

Abstract
Background:
Although targeted therapies have revolutionized the therapeutic landscape of lung adenocarcinomas (LUAD), disease progression on single-agent targeted therapy against known oncogenic drivers is common, and therapeutic options following disease progression are limited. In patients with MDM2 amplification and a concurrent oncogenic driver alteration, we hypothesized that targeting of the tumor suppressor pathway (via restoration of p53 using MDM2 inhibition), and simultaneous targeting of co-occurring MAPK oncogenic pathway might represent a more durably effective therapeutic strategy.
Methods:
We examined genomic next-generation sequencing (NGS) data using the MSK-IMPACT platform to nominate potential targets for combination therapy in LUAD. We investigated the small molecule MDM2 inhibitor milademetan in cell lines and patient-derived xenografts (PDXs) of LUAD with a known driver alteration and MDM2 amplification.
Results:
Of 10,587 patient samples from 7,121 patients with LUAD profiled by NGS, 6% (410/7,121) harbored MDM2 amplification (MDM2amp). MDM2amp was significantly enriched among tumors with driver alterations in METex14 (36%, p<0.001), EGFR (8%, p<0.001), RET (12%, p<0.05), and ALK (10%, p<0.01). The combination of milademetan and the MEK inhibitor trametinib was synergistic in growth inhibition of ECLC5-GLx (TRIM33-RET/MDM2amp), LUAD12c (METex14/KRASG12S/MDM2amp), SW1573 (KRASG12C, TP53 wildtype) and A549 (KRASG12S) cells, and in increasing expression of pro-apoptotic proteins PUMA and BIM. Treatment of ECLC5-GLx and LUAD12c with single agent milademetan increased ERK phosphorylation, consistent with previous data on ERK activation upon MDM2 inhibition. This ERK activation was effectively suppressed by concomitant administration of trametinib. In contrast, ERK phosphorylation induced by milademetan was not suppressed by concurrent RET inhibition using selpercatinib (in ECLC5-GLx) or MET inhibition using capmatinib (in LUAD12c). In vivo, combination milademetan and trametinib was more effective than either agent alone in ECLC5-GLx, LX-285 (EGFRex19del/MDM2amp), L13BS1 (METex14/MDM2amp) and A549 (KRASG12S, TP53 wildtype).
Conclusion:
Combined MDM2/MEK inhibition demonstrates efficacy across multiple patient-derived LUAD models harboring MDM2amp and concurrent oncogenic drivers. This combination, potentially applicable to LUADs with a wide variety of oncogenic driver mutations and kinase fusions activating the MAPK pathway, has evident clinical implications and will be investigated as part of a planned phase 1/2 clinical trial.
Keywords: MDM2, TP53, Lung adenocarcinoma, Targeted therapy, Combination therapy
Introduction
Over the past decade, several targeted therapies directly inhibiting oncogenic drivers have been approved for patients with lung adenocarcinomas (LUAD). While tyrosine kinase inhibitors (TKIs) and other small molecule inhibitors have transformed the care of patients with LUAD driven by receptor tyrosine kinase (RTK) alterations or KRASG12C, acquired resistance to these agents is essentially universal, and options are limited following progression on targeted therapy1–8. After available targeted agents have been exhausted, patients are typically treated with non-targeted, systemic cytotoxic regimens with suboptimal outcomes9. LUAD remains a leading cause of cancer mortality. There is therefore an unmet need to find new therapeutic strategies for patients with LUAD.
The tumor suppressor p53 inhibits the development of malignancy through regulating cell cycle progression, DNA damage repair, and apoptosis. TP53 is the most commonly altered tumor suppressor gene in LUAD10. The most common mechanism of decreased p53 activity in TP53-wildtype in human malignancy occurs through increased activity of murine double minute 2 (MDM2)11. MDM2 amplification (MDM2amp) disrupts p53 activity via ubiquitin-mediated transport for proteasomal degradation through E3-ligase activity12. While several small molecule inhibitors targeting the MDM2-p53 interaction are in early-phase clinical trials, single-agent activity of MDM2 inhibitors have largely been limited13. Single-agent MDM2 inhibition can activate MAPK signaling, which may contribute to the limited activity seen with single-agent MDM2 inhibitors14. Furthermore, MDM2 inhibition in the context of concurrent inhibition of driver alterations in LUAD has not been previously explored.
Since LUADs are largely dependent on MAPK pathway activity—particularly in the context of driver alterations in EGFR, RET, METex14, ALK, or KRAS—we hypothesized that dual inhibition of MDM2 and MEK might be a potent combinatorial strategy for treatment of LUAD with MDM2amp, and that simultaneous targeting of oncogenic and tumor suppressor pathways may lead to more profound responses in patients with concurrent MDM2amp.
Materials and Methods
Materials
Cell culture growth media (DME-F12) and phosphate-buffered saline without calcium or magnesium (PBS) were prepared by the MSK Media Preparation Core Facility. Fetal bovine serum (FBS) was procured from Atlanta Biologicals (Flowery Branch, GA). Antibiotics (Gibco, Cat. 15240062), tissue culture plastics (100mm dishes, 6-well plates, and clear 96-well plates), dimethyl sulfoxide, alamarBlue viability dye, and pClick-IT (Alexa Fluor 647, Cat. C10640 and Annexin-V (Cat. 556454) assays were procured from ThermoFisher (Waltham, MA). SW1573 and H1395 cell lines were procured from American Type Culture Collection (ATCC) (Manassas, VA). H1792 cell line was provided by co-author WWL. A549 cell line was provided by co-author CMR. Black polystyrene and 96-well flat bottom clear plates along with protease and phosphatase inhibitor cocktail, sodium orthovanadate, and radioimmunoprecipitation assay (RIPA) lysis buffer (10X) were procured from EMD-Millipore Sigma (St. Louis, MO). Milademetan was provided by Rain Oncology (Newark, CA). All other small molecule inhibitors were obtained from Selleckchem (Houston, TX) with details provided in Supplemental Table 1. Details of antibodies raised against total or phosphorylated proteins used for Western blotting and immunohistochemistry (IHC) are given in Supplemental Table 2. Precision plus protein kaleidoscope molecular weight markers used for western blotting was procured from Bio-Rad (Hercules, CA). Films for Western Blot film developing were procured from Ewen Parker X-Ray (East Orange, NJ). RNA and DNA isolation kits were procured from Qiagen (Germantown, MD).
Generation of Patient-Derived Xenograft Models and Cell Lines and Efficacy Studies
Tumor tissue were collected under IRB-approved protocols (Memorial Sloan Kettering Cancer Center protocol #14-091 for LX-285 (passage number 9 used for study) and L13BS1 (passage number 5 used for study). LU-01-0448 was obtained from Contract Research Organization WuXi Clinical (passage number 5 used for study). Tumor tissue was immediately minced, mixed (50:50) with Matrigel (Corning, New York, NY) and implanted subcutaneously in 6–8 weeks old female NOD scid gamma mouse (NSG) mice (Jackson Laboratory, Bar Harbor, ME) to generate patient-derived xenografts (PDX) as previously described15. Mice were monitored daily and models were transplanted in mice three times before being deemed established. PDX tumor histology was confirmed by pathology review of H&E slides, and direct comparison to the corresponding patient biospecimens.
To generate cell lines from fresh PDX tumors, tumors were cut into 5mm pieces and then digested in a cocktail of tumor dissociation enzymes obtained from Miltenyi Biotech (130-095-929) in 5-mL serum-free DMEM:F12 media for 1 hour at 37°C, with vortexing every 10 minutes for 1 hour, according to manufacturer’s instructions. Digested samples were resuspended in 45 mL complete growth media to inactivate the dissociation enzymes, and then cells were pelleted by centrifugation. Lastly, the cells were plated in growth media supplemented with 2% FBS and antibiotics and allowed to propagate for multiple generations, passaging with trypsin when approaching confluence. The PDX and cell line models were generated and propagated in the absence of small molecule inhibitors.
For efficacy studies, fresh PDX tumor samples were minced, mixed with Matrigel, and implanted into a subcutaneous flank of female NSG mice to generate xenografts. For ECLC5-GLx, LUAD12c, and A549, cell line xenografts were generated by implanting 10 million cells into the flanks of the animals. After randomizing into groups of five to eight, tumor-bearing animals were treated with vehicle, milademetan (100 mg/kg once daily [QD] on a 5 d on, 2 d off schedule), trametinib (1mg/kg QD on a 5 d on, 2 d off schedule), or combination of both when tumors reached approximately 100–150mm3 volume. For the LU-01-0448 model, drugs were administered on a 7 d dosing schedule. Drugs were prepared as follows: trametinib was resuspended in 0.5 % hydroxypropyl methylcellulose (HPMC), 0.2% Tween80, pH 8.0 and milademetan was resuspended in 0.5% methylcellulose. Stock solutions were made fresh weekly and stored at 4°C. Tumor size and body weight were measured twice weekly, and tumor volume was calculated using the modified ellipsoid formula as done previously16. Upon dosing cessation for the LUAD12c model, tumor volumes were measured once weekly.
Genomic Characterization of Preclinical Models
Cell lines (ECLC5-GLx) and PDXs (LX-285 and L13BS1) were profiled by the Memorial Sloan Kettering Cancer Center-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) platform, a large panel next-generation sequencing assay designed to detect mutations, copy number alterations, and select fusions involving up to 505 cancer-associated genes17. Paired analysis of PDX tissue or cell line and matched-normal sample was performed to unambiguously identify somatic mutations. MDM2 total copy number was computed using the FACETS algorithm as previously described18. Genomic alterations of interest were also confirmed on PCR, FISH, or targeted next-generation sequencing as appropriate.
Growth and Propagation of Cell Lines
Cell lines were maintained in a humidified incubator infused with 5% carbon dioxide at 37C and sub-cultured when stock flasks reached approximately 75% confluence at a 1:4 dilution. SW1573 cell line was maintained in DMEM:F12 growth medium supplemented by 5% FBS and 1% antibiotics. All other cell lines were maintained in DMEM:F12 growth medium supplemented by 10% FBS and 1% antibiotics. The molecular alteration of interest was confirmed by PCR each time a cell line vial was thawed. Cells were tested for mycoplasma contamination every 6 months using MycoAlert-Plus™ kit procured from Lonza (Morristown, NJ).
Viability drug assays
For viability assays, the cells were plated in 96-well flat bottom, black polystyrene plates at a density of 7500 cells per well for ECLC5-GLx and LUAD12c and 5000 cells per well for SW1573 and H1792 and incubated with compounds for 96 hours. For A549, 5000 cells per well were plated and treated with compound for 72 hours. The relative number of viable cells was determined using alamarBlue viability dye, and fluorescence was measured using a Molecular Devices SpectraMax M2 multimodal plate reader (Ex: 555 nm, Em: 585 nm). Data were analyzed by nonlinear regression and curves fitted using Prism 9 to generate concentration that inhibits 50% values (IC50). For synergy drug studies, data were analyzed using the Combenefit Software presenting the HSA synergy method19,20. All data are expressed relative to control values and an average of two to five independent experiments in which each condition was assayed in at least triplicate.
