

Abstract
Background & Aims:
excessive deposition and crosslinking of extracellular matrix (ECM) increases liver density and stiffness, promotes fibrogenesis and increases resistance to fibrolysis. An emerging therapeutic opportunity in liver fibrosis is to target the composition of the ECM or block pathogenic communication with surrounding cells. However, the type and extent of extracellular changes triggering liver fibrosis depends on the underlying etiology. Our aim was to unveil matrisome genes not dependent on etiology that are clinically relevant to liver fibrosis.
Approach & Results:
we used transcriptomic profiles from liver fibrosis cases of different etiologies, to identify and validate liver fibrosis-specific matrisome genes (LFMGs), and their clinical and biological relevance. Dysregulation patterns and cellular landscapes of LFMGs were further explored in mouse models of liver fibrosis progression and regression by bulk and single-cell RNA sequencing (scRNA-seq). We identified 35 LFMGs, independent of etiology, representing a LFMG signature defining liver fibrosis. Expression of the LFMG signature depended on histological severity and was reduced in regressive livers. Patients with liver fibrosis, even with identical pathological scores, could be subclassified into LFMGLow and LFMGHigh with distinguishable clinical, cellular and molecular features. scRNA-seq revealed that microfibrillar associated protein 4+ (MFAP4+) activated hepatic stellate cells (aHSCs) increased in LFMGHigh patients and were primarily responsible for the LFMG signature expression and dysregulation.
Conclusions:
the MFAP4+ aHSC-derived LFMG signature classifies liver fibrosis patients with distinct clinical and biological characteristics. Our findings unveil hidden information from liver biopsies undetectable using traditionally histologic assessments.
Keywords: extracellular matrix, liver stiffness, microenvironment, molecular subclassification, nonparenchymal cells
GRAPHICAL ABSTRACT
Patients with pre- or post-treatment liver fibrosis, or even with identical histological fibrosis stages, can be subclassified into LFMGLow and LFMGHigh based on the MFAP4+ aHSC LFMG signature. LFMGLow and LFMGHigh patients were characterized with different pathological features, liver stiffness, and intrahepatic molecular and cellular heterogeneity. Our findings unveil hidden information from liver biopsies undetectable using traditionally histologic assessments. This provides additional insight for precise liver fibrosis grading, and the molecular underpinnings of liver fibrosis to define clinical modalities and develop therapeutic opportunities.
INTRODUCTION
Liver fibrosis is a sustained wound-healing response that elicits excessive deposition of extracellular matrix (ECM), and results in cirrhosis and hepatocellular carcinoma (HCC). Common causes of liver fibrosis include metabolic disorders, chronic viral infection, alcohol abuse and autoimmune disease. Recent studies support that liver fibrosis, or even end-stage cirrhosis, are reversible upon suppression or elimination of the underlying disease (1–3). However, removing the causative factor is not always sufficient to ameliorate fibrosis. For instance, ~47% of patients with successful suppression of hepatitis B virus (HBV) do not show significant fibrosis reversal (3), and only ~45% of non-alcoholic steatohepatitis (NASH) patients achieve noticeable fibrolysis after lifestyle modification and weight reduction (2). Further, spontaneous fibrosis reversal upon removal of the etiologic factors is often too slow to prevent life-threatening complications such as decompensated cirrhosis or HCC. Hence, we urgently need effective therapeutic drugs, specifically targeting liver fibrosis, independent of pathogenic factors.
Despite pharmacological approaches to re-quiesce activated hepatic stellate cells (aHSCs) and other profibrogenic effector cells (4, 5), there are limited clinical breakthroughs to date. Alternatively, current efforts gear towards enhancing ECM degradation or delaying deposition to reverse or delay fibrosis (6). The existing definition of ECM includes fibrillar proteins (collagens, glycoproteins, proteoglycans), ECM-affiliated proteins, ECM regulators and secreted factors latent in the ECM. These proteins are collectively known as the “matrisome” (7). During liver fibrogenesis, ECM remodeling occurs in the matrisome components, their covalent intra- and inter-molecular crosslinking, and through shifting the ECM mechanical and chemical microenvironment (8). Aggregation and stabilization of certain ECM proteins (e.g., elastin) enhance density and stiffness in the fibrotic liver, therefore resisting fibrolysis and altering liver homeostasis (9, 10). In addition, stiff ECM signals transmit mechanical forces via ECM receptors, mainly integrins, to recruit and activate profibrogenic effector cells (11, 12). Direct ECM intervention or blockade of communication within the ECM and its surrounding cells, creates a therapeutic opportunity in liver fibrosis (13).
The liver matrisome changes dynamically in response to acute and chronic insults, even preceding histological ECM accumulation. Various stimuli such as viral entry (mainly HBV and hepatitis C virus [HCV]) (14, 15) and alcohol abuse (16), result in transitional ECM remodeling in patients, which do not overtly alter the liver architecture but contribute to injury, inflammation and fibrogenesis. Thus, it is vital to identify matrisome proteins not affected by etiology, to establish the fibrotic ECM gene/protein signature, and develop fibrosis-specific targets. While decellularization increases the efficiency in detecting matrisome proteins, it still leaves trace cytoplasmic and soluble proteins within the matrix, limiting the full view of the fibrotic ECM landscape (17). In addition, directly targeting ECM proteins with monoclonal antibodies fails to show benefit, possibly because a stiff matrix acts as a pseudo-barrier that limits drug delivery (18). Instead, matrisome gene-targeted interventions create an opportunity to eventually remodel the ECM in liver fibrosis to its physiological state.
It is well known that molecular aberrations in tumors greatly limit HCC therapy and prognosis (19). However, whether intrahepatic molecular dysregulation challenges nonmalignant liver fibrosis remains unknown. Our recent study identified matrisome genes associated with HBV-related HCC that classify patients with different clinical prognosis, metabolic activity, cell cycle activation, immune infiltration and tumor purity (20). Here, we sought to identify matrisome genes specifically dysregulated during liver fibrogenesis, regardless of etiology, to subclassify patients with distinct clinical relevance and biological features. We systematically integrated analyses of transcriptomic data from liver fibrosis patients pre- and post-antiviral treatment in our center and from publicly available datasets. Dysregulation patterns, cellular landscapes and biological functions of the matrisome genes of interest were further validated in fibrotic or regressive livers by both bulk RNA sequencing (RNA-seq) and single-cell RNA sequencing (scRNA-seq).
MATERIALS AND METHODS
Patients
Fifty-four formalin-fixed paraffin-embedded (FFPE) liver biopsies from 28 patients were retrospectively obtained from our prospective HBV-related fibrosis/cirrhosis cohort studies (NCT01938781, NCT01938820), which was named the BJFSH cohort. Bulk RNA-seq and data preprocessing of human FFPE liver tissues from the BJFSH cohort were carried out by Shanghai NextCODE Co., Ltd (Shanghai, China) as previously reported (21). All patients in the BJFSH cohort provided written informed consent, and the study was approved by the Ethics Committee of Beijing Friendship Hospital, Capital Medical University (2016-P2-021-04), and conducted in accordance with the Declaration of Helsinki. In addition, publicly available gene expression profiles of human liver fibrosis cases of different etiologies (HBV, HCV, alcohol-associated liver disease (ALD) and NASH) were retrieved from Gene Expression Omnibus (GEO). A total of 892 fibrotic or non-fibrotic liver samples from GEO were included in the study. Detailed information of patients from the BJFSH cohort and GEO are provided in Tables S1–5.
Molecular subclassification based on the liver fibrosis-specific matrisome gene (LFMG) signature
Five public transcriptomic profiles of liver fibrosis (GSE130970, GSE49541, GSE84044, GSE103085, GSE14323) were retrieved to determine differentially expressed matrisome genes (DEMGs) between non-fibrotic (mild) and fibrotic (advanced) livers. The LFMG signature was defined as DEMGs commonly identified in at least 4 out of 5 datasets with different etiologies, that could distinguish non-fibrotic (mild) and fibrotic (advanced) livers under multivariate clustering. Based on the LFMG signature expression, unsupervised hierarchical clustering (HCL) analysis, with the average linkage method and “euclidean” or “canberra” as a distance metric, was performed to subclassify liver fibrosis patients. Patients with the LFMG signature uniformly exhibiting relatively low or high expression level, separated by the root dendrogram in the column of the HCL heatmap, were defined as “LFMGLow” and “LFMGHigh”, respectively (Figure S1). The clinical relevance and molecular features between LFMGLow and LFMGHigh, were systematically analyzed and compared in patients with or without identical fibrosis stages from the BJFSH cohort, and further validated in patients from public datasets.
Mice
The mouse model of carbon tetrachloride (CCl4)-induced liver fibrosis progression and resolution mouse was established as previously (9). Bulk RNA-seq of mouse frozen livers was performed by Biomarker Technologies Co., Ltd (Beijing, China), and scRNA-seq was performed at the University of Illinois at Urbana-Champaign DNA Sequencing Laboratory. Studies were approved by the Ethics Committee of Beijing Friendship Hospital, Capital Medical University, and the IACUC office in University of Illinois at Chicago, and were carried out according to the ARRIVE guidelines.
