

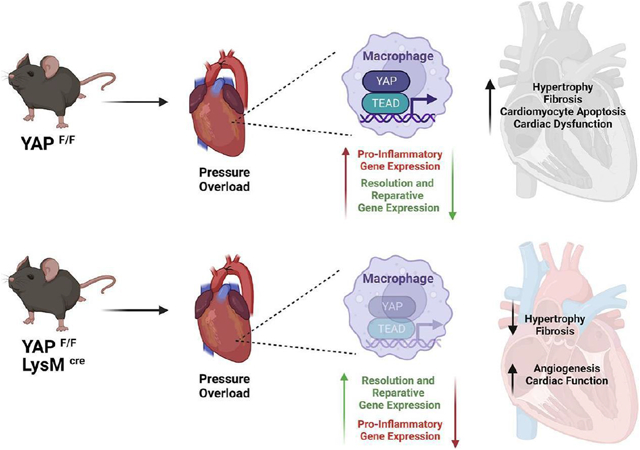
Abstract
Inflammation is an integral component of cardiovascular disease and is thought to contribute to cardiac dysfunction and heart failure. While ischemia-induced inflammation has been extensively studied in the heart, relatively less is known regarding cardiac inflammation during non-ischemic stress. Recent work has implicated a role for Yes-associated protein (YAP) in modulating inflammation in response to ischemic injury; however, whether YAP influences inflammation in the heart during non-ischemic stress is not described. We hypothesized that YAP mediates a pro-inflammatory response during pressure overload (PO)-induced non-ischemic injury, and that targeted YAP inhibition in the myeloid compartment is cardioprotective. In mice, PO elicited myeloid YAP activation, and myeloid-specific YAP knockout mice (YAPF/F;LysMCre) subjected to PO stress had better systolic function, and attenuated pathological remodeling compared to control mice. Inflammatory indicators were also significantly attenuated, while pro-resolving genes including Vegfa were enhanced, in the myocardium, and in isolated macrophages, of myeloid YAP KO mice after PO. Experiments using bone marrow-derived macrophages (BMDMs) from YAP KO and control mice demonstrated that YAP suppression shifted polarization toward a resolving phenotype. We also observed attenuated NLRP3 inflammasome priming and function in YAP deficient BMDMs, as well as in myeloid YAP KO hearts following PO, indicating disruption of inflammasome induction. Finally, we leveraged nanoparticle-mediated delivery of the YAP inhibitor verteporfin and observed attenuated PO-induced pathological remodeling compared to DMSO nanoparticle control treatment. These data implicate myeloid YAP as an important molecular nodal point that facilitates cardiac inflammation and fibrosis during PO stress and suggest that selective inhibition of YAP may prove a novel therapeutic target in non-ischemic heart disease.
Keywords: heart failure, pressure overload, inflammation, fibrosis, macrophage
Graphical Abstract
1. Introduction
Heart failure is a leading cause of morbidity and mortality in the US. Despite advances in therapies to treat the symptoms of heart failure, it remains associated with 50% mortality within 5 years of diagnosis[1]. Thus, a deeper understanding resulting in more effective treatments for this syndrome are desperately needed.
Inflammation occurs in human failing hearts and is thought to drive adverse cardiac remodeling and dysfunction. While initial immune responses can be beneficial, even low-grade chronic inflammation alters the extracellular matrix composition, increases fibrosis, and worsens cardiac function[2]. Recent clinical studies employing targeted inhibition of the cytokine IL-1β have yielded promising results, and have renewed interest in targeting inflammation in heart failure patients[3]. As a leading risk factor for heart failure, hypertension initiates maladaptive changes in immune cell behavior that promote cardiac inflammation, adverse remodeling, and dysfunction. However, the majority of studies investigating cardiac inflammation have focused on ischemic injury models, and far less is known regarding how inflammation is regulated during non-ischemic stress such as left ventricular pressure overload (PO), or how hypertensive inflammation modulates maladaptive cardiac remodeling and heart failure.
The Hippo signaling pathway is fundamental in maintaining adult heart homeostasis and mediates responses to cardiac injury[4]. Most efforts have focused on cardiomyocytes, where activation of Hippo restrains the transcription co-factor Yes-associated protein (YAP) to inhibit proliferation and enhance cell death. Conversely, YAP activation in cardiomyocytes promotes myocardial regeneration and protection against injury[5]. Recent studies have investigated additional cardiac constituents and demonstrated cell-type specificity of YAP function that can mediate opposing outcomes on heart failure[6, 7]. Whereas cardiomyocyte-specific deletion of YAP exacerbates cardiac injury and heart failure[8, 9], fibroblast targeted inhibition of YAP afforded benefit against pathological cardiac remodeling and dysfunction that develops in response to myocardial infarction[7, 10]. Evidence exists linking YAP to inflammation and immune cell function; however, results are somewhat discrepant, and likely cell-type and context-dependent[11, 12]. For example, YAP has been shown to dampen the anti-viral response through antagonizing IRF3 function, thereby increasing susceptibility to viral infection[13]. In contrast, separate studies have found that YAP can promote macrophage polarization toward a pro-inflammatory “M1-like” phenotype in mouse models of atherosclerosis, inflammatory bowel disease, and myocardial infarction[12, 14, 15]. Importantly, whether YAP function in myeloid cells modulates cardiac inflammation or macrophage polarization during non-ischemic heart failure stress has not been examined.
2. Results
2.1. Genetic disruption of myeloid YAP confers cardiac benefit in response to chronic pressure overload stress
We first determined the activation status of myeloid YAP within the pressure overloaded myocardium. CD11b-positive myeloid cells had significantly increased YAP mRNA, as well as increased YAP target gene expression (Ctgf, Cyr61) indicating enhanced YAP activation, 1 week after PO compared to sham operated mice (Figure 1A). Myeloid-specific YAP knockout mice were generated using LysM-Cre (YAPF/F;LysMCre) (Supplemental Figure 1A) and subjected to TAC. Following 4 weeks PO, YAPF/F;LysMCre mice had better systolic function (LVEF%) compared to control YAPF/F mice (Figure 1B, Supplemental Table 1). While we observed no significant difference in LV mass between genotypes (Figure 1C), additional markers of cardiac hypertrophy, including septal wall thickness, individual cardiomyocyte size, and expression of fetal genes Nppa and Nppb, were attenuated in YAPF/F;LysMCre hearts compared to controls after 4 weeks PO (Figure 1D-G, Supplemental Table 1). Reduced cardiomyocyte apoptosis, as determined by troponin-T and TUNEL double-positive staining, was also observed in YAPF/F;LysMCre mice compared to controls after PO (Figure 1H,I) suggesting a cardiac benefit.
Figure 1. Myeloid-targeted YAP deletion improves cardiac function and attenuates cardiac pathology after chronic pressure overload.

