

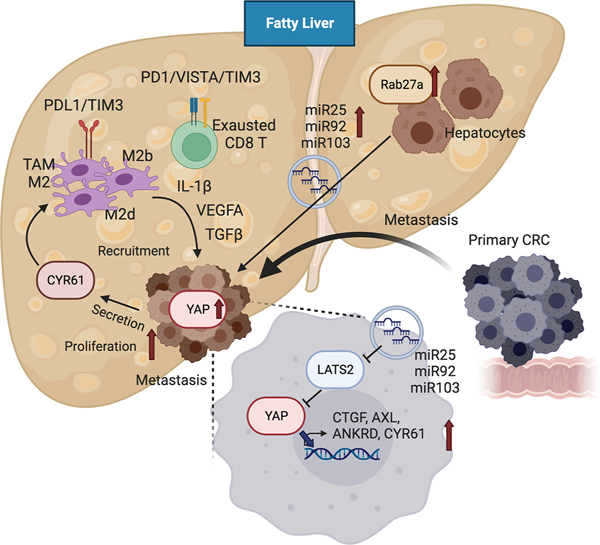
SUMMARY:
Liver metastasis is a major cause of death in patients with colorectal cancer (CRC). Fatty liver promotes liver metastasis, but the underlying mechanism remains unclear. We demonstrated that hepatocyte-derived extracellular vesicles (EVs) in fatty liver enhanced the progression of CRC liver metastasis by promoting oncogenic Yes-associated protein (YAP) signaling and an immunosuppressive microenvironment. Fatty liver upregulated Rab27a expression, which facilitated EV production from hepatocytes. In the liver, these EVs transferred YAP signaling-regulating microRNAs to cancer cells to augment YAP activity by suppressing LATS2. Increased YAP activity in CRC liver metastasis with fatty liver promoted cancer cell growth and an immunosuppressive microenvironment by M2 macrophage infiltration through CYR61 production. Patients with CRC liver metastasis and fatty liver had elevated nuclear YAP expression, CYR61 expression, and M2 macrophage infiltration. Our data indicate that fatty liver-induced EV-microRNAs, YAP signaling, and an immunosuppressive microenvironment promote the growth of CRC liver metastasis.
Keywords: colon cancer, CYR61, exosome, high-fat diet, liver metastasis, M2 macrophage, microRNA, non-alcoholic fatty liver disease, palmitate, Rab27a
Graphical Abstract
The eTOC blurb:
Liver metastasis is a major cause of death in patients with colorectal cancer (CRC). Wang et al. demonstrate that fatty liver enhances the production of hepatocyte-derived extracellular vesicles that promote the progression of CRC liver metastasis by augmenting Yes-associated protein signaling and the immunosuppressive tumor microenvironment.
Colorectal cancer (CRC) is the third most common malignancy and the second leading cause of cancer-related death, accounting for about 900,000 deaths per year worldwide1. The liver is the most common metastatic site for CRC, owing to the unique anatomical link between the intestine and the liver through the portal vein. The liver-specific metabolic and immune microenvironment also supports CRC liver metastasis. Ultimately, 70% of patients with CRC will develop liver metastasis, which is the major cause of death2,3.
Obesity and non-alcoholic fatty liver disease (NAFLD) are the significant risk factors for CRC4. More than 650 million adults are obese worldwide, and the increased prevalence of NAFLD is linked to the obesity epidemic. A growing body of epidemiological evidence indicates that fatty liver increases the occurrence of CRC liver metastasis and the local recurrence after resection of CRC liver metastases, thereby worsening prognosis5–15. Likewise, fatty liver augments metastatic liver tumor growth in animal models of CRC liver metastasis by altering the hepatic inflammatory and immune microenvironment16. These findings suggest metastatic mechanisms may differ owing to heterogeneity of the tumor microenvironment (TME) in fatty liver, which may explain diverse responses to cancer therapies among patients. As such, disease management for liver metastasis may differ in patients with or without fatty liver. There is an urgent need to understand the molecular mechanisms of metastasis in patients with fatty liver to manage those patients effectively.
Extracellular vesicles (EVs) contain bioactive macromolecules, such as microRNAs (miRNAs), lipids, and proteins. EV production and secretion into the extracellular space is regulated by ceramide synthesis via neutral sphingomyelinase and Rab proteins such as Rab27a and Rab27b17,18. Rab27a mediates EV production in melanoma and its metastasis, and is important for EV secretion enhanced by lipid overload19–21. In NAFLD, hepatocyte-derived EV production is increased along with altered miRNA contents22. Previous studies showed that primary tumor-derived EVs travel to the liver and facilitate pre-metastatic niche formation and pro-metastatic inflammatory responses23–27. However, the contribution of EVs produced in the liver, particularly under NAFLD conditions, toward the formation of a pre- and pro-metastatic liver environment that predisposes to CRC liver metastasis has not been explored.
Yes-associated protein (YAP), a transcriptional co-regulator, is the effector of the Hippo pathway, which regulates the activity of pro-cancerous transcription factors, such as transcriptional enhanced associate domains (TEADs)28. When YAP is phosphorylated by the mammalian Ste20-like kinases 1/2 (MST1/2) and by the large tumor suppressor kinases 1/2 (LATS1/2), YAP is localized in cytoplasm and inhibits its activity by proteasomal degradation29. In contrast, unphosphorylated YAP is localized to the nucleus and activates TEAD transcription factors, promoting gene expression for cancer growth, invasion, and metastasis29. G protein-coupled receptors, mechanical cues, cell adhesion, and integrin signaling regulate YAP activity in a phosphorylation-dependent manner, but YAP activity is also regulated post-transcriptionally by miRNAs30,31. Pro-cancerous YAP activity intrinsically contributes to cancer cell behavior, but it also affects surrounding immune cells. Loss of LATS1/2 kinases enhances anti-cancer immunity32, but high YAP activity induces an immunosuppressive microenvironment by recruiting M2 tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells or by upregulating programmed cell death 1 ligand (PD-L1) expression33–37.
In the present study, we reveal that fatty liver produces EVs that contain pro-carcinogenic miRNAs and creates a pre-metastatic and pro-metastatic liver microenvironment that predisposes to CRC liver metastasis. Our results indicate that EV transfer of pro-carcinogenic miRNAs from fatty liver hepatocytes to metastatic cancer cells increases YAP activity by inhibiting LATS2. We show that heightened YAP activity promotes CRC liver metastatic growth by enhancing M2-TAM recruitment and CD8 T cell exhaustion to create an immunosuppressive microenvironment. Thus, NAFLD likely generates a complex, metastatic TME, contributing to CRC liver metastasis. Further, the recent increase in global burden of NAFLD may explain the diverse responses to cancer therapies for patients with CRC and liver metastasis.
RESULTS
Increased Extracellular Vesicle Release by Fatty Liver Enhances Metastatic Tumor Growth in the Liver
To investigate whether fatty liver affects metastatic CRC growth in the liver, wild-type (WT) mice were fed a low-fat diet (LFD) or a high-fat diet (HFD) for 6 weeks followed by splenic injection of syngeneic MC38 CRC cells for an additional 2 weeks to establish a CRC liver metastasis model with or without fatty liver (Figure 1A). The HFD caused an increase in the number and size of metastatic tumors along with increased serum EV particles (Figure 1A–C). The distribution of EV particle sizes was similar from sera of LFD-fed and HFD-fed mice (Figure S1A and S1B). As EVs promote cancer growth38,39, we investigated the effect of serum EVs from HFD-fed mice in CRC cells. Proliferation, migration, and invasion of MC38 cells were enhanced by treatment with serum EVs from LFD-fed mice compared with cells without EV treatment (Figure 1D–1F). The cancer aggressiveness was further enhanced when treated with EVs from HFD-fed mice (Figure 1D–1F). Further, in human samples, the number of serum EVs from patients with NAFLD was greater than healthy controls, and correlated with liver fat content (Figure 1C and S1C). These EVs increased the growth, migration, and invasion of HCT116 cells, suggesting that EVs from patients with NAFLD promote aggressive CRC (Figure 1D–1F, Table S1). Because serum EVs contain liver-derived EVs (Figure S1D) and fatty liver increases EV production22,40,41, we hypothesized that fatty liver-derived EVs enhance metastatic tumor growth. We examined EVs from fat-laden hepatocytes by extraction of EVs from culture supernatants of mouse primary hepatocytes (PHCs) and Huh7 cells treated with palmitate (PA), one of the most abundant free fatty acids in patients with NAFLD. Similar to serum EVs, EVs from hepatocytes expressed the EV-specific markers CD9, CD63, and TSG101, and hepatocyte markers (Figure S1D). PA treatment increased EV secretion from PHCs and Huh7 cells (Figure S1E and S1F). The distribution of EV particle sizes was similar between serum EVs and hepatocyte EVs (Figure S1A and S1E). We confirmed that EVs from PHCs and Huh7 cells were taken up by mouse CRC MC38 cells and human CRC HCT116 cells, respectively (Figure S1G). In line with our serum EV experiments, fat-laden hepatocyte-derived EVs also augmented proliferation, migration, and invasion of mouse and human CRC cells (Figure S1H and S1I).
Figure 1. Increased Extracellular Vesicle Release by Fatty Liver Enhances Metastatic Tumor Growth in the Liver.
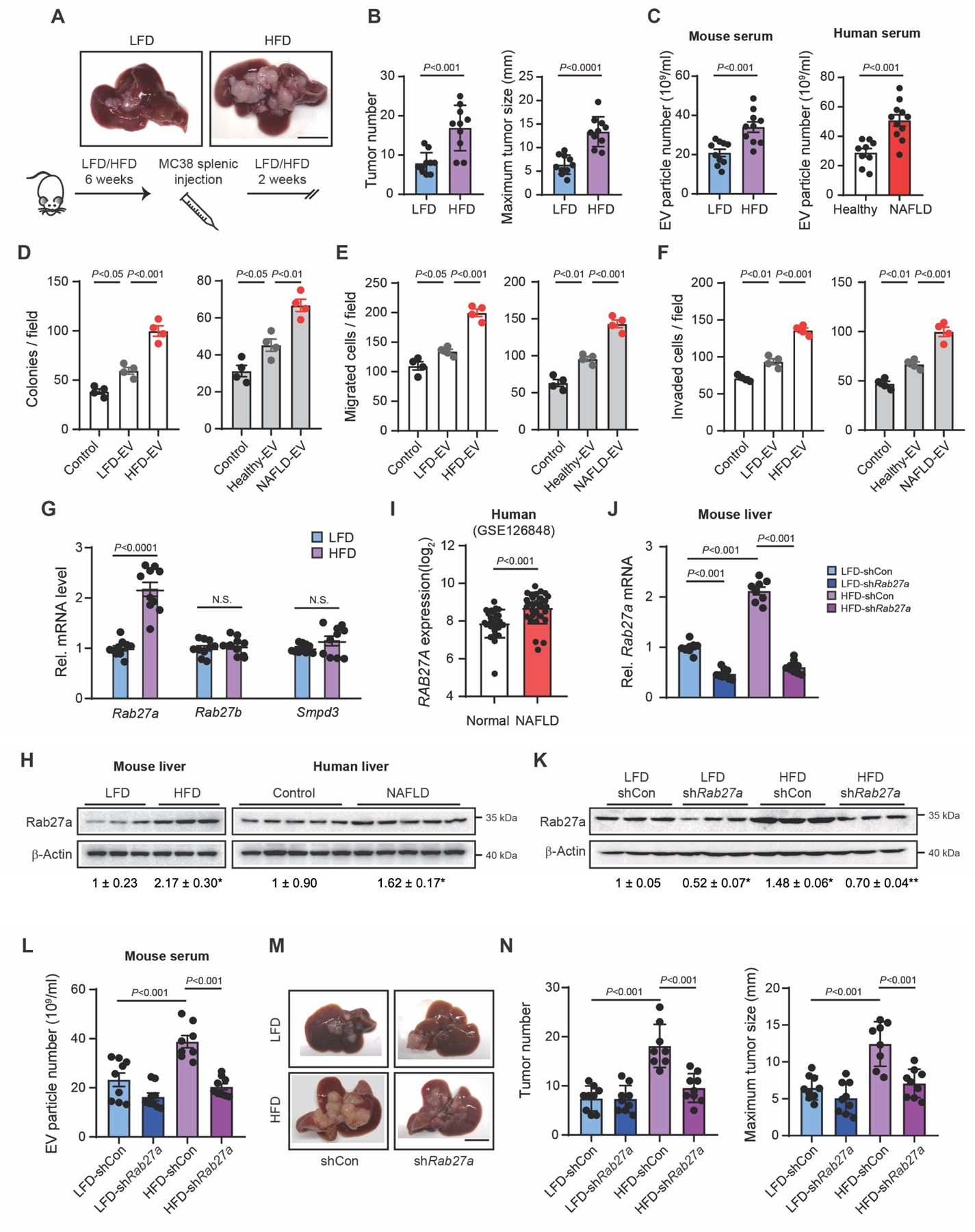
(A) Protocol for the HFD-induced CRC liver metastasis model. Mice were fed a HFD or a LFD for 8 weeks. MC38 cells were injected into the spleen two weeks before euthanasia. Representative macroscopic images. Scale bar, 1 cm.
(B) Tumor number(Left). Maximum tumor size measured as the largest diameter of the largest tumor(right) (n=10/group).
(C) EV particle numbers in the sera of LFD-fed and HFD-fed mice inoculated with MC38 cells (n=10/group)(left) and in the sera of healthy subjects (n=9) or patients with NAFLD (n=11)(right).
(D) MC38 cells were treated with EVs from sera of LFD-fed and HFD-fed mice(left). HCT116 cells were treated with EVs from healthy controls and patients with NAFLD(right). Colony-forming assay (n=4/group). Control indicates cells without EV treatment.
(E,F) In vitro migration and invasion assay. MC38 cells (left) and HCT116 cells (right) treated with EVs were placed in the upper chamber, and the number of cells that migrated and invaded to the lower chamber were quantified (n=4/group). Control indicates cells without EV treatment.
(G) The expression of Rab27a, Rab27b, and Smpd3 in liver extracts of mice fed with a LFD or a HFD for 8 weeks (n=10/group).
(H) Immunoblots and quantifications of liver lysates from (G)(left). Hepatic RAB27A protein expression in healthy controls or patients with NAFLD(right).
(I) Comparison of hepatic RAB27A expression between healthy controls (n=26) and patients with NAFLD (n=31) from GSE126848.
(J-N) After 6 weeks of LFD or HFD feeding, adenovirus expressing short hairpin RNA (shRNA) for Rab27a (shRab27a) or a scrambled-shRNA control (shCon) was administered intravenously to mice. MC38 cells were injected into the spleen 48 hours after adenoviral vector administration (n=8–9/group).
(J) Hepatic Rab27a mRNA expression in non-tumor liver tissues.
(K) Immunoblots and quantifications for Rab27a.
(L) EV particle numbers in mouse serum.
(M) Macroscopic images. Scale bar, 1 cm.
(N) Number of metastatic tumors, and maximum tumor size.
Data shown as mean ± SD (B,I,N) or mean ± SEM (C-G,J,L). Significance determined by two-tailed Student’s t-test (B,C,G,I) or one-way analysis of variance (ANOVA) with Tukey’s post hoc analysis (D-F,H,J-L,N). N.S., not significant. *P<0.05 versus LFD or control (H). *P<0.05 versus LFD-shCon; **P<0.01 versus HFD-shCon. (K).
Extracellular Vesicles Produced in Fatty Liver Aggravate Metastatic Liver Tumor Growth
EV production and secretion are regulated by Rab27a/b and neutral sphingomyelinase17. Rab27a was dramatically upregulated compared with Rab27b or Smpd3 in the liver of HFD-fed mice and PA-treated liver cells (Figure 1G, 1H, and S1J). RAB27A expression was also upregulated in the liver of patients with NAFLD (Figure 1H and 1I). This led us to hypothesize that Rab27a upregulated in fatty liver regulates EV secretion. Rab27a silencing inhibited PA-induced EV secretion in vitro (Figure S1K and S1L). To investigate whether fatty liver-derived EVs promote liver metastasis in vivo, we silenced Rab27a expression in the liver by adenoviral vector (Figure 1J and 1K). To avoid the effect of Rab27a and EVs on the development of NAFLD, we inhibited EV secretion by silencing Rab27a at the time of fatty liver establishment (6 weeks after HFD feeding and two days before CRC inoculation) (Figure S1M and S1N). Hepatocytes, but not tumor cells, were infected with green fluorescent protein (GFP)-tagged adenoviral vectors (Figure S1O). Two days after Rab27a silencing, EV production was inhibited; its low production level was maintained until the end of the experiment (Figure S1P, S1Q, 1K, and 1L). Augmented metastatic liver tumor growth in HFD-fed mice was inhibited by Rab27a silencing, whereas inhibition was not seen in LFD-fed mice (Figure 1M and 1N). This was also confirmed by the liver metastasis model with another syngeneic CMT93 CRC cells (Figure S1R). In contrast, Rab27a overexpression and exogenous injections of PA-induced hepatocyte EV enhanced CRC liver metastasis (Figure S1S–U). These results indicate that the increased Rab27a expression in fatty liver facilitated liver metastasis, likely through Rab27a-mediated EV production.
MiRNAs Are the Functional Extracellular Vesicle Contents That Aggravate Colorectal Cancer Growth in Fatty Liver
Next, we wanted to know which EV contents are important for the increase in CRC liver metastasis in patients with fatty liver. We performed quantitative miRNA polymerase chain reaction (PCR) arrays using EVs from sera of healthy individuals and patients with NAFLD, LFD-fed and HFD-fed mice, and LFD-fed and HFD-fed mice bearing liver metastasis, and from hepatocytes of HFD-fed mice and PA-treated mouse hepatocytes (Figure 2A). Using these five cohorts, we determined that six miRNAs were commonly upregulated. miR-103, miR-25, and miR-92a were the top three differentially expressed miRNAs contained mainly in hepatocyte-derived EVs (Figure S2A and S2B). The data indicate that fatty liver hepatocyte- and PA-treated hepatocyte-derived EVs commonly contain pro-oncogenic miRNAs. Therefore, we used EVs from PA-treated hepatocytes to study the oncogenic role of fatty liver-derived EVs in subsequent experiments. Next, we investigated the functional role of miR-25, miR-92, and miR-103 in CRC cells. PA treatment upregulated miR-25, miR-92, and miR-103 expression in EVs from mouse hepatocytes and Huh7 cells (Figure S2C). These miRNAs were taken up by MC38 and HCT116 cells treated with EVs from PA-treated cells (Figure S2D). Proliferation, migration, and invasion were increased in MC38 and HCT116 cells treated with miR-25, miR-92, and miR-103 mimics (Figure 2B, 2C, S2E, and S2F). A tumorigenic effect of EVs from PA-treated hepatocytes was reduced by combinations of two or three miRNA inhibition in EVs (Figure 2D, 2E, and S2H–S2M). Also, the effect of EVs from fatty liver hepatocytes was suppressed by a combined inhibition of miR-25, miR-92, and miR-103 (Figure S2N–P). These findings support the notion that miR-25, miR-92, and miR-103 are the oncogenic EV players in fatty liver.
