

Abstract
Background:
Flucloxacillin (FLX)-induced liver injury is immune-mediated and highly associated to HLA-B*57:01 expression. Host factors leading to drug-induced liver injury are not yet well understood.
Objective:
Characterize in vivo immune mechanisms determining the development of CD8+ T cells reactive to FLX in animals expressing the risk human leukocyte antigen (HLA) allotype.
Methods:
HLA-B*57:01 transgenic mice (Tg) or Tg strains with H2-KbDb knockout (Tg/KO) or H2-KbDb/PD-1 double knockout (Tg/DKO) were treated with drug and/or anti-CD4 antibody. Drug-induced liver injury was evaluated on the basis of liver enzyme and histologic changes at day 10 of treatment. FLX-reactive CD8+ T cells were characterized in vitro by release of effector molecules on drug restimulation, gene expression, and flow cytometry analysis, and functionality tested for hepatic cytotoxicity.
Results:
CD8+ T-cell responses to FLX in Tg were dependent on both HLA and mouse major histocompatibility complex I presentation and in vivo priming. Eliminating H2-KbDb in Tg/KO to allow exclusive presentation of FLX by HLA resulted in a less robust drug-specific CD8+T-cell response unless CD4+ cells, including regulatory T cells, were depleted. Treatment of Tg/KO with anti-CD4 antibody and FLX led to subclinical liver inflammation associated with an increase in PD1+CD8+ T cells in the lymphoid organs and liver. Impaired PD-1 expression in Tg/DKO led to liver histopathologic and transcriptional alterations but without hepatic enzyme elevations. Moreover, effector lymphocytes accumulated in the liver and showed FLX-dependent hepatic cytotoxicity in vitro when tolerogenic liver cells were depleted.
Conclusions:
In our in vivo models, FLX primes CD8+ T cells to recognize drug presented by HLA-B*57:01 and murine major histocompatibility complex I. HLA-B*57:01–dependent CD8+ T-cell reaction to FLX is limited by the presence of CD4+ cells, presumably regulatory T cells, and PD-1 expression. Tolerogenic hepatic cells limit clinical disease through PD-L1 or additional unexplored mechanisms.
Keywords: Flucloxacillin, HLA-B*57:01 transgenic mice, drug-induced liver injury, tolerance
Adverse reactions to antibiotics are primarily immune-mediated events that occur at significant frequency and can have severe health consequences to the affected patients.1,2 Understanding the mechanisms of adverse reactions to antibiotics, therefore, is critical for public health management to ensure appropriate antibiotic usage and to avoid withdrawal of effective products under development because of side effects.3 In particular, flucloxacillin (FLX), a narrow-spectrum β-lactam antibiotic prescribed to combat gram-positive bacterial infections (ie, Staphylococcus) causes cholestatic hepatitis resulting in liver inflammation and injury.4–6 Genome-wide association studies established a significant linkage between human leukocyte antigen (HLA) alleles B*57:01 and B*57:03, and FLX–drug-induced liver injury (FLX-DILI), with 83% of FLX-DILI cases carrying the B*57 alleles.7,8 The frequency of FLX-DILI varies between 1 to 11 cases for every 10,000 FLX-treated patients depending on treatment length, sex, and age.9 The idiosyncratic nature of its clinical manifestations suggests that other host and environmental factors might play a role in controlling liver injury in addition to HLA.
The detection of liver-infiltrating, FLX-reactive CD8+ T lymphocytes in patients with DILI supports the idea that liver pathogenesis is immune mediated.4,10,11 Although 2 models have been proposed to explain FLX presentation by HLA-B*57:01,12,13 the hepatotoxic mechanism of FLX-reactive T cells is not yet clearly understood. Studies with human peripheral blood mononuclear cells (PBMCs) demonstrated that FLX-reactive CD8+ and CD4+ T cells could be enriched from both drug-naive HLA-B*57:01+ and HLA-B*57:01− healthy subjects when cultured in vitro with the drug.14–17 Also, drug-reactive CD8+ T cells were enriched in PBMCs from HLA-B*57:01+ patients experiencing liver injury,15,16 but not PBMCs from HLA-B*57:01− individuals. Surprisingly, the high frequency of drug-reactive cells measured in vitro contrasts with the low clinical incidence of FLX-DILI, implicating in vivo tolerogenic mechanisms in the control of the disease onset.
Early mouse models developed to study FLX-induced liver injury18,19 did not address the role of HLA during transient DILI due to the lack of HLA expression in murine cells. The generation of transgenic mice expressing HLA-B*57:01 has been instrumental in helping us gain knowledge on the immune response triggered by abacavir (ABC)20–23 and on the immunogenicity of FLX-haptenated peptides or human serum albumin.24,25 Although FLX can be presented by non–HLA-B*57:01 molecules,14,15,18,19 we chose to focus on HLA-B*57:01 using Tg mice to elucidate the mechanisms of drug presentation and subsequent immune activation by the human risk allele with a higher association with FLX-DILI. We confirmed that murine MHC class I (mMHC-I) alleles, in addition to HLA-B*57:01, can prime FLX-reactive T cells. We subsequently studied the role of HLA-B*57:01 in FLX-specific presentation to T cells in recently developed mouse strains exclusively expressing HLA-B*57:01. We demonstrated that the robustness of CD8+ T-cell responses to the drug in vivo was dependent on the absence of CD4+ T cells (including regulatory T [Treg] cells) and programmed cell death 1 (PD-1) expression. Infiltrates of drug-reactive cells in livers of HLA-transgenic animals lacking mMHC-I and PD1, and treated with anti-CD4 antibody (aCD4Ab) and FLX, were associated with histopathologic changes, although insufficient to cause DILI. Drug-specific infiltrating cells, however, were cytotoxic to HLA-positive hepatocytes in vitro, suggesting that liver tolerance rather than clonal deletion may prevent liver injury in vivo.
METHODS
Mice
HLA-B*57:01/H2-Dd (a3) transgenic (Tg) and HLA-B*57:01 Tg/H2-KbDb knockout mice (Tg/KO) were generated as previously described.20,24 HLA-B*57:01 Tg/H2-KbDb KO/PD-1 KO mice (Tg/DKO) were generated by backcrossing the Tg/KO to Pdcd1tm1.1Shr/J mice (The Jackson Laboratory, Bar Harbor, Maine). Wild-type mice (WT) were negative for the transgene and expressed intact mMHC-I. Experimental mice were female unless otherwise indicated, and were older than 12 weeks. Mice were bred and housed under specific-pathogen–free conditions in the Association for Assessment and Accreditation of Laboratory Animal Care International (aka AAALAC)-accredited animal facility of the US Food and Drug Administration (FDA)’s Division of Veterinary Medicine (Silver Spring, Md) in compliance with the Institutional Animal Care and Use Committee regulations of the FDA (protocol 2017–63).
In vitro cell culture assays
In vitro T-cell responses to drug were measured in cultures of purified CD8+ T lymphocytes from spleen or lymph nodes (LNs) or total splenocytes as described in this article’s Methods section in the Online Repository at www.jacionline.org.
In vivo drug sensitization/gavage treatment
Retinoic acid ([RA] MP Biomedicals, Santa Ana, Calif); 100 μg in =0 μL of 100% dimethyl sulfoxide ([DMSO] Sigma, St Louis, Mo) with or without FLX (50 mg in 100 μL of 70% of DMSO; Altamedics Gmbh, Cologne, Nordrhein-Westfalen, Germany) were topically administered on days 1–3 (sensitization). FLX (2.3 mg/250 μL of water for injection) was provided by oral gavage on days 8–9. aCD4Ab (a-CD4 Ab, clone GK1.5; BioXcell, Lebanon, NH) was injected intraperitoneally at 0.25 mg per dose on days −3, 1, 4, and 7. Preparation of reagents is described in the Methods section in the Online Repository.
Blood cell phenotype analysis and expression of HLA and H2-Kb molecules
Blood was collected into MiniCollect Z Serum Separator tubes (Greiner Bio-One, Frickenhausen, Germany) and analyzed by flow cytometry on red blood cell lysis (ammonium–chloride–potassium lysing buffer [Gibco, Thermo Fisher Scientific, Waltham, Mass]).20
Liver enzyme levels in serum
Blood was collected at the time of euthanasia by cardiac puncture into MiniCollect tubes as recommended. Alanine transaminase (ALT) levels were measured using commercially available assay kits (BioAssay Systems, Hayward, Calif).
Histology
Liver sections were submerged into 10% neutral buffered formalin, processed into paraffin blocks, and sectioned at 5 μm onto positively charged slides. Hematoxylin and eosin (H&E) staining was performed by Histoserv (Germantown, Md).
Gene expression analysis
Liver biopsy samples were immediately submerged in TRIzol (Invitrogen; Thermo Fisher Scientific, Waltham, Mass) on excision, flash-frozen, and stored at −80°C until processing. Sample processing was conducted as previously described.20
Flow cytometry
Immune cell subset analysis by intracellular and/or surface marker staining20 was performed using antibodies as described in the Methods section in the Online Repository. Intracellular staining was performed using the BD Cytofix/Cytoperm Plus kit (BD Biosciences, Franklin Lakes, NJ) as per the manufacturer’s instructions.
Isolation of primary hepatocyte
Mouse hepatocytes were isolated using either collagenase type I digestion (Sigma-Aldrich, St Louis, Mo) or enzymes from the mouse liver dissociation kit (Miltenyi Biotec, Bergisch Gladbach, Germany). Details of both protocols are provided in in the Methods section in the Online Repository.
Isolation of nonparenchymal cells and liver leukocytes
Nonparenchymal cell (NPC) isolation was performed upon perfusing the liver with 5–10 mL of 1 × PBS using a peristaltic pump. NPC suspensions were obtained with the mouse liver dissociation kit (Miltenyi Biotec) in an Octo Dissociator with Heaters as per the manufacturer’s instructions. Liver leukocytes were subsequently pelleted in 37.5% Percoll (Cytiva, Marlborough, Mass) solution, washed, and resuspended in DMEM-10 (Gibco).
Cytotoxicity assay
Primary hepatocytes and liver leukocytes or NPC cultures with or without FLX were prepared and maintained in an xCELLigence real-time cell analysis platform (Agilent Technologies, Santa Clara, Calif) as indicated in the Methods section in the Online Repository. Cytolysis was calculated as follows: % Cytolysis = [(Normalized Cell Indexno effector − Normalized Cell Indexeffector)/Normalized Cell Indexno effector)] × 100.
