

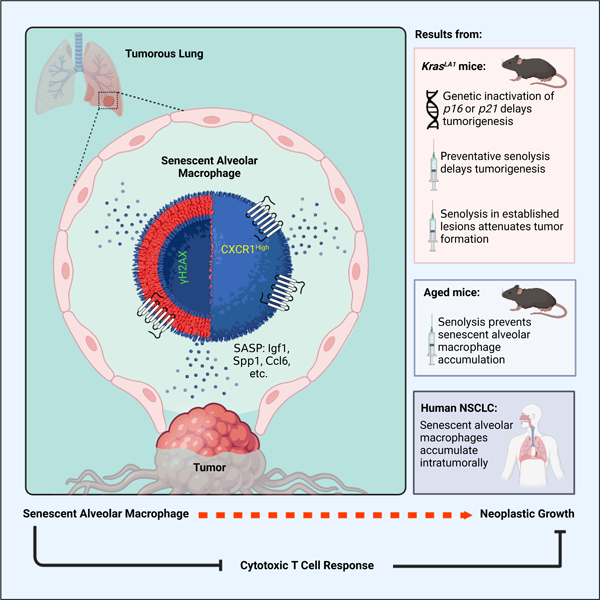
Summary
Senescent cells play relevant but context-dependent roles during tumorigenesis. Here, in an oncogenic Kras-driven lung cancer mouse model, we found that senescent cells, specifically alveolar macrophages, accumulate early in neoplasia. These macrophages have upregulated expression of p16INK4a and Cxcr1, are distinct from previously defined subsets and sensitive to senolytic interventions, and suppress cytotoxic T cell responses. Their removal attenuates adenoma development and progression in mice, indicating their tumorigenesis-promoting role. Importantly, we found that alveolar macrophages with these properties increase with normal aging in mouse lung and in human lung adenocarcinoma in situ. Collectively, our study indicates that a subset of tissue resident macrophages can support neoplastic transformation through altering their local microenvironment, suggesting that therapeutic interventions targeting senescent macrophages may attenuate lung cancer progression during early stages of disease.
Keywords: Cellular Senescence, Alveolar Macrophages, Senolytic, Aging, Cytotoxic T cells
Graphical Abstract
eTOC Blurb
Prieto et al. causally implicate the cellular senescence program in the innate component of the immune system. Senescent alveolar macrophages with tumor-promoting qualities accumulate early in Kras-driven neoplasia, promoting tumorigenesis by adjusting the antitumor T cell response. These senescent macrophages also accumulate with natural age and occur in human NSCLC.
Introduction
Cellular senescence, a hallmark of aging and cancer, is a dynamic biological program that stably arrests the proliferation of damaged cells, including those with excessive oncogenic signaling1,2. Oncogene-induced senescence (OIS) exit from the cell cycle is governed by the activity of tumor suppressor genes, including p53 and p16Ink4a (hereafter referred to p16), thereby preventing these impaired cells from progressing to tumors1,3,4. Additionally, senescent cells develop a secretome, referred to as the senescence-associated secretory phenotype or SASP, that can differ depending on the underlying cellular stressor5. The SASP consists of various cytokines, chemokines, proteases, and other biologically active molecules that functionally recruit cells of the immune system, which, in turn, can target senescent cells for elimination6,7. While senescence may intrinsically function as a protective program against tumorigenesis, senescent cell accumulation can be deleterious to tissue structure and function, likely in part due to SASP production, in promoting many age-related conditions, including cancer. To this end, co-injection of cancer cells with senescent cells in immunocompromised mice results in accelerated tumor growth compared to cancer cell injection alone, indicating that senescent cell-extrinsic factors can promote tumorigenesis8–10.
Senescent cells in vivo have been difficult to identify and characterize, as there is no unique marker established, despite extensive investigation. Additionally, senescent cell characteristics can vary depending on cell type, underlying senescence-inducing stressor, or duration of senescence arrest5,11. Commonly used tools rely on the elevated expression of the cyclin-dependent kinase inhibitors p16 or p21Cip1 (Cdkn1a; hereafter p21) in senescence from both murine and human tissues12–15. Notably, p16-expressing cells are observed in cancer mouse models, including transgenic, transplanted, and spontaneous tumors, and accumulate during the nascent steps of tumorigenesis13. However, it is poorly understood whether and how these cells directly influence tumor formation. To elucidate the role of p16-positive senescent cells in vivo, we have engineered a transgenic mouse model, termed INK-ATTAC, that allows for the selective elimination of p16-positive senescent cells upon the administration of the dimerizing agent AP20187 (hereafter AP)14,16. INK-ATTAC transgenic mice express drug-inducible Caspase 8 under the control of a minimal p16 promoter fragment that is highly active in senescent cells. In INK-ATTAC mouse embryonic fibroblasts (MEFs) immortalized by oncogenic SV40 expression, we found that despite very high p16 levels, the INK-ATTAC transgene is not induced, and AP treatment is unable to eliminate these cells16. These results suggested that neoplastic cells are unlikely to be targeted through AP-mediated elimination through the INK-ATTAC transgene.
Using INK-ATTAC, we have found that removing naturally occurring p16-positive cells from aged mice delays the onset of spontaneous cancer without changing their lifetime predisposition to tumors16. Here, we sought a deeper understanding of this phenomenon by crossing INK-ATTAC transgenic mice to an oncogenic Kras-driven mouse model of lung adenoma formation. In agreement with our naturally aged tumor study, selective senescent cell removal through both genetic and pharmacological approaches (senolytic) from Kras mice delays tumorigenesis. To our surprise, we found that global depletion of the tumor suppressor p16 in Kras mice also suppressed lung adenoma formation, suggesting that senescence occurring outside of neoplastic cells was impacting tumor initiation. Importantly, we identify a subset of tissue-resident macrophages that acquire senescence-associated alterations during oncogene-driven spontaneous tumorigenesis that promote tumor initiation and suppress cytotoxic T cell accumulation. Importantly, macrophages with these features also occur during normative aging in the mouse lung and in human adenocarcinoma in situ. Our results suggest that targeting macrophages that become senescent in response to neoplastic changes could delay the early stages of tumorigenesis.
Results
Senescent cells accumulate after somatic activation of oncogenic Kras
To mechanistically evaluate how senescence impacts neoplasia, we aimed to identify a tumor-prone mouse model that develops senescent cells reproducibly and reliably. KrasLA1 mice (hereafter Kras) are predisposed to early-onset lung tumorigenesis after spontaneous somatic recombination of an allele containing an oncogenic KrasG12D mutation17. Lung adenomas begin to form within the first week after birth and increase in size and multiplicity with age17. This model differs from other transgenic lines as it recapitulates the somatic oncogene activation observed in human non-small cell lung cancer (NSCLC) and results from random recombination events in vivo17. To evaluate whether senescent cells are generated in these animals, we micro-dissected lung lesions and macroscopically normal-appearing, non-dysplastic tissue from 8-week-old Kras mice. As a control, we also collected lung tissue from wild-type (WT) mice. We performed quantitative reverse transcription PCR (RT-qPCR) for well-established biomarkers of senescence, including pro-inflammatory factors that are linked SASP on RNA extracted from these tissues. We observed an increased expression of the cell cycle regulators p16, p19Arf (hereafter referred to p19), and p21 in the isolated lesions. Additionally, the oncogene-induced senescence marker Dec118 and pro-inflammatory SASP factors Il6, Tnfα, Tgfβ, and Mmp10 were also elevated in Kras mice (Figure 1A). We extended these results to the single cell level by employing RNA in situ hybridization for p16 and p21 transcripts in lung sections. We found that lesions from Kras mice had many cells positive for these genes, including cells positive for both markers (Figure 1B). This feature was absent from WT (Figure 1B) and ‘normal’ lung tissue from Kras mice. Next, we assessed where senescent cells were localized during lesion development. We stained lung sections for senescence-associated β-galactosidase (SA-β-Gal) activity, a classical hallmark of senescence that can be observed in vivo. We observed a significant accumulation of SA-β-Gal positive cells across all stages (hyperplasia, adenoma, and adenocarcinoma) of lesion development with a higher amount of SA-β-Gal positive cells in lesions representative of hyperplasia relative to the ‘normal’ area of Kras mice or WT lung tissue (Figure 1C). Collectively, the elevated expression of senescence-associated transcripts, as well as the accumulation of SA-β-Gal positive cells, provide evidence indicating that senescent cells accumulate in Kras lung lesions early during tumorigenesis.
Figure 1: Proinflammatory senescent cells accumulate in the lungs of KrasLA1 mice.
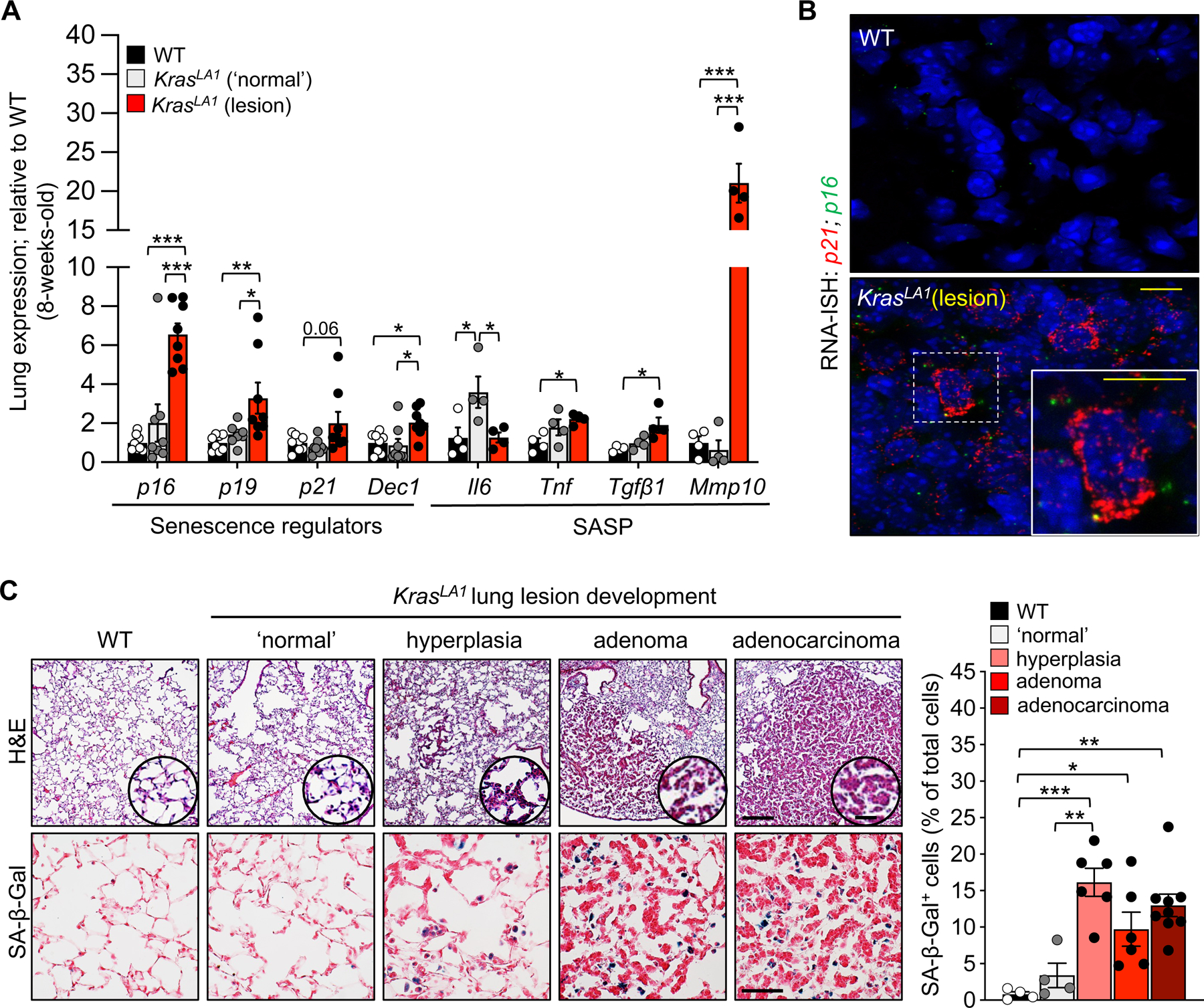
(A) Senescence-associated gene expression in 8-week-old wild-type (WT), KrasLA1 ‘normal’ (no lesion), and KrasLA1 lung lesion areas.
