

SUMMARY
Glial cells and central nervous system (CNS)-infiltrating leukocytes contribute to multiple sclerosis (MS). However, the networks that govern crosstalk among these ontologically distinct populations remain unclear. Here we show, in mice and humans, that CNS-resident astrocytes and infiltrating CD44hiCD4+ T cells generated interleukin-3 (IL-3) while microglia and recruited myeloid cells expressed interleukin-3 receptor-ɑ (IL-3Rɑ). Astrocytic and T cell IL-3 elicited an immune migratory and chemotactic program by IL-3Rɑ+ myeloid cells that enhanced CNS immune cell infiltration, exacerbating MS and its preclinical model. Multiregional snRNA-seq of human CNS tissue revealed the appearance of IL3RA-expressing myeloid cells with chemotactic programing in MS plaques. IL3RA expression by plaque myeloid cells and IL-3 amount in the cerebrospinal fluid predicted myeloid and T cell abundance in the CNS and correlated with MS severity. Our findings establish IL-3:IL-3RA as a glial-peripheral immune network that prompts immune cell recruitment to the CNS and worsens MS.
Keywords: Interleukin-3, multiple sclerosis, chemokine, microglia, astrocyte, monocyte, neuroinflammation, recruitment
Graphical Abstract
eTOC Blurb
IL-3 is a multifunction cytokine who’s function in neuroinflammation is presently unclear. Kiss et al. reveal that astrocyte- and T cell-sourced IL-3 programs myeloid cells to exacerbate neuroinflammation by inciting a cellular recruitment program that drives the accrual of immune cells in the CNS, worsening MS and its preclinical model.
INTRODUCTION
Multiple Sclerosis (MS) is a chronic neuroinflammatory disease that affects young adults and remains without cure1,2. Instigated by autoimmune reactivity to myelin, MS is clinically characterized by episodes of neurologic disability lasting days or weeks. Clinical symptoms often progress over decades and eventually lead to impaired mobility, reduced cognition, and ultimately paralysis and early death. Pathologically, MS presents as inflammatory neuropil and demyelinated lesions throughout the central nervous system (CNS), which are composed of peripherally derived immune cell infiltrates and resident glial cells including astrocytes and microglia. The presence of ontologically distinct immune and glial cell populations – astrocytes deriving from embryonic neuronal progenitors, microglia from primitive yolk sac hematopoiesis, and peripheral innate and adaptive immune cells from adult bone marrow (BM) hematopoiesis – necessitates tightly controlled communication systems to orchestrate cellular function in this complex inflammatory milieu. Immune cell infiltrates and resident glial cell populations are consequential to MS3; however, the communication axes that mediate peripheral-central crosstalk in the diseased CNS remain ill-defined, despite their therapeutic potential2,4.
Interleukin-3 (IL-3) is a multifaceted cytokine and growth factor implicated in inflammatory and autoimmune diseases5,6. A member of the colony-stimulating factor family, IL-3 has been linked to immune disorders including sepsis7, atherosclerosis8, lupus nephritis9, and arthritis10. These studies have mostly focused on IL-3’s function as a hematopoietic growth factor and an endocrine stimulant of monocytosis, while IL-3’s impact beyond its action on BM and splenic hematopoietic stem and progenitor cells is less clear. IL-3’s contributions to brain health and disease have remained largely unknown. Recently, we have demonstrated a fundamental, protective role for IL-3 in Alzheimer’s disease (AD) where it instigates clearance of beta Amyloid (Aβ) and tau by microglia11. Yet the function of IL-3 in other neuroinflammatory contexts, including CNS autoimmunity, MS, and its murine model experimental autoimmune encephalomyelitis (EAE), has remained undefined. While some reports suggest IL-3 may play a role in demyelinating animal models, these studies have employed non-specific tools producing contradictory results12,13, or used artificial overexpression systems14-16. For example, the consequential cell populations that generate the cytokine or functionally respond to it in the inflamed CNS remain unknown, and this axis’s role in the CNS of humans with MS is uncertain. The communication systems amongst resident glial cells and infiltrating immune cells in the inflamed CNS are unresolved, despite being an emerging node for therapeutic intervention17. Therefore, there is a clear need to examine, define, and resolve the IL-3:IL-3Rɑ dyad in mice and humans using contemporary tools to advance our fundamental understanding of the disease and therapeutic applications.
Here we report, in human MS and its murine model, that CNS-resident astrocytes and infiltrating CD44hiCD4+ T cells produce IL-3 which instigates a migratory and chemotactic program by IL-3Rɑ-expressing CNS-resident microglia and peripherally-derived myeloid cells that perpetuates immune cell recruitment, CNS inflammation, clinical and pathological severity. In humans, cerebrospinal fluid (CSF) IL-3 amount predicted clinical MS diagnosis and severity. In mice, deletion of Il3 or Il3rɑ globally or specifically from their relevant cellular sources improved EAE and neural demyelination and restricted immune cell recruitment to the CNS by dampening myeloid-derived chemotactic cues. In humans, regionally defined single-nuclei RNA sequencing (snRNA-seq) of white and grey matter tissue from control individuals and MS patients and pathologic plaque tissue of MS patients demonstrated that IL3RA-expressing myeloid cells represent a unique subset that accumulate in MS plaques and are endowed with immune cell migration and chemotactic properties. Further, plaque myeloid cell IL3RA expression and CSF IL-3 amount associated with immune cell recruitment to the CNS of MS patients. These findings define IL-3 as a critical mediator of central-peripheral immune crosstalk and a regulator of CNS myeloid cell programing and neuroinflammation in MS.
RESULTS
IL-3 associates with human RRMS and exacerbates spinal cord inflammation, demyelination, and clinical severity in EAE
Given our limited knowledge of IL-3 in neuroinflammation, we tested if IL-3 is relevant in human MS. To do so, we measured the cytokine in the CSF, a disease-relevant compartment that sensitively reflects immunogenic processes occurring in the CNS18,19, of 36 patients with clinically diagnosed relapsing remitting MS (RRMS) and 35 age-matched unaffected controls. We found that, relative to control donors, RRMS patients had elevated IL-3 in their CSF (Fig 1A). In controls and RRMS patients, CSF IL-3 amount did not depend on sex (Fig S1A) and among RRMS patients, IL-3 did not depend on whether CSF was collected during phases of remission or relapse (Fig S1B). We then analyzed a separate cohort of patients with clinically isolated syndrome (CIS) who experienced one symptomatic episode of MS and observed that CSF IL-3 was increased in patients who converted to fully diagnosed MS (according to the Poser criteria) compared to individuals who did not convert to a clinical MS diagnosis (non-converters) over a >2-year follow up (Fig 1A). Again, CSF IL-3 was not influenced by sex among converters and non-converters (Fig S1C). Together, these findings put forth a role for IL-3 in the CNS of humans with MS.
Figure 1. Interleukin-3 associates with human RRMS and exacerbates spinal cord inflammation, demyelination, and EAE.
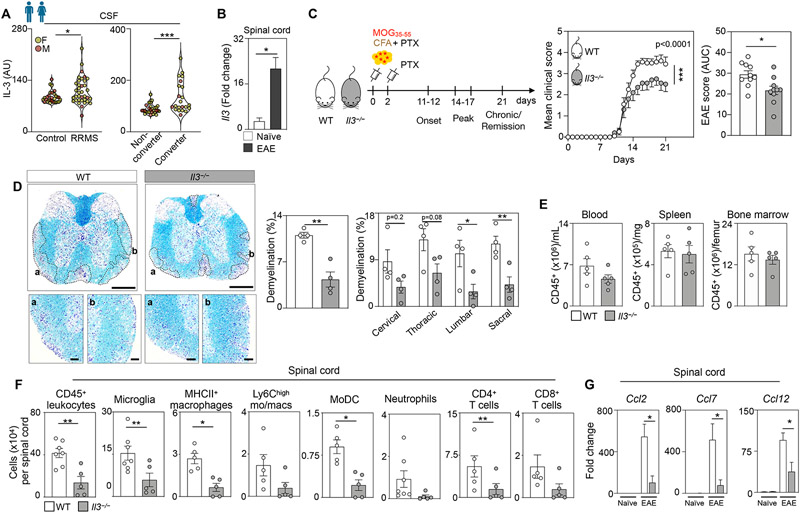
(A) Left panel - IL-3 amount in the CSF of male and female unaffected control subjects and patients with RRMS. Right panel - Baseline CSF IL-3 amount in male and female non-converters and patients who converted to MS diagnosis over a 2 year follow up (n=29 controls, 36 RRMS patients, 28 non-converters, 22 converters; Mann–Whitney U-tests).
(B) Il3 gene expression in the spinal cord in WT EAE mice at disease peak and naive mice (n=4-5 mice/group; Mann–Whitney U-test).
(C) Schematic diagram of the experimental design. EAE was induced in WT and Il3−/− mice by administering myelin oligodendrocyte glycoprotein (MOG35-55) peptide emulsified in complete CFA by subcutaneous injection on day 0 and PTX by intraperitoneal injections on days 0 and 2. Mean clinical disease scores and corresponding AUC analysis of WT and Il3−/− mice over the course of 21 dpi (n=10 mice/group; two-way ANOVA and Mann–Whitney U-test).
(D) Representative histological sections of 4 mice per group and quantification of demyelinated area in diseased spinal cords from WT and Il3−/− mice stained for myelin by Luxol fast blue and counterstained with Cresyl Echt Violet Solution at disease peak. Scale bars represent 500 μm for overview images and 100 μm for the inset images (n=4 mice/group; two-way ANOVA and Mann–Whitney U-test).
(E) Quantification of CD45+ leukocyte numbers in the blood, spleen and bone marrow of WT and Il3−/− mice at the disease peak (n=5 mice/group).
(F) Quantification of leukocyte subsets in the spinal cord of WT and Il3−/− mice at the disease peak (n=5-7 mice/group; Mann–Whitney U-tests).
(G) qPCR analysis of chemokine transcript expression in the spinal cord of healthy and EAE WT and Il3−/− mice (n=4-5 mice/group; one-way ANOVA).
Mean±s.e.m., *p<0.05, **p<0.01, ***p<0.001.
CSF, cerebrospinal fluid; RRMS, relapsing remitting multiple sclerosis; AU, arbitrary units; IL-3, interleukin-3; WT, wildtype; EAE, experimental autoimmune encephalomyelitis; CFA, Freund’s adjuvant; PTX, pertussis toxin; MOG, myelin oligodendrocyte glycoprotein; AUC, area under the curve; dpi, days post immunization.
See also Figures S1 and S2.
To begin mechanistically exploring the function of IL-3 during MS-like neuro-inflammation and -degeneration, we adopted the murine model of MS, experimental autoimmune encephalomyelitis (EAE), in which mice are injected with myelin oligodendrocyte glycoprotein suspended in complete Freund’s adjuvant (MOG35-55/CFA) and pertussis toxin (PTX) causing spinal cord (SC) inflammation, impaired neuromuscular and sensory function, demyelination, and paralysis20. First, we measured Il3 expression in relevant organs of naïve and EAE mice at peak disease. While EAE did not change Il3 expression in the lymph nodes or spleen (Fig S1D), Il3 was significantly higher in the SC of EAE mice relative to healthy controls (Fig 1B). To test if IL-3 has a functional role in EAE, we induced EAE in WT and IL-3-deficient (Il3−/−) mice and monitored clinical disease development over 21 days post immunization (dpi). Clinical assessment revealed that despite similar disease onset, Il3−/− mice had improved clinical scores and were profoundly protected from disease severity and paralysis (Fig 1C). In MS and EAE, paralysis caused by axonal damage and neuro-muscular degeneration is attributed to neuronal demyelination1,21. To determine IL-3’s impact on myelination, we stained SC sections of diseased WT and Il3−/− mice with Luxol fast blue and quantified neuropil and demyelinated areas (Fig 1D). This analysis uncovered substantially reduced demyelination throughout the SC of Il3−/− mice affecting the cervical, thoracic, lumbar, and sacral regions (Fig 1D). These data suggest that IL-3 contributes to demyelination and worsens neuronal integrity, aggravating EAE severity.
We next tested the immunogenic and encephalitogenic functions of IL-3 in EAE. Improved clinical outcomes and reduced disease severity in Il3−/− mice may be due to either alterations in peripheral immune priming in medullary organs or changes in the effector organ, the CNS, that diminish immune cell infiltrates and neuroinflammation. To test the former, we first enumerated peripheral immune cells and found equivalent numbers of T cells, B cells, neutrophils, and monocytes in the blood, spleen, and BM of diseased WT and Il3−/− mice (Fig 1E and Fig S1E and F). Pursuing immune priming more specifically, we measured CD4+ TH cell abundance and proliferation in the spleen and lymph nodes and found that Il3 deletion did not influence these parameters (Fig S2A and B). We next evaluated pro-inflammatory IFN-γ+, IL-17A+, IFN-γ+IL-17A+, and GM-CSF+ TH cell subsets in the spleen and lymph nodes and again did not find alterations in Il3−/− mice (Fig S2C and D). Further, we also observed that IL-3 did not change the amount of resident, migratory, and monocyte-derived dendritic cells (MoDCs) in spleen and lymph nodes nor influence their expression of the co-stimulatory molecules MHCII, CD80, CD86, and OX40L (Fig S2 E-H). To expand on this analysis, we performed ex vivo splenocyte recall response assays to increasing concentrations of MOG35-55 and found that IL-3 deficiency did not influence the capacity of T cells to proliferate or produce cytokines (Fig 2S I-J). All together, these results show IL-3 is dispensable for peripheral priming of myeloid-reactive CD4+ T cells in EAE.