Annexin-V and Click-IT Assays
Annexin-V apoptotic assay (BD Biosciences, NJ) was conducted as per manufacture instructions. Briefly, ECLC5-GLx cells were plated a density of 2 million cells per 100mm dish and serum starved with 0.5% FBS supplemented with 1% antibiotics for 24h the next day after plating. After treatment with either 100nM milademetan, 100nM trametinib, or combination for 48h, cells were harvested using trypsin and then washed twice with cold PBS, and then resuspended in Annexin V Binding Buffer at a concentration of 1×106 cells/mL. After addition of APC Annexin V, cells were incubated for 15 minutes at room temperature. Cells were then analyzed via flow cytometry. Using the same conditions as done for the Annexin V assay, for the cell cycle Click-iT assay (ThermoFisher), EdU was added to cell culture medium at a concentration of 10μM for 2 hours. Cells were harvested using trypsin. After washing cells with PBS, cells were resuspended at a concentration of 1×106 cells/mL in Click-iT fixative, and incubated for 15 minutes. After permeabilization with Click-iT saponin-based reagent, cells were resuspended in the Click-iT reaction cocktail and analyzed by flow cytometry.
Preparation of Whole-Cell Extracts and Western Blotting
Protein levels and phosphorylation state were detected by western blotting. Samples from cell lines were lysed in RIPA assay lysis buffer diluted to 1X in double distilled water containing phosphatase and protease inhibitors and 1ug/mL sodium orthovanadate. For samples derived from PDX models, tumor pieces were dissociated using gentleMACS Dissociator (Auburn, CA). Protein concentration was quantified using using Bradford assay (Biorad, Cat. 5000006). Lysates were denatured in 2X Laemli sample buffer at 55C for 10 minutes, resolved on 4% to 12% Bolt gels (ThermoFischer), and transferred onto polyvinylidene fluoride membranes. Membranes were blocked in 3% bovine serum albumin (Sigma) in Tris-buffered saline supplemented with 0.1% Tween20 (ThermoFisher) (vol/vol) for 1 hour at room temperature and probed with primary antibodies with specificity as outlined in Supplemental Table 2. Bound antibodies were detected with peroxidase-labeled goat antibody raised to mouse or rabbit IgG (R&D Systems, Minneapolis, MN) and imaged with enhanced chemiluminescence western blotting detection reagent (GE Healthcare). Images were captured on radiograph films. Experiments were repeated at least two times from independently prepared samples.
Phosphoproteome Profiling Arrays
The human proteome profiling array system (R&D Systems) contains duplicate validated positive and negative controls and capture antibodies that can simultaneously detect the phosphorylation state of 49 human receptor tyrosine kinases (Proteome Profiler Human Phospho-RTK Array Kit). A total of 2 million cells were plated in 10-cm dishes and grown for 24 hours. Cells were deprived of serum by culturing for 24 hours in growth media supplemented with 0.05% FBS and then treated with milademetan, trametinib, or combination for 48 hours; detection of protein phosphorylation was carried out according to the manufacturer’s instructions. Briefly, the array membranes were blocked, incubated with 600μg total cellular protein per array overnight at 4C on a rocking platform, washed, and incubated with phosphospecific detection antibodies. Captured phosphorylated proteins were detected by enhanced chemiluminescence and imaged on X-ray films.
RT-PCR
RNA was extracted using a Qiagen RNA mini kit, and complementary DNAs (cDNAs) were synthesized using SuperScript IV VILO (ThermoFisher Scientific, Cat. 11754050) according to the manufacturer’s instructions. PrimeSTAR polymerase was used (Cat. R040, Takara, CA). The following primers were used: GAPDH Fw 5′-GGC GCT GAG TAC GTC GTG GAG TCC-3′, GAPDH Rv 5′-AAA GTT GTC ATG GAT GAC CTT GG-3′. TRIM33 exon 14 Fw 5′-AGC AAG AAC CTG GGA CTG AAG ATG-3′, RET exon 12 Rv 5′-TGC TCT GCC TTT CAG ATG GAA GG-3′. MET exon 13 Fw 5-CTTCAACCGTCCTTGGAAAA-3’ MET exon 15 Rv 5’-GCTACTGGGCCCAATCACTA-3’.
RNAseq alignment and quantification
Differential gene expression analysis, principle component analysis, and transcript per million (TPM) normalization by size factors, were done from Kallisto output files using Sleuth v0.30.0 run in gene mode21. Differentially expressed genes were identified using the Wald test. Significant genes were called if the False Discovery Rates (q value) adjusted by the Benjamini-Hochberg method was less than 0.05, and beta (Sleuth-based estimation of log2 fold change) greater than 0.58, which approximately correlated to a log2 fold change of 1.5 in our data. Raw unfiltered differentially expressed gene sets are presented in Supplemental Table 3.
Pathway enrichment analyses
Gene set enrichment analysis (GSEA)22 was performed on full sets of gene expression data across the previously mentioned comparisons. Genes were ranked on p value scores computed as −log10(p value)*(sign of beta). Gene set annotations were taken from Molecular Signatures Database (MSigDB v7.0.1)23. The significance level of enrichment was evaluated using permutation test and the p value was adjusted by Benjamini-Hochberg procedure. Any enriched gene sets with adjusted p value ≤ 0.1 were regarded as significant. This analysis was conducted using ClusterProfiler R package24.
MIC-1 ELISA assay
MIC-1 (GDF15) was detected in plasma of mice (Cat, DGD150, Quantikine ELISA kit, R&D Systems) according to manufacturer instructions. Briefly, after adding the assay diluent to each well, 50 μL of each sample was added to each well and incubated at room temperature for 2 hours on an orbital microplate shaker. Each well was then washed and aspirated as per instructions. 200 μL of GDF15 conjugate was added to each well and incubated at 1 hour at room temperature. Each well was then washed and aspirated as per instructions. 200 μL of substrate solution was then added to each well, followed by 30-minute incubation, and then 50 200 μL of Stop Solution was added to each well. Plates were then read using a microplate reader set to 450 nm.
Tissue microarrays
Tissue microarrays (TMAs) were constructed in the pathology core lab of Precision Pathology Center using an automated TMA Grand Master (3DHistech) and TMA Control software (Version 2.4). TMAs were designed and constructed using archival paraffin-embedded lung cancer tissue samples (N=119) retrieved from the files of the Department of Pathology, Memorial Sloan Kettering Cancer, New York, NY. Histology sections were reviewed by a pathologist and most representative areas to be cored were selected and marked on the H&E slides.
Immunohistochemistry
Immunohistochemistry (IHC) was performed on BOND RX platform from Leica Biosystems using standard protocols with Epitope retrieval solution ER2 and Polymer Refine Detection Kit catalog #DS9800. The MDM2 antibody clone used was OP46–100 from Sigma. Quantification was performed by a pathologist (co-author UB).
Statistical Analysis
For animal studies, area under curve (AUC) analysis was used to compare the average tumor volume between groups. T0 (time 0) was defined as treatment start. We normalized mouse volumes by subtracting the volume at T0 for each mouse, analyzing the growth from treatment start. The aucVardiTest package in R was used to calculate AUC and tumor growth curves were compared using the test proposed by Vardi et al25, a permutation test for comparing growth curves across two groups under dependent right censoring. Data were plotted in Prism and raw p-values were computed in the aucVardiTest package in R. P-value less than 0.05 was considered significant.
Results
MDM2 amplification is present in 6% of lung adenocarcinomas and over-represented in patients with driver alterations in METex14, EGFR, RET, or ALK
Among 10,587 lung adenocarcinomas samples from 7,121 individual patients whose tumors underwent broad next-generation sequencing using MSK-IMPACT, MDM2amp was found in 410 (6%) patients (Figure 1A). MDM2amp was mutually exclusive with TP53 mutations (Figure 1A) (p<0.001). MDM2amp were over-represented among several driver alterations. More than one-third of samples with MET exon 14 splice variant (METex14) harbored co-occurring MDM2amp (36%, p<0.001) (Figure 1B–C). Samples with RET rearrangements (12%, p=0.006), EGFR sensitizing mutations (8%, p<0.001), and ALK rearrangements (10%, p=0.002) were also significantly enriched for MDM2amp. Although the co-occurrence pattern for samples with KRAS mutations were not statistically significant for enrichment in MDM2amp, 104 KRAS-mutant tumors exhibited MDM2amp. Among 10 patients with METex14 alterations and MDM2amp that had pre-treatment and post-treatment (recurrent) samples, the MDM2amp was almost always seen at diagnosis (N=9) and rarely represented an acquired alteration (N=1) (Figure 1D). Among 24 patients with EGFR mutations and MDM2amp who had pre-treatment and post-treatment samples, the MDM2amp was almost always seen at diagnosis (N=20) and rarely represented an acquired alteration (N=4) (Figure 1E). There was an insufficient number of patients who underwent pre-post treatment biopsies with RET or ALK alterations and concurrent MDM2amp. Altogether, these results demonstrate that MDM2amp is a relatively common oncogenic driver alteration in LUAD, and that MDM2amp are over-represented in patients with specific driver alterations, and are usually detected at diagnosis.
Figure 1. MDM2 amplification in lung adenocarcinoma and its co-ocurrence with other driver alterations.