(B) Representative images of RNA in situ hybridization (RNA-ISH) to Cdkn1a (p21) and Cdkn2a (p16).
(C) Representative images of hematoxylin and eosin (upper, left) and senescence-associated β-galactosidase (SA-β-gal, lower, left) staining in WT lung and throughout KrasLA1 lung lesion development. Quantification of SA-β-gal staining (right).
Scale bars: [10μm (inset 5μm) B], [H&E 200μm (inset 25μm), SA-β-Gal 50μm C]. Data are means ± SEM. *P<0.05; **P<0.01; ***P<0.001; (one-way ANOVA with Tukey’s multiple comparison test in A, C).
Clearance of p16-positive senescent cells delay tumorigenesis
We next sought to address the mechanistic contribution of senescent cells to lung lesion formation. One approach to study this involves inactivating p16, a potent tumor suppressor and effector of senescence, from Kras mice to assess whether bypass of senescence impacts lesion development. As expected, wild-type, p16 knock-out mice had no lung lesions at eight weeks of age (data not shown). Surprisingly, knocking out p16 in the germline of Kras (Kras;p16KO) mice resulted in fewer lung lesions at eight weeks of age (Figure S1A), suggesting that p16-expressing cells may promote early lesion formation in Kras mice.
To mechanistically determine whether p16-positive senescent cells participate in lesion formation, we bred Kras mice to INK-ATTAC (hereafter ATTAC) transgenic mice to create cohorts of control (WT, ATTAC, and Kras) and experimental animals (ATTAC;Kras). To selectively remove p16-expressing senescent cells, we treated all mice twice per week with AP starting within the first week of birth until they reached eight weeks of age (Figure 2A). Kras mice lacking the ATTAC transgene reliably and repeatedly developed macroscopic lesions after eight weeks (Figure 2B). Treating ATTAC;Kras with AP from birth resulted in significantly fewer lesions (Figures 2B, 2C). WT and ATTAC mice treated with AP had no observable lesions (data not shown). Lesions that formed in Kras;p16KO or AP-treated ATTAC;Kras mice consistently had a reduced histopathology score reflective of lower cellularity and fewer papillary structures (Figure 2D), suggesting that senescent cell accumulation promotes tumor progression. As expected, we found that AP triggers cell death (Figure S1B) and attenuates the accumulation of SA-β-Gal positive cells specifically in the lesion area of Kras mice (Figure 2E). We also found that the number of cells positive for the senescence marker p21 was significantly reduced in AP-treated ATTAC;Kras lesions as detected by immunofluorescence staining (Figure 2F). Collectively, these results suggest that p16-expressing senescent cells promote adenoma formation and their clearance can attenuate the number and severity of those that form.
Figure 2: Removal of senescent cells in Kras mice delays tumorigenesis.
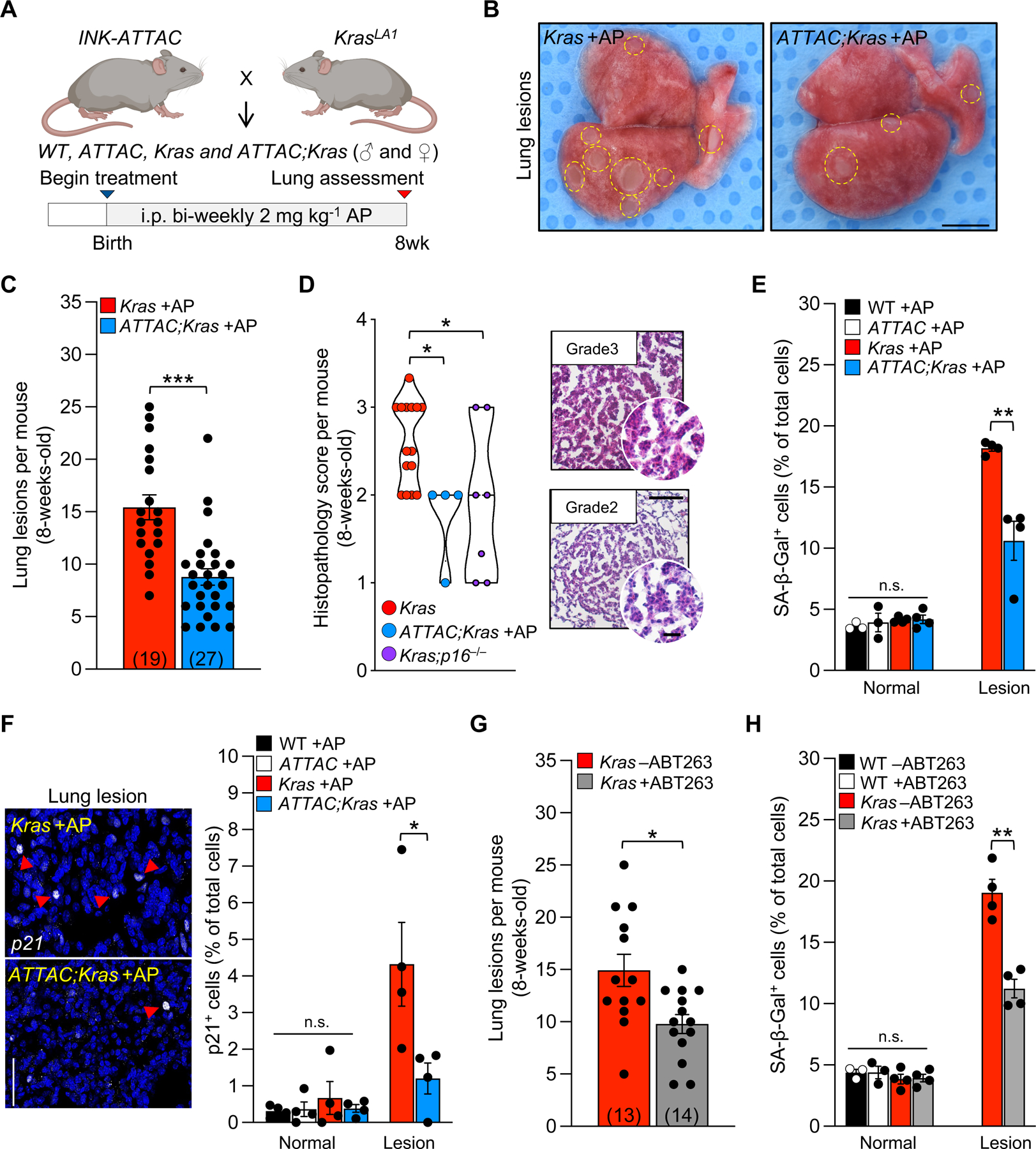
(A) Study design for the ‘prevention’ strategy to remove of senescent cells in ATTAC;Kras mice from birth to eight weeks.
(B) Representative images of lung lesions.
(C) Number of lung lesions per mouse. The number of mice per group are indicated in the parenthesis.
(D) Histopathology score (left) and representative images (right) of H&E-stained lung lesions.
(E) Percentage of SA-β-Gal+ cells in normal and lesion area.
(F) Representative images (left) and quantification (right) of immunostaining for p21.
(G) Number of lung lesions in mice treated with vehicle (−ABT263) or ABT263 (+ABT263). The number of mice per group are indicated in the parenthesis.
(H) Frequency of SA-β-Gal+ cells after vehicle or ABT263 treatment.
Scale bars: 10mm (B), [100μm, inset 20μm (D)], 50μm (F). Data are means ± SEM. ns, non-significant. *P<0.05; **P<0.01; ***P<0.001; [unpaired two-tailed Mann-Whitney test in D, one-way ANOVA with Tukey’s multiple comparison test in E, F, H (normal area), and unpaired two-tailed Student’s t test in C, and E, F, H (lesion area)].
See Figure S1.
Next, we tested if pharmacological elimination of senescent cells, termed senolysis, could also attenuate adenoma formation. We chose to use ABT263 (navitoclax), a potent small-molecule inhibitor of BCL2 family members19, which has previously been used to target senescent cells in in mouse models of atherosclerosis and neurodegeneration20,21. Cancer cells, like senescent cells, exhibit increased expression of various BCL2 family member pro-survival proteins to resist apoptosis, which makes them potentially sensitive for elimination with ABT26322. To alleviate the possibility of mistargeting these cancer cells, we treated animals with a lower dose of ABT263 compared to previous oncolytic studies19,22. WT and Kras mice were treated with ABT263 beginning at weaning age. Similar to AP treatment in ATTAC;Kras mice, ABT263 reduced the number of adenomas that formed (Figure 2G). ABT263 treatment caused cell death in Kras lung tissue (Figure S1C) to a similar extent as transgenic senescent cell clearance. ABT263 also attenuated the accumulation of SA-β-Gal positive cells in lung lesions (Figure 2H). To validate that ABT263 was selectively targeting senescent cells and not pre-neoplastic cells, we treated ATTAC;Kras mice with a combination of both ABT263 and AP. We found that the combination of both agents did not further reduce adenoma number beyond that of each agent individually (Figure S1D). Additionally, treatment of Kras;p16KO mice with ABT263 also was unable to further reduce the number of adenomas (Figure S1E). The absence of any additive effects in these scenarios supports the hypothesis that AP and ABT263 may be impacting the same cell population.