Data availability statement
Raw data from bulk RNA-seq of human FFPE liver biopsies was deposited in the National Genomics Data Center database (22) and are publicly available (accession number PRJCA010948). Raw and processed data from bulk RNA-seq of frozen mouse liver tissues and scRNA-seq of mouse primary liver cells were deposited in NCBI’s GEO database under accession number GSE199392 and “pending”, respectively. All processed gene expression profiles available from GEO database and R codes for scRNA-seq and BayesPrism deconvolution (23) analyses were deposited in the figshare platform (DOI: 10.6084/m9.figshare.22002707).
Statistical analysis
Unpaired student’s t test or Mann–Whitney U test were used to compare continuous variables between independent groups. Paired student’s t test or Wilcoxon matched-pairs signed rank test were used to compare continuous variables between paired groups. Comparisons among more than two groups were performed using one-way ANOVA followed by Tukey’s multiple comparisons test or Kruskal-Wallis test. Fisher’s exact or Chi-square trend test was used to compare categorical variables between any two groups. A p<0.05 was considered statistically significant. R 3.6.3, GraphPad Prism 7 (GraphPad Software Inc., CA, USA), and SangerBox tool (http://sangerbox.com/) were used for statistical and bioinformatic analyses.
Details for additional experiments and analyses are included in the Supporting document.
RESULTS
A matrisome gene-based risk signature characterizes liver fibrosis regardless of etiology
To identify LFMGs independent of etiology, liver fibrosis-related transcriptomic profiles (GSE84044, GSE14323, GSE49541, GSE103580, GSE130970) were retrieved as derivation transcriptomic datasets (Table S1). In total, 33 upregulated and 2 downregulated LFMGs were identified (Figure 1A–B and Table S6). Their dysregulation pattern during liver fibrogenesis was almost fully conserved between humans and mice (Figure S2). These LFMGs were highly interconnected with 161 potential interactions (Figure 1C), suggesting a potential role in liver fibrosis. Specifically, LFMGs encoding proteoglycans, ECM glycoproteins and ECM regulators, were predicted to directly interact with LFMGs encoding structural collagens (Figure 1C). Subsequent functional enrichment analysis revealed that the LFMG regulatory network was involved in fibrogenesis-, proliferation- or immune-related signaling pathways (Figure 1D).
Figure 1. Identification of the LFMG signature and its ability to determine liver fibrosis independent of etiology.
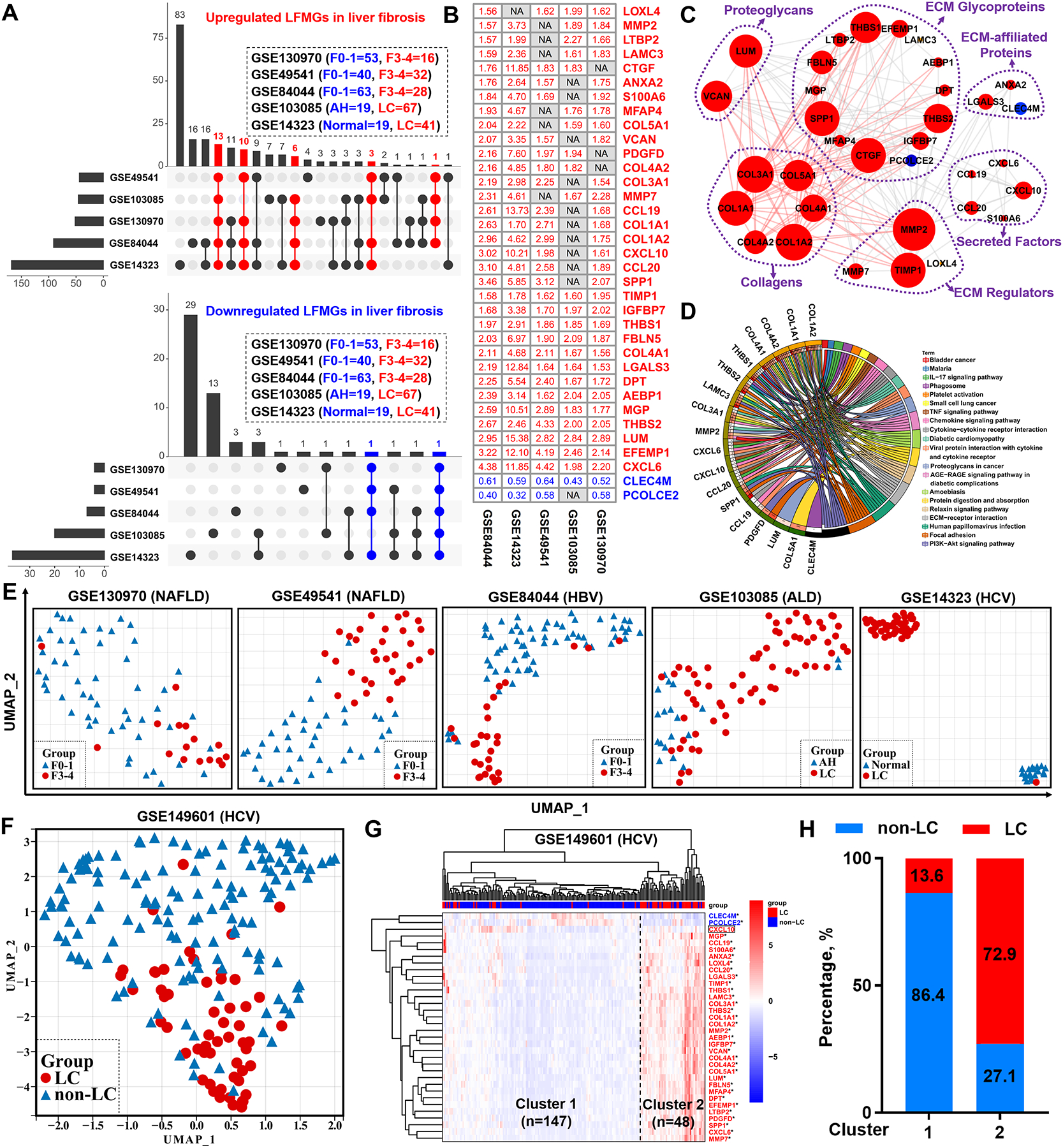
(A) UpSet diagrams of upregulated (red) and downregulated (blue) matrisome genes during liver fibrogenesis. Vertical and horizontal axes represent the number and distribution of DEMGs in the derivation datasets (GSE130970, GSE49541, GSE84044, GSE103085, GSE14323). (B) Fold-change value of the LFMG signature expression between non-fibrotic (mild) and fibrotic (advanced) samples from each derivation dataset (NA: undetectable, red: upregulated, blue: downregulated). (C) LFMG regulatory network. Node represents LFMG. Edge between two nodes represents potential interaction. Edges connecting collagen genes and their first neighbors are highlighted in red. Red indicates upregulated and blue downregulated. (D) Circos plot of the relationships between LFMGs (left) and their enriched KEGG pathways (right). Color-coded ribbons link LFMGs and their corresponding KEGG pathways. Statistical p values of the functional enrichment analysis are color-coded (left outer circle). Darker orange indicates higher significance and darker green lower significance. (E) UMAP plots of all non-fibrotic (mild) and fibrotic (advanced) patients from the derivation datasets. Grouped patients are color- and shape-coded. (F) UMAP plot and (G) unsupervised HCL clustering of all non-cirrhotic and cirrhotic patients from the test dataset (GSE149601). Grouped patients in UMAP plot are color- and shape-coded. In the heatmap, darker blue indicates lower expression and darker red higher expression. LFMG not significantly expressed between two groups of patients is enclosed in a rectangle. *p<0.05 and fold-change >1.5. (H) Proportions of non-cirrhotic (blue) and cirrhotic (red) patients from GSE149601 dataset in Cluster 1 or 2, grouped by HCL analysis. AH, alcoholic hepatitis; ALD, alcohol-associated liver disease; LC, liver cirrhosis; non-LC, non-liver cirrhosis.
Next, we tested the ability of the LFMGs, as a combined signature (the LFMG signature), to discriminate patients. When dimensionality reduction was performed, the LFMG signature could clearly classify patients from all derivation datasets into two distinct clusters, respectively (Figure 1E). Diagnostic robustness of the LFMG signature was further validated in an independent liver fibrosis transcriptomic dataset (GSE149601), using uniform manifold approximation and projection (UMAP) and HCL clustering (Figure 1F–G). The diagnostic accuracy of non-cirrhotic and cirrhotic patients in GSE149601, upon HCL clustering based on the LFMG signature, was 86.4% and 72.9%, respectively (Figure 1H). Of note, the LFMG signature could even identify liver fibrosis when samples with different etiologies, from microarray or RNA-seq platforms, were merged after normalization to the average expression level of each LFMG within one study (Figure S3). As mentioned above, acute or chronic liver injury elicit transitional responses in some ECM genes (14–16), however the expression pattern of LFMGs in patients with different etiologies (GSE84044, GSE130970, GSE48452, GSE103580) was almost unaffected by acute or chronic insults preceding fibrogenesis (Figure S4).
Taken together, we identified a total of 35 LFMGs independent of etiology, whose expression patterns are conserved across species during liver fibrogenesis, which can be used as a LFMG signature characterizing liver fibrosis with robust diagnostic accuracy.