RESULTS
FLX-reactive CD8+ T cells are enriched in spleen of FLX-treated Tg animals
Splenic CD8+ T cells from drug-naive Tg and WT animals were cultured in the presence of FLX or ABC (used as positive control) (Fig 1, A). As reported earlier,20 ABC-treated T cells produced high levels of IFN-γ and granzyme B (GZMB) within the first cycle of stimulation, unlike those treated with FLX, that required 2–3 cycles. Cells from WT mice were not reactive to drug stimulation for 28 days. We then asked whether in vivo priming of the mice with drug could enhance detection of FLX-reactive cells in vitro. Cells from mice treated orally with the human-equivalent dose of FLX did not respond to drug reexposure in vitro (data not shown). Thus, we adopted a RA skin sensitization/oral gavage strategy to administer FLX, which was previously used to generate drug-reactive cells in an MHC-II KO, CD4+ T-cell–depleted, non-HLA–expressing mice.18 This treatment strategy promotes hapten formation, T-cell activation, and migration to the gut–liver axis. Priming Tg animals with FLX in vivo accelerated secretion of IFN-γ by splenic CD8+ T cells when restimulated with drug in vitro. Increased IFN-γ was measurable starting at day 5 of culture as opposed to at day 12 for cultures of cells from drug-naive animals (Fig 1, B). The addition of FLX to cocultures improved the magnitude of released IFN-γ by T cells compared to cultures where FLX was washed off after pulsing the feeder cells (see Fig E1 in the Online Repository at www.jacionline.org). FLX-reactive T cells exhibited morphologic changes associated with cell activation (Fig 1, C). These data indicate that oral administration of FLX after skin sensitization elicits a systemic drug-reactive CD8+ T-cell response in immunocompetent Tg mice.
FIG 1.
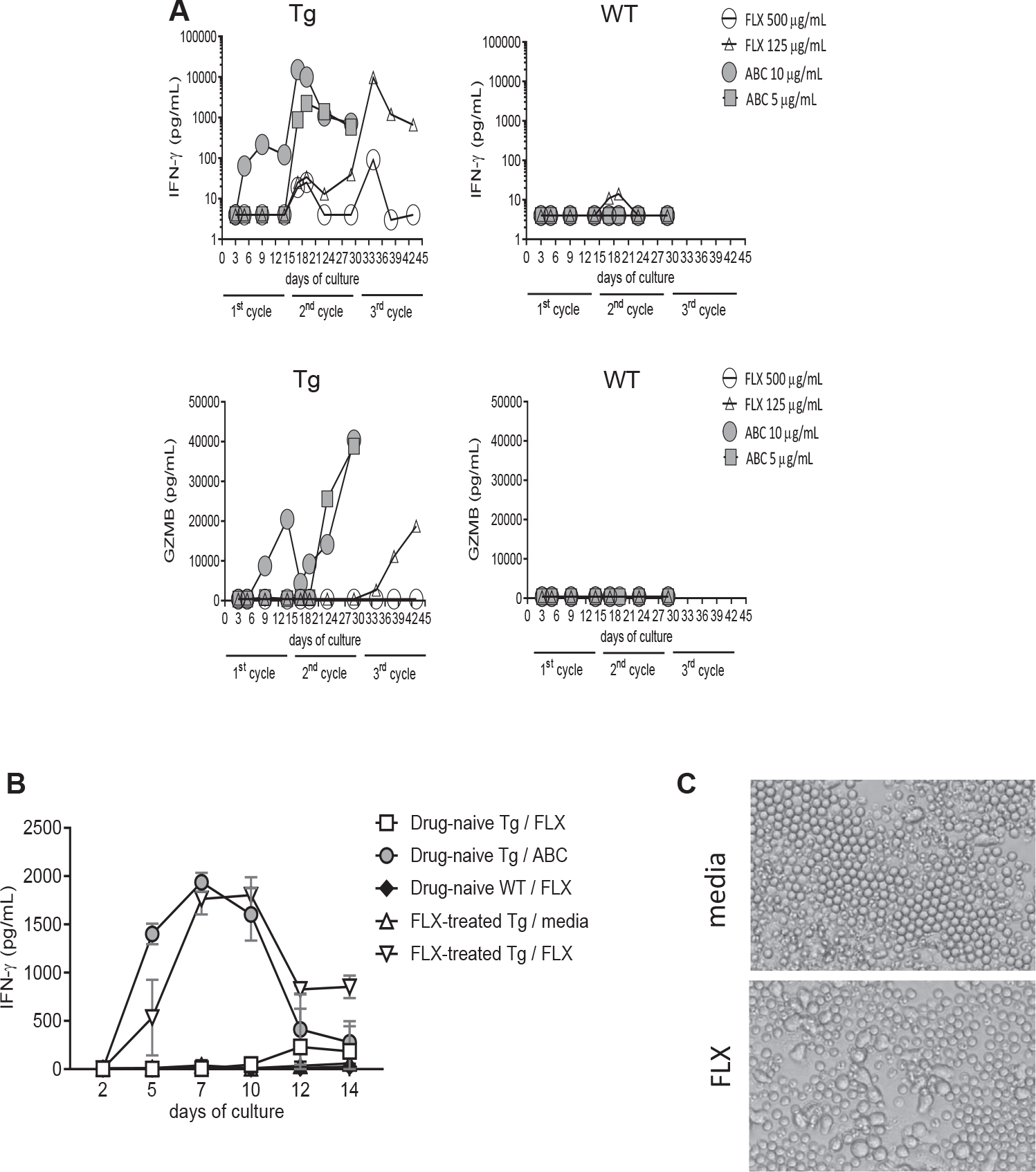
Drug-reactive CD8+ T-cell responses are enriched in FLX-primed Tg mice. CD8+ T cells were isolated from spleens of drug-naive HLA-B*57:01+ (Tg) and WT control animals (A and B) or FLX-treated Tg mice (B) and cultured with irradiated autologous feeders at a 1:2 ratio in the presence or absence of FLX or ABC (125 μg/mL of FLX and 10 μg/mL of ABC in (B)). IFN-γ and GZMB levels were measured at different time points by ELISA. (A) Data shown are from 1 representative of 2 independent experiments. (B) IFN-γ values are presented as means ± SEMs of results from 3 animals per group. (C) Cell morphology changes observed by optical microscopy (20×) at day 4 of culture of CD8+ splenocytes from a representative FLX-primed animal, in the absence or presence of 125 μg/mL of FLX.
Generation of FLX-reactive CD8+ T cells is driven by both HLA-B*57:01 and mMHC-I
T-cell reactivity to FLX has been shown in mice lacking HLA expression,18 suggesting that mMHC-I plays a role in FLX presentation. As such, the Tg strain used here has the potential to present drug via both HLA-B*57:01 and mMHC-I. We isolated CD8+ T cells from the LNs (Fig 2, A) and spleens (Fig 2, B) of untreated Tg or FLX-primed Tg and WT animals. Within 2 days of coculture with drug-pulsed feeder cells, LN CD8+ T cells from drug-primed Tg mice stimulated with 250 μg/mL of FLX showed low but significantly increased levels of IFN-γ in the culture supernatant compared to those from untreated Tg or drug-treated WT mice (Fig 2, A, left). By day 4–5, IFN-γ was detected in cultures of FLX-treated WT animals, although at significantly lower yields than in cultures of the Tg counterparts (Fig 2, A, center and right). The time delay in T-cell activation was also observed in splenocytes (Fig 2, B).
FIG 2.
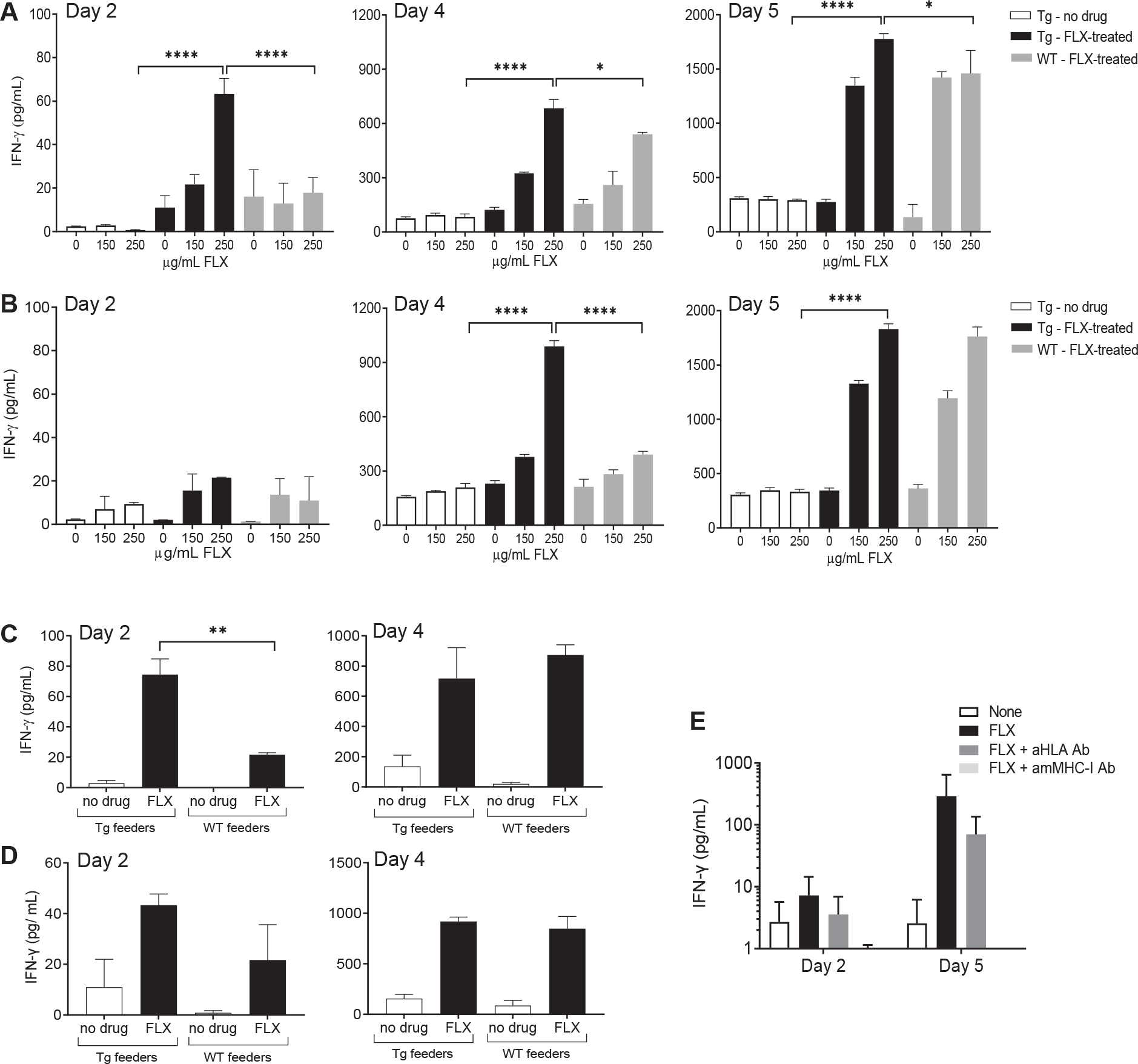
CD8+ T-cell reactivity to FLX is driven by both HLA-B*57:01 and mouse MHC-I molecules following different kinetics. (A and B) CD8+ T cells from LNs (A) and spleens (B) of untreated Tg mice or FLX-treated Tg and WT mice and cultured with irradiated autologous splenocytes at 1:2 ratio for up to 5 days. Feeders were incubated overnight with the indicated drug concentration before irradiation. FLX was also added to cocultures at the same concentration. IFN-γ release was measured by ELISA. Data represent means ± SEMs of values from 3 animals per group. (C and D) CD8+ T cells isolated from LN (C) or spleen (D) of FLX-treated animals were cultured with autologous (Tg) or WT feeders under conditions indicated in the graph. FLX was provided at a concentration of 150 μg/mL. IFN-γ levels in culture supernatants were measured by ELISA at days 2 and 4 of culture. (E) Splenic CD8+ T cells from drug-treated Tg mice were cultured with 150 μg/mL of FLX in vitro with autologous feeders in the presence or absence of 10 μg/mL anti-HLA or anti-mouse MHC-I (H2-Kb and H2-Db) antibodies. IFN-γ concentration in culture supernatants was measured by ELISA. Values represent means + SEMs of 3 animals per group. *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001,1-way ANOVA.