With the surprising observation that Kras;p16KO mice had reduced adenoma formation, we were curious if impacting other cell cycle regulators involved in senescence, namely p21, would impact tumor predisposition. p21 knock-out mice had no lung lesions at eight weeks of age (data not shown) and Kras;p21KO mice exhibited fewer adenomas than Kras mice (Figure S1F). Furthermore, we found that treating Kras;p21KO mice with ABT263 had no additional reduction in adenoma number (Figure S1G). Taken together, these findings highlight senescent cells as a contributor to Kras mediated tumorigenesis and suggest that clearance of these cells may be a new therapeutic strategy to suppress the early stages of cancer.
Senescent alveolar macrophages have tumor-promoting qualities
We next sought to identify the cell types prone to senescence in Kras mice. We relied on SA-β-Gal staining coupled to high resolution imaging via transmission electron microscopy (TEM). We have successfully used this approach in other tissue types16,21 to allow for morphological cellular identification cells containing X-Gal crystals. In Kras mice, we found that various cell types, including immune, epithelial, and stromal cells (as defined by their cellular morphology and location in the lung; see Material and Methods for additional details) contained crystals (Figure 3A). In ATTAC;Kras mice, we found that AP treatment significantly reduced the number of X-Gal-containing immune cells (Figure 3B), suggesting that these may be the pro-tumorigenic senescent cell type. Indeed, tumor-associated immune cells have been shown to influence tumor initiation and their accumulation is a hallmark of the cancer microenvironment23–25.
Figure 3: Single-cell transcriptional profiling of Kras lungs identifies signatures of cellular senescence in p16-expressing macrophages.
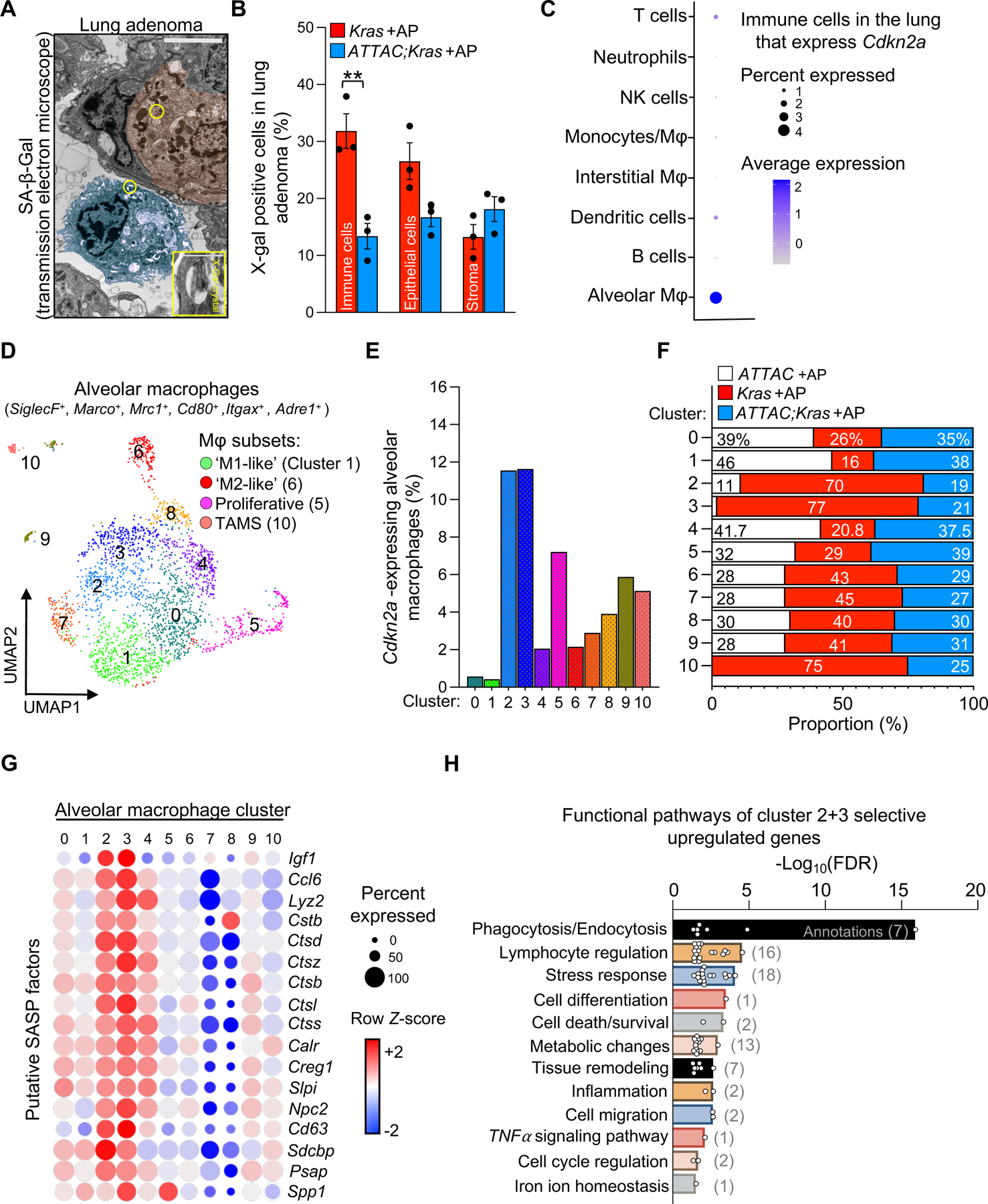
(A) Transmission electron micrograph of an X-Gal+ crystal in an epithelial cell (red) and immune cell (blue) in an 8-week-old Kras mouse.
(B) Percentage of X-gal+ immune, epithelial, and stromal cells in lesions of eight-week-old Kras and ATTAC;Kras mice treated with AP from birth.
(C) Cdkn2a (p16) expression in immune cell types from eight-week-old ATTAC +AP, Kras +AP, and ATTAC;Kras +AP mice.
(D) UMAP plot displaying 11 cell clusters of alveolar macrophages.
(E) Percentage of alveolar macrophages expressing Cdkn2a (p16) in each cluster.
(F) Proportion of alveolar macrophages from each genotype per cluster.
(G) Bubble plot of alveolar macrophages displaying putative SASP factors enriched in cluster 2/3.
(H) Selected functional annotation pathways that are enriched in cluster 2/3.
Scale bar: [5μm (inset 1μm) A]. Data are means ± SEM. ns; non-significant. *P<0.05; **P<0.01; ***P<0.001; (Unpaired two-tailed Student’s t test in B).
To gain further insight into the senescent cell types arising during Kras induced adenoma formation, we conducted unbiased single-cell RNA-sequencing (scRNA-seq) on lungs of eight-week-old ATTAC, Kras, and ATTAC;Kras mice all treated with AP from birth (Figures S2A, S2B). We began the scRNA-seq analysis by exploring which cell populations express p16 (Figures 3C, S2C). Notably, we found that tissue resident alveolar macrophages (AMs) defined as Siglec-F+, Marco+, Mrc1+, Cd80+, Itgax+, and Adre1+ had the highest proportion and level of Cdkn2a expression (Figure 3C). By contrast, other types of macrophages, including monocyte-derived macrophages, and immune cells, including dendritic cells, T lymphocytes, NK cells, monocytes, neutrophils, and B lymphocytes, had much lower per-cell Cdkn2a levels and overall proportion of Cdkn2a-positive cells (Figure 3C). Furthermore, it was a rare population of epithelial or stromal cells that expressed Cdkn2a (Figure S2C). As it has been reported that macrophages can be both p16-expressing and SA-β-Gal+ under homeostatic conditions26–28, we carefully evaluated whether the INK-ATTAC transgene globally eliminates non-senescent macrophages. To assess this, we examined cell survival and death in vitro using both bone-marrow derived macrophages and peritoneal macrophages isolated from three-month-old wild-type and ATTAC mice. These isolated macrophage populations were treated with varying concentrations of AP for 48 hours. We included treatment with clodronate-containing liposomes29,30 as a positive control for macrophage depletion. We found that neither macrophage population is sensitive to AP-mediated cell elimination, and therefore we deem it unlikely that macrophages are globally targeted by ATTAC-mediated cell elimination (Figures S3A–C). Indeed, in lung lesions, the overall macrophage population defined by the pan-marker F4/80 was not changed after AP treatment in ATTAC;Kras mice (Figure S3D). Taken together, these results suggested that p16-positive AMs represent a specific subpopulation of macrophages that can be targeted by ATTAC mediated senolysis.
Consistent with reports of macrophage phenotypic plasticity, unsupervised clustering of scRNA-seq revealed 11 AM cell clusters (Figures 3D, S4A). These include transcriptional profiles representative of ‘classical’ M1-like31 phenotype (cluster 1), ‘alternatively-activated’ M2-like31 macrophages (cluster 6), as well as immunosuppressive, tumor-associated macrophages (cluster 10) or TAMS32 and macrophages with a proliferation signature (cluster 5; Figure S4B). We noticed that the accumulation of p16-expressing AMs was found in clusters (clusters 2 and 3) that, at the transcript level, did not match previously defined macrophage phenotypes (Figures 3E, S4B). Notably, a higher proportion of p16-expressing AMs in cluster 2/3 came from Kras +AP mice compared to wild-type control (ATTAC +AP) or ATTAC;Kras +AP (Figure 3F). There were also fewer TAMS (cluster 10) in ATTAC;Kras AP treated mice (Figure 3F), which nicely concurs with the lower lesion numbers observed in these mice (Figure 2C). We also found that cluster 2/3 AMs express several putative SASP factors, including Spp1, Igf1, Ctsd, and Ctsb (Figure 3G) that have known functions in degrading the extracellular matrix33–35, suggesting that the subset of p16-positive AMs from clusters number 2/3 may be a preferential target of AP treatment to delay tumorigenesis.
To further explore what distinguish AMs in cluster 2/3 from the other subsets, we examined their transcriptional profile compared to the remaining clusters. Using functional annotation analyses, we identified the significantly enriched annotations that were subsequently categorized into functional pathways (Figure 3H). These annotations included genes associated with phagocytosis (Fcgr2b, Fcer1g, and Fcgr3), lymphocyte regulation (Tyrobp, Tgfbr2, and Lgmn), and metabolic processes (Gstm1, Npc2, Hexa, Fabp4, Fabp5, Msmo1, Ch25h, Lipa, Psap, Myo5a, Soat1) (Figures 3H, S4C). Notably, cluster 2/3 AMs also exhibited patterns consistent with cellular senescence, with significant enrichment for genes involved in the negative regulation of cell proliferation, including Ctnnb1, a key regulator of self-renewal in AMs36 and genes that have functions in response to stress, inflammation, and the tumor necrosis factor (TNF) signaling pathway (Figures 3H, S4C), which may stimulate the recruitment of inflammatory cells37,38. Collectively, our results indicate that a subset of lung tissue-resident macrophages present senescence and tumor-promoting qualities during lung lesion development.
