may help the patient receive full doses of chemotherapy. Finally, it has previously been reported that MDR-1 and pgp-1 amplification, which is commonly seen in HSTCL(63), can drive chemoresistance, underpinning rationale for the assessment of non-traditional chemotherapeutics or targeted therapies in future studies; several case reports have suggested alemtuzumab, a CD52-targeting monoclonal antibody, and newer agents such as pralatrexate or the histone deacetylase inhibitor romidepsin may have efficacy in patients with HSTCL(64, 65). Overall, treatment of patients with HSTCL remains a significant challenge with a paucity of data; with the available data(61), a reasonable approach would be to consider induction with ifosfamide, carboplatin, etoposide (ICE), ifosfamide, etoposide, high-dose cytarabine (IVAC), or other platinum-containing high dose regimens; if patients are unable to tolerate this, then CHOP with etoposide (CHOEP or EPOCH) should be considered (Table 2). Preferably, patients should undergo consolidative HCT in patients upon first CR. While either autologous or allogeneic HCT can be considered, there is low-quality evidence that allogeneic HCT may be preferable in eligible patients.
Subcutaneous panniculitis-like T-cell lymphoma and primary cutaneous ɣδ T-cell lymphoma
SPTCL and PCGD-TCL are rare primary cutaneous T-cell lymphomas, which were previously both considered under the category of “subcutaneous panniculitis-like T-cell lymphoma” until the 2008 WHO classification of lymphoid neoplasms(66). SPTCL was initially described in 1991 in a case series of patients with T-cell lymphoma in the subcutaneous adipose tissue, 75% of whom had HPS(67). Later studies identified that these cutaneous T-cell lymphomas arise from distinct cells of origin, which are associated with a more indolent (TCRαβ phenotype) or aggressive (TCRɣδ phenotype) disease course(68). Approximately 20% of patients with SPTCL have a concurrent autoimmune disease, most commonly systemic lupus erythematosus(69, 70); this can be a challenging since lupus erythematosus panniculitis (LEP) can appear strikingly similar, and biopsies require expert dermatopathology review for distinction(73, 74). It is possible that SPTCL and LEP may exist on a spectrum, but in SPTCL, malignant CD8 cytotoxic T-cells expressing CCR5 migrate to adipose tissue, which expresses CCR5 ligands(71). There, the cytotoxic T-cells disrupt adipose membranes, sparing the dermis and epidermis, leading to panniculitis. PCGD-TCL, conversely, is more heterogeneous and aggressive. Similar to HSTCL, PCDG-TCL arises from ɣδ-T-cells; recent studies have demonstrated that these can either be the Vδ1 or Vδ2 subtype leading to superficial epitheliotropic lymphomas or panniculitic lymphomas, respectively, based on their tissue tropism; furthermore, the Vδ2 subtype expresses a cytokine profile consistent with what is seen in HPS(72).
Despite both involving the skin, SPTCL and PCGD-TCL are clinically very different diseases. SPTCL is seen in younger patients (median age 36–38), though can present across a wide age range, while the median age of diagnosis for PCGD-TCL is 59–61 years(70, 73, 74). Both have a female predominance and are characterized by multifocal skin nodules; however, nodules in SPTCL are adipotropic, erythematous, and often painless with a waxing and waning course, whereas nearly half of patients with PCGD-TCL have ulcerative lesions, which can be painful and rapidly progressive. Although PCGD-TCL classically has dermal and epidermal tropism, underpinning the ulcerative phenotype, panniculitic subtypes can be seen(72, 75). Moreover, PCGD-TCL has a lower limb predominance, whereas SPTCL can be distributed along upper or lower extremities or truncally. Diagnosis of either entity is usually made with a skin punch biopsy, though it should be noted that older lesions in SPTCL may exhibit granulomatous inflammation, lipomembranous fat necrosis and/or fibrosis rather than lymphoma. B symptoms are reported in 60–65% of patients with either SPTCL or PCGD-TCL, though HPS is significantly more frequently seen in patients with PCGD-TCL (approximately 50% vs less than 20% in SPTCL)(68, 70). In patients with HPS, typical markers of inflammation, such as hypofibrinogenemia, transaminase elevation, elevated LDH, cytopenias, hyperferritinemia, and hypertriglyceridemia can be seen. For diagnosis of hemophagocytic lymphohistiocytosis (HLH), biopsy specimen should ideally be obtained from the bone marrow, spleen, lymph node, or liver rather than the skin(76, 77). While extracutaneous involvement is uncommon in SPTCL, it can be seen with progressive PCGD-TCL and can involve sites including the lung, thyroid, breast, testis, and mucosa(75).