We then turned our attention to the effector organ. EAE and MS are characterized by recruitment of peripheral immune cells to the CNS that contribute to an inflammatory milieu and aggravate disease severity22,23. We found that IL-3 deficiency significantly reduced the number of CD45+ leukocytes in the SC at peak disease (Fig 1F). Flow cytometric analysis of the SC revealed that Il3−/− mice had reductions in important EAE-relevant immune cell populations, including microglia, and peripherally-derived MHCII+ macrophages, MoDCs, and CD4+ T cells (Fig 1F and Fig S1G), proposing that IL-3 plays a role in immune cell recruitment to the CNS. To assess the impact of IL-3 on immune cell recruitment more carefully, we measured key chemoattractants. In WT animals, EAE robustly augmented the expression of Ccl2, Ccl7, and Ccl12 in the SC (Fig 1G). In Il3−/− mice meanwhile, the generation of this chemotaxis program was significantly blunted (Fig 1G). These findings suggest IL-3 plays a pathogenic role in the SC by driving local inflammation and immune cell recruitment during the effector phase of EAE, leading to paralysis and disability without overt influence on peripheral leukocyte priming, abundance, or function.
Astrocytic IL-3 potentiates EAE
Having established a pathogenic role for IL-3 in EAE, we next sought the relevant cell types that generate the cytokine. Flow cytometric analysis of WT EAE mice suggested that both hematopoietic and non-hematopoietic cells produce IL-3 in the SC (Fig 2A). Among CD45+ hematopoietic cells, we identified CD44hiCD4+ T cells as sources of IL-3 and among non-hematopoietic cells, we found that GFAP+ACSA-2+ astrocytes abundantly produce IL-3 (Fig 2A). Robust Il3 transcript expression was confirmed in CD44hiCD4+ T cells and astrocytes sorted from the diseased SC (Fig S3A). To further verify IL-3 generation by astrocytes, we performed immunofluorescent imaging and found that IL-3-producing cells overlapped with GFAP, an astrocyte marker, in the diseased SC (Fig 2B and Fig S3B-C). Quantification of our flow cytometric data revealed that CD4+ T cells and astrocytes produced IL-3 in approximately equivalent proportions and that these populations accounted for nearly all the IL-3 generated in the SC of EAE mice (Fig 2C). Critically, these findings identify two local sources of IL-3 in the SC; however, their relative contributions to neuroinflammation and disease severity remained unknown.
Figure 2. CD44hi CD4+ T cells and astrocytes generate IL-3 in the CNS and astrocytic IL-3 potentiates EAE.
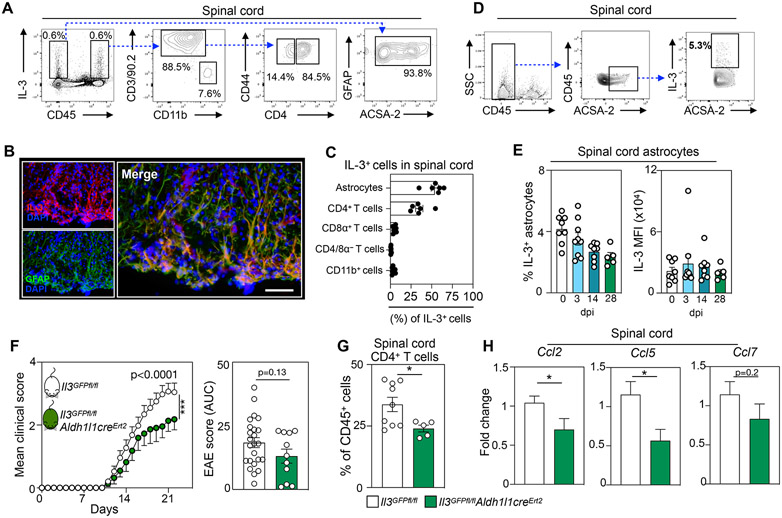
(A) Flow cytometric analysis of IL-3-producing cells in the spinal cords of WT mice at the peak of EAE.
(B) Representative immunofluorescent images of the spinal cord of 4 mice showing IL-3 co-localization with the astrocyte marker GFAP in WT EAE mice at peak disease. Scale bar represents 50 μm.
(C) Flow cytometric quantification of IL-3-producing cells in the spinal cord of WT EAE mice at disease peak (n=6 mice).
(D) Flow cytometric gating strategy identifying IL-3-producing astrocytes in the spinal cord.
(E) Quantification of IL-3 production by spinal cord astrocytes during the course of EAE development (n=5-9 mice/group).
(F) Mean clinical disease scores and corresponding AUC analysis of Il3GFPfl/fl and Il3GFPfl/flAldh1l1creErt2 male and female mice injected with tamoxifen and induced with EAE (n=11-24 mice/group; two-way ANOVA and Mann–Whitney U-test).
(G) Flow cytometric quantification of infiltrating CD4+ T cells in the spinal cord of Il3GFPfl/fl and Il3GFPfl/flAldh1l1creErt2 mice injected with tamoxifen and induced with EAE (n=5-9 mice/group; Mann–Whitney U-test).
(H) qPCR analysis of chemokine transcript expression in the spinal cord of Il3GFPfl/fl and Il3GFPfl/flAldh1l1creErt2 mice injected with tamoxifen and exposed to EAE (n=9-13 mice/group; Mann–Whitney U-tests).
Mean±s.e.m., *p<0.05, **p<0.01, ***p<0.001.
WT, wildtype; EAE, experimental autoimmune encephalomyelitis; GFAP, glial fibrillary acidic protein; DAPI, 4′,6-diamidino-2-phenylindole; AUC, area under the curve; dpi, days post immunization; MFI, mean fluorescence intensity.
See also Figure S3.
To delineate the relevance of these disparate cellular sources of IL-3, we first sought to decipher the contribution of astrocytic IL-3 to CNS inflammation and EAE. Quantification of astrocyte IL-3 production in healthy animals and at various stages of EAE suggested that IL-3+ astrocytes comprise ~5% of the astrocytic pool in the SC (Fig 2D) and that the abundance of IL-3+ astrocytes, and their relative generation of the cytokine, remained unchanged in EAE animals at 3, 14, and 28 dpi (Fig 2E). In agreement, imaging of the EAE SC did not reveal a difference in the abundance of IL-3+GFAP+ astrocytes in demyelinated plaques relative to adjacent unaffected tissue (Fig S3D). To test the specific contribution of astrocyte-sourced IL-3 to EAE, we crossed Il3GFPfl/fl mice with Aldh1l1creErt2 mice to generate tamoxifen-inducible astrocyte-specific IL-3 deficient animals (Il3GFPfl/flAldh1l1creErt2). We then used a tamoxifen injection strategy to delete astrocytic IL-3 in adult mice prior to EAE induction (Fig S3E)11. Tamoxifen injected Il3GFPfl/flAldh1l1creErt2 EAE mice had robust deletion of astrocytic, but not T cell, IL-3 and reduced total IL-3 amount in SC homogenate but not the plasma (Fig S3F-H). Compared to tamoxifen-injected Il3GFPfl/fl littermate controls and beginning at early disease stages, astrocyte-specific Il3 deletion significantly improved clinical disease score and ameliorated paralysis (Fig 2F), a phenotype we observed in both male and female mice (Fig S3I). Moreover, relative to control Il3GFPfl/fl mice, diseased Il3GFPfl/flAldh1l1creErt2 mice had fewer CD4+ T cell infiltrates in the SC (Fig 2G), lower expression of the chemokines Ccl2 and Ccl5, and tendency towards reduced Ccl7 in the SC (Fig 2H). Together, these data reveal that astrocytic IL-3 plays a pathogenic role in CNS inflammation in EAE.
IL-3+CD44hiCD4+ TH cells worsen EAE and spinal cord inflammation
Given that MS is a T cell-mediated CNS autoimmune disease driven by encephalitogenic CD4+ T cells and that we identified CD44hiCD4+ T cells as hematopoietic producers of IL-3, we next assessed whether IL-3 generated by these cells impacts EAE. First, we monitored the kinetics and phenotype of IL-3+ CD44hiCD4+ T cells in the periphery and CNS during EAE. Using our dual IL-3-GFP reporter and floxed mice (Il3GFPfl/fl), we quantified GFP+CD44hiCD4+ T cells in peripheral tissues and the SC at 8,12,16, and 22 days post EAE induction. The number of IL3GFP+CD44hiCD4+ T cells peaked at 8 dpi in the blood, BM, spleen, and lymph nodes, revealing the generation and systemic mobilization of these cells during early disease stages (Fig 3A). In the SC, we observed notable recruitment of IL3GFP+CD44hiCD4+ T cells, peaking at day 12 and remaining elevated throughout later disease timepoints (Fig 3A). In the SC, IL3GFP+CD44hiCD4+ T cells expressed the homing molecule CD62L (Fig S3J) suggesting that they were antigen-experienced effectors, and their abundance was heightened at all grades of clinical severity (Fig S3K). We next tested IL-3 production by specific T helper (TH) cell lineages in the diseased SC and found that the cytokine is broadly generated by TH1, TH17, IL-17A+IFN-γ+, and GM-CSF+ cells (Fig 3B) indicating that IL-3 is produced by many TH cell types in the CNS. These findings demonstrate the systemic mobilization of IL-3 producing effector T cells and their recruitment to the CNS during EAE.
Figure 3. IL-3 production by CD4+ CD44hi effector TH cells promotes EAE development.

(A) Flow cytometric quantification of IL-3GFP+CD44hiCD4+ T cells in the blood, bone marrow, spleen, lymph nodes, and spinal cord throughout the course of EAE (n=3-7 mice/ group; two-way ANOVA).
(B) Flow cytometric analysis of IL-3+ cell frequency among TH1 (IFN-γ+), TH17(IL-17A+), IL-17A+IFN-γ+ (IFN-γ+ IL-17A+), GM-CSF+ (GM-CSF+ IFN-γ+ IL-17A−), and other (GM-CSF− IFN-γ− IL-17A−) CD44hi CD4+ TH cells in the spinal cord at the peak of EAE (n=6 mice).
(C) Mean clinical scores and corresponding AUC analysis of Il3GFPfl/fl and Il3GFPfl/flCd4cre male and female mice exposed to EAE (n=14-19 mice/group; two-way ANOVA and Mann–Whitney U-test).
(D) Quantification of leukocyte subsets in the spinal cord of Il3GFPfl/fl and Il3GFPfl/flCd4cre mice at the disease peak by flow cytometry (n=8-12 mice/group; Mann–Whitney U-tests).
(E) qPCR analysis of chemokine transcript expression in the spinal cord of Il3GFPfl/fl and Il3GFPfl/flCd4cre mice subjected to EAE (n=6-12 mice/group; Mann–Whitney U-tests).
Mean±s.e.m., *p<0.05, **p<0.01, ***p<0.001.
EAE, experimental autoimmune encephalomyelitis; AUC, area under the curve; dpi, days post immunization. See also Figure S3.
We next explored whether IL-3 serves as a crucial pathogenic T cell-derived factor in EAE. To generate T cell-specific IL-3 deficient mice, we crossed Il3GFPfl/fl with Cd4cre mice and subjected the resulting Il3GFPfl/flCd4cre animals, along with Il3GFPfl/fl littermate controls, to EAE. First, we confirmed Il3 transcript deletion in T cells, but not astrocytes, sorted from the SC of diseased Il3GFPfl/flCd4cre mice (Fig S3L). Moreover, flow cytometry demonstrated a robust reduction in IL-3 production by T cells in the SC, spleen, and lymph node of diseased Il3GFPfl/flCd4cre mice (Fig S3M). Together, this resulted in lower IL-3 amount in SC homogenate but not plasma of Il3GFPfl/flCd4cre mice (Fig S3N). After inducing EAE in Il3GFPfl/fl and Il3GFPfl/flCd4cre mice, we found that despite equivalent disease onset, T cell-specific Il3 deletion protected against EAE development, clinical symptoms, and paralysis (Fig 3C). Importantly, we found that both male and female Il3GFPfl/flCd4cre mice were protected from disease (Fig S3O). Flow cytometric enumeration analysis of the SC revealed a blunting of immune cell infiltrates in Il3GFPfl/flCd4cre mice at disease peak (Fig 3D). Further, gene expression analysis revealed reduced Ccl5 and a tendency towards reduced Ccl2 and Ccl7 in the SC of Il3GFPfl/flCd4cre mice (Fig 3E). Together, these data identify CD44hiCD4+ TH cells as pathogenic hematopoietic sources of IL-3 that potentiate SC inflammation and worsen EAE severity.
CNS myeloid cells express IL-3Rɑ and exacerbate spinal cord inflammation, demyelination, and chemokine generation
Having identified T cells and astrocytes as relevant pathogenic sources of IL-3 in EAE, we next sought the cells that respond to the cytokine. IL-3Rɑ (also known as CD123) is IL-3’s specific receptor, and immunofluorescent imaging suggested that CD11b+ myeloid cells express IL-3Rɑ in the diseased SC (Fig 4A). To explore this, we adopted a stringent flow cytometry gating strategy to identify the specific myeloid cells that express IL-3Rɑ in the SC (Fig 4B). We found that during EAE resident microglia augmented IL-3Rɑ becoming responsive to IL-3 (Fig S4A) and that among myeloid populations infiltrating the SC, MHCII+ macrophages, Ly6Chi monocytes and macrophages, and MoDCs are major expressors of IL-3Rɑ (Fig 4C). We confirmed these data at the transcriptional level by measuring Il3rɑ mRNA expression in myeloid cells sorted from the diseased SC (Fig 4D). The abundance of IL-3Rɑ-expressing microglia, MHCII+ macrophages, Ly6Chi monocytes and macrophages, and MoDCs in the SC increased in EAE and correlated with clinical disease severity (Fig S4B).
Figure 4. Spinal cord myeloid cells express IL-3Rɑ and aggravate EAE by instigating immune cell recruitment to the CNS.

(A) Representative immunofluorescent images of IL-3Rɑ (CD123) and CD11b on spinal cord sections of 4 WT EAE mice at disease peak. Scale bar is 100 μm.
(B) Flow plots showing the cell gating strategy for leukocytes and non-leukocytes in the spinal cords of WT EAE mice at peak disease.
