

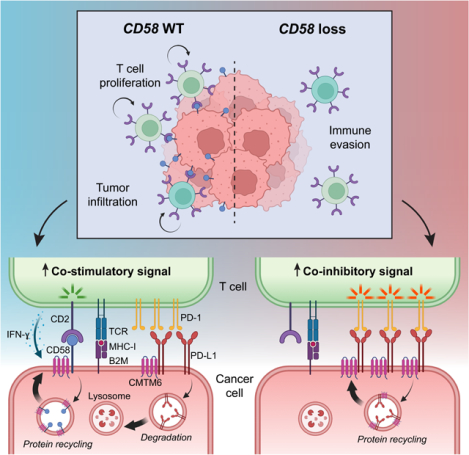
SUMMARY
The cell-autonomous balance of immune-inhibitory and -stimulatory signals is a critical process in cancer immune evasion. Using patient-derived co-cultures, humanized mouse models, and single cell RNA-sequencing of patient melanomas biopsied before and on immune checkpoint blockade, we find that intact cancer cell-intrinsic expression of CD58 and ligation to CD2 is required for anti-tumor immunity and is predictive of treatment response. Defects in this axis promote immune evasion through diminished T cell activation, impaired intratumoral T cell infiltration and proliferation, and concurrently increased PD-L1 protein stabilization. Through CRISPR-Cas9 and proteomics screens, we identify and validate CMTM6 as critical for CD58 stability and upregulation of PD-L1 upon CD58 loss. Competition between CD58 and PD-L1 for CMTM6 binding determines their rate of endosomal recycling over lysosomal degradation. Overall, we describe an underappreciated yet critical axis of cancer immunity and provide a molecular basis for how cancer cells balance immune inhibitory and stimulatory cues.
Graphical Abstract
INTRODUCTION
Immune checkpoint blockade (ICB) therapies, such as anti-PD-1 antibodies, enable CD8+ T cell-mediated anti-tumor immunity and produce durable responses in a portion of patients with metastatic melanoma and other cancers1,2; however, most patients are either intrinsically resistant or acquire resistance after an initial response. While there are a few clinically validated mechanisms of ICB resistance, including genomic alterations that result in impaired antigen presentation or mutations in the IFN-ɣ-JAK-STAT pathway3–6, no clear mechanism of cancer immune evasion is identified in most patients.
Using single-cell RNA-sequencing (scRNA-seq) in melanoma patients and functional screens in patient models, we recently found that cancer cell-autonomous downregulation of CD58 (also known as lymphocyte adhesion factor 3, LFA-3) is associated with resistance to ICB and T cell exclusion7–9; however, the underlying mechanisms are poorly understood. CD58 is physiologically expressed on antigen presenting cells, where it ligates with its receptor CD2 on T cells to deliver “signal 2” following T cell receptor (TCR) and MHC-presented antigen engagement10. CD58 further functions as an adhesion molecule, facilitating the initial binding of effector T cells11–13. CD58 exists in two isoforms via RNA splicing, a glycosylphosphatidylinositol (GPI)-anchored form and a transmembrane (TM) form (hereto referred to as CD58-GPI and CD58-TM, respectively)14–17, though a functional difference between the two isoforms is not well established. The role of CD58 in cancer remains poorly understood, in part due to the lack of a known mouse homolog. Thus, syngeneic models frequently used to study tumor-immune interactions and ICB responses have limited utility in studying the CD58-CD2 axis.
Here, we utilized scRNA-seq analysis of ICB-treated patient samples, in vivo modeling, genetic and protein screening approaches, and biochemical interrogation of CD58 protein interactions to determine the mechanisms by which CD58 loss or downregulation contributes to cancer immune evasion and ICB resistance. We show that intact cancer cell CD58 expression is required for effective T cell mediated anti-tumor responses, and that loss of CD58 confers cancer immune evasion through multiple mechanisms, including impaired activation and proliferation of T cells; reduced tumor lytic capacity, tumor infiltration, and intratumoral T cell expansion; and, interestingly, concurrent upregulation of co-inhibitory PD-L1. We further discovered co-regulation of co-stimulatory (CD58) and co-inhibitory (PD-L1) cancer cell signals by the stabilizing molecule CMTM6. Altogether, this study emphasizes the importance of cancer-cell-intrinsic expression of CD58 for cancer immunity.
RESULTS
Intact cancer cell CD58 expression is necessary for anti-tumor immunity
Prior reports suggest that CD58 downregulation or loss is associated with cancer immune evasion and resistance to ICB in melanoma or CAR T cell therapy in lymphoma7–9,18; however, the underlying mechanisms are poorly understood. Analysis of the Pan-Cancer Atlas indicated that baseline CD58 expression is variable in cutaneous melanoma and virtually absent in uveal melanoma, a rare subtype of melanoma that arises in the eye and has an extremely low response rate to immunotherapies (Fig. S1A), supporting the notion that cancer immune evasion is associated with low CD58 expression. To substantiate these associations in a controlled treatment context, we leveraged scRNA-seq of melanoma patients who underwent matched pre- and early on-ICB-treatment tumor biopsies along with clinical response data (Fig. 1A)19. We determined the strength of gene expression signatures associated with CD58 protein abundance and paucity (up- and downregulated) from our previously published scRNA-/CITE-seq data8 (Table S1). We found higher expression of the CD58 up-signature both at baseline and on-treatment in responders (R) compared to non-responders (NR) (Fig. 1B). In contrast, the CD58 down-signature was lower in responders pre-treatment, and this difference was amplified upon ICB treatment (Fig. S1B). Thus, patient data indicated that cancer-cell-intrinsic expression of CD58 is associated with effective anti-tumor immunity. Analysis of additional patient melanoma scRNA-seq datasets7,20 revealed that the majority of tumor-infiltrating lymphocytes (TILs) maintained high CD2 expression independent of their differentiation state, treatment exposure, or treatment response, suggesting that defects in the CD58-CD2 axis are driven mainly by CD58 loss or downregulation rather than changes in CD2 expression (Fig. S1C–D).
Figure 1. Intact cancer cell CD58 and T cell CD2 signaling is required for anti-tumor immunity.
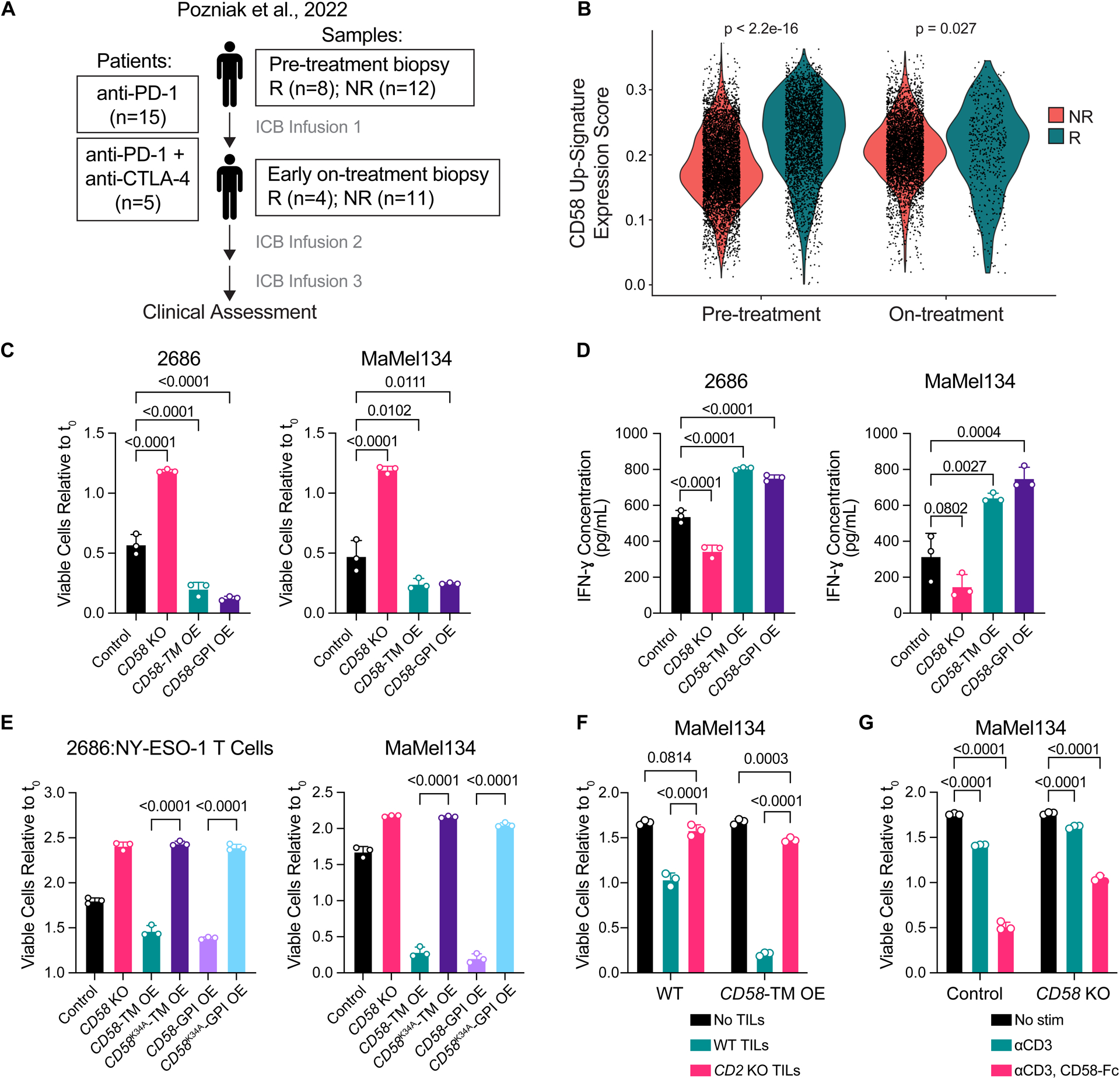
(A) Stage III/IV (AJCC 8th edition) melanoma patients were treated with nivolumab anti-PD-1 ± ipilimumab anti-CTLA-4. Cutaneous, subcutaneous, or lymph node metastases were biopsied prior to initial treatment and 2–3 weeks later shortly prior to the second treatment cycle. Clinical response (R, responders = complete or partial remission; NR, non-responders = stable or progressive disease) was assessed using RECISTv1.1 best overall response criteria.
(B) CD58 up-signature expression scores for malignant cells identified in scRNA-seq analysis of patient melanoma pre- and on-ICB-treatment biopsies from responders versus non-responders. (C-D) Fold change in number of viable 2686 and MaMel134 control, CD58 KO, and CD58-TM or CD58-GPI OE cells after 48 h or 72 h co-culture, respectively, with autologous TILs (C), and IFN-ɣ concentration within cleared media collected from co-cultures (D).
(E) Fold change in number of viable 2686 and MaMel134 control, CD58 KO, CD58-TM OE, CD58K34A-TM OE, CD58-GPI OE, or CD58K34A-GPI OE cells after 48 h co-culture with engineered NY-ESO-1-specific T cells or 72 h with autologous TILs, respectively.
(F) Fold change in number of viable MaMel134 control or CD58-TM OE cells after 48 h co-culture with or without WT or CD2 KO autologous TILs.
(G) MaMel134 TILs were stimulated for 48 h with 1 μg/mL OKT3 +/− 2 μg/mL CD58-Fc chimera prior to co-culturing with autologous melanoma cells; fold change in viable CD58 WT or KO melanoma cells shown after 48 h of co-culture.
Experiments performed in triplicate, with representative experiment shown of at least two independent experiments each (C-G). Statistical analysis performed using Wilcoxon rank sum test (B), one-way ANOVA with Tukey’s multiple comparisons test (C-E), and two-way ANOVA with Tukey’s multiple comparisons test (F,G). Data represent mean ± SD. Top and bottom of violin plots indicate minimum and maximum, respectively, and width represents frequency of values (B).
To mechanistically dissect the role of CD58 cancer cell expression in T cell-mediated tumor lysis, we used patient-derived co-cultures composed of melanoma cells and autologous, ex vivo expanded TILs. We generated otherwise isogenic cell lines with CD58 knockout (KO) followed by rescue with overexpression (OE) of either CD58-TM or CD58-GPI open reading frames (ORFs) (Fig. S2A), followed by TIL co-culture. CD58 loss resulted in significantly reduced tumor lysis and IFN-ɣ production by T cells, while rescue with either CD58-TM or CD58-GPI re-sensitized cancer cells to T cell-mediated tumor killing and rescued IFN-ɣ production (Fig. 1C–D, Fig. S2B).
To exclude the possibility that CD58 loss confers immune evasion through non-specific effects, we engineered human peripheral blood mononuclear cells (PBMCs) to express a T cell receptor (TCR) with high affinity against the common cancer testis antigen NY-ESO-121 (Fig. S2C), which is frequently expressed and presented in melanomas with the HLA-A*2:01 allele, including in one of our cell line models, 2686. In this engineered co-culture system, loss of either CD58 or B2M (which is required for MHC class I antigen presentation) in 2686 cells conferred resistance to NY-ESO-1-TCR T cells (Fig. S2D), thus confirming that the CD58 phenotype was mediated by a specific TCR/epitope interaction. Together, these results show that cancer cell expression of CD58 determines tumor sensitivity by modulating cytotoxic TIL activity in a TCR-specific/epitope-dependent manner.
Cancer cell-intrinsic expression of CD58 promotes T cell responses in a CD2-dependent manner
We next sought to establish that immune modulation by cancer cell autonomous CD58 expression occurs specifically through interaction with CD2 expressed on T cells. While re-expression of CD58-TM or CD58-GPI rescued sensitivity of CD58 KO cells to co-culture with T cells, addition of either a CD2- or CD58- blocking antibody (but not IgG control) abrogated this re-sensitization (Fig. S2E–F). Orthogonally, we used site-directed mutagenesis to create K34A mutated CD58K34A-TM and CD58K34A-GPI ORFs, which show impaired binding to CD222. We rescued CD58 KO cells with CD58WT-TM, CD58WT-GPI, CD58K34A-TM, or CD58K34A-GPI ORFs, followed by co-culture with autologous TILs or NY-ESO-1-TCR T cells (Fig. S2G–H). While overexpression of WT CD58 constructs rescued sensitivity to killing by TILs and IFN-ɣ production, CD58K34A-TM or CD58K34A-GPI ORFs did not (Fig. 1E, Fig. S2I). Next, we used CRISPR-Cas9 to knock out CD2 on TILs (Fig. S2J), and co-cultured WT or CD2 KO TILs with parental or CD58-TM OE cancer cells. Compared to WT TILs, CD2 KO TILs demonstrated minimal cytotoxicity against either parental or CD58-TM OE cells (Fig. 1F).
Next, we considered whether TILs can be stimulated via CD2 to enhance their cytotoxic activity. We used a recombinant CD58-Fc chimera homodimer, which, in combination with CD3 stimulation, was sufficient to increase TIL proliferation and IL-2 production in vitro, and significantly more so than CD3 stimulation alone or in combination with CD28 stimulation (Fig. S2K–L). Notably, cells receiving co-stimulation from the CD58-Fc chimera (compared to TILs activated via CD3 only) more efficiently killed parental cancer cells (as well as CD58 KO cells to a lesser extent, likely due to bystander cytokine activity) (Fig. 1G). We found no expression of CD48, an alternative CD2 ligand23, on multiple melanoma cell lines; thus, CD48 is unlikely to play an important role in this context (Fig. S2M). Together, these results show that an intact CD58-CD2 axis is necessary for efficient TIL-mediated lysis of cancer cells, and that disruption of this specific ligand-receptor interaction confers cancer immune evasion.
Loss of CD58 confers cancer immune evasion via impaired intratumoral T cell infiltration and proliferation
A major impediment to effective anti-tumor immunity and response to ICB is poor or complete lack of tumor infiltration by CD8+ T cells24,25. We therefore sought to examine whether CD58 loss could also impair T cell infiltration of tumors in vivo. Because there is no known mouse homolog of CD58, we established a partly humanized mouse model using NOG mice (NOD.Cg-Prkdcscid II2rgtm1Sug/JicTac) with transgenic expression of human interleukin 2 (hIL2), which is required for T cell survival (Fig. S3A). In these animals, we subcutaneously implanted CD58 WT patient-derived melanoma cells alongside CD58 KO or CD58-TM OE cells as bilateral flank tumors followed by adoptive cell transfer (ACT) with autologous TILs (or PBS control) (Fig 2A, S3B). CD58 KO tumors were highly resistant to ACT (with no responders among eight animals), while half of CD58 WT tumors demonstrated either a complete or partial response to ACT (Fig. 2B–C), suggesting that CD58 loss drives immune evasion in vivo. Interestingly, untreated CD58 KO tumors grew faster than their WT counterparts, which we previously have not observed in vitro8, suggesting additional effects in vivo by which CD58 loss may accelerate tumor growth.
Figure 2. Loss of CD58 confers cancer immune evasion via impaired intratumoral T cell infiltration and proliferation.
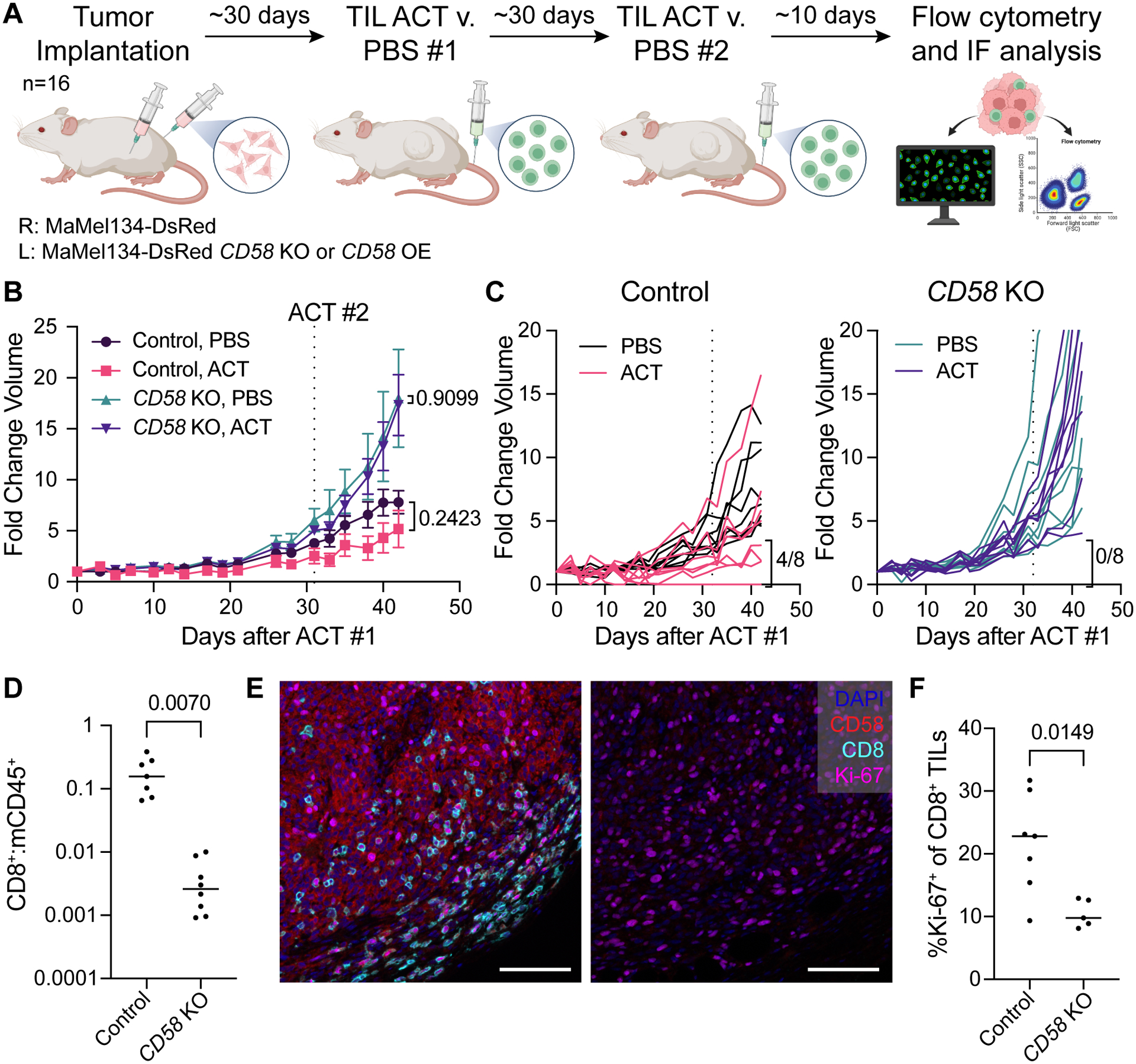
(A) Experimental design of in vivo study of CD58 loss and re-expression in melanoma tumors. MaMel134 NLS-dsRed-expressing parental and CD58 KO or CD58-TM OE melanoma cells were implanted in NOG mice as bilateral subcutaneous flank injections, followed by two treatments with ACT of autologous TILs or PBS control (n=8 per treatment group).