These results, however, did not determine whether the bulk of the response observed in Tg is driven by mMHC-I or HLA. To answer this question, LN (Fig 2, C) and splenic (Fig 2, D) CD8+ T cells from FLX-primed Tg were cocultured with drug-pulsed autologous or WT feeders, with or without FLX. At day 2, low levels of IFN-γ were detected in cocultures of CD8+ T cells with autologous feeders, but higher than with WT feeders. At day 4, though, IFN-γ yields became comparable, confirming that although mMHC-I could support the T-cell reactivity to FLX, the presence of the transgene allowed cells to react earlier (day 2) to in vitro restimulation. To further assess the role of the HLA in this response, we blocked antigen presentation with anti–HLA-B/C or anti–H2-Kb/H2-Db antibodies. IFN-γ production was slightly decreased by anti–HLA-B/C antibody, although not to statistically significant levels, in contrast to the absolute interference by anti–H2-Kb/H2-Db antibody (Fig 2, E). These data confirm the ability of mMHC-I to present FLX to CD8+ T cells, although the early CD8+ T-cell activation may be HLA dependent in Tg mice. Because FLX presentation by HLA-B*57:01 was masked by mMHC-I, we opted to repeat the experiments in a new mouse strain derived from backcrossing Tg with H2-KbDb KO mice (Tg/KO).24
Lack of H2-KbDb Tg/KO results in lower numbers of systemic CD8+ T cells and weaker drug-specific responses
Splenic CD8+ T cells from FLX-treated Tg/KO animals cocultured with drug-pulsed autologous feeders and FLX did not secrete detectable IFN-γ until day 7 (Fig 3, A). In general, cells from female mice released higher levels of IFN-γ than those from males; indeed, 67% of all female samples had ≥1000 pg/mL IFN-γ compared to 17% of all male samples. Addition of anti-HLA antibody in the culture reduced cytokine production by at least 50% in all the animals at some time point (Fig 3, B), indicating a role of HLA in FLX presentation to CD8+ T cells. More pronounced effects occurred in cultures with higher IFN-γ production, and slightly different kinetics were observed for each individual animal.
FIG 3.
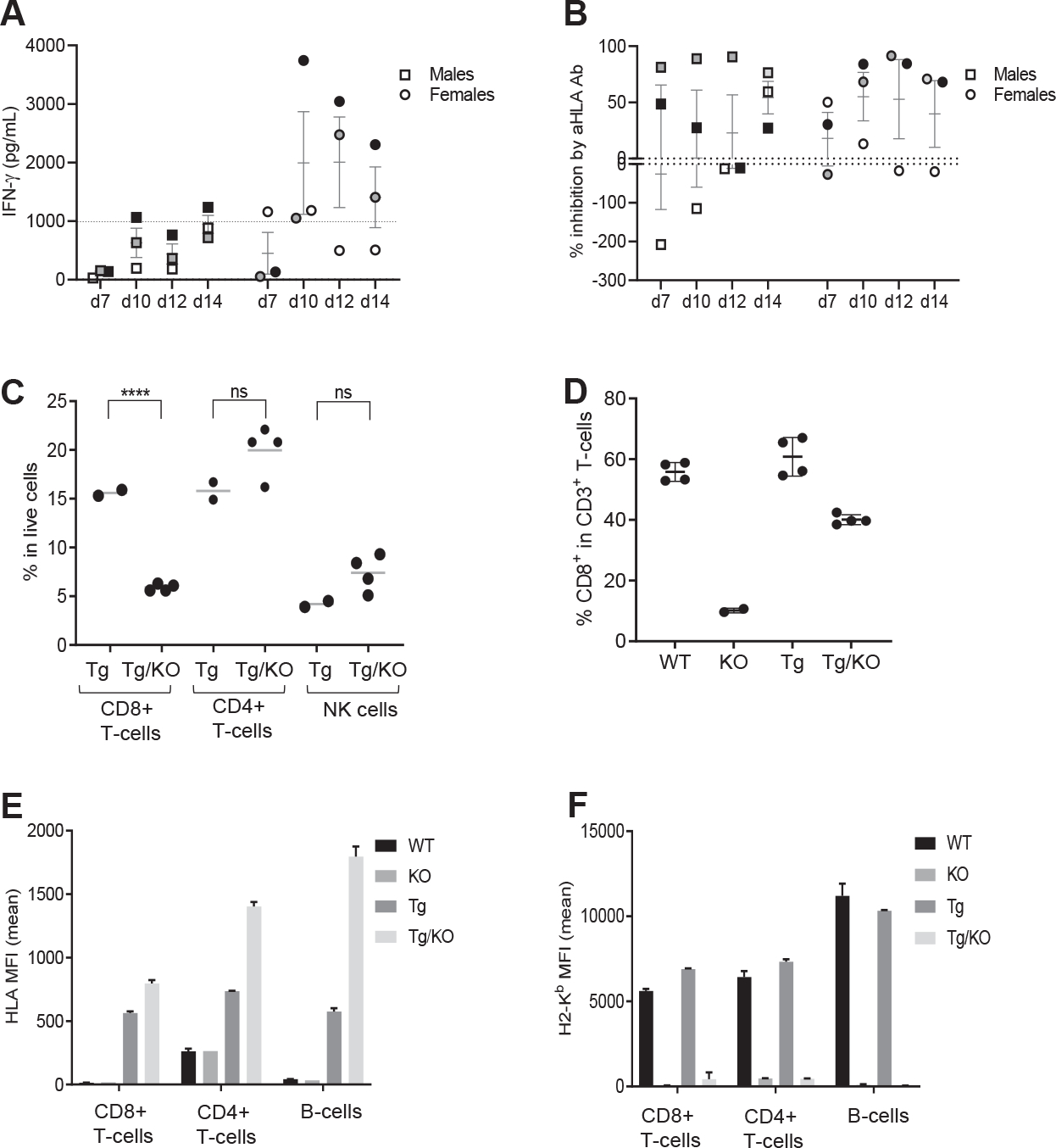
CD8+ T cells from FLX primed Tg/KO cells have a delayed HLA-dependent response to FLX in vitro due to decreased levels of CD8+ T cells influenced by lack of H2-KbDb. (A and B) Splenic CD8+ T cells from FLX-primed Tg/KO animals (males and females, n = 3 each) were cocultured with drug-pulsed irradiated autologous feeders (matching sex) and 250 μg/mL of FLX for 14 days. (A) IFN-γ levels in culture supernatants were measured by ELISA at indicated time points. (B) Inhibition of IFN-γ release was represented as percentage of cytokine level between cultures pretreated with or without anti-HLA antibody. (C) Percentage of CD8+ and CD4+ T lymphocytes and NK cells in spleen of Tg and Tg/KO mice. (D) Percentage of splenic CD8+ T lymphocytes in total CD3+ T cells in mice of different backgrounds. (E and F) MFI for HLA B/C (E) and H2-Kb (F) molecules expressed in blood lymphocyte populations of different mouse strains. ****P ≤ .0001, unpaired t test. NK, Natural killer.
Of note, drug-naive Tg/KO animals had approximately 3 times less total splenic CD8+ T cells than Tg, while CD4+ T-cell and natural killer cell levels were comparable (Fig 3, C). Low frequency of CD8+ T cells could be associated with the lack of mMHC-I expression26 (Fig 3, D). In addition, although HLA expression was enhanced 0.5–3-fold in Tg/KO versus Tg cells (Fig 3, E), the Tg/KO HLA mean fluorescence intensity (MFI) was 4–8.5 times lower than the H2-Kb MFI on Tg or WT cells (Fig 3, F). The difference in the MFI of class I molecules could be attributed to the density of HLA versus H2-Kb molecules on the cell surface in different strains. Although FLX-modified epitopes are presented exclusively by HLA in Tg/KO, the lower expression of overall class I molecules together with the lower frequencyof CD8+ T cells observed in Tg/KO may limit the number of FLX-primed clones in circulation, resulting in a weaker/delayed IFN-γ response after in vitro restimulation of the cells.
