

Abstract
All of Us is a biorepository aiming to advance biomedical research by providing various types of data in diverse human populations. Here we present a demonstration project validating the program’s genomic data in 98,622 participants. We sought to replicate known genetic associations for three diseases (atrial fibrillation [AF], coronary artery disease, type 2 diabetes [T2D]) and two quantitative traits (height and low-density lipoprotein [LDL]) by conducting common and rare variant analyses. We identified one known risk locus for AF, five loci for T2D, 143 loci for height, and nine loci for LDL. In gene-based burden tests for rare loss-of-function variants, we replicated associations between TTN and AF, GIGYF1 and T2D, ADAMTS17, ACAN, NPR2 and height, APOB, LDLR, PCSK9 and LDL. Our results are consistent with previous literature, indicating that the All of Us program is a reliable resource for advancing the understanding of complex diseases in diverse human populations.
The All of Us Research Program (All of Us) is a prospective cohort study launched in 2018 with the goal of improving population-based research and advancing understanding of human disease. To this end, All of Us plans to enroll at least 1 million individuals living in the United States and collect large-scale electronic health record (EHR) data, laboratory and physical measurements, survey responses, and genomic data.1 As of March 2022, All of Us has released whole-genome sequencing data for 98,622 participants and genotype array data for 165,208 participants. All enrolled individuals have provided written informed consent to the program. In order to validate the quality of the genomic data, All of Us launched demonstration projects aimed at replicating well-established genetic findings within the All of Us dataset. Approval to use the dataset for the specified demonstration projects was obtained from the All of Us Institutional Review Board.
To date, large-scale genome-wide association studies (GWAS) have identified hundreds of risk loci across the human genome for cardiometabolic traits.2–6 In the present study, we analyzed common and rare variants from whole-genome sequencing data in the C2021Q3R6 database version of 98,622 participants. The goal of the current project was to ensure the validity of the All of Us dataset, by replicating established genetic associations for five cardiometabolic traits, including atrial fibrillation (AF), coronary artery disease (CAD), type 2 diabetes (T2D), height, and low-density lipoprotein (LDL).
We modified a previously described phenotyping algorithm7 comprising International Classification of Diseases (ICD) codes, self-reported personal medical history, and procedure and operation codes to define AF in the All of Us dataset. For CAD and T2D, we used published phenotyping algorithms obtained from the Electronic Medical Records and Genomics (eMERGE) network, which have been implemented in All of Us’ phenotype library.8,9 Height and LDL were extracted from the program’s physical measurements and EHR data, respectively. Detailed phenotyping strategies were included in Supplemental Methods and Table S1. After removing participants who did not pass the sample quality control (QC) procedures (Supplemental Methods), we identified 98,564 participants with a mean age of 51.31 (standard deviation [SD] 16.87) at enrollment. Of those, 38,263 (38.82%) were male, and 50,213 (50.94%) were not genetically determined to be of European descent. Characteristics of participants are presented in Table 1 and Table S2. After applying the phenotyping algorithms to the All of Us database, we defined 5,120 (5.19%) AF, 3,544 (3.60%) CAD, and 8,557 (8.68%) T2D patients. Furthermore, we identified 34,538 (35.04%) samples with LDL measurements ascertained from the EHR and 94,842 (96.22%) samples with the All of Us measured height available. Using these data, we tested associations between genetic variants and phenotypes, and compared our results to previously published GWAS by estimating genetic correlation.
Table 1.
Characteristics of participants in the present study
| N=98,564 Mean (SD) or N (%) |
|
|---|---|
| Age at enrollment | 51.31 (16.87) |
| Sex (male) | 38,263 (38.82%) |
| Ancestry (genetically determined) | |
| European | 48,351 (49.06%) |
| African | 23,048 (23.38%) |
| Latino/Admixed American | 15,072 (15.29%) |
| Othera | 8,842 (8.97%) |
| East Asian | 2,114 (2.14%) |
| South Asian | 968 (0.98%) |
| Middle Eastern | 169 (0.17%) |
| Race (self-reported) | |
| White | 51,245 (51.99%) |
| Black or African American | 21,682 (22.00%) |
| Asian | 3,063 (3.11%) |
| More than one population | 1,689 (1.71%) |
| Middle Eastern or North African | 557 (0.57%) |
| Native Hawaiian or Other Pacific Islander | 86 (0.09%) |
| Not availableb | 20,242 (20.54%) |
| Ethnicity (self-reported) | |
| Not Hispanic or Latino | 76,228 (77.34%) |
| Hispanic or Latino | 19,431 (19.71%) |
| Not availablec | 2,905 (2.95%) |
| Atrial fibrillation | 5,120 (5.19%) |
| Coronary artery disease | 3,544 (3.60%) |
| Type 2 diabetes | 8,557 (8.68%) |
| N=34,538 | |
| Low-density lipoprotein (mg/dL) | 105.77 (37.27) |
| N=94,842 | |
| Body height (cm) | 167.67 (9.86) |
Ancestry - other: not belonging to one of the other ancestries or is an admixture.
