

Abstract
OBJECTIVE
To assess for TDP-43 deposits in brains with and without a LRRK2 G2019S mutation.
BACKGROUND
LRRK2 G2019S mutations have been associated with parkinsonism and a wide range of pathological findings. There are no systematic studies examining the frequency and extent of TDP-43 deposits in neuropathological samples from LRRK2 G2019S carriers.
METHODS
Twelve brains with LRRK2 G2019S mutations were available for study from the New York Brain Bank at Columbia University; 11 of those had samples available for TDP-43 immunostaining. Clinical, demographic and pathological data are reported for 11 brains with a LRRK2 G2019S mutation and compared to 11 brains without GBA1 or LRRK2 G2019S mutation with a pathologic diagnosis of PD or DLBD. They were frequency matched by age, gender, parkinsonism age of onset and disease duration.
RESULTS
TDP-43 aggregates were present in 73% (n=8) of brains with a LRRK2 mutation and 18% (n=2) of brains without a LRRK2 mutation (p=0.03). In one brain with a LRRK2 mutation, TDP-43 proteinopathy was the primary neuropathological change.
CONCLUSIONS
Extra-nuclear TDP-43 aggregates are observed with greater frequency in LRRK2 G2019S autopsies compared to PD cases without a LRRK2 G2019S mutation. The association between LRRK2 and TDP-43 should be further explored.
INTRODUCTION
The LRRK2 G2019S mutation is present in 1–4% of all Parkinson’s disease (PD) cases and is more common in selected populations, namely North African Berbers (37% in familial cases and 41% in sporadic cases) and Ashkenazi Jews (23% in familial cases and 13% in sporadic cases) (1–4). Penetrance estimates are variable ranging from 25%−100% (3–5). A key striking feature of LRRK2-associated PD is the variability of neuropathologic findings even from related individuals, especially with regards to the presence of Lewy bodies (LB). When examined for other neuropathologic findings, some LRRK2 cases without alpha-synuclein or phosphorylated tau deposition have been found to have transactivation-response DNA binding protein of 43kDa (TDP-43) containing cytoplasmic inclusions (6–8).
TDP-43, which is a RNA/DNA binding intranuclear protein that regulates mRNA splicing, translation, transportation and degradation (9), is found mislocalized in the cytoplasm in the form of inclusions in many neurodegenerative diseases (10–15). TDP-43 was first found in ubiquitin positive inclusions in the central nervous system in cases of amyotrophic lateral sclerosis (ALS) and frontotemporal lobar dementia (FTLD) (9, 16). Subsequently, cytoplasmic TDP-43-positive inclusions were also found to be a key neuropathological features of the newly recognized neuropathological entity called limbic predominant age related TDP-43 encephalopathy neuropathological change (LATE-NC), which is frequently seen in Lewy body disorders (LBD) and Alzheimer’s disease (AD) (17).
The frequency of TDP-43-associated pathology in LRRK2 PD cases is unclear since most autopsy reports have not included TDP-43 immunostaining. In the literature, five cases with LRRK2 pathogenic variants are described as having TDP-43-positive cytoplasmic inclusions and neurites (7, 8, 18, 19).
In this case series, we examined our collection of LRRK2 brains to better understand the frequency of TDP-43 pathology in cases with a LRRK2 G2019S mutation. We compared brains with a LRRK2 mutation to brains with a pathologic diagnosis of PD or diffuse Lewy body disease (DLBD) without LRRK2 or GBA1 mutations to see if TDP-43 pathology is unique to LRRK2 carriers.
METHODS
Materials and methods
All brains were processed using the standardized protocol as outlined previously (20). In brief, fresh brains were divided by a sagittal cut through the corpus callosum and brainstem; one half brain is processed for banking of fresh-frozen samples for research and the contralateral half is immersed in formalin and processed for neuropathologic evaluation. Determination of PD and Braak neuropathologic stage was performed according to published criteria (21–23).