(C) Flow cytometric analysis and quantification of IL-3Rɑ expression by spinal cord cells at peak disease (n=9 mice).
(D) qPCR analysis of Il3rɑ expression by myeloid cells sorted from the spinal cord at disease peak (n=4-5 mice).
(E) Mean clinical score and corresponding AUC analysis of WT and Il3rɑ−/− mice during EAE (n=8-12 mice/group; two-way ANOVA and Mann–Whitney U-test).
(F) Representative histological sections of demyelination in diseased spinal cords from 5 WT and 5 Il3rɑ−/− mice stained for myelin by Luxol fast blue and counterstained with Cresyl Echt Violet Solution at disease peak. Scale bars represent 500 μm for overview images and 100 μm for the inset images.
(G) Quantification of demyelination area in diseased spinal cords from WT and Il3rɑ−/− mice (n=5 mice/group; two-way ANOVA and Mann–Whitney U-test).
(H) Flow cytometric analysis of spinal cord CD45+ leukocyte subsets in WT and Il3rɑ−/− mice at peak disease (n=5-6 mice/group; Mann–Whitney U-tests).
(I) Transcript expression analysis in myeloid cells sorted from the spinal cord of WT EAE mice at the peak of disease and stimulated with rIL-3 (n=5 group; Mann–Whitney U-tests).
(J) Measurement of CCL2 in the media of monocyte cultures exposed to rIL-3 (n=5 group; Mann–Whitney U-test).
(K) Enumeration of immune cells in the brain of mice 24 hours after stereotactic injection of 0, 1, 10, or 100 ng of rIL-3 (n=3-8 mice/group; one-way ANOVA).
Mean±s.e.m., *p<0.05, **p<0.01, ***p<0.001.
WT, wildtype; EAE, experimental autoimmune encephalomyelitis; MFI, mean fluorescence intensity; rIL-3, recombinant interleukin-3.
See also Figure S4.
To test for a consequential role of IL-3Rɑ in EAE we designed a CRISPR-Cas9 nuclease-based editing strategy with dual guide RNAs (gRNAs) to delete exons 2-11 of the endogenous Il3rɑ sequence, thus generating Il3rɑ−/− mice (Fig S4C). First, we confirmed successful deletion of IL-3Rɑ (Fig S4D and E), then subjected WT and Il3rɑ−/− mice to EAE and monitored clinical disease severity. We found that Il3ra deletion significantly improved clinical symptoms and paralysis (Fig 4E). Luxol fast blue staining of SC sections (Fig 4F) revealed profoundly reduced demyelination in the cervical, thoracic, and lumbar regions of Il3rɑ−/− mice (Fig 4G). We then measured immune cell infiltrates to the SC of WT and Il3rɑ−/− mice and found that Il3rɑ deletion robustly limited total CD45+ leukocytes, MHCII+ macrophages, and MoDCs while neutrophils and CD4+ T cells tended to decrease (Fig 4H). Together, these observations confirm that IL-3-sensing myeloid cells play an essential and detrimental role in neuroinflammation and EAE.
Next, we aimed to better understand how IL-3 influences CNS myeloid cell programing and function. First, we sorted CD45+CD11b+ monocytes from the BM of WT mice and stimulated them with recombinant IL-3 (rIL-3) ex vivo. We found that rIL-3 incited expression of genes indicative of differentiation and maturation into myeloid antigen-presenting cells (APCs) including Arg1, Mrc1, Zbtb46, CD209 and Mertk while reducing the monocytic genes Ly6c2 and C5ar1 (Fig S4F). Likewise, flow cytometry analysis showed that IL-3 signaled via IL-3Rɑ to promote monocyte differentiation into MHCII+CD86+CD80+CD11c+Ly6C− APCs (Fig S4G and H). Given our data on IL-3’s role in immune cell abundance and recruitment to the CNS, we hypothesized that IL-3 stimulates the generation of chemotactic and migratory cues by IL-3Rɑ+ myeloid cells in the SC. To test this, we sorted microglia, MoDCs, MHCII+ macrophages, and neutrophils from the SC of EAE mice and stimulated them ex vivo with rIL-3 which robustly increased the expression of the chemokines Ccl2, Ccl7, and Ccl12 (Fig 4I). rIL-3 treatment also raised CCL2 protein abundance in the culture media (Fig 4J). To expand on this, we polarized splenic T cells into TH1, TH17(β), or TH17(23) subsets, co-cultured them with WT or Il3rɑ−/− BM monocytes, and found that IL-3Rɑ deficiency decreased T cell-induced CCL2 generation (Fig S4I). To specifically test IL-3’s ability to stimulate immune cell recruitment to the CNS in vivo, we stereotactically injected mice with saline, 1, 10, or 100ng of rIL-3 into their striatum (Fig 4K). Stereotactic delivery of rIL-3 robustly induced Ly6Chi monocyte, MHCII+ macrophage, MoDC, neutrophil, and CD4+ T cell, but not CD8+ T cell or B cell, recruitment to the CNS in a dose dependent manner (Fig 4K and S4J). Taken together, these findings demonstrate that IL-3 promotes the differentiation of monocyte-derived APCs and the generation of chemotactic cues by resident and recruited IL-3Rɑ+ myeloid cells which stimulate further infiltration of relevant immune cells to the CNS, thereby exacerbating neuroinflammation and EAE.
IL-3:IL-3RA promotes accrual of migratory and chemotactic myeloid cell subsets in human MS plaques
Having established a role for the IL-3: IL-3Rɑ dyad in a murine model of MS, we returned our attention to humans. For an in-depth regional analysis of cell dynamics in the CNS of MS patients, we performed snRNA-seq on dissected regions of human brain tissue. We separately sequenced white and gray matter (WM and GM) tissue from six unaffected control subjects and six MS patients along with demyelinated plaques from the MS patients (Fig 5A). Across all tissues and diagnoses, a total of 77,490 nuclei were sequenced which clustered into all major CNS cell types including myeloid cells, T cells, astrocytes, oligodendrocytes, oligodendrocyte progenitor cells (OPCs), endothelial cells (ECs), vascular leptomeningeal cells (VLMCs), and neurons (multiple clusters of GABAergic and glutamatergic neurons) (Fig 5B). First, we quantified cell abundance across tissue regions in control subjects and MS patients. As expected, we found more neurons in GM relative to WM, and more oligodendrocytes in WM (Fig S5A). We then assessed the cellular composition of MS plaques, which typically evolve in WM, and found few neurons and oligodendrocytes (Fig S5A) but more astrocytes, T cells, and myeloid cells relative to other regions and tissue from control subjects (Fig 5C), reflective of the inflammatory and immunogenic milieu of MS plaques in the human CNS. To better understand how CNS myeloid cells are reprogramed in plaques, we compared myeloid cells isolated from plaques to those from adjacent unaffected WM tissue in MS patients. Transcriptional analysis revealed that myeloid cells in MS plaques profoundly induced pathways associated with immune cell migration, recruitment, and activation (Fig 5D, left), and upregulated chemotactic genes including CCL2, CCL3, CCL4, CCL5, CXCL2, CXCL12, CXCL13, and CXCL16 and their associated receptors CCR1, CCR2, CCR5, CXCR4, and CX3CR1 (Fig 5D, right). These data affirm the inflammatory nature of myeloid cells in the MS plaque microenvironment and demonstrate their activation of chemotactic and immune cell recruitment processes.
Figure 5. IL-3:IL-3RA promotes migratory and chemotactic myeloid cell subsets in CNS plaques of MS patients.

(A) Schematic of human study and snRNAseq analysis of white matter (WM) and gray matter (GM) from unaffected control subjects and MS patients along with plaques from MS patients.
(B) Umap visualization of all CNS cells across all tissue regions and subjects.
(C) Proportion of astrocytes, T cells, and myeloid cells in white matter, grey matter, and plaque of control subjects and MS patients (one-way ANOVA).
(D) Pathway analysis of genes enriched in myeloid cells and expression of chemotactic genes and receptors in myeloid cells deriving from the plaques vs. adjacent white matter of MS patients.
(E) Umap visualizations of IL3RA-expressing myeloid cells across tissue regions and subjects.
(F) Proportion of IL3RA-expressing myeloid cells (left) and relative myeloid IL3RA expression (right) across tissue regions and disease states (one-way ANOVA).
(G) Umap visualization of myeloid clusters and IL3RA expression in MS plaques.
(H) Volcano plot of up and down regulated genes in MS plaque myeloid cluster 2 and GO Biological pathway analysis of upregulated genes.
(I) Proportion of IL3RA expressing myeloid cells (left) and IL3RA expression (right) among myeloid clusters of MS plaques.
(J) Volcano plot of genes enriched in IL3RA expressing and non-expressing plaque myeloid cells and pathway analysis of genes upregulated in IL3RA expressing plaque myeloid cells.
(K) Correlation of plaque myeloid IL3RA expression with the proportion of plaque myeloid and T cells (Spearman correlation).
(L) Correlation of CSF IL-3 amount with CSF mononuclear cell abundance in MS patients (Spearman correlation).
n=6 unaffected control and 6 MS patients; except L n=28 MS patients
Mean±s.e.m., *p<0.05, **p<0.01, ***p<0.001.
WM, white matter; GM, grey matter; MS, multiple sclerosis; CNS, central nervous system; Umap, Uniform manifold approximation and projection; ECs, endothelial cells; VLMCs, vascular leptomeningeal cells; OPC, oligodendrocyte progenitor cells; AU, arbitrary unit.
See also Figure S5.
We next evaluated IL3RA expression across all CNS cell types, tissular regions, and diagnoses. In unaffected subjects and MS patients, we found that IL3RA was expressed widely by all CNS cells in WM and GM and in the plaques of MS patients (Fig S5B and C). Although this suggests broad functionality of IL3RA in the CNS, we focused on myeloid cells given their importance to MS plaque pathology and neuroinflammation and based on our findings in mice. Consistent with our murine data, IL3RA was expressed by myeloid cells in the WM and GM of control subjects and MS patients and in the plaques of MS patients (Fig 5E and S5D). Enumerating IL3RA-expressing cells (IL3RA+) and measuring IL3RA showed that expression is significantly elevated in GM, WM, and plaque myeloid cells of MS patients relative to unaffected controls (Fig 5F). Importantly, IL3RA expression in all other CNS cell types remained unaltered in MS patients (Fig S5E). Further, we corroborated our data in a separate scRNA-seq dataset from a distinct cohort of human subjects24, and found that amongst CSF leukocytes, myeloid cells express IL3RA most abundantly and myeloid IL3RA expression was increased in MS patients (Fig S5G). These data suggest that the IL-3:IL-3RA axis is specifically and dynamically modulated in CNS myeloid cells during MS and may serve a critical function.
To further resolve myeloid cell functions in the relevant tissular microenvironment, we performed subclustering of myeloid cells in MS plaques and identified five clusters (Fig 5G). To assess the properties of each cluster, we conducted pathway analysis on enriched genes. This uncovered distinct functional characteristics for each cluster, including plaque myeloid clusters programmed for cytokine production (cluster 1), migration and chemotaxis (cluster 2), synapse pruning (cluster 3), phagocytosis (cluster 4), and synapse organization/brain development (cluster 5) (Fig S5H). We then quantified IL3RA+ cells and IL3RA expression in each cluster and found that cluster 2, wired for myeloid cell migration, matrix remodeling, and chemotaxis (Fig 5H), was solely and specifically enriched for IL3RA (Fig 5I). In agreement with our murine findings (Fig S4F), MRC1 and MERTK, IL-3-controlled myeloid APC differentiation genes, were upregulated in cluster 2 while C5AR1, an IL-3-controlled undifferentiated monocytic marker, was downregulated (Fig 5H). This unbiased clustering revealed cluster 2 as enriched in IL3RA-expressing plaque myeloid cells programmed for immune cell recruitment and chemotaxis and is relevant to MS.
Next, to acquire a more targeted understanding of IL-3RA-controlled cell function and given our murine findings, we interrogated upregulated and downregulated genes and pathways in IL3RA-expressing myeloid cells residing in plaques. We found that IL3RA-expressing myeloid cells were enriched for chemotactic and migratory genes including CX3CR1, P2RY12, USP9Y, MAP4K1, and CCL3 and programs associated with cell migration and chemotaxis (Fig 5J). Further, the most highly downregulated genes in IL3RA+ plaque myeloid cells, in comparison to IL3RA− cells, were FKBP5, a chaperone of the glucocorticoid receptor, and KCNMA1, an ion channel subunit critical to neurotransmitter release, and loss of function of these genes is strongly associated with demyelinating disease, neuromyelitis, and MS25,26. Together, these findings demonstrate that the CNS of MS patients, particularly in plaques, has more IL3RA-expressing myeloid cells and that IL-3RA plays an important role in the generation of antigen-presenting myeloid cells and their instigation of immune cell trafficking, chemotaxis, and migration programs.
To assess whether IL-3:IL-3RA associates with immune cell recruitment to the CNS in human MS, we correlated IL3RA expression by plaque myeloid cells with myeloid and T cell abundance in the plaques. We found a significant and positive correlation between plaque myeloid IL3RA expression and the number of plaque T cells and myeloid cells (Fig 5K). Finally, we returned to our human cohort in which we measured CSF IL-3 and found that IL-3 in the CSF of RRMS patients correlated positively and significantly with CSF mononuclear cell numbers (Fig 5L). In sum, these data support the hypothesis that IL-3:IL-3RA signaling mediates communication amongst ontologically distinct immune cell subsets in human MS plaques and through the induction of chemotactic and recruitment programs, perpetuates neuroinflammation and disease severity (Fig S5I).