Race - not available: participants who skipped this survey question or self-reported as “None Indicated”, “None of these”, or “I prefer not to answer” were included in this category.
Ethnicity - not available: participants who skipped this question or selected “None Of These” or “Prefer Not To Answer” were included in this category.
For common variants (minor allele frequency [MAF] > 1%), we used a whole-genome regression approach implemented in the REGENIE10 software to test the association between each phenotype and individual single nucleotide variants (SNVs) assuming an additive genetic model, adjusting for age (enrollment age for disease phenotypes, measurement age for continuous traits), sex, and top 20 principal components of ancestry. For binary traits, we also accounted for case-control imbalance using the saddle point approximation (SPA) method11 implemented in REGENIE. We identified one genome-wide significant (P < 5×10−8) risk locus (defined as 500kb upstream and downstream of the lead SNV) for AF (Table 2, Figure 1a, and Figure S2) upstream of PITX2, an established susceptibility locus for AF.2 Pitx2 is critical for specification of cardiac symmetry, myocardial sleeve development in the pulmonary veins, and suppression of a default sinus node in the left atrium.12,13 We did not observe any inflation in the present AF GWAS (genomic inflation factor [λgc]=1.05, LDSC intercept=1.03, [s.e. 0.01]). The genetic correlation between the All of Us GWAS and a prior AF GWAS14 was 1.02 (s.e. 0.29), estimated using LD score regression (LDSC).15 No genome-wide significant signals were identified for CAD (Figure S1). We, however, noted that the most significant locus was at chromosome 9 (lead SNV=rs10811656, OR=1.14 [1.08–1.19], P-value=6.48×10−7) near CDKN2B-AS1, which has been reported to be associated with CAD in prior studies.16 The All of Us CAD GWAS did not demonstrate any inflation (λgc=1.04, LDSC intercept=1.03 [s.e. 0.01]) and has a genetic correlation of 0.89 (s.e. 0.39) with a previous CAD GWAS.16 For T2D, we identified five genome-wide significant loci (Table 2, Figure 1b, and Figure S3) near HFE, CDKN2B, TCF7L2, CCND2, and FTO. HFE has been linked to glycated hemoglobin (HbA1c) levels in a previous report.17 CDKN2B, TCF7L2, CCND2, and FTO have been reported to be associated with T2D.18–20 Minimal genomic inflation was observed (λgc=1.11) in the current GWAS, which was likely due to polygenicity rather than population stratification, as indicated by its LDSC intercept (1.03, s.e. 0.01). The genetic correlation between the All of Us GWAS and a prior T2D GWAS18 was 0.77 (s.e. 0.10).
Table 2.
Top associations between phenotypes and common variants
| Phenotypes | Genome position (GRCh38) | Effect allele (Other allele) | Effect allele frequency | Effect size [95% CI] | P-value | Mapped gene | Additional independent SNVs (P-value in conditional analysis) |
|---|---|---|---|---|---|---|---|
| Disease | |||||||
| Atrial Fibrillation | chr4:110743002 | A (G) | 0.15 | 1.26 [1.18, 1.34] | 2.40×10−13 | PITX2 | No |
| Type 2 Diabetes | chr6:26276061 | G (T) | 0.04 | 0.79 [0.73, 0.86] | 4.22×10−8 | HFE | No |
| chr9:22136441 | C (G) | 0.25 | 1.11 [1.07, 1.16] | 3.39×10−8 | CDKN2B | No | |
| chr10:113039134 | A (T) | 0.25 | 1.19 [1.15, 1.24] | 4.87×10−20 | TCF7L2 | No | |
| chr12:4275678 | G (T) | 0.01 | 0.66 [0.57, 0.77] | 4.23×10−8 | CCND2 | No | |
| chr16:53773852 | G (A) | 0.46 | 1.12 [1.08, 1.16] | 1.33×10−11 | FTO | No | |
| Quantitative traits | |||||||
| Low-density lipoprotein | chr1:55055522 | T (C) | 0.10 | −0.09 [−0.11, −0.06] | 1.71×10−12 | PCSK9 | No |
| chr1:109274968 | T (G) | 0.22 | −0.12 [−0.13, −0.10] | 3.49×10−38 | CELSR2 | No | |
| chr2:21040767 | G (T) | 0.83 | 0.10 [0.08, 0.12] | 1.59×10−21 | APOB | No | |
| chr2:21072960 | A (G) | 0.31 | 0.07 [0.06, 0.09] | 7.75×10−20 | TDRD15 | Yes (6.23×10−13) | |
| chr5:75360714 | C (T) | 0.38 | 0.06 [0.04, 0.07] | 1.18×10−12 | HMGCR | No | |
| chr6:160589086 | G (A) | 0.05 | 0.12 [0.09, 0.16] | 2.94×10−12 | LPA | No | |
| chr19:11085680 | A (AC) | 0.11 | −0.13 [−0.15, −0.10] | 3.73×10−27 | LDLR | No | |
| chr19:44908684 | C (T) | 0.14 | 0.18 [0.16, 0.20] | 9.51×10−62 | APOE | Yes (6.62×10−39) | |
| chr19:44908822 | T (C) | 0.08 | −0.40 [−0.42, −0.37] | 2.03×10−181 | APOE | No | |
| chr19:44935906 | G (C) | 0.23 | −0.03 [−0.05, −0.02] | 1.46×10−4 | APOC1P1, APOC4 | Yes (1.02×10−9) | |
Common variants: variants with a minor allele frequency (MAF) > 1%. Effect size: Odds Ratios (OR) for binary traits, beta for quantitative traits. CI: confidence interval. Mapped gene: variants were mapped to either the closest or trait-associated genes located within 500kb around the variant. Additional independent SNVs: SNVs that were independently significantly associated with the trait, identified in conditional analyses.