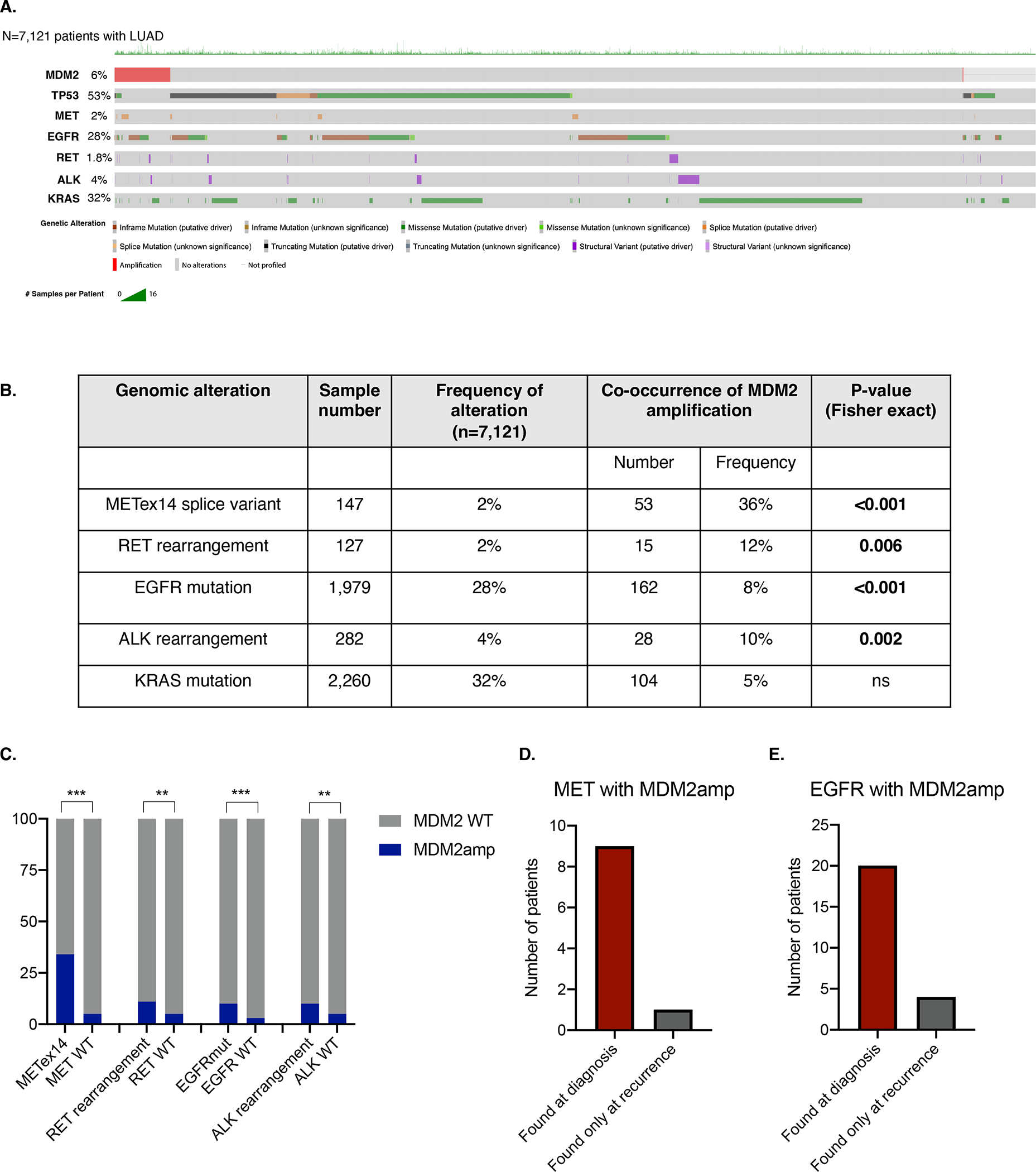
A. Oncoprint of N=7,121 patients with lung adenocarcinoma that underwent large-panel next-generation sequencing with the MSK-IMPACT platform. Color codes for each panel are indicated in the key. B. Frequency of co-occurrence of MDM2 amplification in patients with oncogenic drivers in MET exon 14 splice variant, RET rearrangements, EGFR sensitizing mutations, ALK rearrangements, and KRAS mutations. Significance of co-occurrence computed with Fischer exact test. C. Visual representation of data represented in panel B. D. Analysis of patients with MET exon 14 splice variant and E. EGFR mutations and concurrent MDM2 amplification who underwent biopsy of lung adenocarcinoma at diagnosis and at recurrence. ***p<0.001, **p<0.01, *p<0.05. MDM2amp; MDM2amplification.
Combination of MDM2 and MEK inhibition is synergistic in vitro
The patient-derived models obtained upon resistance to TKI used in our study are described in Figure 2A. The first model (ECLC5) was derived from a patient with stage IV lung adenocarcinoma with a TRIM33-RET fusion. The patient was treated with cabozantinib and achieved a partial response, but developed progressive disease after 7.5 months of therapy26. A biopsy at the time of disease progression revealed MDM2amp (total copy number 12). A patient-derived cell line was derived from the biopsy obtained at the time of progressive disease. This ECLC5 cell line was transduced with GFP-Luciferase and designated the derived cell line ECLC5-GLx. The second model was derived from a patient with stage IV lung adenocarcinoma with METex14 splice variant (LUAD12c)27. The patient was treated with a combination of crizotinib and a MET bivalent antibody and achieved a partial response. After 11 months of therapy, the patient developed resistance and was found to have an acquired KRASG12S alteration27. MDM2amp was present both at diagnosis (total copy number 14) and acquired resistant sample. Another METex14 model L13BS1 was derived from a patient with stage IV lung adenocarcinoma with acquired resistance to crizotinib after 7 months of therapy. MDM2amp (total copy number 12) was present in both diagnosis and acquired resistance sample. The next model (LX-285) was derived from a patient with EGFRex19-deleted stage IV lung adenocarcinoma who was treated with erlotinib for 3 years, and developed an acquired T790M mutation upon resistance to erlotinib. MDM2amp was present in the acquired resistance sample (total copy number 12). At the time of the patient’s diagnosis, MDM2amp status was not assessed and tissue has been exhausted, so it is unknown if the MDM2amp was acquired. We also included a model with EML4-ALK fusion and MDM2amp (total copy number 6) (LU-01-0448), which was procured at diagnosis from a 56-year-old female with stage II LUAD that was treatment-naïve. In addition to these patient-derived models, we also included the H1395 lung adenocarcinoma model with BRAFG469A mutation and MDM2amp28. The SW1573 cell line (KRASG12C) was a top hit on the Cancer Dependency Map portal for MDM2 vulnerability using a CRISPR screen, and was therefore included (TP53 wildtype, CDK2NA homozygous deletion, and no MDM2amp). A549 cell line (KRASG12S) was also included as an additional TP53-wild type LUAD model. For comparator purposes, we included two TP53-mutant cell lines, H1792 (KRASG12C, MDM2amp, TP53mut), and LUAD-0002AS1 (KIF5B-RET, TP53mut). Expression of MDM2 is shown in Figure 2B. Protein expression for MDM2 was evident in all patient-derived models with MDM2amp. Interestingly, SW1573, despite not harboring MDM2amp, had relatively high expression of MDM2 (Figure 2B).
Figure 2. Generation of patient-derived xenografts with concurrent MDM2amplification and concurrent driver alterations.
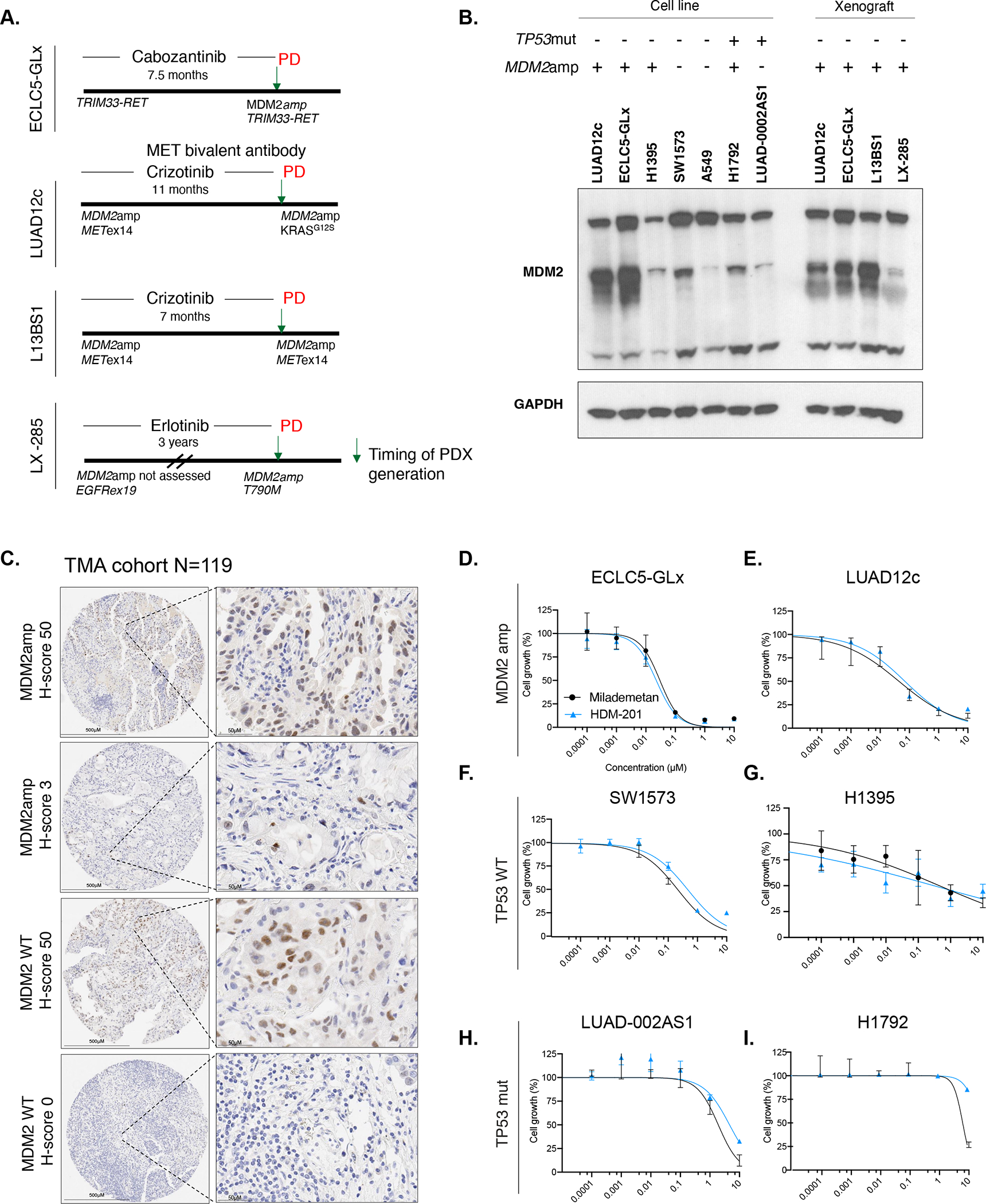
A. Timeline representation of patient-derived models used in the study. Timeline indicates whether the MDM2amplification was present at diagnosis or acquired, as well as the occurrence of relevant acquired co-alterations. Timeline also indicates the time on oral targeted therapy prior to generation of the model. PD indicates progression of disease and arrow represents time of generation of the model. B. Western blot showing MDM2 expression in MDM2amp and TP53mut cell lines as well as patient-derived xenografts generated from mouse tumors (for patient derived cell lines ECLC5-GLx and LUAD12c, in addition to MDM2 expression in the cell line, MDM2 expression was also tested in the untreated xenograft tumors at 7 days). All three isoforms of MDM2 are shown, with GAPDH used as a loading control. At least two independent experiments were conducted. C. Representative images of immunohistochemistry of MDM2 expression according to MDM2 genotype. D. Single-agent activity of MDM2 small molecule inhibitors HDM-201 and milademetan in MDM2-amplified models ECLC5-GLx and E. LUAD12c F. TP53 wildtype models SW1573 and G. H1395 and TP53-mutant models H. LUAD-0002AS1 and I. H1792. Values are expressed relative to the vehicle-treated control (100%). Data were analyzed by nonlinear regression to determine IC50 for inhibition of growth (see Supplemental Table 4 for IC50 values). Results represent the mean ± SD of three replicate determinations in one experiment and repeated in an independent experiment. **p<0.01. MDM2amp; MDM2 amplification. TP53mut; TP53-mutant.