There are histological differences that can help distinguish SPTCL and PCGD-TCL (Table 1). SPTCL is characterized by a lobular panniculitis-like infiltrate on a background of fat necrosis and histiocytes, occasionally exhibiting hemophagocytosis. Neoplastic cytotoxic T-cells rim adipocytes, which is characteristic of SPTCL. Moreover, plasma cells, lymphoid follicles, and hyaline lipomembranous fat necrosis are less obvious, which can be useful to help distinguish SPTCL from LEP(78, 79). PCGD-TCL is characterized by medium to large lymphocytes, rarely with blastoid morphology, which are distributed either epidermally, dermally, or subcutaneously(73, 80, 81). Neoplastic cells in SPTCL typically stain positive for CD3, CD8, granzyme B, TIA-1, and perforin and negative for CD56 and CD30, whereas PCGD-TCL is characterized by cells that are positive for CD3, CD56, granzyme B, TIA-1, and perforin and negative for CD8.
Biallelic HAVCR2 (TIM-3) germline mutations are frequently seen in sporadic SPTCL(82, 83, 84). Aberrant loss of TIM-3 expression can result in immune activation and excessive cytokine release, possibly driving the HPS phenotype in a subset of patients(83). Other recurrent somatic mutations in immune response genes (ASXL1, JAK2, PIAS3, PLCG2) and epigenetic regulators (KMT2D, KMT2C, NUP98) have also been identified in SPTCL(84). Notably, SPTCL without HAVCR2 mutations has an increase in expression of genes associated with lymphocyte homing and immune regulation. This may have prognostic relevance as well, as younger patients and mutated HAVCR2 are more likely to have HPS and worse outcomes(84). PCGD-TCL often harbors recurrent translocations at breakpoints involving 9p21, 14q11.2, 14q32.1, or 16q23.1, which involve critical T-cell regulatory genes including BCL11B and TCL(85). Interestingly, although the Vδ1 or Vδ2 subtypes of PCGD-TCL have clinically distinct phenotypes, their mutational landscape is similar, involving MAPK signaling, JAK/STAT signaling, chromatin modification, and consensus cancer genes (CDKN2A, IDH2, TP53)(72).
Generally, SPTCL is considered to have an excellent prognosis with a 5-year OS over 80%, though patients who experience HPS have worse outcomes with a 5-year OS of 46%(70). Conversely, patients with PCGD-TCL have a 5-year OS of only 11%(70). Of note, ɣδ mycosis fungoides (MF) may exist on a spectrum with PCGD-TCL. ɣδ MF usually behaves indolently, but a subset of patients may experience PCGD-TCL-like progression, which has a clinical, mutational and gene expression profile identical to de novo PCGD-TCL and has equally dismal outcomes after the phenotypic switch(72).
Willemze et al. found that CR/PR could be achieved in 71% and 88% of patients with SPTCL treated with CHOP-like therapy or immunomodulatory therapy (i.e., cyclosporine), respectively(70). This has been more recently corroborated in a retrospective study showing that either chemotherapy or immunomodulatory therapy can achieve high response rates, and although relapse is common, it did not significantly affect prognosis(74). Notably, cyclosporine achieved responses in 94% of patients, and methotrexate showed responses in 100% of patients when used in the first-line, suggesting that these lower-intensity therapies should be considered frontline for patients without severe disease or HLH (Table 2). Nevertheless, patients with more severe disease and/or HLH often still required chemotherapy with consolidative HCT.
Data on appropriate treatment for PCGD-TCL are sparser. In the study by Willemze and colleagues, of 14 patients with PCGD-TCL treated with CHOP-like chemotherapy, CR was only observed in 3 patients, with frequent progression to visceral involvement and HPS, though outcomes were similar whether or not patients had HPS(70). The use of brentuximab vedotin for CD30-expressing PCGD-TCL has only been reported in case series, and though this may have some efficacy, it warrants further investigation(86, 87). Ultimately, in patients with PCGD-TCL, consolidation with allogeneic stem cell transplantation should be considered in eligible patients. In a small retrospective series of 10 patients with PCGD-TCL and 4 with refractory SPTCL, 7 underwent allogeneic HCT after intensive chemotherapy with CHOP with or without etoposide. Of these patients, 4 were alive at up to 5.9 years after transplant, 3 of whom had PCGD-TCL(88). More recently, real-world data was presented on 48 patients with PCGD-TCL(89); although front-line treatment was very heterogeneous, consolidative HCT was performed in 7 patients, of which 6 were allogeneic. Patients who underwent consolidative HCT in first CR had significantly improved OS. There is no consensus or guidance on initial therapy for this rare disease, but most experts would agree that treatment should be commensurate to the acuity and aggressiveness of the initial presentation. Nevertheless, at present, the limited data suggest that regardless of induction chemotherapy, consolidative HCT is critical for improvement in long-term outcomes (Table 2).