Figure 1.
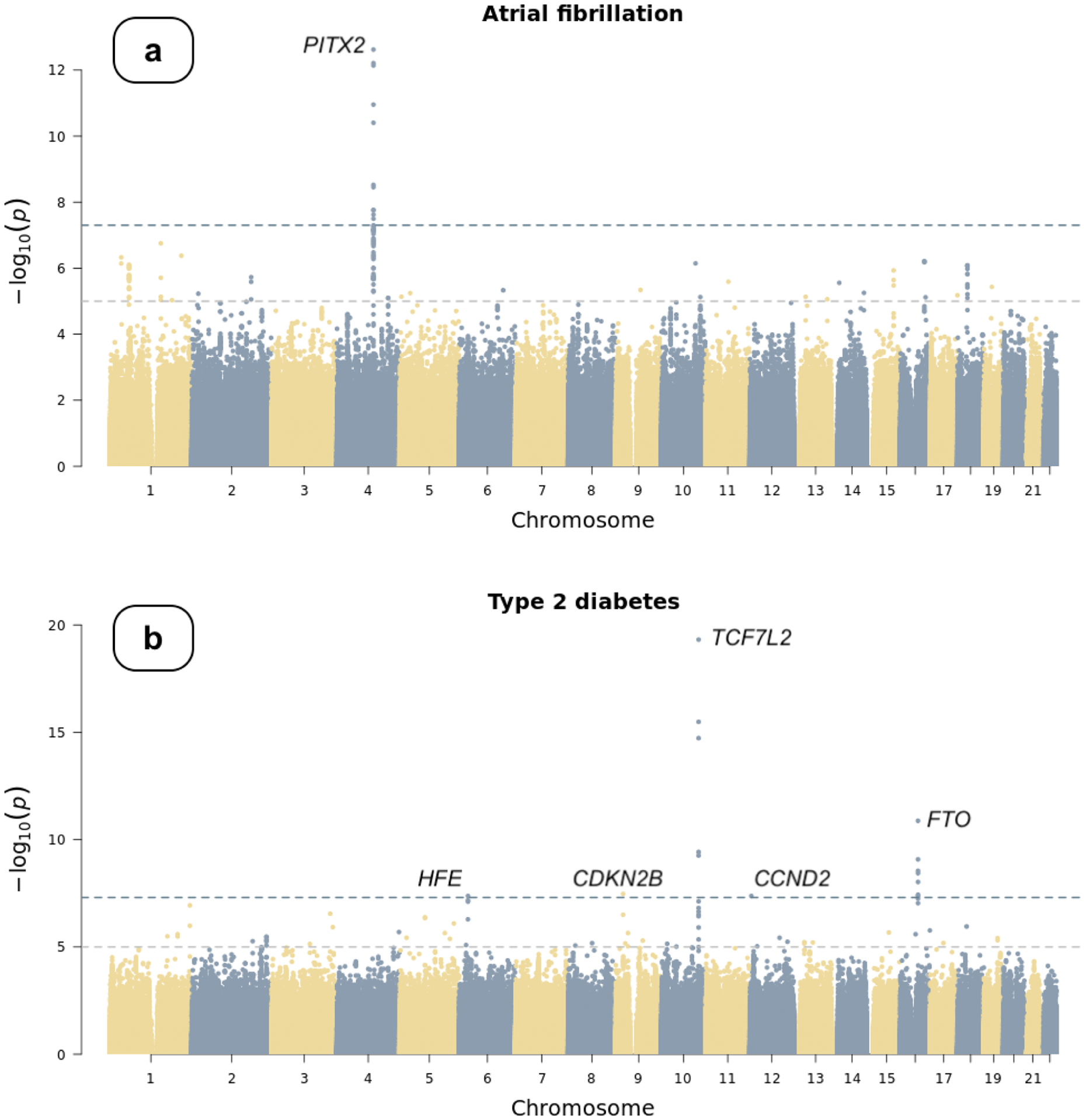
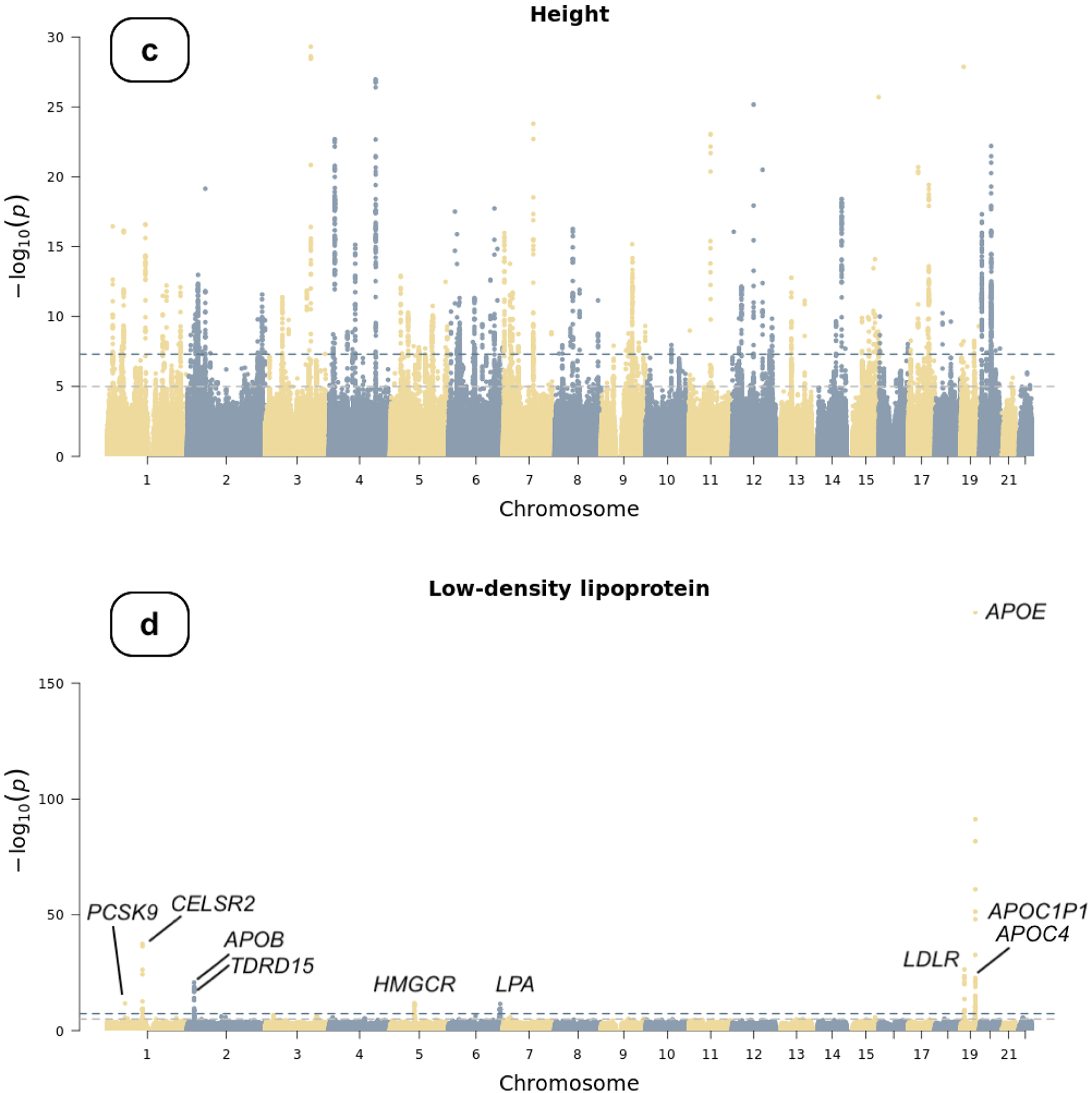
Manhattan plots of genome-wide association studies
Chromosomal variant positions are plotted on the x-axis. The −log10(P values) are plotted on the y-axis. The genome-wide significance threshold (5×10−8) is indicated by the horizontal dotted line. Panels display associations with (a) atrial fibrillation, (b) type 2 diabetes, (c) height, and (d) low-density lipoprotein (LDL). Height and LDL were rank-based inverse normal transformed prior to association testing (see text).