We next assessed MDM2 expression in a cohort of 119 NSCLC samples included on a tissue microarray (TMA). Patient characteristics for this cohort are described in Supplemental Table 3. In this cohort, 6 patients had MDM2amp (5%), similar to what was observed in the MSK-IMPACT analysis, and the median total copy number for MDM2-amplified cases was 6.8 copies (range 3–16). Samples without MDM2 amplification were still observed to express MDM2 (N=26, 22%), with a representative example in Figure 2C (MDM2 WT, H-score 50), suggesting that a strategy geared towards MDM2 inhibition may not only be relevant to patients with MDM2amp.
We next assessed the single-agent activity of two small molecule MDM2 inhibitors milademetan and HDM-201 in models for which cell lines were derived (Supplemental Table 4). The MDM2-amplified patient-derived models ECLC5-GLx (Figure 2D) and LUAD12c (Figure 2E) exhibited marked sensitivity to single-agent MDM2 inhibition with IC50 in the 20–40nM range. The non-amplified TP53 wildtype models SW1573 and H1395 displayed moderate sensitivity to MDM2 inhibition in the 112–450nM range (Figure 2F–G). In contrast, but as expected, TP53-mutant models LUAD-0002AS1 and H1792 were relatively insensitive to MDM2 inhibition (Figure 2H–I). Individual IC50 values are shown in Supplemental Table 4. Given the limited clinical activity of single-agent MDM2 inhibition29, previous reports of MAPK signalling activation with single-agent MDM2 inhibition14, and demonstrated safety in early phase clinical trials with combination MDM2/MEK inhibition30–32, we evaluated the efficacy of combined MDM2/MEK inhibition on cell growth. For all subsequent studies, we focused on milademetan given reports of improved safety profiles compared to other MDM2 inhibitors33. Using the Combenefit analysis34, wherein synergy is defined by a score is ≥235, the combination of milademetan and trametinib was synergistic and induced a dose response shift in ECLC5-GLx (Figure 3A–C), LUAD12c (Figure 3D–F), and SW1573 (Figure 3G–I), but not in TP53-mutant H1792 (Figure 3J–L). We also observed synergistic activity of milademetan in combination with trametinib in H1395 and A549 cell lines (Supplemental Figures 1A–B). Taken together, these results suggest that the combination of MDM2/MEK inhibition was synergistic in vitro both in MDM2amp and MDM2 WT models.
Figure 3. MDM2 inhibition with milademetan and MEK inhibition with trametinib is synergistic in models of lung adenocarcinoma.

A. Synergy matrix for ECLC5-GLx. The synergy matrices are synergy scores, calculated according to the HSA synergy model (which measures whether the expected combination effect equals to the higher effect of individual drugs). The larger numeral in each box is the synergy score; negative values indicate antagonism. The number below the synergy score is the standard deviation. Boxes are coloured blue if the synergy score significant by t-test. B. Milademetan dose-response shift observed in the presence of increasing concentrations of trametinib C. Dose response shift observed for trametinib in the presence of increasing concentrations of milademetan. D. Synergy matrix and E-F. Dose response shifts for LUAD12c. G. Synergy matrix and H-I. Dose response shifts for SW1573. J. Synergy matrix and K-L. Dose response shifts for H1792. The number of biologic replicates (N) is indicated at the top right of the matrix. At least two independent synergy experiments were conducted. ***p<0.001, **p<0.01, *p<0.05. nM; nano molar.
ERK activation following MDM2 inhibition can be suppressed by co-targeting MEK
In order to assess the mechanisms of synergistic MDM2/MEK inhibition in vitro, we assessed protein expression upon treatment with single-agent milademetan, trametinib, or the combination at 6, 24, and 48 hours. Treatment of ECLC5-GLx with milademetan increased ERK phosphorylation, confirming the previous observation of ERK activation upon MDM2 inhibition14 (Figure 4A). As expected, DUSP6, a surrogate for MAPK activation36, was also increased with milademetan. This phenomenon of MAPK signalling activation was suppressed by concurrent milademetan and MEK inhibition using trametinib (Figure 4A). In contrast, ERK phosphorylation was not suppressed by concurrent milademetan and RET inhibition using selpercatinib (Figure 4B), even though this model was sensitive to single-agent selpercatinib (Supplemental Figure 2A) (Supplemental Table 4). Trametinib caused an increase in p27 (Kip1) cell cycle arrest marker, which was also apparent with the combination, while milademetan caused an increase in p21 (Cip1) cell cycle arrest marker (Figure 4A). Interestingly, milademetan caused increased expression of cyclin D1—previously described as a resistance mechanism to single-agent MDM2 inhibition37—but this induction was inhibited in the combination with trametinib (Figure 4A). E2F1, a p53-independent target of MDM2 and cell cycle progression marker, was decreased by milademetan and trametinib individually, which translated to a more profound decrease with the combination compared to either agent alone (Figure 4A). Increased expression of phosphorylated p53 was observed with single-agent milademetan (Figure 4A). BIM—a pro-apoptotic protein—was increased in the combination driven by trametinib, while there was a synergistic increase in the expression of the p53-dependent proapoptotic protein PUMA with the combination (Figure 4A). Similar results were observed in the LUAD12c cell line, particularly, suppression of pERK with the combination with trametinib (Figure 4C) not achieved by combination with capmatinib (Figure 4D) (capmatinib-resistant cell line) (Supplemental Figure 2B) (Supplemental Table 4) and the combination with trametinib also caused increase of PUMA expression (Figure 4C). In SW1573, we did not observe increased pERK with single-agent milademetan (Figure 4E), likely due to high basal ERK expression and saturation of ERK in this KRAS-dependent cell line. pERK was better suppressed by trametinib (Figure 4E) compared to sotorasib (Figure 4F) in this sotorasib-resistant cell line (Supplemental Figure 2C) (Supplemental Table 4). Combination treatment showed enhancements in p21, p27, PUMA, and BIM mirroring observations in ECLC5-GLx and LUAD12c cell lines (Figure 4E). In A549, a second KRAS-mutant (KRASG12S), TP53-wildtype cell line without MDM2amp, similar to what was observed in SW1573, increased pERK phosphorylation was not observed with single-agent milademetan (Supplemental Figure 3). We also observed increased PUMA expression with the combination of milademetan and trametinib compared to either agent alone (Supplemental Figure 3). Together these results suggest that the synergistic activity observed were a result of MAPK suppression driven by trametinib, and increase in PUMA.
Figure 4. Combination of milademetan and trametinib causes increased apoptosis compared to either agent alone; ERK activation following MDM2 inhibition can be suppressed by co-targeting MEK.
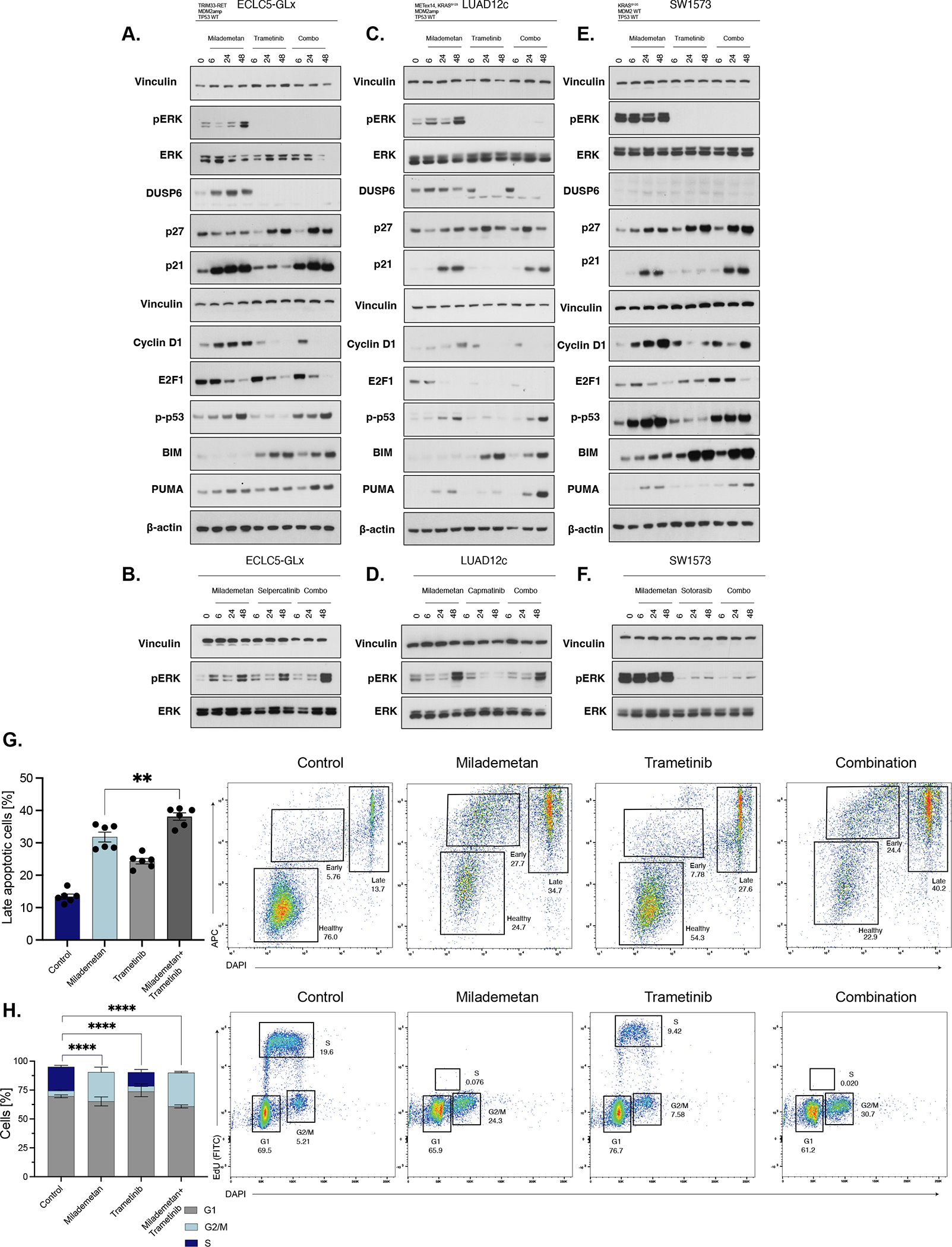
A. Western blot of ECLC5-GLx cells treated with milademetan (100nM), trametinib (100nM) or combination milademetan and trametinib (100 nM each) of at 6, 24, and 48h. B. Western blot of ECLC5-GLx cells treated with milademetan (100 nM) selpercatinib (100 nM) or combination of milademetan and selpercatinib (100 nM each) of at 6, 24, and 48h. C. Western blot of LUAD12c cells treated with milademetan (100 nM), trametinib (20 nM) or of combination milademetan and trametinib (50 nM milademetan, 10 nM of trametinib) at 6, 24, and 48h. D. Western blot of LUAD12c cells treated with milademetan (100nM), capmatinib (100nM) or combination of milademetan and capmatinib (100 nM milademetan, 100 nM of trametinib) at 6, 24, and 48h. E. Western blot of SW1573 cells treated with milademetan (100 nM), trametinib (100 nM) or combination of milademetan and trametinib (100 nM each of at 6, 24, and 48h. F. Western blot of SW1573 cells treated with milademetan (100 nM), sotorasib (100 nM) or combination of milademetan and sotorasib (100 nM each of at 6, 24, and 48h. Vinculin and β-actin used as a Western blot loading control. G. Apoptosis following milademetan (100nM), trametinib (100nM) or combination milademetan and trametinib (100nM each) in ECLC5-GLx measured by Annexin-V and DAPI staining and analyzed by flow cytometry at 48h. Bar chart illustrates cells in the late stage of apoptosis. Representative flow cytometry images shown (n=6, ±SEM, 1-way ANOVA and multiple comparison testing, data represents 3 biological replicates from 2 independent experiments). H. Cell cycle analysis following milademetan (100nM), trametinib (100nM), or combination milademetan and trametinib (100nM each) at 48h measured by Click-iT EdU and DAPI and analyzed by flow cytometry. Bar chart illustrates percentage of cells in the indicated cycle phase. Representative flow cytometry images shown (n=6, ±SEM, 1-way ANOVA and multiple comparison testing, data represents 3 biological replicates from 2 independent experiments). G1 phase; growth or gap-1 phase. G2_M phase; G2-M DNA damage checkpoint. S-phase; synthesis phase. ****p<0.0001, **p<0.01.