(A) Expression of YAP and YAP target gene mRNA in myeloid cells (CD11b+) isolated from left ventricle (LV) tissue 7 days after TAC. (B-D) Postmortem analysis of LV mass normalized to tibia length (TL), and wheat germ agglutinin (WGA) staining to determine cardiac hypertrophy in control and myeloid YAP deficient mice after 4 weeks TAC. (E and F) Quantitative PCR using RNA isolated from LV tissue to measure fetal gene expression in control and myeloid YAP deficient mice after 4 weeks TAC. (G and H) TUNEL-positive (red) cardiomyocytes (cTNT+; green) were determined in control and myeloid YAP deficient mice after 4-weeks TAC. Right panel shows increased magnification of yellow border; white arrow indicates TUNEL-positive cardiomyocyte. Scale bar, 50 μm. N = 3-14 mice/group. All data are presented as mean ± SEM. P values were determined by unpaired, two-tailed Student t-test, or ordinary one-way ANOVA with multiple comparison. *P<0.05, **P<0.01, ***P<0.001, ns = not significant.
Inflammatory processes are intimately tied to cardiac fibroblast activation and the progression of fibrosis, which contributes to pathological remodeling[2]. We observed less fibrosis in YAPF/F;LysMCre mice after PO stress as determined by collagen deposition in the myocardium (Figure 2A,B). Immunofluorescence experiments indicated an attenuation of αSMA-positive myofibroblast presence in YAPF/F;LysMCre hearts compared to controls after PO (Figure 2C,D). The mRNA expression of fibrosis-related genes (Col1a1, Col3a1, Postn) was also attenuated in YAP deficient mice following PO stress compared to control mice (Figure 2E-G). Notably, we found that expression of Vegfa was augmented in YAPF/F;LysMCre hearts after PO and this correlated with increased ratio of isolectin B4+ endothelial cells to cardiomyocytes (Figure 2H-J). Determination of inflammation-related gene expression revealed the suppression of multiple pro-inflammatory genes (e.g. Il-1b, Il-6, Ccr2) in myeloid YAP deficient mice (Figure 3A-F). Conversely, established markers of resolution-associated genes (Arg1, Il-10) were enhanced in YAPF/F;LysMCre myocardium compared to control mice after PO (Figure 3G,H). In addition, we found that the activity of caspase-1, a key effector of the inflammasome that processes pro-IL-1β and pro-IL-18 to mature forms, was repressed in YAPF/F;LysMCre hearts following PO (Figure 3I).
Figure 2. Myeloid-targeted YAP deletion attenuates pressure overload-induced cardiac fibrosis.

(A-B) Collagen deposition was determined by picrosirius red (PSR) staining in control and myeloid YAP deficient LV tissue after 4 weeks TAC. (C-D) αSMA-positive staining (green), cTNT (red), and DAPI (blue) were determined in control and myeloid YAP deficient LV tissue following 4 weeks TAC. (E-G) Quantitative PCR using RNA isolated from LV tissue was performed to measure expression of fibrosis related genes in control and myeloid YAP deleted mice. (H-I) LV tissue was stained for isolectin B4 (green), WGA (red), and DAPI (blue) to determine capillary density in control and myeloid YAP deficient mice after 4 weeks TAC. (J) Expression of Vegfa mRNA was measured in LV tissue of control and myeloid YAP deleted mice. Scale bar, 100 μm. N = 3-10 mice/group. All data are presented as mean ± SEM. P values were determined by ordinary one-way ANOVA with multiple comparison. *P<0.05, **P<0.01, ***P<0.001, ns = not significant.
Figure 3. Cardiac inflammatory markers are altered in myeloid YAP deficient mice.

(A-F) Quantitative PCR using RNA isolated from LV tissue was performed to determine pro-inflammatory gene expression in control and myeloid YAP deleted mice after 4 weeks TAC. (G-H) Expression of resolving-associated genes in LV was determined by qPCR in control and myeloid YAP deficient mice. (I) Activity of caspase-1 in LV tissue was measured in control and myeloid YAP deleted mice. N = 3-6 mice/group. All data are presented as mean ± SEM. P values were determined by ordinary one-way ANOVA with multiple comparison. *P<0.05, **P<0.01, ***P<0.001, ns = not significant.
2.2. Myeloid YAP inhibition improves cardiac phenotype after acute pressure overload stress
To investigate the effect of myeloid YAP deletion at an earlier time point prior to the onset of cardiac dysfunction, a second cohort of mice was characterized at 1-week post-TAC. We observed a significant attenuation of cardiomyocyte enlargement as well as collagen deposition in YAPF/F;LysMCre hearts compared to controls after 1-week TAC (Supplemental Figure 1B-F). The improved phenotype was associated with reduced expression of genes related to fibrosis, inflammation, the fetal gene program, and metabolic substrate utilization switching in the YAPF/F;LysMCre hearts after 1-week PO (Supplemental Figure 2A-F). Examination of myocardial Hippo-Yap pathway activation, as well as potential changes in cardiac autophagy/mitophagy-related proteins, did not reveal differences between control and myeloid YAP deficient mice at sham or 1-week PO (Supplemental Figure 3A-J). We next determined immune cell abundance in the myocardium of YAPF/F;LysMCre and control mice after 1-week PO. Immunostaining of heart sections revealed a reduced number of CD68+ macrophages, as well as reduced CCR2+ cells, present in YAPF/F;LysMCre hearts compared to control after TAC (Supplemental Figure 4A-D). Importantly, co-staining revealed that the majority of the CCR2+ cells we detected were indeed macrophages (Supplemental Figure 4E). The relative amounts of monocytes, macrophages, and neutrophils within peripheral blood were similar between YAPF/F;LysMCre and control mice after TAC (Supplemental Figure 4F), suggesting that the attenuated presence of inflammatory macrophages in YAPF/F;LysMCre hearts may be due in part to impaired recruitment. To better characterize the myeloid population within the myocardium after stress, we isolated macrophage and neutrophil subsets from LV tissues of control and myeloid Yap deficient mice (Supplemental Figure 5A). Yap deficient macrophages isolated from PO hearts had significantly reduced expression of multiple pro-inflammatory genes (Figure 4A-E). On the other hand, Yap deficient macrophages had increased expression levels of pro-resolving genes (e.g. Arg1, Ym1) as well as the pro-angiogenic factor Vegfa (Figure 4M-P). We also examined genes related to migration/chemotaxis and found that their expression was generally reduced in the Yap deficient macrophage populations isolated from PO hearts compared to controls, including a significant reduction of Cxcl2, Ccl2, and Itgal, as well as a trend toward decreased expression of Ccr2, Itgam, and Itgb2 (Figure 4F-L). We also isolated LV neutrophils, and somewhat unexpectedly, did not observe a significant reduction in neutrophil Yap mRNA levels, inflammatory (IL-1b, IL-6, Tnf), or chemotactic genes (Ccl5, Cxcr2), or a difference in the number of neutrophils (Ly6G+) present in the myocardium of YAPF/F;LysMCre mice compared to controls after PO (Supplemental Figure 5B-I). Taken together, these results demonstrate myeloid YAP activation within the myocardium during early PO stress, and selective deletion of myeloid YAP affords improved cardiac function and attenuated fibrotic and inflammatory remodeling, which is likely mediated through altered macrophage recruitment and function.
Figure 4. Characterization of macrophages isolated from hearts of control and myeloid YAP deficient mice following pressure overload stress.

Quantitative PCR using RNA isolated from myocardial flow-sorted macrophages was performed to determine pro-inflammatory (A-E), migration/chemotaxis-related (F-L), and pro-resolving (M-P) gene expression in control and myeloid YAP deleted mice after 1-week TAC. N = 4 mice/group. All data are presented as mean ± SEM. P values were determined by unpaired, two-tailed Student t-test. *P<0.05, **P<0.01, ns = not significant.