Figure 2. MiRNAs Are the Functional Extracellular Vesicle Contents That Aggravate Colorectal Cancer Growth in Fatty Liver.
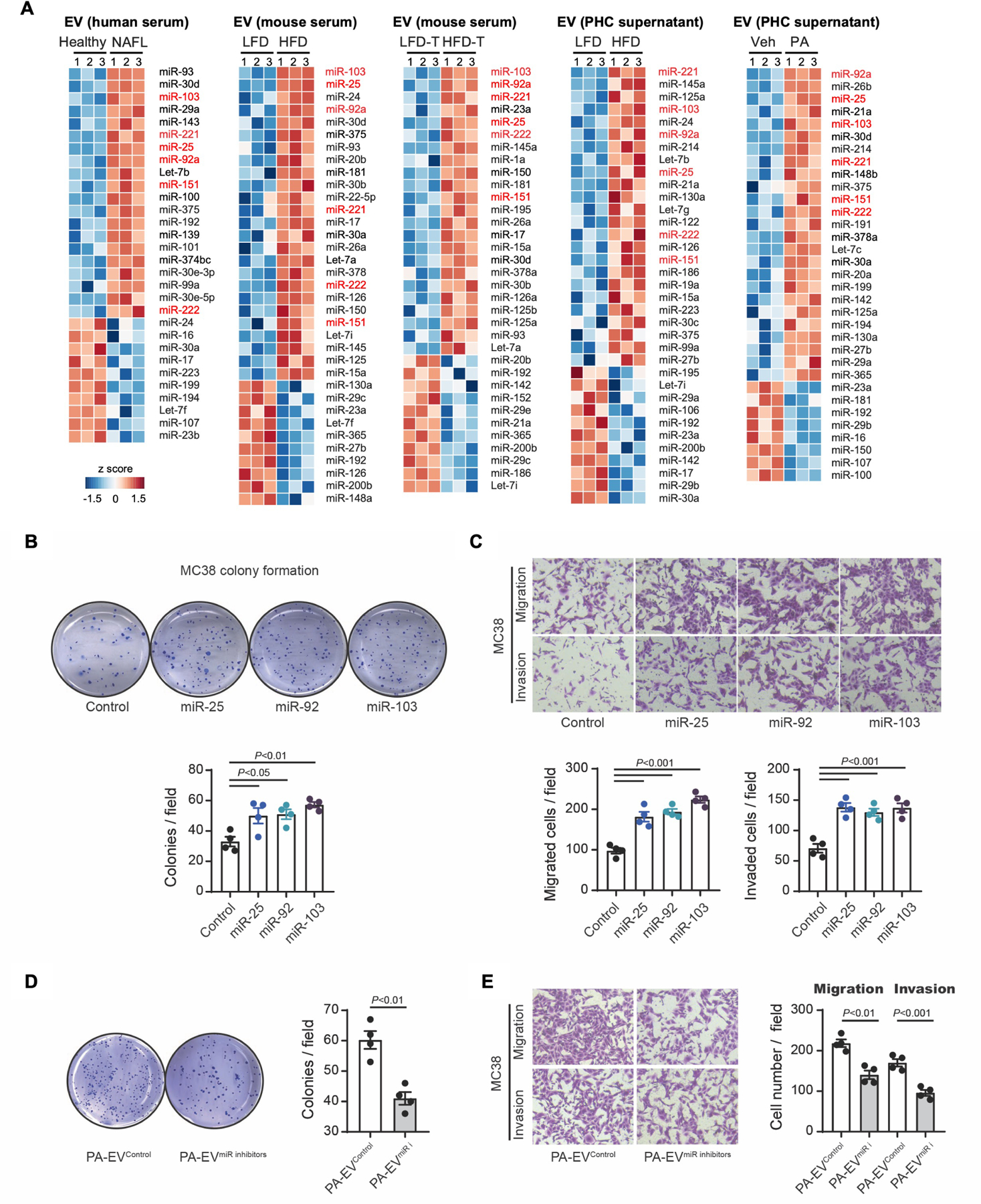
(A) A heatmap of miRNAs. MiRNA PCR array for (1) EVs from the sera of healthy controls and patients with NAFLD, (2) EVs from mouse sera (8-week-LFD or HFD feeding), (3) EVs from sera of metastatic tumor-bearing mice fed a LFD or HFD, (4) EVs from primary hepatocytes (PHCs) of LFD or HFD-fed mice, and (5) EVs from vehicle (Veh)-treated or PA-treated mouse PHCs. The heatmaps illustrate the log2 (fold change) values. The heatmap diagram highlights six common miRNAs. miR-103, miR-25, and miR-92a are the top three differentially expressed miRNAs.
(B) Effect of miR-25, miR-92, and miR-103 on colony formation. Representative images from the colony-forming assay of MC38 cells(upper), and the average colony numbers per field(lower) (n=4/group).
(C) Transwell migration and invasion assay. MC38 cells were transfected with 50 nM control, miR-25, miR-92, or miR-103 mimics. Representative pictures are shown(upper). Quantification of migrated and invaded cells(lower) (n=4/group).
(D,E) Effect of EVs with compound inhibition of miR-25, miR-92, and miR-103. Mouse PHCs were transfected with a combination of three antagomiRs or a control (100 nM each) for 48 hours. Then, cells were treated with 400 μM PA for an additional 24 hours. EVs were collected from the cells transfected with antagomiRs (PA-EVmiR-i) or a control (PA-EVControl).
(D) Colony-forming assay. MC38 cells were treated with PA-EVmiR-i or PA-EVControl (n=4/group).
(E) Transwell migration and invasion assay. MC38 cells were treated with PA-EVmiR-i or PA-EVControl for 48 hours and then placed in the top chamber, and the migration and invasion to the lower chamber were quantified (n=4/group).
Data shown as mean ± SEM (B-E). Significance determined by one-way analysis of variance (ANOVA) with Tukey’s post hoc analysis (B,C) or two-tailed Student’s t-test (D,E).
Extracellular Vesicles Containing miRNAs Regulate Pro-tumorigenic YAP Signaling in Colorectal Cancer Cells
To seek the direct target of EV-miRNAs, we combined three target-gene prediction algorithms, TargetScan, miRDB, and PicTar, and selected tumor suppressor genes predicted frequently in both humans and mice for miR-25, miR-92, and miR-103, based on the Gene Ontology and KEGG Pathway databases (Figure 3A) 42,43. Among the predicted tumor suppressor genes, expression of LATS2, a negative regulator of YAP signaling, was significantly downregulated (Figure 3B and S3A), and expression of nuclear and phospho-YAP proteins was increased and decreased, respectively, in MC38 and HCT116 cells treated with miR-25, miR-92, and miR-103 mimics (Figure 3C, S3B, and S3C). Our reporter assays and mutagenesis studies confirmed that miR-25, miR-92, and miR-103 bind to the 3′-UTR region of Lats2 to regulate Lats2 expression in MC38 cells (Figure 3D and 3E). Consistently, EVs from PA-treated cells reduced LATS2 and increased nuclear YAP expression, whereas the compound inhibition of miR-25, miR-92, and miR-103 in EVs increased LATS2 and reduced nuclear YAP expression in MC38 and HCT116 cells (Figure 3F, 3G, S3D, and S3E). Also, YAP nuclear translocation by EVs from fatty liver hepatocytes was suppressed by a combined inhibition of miR-25, miR-92, and miR-103 (Figure S3F). Our data suggest that miR-25, miR-92, and miR-103 in EVs promote YAP activation through LATS2 downregulation.
Figure 3. Extracellular vesicle-miRNAs Promote YAP Activation in Colorectal Cancer Cells.
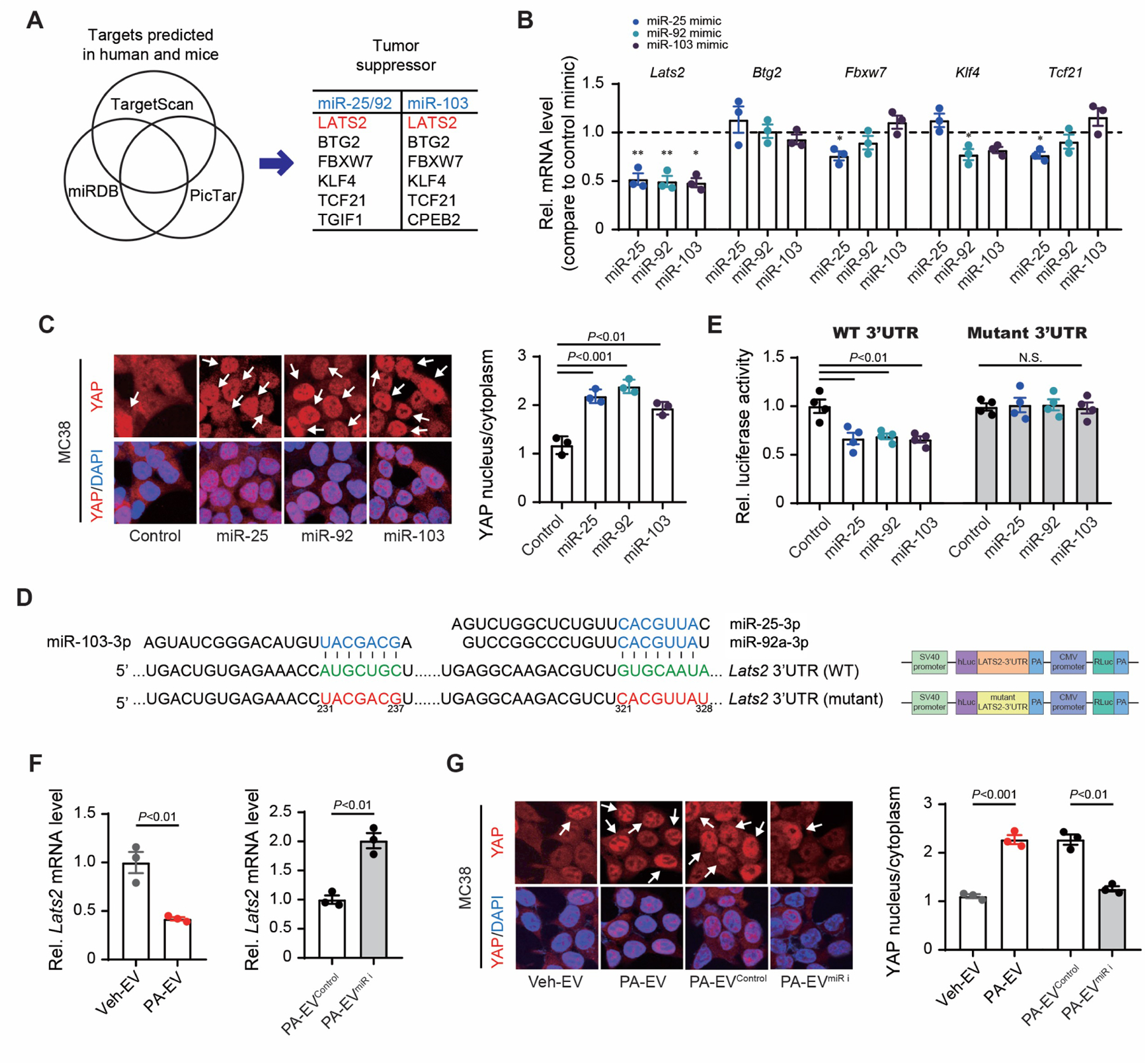
(A) Venn diagram comparing three target-gene prediction algorithms by TargetScan, miRDB, and PicTar. Among the target genes predicted in humans and mice, tumor suppressor genes based on Gene Ontology and the KEGG Pathway were presented in the table.
(B) Quantitative real-time PCR (qRT-PCR) assay for the predicted target genes of miR-25/92 and miR-103. MC38 cells were transfected with 50 nM miR-92, miR-25, and miR-103 mimics (n=3/group).
(C) Effects of miR-25, miR-92, and miR-103 on YAP localization in MC38 cells. Representative immunofluorescent images(left). Quantification of nuclear/cytoplasmic YAP ratios(right) (n=3/group). DAPI, 4′,6-diamidino-2-phenylindole.
(D) Vector constructs of WT and mutant pEZX-LATS2-3--UTR.
(E) LATS2-3′-UTR luciferase activity assay using MC38 cells transfected with control, miR-25, miR-92, or miR-103 mimics for 48 hours (n=4/group).
(F) qRT-PCR assay for Lats2 in lysates of MC38 cells treated with EVs. EVs were obtained from supernatants of hepatocytes treated with vehicle (Veh-EV), PA (PA-EV)(left), or hepatocytes transfected with antagomiRs for miR-25, miR-92, and miR-103 (PA-EVmiR-i) or a control (PA-EVControl) followed by PA treatment(right) (n=3/group).
(G) Effects of EVs on YAP localization in MC38 cells. MC38 cells were treated with Veh-EV, PA-EV, PA-EVmiR-i, or PA-EVControl. Representative immunofluorescent images(left). Quantification of nuclear/cytoplasmic YAP ratios(right) (n=3/group).
Data shown as mean ± SEM (B,C,E-G). Significance determined by two-tailed Student’s t-test (B,F,G) or one-way analysis of variance (ANOVA) with Tukey’s post hoc analysis (C,E). *P<0.05 and **P<0.01 versus control (B). N.S., not significant.
See also Figure S3.
YAP Activity Is Increased in Colorectal Cancer Liver Metastasis Enhanced by Non-Alcoholic Fatty Liver Disease
We then examined the gene expression profile of metastatic tumors in HFD-fed and LFD-fed mice. Gene set enrichment analysis (GSEA) demonstrated that YAP and oncogenic signatures, such as epithelial-mesenchymal transition, cell cycle, angiogenesis, E2F, MYC targets, transforming growth factor beta (TGF-β), Kras signaling, and angiogenesis, were enriched in tumors from HFD-fed mice (Figure 4A, S4A, and S4B), supporting an increased YAP activity and cancer aggressiveness by fatty liver. To test our hypothesis of EV regulation of YAP activity in fatty liver, we examined YAP nuclear localization and YAP target gene expression. In metastatic tumors, HFD feeding enhanced YAP nuclear localization, increased expression of YAP and its target genes (Ankrd1, Axl1, Ccn2, and Ccn1), and downregulated expression of Lats2, but not Lats1 (Figure 4B–D, and S4C). We also examined the role of EVs in YAP expression by silencing of Rab27a in hepatocytes in vivo. Rab27a silencing reversed the decreased Lats2 expression and increased nuclear YAP and YAP target gene expression in metastatic tumors of HFD-fed mice (Figure 4E and 4F), corroborating our hypothesis of EV regulation of YAP activity in metastatic tumors in fatty liver.
Figure 4. YAP Activity Contributes to Colorectal Cancer Liver Metastasis Enhanced by Non-Alcoholic Fatty Liver Disease.
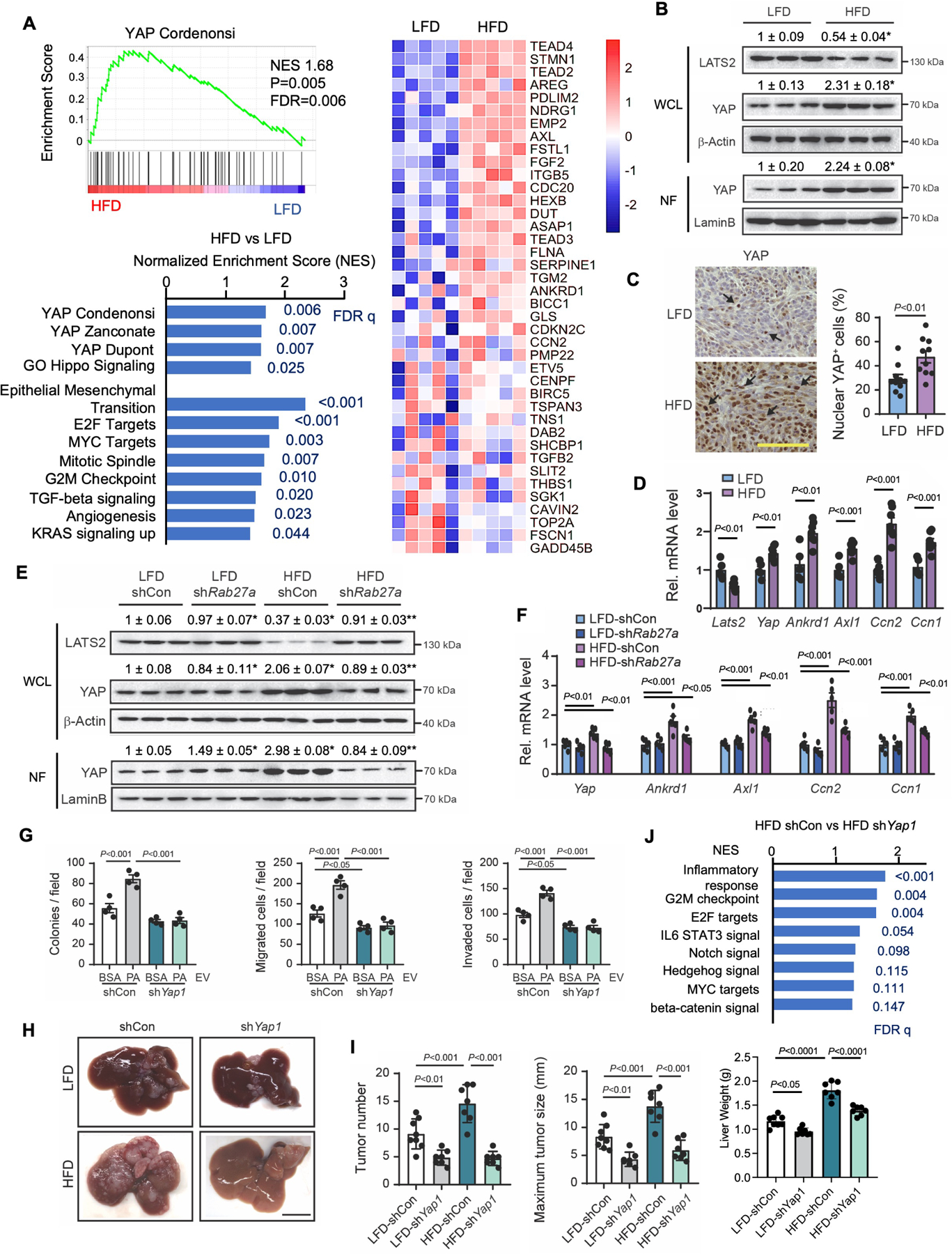
(A) RNA-seq for tumor samples from Figure 1A (n=5/group). GSEA for YAP target and oncogenic gene sets in tumors from LFD-fed and HFD-fed mice. A heatmap of YAP target genes. NES, normalized enrichment score; FDR, false discovery rate.
(B) Immunoblots and quantification of whole cell lysates (WCL) and nuclear fractions (NF) from tumors in Figure 1A.