(B-C) Fold change volume of control and CD58 KO tumors following initial ACT treatment or PBS control, with individual tumors shown in (C). For each group, individual tumors with partial or complete response to therapy, defined as <4-fold change in volume from initial ACT treatment to endpoint, are indicated.
(D) Ratio of human CD8+ cells to mouse CD45+ immune cells within ACT-treated control and CD58 KO tumors.
(E) Representative multiplexed immunofluorescence of FFPE tissue sections from mouse tumors staining for DAPI, CD58, CD8, and Ki-67. Scale bar = 100 μm.
(F) Percent of CD8+ TILs within ACT-treated control and CD58 KO tumors that express Ki-67. Statistical analysis performed using unpaired (B – PBS v. ACT) and paired (B – Control v. KO - D, F) two-sided T-tests. Line at median. Data represent mean ± SEM (B).
We analyzed tumors by flow cytometry for T cell infiltration (Fig. S3C–E) and found that CD58 KO tumors showed ~100 fold lower infiltration by CD8+ TILs compared to CD58 WT tumors, which was consistent with multiplexed immunofluorescence (IF) of tissue sections (Fig. 2D, E). We further determined that reduced TIL abundance in CD58 KO tumors was not simply a function of larger tumor size (Fig. S3F). Furthermore, among tumor-infiltrating T cells, those from CD58 KO tumors showed a significantly reduced proliferation rate compared to those from CD58 WT tumors (Fig. 2F). In contrast, in CD58-TM OE tumors, both infiltration and subsequent proliferation of T cells were rescued (Fig. S3G–H).
To further dissect the kinetics of T cell infiltration, we determined T cell abundances in tumor-bearing (CD58 WT or KO) animals shortly after ACT (7 days). We found reduction in both total CD8+ and granzyme B+CD8+ T cell infiltration in CD58 KO tumors at this early time point (Fig. S4A–D). Similarly, tumor-bearing animals treated with a CD58-blocking antibody had reduced T cell infiltration and differentiation (Fig. S4E–I). In timecourse experiments, we immunophenotyped T cells from tumors (WT, CD58 KO or CD58-TM OE) and spleens 5, 12, and 19 days after ACT (Fig. S4J). Consistent with results above, CD58 KO tumors showed scant tumor infiltration by T cells across all time points (Fig. S4K); instead, T cells were primarily found in the spleen (Fig. S4L). In contrast, WT tumors showed progressive infiltration with T cells over time, and CD58-TM rescued tumors showed an initial peak in T cell infiltration on day 12 (Fig. S4K). These kinetics were accompanied by progressive naïve to memory differentiation and an increased fraction of PD-1+CD8+ T cells in WT and CD58-TM OE tumors, but not in CD58 KO tumors, as well as granzyme B maintenance only in T cells from WT tumors (Fig. S4M–P). In contrast, PD-1 and granzyme B expression were not maintained in T cells from spleens of mice with or without xenografts, indicating a tumor-specific response as opposed to an MHC mismatch response (Fig. S4Q–R). Together, these results demonstrate that cancer cell-intrinsic expression of CD58 is critical for early entry of T cells into the tumor and intra-tumoral differentiation, consistent with in silico predictions in ICB-resistant melanoma patients.
Concurrent upregulation of PD-L1 in CD58 loss contributes to cancer immune evasion
We previously noted that CD58 loss is sufficient to promote concurrent upregulation of PD-L18, but the underlying mechanisms and functional role of this co-regulation remain unknown. To address this, we first used CD58 KO cell lines rescued with either CD58-TM or CD58-GPI, followed by IFN-ɣ stimulation and flow-cytometric or total protein evaluation of PD-L1. While CD58 loss resulted in increased PD-L1 surface and total protein abundance, rescue with either CD58 isoform abrogated concurrent PD-L1 increases, even suppressing PD-L1 to below baseline levels (Fig. 3A–C, Fig. S5A–B). CD58 KO cells rescued with K34A-mutated CD58 ORFs were also able to downregulate PD-L1 expression, suggesting that this CD2-binding site is not necessary for CD58/PD-L1 co-regulation. Importantly, cell surface expression of MHC I protein remained unchanged in both CD58 null cells and with rescue of either isoform of CD58 (Fig. 3D, Fig. S5C), indicating that neither resistance nor rescued sensitivity to TIL co-culture (Fig. 1C) were due to altered antigen presentation.
Figure 3. Concurrent upregulation of PD-L1 in CD58 loss contributes to cancer immune evasion.
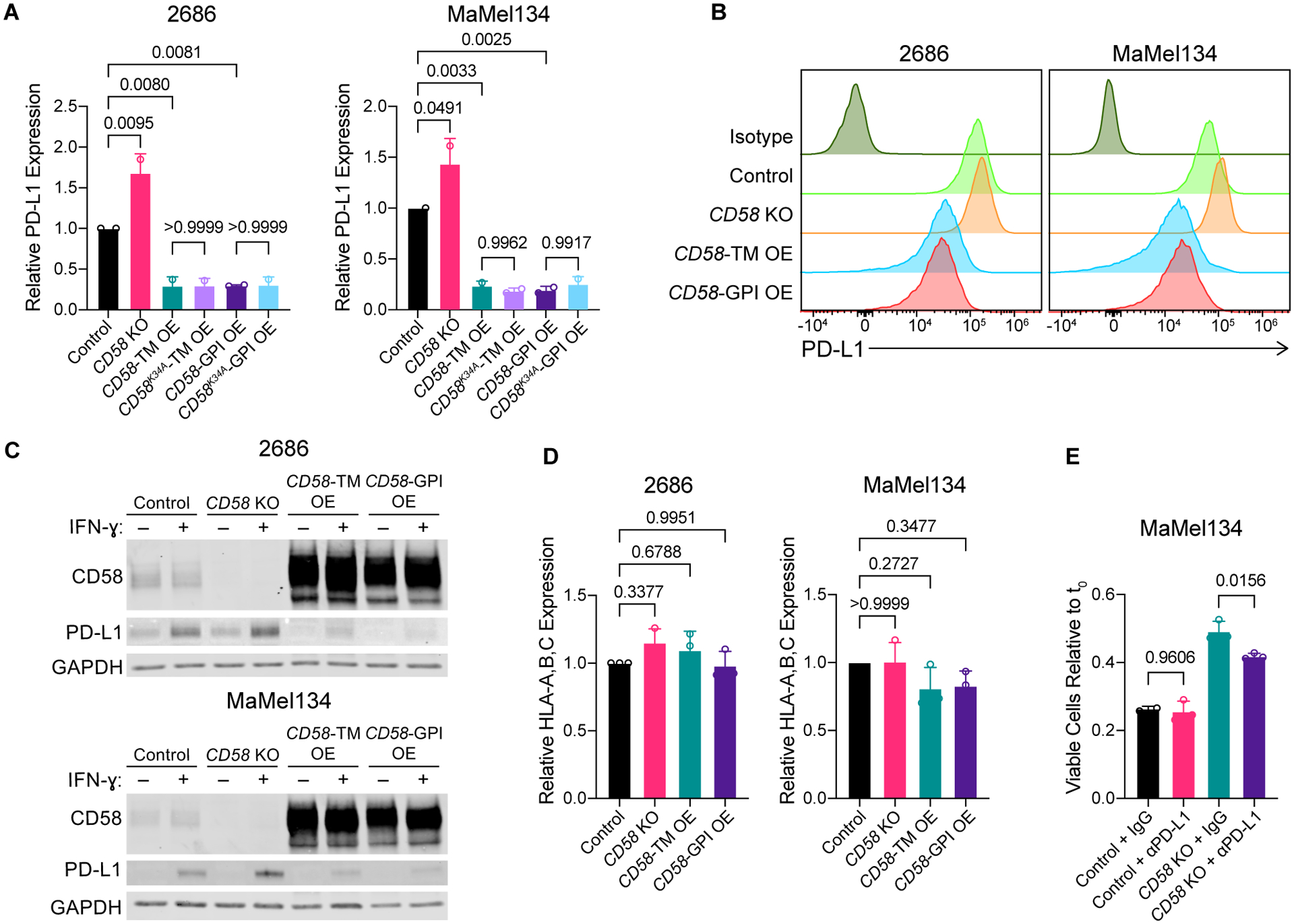
(A-C) Surface (A-B) and whole protein (C) PD-L1 expression in 2686 and MaMel134 control, CD58 KO, CD58-TM, CD58K34A-TM, CD58-GPI, and CD58K34A-GPI OE cells following 72 h stimulation with 10 ng/mL IFN-ɣ assessed by flow cytometry and immunoblotting, respectively.
(D) As in (A), but showing HLA-A,B,C expression.
(E) Fold change in number of viable MaMel134 control and CD58 KO cells after 24 h co-culture with pre-stimulated autologous TILs in the presence of 10 μg/mL anti-PD-L1 (B7-H1) blocking antibody or IgG isotype control. TILs were stimulated overnight with 1 μg/mL anti-CD3 (OKT3) antibody prior to co-culture with melanoma cells.
Experiments performed in duplicate (A, D) or triplicate (E). Independent experiments shown in (A, D) and representative experiment shown from at least two independent experiments in (B, C, E). Statistical analysis performed using one-way ANOVA with Tukey’s multiple comparisons test. Data represent mean ± SD.
See also Figure S5.
To determine whether increased PD-L1 contributes to cancer immune evasion in CD58 loss, we next performed co-culture experiments of CD58 KO cancer cells with or without pharmacological PD-L1 blockade. For this purpose, we stimulated CD8+ autologous TILs using anti-CD3 antibodies in vitro to produce PD-1+ TILs (Fig. S5D). When co-cultured with PD-1+ TILs in the presence of an anti-PD-L1 blocking antibody, CD58 KO melanoma cells demonstrated increased sensitivity to TIL-mediated killing, whereas control cells remained unaffected (Fig. 3E). Thus, elevated PD-L1 expression in CD58 loss contributes to immune evasion, adding to the mechanisms through which CD58 loss or downregulation may contribute to ICB resistance.
Genome-scale CRISPR-Cas9 screen identifies CMTM6 as a positive regulator of CD58
To begin dissecting mechanisms of CD58/PD-L1 co-regulation, we first sought to identify regulators of CD58. First, we performed a genome-wide CRISPR-Cas9 loss-of-function screen combined with fluorescence-activated cell sorting (FACS) (Fig. 4A; Fig. S5E–I)26. The premise of this screen is that perturbations enriched in the cell population with low CD58 protein (CD58lo) may be physiological positive regulators of CD58. Eleven gene targets were consistently enriched within CD58lo cells compared to control middle CD58-expressing (CD58mi) cells (Fig. 4B,C, Fig. S5J, Tables S2,3). As expected, the top “hit” in this screen was knockout of CD58 itself; other expected hits included PIGV and PGAP2, which are both involved in GPI anchor formation, further confirming the robustness of this screen. Interestingly, among the top hits was CMTM6, which has recently been described as an important regulator of PD-L1 maintenance27,28.
Figure 4. Genome-scale CRISPR-Cas9 screen identifies CMTM6 as a positive regulator of CD58.
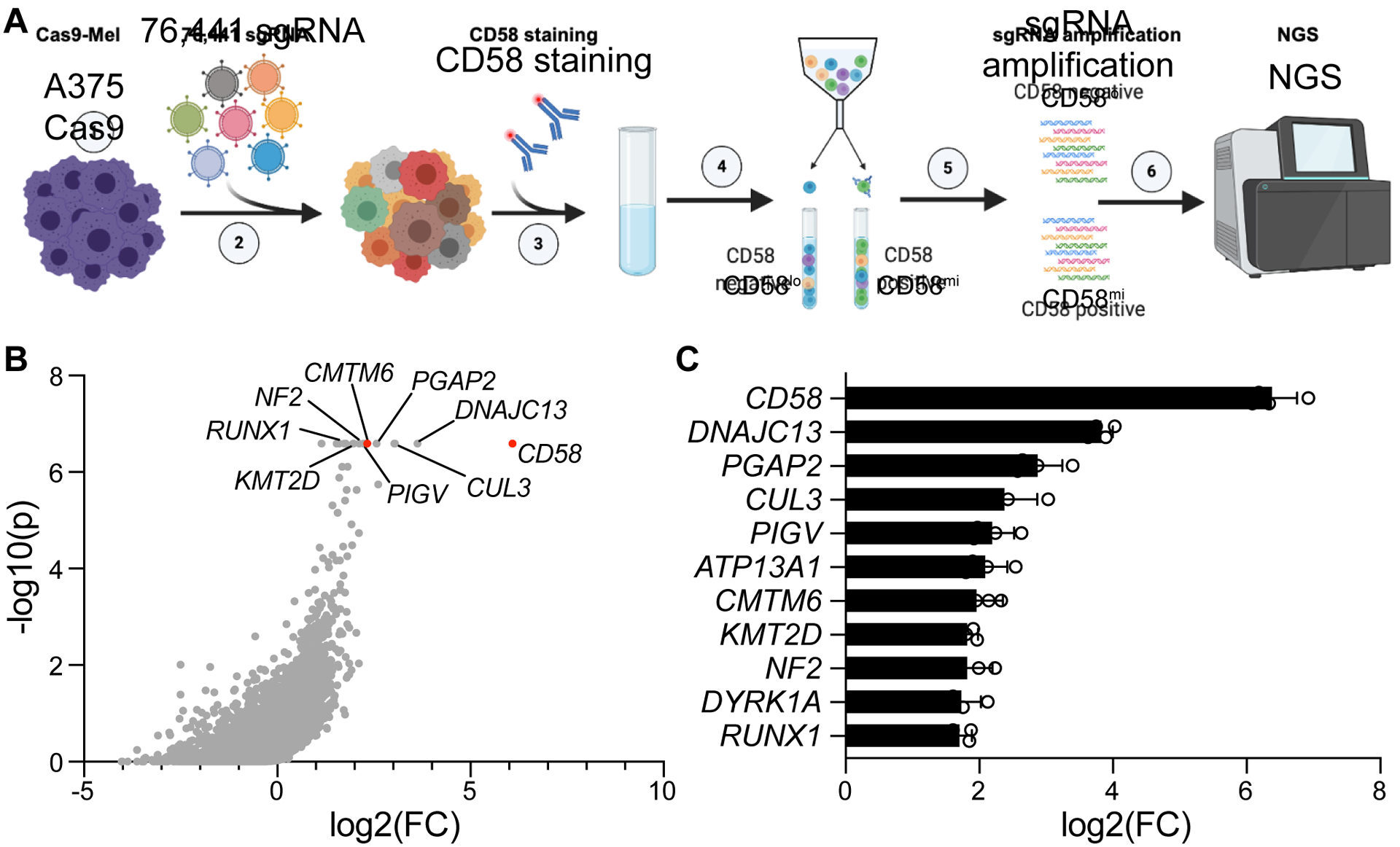
(A) Experimental design of whole genome-wide CRISPR-Cas9 KO screen to identify positive regulators of CD58. Screen was performed independently twice, with two technical replicates each.
(B) Distribution of CRISPR-Cas9 target gene enrichment within the CD58lo population compared to the CD58mi population from an example replicate, showing log2 fold change of gene enrichment and P-value for enrichment calculated from a negative binomial model as determined by the Model-based Analysis of Genome-wide CRISPR-Cas9 Knockout (MAGeCK) algorithm48.
(C) Genes identified as positive regulators of CD58 (criterion of FDR<0.25 across all four replicates), ranked by log2 fold change enrichment of targeting sgRNAs in CD58lo versus control CD58mi population across replicates.
Data represent mean ± SD.
CMTM6 binds to and promotes protein stability of CD58 via endosomal recycling
To complement the functional genetic approach, we also performed mass spectrometry analysis of CD58 immunoprecipitation lysates (IP-MS) to identify binding partners of protein CD58. Across three biological replicates in two different human melanoma cell lines (A375 and 2686), CMTM6 was consistently identified as a top interaction partner of CD58 (Fig. 5A–B, Fig. S6A–B, Table S4). Notably, we did not identify a direct genetic or protein interaction between CD58 and PD-L1 in either the CRISPR-Cas9 or the IP-MS screen, suggesting that their co-regulation is controlled by an alternative mediator. Because CMTM6 was nominated as a regulator of CD58 across screens, and given its involvement in PD-L1 surface protein maintenance, we chose to further investigate its role as such a potential regulator. First, we validated that CMTM6 co-immunoprecipitated with CD58 in protein lysates used for IP-MS (Fig. 5B). Second, we generated CMTM6 KO melanoma cell lines and showed that CMTM6 loss led to decreased CD58 and PD-L1 surface and total protein abundance in multiple models compared to otherwise isogenic controls (Fig. 5C–E, Fig. S6C,D). Rescue of CMTM6 KO with a CMTM6 ORF was sufficient to recover expression of protein CD58 and PD-L1. In contrast, CD58 mRNA levels were unaffected by perturbations of CMTM6, demonstrating that CMTM6 regulates CD58 on a protein level but not a transcript level (Fig. 5F, Fig. S6E).
Figure 5. CMTM6 binds to and promotes protein stability of CD58 via endosomal recycling.
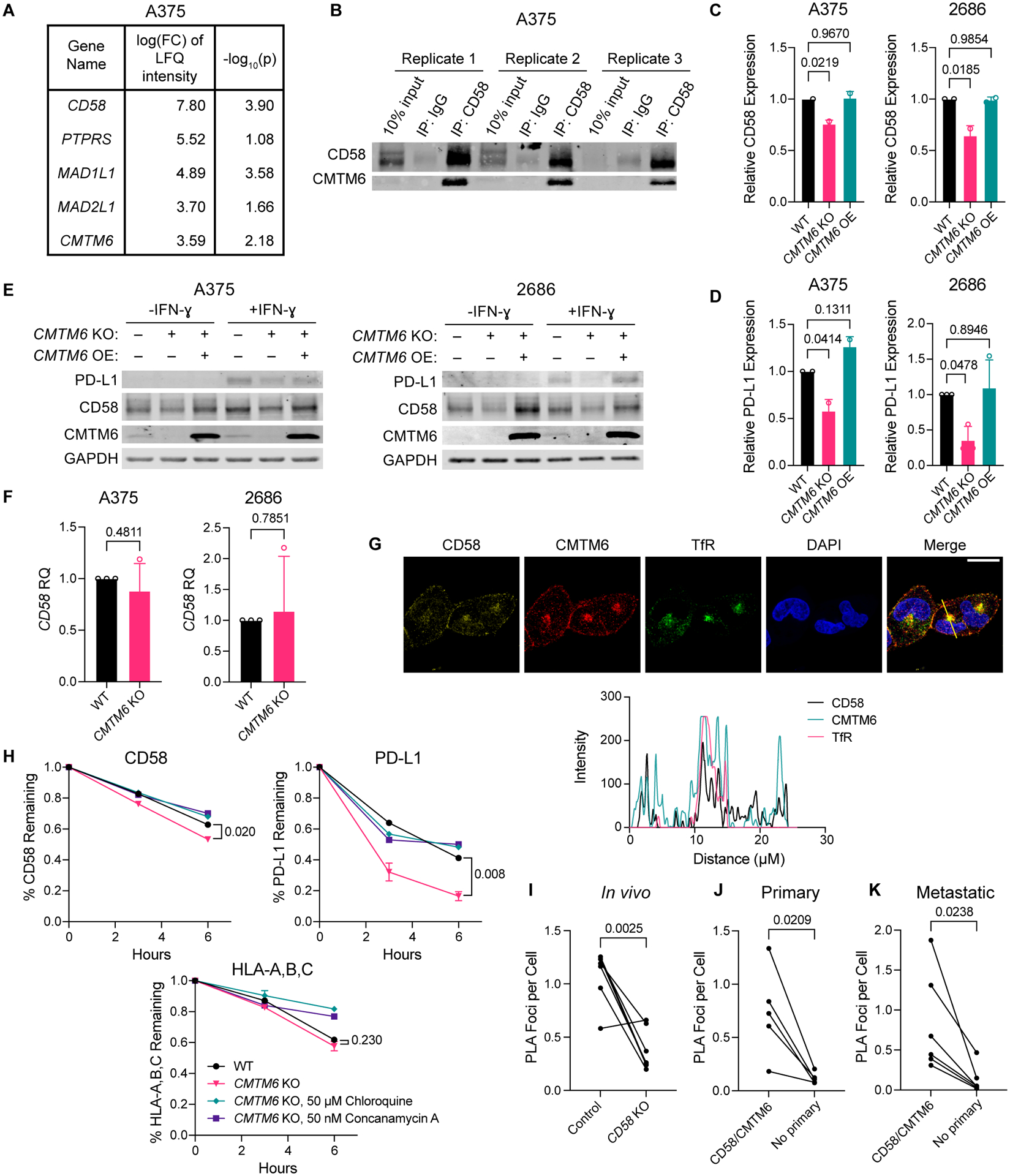
(A) Genes whose encoded proteins were enriched in CD58 IP versus IgG isotype IP lysates from A375 melanoma cells by mass spectrometry analysis. Inclusion criteria: FDR<0.05, log2 FC of LFQ intensity value >3, and an average spectral count within CRAPome database <2 to exclude background contaminants49. Experiment was performed with three biological replicates.