2.3. YAP modulates macrophage gene expression and facilitates NLRP3 inflammasome function
To further investigate the mechanism underlying the observed cardiac benefit of myeloid YAP deletion, we isolated primary bone marrow-derived macrophages (BMDMs) from wild-type C57BL6/J mice. Treatment of BMDMs with either LPS or recombinant IL-1β caused an increase in total YAP protein levels. The amount of phosphorylated YAP was not significantly changed by either treatment resulting in a decreased phospho-YAP to total YAP ratio (Figure 5A-C). TEAD-luciferase reporter assays indicated that LPS treatment induced YAP transcriptional activation, which was highly sensitive to verteporfin, a pharmacological inhibitor of YAP-TEAD function[16] (Figure 5D). These data demonstrate the activation of macrophage YAP by pro-inflammatory stimuli, consistent with previous reports[12, 14]. We then isolated and stimulated BMDMs from YAPF/F;LysMCre and control mice using LPS to induce an “M1-like” polarization or IL-4 to elicit an “M2-like” phenotype. In YAP deficient cells, LPS induced upregulation of pro-inflammatory cytokines and chemokines was significantly attenuated compared to control BMDMs (Figure 6A-G). On the other hand, genes induced by IL-4 treatment and typically associated with resolution of inflammatory processes and wound healing were further enhanced in BMDMs from YAPF/F;LysMCre mice compared to controls (Figure 6H-L). Moreover, expression of active YAPS127A in the macrophage-like cell line RAW264.7 increased expression of pro-inflammatory genes (Supplemental Figure 6A-F).
Figure 5. YAP activation in macrophages.

(A) Representative western blot demonstrating the upregulation of YAP in bone marrow-derived macrophages following stimulation with LPS (100 ng/mL) or IL-1β (10 ng/mL). (B-C) Quantitation of results shown in panel A. (D) Luciferase reporter assay determined YAP activation in RAW264.7 cells following LPS treatment, in the presence or absence of verteporfin (VP; 1μM). N = 3 experimental replicates. All data are presented as mean ± SEM. P values were determined by ordinary one-way ANOVA with multiple comparison. **P<0.01, ***P<0.001, ns = not significant.
Figure 6. YAP deletion influences the polarization of bone marrow-derived macrophages.

(A-G) Bone marrow-derived macrophages (BMDMs) were prepared from control and myeloid YAP deficient mice and stimulated with LPS (100 ng/mL). Gene expression was measured by qPCR. (H-L) BMDMs from control or myeloid YAP deficient mice were treated with recombinant IL-4 (20 ng/mL). Gene expression was measured by qPCR. N = 3-4 experimental replicates. All data are presented as mean ± SEM. P values were determined by ordinary one-way ANOVA with multiple comparison. *P<0.05, **P<0.01, ***P<0.001, ns = not significant.
The NLRP3 inflammasome is active in myeloid cells and contributes to myocardial inflammation during injury[17]. To initialize inflammasome activation, transcription of essential components, including NLRP3, are upregulated in what is referred to as priming. Next, inflammasome components form a complex, leading to the activation of caspase-1, and the subsequent cleavage and extracellular release of IL-1β and IL-18 to promote inflammation. Using BMDMs from YAPF/F;LysMCre and control mice, we observed a significant attenuation of NLRP3 mRNA and protein induction by LPS in the YAP deficient cells (Figure 7A,B). Treatment of BMDMs with LPS followed by nigericin, a combination that elicits inflammasome priming and activation, triggered caspase-1 activation and the accumulation of IL-1β and IL-18 protein in the culture media (Figure 7C-E). These responses were significantly blunted in BMDMs isolated from YAPF/F;LysMCre mice, indicating that YAP inhibition dampens NLRP3 inflammasome function in macrophages.
Figure 7. YAP deletion impairs NLRP3 inflammasome function in macrophages.

(A-B) LPS (100 ng/mL) induced Nlrp3 mRNA and protein levels were determined in control and YAP deficient bone marrow-derived macrophages (BMDMs). (C) Caspase-1 activity was induced by LPS (100 ng/mL) and nigericin (10 μM) stimulation in control and YAP deficient BMDMs. (D-E) BMDMs were treated with LPS and nigericin to induce NLRP3 inflammasome activation and soluble IL-1β and IL-18 protein were detected in the culture media. N = 3-4 experimental replicates. All data are presented as mean ± SEM. P values were determined by ordinary one-way ANOVA with multiple comparison. *P<0.05, **P<0.01, ***P<0.001, nd = not detectable.
To further investigate the transcriptional regulation of key components of the inflammasome, NLRP3 and IL-1β, we determined YAP promoter occupancy of each gene based on conserved cis TEAD recognition motifs[12]. ChIP experiments performed in RAW264.7 cells demonstrated the enrichment of endogenous YAP at 2 separate regions within the NLRP3 promoter, as well as 2 regions of the IL-1β promoter (Figure 8A-C, F-H). This response was further enhanced following the expression of exogenous active YAP (Figure 8A-C, F-H). We next generated luciferase reporter constructs by cloning endogenous promoter regions of the NLRP3 and IL-1β mouse genes. The co-transfection of each reporter construct with active YAP elicited significant increases in reporter gene expression compared to GFP controls (Figure 8D,E,I,J). Importantly, these YAP-induced transcriptional responses were sensitive to concomitant treatment with verteporfin, providing further evidence of TEAD involvement (Figure 8D,E,I,J).
Figure 8. YAP promotes the transcription of inflammasome related genes.

(A and F) Schemas representing the mouse Nlrp3 and Il-1b gene proximal promoters. Transcription start sites (TSS) and conserved TEAD motifs (grey boxes) are shown. (B-C) Chromatin immunoprecipitation (ChIP) assays were carried out using RAW264.7 cells transfected with GFP or YAP expression vectors and anti-YAP antibody. Separate primer sets (P1 or P2) were used to detect distinct regions of the Nlrp3 promoter. (D-E) HEK293 cells were transfected with luciferase reporter constructs cloned from 2 separate regions of the Nlrp3 promoter (L1 or L2) in combination with GFP or YAP plasmids, in the presence or absence of verteporfin (VP; 1 μM). (G-H) ChIP assays performed in RAW264.7 cells using 2 different primer sets (P1 or P2) to detect distinct regions of the Il-1b promoter. (I-J) Luciferase assays conducted in HEK293 cells using reporter constructs generated from 2 separate regions of the Il-lb promoter (L1 or L2). N = 3-4 experimental replicates. All data are presented as mean ± SEM. P values were determined by ordinary one-way ANOVA with multiple comparison. *P<0.05, **P<0.01, ***P<0.001.
2.4. Pharmacological YAP inhibition attenuates maladaptive cardiac remodeling in response to pressure overload stress
Next, we sought to test whether pharmacological inhibition of YAP could shift macrophage polarization and provide cardiac benefit in vivo. RAW264.7 cells were pretreated with verteporfin or vehicle control, followed by stimulation using LPS or IL-4 to provoke inflammatory and resolving gene expression, respectively. Inhibition of YAP significantly blunted LPS-induced cytokines/chemokines, while augmenting expression of resolution associated genes, Arg1 and Ym1 (Supplemental Figure 7A-E). Moreover, we observed increased VEGFA protein following macrophage YAP inhibition (Supplemental Figure 7F,G) suggesting that YAP-mediated suppression of Vegfa expression is cell autonomous.