Conclusion
Over the last several decades, there have been significant advances in the field of MTNKN, including expanding therapeutic options and updates in disease classifications(4, 5). Unfortunately, data on the utility of many novel agents are limited to case reports in the majority of MTNKN due to their rarity, which often precludes them from being heavily represented in prospective clinical trials. This has posed a significant challenge to clinicians and patients because many of these MTNKN are treated based off anecdotal experience or by extrapolating data from other biologic entities. As such, outcomes for many rare T-cell lymphomas, especially EATL, MEITL, HSPTCL, and PCGD-TCL, are still dismal with 3-year OS under 50%(26). Moreover, relative inexperience in the diagnostic evaluation of different MTNKN can lead to delays in care or sub-optimal treatment strategies; two groups have independently reported that referrals to tertiary or “expert” centers for MTNKN result in diagnostic reclassification in up to approximately one-third of cases(90, 91), often with implications in therapeutic decision-making. Thus, it is critical that patients with MTNKN have their pathology reviewed at a center with expertise in these diseases.
Nevertheless, retrospective studies and small case series have provided some evidence base for current clinical practices, such as the use of brentuximab vedotin in EATL(38) or consolidative allogeneic HCT in patients with PCGD-TCL(89). Moreover, our understanding of disease biology continues to improve through the use of genomic profiling(92). These technologies have allowed for the identification of multiple potentially targetable pathways across these rare lymphomas, including JAK/STAT signaling, PI3K signaling, and epigenetic pathways, all of which are areas under active investigation. Thus, although MTNKN display extensive disease heterogeneity, accumulating molecular data will hopefully refine these entities and help identify more personalized approaches to treatment.
Funding
RSB is supported by the Hematopoiesis Training Program grant at the University of Pennsylvania (T32DK07780) from the National Institutes of Health (NIH).
Conflicts of Interest
RSB receives consultancy fees from Alva10. SKB receives or has received honoraria/consultancy fees from Acrotech, Affimed, Daiichi Sankyo, Janssen, Kyowa Kirin, and Seagen. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73(1):17–48. [DOI] [PubMed] [Google Scholar]
- 2.Vose J, Armitage J, Weisenburger D, International TCLP. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008;26(25):4124–30. [DOI] [PubMed] [Google Scholar]
- 3.Bellei M, Foss FM, Shustov AR, Horwitz SM, Marcheselli L, Kim WS, et al. The outcome of peripheral T-cell lymphoma patients failing first-line therapy: a report from the prospective, International T-Cell Project. Haematologica. 2018;103(7):1191–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IBO, Berti E, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia. 2022;36(7):1720–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campo E, Jaffe ES, Cook JR, Quintanilla-Martinez L, Swerdlow SH, Anderson KC, et al. The International Consensus Classification of Mature Lymphoid Neoplasms: a report from the Clinical Advisory Committee. Blood. 2022;140(11):1229–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmitz N, Trumper L, Ziepert M, Nickelsen M, Ho AD, Metzner B, et al. Treatment and prognosis of mature T-cell and NK-cell lymphoma: an analysis of patients with T-cell lymphoma treated in studies of the German High-Grade Non-Hodgkin Lymphoma Study Group. Blood. 2010;116(18):3418–25. [DOI] [PubMed] [Google Scholar]
- 7.Ellin F, Landstrom J, Jerkeman M, Relander T. Real-world data on prognostic factors and treatment in peripheral T-cell lymphomas: a study from the Swedish Lymphoma Registry. Blood. 2014;124(10):1570–7. [DOI] [PubMed] [Google Scholar]