(B) Immunoblotting for CD58 and CMTM6 of A375 CD58 IP lysates used for IP-MS analysis. (C-D) Cell surface expression of CD58 (C) and PD-L1 (D) in A375 and 2686 WT, CMTM6 KO, and CMTM6 OE cells as assessed by flow cytometry.
(E) Immunoblotting for PD-L1, CD58, and CMTM6 in A375 and 2686 WT, CMTM6 KO, and CMTM6 OE cells after 72 h with or without 10 ng/mL IFN-ɣ.
(F) Relative gene expression of CD58 in A375 and 2686 WT or CMTM6 KO cells.
(G) 2686 WT cells were fixed and stained for CD58, CMTM6, and transferrin receptor (TfR), a marker of recycling endosomes, and analyzed by confocal microscopy. Profile plots of relative fluorescence intensity along yellow line shown at bottom. Scale bar = 20 μm.
(H) 2686 WT and CMTM6 KO cells were stained for cell surface CD58, PD-L1, and HLA-A,B,C with fluorophore-conjugated antibodies following 72 h with 10 ng/mL IFN-ɣ stimulation, and then incubated at 37 °C for 3–6 h to allow for recycling of antibody-bound cell surface proteins in the presence or absence of lysosomal inhibitors chloroquine or concanamycin A. Remaining CD58, PD-L1, and HLA-A,B,C expression was assessed by flow cytometry.
(I-K) Number of PLA foci representing CD58/CMTM6 interactions per cell in FFPE sections of CD58 WT versus KO tumors from mice shown in Figure 2A (I; n=7 each) and from patient primary (J; n=5) and metastatic (K; n=6) melanoma samples.
Experiments performed in duplicate, with independent experiments shown (C-D). Representative images shown from two independent experiments (E, G). Experiments performed with four technical replicates, with three independent experiments shown (F). Experiments performed in duplicate, with representative experiment shown from two independent experiments (H). Statistical analysis performed using one-way ANOVA with Tukey’s multiple comparisons test (C, D) and two-sided unpaired (F, H) or paired (I-K) T-tests. Data represent mean ± SD.
Given the role of CMTM6 in shuttling PD-L1 to the recycling endosome, we next investigated the subcellular localization of both CD58 and CMTM6. In IF confocal imaging, CD58 colocalized with CMTM6 both in recycling endosomes (marked by expression of transferrin receptor (TfR)) and the cell membrane (Fig. 5G, Fig. S6F). Furthermore, CMTM6 is known to shuttle PD-L1 to recycling endosomes to prevent its degradation in lysosomes, and so we reasoned that CMTM6 might regulate CD58 similarly. Indeed, in a flow-cytometry based degradation assay, we found that both CD58 and PD-L1, but not MHC class I, were degraded at higher rates in CMTM6 KO compared to isogenic parental cells, and that this degradation could be restricted by lysosomal inhibitors (Fig. 5H). Next, using proximity ligation assays (PLA) in melanoma xenografts and patient tumors (both primary and metastatic melanoma lesions), we showed that CMTM6 and CD58 maintained close physical proximity in situ (Fig. 5I–K, Fig. S6G,H). We therefore propose that, similar to its stabilization of PD-L1, CMTM6 binds to CD58 and promotes its recycling from the cell surface to endosomes, thus preventing its lysosomal degradation. In line with this finding, another positive regulator of CD58 that we identified in our CRISPR-Cas9 KO screen was DNAJC13, whose encoded protein plays a role in mediating membrane trafficking from early to recycling endosomes29.
CMTM6 is necessary for and enables an increase of PD-L1 in cells with CD58 loss
To determine whether CMTM6 is necessary for the concurrent increase in PD-L1 upon CD58 loss, we generated CD58/CMTM6 double knockout (DKO) cell lines (Fig. S7A). While CD58 KO resulted in increased PD-L1 expression, CD58/CMTM6 DKO did not; in contrast, re-expression of CMTM6 in CD58/CMTM6 DKO lines rescued concurrent PD-L1 upregulation (Fig. 6A, Fig. S7B). Additionally, given the role of CMTM6 in regulating both PD-L1 and CD58, we reasoned that rather than interacting directly, PD-L1 and CD58 might undergo co-regulation via CMTM6. To test this hypothesis, we first performed co-IP of CMTM6 or CD58 pulldowns followed by probing for CMTM6, CD58, and PD-L1. While CMTM6 bound to both CD58 and PD-L1 (Fig. 6B), CD58 bound only to CMTM6 and not PD-L1 (Fig. 6C). Furthermore, CD58 loss had no impact on CD274 (PD-L1) mRNA expression, suggesting that CD58/PD-L1 co-regulation occurs on a protein level (Fig. 6D, Fig S7C).
Figure 6: CMTM6 is necessary for and enables an increase of PD-L1 in cells with CD58 loss.
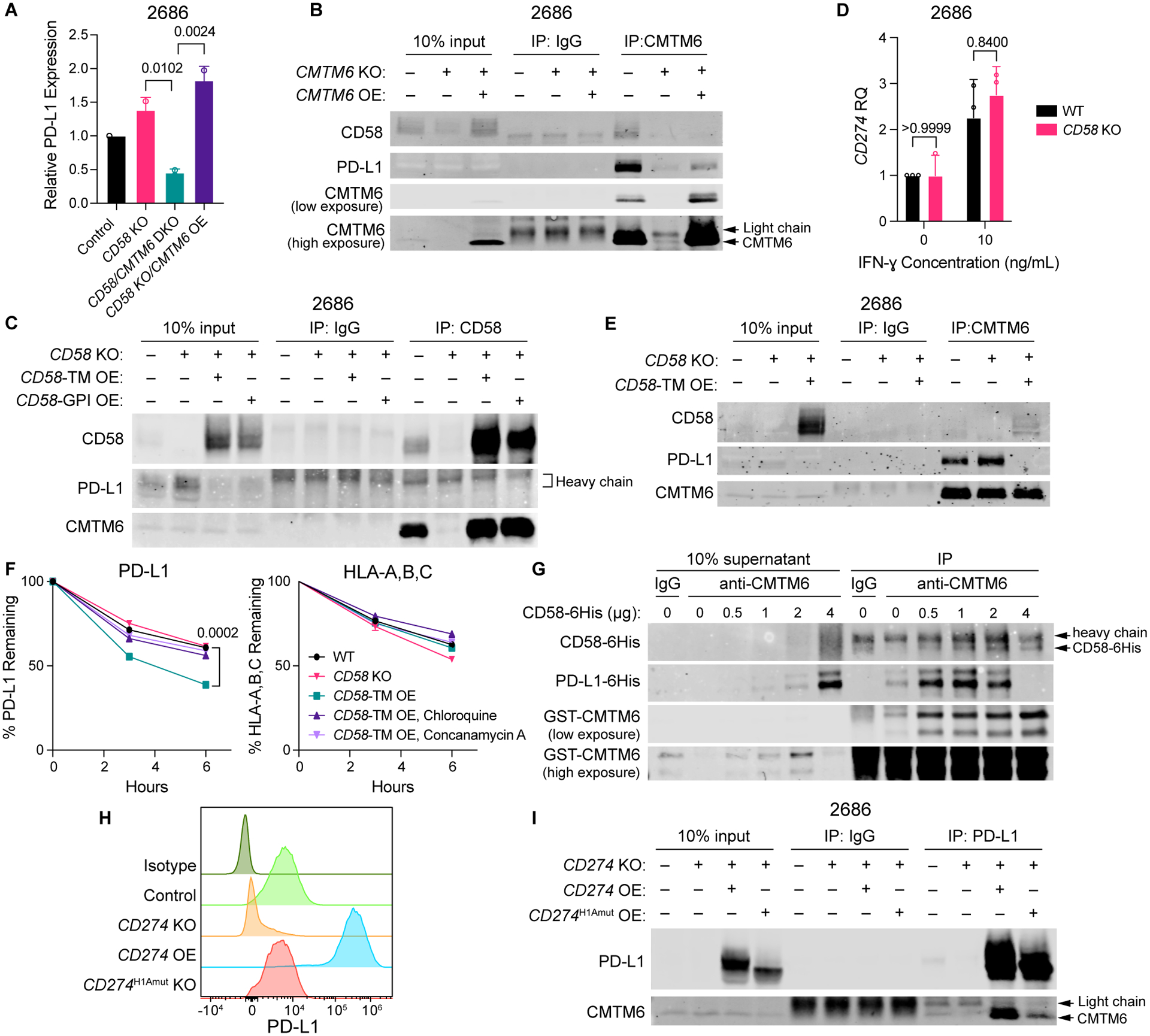
(A) Flow cytometry analysis of surface expression of PD-L1 in 2686 control, CD58 KO, CD58/CMTM6 DKO, and CD58 KO/CMTM6 OE cells after 72 h with 10 ng/mL IFN-ɣ .
(B) Co-IP of PD-L1 and CD58 with CMTM6 pulldown in 2686 WT, CMTM6 KO, and CMTM6 OE cells after 72 h with 10 ng/mL IFN-ɣ.
(C) Co-IP of CMTM6 and PD-L1 with CD58 pulldown in 2686 WT, CD58 KO, CD58-TM, and CD58GPI OE cells after 72 h with 10 ng/mL IFN-ɣ.
(D) Relative gene expression of CD274 in 2686 WT and CD58 KO cells after 72 h with or without 10 ng/ml IFN-ɣ.
(E) Co-IP of CD58 and PD-L1 with CMTM6 pulldown in 2686 WT, CD58 KO, and CD58-TM OE cells after 72 h with 10 ng/mL IFN-ɣ.
(F) Remaining PD-L1 and HLA-A,B,C in 2686 WT, CD58 KO, and CD58-TM OE cells (pre-stimulated with 10 ng/mL IFN-ɣ for 72 h) after 3 and 6 hours of incubation at 37 °C post-staining with fluorophore-conjugated antibodies, in the presence or absence of chloroquine or concanamycin A, as assessed by flow cytometry.
(G) Co-IP of PD-L1–6His and CD58–6His protein with GST-CMTM6 protein from a mixture of 4 μg PD-L1–6His, 1 μg GST-CMTM6, and increasing amounts of CD58–6His purified recombinant proteins.
(H) Surface expression of PD-L1 in 2686 control, CD274 KO, CD274 OE, and CD274H1Amut OE cells after 72 h with 10 ng/mL IFN-ɣ. Counts normalized to mode.
(I) Co-IP of CMTM6 with PD-L1 pulldown in 2686 control, CD274 KO, CD274 OE, and CD274H1Amut OE cells after 72 h 10 ng/mL with IFN-ɣ.
Representative blot shown from two independent experiments (B, C, E, G, I). Experiments performed in duplicate (A, F) or with four technical replicates (D), with independent experiments (A, D) or representative experiment (F) shown. Statistical analysis performed using two-sided T-test (D, F) and one-way ANOVA with Tukey’s multiple comparisons testing (A). Data represent mean ± SD.See also Figure S7.
Interestingly, loss of CD58 increased co-IP of PD-L1 with CMTM6, while CD58-TM OE resulted in reduced CMTM6/PD-L1 binding (Fig. 6E). Thus, we reasoned that CD58 regulates PD-L1 expression by enabling its degradation through competition for CMTM6. Indeed, CD58-TM OE resulted in more rapid PD-L1 protein degradation, which was rescued by addition of lysosomal inhibitors (Fig. 6F). We next tested whether CD58 protein directly interferes with PD-L1-CMTM6 interactions using recombinant polyhistidine-tagged CD58 and PD-L1, and GST-tagged CMTM6 protein. After establishing direct interaction between CD58 and CMTM6, we found that increasing the amount of CD58–6His led to reduced pulldown of PD-L1–6His with GST-CMTM6 (Fig. 6G; Fig. S7D). Together, these results suggest that loss of CD58 results in increased PD-L1 levels by stabilizing CMTM6-PD-L1 Interactions and therefore reducing lysosomal PD-L1 degradation.
Both extracellular loops in the MARVEL domain in CMTM6 are required for binding of CD58 or PD-L1
We sought to identify respective binding sites on CMTM6 for PD-L1 and CD58. A recent study demonstrated that an N-terminal domain (spanning amino acids (aa) 20–32) is necessary for stabilization of PD-L1 at the cell membrane, and that addition of an antibody (clone H1A) targeting this epitope reduces PD-L1 levels by putatively disrupting interactions with CMTM630. We generated cell lines with KO of CD274 (which encodes PD-L1) and rescued these with an ORF encoding either WT CD274 or a mutated form in which the coding region for aa 20–32 was scrambled (CD274H1Amut). Overexpression of CD274H1Amut failed to completely rescue PD-L1 surface or total protein expression (Fig. 6H, S7E–G) and demonstrated impaired binding to CMTM6 (Fig. 6I). Additionally, rescue with WT CD274, but not CD274H1Amut, disrupted CD58-CMTM6 interactions (Fig. S7H,I). Furthermore, increased PD-L1 protein abundance downregulated surface CD58 expression and abrogated CD58/CMTM6 interactions in in vitro protein binding assays (Fig. S7J–K). Together, these data validate the CMTM6-binding region in PD-L1, and further support the notion that competition for CMTM6 by CD58 and PD-L1 is important in the co-regulation of these proteins.
CMTM6 is mainly comprised of a large, conserved MARVEL domain containing four transmembrane helices (Fig. 7A), with N’ and C’ intracellular tails of unknown structure. Because CMTM6 bound to both CD58-TM and -GPI isoforms (Fig. 6C), as well as PD-L1 through interaction with its extracellular domain, we reasoned that the extracellular loops (EC1 and EC2) within the MARVEL domain are essential for CMTM6 interactions with both binding partners. We synthesized several CMTM6 mutant proteins, including single- and double-mutants of EC1 and EC2, which we replaced with homologous sequences from murine CMTM6 that shuttle to the cell membrane, but do not bind human CD58 or PD-L1 (Fig. 7B–C). We rescued CMTM6 KO melanoma cell lines with these mutants (Fig. 7D) and validated that the EC1 and EC2 mutants were properly expressed at the cell surface and recycling endosomes (while the N’ and C’ truncated forms were poorly expressed and trapped in the cytosol) (Fig. 7E). We found that single EC1 and EC2 mutants retained intact binding of CMTM6 to both CD58 and PD-L1, but combined EC1/EC2 double-mutants led to decreased binding of CMTM6 to either protein (Fig. 7F). Thus, the extracellular loops within the MARVEL domain appear to play an important role in binding to PD-L1 and CD58.
Figure 7: Both extracellular loops in the MARVEL domain in CMTM6 are required for binding of CD58 or PD-L1.
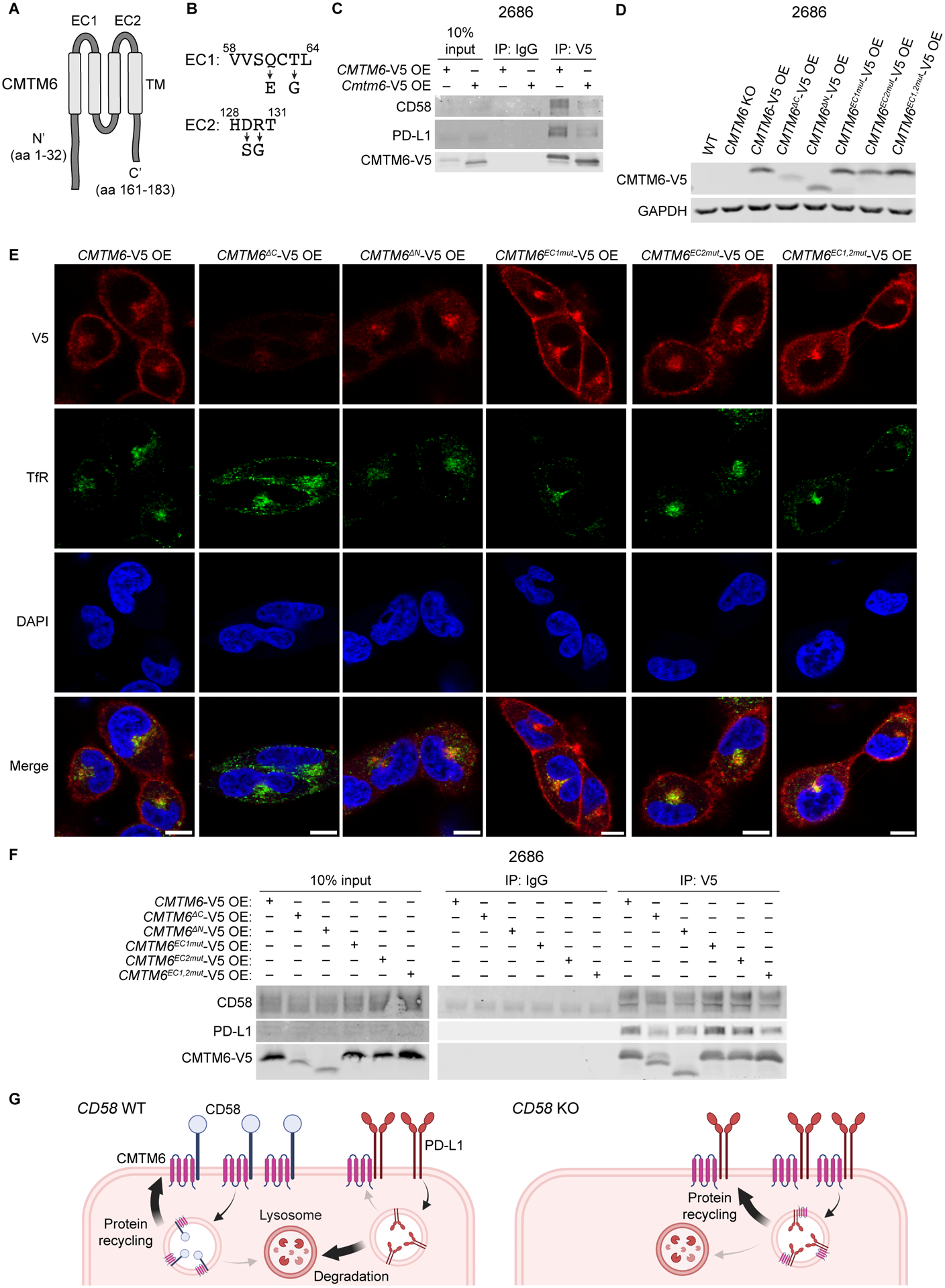
(A) AlphaFold prediction for human CMTM6 protein structure includes transmembrane (TM) MARVEL domain, two extracellular (EC) loops, and intracellular N’ and C’ tails50,51.
(B) Altered amino acid sequences within extracellular loops of CMTM6 for EC1 and EC2 mutants derived from homologous regions in murine CMTM6.
(C) Co-IP of PD-L1 and CD58 with human or mouse CMTM6 using anti-V5 IP in 2686 CMTM6-V5 OE and Cmtm6-V5 OE cells after 72h with 10 ng/mL IFN-ɣ.
(D) Immunoblotting for V5-tagged protein in 2686 WT, CMTM6 KO, CMTM6-V5 OE, and shown CMTM6 mutant, V5-tagged protein-expressing cells.
(E) 2686 V5-tagged CMTM6 WT and mutant-expressing cells were fixed and stained for V5 and transferrin receptor (TfR) and analyzed by confocal microscopy. Scale bars = 10 μm.
(F) Co-IP of PD-L1 and CD58 with V5-tagged protein in 2686 V5-tagged CMTM6 WT and mutant-expressing cells after 72h with 10 ng/mL IFN-ɣ.
(G) Proposed model of PD-L1 regulation by CD58.
Representative images shown from two independent experiments (C, E, F).
Together, these data support a model in which both CD58 and PD-L1 require and directly bind to CMTM6, likely via its extracellular loops. When CD58 expression is lost, additional PD-L1 protein binds to released CMTM6 and is stabilized at the cell surface through increased recycling and reduced lysosomal degradation (Fig. 7G).
DISCUSSION
Here, we find that an intact CD58-CD2 axis is necessary for effective anti-tumor immunity, and that its absence results in immune resistance via multiple mechanisms, including impaired T cell activation, inhibited T cell entry and proliferation within tumors, and concurrent upregulation of PD-L1. We demonstrate that these mechanisms are orthogonal to clinically validated mechanisms of resistance to immunotherapies, such as impaired antigen presentation4,5 or IFN-ɣ-JAK-STAT signaling3,6. CD58 loss has previously been implicated in immune escape in hematological malignancies31–34 and has recently been identified as a potential mechanism of resistance to CD19-targeting CAR T cells in patients with diffuse large B cell lymphoma (DLBCL)18. Together, this evidence suggests that cancer-cell-intrinsic expression of CD58 may play an important role in cancer immune evasion across different lineages and types of immunotherapies; thus, our study provides important mechanistic insights into the underpinnings of this axis.