We then formulated PLGA nanoparticles loaded with either verteporfin or DMSO vehicle[18]. We first used nanoparticles loaded with fluorescent dye to monitor cell uptake and track in vivo distribution. We observed selectivity in cell culture by immunofluorescence (Supplemental Figure 8A). In vivo analysis by flow cytometry indicated nanoparticle selectivity for macrophages and neutrophils in both heart and splenic tissue (Supplemental Figure 8B,C). Importantly, these nanoparticles avoided uptake by cardiomyocytes at all time points, and remained detectable in myeloid cells after 72 hours (Supplemental Figure 8B,C).
We then subjected wild type C57BL6/J mice to TAC, and randomized mice to receive either DMSO control (NP-DMSO) or verteporfin-loaded (NP-VP) nanoparticle administration every 3 days beginning 1 day after surgery. After 2 weeks, echo was performed, and mice were euthanized for analysis. There was no difference in cardiac function or hypertrophy between treatment groups at this relatively early timepoint that precedes dysfunction (Supplemental Table 2, Supplemental Figure 9). However, we noted attenuated cardiac fibrosis, and increased capillary density, in mice administered NP-VP compared to NP-DMSO (Figure 9A,B). Additionally, we observed reduced cardiomyocyte apoptosis and attenuated pro-inflammatory and fibrotic gene expression in myocardium of NP-VP treated mice (Figure 9C,D). We measured IL-1β protein levels in blood after 2-week TAC and found that mice administered NP-VP had reduced serum IL-1β compared to NP-DMSO controls (Figure 9E). Finally, we detected increased Vegfa mRNA as well as VEGF protein in myocardium of NP-VP treated mice compared to NP-DMSO controls (Figure 9F,G). Together these results indicate that treatment of verteporfin-loaded nanoparticles antagonized PO-induced pathological cardiac remodeling.
Figure 9. Effect of pharmacological YAP inhibition on pathological cardiac remodeling caused by pressure overload.

Wild type mice were subjected to TAC surgery and administered control DMSO-loaded nanoparticles (NP-DMSO) or verteporfin-loaded nanoparticles (NP-VP) for 2 weeks. (A) Collagen deposition was determined by PSR staining after TAC. (B) Capillary density was determined by isolectin B4 (IB4; green), WGA (red), and DAPI (blue) co-staining in LV tissue after TAC. (C) Apoptotic cardiomyocytes were determined by TUNEL assay with cTNT counterstaining after TAC. (D) Quantitative PCR was performed using RNA isolated from LV tissue after TAC. (E) The concentration of IL-1β protein was measured by ELISA in blood serum collected 2 weeks after TAC. (F) qPCR was performed using RNA from LV tissue after TAC. (G) VEGF protein was measured by ELISA in LV tissue. Scale bar, 100 μm. N = 3-4 mice/group. All data are presented as mean ± SEM. P values were determined by unpaired, two-tailed Student t-test. *P<0.05, **P<0.01.
3. Discussion
Despite being a sterile environment, myocardial inflammation is triggered by non-ischemic stress such as sustained pressure overload[19], and is thought to contribute to maladaptive remodeling of the heart. Elegant studies employing single cell RNA sequencing have begun to define the inflammatory milieu of the PO mouse heart[20, 21]; however, our understanding of the regulatory processes involved in orchestrating this response remains incomplete. Macrophages are the most abundant immune cells recruited to the heart during the early phase of PO stress, and are highly inflammatory soon after injury[22-24]. Moreover, preclinical studies have demonstrated that targeting macrophages to alter their polarization and limit their pro-inflammatory function can be cardioprotective[25, 26]. In the current study, we identified the transcriptional cofactor YAP as an important modulator of macrophage function, thereby influencing the heart’s response to PO. Our results demonstrate that inhibition of YAP in the myeloid compartment affords cardiac benefit. We predict this is due to reduced recruitment of pro-inflammatory macrophages (i.e. CCR2+) and attenuated pro-inflammatory cytokine/chemokine production, as we observed attenuation of multiple factors (e.g. IL-1β, IL-6) known to promote cardiac injury and dysfunction. In addition, YAP inhibition enhanced the expression of resolution-related genes including Vegfa, which correlated with increased endothelial cell density following PO stress. The ratio of capillaries to cardiomyocytes is an important determinant of heart function and heart failure progression[27], and VEGFA is critical for the formation of new vessels[28]; therefore, we believe this mechanism likely also provides benefit stemming from myeloid YAP suppression.
Inflammation is an attractive process for the potential mitigation of cardiac pathology, yet global macrophage depletion leads to worsened outcomes after cardiac injury and more nuance is required for therapeutic benefit[29]. To this end, studies targeting macrophage subsets to modify their pro-inflammatory capacity have demonstrated cardioprotection in the setting of ischemic stress [23, 24, 26, 30, 31]. Recruited macrophages are monocyte-derived, largely CCR2-positive, and contribute to the inflammatory milieu through cytokine/chemokine secretion, which exacerbates myocardial damage and facilitates fibroblast activation, extracellular matrix breakdown, cell death, and impaired angiogenesis in the heart[32]. On the other hand, macrophages are also needed for proper resolution of inflammation and tissue repair. Increasing evidence indicates that cardiac resident macrophages serve a protective function against injury and are fundamental for heart homeostasis and wound healing responses including the maintenance proper electrical conduction, and disposal of waste secreted from cardiomyocytes [33-37]. Our approach to inhibit YAP using LysMCre mice is unlikely to target resident cardiac macrophages, and whether YAP function differs between macrophage subtypes remains an unanswered question. However, because YAP can control both pro-inflammatory behavior, including CCR2 expression and cardiac macrophage abundance after PO, as well as VEGFA expression and capillary density to regulate wound healing, this transcription cofactor may be seen as an attractive target for manipulation. Based on this hypothesis, we employed nanoparticle-mediated delivery of the YAP inhibitor verteporfin in mice subjected to PO stress, similar to prior work targeting YAP in pulmonary hypertension[38]. Our results demonstrate that selective pharmacological targeting of YAP inhibitor to myeloid cells, while avoiding cardiomyocyte uptake in vivo, antagonized myocardial inflammation and fibrosis, hallmarks of pathological remodeling, and encouraged angiogenic responses, suggesting therapeutic potential of this approach.