DISCUSSION
Inflammatory networks, glial cells, and infiltrating immune cells play consequential roles in neuroinflammation. However, little is known about the communication systems that coordinate cellular function and crosstalk in this diverse CNS ecosystem. Here, we have reported on IL-3-mediated central-peripheral immune communication in MS and CNS inflammation and position IL-3 as a pathogenic effector cytokine-growth factor. Our study identified both CNS-resident and infiltrating cells that generated IL-3 and responded to it. This integrated bidirectional network, we reveal, has a fundamental consequence on the programing and function of myeloid cell subsets in the CNS and in the clinical and pathological severity of MS.
Our data showed that resident astrocytes and infiltrated CD44hiCD4+ effector T cells are the relevant sources of IL-3 in the inflamed CNS and that both microglia and peripherally-derived myeloid cells (MHCII+ macrophages, MoDCs, and monocytes) respond to IL-3 by expressing IL-3Rɑ. IL-3 signaling instigated a chemotactic program by IL-3Rɑ+ myeloid cells that perpetuated immune cell recruitment to the CNS and worsened clinical severity. In humans, we found that IL-3 is present in the CSF and correlated with disease diagnosis and severity. Region-specific snRNAseq of brain tissue from control subjects and MS patients revealed augmentation of IL3RA by myeloid cells in white and grey matter and plaque tissue of MS patients and that IL3RA-expressing myeloid cells in the MS plaque represent a migratory and chemotactic subset that instigated immune cell recruitment to the CNS. Together, these findings identify IL-3:IL-3RA as a pathogenic axis that mediates bidirectional communication between peripherally derived immune cells and resident CNS glial cells, thereby exacerbating MS and neuroinflammation.
Our results begin to resolve IL-3 in CNS health and disease. Since its discovery decades ago, IL-3 has been associated with multiple inflammatory diseases; however, its role in the CNS and neuroinflammation has remained vastly understudied. Prior work from our group has established IL-3 involvement in Alzheimer’s disease (AD)11. In this context, IL-3 safeguards against AD by inciting microglial clustering and clearance of Aβ and tau. Here, we report that IL-3 is detrimental in MS and EAE, neuroinflammatory conditions defined by substantial recruitment of peripheral immune cells to the CNS where they contribute to disease pathology and severity. Our results show that IL-3-stimulated chemotactic and migratory programs aggravate neuroinflammation and MS severity. Yet, in AD, in which microglial migration towards and clearance of Aβ and tau resolve pathology, IL-3 ameliorates disease. These findings underscore the critical nuances of balanced inflammation and IL-3 in neurological disease: where instigation can be protective in one setting and harmful in another.
The fundamental actions of IL-3 in MS have, until now, been unclear. MS presents more often in females and the IL3RA gene is encoded on the human X chromosome, perhaps indicating that homozygosity and/or increased expression of this gene contributes to disease development. Indeed, our data demonstrate that in comparatively healthy grey and white matter tissue of MS patients, which are devoid of MS plaques, IL3RA expression is elevated relative to corresponding tissue from control subjects. Although prior research has suggested IL-3 may be involved in demyelinating disease12,13, these studies have been limited and reported directly contradictory findings. Here we firmly establish a fundamental and pathogenic role for this cytokine in autoimmune neuroinflammation and MS, using generated and cell-specific murine models, multiregional snRNA-seq of the human CNS, and contemporary tools.
We position IL-3 as a paracrine modulator of myeloid cell recruitment and function through its local activities in the CNS. Myelopoiesis is heightened in MS and EAE27; however, we found that in MS, unlike in other inflammatory settings6, IL-3 does not serve as an endocrine amplifier of monocyte generation or hematopoiesis in distal compartments. Likewise, we did not find an impact of IL-3 on peripheral immune priming. The effects of IL-3 in MS and EAE appear to be local, and our data identified the effector organ, the CNS, as the consequential site for IL-3 production and action and reveal multifunctionality of the cytokine beyond leukocyte generation.
We present astrocytes and pro-inflammatory T cells as the two consequential sources of IL-3 in the CNS during EAE and employ cell-specific models to demonstrate their relevance. We characterize CD4+ TH cell subsets that generate IL-3 in the inflamed CNS and show co-production of GM-CSF, IFN-γ, and IL-17A. IL-3 generation by CD4+ T cells might be directed by a pathogenic effector program or by inducible factors downstream of TCR and costimulatory signaling, but this requires formal evaluation in future studies28,29. Ultimately, our data reveal IL-3 as part of a pathogenic T cell signature. Further work will also be required to better characterize IL-3-producing astrocytes, including testing their transcriptional, metabolic, and biological programing and regional specificity in the CNS. Our observations that astrocytes generate IL-3 in the healthy CNS and that the cytokine is present in the CSF of healthy humans, raises questions about its role in brain homeostatic functioning. Additionally, our data identified multiple CNS cell types that express IL3RA in health and disease. Future studies will need to investigate IL-3 in brain health, development, and aging, and systematically resolve the role of IL-3RA in diverse CNS cells including neurons, oligodendrocytes, and endothelial cells, among others.
MS currently has no cure and patients are susceptible to developing severe disability and have shorter life expectancies. Targeting glial-peripheral signaling may be an effective therapeutic strategy17; and our data propose focusing on IL-3-mediated communication to curb immune cell infiltration, demyelination, and clinical symptoms. Biologics and small molecules targeting IL-3 signaling have been used in cancer therapy, and their application to MS treatment merits further exploration30-32. Whether targeting IL-3 or IL-3RA by pharmacological, biological, or genetic means, or identifying more appropriate downstream targets in the IL-3 axis, this pathway may offer an additional vista in drug development for MS and other neuroinflammatory conditions. Altogether, our data resolve IL-3 signaling in MS and identify the cytokine as a multifaceted actor that coordinates bidirectional communication amongst resident glia and infiltrating immune cells during CNS autoimmune inflammation.
LIMITATIONS OF THE STUDY
Our study elevates IL-3 as a critical mediator of neuroinflammation in MS and its preclinical model. A limitation of our study is that we use a murine model of progressive MS that does not fully recapitulate the relapse and remitting features of clinical MS. Further investigations into the role of IL-3 and its therapeutic application in preclinical models of RRMS are warranted. Further, the stimuli and factors that induce astrocytic IL-3 production are unclear and a granular investigation into the functional attributes of IL-3 producing astrocytes deserves investigation. Our snRNAseq analysis of human brain tissue was unable to identify IL-3 producing cells. This is common in astrocyte profiling and likely because the depth of our sequencing was not sufficient to detect this rare and quickly degraded transcript and that transcription of Il3 occurs in discrete temporal windows dependent on stimuli and environment. Additional technologies that detect and profile rare astrocytic populations are becoming available (such as focused interrogation of cells by nucleic acid detection and sequencing, FIND-seq33) and can be used to interrogate IL-3 producing astrocytes. Finally, our data suggest that many cells in the CNS respond to IL-3. The contribution of IL3RA expressing non-myeloid cells, such as neurons, oligodendrocytes, or endothelial cells, to MS pathology was not explored here and should be tested.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Cameron S. McAlpine (Cameron.mcalpine@mssm.edu).
Materials availability
Mouse lines generated in this study are available from the lead contact upon request. Distribution of mouse lines may require a material transfer agreement (MTA).
Data and code availability
Single nuclear RNA-seq data have been deposited at GEO under the accession GSE227781 and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-CD45 BUV737 (clone 30-F11) | BD Biosciences | Cat# 748371 |
| anti-CD45 BV711 (clone 30-F11) | BioLegend | Cat# 103147 |
| anti-CD3 (clone 17A2) FITC | BioLegend | Cat# 100204 |
| anti-CD3ε (clone 145-2C11) PE | BioLegend | Cat# 100308 |
| anti-CD4 (clone RM4-5) FITC | BioLegend | Cat# 100510 |
| anti-CD4 (clone RM4-5) PE-Cyanine7 | BioLegend | Cat# 100528 |
| anti-CD8a (clone 53–6.7) Alexa Fluor 700 | BioLegend | Cat# 100730 |
| anti-CD90.2 (clone 53-2.1) PE | BioLegend | Cat# 140308 |
| anti-CD19 (clone 6D5) | BioLegend | Cat# 115538 |
| anti-B220 (clone RA3-6B2) BV785 | BioLegend | Cat# 103246 |
| anti-Ly6G (clone 1A8) BV421 | BioLegend | Cat# 127645 |
| anti-Ly-6C (AL-21) BV605 | BD Biosciences | Cat# 563011 |
| anti-I-A/I-E (MHCII; clone M5/114.15.2) Alexa Fluor 700 | BioLegend | Cat# 107622 |
| anti-CD11b (clone M1/70) APC/Cyanine7 | BioLegend | Cat# 101226 |
| anti-CD11c (clone HL3) PerCP-Cy5.5 | BD Biosciences | Cat# 560584 |
| anti-CD44 (clone IM7) PE-Cyanine7 | BioLegend | Cat# 103030 |
| anti-CD62L (clone MEL-14) Alexa Fluor 700 | BioLegend | Cat# 104426 |
| anti-CD64 (clone X54-5/7.1) PE | BioLegend | Cat# 139304 |
| anti-CD80 (clone 16-10A1) FITC | BioLegend | Cat# 104706 |
| anti-CD86 (clone GL-1) PE/Cyanine7 | BioLegend | Cat# 105014 |
| anti-OX40L (clone RM134L) APC | BioLegend | Cat# 108812 |
| anti-CD69 (clone H1.2F3) BV711 | BioLegend | Cat# 104537 |
| anti-CD49d (integrin ɑ4; clone R1-2) PE | BioLegend | Cat# 103608 |
| Anti-Ly6G (clone 1AB-Ly6g) PerCP/eFluor710 | Thermo Fisher Sci. | Cat# 46-9668-82 |
| Anti-I-A/I-E (MHCII; clone M5/114.15.2) BUV495 | BD Biosciences | Cat# 750281 |
| anti-Ki67 (clone SolA15) PE | ThermoFisher Sci. | 12-5698-82 |
| anti-IL-3Rɑ (CD123; clone REA114) | Miltenyi Biotec | Cat# 130-102-583 |
| anti-F4/80 (clone BM8) APC/Cyanine7 | BioLegend | Cat# 123118 |
| anti-CD115 (CSF-1R; clone AFS98) BV605 | BioLegend | Cat# 135517 |
| anti-CX3CR1 (clone SA011F11) PE-Cyanine7 | BioLegend | Cat# 149016 |
| anti-IL-3 (clone MP2-8F8) | BD Biosciences | Cat# 554383 |
| anti-IFNγ (clone XMG1.2) BV605 | BioLegend | Cat# 505840 |
| anti-IL-17A (clone TC11-18H10.1) APC/Cyanine7 | BioLegend | Cat# 506922 |
| anti-GM-CSF (clone MP1-22E9) BV421 | BD Biosciences | Cat# 564747 |
| anti-CD11b (clone M1/70) BUV805 | BD Biosciences | Cat# 741934 |
| anti-ACSA2 (clone REA969) APC | Miltenyi Biotec | Cat# 130-116-245 |
| anti-CD19 (clone 1D3) BUV661 | BD Biosciences | Cat# 612971 |
| anti-CD8 (clone 53-6.7) PE-Dazzle594 | BioLegend | Cat# 100762 |
| anti-IL3 | BioLegend | Cat# 503902 |
| AF488 anti-GFAP | eBioscience | Cat# 53-9892 |
| Anti-IL3Rɑ | US Biological | Cat# 141039 |
| anti-CD11b | BD Biosciences | Cat# 550282 |
| anti-rabbit IgG | Vector Laboratories | Cat# BA-1000 |
| anti-rat IgG | Thermo Fisher Scientific | Cat# A-11006 |
| anti-rat IgG | Vector Laboratories | Cat# BA-4001 |
| Mouse anti-IL4 | BioLegend | Cat# 504122 |
| Mouse anti-CD28 | Thermo Fisher Sci. | Cat# 16-0281-82 |
| Mouse anti-CD3ε | Thermo Fisher Sci. | Cat# 16-0031-82 |
| Mouse anti-IL12 | Thermo Fisher Sci. | Cat# 45-7123-80 |
| Mouse anti- IFNy | BioLegend | Cat# 513208 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Human brain tissue | Human Brain and Spinal Fluid Resource Center, UCLA | N/A |
| Human CSF samples | Department of Neurology of the University Hospital Basel | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| DAPI | Thermoscientific | Cat# 62248 |
| IL-3 | Peprotech | Cat# 213-13 |
| IL-12 | Peprotech | Cat#210-12 |
| TGF-β1 | Peprotech | Cat# 100-21 |
| IL-6 | Peprotech | Cat# 216-16 |
| IL-23 | Peprotech | Cat# 200-23 |
| IL-2 | Peprotech | Cat# 212-12 |
| IL-1β | BioLegend | Cat# 575102 |
| Tamoxifen | Sigma Aldrich | T5648 |
| Critical commercial assays | ||
| RNeasy Mini kit | Qiagen | Cat# 74104 |
| RNeasy Micro Kit | Qiagen | Cat# 74004 |
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher | Cat# 4368814 |
| MOG35-55/CFA Emulsion PTX Hooke Kit™ | Hooke Laboratories | Cat# EK-2110 |
| Luxol Fast Blue Stain Kit | Abcam | Cat# ab150675 |
| IL-3 ELISA kit | Boster Biological | Cat# EK0403 |
| IFNγ ELISA kit | R&D Systems | Cat# MIF00 |
| IL17A ELISA kit | R&D Systems | Cat# M1700 |
| GM-CSF ELISA kit | R&D Systems | Cat# MGM00 |
| CCL2 ELISA Kit | R&D Systems | Cat# MJE00B |
| Zombie Aqua Fixable Viability Kit | BioLegend | 423102 |
| eBioscience FoxP3/ Transcription Factor Staining Buffer Set | Thermo Fisher Sci. | 00-5523-00 |
| Fixation/Permeabilization Kit with BD GolgiPlug | BD Biosciences | 555028 |
| CountBright Absolute Counting Beads | Thermo Fisher Sci. | C36950 |
| Monocyte Isolation Kit | Miltenyi Biotec | 130-100-629 |
| Chromium Single Cell 3’ Reagent Kits v3 | 10x Genomics | Cat# 1000075 |
| Chromium i7 Sample Index Plate | 10x Genomics | Cat# 220103 |
| Kapa Library Quantification Kit | KAPA Biosystems | Cat# KK4873 |
| Tapestation D5000 ScreenTape | Agilent Technologies | Cat# 5067-5588 |
| Tapestation D5000 Reagents | Agilent Technologies | Cat# 5067-5589 |
| Deposited data | ||
| Single nuclear RNA-seq | Gene Expression omnibus | GSE227781 |
| Experimental models: Cell lines | ||
| Experimental models: Organisms/strains | ||
| C57BL/6J | The Jackson Laboratories | Strain# 000664 |
| Aldh1l1cre Ert2 | The Jackson Laboratories | Strain# 031008 |
| Cd4cre | The Jackson Laboratories | Strain# 022071 |
| Il3 −/− | The Jackson Laboratories | Strain# 026277 |
| IL3 GFPfl/fl | McAlpine lab | N/A |
| Il3rɑ −/− | McAlpine lab | N/A |
| Oligonucleotides | ||
| Il3 (Mm00439631_m1) | Thermo Fisher | Cat# 4331182 |
| Ccl2 (Mm00441242_m1) | Thermo Fisher | Cat# 4331182 |
| Ccl5 (Mm01302427_m1) | Thermo Fisher | Cat# 4331182 |
| Ccl7 (Mm00443113_m1) | Thermo Fisher | Cat# 4331182 |
| Ccl12 (Mm01617100_m1) | Thermo Fisher | Cat# 4331182 |
| Il3rɑ (Mm00434273_m1) | Thermo Fisher | Cat# 4331182 |
| Csf2rb (Mm00655745_m1) | Thermo Fisher | Cat# 4331182 |
| Ccr7 (Mm99999130_s1) | Thermo Fisher | Cat# 4331182 |
| Flt3 (Mm00439016_m1) | Thermo Fisher | Cat# 4331182 |
| Kit (Mm00445212_m1) | Thermo Fisher | Cat# 4331182 |
| Spp1 (Mm00436767_m1) | Thermo Fisher | Cat# 4331182 |
| C5ar1 (Mm00500292_s1) | Thermo Fisher | Cat# 4331182 |
| Irf4 (Mm00516431_m1) | Thermo Fisher | Cat# 4331182 |
| Ly6c2 (Mm00841873_m1) | Thermo Fisher | Cat# 4331182 |
| Cd209a (Mm00460067_m1) | Thermo Fisher | Cat# 4331182 |
| Fcgr1 (Mm00438874_m1) | Thermo Fisher | Cat# 4331182 |
| Zbtb46 (Mm00511327_m1) | Thermo Fisher | Cat# 4331182 |
| Actb (Mm00607939_s1) | Thermo Fisher | Cat# 4331182 |
| Recombinant DNA | ||
| Software and algorithms | ||
| NDP.view 2 | Hamamatsu Photonics | www.hamamatsu.com |
| FlowJo v10 | FlowJo | www.flowjo.com |
| GraphPad Prism v9 | GraphPad Software | www.graphpad.com |
| BioRender | BioRender | www.biorender.com |
| SomaScan | SomaLogic Inc | www.somalogic.com |
| Cell Ranger | 10x Genomics | v7.0.0 |
| pegasus | https://github.com/lilab-bcb/pegasus | v1.7.0 |
| scanpy | https://github.com/scverse/scanpy | v1.8.2 |
| anndata | https://github.com/scverse/anndata | v0.8.0 |
| numpy | https://github.com/numpy/numpy | v1.21.5 |
| scipy | https://github.com/scipy/scipy | v1.8.0 |
| pandas | https://github.com/pandas-dev/pandas | v1.4.1 |
| umap | https://github.com/lmcinnes/umap | v0.5.2 |
| scikit-learn | https://github.com/scikit-learn/scikit-learn | v1.0.2 |
| statsmodels | https://github.com/statsmodels/statsmodels | v0.13.2 |
| python-igraph | https://github.com/igraph/python-igraph | v0.9.9 |
| Other | ||
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice.