Senescent alveolar macrophages accumulate in tumorous lung
To functionally validate if the subsets of p16-expressing AMs identified through scRNA-seq accumulate in Kras tissue, we screened for potential cell-surface biomarkers that could selectively distinguish these cells from the other alveolar macrophages. We first identified 1191 cell surface genes that were part of the GO annotation ‘cell surface’ (GO: 009986) or all its derived terms. We then assessed whether any of these genes are selectively induced in cluster 2/3 from our scRNA-seq analysis. We found that 17 cell surface proteins are induced in cluster 2/3 with lower or no expression in the other AM subsets (Figure 4A). Among these 17 genes, we noted Cxcr1, a chemokine receptor with a high affinity for interleukin-8 (IL-8) that mediates immune and inflammatory responses39, was highly enriched in cluster 2/3 (Figures 4A, 4B). CXCR1 has been implicated in the pathophysiology of tumor progression in many cancers, including human NSCLC40 and can be targeted as a drug-receptor41. With these observations in mind, we tested whether AMs of Kras lung tissue express CXCR1. Co-immunostaining for lung tissue resident macrophage marker, SIGLEC-F, and CXCR1 revealed a subset of AMs positive for both markers. Notably, we found that a significant proportion of SIGLEC-F+ and CXCR1High AMs accumulate in Kras lesions relative to young, 8-week-old, WT lung or ‘normal’ non-tumorous areas of the Kras lung (Figure 4C).
Figure 4: Senescent alveolar macrophages accumulate in lesions of Kras mice and with advancing age in ATTAC mice.
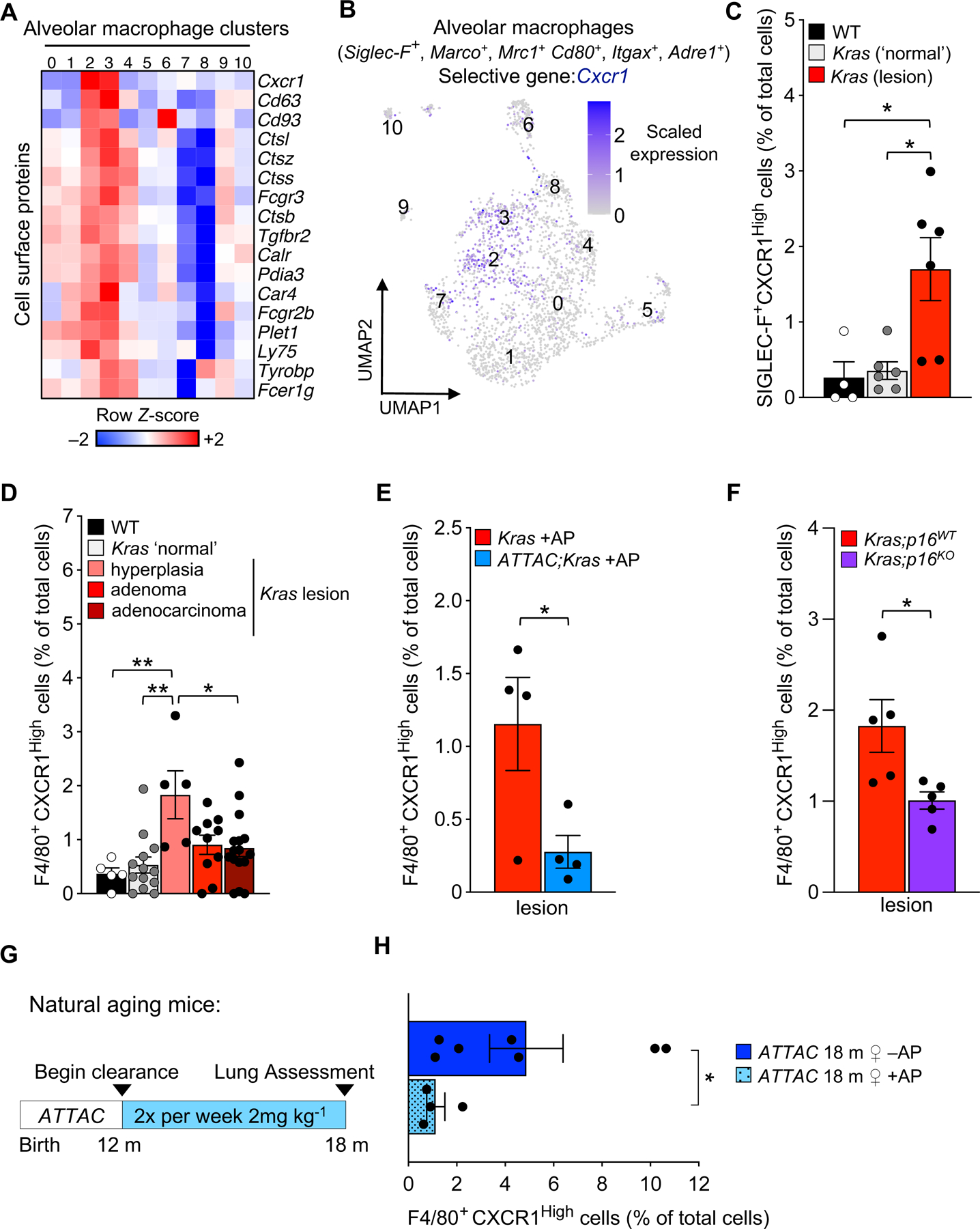
(A) Heatmap of 17 cell surface genes enriched in clusters 2/3 of alveolar macrophages from eight-week-old ATTAC +AP, Kras +AP, and ATTAC;Kras +AP mice.
(B) UMAP plot of Cxcr1 expression in alveolar macrophages.
(C) Immunofluorescence quantification of CXCR1High tissue-resident macrophages (SIGLEC-F+) in the lungs of wildtype (WT) and Kras mice.
(D-F) Immunofluorescence quantification of CXCR1High macrophages (F4/80+) throughout lesion development in Kras mice (D), with AP treatment from birth to eight weeks in Kras and ATTAC;Kras mice (E), and with genetic inactivation of p16 in Kras mice (F).
(G) Study design for the removal of senescent cells in naturally aged INK-ATTAC mice.
(H) Immunofluorescence quantification of CXCR1High macrophages (F4/80+) in the lungs of naturally aged 18-month-old INK-ATTAC female mice treated with vehicle or AP.
Data are means ± SEM. *P<0.05; **P<0.01; ***P<0.001; (one-way ANOVA with Tukey’s multiple comparison test in C, D, Unpaired two-tailed Student’s t test in E, F and Mann-Whitney test in H).
Next, we set out to assess when CXCR1High macrophages began accumulating during lung lesion development. We stained for CXCR1 and F4/80 macrophages across multiple lesions that represented the different histopathology stages of hyperplasia, adenoma, and adenocarcinoma in Kras mice. We found that F4/80+ CXCR1High cells start to accumulate early on during lesion formation, with a notable proportion of these cells present in hyperplastic lesions (Figure 4D).
To further validate our scRNA-seq analysis, we assessed whether AP treatment in ATTAC;Kras mice reduces the frequency of F4/80+ CXCR1High cells. We found a higher proportion of cells were positive for F4/80 and CXCR1High in Kras +AP lesions than in AP-treated ATTAC;Kras lesions (Figure 4E). To extend our analysis, we reasoned that if AP treatment delays the accumulation of CXCR1High AMs, prevention of the senescence program with p16 knockout in Kras lungs should also prevent or delay their accumulation. Accordingly, we found significantly fewer F4/80+ CXCR1High cells in Kras;p16KO lesions compared to Kras;p16WT lesions (Figure 4F). Altogether, our results indicate that CXCR1High can be used as a tool to distinguish senescent alveolar macrophages from others, and that this subset of AMs accumulate in the early stages of lesion development. Importantly, these observations reinforce our hypothesis that a subset of alveolar macrophages with senescent qualities accumulate in Kras-tumorous lung.
Senescent alveolar macrophages increase in aged lung
A distinguishing characteristic of senescent cells is that they accumulate with age in tissues and organs, including the lung16. We observed less than one percent of CXCR1High macrophages in the lungs of 8-week-old WT mice (Figures 4C, 4D). To explore if this population increased with otherwise normal advancing age and was sensitive to senescence clearing strategies, we used 18-month-old female ATTAC mice that had been treated biweekly with vehicle or AP starting at 12 months of age to eliminate p16-expressing senescent cells (Figure 4G). From this analysis, we found that the percentage of F4/80+ CXCR1High cells increased with age in vehicle treated ATTAC females and that AP treatment prevented this accumulation (Figure 4H). These results suggest that CXCR1High AMs accumulate with normative aging and can be targeted with ATTAC-mediated clearance.
To further corroborate whether CXCR1High could be used as a biomarker for senescence-associated changes in macrophages, we tested if AMs, defined by SIGLEC-F, co-stain with CXCR1 and γH2AX, which is present at sites of DNA damage and has been used as an indicator of senescence42. We found that Kras lung lesions contained SIGLEC-F+ CXCR1High AMs with nuclear phospho-γH2AX foci (Figure 5A), which further supports the idea that this subset of AMs displays multiple senescence-associated alterations.
Figure 5: Targeted killing of senescent alveolar macrophages in established disease delays tumor formation in Kras mice.