(C) Representative images of immunohistochemistry for YAP in metastatic liver tumors in mice fed a LFD or HFD (n=10/group). Quantification of nuclear YAP+ cells. Scale bar, 100 μm.
(D) qRT-PCR assays for Lats2, Yap, and YAP target genes (Ankrd1, Axl1, Ccn2, and Ccn1) in metastatic liver tumors in mice fed a LFD (n=5) or HFD (n=7).
(E) Representative immunoblot images and quantifications for LATS2 and YAP in metastatic tumors. Samples were from Figure 1K. Adenovirus expressing shRab27a or shCon was administered into the tail vein before splenic injection of MC38 cells.
(F) qRT-PCR assays for Yap and its target genes (Ankrd1, Axl1, Ccn2, and Ccn1) in metastatic liver tumors.
(G) The effect of Yap1 knockdown in MC38 cells on colony formation, migration, and invasion (n=4 per group). MC38 cells with shCon or shRNA for Yap1 (shYap1) were treated with EVs from supernatant of vehicle-treated (bovine serum albumin, BSA) or PA-treated PHCs.
(H-J) After 6 weeks of LFD or HFD feeding, MC38 cells with shCon or shYap1 were injected into the spleens of mice (n=7–8/group).
(H) Representative macroscopic images of liver metastases. Scale bar, 1 cm.
(I) Number of tumors, maximum tumor size (mm), and liver weights (g).
(J) GSEA oncogenic gene sets in tumors from control and Yap-silenced tumors.
Data shown as mean ± SEM (C,D,F,G) or mean ± SD (I). Significance determined by two-tailed Student’s t-test (C,D) or oneway ANOVA with Tukey’s post hoc analysis (B, E-G, and I). *P<0.05 versus LFD (B). *P<0.05 versus LFD-shCon; **P<0.01 versus HFD-shCon. (E). N.S., not significant.
See also Figure S4.
YAP Signaling Promotes Metastatic Tumor Growth and an Immunosuppressive Environment in Fatty Liver
Because EV production is associated with YAP activity in liver metastasis with fatty liver (Figure 4E and 4F), we investigated the role of YAP in the pro-tumorigenic effect of EVs derived from fat-laden hepatocytes. Proliferation, migration, and invasion were increased in CRC cells treated with EVs from PA-treated hepatocytes, but these features were diminished when Yap1 was silenced (Figure 4G and S4D–S4F). We then investigated the role of YAP in liver metastasis in vivo. Control and Yap1-silenced MC38 cells were inoculated into mice after 6 weeks of HFD feeding. Silencing of Yap1 in CRC in both LFD-fed and HFD-fed mice inhibited tumor growth and lowered oncogenic gene signatures (e.g., MYC targets, cell cycle, interleukin-6, Notch, and β-catenin signaling) (Figure 4H–4J) without affecting fatty liver (Figure S4G). Our data indicate that YAP promotes CRC malignant features regardless of fatty liver. As reported16, fatty liver creates an immunosuppressive TME by infiltration of M2-TAMs. Yap1 knockdown in cancer cells reduced M2-TAM infiltration increased by fatty liver (Figure 5A and S5A). We then investigated whether humoral mediators induced by YAP contribute to the cancer cell-TAM interplay. Liver macrophages co-cultured with WT MC38 cells increased liver macrophage migration compared with MC38 cells without co-culture, whereas Yap1 knockdown in MC38 cells reduced liver macrophage migration (Figure 5B). Similarly, M2 polarization of macrophages was facilitated by co-culture with WT MC38 cells, whereas Yap1 knockdown in MC38 cells did not (Figure 5C). Because Yap1 knockdown in LFD-fed mice did not affect M2-TAM infiltration, we concluded that YAP activity in cancer cells promotes M2-TAM infiltration only in the fatty liver TME. Taken together, pro-metastatic YAP activity could contribute to two effects: the first is the proliferation, migration, and invasion of cancer cells; the second is the cancer cell-TME interplay—the infiltration of TAMs and the polarization of M2 macrophages—through humoral factor secretion. Importantly, these effects of YAP result from EVs released from fat-laden hepatocytes.
Figure 5. CYR61 Is the Critical Factor for YAP-Mediated Liver Metastasis by Induction of an Immunosuppressive Tumor Microenvironment.
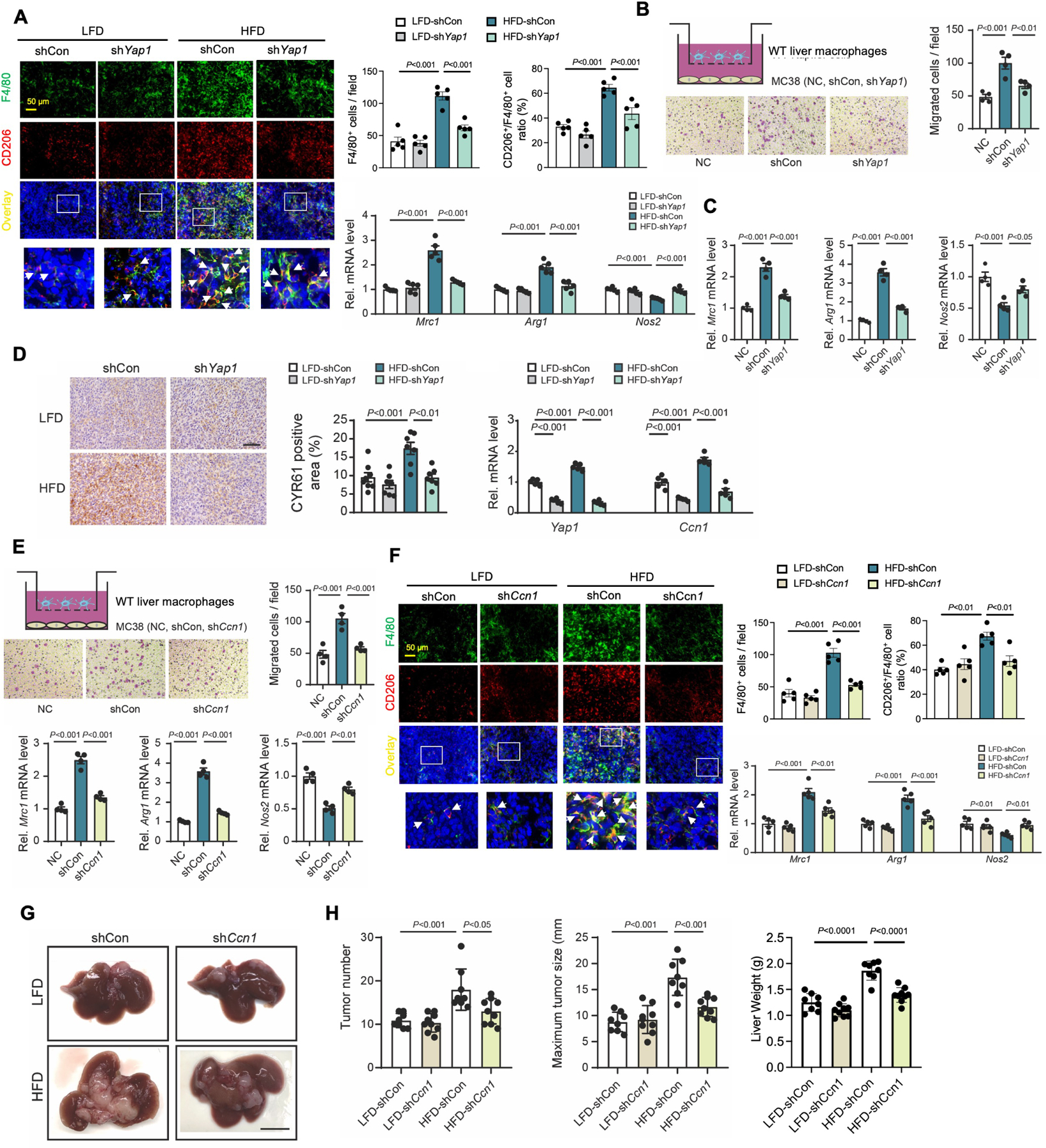
(A) Co-localization of F4/80 and CD206 in tumors from Figure 4H. Representative immunofluorescent images. Quantification of F4/80+ cells and ratio of CD206+/F4/80+. qRT-PCR assays for Mrc1 (CD206), Arg1 (arginase 1), and Nos2 (iNOS) in tumors (n=5/group).
(B) Liver macrophage migration assay. WT liver macrophages were placed in the upper chamber. Culture media only (negative control, NC) or MC38 cells stably overexpressing shCon or shYap1 were seeded in the lower chamber. Representative images (left). Quantification of migrated cells (right) (n=4/group).
(C) qRT-PCR assays for Mrc1, Arg1, and Nos2 in liver macrophages. Liver macrophages were treated with media only (NC) or co-cultured with MC38 cells (shCon or shYap1) (n=4/group).
(D) Representative images of immunohistochemistry for CYR61 in tumors from Figure 4H (LFD-shCon, LFD-shYap1, HFD-shCon, and HFD-shYap1). Scale bar, 100 μm. Quantification of CYR61-positive area. qRT-PCR assays for Yap1 and Ccn1 (n=5/group).
(E) Liver macrophage migration assay. WT liver macrophages were placed in the upper chamber. Culture media only (NC) or MC38 cells with shCon or shRNA for Ccn1 (shCcn1) were seeded in the lower chamber. Representative images (left). Quantification of migrated cells (right) (n=4/group). qRT-PCR assays for Mrc1, Arg1, and Nos2 in liver macrophages. Liver macrophages were treated with media only (NC) or co-cultured with MC38 cells (shCon or shCcn1) (n=4/group).
(F-H) MC38 cells with shCon or shCcn1 (LFD-shCon, LFD-shCcn1, HFD-shCon, and HFD-shCcn1) were inoculated in LFD-fed or HFD-fed mice.
(F) Co-localization of F4/80 and CD206 in tumors. Representative immunofluorescent images (left). Quantification of F4/80+ cells, and ratio of CD206+/F4/80+ (n=5/group)(right). qRT-PCR assays for Mrc1, Arg1, and Nos2 in tumors (n=5/group).
(G) Macroscopic pictures of livers (n=8=9/group). Scale bar, 1 cm.
(H) Number of tumors. Maximum tumor size (mm). Liver weights (g).
Data shown as mean ± SEM (A-F) or mean ± SD (H). Significance determined by one-way analysis of variance (ANOVA) with Tukey’s post hoc analysis (A-F,H).
See also Figure S5.
CYR61 Is the Critical Factor for YAP-mediated Liver Metastasis by Induction of an Immunosuppressive Tumor Microenvironment
Our in vitro experiments suggested that YAP-induced humoral factor(s) contributed to immunosuppressive M2-TAM infiltration. In Yap1-silenced tumors, YAP target molecules, such as Ccn1, Ankrd1, Axl, and Ccn2, were downregulated (Figure 5D and S5B). Among those, CYR61, encoded by Ccn1, is a secreted protein and a chemoattractant 44. The CYR61 protein expression was upregulated in tumors compared to non-tumor liver tissues and it was further increased in tumors with fatty liver (Figure S5C). To investigate the role of CYR61 in cancer cells toward macrophage activity, the Ccn1 gene was silenced in MC38 cells, and liver macrophages were co-cultured with MC38 cells. When Ccn1 was silenced in MC38 cells, liver macrophage migration and M2 polarization of macrophages were inhibited (Figure 5E). In vivo experiments corroborated our in vitro data, showing that increased M2-TAM infiltration and M2 macrophage gene expression in fatty liver were inhibited when Ccn1 was silenced in cancer cells (Figure 5F and S5D). Notably, Ccn1 silencing in cancer cells reduced tumor growth only in mice with fatty liver (Figure 5G and 5H) but did not affect the degree of fatty liver (Figure S5E). Unlike Yap1 knockdown, Ccn1 knockdown in cancer cells did not affect tumor growth and TAM infiltration in LFD-fed mice in vivo or CRC behavior (colony formation, migration, and invasion) in vitro (Figure S5F). In contrast, Ccn1 overexpression increased tumor growth and M2-TAM infiltration in vivo whereas it increased CRC cell migration and invasion, but not proliferation, in vitro (Figure S5G–J). These results suggest that CYR61 mediates the migration of CRC cells and M2-TAMs, contributing to tumor growth in fatty liver. We also examined the role of EVs in M2-TAMs and inflammasome components that we previously reported to play a role in liver metastasis with fatty liver using hepatic Rab27a-silenced mice16. Rab27a silencing reduced the increased inflammasome-related molecules and M2-TAMs in metastases of HFD-fed mice (Figure S5K and S5L), supporting the role of EVs in inflammasome activity and M2-TAM recruitment in the fatty liver TME.
High-Fat Diet and YAP Signaling Contribute to an Immunosuppressive Tumor Microenvironment
To further investigate heterogeneity of M2-TAMs, we employed single-cell RNA sequencing (scRNA-seq) analysis of immune cells in metastatic tumors. Unsupervised clustering identified 22 clusters (Figure 6A and S6A–S6E). Annotation of the clusters with the expression of known markers revealed two M1 and four M2 TAM subpopulations and two distinct CD8 T cell populations45–50. The proportion of total M2-TAMs was not changed by HFD feeding (Figure 6A and 6B). We re-clustered M2-TAMs and determined enriched M2a- and M2b-like subpopulations in HFD-fed mice (Figure 6C, S6F and S6G). Analysis of differentially expressed genes revealed significant alterations in M2-TAMs from HFD-fed mice (Figure 6D, 6E, and S6H). The CYR61 receptor component integrin αv was abundantly expressed in M2-TAMs, but another receptor component, integrin β5, was enriched in M2b- and M2c-like subsets and enriched by HFD feeding (Figure 6C–6E). Expression of Vegfa and Tgfb3 increased in M2b- and M2d-like subsets; Tgfbi and Cxcl16 were enriched in M2a- and M2b-like subsets; Il1b and Havcr2 (Tim3) were enriched in the M2b-like subset; Cd274 (PD-L1) was enriched in the M2d-like subset; and Vsir (VISTA) was enriched in the M2c-like subset in HFD-fed mice (Figure 6E). In addition to M2-TAMs, scar- and lipid-associated macrophages (SAM/LAM) could be important to support tumor growth in NAFLD51,52. Based on the reanalysis of the published data (GSE157600)53, SAM/LAM are the major TAM subset that contains M2 type macrophages in liver metastasis (Figure S6I–N). Our scRNA-seq data revealed all M2 subpopulations are SAM/LAM (Figure S6O–Q). PD1+ CD8 T cells expressed various immunosuppressive genes (Pdcd1lg2 [PD-L2], Ctla4, Lag3, and Cxcr6) enriched in TMEs associated with a HFD (Figure 6F). HFD feeding increased the expression of immunosuppressive genes (Cd274, Havcr2 [Tim3], and Vsir) in PD1− CD8 T cells (Figure 6F). While not increased in mice fed a HFD, Tox expression was higher in PD1+ CD8 T cells than in PD1− CD8 T cells. We then examined the role of YAP in cancer cells in relation to the immune TME in fatty liver. Yap1 silencing in cancer cells reduced Il1b and Cxcl16 expression in M2a-like and M2b-like subsets and reduced Vegfa and Cd274 expression in the M2d-like subset (Figure 6G). Yap1 silencing in cancer cells also reduced the expression of Pdcd1, Pdcd1lg2, Havcr2, and Cxcr6 in CD8 T cells. These data indicate that fatty liver enhanced the immunosuppressive TME, including M2-TAMs, SAM/LAM, and CD8 T cells, partially mediated by tumor-derived YAP activity.
Figure 6. Non-Alcoholic Fatty Liver Disease Contributes to Tumor-Promoting Tumor-associated Macrophages and CD8 T Cell Phenotypes in the Tumor Microenvironment of Colorectal Cancer Liver Metastasis.
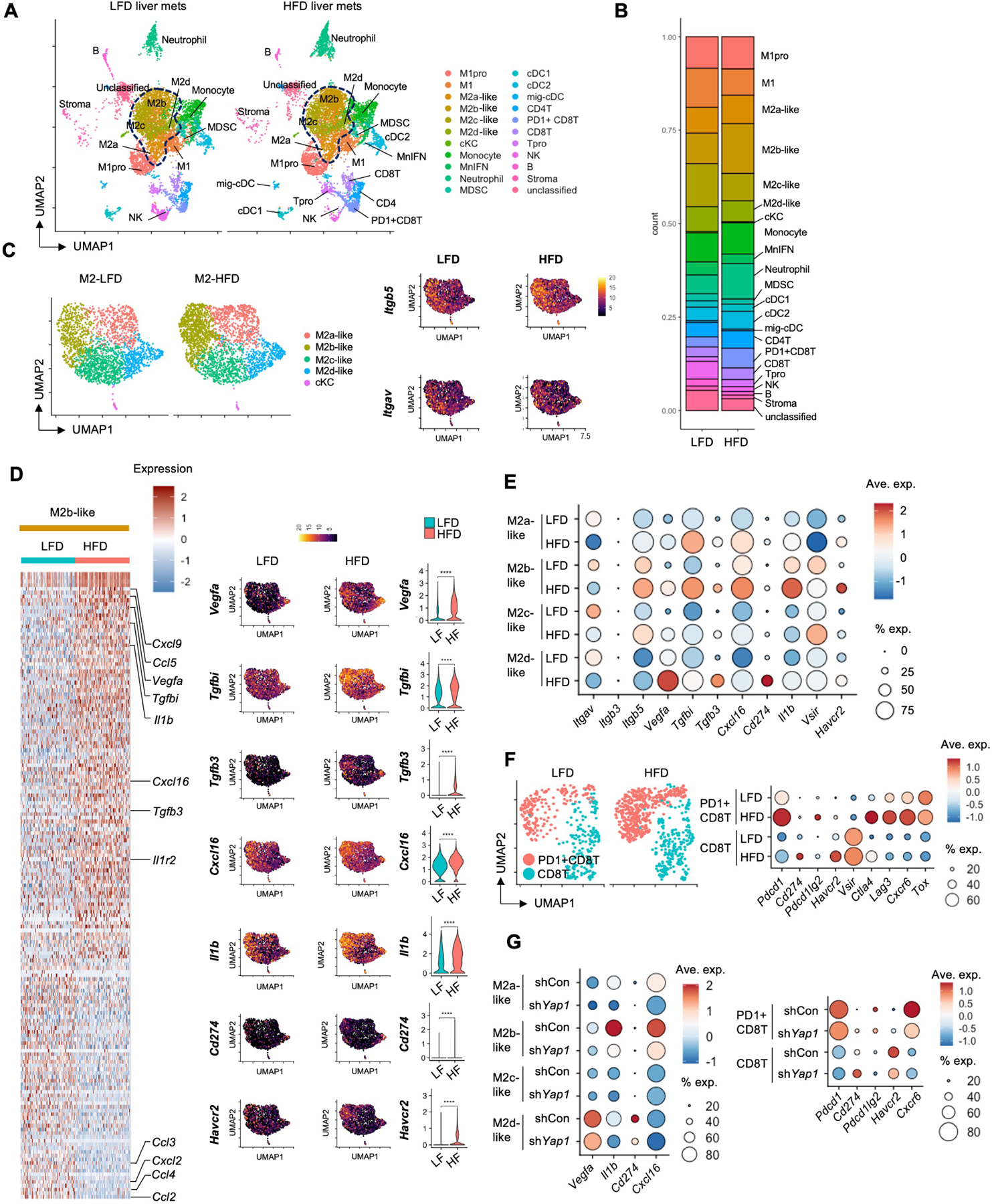
(A) Determination of tumor-infiltrating immune cell populations. Uniform manifold approximation and projection (UMAP) of single-cell RNA-seq (scRNA-seq) from 15,141 CD45+ cells showing 22 clusters determined by integrated analysis, colored by cluster. Cells were from metastatic liver tumors of LFD-fed and HFD-fed mice (n=3/group).