Previous work has suggested that activation of the MAPK pathway by single-agent milademetan was caused by p53-dependent increase in reactive oxygen species, which in turn caused phosphorylation of receptor tyrosine kinases (RTKs)14. In order to explore any possible RTK activation caused by MDM2 inhibition in our cell lines, we performed a phospho-RTK array. In ECLC5-GLx, single-agent milademetan caused increase in EGFR (Supplemental Figure 4A), while in LUAD12c, single-agent milademetan caused an increase in FGFR4 (Supplemental Figure 4B). In line with the observation that milademetan-induced MAPK signalling was not observed in SW1573, we did not observe phosphorylation of RTKs in this cell line (Supplemental Figure 4C). These results suggest that activated RTKs by MDM2 inhibition may be different in cancers with distinct genomic backgrounds.
Apoptotic profiling by flow cytometry revealed that the combination of milademetan and trametinib induced a higher proportion of late apoptotic cells compared to the control and either agent alone (Figure 4G), consistent with the enhanced expression of PUMA and BIM observed by western blotting. Cell cycle profiling revealed that cell cycle arrest as measured by the proportion of cells in the synthesis phase (S-phase) of the cell cycle was primarily driven by single-agent milademetan (Figure 4H).
Taken together, these results demonstrate synergistic activity of MDM2 and MEK inhibition across different lung adenocarcinoma cell lines with diverse oncogenic driver backgrounds, and ERK activation following MDM2 inhibition can be suppressed by co-targeting MEK. Importantly, the combination of MDM2 and MEK inhibition increased apoptosis, beyond that achieved by either agent alone.
Transcriptomic profiling reveals suppression of milademetan-induced activation of the MAPK pathway by trametinib
We next performed transcriptomic analysis to better understand the mechanisms potentially responsible for combinatorial efficacy of milademetan and trametinib. In the ECLC5-GLx cell line, relative to the untreated negative control, treatment with milademetan induced increased expression of LATS2, a tumor suppressor which augments p53-mediated apoptosis38, as well as increased expression of PCSK5 which also promotes apoptosis39 (Figure 5A). We observed decreased expression of E2F1 in milademetan, pointing to increased cell cycle arrest (Figure 5A), also observed on western blot (Figure 4A). We also observed increased expression of genes related to MAPK pathway activation, such as USP1840 and IFR941 (Figure 5A). Pathways related to MAPK signalling (HALLMARK_KRAS_SIGNALLING_UP) and apoptosis were increased in ECLC5-GLx cells treated with milademetan (Figure 5B). In contrast, pathways related to cell cycle progression and MYC targets were decreased with milademetan (Figure 5B). As expected, relative to the untreated control, trametinib induced decreased expression of DUSP6 and SPRY4 genes (Figure 5C), also seen on western blot for DUSP6 (Figure 4A). Pathway analysis showed decreased MAPK signalling with trametinib (HALLMARK_KRAS_SIGNALLING_DOWN) (Figure 5D). This relative decreased transcriptional output of the MAPK pathway (decreased DUSP6 and SPRY4) was also observed with the combination (Figure 5E), also seen on western blot for DUSP6 (Figure 4A). Decreased expression of E2F1 and increased expression of PCSK5 was also seen in the combination (Figure 5E). Pathway analysis showed enrichment of p53-related pathways, and negative enrichment of MAPK signalling pathways (Figure 5F) in the combination relative to untreated control.
Figure 5. Transcriptomic characterization of effects of combination milademetan and trametinib.
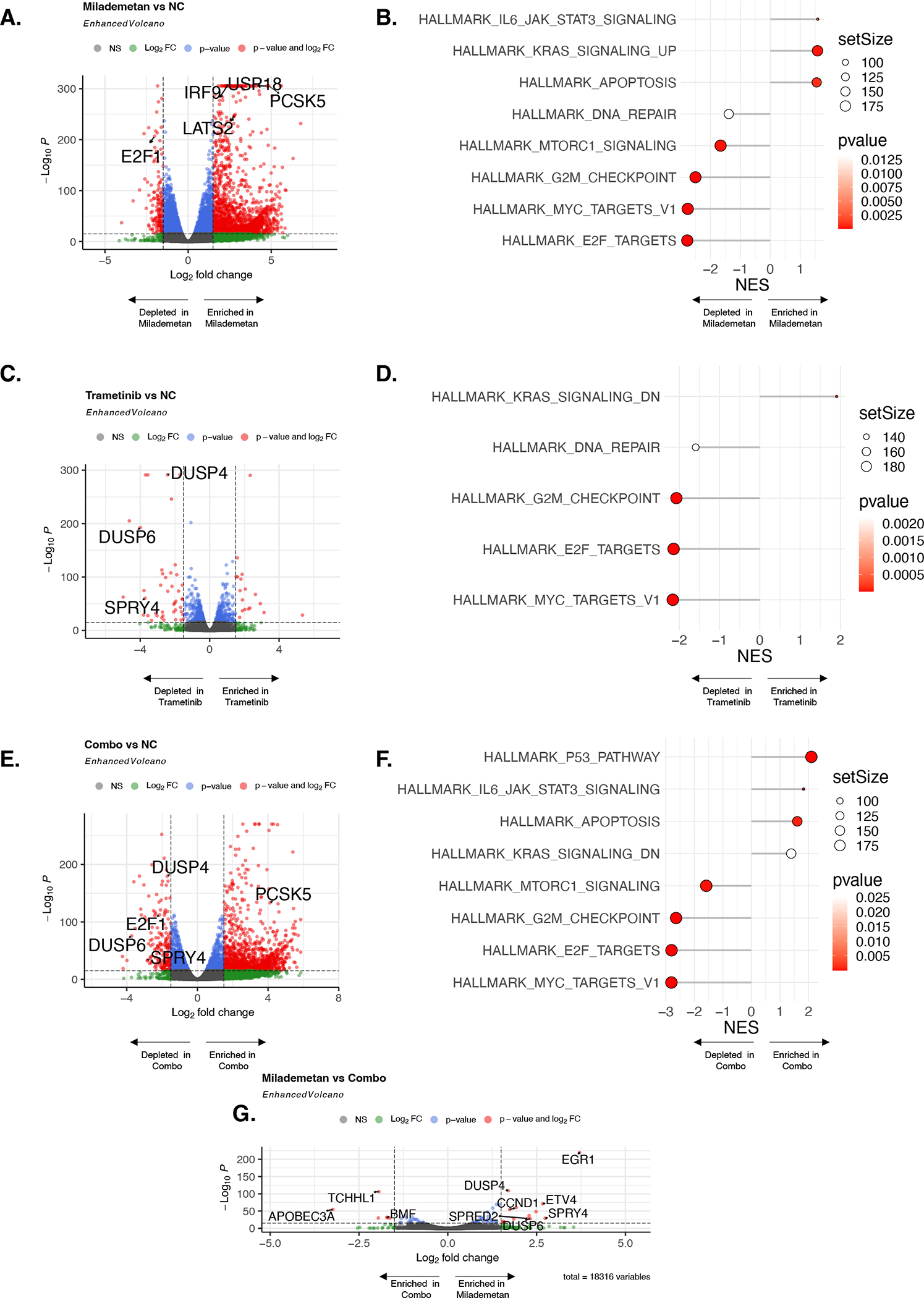
ECLC5-GLx cells were treated with milademetan (100 nM), trametinib (100 nM), or combination (100 nM each) for 48h. 4 biological replicates from 4 independent experiments were pooled for RNA sequencing. A. Volcano plot for milademetan vs untreated control (NC; negative control) showing differentially expressed genes (DEG). Grey dots represent non-significant value; green dots represent DEG significant by p-value but not by fold-change threshold; blue dots represent DEG significant by fold-change threshold but not by p-value; red dots represent significant DEG by both fold-change and p-value threshold. B. Pathway enrichment score for milademetan relative to the untreated control. C. Volcano plot for trametinib vs untreated control. D. Pathway enrichment score for trametinib relative to the untreated control. E. Volcano plot for combination vs untreated control F. Pathway enrichment score for combination vs untreated control. G. Volcano plot for DEG for milademetan condition relative to combination condition. Significant p-values corrected for multiple hypothesis testing. NES; normalized enrichment score.
To untangle the effect of milademetan compared to trametinib relative to the combination, we performed an additional comparison of differentially expressed genes in milademetan vs. the combination. Relative to the combination, milademetan was associated with increase in MAPK signalling-related gene expression (increased expression of DUSP4 and DUSP6), but relative decreased expression in the combination (Figure 5G). Interestingly, CCND1 was also increased with milademetan (Figure 5G), but relatively decreased in the combination, as observed in the proteomic analyses in Figure 4A. CCND1 over-expression has previously been described as a mechanism of resistance to single-agent MDM2 inhibitors42, suggesting that this resistance mechanism could be overcome with a combinatorial strategy. Individual gene counts of interest according to condition are presented in Supplemental Figure 5. We observed similar patterns of gene expression according to treatment condition in a second cell line LUAD12c (LATS2 and PCSK5, Supplemental Figure 6A–B), cell cycle arrest gene E2F1 (Supplemental Figure 6C), MAPK signalling genes DUSP6 and DUSP4 (Supplemental Figure 6D–E), and CCND1 (Supplemental Figure 6F)). Taken together these results suggest that the synergistic activity of milademetan and trametinib is possibly driven by suppression of the milademetan-induced activation of the MAPK pathway by trametinib, and complementary contributions of pro-apoptotic pathway activation and cell cycle inhibition.