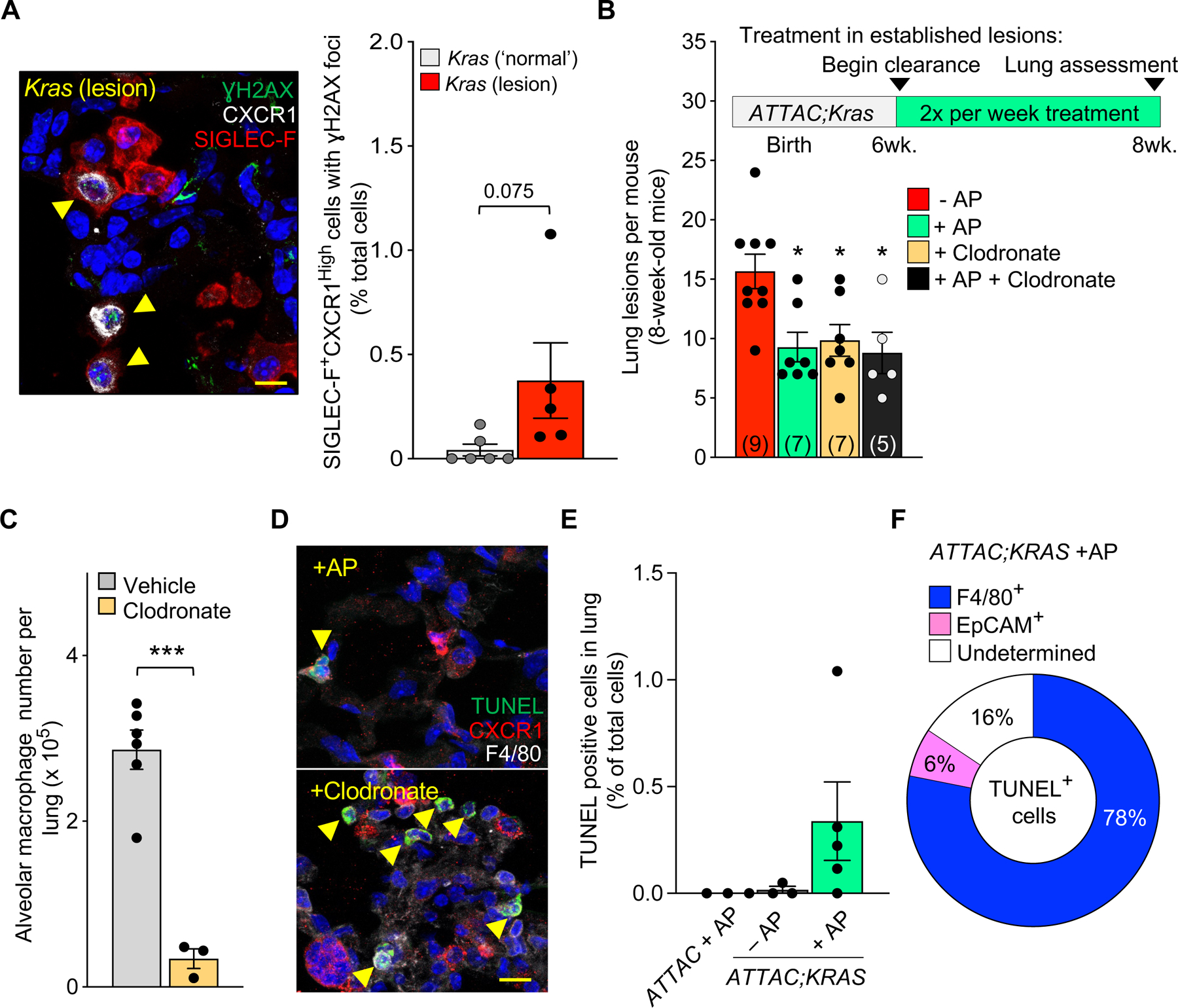
(A) Immunofluorescence image (left, yellow arrows) and quantification (right) of γH2AX+ CXCR1High SIGLEC-F+ cells in lungs of Kras mice.
(B) Study design for the ‘intervention’ strategy to remove senescent cells or macrophages in established disease and the number of lung lesions per mouse after treatment.
(C) Quantification of alveolar macrophages via flow cytometry after vehicle or clodronate treatment.
(D) Immunofluorescence image showing TUNEL+ CXCR1HIgh F4/80+ cells in Kras lung lesions after AP or clodronate treatment.
(E) Immunofluorescence quantification of TUNEL+ cells in the lungs of ATTAC;Kras mice after vehicle or AP treatment.
(F) Percentage of cell types that are TUNEL+ after AP treatment.
Scale bar: 10μm (A, E). Data are means ± SEM. *P<0.05; **P<0.01; ***P<0.001; (Unpaired two-tailed Student’s t test in A, F, one-way ANOVA with Tukey’s multiple comparison test in B (compared to −AP control), C.
See Figure S5.
Clearance of senescent alveolar macrophages delays lung tumorigenesis
Having observed that senescent alveolar CXCR1+ macrophages accumulate in WT-aged lungs and in neoplastic lesions of young Kras mice, we wanted to explore if treatment to remove these cells after hyperplasia has occurred would attenuate tumor development. To test this, we designed a new ‘intervention’ strategy, in which rather than beginning clearance as senescent cells arise from birth (‘prevention’), we started treatment in already established lesions of six-week-old ATTAC;Kras mice. In addition to AP treatment, we used clodronate-containing liposomes to non-selectively target macrophages30. Clodronate was used alone or in addition to AP treatment (Figure 5B). Similar to our ‘prevention’ strategy, we found that short-term AP treatment significantly attenuates lesion formation in ATTAC;Kras mice (Figure 5B). Likewise, we found that non-selective elimination of AMs with clodronate (Figure 5C) also reduced the number of lesions to the same extent as with AP treatment (Figure 5B). Co-treatment had no further effect on lesion formation (Figure 5B), suggesting that clodronate and AP target similar cell populations in ATTAC;Kras mice. In these animals, we assessed for cellular death, defined as being TUNEL-positive, along with CXCR1 and the pan-macrophage marker F4/80. We found that AP treatment targets CXCR1High macrophages, whereas clodronate had many more TUNEL-positive cells, including those lacking CXCR1 (Figure 5D). We confirmed that clodronate by itself does not function as a senolytic in vitro (Figure S5). Given the importance of AMs for normal lung function, we stress that a pan-killing macrophage strategy is not ideal. However, selective clearance of the small subset of seemingly senescent tumor-promoting macrophages is sufficient to delay tumor formation. We found no evidence of TUNEL-positive cells in healthy lungs from young WT ATTAC mice treated with AP (Figure 5E). In the lungs of ATTAC;Kras mice, we found very little evidence of TUNEL-positive cells after vehicle treatment, whereas after AP, a modest percentage of cells were positive for TUNEL staining (Figure 5E). We note that most of these cells were F4/80+ and only a small fraction was Epcam+ (Figure 5F), an epithelial marker that is upregulated in proliferative cancer cells43. Altogether, our results indicate that cellular senescence occurs in a subset of AMs within the innate immune system that are causally implicated in Kras-driven lung tumorigenesis.
Senescent cell clearance during lung tumorigesis promotes cytotoxic T cell accumulation
To understand the pro-tumorigenic role of senescent cells during tumorigenesis, we tested if the removal of senescent cells impacts the antitumor T cell response. We began the analysis using our unbiased scRNA-seq dataset focusing on T cells, defined by the expression of classical T cell markers Cd3d, Cd3e, Cd3g, and Trac. Unsupervised clustering from our scRNA-seq analysis revealed 13 clusters of T cells (Figure 6A). These include subsets with a transcriptional profile indicative of CD4+ T cells (naïve-like, T-reg, and cytotoxic cells) as well as subsets of CD8+ T cells (naïve-like, memory, cytotoxic, proliferative cytotoxic cells), and γδ T cells (Figures 6A, 6B, S6A). We noticed that the highest proportion of p16-expressing T cells occurs in cluster 12. Unlike p16-expressing AM cluster 2/3 (Figure S4B), T cell cluster 12 expressed high levels of cell cycle progression genes and therefore represents actively proliferating cells (Figures S6B–D). We then assessed whether the proportion of any T cell subsets, such as cytotoxic T lymphocytes (CTL), are adapted by senescent cell removal. We found that lungs from tumor-bearing Kras +AP mice contained fewer CD8+ CTLs (cluster 5 and 12), while ATTAC;Kras +AP lungs harbored more of these cells (Figure 6C). This suggests that senescent cells could be counteracting CTL expansion or infiltration. This mechanism appears independent of CD4+ Tregs (cluster 8), as their numbers did not change with senescent cell removal (Figure 6C). To critically test if properties present in senescent AMs, including the expression of Gpnmb, which can inhibit T cell activation44, and Igf1, which plays an important role in the development and survival of T cells45 (Figure S4C), could influence CTL responses, we performed an in-depth flow-cytometry analysis on Kras and ATTAC;Kras mice treated with our AP ‘intervention’ strategy (Figure 6D). We found that AP-mediated senescent cell clearance did not change the distribution of most T cell subsets including CD4+ FOXP3+ CD25+ Tregs (Figure S7). However, we found an increase in the proportion of Perforin (PRF) expressing CD8+ T cells, and an accumulation of PRF+ CD4+ T cells and double positive Granzyme B (GZMB+) PRF+ CD4+ subsets in ATTAC;Kras mice after AP treatment (Figure 6F). These results indicate that senescent cells, including senescent AMs, modulate T cells antitumor response by inhibiting the accumulation of cytotoxic T cells, thereby facilitating lesion formation.
Figure 6: Senescent cell clearance facilitates cytotoxic T cells accumulation.
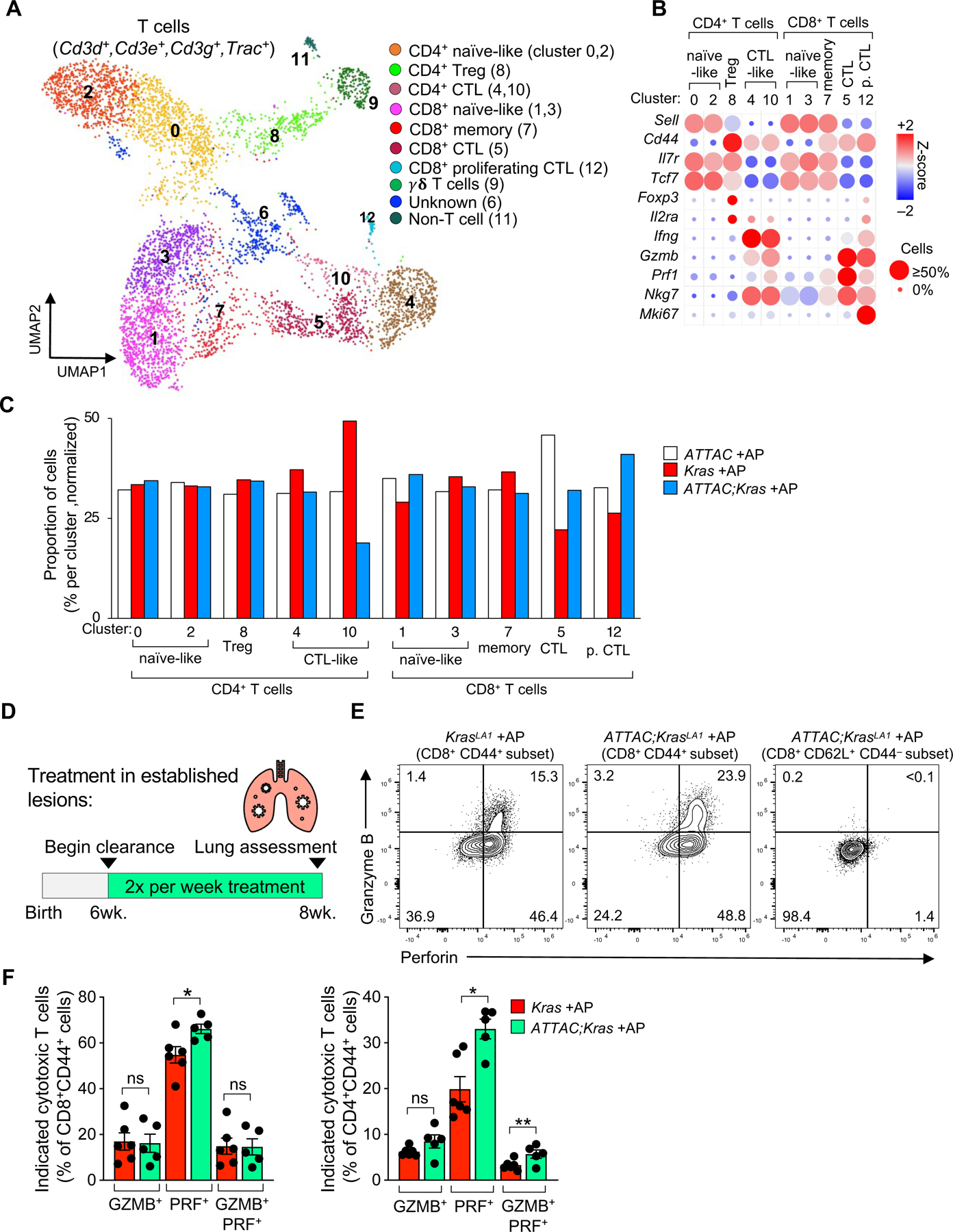
(A) UMAP plot displaying 13 cell clusters of T cells from eight-week-old ATTAC, Kras, and ATTAC;Kras mice treated with AP from birth.