Wild-type C57BL/6J, Aldh1l1creErt2, and Cd4cre mice were purchased from the Jackson Laboratory. C57BL/6J mice were also bred in-house. Il3−/− mice were bred in house7,34. IL3GFPfl/fl mice were bred in house and their generation has been previously described11. Age- and sex- matched mice were used. Where appropriate, mice were randomly assigned to interventions. Littermate controls were used in all cell-specific genetic deletion studies. Experiments were initiated when mice were 10-12 weeks old. All mice had free access to food and water. For EAE experiments, mice were single housed. All animal protocols were approved by the Animal Review Committee at the Massachusetts General Hospital (protocol nos. 2011N000035 and 2015N000044) and/or the Icahn School of Medicine at Mount Sinai (protocol nos. PROTO202100023 and PROTO202000262) and were in compliance with relevant ethical regulations.
CRISPR-Cas9 generation of Il3rɑ−/− mice.
To generate Il3ra−/− mice on the C57BL/6J background, two SpCas9 guide RNAs (gRNAs) were designed to target genomic regions within the first intron and 3’ of the stop codon of the mouse Il3rɑ gene using on-target and off-target prediction software 35,36. The on-target activities of candidate gRNAs were tested by microinjection of ribonucleoprotein (RNP) complexes comprised of TrueCut Cas9 v2 (ThermoFisher) and synthetic gRNAs (Synthego) into mouse zygotes. All microinjections were performed at the Genome Modification Facility (Harvard University). Injected zygotes developed to the blastocyst stage prior to undergoing genomic DNA extraction. To confirm the genome editing efficiencies of candidate gRNAs at the mouse Il3ra locus, the target genomic regions were amplified by PCR using the following forward and reverse amplicon primers: oBK8664 (forward; 5’-GATGATGTCATTCTCACCCCCAGATGTC-3’) and oBK8667 (reverse; 5’-TGCAGGTTCTGGATGGGCGTGGTC-3’) to sequence intron 1 targets for Il3ra; and oBK8668 (forward; 5’-GGACAGGAAGTGACACTGGGGGTCAG-3’) and oBK8671 (reverse; 5’-GCAATCCCTCTGTCTCAGCTCCTG-3’) to sequence stop codon targets for Il3ra. Amplicons were sent for Sanger sequencing and the approximate level of on-target activity was determined using ICE37. The most effective gRNAs of each pair examined (mIl3ra-1: spacer sequence: GGGACCAATGATGTCACCTA, and PAM: GGG; and mIL3ra-STOP-2: spacer sequence: AGACGCCTGAGAACTGTGTG, and PAM: GGG, which target the first intron and 3’ of the stop codon of the Il3rɑ gene, respectively) were then used for microinjections to generate Il3ra−/− mice by excision of a 7712 bp region of genomic DNA spanning Il3ra introns 2-11 (Fig S4C). Injected embryos were implanted into pseudopregnant recipients, and Il3rɑ gene-targeted mice were genotyped at 3 weeks of age. To genotype mice, genomic DNA was extracted from tail snips in 200 μL of tail lysis buffer (100 mM Tris-HCl, 200mM NaCl, 5mM EDTA, 0.05% SDS, 12.5 mM DTT, 1.4 μg/μl Proteinase K (New England Biolabs) via ~16-hour incubation at 55 °C. Lysates were cleaned up using 0.7x paramagnetic beads prepared as previously described38,39. Subsequent Il3ra−/− mice were genotyped by PCR using the following forward and reverse amplicon primers: oBK8665 (forward; 5’- CCCTAAGCTCTTCCCTTCTTGTTGGC-3’) and oBK8670 (reverse; 5’-CCTTCAGAGCCCCACTTCCTGTCGAAG-3’) to detect the Il3ra ablated allele; and oBK8668 (forward; 5’- GGACAGGAAGTGACACTGGGGGTCAG-3’) and oKAC236 (reverse; 5’- CCAGAAGGAACCCGAGCTTCATC-3’) to detect the wild-type allele. Deletion of the targeted Il3ra genomic locus was confirmed by Sanger sequencing using the following primers: oKAC233 (forward; 5’-GGACCATGACAGGAACCAGAAGC-3’) and oKAC237 (reverse; 5’-GTTACAACACCTAGAAGTAGTACCTCCTC-3’). One founder mouse with successful deletion of Il3ra exons 2-11 was selected for further breeding in-house with C57BL/6J mice.
Human CSF proteomic subjects.
CSF samples were collected from study participants in the Department of Neurology of the University Hospital Basel. Participants were characterized as (i) healthy controls with no evidence of neurological disease (20 females and 15 males, age: 43.5±2.1 years), (ii) patients with clinical defined RRMS (29 females and 7 males, age: 36.4±1.6 years), or (iii) patients that have experienced a single clinical episode suggestive of MS, known as clinically isolated syndrome (CIS). CIS patients were further differentiated into those who developed clinically defined MS (converters, 17 females and 5 males, age: 36.4±2.0), according to the Poser criteria, during follow-up of at least 2 years, and those that remained stable (non-converters, 21 females and 11 males, age: 31.3±2.1). Patients with neuromyelitis optica, a history of progressive disease, active systemic infection, or undergoing steroid treatment at the time of sampling, were excluded from the study. Written informed consent was obtained from all participants according to the Declaration of Helskinki. Ethical approval was obtained by local ethics and IRB committee for use of deidentified samples. For RRMS patients, some samples were collected during remission (58% of RRMS samples) and others during a relapse (42% of RRMS samples).
Human snRNAseq subjects.
Twelve fresh frozen brain tissue specimens, consisting of 6 MS cases (4 males and 2 females, age: 64±6) and 6 unaffected controls (4 males and 2 females, age: 63±3.3), were obtained through the NIH NeuroBiobank from the Human Brain and Spinal Fluid Resource Center, UCLA. All neuropsychological, diagnostic, and autopsy protocols were approved by the respective Institutional Review Boards. For the cases, each specimen contained a visible plaque and adjacent normal-appearing white matter (NAWM) and normal-appearing gray matter (NAGM). The unaffected control specimens consisted of NAWM and NAGM.
METHOD DETAILS
In Vivo Interventions
EAE induction and scoring.
EAE was induced using the MOG35-55/CFA Emulsion PTX Hooke Kit™ (Cat. No. EK-2110; Hooke Laboratories, Inc.). Briefly, mice were subcutaneously injected in the flank with 100 μg myelin oligodendrocyte glycoprotein peptide residues 35-55 (MOG35-55: MEVGWYRSPFSRVVHLYRNGK) emulsified in complete Freund’s adjuvant (CFA) containing 2-5 mg/mL of heat-killed Mycobacterium tuberculosis H37Ra on day 0. Mice also received between 80-100 ng pertussis toxin (PTX) in PBS via intraperitoneal injection on days 0 and 2. Mice were monitored and scored daily for disease signs according to the following 5-point clinical scoring criteria: 0: no disease; 0.5: tail weakness/partial limp tail; 1.0: full limp tail; 1.5: mild gait impairment; 2.0: severe gait impairment/hindlimb paresis; 2.5: partial hindlimb paralysis; 3.0: hindlimb paralysis/legs unable to paddle past hip; 3.5: hindlimb paralysis and forelimb paresis; 4.0: hindlimb paralysis and partial forelimb paralysis; 5.0: moribund or death.
Tamoxifen injection.
Tamoxifen (20 mg/ml, Sigma Aldrich) was prepared in corn oil and allowed to dissolve at 37 °C overnight while shaking. Mice were injected i.p. with 2 mg tamoxifen on days indicated in Fig 3E.
Intracranial injections.
Striatal injections were performed as previously described40-42. Mice were anaesthetized with 3% isoflurane; the head was shaved and placed in a stereotactic frame. A midline incision exposed the skull and a burr hole was drilled above the left striatum. A 1μL injection of murine recombinant IL-3 (rIL-3; 10 ng; Peprotech) was administered with a pulled glass capillary using the stereotactic coordinates: Anterior/Posterior, +1; Medial/Lateral, −2; and Dorsal/Ventral, −2.5. A 1 μL injection of sterile saline was used as a control. Postinjection, the capillary was held in place for one minute to limit backflow. The scalp was sutured, and animals were sacrificed 24 hours later.
Cells
Cell isolation.
Peripheral blood was collected by retro-orbital bleeding, and erythrocytes were lysed in RBC lysis buffer (BioLegend, San Diego, CA). The spleen (SP), draining lymph nodes (LN), femurs, tibia, brain, and spinal cord (SC) were collected on ice after 20 ml vascular perfusion with cold PBS. Minced SP and LN and flushed bones were strained through 40 μm-nylon mesh (BD Biosciences) with PBS and further subjected to RBC lysis to generate single-cell suspensions of splenocytes, lymph nodes, and bone marrow (BM). Naive and inflamed SC were carefully removed by hydraulic extrusion with PBS and an 18 G needle. Fresh brains from intracranial injection experiments were dissected into 2 hemispheres and the injection site isolated for analysis. CNS tissue was minced and digested for 20 minutes in a thermomixer at 1000 rpm and 37°C in a 1 ml volume digestion mix of 450 U/ml collagenase I, 125 U/ml collagenase XI, 60 U/ml DNase I, 60 U/ml hyaluronidase, and 20 mM HEPES (Sigma-Aldrich) in PBS. After digestion, SC suspensions were strained using 70 μm-nylon mesh (BD Biosciences), mixed with a 30% Percoll/PBS solution, and a 70% Percoll/PBS solution was underlaid via disposable glass Pasteur pipette prior to centrifugation for 20 minutes at 690 x g and 25-26°C with the acceleration and brake remaining off. The upper cell fraction and mononuclear cell interface were collected and washed with room temperature PBS before downstream application.
Flow Cytometry and cell sorting.