(B) Stacked bar plots depict the proportion of immune cell types in metastatic liver tumors of LFD-fed and HFD-fed mice.
(C) Re-clustered M2 macrophage subpopulations. Expression of CYR61 receptors Itgb5 and Itgav.
(D) Heatmap of the M2b-like subpopulation is shown at a single-cell level. UMAPs and violin plots for Vegfa, Tgfbi, Tgfb3, Cxcl16, Il1b, Cd274, and Havcr2 expression. ****P<0.0001.
(E) Dot plot for expression of key tumor-promoting and immunomodulatory genes (columns) by specific M2 subpopulations (rows). Dot size represents the cell fraction within the M2 subpopulations. Fill color indicates average expression (ave. exp.).
(F) Re-clustering of CD8 T cells with and without Pdcd1 expression. Dot plot for expression of key immunomodulatory genes (columns) by CD8 T cell subpopulations (rows).
(G) scRNA-seq of CD45+ cells from control and Yap1-silenced tumors of HFD-fed mice (n=3/group). Dot plot for expression of key tumor-promoting and immunomodulatory genes (columns) by specific M2 and CD8 T cell subpopulations (rows).
See also Figure S6.
Increased YAP Activity and an Immunosuppressive Tumor Microenvironment in Patients with Colorectal Cancer Liver Metastasis and Non-Alcoholic Fatty Liver Disease
To enhance the clinical implications of our study, we examined tissue samples from patients with CRC liver metastasis with or without fatty liver. The clinical characteristics demonstrated no significant differences in gender, age, ethnicity, and history of chemotherapy between the groups of patients with or without fatty liver. Among several analyzed parameters, only body mass index was higher in the fatty liver group than in the non-fatty liver group (Table S2). Cancer cells were occasionally positive for nuclear YAP and cytoplasmic CYR61 in patients without fatty liver, whereas nuclear YAP and cytoplasmic CYR61 were abundant in patients with fatty liver (Figure 7A). M2-TAMs accumulated more in metastatic tumors in patients with fatty liver compared with those without fatty liver (Figure 7B). To further investigate the immune microenvironment in patients with CRC liver metastasis with or without fatty liver, imaging mass cytometry (IMC) was used for multiplex tissue imaging with metal-conjugated antibodies. Spatial analysis of 29 protein targets was performed using a tissue microarray (TMA) comprising samples from 13 patients with CRC liver metastasis with steatosis and 17 patients with CRC liver metastasis without steatosis (Figure 7C, Table S3). We mapped liver- and TME-constituting cell populations, including hepatocytes, immune cells, stromal cells, and tumor cells (Figure S7A–S7D). The immune cells were re-clustered to T cells, B cells, TAMs, and other myeloid and lymphoid cell populations (Figure 7D, S7E–S7G). TAMs were divided into M1, M2, and other subsets (Figure 7E and 7F). The M2-TAMs were increased in patients with NAFLD. Immune checkpoint molecules (e.g., PD-L1 and galectin-9) were enriched in the M2-TAMs, and TIM3 expression was higher in patients with NAFLD than in patients with healthy livers (Figure 7F). CD8 T cells were enriched in tumors of patients with NAFLD (Figure 7G). PD-1 and PD-L1 in regulatory T cells were higher in patients with NAFLD, and VISTA and TIM3 were higher in all T cell subsets from patients with NAFLD (Figure 7H). Because our TMA panel consisted of both primary and metastatic tumor tissues, we examined the expression of YAP and Ki67 in both primary and metastatic sites. Intriguingly, primary CRC samples from patients with NAFLD had higher YAP and Ki67 expression than patients with healthy livers, and metastatic tumors had higher expression than primary tumors (Figure 7I). We also examined the spatial analysis by measuring the distance between immune cells and metastatic liver tumors. M2-TAMs were located closer to YAP-positive tumors than to YAP-negative tumors (Figure 7J). Conversely, the location of CD8 T cells was more distant to YAP-positive tumors than to YAP-negative tumors. These findings indicate that immunosuppressive M2-TAMs and T cells were enriched in patients with NAFLD, and YAP expression in cancer cells could affect the distance between immune cells and cancer cells, which could regulate the aggressiveness of metastatic tumors. Intriguingly, the IMC analysis of immune cells on primary CRC revealed increased M2-TAMs and immune checkpoint molecules were observed in Stage IV primary CRC with NAFLD (Figure S7H–Q). While T cell subsets were not changed by NAFLD, immune checkpoint molecules were increased in regulatory, CD4 and CD8 T cells (Figure S7R and S7S). The distances of both M2-TAMs and CD8 T cells to YAP-positive tumors were larger than those to YAP-negative tumors (Figure S7T). These findings suggest NAFLD also changes immune TME in the primary site.
Figure 7. Increased YAP Activity and Immunosuppressive Tumor Microenvironment in Colorectal Cancer Liver Metastasis Patients with Non-Alcoholic Fatty Liver Disease.
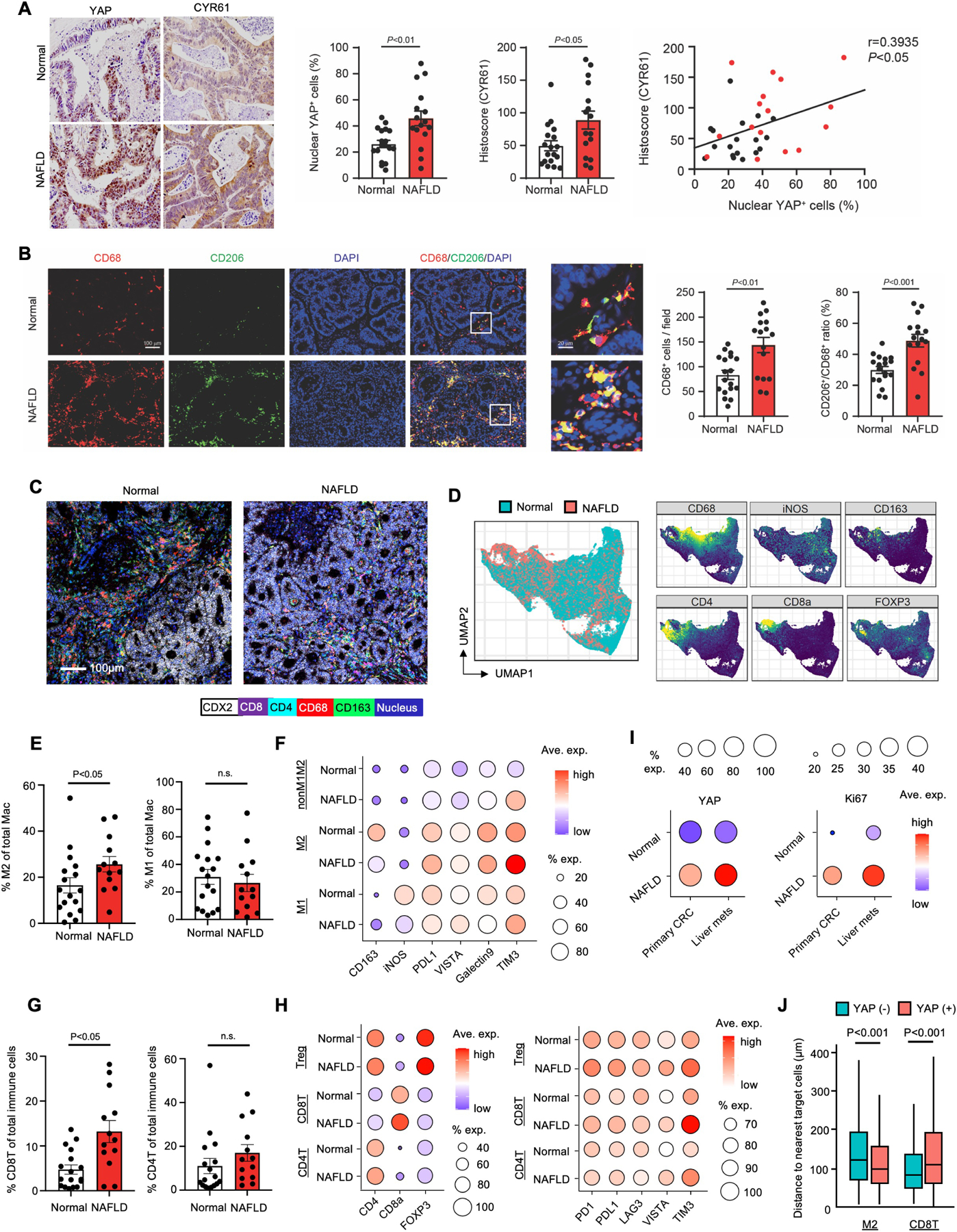
(A) Representative immunohistochemistry images of YAP and CYR61 in metastatic liver tumors of patients with NAFLD (n=16) or without NAFLD (n=18; Normal). Quantification of nuclear YAP+ cells and CYR61-positive areas in human CRC liver metastasis. The correlation between nuclear YAP+ cells and the CYR61-positive areas (right).
(B) Co-localization of CD68 and CD206 in metastatic tumors of patients with NAFLD or without NAFLD (Normal). Representative immunofluorescent images. Quantification of CD68+ cells. Ratio of CD206+/CD68+ cells in human CRC liver metastasis.
(C-H) IMC for TMA comprising CRC liver metastasis patients with NAFLD (n=13) and without NAFLD (n=17; Normal).
(C) Representative IMC images for metastatic liver tumors for CDX2, CD8, CD4, CD68, and CD163 expression.
(D) UMAP of IMC from 147,328 immune cells, colored to distinguish normal and NAFLD conditions (left). UMAPs of expression of CD68, iNOS, CD163, CD8, CD4, and FOXP3 (right).
(E) Quantification of CD163-expressing (M2) and iNOS-expressing (M1) macrophages (Mac).
(F) Dot plot for expression of key cluster-identification and immunomodulatory molecules (columns) by macrophage subpopulations from patients with or without NAFLD (rows). Dot size represents the cell fraction within each cell population. Fill color indicates average expression.
(G) Quantification of CD8 and CD4 T cells.
(H) Dot plot for expression of key cluster-identification and immunomodulatory molecules (columns) by T cell subpopulations in patients with or without NAFLD (rows). Dot size represents the cell fraction within each cell population. Fill color indicates average expression.
(I) Dot plot for YAP and Ki67 expression in primary CRC and metastatic tumors (columns) in patients with or without NAFLD (rows). Dot size represents the cell fraction within each cell population. Fill color indicates average expression.
(J) Spatial analysis of IMC to measure the distances between immune cells (M2 macrophages or CD8 T cells) and metastatic liver tumor cells with and without YAP expression (n=146,429 cells). Data shown as mean ± SEM (A,B,E,G) or mean ± SD (J). Significance determined by two-tailed Student’s t-test (A,B,G,J) or Mann-Whitney U test (E).
DISCUSSION
Recent evidence has demonstrated that NAFLD is more tightly associated with a risk of primary cancers of liver, breast, prostate, pancreas, and colorectum than with a risk of obesity alone4. As 25% of adults in the United States have NAFLD and its prevalence is increasing 54, NAFLD-associated cancers are a significant health issue. The incidence and recurrence rates for CRC liver metastasis is higher in patients with fatty liver than in patients with non-fatty liver5–15. These data suggest that the underlying liver microenvironment could induce pre- and pro-metastatic cues for CRC liver metastasis. While the tumorigenic role of tumor EVs is well-studied, the effect of EVs derived from metastatic organs is poorly understood. We found that fatty liver-derived EVs create the pre-metastatic liver microenvironment. Within the liver, the oncogenic miRNAs in these EVs augment YAP activity in cancer cells by suppressing LATS2. Increased YAP activity intrinsically enhances the malignant potential of cancer cells and creates an immunosuppressive TME through CYR61 induction. The scRNA-seq analysis identified M2-TAMs with higher tumor-promoting factors (e.g., Vegfa and Il-1b) and CD8 T cells with immunomodulatory molecules (e.g., Pdcd1, Cd274, Pdcd1l2, and Havcr2) in HFD-fed mice bearing CRC liver metastasis. The IMC study using human specimens revealed immunosuppressive M2-TAM- and CD8 T cell-enriched TMEs in patients with CRC liver metastasis and fatty liver. Our animal model fed a HFD develop simple steatosis, which is an early stage NAFLD and much more common than non-alcoholic steatohepatitis in humans. Our data suggest that patients with NAFLD, even at the simple steatosis stage, develop a pro-metastatic TME, which is distinct from patients without fatty liver, and may lead to aggressive metastasis growth, therapeutic resistance, and worsening survival rates.
The liver has a unique anatomical relationship with the intestine through the portal vein. Because of intestine-derived factors, such as food- and gut microbiome-derived products, the liver is tolerogenic to those intestinal factors to prevent an immune reaction, which may lead to a favorable landscape for cancer growth and metastasis. Recent reports demonstrated liver metastasis affects systemic immunity by diminishing circulating and liver CD8 T cells, which reduced the efficacy of immunotherapy53,55. The studies showed liver metastasis creates an immunosuppressive microenvironment with M2 characteristics. These data corroborate our study showing increased M2 polarization in patients with liver metastasis and fatty liver. Another report showed HFD feeding induces a systemic immunosuppressive landscape by inhibition of CD8 T cell functions, enhancing subcutaneous CRC growth45. Systemic fat overload induces metabolic re-programming of both cancer and CD8 T cells, creating a favorable environment for cancer growth. A more recent study showed that exhausted CD8 T cells with expression of PD-1 and CXCR6 enriched in liver cancer patients with NAFLD enhance resistance to immune-checkpoint inhibitors56. Our present study shows that CD8 T cells highly express PD-1 and immunomodulatory molecules in fatty liver, suggesting these CD8 T cells have lower anti-cancer capability. These studies suggest an immunosuppressive pro-metastatic TME in fatty liver.
Previous seminal works demonstrated that pro-tumorigenic EVs from primary cancers help create a pre-metastatic niche in the liver23–27. Primary pancreatic cancer-derived EVs prime liver macrophages by delivering macrophage migration inhibitory factor for TGF-β production; this TGF-β creates a fibrotic TME by induction of fibronectin in hepatic stellate cells27. Tumor-derived EVs also create pre-metastatic niches in breast cancer lung metastasis and pancreatic cancer liver metastasis through integrin α6β4 and integrin αvβ1 signaling, respectively26. In addition, EV delivering of EGFR to liver stroma cells promotes gastric cancer liver metastasis25. These studies demonstrate a novel concept of primary cancer cell-derived EVs affecting organotropic metastasis through their functional cargo. Our study proposes that pre- and pro-metastatic niches can be created by metabolic abnormalities in the metastatic organ itself through EV signaling. Our IMC data showed that YAP expression and the TME are altered in both primary and metastatic tumors of patients with NAFLD, indicating that some pro-metastatic signals from fatty liver affect the primary cancer, thus promoting its metastatic potential. This, however, requires further investigation.
The Hippo/YAP pathway is frequently activated in human cancers31. YAP binding to TEAD transcription factors facilitates cancer cell growth, invasion, and metastasis. Increased nuclear YAP and TEAD expression in cancers correlate with poor survival and resistance to chemotherapy, radiotherapy, and immunotherapy. In addition to YAP in cancer cells, YAP in peritumoral hepatocytes and TME-constituting cells also regulates tumor growth in the liver57. YAP controls TAM activity and infiltration of M2-TAM and myeloid-derived suppressor cells in many cancers33,34,58–60. YAP also contributes to evasion from T cell-mediated anti-tumor immunity in melanoma and head and neck cancer with the expression of PD-L1 or by regulatory T cell activity37,61–63. These findings indicate the multifactorial roles of YAP in cancer biology. In our present study, YAP activity in cancer cells increased CYR61 production in fatty liver, recruiting TAMs, which include SAM/LAM, to cancer lesions and polarizing them to a M2 phenotype. Approximately 50% of human liver metastasis samples in the present study had fatty liver. Fatty liver correlates well with nuclear YAP expression with M2-TAM infiltration in both primary and metastatic sites. Considering the general disease rate of NAFLD is ~25%, patients with CRC liver metastasis had higher rates of NAFLD, suggesting patients with NAFLD were vulnerable to CRC liver metastasis, likely through modulation of the TME to cancer-favorable conditions.
In summary, our study demonstrates that the pre- and pro-metastatic liver environment of fatty liver is induced by pro-carcinogenic EVs and results in an immunosuppressive TME, in which YAP plays an important role. Our study provides new insight into distinct liver TMEs in patients with and without fatty liver, which may contribute to the aggressiveness of metastatic tumors and weak responses to anti-cancer therapy in patients with fatty liver. Additional studies are warranted to develop precision medicine for treating patients with CRC and liver metastasis.
Limitations of Study
Our study revealed the pro-metastatic function of fatty liver-derived EVs within the liver. However, we did not address the effect of these EVs in distant sites, including primary cancers. Because our IMC study for human Stage IV CRC suggested primary CRC is more metastatic in patients with NAFLD, signals from fatty liver, including EVs, are likely involved in metastasis. In patients with NAFLD, EVs from adipose tissues and blood cells may also contribute to the circulating EV pool and affect metastasis. In addition, broader ranges of miRNAs in EVs or other EV contents, such as lipids and proteins may have pro-metastatic effects. While we determined the immunosuppressive phenotype of TAMs and T cells in fatty liver TME, we did not address the functional roles of these cells using macrophage-deleted or immunocompromised T and B cell-deficient mice experimentally. To address these limitations, further studies are warranted.
STAR★Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ekihiro Seki (Ekihiro.Seki@cshs.org).
Materials Availability
Plasmids generated in this study are available from the lead contact without restriction.
Data and Code Availability
The raw and processed sequencing data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through Bioproject PRJNA836911 (Bulk RNA sequencing: GSE227913 and single-cell RNA sequencing: GSE227914). Source Data is available in Data S1.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
MC38, CMT93, HCT116, Huh7, and HEK293T cells were cultured in normal Dulbecco’s modified Eagle medium (DMEM) without pyruvate and supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. MC38 cells are from a female mouse, CMT93 cells are from a male mouse, and HEK293T cells are from a human female. HCT116 and Huh7 cells are from human males. All cells were cultured at 37°C in a humidified 5% CO2 incubator. All FBS was heat-inactivated prior to use. All cell lines were free of mycoplasma and were authenticated by the ATCC.