(B) Bubble plot of signature genes to identify T cell subsets.
(C) Percentage of T cell subsets by genotype in each cluster. Cell numbers were normalized across group for this analysis.
(D) Study design for the ‘intervention’ strategy to remove senescent cells in established Kras-driven lesions.
(E) Gating strategy for flow analysis of cytotoxic T cells defined as Granzyme B positive, Perforin (PRF) positive or double positive T cells.
(F) Flow cytometric quantification of the indicated cytotoxic T cell subsets after senescent cell clearance in established lesions.
Data are means ± SEM. *P<0.05 (Unpaired two-tailed Student’s t test in F).
Senescent p16-expressing macrophages accumulate in the early stages of human lung cancer
To determine if our findings have clinical relevance in human malignancy, we investigate whether AMs with high expression of CXCR1 exist in lung tumors. Since our preclinical Kras mouse model recapitulates the early stages observed in human non-small cell lung cancer (NSCLC), we performed our analysis using patient samples derived from treatment-naïve early-stage NSCLC adenocarcinoma in situ (AIS) (Figure 7A). Fortunately, our clinical samples contained areas of ‘normal’ looking lung adjacent to the tumor (Figure 7B). Co-immunostaining for CXCR1 and the macrophage marker CD68 revealed that these macrophages exist in human AIS, and at higher proportions in the intratumoral area compared to the tumor adjacent ‘normal’ area (Figure 7C). In support of our mouse studies that these cells represent macrophages with senescence-associated alterations, triple-immunostaining for CD68, CXCR1, and p16 identified CXCR1High macrophages containing nuclear p16 that accumulate intratumorally (Figures 7D, 7E). Altogether, these observations in human NSCLC AIS support the hypothesis that oncogenic alterations hijack immune cells from the tumor microenvironment to promote senescence-driven tumorigenesis.
Figure 7: Senescent p16-expressing macrophages accumulate in the early stage of human non-small cell lung cancer (NSCLC).
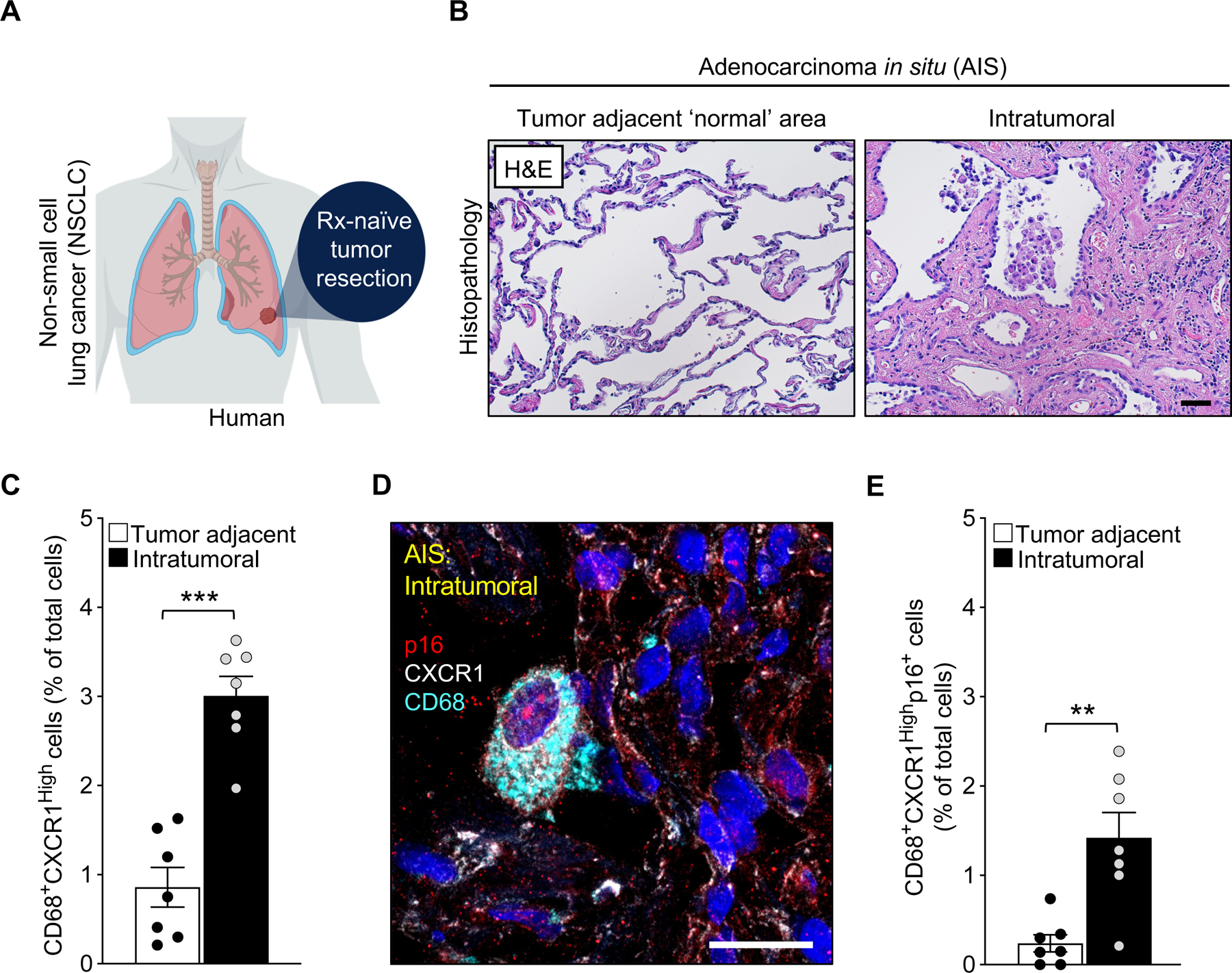
(A) Schematic of human NSCLC sample collection.
(B) Representative images of tumor adjacent area and intratumoral area of adenocarcinoma in situ (AIS) stained with hematoxylin and eosin.
(C) Immunofluorescence quantification of CD68+ CXCR1High cells in AIS samples.
(D) Representative image showing the expression of nuclear p16, CXCR1, and CD68 in intratumoral cells of AIS samples.
(E) Quantification of nuclear p16+ CD68+ CXCR1High cells in the indicated area of AIS.
Scale bars: 50μm in B, 20 μm in D). Data are means ± SEM. *P<0.05; **P<0.01; ***P<0.001; (Unpaired two-tailed Student’s t test in C, E).
Discussion
In previous work, we demonstrated that the continuous elimination of p16-positive senescent cells extends the healthy lifespan of naturally aged mice and delays spontaneous tumorigenesis16. In this study, we define the mechanistic contribution of senescent cells to spontaneous Kras-driven tumorigenesis. We use genetic or pharmacological senolysis in a preclinical Kras-driven murine cancer model to implicate a subset of p16-expressing AMs with signatures reminiscent of cellular senescence that suppress the antitumor T cell response during lung tumorigenesis. Clinically, we observe macrophages with similar characteristics that accumulate in tumors from patients with non-small cell lung cancer (NSCLC).
Consistent with our murine Kras model, recent work in human patients with treatment-naïve NSCLC lesions and an orthotopic mouse model that uses KrasG12D and p53 null (KP) epithelial cells has also implicated the totality of tissue-resident macrophages as drivers of lung tumorigenesis 46. Here, we focus on a specific subset of AMs, notably defined by p16 and Cxcr1 expression, with senescence-like properties. Targeted removal of these cells via the transgenic ATTAC system in mice attenuated Kras-driven lung tumorigenesis. Other studies26,27 have suggested that non-senescent macrophages can express signatures of cellular senescence, such as high expression of the cell-cycle regulator p16, which could make them susceptible to elimination via the ATTAC transgene. Importantly, we find that AP treatment of non-senescent ATTAC macrophages has no effect on cell survival (Figure S3). Additionally, AP-mediated killing in vivo is infrequent and specific to this subset of macrophages. These observations strengthen the hypothesis that a subset of p16-expressing macrophages drive lung tumorigenesis. In this study, proliferating T cells exhibited high levels of Cdkn2a, but their numbers are not changed upon AP treatment (Figures 6, S6), suggesting that ‘non-senescent’ p16-expressing cells are not targeted in INK-ATTAC transgenic mice. This is consistent with previous work in which we showed that SV40 immortalized cells highly express p16 but are not killed upon AP treatment16. Together, these results suggest it is unlikely that AP is killing proliferating p16-expressing cells.
In cancer, the complex and paradoxical roles of senescence are influenced by the induction mode and cell type of origin. Meaningful clues defining why senescent cells have such heterogeneous functions are inferred by how long they linger in organs and tissues5. Here, we implicate the cellular senescence program in the innate component of the immune system. Our study indicates that a subset of lung tissue-resident macrophages with multiple senescent traits, including the expression of p16 and CXCR1, accumulate with normative aging and during neoplastic growth. We found that this subset of macrophages persists throughout different stages of Kras-driven tumorigenesis and suppress the recruitment (or proliferation) of cytotoxic T-cells. Importantly, we also found that senescent alveolar macrophages exist in the early stage of treatment-naive human NSCLCs. However, further research is needed to fully characterize the properties specific to these cells, to understand what drives their acquisition of these senescence-associated changes, develop interventional strategies that specifically target senescent cell types of interest, and to explore how they may promote age-related tumorigenesis in late life.