We applied rank-based inverse normal transformation (INT) to body height and LDL cholesterol prior to association testing. Using data from 94,842 participants, we identified 143 genome-wide significant loci in the height GWAS (Figure 1c, Table S3). Additionally, we identified 10 secondary independent SNVs at these loci in a conditional analysis using the GCTA software21 (Figure 1c). The genetic correlation between the All of Us GWAS and a previously published height GWAS6 was 0.96 (s.e. 0.02). Although the genomic inflation factor of the All of Us GWAS was relatively high (λgc=1.33), it was likely due to polygenicity as implicated by its LDSC intercept (1.06, s.e. 0.02) and as height is a highly polygenic trait.22 For LDL cholesterol, we identified seven genome-wide significant loci (Table 2, Figure 1d, and Figure S4) in 34,538 participants implicating PCSK9, CELSR2, APOB, HMGCR, LPA, LDLR, and APOE gene regions. Three additional independent SNVs were identified in a conditional analysis, implicating TDRD15, APOE, and APOC1P1/APOC4 (Table 2). Genetic variants at the gene encoding proprotein convertase subtilisin/kexin type 9 (PCSK9), the gene encoding cadherin EGF LAG seven-pass G-type receptor 2 (CELSR2), the apolipoprotein B and E genes (APOB and APOE), the gene encoding 3-Hydroxy-3-Methylglutaryl-CoA reductase (HMGCR), the LDL receptor gene (LDLR), and the gene encoding tudor domain containing 15 (TDRD15) have been consistently associated with LDL cholesterol levels.23–26 We did not observe any inflation in the current GWAS (λgc=1.02, LDSC intercept=1.01 [s.e. 0.01]). The genetic correlation between the All of Us LDL GWAS and a prior GWAS of LDL27 was 0.99 (s.e. 0.26). We compared the summary statistics of the lead common genetic variants identified in the present study to those in the corresponding reference GWAS (Table S4) and vice versa (Table S5–9).
We then sought to replicate known rare variant (MAF < 0.1% and Population Maximum MAF < 0.1%) associations, including those between TTN and AF, LPL and CAD, GIGYF1 and T2D, ADAMTS10, ADAMTS17, ACAN, NPR2 and height, APOB, LDLR, PCSK9 and LDL.28 We first counted the number of sequenced participants who were carriers of high-confidence loss-of-function (LoF) variants of these genes. LoF variants predicted by Loss-of-Function Transcript Effect Estimator (LOFTEE)29 can disrupt the function of protein-coding genes and thus may have functional impacts on phenotypes that are associated with these genes. Genes with < 20 carriers were removed from the analysis per All of Us’ Data and Dissemination Policy [https://www.researchallofus.org/data-tools/data-access/]. We performed gene-based burden tests as implemented in REGENIE,10 adjusting for age (enrollment age for disease phenotypes, measurement age for continuous traits), sex, top 20 principal components of ancestry, and case-control imbalance using the SPA method for binary traits. We observed significant associations in the All of Us data release for each of the previously reported genes and phenotypes (Figure 2). For example, 1 unit increase in the burden of LoF variants within TTN was associated with 2.23 [1.65, 2.72] (P-value=5.05×10−8) times odds of diagnosing with AF. Likewise, 1 unit increase in the burden of LoF variants within GIGYF1 was associated with 9.03 [3.32, 24.53] (P-value=2.39×10−5) times odds of diagnosing with T2D, and 1 unit increase in the burden of LoF variants within APOB was associated with 1.55 [1.23, 1.90] unit decrease in the INT transformed LDL level. These associations showed the same directional effects and similar effect sizes compared to results from a recently published study using whole-exome sequencing data in the UK Biobank28 (Figure 2).
Figure 2.