Depletion of CD4+ T cells, including Treg cells, induces mild liver inflammation and enrichment of infiltrating drug-reactive PD-1+CD8+ T cells
In humans, FLX-induced liver injury manifests with hepatic enzyme elevation because of liver damage.4,7 Although FLX-DILI has been linked to HLA-B*57:01, its frequency is very low,7–9 which is thought to be in part due to robust immunoregulatory mechanisms. Tg/KO treated with FLX did not show signs of liver inflammation or disease (Fig 4) despite developing FLX-reactive CD8+ T cells by day 10 of treatment. In an attempt to break hepatic tolerance to FLX, we depleted the animals of CD4+ T cells (including Treg cells) before and during drug treatment. Anti-CD4 antibody in combination with FLX did not lead to either ALT elevation (Fig 4, A) or hepatic histologic change (Fig 4, B). However, gene expression analysis revealed mild liver inflammation, characterized by increased transcription of several inflammatory genes, including T-cell markers and effector molecules (Cd8, Infg, Gzmb1, Nfatc2, Pd1), calgranulin S100a9, antigen-presenting cell markers (Xcr1, Cd11b, Cd38), chemokines and receptors (Cxcr2, Cxcl10), oxidative stress pathways (Nos2), apoptotic molecules (Fas), and Akrb10 (Fig 4, C). Although the expression of these genes was elevated in liver of animals treated with either aCD4Ab alone or in combination with drug, the magnitude of the inflammatory response was amplified in mice treated with FLX.
FIG 4.
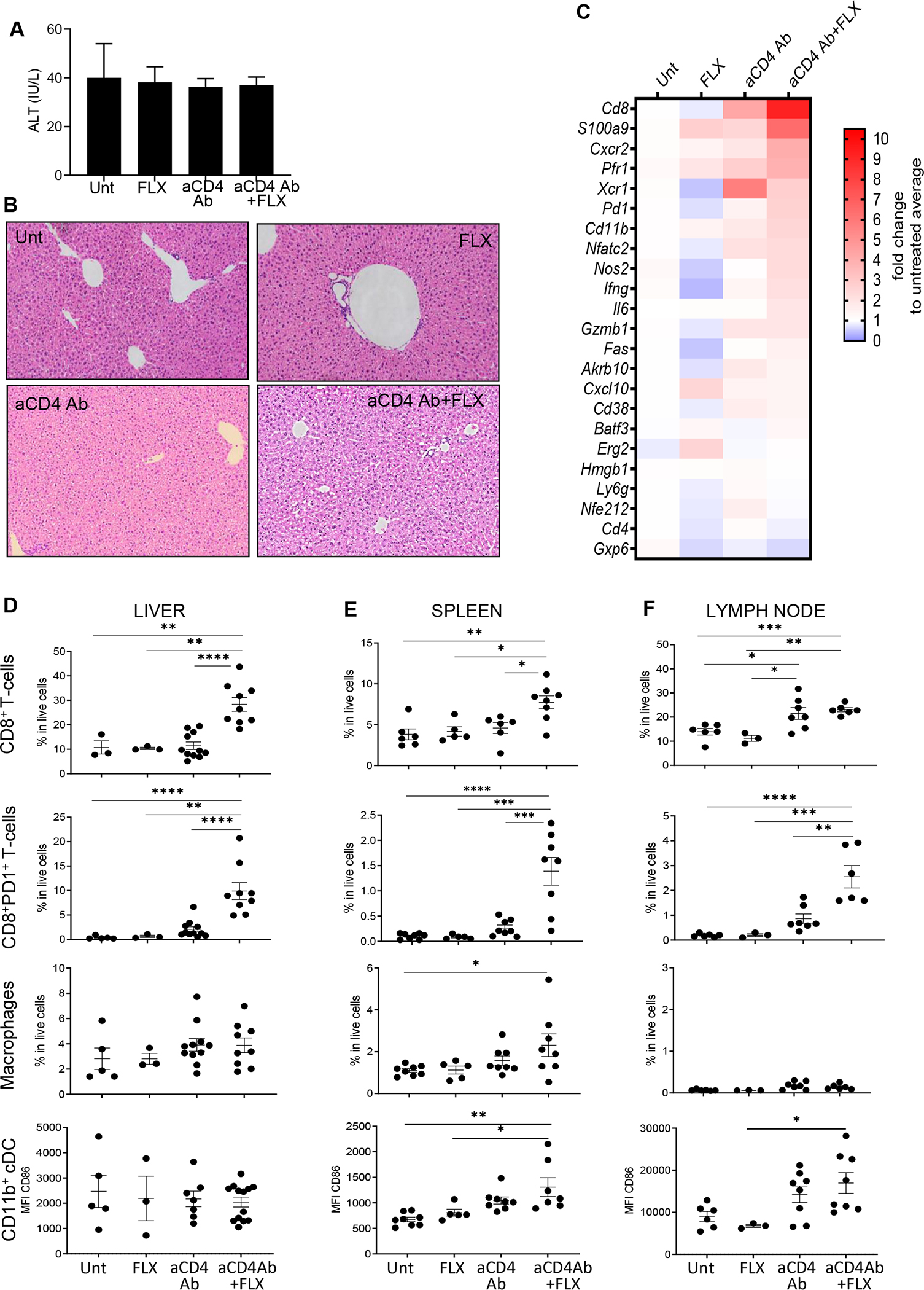
FLX treatment of Tg/KO mice leads to mild liver inflammation if CD4+ T cells, including Treg cells, are depleted. FLX was administered to Tg/KO mice with or without aCD4Ab. All animals were treated with RA. At day 10 after initiation of drug treatment, liver inflammation was evaluated by serum levels of ALTs (A), H&E staining of fixed liver sections (representative mouse per group) (B), and gene expression analysis of perfused liver by real-time PCR (geomean of n = 3–6 mice per group) (C). (D) Infiltrating liver leukocyte populations were isolated from perfused livers and characterized by FACS. Spleen (E) and LN (F) cell suspensions were obtained and analyzed with the same FACS antibody panel (n = 3–9 mice per group). Dead cells were excluded from analysis. Macrophages are CD11bhiF4/80+; cDC CD11b+ are CD19−CD11c+CD8− cells (see gating strategy in Fig E5). *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001, 1-way ANOVA. FACS, Fluorescence-activated cell sorting.
CD8+ T cells were enriched in the liver of animals treated with aCD4Ab 1 FLX, especially PD-1+CD8+ T cells, averaging 34% of the total CD8+ T-cell subset compared to 18% in the aCD4Ab control group or 5% within the untreated mice (Fig 4, D, top).
A higher frequency of PD-1+CD8+ T cells was also detected in the spleens and LNs of aCD4Ab + FLX–treated animals (Fig 4, E and F), indicating that the effect of FLX on CD8+ T-cell activation was systemic. Anti–CD4Ab + FLX treatment slightly increased the number of splenic macrophages (Fig 4, E). Although the frequency of conventional dendritic cell (DC) subsets was not altered by any of the treatments (data not shown), depletion of CD4+ cells had an impact on expression of DC maturation markers, such as CD86. This occurred in lymphoid organs but not in liver (Fig 4, D–F), implicating the lack of DC maturation in maintaining suboptimal T-cell activation in the liver.
Interference with PD-1 expression increases liver inflammation and FLX-reactive CD8+ T cells without breaking liver tolerance
We hypothesize thatPD-1expression by FLX-reactiveCD8+ T cells of Tg/KO may prevent the expansion of the effector immune response, especially in the liver, where resident cells such as liver sinusoidal endothelial cells (LSECs) and Kupffer cells (KCs) express high levels of programmed cell death ligand 1 (PD-L1).27,28 Therefore, we generated a double-knockout mouse strain lacking PD-1 expression (Tg/DKO) from the Tg/KO. PD-L1 expression was evaluated on liver cells (see Fig E2 in the Online Repository at www.jacionline.org). Drug-naive Tg/DKO animals had higher levels of circulating CD8+ T cells than Tg/KO animals (P = .0408) (Fig 5, A) and inflammation (see Fig E3 in the Online Repository). Splenocytes isolated from aCD4Ab + FLX–treated Tg/DKO animals, unlike those from similarly treated Tg/KO, were enriched in IFN-γ+CD8+ and GZMB+CD8+ T cells (Fig 5, B and C). These results were supported by early (day 2) measurable levels of IFN-γ in Tg/DKO splenocyte cultures supplemented with the drug (Fig 5, D). Animals that received FLX had enlarged gallbladders compared to the other control groups (Fig 5, E). The liver parenchyma of several Tg/DKO from the aCD4Ab + FLX group was inflamed, as evidenced by the presence of cell swelling, hepatocyte apoptosis, and hepatocyte dropout near the central vein endothelium, consistent with zone 3 inflammation, in addition to periportal inflammation with portal vein endotheliitis (Fig 5, F, and see Fig E4 in the Online Repository). Lymphoid foci and microgranulomas were more abundant compared to the aCD4Ab + FLX–treated Tg/KO (Fig 4, D) and Tg/DKO control groups (Fig 5, F). ALT levels remained unchanged at day 10 of drug treatment (Fig 5, G). Histologic changes correlated with an increase in RNA expression of genes related to immune response and inflammation, especially Tnfa, Il1b, Il-6 and Cxcr2 (Fig 5, H), which have been associated with cholestasis in previous studies.29,30
FIG 5.