While generally thought to stimulate transcription, YAP has been shown to repress gene expression in certain contexts[39, 40]. Our current work demonstrates that YAP inhibition in macrophages results in the attenuation of pro-inflammatory gene expression while augmenting expression of genes associated with resolution and wound healing; findings that are consistent with prior reports[12, 14, 15]. Of particular interest is the ability of YAP to negatively regulate expression of Vegfa and the observed increase in VEGFA protein following inhibition of YAP in vivo and in vitro. Previous work has implicated several established epigenetic regulators as important partners of YAP to mediate transcriptional repression including HDAC3-NCoR, NuRD complex, and p53[12, 15, 39]. However, the precise mechanism underlying the suppression of macrophage VEGFA expression by YAP, and the potential necessity of TEAD for this negative regulation, will be explored further in future studies. Myeloid YAP inhibition also decreased NLRP3 inflammasome activity. Genetic and pharmacological inhibition of the NLRP3 inflammasome afford cardiac protection against injury and dysfunctional[41, 42]. Our results indicate that macrophage YAP can promote inflammasome function by facilitating the upregulation of NLRP3, the subsequent activation of caspase-1, and the processing of its critical substrate, IL-1β. This is consistent with a previous report demonstrating positive regulation of NLRP3 by YAP through direct interaction and prevention of NLRP3 proteasomal degradation[43]. While our data demonstrate transcriptional regulation of NLRP3 by YAP-TEAD, we cannot rule out additional reinforcement of NLRP3 protein through enhanced stability. IL-1β is upregulated in failing hearts and known to elicit deleterious effects in stressed myocardium[32]. Findings from recent clinical trials aimed at neutralizing IL-1β in patients provide additional clinical relevance to the modulation of pathways, e.g. YAP, that regulate its maturation and bioavailability[3, 44].
Recent work from our group and others has begun to elucidate cell type specific functions of Hippo-YAP signaling in resident cardiac cell types, as well as non-resident cells that traffic the heart. Whereas inhibition of YAP in cardiomyocytes worsened heart function following PO or MI stress[8, 9, 45], disruption of YAP in cardiac fibroblasts provided protection against adverse remodeling and heart failure[7, 10]. Our current study demonstrates the cardiac benefit of YAP inhibition in myeloid cells during PO, a result consistent with previous work using myeloid YAP/TAZ deficient mice in an MI injury model[12]. Importantly, Hippo-YAP signaling was not altered in the myocardium of myeloid YAP deficient mice at baseline or during PO stress. Based on these findings, we here leveraged a delivery approach that would avoid cardiomyocyte targeting of the YAP inhibitor verteporfin and prevent deleterious off-target effects. Indeed, we detected minimal nanoparticle uptake in cardiomyocytes in vivo, and observed attenuated cardiac pathology in mice administered verteporfin-loaded nanoparticles after the onset of PO stress. These encouraging results provide rationale for testing this approach in additional models of cardiac stress. Together, these findings highlight the effectiveness of blocking myeloid YAP function, and the importance of cell type specificity when targeting Hippo-YAP signaling as a potential therapeutic intervention for heart disease.
Intercellular communication also influences cardiac inflammation and wound healing responses to stress. Through modulation of YAP in the myeloid compartment, we observed altered fibrosis, cardiomyocyte apoptosis, and blood vessel density, which may be mediated through paracrine effects upon cardiac fibroblasts, myocytes, and endothelial cells. Macrophages have been shown to be indispensable for angiogenic responses that are critical for heart regeneration and repair[46]. We propose that negative regulation of VEGFA by macrophage YAP represents an important paracrine node to modulate angiogenesis during PO. Identification of additional factors downstream of YAP to regulate non-cell autonomous effects (e.g. cardiomyocyte apoptosis) remains an area of future investigation.
In summary, we show that inhibition of myeloid YAP attenuates multiple aspects of pathological cardiac remodeling including the recruitment of pro-inflammatory cells, myocardial fibrosis, and cardiomyocyte apoptosis. YAP inhibition alters macrophage functional responses and dampens pro-inflammatory gene expression while augmenting transcription of genes associated with wound healing and angiogenesis. Specifically, YAP deficient macrophages have impaired NLRP3 inflammasome function and decreased production of IL-1β, among other pro-inflammatory cytokines. Conversely, cardiac VEGFA was increased in mice with targeted YAP inhibition. We propose that this combination of macrophage-derived effects work collectively to antagonize pathological remodeling of the heart and the transition to heart failure, thereby positioning myeloid YAP as an intriguing therapeutic target for continued investigation in heart disease.
3.1. Study Limitations
Although macrophages contribute to myocardial inflammation and subsequent dysfunction, other immune cell types also play important roles. Neutrophils are recruited to the pressure overloaded heart and contribute to dysfunction through a Wnt5a-mediated mechanism[47]. In this LysMCre model we did not observe a significant reduction in YAP expression in neutrophils isolated from pressure overloaded myocardium, and thus we cannot infer neutrophil YAP function in this context and believe that future studies using neutrophil-specific Cre expression are warranted. The recruitment of T cells to the myocardium also occurs after PO stress and influences remodeling and impaired heart function[48, 49]. While we did not observe differences in the abundance of these cell types in myeloid YAP deficient mice, we cannot rule out their altered function in vivo, which could result from manipulating the myeloid compartment, and is a limitation of this study. In addition, while our nanoparticle experiments demonstrate cardiac benefit and show selective uptake in cells from heart and spleen, we did not determine the efficiency of YAP inhibition using this approach in vivo because of the technical challenges associated with doing so. Finally, we employed pressure overload to induce adverse remodeling and cardiac dysfunction. Evaluation of myeloid YAP deficient mice in additional injury models may provide a more complete characterization of YAP in these immune cells and its contribution to heart failure progression.
4. Materials and Methods
Additional information on materials and methods is included in the Supplemental Appendix.
4.1. Mice.
Conditional Yap1 allele floxed mice[50] were crossed with myeloid (LysM) Cre recombinase transgenic mice [51] to generate conditional YAP deleted mice. Wild-type mice (stock #000664) and LysM-Cre mice (stock #004781) were purchased from The Jackson Laboratories. All mice were C57BL/6J background. Eight- to ten-week-old mice were used for these studies. Male and female age and gender matched littermate controls (YapF/F) were used for all experiments. All mouse studies were performed blinded to genotype. Mice were housed in a temperature-controlled environment with 12-hour light/dark cycles where they received food and water ad libitum. All protocols concerning the use of animals were approved by the Institutional Animal Care and Use Committee at Rutgers, The State University of New Jersey.
4.2. Transverse aortic constriction.
The method for imposing pressure overload in mice has been described previously[52]. Briefly, mice were subcutaneously injected with a small volume of bupivacaine, anesthetized with pentobarbital (60 mg/kg, i.p.), and mechanically ventilated. The left chest was opened at the second intercostal space. Aortic constriction was achieved by ligation of the transverse thoracic aorta between the innominate artery and left common carotid artery with a 27-gauge needle using a 7-0 braided polyester suture. Sham operation was performed without constricting the aorta. Mice with a pressure gradient < 50 mmHg after TAC surgery were excluded from the study.
4.3. Echocardiography.
Mice were anesthetized using isoflurane (3% induction, 1-2% maintenance), and echocardiography was performed as described previously[53], using a 30-MHz linear ultrasound transducer. Two-dimensional guided M-mode measurements of LV internal diameter were obtained from at least three beats and then averaged. LV end-diastolic dimension (LVIDd) was measured at the time of the apparent maximal LV diastolic dimension, and LV end-systolic dimension (LVIDs) was measured at the time of the most anterior systolic excursion of the posterior wall.
4.4. ELISA.
IL-1β and IL-18 protein were measured using commercially available kits according to the manufacturer’s instructions (Invitrogen #BMS6002, #BMS618-3). Mouse VEGF protein was measured according to manufacturer’s instructions (R&D Systems, #MMV00).