Histochemical staining protocols
In addition to sections stained with Luxol-fast blue and counterstained with hematoxylin and eosin, sections from selected blocks were separately immunostained for alpha-synuclein and TDP-43 (supplementary table 1) using an automated immunostaining platform that utilizes a 3,3’-diaminobenzidine-based immunostaining method. Hematoxylin counterstains were performed.
TDP-43 immunohistochemical stains were performed on sections of the amygdala, hippocampal formation at the level of the lateral geniculate body and superior frontal cortex (Brodmann area 9) for determination of LATE-NC staging (24). Additionally, TDP-43 immunohistochemical stains were performed on the midbrain.
Clinical information and case selection
All brains were screened as previously described for the LRRK2 G2019S and 10 GBA1 mutations and variants (25, 26). Of the twelve cases available with a LRRK2 G2019S mutation, eleven cases had samples available with TDP-43 staining. The cases without a LRRK2 or GBA1 mutation were selected if they had a pathologic diagnosis of PD or DLBD; cases were selected in a blinded fashion regarding TDP-43 pathology. Cases with and without a LRRK2 mutation were matched to the best of our ability by gender, age of onset and disease duration. Clinical information was obtained by chart review of medical records from Columbia University and outside records, if applicable. All cases were clinically diagnosed with PD by a movement disorders neurologist. Demographics were recorded for each patient reviewed.
Statistical analysis
To determine the significance of TDP-43 deposition in cases with and without a LRRK2 G2019S mutation, Fisher Exact analysis was performed using SPSS version 28.0.
RESULTS
The demographics and clinical characteristics of all the cases reviewed are presented in Table 1. By design, there was no difference between the groups in age, gender and disease duration.
Table 1:
Demographics
| Cases with LRRK2 G2019S mutation | PD cases without LRRK2 or GBA1 mutations | |
|---|---|---|
| Number of cases (n) | 11 | 11 |
| Female gender % (n) | 36% (4) | 45% (5) |
| Parkinsonism age of onset (mean; years-of-age) | 61 ± 11.2 | 56.9 ± 9.4 |
| Mean disease duration (mean; years ± SD) | 18.6 ± 7.5 | 20.8 ± 6.9 |
| Age of death (mean; years of age ± SD) | 79.3 ± 8.6 | 77.6 ± 8 |
TDP-43 extra-nuclear deposits were present in 8 of 11 brains (73%) with a LRRK2 mutation and in 2 of 11 (18%) non-carrier PD brains (p=0.03, Fisher Exact). The deposits in the LRRK2 brains were seen in three different patterns:
TDP-43 dystrophic neurites (DN) in the amygdala were present in all positively stained brains (n = 8), and among them five exclusively in the amygdala (Figure 1A).
Two brains had TDP-43 DN in the substantia nigra (SN) (Figure 1B).
One patient had FTLD Type C with neuronal loss and gliosis primarily in the frontal lobe and aberrant TDP-43 staining mainly in the prefrontal, motor and temporal cortices. Neuronal cytoplasmic inclusions (NCI) were seen in the frontal lobe, hippocampus and amygdala (Figure 1C).