Mice
Six-week-old C57BL/6J male mice were purchased from Jackson Laboratories. For all experiments, 8-week-old mice were randomly assigned to a LFD (PicoLab Rodent Diet 20 5053; LabDiet) or a HFD (60% of calories from fat; Bio-Serv, Cat# S3282) for 6–8 weeks. All mouse colonies and experimental mice were maintained in the same animal facility at Cedars-Sinai Medical Center and housed in specific pathogen-free conditions. All studies were performed in accordance with the National Institutes of Health recommendations outlined in the Guide for the Care and Use of Laboratory Animals. All animal experiment protocols were approved by the Cedars-Sinai Medical Center Institutional Animal Care and Use Committee and the Medical Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology.
Patient Cohorts
Biospecimens (tissue and blood) were collected from patients with NAFLD, patients with CRC liver metastasis and NAFLD, and patients with normal livers undergoing a surgical resection or tissue biopsy at Cedars-Sinai Medical Center. Some blood samples were obtained from healthy volunteers. All patients and volunteers from whom biospecimens were obtained provided informed consent through the Cedars-Sinai Medical Center Institutional Review Board-approved biospecimen collection and analysis protocol. Clinical, demographic, and pathologic data were identified by retrospective review of electronic medical records (Table S1–S3).
METHOD DETAILS
Hepatocyte and Liver Macrophage Isolation and Treatment
C57BL/6J mice (Jackson Laboratory) were used to isolate PHCs using the in situ collagenase perfusion method described previously64. Briefly, PHCs were isolated by collagenase in situ perfusion of livers, followed by 50 x g low speed centrifugation for 3 times to wash cells and separate hepatocytes from non-parenchymal cells. Mouse PHCs in DMEM containing 10% FBS and 1% antibiotic-antimycotic (Thermo Fisher Scientific, Cat# 15240062) were seeded on plates coated with rat collagen I (Corning, Cat# 354236). Primary liver macrophages were isolated from C57BL/6J mice using perfusion with collagenase D (Sigma-Aldrich, Cat# 11088882001) and pronase E (Sigma-Aldrich, Cat# 10165921001), followed by Nycodenz density gradient centrifugation (Accurate Chemical and Scientific Corporation, Cat# AN1002424), as previously described16,65. Briefly, Primary mouse Kupffer cells will be isolated by a 2-step pronase-collagenase in situ perfusion and a 12.5% Nycodenz density gradient centrifugation followed by magnetic cell sorting system using anti-CD11b and F4/80-microbeads (Miltenyi Biotec). Liver macrophages were cultured in DMEM with 10% FBS and 1% antibiotic-antimycotic. In the in vitro experiments, liver macrophages represent liver resident Kupffer cells from normal non-tumor livers.
Mouse PHCs were seeded in 6-well plates and transfected with Silencer Rab27a siRNA (Thermo Fisher Scientific, Cat# AM16706) or Silencer Rab27b siRNA (Thermo Fisher Scientific, Cat# AM16708) using Lipofectamine 3000 transfection reagent (Thermo Fisher Scientific, Cat# L3000015). Silencer Negative Control siRNA (Thermo Fisher Scientific, Cat# AM4638) was used as a negative control. The optimized final concentration of siRNA in the culture medium was 100 nM. Cells and culture media were harvested 48 hours after siRNA transfection. For siRNA and PA combination treatment experiments, PHCs were first transfected with siRNA for 48 hours and then treated with PA (final concentration of 400 μM) for 24 hours. The culture medium was then collected for EV isolation.
Extracellular Vesicle Extraction
EVs were isolated from culture supernatant as follows. Mouse PHCs or Huh7 cells were cultured in DMEM with vehicle or PA (400 μM) for 24 hours, and then culture supernatant was collected for EV isolation. The medium was centrifuged at 300 ×g for 10 minutes to remove whole cells and then at 2000 ×g for 10 minutes to remove debris and apoptotic bodies. The resulting medium was applied to a 0.22 μm vacuum filtration unit (Sigma-Aldrich, Cat# S2GPU05RE) to eliminate large EVs, and then the medium was centrifuged at 100,000 ×g for 70 minutes66,67. Pellets were resuspended in PBS and centrifuged a second time at 100,000 ×g for 70 minutes. The final pellet was resuspended in PBS. The protein concentration of the EV samples was evaluated using a bicinchoninic acid protein assay kit (Thermo Fisher Scientific, Cat# 23225). For some experiments, EVs were isolated from the supernatant of mouse PHCs or Huh7 cells with inhibition of miR-25, miR-92, and/or miR-103 by transfecting with single or combinations of two or three antagomiRs for miR-25, miR-92, and miR-103 or a control antagomiR (100 nM each, Bioneer) for 48 hours followed by vehicle or PA (400 μM) treatment for an additional 24 hours. In these EVs, EV contents of miR-25, miR-92, and miR-103 were decreased. EVs were isolated from serum as follows. Human sera were collected from patients with NAFLD (n=11) and healthy volunteers (n=9) and stored at −80°C. To isolate the EVs, frozen human and mouse sera were thawed in a 37°C water bath and applied to a 0.22 μm filter to remove large EVs. EV isolation was performed using an EV isolation kit for serum (Qiagen, Cat# 77023) in accordance with the manufacturer’s instructions. The resulting pellets were resuspended in PBS. The EVs were stored at −80°C for long-term storage and thawed on ice before use.
To count the number of EVs, isolated EVs were analyzed by tunable resistive pulse sensing on a qNano instrument (Izon Science). First, the qNano instrument was calibrated in accordance with the manufacturer’s recommendations67. Subsequently, the NP100 nanopore membrane was fixed on the instrument and stretched to 49 mm. After applying the EV samples to the membrane, the voltage was set between 0.1–0.4 V to reach a stable 120 nA current. The EV samples were counted for 5 minutes, and at least 500 events were recorded to measure particle numbers. All measurements were calibrated using known concentrations of beads (Izon Science, Cat# CPC100B) with a diameter of 110 nm. Data processing and analysis were performed using the Izon Control Suite software v2.2 (Izon Science).
For PKH26 and PKH67 labeling of EVs, EVs pellets were resuspended in diluent C with PKH26 and PKH67 labeling reagent (Sigma-Aldrich, Cat# MIDI26 and MIDI67) and incubated for 20 minutes at room temperature. Labeled EVs were ultracentrifuged at 100,000 ×g for 70 minutes and washed with PBS. The ultracentrifugation was repeated, and the EVs in the pellet were resuspended in PBS for further analysis.
Mouse Liver Metastasis Models
Splenic injection of MC38 cells (C57BL/6 background, CRC cell line, p53 mutated) and CMT93 cells (C57BL/6 background, CRC cell line, p53 intact) were used to establish a pre-clinical murine model of hepatic metastasis16,68. Briefly, trypsinized CRC cells were resuspended in cold phosphate-buffered saline (PBS) to a final concentration of 106/ml. Mice were anesthetized by isoflurane and a laparotomy was performed on the left abdomen. Then, 100 μl CRC cells (MC38 cells 1×105 cells/mouse and CMT93 cells 1×106 cells/mouse) were injected slowly into the spleen. Mice were euthanized at the study end point (two weeks after CRC cell inoculation) for analysis and sampling. We evaluated metastatic liver tumors macroscopically by counting visible tumor numbers and measuring the maximal size of tumors. Tumor and non-tumor liver tissues were separated macroscopically and used for mRNA, protein, and histological analyses.
In Vivo Adenoviral Vector Treatment
Rab27a-shRNA adenovirus (Vector Biolabs, Cat# shADV-269969), Rab27a adenovirus (Vector Biolabs, Cat# ADV-269969), CYR61 adenovirus (Vector Biolabs, Cat# ADV-256579), and scrambled-shRNA adenovirus (Vector Biolabs, Cat# 1122) were amplified in the HEK293T cells and purified with the Adeno-X™ Maxi Purification Kit (Takara Bio, Cat# 631532). The purified adenoviruses were titered with the Adeno-X™ Rapid Titer Kit (Takara Bio, Cat# 632250) in accordance with the manufacturer’s instructions. Then, 1×109 pfu adenovirus in 100μl PBS was injected into each mouse via the tail vein. The mice were injected with CRC cells into the spleen 48 hours after adenovirus treatment (Figure S1M).
In Vivo EV Treatment
EVs were isolated from culture supernatant of mouse PHCs cultured in DMEM with vehicle or PA (400 μM) for 24 hours as described above. 100 μg EVs in 100μl PBS was injected into each mouse via the tail vein on day 0, 5, and 10 after the splenic injection of CRC cells (Figure S1T)26,27,69,70.
Cell Culture
For EV treatment experiments, MC38 or HCT116 cells were seeded in 6-well plates at 30–50% confluence and then treated with EVs derived from the culture media of primary mouse hepatocytes or Huh7 cells, or EVs extracted from serum of patients or mice at 100 μg/ml. Total RNA and protein were extracted from MC38 and HCT116 cells 48 hours after EV treatment. For the migration and invasion assay, MC38 cells were trypsinized and resuspended 48 hours after EV treatment and then seeded in transwell chambers to evaluate migration and invasion. For colony formation assay, MC38 cells were trypsinized and resuspended 48 hours after EV treatment and then seeded on the plate to evaluate colony formation as the method described below. For miRNA treatment experiments, MC38 and HCT116 cells were seeded in 6-well plates or chamber slides at 50–70% confluency and transfected with 50 nM miRNA mimics (miR-25, miR-92, miR-103, or control) using Lipofectamine 3000 transfection reagent (Thermo Fisher Scientific, Cat# L3000015) for 48 hours. To evaluate migration, invasion, or colony formation after miRNA treatment, cells were trypsinized, counted, and seeded in transwell chambers or on the plate for colony formation assay for further analysis. To examine gene expression, cells were collected for RNA extraction.
Cells with Stable Gene Silencing
Prior to stable transfection, we created a kill curve to establish that 1 μg/ml puromycin (Thermo Fisher Scientific, Cat# A1113803) was the most appropriate concentration required to kill untransfected MC38 cells. MC38 cells were seeded in 6-well plates at 50–70% confluence and transfected using Lipofectamine 3000 transfection reagent with Yap1 shRNA plasmid (Santa Cruz Biotechnology, Cat# sc-38638-SH) or Ccn1 shRNA plasmid (Santa Cruz Biotechnology, Cat# sc-39332-SH), which both contain a puromycin-resistance gene. A plasmid encoding a scrambled-shRNA sequence was used as a control (Santa Cruz, Cat# sc-108060), as it will not cause degradation of specific cellular messages. The cells were washed with PBS 48 hours after transfection and passaged at different dilutions (1:10, 1:20, 1:50, 1:100, 1:200, and 1:500) in DMEM with 10% FBS and 1 μg/ml puromycin. The puromycin-containing medium was changed every 3 days for 2–3 weeks of cell culture. Cell colony formation was carefully monitored, and single colonies were transferred into 96-well plates. After cell passage, Yap1 and Ccn1 mRNA expression was evaluated in each well, and we chose the cells with the most effective gene silencing for further use.
In Vitro Migration and Invasion Assay
Cell migration and invasion were measured using transwell polycarbonate-membrane cell culture inserts (Corning, Cat# CLS3422) and BioCoat Matrigel Invasion Chambers with 8.0 μm PET Membrane (Corning, Cat# 354480), respectively. Cells were harvested after treatments, counted, and resuspended in serum-free DMEM. Then, 104 cells (HCT116 or MC38) or 5 × 103 cells (liver macrophages) were seeded in transwell chambers with or without Matrigel, and DMEM with 10% FBS was added into the lower chamber. The cells were washed with PBS and fixed in 4% paraformaldehyde 16–24 hours after seeding. The cells on the upper polycarbonate membranes were gently wiped with cotton tips. The migrating and invading cells were stained with hematoxylin and eosin. For each independent experiment, four inserts per condition were used, and eight fields were selected randomly under a 10× objective lens on a DMi8 inverted microscope (Leica). The number of invading cells in each image were counted using the “Analyze Particles” function of ImageJ.
In Vitro Colony Formation Assay
Sterilized 1.2% and 0.6% low-molecular-weight agar (LMWA) solutions were prepared in deionized water. Then, 1.2% LMWA and 2× DMEM were mixed with 20% FBS at a 1:1 ratio, and 2 ml of this mixture was added to each well of a 6-well plate. After the agar solidified, 0.6% LMWA and 2× culture medium were mixed with cells and added to the 6-well plates. The plates were placed on a flat surface at 4°C for 15 minutes to solidify the mixture. The final concentration of the cells in each well was 2 × 103/well. After 2–3 weeks of incubation at 37°C and 5% CO2, the cell colonies were fixed with 4% paraformaldehyde for 10 minutes and stained with 0.05% crystal violet solution. The colonies with diameter > 1 mm were counted using ImageJ software.
RNA Extraction, qPCR, and qPCR Array
Total RNA was isolated from cells or tissues with TRIzol (Thermo Fisher Scientific, Cat# 15596018) in accordance with the manufacturer’s instructions. The reverse transcription reactions were performed using iScript Reverse Transcription Supermix (Bio-Rad Laboratories, Cat# 1708841). Then, qRT-PCR was conducted in 96-well plates using iTaq Universal SYBR Green Supermix (Bio-Rad Laboratories, Cat# 1725122) on the CFX96 real-time system (Bio-Rad Laboratories). For miRNA PCR quantification, total RNA was reverse-transcribed into cDNA using a miScript II RT Kit (Qiagen, Cat# 218161) and was analyzed on the CFX96 system using a miScript SYBR Green PCR Kit (Qiagen, Cat# 218075). The comparative CT method was used to calculate the fold change of gene expression. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a housekeeping gene for detection of mRNA expression, and RNU6 was used as an internal control for detection of miRNA expression. Primer sequences are listed in the table.
Total RNA from human and mouse serum EVs was extracted using an RNeasy Mini Kit (Qiagen, Cat# 74104) in accordance with the manufacturer’s instructions. The concentration and quality of the RNA samples were evaluated by absorbance at OD 260/280 nm and OD 230/280 nm using a NanoDrop 2000 (Thermo Fisher Scientific). Then, the total RNA from each sample was diluted to the same concentration to facilitate the following steps. To detect miRNA expression in EVs, the total RNA was reverse-transcribed to cDNA using a miScript II RT Kit (Qiagen, Cat# 218161). In each reaction tube, 1 μg RNA was added to the reaction mixture and incubated at 37°C for 60 minutes and 95°C for 5 minutes, and then the sample was cooled to 4°C. The cDNA samples were diluted 1:10 in nuclease-free water before addition to the PCR array. To identify the candidate miRNAs in EVs, we performed a mouse and human liver Pathway-Focused miScript miRNA PCR Array (Qiagen Cat# 331221) with a miScript SYBR Green PCR Kit (Qiagen, Cat# 218075) on the CFX-96 real-time system. The quantification cycle values (Cq) were calculated using CFX Maestro Software V4.1 (Bio-Rad Laboratories). Raw Cq values were uploaded to Qiagen PCR Array tools for subsequent analysis. RNU6 was determined automatically as the most stable reference miRNA in the array by the Qiagen PCR Array tools.
Luciferase Reporter Assay
WT and mutant LATS2 3′-UTR sequences were constructed and added to the pEZX-MT06 plasmid containing dual-luciferase reporter genes (GeneCopoeia, Cat# MmiT031281-MT06 and MmiT031281-MT06-01). The MC38 cells were seeded in 6-well plates and transfected with the plasmids. After transfection for 24 hours, the cells were treated with miR-25, miR-92a, or miR-103 mimics or a control miRNA (50 nM each) for 48 hours. Afterward, the cell culture medium was collected, and the relative luciferase activity (Gaussia luciferase/secreted alkaline phosphatase ratio) was evaluated using a Luc-Pair Duo-Luciferase Assay Kit 2.0 (GeneCopoeia, Cat# LF001).
Protein Extraction and Western Blotting
Cell and tissue samples were harvested and homogenized in a lysis buffer comprising 50 mM Tris-HCl pH 8.0, 5 mM EDTA, 150 mM NaCl, 0.5% Nonidet P-40, 0.5 mM phenylmethylsulfonyl fluoride, and 0.5 mM dithiothreitol for 30 minutes at 4°C. The protein concentration was determined using a bicinchoninic acid protein assay kit (Thermo Fisher Scientific). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis was used to fractionate 30 μg protein mixed with loading buffer, and the separated samples were transferred to a nitrocellulose membrane. The membrane was blocked with 5% BSA/PBS at room temperature for 1 hour, incubated with primary antibody at 4°C overnight, and then incubated with a secondary antibody conjugated to horseradish peroxidase. Antibody binding was visualized using the enhanced ECL system (Thermo Fisher Scientific, Cat# 32109). The band intensity was quantified using ImageJ software (National Institutes of Health).
Histology
All tissues were fixed in 10% PBS-buffered formalin and embedded in paraffin. The slides were deparaffinized with xylene and dehydrated in gradient ethanol. Antigen retrieval was performed with sodium citrate buffer using a heat-induced method. Mouse monoclonal anti-YAP and rabbit monoclonal anti-CYR61 were used for immunohistochemistry staining on mouse and human tissue slides. For YAP staining of mouse samples, the Mouse on Mouse (M.O.M.) kit (Vector Laboratories, Cat# BMK2202) was used in accordance with the manufacturer’s instructions. After primary antibody incubation, the slides were incubated with horseradish peroxidase-conjugated secondary antibody for 1 hour at room temperature. The ABC Peroxidase Standard Staining Kit and a 3,3′-diaminobenzidine peroxidase substrate were used to visualize the YAP and CYR61 signals on the slides. Images were captured on a DMi8 inverted microscope (Leica) and analyzed by ImageJ software. For Oil Red O staining, frozen liver sections were air-dried at room temperature for 60 minutes and fixed in 10% formalin-PBS for 10 minutes. The fixed sections were rinsed with 60% isopropanol and stained with freshly prepared Oil Red O working solution for 15 minutes. After washing with distilled water, the slides were mounted with aqueous mounting medium. Images were captured on a DMi8 inverted microscope (Leica) and analyzed by ImageJ software. Expression levels were scored based on staining intensity and area of tumor cells using a weighted histoscore calculated from the sum of (1 × % weak staining) + (2 × % moderate staining) + (3 × % strong staining) using ImageJ software 71.
Immunofluorescence Staining
MC38 and HCT116 cells were seeded in slide chambers and treated with indicated stimuli. After routine fixing and blocking, cells were incubated with anti-YAP antibody (1:100 in PBS) at 4°C overnight, followed by Alexa Fluor 488-conjugated secondary antibody for 1 hour at room temperature. Images were captured using a Zeiss LSM800 microscope and analyzed by ImageJ software. F4/80 and CD206 immunofluorescence staining were performed on mouse frozen tissues. The Alexa Fluor 488-conjugated and Alexa Fluor 568-conjugated secondary antibodies were used to visualize the F4/80 and CD206 signals, respectively.