4.5. Cell culture and reagents.
The RAW264.7 cell line was purchased from ATCC (TIB-71) and maintained according to established protocols[54]. RAW264.7 cells were transfected with eGFP (Addgene #13031) or YAPS127A (Addgene #27370). For bone marrow derived macrophage (BMDM) experiments, bone marrow cells were isolated from adult YapF/F;LysM-Cre and control YapF/F mice[55]. Cells were cultured in complete RPMI medium supplemented with 20ng/mL recombinant M-CSF (PeproTech) for 5-7 days. Cells were serum starved 24 hours prior to stimulation. Mouse recombinant IL-4 was purchased from R&D Systems. Lipopolysaccharide (LPS) and verteporfin were purchased from Sigma.
4.6. Statistical analysis.
All data are reported as mean ± standard error of the mean (SEM). P values were determined by unpaired, two-tailed Student t-test to evaluate the difference in means between two groups. Evaluation between three or more groups was done using ordinary one-way analysis of variance (ANOVA). Post-hoc multiple pairwise comparisons were performed using Tukey’s test. The normality of continuous variables was determined using the Shapiro-Wilk test. Statistical analyses were performed using Graph Pad Prism 9. A P value < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgements.
The authors thank Andreas Ivessa for histology assistance and Sukhwinder Singh for assistance with flow cytometry.
Sources of Funding.
This work was supported by funding from the National Institutes of Health (R01HL157483 and R01HL127339 to DPD; F31HL162545 to JF) and the American Heart Association (20TPA35490150 to DPD).
Abbreviations:
- BMDM
bone marrow derived macrophage
- ChIP
chromatin immunoprecipitation
- IL-1β
interleukin 1 beta
- IL-4
interleukin 4
- IL-6
interleukin 6
- IL-18
interleukin 18
- LPS
lipopolysaccharide
- NLRP3
NOD-LRR- and pyrin domain-containing protein 3
- PLGA
poly lactic-co-glycolic acid
- PO
pressure overload
- TAC
transverse aortic constriction
- TEAD
TEA family transcription factor
- VEGF
vascular endothelial growth factor
- YAP
Yes-associated protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest. Nothing to disclose.
References
- [1].Tsao CW, Aday AW, Almarzooq ZI, Anderson CAM, Arora P, Avery CL, Baker-Smith CM, Beaton AZ, Boehme AK, Buxton AE, Commodore-Mensah Y, Elkind MSV, Evenson KR, Eze-Nliam C, Fugar S, Generoso G, Heard DG, Hiremath S, Ho JE, Kalani R, Kazi DS, Ko D, Levine DA, Liu J, Ma J, Magnani JW, Michos ED, Mussolino ME, Navaneethan SD, Parikh NI, Poudel R, Rezk-Hanna M, Roth GA, Shah NS, St-Onge MP, Thacker EL, Virani SS, Voeks JH, Wang NY, Wong ND, Wong SS, Yaffe K, Martin SS, American E Heart Association Council on, C. Prevention Statistics, S. Stroke Statistics, Heart Disease and Stroke Statistics-2023 Update: A Report From the American Heart Association, Circulation (2023). [DOI] [PubMed] [Google Scholar]
- [2].Frangogiannis NG, The inflammatory response in myocardial injury, repair, and remodelling, Nature reviews. Cardiology 11(5) (2014) 255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ, Group CT, Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease, N Engl J Med 377(12) (2017) 1119–1131. [DOI] [PubMed] [Google Scholar]
- [4].Zhou Q, Li L, Zhao B, Guan KL, The hippo pathway in heart development, regeneration, and diseases, Circ Res 116(8) (2015) 1431–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Heallen T, Morikawa Y, Leach J, Tao G, Willerson JT, Johnson RL, Martin JF, Hippo signaling impedes adult heart regeneration, Development 140(23) (2013) 4683–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ramjee V, Li D, Manderfield LJ, Liu F, Engleka KA, Aghajanian H, Rodell CB, Lu W, Ho V, Wang T, Li L, Singh A, Cibi DM, Burdick JA, Singh MK, Jain R, Epstein JA, Epicardial YAP/TAZ orchestrate an immunosuppressive response following myocardial infarction, J Clin Invest 127(3) (2017) 899–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Francisco J, Zhang Y, Jeong JI, Mizushima W, Ikeda S, Ivessa A, Oka S, Zhai P, Tallquist MD, Del Re DP, Blockade of Fibroblast YAP Attenuates Cardiac Fibrosis and Dysfunction Through MRTF-A Inhibition, JACC Basic Transl Sci 5(9) (2020) 931–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Del Re DP, Yang Y, Nakano N, Cho J, Zhai P, Yamamoto T, Zhang N, Yabuta N, Nojima H, Pan D, Sadoshima J, Yes-associated protein isoform 1 (Yap1) promotes cardiomyocyte survival and growth to protect against myocardial ischemic injury, J Biol Chem 288(6) (2013) 3977–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Byun J, Del Re DP, Zhai P, Ikeda S, Shirakabe A, Mizushima W, Miyamoto S, Brown JH, Sadoshima J, Yes-associated protein (YAP) mediates adaptive cardiac hypertrophy in response to pressure overload, J Biol Chem 294(10) (2019) 3603–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mia MM, Cibi DM, Binte Abdul Ghani SA, Singh A, Tee N, Sivakumar V, Bogireddi H, Cook SA, Mao J, Singh MK, Loss of Yap/taz in cardiac fibroblasts attenuates adverse remodeling and improves cardiac function, Cardiovasc Res (2021). [DOI] [PubMed] [Google Scholar]
- [11].Zhang Q, Meng F, Chen S, Plouffe SW, Wu S, Liu S, Li X, Zhou R, Wang J, Zhao B, Liu J, Qin J, Zou J, Feng XH, Guan KL, Xu P, Hippo signalling governs cytosolic nucleic acid sensing through YAP/TAZ-mediated TBK1 blockade, Nat Cell Biol 19(4) (2017) 362–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mia MM, Cibi DM, Abdul Ghani SAB, Song W, Tee N, Ghosh S, Mao J, Olson EN, Singh MK, YAP/TAZ deficiency reprograms macrophage phenotype and improves infarct healing and cardiac function after myocardial infarction, PLoS Biol 18(12) (2020) e3000941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wang S, Xie F, Chu F, Zhang Z, Yang B, Dai T, Gao L, Wang L, Ling L, Jia J, van Dam H, Jin J, Zhang L, Zhou F, YAP antagonizes innate antiviral immunity and is targeted for lysosomal degradation through IKKvarepsilon-mediated phosphorylation, Nat Immunol 18(7) (2017) 733–743. [DOI] [PubMed] [Google Scholar]
- [14].Liu M, Yan M, Lv H, Wang B, Lv X, Zhang H, Xiang S, Du J, Liu T, Tian Y, Zhang X, Zhou F, Cheng T, Zhu Y, Jiang H, Cao Y, Ai D, Macrophage K63-Linked Ubiquitination of YAP Promotes Its Nuclear Localization and Exacerbates Atherosclerosis, Cell reports 32(5) (2020) 107990. [DOI] [PubMed] [Google Scholar]