The LFMG signature expression depends on histological severity and is reduced in regressive liver fibrosis
To determine if the expression of the LFMG signature changed gradually during progression of liver fibrosis, soft clustering analysis of LFMGs was performed in the GSE84044 and GSE130970 datasets. As shown in Figure 2A, LFMGs were gradually dysregulated with fibrosis stage, highlighting that the LFMG signature depends on histopathological severity of liver fibrosis. We next asked whether the LFMG signature was involved in regression of liver fibrosis. First, mouse models of fibrosis regression were analyzed. The LFMG signature was reduced during early spontaneous fibrosis resolution in mice (Figure S2B). Second, HBV fibrotic patients from the BJFSH cohort biopsied at baseline and at 78 and 260 weeks of antiviral treatment were analyzed (Table S4–5). Patients were diagnosed as “regressive” or “non-regressive” after treatment based on the pathological scoring criteria shown in Figure 2B–C. Bulk RNA-seq of FFPE liver biopsies showed that after 78 weeks of antiviral treatment, expression of LFMGs significantly decreased in regressive patients but there was no change in non-regressive patients; after 260 weeks of treatment, expression of LFMGs was significantly reduced in both groups of patients, although the pathological diagnosis of non-regressive patients did not improve; by contrast, regressive patients showed faster and greater extent of recovery than non-regressive patients (Figure 2D–E). Collectively, expression of the LFMG signature depends on the histological severity of fibrosis and decreases in liver fibrosis regression.
Figure 2. Expression pattern of the LFMG signature in liver fibrosis regressive or non-regressive patients from the BJFSH cohort during continuous antiviral therapy.
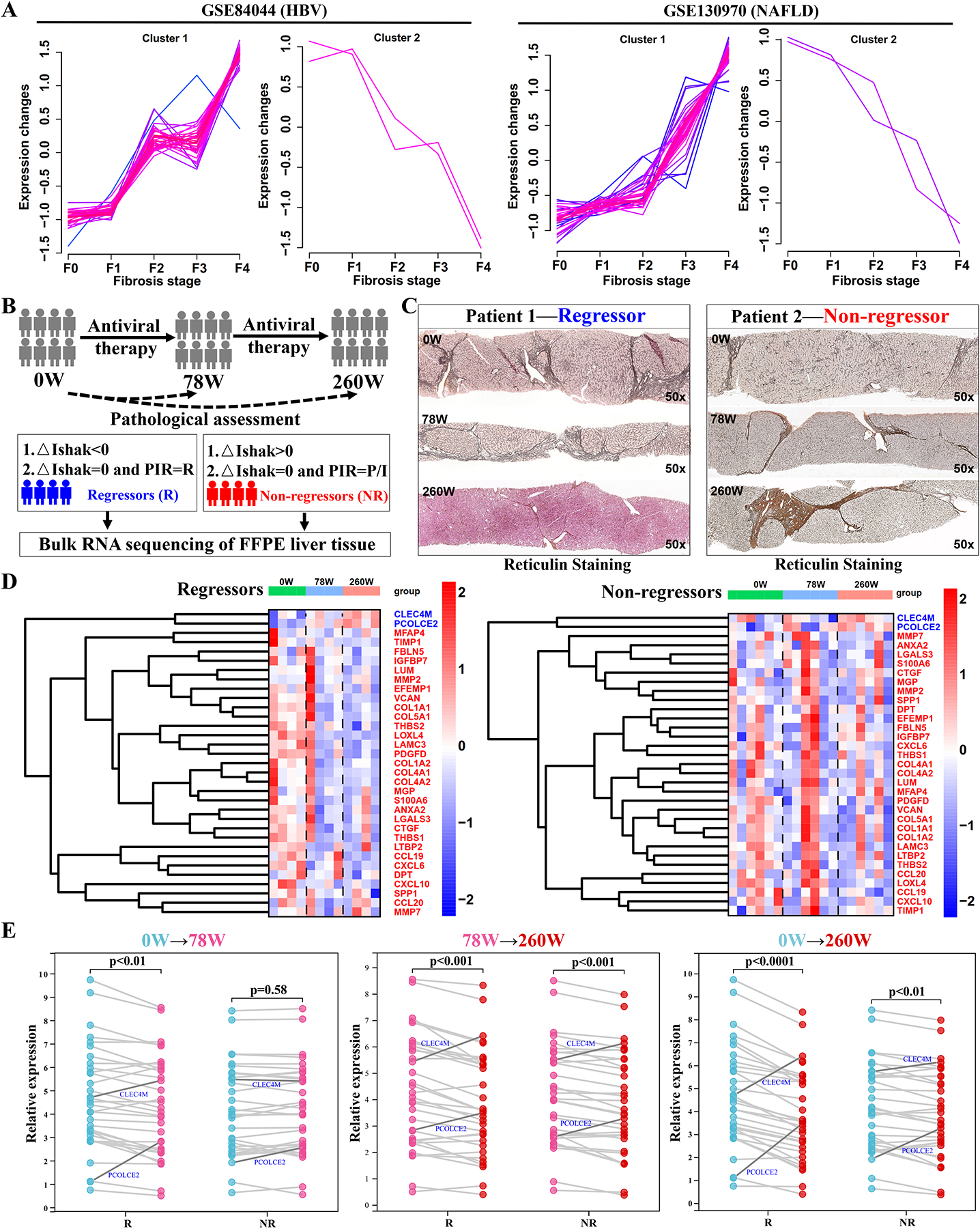
(A) Soft clustering of LFMGs expression during liver fibrosis progression in GSE84044 and GSE130970 datasets. Color code represents membership values consistency of expression profiles within a given cluster. Pink lines correspond to genes with higher membership value. Blue lines correspond to genes with lower membership value. (B) Schematic diagram of definitions of liver fibrosis regression and non-regression. Liver fibrosis patients who received no treatment, 78 or 260 weeks of antiviral treatment from the BJFSH cohort are labeled as 0W, 78W or 260W, respectively. △Ishak=Ishakpost-treatment-Ishakpre-treatment. (C) Dynamic histopathological variation of liver biopsies by reticulin staining from regressive (Patient 1) and non-regressive (Patient 2) liver fibrosis patients, with three liver biopsies from treatment-naïve, 78 and 260 weeks of antiviral treatment (magnification: 50x). (D) Heatmap of the dynamic LFMG signature expression in regressive and non-regressive liver fibrosis patients biopsied three times. Expression is scaled as a distribution with mean=0 and SD=1. Darker red indicates higher expression; darker blue indicates lower expression. COL3A1 and AEBP1 were undetectable in liver samples from the BJFSH cohort. (E) Comparison of LFMGs expression between paired liver biopsies from regressive and non-regressive liver fibrosis patients. Expression levels were averaged among patients and log2-transformed. Patients with no treatment, 78 and 260 weeks of antiviral therapy are color-coded. LFMGs downregulated in liver fibrogenesis but upregulated in regressive liver (PCOLCE2 and CLEC4M) are highlighted in blue. NR, non-regressor; R, regressor.
The LFMG signature molecularly subclassifies liver fibrosis patients with or without identical fibrosis stage
Given that members of the LFMG signature exhibited consistent expression pattern during liver fibrogenesis, we speculated that the signature could also subclassify liver fibrosis patients. As expected, unsupervised HCL based on the LFMG signature, identified two distinct subgroups (LFMGLow and LFMGHigh) in treatment-naïve patients and in patients after 78-week or 260-week antiviral treatment from the BJFSH cohort (Figure 3A). The subclassification potential of the LFMG signature was further tested and replicated in additional publicly available datasets (GSE84044, GSE130970, GSE193080, GSE193066) (Figure S5–6). By contrast, the LFMG signature subclassified patients better than the histological fibrosis stage (Figure 3B and S5–6). Of note, even patients with equal fibrosis stage from the BJFSH cohort could be subclassified based on the LFMG signature (Figure S7). We next broadened the LFMG signature subclassification to patients with identical fibrosis stage from publicly available datasets and confirmed the findings from the BJFSH cohort (Figure S8–12). Accordingly, the LFMG signature showed potential to subclassify liver fibrosis patients, and even patients with identical fibrosis stage.
Figure 3. Subclassification of liver fibrosis patients from the BJFSH cohort and comparison of histological severity and liver stiffness.