Bulk RNA Sequencing
Total RNA was extracted from metastatic liver tumor tissues using TRIzol and purified using a NucleoSpin RNA kit in accordance with the manufacturer’s instructions. RNA purity, concentration, and integrity were evaluated with an Agilent 2100 BioAnalyzer (Agilent Technologies). The BGI performed the RNA sequencing. RNA libraries were prepared with poly A selection and sequenced using a DNBSEQ-G400 by standard DNBSEQ mRNA library protocols.
Flow Cytometry and Staining
Cells were seeded in 24-well plates at 30–50% confluency and cultured in serum-free DMEM. Then, the cells were treated with PKH26- or PKH67-labeled EVs for 24 hours. The treated cells were suspended in a buffer containing propidium iodide for 30 minutes and analyzed using flow cytometry (BD Bioscience, Accuri™ C6).
Transmission Electron Microscopy
A PBS-diluted EV sample was fixed 1:1 (v/v) in 2% glutaraldehyde (Sigma-Aldrich) for 30 minutes at room temperature. Then, 5 µL of the fixed sample was loaded onto a 200× mesh carbon-coated copper grid and incubated for 10 minutes. The copper grid was washed with distilled water and the excess liquid was carefully removed from the grid. Next, the grid was placed on 30 µL of 1.5% uranyl acetate for 10 minutes, and the grid was examined by electron microscopy using a transmission electron microscope (JEM 1200 EX; JEOL) at the University of California, Los Angeles, USA. The images of EVs were captured with an AMT digital camera for data acquisition.
Single-cell RNA Sequencing
Tumor-infiltrating immune cells were isolated by dissociation of MC38 liver metastatic tumors separated from non-tumor liver tissues, followed by positive selection using anti-CD45 antibodies conjugated to magnetic beads (Miltenyi Biotec) for single-cell analysis. Cells from individual mice were barcoded using TotalSeq cell hashing antibodies. The barcoded tumor- infiltrating CD45+ cells from three different mice were pooled for each group (two different diet conditions [HFD and LFD]; three samples were prepared per diet condition). Single-cell RNA-seq libraries were prepared per the 10x Genomics Single Cell 3’ v 3.1 Reasgent Kits User Guide, and the Chromium Controller, X, or Connect was used. For a target recovery of 10,000 single cells, 12,000–13,000 live cells were loaded onto a Chromium Controller (10x Genomics). Barcoded sequencing libraries were quantified by quantitative PCR using the Collibri Library Quantification Kit (Thermo Fisher Scientific), Fragment analysis was performed on the 4200 TapeStation (Agilent Techenologies), The libraries of samples were sequenced on a NovaSeq 6000 sequencer in accordance with the Single Cell 3’ v 3.1 Reagent Kits User Guide, with a sequencing depth of ~40,000 reads/cell.
Imaging Mass Cytometry and Staining
IMC was performed on formalin-fixed, paraffin-embedded tissue sections of a TMA comprising primary CRC, CRC liver metastasis, and adjacent non-cancer liver tissues from 17 patients without NAFLD and 13 patients with NAFLD (Table S3). Formalin-fixed, paraffin-embedded tissue sections were heated at 65°C for 90 minutes and then processed for antigen retrieval by immersion in xylene for 20 minutes, followed by 100% ethanol, 95% ethanol, 80% ethanol, and 70% ethanol for 5 minutes each. The tissue sections were rinsed for 5 minutes in water and then immersed in Tris-EDTA pH 9 solution with Tween 20 at 95°C for 30 minutes. The slides were cooled in the solution for an additional 30 minutes; then the slides were rinsed in water and in PBS for 10 minutes each. The sections were blocked with a 3% BSA-PBS solution for 45 minutes at room temperature. Metal-conjugated antibodies were obtained from Fluidigm; commercially available antibodies with custom conjugations were created using the Fluidigm Maxpar Antibody Labeling Kit (Table S4). Blocking buffer was replaced with the metal-conjugated antibody staining cocktail, and the sections were incubated at 4°C overnight in a hydration chamber. The following day, the slides were washed twice for 8 minutes in a solution of 0.1% Triton X-100 in PBS and then washed twice for 8 minutes in PBS. The sections were stained with iridium-191/iridium-193 nuclear stain for 40 minutes at room temperature, and the slides were quickly rinsed with water and dried completely at room temperature. The tissue sections were ablated and measured by imaging mass cytometry using a Fluidigm Hyperion imaging system. Image analysis was performed with MCD Viewer software (Fluidigm) and the histoCAT package.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analyses
Statistical significance was assessed using GraphPad Prism version 9.3.1 for mac OS (GraphPad Software). Differences between two groups were determined using a two-sample t-test or the nonparametric Mann-Whitney U test if the normality assumption was violated and the sample size was small. Differences among multiple groups were determined using one-way ANOVA or the nonparametric Kruskal-Wallis test followed by Tukey’s post hoc analysis. Patient data are presented as frequency (percentage, %) for categorical variables and mean (± standard deviation, SD) or median (interquartile range, IQR) for continuous variables, as appropriate. Patient characteristics were compared between groups of interest using two-sample t-tests or Mann-Whitney tests for continuous variables and the chi-squared test or Fisher’s exact test for categorical variables, as appropriate. p-values < 0.05 were considered statistically significant. No data was excluded from this manuscript and can be found online under Data S1 - Source Data. Sample sizes were not predetermined using statistical methods.
Bulk RNA Sequencing Analysis
STAR (2.7.3a) was used to align sequencing reads to the reference mouse genome (GRCm38/mm10) via Partek Flow software. The relative gene expression read counts were mapped using Partek E/M to quantify to the annotation model, mm10 (Ensembl Transcripts release 99). DESeq2 was used to conduct differential gene expression analysis using read counts and the median of ratios method. The P-values of multiple tests were adjusted using the Benjamini-Hochberg method, and the significance level was designated as FDR < 0.01 and log2 (fold change) > 1 or −1. GSEA was performed using GSEA software (Broad Institute)72,73.
Single-cell RNA Sequencing Analysis
Sequencing libraries were loaded onto an Illumina HiSeq system with sequencing settings recommended by 10x Genomics. Sequencing was performed by the Applied Genomics, Computation & Translational Core at Cedars-Sinai Medical Center. The demultiplexed raw reads were aligned to the transcriptome using STAR (version 2.5.1) with default parameters, using mouse mm10 transcriptome reference from Ensembl version 84 annotation, containing all protein coding and long non-coding RNA genes. Expression counts for each gene in all samples were collapsed and normalized to unique molecular identifier (UMI) counts using Cell Ranger software version 4.0.0 (10x Genomics). The processed data contains a large digital expression matrix with cell barcodes as rows and gene identities as columns. The processed matrix were loaded into the R with the Seurat package v4.0.5 in accordance with the introductory vignettes workflow by Seurat using their default parameters. Outlier cells were filtered using library size, number of expressed genes, and mitochondrial proportion (nFeature_RNA > 200, nFeature_RNA < 7500, and percent.mt < 10). Public data (GSE157600) was collected from NCBI Gene Expression Omnibus to obtain the liver samples with or without tumor53. In the outlier filtering steps, lenient criterion on mitochondrial proportion (percent.mt <25) was applied to preserve the sample size. The counts from Cell Ranger were normalized and log2 transformed using the NormalizeData function of the Seurat package. Samples were integrated, and principal component analysis was performed by RunPCA dimensionality reduction, followed by RunUMAP. The first 20 principal components were used for clustering, with 1.0 set as the resolution parameter. In both public and our own data, some clusters were combined by gene expression similarity. The protein tyrosine phosphatase receptor type C (Ptprc, CD45) >1 parameter was used to filter our targeted myeloid cells, and macrophage UMAP was created by cell-identification criteria. Differential gene expression was assessed using the FindMarkers of the Seurat default function.
Imaging Mass Cytometry Analysis
Raw IMC data were processed to obtain single-cell masks (segmentation) and average protein expression per cell using the IMC Segmentation pipeline (https://github.com/BodenmillerGroup/ImcSegmentationPipeline). Briefly, cell segmentation was performed using Ilastik. Select channels were chosen for Ilastik training to identify nuclei (DNA1/DNA2/HistoneH3), cell cytosol (αSMA/CK19/HLAABC/HLADR/HepPar1/CD68), and background areas. Average protein and morphological measurements were taken for each cell using Python and CellProfiler.
Further IMC processing was performed using R packages. Using the output from the IMC Segmentation pipeline, cells larger than 1000 pixels (µm2) and smaller than 15 pixels were removed. Images with fewer than 500 cells and patient samples with fewer than 5,000 cells were also removed. Single-cell protein measurements were arcsinh transformed using a co-factor of 5 and censored at the 99th percentile. Cell phenotyping was performed using PhenoGraph and plotted using UMAP. Two levels of PhenoGraph were performed using cell phenotyping and functional proteins to obtain major cell types (immune, liver, stroma, and tumor) and immune-functional subtypes by manual curation of PhenoGraph clusters. Spatial regions with high liver-cell density were identified and excluded from analysis to obtain metastatic tumor regions from liver metastasis tissue samples. A cell was defined as being in a high liver-density zone if the mean distance of its five nearest liver cells (censored at 100 µm) was less than 30 µm. Single-cell measurements and patient-level measurements were compared using two-tailed Student’s t-test or Mann-Whitney U test.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-CD9 | Santa Cruz Biotechnology | Cat# sc-13118, RRID: AB_631471 |
| Mouse monoclonal anti-CD63 | Santa Cruz Biotechnology | Cat# sc-5275, RRID: AB_627877 |
| Goat polyclonal anti-HSP70 | Santa Cruz Biotechnology | Cat# sc-1060, RRID: AB_631685 |
| Mouse monoclonal anti-TSG101 | Santa Cruz Biotechnology | Cat# sc-7964, RRID: AB_671392 |
| Mouse monoclonal anti-GM130 | Santa Cruz Biotechnology | Cat# sc-55591, RRID: AB_1124984 |
| Rabbit polyclonal anti-cytochrome c | Thermo Fisher Scientific | Cat# 10993–1-AP, RRID: AB_2090467 |
| Mouse monoclonal anti-actin | Sigma-Aldrich | Cat# A5441, RRID: AB_476744 |
| Mouse monoclonal anti-Rab27a | Santa Cruz Biotechnology | Cat# sc-74586, RRID: AB_2176724 |
| Rabbit monoclonal anti-LATS2 (D83D6) | Cell Signaling Technology | Cat# 5888S, RRID: AB_10835233 |
| Mouse monoclonal anti-YAP | Santa Cruz Biotechnology | Cat# sc-101199, RRID: AB_1131430 |
| Rabbit monoclonal anti-phospho-YAP (Ser127) | Cell Signaling Technology | Cat# 13008, RRID: AB_2650553 |
| Mouse monoclonal anti-lamin B1 | Santa Cruz Biotechnology | Cat# sc-374015, RRID: AB_10947408 |
| Mouse monoclonal anti-CYR61 | Novus Biologicals | NB100-356, RRID: AB_10000986 |
| Rat monoclonal anti-F4/80 | Thermo Fisher Scientific | Cat# 14–4801, RRID: AB_2314387 |
| Mouse monoclonal anti-human CD206 | R&D Systems | Cat# MAB25341, RRID: AB_10890782 |
| Goat polyclonal anti-mouse CD206 | R&D Systems | Cat# AF2535, RRID: AB_2063012 |
| Rabbit monoclonal anti-CD68 | Abcam | Cat# ab213363, RRID: AB_2801637 |
| Goat anti-rabbit IgG-biotin | Thermo Fisher Scientific | Cat# 31820, RRID: AB_228340 |
| Goat anti-mouse IgG-biotin | BD Bioscience | Cat# 553999, RRID: AB_395196 |
| Donkey anti-goat antibody-Alexa 568 | Thermo Fisher Scientific | Cat# A11057, RRID: AB_2534104 |
| Goat anti-mouse antibody-Alexa 488 | Thermo Fisher Scientific | Cat# A11001, RRID: AB_2534069 |
| Goat anti-Rabbit antibody-Alexa 568 | Thermo Fisher Scientific | Cat# A11011, RRID: AB_143157 |
| Donkey anti-rat antibody-Alexa 488 | Thermo Fisher Scientific | Cat# A21208, RRID: AB_2535794 |
| Mouse monoclonal anti-αSMA, Clone 1A4 | Novus Biologicals | Cat# NBP2-34522, RRID: N/A |
| Mouse monoclonal anti-CD31, Clone C31.3 | Abcam | Cat# ab187377, RRID: AB_2756834 |
| Mouse monoclonal anti-galectin-9, Clone 1G3 | EMD Millipore | Cat# MABT833, RRID: N/A |
| Rabbit monoclonal anti-YAP1, Clone EP1674Y | Abcam | Cat# ab52771, RRID: AB_2219141 |
| Mouse monoclonal anti-CD14, Clone EPR3653 | Fluidigm | Cat# 3144025D, RRID: N/A |
| Mouse monoclonal anti-CK19, Clone RCK108 | Abcam | Cat# ab9221, RRID: AB_307088 |
| Mouse monoclonal anti-CD163, Clone EDHu-1 | Fluidigm | Cat# 3147021D, RRID: AB_2892115 |
| Rabbit polyclonal anti-NKG2D | Novus Biologicals | Cat# NBP2-43645, RRID: N/A |
| Rabbit monoclonal anti-PDL1, Clone 28–8 | Abcam | Cat# ab205921, RRID: AB_2687878 |
| Sheep polyclonal anti-FAP antibody | R&D Systems | Cat# AF3715, RRID: AB_2102369 |
| Mouse monoclonal anti-CD11c, Clone ITGAX/1242 | Abcam | Cat# ab212508, RRID: N/A |
| Rabbit monoclonal anti-LAG3, Clone D2G40 | Fluidigm | Cat# 3153028D, RRID: N/A |
| Mouse monoclonal anti-HepPar1, Clone V3109 | NSJ Bioreagents | Cat# V3109SAF, RRID: N/A |
| Mouse monoclonal anti-FoxP3, Clone 236A/E7 | Fluidigm | Cat# 3155016D, RRID: N/A |
| Mouse monoclonal anti-CD4, Clone EPR6855 | Fluidigm | Cat# 3156036D, RRID: N/A |
| Mouse monoclonal anti-CD68, Clone KP1 | Fluidigm | Cat# 3159035D, RRID: AB_2810859 |
| Rabbit monoclonal anti-VISTA, Clone D1L2G | Fluidigm | Cat# 3160025D, RRID: AB_2811070 |
| Mouse monoclonal anti-CD20, Clone H1 | Fluidigm | Cat# 3161029D, RRID: AB_2811016 |
| Mouse monoclonal anti-CD8a, Clone C8/144B | Fluidigm | Cat# 3162034D, RRID: AB_2811053 |
| Rabbit monoclonal anti-TIM3, Clone D5D5R | Cell Signaling Technology | Cat# 45208, RRID: AB_2716862 |
| Mouse monoclonal anti-PD1, Clone NAT105 | Abcam | Cat# ab201811, RRID: N/A |
| Rabbit monoclonal anti-iNOS, Clone SP126 | Abcam | Cat# ab239990, RRID: N/A |