We show that CD58 is crucial for infiltration of tumors by T cells, likely via its adhesive properties when ligated with CD2, and that increased CD58 co-stimulatory signaling via CD2 can enhance T cell activation and cytotoxicity. Our findings are consistent with recent work indicating that CD58-CD2 interactions amplify T cell signaling by forming the outer edge “corolla” of the immune synapse, and that corolla formation is highly sensitive to CD58 density35. Co-stimulatory signals have also recently been shown to be necessary for expansion and effector differentiation of stem-like CD8+ TILs36. Furthermore, while CD2 and CD28 co-stimulation are comparable in their ability to promote proliferation, cytokine production, and effector differentiation in naïve T cells, CD2 stimulation more efficiently maintains a pool of progenitor-like T cells that are important for response to PD-1 blockade37–40. We show that the majority of TILs in patients with melanoma maintain CD2 expression. Thus, selective restoration of this axis within the tumor microenvironment may serve as a promising therapeutic approach in tumors that acquire immune resistance through cancer cell-intrinsic CD58 downregulation. For example, a recent study found that engineered CAR T cells coupled with a CD2 receptor may overcome resistance due to CD58 loss in an in vivo model of DLBCL18. Additionally, in solid tumors, trispecific antibodies were recently shown to be effective in targeting CD28+ T cells against HER2+ breast cancer in vivo41; similar antibodies might be engineered to instead stimulate T cells via CD2 in a targeted manner.
How cancer cells balance potential co-stimulatory signals – which in certain contexts may confer a fitness advantage42 – and co-inhibitory signals remains obscure, but is fundamental to our understanding of tumor-immune interactions and the efficacy of cancer immunotherapies. Here, we show that loss of CD58 results in increased PD-L1 protein abundance, thus leading to a dual setback to T cell mediated anti-tumor immunity: lack of stimulation by CD58 and increased inhibition by PD-L1. Through unbiased CRISPR-Cas9 KO and IP-MS screens, we identify CMTM6, previously shown to be required for PD-L1 maintenance27,28, as a key regulator of protein CD58. Our data support a model wherein CD58 and PD-L1 compete for CMTM6 for protein maintenance in the cell membrane and recycling endosomes instead of lysosomal degradation, resulting in upregulation of PD-L1 in the context of CD58 loss. Interestingly, PD-1-PD-L1 engagement has itself been shown to be localized at the corolla formed by CD58-CD2 ligation in the immune synapse in model systems, thereby decreasing CD2 signal amplification in T cells35. Thus, CD58’s regulation of PD-L1 may further support the maintenance of intact CD58-CD2 axis signaling, as it prevents buffering of this interaction by PD-1-PD-L1 ligation.
In patients with melanoma, non-small cell lung cancer, and colorectal cancer, CMTM6 protein abundance has been associated with increased T cell abundance in tumor biopsies43–45, but its role as a predictive biomarker remains unclear46, likely due to its dual role in co-regulating PD-L1 and CD58 demonstrated here. While CMTM6’s protein regulation of CD58 cannot explain the transcriptional downregulation of CD58 that we observe in ICB-resistant patient melanomas, CMTM6 itself may be a target for therapeutically manipulating co-stimulatory/inhibitory signals. Thus, dissecting the specific interactions between CD58 and PD-L1 with CMTM6 is essential for the development of such therapeutics. Importantly, we show that CMTM6’s two extracellular loops are required for binding to both CD58 and PD-L1, while on PD-L1, a short N-terminal, extracellular domain is necessary for binding to CMTM6. Antibodies targeting these domains – for example, H1A on PD-L130 – therefore represent a potential therapeutic opportunity to destabilize PD-L1. Furthermore, therapeutic restoration of CD58 may be tractable, as our human data suggest that tumors with resistance to ICB exhibit downregulation rather than genetic loss of CD58. However, further work is needed to uncover the structure of CMTM6 and define its exact binding to CD58 and PD-L1 in order to better inform therapeutic design.
Our work provides a mechanistic basis for how co-inhibitory and co-stimulatory proteins are co-regulated in cancer. While we focused on the interaction of CD58 and PD-L1, this raises the broader conceptual question of how perturbation (pharmacological or genetic) of one checkpoint influences that of others in the same cell. Systematic approaches, such as combinatorial Perturb-CITE-seq of human models8, may provide important insights and inform rationale for the next generation of combinations of cancer immunotherapies.
In summary, our work contributes three major advances. First, we identify a central and necessary function of the CD58-CD2 axis in anti-tumor immunity and demonstrate that defects in this interaction confer resistance to T cell-mediated killing by multiple mechanisms. Second, we find that CMTM6 is a critical regulator of the yin and yang of co-stimulatory and co-inhibitory cues by cancer cells. Third, we present the molecular basis for clinically relevant roles of CD58 loss in conferring resistance to ICB, which may pave the way for rational development of therapies overcoming this resistance mechanism.
Limitations of the Study
We have shown across multiple patient melanoma cohorts treated with ICB that CD58 loss or downregulation is associated with treatment resistance. However, we have focused on the regulation of CD58 only at a protein level by CMTM6. In both our own genetic screening and in a study of B cell acute lymphoblastic leukemia47, the transcription factor RUNX1 was identified as a positive regulator of CD58. Further research into RUNX1 and other potential transcriptional regulators of CD58 may help further inform our understanding of how CD58 expression is lost in melanoma in the context of immune evasion and ICB resistance. Additionally, while we have shown that CD58 is essential for tumor infiltration by CD8+ T cells in vivo, our experiments do not explain why intratumoral T cell numbers are lower in CD58 KO tumors. CD58 may either facilitate initial infiltration into the tumor, or expansion of T cell populations once in the tumor, or some combination of the two.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Benjamin Izar (bi2175@cumc.columbia.edu).
Materials availability
Materials generated in this study are available upon request to the lead contact
Data and code availability
CRISPR-Cas9 KO screen data has been deposited at GEO and is publicly available as of the date of publication. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository52 and is publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper analyzes existing, publicly available data. The accession numbers for these datasets are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal mouse anti-ARF6 AF488 (clone 3A-1) | Santa Cruz Biotechnology | Cat#sc-7971 AF488 |
| Monoclonal mouse anti-Calnexin AF488 (clone AF18) | ThermoFisher Scientific | Cat#MA3–027-A488 |
| Monoclonal mouse anti-GM130 AF488 (clone 35/GM130) | BD Biosciences | Cat#560257 |
| Monoclonal mouse anti-TfR AF488 (clone #29806) | R&D Systems | Cat#FAB2474G-100UG |
| Polyclonal rabbit anti-TGN46 AF488 | Novus Biologicals | Cat#NBP1–49643AF488 |
| Polyclonal sheep anti-EEA1 AF488 | R&D Systems | Cat#IC8047G |
| Polyclonal sheep anti-LAMP1 AF488 | R&D Systems | Cat#IC7985G |
| Polyclonal goat anti-Mouse IgG (H+L) AF594 | ThermoFisher Scientific | Cat#A-11032 |
| Polyclonal goat anti-Rabbit (gG (H+L) AF647 | ThermoFisher Scientific | Cat#A-21245 |
| Monoclonal mouse anti-CD8 (clone 4B11) | Leica | Cat#CD8–4B11-L-CE |
| Monoclonal mouse anti-CD58 (clone TS2/9) | ThermoFisher Scientific | Cat#MA5800 |
| Monoclonal mouse anti-Ki-67 (MIB-1) | Nordic MUbio | Cat#KI500 |
| Monoclonal anti-mouse CD16/32 (clone 93) | Biolegend | Cat#101302 |
| Monoclonal mouse anti-CD2 BV605 (clone RPA-2.10) | Biolegend | Cat#300223 |
| Monoclonal mouse anti-CD279 (PD-1) APC (clone EH12.2H7) | Biolegend | Cat#329908 |
| Monoclonal mouse anti-CD3 PE-Cy7 (clone HIT3a) | Biolegend | Cat#300316 |
| Monoclonal mouse anti-CD4 AF700 (clone RPA-T4) | Biolegend | Cat#300526 |
| Monoclonal mouse anti-CD45RA Brilliant Violet 711 (clone HI100) | Biolegend | Cat#304137 |
| Monoclonal mouse anti-CD45RO Brilliant Violet 570 (clone UCHL1) | Biolegend | Cat#304226 |
| Monoclonal mouse anti-CD48 FITC (clone BJ40) | Biolegend | Cat#336705 |
| Monoclonal mouse anti-CD58 APC (clone TS2/9) | Biolegend | Cat#330918 |
| Monoclonal mouse anti-CD58 BV421 (clone 1C3) | BD Biosciences | Cat#564363 |
| Monoclonal mouse anti-CD58 PE-Cy5 (clone TS2/9) | Biolegend | Cat#330909 |
| Monoclonal mouse anti-CD8 AF488 (clone HIT8a) | Biolegend | Cat#300916 |
| Monoclonal mouse anti-Granzyme B PE-CF594 (clone GB11) | BD Biosciences | Cat#562462 |
| Monoclonal mouse anti-human CD45 Pacific Blue (clone HI30) | Biolegend | Cat#304022 |
| Monoclonal mouse anti-HLA A,B,C BV605 (clone W6/32) | Biolegend | Cat#311432 |
| Monoclonal mouse anti-PD-L1 APC (clone 29E.2A3) | Biolegend | Cat#329708 |
| Monoclonal mouse anti-PD-L1 BV785 (clone 29E.2A3) | Biolegend | Cat#329736 |
| Monoclonal rat anti-Ki-67 eFluor506 (clone SolA15) | ThermoFisher Scientific | Cat#69-5698-82 |
| Monoclonal rat anti-mouse CD45 BV510 (clone 30-F11) | Biolegend | Cat#103138 |
| Monoclonal mouse anti-CD58 (clone TS2/9) | Biolegend | Cat#330923 |
| Monoclonal rat anti-CD2 (LO-CD2b) | Laboratory of Megan Sykes | n/a |
| Monoclonal mouse anti-CD28 (clone CD28.2) | ThermoFisher Scientific | Cat#16-0289-81 |
| Monoclonal mouse anti-CD3 (clone OKT3) | Miltenyi Biotec | Cat#130-093-387 |
| Monoclonal mouse anti-PD-L1 (clone 29E.2A3) | Bio X Cell | Cat#BE0285R001MG |
| Mouse IgG2b isotype control (clone MPC-11) | Bio X Cell | Cat#BE0086R001MG |
| Mouse IgG4, κ isotype control (clone QA16A15) | Biolegend | Cat#403701 |
| Rat IgG2b negative control (clone LTF-2) | EMD Millipore | Cat#MABF1076Z |
| APC Mouse IgG1, κ Isotype Ctrl Antibody (clone MOPC-21) | Biolegend | Cat#981806 |
| APC Mouse IgG2b, κ Isotype Ctrl Antibody (clone MPC-11) | Biolegend | Cat#400319 |
| Brilliant Violet 421™ Mouse IgG2a, κ Isotype Ctrl Antibody (clone MOPC-173) | Biolegend | Cat#400259 |
| Brilliant Violet 605™ Mouse IgG2a, κ Isotype Ctrl Antibody (clone MOPC-173) | Biolegend | Cat#400269 |
| Brilliant Violet 785™ Mouse IgG2b, κ Isotype Ctrl Antibody (clone MPC-11) | Biolegend | Cat#400355 |
| FITC Mouse IgG1, κ Isotype Ctrl (clone MOPC-21) | Biolegend | Cat#981802 |
| Monoclonal mouse anti-CD58 (clone TS2/9) | Abcam | Cat#ab171087 |
| Monoclonal mouse anti-His (clone 27E8) | Cell Signaling Technology | Cat#2366 |
| Monoclonal mouse anti-PD-L1 (clone 405.9A11) | Cell Signaling Technology | Cat#29122 |
| Polyclonal rabbit anti-CMTM6 | MilliporeSigma | Cat#HPA026980 |
| Polyclonal rabbit anti-CMTM6 | Invitrogen | Cat#PA5–55472 |
| Monoclonal rabbit anti-PD-L1 (clone E1L3N) | Cell Signaling Technology | Cat#13684 |
| Monoclonal rabbit anti-V5 (clone D3H8Q) | Cell Signaling Technology | Cat#13202 |
| Monoclonal rabbit anti-CD58 (clone EP15041) | Abcam | Cat#ab196648 |
| Monoclonal rabbit anti-GAPDH (clone 14C10) | Cell Signaling Technology | Cat#2118 |
| IRDye 680RD Goat anti-Mouse IgG (H+L) | Li-Cor | Cat#926–68070 |
| IRDye 680RD Goat anti-Rabbit IgG (H+L) | Li-Cor | Cat#926–68071 |
| IRDye 800CW Goat anti-Mouse IgG (H+L) | Li-Cor | Cat#926–32210 |
| Bacterial and virus strains | ||
| Human CRISPR Knockout Pooled Library (Brunello) | Doench et al.26 | Addgene Virus #73178-LV |
| Chemicals, peptides, and recombinant proteins | ||
| Accutase | Innovative Cell Technologies | Cat#AT104 |
| ACK Lysing Buffer | Quality Biological | Cat#118-156-101 |
| BOND Epitope Retrieval Solution 1 | Leica Biosystems | Cat#AR9961 |
| BONE Epitope Retrieval Solution 2 | Leica Biosystems | Cat#AR9640 |
| Cas9-NLS purified protein | QB3 MacroLab | N/A |
| CellEvent Caspase-3/7 Detection Reagent | ThermoFisher Scientific | Cat#C10423 |
| CFSE | ThermoFisher Scientific | Cat#C34570 |
| Chloroquine diphosphate | Sigma Aldrich | Cat#C6628–25G |
| Collagenase D | Sigma Aldrich | Cat#11088866001 |
| Concanamycin A | Santa Cruz Biotechnology | Cat#sc-202111 |
| Digitonin | Sigma-Aldrich | Cat#D141 |
| DNase I | Sigma Aldrich | Cat#10104159001 |
| Ex Taq DNA Polymerase | TaKaRa | Cat#RR001C |
| Gateway BP Clonase II Enzyme Mix | ThermoFisher Scientific | Cat#11789020 |
| Gateway LR Clonase iI Enzyme Mix | ThermoFisher Scientific | Cat#11791020 |
| Halt Protease and Phosphatase Inhibitor Cocktail | ThermoFisher Scientific | Cat#76445 |
| Human IL-2 | Chiron | Cat#53905-991-01 |
| Insulin-Transferrin-Selenium | ThermoFisher Scientific | Cat#41400–045 |
| Laemmli SDS-Sample Buffer | Boston BioProducts | Cat#BP-111R |
| Matrigel Basement Membrane Matrix, LDEV Free | Corning | Cat#354234 |
| Matrigel Matrix High Concentration, Phenol-Red Free, LDEVfree | Corning | Cat#354262 |
| Opti-MEM I Reduced Serum Medium | ThermoFisher Scientific | Cat#31985062 |
| Polybrene Infection/Transfection Reagent | Sigma-Aldirch | Cat#TR-1003 |
| Recombinant CD58 His-tag protein | VWR | Cat#10797–676 |
| Recombinant CD58/LFA-3 Fc chimera protein | R&D Systems | Cat#10068-CD-050 |
| Recombinant human CMTM6 GST (N-term) protein | Novus Biologicals | Cat#H00054918-P01 |
| Recombinant human IFN-gamma | Abcam | Cat#ab9659 |
| Recombinant human PD-L1/B7-H1 His-tag protein | R&D Systems | Cat#9049-B7–100 |
| RIPA buffer | Sigma Aldrich | Cat#R0278 |
| TaqMan Fast Advanced Master Mix | ThermoFisher Scientific | Cat#4444557 |
| TransIT-LT1 Transfection Reagent | Mirus Bio | Cat#MIR 2300 |
| Zombie NIR | Biolegend | Cat#423105 |
| Zombie Violet | Biolegend | Cat#423113 |
| Critical commercial assays | ||
| Duolink In Situ Detection Reagents FarRed | Sigma-Aldrich | Cat#DUO92013 |
| IFN-gamma ELISA kit | Biolegend | Cat#430104 |
| IL-2 ELISA kit | Biolegend | Cat#431804 |
| QuikChange XL Site-Directed Mutagenesis Kit | Agilent | Cat#200516 |
| SF Cell Line Kit 4D X Kit | Lonza | Cat#197174 |
| Opal 6-Plex Detection Kit - for Whole Slide Imaging | Akoya Biosciences | Cat#NEL871001KT |
| ACTB TaqMan Gene Expression Assay | ThermoFisher Scientific | Assay ID: Hs99999903_m1 |
| CD274 TaqMan Gene Expression Assay | ThermoFisher Scientific | Assay ID: Hs00204257_m1 |
| CD58 TaqMan Gene Expression Assay | ThermoFisher Scientific | Assay ID: Hs01560660_m1 |
| GAPDH TaqMan Gene Expression Assay | ThermoFisher Scientific | Assay ID: Hs02786624 |
| Deposited data | ||
| A375 CD58 CRISPR/Cas9 KO screen data | This paper | GEO: GSE225235 |
| Melanoma scRNA-seq | Sade-Feldman et al.20 | GEO: GSE120575 |
| Melanoma scRNA-seq | Jerby-Arnon et al.7 | GEO: GSE115978 |
| Melanoma scRNA-seq | Pozniak et al.19 | EGA: EGAS00001006488 |
| A375 and 2686 CD58 IP mass spectrometry data | This paper | Proteome Xchange: PXD039710 |
| Experimental models: Cell lines | ||
| Human: 2686 | MD Anderson Cancer Center | N/A |
| Human: MaMel134 | UK-Essen | N/A |
| Human: A375 | ATCC | CRL-1619 |
| Human: WM852 | Rockland | WM852-01-0001 |
| Human: Jurkat | ATCC | TIB-152 |
| Human: HEK293T | ATCC | CRL-3216 |
| Experimental models: Organisms/strains | ||
| Mouse: NOD.Cg-Prkdcscid II2rgtm1Sug Tg(CMV-IL2)4–2Jic/JicTac | Taconic | Taconic: 13440-F |
| Mouse: NOD.Cg-Prkdcscid II2rgtm1Wjl/SzJ | The Jackson Laboratory | JAX: 005557 |
| Biological samples | ||
| Human 2686 TILs | MD Anderson Cancer Center | N/A |
| Human MaMel134 TILs | UK-Essen | N/A |
| Patient melanoma FFPE blocks | New York Presbyterian Hospital/Columbia University Irving Medical Center | N/A |
| Oligonucleotides | ||
| QuikChange CD58 K34A F: TTTTCCAGTTCTGCAACTGCATCCTTTTGTTTTTTCCATAGGACCTCIIIIAA | This paper | N/A |
| QuikChange CD58 K34A R: TTAAAAGAGGTCCTATGGAAAAAACAAAAGGATGCAGTTG CAGAACT GGAAAA | This paper | N/A |
| P5 and P7 primers (See Table S5) | Doench et al. | N/A |
| CD58 crRNA: AATGCTCTGGTATCATGCAT | This paper | N/A |
| CD274 crRNA: ACTGCTTGTCCAGATGACTT | This paper | N/A |
| B2M crRNA: GAGTAGCGCGAGCACAGCTA | This paper | N/A |
| CD2 crRNA: CGATGATCAGGATATCTACA | This paper | N/A |
| CMTM6 crRNA: GGTGTACAGCCCCACTACGG | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: pLEX307 CD58 TM | This paper | N/A |
| Plasmid: pLEX307_CD58_GPI | This paper | N/A |
| Plasmid: pLEX307_CD58_TM_K34A | This paper | N/A |
| Plasmid: pLEX307_CD58_GPI_K34A | This paper | N/A |
| Plasmid: pLEX307_CMTM6 | This paper | N/A |
| Plasmid: pLEX307_CMTM6_V5 | This paper | N/A |
| Plasmid: pLEX307_CMTM6_Ctr_V5 | This paper | N/A |
| Plasmid: pLEX307_CMTM6_Ntr_V5 | This paper | N/A |
| Plasmid: pLEX307_CMTM6_EC1mut_V5 | This paper | N/A |
| Plasmid: pLEX307_CMTM6_EC2mut_V5 | This paper | N/A |
| Plasmid: pLEX307_CMTM6_EC1&2mut_V5 | This paper | N/A |
| Plasmid: pLX311_CD274 | This paper | N/A |
| Plasmid: pLX311_CD274_H1Amut | This paper | N/A |
| Plasmid: pXPR-011 | Doench et al.55 | Addgene #59702 |
| Plasmid: pLEX_307 | Laboratory of David Root | Addgene #41392 |
| Plasmid: pLX_TRC311 | Laboratory of John Doench | Addgene #113668 |
| Plasmid: pDONR221 | ThermoFisher Scientific | Cat#12536017 |
| Plasmid: pxsPAX2 | Laboratory of Dider Trono | Addgene #12260 |
| Plasmid: pMD2.G | Laboratory of Dider Trono | Addgene #12259 |
| Plasmid: pLX_311-Cas9 | Doench et al.55 | Addgene #96924 |
| Software and algorithms | ||
| ImageJ | Schneider et al.58 | https://imagej.net |
| FlowJo v10.8.1 | BD Biosciences | https://www.flowjo.com |
| CellProfiler v4.2.1 | Stirling et al.59 | https://cellprofiler.org |
| Prism v9.5.0 | GraphPad | https://www.graphpad.com |
| MAGeCK v0.5.9.2 | Li et al.48 | https://sourceforge.net/p/mageck/wiki/Home/ |
| InForm v2.5.1 | Akoya Biosciences | https://www.akoyabio.com/phenoimager/software/inform-tissue-finder/ |
| Image Studio Lite v5.2.5 | Li-Cor | https://www.licor.com/bio/image-studio/ |
| MaxQuant v2.0.1.0 | Cox and Mann66 | https://www.maxquant.org/ |
| Perseus | Tyanova et al.67 | https://maxquant.net/perseus/ |
| Andromeda | Cox et al.65 | https://bioinformaticshome.com/tools/proteomics/descriptions/Andromeda.html |
| SnapGene 6.0.5 | Dotmatics | https://www.snapgene.com |
| R v4.1.1 | The R Project | https://www.rproject.org |
| AUCell v1.20.2 | Aibar et al.63 | https://bioconductor.org/packages/release/bioc/html/AUCell.html |
| Seurat v4.1.0 | Hao et al.64 | https://satijalab.org/seurat/ |
| ZEN v3.4.91 | Zeiss | https://www.zeiss.com/microscopy/en/products/software/zeisszen.html |
| Other | ||
| SPRIselect beads | Beckman Coulter | Cat# B23318 |
| Protein A Dynabeads | ThermoFisher Scientific | Cat#10006D |
| Protein G Dynabeads | ThermoFisher Scientific | Cat#10003D |
| 8 well chambered cover glass | Cellvis | Cat#C8–1.5H-N |
EXPERIMENTAL MODEL AND STUDY PARTICIPANTS
Mice
All mouse experiments were performed at Columbia University following the Institutional Animal Care and Use Committee (IACUC) protocol #AC-AABE6570 using 6–10 week-old female NOD.Cg-Prkdcscid Il2rgtm1Sug Tg(CMV-IL2)4–2Jic/JicTac (hIL-2 NOG, Taconic, 13440-F) and NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG, The Jackson Laboratory 005557). All mice were maintained under pathogen-free conditions. Animals were randomly assigned to experimental groups.