- [15].Zhou X, Li W, Wang S, Zhang P, Wang Q, Xiao J, Zhang C, Zheng X, Xu X, Xue S, Hui L, Ji H, Wei B, Wang H, YAP Aggravates Inflammatory Bowel Disease by Regulating M1/M2 Macrophage Polarization and Gut Microbial Homeostasis, Cell reports 27(4) (2019) 1176–1189 e5. [DOI] [PubMed] [Google Scholar]
- [16].Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, Liu JO, Pan D, Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP, Genes Dev 26(12) (2012) 1300–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN, Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease, Physiol Rev 99(4) (2019) 1765–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hu CM, Fang RH, Wang KC, Luk BT, Thamphiwatana S, Dehaini D, Nguyen P, Angsantikul P, Wen CH, Kroll AV, Carpenter C, Ramesh M, Qu V, Patel SH, Zhu J, Shi W, Hofman FM, Chen TC, Gao W, Zhang K, Chien S, Zhang L, Nanoparticle biointerfacing by platelet membrane cloaking, Nature 526(7571) (2015) 118–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shioi T, Matsumori A, Kihara Y, Inoko M, Ono K, Iwanaga Y, Yamada T, Iwasaki A, Matsushima K, Sasayama S, Increased expression of interleukin-1 beta and monocyte chemotactic and activating factor/monocyte chemoattractant protein-1 in the hypertrophied and failing heart with pressure overload, Circ Res 81(5) (1997) 664–71. [DOI] [PubMed] [Google Scholar]
- [20].Martini E, Kunderfranco P, Peano C, Carullo P, Cremonesi M, Schorn T, Carriero R, Termanini A, Colombo FS, Jachetti E, Panico C, Faggian G, Fumero A, Torracca L, Molgora M, Cibella J, Pagiatakis C, Brummelman J, Alvisi G, Mazza EMC, Colombo MP, Lugli E, Condorelli G, Kallikourdis M, Single-Cell Sequencing of Mouse Heart Immune Infiltrate in Pressure Overload-Driven Heart Failure Reveals Extent of Immune Activation, Circulation 140(25) (2019) 2089–2107. [DOI] [PubMed] [Google Scholar]
- [21].Li Y, Ren P, Dawson A, Vasquez HG, Ageedi W, Zhang C, Luo W, Chen R, Li Y, Kim S, Lu HS, Cassis LA, Coselli JS, Daugherty A, Shen YH, LeMaire SA, Single-Cell Transcriptome Analysis Reveals Dynamic Cell Populations and Differential Gene Expression Patterns in Control and Aneurysmal Human Aortic Tissue, Circulation 142(14) (2020) 1374–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ, The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions, J Exp Med 204(12) (2007) 3037–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Patel B, Bansal SS, Ismahil MA, Hamid T, Rokosh G, Mack M, Prabhu SD, CCR2(+) Monocyte-Derived Infiltrating Macrophages Are Required for Adverse Cardiac Remodeling During Pressure Overload, JACC Basic Transl Sci 3(2) (2018) 230–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Liao X, Shen Y, Zhang R, Sugi K, Vasudevan NT, Alaiti MA, Sweet DR, Zhou L, Qing Y, Gerson SL, Fu C, Wynshaw-Boris A, Hu R, Schwartz MA, Fujioka H, Richardson B, Cameron MJ, Hayashi H, Stamler JS, Jain MK, Distinct roles of resident and nonresident macrophages in nonischemic cardiomyopathy, Proc Natl Acad Sci U S A 115(20) (2018) E4661–E4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Leuschner F, Dutta P, Gorbatov R, Novobrantseva TI, Donahoe JS, Courties G, Lee KM, Kim JI, Markmann JF, Marinelli B, Panizzi P, Lee WW, Iwamoto Y, Milstein S, Epstein-Barash H, Cantley W, Wong J, Cortez-Retamozo V, Newton A, Love K, Libby P, Pittet MJ, Swirski FK, Koteliansky V, Langer R, Weissleder R, Anderson DG, Nahrendorf M, Therapeutic siRNA silencing in inflammatory monocytes in mice, Nat Biotechnol 29(11) (2011) 1005–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Courties G, Heidt T, Sebas M, Iwamoto Y, Jeon D, Truelove J, Tricot B, Wojtkiewicz G, Dutta P, Sager HB, Borodovsky A, Novobrantseva T, Klebanov B, Fitzgerald K, Anderson DG, Libby P, Swirski FK, Weissleder R, Nahrendorf M, In vivo silencing of the transcription factor IRF5 reprograms the macrophage phenotype and improves infarct healing, J Am Coll Cardiol 63(15) (2014) 1556–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K, Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure, J Clin Invest 115(8) (2005) 2108–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Taimeh Z, Loughran J, Birks EJ, Bolli R, Vascular endothelial growth factor in heart failure, Nature reviews. Cardiology 10(9) (2013) 519–30. [DOI] [PubMed] [Google Scholar]
- [29].van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJ, Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice, Am J Pathol 170(3) (2007) 818–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Majmudar MD, Keliher EJ, Heidt T, Leuschner F, Truelove J, Sena BF, Gorbatov R, Iwamoto Y, Dutta P, Wojtkiewicz G, Courties G, Sebas M, Borodovsky A, Fitzgerald K, Nolte MW, Dickneite G, Chen JW, Anderson DG, Swirski FK, Weissleder R, Nahrendorf M, Monocyte-directed RNAi targeting CCR2 improves infarct healing in atherosclerosis-prone mice, Circulation 127(20) (2013) 2038–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kaikita K, Hayasaki T, Okuma T, Kuziel WA, Ogawa H, Takeya M, Targeted deletion of CC chemokine receptor 2 attenuates left ventricular remodeling after experimental myocardial infarction, Am J Pathol 165(2) (2004) 439–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Prabhu SD, Frangogiannis NG, The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis, Circ Res 119(1) (2016) 91–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, Hucker WJ, Wulfers EM, Seemann G, Courties G, Iwamoto Y, Sun Y, Savol AJ, Sager HB, Lavine KJ, Fishbein GA, Capen DE, Da Silva N, Miquerol L, Wakimoto H, Seidman CE, Seidman JG, Sadreyev RI, Naxerova K, Mitchell RN, Brown D, Libby P, Weissleder R, Swirski FK, Kohl P, Vinegoni C, Milan DJ, Ellinor PT, Nahrendorf M, Macrophages Facilitate Electrical Conduction in the Heart, Cell 169(3) (2017) 510–522 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nicolas-Avila JA, Lechuga-Vieco AV, Esteban-Martinez L, Sanchez-Diaz M, Diaz-Garcia E, Santiago DJ, Rubio-Ponce A, Li JL, Balachander A, Quintana JA, Martinez-de-Mena R, Castejon-Vega B, Pun-Garcia A, Traves PG, Bonzon-Kulichenko E, Garcia-Marques F, Cusso L, N AG, Gonzalez-Guerra A, Roche-Molina M, Martin-Salamanca S, Crainiciuc G, Guzman G, Larrazabal J, Herrero-Galan E, Alegre-Cebollada J, Lemke G, Rothlin CV, Jimenez-Borreguero LJ, Reyes G, Castrillo A, Desco M, Munoz-Canoves P, Ibanez B, Torres M, Ng LG, Priori SG, Bueno H, Vazquez J, Cordero MD, Bernal JA, Enriquez JA, Hidalgo A, A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart, Cell 183(1) (2020) 94–109 e23. [DOI] [PubMed] [Google Scholar]