| Rabbit monoclonal anti-CDX2, Clone 2951R | Novus Biologicals | Cat# NBP3-08738, RRID: N/A |
| Rabbit polyclonal anti-CD3 | Fluidigm | Cat# 3170019D, RRID: AB_2811048 |
| Mouse monoclonal anti-CD15, Clone HI98 | Biolegend | Cat# 301902, RRID: AB_314194 |
| Rabbit monoclonal anti-CD11b, Clone EPR1344 | Abcam | Cat# ab209970, RRID: N/A |
| Mouse monoclonal anti-Ki67, Clone B56 | BD Biosciences | Cat# 556003, RRID: AB_396287 |
| Rabbit monoclonal anti-histone H3, Clone EPR16987 | Abcam | Cat# ab238971, RRID: N/A |
| Rabbit monoclonal anti-CD453, Clone D9M8I | Cell Signaling Technology | Cat# 47937, RRID: N/A |
| Mouse monoclonal anti-HLA-ABC, Clone EMR8-5 | Abcam | Cat# ab70328, RRID: AB_1269092 |
| Mouse monoclonal anti-HLA-DR, Clone L243 | Fluidigm | Cat# 3174023D, RRID: AB_2811059 |
| – | – | – |
| Bacterial and virus strains | ||
| Scrambled-shRNA with GFP adenovirus | Vector Biolabs | Cat# 1122 |
| Mouse RAB27A shRNA silencing adenovirus | Vector Biolabs | Cat# shADV-269969 |
| Mouse RAB27A adenovirus | Vector Biolabs | Cat# ADV-269969 |
| Mouse GFP-CYR61 adenovirus | Vector Biolabs | Cat# ADV-256579 |
| – | – | – |
| Biological samples | – | – |
| Human CRC liver metastasis samples | Cedars-Sinai Medical Center | N/A |
| – | – | – |
| – | – | – |
| – | – | – |
| – | – | – |
| Chemicals, peptides, and recombinant proteins | ||
| Palmitate | Sigma-Aldrich | Cat# P9767 |
| TRIzol | Thermo Fisher Scientific | Cat# 15596–018 |
| DAPI | Thermo Fisher Scientific | Cat# D1306 |
| Oil Red O | VWR | Cat# AAA12989-14 |
| DMEM, high glucose | Thermo Fisher Scientific | Cat# 11965118 |
| 1x DPBS | Thermo Fisher Scientific | Cat# 21600069 |
| Fetal bovine serum | Sigma-Aldrich | Cat# F0926 |
| Fetal bovine serum, exosome-depleted | Thermo Fisher Scientific | Cat# A2720801 |
| Trypsin EDTA | Thermo Fisher Scientific | Cat# 15400054 |
| Bovine serum albumin | Gold Biotechnology | Cat# A421-100 |
| Puromycin dihydrochloride | Thermo Fisher Scientific | Cat# A1113803 |
| Antibiotic-antimycotic | Thermo Fisher Scientific | Cat# 15240–062 |
| Lipofectamine 3000 | Thermo Fisher Scientific | Cat# L3000015 |
| UltraPure low melting point agarose | Thermo Fisher Scientific | Cat# 16520050 |
| Rat collagen I | Corning | Cat# 354236 |
| Collagenase D | Roche | Cat# 11088866001 |
| Pronase E | Sigma-Aldrich | Cat# 10165921001 |
| Nycodenz | Axis-Shield | Cat# Axs-1002424 |
| – | – | – |
| – | – | – |
| Critical commercial assays | ||
| RNeasy Mini Kit | Qiagen | Cat# 74104 |
| iScript Reverse Transcription Supermix | Bio-Rad Laboratories | Cat# 1708841 |
| iTaq Universal SYBR Green Supermix | Bio-Rad Laboratories | Cat# 1725122 |
| Exosome Isolation Reagent (serum) | Thermo Fisher Scientific | Cat# 4478360 |
| Exosome Isolation Reagent (culture media) | Thermo Fisher Scientific | Cat# 4478359 |
| miScript II RT Kit | Qiagen | Cat# 218161 |
| miScript SYBR Green PCR Kit | Qiagen | Cat# 218075 |
| miScript miRNA PCR Array Human Liver miFinder | Qiagen | Cat# 331221 |
| miScript miRNA PCR Array Mouse Liver miFinder | Qiagen | Cat# 331221 |
| miScript PreAMP PCR Kit | Qiagen | Cat# 331452 |
| exoRNeasy Serum/Plasma Starter Kit | Qiagen | Cat# 77023 |
| exoRNeasy Midi Kit (50) | Qiagen | Cat# 77144 |
| PKH67 Green Fluorescent Cell Linker | Sigma-Aldrich | Cat# MIDI67 |
| PKH26 Red Fluorescent Cell Linker | Sigma-Aldrich | Cat# MIDI26 |
| Nuclear Extraction Kit | Abcam | Cat# ab113474 |
| SuperSignal West Femto Maximum Sensitivity Substrate | Thermo Fisher Scientific | Cat# PI34096 |
| DAB Substrate Kit, Peroxidase | Vector Laboratories | Cat# SK4100 |
| M.O.M. (Mouse on Mouse) Immunodetection Kit | Vector Laboratories | Cat# BMK2202 |
| ABC Peroxidase Standard Staining Kit | Thermo Fisher Scientific | Cat# 32020 |
| Luc-Pair Duo-Luciferase Assay Kit 2.0 | GeneCopoeia | Cat# LF001 |
| Transwell polycarbonate-membrane cell culture insert | Corning | Cat# CLS3422 |
| BioCoat Matrigel Invasion Chambers with 8.0 μm PET Membrane | Corning | Cat# 354480 |
| Bicinchoninic acid protein assay kit | Thermo Fisher Scientific | Cat# 23225 |
| – | – | – |
| Deposited data | ||
| Raw and processed sequencing data | This paper | NCBI Bioproject: GEO: GSE227913; GEO: GSE227914 |
| scRNAseq data | Yu et al. 53 | GSE157600 |
| Data S1. Unprocessed source data underlying all blots and graphs | This paper | N/A |
| – | – | – |
| – | – | – |
| Experimental models: Cell lines | ||
| Mouse MC38 colorectal adenocarcinoma | Gift from Michael Karin 64 | RRID: CVCL_B288 |
| Mouse CMT93 colorectal adenocarcinoma | ATCC | Cat# CCL223, RRID: CVCL_1986 |
| Human HCT116 colon cancer | ATCC | Cat# CCL-247, RRID: CVCL_0291 |
| Human Huh7 hepatocellular carcinoma | CLS | Cat# 300156/p7178_HuH7, RRID: CVCL_0336 |
| Human HEK293T | ATCC | Cat# CRL-3216, RRID: CVCL_0063 |
| – | – | – |
| Experimental models: Organisms/strains | ||
| C57BL/6J | The Jackson Laboratory | – |
| – | – | – |
| – | – | – |
| – | – | – |
| – | – | – |
| – | – | – |
| Oligonucleotides | ||
| Human miR-25-3p primers Forward: CATTGCACTTGTCTCGGTCTGA Reverse: CTCAACTGGTGTCGTGGAGTCGG |
Integrated DNA Technologies | N/A |
| Human miR-92a-3p primers Forward: TATTGCACTTGTCCCGGCCTG Reverse: CTCAACTGGTGTCGTGGAGTCGG |
Integrated DNA Technologies | N/A |
| Human miR-103-3p primers Forward: AGCAGCATTGTACAGGGCTATGA Reverse: CTCAACTGGTGTCGTGGAGTCGG |
Integrated DNA Technologies | N/A |
| Human RNU6-1 primers Forward: CGCAAGGATGACACGCAAATTC Reverse: CTCAACTGGTGTCGTGGAGTCGG |
Integrated DNA Technologies | N/A |
| Mouse miR-25-3p primers Forward: CATTGCACTTGTCTCGGTCTGA Reverse: CTCAACTGGTGTCGTGGAGTCGG |
Integrated DNA Technologies | N/A |
| Mouse miR-92a-3p primers Forward: TATTGCACTTGTCCCGGCCTG Reverse: CTCAACTGGTGTCGTGGAGTCGG |
Integrated DNA Technologies | N/A |
| Mouse miR-103-3p primers Forward: AGCAGCATTGTACAGGGCTATGA Reverse: CTCAACTGGTGTCGTGGAGTCGG |
Integrated DNA Technologies | N/A |
| Mouse RNU6-1 primers Forward: TGACACGCAAATTCGTGAAG Reverse: CTCAACTGGTGTCGTGGAGTCGG |
Integrated DNA Technologies | N/A |
| Mouse miR-151a-3p primers Forward: CTAGACTGAGGCTCCTTGAGG Reverse: CTCAACTGGTGTCGTGGAGTCGG |
Integrated DNA Technologies | N/A |
| Mouse miR-222-3p primers Forward: CTCAGTAGCCAGTGTAGATCC Reverse: CTCAACTGGTGTCGTGGAGTCGG |
Integrated DNA Technologies | N/A |
| Mouse miR-221-3p primers Forward: AGCTACATTGTCTGCTGGGTTTC Reverse: CTCAACTGGTGTCGTGGAGTCGG |
Integrated DNA Technologies | N/A |
| Human ANKRD1 primers Forward: AGAGGCACGTCTGTGTTTTGG Reverse: ATCTTGGCCCCTGAGTTTACC |
Integrated DNA Technologies | N/A |
| Human AXL primers Forward: GTGGGCAACCCAGGGAATATC Reverse: GTACTGTCCCGTGTCGGAAAG |
Integrated DNA Technologies | N/A |
| Human CTGF primers Forward: ACCGACTGGAAGACACGTTTG Reverse: CCAGGTCAGCTTCGCAAGG |
Integrated DNA Technologies | N/A |
| Human CYR61 primers Forward: ACCGCTCTGAAGGGGATCT Reverse: ACTGATGTTTACAGTTGGGCTG |
Integrated DNA Technologies | N/A |
| Human GAPDH primers Forward: GGAGCGAGATCCCTCCAAAAT Reverse: GGCTGTTGTCATACTTCTCATGG |
Integrated DNA Technologies | N/A |
| Human LATS2 primers Forward: ACCCCAAAGTTCGGACCTTAT Reverse: CATTTGCCGGTTCACTTCTGC |
Integrated DNA Technologies | N/A |
| Human Rab27a primers Forward: GCTTTGGGAGACTCTGGTGTA Reverse: TCAATGCCCACTGTTGTGATAAA |
Integrated DNA Technologies | N/A |
| Human Rab27b primers Forward: TAGACTTTCGGGAAAAACGTGTG Reverse: AGAAGCTCTGTTGACTGGTGA |
Integrated DNA Technologies | N/A |
| Human SMPD3 Forward: GCTGCCCTTTGCGTTTCTC Reverse: TCCAGCCGTGAATAGATGTAGG |
Integrated DNA Technologies | N/A |
| Human YAP1 primers Forward: TAGCCCTGCGTAGCCAGTTA Reverse: TCATGCTTAGTCCACTGTCTGT |
Integrated DNA Technologies | N/A |
| Mouse ARG1 primers Forward: TTTTTCCAGCAGACCAGCTT Reverse: AGAGATTATCGGAGCGCCTT |
Integrated DNA Technologies | N/A |
| Mouse AXL primers Forward: GGAACCCAGGGAATATCACAGG Reverse: AGTTCTAGGATCTGTCCATCTCG |
Integrated DNA Technologies | N/A |
| Mouse ANKRD1 primers Forward: AAACGGACGGCACTCCACCG Reverse: CGCTGTGCTGAGAAGCTTGTCT |
Integrated DNA Technologies | N/A |
| Mouse BTG2 primers Forward: ATGAGCCACGGGAAGAGAAC Reverse: GCCCTACTGAAAACCTTGAGTC |
Integrated DNA Technologies | N/A |
| Mouse CPEB2 primers Forward: TTGGAGCAACCATCAGAACAG Reverse: TACCCGAGAACGGCTTCTTCA |
Integrated DNA Technologies | N/A |
| Mouse CTGF primers Forward: GGCCTCTTCTGCGATTTCG Reverse: GCAGCTTGACCCTTCTCGG |
Integrated DNA Technologies | N/A |
| Mouse CYR61 primers Forward: TAAGGTCTGCGCTAAACAACTC Reverse: CAGATCCCTTTCAGAGCGGT |
Integrated DNA Technologies | N/A |
| Mouse FBXW7 primers Forward: GTTCCGCTGCCTAATCTTCCT Reverse: CCCTTCAGGGATTCTGTGCC |
Integrated DNA Technologies | N/A |
| Mouse KLF4 primers Forward: GGCGAGTCTGACATGGCTG Reverse: GCTGGACGCAGTGTCTTCTC |
Integrated DNA Technologies | N/A |
| Mouse GAPDH primers Forward: TGGATTTGGACGCATTGGTC Reverse: TTTGCACTGGTACGTGTTGAT |
Integrated DNA Technologies | N/A |
| Mouse LATS2 primers Forward: GGACCCCAGGAATGAGCAG Reverse: CCCTCGTAGTTTGCACCACC |
Integrated DNA Technologies | N/A |
| Mouse LATS1 primers Forward: AATTTGGCACACATCATAAAGCC Reverse: ACGAGGGTCTTGGTAACTCATT |
Integrated DNA Technologies | N/A |
| Mouse MRC2 primers Forward: CCTGGCTGAAGATGAGGAAG Reverse: GTCTTGCTTCTCGGGGGACT |
Integrated DNA Technologies | N/A |
| Mouse NOS2 primers Forward: ACATCGACCCGTCCACAGTAT Reverse: CAGAGGGGTAGGCTTGTCTC |
Integrated DNA Technologies | N/A |
| Mouse IL-4 primers Forward: GGTCTCAACCCCCAGCTAGT Reverse: GCCGATGATCTCTCTCAAGTGAT |
Integrated DNA Technologies | N/A |
| Mouse IL-13 primers Forward: CACACAAGACCAGACTCCCC Reverse: TCTGGGTCCTGTAGATGGCAAT |
Integrated DNA Technologies | N/A |
| Mouse CD163 primers Forward: TGGGTGGGGAAAGCATAACT Reverse: AAGTTGTCGTCACACACCGT |
Integrated DNA Technologies | N/A |
| Mouse TGF-b1 primers Forward: CAGCTCCTCATCGTGTTGGTG Reverse: GCACATACAAATGGCCTGTCTC |
Integrated DNA Technologies | N/A |
| Mouse IFN-g primers Forward: ATGAACGCTACACACTGCATC Reverse: CCATCCTTTTGCCAGTTCCTC |
Integrated DNA Technologies | N/A |
| Mouse Rab27a primers Forward: TCGGATGGAGATTACGATTACCT Reverse: TTTTCCCTGAAATCAATGCCCA |
Integrated DNA Technologies | N/A |
| Mouse Rab27b primers Forward: TGGCTGAAAAATATGGCATACCA Reverse: CCAGAAGCGTTTCCACTGACT |
Integrated DNA Technologies | N/A |
| Mouse SMPD3 primers Forward: ACACGACCCCCTTTCCTAATA Reverse: GGCGCTTCTCATAGGTGGTG |
Integrated DNA Technologies | N/A |
| Mouse TCF21 primers Forward: CTCCCTGAAAGTGGACTCCAA Reverse: CGGGCTTTTCTTAGTGGGC |
Integrated DNA Technologies | N/A |
| Mouse TGIF1 primers Forward: TGAAGCTGCAATGGGTATCAAA Reverse: GGTATGGCAGATCACTGACGG |
Integrated DNA Technologies | N/A |
| Mouse YAP1 primers Forward: ACCCTCGTTTTGCCATGAAC Reverse: TGTGCTGGGATTGATATTCCGTA |
Integrated DNA Technologies | N/A |
| Human miR-25-3p mimic 5′CAUUGCACUUGUCUCGGUCUGA | Bioland Scientific LLC | N/A |
| Human miR-92a-3p mimic 5′UAUUGCACUUGUCCCGGCCUGU | Bioland Scientific LLC | N/A |
| Human miR-103-3p mimic 5′AGCAGCAUUGUACAGGGCUAUGA | Bioland Scientific LLC | N/A |
| Mouse miR-25-3p mimic 5′CAUUGCACUUGUCUCGGUCUGA | Bioland Scientific LLC | N/A |
| Mouse miR-92a-3p mimic 5′UAUUGCACUUGUCCCGGCCUG | Bioland Scientific LLC | N/A |
| Mouse miR-103-3p mimic 5′AGCAGCAUUGUACAGGGCUAUGA | Bioland Scientific LLC | N/A |
| Human miR-25-3p antisense (antagomiR) | Bioneer | N/A |
| Human miR-92a-3p antisense (antagomiR) | Bioneer | N/A |
| Human miR-103-3p antisense (antagomiR) | Bioneer | N/A |
| Mouse Rab27a siRNA | Thermo Fisher Scientific | Cat# AM16706, Assay ID 161926 |
| Mouse Rab27b siRNA | Thermo Fisher Scientific | Cat# AM16706, Assay ID 174906 |
| Negative Control siRNA | Thermo Fisher Scientific | Cat# AM4638 |
| – | – | – |
| Recombinant DNA | ||
| Yap1 shRNA plasmid (mouse) | Santa Cruz Biotechnology | Cat# sc-38638-SH |
| Ccn1 shRNA plasmid (mouse) | Santa Cruz Biotechnology | Cat# sc-39332-SH |
| Control shRNA plasmid (mouse) | Santa Cruz Biotechnology | Cat# sc-108060 |
| miRNA 3′-UTR target expression clone for mouse Lats2 (NM_015771.2) | GeneCopoeia | Cat# MmiT031281-MT06 |
| Custom mutant (2 sites) miRNA 3′-UTR target for mouse Lats2 (NM_015771.2) | GeneCopoeia | Cat# CS-MmiT031281-MT06-01 |
| miRNA target clone control vector for pEZX-MT06 | GeneCopoeia | Cat# CmiT000001-MT06 |
| – | – | – |
| Software and algorithms | ||
| ImageJ | https://imagej.nih.gov/ij/ | N/A |
| GraphPad Prism 9.0 | GraphPad Software | N/A |
| TargetScan | http://www.targetscan.org | N/A |
| miRDB | http://mirdb.org | – |
| PicTar | https://pictar.mdc-berlin.de | – |
| FlowJo | BD Bioscience | – |
| R 4.1.0 | R Core Team | https://www.r-project.org |
| Rphenograph (R package) | Levin et al. 65 | https://github.com/JinmiaoChenLab/Rphenograph |
| Tidyverse (R package) | https://www.tidyverse.org/ | – |
| Data.table (R package) | https://rdatatable.gitlab.io/data.table/index.html | – |
| Pheatmap (R package) | https://github.com/raivokoldse/pheatmap | – |
| Spatstat (R package) | https://spatstat.org/ | – |
| Rstudio | Rstudio Team, 2020 | http://www.rstudio.com |
| MCD Viewer | Fluidigm, 2021 | https://www.fluidigm.com/products-services/software |
| HistocatS | Schapiro et al. 66 | https://github.com/BodenmillerGroup/histoCAT |
| STAR 2.7.3a | Dobin et al. 67 | https://github.com/alexdobin/STAR |
| GSEA | Broad Institute | https://www.gsea-msigdb.org/gsea/index.jsp |
| Cell Ranger 6.0.0 | 10xGenomics | http://10xgenomics.com/ |
| Seurat package 4.0.5 | Stuart et al. 68 | https://satijalab.org/seurat/ |
| IMCSegmentationPipeline | https://github.com/BodenmillerGroup/ImcSegmentationPipeline | – |
| – | – | – |
| – | – | – |
| Other | ||
| Diet (Normal Chow) | LabDiet | Cat# 3005740–220 |
| HFD | Bio-Serv | Cat# S3282 |
| – | – | – |
| – | – | – |
| – | – | – |
Highlights:
Fatty liver upregulates hepatic Rab27a expression, promoting EV production
Fatty liver-derived EVs enhance YAP activity in CRC liver metastasis
Enhanced YAP induces CYR61 expression in CRC liver metastasis
CYR61 modulates the immunosuppressive environment in CRC liver metastasis
ACKNOWLEDGMENTS
This work is supported by the National Institutes of Health (P01CA233452 to E.S., N.B., S.P., E.P., and S.C.L.; R21AA025841 to E.S. and S.C.L.; R01DK085252 to E.S.), the Center for Integrated Research in Cancer and Lifestyle Award (to E.S.), and a Project Acceleration Award by the Cedars-Sinai Cancer Center at Cedars-Sinai Medical Center (to E.S. and S.P.).