Figure 1 Legend:
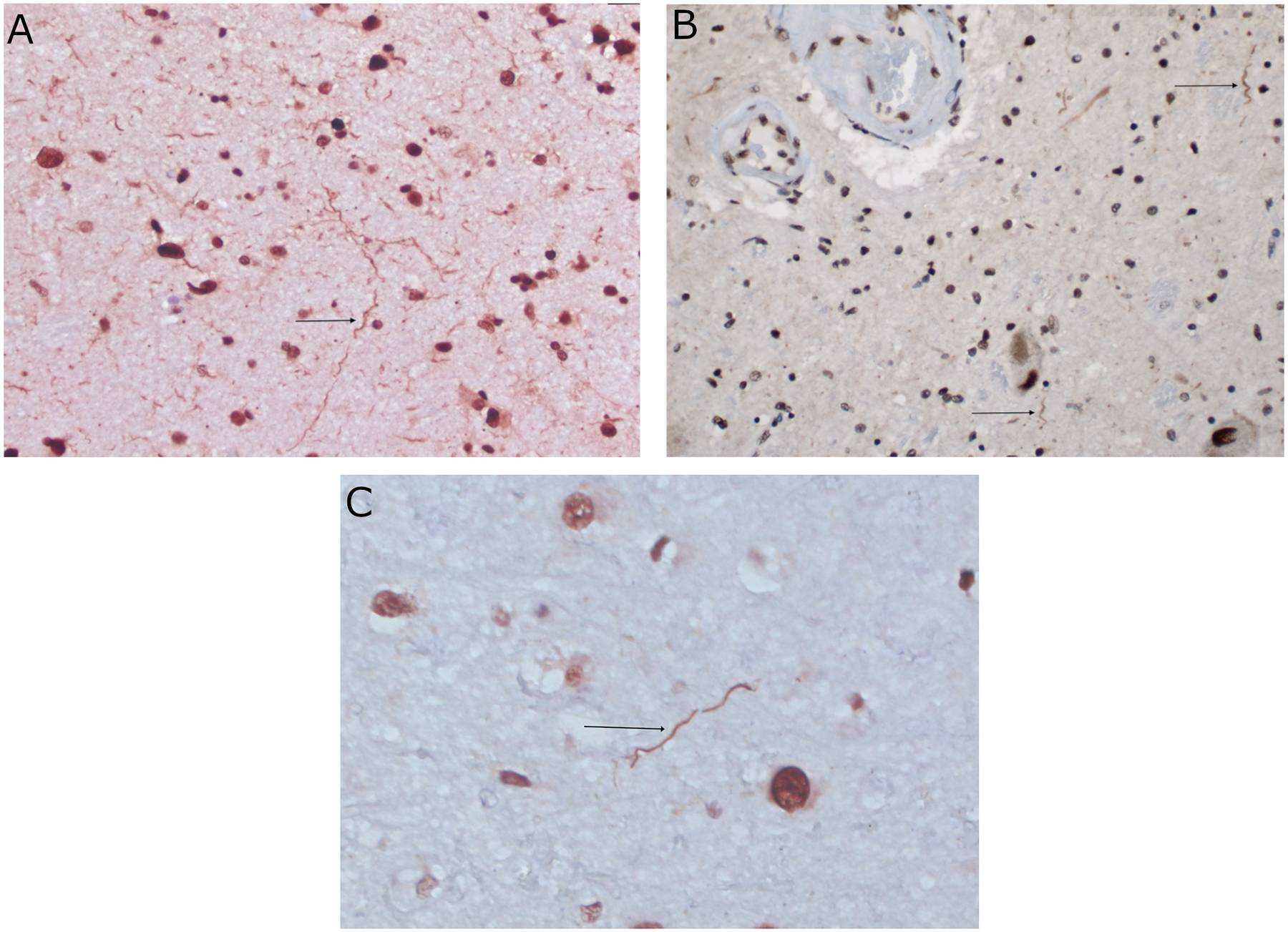
Photomicrographs of immunohistochemical stains against non-phosphorylated TDP-43. A: Arrow pointing to dystrophic neurites in amygdala of case 8. Original magnification 400X; TDP-43. B: Arrows pointing to dystrophic neurites in substantia nigra of case 1. Original magnification 400X; TDP-43. C: Arrow pointing to a long, thick dystrophic neurite in the motor cortex of case 7 with a pathologic diagnosis of FTLD type C. Original magnification 630X; TDP-43.
For the non-carrier brains, one brain had NCI in the hippocampus and the amygdala. Another brain had DN in the frontal cortex, hippocampus and amygdala. None of the non-carrier brains had TDP-43 deposition in the midbrain.
A summary of the distribution of TDP-43, alpha-synuclein, tau and amyloid deposits for cases with a LRRK2 G2019S mutation are summarized in Supplementary Table 2 and non-carrier brains are summarized in Supplementary Table 3.