- 8.Horwitz S, O’Connor OA, Pro B, Trumper L, Iyer S, Advani R, et al. The ECHELON-2 Trial: 5-year results of a randomized, phase III study of brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma. Ann Oncol. 2022;33(3):288–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gough KR, Read AE, Naish JM. Intestinal reticulosis as a complication of idiopathic steatorrhoea. Gut. 1962;3(3):232–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Isaacson PG, Du MQ. Gastrointestinal lymphoma: where morphology meets molecular biology. J Pathol. 2005;205(2):255–74. [DOI] [PubMed] [Google Scholar]
- 11.Gale J, Simmonds PD, Mead GM, Sweetenham JW, Wright DH. Enteropathy-type intestinal T-cell lymphoma: clinical features and treatment of 31 patients in a single center. J Clin Oncol. 2000;18(4):795–803. [DOI] [PubMed] [Google Scholar]
- 12.Lee MY, Tsou MH, Tan TD, Lu MC. Clinicopathological analysis of T-cell lymphoma in Taiwan according to WHO classification: high incidence of enteropathy-type intestinal T-cell lymphoma. Eur J Haematol. 2005;75(3):221–6. [DOI] [PubMed] [Google Scholar]
- 13.Chott A, Haedicke W, Mosberger I, Fodinger M, Winkler K, Mannhalter C, et al. Most CD56+ intestinal lymphomas are CD8+CD5-T-cell lymphomas of monomorphic small to medium size histology. Am J Pathol. 1998;153(5):1483–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cellier C, Delabesse E, Helmer C, Patey N, Matuchansky C, Jabri B, et al. Refractory sprue, coeliac disease, and enteropathy-associated T-cell lymphoma. French Coeliac Disease Study Group. Lancet. 2000;356(9225):203–8. [DOI] [PubMed] [Google Scholar]
- 15.Mention JJ, Ben Ahmed M, Begue B, Barbe U, Verkarre V, Asnafi V, et al. Interleukin 15: a key to disrupted intraepithelial lymphocyte homeostasis and lymphomagenesis in celiac disease. Gastroenterology. 2003;125(3):730–45. [DOI] [PubMed] [Google Scholar]
- 16.Di Sabatino A, Ciccocioppo R, Cupelli F, Cinque B, Millimaggi D, Clarkson MM, et al. Epithelium derived interleukin 15 regulates intraepithelial lymphocyte Th1 cytokine production, cytotoxicity, and survival in coeliac disease. Gut. 2006;55(4):469–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malamut G, El Machhour R, Montcuquet N, Martin-Lanneree S, Dusanter-Fourt I, Verkarre V, et al. IL-15 triggers an antiapoptotic pathway in human intraepithelial lymphocytes that is a potential new target in celiac disease-associated inflammation and lymphomagenesis. The Journal of clinical investigation. 2010;120(6):2131–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deleeuw RJ, Zettl A, Klinker E, Haralambieva E, Trottier M, Chari R, et al. Whole-genome analysis and HLA genotyping of enteropathy-type T-cell lymphoma reveals 2 distinct lymphoma subtypes. Gastroenterology. 2007;132(5):1902–11. [DOI] [PubMed] [Google Scholar]
- 19.Delabie J, Holte H, Vose JM, Ullrich F, Jaffe ES, Savage KJ, et al. Enteropathy-associated T-cell lymphoma: clinical and histological findings from the international peripheral T-cell lymphoma project. Blood. 2011;118(1):148–55. [DOI] [PubMed] [Google Scholar]
- 20.Al-Toma A, Verbeek WH, Hadithi M, von Blomberg BM, Mulder CJ. Survival in refractory coeliac disease and enteropathy-associated T-cell lymphoma: retrospective evaluation of single-centre experience. Gut. 2007;56(10):1373–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malamut G, Chandesris O, Verkarre V, Meresse B, Callens C, Macintyre E, et al. Enteropathy associated T cell lymphoma in celiac disease: a large retrospective study. Dig Liver Dis. 2013;45(5):377–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verbeek WH, Van De Water JM, Al-Toma A, Oudejans JJ, Mulder CJ, Coupe VM. Incidence of enteropathy--associated T-cell lymphoma: a nation-wide study of a population-based registry in The Netherlands. Scand J Gastroenterol. 2008;43(11):1322–8. [DOI] [PubMed] [Google Scholar]
- 23.Nicolae A, Xi L, Pham TH, Pham TA, Navarro W, Meeker HG, et al. Mutations in the JAK/STAT and RAS signaling pathways are common in intestinal T-cell lymphomas. Leukemia. 2016;30(11):2245–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Di Sabatino A, Biagi F, Gobbi PG, Corazza GR. How I treat enteropathy-associated T-cell lymphoma. Blood. 2012;119(11):2458–68. [DOI] [PubMed] [Google Scholar]
- 25.Novakovic BJ, Novakovic S, Frkovic-Grazio S. A single-center report on clinical features and treatment response in patients with intestinal T cell non-Hodgkin’s lymphomas. Oncol Rep. 2006;16(1):191–5. [PubMed] [Google Scholar]