For decades, much of our knowledge of senescent cell biology came from in vitro cell culture experiments without much understanding of how senescent cells affect health and disease in vivo. In this study, we provide meaningful in vivo insights into the function of the senescence effectors, p16 and p21, during lung tumorigenesis. Strikingly, we found that precluding cells from engaging with tumor suppressors p16 or p21 in the early stages of cancer delays tumor formation, supporting the hypothesis that p16- or p21- driven cellular senescence in the tissue microenvironment creates a permissive environment for neoplasia. Continued study into the function of the tumor suppressor genes, p16 and p21, specifically in immune cells in pathological contexts, will be critical to understanding how targeting these cells may be protective from disease. To this end, our study defines a subset of innate-immune cells that acquire senescence-like properties and suggests that their targeted removal in the early stages of pathology is a plausible therapeutic intervention to delay tumor formation.
STAR METHODS
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Darren J. Baker (baker.darren@mayo.edu).
Materials availability
As described in the key resources table, cell lines are available from ATCC or on request. Mouse models are available from The Jackson laboratory or on request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-p21 [EPR18021] | Abcam | Cat# ab188224 |
| Rat monoclonal anti-F4/80 (BM8.1) | Cell Signaling | Cat# 71299 |
| Rabbit polyclonal anti-CXCR1 (used in mouse lung) | Invitrogen | Cat# PA5-95749 |
| Mouse monoclonal anti-phospho-Histone H2AX (Ser139), clone JBW301 | Millipore Sigma | Cat# 05-636 |
| Rat anti-mouse CD45, clone 30-F11 | BioLegend | Cat #103101 |
| Live/Dead fixable blue dead cell stain | Invitrogen | Cat# L34961 |
| Rat anti-CD11b, clone M1/70 | BD biosciences | Cat# 612977 |
| Rat anti-mouse/human CD44, clone IM7 | BioLegend | Cat# 103040 |
| Rat anti-mouse CD4, clone RM4-5 | BD biosciences | Cat# 558107 |
| Rat anti-mouse CD25, clone PC61 | BD biosciences | Cat# 566202 |
| Rat anti-mouse CD8a, clone 53-6.7 | BioLegend | Cat# 100750 |
| Mouse IgG1 anti-mouse/rat/human FOXP3 | BioLegend | Cat# 320007 |
| Rat anti-mouse Perforin, clone S16009A | BioLegend | Cat# 154315 |
| Rat anti-mouse CD62L, clone MEL-14 | BioLegend | Cat# 104410 |
| Rat anti-human/mouse Granzyme B, clone QA18A28 | BioLegend | Cat# 396409 |
| Armenian Hamster anti-mouse TCRβ chain, clone H57-597 | BioLegend | Cat# 109219 |
| Rat anti-mouse CD16/CD32 (Mouse BD Fc block), clone 2.4G2 (RUO) | BD biosciences | Cat# 553142 |
| Rat anti-mouse CD170 (Siglec-F), clone S17007L | BioLegend | Cat# 155505 |
| Armenian Hamster anti-mouse CD11c, clone N418 | BioLegend | Cat# 117321 |
| Rat anti-mouse/human CD11b, clone M1/70 | BioLegend | Cat# 101201 |
| Rat anti-mouse MERTK (MER), clone 2B10C42 | BioLegend | Cat# 151502 |
| Mouse IgG1 anti-mouse CD64, clone X54-5/7.1 | BioLegend | Cat# 139303 |
| Rat anti-mouse Ly-6G, clone 1A8 | BioLegend | Cat# 127601 |
| Rat anti-mouse I-A/I-E, clone M5/114.15.2 | BioLegend | Cat# 107601 |
| Rat anti-mouse Ly-6C, clone HK1.4 | BioLegend | Cat# 128015 |
| Mouse anti-human CD68, clone KP1 | Agilent Dako | Cat # GA60961-2 |
| Mouse anti-human CXCR1, clone 42705 | R&D systems | Cat# MAB330-SP |
| Rabbit anti-human CDKN2A, clone EPR1473 | Abcam | Cat# ab108349 |
| Bacterial and virus strains | ||
| Not applicable | ||
| Biological samples | ||
| Human non-small cell lung cancer tumor sections | Mayo Clinic Tissue Registry | IRB# 22-010460 |
| Chemicals, peptides, and recombinant proteins | ||
| AP20187 B/B homodimerizer | Takara | Cat# 635059 |
| ABT263 | Cayman | Cat# 923564-51-6 |
| Phosal 50 PG | Lipoid | Cat# NC0130871 |
| PEG400 | Sigma | Cat# 91893 |
| M-CSF | R&D systems | Cat# 416-ML-010/CF |
| Phenazine methosulfate | Sigma | Cat# P9625 |
| CellTiter 96 AQueous MTS Reagent | Promega | Cat# G112 |
| Antigen Unmasking Solution, Tris-based | Vector Laboratories | Cat# H-3301 |
| Nuclear Fast Read | Vector Laboratories | Cat# H-3403 |
| Red blood cell lysis solution | MACS Miltenyi Biotec | Cat# 130-094-183 |
| Liposomal clodronate | Clodrosome | Cat# SKU CLD-8909 |
| Critical commercial assays | ||
| In Situ Cell Death Detection Kit, Fluorescein | Roche | Cat# 11684795910 |
| RNAscope Multiplex Fluorescent Detection Reagents v2 | Advanced Cell Diagnostics Inc | Cat# 323110 |
| RNAscope H2O2 and protease Reagents | Advanced Cell Diagnostics Inc | Cat# 322381 |
| SA-b-Gal assay | Cell Signaling | Cat# 9860S |
| Lung Dissociation Kit, mouse | MACS Miltenyi Biotec | Cat# 130-095-927 |
| eBioscience™ Foxp3 / Transcription Factor Staining Buffer Set | Invitrogen | Cat# 00-5523-00 |
| Deposited data | ||
| 10x RNA Seq for Mouse Lung Tumorigenic Tissue | This paper | GEO: GSE201247 |
| Experimental models: Cell lines | ||
| Human lung fibroblasts, IMR-90 cells | ATCC | Cat# CCL-186 |
| Primary peritoneal macrophages | This paper | N/A |
| Monocyte-derived macrophages | This paper | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: B6.129S2-Krastm2Tyj/Nci[17], also known as KrasLA1 | MMHCC (NCI Frederick) | Strain# 01BM2 |
| Mouse: p16 knockout | Baker et al.,48 | |
| Mouse: p21 knockout | Jackson Lab | Strain# 003263 |
| Mouse: INK-ATTAC | Baker et al.,16 | |
| Oligonucleotides | ||
| Dec1 forward 5’- GGCGGGGAATAAAACGGAGCGA −3’, reverse 5’ CCTCACGGGCACAAGTCTGGAA – 3’ | This paper | |
| Il-1a forward 5’ – TCAACCAAACTATATATCAGGATGTGG – 3’, reverse 5’ – CGAGTAGGCATACATGTCAAATTTTAC – 3’; | This paper | |
| Il-6 forward 5’ – TAGTCCTTCCTACCCCAATTTCC – 3’, reverse 5’ – TTGGTCCTTAGCCACTCCTTC – 3’ | This paper | |
| Tnfa forward 5’ – TGTGCTCAGAGCTTTCAACAA – 3’, reverse 5’ – CTTGATGGTGGTGCATGAGA – 3’; | This paper | |
| Tgfb forward 5’ – CCGAATGTCTGACGTATTGAAGA – 3’, reverse 5’ – GCGGACTACTATGCTAAAGAGG – 3’ | This paper | |
| Tbp forward 5’ – ACCGTGAATCTTGGCTGTAAAC – 3’, reverse 5’ – GCAGCAAATCGCTTGGGATTA – 3’ | This paper | |
| p16Ink4a forward 5’ – CCCAACGCCCCGAACT – 3’ reverse 5’ - GCAGAAGAGCTGCTACGTGAA – 3’ | Baker et al., 16,20,21,48 | |
| p21Cip/Kip forward 5’ - GTCCAATCCTGGTGATGTCC – 3’ reverse 5’ –GTTTTCGGCCCTGAGATGT – 3’ | Baker et al.,16,20,21,48 | |
| P19Arf forward 5’ - GCCGCACCGGAATCCT – 3’ reverse 5’ – TTGAGCAGAAGAGCTGCTACGT – 3’ | Baker et al.,16,20,21,48 | |
| Recombinant DNA | ||
| Not applicable | ||
| Software and algorithms | ||
| GraphPad Prism 9 (version 9.3.1) | https://www.graphpad.com/ | |
| FlowJo 10.2 (Tree Star) | https://www.flowjo.com/solutions/flowjo/downloads/previous-versions | |
| Fiji from ImageJ (version 1.52n) | https://imagej.net/software/fiji/ | |
| Other | ||
Data and code availability
Raw and processed data for single-cell RNA-seq have been deposited at Gene Expression Omnibus GSE201247.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Materials and methods
Mouse strains and drug treatment
B6.129S2-Krastm2Tyj/Nci17, also known as KrasLA1, and p16Ink4a knockout mice47 have been generated previously and were originally obtained from the MMHCC (NCI Frederick) as previously described48,49. These mice were bred to C57BL/6 mice for at least 5 generations. Male p16Ink4a knockout male mice were bred initially to K-rasLA1 female mice. Subsequent offspring were inter-crossed to generate cohorts of wild-type (WT), p16 knockout (KO), Kras;p16WTand Kras;p16KO mice for littermate controls. A similar strategy was used to make cohorts of Kras;p21KO mice (Jax strain #003263) after breeding p21KO mice to C57BL/6 for at least 5 generations. C57BL/6 ATTAC transgenic mice were generated as previously described14,16. Male ATTAC mice were bred to KrasLA1 females to generate cohorts of WT, ATTAC, Kras and ATTAC;Kras mice. All mice from this cohort were assigned to receive AP20187 (AP; B/B homodimerizer; Clontech/Takara) twice a week beginning within the first week of birth. Dosing of AP was 2 mg per kg body weight. Senolytic intervention was performed in C57BL/6 wild-type and KrasLA1 mice. At weaning age (~3 weeks of age), mice were assigned to receive either ABT263 (Cayman #923564–51-6) or vehicle (Phosal 50 PG Lipoid #NC0130871, 60%; PEG400, Sigma #91893, 30%; ethanol, 10%). ABT263 was administered by oral gavage at a dose of 50 mg per kg body weight on a repeating regimen for five consecutive days during weeks 3 and 6 of age. Both male and female mice were used for experiments. All mice were on a pure C57BL/6 genetic background and no data was excluded. Mice were housed in a 12h:12h light:dark cycle environment in pathogen-free barrier conditions as described in detail14,16. Compliance with relevant ethical regulations and all animal procedures were reviewed and approved by the Mayo Clinic Institutional Animal Care and Use Committee.