Single cell suspensions were stained in PBS supplemented with sterile 2% FBS and 0.5% BSA. The following monoclonal antibodies were used for flow cytometric analysis: anti-CD45 (clone 30-F11), anti-CD3 (clone 17A2), anti-CD3ε (clone 145-2C11), anti-CD4 (clone RM4-5), anti-CD8a (clone 53–6.7), anti-CD90.2 (clone 53-2.1), anti-CD19 (clone 6D5), anti-B220 (clone RA3-6B2), anti-Ly6G (clone 1A8), anti-Ly-6C (AL-21), anti-I-A/I-E (MHCII; clone M5/114.15.2), anti-CD11b (clone M1/70), anti-CD11c (clone HL3), anti-CD44 (clone IM7), anti-CD62L (clone MEL-14), anti-CD64 (clone X54-5/7.1), anti-CD80 (clone 16-10A1), anti-CD86 (clone GL-1), anti-OX40L (clone RM134L), anti-CD69 (clone H1.2F3), anti-CD49d (integrin ɑ4; clone R1-2), anti-CD29 (integrin β1; clone HMβ1-1), anti-CXCR3 (CD183; clone CXCR3-173), anti-CCR6 (clone 29-2L17), anti-IL-3Rɑ (CD123; clone REA114), anti-F4/80 (clone BM8), anti-CD115 (CSF-1R; clone AFS98), anti-CX3CR1 (clone SA011F11), anti-IL-3 (clone MP2-8F8), anti-IFNγ (clone XMG1.2), anti-IL-17A (clone TC11-18H10.1), anti-GM-CSF (clone MP1-22E9), and anti-Ki67 (clone SolA15). All antibodies were used at a 1:200 dilution except for anti-IL-3 (1:166) and anti-CD123 (1:10). Antibodies were purchased from either BioLegend, BD Biosciences, eBioscience, or Miltenyi Biotec (Sunnyvale, CA). Viable cells were identified as unstained cells with Zombie Aqua™ (1:500-1:1000 dilution; BioLegend). For intracellular cytokine staining and analysis, cell suspensions were stimulated in 2% FBS RPMI-1640 medium with 50 ng/ml PMA and 500 ng/ml ionomycin (Sigma-Aldrich) in the presence of GolgiStop and GolgiPlug (each 1:2000; BD Biosciences) for 3.5-4 hours at 37°C, 5% CO2 prior to viability staining with ZombieAqua plus Fc block with TruStain FcX™ (BioLegend) for 15 minutes at room temperature, extracellular antibody staining for 20 minutes at room temperature, fixation with BD Cytofix™ for 20 minutes at 4°C, and permeabilization for 30 minutes with BD Perm/Wash Buffer (BD Biosciences). Intracellular cytokine staining was performed in BD Perm/Wash Buffer for 45 minutes at room temperature. For Ki67 proliferation staining and analysis, cells were subject to viability staining with ZombieAqua™ plus Fc block with TruStain FcX™ (BioLegend) for 15 minutes at room temperature and extracellular antibody staining for 20 minutes at room temperature prior to fixation/permeabilization and intranuclear staining with the eBioscience™ Foxp3/Transcription. Factor Staining Buffer Set according to the manufacturer’s instructions (ThermoFisher Scientific). Data were acquired on a BD LSRII and an Cytek Aurora Spectral cytometer and analyzed with FlowJo software (Tree Star, Ashland, OR). Cell sorting was conducted on a FACS Aria II or a Cytek Aurora cell sorter.
Ex vivo cell cultures.
IL-3 production by tissular T cells.
To analyze IL-3 production in T cells isolated from tissue, single-cell suspensions of roughly 2-5x106 cells per well were stimulated in 96-well flat-bottom plates in complete media for 20-24 hr in the presence of plate-bound anti-CD3ε (10 μg/ml) and soluble anti-CD28 (10 μg/ml) prior to intracellular cytokine staining (see staining strategies under Flow cytometry above).
T cell recall response.
To analyze T cell proliferation and cytokine production in Il3+/+ or Il3−/− mice in the peripheral compartment at the preclinical disease stage, spleens (n=3/group) were isolated on 8 dpi after subcutaneous MOG35-55/CFA injection and cultured in the presence of increasing concentrations of MOG35-55 for 16 hours at 2-5x105 cells per well (with technical duplicates) prior to flow cytometry analysis and supernatants were collected for ELISA.
T cell/monocyte co-culture.
For ex vivo T cell polarizations and monocyte co-culture, splenic CD4+ T cells were isolated and polarized for differentiation into TH subsets by exposing cells to the following cytokine cocktails: TH1: IL-12 (20 ng/ml) and anti-IL-4 (10 μg/ml); “nonpathogenic” TH17 (referred to as Th17(β)): recombinant human TGF-β1 (2 ng/ml; PeproTech), IL-6 (25 ng/ml), and anti-IL-4, anti-IL-12/23, and anti-IFN-γ (10 μg/ml each); and “pathogenic” TH17 (referred to as Th17(23)): IL-23 (20 ng/ml), IL-1β (20 ng/ml), IL-6 (25 ng/ml), and anti-IL-4 and anti-IFN-γ (10 μg/ml each). After the 48 hr rest phase in IL-2, the indicated CD4+ TH cell populations (1 × 105 cells/well for TH1, TH17(β) and TH17(23)) were co-cultured with freshly isolated BM monocytes (1 × 105 cells/well), plate-bound anti-CD3ε (2 μg/ ml), soluble anti-CD28 (2 μg/ml) and TH-polarizing cytokines for 24 hr in flat-bottom 96-well plates. Cell culture supernatant was collected for ELISA to assess CCL2 production.
SC myeloid cell sorting and qPCR.
To analyze gene expression of myeloid cell chemoattractants from leukocytes in the inflamed spinal cord at the peak of disease (15 dpi), sorted leukocyte subsets (1x104 cells per well) were stimulated in 96-well flat-bottom plates with or without recombinant murine IL-3 (50 ng/ml; PeproTech) for 16 hours and subjected to mRNA isolation and qPCR as described below.
Mouse bone marrow (BM) monocyte culture.
Femurs and tibias were collected from mice as described and processed into single-cell suspensions. BM monocytes were isolated by MACS magnetic depletion of non-monocytes via the Monocyte Isolation Kit (BM) (Miltenyi Biotec). BM monocytes were washed in PBS and further cultured in complete media at a concentration of 0.5-1x106 cells/ml in flat-bottom 96-well plates for up to 5 days recombinant murine IL-3 (PeproTech) at 20 ng/ml. Cells were then analyzed via flow cytometry or mRNA was isolated for downstream qPCR analysis.
Histopathology.
Tissue processing and staining.
Mice were euthanized under isoflurane anesthesia, perfused intracardially with 20 ml ice-cold PBS, and spinal columns were collected on-ice. Spinal cords were carefully removed by hydraulic extrusion with PBS and an 18 G needle (320). Then, spinal cords were divided into four sections and embedded in Tissue-Tek O.C.T. compound (Sakura Finetek, Torrance, CA), frozen in 2-Methylbutane (Fisher Scientific, Fair Lawn, NJ), cooled with dry ice, and serial frozen sections were prepared (7-μm slices).
Spinal cord demyelination analysis by Luxol fast blue (LFB) staining.
Demyelination of spinal cords was analyzed by staining with LFB according to Luxol Fast Blue Stain Kit (Myelin Stain) manufacturer instructions (Cat. No. ab150675; Abcam) and counterstained with Cresyl Echt Violet Solution. Demyelinated areas were scanned using a digital slide scanner (NanoZoomer 2.0RS; Hamamatsu Photonics KK, Japan) and processed and analyzed using NDP.view 2 viewing software (Hamamatsu Photonics KK, Japan). Briefly, normalized demyelinated area (μm2) was evaluated for each spinal cord vertebral region (i.e., cervical, thoracic, lumbar, and sacral) by normalizing the total area of gated white matter demyelinated regions of interest (ROIs) within a given section to the total area of each spinal cord tissue section analyzed. Percent demyelination for the spinal cord of each mouse was calculated as follows: Percent demyelination = [Sum of the area of gated white matter demyelinated ROIs] ÷ [Sum of the total area of spinal cord tissue section analyzed] × 100.
Immunohistology for IL-3-producing cells and IL-3Rɑ+ cells in the central nervous system.
An anti-IL3 antibody (1:5, 503902, MP2-8F8, BioLegend) followed by a biotinylated rabbit anti-rat IgG secondary antibody and streptavidin DyLight 594 (1:100, BA-4001 and 1:600, SA-5594, Vector Laboratories) were used for IL-3 detection. For co-localization of GFAP, AF488-GFAP antibody (1:50, 53-9892, eBioscience) was incubated at 4°C overnight. For co-localization of IL-3Rɑ with CD11b, an anti-Interleukin 3 Receptor Alpha antibody (1:50, 141039, US Biological) followed by an anti-CD11b antibody (1:25, 550282, M1/70, BD Biosciences) were incubated at 4°C overnight after blocking with 4% goat serum in PBS. A biotinylated goat anti-rabbit IgG secondary antibody (1:100, BA-1000, Vector Laboratories) and streptavidin DyLight 594 were used for IL-3Rɑ and a goat anti-rat IgG secondary antibody, Alexa Fluor 488 (1:100, A-11006, Thermo Fisher Scientific) were used for CD11b. All sections were counterstained with DAPI (1:3000, D21490, Thermo Fisher Scientific) and the slides were scanned using a digital slide scanner (NanoZoomer 2.0RS; Hamamatsu Photonics KK, Japan) and processed and analyzed using NDP.view 2 viewing software (Hamamatsu Photonics KK, Japan).
Molecular Biology
Quantitative PCR.
Mouse total RNA was isolated using the RNeasy Mini Kit (Qiagen, Venlo, Netherlands) and ~10 mg tissue/sample according to the manufacturer’s instructions. cDNA was generated from 1 μg of total RNA per sample using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). For cells, 5 × 103 Ly6Chigh monocytes, moDC, MHCIIhigh macrophages, neutrophils, and microglia were sorted from inflamed spinal cord or harvested from cultured cells, and total RNA was extracted using the RNeasy Micro Kit (Qiagen) followed by cDNA transcription as described above. Quantitative PCR (qPCR) was performed using following TaqMan primers and their corresponding assay ID (Applied Biosystems): Il3 (Mm00439631_m1), Ccl2 (Mm00441242_m1), Ccl5 (Mm01302427_m1), Ccl7 (Mm00443113_m1), Ccl12 (Mm01617100_m1), Il3rɑ (Mm00434273_m1), Csf2rb (Mm00655745_m1), Ccr7 (Mm99999130_s1), Flt3 (Mm00439016_m1), Kit (Mm00445212_m1), Spp1 (Mm00436767_m1), C5ar1 (Mm00500292_s1), Irf4 (Mm00516431_m1), Ly6c2 (Mm00841873_m1), Cd209a (Mm00460067_m1), Fcgr1 (Mm00438874_m1), Zbtb46 (Mm00511327_m1), and Actb (Cat. No. 4352341E; assay ID: Mm00607939_s1) as an endogenous control. qPCR was run on a 7500 thermal cycler (Applied Biosystems), and data were quantified with the 2−ΔΔCt method.
ELISA.
IL-3 was measured in serum, spinal cord tissue and cell culture supernatants with the Mouse IL-3 ELISA Kit (Boster Biological). Specifically for spinal cord tissue ELISA measurements, ~10 mg tissue/sample was homogenized in 0.3 ml RIPA buffer and left to sit for 10 minutes on ice, prior to microcentrifugation at 10,000 rpm for 8 minutes and collection of the supernatant. CCL2, IFNγ, IL17A, and GM-CSF were measured in culture media using ELISAs from R&D Systems and the manufacturer’s instructions.
Human CSF proteomics
CSF samples were centrifuged at 400g for 10 minutes at room temperature. Cell-free supernatant was isolated and stored at −80°C until proteomics analysis, within 2 hours of collection. Leukocyte concentration was determined as per standard laboratory procedures (cells/μL). IL-3 concentration in CSF was determined using the SomaScan® platform from SomaLogic Inc, Boulder, Co., as previously described43. Data are presented in arbitrary units.
Human snRNAseq
Isolation and fluorescence-activated nuclear sorting (FANS) of nuclei for frozen human postmortem brain.
Samples were processed in batches consisting of 2 individuals; 1 case and 1 control. For each case, we processed 3 dissections; 1 of gray matter, 1 white matter, and 1 containing an MS plaque. For each Control we processed 2 dissections; 1 gray matter and 1 white matter. Approximately 25 mg of frozen postmortem human brain tissue from each dissection was homogenized in cold lysis buffer (0.32M Sucrose, 5 mM CaCl2, 3 mM Magnesium acetate, 0.1 mM, EDTA, 10 mM Tris-HCl, pH8, 1 mM DTT, 0.1% Triton X-100) and filtered through a 40 μm cell strainer. All buffers were supplemented with RNAse inhibitors (Takara). The flow-through was underlaid with sucrose solution (1.8 M Sucrose, 3 mM Magnesium acetate, 1 mM DTT, 10 mM Tris-HCl, pH8) and centrifuged at 107,000 g for 1 hour at 4°C. Pellets were resuspended in PBS supplemented with 0.5% bovine serum albumin (BSA). Prior to FANS, volumes were brought up to 500 μl with PBS and DAPI (Thermoscientific) added to a final concentration of 1 μg/ml. DAPI-positive nuclei were sorted into tubes pre-coated with 5% BSA using a FACSAria flow cytometer (BD Biosciences).
Single-nuclei gene expression library preparation.
Following FANS, nuclei were quantified (Countess II, Life Technologies) and 8,000 from each sample loaded on a single 10x lane using single cell 3’ v3 reagents (10x Genomics). cDNA libraries were prepared according to the manufacturer’s protocol. Libraries were sequenced at New York Genome Center using the Novaseq platform (Illumina).
Quantification of single-nuclei gene expression data.
We used the cellranger (v.7.0.0) pipeline for preprocessing of the single-cell sequencing data. We aligned the fastq reads to the reference genome hg38, quantified the transcriptomic reads, and called cell barcodes to generate a feature-count-by-barcode matrix for subsequent analysis.