Combination of MDM2 and MEK inhibition is effective in patient-derived xenograft lung adenocarcinoma models
We next assessed the combinatorial activity of milademetan and trametinib in vivo in patient-derived models (Figure 2A). First, in ECLC5-GLx, a model with MDM2amp and TRIM33-RET fusion with TP53 wildtype status, combined milademetan and trametinib was more effective at reducing tumor volume compared to either agent alone (p=0.0068 compared to milademetan and p=0.0112 compared to trametinib) (Figure 6A). In LX-285 model with EGFRex19 deletion, acquired T790M mutation, MDM2amp, and TP53 wildtype status, the combination of milademetan and trametinib was more effective at reducing tumor volume compared to either agent alone (p=0.0068 compared to milademetan and p=0.0094 compared to trametinib) (Figure 6B). In LUAD12c, a model with MDM2amp and METex14, KRASG12S and TP53 wildtype status, single-agent milademetan was effective in reducing tumor volume, despite the presence of two other oncogenic driver alterations in MET and KRAS (Figure 6C). Notably, this model is resistant to capmatinib (Supplemental Figure 2B) (Supplemental Table 4), the current standard-of-care in METex14-mutant cancers, and was also resistant to crizotinib as previously published27. However, long-term tumor growth post-dosing cessation was suppressed in the combination group (Figure 6C). In a second METex14 model L13BS1, the combination of milademetan and trametinib was more effective than either agent alone at reducing tumor growth (p=0.0086 compared to milademetan, 0.0072 compared to trametinib) (Figure 6D). In the EML4-ALK model, LU-01-0448, combination of milademetan and trametinib was also effective, (Supplemental Figure 7A), and lower dose of milademetan (50mg/kg) with trametinib was also more effective compared to either agent alone (Supplemental Figure 6E). Plasma concentration of the secreted protein GDF15 (also known as MIC-1), a p53 gene target and pharmacodynamic marker of p53 activation, was also higher in the combination milademetan and trametinib group (Supplemental Figure 7B–C), consistent with enhanced p53 activation. Combination of milademetan and trametinib was more effective than either agent alone in A549 cell line (KRASG12S) (Figure 6F). Consistent with enhanced apoptosis, we observed increased PUMA expression in ECLC5-GLx and in LU-01-0448 with combination milademetan and trametinib treatment (Supplemental Figure 7D–E). No signs of toxicity were observed in mice treated with the combination of trametinib and milademetan, as per mouse weight measurements showing no weight loss as compared to either therapy alone or the control group (Supplemental Figure 8A–F).
Figure 6. Combination of milademetan and trametinib is effective in patient-derived models of MDM2 amplification with concurrent driver alterations.

Lung adenocarcinoma models were treated with the indicated dose of either vehicle, milademetan, trametinib, or combination daily on a 5d schedule (5–8 mice/group). The tumor volume over time is shown. P-values for each comparison (Milademetan vs. Trametinib; Trametinib vs. Combination; and Milademetan vs. Combination) shown in the table below growth curves for each model. The aucVardiTest package in R was used to calculate AUC and tumor growth curves were compared using the test proposed by Vardi et al25, a permutation test for comparing growth curves across two groups under dependent right censoring. A. ECLC5-GLx patient-derived cell line xenograft model (TRIM33-RET fusion, MDM2amp, TP53 wildtype status). B. LX-285 patient-derived xenograft model (EGFRex19 deletion, T790M mutation, MDM2amp, TP53 wildtype). C. LUAD12c patient-derived cell line xenograft model (METex14, KRASG12S, TP53 wildtype). D. L13-BS1 patient-derived xenograft model (METex14 splice variant, MDM2amp, TP53 wildtype. E. LU-01-0448 patient-derived xenograft (EML4/ALK fusion, MDM2amp, TP53 wildtype) (treated on a 7d schedule) and F. A549 cell line xenograft (KRASG12S, TP53 wildtype). Results shown represent mean ± SEM. For LUAD12c, dosing of milademetan and milademetan+trametinib groups stopped at 113 days (indicated on graph with dashed line) and animals were observed and tumors measured post cessation of dosing. Administration of the combination had no adverse effect of animal weight (Supplemental Figure 8). Mila; milademetan. Tram; trametinib. Combo; combination.
Discussion
With broad use of comprehensive molecular profiling in LUAD, there is a growing identification of patients with more than one targetable alteration at diagnosis, thereby opening the possibility of combination approaches to extend time on and response to targeted therapy. Identifying the most appropriate combinations in patients with multiple alterations remains a significant challenge. A recent study aimed to match patients with a combinatorial approach according to molecular profiling and suggested that rationally selected, molecularly-guided targeted therapy combinations could be associated with improved disease control and overall survival43. However, a recent systematic review revealed that the majority of clinical trials evaluating combination targeted therapy lacked preclinical evidence supporting the combination in the given indication, and that most combination clinical trials were performed in the genomic biomarker-unselected setting44.
Our study provides preclinical evidence across multiple models that MDM2amp is a targetable alteration in the context of LUAD with defined molecular drivers. We found that 6% of patients with LUAD harboured MDM2amp, and that these amplifications were mostly mutually exclusive with TP53 mutations. MDM2amp was significantly over-represented among samples with specific concurrent driver alterations, notably including METex14, RET, EGFR, and ALK. These oncogenic drivers share the property of aberrant activation of the MAPK mitogenic signaling, a pathway also driven by oncogenic KRAS. The combination of small molecule MDM2 inhibitor milademetan and MEK inhibitor trametinib was synergistic against MDM2-amplified cell lines as well as a cell line with high MDM2 expression, and the combination increased expression of pro-apoptotic proteins PUMA and BIM, beyond that achieved by either agent alone. Moreover, we showed that MDM2/MEK combination induced increased GDF15, a downstream marker of tp53 activation and a biomarker used in clinical trials employing MDM2 inhibition45. Treatment of cell lines with RTK drivers with milademetan induced ERK phosphorylation, confirming a previous report of ERK activation upon MDM2 inhibition14; this potentially compensatory response could be entirely abrogated by MEK inhibition. In vivo, combination milademetan and trametinib was more effective than either agent alone in achieving long-term tumor control in most models. We found consistent efficacy of MDM2/MEK inhibition across multiple patient-derived LUAD models harboring MDM2amp and across different backgrounds of oncogenic drivers. This combination would be of potential clinical utility for LUADs with a wide variety of oncogenic driver mutations and kinase fusions driving MAPK signalling.
MDM2amp have been described in several malignancies including breast, gastrointestinal cancers and de-differentiated liposarcoma46,47,48. Several small molecule inhibitors of MDM2 have been developed to restore wildtype TP53 function – disappointingly however, the results of early phase trials have been limited by modest clinical activity13. The limited efficacy could be due to the paradoxical activation of the MAPK pathway with single-agent MDM2 inhibitors14, particularly relevant in lung adenocarcinomas, where MAPK pathway activation is frequent and associated with poor prognosis in cases with or without a defined oncogenic driver alteration49–52. Previous work reported that this paradoxical MAPK pathway activation was driven by p53-mediated generation of reactive species, which in turn phosphorylate RTKs14. Our work has elucidated that these activated RTKs may be different in cancers with distinct genomic backgrounds; hence the potential utility of focusing on the convergent downstream signaling pathway by employing a MEK inhibitor. In the context of KRAS-mutated gastrointestinal cancers, Wang et al also recently reported that p53 interacted with ERK2 through a hydrophobic interaction and disruption of this complex via trametinib was lethal to KRAS-mutant cells53.
Milademetan is an oral small molecule inhibitor of the MDM2-p53 complex that reactivates p53. Despite reports of high toxicity with single-agent MDM2 inhibitors, the intermittent dosing schedule reported as part of a phase 1 trial was associated with lower rates of dose limiting toxicities, most frequently thrombocytopenia33,54. Notably thrombocytopenia is rarely seen with MEK inhibition, supporting potential combinatorial dosing.
Our results demonstrate that the combination of MDM2 inhibition with milademetan and MEK inhibition should be investigated as a targeted therapeutic strategy in patients with LUAD demonstrating potential MDM2 dependence and a concurrent driver alteration. These findings are not only limited to patients with MDM2amp, but may also be applicable to patients without MDM2amp but with higher levels of MDM2 expression. For example, we observed synergistic activity of the combination in the MDM2-wildtype, KRASG12C-altered SW1573 cell line which is resistant to direct inhibition of KRASG12C, and also in A549 (KRASG12S alteration with currently no approved targeted therapy). Interestingly, these models both exhibit deletion of CDKN2A, a known negative regulator of MDM255. CDKN2A loss abolishes p14ARF inhibition of MDM2, leading to MDM2-mediated p53 degradation55. Therefore, as also previously demonstrated, an MDM2-inhibitor-directed strategy may also be applicable to patients with CDKN2A deletions, which occur in almost 20% of lung adenocarinomas56 and also associated with poor prognosis57. Employing a combinatorial approach in lung cancers with a targetable alteration using a MEK inhibitor may also be useful in situations of acquired resistance to RTK inhibitors. A strategy employing MEK inhibition also has the added advantage of previously demonstrated safety in early phase trials31,32.
Our study is the first to study the combination of MDM2/MEK inhibition in models of lung adenocarcinoma across a diversity of concurrent oncogenic alterations in LUAD, demonstrating therapeutic benefit in a wide variety of patient-derived xenografts with distinct mutational profiles. This study provides a strong biological rationale to study an MDM2-MEK inhibitor combination in a clinical trial for patients with LUAD and a driver alteration with TP53 wildtype status and MDM2amp, particularly of interest in cases of acquired resistance to targeted therapy, for which targeted therapy options are limited. In addition, our data in SW1573 and A549 models support the study of this combination in patients with a driver alteration and TP53 wildtype status. MDM2 inhibitors are being tested in clinical trials54,58, but none are investigating a combination with MEK inhibition in oncogene-driven LUAD. This combination strategy of MDM2/MEK inhibition could offer a new therapeutic approach outside conventional chemotherapy in a substantial subpopulation of patients with disease progression on targeted therapy. As a result of our findings, a phase 1/2 clinical trial investigating the use of milademetan in combination with MEK inhibition in patients with TP53 wildtype/MDM2amp LUAD with concurrent driver alterations in EGFR, METex14, RET, ALK, or KRAS who progress on standard targeted therapy is currently planned.
Supplementary Material
Figure S1. Synergy matrices for A. H1395 (BRAFG469A, MDM2amp, and TP53 wildtype) and B. A549 (KRASG12S, TP53 wildtype).
Figure S2. A. Single-agent activity of Selpercatinib RET inhibitor in ECLC5-GLx patient-derived cell line (TRIM33-RET, MDM2amp) B. Single-agent activity of Capmatinib MET inhibitor patient-derived cell line in LUAD12c cell line (METex14, KRASG12S, MDM2amp) C. Single-agent activity of Sotorasib KRAS inhibitor in SW1573 cell line (KRASG12C). Values are expressed relative to the vehicle-treated control (100%). Data were analyzed by nonlinear regression to determine IC50 for inhibition of growth (see Supplemental Table 4 for IC50 values). Results represent the mean ± SD of three replicate determinations in one experiment and repeated in an independent experiment.