Cell lines
A375 (female) cells, Jurkats (male), and HEK293T (female) cells were purchased from ATCC, and WM852 (male) cells were purchased from Rockland Inc. Patient-derived melanoma line 2686 and matched TILs were previously derived (under institutional review board protocol no. 2004–0069) and provided by MD Anderson Cancer Center. Patient-derived melanoma cell line MaMel134 and matched TILs were provided by UK-Essen. Buffy coats were purchased from the New York Blood Center or provided by Brigham and Women’s Hospital and used for production of engineered T cells. A375 and HEK293T cells were cultured in DMEM medium supplemented with 10% heat-inactivated fetal bovine serum (D10). All other cell lines were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum, GlutaMax, 10 mM HEPES, 10 mg/L insulin, 5.5 mg/L transferrin, 6.7 μg/L sodium, and 55 μM 2-mercaptoethanol (all from Thermo Fisher Scientific). TILs were cultured in T cell media (RPMI 1640 medium supplemented with 10% human AB serum (Fisher), GlutaMax, 10 mM HEPES, 100 IU/mL penicillin and 100 μg/mL streptomycin (Thermo)), supplemented with clinical grade 300–3000 IU/mL human IL-2 (Proleukin, Chiron). All cells were cultured at 37 °C and 5% CO2 in a humidified incubator. Cell lines were tested routinely for mycoplasma contamination using PlasmoTest (InvivoGen).
Patient samples
Patient tissue specimens were collected under IRB approved protocols at New York Presbyterian Hospital/Columbia University Medical Center (AAAT7416, AAAT2278). All procedures performed on patient samples were in accordance with the ethical standards of the IRB and the Helsinki Declaration and its later amendments. Informed consent for tissue usage was obtained in the patient enrollment to the clinical protocol.
METHOD DETAILS
Generation of NY-ESO-1 TCR T cells
Primary human cells were isolated from blood collars provided by Brigham and Women’s Hospital. Human blood mononuclear cells were isolated by Ficoll-Paque gradient centrifugation using SepMate isolation tubes (StemCell #85450). CD8+ T cells were then isolated by negative selection using the EasySep CD8 isolation kit (StemCell) according to manufacturer’s instructions. T cells were activated with CD3/CD28 Dynabeads (Thermofisher) at a 1:1 ratio for 48 hours in media supplemented with 30U/mL of IL-2. NY-ESO-1 TCR was introduced as described previously21,53. Briefly, the NY-ESO-1 construct was introduced into the endogenous TRAC locus by homology-directed repair. T cells were electroporated with a ribonucleoprotein complex (RNP) of Cas9 protein with a bound TRAC guide RNA (gRNA) as well the NY-ESO-1 TCR construct. The TRAC gRNA (60mM) was assembled by incubating equimolar quantities of TRAC crispr RNA (crRNA) and trans-activating crispr RNA (tracrRNA) at 95oC for 5 minutes. Then, the resulting gRNA was assembled into RNPs by incubation with an equal volume of Cas9-NLS protein (20 mM) at 37 oC for 15 min. CD8 T cells (1×106) were electroporated with Cas9-TRAC gRNA RNPs and 2.5 mg of single-stranded NY-ESO-1 TCR DNA template. The ssDNA template was synthesized using GuideIt Long ssDNA kit (Takara) according to the manufacturer’s instructions. Electroporated T cells were cultured overnight and then re-activated with Dynabeads at a 1:1 ratio. T cells were expanded for 10 days in media containing IL-2 and then sorted based on expression of CD3 and NY-ESO-1 TCR using the HA epitope tag attached to the N-terminus of the mature NY-ESO-1 TCR a chain. Sorted T cells were expanded, frozen in aliquots, and stored at −80oC until use.
Expansion of autologous TILs or NY-ESO-1 TCR T cells
Patient-derived TILs and human CRISPR-Cas9 NY-ESO-1 TCR knock-in CD8+ T cells were expanded using a rapid-expansion protocol (REP) as previously described8. For expansion, PBMCs were isolated from three different donors and used as feeder cells as follows. After isolation of PBMCs via Ficoll gradient centrifugation (Ficoll-Paque Premium, Cytiva), red blood cells were lysed using ACK-lysis buffer (Quality Biological), and PBMCs were washed once in PBS and then resuspended in AIM-V media (Thermo) before irradiating with 5,000 rad. On day 0, 5e5-1e6 TILs or edited CD8 T cells were added to 100e6 irradiated feeder cells in a G-Rex 10 bottle (Wilson Wolf) in REP media (1:1 mixture of AIM-V media and T cell media) supplemented with a final concentration of 30 ng/mL anti-CD3 antibody (OKT3) (Miltenyi) and 3,000 IU/mL human recombinant IL-2 (Proleukin, Chiron). On day 2, 3,000 IU/ml human recombinant IL-2 was added. On day 5, fresh media and IL-2 were added by half-media exchange. On day 7, cells were counted and >60e6 cells were propagated to a G-Rex 100 bottle in a total volume of 600 mL AIM-V with 3,000 IU/mL IL-2. On day 10, additional 400 mL AIM-V with 3000 IU/mL IL-2 was added. On day 12 additional 3000 IU/mL IL-2 were added. On day 14 expansion was complete and the expanded cells were collected, cryopreserved, and stored in liquid nitrogen until use in in vitro or in vivo experiments.
CRISPR-Cas9 KO cell line generation
Virus-free KO human melanoma cell lines and TILs were generated by nucleofection of Cas9 ribonucleoproteins (RNPs) using the SF Cell Line kit (Lonza). TILs were pre-stimulated on culture dishes coated with 100 ng/mL anti-CD3 (OKT3) antibodies overnight. Target sequences for CD58, CD274, B2M, CD2, and CMTM6 were selected from CRISPick26,54 (see Table S5 for crRNA sequences). Briefly, equimolar ratios of CRISPR RNA (crRNA; IDT) and transactivating CRISPR RNA (tracrRNA; IDT) were incubated at 95 °C for 5 min and then cooled to room temperature to form gRNA. Recombinant Cas9 enzyme (MacroLab) was incubated with gRNA at a 1:10 molar ratio at 37 °C for 15 min to form RNP complexes. Melanoma cells were resuspended at a cell density of 100,000 cells in 20 μL SF electroporation buffer with supplement (Lonza) and TILs were resuspended at a cell density of 1 million cells in 20 μL P3 electroporation buffer with supplement (Lonza) and then combined with 3 μL of RNP mixture and nucleofected using program DJ-110 (melanoma) or EH100 (TILs) on a 4D Nucleofector (Lonza). Melanoma cells were immediately recovered in full melanoma media in 12-well plates. TILs were left to incubate for 15 min at 37 °C prior to transfer to a 24 well recovery plate containing 300 IU/mL hIL-2. Pure cancer KO populations were generated by cell surface staining and sorting by FACS as described below. CD2 editing in TILs achieved >95% KO and required no sorting. Prior to co-culture, TILs were rested for 10 days and unedited TILs from the initial stimulation step were cultured in parallel and used as controls.
CD58, CMTM6, and CD274 OE construct cloning
CD58, CMTM6, CMTM6ΔC, CMTM6 ΔN, CMTM6EC1mut, CMTM6EC2mut, CMTM6EC1,2mut, CD274, and CD274H1Amut open reading frames (ORFs) with a 5’ Kozak sequence were flanked by attB cloning sites and ordered as gene blocks (GeneWiz). CMTM6 ΔC and CMTM6 ΔN constructs are missing aa 130 and aa 161–183, respectively. CD274H1Amut construct has coding region for aa 20–32 replaced with CCGGTTAAAGATACAGTCTATGAATTAGTTTATACCGTT. Gene blocks were cloned into pDONR™221 (Thermo) to create an entry clone using the Gateway Cloning system (Thermo), and then cloned into the constitutive expression vector pLEX_307 (gift from David Root; Addgene #41392) or pLX_TRC311 (gift from John Doench; Addgene #113668). CD58K34A OE plasmids were generated through site-directed mutagenesis of existing CD58 ORF plasmids using the QuikChange XL Site-Directed Mutagenesis Kit (Agilent Technologies) per manufacturer protocol (see Table S5 for primer sequences). Plasmid insert validation was performed via Sanger sequencing (GeneWiz). Whole plasmid sequencing validation of empty vectors was performed by the Massachusetts General Hospital Center for Computation and Integrative Biology DNA Core. All plasmid design and Sanger sequencing analysis was performed using Snapgene 6.0.5 (Dotmatics).
Lentivirus production
HEK293T cells were seeded in 6-well plates in D10 media. The following day, 3 μL TransIT-LT1 transfection reagent (Mirus Bio) was mixed with 15 μL Opti-MEM (Thermo) and incubated for 5 min at room temperature (RT). A mixture of 500 ng expression plasmid, 500 ng packaging plasmid psPAX2 (gift from Didier Trono, Addgene #12260), and 250 ng envelope plasmid pMD2.G (gift from Didier Trono, Addgene #12259) was prepared to a final volume of 37.5 μL in Opti-MEM. Transfection reagent mix was combined with plasmid mix, incubated for 30 min at RT, and then added dropwise to HEK293T cells. After 24 h, cell media was replaced with DMEM supplemented with 20% FBS. Cell media was collected and replaced after 48 h and 72 h and filtered through a 0.45 μm syringe filter (Thermo). Lentivirus was stored at −80 °C.
Transduction of CD58, CMTM6, and PD-L1 ORF constructs
Human melanoma cells were seeded in 6-well plates with 0.1–1 mL lentivirus in a final volume of 3 mL with 4 μg/mL polybrene (Millipore). Cells were spun at 1,000 x g for 2 h at 30 °C, and an additional 3 mL media was added to each well after spinning. Cells were incubated overnight at 37 °C and 5% CO2 in a humidified incubator. The next day, infected and non-infected cells were seeded across two wells each in a 6-well plate at equal seeding density to monitor transduction efficiency, with all remaining cells grown up in appropriate sized flasks. 2 days after transduction, one well each of cells was put into puromycin or blasticidin selection (Thermo) at following concentrations: Puromycin - 0.75 μg/mL A375 and 2686, 0.5 μg/mL for MaMel134, and 1 μg/mL for WM852; and Blasticidin - 4 μg/mL for A375 and 2 μg/mL for 2686. Once all non-infected cells were dead, transduction efficiency was calculated as the number of infected cells surviving in selection divided by the number of infected cells growing without selection.
Co-culture assays
Melanoma target cells previously transduced to express nuclear localization signal (NLS)-dsRed were co-cultured with matched TILs or NY-ESO-1-targeting engineered T cells as previously described8. Briefly, melanoma target cells were seeded in a black-walled 96-well plate (Corning) at a cell density of 5,000 or 10,000 cells/well. After 16 h, culture medium was replaced with 100 μL full melanoma medium with 4 μM Caspase-3/7 activity dye (CellEvent, Thermo Fisher Scientific). Plates were then imaged on a Celigo Imaging Cytometer (Nexcelom) for a t0 count of viable target cells. Two days prior to co-culture, TILs were thawed into complete TIL media with 3000 IU/mL IL-2. TILs were collected and resuspended in complete melanoma medium and added to melanoma cells in 100 μL per well at increasing TIL:target ratios. After 24 h, 48 h, and 72 h, plates were imaged again to track viable melanoma cell growth. At the end of co-culture, plates were spun at 400 x g for 5 min and cleared culture media was collected for cytokine analysis by ELISA described below. Cell counts were performed using Celigo Imaging Cytometer software. Viable melanoma cells were defined as DsRed+ cells that were not Caspase-3/7+, with a size threshold to exclude debris. Viable melanoma cell counts were then normalized to respective t0 counts. Where indicated, target cells were pretreated overnight with 10 μg/mL anti-CD58 (TS2/9; Biolegend) blocking antibody or 10 μg/mL anti-PD-L1 (29E.2A3) blocking antibody or IgG isotype (MPC-11) control, and treated again upon addition of T cells. T cells were pre-treated with 200 ng/mL anti-CD2 (LO-CD2b) blocking antibody or IgG isotype control (LTF-2) overnight and then again upon addition to target cells, where indicated. For PD-L1 blocking co-culture experiments, TILs were cultured overnight on plates coated with 1 μg/mL anti-CD3 (OKT3) activating antibody prior to addition to target cells. TILs were then assayed for PD-1 expression by flow cytometry, as described below.
Flow cytometry
For in vitro analysis of cell surface markers, cells were detached with Accutase (Innovative Cell Technologies, Inc.) as needed and washed with PBS. Cells were first stained with Zombie-NIR Fixable Viability Dye (Biolegend) for 15 min at RT in the dark and washed once with PBS, followed by staining with anti-CD58 APC (TS2/9), anti-PD-L1 BV785 (29E.2A3), anti-HLA-ABC BV605 (W6/32), anti-PD-1 APC (EH12.2H7), anti-CD2 BV605 (RPA-2.10), anti-CD58 BV421 (1C3), and/or anti-CD48 FITC (BJ40) antibodies (see Key Resources Table for antibodies) in FACS buffer (2% FBS, 2 mM EDTA in PBS) for 20 min on ice in the dark. Cells were washed 2X with FACS buffer and then fixed with Fixation Buffer (Biolegend), washed 2X with FACS buffer, and analyzed on an Aurora 3-laser spectral cytometer (Cytek). Cells stained with isotype control antibodies were used as negative controls. All analysis of flow cytometry data was performed using FlowJo v10.8.1(BD). For quantification of expression, median fluorescence intensity values were used.
ELISAs
IL-2 and IFN-ɣ levels were detected in cleared cell media using the ELISA MAX™ Deluxe ELISA kits (Biologend) per manufacturer protocol. Absorbance values were measured using a Synergy H1 plate reader (Biotek).
T cell co-stimulation proliferation assay
24-well tissue culture-treated plates were pre-coated with 1 μg/mL anti-CD3 (OKT3) antibody (Miltenyi Biotec) in 1 mL PBS per well, +/− 1 μg/mL anti-CD28 (CD28.2) antibody (eBioscience/Thermo) or recombinant human CD58-Fc chimera protein (R&D Systems) at 37 °C for 2 hours. TILs were stained with 1 μM CFSE (Thermo) in PBS for 1 min at 37 °C at a cell density of 5 million cells/mL and then washed with complete media. Cells were then seeded in pre-coated plates at a density of 300,000 cells/well in 1 mL TIL media. Cleared cell media was collected after 24 and 48 h to use for ELISAs. Five days after seeding, TILs were collected, stained with Zombie-NIR (Biolegend), fixed with Fixation Buffer (Biolegend), and analyzed for cell proliferation by flow cytometry.
MaMel134 in vivo melanoma xenograft and ACT, tumor processing, and analysis
Female NOD.Cg-Prkdcscid Il2rgtm1Sug Tg(CMV-IL2)4–2Jic/JicTac mice (NOG hIL-2; Taconic) were implanted with 2 million each MaMel134 DsRed control and MaMel134 DsRed CD58 KO or CD58-TM OE cells resuspended in 25% Matrigel High Concentration Matrix (Corning) via bilateral subcutaneous flank injections. Mice were weighed once a week and tumors were measured 2–3 times/week using digital calipers. Three days prior to ACT/PBS injection, cryopreserved TILs were thawed and cultured in T cell media supplemented with 3000 IU/ml human recombinant IL-2. Once tumors were palpable for all mice (~1 month), 8 mice from each cohort were injected with 5 million MaMel134 TILs in 200 μL PBS by tail vein injection, with the remaining half of each cohort receiving 200 μL PBS control. Submandibular blood collection was performed once a week to monitor circulating TILs by flow cytometry. Due to unclear responses to ACT, one month after initial ACT/PBS injections, a second round of ACT or PBS was administered. Once largest tumor pairs reached a summed diameter of 20 mm, mice were sacrificed, and tumors were harvested. Tumors were weighed and when possible a portion was fixed in 10% formalin and embedded in paraffin. The remainder was dissociated with digestion media (collagenase D and DNase I (both Roche, Sigma-Aldrich) in PBS) and filtered through 70 μM cell strainers to produce a single cell suspension for flow analysis. Cells were incubated in ACK Lysing Buffer (Quality Biological) for 1 min at RT and washed with PBS; ACK lysis was repeated as necessary. Cells were stained with Zombie-NIR followed by Fc block with anti-mouse CD16/32 antibody (Biolegend). Cells were then stained for cell surface markers as described above using anti-human CD45 PacBlue (HI30), anti-mouse CD45 BV510 (30-F11), anti-CD8 AF488 (HIT8a), anti-CD58 PE-Cy5 (TS2/9), anti-CD3 PE-Cy7 (HIT3a), and anti-CD4 AF700 (RPA-T4) antibodies. Ki-67 stained using the Foxp3/Transcription Factor Staining Kit (eBioscience, Thermo) per manufacturer protocol with an anti-Ki-67 eF506 (SolA15) antibody (see Key Resources Table for antibodies). FMO control was prepared for Ki-67. CD8+:mCD45+ metric was used to assess tumor infiltration by T cells in order to control for differences in tumor size, with assumption that mCD45 infiltration is not impacted by human CD58 expression. Samples with fewer than 40 CD8+ T cells were excluded from downstream phenotyping analysis. Tumor volumes were calculated as (Tumor width x Tumor width x Tumor length)/2. Fold change growth was calculated using the average of the three tumor measurements immediately before, after, and on the day of the first ACT/PBS injection as t0.
A375 in vivo melanoma xenograft and ACT, tumor processing, and analysis
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (Jackson Laboratory) were implanted with 1 million each A375 WT and CD58 KO cells resuspended in 50% Matrigel (Corning) via bilateral subcutaneous flank injections. After 10 days, mice were treated with ACT of 2 million NY-ESO-1 TCR T cells (prepared as described above) in 200 μL PBS by tail vein injection or PBS. Mice were dosed with 100,000 IU human IL-2 by IP injection on days 0–3 after ACT and then twice per week. Mice were weighed once a week and tumors were measured 2–3 times/week using digital calipers. Mice were euthanized 7 days after ACT when tumor pairs reached a summed diameter of 20 mm; tumors were collected, digested, and processed for flow as described above. An additional cohort of mice were implanted with WT cells only as unilateral injections and dosed with 100 μg anti-CD58 (TS2/9; Biolegend) antibody or isotype control (QA16A15; Biolegend) on days 2, 5, 8, 11, and 14 after ACT. Mice were euthanized 15 days after ACT when tumors reached 20 mm in diameter; tumors were again collected, digested, and processed for flow as described above. Additional anti-granzyme B PE-CF594 (GB11), anti-PD-1 APC (293.2A3), anti-CD45RA Brilliant Violet 711 (HI100), and anti-CD45RO Brilliant Violet 570 (UCHL1) antibodies were used.