- 26.Foss FM, Horwitz SM, Civallero M, Bellei M, Marcheselli L, Kim WS, et al. Incidence and outcomes of rare T cell lymphomas from the T Cell Project: hepatosplenic, enteropathy associated and peripheral gamma delta T cell lymphomas. Am J Hematol. 2020;95(2):151–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Baaij LR, Berkhof J, van de Water JM, Sieniawski MK, Radersma M, Verbeek WH, et al. A New and Validated Clinical Prognostic Model (EPI) for Enteropathy-Associated T-cell Lymphoma. Clin Cancer Res. 2015;21(13):3013–9. [DOI] [PubMed] [Google Scholar]
- 28.Domizio P, Owen RA, Shepherd NA, Talbot IC, Norton AJ. Primary lymphoma of the small intestine. A clinicopathological study of 119 cases. Am J Surg Pathol. 1993;17(5):429–42. [DOI] [PubMed] [Google Scholar]
- 29.Chan TSY, Lee E, Khong PL, Tse EWC, Kwong YL. Positron emission tomography computed tomography features of monomorphic epitheliotropic intestinal T-cell lymphoma. Hematology. 2018;23(1):10–6. [DOI] [PubMed] [Google Scholar]
- 30.Lesmes JA, Poveda J. Primary gastrointestinal T-cell lymphomas: concepts and diagnostic insights. Diagnostic Histopathology. 2021;27(2):57–61. [Google Scholar]
- 31.Green PH, Jabri B. Coeliac disease. Lancet. 2003;362(9381):383–91. [DOI] [PubMed] [Google Scholar]
- 32.Al-Toma A, Goerres MS, Meijer JW, Pena AS, Crusius JB, Mulder CJ. Human leukocyte antigen-DQ2 homozygosity and the development of refractory celiac disease and enteropathy-associated T-cell lymphoma. Clin Gastroenterol Hepatol. 2006;4(3):315–9. [DOI] [PubMed] [Google Scholar]
- 33.Moffitt AB, Ondrejka SL, McKinney M, Rempel RE, Goodlad JR, Teh CH, et al. Enteropathy-associated T cell lymphoma subtypes are characterized by loss of function of SETD2. The Journal of experimental medicine. 2017;214(5):1371–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang D, Lim JQ, Cheah DMZ, Kahliab K, Laurensia Y, Pang JWL, et al. Whole-genome sequencing reveals potent therapeutic strategy for monomorphic epitheliotropic intestinal T-cell lymphoma. Blood Adv. 2020;4(19):4769–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sieniawski M, Angamuthu N, Boyd K, Chasty R, Davies J, Forsyth P, et al. Evaluation of enteropathy-associated T-cell lymphoma comparing standard therapies with a novel regimen including autologous stem cell transplantation. Blood. 2010;115(18):3664–70. [DOI] [PubMed] [Google Scholar]
- 36.Phillips EH, Lannon MM, Lopes A, Chadwick H, Jones G, Sieniawski M, et al. High-dose chemotherapy and autologous stem cell transplantation in enteropathy-associated and other aggressive T-cell lymphomas: a UK NCRI/Cancer Research UK Phase II Study. Bone Marrow Transplant. 2019;54(3):465–8. [DOI] [PubMed] [Google Scholar]
- 37.Jantunen E, Boumendil A, Finel H, Luan JJ, Johnson P, Rambaldi A, et al. Autologous stem cell transplantation for enteropathy-associated T-cell lymphoma: a retrospective study by the EBMT. Blood. 2013;121(13):2529–32. [DOI] [PubMed] [Google Scholar]
- 38.Sibon D, Khater S, Bruneau J, Brouzes C, Lhermitte L, Molina TJ, et al. The Eatl-001 Trial: Results of a Phase 2 Study of Brentuximab Vedotin and CHP Followed By Consolidation with High-Dose Therapy - Autologous Stem-Cell Transplantation (HDT-ASCT) in the Frontline Treatment of Patients with Enteropathy-Associated T-Cell Lymphoma. Blood. 2021;138(Supplement 1):136-.33684939 [Google Scholar]
- 39.d’Amore F, Relander T, Lauritzsen GF, Jantunen E, Hagberg H, Anderson H, et al. Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J Clin Oncol. 2012;30(25):3093–9. [DOI] [PubMed] [Google Scholar]
- 40.Yi JH, Lee GW, Do YR, Jung HR, Hong JY, Yoon DH, et al. Multicenter retrospective analysis of the clinicopathologic features of monomorphic epitheliotropic intestinal T-cell lymphoma. Ann Hematol. 2019;98(11):2541–50. [DOI] [PubMed] [Google Scholar]
- 41.Tse E, Gill H, Loong F, Kim SJ, Ng SB, Tang T, et al. Type II enteropathy-associated T-cell lymphoma: a multicenter analysis from the Asia Lymphoma Study Group. Am J Hematol. 2012;87(7):663–8. [DOI] [PubMed] [Google Scholar]
- 42.Farcet JP, Gaulard P, Marolleau JP, Le Couedic JP, Henni T, Gourdin MF, et al. Hepatosplenic T-cell lymphoma: sinusal/sinusoidal localization of malignant cells expressing the T-cell receptor gamma delta. Blood. 1990;75(11):2213–9. [PubMed] [Google Scholar]