The patient with FTLD type C clinically had levodopa responsive parkinsonism. This patient had an 18-year clinical course with initial symptoms of slowness of movement and cognitive changes. She was clinically diagnosed with PD and had a significant response to levodopa with motor fluctuations addressed with a dopamine agonist. She had a rest tremor in her left leg, and ultimately developed motor fluctuations with sudden offs that manifested as anxiety, panic, urinary urgency, rigidity and pain. Clinical histories for the LRRK2 cases are summarized in the supplementary material section.
DISCUSSION
In this case series, TDP-43 pathology was assessed in 22 postmortem brains from patients with a clinical diagnosis of parkinsonism and we found that TDP-43 pathology is more common in brains with a LRRK2 mutation. TDP-43 deposits were more common in LRRK2 G2019S brains (73% versus 18%, p= 0.03, Fisher Exact), and among the LRRK2 brains TDP-43 deposits were heterogenous. Specifically, one case had TDP-43 as the primary pathology (FTLD type C), all had DN in the amygdala and two cases had DN in the SN without any NCI. For the cases without a LRRK2 mutation, one case had DN in the amygdala and hippocampus and the other case had NCI in the frontal cortex.
The phenotype of TDP-43 pathology in the SN in the absence of LB in LRRK2 PD may be parkinsonism. The two LRRK2 cases reported in the literature with these pathologic features, and case 2 from our series, had levodopa responsive parkinsonism with resting tremor, bradykinesia and rigidity (6, 19). These patients are clinically indistinguishable from idiopathic PD. None of our cases without a LRRK2 mutation had TDP-43 pathology in the SN. In the literature, one case without any genetic mutations and a clinical history of PD was reported to exclusively have TDP-43 proteinopathy and neuronal loss in the SN (27).
The frequency of TDP-43 pathology reported in PD is variable and is not routinely screened for on autopsy. 18% of our cases without a LRRK2 mutation had TDP-43 pathology. This is similar to what has been reported in autopsy studies where TDP-43 pathology was seen in 7–24% of cases (14, 28). To our knowledge this is the first study to look at the frequency in cases with a LRRK2 mutation where a much higher percentage of cases (73%) had TDP-43 aggregates. Only one additional case in the literature has been described of a patient with FTLD and a LRRK2 G2019S mutation(29). Clinically, the patient had postural and action tremors, but no signs of parkinsonism. It is unclear why these cases had more incident TDP-43 pathology, but LRRK2 mutations may play a role in TDP-43 aggregation.
In conclusion, this is the largest series to date investigating the association between LRRK2 mutations and TDP-43 aggregation. While all participants in the current series were clinically diagnosed with PD, it is estimated that most carriers of the G2019S mutation will never develop PD(3). The heterogeneity of the pathology in brains with LRRK2 mutations may raise the question of the causality of the mutation in the neurodegenerative process; however, the significant increase in frequency of TDP-43 aggregates in LRRK2 cases and the presence of tau in all our cases (regardless of LRRK2 mutation status) supports the causative role it may play specifically for TDP-43 aggregates. Although the other co-pathologies present in all our cases make it challenging to propose a causal role for TDP-43 pathology, it is tempting to postulate that TDP-43 might be pathogenic in our case with FTLD type C where it is the primary pathology. The small size of our cohort with limited clinical history prevents us from making conclusions regarding the clinical phenotype of TDP-43 proteinopathy. Our genetic data is limited to LRRK2 and GBA1 and future work will look into other genetic markers such as TMEM106B and GRN for our FTLD case and APOE for the cases with AD pathology. In this case series, we were limited by having only non-phosphorylated TDP-43 analysis. Given that LRRK2 G2019S is associated with increased kinase activity(30) and given the many phosphorylation sites on TDP-43(31) future studies should compare the level of phosphorylation of TDP-43 in LRRK2 G2019S brains to idiopathic PD. We also suspect that there may be seeding between TDP-43 and synuclein. Colocalization was observed in the autopsy series reported by Uemura and colleagues (17) and future studies would use double immunostaining to evaluate for this in LRRK2 brains. The presence of TDP-43 in LBD underscores the need to screen for it in postmortem brains of patients who have a history of parkinsonism, especially if they have a LRRK2 mutation. Further investigation is needed to better understand the potential biological link between LRRK2 mutations and TDP-43 aggregation.