2686 in vivo melanoma xenograft and ACT timecourse
Female NOD.Cg-Prkdcscid Il2rgtm1Sug Tg(CMV-IL2)4–2Jic/JicTac mice (NOG hIL-2; Taconic) were implanted with 5 million 2686 DsRed control, CD58, or CD58-TM OE cells resuspended in 25% Matrigel High Concentration Matrix (Corning) via subcutaneous flank injections. Additional control mice without tumor xenografts were used to control for potential MHC mismatch. Mice were weighed once a week and tumors were measured 2–3 times/week using digital calipers. Once tumors reached a mean diameter of ~4 mm, mice were injected with 3 million NY-ESO-1 TCR T cells in 200 μL PBS by tail vein injection or 200 μL PBS control. At days 5, 12, and 19 after ACT injection, mice were sacrificed, and blood, spleen, and tumors were harvested. Tumors were processed as above. Spleens were dissociated by mashing with the end of a 1 mL syringe plunger against a 70 μm cell strainer and rinsing with PBS. Blood was collected by cardiac puncture. Splenic and blood cells were incubated in ACK Lysing Buffer (Quality Biological) for 1 min at RT and washed with PBS; ACK lysis was repeated as necessary. All single cell suspensions were stained as described above. FMO controls were prepared for CD45RO, CD45RA, PD-1, and granzyme B.
Multiplexed immunofluorescence
Multiplexed immunofluorescence staining of animal tumors was performed using CD8 (4B11), CD58 (TS2/9; Thermo), and Ki-67 (MIB-1) antibodies (see Key Resources Table for antibodies) with the Opal 7-colour IHC kit (Akoya Bioscience) on a Leica Bond RX automated stainer (Leica Biosystems). FFPE tissue sections (5 μm) were baked for 2 h at 60 °C, followed by automatic deparaffinization, rehydration, and antigen retrieval in BOND Epitope Retrieval Solution 2, pH 9 (Leica Biosystems) for 30 min at 95 °C. Immunofluorescence staining with Opal and tyramide signal amplification (TSA) were performed in six cycles. In each cycle, the tissue was incubated sequentially with a primary antibody for 30 min at room temperature, the secondary antibody conjugated to polymeric horseradish peroxidase (HRP), an Opal fluorophore in TSA buffer, and BOND Epitope Retrieval Solution 1, pH 6 (Leica Biosystems) for 20 min at 95 °C to strip the tissue-bound primary-secondary antibody complexes before the next staining cycle. After nuclear counterstaining with DAPI, slides were coverslipped with Vectrashield HardSet Antifade mounting medium (Vector Laboratories) and 12–15 areas per slide were imaged using the Vectra 3 automated multispectral microscope (Akoya/PerkinElmer) with Vectra 3.0.5 software. Regions of interest were chosen for multispectral imaging (MSI) at 20X magnification and spectral unmixing using InForm v2.4.6 software (Akoya). Demultiplexed images were exported as 32-bit TIFF files for further analysis.
SDS-PAGE and immunoblotting
For each blot, melanoma cells were stimulated with or without 10 ng/mL IFN-ɣ (Abcam) for 72 h and detached and collected using 0.05% Trypsin (Thermo). Cells were lysed in RIPA buffer (Sigma) with 10X concentration of Halt™ Protease and Phosphatase Inhibitor Cocktail (Thermo) for 30 min on ice, and spun at 10,000 rpm for 15 min at 4 °C in a microcentrifuge. Supernatants were collected and protein was quantified using the Pierce™ BCA Protein Assay Kit (Thermo). Cell lysates were brought to the desired concentration in RIPA buffer with 1X Laemmli SDS-Sample Buffer (Boston Bioproducts Inc.) and boiled at 95 °C for 10 min. Lysates were ran on 10% TGX™ FastCast acrylamide gels (BioRad) in tris-glycine SDS running buffer and transferred onto PVDF membranes using the iBlot 2 Dry Blotting system (Thermo). Blots were blocked with 1X Western Blocking Reagent (Sigma) in trisbuffered saline (TBS) for 1 hour and then probed overnight with anti-CD58 (TS2/9 or EP15041), anti-PD-L1 (405.9A11 or E1L3N), anti-CMTM6, anti-V5 (D3H8Q), anti-GAPDH (14C10), or anti-His (27E8) diluted in TBS with 0.1% Tween-20 (TBS-T; Sigma) at 4 °C with rotation (see Key Resources Table for antibodies). Blots were then washed 3X with TBS-T and probed with fluorophore-conjugated IRDye secondary antibodies (Li-Cor) for 1 h at RT with rocking and washed 3X with TBS-T. All blots were developed on the Odyssey CLx Imaging system (Li-Cor) and analyzed in Image Studio Lite v5.2.5 (Li-Cor).
A375 Cas9 transduction
pLX_311-Cas9 lentivirus was produced via transfection of HEK293T cells as described above (plasmid gift of John Doench, William Hahn, and David Root; Addgene #9692455). A375 WT cells were seeded in 12-well plates with 1 million cells per well with increasing volumes of pLX_311-Cas9 lentivirus to a final volume of 2 mL with 4 μg/mL polybrene. Cells were spun at 1,000 x g for 2 h at 30 °C, and 2 mL media was added to each well after spinning. Cells were then incubated at 37 °C, 5% CO2 in a humidified incubator overnight. The next morning, 100,000 transduced cells and non-transduced cells were seeded at equal densities across two wells each per condition of a 6-well plate. The next day, cells were put into selection with 4 μg/mL blasticidin (Thermo), and at the end of selection transduction efficiency was calculated as above.
Cas9 activity assay
To measure Cas9 editing efficiency, Cas9-expressing A375 cells were transduced with lentivirus containing expression plasmid pXPR-011 (gift of John Doench and David Root; Addgene #59702), which encodes for enhanced green fluorescent protein (EGFP) and a gRNA targeting EGFP55. As a no-editing control, WT A375 cells were also transduced. Transduced cells were put into 0.75 μg/mL puromycin selection. At the end of selection, EGFP expression of transduced cells was assessed by flow cytometry on an Aurora spectral cytometer (Cytek). Cas9 activity was measured as the percentage of EGFP-negative cells, using no-editing control cells as gating controls.
Brunello library titration
A375 Cas9-expressing cells were seeded in 12-well plates at a density of 1 million cells per well with increasing volumes of lentivirus (1.02×107 transduction units/mL) carrying the human CRISPR KO pooled library Brunello in a lentiGuide-Puro backbone (gift from David Root and John Doench; Addgene #73178)26 to a final volume of 2 mL with 4 μg/mL polybrene. Cells were spun at 1,000 x g for 2 h at 30 °C, and 2 mL media was added to each well after spinning, keeping cells in 4 μg/mL blasticidin selection. Cells were then incubated at 37 °C, 5% CO2 in a humidified incubator overnight. The next morning, 200,000 transduced cells and non-transduced cells were seeded across two wells each per condition of a 6-well plate. The next day, cells were put into selection with 0.75 μg/mL puromycin, and at the end of selection transduction efficiency was calculated as above. Cells with a transduction efficiency of ~20% were selected, resulting in an MOI of 0.3 and therefore a single integration rate of 95%.
Anti-CD58-APC antibody titration
The anti-CD58-APC (TS2/9) antibody (Biolegend) used for the CRISPR-Cas9 KO screen was titrated by staining pooled A375 WT and CD58 KO cells as above with Zombie-Violet followed by increasing concentrations of anti-CD58-APC antibody. Cells were then fixed and analyzed on the same Influx (BD) sorter used for screen sorting. For each condition, CD58-negative and CD58-positive populations were gated the median fluorescence intensity of APC fluorescence was calculated. The separation index for each antibody concentration was then calculated as follows:
The lowest concentration that gave the relatively highest separation index was chosen and used for the screen.
Transduction of Brunello library
To reach a desired representation of ~500 cells/sgRNA, with a transduction efficiency of 20%, 220 million A375 Cas9-expressing cells were seeded across 12-well plates at a density of 1 million cells/mL with 30 μL Brunello virus per well to a final volume of 2 mL with 4 μg/mL polybrene. Cells were spun at 1,000 x g for 2 h at 30 °C. After spinning, 2 mL fresh media was added to each well and cells were incubated overnight at 37 °C, 5% CO2 in a humidified incubator. One day after transduction, cells were expanded up into T175 flasks. At two days post-transduction, cells were put into selection with 0.75 μg/mL puromycin, and cells were continuously expanded up until sorting, keeping in puromycin and blasticidin throughout to maintain Cas9 and sgRNA expression. Two biological replicates were performed (labeled 1 and 2 in Table S2–3).
A375 Cas9 Brunello CD58 FACS
10 days after transduction with Brunello virus, sgRNA library- and Cas9-expressing A375 cells were detached with Accutase, washed with PBS, and split into two technical replicates. For each replicate, 130 million cells were transferred to 96 well V-bottom storage plates (Corning) with 1 million cells/well. Cells were first stained with Zombie-Violet (Biolegend) for 15 min at RT and washed 1X. Cells were then stained with anti-CD58-APC (TS2/9) antibody (Biolegend) in FACS buffer for 20 min on ice and washed 2X. Cells were fixed with fixation buffer and incubated for 20 min at RT and washed 2X. Cells were then resuspended in 100 μL FACS buffer/well and pooled together to use for sorting. Cells were sorted on an Influx (BD) cell sorter. A375 Cas9 cells without sgRNA expression and A375 CD58 KO cells were used for gating controls. Cells with the lowest 5% of CD58 expression were collected as the target population, alongside cells within ~1 standard deviation above and below the median as controls. Sorted cells were kept in FACS buffer and stored at 4 °C overnight. Unsorted cells were also reserved to be used for essential gene dropout analysis. Two technical replicates were performed per biological replicates (labeled 1A, 1B, 2A, and 2B in Table S2–3).
A375 Cas9 Brunello genomic DNA (gDNA) extraction
gDNA was collected from sorted and unsorted cells using a DNeasy Blood and Tissue Kit (Qiagen). Cells were first pelleted and split across multiple microcentrifuge tubes with a maximum of 2.5 million cells/tube. Cells were resuspended in 200 μL PBS, to which 20 μL proteinase K and 200 μL Buffer AL were added. Cells were then incubated overnight at 65 °C with shaking at 1000 rpm to remove formaldehyde crosslinking. The next morning, gDNA was extracted from cell lysates per manufacturer protocol. Viral DNA was also extracted from 2 mL Brunello virus using the QIAamp Micro DNA kit (Qiagen).
sgRNA PCR amplification and clean up
sgRNA sequences were amplified from gDNA by PCR using Ex Taq DNA polymerase (TaKaRa). PCR mixes were prepared with 10 μL 10X reaction buffer, 8 μL dNTP, 5 μL DMSO, 0.5 μL equimolar mix of staggered P5 primers (100 μM), 1.5 μL Ex Taq polymerase, 5 μL unique P7 primer (10 μM) per condition, and a maximum of 10 μg gDNA to a final volume of 100 μL (see Table S5 for primer sequences). PCR reactions were cycled at 95 °C for 1 minute; followed by a total 28 cycles of 95 °C for 30 sec, 53 °C for 30 sec, and 72 °C for 30 sec; 72 °C for 10 minutes; and then finally 4 °C. 25 μL from each PCR product was pooled and loaded onto a 1.5% agarose gel (Bio-Rad) and ran at 100V. The ~350 bp PCR product was extracted using the Zymoclean Gel DNA Recovery Kit (Zymo) per manufacturer protocol. To remove all buffer salts, a final inclusive cleanup was performed using SPRI beads (Agilent). SPRI beads were added to PCR product at a 1.2X ratio and incubated for 5 min. After placing on a magnet for two minutes, beads were washed 2X with 80% ethanol and resuspended in TE buffer and quantified on a TapeStation (Agilent). Final prepared sgRNA library was sequenced on a NextSeq 550 sequencer (Illumina).
Co-immunoprecipitation
Co-immunoprecipitation protocol adapted from Lee et al.56 For each IP, 5 million melanoma cells were stimulated with or without 10 ng/ml IFN-ɣ for 72 h and detached using 0.05% Trypsin and pelleted. Cells were then lysed in 1% digitonin lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 5 mM EDTA, 1% digitonin, 10X Halt™ Protease and Phosphatase Inhibitor Cocktail (Thermo)) for 30 min at 4 °C with rotation and then spun at 13,000 x g for 10 min. Supernatant was collected and lysates were then diluted 2X to 0.5% digitonin. Lysates were incubated with 1 or 2 μg anti-CD58 (TS2/9; Abcam), anti-CMTM6, anti-PD-L1 (405.9A11), or anti-V5 (D3H8Q) antibody (see Key Resources Table for antibodies) or IgG isotype control for 3 h at 4 °C with rotation. Antigen-antibody mixtures were then used to resuspend 10 μL cleared Protein A or Protein G Dynabeads (Thermo) and incubated for 2 h at 4 °C with rotation. Samples were washed 4 times with wash buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 5 mM EDTA, 0.1% digitonin). Samples were then either flash frozen on dry ice for MS analysis, or resuspended in 1X Laemmli SDS-Sample Buffer (Boston BioProducts) and boiled at 95 °C for 10 min to elute off protein for SDS-PAGE as described above. For MS analysis, three replicates were independently prepared of CD58 and IgG control pulldowns.
On-bead digestion of samples for mass spectrometry
Proteins bound to magnetic beads were washed five times with 200 μl of 50 mM ammonium bicarbonate and subjected to disulfide bond reduction with 5 mM TECP (RT, 30 min) and alkylation with 10 mM iodoacetamide (RT, 30 min in the dark). Excess iodoacetamide was quenched with 5 mM DTT (RT, 15 min). Proteins bound on beads were digested overnight at 37°C with 1 μg of trypsin/LysC mix. The next day, digested peptides were collected in a new microfuge tube and digestion was stopped by the addition of 1% TFA (final v/v), and centrifuged at 14,000 g for 10 min at room temperature. Cleared digested peptides were desalted on SDB-RP Stage-Tip and dried in a speed-vac. Peptides were dissolved in 3% acetonitrile/0.1% formic acid.
Liquid chromatography with tandem mass spectrometry (LC-MS/MS)
Peptides were separated within 80 min at a flow rate of 400 nl/min on a reversed-phase C18 column with an integrated CaptiveSpray Emitter (25 cm x 75μm, 1.6 μm, IonOpticks). Mobile phases A and B were with 0.1% formic acid in water and 0.1% formic acid in ACN. The fraction of B was linearly increased from 2 to 23% within 70 min, followed by an increase to 35% within 10 min and a further increase to 80% before re-equilibration. The timsTOF Pro was operated in PASEF mode57 with the following settings: Mass Range 100 to 1700m/z, 1/K0 Start 0.6 V·s/cm2, End 1.6 V·s/cm2, Ramp time 100ms, Lock Duty Cycle to 100%, Capillary Voltage 1600V, Dry Gas 3 l/min, Dry Temp 200°C, PASEF settings: 10 MSMS Frames (1.16 seconds duty cycle), charge range 0–5, active exclusion for 0.4 min, Target intensity 20000, Intensity threshold 2500, CID collision energy 59 eV. A polygon filter was applied to the m/z and ion mobility plane to select features most likely representing peptide precursors rather than singly charged background ions.
RT-qPCR
Melanoma cells were stimulated with or without 10 ng/mL IFN-ɣ for 72 h and then detached using 0.05% Trypsin and pelleted. Total RNA was collected from cell pellets using an RNeasy Mini kit (Qiagen). cDNA was then generated using the iScript™ cDNA Synthesis Kit (BioRad), and qPCR was performed using TaqMan Fast Advanced Master Mix (Applied Biosystems) in combination with TaqMan Gene Expression Assays (see Key Resources Table for assay IDs) per manufacturer protocol on a ViiA 7 RT-PCR system (Applied Biosystems). Gene expression for CD58 and CD274 was normalized to ACTB and GAPDH expression, and compared to WT, unstimulated cells.
Immunocytochemistry
2686 melanoma cells were seeded in 8-well chamber slides with #1.5 coverglass bottom (CellVis) at a density of 20,000 cells per chamber with or without 10 ng/mL IFN-ɣ and incubated overnight at 37 °C, 5% CO2 in a humidified incubator. The following day, cells were fixed in 4% paraformaldehyde (Fisher Scientific) for 15 min at RT. Cells were washed 2X with PBS and permeabilized with 0.25% Triton X-100 (Fisher Scientific) for 5 min at RT. Cells were washed 2X and then blocked with 1% BSA (Corning) for 1 h at RT. Cells were washed 2X and treated with freshly made 1 mg/mL NaBH4 in PBS for 7 min at RT. Cells were washed 3X and then stained with primary anti-CMTM6 or anti-CD58 (TS2/9; Thermo) antibody prepared in 1% BSA for 1 h at RT. Cells were washed 3X and then stained with secondary anti-mouse AF594 or anti-rabbit AF647 antibody for 30 min at RT. Cells were washed 3X and then optionally stained with fluorophore-conjugated anti-ARF6, anti-Calnexin, anti-GM130, anti-TfR, anti-TGN46, anti-EEA1, or anti-LAMP1 AF488 antibody for 1 h at RT. Cells were washed 3X and stained with Hoechst 33342 (Thermo) for 5 min at RT. Cells were washed and then imaged using an inverted LSM 900 single point scanning confocal microscope (Zeiss) attached to Axio Observer motorized base (Zeiss) and Plan-Apochromat 63X/1.40 N.A oil immersion lens (Zeiss) with 405 nm, 488 nm, and 640 nm laser lines. Images were collected by frame scanning bi-directionally at 2.06 μs/pixel with 4-line averaging, 0.44 μm pixel size, and 1 AU pinhole size using Immersol 518F immersion oil (Zeiss) on ZEN 3.4.91 software (Zeiss) and saved as czi files. All image analysis was performed in ImageJ58.
Degradation assay
2686 melanoma cells were assayed for degradation of CD58, PD-L1, and HLA-A,B,C as previously described28. Briefly, cells were cultured for 72 h with or without 10 ng/mL IFN-ɣ and then stained with anti-CD58 APC (TS2/9), anti-PD-L1 BV785 (29E.2A3), and anti-HLA-A,B,C BV605 (W6/32) conjugated antibodies for 1 h on ice. After washing, cells were replated in complete medium and incubated at 37 °C for 3–6 h in the presence or absence of 50 μM chloroquine or 50 nM concanamycin A. Cells were collected at each timepoint with Accutase, washed, stained with Zombie-NIR, fixed as described previously, and analyzed on an Aurora spectral cytometer (Cytek). Degradation of CD58, PD-L1, and HLA-A,B,C was measured by a decline in fluorescence intensity over time.
Proximity ligation amplification
PLA analysis was performed on animal and deidentified patient melanoma FFPE sections using the Duolink In Situ kit (Sigma Aldrich). FFPE sections (5 μm) were first deparaffinized, and antigen retrieval was performed in a 95 °C sous-vide bath in eBioscience IHC Antigen Retrieval Solution, pH 9 (ThermoFisher) for 30 min. Sections were then washed in TBS and permeabilized in 0.25% Triton X-100 for 10 min. Sections were washed and photobleached on LED light in bleaching solution (25 mL PBS, 4.5 mL 30% H2O2, 0.08 mL 10N NaOH) for 30 min. Sections were then blocked for 1 h at 37 °C using the Duolink blocking buffer, and further processed according to the vendor protocol. Sections were stained with anti-CMTM6 rabbit (Millipore Sigma) and anti-CD58 mouse (TS2/9; Thermo) (see Key Resources Table for antibodies) antibodies overnight at 4 °C followed by Duolink In Situ PLA Anti-Rabbit PLUS and Anti-Mouse MINUS probes. Slides were mounted using DAPI Fluoromount-G Mounting Medium (SouthernBiotech) and then imaged on a LSM 900 confocal microscope (Zeiss) as described for ICC above using 405 nm and 640 laser lines. Z-stacks were collected at 1 μm intervals over 4 μm range. Stacks were edited in ImageJ to generate maximum intensity Z-projections with smoothing. Z-projection images were analyzed using CellProfiler 4.2.159 to count the number of nuclei and PLA foci. Nuclei were identified on DAPI channel images using the Otsu method, with declumping performed by shape. PLA foci were identified on PLA channel images using the robust background method, with declumping performed by shape. Size restrictions were entered for nuclei and PLA identification to improve accuracy. Number of nuclei and PLA were then counted to generate the PLA per nuclei statistic. Minimum of 500 nuclei were counted per slide.