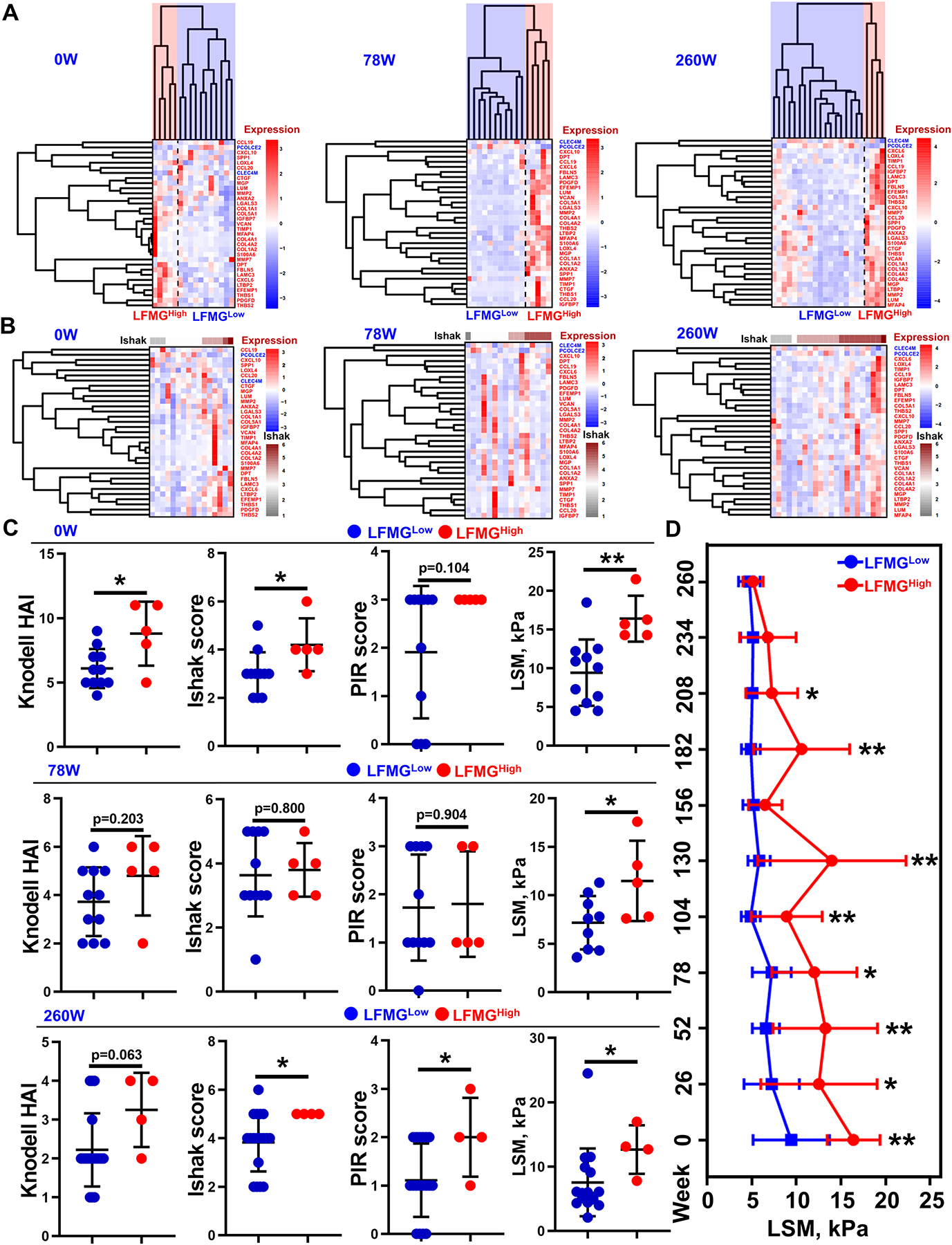
Heatmaps of subgroups of liver fibrosis patients treatment-naïve or on antiviral treatment for 78 or 260 weeks from the BJFSH cohort, based on (A) the LFMG signature expression upon HCL clustering or (B) histological fibrosis stage. Expression is scaled as a distribution with mean=0 and SD=1. The darker the blue, the lower the expression; the darker the red, the higher the expression. Higher Ishak score is coded as dark red and the lower Ishak score is coded as grey. (C) Comparisons of Knodell HAI, Ishak score, PIR score and liver stiffness measurement (LSM) values between subgroups of patients pre- or post-treatment. *p<0.05 and **p<0.01 vs LFMGLow patients. (D) Time-course changes of LSM value between LFMGLow and LFMGHigh patients from the BJFSH cohort at baseline. LSM values were measured every 26 weeks up to 260 weeks of antiviral treatment. LSM values were systematically compared between LFMGLow and LFMGHigh patients at each time point by unpaired Student’s t test or Mann–Whitney U test. *p<0.05 and **p<0.01. HAI, histological activity index; LSM, liver stiffness measurement; PIR, progressive, indeterminate and predominately regressive.
Subgroups of liver fibrosis patients identified by the LFMG signature exhibit different clinical, cellular and molecular features.
To further understand the LFMG signature-based subclassification, we evaluated clinical and biological differences between LFMGLow and LFMGHigh patients. First, serum biochemical, histologic indexes and liver stiffness were systematically compared. In the BJFSH cohort, serum alkaline phosphatase activity was higher in LFMGHigh patients at baseline (p<0.05), and serum alanine transaminase or gamma-glutamyl transpeptidase activities (p<0.05) were higher in LFMGHigh patients after 260 weeks of antiviral treatment, compared to their corresponding LFMGLow patients, while the remainder serum biochemical parameters remained unchanged between treatment-naïve or post-treatment patients (Table S7–9). Histologic inflammation activity or NASH indexes were elevated in LFMGHigh patients from the BJFSH cohort at baseline, after 260 weeks of antiviral treatment and in publicly available datasets (GSE84044, GSE130970, GSE193066) (Figure 3C and S4–5). In patients with equal fibrosis stage from the BJFSH cohort and publicly available datasets, inflammation activity and NASH indexes were consistently comparable between both subgroups (Figure S7–12). Importantly, LFMGHigh patients from the BJFSH cohort at baseline, showed significantly increased fibrosis stage over LFMGLow patients (Figure 3C), which was further confirmed in publicly available datasets (GSE84044, GSE130970, GSE193080, GSE193066) (Figure S5–6). In agreement with the histological fibrosis stage, LFMGHigh patients from the BJFSH cohort, at baseline and post-treatment, exhibited increased liver stiffness over LFMGLow patients (Figure 3C). More importantly, differences in liver stiffness between subgroups at baseline did not disappear until after the 260-week antiviral treatment (Figure 3D).
Second, liver infiltrating immune cells and fibroblasts, between subgroups of liver fibrosis patients from the BJFSH cohort and publicly available datasets, were analyzed and compared by the MCPcounter algorithm (24). B cells, NK cells, CD8 T cells, T cells, cytotoxic lymphocytes, myeloid dendritic cells and endothelial cells, increased in LFMGHigh compared to LFMGLow patients, with or without similar fibrosis stage (p<0.05 for meta-analysis), although differences were not consistent among studies (Figure 4A and S13–14). However, increased fibroblasts or decreased neutrophils were found in LFMGHigh compared to LFMGLow patients, with varying fibrosis stages, in all datasets (Figure 4A–B and S13). Even in patients with identical fibrosis stage, LFMGHigh patients had higher abundance of fibroblasts than LFMGLow patients, which occurred in all studies (Figure S14). Next, we analyzed HSC activation markers. ACTA2 mRNA was significantly upregulated in LFMGHigh compared to LFMGLow patients, with or without equal fibrosis stage, from the BJFSH cohort and all publicly available datasets (Figure 4C and S15). Although patients from the two subgroups shared comparable histopathological fibrosis score, ACTA2 immunostaining revealed that LFMGHigh patients from the BJFSH cohort, pre- or post-treatment, had more activated HSCs in the fibrous septal region than LFMGLow patients (Figure 4D).
Figure 4. Comparison of liver-infiltrating immune cells, fibroblasts and HSC activity between LFMGLow and LFMGHigh patients from the BJFSH cohort.