Clustering, annotation, and visualization of single-nuclei data.
We used the pegasus (v1.7.0)44 pipeline for QC and clustering, and scanpy (v1.9.1)45 pipeline for visualization of the data. We first aggregated all 30 single-cell libraries and performed QC. We filtered out cells expressing less than 500 genes, less than 500 UMIs, and having mitochondrial gene fraction greater than 10 percent. After QC, we retained 77,490 cells for downstream analysis. We also filtered out genes expressed in less than 0.05% of all cells and retained 27,396 genes for analysis. The log-normalized counts were subjected to dimensionality reduction, and the first 25 PCs were used for clustering analysis. Since the single-cell libraries were prepared in 6 batches, we used Harmony46 to correct for potential batch effect. We used Scrublet to filter out 2,315 cells predicted to be potential doublets from the data. Clustered cells were annotated based on marker genes defined in the pegasus pipeline. We used cell markers defined in both “human_brain” and “human_immune” to annotate 18 clusters. We used the Mann-Whitney U test for differential expression analysis by condition or clusters. For the cluster-based differential tests, we compared cells within the cluster with the rest of the cells. To count the cells expressing IL3RA (IL3RA+ cells), we binarized the expression of IL3RA by cells expressing log-normalized expression greater than 1 (Fig S5B).
QUANTIFICATION AND STATISTICAL ANALYSIS
Results are shown as mean ± s.e.m. Statistical analysis was performed using GraphPad Prism 7 (Graphpad Software). Statistical tests included unpaired, two-tailed non-parametric Mann–Whitney U-tests (when Gaussian distribution was not assumed). For multiple comparisons, a non-parametric multiple-comparisons test comparing the mean rank of each group (when Gaussian distribution was not assumed) was used, or one- or two-way ANOVAs followed by Tukey’s test were used. For correlation analysis, correlation was computed using Spearman correlation or Pearson correlation coefficients. P values of 0.05 or less were considered to denote significance. Each experiment was repeated independently at least three times with similar results.
Supplementary Material
Highlights.
IL-3 is elevated in the CSF of MS patients and associates with disease severity
IL-3 deletion protects against neuroinflammation and disease in a mouse model of MS
In the CNS, astrocytes and T cells produce IL-3
In MS, IL-3 programs IL3RA-expressing myeloid cells to generate chemotactic cues
ACKNOWLEDGEMENTS
We thank the HCI-CRM Flow Cytometry Core Facility at the Massachusetts General Hospital and the Flow Cytometry CoRE at the Icahn School of Medicine at Mount Sinai for assistance with cell sorting. We thank the Human Immune Monitoring Center at the Icahn School of Medicine at Mount Sinai for assistance in validation analysis. We thank K. Joyes for manuscript copy editing. This work was funded by the an Erwin Schrödinger Postdoctoral Fellowship J4645 by the Austrian Science Fund (to M.G.K.); National Institutes of Health (NIH) Ruth L. Kirschstein National Research Service Award Individual Predoctoral Fellowship F31HL147364 (to J.E.M.); NIH R35 HL139598, NIH R01 NS127808, and NIH R01 NS108419 (to M.N.); an MGH ECOR Howard M. Goodman Award (to B.P.K); NIH R01AG065582, R01AG067025, R01AG050986 (to P.R.); NIH National Center for Advancing Translational Sciences UL1TR001873; Cure Alzheimer’s Fund, NIH R35 HL135752, P01 HL131478, and the Patricia and Scott Eston MGH Research Scholar (to F.K.S.); NIH P01 HL142494 (to M.N., B.P.K., and F.K.S.); NIH K99/R00 HL151750, R01 HL158534, and the Cure Alzheimer’s Fund (to C.S.M.).
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
B.P.K. is an inventor on patents and/or patent applications filed by Mass General Brigham that describe genome engineering technologies. B.P.K. is a consultant for EcoR1 capital, and is an advisor to Acrigen Biosciences, Life Edit Therapeutics, and Prime Medicine.
References
- 1.Reich DS, Lucchinetti CF, and Calabresi PA (2018). Multiple Sclerosis. N. Engl. J. Med 378, 169–180. 10.1056/NEJMra1401483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baecher-Allan C, Kaskow BJ, and Weiner HL (2018). Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 97, 742–768. 10.1016/j.neuron.2018.01.021. [DOI] [PubMed] [Google Scholar]
- 3.Lassmann H (2018). Multiple Sclerosis Pathology. Cold Spring Harb. Perspect. Med 8. 10.1101/cshperspect.a028936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McGinley MP, Goldschmidt CH, and Rae-Grant AD (2021). Diagnosis and Treatment of Multiple Sclerosis: A Review. JAMA 325, 765–779. 10.1001/jama.2020.26858. [DOI] [PubMed] [Google Scholar]
- 5.Dougan M, Dranoff G, and Dougan SK (2019). GM-CSF, IL-3, and IL-5 Family of Cytokines: Regulators of Inflammation. Immunity 50, 796–811. 10.1016/j.immuni.2019.03.022. [DOI] [PubMed] [Google Scholar]
- 6.Mindur JE, and Swirski FK (2019). Growth factors as immunotherapeutic targets in cardiovascular disease. Arterioscler. Thromb. Vasc. Biol 39, 1275–1287. 10.1161/ATVBAHA.119.311994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weber GF, Chousterman BG, He S, Fenn AM, Nairz M, Anzai A, Brenner T, Uhle F, Iwamoto Y, Robbins CS, et al. (2015). Interleukin-3 amplifies acute inflammation and is a potential therapeutic target in sepsis. Science (80-. ). 347, 1260–1265. 10.1126/science.aaa4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robbins CS, Chudnovskiy A, Rauch PJ, Figueiredo JL, Iwamoto Y, Gorbatov R, Etzrodt M, Weber GF, Ueno T, Van Rooijen N, et al. (2012). Extramedullary hematopoiesis generates Ly-6C high monocytes that infiltrate atherosclerotic lesions. Circulation 125, 364–374. 10.1161/CIRCULATIONAHA.111.061986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Renner K, Hermann FJ, Schmidbauer K, Talke Y, Rodriguez Gomez M, Schiechl G, Schlossmann J, Brühl H, Anders H-J, and Mack M (2015). IL-3 contributes to development of lupus nephritis in MRL/lpr mice. Kidney Int. 88, 1088–1098. 10.1038/ki.2015.196. [DOI] [PubMed] [Google Scholar]
- 10.Srivastava RK, Tomar GB, Barhanpurkar AP, Gupta N, Pote ST, Mishra GC, and Wani MR (2011). IL-3 attenuates collagen-induced arthritis by modulating the development of Foxp3+ regulatory T cells. J. Immunol 186, 2262–2272. 10.4049/jimmunol.1002691. [DOI] [PubMed] [Google Scholar]
- 11.McAlpine CS, Park J, Griciuc A, Kim E, Choi SH, Iwamoto Y, Kiss MG, Christie KA, Vinegoni C, Poller WC, et al. (2021). Astrocytic interleukin-3 programs microglia and limits Alzheimer’s disease. Nature 595, 701–706. 10.1038/s41586-021-03734-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Renner K, Hellerbrand S, Hermann F, Riedhammer C, Talke Y, Schiechl G, Gomez MR, Kutzi S, Halbritter D, Goebel N, et al. (2016). IL-3 promotes the development of experimental autoimmune encephalitis. JCI Insight. 10.1172/jci.insight.87157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee PW, Xin MK, Pei W, Yang Y, and Lovett-Racke AE (2018). IL-3 Is a Marker of Encephalitogenic T Cells, but Not Essential for CNS Autoimmunity. Front. Immunol 9, 1255. 10.3389/fimmu.2018.01255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chiang CS, Powell HC, Gold LH, Samimi A, and Campbell IL (1996). Macrophage/microglial-mediated primary demyelination and motor disease induced by the central nervous system production of interleukin-3 in transgenic mice. J. Clin. Invest 97, 1512–1524. 10.1172/JCI118574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Powell HC, Garrett RS, Brett FM, Chiang CS, Chen E, Masliah E, and Campbell IL (1999). Response of glia, mast cells and the blood brain barrier, in transgenic mice expressing interleukin-3 in astrocytes, an experimental model for CNS demyelination. Brain Pathol. 9, 219–235. 10.1111/j.1750-3639.1999.tb00220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chavany C, Vicario-Abejón C, Miller G, and Jendoubi M (1998). Transgenic mice for interleukin 3 develop motor neuron degeneration associated with autoimmune reaction against spinal cord motor neurons. Proc. Natl. Acad. Sci. U. S. A 95, 11354–11359. 10.1073/pnas.95.19.11354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Linnerbauer M, Wheeler MA, and Quintana FJ (2020). Astrocyte Crosstalk in CNS Inflammation. Neuron. 10.1016/j.neuron.2020.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mosleth EF, Vedeler CA, Liland KH, McLeod A, Bringeland GH, Kroondijk L, Berven FS, Lysenko A, Rawlings CJ, Eid KE-H, et al. (2021). Cerebrospinal fluid proteome shows disrupted neuronal development in multiple sclerosis. Sci. Rep 11, 4087. 10.1038/s41598-021-82388-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stangel M, Fredrikson S, Meinl E, Petzold A, Stüve O, and Tumani H (2013). The utility of cerebrospinal fluid analysis in patients with multiple sclerosis. Nat. Rev. Neurol 9, 267–276. 10.1038/nrneurol.2013.41. [DOI] [PubMed] [Google Scholar]
- 20.Mendel I, Kerlero de Rosbo N, and Ben-Nun A (1995). A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: fine specificity and T cell receptor V beta expression of encephalitogenic T cells. Eur. J. Immunol 25, 1951–1959. 10.1002/eji.1830250723. [DOI] [PubMed] [Google Scholar]
- 21.Constantinescu CS, Farooqi N, O’Brien K, and Gran B (2011). Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol 164, 1079–1106. 10.1111/j.1476-5381.2011.01302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caravagna C, Jaouën A, Desplat-Jégo S, Fenrich KK, Bergot E, Luche H, Grenot P, Rougon G, Malissen M, and Debarbieux F (2018). Diversity of innate immune cell subsets across spatial and temporal scales in an EAE mouse model. Sci. Rep 8, 5146. 10.1038/s41598-018-22872-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greiner T, and Kipp M (2021). What Guides Peripheral Immune Cells into the Central Nervous System? Cells 10. 10.3390/cells10082041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schafflick D, Xu CA, Hartlehnert M, Cole M, Schulte-Mecklenbeck A, Lautwein T, Wolbert J, Heming M, Meuth SG, Kuhlmann T, et al. (2020). Integrated single cell analysis of blood and cerebrospinal fluid leukocytes in multiple sclerosis. Nat. Commun 11, 247. 10.1038/s41467-019-14118-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bechmann L, Busse K, Stoppe M, Cotte S, Ettrich B, and Then Bergh F (2014). Corticosteroid receptor expression and in vivo glucocorticoid sensitivity in multiple sclerosis. J. Neuroimmunol 276, 159–165. 10.1016/j.jneuroim.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 26.Umeton R, Bellucci G, Bigi R, Romano S, Buscarinu MC, Reniè R, Rinaldi V, Pizzolato Umeton R, Morena E, Romano C, et al. (2022). Multiple sclerosis genetic and non-genetic factors interact through the transient transcriptome. Sci. Rep 12, 7536. 10.1038/s41598-022-11444-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi K, Li H, Chang T, He W, Kong Y, Qi C, Li R, Huang H, Zhu Z, Zheng P, et al. (2022). Bone marrow hematopoiesis drives multiple sclerosis progression. Cell 185, 2234–2247.e17. 10.1016/j.cell.2022.05.020. [DOI] [PubMed] [Google Scholar]
- 28.Krishnarajah S, and Becher B (2022). T(H) Cells and Cytokines in Encephalitogenic Disorders. Front. Immunol 13, 822919. 10.3389/fimmu.2022.822919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wagner CA, Roqué PJ, and Goverman JM (2020). Pathogenic T cell cytokines in multiple sclerosis. J. Exp. Med 217. 10.1084/jem.20190460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palmeri S, Leonardi V, Danova M, Porta C, Ferrari S, Fincato G, and Citarrella P (1999). Prospective, randomized trial of sequential interleukin-3 and granulocyte- or granulocyte-macrophage colony-stimulating factor after standard-dose chemotherapy in cancer patients. Haematologica 84, 1016–1023. [PubMed] [Google Scholar]