In vitro binding assay
Recombinant GST-CMTM6 (Abnova) was combined with CD58–6His (VWR) and/or PD-L1–6His (Novus Biologicals) in 200 μL equilibrium buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 5 mM EDTA, or 20 mM Tris-HCl, pH 7.5, 250 mM NaCl). Where indicated, gradient of CD58–6His or PD-L1–6His was added. Mixtures were incubated for 2.5 h at 4 °C with rotation. Mixtures were then processed as above in co-immunoprecipitation experiments beginning with incubation with antibody. Washes were performed using equilibrium buffer with 0.1% digitonin.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details for experiments can be found in figure legends and figures. Statistical analysis was performed using GraphPad Prism 9.5.0.
TCGA analysis
Analysis of TCGA PanCancer Atlas uveal and skin cutaneous melanoma RNA-seq datasets for CD58 mRNA expression was performed via cBioPortal60,61. P-value was calculated using the Wilcoxon Rank-Sum test.
CD58 signature generation and analysis
Transcriptome data from scRNA-seq and proteome data from CITE-seq from melanoma cells after co-culture with autologous TILs8 were first integrated for further comparison using totalVI62. Briefly, UMI counts originating from transcripts in scRNA-seq and UMI counts originating from proteins in CITE-seq were combined to learn a joint latent space where each cell is modeled using both modalities. This joint latent space was used to calculate normalized and denoised expression values for each transcript and protein enabling direct comparison of the expression values across the two modalities. A Pearson’s correlation coefficient was calculated for the totalVI imputed expression values between the CD58 transcript and CD58 protein and all other transcripts and proteins. A threshold was applied to the distribution of correlation coefficient’s to only include significant correlations. The transcripts and genes passing these thresholds are referred to as the “Up” programs when they are significantly correlated with CD58 protein expression and the “Down” programs when they are significantly anti-correlated with CD58 protein expression. The identification of malignant cells from the scRNA-seq cohort of metastatic melanoma lesions was performed as described in the Pozniak et al. study19. The samples in which malignant cells were present were acquired from 20 patients with 35 samples (20 pre- and 15 early on-treatment), of which 12 samples were matched. 23 samples were from Non-Responders (12 pre- and 11 early on-treatment) and 12 samples from Responders (8 pre- and 4 early on-treatment). The gene signature scores were measured using the AUCell v1.20.263 package in R and compared between responders and non-responders using Wilcoxon test.
scRNA-seq analysis of external melanoma scRNA-seq cohorts
scRNA-seq datasets were obtained from GEO (GSE1159787) and the Single Cell Portal (SCP39820) and loaded into Seurat v4.1.064 in R v4.1.1 (https://www.r-project.org/). CD8+ T cells were separated into TOX+ and TCF7+CD8+ T cells using a >0.5 threshold applied to the normalized gene expression of the two markers genes. Cells that were positive for both markers were excluded. P-values were calculated using the Wilcoxon Rank-Sum test.
CRISPR-Cas9 KO screen analysis
Data was analyzed using MAGeCK Version 0.5.9.2. Briefly, sgRNA counts by condition were generated using mageck count and compared with mageck test using default parameters. Comparisons between FACS gates identified potential positive regulators of CD58. Inclusion criterium for defined positive regulators of CD58 was <0.25 FDR for enrichment of a gene target in CD58lo v. CD58mi cells across all four replicates of the screen.
LC-MS/MS data analysis
Acquired PASEF raw files were analyzed using the MaxQuant environment v.2.0.1.0 and Andromeda for database searches at default settings with a few modifications65,66. The default is used for first search tolerance and main search tolerance (20 ppm and 4.5 ppm, respectively). MaxQuant was set up to search with the reference human proteome database downloaded from UniProt. MaxQuant performed the search trypsin digestion with up to 2 missed cleavages. Peptide, site, and protein false discovery rates (FDR) were all set to 1% with a minimum of 1 peptide needed for identification; label free quantitation (LFQ) was performed with a minimum ratio count of 1. The following modifications were used for protein identification and quantification: Carbamidomethylation of cysteine residues (+57.021 Da) was set as static modifications, while the oxidation of methionine residues (+15.995 Da), deamidation (+0.984) on asparagine and glutamine were set as a variable modification. Results obtained from MaxQuant, were imported into Perseus v.1.6.15.067 for t-test statistical analysis (FDR<0.05) to identify proteins demonstrating statistically significant changes in abundance. Inclusion criteria for selection as an interactor with CD58: FDR<0.05, log2(foldchange) of LFQ intensity value >3, average spectral count within CRAPome database <2 to exclude background contaminants49.
Supplementary Material
Table S1. CD58 up- and down-signature programs, Related to Figure 1
Table S2. MAGeCK summary of gene KO enrichment from A375 genome-wide CRISPR-Cas9 screen to identify regulators of CD58, Related to Figure 4
Table S3. MAGeCK summary of sgRNA enrichment from A375 genome-wide CRISPR-Cas9 screen to identify regulators of CD58, Related to Figure 4
Table S4. Mass spectrometry of IgG v. CD58 IP significantly changed candidates, Related to Figure 5
eTOC blurb.
Ho et al. identify a critical role for defects in the CD58-CD2 axis as drivers of immune evasion and impaired intra-tumoral T cell infiltration. CD58 and PD-L1 compete for CMTM6 for protein stabilization, thus representing an additional mechanism for balancing immune-stimulatory and - inhibitory signals.
Highlights.
Loss of CD58 expression confers immune evasion through multiple mechanisms.
CD58 and PD-L1 are co-regulated in cancer cells.
Genome-scale screens identify CMTM6 as important regulator of CD58.
CD58 and PD-L1 compete for CMTM6 for protein stability.
Acknowledgments
P.H., Z.W., L.C., M.M., and E.K.M. are supported by Training Grant T32GM007367. J.P. received financial support from the Marie Curie Individual Fellowship (H2020-MSCA-IF-2019, #896897). B.I. is supported by National Institute of Health/National Cancer Institute (NIH/NCI) grants K08CA222663, R21CA263381, R37CA258829, and R01CA266446; the American Cancer Society Research Scholar Grant 21–104-01-IBCD; the Burroughs Wellcome Fund Career Award for Medical Scientists; a Tara Miller Melanoma Research Alliance Young Investigator Award; the Louis V. Gerstner, Jr. Scholars Program; and the V Foundation Scholars Award. K.W.W. is supported by NIH grants R01CA238039 and R01CA25159. C.A. was supported by a TL1 award. This work was supported by Herbert Irving Comprehensive Cancer Center core facility grants (P30CA013696, S10OD020056). This work was partly supported by the Grand Challenge Program of VIB (Pointillism), FWO (#G0C530N and G070622N), Stichting tegen kanker (FAF-F/2018/1265), and KU Leuven (C1 grant) to J-C.M. Results shown here are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. Figures and graphical abstract created with BioRender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
B.I. is a consultant for or received honoraria from Volastra Therapeutics, Johnson & Johnson/Janssen, Novartis, Eisai, AstraZeneca and Merck, and has received research funding to Columbia University from Agenus, Alkermes, Arcus Biosciences, Checkmate Pharmaceuticals, Compugen, Immunocore, and Synthekine. K.W.W. serves on the scientific advisory board of T-Scan Therapeutics, SQZ Biotech, Nextechinvest and receives sponsored research funding from Novartis. He is a co-founder of Immunitas, a biotech company. None of these represent a conflict of interest pertaining to the presented work. P.H., J.C.M., and B.I. filed a patent pertaining to the presented work.
References
- 1.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph R, Weber JS, et al. (2019). Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Annals of Oncology: Official Journal of the European Society for Medical Oncology 30, 582–588. 10.1093/annonc/mdz011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob J-J, Rutkowski P, Lao CD, Cowey CL, Schadendorf D, Wagstaff J, Dummer R, et al. (2019). Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. The New England Journal of Medicine 381, 1535–1546. 10.1056/NEJMoa1910836. [DOI] [PubMed] [Google Scholar]
- 3.Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. (2016). Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 167, 397–404 e399. 10.1016/j.cell.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. (2016). Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med 375, 819–829. 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane J-P, Bjorgaard SL, Hammond MR, Vitzthum H, Blackmon SM, et al. (2017). Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nature Communications 8, 1136. 10.1038/s41467017-01062-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kalbasi A, Tariveranmoshabad M, Hakimi K, Kremer S, Campbell KM, Funes JM, Vega-Crespo A, Parisi G, Champekar A, Nguyen C, et al. (2020). Uncoupling interferon signaling and antigen presentation to overcome immunotherapy resistance due to JAK1 loss in melanoma. Sci Transl Med 12. 10.1126/scitranslmed.abb0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su M-J, Melms JC, Leeson R, Kanodia A, Mei S, Lin J-R, et al. (2018). A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175, 984–997.e924. 10.1016/j.cell.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frangieh CJ, Melms JC, Thakore PI, Geiger-Schuller KR, Ho P, Luoma AM, Cleary B, Jerby-Arnon L, Malu S, Cuoco MS, et al. (2021). Multimodal pooled Perturb-CITE-seq screens in patient models define mechanisms of cancer immune evasion. Nat Genet 53, 332–341. 10.1038/s41588-021-00779-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y, Fan JL, Melms JC, Amin AD, Georgis Y, Barrera I, Ho P, Tagore S, AbrilRodríguez G, He S, et al. (2023). Multimodal single-cell and whole-genome sequencing of small, frozen clinical specimens. Nature Genetics 55, 19–25. 10.1038/s41588-022-01268-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tiefenthaler G, Hünig T, Dustin ML, Springer TA, and Meuer SC (1987). Purified lymphocyte function-associated antigen-3 and T11 target structure are active in CD2-mediated T cell stimulation. European Journal of Immunology 17, 1847–1850. 10.1002/eji.1830171227. [DOI] [PubMed] [Google Scholar]
- 11.Shaw S, Ginther Luce GE, Quinones R, Gress RE, Springer TA, and Sanders ME (1986). Two antigen-independent adhesion pathways used by human cytotoxic T-cell clones. Nature 323, 262–264. 10.1038/323262a0. [DOI] [PubMed] [Google Scholar]
- 12.Dustin ML, Sanders ME, Shaw S, and Springer TA (1987). Purified lymphocyte function-associated antigen 3 binds to CD2 and mediates T lymphocyte adhesion. The Journal of Experimental Medicine 165, 677–692. 10.1084/jem.165.3.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Selvaraj P, Plunkett ML, Dustin M, Sanders ME, Shaw S, and Springer TA (1987). The T lymphocyte glycoprotein CD2 binds the cell surface ligand LFA-3. Nature 326, 400–403. 10.1038/326400a0. [DOI] [PubMed] [Google Scholar]
- 14.Seed B (1987). An LFA-3 cDNA encodes a phospholipid-linked membrane protein homologous to its receptor CD2. Nature 329, 840–842. 10.1038/329840a0. [DOI] [PubMed] [Google Scholar]
- 15.Wallner BP, Frey AZ, Tizard R, Mattaliano RJ, Hession C, Sanders ME, Dustin ML, and Springer TA (1987). Primary structure of lymphocyte function-associated antigen 3 (LFA-3). The ligand of the T lymphocyte CD2 glycoprotein. The Journal of Experimental Medicine 166, 923–932. 10.1084/jem.166.4.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ariel O, Levi Y, and Hollander N (2009). Signal transduction by CD58: the transmembrane isoform transmits signals outside lipid rafts independently of the GPI-anchored isoform. Cellular Signalling 21, 1100–1108. 10.1016/j.cellsig.2009.02.022. [DOI] [PubMed] [Google Scholar]
- 17.Ariel O, Kukulansky T, Raz N, and Hollander N (2004). Distinct membrane localization and kinase association of the two isoforms of CD58. Cellular Signalling 16, 667–673. 10.1016/j.cellsig.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 18.Majzner RG, Frank MJ, Mount C, Tousley A, Kurtz DM, Sworder B, Murphy KA, Manousopoulou A, Kohler K, Rotiroti MC, et al. (2020). CD58 Aberrations Limit Durable Responses to CD19 CAR in Large B Cell Lymphoma Patients Treated with Axicabtagene Ciloleucel but Can be Overcome through Novel CAR Engineering. Blood 136, 53–54. 10.1182/blood-2020-139605. [DOI] [Google Scholar]
- 19.Pozniak J, Pedri D, Landeloos E, Herck YV, Antoranz A, Karras P, Nowosad A, Makhzami S, Bervoets G, Dewaele M, et al. (2022). A TCF4/BRD4-dependent regulatory network confers cross-resistance to targeted and immune checkpoint therapy in melanoma. bioRxiv. 10.1101/2022.08.11.502598. [DOI] [Google Scholar]
- 20.Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, et al. (2018). Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 175, 998–1013 e1020. 10.1016/j.cell.2018.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mathewson ND, Ashenberg O, Tirosh I, Gritsch S, Perez EM, Marx S, Jerby-Arnon L, Chanoch-Myers R, Hara T, Richman AR, et al. (2021). Inhibitory CD161 receptor identified in glioma-infiltrating T cells by single-cell analysis. Cell 184, 1281–1298 e1226. 10.1016/j.cell.2021.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Osborn L, Day ES, Miller GT, Karpusas M, Tizard R, Meuer SC, and Hochman PS (1995). Amino acid residues required for binding of lymphocyte function-associated antigen 3 (CD58) to its counter-receptor CD2. The Journal of Experimental Medicine 181, 429–434. 10.1084/jem.181.1.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arulanandam AR, Moingeon P, Concino MF, Recny MA, Kato K, Yagita H, Koyasu S, and Reinherz EL (1993). A soluble multimeric recombinant CD2 protein identifies CD48 as a low affinity ligand for human CD2: divergence of CD2 ligands during the evolution of humans and mice. Journal of Experimental Medicine 177, 1439–1450. 10.1084/jem.177.5.1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, and Gajewski TF (2009). Chemokine Expression in Melanoma Metastases Associated with CD8+ T-Cell Recruitment. Cancer Research 69, 3077–3085. 10.1158/0008-5472.CAN-08-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34, 184–191. 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mezzadra R, Sun C, Jae LT, Gomez-Eerland R, de Vries E, Wu W, Logtenberg MEW, Slagter M, Rozeman EA, Hofland I, et al. (2017). Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature 549, 106–110. 10.1038/nature23669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burr ML, Sparbier CE, Chan Y-C, Williamson JC, Woods K, Beavis PA, Lam EYN, Henderson MA, Bell CC, Stolzenburg S, et al. (2017). CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature 549, 101–105. 10.1038/nature23643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fujibayashi A, Taguchi T, Misaki R, Ohtani M, Dohmae N, Takio K, Yamada M, Gu J, Yamakami M, Fukuda M, et al. (2008). Human RME-8 is involved in membrane trafficking through early endosomes. Cell Struct Funct 33, 35–50. 10.1247/csf.07045. [DOI] [PubMed] [Google Scholar]
- 30.Tu X, Qin B, Zhang Y, Zhang C, Kahila M, Nowsheen S, Yin P, Yuan J, Pei H, Li H, et al. (2019). PD-L1 (B7-H1) Competes with the RNA Exosome to Regulate the DNA Damage Response and Can Be Targeted to Sensitize to Radiation or Chemotherapy. Molecular Cell 74, 1215–1226.e1214. 10.1016/j.molcel.2019.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Billaud M, Rousset F, Calender A, Cordier M, Aubry JP, Laisse V, and Lenoir GM (1990). Low expression of lymphocyte function-associated antigen (LFA)-1 and LFA-3 adhesion molecules is a common trait in Burkitt’s lymphoma associated with and not associated with Epstein-Barr virus. Blood 75, 1827–1833. 10.1182/blood.V75.9.1827.1827. [DOI] [PubMed] [Google Scholar]
- 32.Challa-Malladi M, Lieu YK, Califano O, Holmes AB, Bhagat G, Murty VV, Dominguez-Sola D, Pasqualucci L, and Dalla-Favera R (2011). Combined genetic inactivation of β2-Microglobulin and CD58 reveals frequent escape from immune recognition in diffuse large B cell lymphoma. Cancer Cell 20, 728–740. 10.1016/j.ccr.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schneider M, Schneider S, Zühlke-Jenisch R, Klapper W, Sundström C, Hartmann S, Hansmann M-L, Siebert R, Küppers R, and Giefing M (2015). Alterations of the CD58 gene in classical Hodgkin lymphoma. Genes, Chromosomes & Cancer 54, 638–645. 10.1002/gcc.22276. [DOI] [PubMed] [Google Scholar]
- 34.Abdul Razak FR, Diepstra A, Visser L, and van den Berg A (2016). CD58 mutations are common in Hodgkin lymphoma cell lines and loss of CD58 expression in tumor cells occurs in Hodgkin lymphoma patients who relapse. Genes and Immunity 17, 363–366. 10.1038/gene.2016.30. [DOI] [PubMed] [Google Scholar]
- 35.Demetriou P, Abu-Shah E, Valvo S, McCuaig S, Mayya V, Kvalvaag A, Starkey T, Korobchevskaya K, Lee LYW, Friedrich M, et al. (2020). A dynamic CD2-rich compartment at the outer edge of the immunological synapse boosts and integrates signals. Nature Immunology 21, 1232–1243. 10.1038/s41590-020-0770-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prokhnevska N, Cardenas MA, Valanparambil RM, Sobierajska E, Barwick BG, Jansen C, Reyes Moon A, Gregorova P, delBalzo L, Greenwald R, et al. (2023). CD8+ T cell activation in cancer comprises an initial activation phase in lymph nodes followed by effector differentiation within the tumor. Immunity 56, 107–124.e105. 10.1016/j.immuni.2022.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schluck M, Eggermont LJ, Weiden J, Popelier C, Weiss L, Pilzecker B, Kolder S, Heinemans A, Rodriguez Mogeda C, Verdoes M, et al. (2022). Dictating Phenotype, Function, and Fate of Human T Cells with Co-Stimulatory Antibodies Presented by Filamentous Immune Cell Mimics. Advanced Therapeutics. 10.1002/adtp.202200019. [DOI] [Google Scholar]
- 38.Leitner J, Herndler-Brandstetter D, Zlabinger GJ, Grubeck-Loebenstein B, and Steinberger P (2015). CD58/CD2 Is the Primary Costimulatory Pathway in Human CD28-CD8+ T Cells. Journal of Immunology (Baltimore, Md.: 1950) 195, 477–487. 10.4049/jimmunol.1401917. [DOI] [PubMed] [Google Scholar]