Figure S3. Western blot analysis for A549 cell line at 0 hours, 6hours, 24hours, and 48hours with milademetan 100nM, trametinib 50nM, or combination. GAPDH used as a loading control.
Figure S4. Phospho-RTK arrays for control and milademetan (100nm) at 48h A. ECLC5-GLx (TRIM33-RET, MDM2amp, TP53 wildtype), B. LUAD12c (METex14, KRASG12S, MDM2amp, TP53 wildtype), and C. SW1573 cell line (KRASG12C, TP53 wildtype).
Figure S5. Individual gene counts by RNAsequencing according to treatment condition in ECLC5-GLx cell line (TRIM33-RET, MDM2amp, TP53 wildtype). Gene of interest presented on top of each graph. A. LATS2 B. PCKS5 C. E2F1 D. USP18 E. IRF9 F. DUSP6 G. DUSP4 H. CCND1. Results represent 4 biological replicates for each condition. NC; untreated. Mila; milademetan 100nM. Tram; Trametinib 100nM. Combo; combination milademetan and trametinib 100nM each. *p<0.05 by Wilcox Test.
Figure S6. Individual gene counts by RNAsequencing according to treatment condition in LUAD12c cell line (METex14, KRASG12S, MDM2amp, TP53 wildtype). Cells treated with 100nM milademetan, 20 nM trametinib, or combination 100 nM milademetan and 10 nM trametinib. 4 biological replicates from 4 independent experiments were pooled for RNA sequencing. Gene of interest presented on top of each graph. A. LATS2 B. PCKS5 C. E2F1 D. DUSP6 E. DUSP4 F. CCND1. Results represent 4 biological replicates for each condition. NC; untreated. Mila; milademetan 100nM. Tram; Trametinib 100nM. Combo; combination milademetan and trametinib 100nM each. *p<0.05 by Wilcox Test.
Figure S7. A. Lung adenocarcinoma model LU-01-448 model (EML4/ALK, MDM2amp, TP53 wildtype) was treated with the indicated dose of either vehicle, milademetan, trametinib, or combination daily on a 7d schedule (8 mice/group). Dosing cessation indicated with a dashed line. GDF15 fold change at B. post-first dose in hours relative to baseline and C. after 7-days of dosing according to each treatment group relative to baseline. D. Western blot on protein lysates obtained from mice engrafted with ECLC5-GLx tumors treated with vehicle, milademetan (100mg/kg), trametinib (1mg/kg) or combination (milademetan 100mg/kg and trametinib 1mg/kg) at 7d post first dose. 3 mice/group, mouse number indicated on the top. E. Western blot on protein lysates obtained from mice engrafted with LU-01-0448 tumors treated with vehicle, milademetan (100mg/kg), trametinib (1mg/kg) or combination (milademetan 100mg/kg and trametinib 1mg/kg) after dosing on day 7. 3 mice/group, mouse number indicated on the top. Vinculin and β-actin used as Western blot loading controls.
Figure S8. Tumor-bearing animal weights in grams during course of treatment for all models evaluated in the study.
Acknowledgements:
The authors wish to thank Jenna-Marie Dix and Marissa Mattar for administrative support. The authors also wish to thank Clare Wilhelm for editorial support. The authors also wish to thank Dr. Audrey Mauguen from the MSK Biostatistics Department for assistance with in vivo statistical analyses.
Funding information:
AE effort funded by: Canadian Institutes of Health Research Fellowship, the Royal College of Physicians and Surgeons of Canada Detweiler Traveling Fellowship, the Henry R. Shibata Cedar’s Cancer Centre Fellowship, and The Ning Zhao & Ge Li Family Initiative for Lung Cancer Research and New Therapies. This study was funded partially by: Rain Oncology Inc., the National Institute of Health P01-CA129243-12 and the National Cancer Institute Cancer Center Core Grant P30-CA008748. Dr. Drilon was supported by the National Cancer Institute/National Institute of Health RO1 grant 1R01CA251591-01A1.
Footnotes
Conflicts of Interest:
AE reports speaking fees from Merck. CMV reports consulting fees from Paige AI. AD reports Honoraria/Advisory Boards: Ignyta, Genentech, Roche, MORE Health, AXIS, Loxo, Eli Lilly and Company, Bayer, AbbVie, EPG Health, Takeda, Ariad, Millenium, 14ner, Elevation Oncology, Harborside Nexus, TP Therapeutics, ArcherDX, Liberum, AstraZeneca, Monopteros, RV More, Pfizer, Novartis, Ology, Blueprint Medicines, EMD Serono, Amgen, Helsinn, Medendi, TouchIME, BeiGene, Repare RX, Janssen, BergenBio, Nuvalent, Entos, Hengrui Therapeutics, Merus, Treeline Bio, Exelixis, Chugai Pharmaceutical, Prelude, Tyra Biosciences, Remedica Ltd, Applied Pharmaceutical Science, Inc., Verastem, mBrace, Treeline, MonteRosa, AXIS, EcoR1; Associated Research Paid to Institution: Pfizer, Exelixis, GlaxoSmithKlein, Teva, Taiho, PharmaMar; Royalties: Wolters Kluwer; Other (Food/Beverage): Merck, Puma, Merus, Boehringer Ingelheim CME Honoraria: Medscape, OncLive, PeerVoice, Physicians Education Resources, Targeted Oncology, Research to Practice, Axis, Peerview Institute, Paradigm Medical Communications, WebMD, MJH Life Sciences, AXIS, EPG Health, JNCC/Harborside, I3 Health. GJR reports institutional research support from Mirati, Lilly, Takeda, Merck, Roche, Pfizer, and Novartis. WWL is a scientific advisor for Hyperbio Therapeutics. MGK reports honoraria from AstraZeneca and Pfizer. VGT and RCD are employees and shareholders of Rain Oncology Inc. CMR has consulted with AbbVie, Amgen, Astra Zeneca, D2G, Daiichi Sankyo, Epizyme, Genentech/Roche, Ipsen, Jazz, Kowa, Lilly, Merck, and Syros, and serves on the scientific advisory boards of Bridge Medicines, Earli, and Harpoon Therapeutics. Romel Somwar has received research grants from Merus, LOXO Oncology and Elevation Oncology Inc. and Helsinn. ML has received honoraria for advisory board participation from Merck, AstraZeneca, Bristol Myers Squibb, Blueprint Medicines, Janssen Pharmaceuticals, Takeda Pharmaceuticals, Lilly Oncology, LOXO Oncology, Bayer, ADC Therapeutics, Riken Genesis/Sysmex, and PaigeAI, and research support from Rain Oncology, LOXO Oncology, Merus, Elevation Oncology, Helsinn Therapeutics, and ADC Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19(8):2240–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosen EY, Won HH, Zheng Y, et al. The evolution of RET inhibitor resistance in RET-driven lung and thyroid cancers. Nat Commun. 2022;13(1):1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jänne PA, Riely GJ, Gadgeel SM, et al. Adagrasib in Non–Small-Cell Lung Cancer Harboring a KRASG12C Mutation. New England Journal of Medicine. 2022;387(2):120–131. [DOI] [PubMed] [Google Scholar]
- 4.Skoulidis F, Li BT, Dy GK, et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. New England Journal of Medicine. 2021;384(25):2371–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drilon A, Oxnard GR, Tan DSW, et al. Efficacy of Selpercatinib in RET Fusion–Positive Non–Small-Cell Lung Cancer. New England Journal of Medicine. 2020;383(9):813–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paik PK, Felip E, Veillon R, et al. Tepotinib in Non–Small-Cell Lung Cancer with MET Exon 14 Skipping Mutations. New England Journal of Medicine. 2020;383(10):931–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hong DS, Fakih MG, Strickler JH, et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. New England Journal of Medicine. 2020;383(13):1207–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schoenfeld AJ, Chan JM, Kubota D, et al. Tumor Analyses Reveal Squamous Transformation and Off-Target Alterations As Early Resistance Mechanisms to First-line Osimertinib in EGFR-Mutant Lung Cancer. Clin Cancer Res. 2020;26(11):2654–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garon EB, Ciuleanu TE, Arrieta O, et al. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): a multicentre, double-blind, randomised phase 3 trial. Lancet. 2014;384(9944):665–673. [DOI] [PubMed] [Google Scholar]
- 10.Jordan EJ, Kim HR, Arcila ME, et al. Prospective Comprehensive Molecular Characterization of Lung Adenocarcinomas for Efficient Patient Matching to Approved and Emerging Therapies. Cancer Discovery. 2017;7(6):596–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chène P Inhibiting the p53–MDM2 interaction: an important target for cancer therapy. Nature Reviews Cancer. 2003;3(2):102–109. [DOI] [PubMed] [Google Scholar]
- 12.Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69(7):1237–1245. [DOI] [PubMed] [Google Scholar]
- 13.Zhu H, Gao H, Ji Y, et al. Targeting p53–MDM2 interaction by small-molecule inhibitors: learning from MDM2 inhibitors in clinical trials. Journal of Hematology & Oncology. 2022;15(1):91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roy S, Laroche-Clary A, Verbeke S, Derieppe MA, Italiano A. MDM2 Antagonists Induce a Paradoxical Activation of Erk1/2 through a P53-Dependent Mechanism in Dedifferentiated Liposarcomas: Implications for Combinatorial Strategies. Cancers (Basel). 2020;12(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mattar M, McCarthy CR, Kulick AR, Qeriqi B, Guzman S, de Stanchina E. Establishing and Maintaining an Extensive Library of Patient-Derived Xenograft Models. Front Oncol. 2018;8:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Odintsov I, Mattar MS, Lui AJW, et al. Novel Preclinical Patient-Derived Lung Cancer Models Reveal Inhibition of HER3 and MTOR Signaling as Therapeutic Strategies for NRG1 Fusion-Positive Cancers. J Thorac Oncol. 2021;16(7):1149–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn. 2015;17(3):251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arora A, Shen R, Seshan VE. FACETS: Fraction and Allele-Specific Copy Number Estimates from Tumor Sequencing. Methods Mol Biol. 2022;2493:89–105. [DOI] [PubMed] [Google Scholar]
- 19.Di Veroli GY, Fornari C, Wang D, et al. Combenefit: an interactive platform for the analysis and visualization of drug combinations. Bioinformatics. 2016;32(18):2866–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berenbaum MC. What is synergy? Pharmacol Rev. 1989;41(2):93–141. [PubMed] [Google Scholar]
- 21.Pimentel H, Bray NL, Puente S, Melsted P, Pachter L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat Methods. 2017;14(7):687–690. [DOI] [PubMed] [Google Scholar]
- 22.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27(12):1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16(5):284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vardi Y, Ying Z, Zhang CH. Two-sample tests for growth curves under dependent right censoring. Biometrika. 2001;88(4):949–960. [Google Scholar]