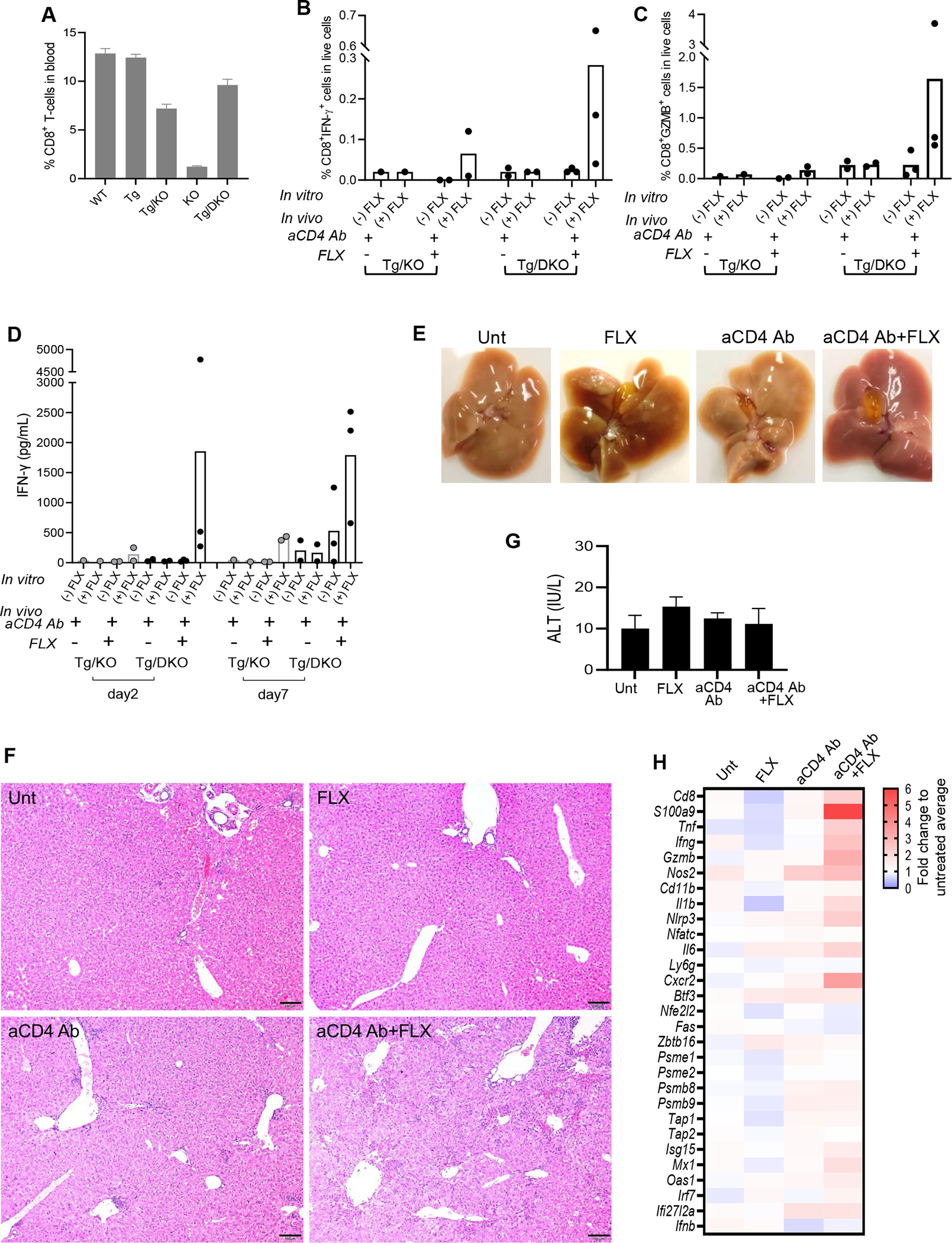
Enhanced drug reaction and liver histopathology in Tg/DKO mice after aCD4Ab + FLX treatment. (A) Flow cytometry analysis of CD8+ T cells in blood of drug-naive animals of different strains. (B-D) Tg/KO or Tg/DKO animals (n = 2–3 per group) were treated with aCD4Ab or aCD4Ab + FLX. Frequency of splenic IFN-γ+CD8+ (B) and GZMB+CD8+ (C) T cells was measured by flow cytometry analysis after day 10 of drug in vivo treatment and subsequent culture in the presence or absence of 250 μg/mL of FLX for 16 hours before adding brefeldin A for 4 hours. (D) IFN-γ levels in culture supernatant of total splenocytes restimulated with or without 250 μg/mL of FLX for 2 or 5 days. (E-H) Tg/DKO mice were treated as described in Methods. (E) Liver and gallbladder at euthanasia (1 representative animal). (F) H&E staining of fixed liver sections (scale bar 5 100 μm) (1 representative mouse per group). Additional sections showed more foci of inflammation in aCD4Ab–treated mice than untreated or FLX control groups, but inflammation and hepatocellular injury were most prominent in the aCD4Ab + FLX group (Fig E4). (G) Serum ALT at day 10 of treatment (n = 3 per group). (H) Gene expression analysis of perfused liver by real-time PCR (geomean of n = 3–12 mice per group).
The frequency of CD8+ T cells with an effector/effector memory phenotype (CD44+CD62L−) was increased in the spleen of Tg/DKO animals treated with aCD4Ab + FLX compared to control groups (Fig 6, A). CD44+CD62L−CD8+ T cells were also found elevated in the liver of aCD4Ab- and aCD4Ab + FLX–treated animals (Fig 6, B). The combination of aCD4Ab + FLX led to an increase of macrophages in spleen but not liver without affecting CD11b+ DC levels (Fig 6, C and D), mimicking our previous observations in Tg/KO. Similarly, the expression levels of CD86 were elevated on CD11b+ DC isolated from the spleen but not liver of aCD4Ab + FLX animals (Fig 6, E and F). Collectively these results show that although PD-1 plays a role in limiting the expansion of FLX-reactive cells in Tg/KO, PD-1 is not a critical factor in maintaining liver tolerance in vivo, as shown in Tg/DKO.
FIG 6.
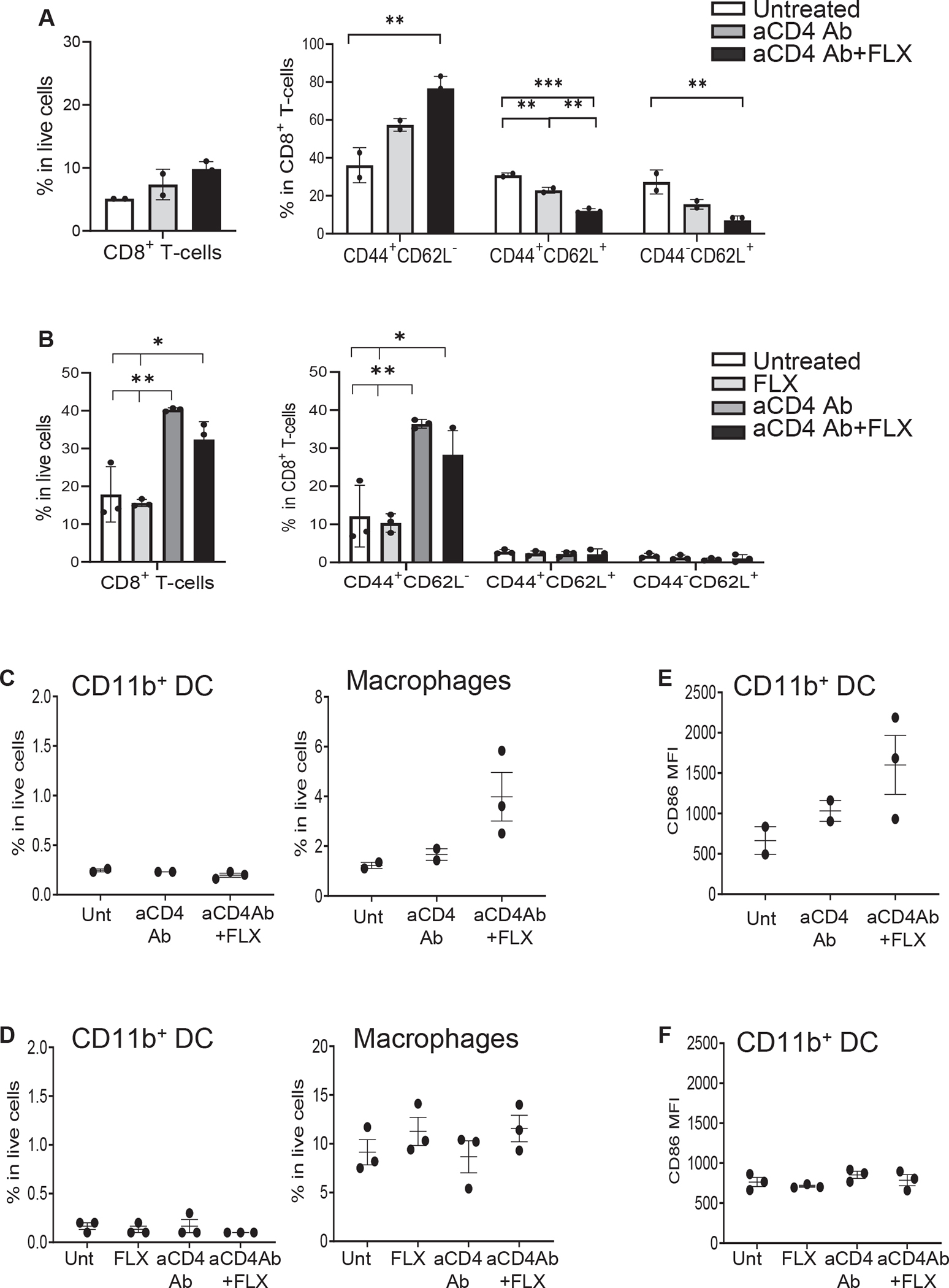
Leukocyte profiling in spleen and liver of Tg/DKO mice. Animals received FLX and/or aCD4Ab. Livers were perfused before cell isolation. (A and B) Flow cytometry analysis of CD8+ T-cell subsets obtained from spleen (A) or liver (B) of Tg/DKO mice. (C and D) Frequency of DC (CD11b+CD11c+CD8−) and macrophage (F4/80+CD11b+) in spleen (C) and liver (D). (E and F) MFI of CD86+CD11b+ DCs isolated from spleen (E) and liver (F) of the same animals in (C) and (D), respectively.
In vitro FLX-reactive CD8+ T cells hepatotoxicity is dependent on KCs and LSECs
To address whether FLX-reactive cells play a role in hepatotoxicity, we examined the cytotoxicity of purified leukocytes from perfused livers of aCD4Ab + FLX–treated mice on primary hepatocytes. Effector cells from both Tg/KO and Tg/DKO treated with aCD4Ab + FLX (EaCD4Ab+FLX) quickly killed hepatocyte targets of aCD4Ab + FLX–treated mice (TaCD4Ab+FLX) on restimulation with drug in vitro (Fig 7, A and B, left). The cytotoxicity of the EaCD4Ab+FLX cells was drug concentration dependent, and although its intensity varied among independent experiments, drug specificity was consistent compared to unstimulated cells in both Tg/DKO and Tg/KO. EaCD4Ab+FLX from Tg/DKO animals showed low levels of hepatotoxicity without restimulation. Unlike EaCD4Ab+FLX from Tg/KO, those from Tg/DKO were cytotoxic to hepatocytes of drug-naive mice (TUnt) showing enhanced effector function when soluble FLX was added to the cultures (Fig 7, A, right). The lack of HLA expression on hepatocytes impaired EaCD4Ab+FLX cytotoxicity (Fig 7, B, right).
FIG 7.