- 39.Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, Sasmal DK, Huang J, Kim JM, Mellman I, and Vale RD (2017). T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science (New York, N.Y.) 355, 1428–1433. 10.1126/science.aaf1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, Konieczny BT, Daugherty CZ, Koenig L, Yu K, et al. (2017). Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science (New York, N.Y.) 355, 1423–1427. 10.1126/science.aaf0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seung E, Xing Z, Wu L, Rao E, Cortez-Retamozo V, Ospina B, Chen L, Beil C, Song Z, Zhang B, et al. (2022). A trispecific antibody targeting HER2 and T cells inhibits breast cancer growth via CD4 cells. Nature. 10.1038/s41586-022-04439-0. [DOI] [PubMed] [Google Scholar]
- 42.Xu S, Wen Z, Jiang Q, Zhu L, Feng S, Zhao Y, Wu J, Dong Q, Mao J, and Zhu Y (2015). CD58, a novel surface marker, promotes self-renewal of tumor-initiating cells in colorectal cancer. Oncogene 34, 1520–1531. 10.1038/onc.2014.95. [DOI] [PubMed] [Google Scholar]
- 43.Martinez-Morilla S, Zugazagoitia J, Wong PF, Kluger HM, and Rimm DL (2020). Quantitative analysis of CMTM6 expression in tumor microenvironment in metastatic melanoma and association with outcome on immunotherapy. Oncoimmunology 10, 1864909. 10.1080/2162402X.2020.1864909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zugazagoitia J, Liu Y, Toki M, McGuire J, Ahmed FS, Henick BS, Gupta R, Gettinger SN, Herbst RS, Schalper KA, and Rimm DL (2019). Quantitative Assessment of CMTM6 in the Tumor Microenvironment and Association with Response to PD-1 Pathway Blockade in Advanced-Stage Non–Small Cell Lung Cancer. Journal of Thoracic Oncology 14, 2084–2096. 10.1016/j.jtho.2019.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peng Q-H, Wang C-H, Chen H-M, Zhang R-X, Pan Z-Z, Lu Z-H, Wang G-Y, Yue X, Huang W, and Liu R-Y (2021). CMTM6 and PD-L1 coexpression is associated with an active immune microenvironment and a favorable prognosis in colorectal cancer. Journal for ImmunoTherapy of Cancer 9. 10.1136/jitc-2020-001638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao Y, Zhang M, Pu H, Guo S, Zhang S, and Wang Y (2020). Prognostic Implications of PanCancer CMTM6 Expression and Its Relationship with the Immune Microenvironment. Front Oncol 10, 585961. 10.3389/fonc.2020.585961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Y, Moriyama T, Yoshimura S, Zhao X, Li Z, Yang X, Paietta E, Litzow MR, Konopleva M, Yu J, et al. (2022). PAX5 epigenetically orchestrates CD58 transcription and modulates blinatumomab response in acute lymphoblastic leukemia. Sci Adv 8, eadd6403. 10.1126/sciadv.add6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR-Cas9 knockout screens. Genome Biology 15. 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY, et al. (2013). The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat Methods 10, 730–736. 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, Yuan D, Stroe O, Wood G, Laydon A, et al. (2022). AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Research 50, D439–D444. 10.1093/nar/gkab1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perez-Riverol Y, Bai J, Bandla C, Garcia-Seisdedos D, Hewapathirana S, Kamatchinathan S, Kundu DJ, Prakash A, Frericks-Zipper A, Eisenacher M, et al. (2022). The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res 50, D543–D552. 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roth TL, Puig-Saus C, Yu R, Shifrut E, Carnevale J, Li PJ, Hiatt J, Saco J, Krystofinski P, Li H, et al. (2018). Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 559, 405–409. 10.1038/s41586-018-0326-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sanson KR, Hanna RE, Hegde M, Donovan KF, Strand C, Sullender ME, Vaimberg EW, Goodale A, Root DE, Piccioni F, and Doench JG (2018). Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat Commun 9, 5416. 10.1038/s41467-018-07901-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Doench JG, Hartenian E, Graham DB, Tothova Z, Hegde M, Smith I, Sullender M, Ebert BL, Xavier RJ, and Root DE (2014). Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotechnol 32, 1262–1267. 10.1038/nbt.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee Y-R, Kang W, and Kim Y-M (2016). Detection of Interaction Between Toll-Like Receptors and Other Transmembrane Proteins by Co-immunoprecipitation Assay. In Toll-Like Receptors, pp. 107–120. 10.1007/978-1-4939-3335-8_7. [DOI] [PubMed] [Google Scholar]
- 57.Meier F, Brunner AD, Koch S, Koch H, Lubeck M, Krause M, Goedecke N, Decker J, Kosinski T, Park MA, et al. (2018). Online Parallel Accumulation-Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer. Mol Cell Proteomics 17, 2534–2545. 10.1074/mcp.TIR118.000900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stirling DR, Swain-Bowden MJ, Lucas AM, Carpenter AE, Cimini BA, and Goodman A (2021). CellProfiler 4: improvements in speed, utility and usability. BMC Bioinformatics 22. 10.1186/s12859-021-04344-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Science Signaling 6. 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discovery 2, 401–404. 10.1158/2159-8290.Cd-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gayoso A, Steier Z, Lopez R, Regier J, Nazor KL, Streets A, and Yosef N (2021). Joint probabilistic modeling of single-cell multi-omic data with totalVI. Nature Methods 18, 272–282. 10.1038/s41592-020-01050-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine J-C, Geurts P, Aerts J, et al. (2017). SCENIC: single-cell regulatory network inference and clustering. Nature Methods 14, 1083–1086. 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 35733587.e3529. 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, and Mann M (2011). Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10, 1794–1805. 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- 66.Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnology 26, 1367–1372. 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 67.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, and Cox J (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13, 731–740. 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 68.Arulanandam AR, Kister A, McGregor MJ, Wyss DF, Wagner G, and Reinherz EL (1994). Interaction between human CD2 and CD58 involves the major beta sheet surface of each of their respective adhesion domains. The Journal of Experimental Medicine 180, 1861–1871. 10.1084/jem.180.5.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. CD58 up- and down-signature programs, Related to Figure 1
Table S2. MAGeCK summary of gene KO enrichment from A375 genome-wide CRISPR-Cas9 screen to identify regulators of CD58, Related to Figure 4
Table S3. MAGeCK summary of sgRNA enrichment from A375 genome-wide CRISPR-Cas9 screen to identify regulators of CD58, Related to Figure 4
Table S4. Mass spectrometry of IgG v. CD58 IP significantly changed candidates, Related to Figure 5
Data Availability Statement
CRISPR-Cas9 KO screen data has been deposited at GEO and is publicly available as of the date of publication. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository52 and is publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper analyzes existing, publicly available data. The accession numbers for these datasets are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal mouse anti-ARF6 AF488 (clone 3A-1) | Santa Cruz Biotechnology | Cat#sc-7971 AF488 |
| Monoclonal mouse anti-Calnexin AF488 (clone AF18) | ThermoFisher Scientific | Cat#MA3–027-A488 |
| Monoclonal mouse anti-GM130 AF488 (clone 35/GM130) | BD Biosciences | Cat#560257 |
| Monoclonal mouse anti-TfR AF488 (clone #29806) | R&D Systems | Cat#FAB2474G-100UG |
| Polyclonal rabbit anti-TGN46 AF488 | Novus Biologicals | Cat#NBP1–49643AF488 |
| Polyclonal sheep anti-EEA1 AF488 | R&D Systems | Cat#IC8047G |
| Polyclonal sheep anti-LAMP1 AF488 | R&D Systems | Cat#IC7985G |
| Polyclonal goat anti-Mouse IgG (H+L) AF594 | ThermoFisher Scientific | Cat#A-11032 |
| Polyclonal goat anti-Rabbit (gG (H+L) AF647 | ThermoFisher Scientific | Cat#A-21245 |
| Monoclonal mouse anti-CD8 (clone 4B11) | Leica | Cat#CD8–4B11-L-CE |
| Monoclonal mouse anti-CD58 (clone TS2/9) | ThermoFisher Scientific | Cat#MA5800 |
| Monoclonal mouse anti-Ki-67 (MIB-1) | Nordic MUbio | Cat#KI500 |
| Monoclonal anti-mouse CD16/32 (clone 93) | Biolegend | Cat#101302 |
| Monoclonal mouse anti-CD2 BV605 (clone RPA-2.10) | Biolegend | Cat#300223 |
| Monoclonal mouse anti-CD279 (PD-1) APC (clone EH12.2H7) | Biolegend | Cat#329908 |
| Monoclonal mouse anti-CD3 PE-Cy7 (clone HIT3a) | Biolegend | Cat#300316 |
| Monoclonal mouse anti-CD4 AF700 (clone RPA-T4) | Biolegend | Cat#300526 |
| Monoclonal mouse anti-CD45RA Brilliant Violet 711 (clone HI100) | Biolegend | Cat#304137 |
| Monoclonal mouse anti-CD45RO Brilliant Violet 570 (clone UCHL1) | Biolegend | Cat#304226 |
| Monoclonal mouse anti-CD48 FITC (clone BJ40) | Biolegend | Cat#336705 |
| Monoclonal mouse anti-CD58 APC (clone TS2/9) | Biolegend | Cat#330918 |
| Monoclonal mouse anti-CD58 BV421 (clone 1C3) | BD Biosciences | Cat#564363 |
| Monoclonal mouse anti-CD58 PE-Cy5 (clone TS2/9) | Biolegend | Cat#330909 |
| Monoclonal mouse anti-CD8 AF488 (clone HIT8a) | Biolegend | Cat#300916 |
| Monoclonal mouse anti-Granzyme B PE-CF594 (clone GB11) | BD Biosciences | Cat#562462 |
| Monoclonal mouse anti-human CD45 Pacific Blue (clone HI30) | Biolegend | Cat#304022 |
| Monoclonal mouse anti-HLA A,B,C BV605 (clone W6/32) | Biolegend | Cat#311432 |
| Monoclonal mouse anti-PD-L1 APC (clone 29E.2A3) | Biolegend | Cat#329708 |
| Monoclonal mouse anti-PD-L1 BV785 (clone 29E.2A3) | Biolegend | Cat#329736 |
| Monoclonal rat anti-Ki-67 eFluor506 (clone SolA15) | ThermoFisher Scientific | Cat#69-5698-82 |
| Monoclonal rat anti-mouse CD45 BV510 (clone 30-F11) | Biolegend | Cat#103138 |
| Monoclonal mouse anti-CD58 (clone TS2/9) | Biolegend | Cat#330923 |
| Monoclonal rat anti-CD2 (LO-CD2b) | Laboratory of Megan Sykes | n/a |
| Monoclonal mouse anti-CD28 (clone CD28.2) | ThermoFisher Scientific | Cat#16-0289-81 |
| Monoclonal mouse anti-CD3 (clone OKT3) | Miltenyi Biotec | Cat#130-093-387 |
| Monoclonal mouse anti-PD-L1 (clone 29E.2A3) | Bio X Cell | Cat#BE0285R001MG |
| Mouse IgG2b isotype control (clone MPC-11) | Bio X Cell | Cat#BE0086R001MG |
| Mouse IgG4, κ isotype control (clone QA16A15) | Biolegend | Cat#403701 |
| Rat IgG2b negative control (clone LTF-2) | EMD Millipore | Cat#MABF1076Z |
| APC Mouse IgG1, κ Isotype Ctrl Antibody (clone MOPC-21) | Biolegend | Cat#981806 |
| APC Mouse IgG2b, κ Isotype Ctrl Antibody (clone MPC-11) | Biolegend | Cat#400319 |
| Brilliant Violet 421™ Mouse IgG2a, κ Isotype Ctrl Antibody (clone MOPC-173) | Biolegend | Cat#400259 |
| Brilliant Violet 605™ Mouse IgG2a, κ Isotype Ctrl Antibody (clone MOPC-173) | Biolegend | Cat#400269 |
| Brilliant Violet 785™ Mouse IgG2b, κ Isotype Ctrl Antibody (clone MPC-11) | Biolegend | Cat#400355 |
| FITC Mouse IgG1, κ Isotype Ctrl (clone MOPC-21) | Biolegend | Cat#981802 |
| Monoclonal mouse anti-CD58 (clone TS2/9) | Abcam | Cat#ab171087 |
| Monoclonal mouse anti-His (clone 27E8) | Cell Signaling Technology | Cat#2366 |
| Monoclonal mouse anti-PD-L1 (clone 405.9A11) | Cell Signaling Technology | Cat#29122 |
| Polyclonal rabbit anti-CMTM6 | MilliporeSigma | Cat#HPA026980 |
| Polyclonal rabbit anti-CMTM6 | Invitrogen | Cat#PA5–55472 |
| Monoclonal rabbit anti-PD-L1 (clone E1L3N) | Cell Signaling Technology | Cat#13684 |
| Monoclonal rabbit anti-V5 (clone D3H8Q) | Cell Signaling Technology | Cat#13202 |
| Monoclonal rabbit anti-CD58 (clone EP15041) | Abcam | Cat#ab196648 |
| Monoclonal rabbit anti-GAPDH (clone 14C10) | Cell Signaling Technology | Cat#2118 |
| IRDye 680RD Goat anti-Mouse IgG (H+L) | Li-Cor | Cat#926–68070 |
| IRDye 680RD Goat anti-Rabbit IgG (H+L) | Li-Cor | Cat#926–68071 |
| IRDye 800CW Goat anti-Mouse IgG (H+L) | Li-Cor | Cat#926–32210 |
| Bacterial and virus strains | ||
| Human CRISPR Knockout Pooled Library (Brunello) | Doench et al.26 | Addgene Virus #73178-LV |
| Chemicals, peptides, and recombinant proteins | ||
| Accutase | Innovative Cell Technologies | Cat#AT104 |
| ACK Lysing Buffer | Quality Biological | Cat#118-156-101 |
| BOND Epitope Retrieval Solution 1 | Leica Biosystems | Cat#AR9961 |
| BONE Epitope Retrieval Solution 2 | Leica Biosystems | Cat#AR9640 |
| Cas9-NLS purified protein | QB3 MacroLab | N/A |
| CellEvent Caspase-3/7 Detection Reagent | ThermoFisher Scientific | Cat#C10423 |
| CFSE | ThermoFisher Scientific | Cat#C34570 |
| Chloroquine diphosphate | Sigma Aldrich | Cat#C6628–25G |
| Collagenase D | Sigma Aldrich | Cat#11088866001 |
| Concanamycin A | Santa Cruz Biotechnology | Cat#sc-202111 |
| Digitonin | Sigma-Aldrich | Cat#D141 |
| DNase I | Sigma Aldrich | Cat#10104159001 |
| Ex Taq DNA Polymerase | TaKaRa | Cat#RR001C |
| Gateway BP Clonase II Enzyme Mix | ThermoFisher Scientific | Cat#11789020 |
| Gateway LR Clonase iI Enzyme Mix | ThermoFisher Scientific | Cat#11791020 |
| Halt Protease and Phosphatase Inhibitor Cocktail | ThermoFisher Scientific | Cat#76445 |
| Human IL-2 | Chiron | Cat#53905-991-01 |
| Insulin-Transferrin-Selenium | ThermoFisher Scientific | Cat#41400–045 |
| Laemmli SDS-Sample Buffer | Boston BioProducts | Cat#BP-111R |
| Matrigel Basement Membrane Matrix, LDEV Free | Corning | Cat#354234 |
| Matrigel Matrix High Concentration, Phenol-Red Free, LDEVfree | Corning | Cat#354262 |
| Opti-MEM I Reduced Serum Medium | ThermoFisher Scientific | Cat#31985062 |
| Polybrene Infection/Transfection Reagent | Sigma-Aldirch | Cat#TR-1003 |
| Recombinant CD58 His-tag protein | VWR | Cat#10797–676 |
| Recombinant CD58/LFA-3 Fc chimera protein | R&D Systems | Cat#10068-CD-050 |
| Recombinant human CMTM6 GST (N-term) protein | Novus Biologicals | Cat#H00054918-P01 |
| Recombinant human IFN-gamma | Abcam | Cat#ab9659 |
| Recombinant human PD-L1/B7-H1 His-tag protein | R&D Systems | Cat#9049-B7–100 |
| RIPA buffer | Sigma Aldrich | Cat#R0278 |
| TaqMan Fast Advanced Master Mix | ThermoFisher Scientific | Cat#4444557 |
| TransIT-LT1 Transfection Reagent | Mirus Bio | Cat#MIR 2300 |
| Zombie NIR | Biolegend | Cat#423105 |
| Zombie Violet | Biolegend | Cat#423113 |
| Critical commercial assays | ||
| Duolink In Situ Detection Reagents FarRed | Sigma-Aldrich | Cat#DUO92013 |
| IFN-gamma ELISA kit | Biolegend | Cat#430104 |
| IL-2 ELISA kit | Biolegend | Cat#431804 |
| QuikChange XL Site-Directed Mutagenesis Kit | Agilent | Cat#200516 |
| SF Cell Line Kit 4D X Kit | Lonza | Cat#197174 |
| Opal 6-Plex Detection Kit - for Whole Slide Imaging | Akoya Biosciences | Cat#NEL871001KT |
| ACTB TaqMan Gene Expression Assay | ThermoFisher Scientific | Assay ID: Hs99999903_m1 |
| CD274 TaqMan Gene Expression Assay | ThermoFisher Scientific | Assay ID: Hs00204257_m1 |
| CD58 TaqMan Gene Expression Assay | ThermoFisher Scientific | Assay ID: Hs01560660_m1 |
| GAPDH TaqMan Gene Expression Assay | ThermoFisher Scientific | Assay ID: Hs02786624 |
| Deposited data | ||
| A375 CD58 CRISPR/Cas9 KO screen data | This paper | GEO: GSE225235 |
| Melanoma scRNA-seq | Sade-Feldman et al.20 | GEO: GSE120575 |
| Melanoma scRNA-seq | Jerby-Arnon et al.7 | GEO: GSE115978 |
| Melanoma scRNA-seq | Pozniak et al.19 | EGA: EGAS00001006488 |
| A375 and 2686 CD58 IP mass spectrometry data | This paper | Proteome Xchange: PXD039710 |
| Experimental models: Cell lines | ||
| Human: 2686 | MD Anderson Cancer Center | N/A |
| Human: MaMel134 | UK-Essen | N/A |
| Human: A375 | ATCC | CRL-1619 |
| Human: WM852 | Rockland | WM852-01-0001 |
| Human: Jurkat | ATCC | TIB-152 |
| Human: HEK293T | ATCC | CRL-3216 |
| Experimental models: Organisms/strains | ||
| Mouse: NOD.Cg-Prkdcscid II2rgtm1Sug Tg(CMV-IL2)4–2Jic/JicTac | Taconic | Taconic: 13440-F |
| Mouse: NOD.Cg-Prkdcscid II2rgtm1Wjl/SzJ | The Jackson Laboratory | JAX: 005557 |
| Biological samples | ||
| Human 2686 TILs | MD Anderson Cancer Center | N/A |
| Human MaMel134 TILs | UK-Essen | N/A |
| Patient melanoma FFPE blocks | New York Presbyterian Hospital/Columbia University Irving Medical Center | N/A |
| Oligonucleotides | ||
| QuikChange CD58 K34A F: TTTTCCAGTTCTGCAACTGCATCCTTTTGTTTTTTCCATAGGACCTCIIIIAA | This paper | N/A |
| QuikChange CD58 K34A R: TTAAAAGAGGTCCTATGGAAAAAACAAAAGGATGCAGTTG CAGAACT GGAAAA | This paper | N/A |
| P5 and P7 primers (See Table S5) | Doench et al. | N/A |
| CD58 crRNA: AATGCTCTGGTATCATGCAT | This paper | N/A |
| CD274 crRNA: ACTGCTTGTCCAGATGACTT | This paper | N/A |
| B2M crRNA: GAGTAGCGCGAGCACAGCTA | This paper | N/A |
| CD2 crRNA: CGATGATCAGGATATCTACA | This paper | N/A |
| CMTM6 crRNA: GGTGTACAGCCCCACTACGG | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: pLEX307 CD58 TM | This paper | N/A |
| Plasmid: pLEX307_CD58_GPI | This paper | N/A |
| Plasmid: pLEX307_CD58_TM_K34A | This paper | N/A |
| Plasmid: pLEX307_CD58_GPI_K34A | This paper | N/A |
| Plasmid: pLEX307_CMTM6 | This paper | N/A |
| Plasmid: pLEX307_CMTM6_V5 | This paper | N/A |
| Plasmid: pLEX307_CMTM6_Ctr_V5 | This paper | N/A |
| Plasmid: pLEX307_CMTM6_Ntr_V5 | This paper | N/A |
| Plasmid: pLEX307_CMTM6_EC1mut_V5 | This paper | N/A |
| Plasmid: pLEX307_CMTM6_EC2mut_V5 | This paper | N/A |
| Plasmid: pLEX307_CMTM6_EC1&2mut_V5 | This paper | N/A |
| Plasmid: pLX311_CD274 | This paper | N/A |
| Plasmid: pLX311_CD274_H1Amut | This paper | N/A |
| Plasmid: pXPR-011 | Doench et al.55 | Addgene #59702 |
| Plasmid: pLEX_307 | Laboratory of David Root | Addgene #41392 |
| Plasmid: pLX_TRC311 | Laboratory of John Doench | Addgene #113668 |
| Plasmid: pDONR221 | ThermoFisher Scientific | Cat#12536017 |
| Plasmid: pxsPAX2 | Laboratory of Dider Trono | Addgene #12260 |
| Plasmid: pMD2.G | Laboratory of Dider Trono | Addgene #12259 |
| Plasmid: pLX_311-Cas9 | Doench et al.55 | Addgene #96924 |
| Software and algorithms | ||
| ImageJ | Schneider et al.58 | https://imagej.net |
| FlowJo v10.8.1 | BD Biosciences | https://www.flowjo.com |
| CellProfiler v4.2.1 | Stirling et al.59 | https://cellprofiler.org |
| Prism v9.5.0 | GraphPad | https://www.graphpad.com |
| MAGeCK v0.5.9.2 | Li et al.48 | https://sourceforge.net/p/mageck/wiki/Home/ |
| InForm v2.5.1 | Akoya Biosciences | https://www.akoyabio.com/phenoimager/software/inform-tissue-finder/ |
| Image Studio Lite v5.2.5 | Li-Cor | https://www.licor.com/bio/image-studio/ |
| MaxQuant v2.0.1.0 | Cox and Mann66 | https://www.maxquant.org/ |
| Perseus | Tyanova et al.67 | https://maxquant.net/perseus/ |
| Andromeda | Cox et al.65 | https://bioinformaticshome.com/tools/proteomics/descriptions/Andromeda.html |
| SnapGene 6.0.5 | Dotmatics | https://www.snapgene.com |
| R v4.1.1 | The R Project | https://www.rproject.org |
| AUCell v1.20.2 | Aibar et al.63 | https://bioconductor.org/packages/release/bioc/html/AUCell.html |
| Seurat v4.1.0 | Hao et al.64 | https://satijalab.org/seurat/ |
| ZEN v3.4.91 | Zeiss | https://www.zeiss.com/microscopy/en/products/software/zeisszen.html |
| Other | ||
| SPRIselect beads | Beckman Coulter | Cat# B23318 |
| Protein A Dynabeads | ThermoFisher Scientific | Cat#10006D |
| Protein G Dynabeads | ThermoFisher Scientific | Cat#10003D |
| 8 well chambered cover glass | Cellvis | Cat#C8–1.5H-N |