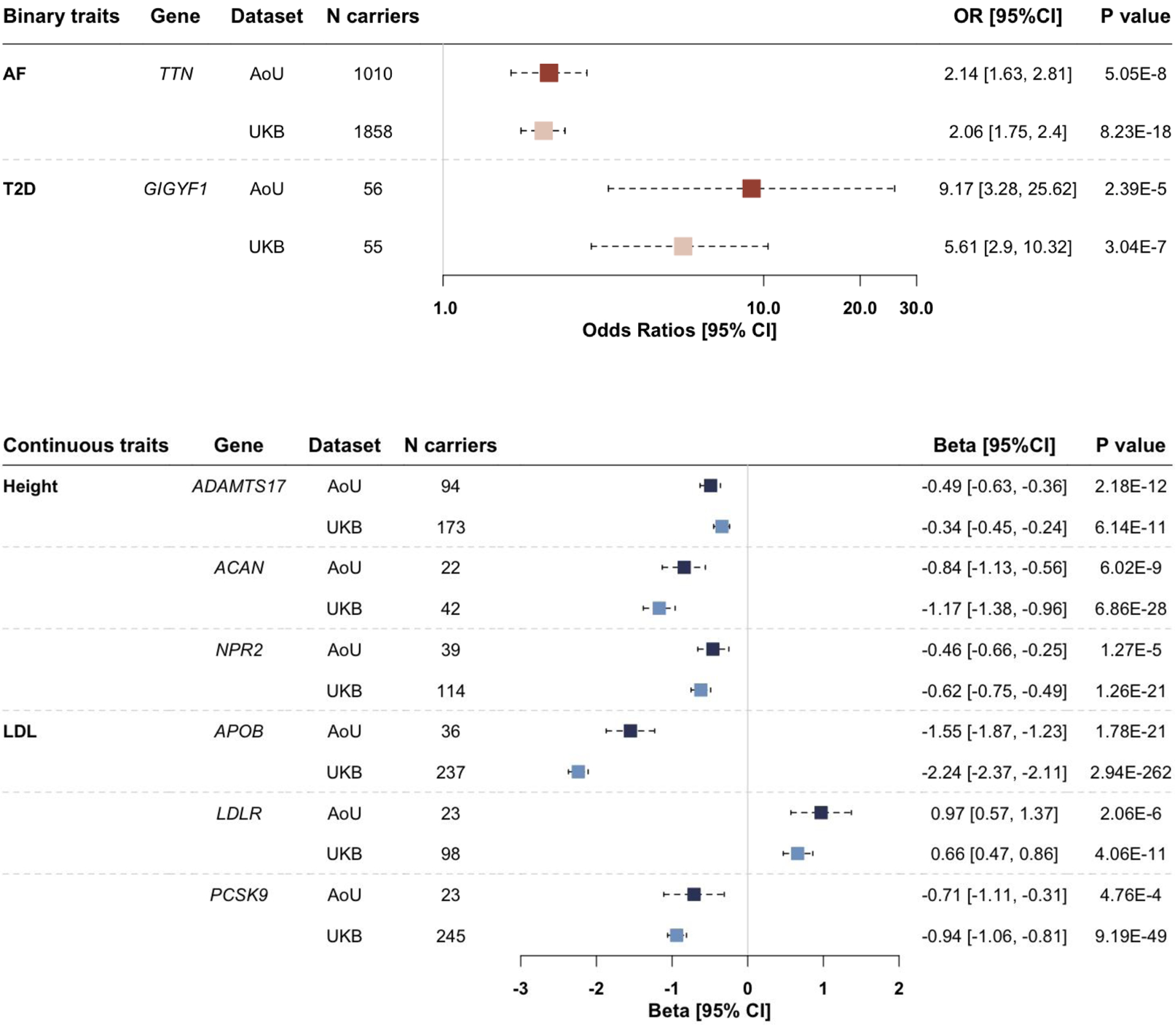
Associations between phenotypes and genes harboring rare variants
Rare variants: rare (minor allele frequency [MAF] < 0.1% and Population Maximum MAF < 0.1%) loss-of-function variants. AF: atrial fibrillation. T2D: type 2 diabetes. LDL: low-density lipoprotein. AoU: the All of Us research program. UKB: UK Biobank. N carriers: number of participants who carry at least one rare variant within each gene. Results are presented in forest plots, with effect sizes (odds ratios [OR] for AF and T2D, betas for height and LDL) and 95% confidence intervals (95% CI). ORs were plotted on a logarithmic scale.
Our study should be interpreted in the context of the design. First, we analyzed samples from all ancestry groups together, which may not fully address population stratification and thus may result in inflated test statistics. However, the whole-genome regression approach has been shown to account for population structure and relatedness and is an established method for analyzing genetic data from diverse populations.10,30 Also, the genomic inflation factors and LDSC intercepts reported in the present study did not indicate inflated test statistics. Second, the disease phenotypes (AF, CAD, and T2D) were defined using electronic health records and self-reported data only, which may result in misclassification and thus limit statistical power. However, the phenotyping algorithms have been validated in previous studies, and the genetic correlation estimates between our GWAS and corresponding previously published large-scale GWAS indicated good consistency. Scatter plots comparing the effect sizes of significantly associated variants reported by the reference GWAS to the effect sizes of those variants in our study showed high correlations (Figure S6), indicating that the potential phenotype misclassification issue did not severely impact the validity of the results. Third, the LDL levels in the present study were not corrected for statin use. More precise testing variables can be obtained if electronic health record data is leveraged to perform this adjustment. However, since the current GWAS has a high genetic correlation with the reference GWAS, we submit that the present analysis suffices for dataset quality validation. Fourth, we only included high-confidence loss-of-function variants in the rare variant analysis, which may not fully represent the associations between genes and phenotypes since other rare variants (e.g., deleterious missense variants) that we did not include may have an impact on phenotypic expression.