Supplementary Material
Acknowledgement:
Data on case 6 was available through the AJ-LRRK2 Study, which was funded by the Michael J. Fox Foundation (Marder, PI). The NY Brain Bank is funded by the Parkinson’s Foundation and ADRC.
Funding Sources for study:
Columbia University Parkinson’s Disease Brain Bank is funded by the Parkinson’s Foundation
Financial Disclosures:
R.N.A. is funded by the NIH, DoD, the Parkinson’s Foundation, and the Michael. J. Fox Foundation. He received consultation fees from Avrobio, Caraway, GSK, Merck, Ono Therapeutics, Takeda Pharmaceutical and Genzyme/Sanofi. The other authors declare no competing interests.
Footnotes
Financial Disclosure/Conflict of Interest concerning the research related to the manuscript: None
References
- 1.Healy DG, Falchi M, O’Sullivan SS, Bonifati V, Durr A, Bressman S, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 2008;7(7):583–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schulte C, Gasser T. Genetic basis of Parkinson’s disease: inheritance, penetrance, and expression. Appl Clin Genet. 2011;4:67–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee AJ, Wang Y, Alcalay RN, Mejia-Santana H, Saunders-Pullman R, Bressman S, et al. Penetrance estimate of LRRK2 p.G2019S mutation in individuals of non-Ashkenazi Jewish ancestry. Mov Disord. 2017;32(10):1432–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lesage S, Ibanez P, Lohmann E, Pollak P, Tison F, Tazir M, et al. G2019S LRRK2 mutation in French and North African families with Parkinson’s disease. Ann Neurol. 2005;58(5):784–7. [DOI] [PubMed] [Google Scholar]
- 5.Marder K, Wang Y, Alcalay RN, Mejia-Santana H, Tang MX, Lee A, et al. Age-specific penetrance of LRRK2 G2019S in the Michael J. Fox Ashkenazi Jewish LRRK2 Consortium. Neurology. 2015;85(1):89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601–7. [DOI] [PubMed] [Google Scholar]
- 7.Wider C, Dickson DW, Wszolek ZK. Leucine-rich repeat kinase 2 gene-associated disease: redefining genotype-phenotype correlation. Neurodegener Dis. 2010;7(1–3):175–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ling H, Kara E, Bandopadhyay R, Hardy J, Holton J, Xiromerisiou G, et al. TDP-43 pathology in a patient carrying G2019S LRRK2 mutation and a novel p.Q124E MAPT. Neurobiol Aging. 2013;34(12):2889 e5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao J, Wang L, Huntley ML, Perry G, Wang X. Pathomechanisms of TDP-43 in neurodegeneration. J Neurochem. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain. 2019;142(6):1503–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kwong LK, Neumann M, Sampathu DM, Lee VM, Trojanowski JQ. TDP-43 proteinopathy: the neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta Neuropathol. 2007;114(1):63–70. [DOI] [PubMed] [Google Scholar]
- 12.Dewan R, Chia R, Ding J, Hickman RA, Stein TD, Abramzon Y, et al. Pathogenic Huntingtin Repeat Expansions in Patients with Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Neuron. 2021;109(3):448–60 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hickman RA, Dewan R, Cortes E, Traynor BJ, Marder K, Vonsattel JP. Amyotrophic lateral sclerosis is over-represented in two Huntington’s disease brain bank cohorts: further evidence to support genetic pleiotropy of pathogenic HTT gene expansion. Acta Neuropathol. 2022;143(1):105–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakashima-Yasuda H, Uryu K, Robinson J, Xie SX, Hurtig H, Duda JE, et al. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol. 2007;114(3):221–9. [DOI] [PubMed] [Google Scholar]