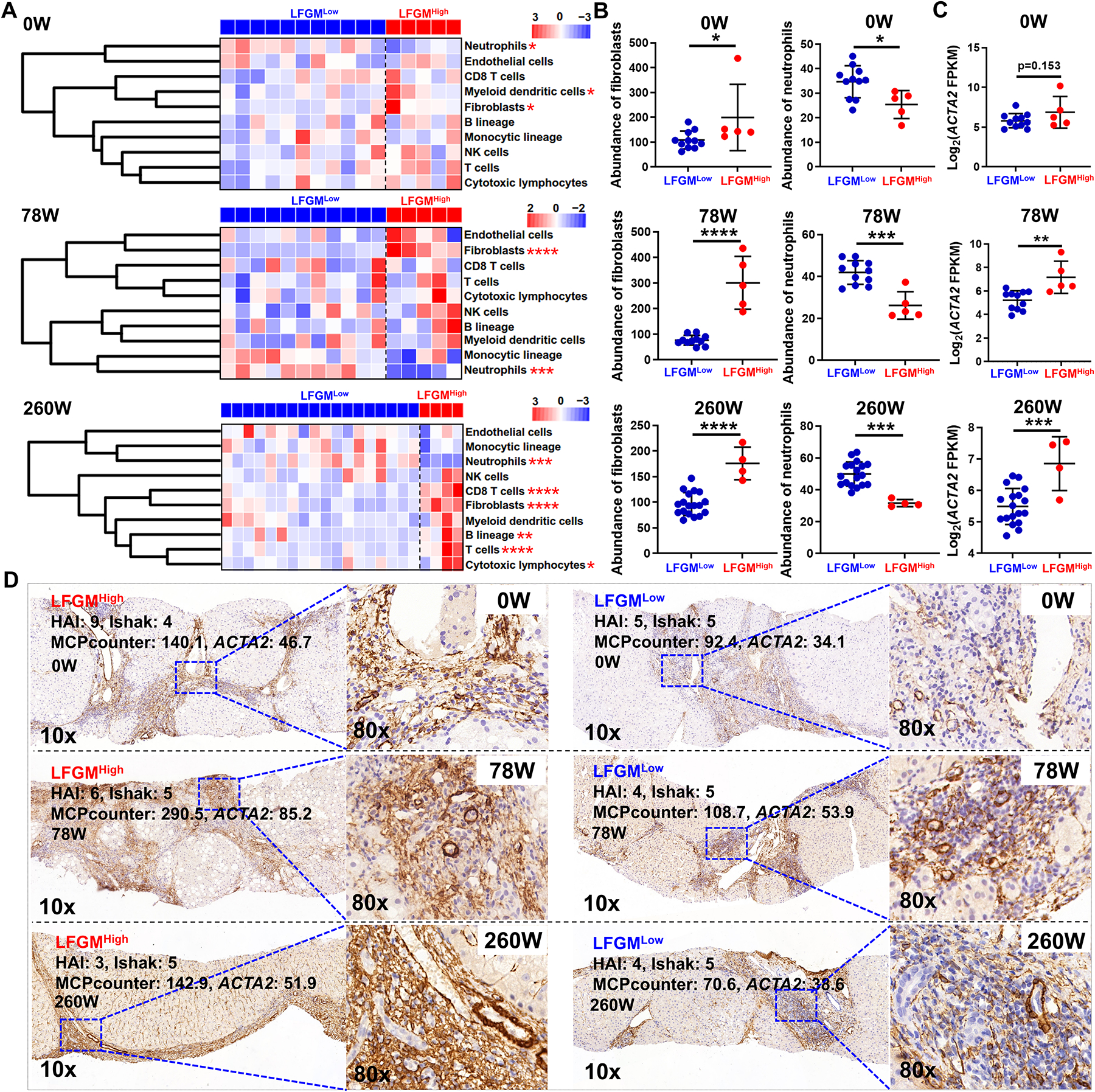
(A) Heatmaps of liver-infiltrating immune cells and stromal cells between subgroups from patients at baseline and after 78 or 260 weeks of antiviral treatment. Expression is scaled as a distribution with mean=0 and SD=1. Darker blue indicates lower abundance; darker red indicates higher abundance. Subgroups of liver fibrosis patients are color-coded. *p<0.05 and **p<0.01 vs LFMGLow. (B) Comparisons of fibroblasts or neutrophils between subgroup of patients from the BJFSH cohort pre- or post-treatment. *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001 vs LFMGLow. (C) Comparison of ACTA2 expression levels detected by bulk RNA-seq between subgroups of patients pre- or post-treatment. **p<0.01 and ***p<0.05 vs LFMGLow. (D) Immunohistochemical staining of ACTA2 in liver biopsies from LFMGLow and LFMGHigh patients with comparable fibrosis stage at baseline or treated for 78 weeks (78W) or 260 weeks (260W) (Magnification: 10x or 80x).
Third, gene set enrichment analysis was performed to identify molecular differences between LFMGLow and LFMGHigh patients. As shown in Figure 5A–C, in patients from the BJFSH cohort at baseline or after antiviral therapy, metabolism-related pathways were not suppressed in LFMGLow patients, while fibrogenesis or proliferation-related signaling was not suppressed in LFMGHigh patients. Functional differences between subgroups, were further verified in patients from all publicly available datasets with or without identical fibrosis stage (figshare, DOI: 10.6084/m9.figshare.22002707). Moreover, differences in metabolic signaling between LFMGLow and LFMGHigh patients from the BJFSH cohort, evened out after 260 weeks of antiviral therapy (Figure 5A–C). In contrast, differences in ECM-cell interaction and proliferation-related signaling between LFMGLow and LFMGHigh patients from the BJFSH cohort, were more apparent after antiviral therapy (Figure 5D–E).
Figure 5. Functional comparison between LFMGLow and LFMGHigh patients from the BJFSH cohort pre- and post-treatment.
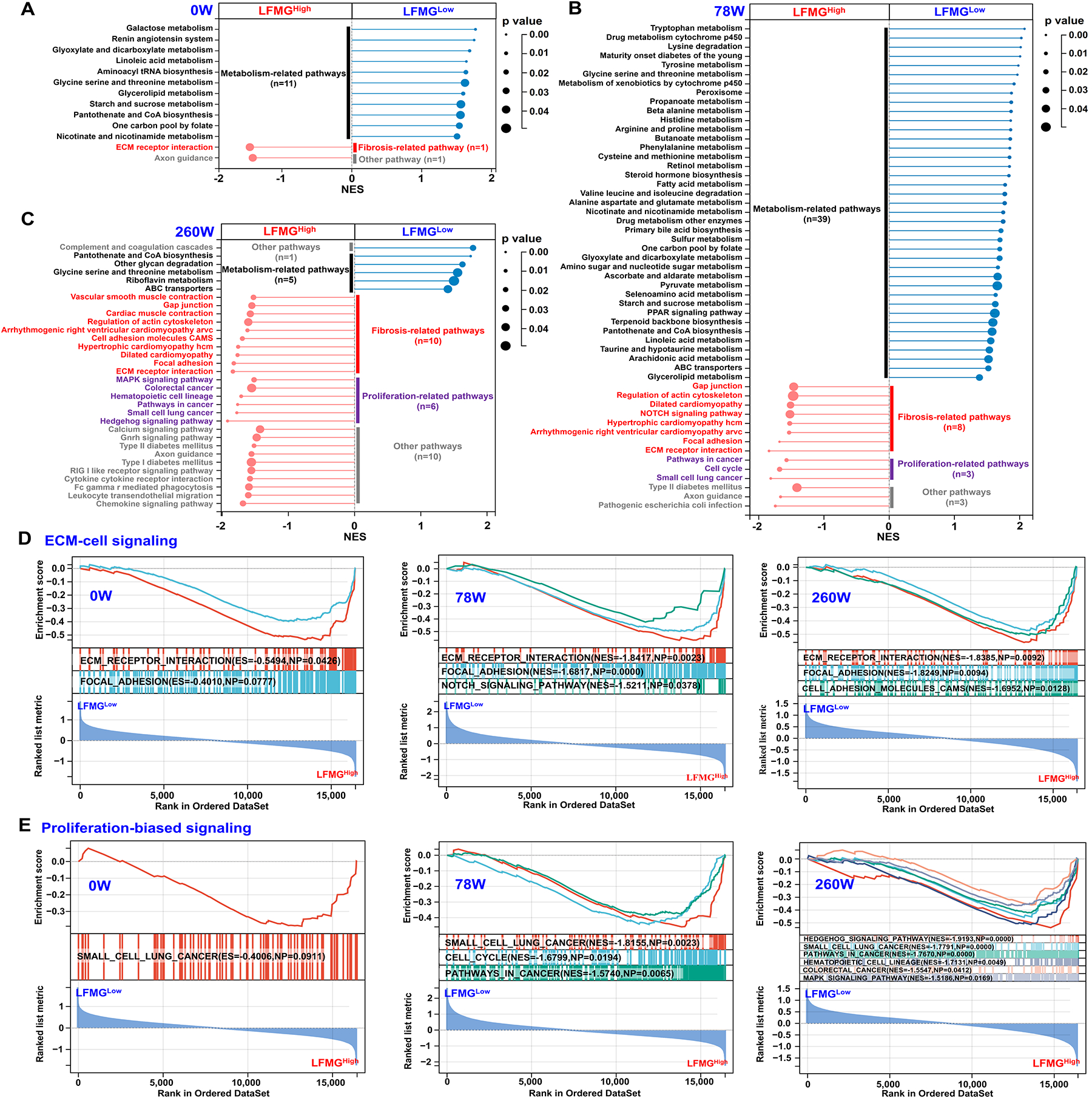
Gene set enrichment analysis revealed suppressed or not suppressed KEGG pathways in LFMGHigh compared to LFMGLow patients from the BJFSH cohort treatment-naïve (0W) (A) and (B) 78 weeks (78 W) or (C) 260 weeks (260W) on antiviral therapy. Circle size represents gene number, color indicates p-value, and line length represents NES. KEGG pathway categories on the left side of the dot plot represent not suppressed signaling in LFMGHigh patients, while KEGG pathway categories on the right side represents not suppressed signaling in LFMGLow patients. Categories of metabolism, fibrosis or proliferation-related pathways and others are color coded. (D and E) Repressive enrichment plots (ECM-cell signaling and proliferation-biased signaling) of gene set enrichment analysis results from LFMGHigh compared to LFMGLow patients at 0W, 78W and 260W. NES, normalized enrichment score.
In summary, LFMGLow and LFMGHigh patients, subclassified by the LFMG signature, have distinct fibrosis stages, liver stiffness, infiltrating immune cells or fibroblasts, and intrahepatic molecular signaling, including metabolism, proliferation and fibrogenesis-related pathways.