- 26.Hayashi T, Odintsov I, Smith RS, et al. RET inhibition in novel patient-derived models of RET-fusion positive lung adenocarcinoma reveals a role for MYC upregulation. Dis Model Mech. 2020;14(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suzawa K, Offin M, Lu D, et al. Activation of KRAS Mediates Resistance to Targeted Therapy in MET Exon 14-mutant Non-small Cell Lung Cancer. Clin Cancer Res. 2019;25(4):1248–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hai J, Sakashita S, Allo G, et al. Inhibiting MDM2-p53 Interaction Suppresses Tumor Growth in Patient-Derived Non-Small Cell Lung Cancer Xenograft Models. J Thorac Oncol. 2015;10(8):1172–1180. [DOI] [PubMed] [Google Scholar]
- 29.Haronikova L, Bonczek O, Zatloukalova P, et al. Resistance mechanisms to inhibitors of p53-MDM2 interactions in cancer therapy: can we overcome them? Cell Mol Biol Lett. 2021;26(1):53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Erba HP, Becker PS, Shami PJ, et al. Phase 1b study of the MDM2 inhibitor AMG 232 with or without trametinib in relapsed/refractory acute myeloid leukemia. Blood Adv. 2019;3(13):1939–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moschos SJ, Sandhu SK, Lewis KD, et al. Phase 1 study of the p53-MDM2 inhibitor AMG 232 combined with trametinib plus dabrafenib or trametinib in patients (Pts) with TP53 wild type (TP53WT) metastatic cutaneous melanoma (MCM). Journal of Clinical Oncology. 2017;35(15_suppl):2575–2575. [Google Scholar]
- 32.de Weger VA, de Jonge M, Langenberg MHG, et al. A phase I study of the HDM2 antagonist SAR405838 combined with the MEK inhibitor pimasertib in patients with advanced solid tumours. Br J Cancer. 2019;120(3):286–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gounder MM, Bauer TM, Schwartz GK, et al. A First-in-Human Phase I Study of Milademetan, an MDM2 Inhibitor, in Patients With Advanced Liposarcoma, Solid Tumors, or Lymphomas. Journal of Clinical Oncology. 2023:JCO.22.01285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Di Veroli GY, Fornari C, Wang D, et al. Combenefit: an interactive platform for the analysis and visualization of drug combinations. Bioinformatics. 2016;32(18):2866–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khandelwal Gilman KA, Han S, Won YW, Putnam CW. Complex interactions of lovastatin with 10 chemotherapeutic drugs: a rigorous evaluation of synergism and antagonism. BMC Cancer. 2021;21(1):356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buffet C, Hecale-Perlemoine K, Bricaire L, et al. DUSP5 and DUSP6, two ERK specific phosphatases, are markers of a higher MAPK signaling activation in BRAF mutated thyroid cancers. PLoS One. 2017;12(9):e0184861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang P, Chen W, Li X, et al. Downregulation of cyclin D1 sensitizes cancer cells to MDM2 antagonist Nutlin-3. Oncotarget. 2016;7(22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aylon Y, Ofir-Rosenfeld Y, Yabuta N, et al. The Lats2 tumor suppressor augments p53-mediated apoptosis by promoting the nuclear proapoptotic function of ASPP1. Genes Dev. 2010;24(21):2420–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jaaks P, Bernasconi M. The proprotein convertase furin in tumour progression. Int J Cancer. 2017;141(4):654–663. [DOI] [PubMed] [Google Scholar]
- 40.Zhao C, Huang R, Zeng Z, et al. Downregulation of USP18 reduces tumor-infiltrating activated dendritic cells in extranodal diffuse large B cell lymphoma patients. Aging (Albany NY). 2021;13(10):14131–14158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharma BR, Karki R, Sundaram B, Wang Y, Vogel P, Kanneganti TD. The Transcription Factor IRF9 Promotes Colorectal Cancer via Modulating the IL-6/STAT3 Signaling Axis. Cancers (Basel). 2022;14(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang P, Chen W, Li X, et al. Downregulation of cyclin D1 sensitizes cancer cells to MDM2 antagonist Nutlin-3. Oncotarget. 2016;7(22):32652–32663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sicklick JK, Kato S, Okamura R, et al. Molecular profiling of advanced malignancies guides first-line N-of-1 treatments in the I-PREDICT treatment-naïve study. Genome Med. 2021;13(1):155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tan AC, Bagley SJ, Wen PY, et al. Systematic review of combinations of targeted or immunotherapy in advanced solid tumors. Journal for ImmunoTherapy of Cancer. 2021;9(7):e002459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guerreiro N, Jullion A, Ferretti S, Fabre C, Meille C. Translational Modeling of Anticancer Efficacy to Predict Clinical Outcomes in a First-in-Human Phase 1 Study of MDM2 Inhibitor HDM201. Aaps j. 2021;23(2):28. [DOI] [PubMed] [Google Scholar]
- 46.Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature. 1993;362(6423):857–860. [DOI] [PubMed] [Google Scholar]
- 47.Quesnel B, Preudhomme C, Fournier J, Fenaux P, Peyrat JP. MDM2 gene amplification in human breast cancer. Eur J Cancer. 1994;30a(7):982–984. [DOI] [PubMed] [Google Scholar]
- 48.Ladanyi M, Lewis R, Jhanwar SC, Gerald W, Huvos AG, Healey JH. MDM2 and CDK4 gene amplification in Ewing’s sarcoma. The Journal of Pathology. 1995;175(2):211–217. [DOI] [PubMed] [Google Scholar]
- 49.Sato H, Schoenfeld AJ, Siau E, et al. MAPK Pathway Alterations Correlate with Poor Survival and Drive Resistance to Therapy in Patients with Lung Cancers Driven by ROS1 Fusions. Clinical Cancer Research. 2020;26(12):2932–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Greenberg AK, Basu S, Hu J, et al. Selective p38 activation in human non-small cell lung cancer. Am J Respir Cell Mol Biol. 2002;26(5):558–564. [DOI] [PubMed] [Google Scholar]
- 51.von Itzstein MS, Drapkin BJ, Minna JD. How lung cancer cells change identity. Elife. 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu D, Nagelberg A, Chow JL, et al. MET Exon 14 Splice-Site Mutations Preferentially Activate KRAS Signaling to Drive Tumourigenesis. Cancers (Basel). 2022;14(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang X, Xie Q, Ji Y, et al. Targeting KRAS-mutant stomach/colorectal tumors by disrupting the ERK2-p53 complex. Cell Rep. 2023;42(1):111972. [DOI] [PubMed] [Google Scholar]
- 54.Gounder MM, Bauer TM, Schwartz GK, et al. 7LBA Late Breaking - Milademetan, an oral MDM2 inhibitor, in well-differentiated/dedifferentiated liposarcoma: results from a phase 1 study in patients with solid tumors or lymphomas. European Journal of Cancer. 2020;138:S3–S4. [Google Scholar]
- 55.Nag S, Qin J, Srivenugopal KS, Wang M, Zhang R. The MDM2-p53 pathway revisited. J Biomed Res. 2013;27(4):254–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu W, Zhuang C, Huang T, et al. Loss of CDKN2A at chromosome 9 has a poor clinical prognosis and promotes lung cancer progression. Molecular Genetics & Genomic Medicine. 2020;8(12):e1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Konopleva M, Martinelli G, Daver N, et al. MDM2 inhibition: an important step forward in cancer therapy. Leukemia. 2020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Synergy matrices for A. H1395 (BRAFG469A, MDM2amp, and TP53 wildtype) and B. A549 (KRASG12S, TP53 wildtype).
Figure S2. A. Single-agent activity of Selpercatinib RET inhibitor in ECLC5-GLx patient-derived cell line (TRIM33-RET, MDM2amp) B. Single-agent activity of Capmatinib MET inhibitor patient-derived cell line in LUAD12c cell line (METex14, KRASG12S, MDM2amp) C. Single-agent activity of Sotorasib KRAS inhibitor in SW1573 cell line (KRASG12C). Values are expressed relative to the vehicle-treated control (100%). Data were analyzed by nonlinear regression to determine IC50 for inhibition of growth (see Supplemental Table 4 for IC50 values). Results represent the mean ± SD of three replicate determinations in one experiment and repeated in an independent experiment.
Figure S3. Western blot analysis for A549 cell line at 0 hours, 6hours, 24hours, and 48hours with milademetan 100nM, trametinib 50nM, or combination. GAPDH used as a loading control.
Figure S4. Phospho-RTK arrays for control and milademetan (100nm) at 48h A. ECLC5-GLx (TRIM33-RET, MDM2amp, TP53 wildtype), B. LUAD12c (METex14, KRASG12S, MDM2amp, TP53 wildtype), and C. SW1573 cell line (KRASG12C, TP53 wildtype).
Figure S5. Individual gene counts by RNAsequencing according to treatment condition in ECLC5-GLx cell line (TRIM33-RET, MDM2amp, TP53 wildtype). Gene of interest presented on top of each graph. A. LATS2 B. PCKS5 C. E2F1 D. USP18 E. IRF9 F. DUSP6 G. DUSP4 H. CCND1. Results represent 4 biological replicates for each condition. NC; untreated. Mila; milademetan 100nM. Tram; Trametinib 100nM. Combo; combination milademetan and trametinib 100nM each. *p<0.05 by Wilcox Test.
Figure S6. Individual gene counts by RNAsequencing according to treatment condition in LUAD12c cell line (METex14, KRASG12S, MDM2amp, TP53 wildtype). Cells treated with 100nM milademetan, 20 nM trametinib, or combination 100 nM milademetan and 10 nM trametinib. 4 biological replicates from 4 independent experiments were pooled for RNA sequencing. Gene of interest presented on top of each graph. A. LATS2 B. PCKS5 C. E2F1 D. DUSP6 E. DUSP4 F. CCND1. Results represent 4 biological replicates for each condition. NC; untreated. Mila; milademetan 100nM. Tram; Trametinib 100nM. Combo; combination milademetan and trametinib 100nM each. *p<0.05 by Wilcox Test.
Figure S7. A. Lung adenocarcinoma model LU-01-448 model (EML4/ALK, MDM2amp, TP53 wildtype) was treated with the indicated dose of either vehicle, milademetan, trametinib, or combination daily on a 7d schedule (8 mice/group). Dosing cessation indicated with a dashed line. GDF15 fold change at B. post-first dose in hours relative to baseline and C. after 7-days of dosing according to each treatment group relative to baseline. D. Western blot on protein lysates obtained from mice engrafted with ECLC5-GLx tumors treated with vehicle, milademetan (100mg/kg), trametinib (1mg/kg) or combination (milademetan 100mg/kg and trametinib 1mg/kg) at 7d post first dose. 3 mice/group, mouse number indicated on the top. E. Western blot on protein lysates obtained from mice engrafted with LU-01-0448 tumors treated with vehicle, milademetan (100mg/kg), trametinib (1mg/kg) or combination (milademetan 100mg/kg and trametinib 1mg/kg) after dosing on day 7. 3 mice/group, mouse number indicated on the top. Vinculin and β-actin used as Western blot loading controls.
Figure S8. Tumor-bearing animal weights in grams during course of treatment for all models evaluated in the study.