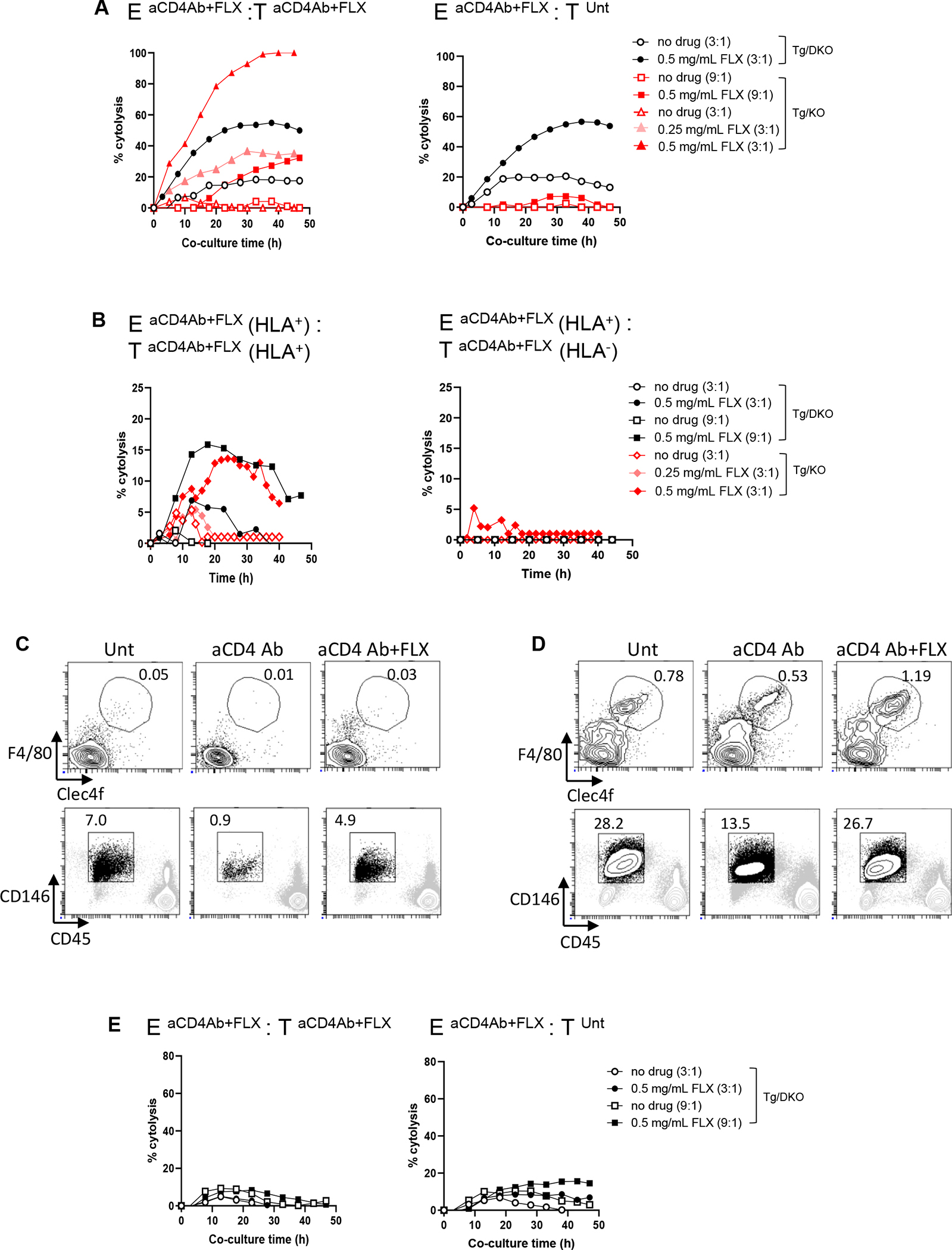
In vitro hepatocyte cytolysis by FLX-reactive leukocytes of mice treated with aCD4Ab + FLX is influenced by drug reexposure, HLA expression, and presence of liver tolerogenic cells. Tg/DKO and Tg/KO animals were treated with aCD4Ab + FLX or left untreated as per our 10-day standard protocol. (A and B) Primary hepatocytes were isolated from perfused livers and seeded in E-plates (target cells [T] at 20,000 cells per well) for 18 hours. Subsequently, liver leukocytes from aCD4Ab + FLX–treated mice (effector cells [E]) were added to cultures at an E:T ratio of 3:1 or 9:1 (E and T from same mouse strain). Superscript annotations correspond to in vivo treatment of animals from which E and T cells were isolated. Selected cocultures were exposed to FLX. Percentage cytolysis was measured as a function of impedance, as indicated in the Methods section in the Online Repository. In (B), T were isolated from HLA+ or HLA− mice from Tg/KO or Tg/DKO strains. E were from HLA+ animals. Different symbols correspond to different animals. (C and D) Frequency of KCs (F4/80+Clec4f+) and LSECs (CD146+CD45−) in cell suspensions of enriched liver leukocytes (C) or bulk NPC (D) from perfused livers of untreated or treated Tg/DKO animals. (E) Cytolysis was evaluated as indicated in (A) but with cocultures of hepatocytes and NPC from perfused livers of Tg/DKO mice.
Liver leukocyte samples did not include KCs (F4/80+Clec4f+) and LSECs (CD45−CD146+) (Fig 7, C), in contrast to suspensions of bulk NPC analyzed before submitting the samples to Percoll gradient separation (Fig 7, D). Bulk NPC from aCD4Ab + FLX–treated Tg/DKO animals, unlike liver leukocytes, were unable to kill hepatocyte targets (Fig 7, E).
These data, taken together, indicate that in vivo FLX-primed and expanded CD8+ T cells are not deleted in the liver and can kill hepatocytes from aCD4Ab + FLX–treated mice depending on the presence of KCs and LSECs. Lack of PD-1 expression predisposes some effector cells to be cytotoxic to drug-naive targets when drug is in solution. Additional studies are needed to understand the in vivo mechanisms responsible for the control exerted by KCs and LSECs on DILI onset.
DISCUSSION
A number of drugs have been described to interact with different HLA alleles and lead to hypersensitivity reactions.31 The development of HLA transgenic animal models is critical to elucidate the mechanisms driving immune-mediated drug hypersensitivity reactions (including FLX-DILI). Here, we characterize FLX immune reactivity in existing and novel HLA-B*57:01 Tg strains and discuss tolerogenic mechanisms that may contribute to restrict drug-responsive CD8+ T cells.
CD8+ T cells obtained from drug-naive Tg animals reacted to FLX after 2–3 cycles of in vitro stimulation demonstrating comparable kinetics to those reported in PBMC cultures from drug-naive healthy HLA-B*57:01+ human donors.14,17 Compared to ABC, murine cell activation with FLX was delayed and less robust, suggesting a more oligoclonal immune response by FLX. FLX primed T cells in vivo, as evidenced by rapid IFN-γ release to drug reexposure in vitro. This was analogous to PBMCs from FLX-treated patients with liver injury that respond to drug when restimulated in vitro, as opposed to cells from drug-tolerant or drug-naive subjects.15
As shown, splenic and LN CD8+ T cells from FLX-treated HLA-B*57:01− mice (WT) also produced IFN-γ in culture. This was not unexpected; FLX-reactive CD8+ T cells were previously reported from drug-treated mice not expressing the human risk allele18 and from PBMC of HLA-B*57:01− human donors prescribed FLX.15 Interestingly, however, cells from WT mice showed a delayed IFN-γ release of at least 48 hours compared to cells from Tg mice, which could be attributed to differences in the mechanisms by which class I molecules present the drug to T cells.32 While Tg T cells showed a rapid response to soluble drug, as observed in HLA-B*57:01+ PBMCs from healthy donors and DILI patients,14–16 the delayed response of WT T cells may rely on drug–haptenated peptide formation dependent on intracellular antigen processing, similar to what was reported for HLA-B*57:01− PBMCs.14 Differences in the IFN-γ levels between Tg and WT cells at day 5 of culture (Fig 2, A and B) may be related to an additive effect of drug presentation by HLA and mMHC-I in Tg mice.
To focus on exclusive presentation of FLX epitopes by HLA-B*57:01,we adopted the Tg/KO strain. The weaker drug response observed in Tg/KO mice compared to Tg mice could be attributed to fewer drug-specific CD8+ T cells due to lower peripheral CD8+ T-cell levels in H2-KbDbKO mice,26,33,34 and a diminished density of total class I molecules. However, studies showing that TCR-Vβ diversity as well as the rate of thymic generation/egress of CD8+ T cells are not affected by the lack of H2-KbDb 26,34 would suggest that the low numbers of circulating FLX-reactive CD8+ T cells in Tg/KO are due to an impairment in the cell maintenance rather than their generation. Factors limiting the expansion of FLX-reactive CD8+ T-cell response could be the absence of FLX-reactive WT (HLA−) T cells acting as helper cells and/or the presence of CD4+ Treg cells.
Treg cells play a key role in the maintenance of T-cell homeostasis, immune tolerance, and T-cell–mediated autoimmunity prevention.35–37 Depletion of CD4+ T cells, including Treg cells, as applied previously in other murine models of drug hypersensitivity18,20,22 led to mild inflammation that was insufficient to trigger hepatocyte injury as measured by ALT release or histopathology. This was in contrast to a nontransgenic mouse model of FLX in which ALTs were transiently elevated but drug presentation and T-cell activation by the murine MHC-I was not addressed.18 In the Tg/KO, treatment with aCD4Ab + FLX increased PD1+ CD8+ T cells both in peripheral lymphatic organs and liver, consistent with TCR engagement and T-cell priming by drug antigens. Lack of CD4+ non-Treg cells did not seem to compromise the generation of CD8+ T cells reactive to drug. Although splenic and LN DCs presenting drug epitopes and expressing costimulatory molecules could contribute to the activation of peripheral CD8+ T cells, liver-infiltrating CD8+ T cells encountered an environment of low costimulation and high immune suppression and were not responsive (Fig 4, D, and Fig E2). Drug-reactive cells activated in lymphoid organs may reach the liver sinusoids from the periphery and get arrested in the epithelium of the vessels or infiltrate the tissue if the organ is under stress or inflammation.28 There, CD8+ T cells will encounter resident cells such as KCs, LSECs, and hepatocytes, biased to enforcing tolerance by the secretion of anti-inflammatory cytokines and expression of inhibitory PD-L1 molecules.27,28,38,39 Additionally, CD8+ T cells will end up tolerized by LSECs presenting antigen in an MHC-I–restricted manner through PD-L1 but not CD80/86 upregulation.27 Thus, in the liver, the constitutive expression of PD-L1 on resident cells could account for the decreased effector function of infiltrating PD1+CD8+ T cells.