Microfibrillar associated protein 4+ (MFAP4+) aHSCs contribute to the LFMG signature dysregulation and are increased in LFMGHigh patients
Next, scRNA-seq of non-parenchymal cells (NPCs) from mice with fibrosis or undergoing resolution was performed, to identify the main cellular source of the LFMG signature and to evaluate its possible dysregulation. After quality control and filtering (Figure S16A), clustering of 32,840 high-quality cells, identified 20 discrete cell populations annotated to 12 cell lineages based on known cell makers (Figure S16B–E and Table S10). The main NPC lineages, were further re-clustered into 5 HSCs (Ccl2+ quiescent HSC [qHSC], Lrrn3+ qHSC, Acta2+ aHSC, Col27a1+ aHSC and Mfap4+ aHSC), 6 infiltrating macrophages and Kupffer cells (Cd63+ Mac, Chil3+ Mac, Eno3+ Mac, Fn1+ Mac, Cd163+ Kupffer and MHC Kupffer) and 6 endothelial cells (ECs) (artery EC, liver sinusoidal endothelial cell 1 [LSEC1], LSEC2, LSEC3, vein EC and lymphatic EC) subtypes, respectively (Figure 6A, S16F and Table S10). We next visualized the expression landscape of the LFMG signature in all NPC subtypes, and found that HSCs, specifically Mfap4+ aHSCs, were the primary cellular source of the LFMG signature and its dysregulation (Figure 6B). The percentage of Mfap4+ aHSCs increased from 3.7% in control (mineral oil) to 36.4% in fibrotic mice, and decreased to 22.9% during resolution (Figure 6C–D). We subsequently performed functional enrichment analysis, based on the top 1,000 genes with the highest abundance in Mfap4+ aHSCs, highlighting distinct biological roles of Mfap4+ compared to Acta2+ aHSCs (Figure 6E), indicating that Mfap4+ aHSC are a newly identified HSC subtype, different from Acta2+ aHSCs.
Figure 6. scRNA-seq and deconvolution analyses of cellular origin of the LFMG signature expression and dysregulation.
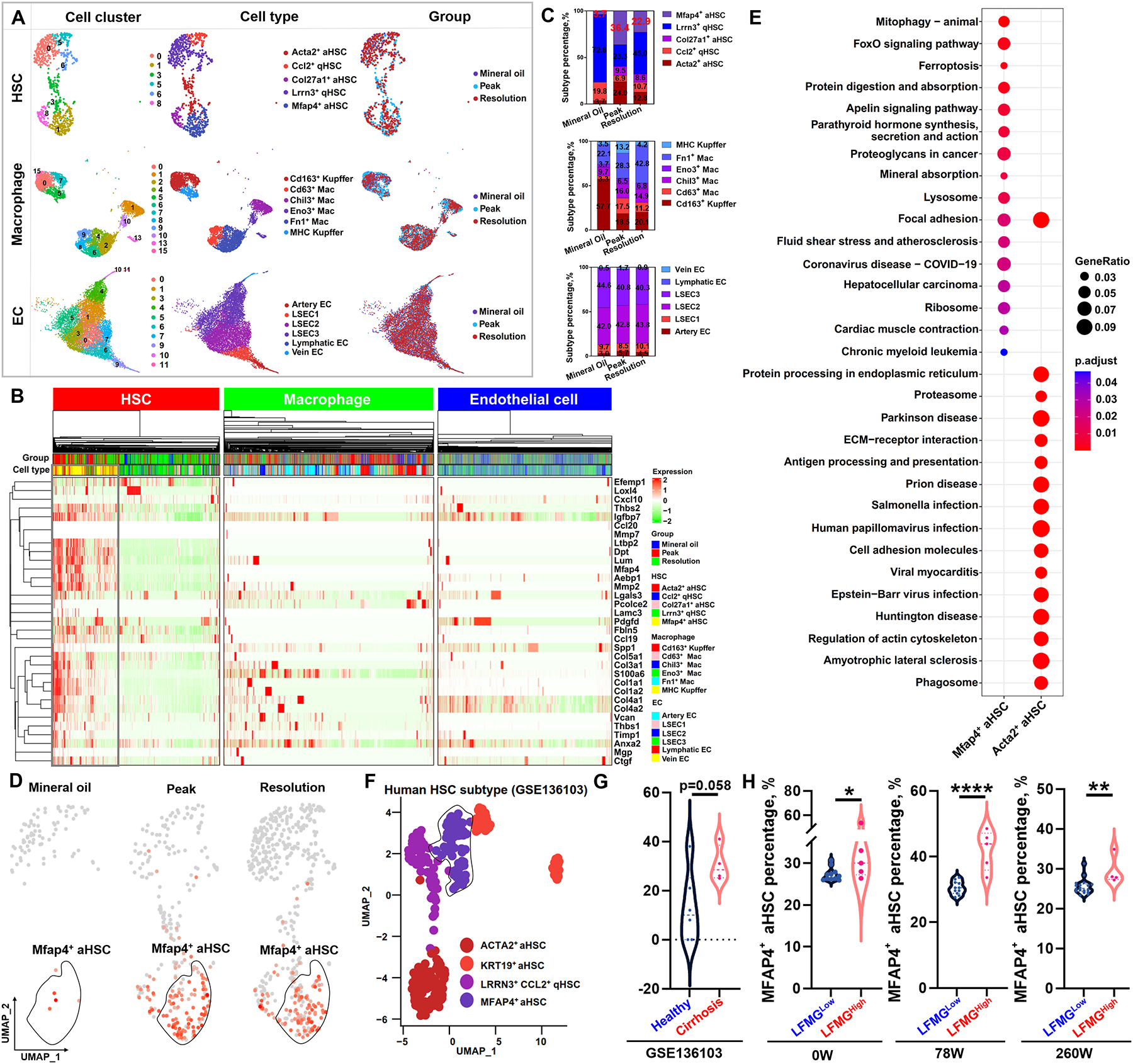
(A) UMAP plots for subset identification of HSCs, infiltrating and resident macrophages, and ECs from NPCs isolated from mice treated with mineral oil (control) or CCl4 (peak fibrosis) or undergoing 1 week of spontaneous resolution. UMAP plots from left to right panels are cell clusters color-coded according to gene expression characteristics, annotated cell types per known markers, and spatial distribution of color-coded cells from mineral oil, peak and resolution, respectively. (B) Complex heatmap shows expression abundance of the LFMG signature among all subtypes of HSCs, macrophages and ECs in the three groups of mice. Expression levels are scaled as a distribution with mean=0 and SD=1. The darker the green, the lower the expression; the darker the red, the higher the expression. Cell subtypes and groups are also color-coded. (C) Comparison of the percentage of HSCs, macrophages and ECs subtypes (color-coded) among mineral oil, peak fibrosis and resolution. (D) UMAP plots of normalized expression of MFAP4 among three groups of mice. The higher the red color intensity, the higher the expression level. (E) Comparison of significantly enriched KEGG pathways between MFAP4+ and ACTA2+ aHSCs based on the top 1,000 genes with the highest expression abundance. Circle size represents gene ratio. Adjusted p value is color-coded. (F) UMAP plot for subset identification of HSCs from public GSE136103 dataset with 4 cirrhotic and 6 healthy human liver samples. HSC subtypes are color-coded. (G) Comparison of MFAP4+ aHSC percentage between healthy and cirrhotic livers from public GSE136103 dataset. (H) Comparison of MFAP4+ aHSC percentage between LFMGLow and LFMGHigh patients from the BJFSH cohort at baseline or after treatment. *p<0.05, **p<0.01 and ***p<0.001.
To investigate whether MFAP4+ aHSCs were responsible for the LFMG signature expression and dysregulation in humans, we re-analyzed a publicly available scRNA-seq dataset (GSE136103) (25, 26), containing CD45− NPCs from 4 cirrhotic and 6 healthy human livers, and identified 4 HSC subtypes (LRRN3+CCL2+ qHSC, ACTA2+ aHSC, KRT19+ aHSC and MFAP4+ aHSC) (Figure 6F and S17). In agreement with findings from mouse models, the percentage of MFAP4+ aHSCs increased in cirrhotic livers compared to healthy controls (p=0.058, Figure 6G). The LFMG signature was mainly expressed and dysregulated in human MFAP4+ aHSCs compared to other HSC subtypes (Figure S18A). Functional analysis also highlighted different biological roles between human MFAP4+ and ACTA2+ aHSCs (Figure S18B).
Next, we deconvolved and compared the distribution of MFAP4+ aHSC fractions between LFMGLow and LFMGHigh patients, using bulk RNA-seq data from the BJFSH cohort and all public datasets via BayesPrism (23), a newly developed Bayesian model to analyze cellular composition from bulk RNA-seq. During this analysis, scRNA-seq (GSE136103) was used as input information. Notably, compared to LFMGLow patients, the proportion of MFAP4+ aHSCs was significantly higher in LFMGHigh patients from the BJFSH cohort, either at baseline or after treatment (Figure 6H). The higher MFAP4+ aHSC proportion in LFMGHigh patients, with or without identical fibrosis stages, was further confirmed in all publicly available datasets (Figure S19).
In summary, analyses of scRNA-seq data from mice and humans identify a novel aHSC subtype (MFAP4+ aHSCs), which is highly responsible for the LFMG signature expression and dysregulation during liver fibrosis. Furthermore, the proportion of MFAP4+ aHSCs is notably increased in LFMGHigh compared to LFMGLow patients.
DISCUSSION
Uncontrolled ECM deposition is a common pathological feature of liver fibrosis, regardless of etiology, and its degradation represents a key step determining restoration of lobular architecture and reversal of liver fibrosis (27). Although collagens are notoriously regarded as the most abundant ECM components in liver fibrosis, it remains unknown whether targeting collagen gene expression is an effective therapy (4). In this study, we unveiled a panel of 35 matrisome genes (LFMGs), gradually dysregulated during liver fibrosis, regardless of etiology, which provide potential matrisome targets for liver fibrosis beyond collagens.