- 15.Koga S, Lin WL, Walton RL, Ross OA, Dickson DW. TDP-43 pathology in multiple system atrophy: colocalization of TDP-43 and alpha-synuclein in glial cytoplasmic inclusions. Neuropathol Appl Neurobiol. 2018;44(7):707–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351(3):602–11. [DOI] [PubMed] [Google Scholar]
- 17.Uemura MT, Robinson JL, Cousins KAQ, Tropea TF, Kargilis DC, McBride JD, et al. Distinct characteristics of limbic-predominant age-related TDP-43 encephalopathy in Lewy body disease. Acta Neuropathol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Covy JP, Yuan W, Waxman EA, Hurtig HI, Van Deerlin VM, Giasson BI. Clinical and pathological characteristics of patients with leucine-rich repeat kinase-2 mutations. Mov Disord. 2009;24(1):32–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sakuwa M, Adachi T, Suzuki Y, Yoshida K, Fukuda H, Miura H, et al. First Japanese autopsy case showing LRRK2 mutation G2019S and TDP-43 proteinopathy. Parkinsonism Relat Disord. 2021;91:85–7. [DOI] [PubMed] [Google Scholar]
- 20.Vonsattel JP, Del Amaya MP, Keller CE. Twenty-first century brain banking. Processing brains for research: the Columbia University methods. Acta Neuropathol. 2008;115(5):509–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Braak H, Del Tredici K, Rüb U, De Vos RA, Steur ENJ, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiology of aging. 2003;24(2):197–211. [DOI] [PubMed] [Google Scholar]
- 22.Gelb DJ, Oliver E, Gilman S. Diagnostic Criteria for Parkinson Disease. Archives of Neurology. 1999;56(1):33–9. [DOI] [PubMed] [Google Scholar]
- 23.Dickson DW. Neuropathology of Parkinson disease. Parkinsonism & related disorders. 2018;46:S30–S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain. 2019;142(6):1503–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alcalay RN, Dinur T, Quinn T, Sakanaka K, Levy O, Waters C, et al. Comparison of Parkinson risk in Ashkenazi Jewish patients with Gaucher disease and GBA heterozygotes. JAMA Neurol. 2014;71(6):752–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alcalay RN, Mirelman A, Saunders-Pullman R, Tang MX, Mejia Santana H, Raymond D, et al. Parkinson disease phenotype in Ashkenazi Jews with and without LRRK2 G2019S mutations. Mov Disord. 2013;28(14):1966–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamashita R, Beck G, Yonenobu Y, Inoue K, Mitsutake A, Ishiura H, et al. TDP-43 Proteinopathy Presenting with Typical Symptoms of Parkinson’s Disease. Mov Disord. 2022;37(7):1561–3. [DOI] [PubMed] [Google Scholar]
- 28.Yokota O, Davidson Y, Arai T, Hasegawa M, Akiyama H, Ishizu H, et al. Effect of topographical distribution of alpha-synuclein pathology on TDP-43 accumulation in Lewy body disease. Acta Neuropathol. 2010;120(6):789–801. [DOI] [PubMed] [Google Scholar]
- 29.Dachsel JC, Ross OA, Mata IF, Kachergus J, Toft M, Cannon A, et al. Lrrk2 G2019S substitution in frontotemporal lobar degeneration with ubiquitin-immunoreactive neuronal inclusions. Acta Neuropathol. 2007;113(5):601–6. [DOI] [PubMed] [Google Scholar]
- 30.Kalogeropulou AF, Purlyte E, Tonelli F, Lange SM, Wightman M, Prescott AR, et al. Impact of 100 LRRK2 variants linked to Parkinson’s disease on kinase activity and microtubule binding. Biochem J. 2022;479(17):1759–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eck RJ, Kraemer BC, Liachko NF. Regulation of TDP-43 phosphorylation in aging and disease. Geroscience. 2021;43(4):1605–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.