- 31.Ballestrero A, Ferrando F, Garuti A, Basta P, Gonella R, Stura P, Mela GS, Sessarego M, Gobbi M, and Patrone F (1999). Comparative effects of three cytokine regimens after high-dose cyclophosphamide: granulocyte colony-stimulating factor, granulocyte-macrophage colony-stimulating factor (GM-CSF), and sequential interleukin-3 and GM-CSF. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol 17, 1296. 10.1200/JCO.1999.17.4.1296. [DOI] [PubMed] [Google Scholar]
- 32.He SZ, Busfield S, Ritchie DS, Hertzberg MS, Durrant S, Lewis ID, Marlton P, McLachlan AJ, Kerridge I, Bradstock KF, et al. (2015). A Phase 1 study of the safety, pharmacokinetics and anti-leukemic activity of the anti-CD123 monoclonal antibody CSL360 in relapsed, refractory or high-risk acute myeloid leukemia. Leuk. Lymphoma 56, 1406–1415. 10.3109/10428194.2014.956316. [DOI] [PubMed] [Google Scholar]
- 33.Clark IC, Wheeler MA, Lee H-G, Li Z, Sanmarco LM, Thaploo S, Polonio CM, Shin SW, Scalisi G, Henry AR, et al. (2023). Identification of astrocyte regulators by nucleic acid cytometry. Nature 614, 326–333. 10.1038/s41586-022-05613-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anzai A, Mindur JE, Halle L, Sano S, Choi JL, He S, McAlpine CS, Chan CT, Kahles F, Valet C, et al. (2019). Self-reactive CD4+ IL-3+ T cells amplify autoimmune inflammation in myocarditis by inciting monocyte chemotaxis. J Exp Med 216, 369–383. 10.1084/jem.20180722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bae S, Park J, and Kim JS (2014). Cas-OFFinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Conant D, Hsiau T, Rossi N, Oki J, Maures T, Waite K, Yang J, Joshi S, Kelso R, Holden K, et al. (2022). Inference of CRISPR Edits from Sanger Trace Data. Cris. J 5, 123–130. 10.1089/crispr.2021.0113. [DOI] [PubMed] [Google Scholar]
- 38.Kleinstiver BP, Sousa AA, Walton RT, Tak YE, Hsu JY, Clement K, Welch MM, Horng JE, Malagon-Lopez J, Scarfò I, et al. (2019). Engineered CRISPR–Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nat. Biotechnol 10.1038/s41587-018-0011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rohland N, and Reich D (2012). Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res. 10.1101/gr.128124.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blond D, Campbell SJ, Butchart AG, Perry VH, and Anthony DC (2002). Differential induction of interleukin-1beta and tumour necrosis factor-alpha may account for specific patterns of leukocyte recruitment in the brain. Brain Res. 958, 89–99. 10.1016/s0006-8993(02)03473-x. [DOI] [PubMed] [Google Scholar]
- 41.McCluskey L, Campbell S, Anthony D, and Allan SM (2008). Inflammatory responses in the rat brain in response to different methods of intra-cerebral administration. J. Neuroimmunol 194, 27–33. 10.1016/j.jneuroim.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 42.Dickens AM, Tovar-Y-Romo LB, Yoo S-W, Trout AL, Bae M, Kanmogne M, Megra B, Williams DW, Witwer KW, Gacias M, et al. (2017). Astrocyte-shed extracellular vesicles regulate the peripheral leukocyte response to inflammatory brain lesions. Sci. Signal 10. 10.1126/scisignal.aai7696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Probert F, Yeo T, Zhou Y, Sealey M, Arora S, Palace J, Claridge TDW, Hillenbrand R, Oechtering J, Leppert D, et al. (2021). Integrative biochemical, proteomics and metabolomics cerebrospinal fluid biomarkers predict clinical conversion to multiple sclerosis. Brain Commun. 3, fcab084. 10.1093/braincomms/fcab084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li B, Gould J, Yang Y, Sarkizova S, Tabaka M, Ashenberg O, Rosen Y, Slyper M, Kowalczyk MS, Villani A-C, et al. (2020). Cumulus provides cloud-based data analysis for large-scale single-cell and single-nucleus RNA-seq. Nat. Methods 17, 793–798. 10.1038/s41592-020-0905-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wolf FA, Angerer P, and Theis FJ (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15. 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh P-R, and Raychaudhuri S (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296. 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single nuclear RNA-seq data have been deposited at GEO under the accession GSE227781 and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-CD45 BUV737 (clone 30-F11) | BD Biosciences | Cat# 748371 |
| anti-CD45 BV711 (clone 30-F11) | BioLegend | Cat# 103147 |
| anti-CD3 (clone 17A2) FITC | BioLegend | Cat# 100204 |
| anti-CD3ε (clone 145-2C11) PE | BioLegend | Cat# 100308 |
| anti-CD4 (clone RM4-5) FITC | BioLegend | Cat# 100510 |
| anti-CD4 (clone RM4-5) PE-Cyanine7 | BioLegend | Cat# 100528 |
| anti-CD8a (clone 53–6.7) Alexa Fluor 700 | BioLegend | Cat# 100730 |
| anti-CD90.2 (clone 53-2.1) PE | BioLegend | Cat# 140308 |
| anti-CD19 (clone 6D5) | BioLegend | Cat# 115538 |
| anti-B220 (clone RA3-6B2) BV785 | BioLegend | Cat# 103246 |
| anti-Ly6G (clone 1A8) BV421 | BioLegend | Cat# 127645 |
| anti-Ly-6C (AL-21) BV605 | BD Biosciences | Cat# 563011 |
| anti-I-A/I-E (MHCII; clone M5/114.15.2) Alexa Fluor 700 | BioLegend | Cat# 107622 |
| anti-CD11b (clone M1/70) APC/Cyanine7 | BioLegend | Cat# 101226 |
| anti-CD11c (clone HL3) PerCP-Cy5.5 | BD Biosciences | Cat# 560584 |
| anti-CD44 (clone IM7) PE-Cyanine7 | BioLegend | Cat# 103030 |
| anti-CD62L (clone MEL-14) Alexa Fluor 700 | BioLegend | Cat# 104426 |
| anti-CD64 (clone X54-5/7.1) PE | BioLegend | Cat# 139304 |
| anti-CD80 (clone 16-10A1) FITC | BioLegend | Cat# 104706 |
| anti-CD86 (clone GL-1) PE/Cyanine7 | BioLegend | Cat# 105014 |
| anti-OX40L (clone RM134L) APC | BioLegend | Cat# 108812 |
| anti-CD69 (clone H1.2F3) BV711 | BioLegend | Cat# 104537 |
| anti-CD49d (integrin ɑ4; clone R1-2) PE | BioLegend | Cat# 103608 |
| Anti-Ly6G (clone 1AB-Ly6g) PerCP/eFluor710 | Thermo Fisher Sci. | Cat# 46-9668-82 |
| Anti-I-A/I-E (MHCII; clone M5/114.15.2) BUV495 | BD Biosciences | Cat# 750281 |
| anti-Ki67 (clone SolA15) PE | ThermoFisher Sci. | 12-5698-82 |
| anti-IL-3Rɑ (CD123; clone REA114) | Miltenyi Biotec | Cat# 130-102-583 |
| anti-F4/80 (clone BM8) APC/Cyanine7 | BioLegend | Cat# 123118 |
| anti-CD115 (CSF-1R; clone AFS98) BV605 | BioLegend | Cat# 135517 |
| anti-CX3CR1 (clone SA011F11) PE-Cyanine7 | BioLegend | Cat# 149016 |
| anti-IL-3 (clone MP2-8F8) | BD Biosciences | Cat# 554383 |
| anti-IFNγ (clone XMG1.2) BV605 | BioLegend | Cat# 505840 |
| anti-IL-17A (clone TC11-18H10.1) APC/Cyanine7 | BioLegend | Cat# 506922 |
| anti-GM-CSF (clone MP1-22E9) BV421 | BD Biosciences | Cat# 564747 |
| anti-CD11b (clone M1/70) BUV805 | BD Biosciences | Cat# 741934 |
| anti-ACSA2 (clone REA969) APC | Miltenyi Biotec | Cat# 130-116-245 |
| anti-CD19 (clone 1D3) BUV661 | BD Biosciences | Cat# 612971 |
| anti-CD8 (clone 53-6.7) PE-Dazzle594 | BioLegend | Cat# 100762 |
| anti-IL3 | BioLegend | Cat# 503902 |
| AF488 anti-GFAP | eBioscience | Cat# 53-9892 |
| Anti-IL3Rɑ | US Biological | Cat# 141039 |
| anti-CD11b | BD Biosciences | Cat# 550282 |
| anti-rabbit IgG | Vector Laboratories | Cat# BA-1000 |
| anti-rat IgG | Thermo Fisher Scientific | Cat# A-11006 |
| anti-rat IgG | Vector Laboratories | Cat# BA-4001 |
| Mouse anti-IL4 | BioLegend | Cat# 504122 |
| Mouse anti-CD28 | Thermo Fisher Sci. | Cat# 16-0281-82 |
| Mouse anti-CD3ε | Thermo Fisher Sci. | Cat# 16-0031-82 |
| Mouse anti-IL12 | Thermo Fisher Sci. | Cat# 45-7123-80 |
| Mouse anti- IFNy | BioLegend | Cat# 513208 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Human brain tissue | Human Brain and Spinal Fluid Resource Center, UCLA | N/A |
| Human CSF samples | Department of Neurology of the University Hospital Basel | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| DAPI | Thermoscientific | Cat# 62248 |
| IL-3 | Peprotech | Cat# 213-13 |
| IL-12 | Peprotech | Cat#210-12 |
| TGF-β1 | Peprotech | Cat# 100-21 |
| IL-6 | Peprotech | Cat# 216-16 |
| IL-23 | Peprotech | Cat# 200-23 |
| IL-2 | Peprotech | Cat# 212-12 |
| IL-1β | BioLegend | Cat# 575102 |
| Tamoxifen | Sigma Aldrich | T5648 |
| Critical commercial assays | ||
| RNeasy Mini kit | Qiagen | Cat# 74104 |
| RNeasy Micro Kit | Qiagen | Cat# 74004 |
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher | Cat# 4368814 |
| MOG35-55/CFA Emulsion PTX Hooke Kit™ | Hooke Laboratories | Cat# EK-2110 |
| Luxol Fast Blue Stain Kit | Abcam | Cat# ab150675 |
| IL-3 ELISA kit | Boster Biological | Cat# EK0403 |
| IFNγ ELISA kit | R&D Systems | Cat# MIF00 |
| IL17A ELISA kit | R&D Systems | Cat# M1700 |
| GM-CSF ELISA kit | R&D Systems | Cat# MGM00 |
| CCL2 ELISA Kit | R&D Systems | Cat# MJE00B |
| Zombie Aqua Fixable Viability Kit | BioLegend | 423102 |
| eBioscience FoxP3/ Transcription Factor Staining Buffer Set | Thermo Fisher Sci. | 00-5523-00 |
| Fixation/Permeabilization Kit with BD GolgiPlug | BD Biosciences | 555028 |
| CountBright Absolute Counting Beads | Thermo Fisher Sci. | C36950 |
| Monocyte Isolation Kit | Miltenyi Biotec | 130-100-629 |
| Chromium Single Cell 3’ Reagent Kits v3 | 10x Genomics | Cat# 1000075 |
| Chromium i7 Sample Index Plate | 10x Genomics | Cat# 220103 |
| Kapa Library Quantification Kit | KAPA Biosystems | Cat# KK4873 |
| Tapestation D5000 ScreenTape | Agilent Technologies | Cat# 5067-5588 |
| Tapestation D5000 Reagents | Agilent Technologies | Cat# 5067-5589 |
| Deposited data | ||
| Single nuclear RNA-seq | Gene Expression omnibus | GSE227781 |
| Experimental models: Cell lines | ||
| Experimental models: Organisms/strains | ||
| C57BL/6J | The Jackson Laboratories | Strain# 000664 |
| Aldh1l1cre Ert2 | The Jackson Laboratories | Strain# 031008 |
| Cd4cre | The Jackson Laboratories | Strain# 022071 |
| Il3 −/− | The Jackson Laboratories | Strain# 026277 |
| IL3 GFPfl/fl | McAlpine lab | N/A |
| Il3rɑ −/− | McAlpine lab | N/A |
| Oligonucleotides | ||
| Il3 (Mm00439631_m1) | Thermo Fisher | Cat# 4331182 |
| Ccl2 (Mm00441242_m1) | Thermo Fisher | Cat# 4331182 |
| Ccl5 (Mm01302427_m1) | Thermo Fisher | Cat# 4331182 |
| Ccl7 (Mm00443113_m1) | Thermo Fisher | Cat# 4331182 |
| Ccl12 (Mm01617100_m1) | Thermo Fisher | Cat# 4331182 |
| Il3rɑ (Mm00434273_m1) | Thermo Fisher | Cat# 4331182 |
| Csf2rb (Mm00655745_m1) | Thermo Fisher | Cat# 4331182 |
| Ccr7 (Mm99999130_s1) | Thermo Fisher | Cat# 4331182 |
| Flt3 (Mm00439016_m1) | Thermo Fisher | Cat# 4331182 |
| Kit (Mm00445212_m1) | Thermo Fisher | Cat# 4331182 |
| Spp1 (Mm00436767_m1) | Thermo Fisher | Cat# 4331182 |
| C5ar1 (Mm00500292_s1) | Thermo Fisher | Cat# 4331182 |
| Irf4 (Mm00516431_m1) | Thermo Fisher | Cat# 4331182 |
| Ly6c2 (Mm00841873_m1) | Thermo Fisher | Cat# 4331182 |
| Cd209a (Mm00460067_m1) | Thermo Fisher | Cat# 4331182 |
| Fcgr1 (Mm00438874_m1) | Thermo Fisher | Cat# 4331182 |
| Zbtb46 (Mm00511327_m1) | Thermo Fisher | Cat# 4331182 |
| Actb (Mm00607939_s1) | Thermo Fisher | Cat# 4331182 |
| Recombinant DNA | ||
| Software and algorithms | ||
| NDP.view 2 | Hamamatsu Photonics | www.hamamatsu.com |
| FlowJo v10 | FlowJo | www.flowjo.com |
| GraphPad Prism v9 | GraphPad Software | www.graphpad.com |
| BioRender | BioRender | www.biorender.com |
| SomaScan | SomaLogic Inc | www.somalogic.com |
| Cell Ranger | 10x Genomics | v7.0.0 |
| pegasus | https://github.com/lilab-bcb/pegasus | v1.7.0 |
| scanpy | https://github.com/scverse/scanpy | v1.8.2 |
| anndata | https://github.com/scverse/anndata | v0.8.0 |
| numpy | https://github.com/numpy/numpy | v1.21.5 |
| scipy | https://github.com/scipy/scipy | v1.8.0 |
| pandas | https://github.com/pandas-dev/pandas | v1.4.1 |
| umap | https://github.com/lmcinnes/umap | v0.5.2 |
| scikit-learn | https://github.com/scikit-learn/scikit-learn | v1.0.2 |
| statsmodels | https://github.com/statsmodels/statsmodels | v0.13.2 |
| python-igraph | https://github.com/igraph/python-igraph | v0.9.9 |
| Other | ||