In conclusion, we replicated known genetic associations in the current release of the All of Us research program, indicating that the dataset is a rich and robust resource for common and rare variant genetic discovery. The results of our analyses support the validity of genetic discovery in this multi-ancestry sample. As more data become available in the coming releases, the use of this dataset will facilitate the advancement of biomedical research in diverse human populations.
Supplementary Material
Acknowledgements
We would like to thank the All of Us research program participants, as this study and the database are possible because of their contributions. All of Us established core values and responsible strategies to sustain public trust in biomedical research. We hope the partnership between the participants and the program will benefit the participants and improve the health of future generations.
Funding Sources
The All of Us Research Program is supported by the National Institutes of Health, Office of the Director: Regional Medical Centers: 1 OT2 OD026549; 1 OT2 OD026554; 1 OT2 OD026557; 1 OT2 OD026556; 1 OT2 OD026550; 1 OT2 OD 026552; 1 OT2 OD026553; 1 OT2 OD026548; 1 OT2 OD026551; 1 OT2 OD026555; IAA #: AOD 16037; Federally Qualified Health Centers: HHSN 263201600085U; Data and Research Center: 5 U2C OD023196; Biobank: 1 U24 OD023121; The Participant Center: U24 OD023176; Participant Technology Systems Center: 1 U24 OD023163; Communications and Engagement: 3 OT2 OD023205; 3 OT2 OD023206; and Community Partners: 1 OT2 OD025277; 3 OT2 OD025315; 1 OT2 OD025337; 1 OT2 OD025276. In addition, the All of Us Research Program would not be possible without the partnership of its participants. Dr. Ellinor is supported by grants from the National Institutes of Health (1RO1HL092577, 1R01HL157635, 1R01HL157635), from the American Heart Association (18SFRN34110082), and from the European Union (MAESTRIA 965286). Dr. Lubitz previously received support from NIH grants R01HL139731 and R01HL157635, and American Heart Association 18SFRN34250007 during this project. Dr. Choi was previously supported by the NHLBI BioData Catalyst Fellows program.
Conflict of Interest
The authors declare no competing non-financial interests but the following competing financial interests: Dr. Ellinor receives sponsored research support from Bayer AG, IBM Research, Bristol Myers Squibb, and Pfizer; he has also served on advisory boards or consulted for Bayer AG and MyoKardia. Dr. Lubitz is a full-time employee of Novartis as of July 18, 2022. Dr. Lubitz has received sponsored research support from Bristol Myers Squibb, Pfizer, Boehringer Ingelheim, Fitbit, Medtronic, Premier, and IBM, and has consulted for Bristol Myers Squibb, Pfizer, Blackstone Life Sciences, and Invitae.
Data Availability
Access to individual-level data from the All of Us research program is available to researchers whose institution has signed a data use agreement with All of Us (https://www.researchallofus.org/register/). All of Us provides a publicly available data browser (https://databrowser.researchallofus.org/) containing aggregate-level participant data for users to explore the available data, including genomic variants. Electronic health records (EHR) data, used for phenotyping, belongs to the registered tier dataset. Whole-genome sequencing data belongs to the controlled tier dataset, which requires additional training to access.
References
- 1.The “All of Us” Research Program | NEJM. Accessed November 1, 2021. https://www.nejm.org/doi/full/10.1056/NEJMsr1809937
- 2.Roselli C, Rienstra M, Ellinor PT. Genetics of Atrial Fibrillation in 2020. Circ Res. 2020;127(1):21–33. doi: 10.1161/CIRCRESAHA.120.316575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McPherson R, Tybjaerg-Hansen A. Genetics of Coronary Artery Disease. Circ Res. 2016;118(4):564–578. doi: 10.1161/CIRCRESAHA.115.306566 [DOI] [PubMed] [Google Scholar]
- 4.Ali O Genetics of type 2 diabetes. World J Diabetes. 2013;4(4):114–123. doi: 10.4239/wjd.v4.i4.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.A large electronic-health-record-based genome-wide study of serum lipids | Nature Genetics. Accessed November 2, 2021. https://www.nature.com/articles/s41588-018-0064-5 [DOI] [PMC free article] [PubMed]
- 6.Yengo L, Sidorenko J, Kemper KE, et al. Meta-analysis of genome-wide association studies for height and body mass index in ~700000 individuals of European ancestry. Hum Mol Genet. 2018;27(20):3641–3649. doi: 10.1093/hmg/ddy271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khurshid S, Choi SH, Weng LC, et al. Frequency of Cardiac Rhythm Abnormalities in a Half Million Adults. Circ Arrhythm Electrophysiol. 2018;11(7):e006273. doi: 10.1161/CIRCEP.118.006273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.MidSouth CDRN - Coronary Heart Disease Algorithm | PheKB. Accessed November 2, 2021. https://phekb.org/phenotype/midsouth-cdrn-coronary-heart-disease-algorithm
- 9.Type 2 Diabetes Mellitus | PheKB. Accessed November 2, 2021. https://www.phekb.org/phenotype/type-2-diabetes-mellitus