Abrogation of PD-1 expression in mice, together with CTLA-4 blocking, has been shown to sustain amodiaquine liver inflammation transiently for several weeks.40 To evaluate the role of PD-1 in FLX tolerance, we administered drug to aCD4Ab-treated Tg/DKO. Mice showed liver parenchymal alterations comparable to those described in the amodiaquine mouse model and in liver sections of patients with FLX hypersensitivity that are characterized by influx of leukocytic cells, especially CD8+ T cells and macrophages, with some hepatocyte damage, evidenced by the presence of apoptotic cells.4,11,40 Additionally, gallbladder enlargement by FLX indicated possible alterations in bile acid metabolism, although cholestasis was not supported by histology or liver enzyme elevations. The higher levels of effector/effector memory CD44+CD62L−CD8+ T cells (TEM) in Tg/DKO were attributed to lymphocyte dysregulation due to the lack of PD-1 molecules.41,42 Liver injury with ALT elevations was not evident despite increased splenic and hepatic TEM, indicating that liver-infiltrating leukocytes were still tolerized. Differences in liver damage between our animal model and what happens in FLX-DILI patients may be attributed to factors such as differences in drug bioavailability, antibiotic metabolism, and adduct formation leading to higher concentration of target drug neoantigens to stimulate T cells in humans; levels of preexisting drug-reactive T cells, perhaps from previous antibiotic priming; bacterial innate signaling from ongoing infection that activates antigen-presenting cell for T-cell priming; immune-inhibitor pathways such as checkpoint molecules and Treg cell function; and/or the duration of drug treatment.
Liver leukocytes isolated from aCD4Ab + FLX–treated Tg/DKO and Tg/KO animals showed hepatocyte killing in vitro when depleted from KCs and LSECs, confirming the coexistence of multiple tolerizing mechanisms exerted by these hepatic resident cells on FLX-reactive CD8+ T cells in vivo. Although hepatocytes can prime naive CD8+ T cells in vitro by acting as nonprofessional antigen-presenting cells,43 the rapid cytotoxic response of effector cells from aCD4Ab + FLX–primed animals to drug restimulation, as well as the lack of toxicity to target cells not expressing class I molecules, suggests drug-epitope presentation by hepatocytes to in vivo drug-primed effectors.
A limitation of drug hypersensitivity animal models is the absence of baseline disease, such as bacterial infection for which FLX is used to treat patients. Considering a role of danger signals in the development of immune-mediated adverse drug reactions,44 low inflammation due to the absence of pathogen-associated and danger-associated molecular patterns (aka PAMP and DAMP) could restrict the upregulation of costimulatory molecules on DC to enhance optimal drug-epitope presentation to the T cells and further activation. Although in vivo FLX-primed cells from both Tg/KO strains were able to quickly kill hepatocytes from drug-treated animals when restimulated in vitro, presumably through recognition of drug epitopes presented by target cells, only Tg/DKO effectors exerted cytotoxicity on drug-naïve targets. Early cytotoxicity in vitro may occur by TCR activation by noncovalently bound drug to HLA or TCR, which is labile and reversible and thus likely less stable than drug-haptenated peptides.11,45 Suboptimal TCR stimulation in the absence of costimulation or/and proinflammatory cytokines may lead to T-cell tolerance or anergy.46,47 Unlike Tg/KO, Tg/DKO had higher bystander liver inflammation associated with lack of PD-1 expression,41,48 which was enhanced by the depletion of CD4+ T cells. This, together with the propensity of PD-1 KO T cells to adopt a TEM phenotype in lymphopenic environments characterized by rapid activation on restimulation,42 could explain that CD44+CD62L− cells from aCD4Ab + FLX–treated Tg/DKO recognizing noncovalently bound drug complexes in a proinflammatory environment (mimicking, perhaps, that derived from PAMP and DAMP in infections) can quickly kill hepatocytes despite being suboptimally activated through the TCR.
Of note, our study does not directly address the mechanisms by which FLX causes DILI in non–HLA-B*57:01, which occurs in approximately 17% of FLX-DILI cases. However, we intentionally chose the drug and HLA pair with the highest chances to result in FLX-DILI to generate a preclinical model with consistent liver inflammation. Determining whether the immune mechanisms we report that lead to FLX-DILI are exclusive for HLA-B*57:01 or common to other HLAs will require follow-up studies.
In summary, several key elements appear necessary to induce FLX-specific CD8+ T-cell responses in mice even when these are only targeted to HLA-B*57:01, the allele associated with FLX-DILI. Depletion of CD4+ T cells, presumably Treg cells, is necessary for T-cell priming, activation, and influx into liver. Lack of Treg cells could lead to DC activation and enhanced expression of costimulatory surface molecules including CD80 and CD86 in spleen and LN antigen-presenting cells,20 but not in the liver. This suggests that part of the suppressive environment is acting to limit DC activation, possibly stabilizing these cells in a tolerizing state. The increase in PD-1+CD8+ T cells in the livers of aCD4Ab + FLX–treated Tg/KO suggests that these cells are recruited to the liver after peripheral activation in the lymphoid tissues and that PD-1 expression may limit T-cell function. Depletion of Treg cells may support T-cell responses to drug in the gut–lymphoid axis through activation of DCs and lack of IL-2 consumption. This most likely occurs during primary T-cell responses to FLX-modified liver self-antigens in draining LN and gut lymphoid tissue. T cells, once activated in a RA-rich environment that imprints gut–liver homing, can migrate back to the liver, where they are either tolerized or deleted. The recovery of cytotoxic activity from hepatic CD8+ T cells on primary hepatocytes in vitro demonstrates that T cells are not depleted but instead suppressed in the liver environment, which includes KC- and LSEC-mediated mechanisms. PD-1 engagement on T cells by PD-L1 on NPC and possibly other checkpoint molecules limit CD8+ T-cell–mediated activation and killing of hepatocytes, as suggested by the suppression of T-cell cytotoxicity by KCs and/or LSECs in a PD-1–deleted background.
We believe that the knowledge gained with murine strains that express only HLA-B*57:01 will be useful to unravel host cell immune response and tolerance mechanisms to other drugs with potential to cause HLA-mediated DILI. Ongoing studies of these models are focused on manipulating host pathways that control hepatic tolerance resulting in sustained FLX-DILI to elucidate the clinical effects exerted by drug-reactive T cells.
Supplementary Material
Key messages.
FLX presentation by HLA-B*57:01 in mice leads to CD8+ T-cell activation with hepatotoxic potential, which is restricted by the presence of liver tolerogenic cells in vivo.
CD4+ cells, presumably Treg cells, and PD-1 expression limit HLA-B*57:01–dependent immune responses to FLX.
HLA-B*57:01 transgenic mice are promising systems to understand DILI and tolerance.
Acknowledgments
Supported by the US Food and Drug Administration (FDA) Intramural Research Program, and in part by a Postgraduate Research Fellowship Award to K.K.S., S.S., and H.A. from the Oak Ridge Institute for Science and Education (ORISE) through an interagency agreement between the US Department of Energy and the FDA.
We thank Janet Woodcock, former director of the Center for Drug Evaluation and Research (CDER, FDA), for her support of this project. We also wish to thank the personnel of the Division of Veterinary Services (FDA) for care of the mice. We thank David Hone, Weiming Ouyang, and Joao Pedras Vasconcelos for critically reading drafts of the report.
Abbreviations used
- ABC
Abacavir
- aCD4Ab
Anti-CD4 antibody
- ALT
Alanine transaminase
- DC
Dendritic cell
- DILI
Drug-induced liver injury
- DMSO
Dimethyl sulfoxide
- FDA
US Food and Drug Administration
- FLX
Flucloxacillin
- GZMB
Granzyme B
- H&E
Hematoxylin and eosin
- HLA
Human leukocyte antigen
- KO
Knockout
- KC
Kupffer cell
- LN
Lymph node
- LSEC
Liver sinusoidal endothelial cell
- MFI
Mean fluorescence intensity
- MHC
Major histocompatibility complex
- mMHC
Murine MHC
- NPC
Nonparenchymal cell
- PBMC
Peripheral blood mononuclear cell
- PD-1
Programmed cell death 1
- PD-L1
Programmed cell death ligand 1
- RA
Retinoic acid
- TEM
Effector memory T
- Tg
HLA-B*57:01 transgenic mice
- Tg/DKO
HLA-B*57:01 transgenic/H2-Kb Db and PD-1 double-KO mice
- Tg/KO
HLA-B*57:01 transgenic/H2-Kb Db KO mice
- Treg
Regulatory T
- WT
Wild type
Footnotes
Disclosure of potential conflict of interest: The authors declare that they have no relevant conflicts of interest.