The LFMGs are robust and reliable. First, they were identified from multiple independent transcriptomic profiles of human liver fibrosis, with many fibrosis and control cases, and further validated in an independent transcriptomic profile. Generally, large samples from different studies would, to a large extent, reduce biases in transcriptomic profiling, resulting from batch effect, sequencing platforms used, differences in tissue dissection and storage, or inconsistency in etiology. Second, their expression pattern was confirmed in mouse models of liver fibrosis in pure C57BL/6J or mixed genetic background, indicating that the dysregulation pattern of LFMGs is conserved across species and is robust despite genetic heterogeneity. Third, these histology severity-dependent LFMGs could be used as a combined signature (the LFMG signature), to distinguish fibrotic from control samples, in derivation and test datasets upon unsupervised clustering. In addition, cross-sectional comparisons by merging samples from different studies, after normalizing the expression levels of LFMGs, clearly grouped case-control samples upon unsupervised clustering. Also, we explored expression patterns of LFMGs in patients of different etiologies preceding fibrosis from publicly available datasets, and confirmed lack of response to acute or chronic insults. Overall, the above evidence supports that the LFMG signature is liver fibrosis-specific and not altered by etiology.
Moreover, this study highlights that the LFMG signature has the potential to assess subtle changes in liver fibrosis from an innovative viewpoint, by incorporating matrisome gene expression features. Despite how appealing non-invasive tests in liver fibrosis are, there are inherent limitations such as variability and inaccuracy, indicating that they should be used as “auxiliary tools for diagnosis” only (28). Certainly, liver biopsy is still the gold standard for staging fibrosis. However, considering that histologic diagnosis is also somewhat subjective, susceptible to biopsy size, sampling disparity and error, and inter- and intra-observer variation, scientists and clinicians are still trying to improve the histologic evaluation of liver fibrosis. The “Beijing Classification” pathological assessment system (3) and qFibrosis (29) are promising practices in histologic optimization and accurate grading of liver fibrosis, especially in fibrosis regression. In our study, the LFMG signature decreased in regressive but not in non-regressive patients after short-term antiviral treatment. With extended treatment time, the LFMG signature expression decreased in non-regressive patients but was slower and more modest compared to regressive patients. Except for assessment of precise changes in paired liver biopsies pre- and post-treatment, the LMFG-signature can also be robustly used to subclassify liver fibrosis patients, even with equal pathological fibrosis stage. These findings indicate that the LFMG signature expression is probably more sensitive and objective to identify subtle ECM remodeling that is imperceptible to the naked eye than the current histological scoring, which would be a promising molecular grading system in liver fibrosis progression, and particularly, in regression.
Moreover, we uncovered the differences in clinical, cellular and molecular features between LFMGLow and LFMGHigh patients graded by the LFMG signature. First, we found that LFMGHigh have more liver stiffness than LFMGLow patients. Although LFMGHigh patients with short-term treatment have comparable histologic fibrosis scores than those of LFMGLow patients, they still had relatively higher liver stiffness. Clinically, increased liver stiffness usually denotes hepatic dysfunction and unfavorable liver-related events (30–32). At the molecular level, ECM stiffness regulates the mechanosensitive Hippo pathway and, in turn, promotes ECM gene expression, further aggravating fibrosis (33). The LFMG signature and ECM stiffness probably constitute a positive-feedback loop exacerbating liver fibrosis, as it may occur in LFMGHigh patients. In addition, the difference in liver stiffness between LFMGHigh and LFMGLow patients at baseline will not disappear until a long-term treatment.
Second, compared to LFMGLow patients, increased number of fibroblasts and decreased neutrophils were observed in LFMGHigh patients. Activated HSCs and portal fibroblasts are the main cell types involved in the LFMG signature expression, ECM remodeling and fibrogenesis (34). Although neutrophils were traditionally considered prototypical inflammatory cells, it is now demonstrated that they alleviate liver inflammation and even fibrosis (35, 36). Therefore, the presence of more fibroblasts but less neutrophils in LFMGHigh patients, suggest less ability to resolve fibrosis after eliminating the underlying etiology.
Third, we observed that metabolism-related signaling, rather than fibrogenesis and proliferation-related signaling, were not suppressed in LFMGLow compared to LFMGHigh patients. Given that lymphocytes, dendritic cells, endothelial cells, fibroblasts and aHSCs increased in LFMGHigh patients, the relatively lower proportion of hepatocytes and higher abundance of ECM fibers in liver biopsies from LFMGHigh patients would be the most likely explanation resulting in functional differences between subgroups. In addition, recent data points out that dysfunctional liver due to acute or chronic damage has higher regenerative capacity due to a suppressed metabolic response and subsequent activation of regenerative hepatocellular proliferation (37). A metabolic response to liver injury initiates regenerative hepatocellular proliferation (38); and long-term metabolic suppression induces compensatory hepatocyte hypertrophy, which may initiate an inflammatory response and even fibrosis (37). Accordingly, we anticipate that LFMGHigh patients probably have greater hepatic dysfunction and pro-fibrogenic activity than LFMGLow patients. This gap between subgroups may become narrower when treatment time is extended, as differences in activities of metabolic signaling decreased after long-term treatment.
Lastly, by scRNA-seq analysis and BayesPrism deconvolution, we investigated the cellular source of the LFMG signature to further understand the pathogenesis of liver fibrosis (23). We identified that MFAP4+ aHSCs, distinct from ACTA2+ aHSCs, are the main contributors to the LFMG signature and its dysregulation in human and mouse liver fibrosis. The higher expression of the LFMG signature in the increased MFAP4+ aHSCs enhances ECM deposition, which explains why LFMGHigh patients have higher liver stiffness. Recently, MFAP4 has attracted attention in the fibrosis field, as it is a microfibril-associated ECM protein, binds collagen, elastin and elastic fiber components including fibrillin-1 and −2, lysyl oxidase as well as desmosine, and is involved in ECM homeostasis (39). Although MFAP4 expression increases in fibrotic livers and sera, is relevant to ECM turnover and fibrosis severity (39–42), the specific role in liver fibrosis has not been elucidated yet.
In conclusion, using an integrated analysis of the BJFSH cohort and publicly available transcriptomic profiles, we identified a robust LFMG signature, specific of fibrosis regardless of etiology. This signature can precisely assess subtle changes of liver fibrosis at the genetic and molecular level, which might be especially important in evaluating regressive fibrosis poorly assessed by existing staging systems (43). The LFMG signature can robustly subclassify liver fibrosis patients with or without identical fibrosis stage into subgroups. Although the clinical, cellular and molecular features of LFMGHigh patients predict worse outcome, the direct relationship of the LFMG signature subclassification and liver fibrosis regression or adverse liver-related events are still unknown, which is a limitation of our current study and requires additional patients enrolled and a longer follow-up. In addition, although LFMG signature subclassification has potential in predicting clinical outcome of liver fibrosis after etiology treatment, its predictive value still requires in-depth comparisons against histology and non-invasive liver stiffness directly in further studies as LFMG signature is strongly associated with liver stiffness. Importantly, we identified that MFAP4+ aHSCs are the main cellular source of the LFMG signature and of the differences LFMGLow and LFMGHigh patients. The roles of MFAP4 itself and of MFAP4+ aHSCs in liver fibrosis deserve further investigation.
Supplementary Material
Financial support statement:
this work was supported by the National Natural Science Foundation of China (82170613 [to W.C.], 81970524 [to H.Y.], 82130018 [to H.Y.], 81800534 [to W.C.]), National Science and Technology Major Project (2018ZX10302204) (to H.Y.), Beijing Talent Fund (2018000021469G202) (to W.C.), and US National Institute of Diabetes and Digestive and Kidney Diseases (R01 DK111677) (to N. N.).
Abbreviations:
- aHSC
activated hepatic stellate cell
- CCl4
carbon tetrachloride
- DEMG
differentially expressed matrisome gene
- EC
endothelial cell
- ECM
extracellular matrix
- FFPE
formalin-fixed and paraffin-embedded
- GEO
Gene Expression Omnibus
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- HCL
hierarchical clustering
- HCV
hepatitis C virus
- HSC
hepatic stellate cell
- LFMG
liver fibrosis-specific matrisome gene
- LSEC
liver sinusoidal endothelial cell
- MFAP4
microfibrillar associated protein 4
- NASH
non-alcoholic steatohepatitis
- NPC
nonparenchymal cell
- qHSC
quiescent HSC
- RNA-seq
RNA sequencing
- scRNA-seq
single cell RNA sequencing
- UMAP
uniform manifold approximation and projection
Footnotes
Conflict of interest: the authors declare no conflict of interest.