- 43.Harris NL, Jaffe ES, Stein H, Banks PM, Chan JK, Cleary ML, et al. A revised European-American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood. 1994;84(5):1361–92. [PubMed] [Google Scholar]
- 44.Ribot JC, Lopes N, Silva-Santos B. gammadelta T cells in tissue physiology and surveillance. Nat Rev Immunol. 2021;21(4):221–32. [DOI] [PubMed] [Google Scholar]
- 45.Krishnan M, Lunning M. Hepatosplenic gamma-delta T-Cell Lymphoma: Who Is on Your Speed Dial? J Oncol Pract. 2019;15(6):307–12. [DOI] [PubMed] [Google Scholar]
- 46.Pro B, Allen P, Behdad A. Hepatosplenic T-cell lymphoma: a rare but challenging entity. Blood. 2020;136(18):2018–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yabe M, Medeiros LJ, Daneshbod Y, Davanlou M, Bueso-Ramos CE, Moran EJ, et al. Hepatosplenic T-cell lymphoma arising in patients with immunodysregulatory disorders: a study of 7 patients who did not receive tumor necrosis factor-alpha inhibitor therapy and literature review. Ann Diagn Pathol. 2017;26:16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yabe M, Medeiros LJ, Tang G, Wang SA, Ahmed S, Nieto Y, et al. Prognostic Factors of Hepatosplenic T-cell Lymphoma: Clinicopathologic Study of 28 Cases. Am J Surg Pathol. 2016;40(5):676–88. [DOI] [PubMed] [Google Scholar]
- 49.Falchook GS, Vega F, Dang NH, Samaniego F, Rodriguez MA, Champlin RE, et al. Hepatosplenic gamma-delta T-cell lymphoma: clinicopathological features and treatment. Ann Oncol. 2009;20(6):1080–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ochenrider MG, Patterson DJ, Aboulafia DM. Hepatosplenic T-cell lymphoma in a young man with Crohn’s disease: case report and literature review. Clin Lymphoma Myeloma Leuk. 2010;10(2):144–8. [DOI] [PubMed] [Google Scholar]
- 51.Kandiel A, Fraser AG, Korelitz BI, Brensinger C, Lewis JD. Increased risk of lymphoma among inflammatory bowel disease patients treated with azathioprine and 6-mercaptopurine. Gut. 2005;54(8):1121–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bernstein CN, Blanchard JF, Kliewer E, Wajda A. Cancer risk in patients with inflammatory bowel disease: a population-based study. Cancer. 2001;91(4):854–62. [DOI] [PubMed] [Google Scholar]
- 53.Belhadj K, Reyes F, Farcet JP, Tilly H, Bastard C, Angonin R, et al. Hepatosplenic gammadelta T-cell lymphoma is a rare clinicopathologic entity with poor outcome: report on a series of 21 patients. Blood. 2003;102(13):4261–9. [DOI] [PubMed] [Google Scholar]
- 54.Weidmann E. Hepatosplenic T cell lymphoma. A review on 45 cases since the first report describing the disease as a distinct lymphoma entity in 1990. Leukemia. 2000;14(6):991–7. [DOI] [PubMed] [Google Scholar]
- 55.Yabe M, Medeiros LJ, Tang G, Wang SA, K PP, Routbort M, et al. Dyspoietic changes associated with hepatosplenic T-cell lymphoma are not a manifestation of a myelodysplastic syndrome: analysis of 25 patients. Hum Pathol. 2016;50:109–17. [DOI] [PubMed] [Google Scholar]
- 56.Nosari A, Oreste PL, Biondi A, Costantini MC, Santoleri L, Intropido L, et al. Hepatosplenic gammadelta T-cell lymphoma: a rare entity mimicking the hemophagocytic syndrome. Am J Hematol. 1999;60(1):61–5. [DOI] [PubMed] [Google Scholar]
- 57.Vega F, Medeiros LJ, Bueso-Ramos C, Jones D, Lai R, Luthra R, et al. Hepatosplenic gamma/delta T-cell lymphoma in bone marrow. A sinusoidal neoplasm with blastic cytologic features. American journal of clinical pathology. 2001;116(3):410–9. [DOI] [PubMed] [Google Scholar]
- 58.Macon WR, Levy NB, Kurtin PJ, Salhany KE, Elkhalifa MY, Casey TT, et al. Hepatosplenic alphabeta T-cell lymphomas: a report of 14 cases and comparison with hepatosplenic gammadelta T-cell lymphomas. Am J Surg Pathol. 2001;25(3):285–96. [DOI] [PubMed] [Google Scholar]