The authors acknowledge the Imaging Mass Cytometry Research Core, the Biobank and Research Pathology Core, and the Applied Genomics, Computation & Translational Research Core at Cedars-Sinai Cancer and Cedars-Sinai Medical Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
M.N. is a member of the advisory board of Altimmune, BI, BMS, Cytodyn, 89BIO, EchoSens, Gilead, GSK, Madrgial, Merck, Novo Nordisk, OWL, Prespecturm, Pfizer, Roche diagnostic and Siemens, Terns and Takeda, received research support from Allergan, Akero, BMS, Gilead, Galectin, Genfit, GSK, Conatus, Corcept, Enanta, Madrigal, Novartis, Novo Nordisk, Shire, Takeda, Terns, Viking and Zydus, and is a stockholder in Anaetos, Rivus Pharma, CIMA, ChronWell and Viking. All other authors declare no competing interests.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
References:
- 1.Keum N, and Giovannucci E (2019). Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nat Rev Gastroenterol Hepatol 16, 713–732. 10.1038/s41575-019-0189-8. [DOI] [PubMed] [Google Scholar]
- 2.Helling TS, and Martin M (2014). Cause of death from liver metastases in colorectal cancer. Ann Surg Oncol 21, 501–506. 10.1245/s10434-013-3297-7. [DOI] [PubMed] [Google Scholar]
- 3.Wang J, Li S, Liu Y, Zhang C, Li H, and Lai B (2020). Metastatic patterns and survival outcomes in patients with stage IV colon cancer: A population-based analysis. Cancer Med 9, 361–373. 10.1002/cam4.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allen AM, Hicks SB, Mara KC, Larson JJ, and Therneau TM (2019). The risk of incident extrahepatic cancers is higher in non-alcoholic fatty liver disease than obesity - A longitudinal cohort study. J Hepatol 71, 1229–1236. 10.1016/j.jhep.2019.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hamady ZZ, Rees M, Welsh FK, Toogood GJ, Prasad KR, John TK, and Lodge JP (2013). Fatty liver disease as a predictor of local recurrence following resection of colorectal liver metastases. Br J Surg 100, 820–826. 10.1002/bjs.9057. [DOI] [PubMed] [Google Scholar]
- 6.Wu K, Zhai MZ, Weltzien EK, Cespedes Feliciano EM, Meyerhardt JA, Giovannucci E, and Caan BJ (2019). Non-alcoholic fatty liver disease and colorectal cancer survival. Cancer Causes Control 30, 165–168. 10.1007/s10552-018-1095-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lv Y, and Zhang HJ (2020). Effect of Non-alcoholic Fatty Liver Disease on the Risk of Synchronous Liver Metastasis: Analysis of 451 Consecutive Patients of Newly Diagnosed Colorectal Cancer. Front Oncol 10, 251. 10.3389/fonc.2020.00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schulz PO, Ferreira FG, Nascimento Mde F, Vieira A, Ribeiro MA, David AI, and Szutan LA (2015). Association of nonalcoholic fatty liver disease and liver cancer. World J Gastroenterol 21, 913–918. 10.3748/wjg.v21.i3.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brouquet A, and Nordlinger B (2013). Metastatic colorectal cancer outcome and fatty liver disease. Nat Rev Gastroenterol Hepatol 10, 266–267. 10.1038/nrgastro.2013.53. [DOI] [PubMed] [Google Scholar]
- 10.Kondo T, Okabayashi K, Hasegawa H, Tsuruta M, Shigeta K, and Kitagawa Y (2016). The impact of hepatic fibrosis on the incidence of liver metastasis from colorectal cancer. Br J Cancer 115, 34–39. 10.1038/bjc.2016.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ocak Duran A, Yildirim A, Inanc M, Karaca H, Berk V, Bozkurt O, Ozaslan E, Ucar M, and Ozkan M (2015). Hepatic steatosis is associated with higher incidence of liver metastasis in patients with metastatic breast cancer; an observational clinical study. J BUON 20, 963–969. [PubMed] [Google Scholar]
- 12.Molla NW, Hassanain MM, Fadel Z, Boucher LM, Madkhali A, Altahan RM, Alrijraji EA, Simoneau EB, Alamri H, Salman A, et al. (2017). Effect of non-alcoholic liver disease on recurrence rate and liver regeneration after liver resection for colorectal liver metastases. Curr Oncol 24, e233–e243. 10.3747/co.24.3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramos E, Torras J, Llado L, Rafecas A, Serrano T, Lopez-Gordo S, Busquets J, and Fabregat J (2016). The influence of steatosis on the short- and long-term results of resection of liver metastases from colorectal carcinoma. HPB (Oxford) 18, 389–396. 10.1016/j.hpb.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amptoulach S, Gross G, and Kalaitzakis E (2015). Differential impact of obesity and diabetes mellitus on survival after liver resection for colorectal cancer metastases. J Surg Res 199, 378–385. 10.1016/j.jss.2015.05.059. [DOI] [PubMed] [Google Scholar]
- 15.Shen Z, Ye Y, Bin L, Yin M, Yang X, Jiang K, and Wang S (2010). Metabolic syndrome is an important factor for the evolution of prognosis of colorectal cancer: survival, recurrence, and liver metastasis. Am J Surg 200, 59–63. 10.1016/j.amjsurg.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 16.Ohashi K, Wang Z, Yang YM, Billet S, Tu W, Pimienta M, Cassel SL, Pandol SJ, Lu SC, Sutterwala FS, et al. (2019). NOD-like receptor C4 Inflammasome Regulates the Growth of Colon Cancer Liver Metastasis in NAFLD. Hepatology 70, 1582–1599. 10.1002/hep.30693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McAndrews KM, and Kalluri R (2019). Mechanisms associated with biogenesis of exosomes in cancer. Mol Cancer 18, 52. 10.1186/s12943-019-0963-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, Moita CF, Schauer K, Hume AN, Freitas RP, et al. (2010). Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol 12, 19–30; sup pp 11–13. 10.1038/ncb2000. [DOI] [PubMed] [Google Scholar]
- 19.Akavia UD, Litvin O, Kim J, Sanchez-Garcia F, Kotliar D, Causton HC, Pochanard P, Mozes E, Garraway LA, and Pe’er D (2010). An integrated approach to uncover drivers of cancer. Cell 143, 1005–1017. 10.1016/j.cell.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, Garcia-Santos G, Ghajar C, et al. (2012). Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med 18, 883–891. 10.1038/nm.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao Y, Zhao MF, Jiang S, Wu J, Liu J, Yuan XW, Shen D, Zhang JZ, Zhou N, He J, et al. (2020). Liver governs adipose remodelling via extracellular vesicles in response to lipid overload. Nat Commun 11, 719. 10.1038/s41467-020-14450-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Povero D, Eguchi A, Li H, Johnson CD, Papouchado BG, Wree A, Messer K, and Feldstein AE (2014). Circulating extracellular vesicles with specific proteome and liver microRNAs are potential biomarkers for liver injury in experimental fatty liver disease. PLoS One 9, e113651. 10.1371/journal.pone.0113651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zeng Z, Li Y, Pan Y, Lan X, Song F, Sun J, Zhou K, Liu X, Ren X, Wang F, et al. (2018). Cancer-derived exosomal miR-25-3p promotes pre-metastatic niche formation by inducing vascular permeability and angiogenesis. Nat Commun 9, 5395. 10.1038/s41467-018-07810-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R, Lovat F, Fadda P, Mao C, Nuovo GJ, et al. (2012). MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci U S A 109, E2110–2116. 10.1073/pnas.1209414109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang H, Deng T, Liu R, Bai M, Zhou L, Wang X, Li S, Wang X, Yang H, Li J, et al. (2017). Exosome-delivered EGFR regulates liver microenvironment to promote gastric cancer liver metastasis. Nat Commun 8, 15016. 10.1038/ncomms15016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, Molina H, Kohsaka S, Di Giannatale A, Ceder S, et al. (2015). Tumour exosome integrins determine organotropic metastasis. Nature 527, 329–335. 10.1038/nature15756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, Becker A, Hoshino A, Mark MT, Molina H, et al. (2015). Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol 17, 816–826. 10.1038/ncb3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Calses PC, Crawford JJ, Lill JR, and Dey A (2019). Hippo Pathway in Cancer: Aberrant Regulation and Therapeutic Opportunities. Trends Cancer 5, 297–307. 10.1016/j.trecan.2019.04.001. [DOI] [PubMed] [Google Scholar]
- 29.Meng Z, Moroishi T, and Guan KL (2016). Mechanisms of Hippo pathway regulation. Genes Dev 30, 1–17. 10.1101/gad.274027.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Samji P, Rajendran MK, Warrier VP, Ganesh A, and Devarajan K (2021). Regulation of Hippo signaling pathway in cancer: A MicroRNA perspective. Cell Signal 78, 109858. 10.1016/j.cellsig.2020.109858. [DOI] [PubMed] [Google Scholar]
- 31.Dey A, Varelas X, and Guan KL (2020). Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat Rev Drug Discov 19, 480–494. 10.1038/s41573-020-0070-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moroishi T, Hayashi T, Pan WW, Fujita Y, Holt MV, Qin J, Carson DA, and Guan KL (2016). The Hippo Pathway Kinases LATS1/2 Suppress Cancer Immunity. Cell 167, 1525–1539 e1517. 10.1016/j.cell.2016.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo X, Zhao Y, Yan H, Yang Y, Shen S, Dai X, Ji X, Ji F, Gong XG, Li L, et al. (2017). Single tumor-initiating cells evade immune clearance by recruiting type II macrophages. Genes Dev 31, 247–259. 10.1101/gad.294348.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang G, Lu X, Dey P, Deng P, Wu CC, Jiang S, Fang Z, Zhao K, Konaparthi R, Hua S, et al. (2016). Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov 6, 80–95. 10.1158/2159-8290.CD-15-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee BS, Park DI, Lee DH, Lee JE, Yeo MK, Park YH, Lim DS, Choi W, Lee DH, Yoo G, et al. (2017). Hippo effector YAP directly regulates the expression of PD-L1 transcripts in EGFR-TKI-resistant lung adenocarcinoma. Biochem Biophys Res Commun 491, 493–499. 10.1016/j.bbrc.2017.07.007. [DOI] [PubMed] [Google Scholar]
- 36.Janse van Rensburg HJ, Azad T, Ling M, Hao Y, Snetsinger B, Khanal P, Minassian LM, Graham CH, Rauh MJ, and Yang X (2018). The Hippo Pathway Component TAZ Promotes Immune Evasion in Human Cancer through PD-L1. Cancer Res 78, 1457–1470. 10.1158/0008-5472.CAN-17-3139. [DOI] [PubMed] [Google Scholar]
- 37.Kim MH, Kim CG, Kim SK, Shin SJ, Choe EA, Park SH, Shin EC, and Kim J (2018). YAP-Induced PD-L1 Expression Drives Immune Evasion in BRAFi-Resistant Melanoma. Cancer Immunol Res 6, 255–266. 10.1158/2326-6066.CIR-17-0320. [DOI] [PubMed] [Google Scholar]
- 38.Zhang H, Deng T, Ge S, Liu Y, Bai M, Zhu K, Fan Q, Li J, Ning T, Tian F, et al. (2019). Exosome circRNA secreted from adipocytes promotes the growth of hepatocellular carcinoma by targeting deubiquitination-related USP7. Oncogene 38, 2844–2859. 10.1038/s41388-018-0619-z. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 39.Lin Q, Zhou CR, Bai MJ, Zhu D, Chen JW, Wang HF, Li MA, Wu C, Li ZR, and Huang MS (2020). Exosome-mediated miRNA delivery promotes liver cancer EMT and metastasis. Am J Transl Res 12, 1080–1095. [PMC free article] [PubMed] [Google Scholar]
- 40.Dasgupta D, Nakao Y, Mauer AS, Thompson JM, Sehrawat TS, Liao CY, Krishnan A, Lucien F, Guo Q, Liu M, et al. (2020). IRE1A Stimulates Hepatocyte-Derived Extracellular Vesicles That Promote Inflammation in Mice With Steatohepatitis. Gastroenterology 159, 1487–1503 e1417. 10.1053/j.gastro.2020.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li J, Liu H, Mauer AS, Lucien F, Raiter A, Bandla H, Mounajjed T, Yin Z, Glaser KJ, Yin M, and Malhi H (2019). Characterization of Cellular Sources and Circulating Levels of Extracellular Vesicles in a Dietary Murine Model of Nonalcoholic Steatohepatitis. Hepatol Commun 3, 1235–1249. 10.1002/hep4.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29. 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kanehisa M, and Goto S (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30. 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bian Z, Peng Y, You Z, Wang Q, Miao Q, Liu Y, Han X, Qiu D, Li Z, and Ma X (2013). CCN1 expression in hepatocytes contributes to macrophage infiltration in nonalcoholic fatty liver disease in mice. J Lipid Res 54, 44–54. 10.1194/jlr.M026013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ringel AE, Drijvers JM, Baker GJ, Catozzi A, Garcia-Canaveras JC, Gassaway BM, Miller BC, Juneja VR, Nguyen TH, Joshi S, et al. (2020). Obesity Shapes Metabolism in the Tumor Microenvironment to Suppress Anti-Tumor Immunity. Cell 183, 1848–1866 e1826. 10.1016/j.cell.2020.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klopfleisch R (2016). Macrophage reaction against biomaterials in the mouse model - Phenotypes, functions and markers. Acta Biomater 43, 3–13. 10.1016/j.actbio.2016.07.003. [DOI] [PubMed] [Google Scholar]
- 47.Roszer T (2015). Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediators Inflamm 2015, 816460. 10.1155/2015/816460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu K, Lin K, Li X, Yuan X, Xu P, Ni P, and Xu D (2020). Redefining Tumor-Associated Macrophage Subpopulations and Functions in the Tumor Microenvironment. Front Immunol 11, 1731. 10.3389/fimmu.2020.01731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anders CB, Lawton TMW, Smith HL, Garret J, Doucette MM, and Ammons MCB (2022). Use of integrated metabolomics, transcriptomics, and signal protein profile to characterize the effector function and associated metabotype of polarized macrophage phenotypes. J Leukoc Biol 111, 667–693. 10.1002/JLB.6A1120-744R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alshetaiwi H, Pervolarakis N, McIntyre LL, Ma D, Nguyen Q, Rath JA, Nee K, Hernandez G, Evans K, Torosian L, et al. (2020). Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Sci Immunol 5. 10.1126/sciimmunol.aay6017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang P, Chen Z, Kuang H, Liu T, Zhu J, Zhou L, Wang Q, Xiong X, Meng Z, Qiu X, et al. (2022). Neuregulin 4 suppresses NASH-HCC development by restraining tumor-prone liver microenvironment. Cell Metab 34, 1359–1376 e1357. 10.1016/j.cmet.2022.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, Lundgren P, Bleriot C, Liu Z, Deczkowska A, et al. (2019). Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell 178, 686–698 e614. 10.1016/j.cell.2019.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu J, Green MD, Li S, Sun Y, Journey SN, Choi JE, Rizvi SM, Qin A, Waninger JJ, Lang X, et al. (2021). Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat Med 27, 152–164. 10.1038/s41591-020-1131-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Truong E, and Noureddin M (2022). The Interplay Between Nonalcoholic Fatty Liver Disease and Kidney Disease. Clin Liver Dis 26, 213–227. 10.1016/j.cld.2022.01.008. [DOI] [PubMed] [Google Scholar]
- 55.Lee JC, Mehdizadeh S, Smith J, Young A, Mufazalov IA, Mowery CT, Daud A, and Bluestone JA (2020). Regulatory T cell control of systemic immunity and immunotherapy response in liver metastasis. Sci Immunol 5. 10.1126/sciimmunol.aba0759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pfister D, Nunez NG, Pinyol R, Govaere O, Pinter M, Szydlowska M, Gupta R, Qiu M, Deczkowska A, Weiner A, et al. (2021). NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 592, 450–456. 10.1038/s41586-021-03362-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moya IM, Castaldo SA, Van den Mooter L, Soheily S, Sansores-Garcia L, Jacobs J, Mannaerts I, Xie J, Verboven E, Hillen H, et al. (2019). Peritumoral activation of the Hippo pathway effectors YAP and TAZ suppresses liver cancer in mice. Science 366, 1029–1034. 10.1126/science.aaw9886. [DOI] [PubMed] [Google Scholar]
- 58.Davra V, Kumar S, Geng K, Calianese D, Mehta D, Gadiyar V, Kasikara C, Lahey KC, Chang YJ, Wichroski M, et al. (2020). Axl and Mertk receptors cooperate to promote breast cancer progression by combined oncogenic signaling and evasion of host anti-tumor immunity. Cancer Res 10.1158/0008-5472.CAN-20-2066. [DOI] [PMC free article] [PubMed]
- 59.Azad T, Nouri K, Janse van Rensburg HJ, Maritan SM, Wu L, Hao Y, Montminy T, Yu J, Khanal P, Mulligan LM, and Yang X (2020). A gain-of-functional screen identifies the Hippo pathway as a central mediator of receptor tyrosine kinases during tumorigenesis. Oncogene 39, 334–355. 10.1038/s41388-019-0988-y. [DOI] [PubMed] [Google Scholar]
- 60.Falcone I, Conciatori F, Bazzichetto C, Bria E, Carbognin L, Malaguti P, Ferretti G, Cognetti F, Milella M, and Ciuffreda L (2020). AXL Receptor in Breast Cancer: Molecular Involvement and Therapeutic Limitations. Int J Mol Sci 21. 10.3390/ijms21228419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miao J, Hsu PC, Yang YL, Xu Z, Dai Y, Wang Y, Chan G, Huang Z, Hu B, Li H, et al. (2017). YAP regulates PD-L1 expression in human NSCLC cells. Oncotarget 8, 114576–114587. 10.18632/oncotarget.23051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stampouloglou E, Cheng N, Federico A, Slaby E, Monti S, Szeto GL, and Varelas X (2020). Yap suppresses T-cell function and infiltration in the tumor microenvironment. PLoS Biol 18, e3000591. 10.1371/journal.pbio.3000591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ni X, Tao J, Barbi J, Chen Q, Park BV, Li Z, Zhang N, Lebid A, Ramaswamy A, Wei P, et al. (2018). YAP Is Essential for Treg-Mediated Suppression of Antitumor Immunity. Cancer Discov 8, 1026–1043. 10.1158/2159-8290.CD-17-1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Q, Kim SY, Matsushita H, Wang Z, Pandyarajan V, Matsuda M, Ohashi K, Tsuchiya T, Roh YS, Kiani C, et al. (2021). Oral administration of PEGylated TLR7 ligand ameliorates alcohol-associated liver disease via the induction of IL-22. Proc Natl Acad Sci U S A 118. 10.1073/pnas.2020868118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang YM, Noureddin M, Liu C, Ohashi K, Kim SY, Ramnath D, Powell EE, Sweet MJ, Roh YS, Hsin IF, et al. (2019). Hyaluronan synthase 2-mediated hyaluronan production mediates Notch1 activation and liver fibrosis. Sci Transl Med 11. 10.1126/scitranslmed.aat9284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Greening DW, Xu R, Ji H, Tauro BJ, and Simpson RJ (2015). A protocol for exosome isolation and characterization: evaluation of ultracentrifugation, density-gradient separation, and immunoaffinity capture methods. Methods Mol Biol 1295, 179–209. 10.1007/978-1-4939-2550-6_15. [DOI] [PubMed] [Google Scholar]
- 67.Minciacchi VR, Spinelli C, Reis-Sobreiro M, Cavallini L, You S, Zandian M, Li X, Mishra R, Chiarugi P, Adam RM, et al. (2017). MYC Mediates Large Oncosome-Induced Fibroblast Reprogramming in Prostate Cancer. Cancer Res 77, 2306–2317. 10.1158/0008-5472.CAN-16-2942. [DOI] [PubMed] [Google Scholar]
- 68.Bhattacharjee S, Hamberger F, Ravichandra A, Miller M, Nair A, Affo S, Filliol A, Chin L, Savage TM, Yin D, et al. (2021). Tumor restriction by type I collagen opposes tumor-promoting effects of cancer-associated fibroblasts. J Clin Invest 131. 10.1172/JCI146987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim J, Lee C, Shin Y, Wang S, Han J, Kim M, Kim JM, Shin SC, Lee BJ, Kim TJ, and Jung Y (2021). sEVs from tonsil-derived mesenchymal stromal cells alleviate activation of hepatic stellate cells and liver fibrosis through miR-486-5p. Mol Ther 29, 1471–1486. 10.1016/j.ymthe.2020.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kang M, Jordan V, Blenkiron C, and Chamley LW (2021). Biodistribution of extracellular vesicles following administration into animals: A systematic review. J Extracell Vesicles 10, e12085. 10.1002/jev2.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cheung EC, DeNicola GM, Nixon C, Blyth K, Labuschagne CF, Tuveson DA, and Vousden KH (2020). Dynamic ROS Control by TIGAR Regulates the Initiation and Progression of Pancreatic Cancer. Cancer Cell 37, 168–182 e164. 10.1016/j.ccell.2019.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34, 267–273. 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw and processed sequencing data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through Bioproject PRJNA836911 (Bulk RNA sequencing: GSE227913 and single-cell RNA sequencing: GSE227914). Source Data is available in Data S1.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.