REFERENCES
- 1.Andrade RJ, Tulkens PM. Hepatic safety of antibiotics used in primary care. J Antimicrob Chemother 2011;66:1431–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goh SJR, Tuomisto JEE, Purcell AW, Mifsud NA, Illing PT. The complexity of T cell–mediated penicillin hypersensitivity reactions. Allergy 2021;76:150–67. [DOI] [PubMed] [Google Scholar]
- 3.Blumenthal KG, Peter JG, Trubiano JA, Phillips EJ. Antibiotic allergy. Lancet 2019;393:183–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olsson R, Wiholm BE, Sand C, Zettergren L, Hultcrantz R, Myrhed M. Liver damage from flucloxacillin, cloxacillin and dicloxacillin. J Hepatol 1992;15:154–61. [DOI] [PubMed] [Google Scholar]
- 5.Russmann S, Kaye JA, Jick SS, Jick H. Risk of cholestatic liver disease associated with flucloxacillin and flucloxacillin prescribing habits in the UK: cohort study using data from the UK General Practice Research Database. Br J Clin Pharmacol 2005;60:76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hussaini SH, O’Brien CS, Despott EJ, Dalton HR. Antibiotic therapy: a major cause of drug-induced jaundice in southwest England. Eur J Gastroenterol Hepatol 2007;19:15–20. [DOI] [PubMed] [Google Scholar]
- 7.Daly AK, Donaldson PT, Bhatnagar P, Shen Y, Pe’er I, Floratos A, et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet 2009;41:816–9. [DOI] [PubMed] [Google Scholar]
- 8.Nicoletti P, Aithal GP, Chamberlain TC, Coulthard S, Alshabeeb M, Grove JI, et al. Drug-induced liver injury due to flucloxacillin: relevance of multiple human leukocyte antigen alleles. Clin Pharmacol Ther 2019;106:245–53. [DOI] [PubMed] [Google Scholar]
- 9.Wing K, Bhaskaran K, Pealing L, Root A, Smeeth L, van Staa TP, et al. Quantification of the risk of liver injury associated with flucloxacillin: a UK population-based cohort study. J Antimicrob Chemother 2017;72:2636–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maria VA, Victorino RM. Diagnostic value of specific T cell reactivity to drugs in 95 cases of drug induced liver injury. Gut 1997;41:534–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wuillemin N, Terracciano L, Beltraminelli H, Schlapbach C, Fontana S, Krähenbühl S, et al. T cells infiltrate the liver and kill hepatocytes in HLA-B*57:01–associated floxacillin-induced liver injury. Am J Pathol 2014;184: 1677–82. [DOI] [PubMed] [Google Scholar]
- 12.Jenkins RE, Meng X, Elliott VL, Kitteringham NR, Pirmohamed M, Park BK. Characterisation of flucloxacillin and 5-hydroxymethyl flucloxacillin haptenated HSA in vitro and in vivo. Proteomics Clin Appl 2009;3:720–9. [DOI] [PubMed] [Google Scholar]
- 13.Pichler WJ, Adam J, Watkins S, Wuillemin N, Yun J, Yerly D. Drug hypersensitivity: how drugs stimulate T cells via pharmacological interaction with immune receptors. Int Arch Allergy Immunol 2015;168:13–24. [DOI] [PubMed] [Google Scholar]
- 14.Wuillemin N, Adam J, Fontana S, Krähenbühl S, Pichler WJ, Yerly D. HLA Haplotype determines hapten or p-i T cell reactivity to flucloxacillin. J Immunol 2013; 190:4956–64. [DOI] [PubMed] [Google Scholar]
- 15.Monshi MM, Faulkner L, Gibson A, Jenkins RE, Farrell J, Earnshaw CJ, et al. Human leukocyte antigen (HLA)-B*57:01–restricted activation of drug-specific T cells provides the immunological basis for flucloxacillin-induced liver injury. Hepatology 2013;57:727–39. [DOI] [PubMed] [Google Scholar]
- 16.Yaseen FS, Saide K, Kim SH, Monshi M, Tailor A, Wood S, et al. Promiscuous T-cell responses to drugs and drug-haptens. J Allergy Clin Immunol 2015;136: 474–6.e8. [DOI] [PubMed] [Google Scholar]
- 17.Faulkner L, Gibson A, Sullivan A, Tailor A, Usui T, Alfirevic A, et al. Detection of primary T cell responses to drugs and chemicals in HLA-typed volunteers: implications for the prediction of drug immunogenicity. Toxicol Sci 2016;154: 416–29. [DOI] [PubMed] [Google Scholar]
- 18.Nattrass R, Faulkner L, Vocanson M, Antoine DJ, Kipar A, Kenna G, et al. Activation of flucloxacillin-specific CD8+ T-cells with the potential to promote hepatocyte cytotoxicity in a mouse model. Toxicol Sci 2015;146:146–56. [DOI] [PubMed] [Google Scholar]
- 19.Takai S, Higuchi S, Yano A, Tsuneyama K, Fukami T, Nakajima M, et al. Involvement of immune- and inflammatory-related factors in flucloxacillin-induced liver injury in mice. J Appl Toxicol 2015;35:142–51. [DOI] [PubMed] [Google Scholar]
- 20.Cardone M, Garcia K, Tilahun ME, Boyd LF, Gebreyohannes S, Yano M, et al. A transgenic mouse model for HLA-B*57:01–linked abacavir drug tolerance and reactivity. J Clin Invest 2018;128:2819–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Susukida T, Aoki S, Kogo K, Fujimori S, Song B, Liu C, et al. Evaluation of immune-mediated idiosyncratic drug toxicity using chimeric HLA transgenic mice. Arch Toxicol 2018;92:1177–88. [DOI] [PubMed] [Google Scholar]
- 22.Susukida T, Kuwahara S, Song B, Kazaoka A, Aoki S, Ito K. Regulation of the immune tolerance system determines the susceptibility to HLA-mediated abacavir-induced skin toxicity. Commun Biol 2021;4:1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song B, Aoki S, Liu C, Susukida T, Ito K. An animal model of abacavir-induced HLA-mediated liver injury. Toxicol Sci 2018;162:713–23. [DOI] [PubMed] [Google Scholar]
- 24.Puig M, Ananthula S, Venna R, Kumar Polumuri S, Mattson E, Walker LM, et al. Alterations in the HLA-B*57:01 immunopeptidome by flucloxacillin and immunogenicity of drug-haptenated peptides. Front Immunol 2020;11:629399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao Y, Song B, Aoki S, Ito K. Conjugation of human serum albumin and flucloxacillin provokes specific immune response in HLA-B*57:01 transgenic mice. Immunol Lett 2022;249:5–11. [DOI] [PubMed] [Google Scholar]
- 26.Sng XYX, Li J, Zareie P, Assmus LM, Lee JKC, Jones CM, et al. The impact of MHC class I dose on development and maintenance of the polyclonal naive CD8+ T cell repertoire. J Immunol 2020;204:3108–16. [DOI] [PubMed] [Google Scholar]
- 27.Diehl L, Schurich A, Grochtmann R, Hegenbarth S, Chen L, Knolle PA. Tolerogenic maturation of liver sinusoidal endothelial cells promotes B7-homolog 1–dependent CD8+ T cell tolerance. Hepatology 2008;47:296–305. [DOI] [PubMed] [Google Scholar]
- 28.Heymann F, Peusquens J, Ludwig-Portugall I, Kohlhepp M, Ergen C, Niemietz P, et al. Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology 2015;62:279–91. [DOI] [PubMed] [Google Scholar]
- 29.Kosters A, Karpen SJ. The role of inflammation in cholestasis: clinical and basic aspects. Semin Liver Dis 2010;30:186–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Konishi T, Schuster RM, Goetzman HS, Caldwell CC, Lentsch AB. Cell-specific regulatory effects of CXCR2 on cholestatic liver injury. Am J Physiol Gastrointest Liver Physiol 2019;317:G773–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y, Deshpande P, Hertzman RJ, Palubinsky AM, Gibson A, Phillips EJ. Genomic risk factors driving immune-mediated delayed drug hypersensitivity reactions. Front Genet 2021;12:641905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.White KD, Chung WH, Hung SI, Mallal S, Phillips EJ. Evolving models of the immunopathogenesis of T cell–mediated drug allergy: the role of host, pathogens, and drug response. J Allergy Clin Immunol 2015;136:219–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pérarnau B, Saron MF, Reina San Martin B, Bervas N, Ong H, Soloski MJ, et al. Single H2Kb, H2Db and double H2KbDb knockout mice: peripheral CD8+ T cell repertoire and anti-lymphocytic choriomeningitis virus cytolytic responses. Eur J Immunol 1999;29:1243–52. [DOI] [PubMed] [Google Scholar]
- 34.Ureta-Vidal A, Firat H, Pérarnau B, Lemonnier FA. Phenotypical and functional characterization of the CD8+ T cell repertoire of HLA-A2.1 transgenic, H-2KbnullDbnull double knockout mice. J Immunol 1999;163:2555–60. [PubMed] [Google Scholar]
- 35.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell 2008;133:775–87. [DOI] [PubMed] [Google Scholar]
- 36.Kalia V, Penny Laura A, Yuzefpolskiy Y, Baumann Florian M, Sarkar S. Quiescence of memory CD8+ T cells is mediated by regulatory t cells through inhibitory receptor CTLA-4. Immunity 2015;42:1116–29. [DOI] [PubMed] [Google Scholar]
- 37.Poitrasson-Riviére M, Bienvenu B, Le Campion A, Bécourt C, Martin B, Lucas B. Regulatory CD4+ T cells are crucial for preventing CD8+ T cell–mediated autoimmunity. J Immunol 2008;180:7294–304. [DOI] [PubMed] [Google Scholar]
- 38.Iwai Y, Terawaki S, Ikegawa M, Okazaki T, Honjo T. PD-1 inhibits antiviral immunity at the effector phase in the liver. J Exp Med 2003;198:39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benseler V, Warren A, Vo M, Holz LE, Tay SS, Le Couteur DG, et al. Hepatocyte entry leads to degradation of autoreactive CD8 T cells. Proc Natl Acad Sci U S A 2011;108:16735–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Metushi IG, Hayes MA, Uetrecht J. Treatment of PD-1−/− mice with amodiaquine and anti-CTLA4 leads to liver injury similar to idiosyncratic liver injury in patients. Hepatology 2015;61:1332–42. [DOI] [PubMed] [Google Scholar]
- 41.Keir ME, Freeman GJ, Sharpe AH. PD-1 regulates self-reactive CD8+ T cell responses to antigen in lymph nodes and tissues. J Immunol 2007;179:5064–70. [DOI] [PubMed] [Google Scholar]
- 42.Charlton JJ, Chatzidakis I, Tsoukatou D, Boumpas DT, Garinis GA, Mamalaki C. Programmed death-1 shapes memory phenotype CD8 T cell subsets in a cell-intrinsic manner. J Immunol 2013;190:6104. [DOI] [PubMed] [Google Scholar]
- 43.Wahl C, Bochtler P, Chen L, Schirmbeck R, Reimann J. B7-H1 on hepatocytes facilitates priming of specific CD8 T cells but limits the specific recall of primed responses. Gastroenterology 2008;135:980–8. [DOI] [PubMed] [Google Scholar]
- 44.Séguin B, Uetrecht J. The danger hypothesis applied to idiosyncratic drug reactions. Curr Opin Allergy Clin Immunol 2003;3:235–42. [DOI] [PubMed] [Google Scholar]
- 45.Pichler WJ, Watkins S, Yerly D. Risk assessment in drug hypersensitivity: detecting small molecules which outsmart the immune system. Front Allergy 2022;3:827893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chikuma S, Terawaki S, Hayashi T, Nabeshima R, Yoshida T, Shibayama S, et al. PD-1–mediated suppression of IL-2 production induces CD8+ T cell anergy in vivo. J Immunol 2009;182:6682. [DOI] [PubMed] [Google Scholar]
- 47.Schietinger A, Greenberg PD. Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol 2014;35:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Affolter T, Llewellyn HP, Bartlett DW, Zong Q, Xia S, Torti V, et al. Inhibition of immune checkpoints PD-1, CTLA-4, and IDO1 coordinately induces immune-mediated liver injury in mice. PLoS One 2019;14:e0217276. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.