REFERENCES
- 1.Marcellin P, Gane E, Buti M, Afdhal N, Sievert W, Jacobson IM, Washington MK, et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. Lancet 2013;381:468–475. [DOI] [PubMed] [Google Scholar]
- 2.Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, Torres-Gonzalez A, Gra-Oramas B, Gonzalez-Fabian L, Friedman SL, et al. Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015;149:367–378 e365; quiz e314–365. [DOI] [PubMed] [Google Scholar]
- 3.Sun Y, Zhou J, Wang L, Wu X, Chen Y, Piao H, Lu L, et al. New classification of liver biopsy assessment for fibrosis in chronic hepatitis B patients before and after treatment. Hepatology 2017;65:1438–1450. [DOI] [PubMed] [Google Scholar]
- 4.Schuppan D, Ashfaq-Khan M, Yang AT, Kim YO. Liver fibrosis: Direct antifibrotic agents and targeted therapies. Matrix Biol 2018;68–69:435–451. [DOI] [PubMed] [Google Scholar]
- 5.Wang FD, Zhou J, Chen EQ. Molecular Mechanisms and Potential New Therapeutic Drugs for Liver Fibrosis. Front Pharmacol 2022;13:787748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ikenaga N, Peng ZW, Vaid KA, Liu SB, Yoshida S, Sverdlov DY, Mikels-Vigdal A, et al. Selective targeting of lysyl oxidase-like 2 (LOXL2) suppresses hepatic fibrosis progression and accelerates its reversal. Gut 2017;66:1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Naba A, Clauser KR, Ding H, Whittaker CA, Carr SA, Hynes RO. The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol 2016;49:10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arteel GE, Naba A. The liver matrisome - looking beyond collagens. JHEP Rep 2020;2:100115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen W, Yan X, Xu A, Sun Y, Wang B, Huang T, Wang H, et al. Dynamics of elastin in liver fibrosis: Accumulates late during progression and degrades slowly in regression. J Cell Physiol 2019;234:22613–22622. [DOI] [PubMed] [Google Scholar]
- 10.Karsdal MA, Nielsen SH, Leeming DJ, Langholm LL, Nielsen MJ, Manon-Jensen T, Siebuhr A, et al. The good and the bad collagens of fibrosis - Their role in signaling and organ function. Adv Drug Deliv Rev 2017;121:43–56. [DOI] [PubMed] [Google Scholar]
- 11.Tschumperlin DJ, Ligresti G, Hilscher MB, Shah VH. Mechanosensing and fibrosis. J Clin Invest 2018;128:74–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Long Y, Niu Y, Liang K, Du Y. Mechanical communication in fibrosis progression. Trends Cell Biol 2022;32:70–90. [DOI] [PubMed] [Google Scholar]
- 13.Yokosaki Y, Nishimichi N. New Therapeutic Targets for Hepatic Fibrosis in the Integrin Family, alpha8beta1 and alpha11beta1, Induced Specifically on Activated Stellate Cells. Int J Mol Sci 2021;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng H, Li X, Niu D, Chen WN. Protein profile in HBx transfected cells: a comparative iTRAQ-coupled 2D LC-MS/MS analysis. J Proteomics 2010;73:1421–1432. [DOI] [PubMed] [Google Scholar]
- 15.Reungoat E, Grigorov B, Zoulim F, Pecheur EI. Molecular Crosstalk between the Hepatitis C Virus and the Extracellular Matrix in Liver Fibrogenesis and Early Carcinogenesis. Cancers (Basel) 2021;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poole LG, Arteel GE. Transitional Remodeling of the Hepatic Extracellular Matrix in Alcohol-Induced Liver Injury. Biomed Res Int 2016;2016:3162670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Solarte David VA, Guiza-Arguello VR, Arango-Rodriguez ML, Sossa CL, Becerra-Bayona SM. Decellularized Tissues for Wound Healing: Towards Closing the Gap Between Scaffold Design and Effective Extracellular Matrix Remodeling. Front Bioeng Biotechnol 2022;10:821852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen W, Yang A, Jia J, Popov YV, Schuppan D, You H. Lysyl Oxidase (LOX) Family Members: Rationale and Their Potential as Therapeutic Targets for Liver Fibrosis. Hepatology 2020;72:729–741. [DOI] [PubMed] [Google Scholar]
- 19.Zhu S, Hoshida Y. Molecular heterogeneity in hepatocellular carcinoma. Hepat Oncol 2018;5:HEP10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen W, Desert R, Ge X, Han H, Song Z, Das S, Athavale D, et al. The Matrisome Genes From Hepatitis B-Related Hepatocellular Carcinoma Unveiled. Hepatol Commun 2021;5:1571–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li R, Yang Z, Shao F, Cheng H, Wen Y, Sun S, Guo W, et al. Multi-omics profiling of primary small cell carcinoma of the esophagus reveals RB1 disruption and additional molecular subtypes. Nat Commun 2021;12:3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Members C-N, Partners. Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res 2022;50:D27–D38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chu T, Wang Z, Pe’er D, Danko CG. Cell type and gene expression deconvolution with BayesPrism enables Bayesian integrative analysis across bulk and single-cell RNA sequencing in oncology. Nat Cancer 2022;3:505–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Becht E, Giraldo NA, Lacroix L, Buttard B, Elarouci N, Petitprez F, Selves J, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol 2016;17:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, Portman JR, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019;575:512–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dobie R, Wilson-Kanamori JR, Henderson BEP, Smith JR, Matchett KP, Portman JR, Wallenborg K, et al. Single-Cell Transcriptomics Uncovers Zonation of Function in the Mesenchyme during Liver Fibrosis. Cell Rep 2019;29:1832–1847 e1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bedossa P Reversibility of hepatitis B virus cirrhosis after therapy: who and why? Liver Int 2015;35 Suppl 1:78–81. [DOI] [PubMed] [Google Scholar]
- 28.Patel K, Sebastiani G. Limitations of non-invasive tests for assessment of liver fibrosis. JHEP Rep 2020;2:100067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu S, Wang Y, Tai DCS, Wang S, Cheng CL, Peng Q, Yan J, et al. qFibrosis: a fully-quantitative innovative method incorporating histological features to facilitate accurate fibrosis scoring in animal model and chronic hepatitis B patients. J Hepatol 2014;61:260–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh S, Fujii LL, Murad MH, Wang Z, Asrani SK, Ehman RL, Kamath PS, et al. Liver stiffness is associated with risk of decompensation, liver cancer, and death in patients with chronic liver diseases: a systematic review and meta-analysis. Clin Gastroenterol Hepatol 2013;11:1573–1584 e1571–1572; quiz e1588–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang J, Li J, Zhou Q, Zhang D, Bi Q, Wu Y, Huang W. Liver stiffness measurement predicted liver-related events and all-cause mortality: A systematic review and nonlinear dose-response meta-analysis. Hepatol Commun 2018;2:467–476. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Koch A, Horn A, Duckers H, Yagmur E, Sanson E, Bruensing J, Buendgens L, et al. Increased liver stiffness denotes hepatic dysfunction and mortality risk in critically ill non-cirrhotic patients at a medical ICU. Crit Care 2011;15:R266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu F, Lagares D, Choi KM, Stopfer L, Marinkovic A, Vrbanac V, Probst CK, et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol 2015;308:L344–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Acharya P, Chouhan K, Weiskirchen S, Weiskirchen R. Cellular Mechanisms of Liver Fibrosis. Front Pharmacol 2021;12:671640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Calvente CJ, Tameda M, Johnson CD, Del Pilar H, Lin YC, Adronikou N, De Mollerat Du Jeu X, et al. Neutrophils contribute to spontaneous resolution of liver inflammation and fibrosis via microRNA-223. J Clin Invest 2019;129:4091–4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guillot A, Tacke F. The Unexpected Role of Neutrophils for Resolving Liver Inflammation by Transmitting MicroRNA-223 to Macrophages. Hepatology 2020;71:749–751. [DOI] [PubMed] [Google Scholar]
- 37.Caldez MJ, Bjorklund M, Kaldis P. Cell cycle regulation in NAFLD: when imbalanced metabolism limits cell division. Hepatol Int 2020;14:463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang J, Rudnick DA. Elucidating the metabolic regulation of liver regeneration. Am J Pathol 2014;184:309–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanaan R, Medlej-Hashim M, Jounblat R, Pilecki B, Sorensen GL. Microfibrillar-associated protein 4 in health and disease. Matrix Biol 2022;111:1–25. [DOI] [PubMed] [Google Scholar]
- 40.Madsen BS, Thiele M, Detlefsen S, Sorensen MD, Kjaergaard M, Moller LS, Rasmussen DN, et al. Prediction of liver fibrosis severity in alcoholic liver disease by human microfibrillar-associated protein 4. Liver Int 2020;40:1701–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bracht T, Molleken C, Ahrens M, Poschmann G, Schlosser A, Eisenacher M, Stuhler K, et al. Evaluation of the biomarker candidate MFAP4 for non-invasive assessment of hepatic fibrosis in hepatitis C patients. J Transl Med 2016;14:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Molleken C, Ahrens M, Schlosser A, Dietz J, Eisenacher M, Meyer HE, Schmiegel W, et al. Direct-acting antivirals-based therapy decreases hepatic fibrosis serum biomarker microfibrillar-associated protein 4 in hepatitis C patients. Clin Mol Hepatol 2019;25:42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kleiner DE. On beyond staging and grading: Liver biopsy evaluation in a posttreatment world. Hepatology 2017;65:1432–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data from bulk RNA-seq of human FFPE liver biopsies was deposited in the National Genomics Data Center database (22) and are publicly available (accession number PRJCA010948). Raw and processed data from bulk RNA-seq of frozen mouse liver tissues and scRNA-seq of mouse primary liver cells were deposited in NCBI’s GEO database under accession number GSE199392 and “pending”, respectively. All processed gene expression profiles available from GEO database and R codes for scRNA-seq and BayesPrism deconvolution (23) analyses were deposited in the figshare platform (DOI: 10.6084/m9.figshare.22002707).