- 59.Nicolae A, Xi L, Pittaluga S, Abdullaev Z, Pack SD, Chen J, et al. Frequent STAT5B mutations in gammadelta hepatosplenic T-cell lymphomas. Leukemia. 2014;28(11):2244–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McKinney M, Moffitt AB, Gaulard P, Travert M, De Leval L, Nicolae A, et al. The Genetic Basis of Hepatosplenic T-cell Lymphoma. Cancer Discov. 2017;7(4):369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Voss MH, Lunning MA, Maragulia JC, Papadopoulos EB, Goldberg J, Zelenetz AD, et al. Intensive induction chemotherapy followed by early high-dose therapy and hematopoietic stem cell transplantation results in improved outcome for patients with hepatosplenic T-cell lymphoma: a single institution experience. Clin Lymphoma Myeloma Leuk. 2013;13(1):8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tanase A, Schmitz N, Stein H, Boumendil A, Finel H, Castagna L, et al. Allogeneic and autologous stem cell transplantation for hepatosplenic T-cell lymphoma: a retrospective study of the EBMT Lymphoma Working Party. Leukemia. 2015;29(3):686–8. [DOI] [PubMed] [Google Scholar]
- 63.Travert M, Huang Y, de Leval L, Martin-Garcia N, Delfau-Larue MH, Berger F, et al. Molecular features of hepatosplenic T-cell lymphoma unravels potential novel therapeutic targets. Blood. 2012;119(24):5795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jaeger G, Bauer F, Brezinschek R, Beham-Schmid C, Mannhalter C, Neumeister P. Hepatosplenic gammadelta T-cell lymphoma successfully treated with a combination of alemtuzumab and cladribine. Ann Oncol. 2008;19(5):1025–6. [DOI] [PubMed] [Google Scholar]
- 65.Amengual JE, Lichtenstein R, Lue J, Sawas A, Deng C, Lichtenstein E, et al. A phase 1 study of romidepsin and pralatrexate reveals marked activity in relapsed and refractory T-cell lymphoma. Blood. 2018;131(4):397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood. 2011;117(19):5019–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gonzalez CL, Medeiros LJ, Braziel RM, Jaffe ES. T-cell lymphoma involving subcutaneous tissue. A clinicopathologic entity commonly associated with hemophagocytic syndrome. Am J Surg Pathol. 1991;15(1):17–27. [DOI] [PubMed] [Google Scholar]
- 68.Toro JR, Liewehr DJ, Pabby N, Sorbara L, Raffeld M, Steinberg SM, et al. Gamma-delta T-cell phenotype is associated with significantly decreased survival in cutaneous T-cell lymphoma. Blood. 2003;101(9):3407–12. [DOI] [PubMed] [Google Scholar]
- 69.Lopez-Lerma I, Penate Y, Gallardo F, Marti RM, Mitxelena J, Bielsa I, et al. Subcutaneous panniculitis-like T-cell lymphoma: Clinical features, therapeutic approach, and outcome in a case series of 16 patients. J Am Acad Dermatol. 2018;79(5):892–8. [DOI] [PubMed] [Google Scholar]
- 70.Willemze R, Jansen PM, Cerroni L, Berti E, Santucci M, Assaf C, et al. Subcutaneous panniculitis-like T-cell lymphoma: definition, classification, and prognostic factors: an EORTC Cutaneous Lymphoma Group Study of 83 cases. Blood. 2008;111(2):838–45. [DOI] [PubMed] [Google Scholar]
- 71.Magro CM, Wang X. CCL5 expression in panniculitic T-cell dyscrasias and its potential role in adipocyte tropism. Am J Dermatopathol. 2013;35(3):332–7. [DOI] [PubMed] [Google Scholar]
- 72.Daniels J, Doukas PG, Escala MEM, Ringbloom KG, Shih DJH, Yang J, et al. Cellular origins and genetic landscape of cutaneous gamma delta T cell lymphomas. Nature communications. 2020;11(1):1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Guitart J, Weisenburger DD, Subtil A, Kim E, Wood G, Duvic M, et al. Cutaneous gammadelta T-cell lymphomas: a spectrum of presentations with overlap with other cytotoxic lymphomas. Am J Surg Pathol. 2012;36(11):1656–65. [DOI] [PubMed] [Google Scholar]
- 74.Guitart J, Mangold AR, Martinez-Escala ME, Walker CJ, Comfere NI, Pulitzer M, et al. Clinical and Pathological Characteristics and Outcomes Among Patients With Subcutaneous Panniculitis-like T-Cell Lymphoma and Related Adipotropic Lymphoproliferative Disorders. JAMA Dermatol. 2022;158(10):1167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Willemze R. Cutaneous lymphomas with a panniculitic presentation. Semin Diagn Pathol. 2017;34(1):36–43. [DOI] [PubMed] [Google Scholar]
- 76.La Rosee P, Horne A, Hines M, von Bahr Greenwood T, Machowicz R, Berliner N, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019;133(23):2465–77. [DOI] [PubMed] [Google Scholar]
- 77.Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–31. [DOI] [PubMed] [Google Scholar]