- [35].Wong NR, Mohan J, Kopecky BJ, Guo S, Du L, Leid J, Feng G, Lokshina I, Dmytrenko O, Luehmann H, Bajpai G, Ewald L, Bell L, Patel N, Bredemeyer A, Weinheimer CJ, Nigro JM, Kovacs A, Morimoto S, Bayguinov PO, Fisher MR, Stump WT, Greenberg M, Fitzpatrick JAJ, Epelman S, Kreisel D, Sah R, Liu Y, Hu H, Lavine KJ, Resident cardiac macrophages mediate adaptive myocardial remodeling, Immunity 54(9) (2021) 2072–2088 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zaman R, Hamidzada H, Kantores C, Wong A, Dick SA, Wang Y, Momen A, Aronoff L, Lin J, Razani B, Mital S, Billia F, Lavine KJ, Nejat S, Epelman S, Selective loss of resident macrophage-derived insulin-like growth factor-1 abolishes adaptive cardiac growth to stress, Immunity 54(9) (2021) 2057–2071 e6. [DOI] [PubMed] [Google Scholar]
- [37].Revelo XS, Parthiban P, Chen C, Barrow F, Fredrickson G, Wang H, Yucel D, Herman A, van Berlo JH, Cardiac Resident Macrophages Prevent Fibrosis and Stimulate Angiogenesis, Circ Res 129(12) (2021) 1086–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Acharya AP, Tang Y, Bertero T, Tai YY, Harvey LD, Woodcock CC, Sun W, Pineda R, Mitash N, Konigshoff M, Little SR, Chan SY, Simultaneous Pharmacologic Inhibition of Yes-Associated Protein 1 and Glutaminase 1 via Inhaled Poly(Lactic-co-Glycolic) Acid-Encapsulated Microparticles Improves Pulmonary Hypertension, J Am Heart Assoc 10(12) (2021) e019091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kim M, Kim T, Johnson RL, Lim DS, Transcriptional co-repressor function of the hippo pathway transducers YAP and TAZ, Cell reports 11(2) (2015) 270–82. [DOI] [PubMed] [Google Scholar]
- [40].Beyer TA, Weiss A, Khomchuk Y, Huang K, Ogunjimi AA, Varelas X, Wrana JL, Switch enhancers interpret TGF-beta and Hippo signaling to control cell fate in human embryonic stem cells, Cell reports 5(6) (2013) 1611–24. [DOI] [PubMed] [Google Scholar]
- [41].Mezzaroma E, Toldo S, Farkas D, Seropian IM, Van Tassell BW, Salloum FN, Kannan HR, Menna AC, Voelkel NF, Abbate A, The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse, Proc Natl Acad Sci U S A 108(49) (2011) 19725–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Marchetti C, Toldo S, Chojnacki J, Mezzaroma E, Liu K, Salloum FN, Nordio A, Carbone S, Mauro AG, Das A, Zalavadia AA, Halquist MS, Federici M, Van Tassell BW, Zhang S, Abbate A, Pharmacologic Inhibition of the NLRP3 Inflammasome Preserves Cardiac Function After Ischemic and Nonischemic Injury in the Mouse, J Cardiovasc Pharmacol 66(1) (2015) 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wang D, Zhang Y, Xu X, Wu J, Peng Y, Li J, Luo R, Huang L, Liu L, Yu S, Zhang N, Lu B, Zhao K, YAP promotes the activation of NLRP3 inflammasome via blocking K27-linked polyubiquitination of NLRP3, Nature communications 12(1) (2021) 2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Van Tassell BW, Canada J, Carbone S, Trankle C, Buckley L, Oddi Erdle C, Abouzaki NA, Dixon D, Kadariya D, Christopher S, Schatz A, Regan J, Viscusi M, Del Buono M, Melchior R, Mankad P, Lu J, Sculthorpe R, Biondi-Zoccai G, Lesnefsky E, Arena R, Abbate A, Interleukin-1 Blockade in Recently Decompensated Systolic Heart Failure: Results From REDHART (Recently Decompensated Heart Failure Anakinra Response Trial), Circ Heart Fail 10(11) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, Porrello ER, Mahmoud AI, Tan W, Shelton JM, Richardson JA, Sadek HA, Bassel-Duby R, Olson EN, Hippo pathway effector Yap promotes cardiac regeneration, Proc Natl Acad Sci U S A 110(34) (2013) 13839–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, Bassel-Duby R, Sadek HA, Olson EN, Macrophages are required for neonatal heart regeneration, J Clin Invest 124(3) (2014) 1382–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wang Y, Sano S, Oshima K, Sano M, Watanabe Y, Katanasaka Y, Yura Y, Jung C, Anzai A, Swirski FK, Gokce N, Walsh K, Wnt5a-Mediated Neutrophil Recruitment Has an Obligatory Role in Pressure Overload-Induced Cardiac Dysfunction, Circulation 140(6) (2019) 487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Salvador AM, Nevers T, Velazquez F, Aronovitz M, Wang B, Abadia Molina A, Jaffe IZ, Karas RH, Blanton RM, Alcaide P, Intercellular Adhesion Molecule 1 Regulates Left Ventricular Leukocyte Infiltration, Cardiac Remodeling, and Function in Pressure Overload-Induced Heart Failure, J Am Heart Assoc 5(3) (2016) e003126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kallikourdis M, Martini E, Carullo P, Sardi C, Roselli G, Greco CM, Vignali D, Riva F, Ormbostad Berre AM, Stolen TO, Fumero A, Faggian G, Di Pasquale E, Elia L, Rumio C, Catalucci D, Papait R, Condorelli G, T cell costimulation blockade blunts pressure overload-induced heart failure, Nature communications 8 (2017) 14680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zhang N, Bai H, David KK, Dong J, Zheng Y, Cai J, Giovannini M, Liu P, Anders RA, Pan D, The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals, Dev Cell 19(1) (2010) 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I, Conditional gene targeting in macrophages and granulocytes using LysMcre mice, Transgenic Res 8(4) (1999) 265–77. [DOI] [PubMed] [Google Scholar]
- [52].Del Re DP, Matsuda T, Zhai P, Gao S, Clark GJ, Van Der Weyden L, Sadoshima J, Proapoptotic Rassf1A/Mst1 signaling in cardiac fibroblasts is protective against pressure overload in mice, J Clin Invest 120(10) (2010) 3555–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Matsuda T, Zhai P, Sciarretta S, Zhang Y, Jeong JI, Ikeda S, Park J, Hsu CP, Tian B, Pan D, Sadoshima J, Del Re DP, NF2 Activates Hippo Signaling and Promotes Ischemia/Reperfusion Injury in the Heart, Circ Res 119(5) (2016) 596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Raschke WC, Baird S, Ralph P, Nakoinz I, Functional macrophage cell lines transformed by Abelson leukemia virus, Cell 15(1) (1978) 261–7. [DOI] [PubMed] [Google Scholar]
- [55].Francisco J, Byun J, Zhang Y, Kalloo OB, Mizushima W, Oka S, Zhai P, Sadoshima J, Del Re DP, The tumor suppressor RASSF1A modulates inflammation and injury in the reperfused murine myocardium, J Biol Chem 294(35) (2019) 13131–13144. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.