- 10.Mbatchou J, Barnard L, Backman J, et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat Genet. 2021;53(7):1097–1103. doi: 10.1038/s41588-021-00870-7 [DOI] [PubMed] [Google Scholar]
- 11.Zhou W, Nielsen JB, Fritsche LG, et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat Genet. 2018;50(9):1335–1341. doi: 10.1038/s41588-018-0184-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ryan AK, Blumberg B, Rodriguez-Esteban C, et al. Pitx2 determines left-right asymmetry of internal organs in vertebrates. Nature. 1998;394(6693):545–551. doi: 10.1038/29004 [DOI] [PubMed] [Google Scholar]
- 13.Tessari A, Pietrobon M, Notte A, et al. Myocardial Pitx2 Differentially Regulates the Left Atrial Identity and Ventricular Asymmetric Remodeling Programs. Circ Res. 2008;102(7):813–822. doi: 10.1161/CIRCRESAHA.107.163188 [DOI] [PubMed] [Google Scholar]
- 14.Roselli C, Chaffin MD, Weng LC, et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat Genet. 2018;50(9):1225–1233. doi: 10.1038/s41588-018-0133-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bulik-Sullivan BK, Loh PR, Finucane HK, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015;47(3):291–295. doi: 10.1038/ng.3211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van der Harst P, Verweij N. Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease. Circ Res. 2018;122(3):433–443. doi: 10.1161/CIRCRESAHA.117.312086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soranzo N, Sanna S, Wheeler E, et al. Common Variants at 10 Genomic Loci Influence Hemoglobin A1C Levels via Glycemic and Nonglycemic Pathways. Diabetes. 2010;59(12):3229–3239. doi: 10.2337/db10-0502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahajan A, Go MJ, Zhang W, et al. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet. 2014;46(3):234–244. doi: 10.1038/ng.2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cook JP, Morris AP. Multi-ethnic genome-wide association study identifies novel locus for type 2 diabetes susceptibility. Eur J Hum Genet EJHG. 2016;24(8):1175–1180. doi: 10.1038/ejhg.2016.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morris AP, Voight BF, Teslovich TM, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44(9):981–990. doi: 10.1038/ng.2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang J, Ferreira T, Morris AP, et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet. 2012;44(4):369–375. doi: 10.1038/ng.2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang J, Benyamin B, McEvoy BP, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42(7):565–569. doi: 10.1038/ng.608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Waterworth DM, Ricketts SL, Song K, et al. Genetic Variants Influencing Circulating Lipid Levels and Risk of Coronary Artery Disease. Arterioscler Thromb Vasc Biol. 2010;30(11):2264–2276. doi: 10.1161/ATVBAHA.109.201020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klarin D, Damrauer SM, Cho K, et al. Genetics of blood lipids among ~300,000 multi-ethnic participants of the Million Veteran Program. Nat Genet. 2018;50(11):1514–1523. doi: 10.1038/s41588-018-0222-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Teslovich TM, Musunuru K, Smith AV, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466(7307):707–713. doi: 10.1038/nature09270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sandhu MS, Waterworth DM, Debenham SL, et al. LDL-cholesterol concentrations: a genome-wide association study. Lancet. 2008;371(9611):483–491. doi: 10.1016/S0140-6736(08)60208-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakaue S, Kanai M, Tanigawa Y, et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat Genet. 2021;53(10):1415–1424. doi: 10.1038/s41588-021-00931-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jurgens SJ, Choi SH, Morrill VN, et al. Analysis of rare genetic variation underlying cardiometabolic diseases and traits among 200,000 individuals in the UK Biobank. Nat Genet. Published online February 17, 2022:1–11. doi: 10.1038/s41588-021-01011-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McLaren W, Gil L, Hunt SE, et al. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17(1):122. doi: 10.1186/s13059-016-0974-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peterson RE, Kuchenbaecker K, Walters RK, et al. Genome-wide Association Studies in Ancestrally Diverse Populations: Opportunities, Methods, Pitfalls, and Recommendations. Cell. 2019;179(3):589–603. doi: 10.1016/j.cell.2019.08.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Access to individual-level data from the All of Us research program is available to researchers whose institution has signed a data use agreement with All of Us (https://www.researchallofus.org/register/). All of Us provides a publicly available data browser (https://databrowser.researchallofus.org/) containing aggregate-level participant data for users to explore the available data, including genomic variants. Electronic health records (EHR) data, used for phenotyping, belongs to the registered tier dataset. Whole-genome sequencing data belongs to the controlled tier dataset, which requires additional training to access.