- 78.LeBlanc RE, Tavallaee M, Kim YH, Kim J. Useful Parameters for Distinguishing Subcutaneous Panniculitis-like T-Cell Lymphoma From Lupus Erythematosus Panniculitis. Am J Surg Pathol. 2016;40(6):745–54. [DOI] [PubMed] [Google Scholar]
- 79.Sitthinamsuwan P, Pattanaprichakul P, Treetipsatit J, Pongpruttipan T, Sukpanichnant S, Pincus LB, et al. Subcutaneous Panniculitis-Like T-Cell Lymphoma Versus Lupus Erythematosus Panniculitis: Distinction by Means of the Periadipocytic Cell Proliferation Index. Am J Dermatopathol. 2018;40(8):567–74. [DOI] [PubMed] [Google Scholar]
- 80.Merrill ED, Agbay R, Miranda RN, Aung PP, Tetzlaff MT, Young KH, et al. Primary Cutaneous T-Cell Lymphomas Showing Gamma-Delta (gammadelta) Phenotype and Predominantly Epidermotropic Pattern are Clinicopathologically Distinct From Classic Primary Cutaneous gammadelta T-Cell Lymphomas. Am J Surg Pathol. 2017;41(2):204–15. [DOI] [PubMed] [Google Scholar]
- 81.Muhsen IN, El Fakih R, Hamadani M, Lazarus HM, Kharfan-Dabaja MA, Aljurf M. Clinical, Diagnostic and Prognostic Characteristics of Primary Cutaneous Gamma Delta T-cell Lymphomas. Clin Hematol Int. 2022;4(1–2):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Polprasert C, Takeuchi Y, Kakiuchi N, Yoshida K, Assanasen T, Sitthi W, et al. Frequent germline mutations of HAVCR2 in sporadic subcutaneous panniculitis-like T-cell lymphoma. Blood Adv. 2019;3(4):588–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gayden T, Sepulveda FE, Khuong-Quang DA, Pratt J, Valera ET, Garrigue A, et al. Germline HAVCR2 mutations altering TIM-3 characterize subcutaneous panniculitis-like T cell lymphomas with hemophagocytic lymphohistiocytic syndrome. Nature genetics. 2018;50(12):1650–7. [DOI] [PubMed] [Google Scholar]
- 84.Koh J, Jang I, Mun S, Lee C, Cha HJ, Oh YH, et al. Genetic profiles of subcutaneous panniculitis-like T-cell lymphoma and clinicopathological impact of HAVCR2 mutations. Blood Adv. 2021;5(20):3919–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yamamoto-Sugitani M, Kuroda J, Shimura Y, Nagoshi H, Chinen Y, Ohshiro M, et al. Comprehensive cytogenetic study of primary cutaneous gamma-delta T-cell lymphoma by means of spectral karyotyping and genome-wide single nucleotide polymorphism array. Cancer Genet. 2012;205(9):459–64. [DOI] [PubMed] [Google Scholar]
- 86.Rubio-Gonzalez B, Zain J, Garcia L, Rosen ST, Querfeld C. Cutaneous Gamma-Delta T-Cell Lymphoma Successfully Treated With Brentuximab Vedotin. JAMA Dermatol. 2016;152(12):1388–90. [DOI] [PubMed] [Google Scholar]
- 87.Talpur R, Chockalingam R, Wang C, Tetzlaff MT, Duvic M. A Single-Center Experience With Brentuximab Vedotin in Gamma Delta T-Cell Lymphoma. Clin Lymphoma Myeloma Leuk. 2016;16(2):e15–9. [DOI] [PubMed] [Google Scholar]
- 88.Gibson JF, Alpdogan O, Subtil A, Girardi M, Wilson LD, Roberts K, et al. Hematopoietic stem cell transplantation for primary cutaneous gammadelta T-cell lymphoma and refractory subcutaneous panniculitis-like T-cell lymphoma. J Am Acad Dermatol. 2015;72(6):1010–5 e5. [DOI] [PubMed] [Google Scholar]
- 89.David KA, Pulitzer M, Guitart J, Martinez-Escala ME, Geller S, Wang Y, et al. Characteristics, Treatment Patterns, and Outcomes in Primary Cutaneous Gamma Delta T Cell Lymphoma (PCGDTCL): A Real World Multi-Institutional Analysis of a Rare Malignancy. Blood. 2019;134(Supplement_1):4028-. [Google Scholar]
- 90.Herrera AF, Crosby-Thompson A, Friedberg JW, Abel GA, Czuczman MS, Gordon LI, et al. Comparison of referring and final pathology for patients with T-cell lymphoma in the National Comprehensive Cancer Network. Cancer. 2014;120(13):1993–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Laurent C, Baron M, Amara N, Haioun C, Dandoit M, Maynadie M, et al. Impact of Expert Pathologic Review of Lymphoma Diagnosis: Study of Patients From the French Lymphopath Network. J Clin Oncol. 2017;35(18):2008–17. [DOI] [PubMed] [Google Scholar]
- 92.de Leval L, Alizadeh AA, Bergsagel PL, Campo E, Davies A, Dogan A, et al. Genomic profiling for clinical decision making in lymphoid neoplasms. Blood. 2022;140(21):2193–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jaffe ES. World Health Organization classification of tumours. Pathology and genetics of tumours of haematopoietic and lymphoid tissues. 2001. [Google Scholar]