Tissue collection and analysis of tumors
Tumor burden and size were assessed using an eyepiece reticle on a standard dissection microscope. Representative lung images were acquired using a digital camera (Nikon D750 FX-format) with a 105mm camera lens (Nikon 105mm f/2.8D AF Micro-Nikkor Lens). Individual lung adenomas with a diameter of at least 2mm and flanking areas of ‘normal’ lung tissue (adjacent tissue not including the adenoma) per mouse were micro-dissected and frozen. The remaining lobes were fixed with 4% paraformaldehyde (PFA) for two hours at 4°C and then transferred to a 30% sucrose solution for 24 hours at 4°C. All samples were O.C.T. (Tissue-Tek #4583) embedded and stored at −80°C. Standard hematoxylin and eosin staining was performed on lung sections. A board-certified pathologist with expertise in lung pathology assisted in the histological evaluation of lung lesions (Dr. Yi). Lung samples were blinded and scored using the following numeric semiquantitative assessment of architectural complexity: 1=flat, 2–3 layers of cells lining the alveolar septa without detached papillary cell clusters; 2= 3–5 layers of cells, with occasional detached papillary cell clusters; 3= thicker, >5 layers or clumped cells with frequent detached papillary cell clusters.
RNA in situ hybridization (RNA-ISH)
RNA-ISH was performed on O.C.T. embedded lung tissues that were sectioned at 10μm thickness following the manufacturer’s protocol (Advanced Cell Diagnostics Inc) for fixed-frozen tissue samples with the following modifications. Samples were treated with hydrogen peroxide for 10 min at room temperature (RT, 25°C) followed by dH2O washes. Target retrieval was performed using a steamer for 5 min followed by a 15 second dH2O wash and letting the samples dry at RT for 5 minutes. Samples were then treated with protease III and incubated for 30 minutes at 40°C followed by dH2O washes and then incubated with the target probes (p16, p21) for 2 h at 40°C. Samples were transfer to 5x SSC buffer and incubated overnight at RT. Subsequently, samples were removed from SSC buffer and placed in washed buffer and then incubated with AMP1 for 30 min at 40°C. This process was repeated for AMP2 and AMP3. Lastly, samples were treated with the appropriate HRP channel and counterstained with DAPI. All samples were stored at 4°C until imaged.
Senescence-associated β-galactosidase staining
SA-β-Gal assay was performed on O.C.T. embedded tissues that were section at 10μm thickness. Staining was performed as previously described16,21 and cell nuclei were counterstained by adding nuclear fast red (Vector Laboratories – H-3403) directly to the samples for 5 minutes at RT followed by dH2O washes, dehydration, clearing, and mounting. Imaging was performed using an Olympus BX53 Fluorescence microscope and DP80 digital camera. Quantification was performed using the Fiji application from ImageJ (version 1.52n).
Senescence-associated β-galactosidase transmission electron microscopy (GAL-TEM)
Localization of X-Gal crystals after SA-β-Gal staining was performed as previously described16,21 with minor modifications for the lung fixation. After sacrifice, lung inflation was performed with 4% PFA through the trachea with a volume estimation of 1.5mL. Lungs were isolated and placed in Trump’s fixative solution overnight at 4°C. This was followed by standard transmission electron microscopy procedures to embed the tissue. Images were acquired and quantified using Jeol 400+ electron microscope with an 80-kV acceleration voltage. Two grids were obtained per tissue, and >100 cells were scanned per grid at a magnification of 12,000x to detect cells with X-gal crystals. To define cell-types, cells were identified based on their location and morphology and grouped into three main categories: immune (cells in the open alveolar space with morphological features resembling phagosomes and thin lamellipodia projections), epithelial [cells with intracellular surfactant-storing lamellar bodies (type II pneumocytes) and cells located on the lining of the subpleural alveolar septa (type I pneumocytes)], and stromal (cells located in the connective tissue). Cells that were not able to be distinguish based on their morphology were not considered for the quantification.
Quantitative RT-PCR
RNA extraction, cDNA synthesis, and RT–qPCR analysis were performed on individual lungs from wild-type, Kras ‘normal’ adjacent non-dysplastic tissue, and lung adenomas (diameter of at least 2mm) from mouse as previously described20,48. Primers used to amplify p16Ink4a, p19Arf, p21 were as previously described16,20,21,48. The following additional primers were used: Dec1 forward 5’- GGCGGGGAATAAAACGGAGCGA −3’, reverse 5’ CCTCACGGGCACAAGTCTGGAA – 3’; Il-1α forward 5’ – TCAACCAAACTATATATCAGGATGTGG – 3’, reverse 5’ – CGAGTAGGCATACATGTCAAATTTTAC – 3’; Il-6 forward 5’ – TAGTCCTTCCTACCCCAATTTCC – 3’, reverse 5’ – TTGGTCCTTAGCCACTCCTTC – 3’; Tnfα forward 5’ – TGTGCTCAGAGCTTTCAACAA – 3’, reverse 5’ – CTTGATGGTGGTGCATGAGA – 3’; Tgfβ forward 5’ – CCGAATGTCTGACGTATTGAAGA – 3’, reverse 5’ – GCGGACTACTATGCTAAAGAGG – 3’; Tbp forward 5’ – ACCGTGAATCTTGGCTGTAAAC – 3’, reverse 5’ – GCAGCAAATCGCTTGGGATTA – 3’. The expression of all genes was normalized first to Tbp.
Immunofluorescence staining and Imaging in mouse samples
10μm thick lung sections were made from O.C.T. embedded tissue and mounted on precleaned microscope slides. Samples were permeabilized with 0.5% Triton in 1x PBS for 20 min at RT and incubated in blocking solution (5% BSA in 1x PBS) for 30 min at RT. Incubation with primary antibodies p21 (Abcam #ab188224; 1:250), F4/80 (Cell Signaling #71299; 1:250), Cxcr1 (Invitrogen #PA5-95749;1;100), Anti-phospho-Histone H2A.X (Ser139) (Millipore Sigma #JBW301; 1:50), and Siglec-F (CD170) (BioLegend #155505; 1:100) was done overnight at 4°C, followed by 1x PBS washes, and a 2 hour incubation with secondary antibodies at RT. Lastly, samples were stained with DAPI and stored at 4°C until imaged. TUNEL staining was done following In Situ manufacture’s protocol (Roche, In Situ Cell Death Detection Kit, Fluorescein #11684795910). Imaging was done using the Zeiss LSM 780 confocal system.
Human NSCLC patient histopathology & immunofluorescence experiments
Samples from patients with treatment-naïve, early-stage lung non-mucinous adenocarcinoma in situ (AIS) were acquired with consent from the tissue registry of Mayo Clinic underneath IRB protocol 22-010460. In brief, 5μm thick lung sections made from paraffin embedded tissue, mounted on precleaned microscope slides were baked at 55°C overnight. Samples were deparaffinized in Xylene x2 for 10 minutes each, dehydrated in a series of ethanol: 100% x2, 95%, 70% for 3 minutes each at RT, and washed in 1x PBS for 5 minutes at RT. Antigen retrieval was done by antigen unmasking solution, Tris-Based (Vector Laboratories #H-3301-250) for 40 min in pressure cooker; once slides were completely cooled, samples were washed with 1x PBS x2 for 5 minutes. Autofluorescence was reduced by adding lipofuscin autofluorescence quencher (TrueBlack #23007) in 70% ethanol for 30 seconds followed by dH2O x3 washes for 5 minutes. Samples were permeabilized with 0.5% Triton in 1x PBS for 20 min at RT and incubated in blocking solution (5% BSA in 1x PBS) for 30 min at RT. Incubation with primary antibodies CD68 (Dako #IR609; 1:100), CXCR1 (R&D #42705; 1:100), and p16 (Abcam #ab108349; 1:100). ) was done overnight at 4°C, followed by 1x PBS washes, and a 2 hour incubation with secondary antibodies at RT. Lastly, samples were stained with DAPI and stored at 4°C until imaged. Imaging was done using the Zeiss LSM 780 confocal system.
Quantification from immunofluorescence images
Quantification of total cells was performed in FIJI (version 2.9.0/1.53t) using an automated counter of Dapi+ nuclei. Scoring of all other stains was performed manually on blinded, max-projected z-stacked TIFs. Data are presented as mean ± S.E.M. with individual data points representing the average counts from 3–5 images per biological sample.
Single-cell suspension
Mice were perfused through the heart with ice-cold Dulbecco’s phosphate-buffered saline (DPBS; pH7.4) until lung lobes were clear. Dissociation of the lobes was performed following the manufacturer’s protocol (MACS, Miltenyi Biotec; Lung Dissociation Kit, mouse’ #130-095-927). Single cell suspension was incubated with red blood cell lysis solution (MACS, Miltenyi Biotec #130-094-183) for 10 min and resuspended with autoMACS buffer rinsing solution (MACS, Miltenyi Biotec #130-091-222).
Flow cytometry
For T cell flow cytometry experiments, lung single cell suspensions were stained with diluted cell surface antibodies (see Key Resource Table) and Live/Dead Fixable Blue Dead Cell Stain (1:500, Invitrogen #L23105). Staining was performed for 30 minutes at 4 C in PBS plus 2% fetal bovine serum (FBS). Cells were washed in PBS/2%FBS and subjected to fixation and permeabilization (Foxp3 / Transcription Factor Staining Buffer Set, eBioscience, #00-5523-00) according to the manufacturer’s instructions. Intracellular antibody staining (see Key Resource Table) was performed for 45 minutes at room temperature. Cells were washed and analyzed on a 5-laser Cytek Aurora (Cytek Biosciences) and FlowJo software (Tree Star). Cells were gated to only include single cells, viable cells and CD45+ CD11B− cells. Remaining cells were gated for TCRb expression indicative of T cells. All following gating strategies are indicated in Figure 6 and Supplementary Figure 7.
For AMs flow cytometry experiment, Fc-gamma receptors were blocked with anti CD16/32 (2.4G2). Cell surfaces were immunolabeled with following cocktails of fluorochrome-conjugated antibodies: (Biolegend) Siglec-F (E50-2440), CD11c (N418), CD11b (M1/70), merTK (2B10c42), CD64 (X54-4/7.1), Ly6G (1A8), I-A/I-E (M5/114.15.2), Ly6C (HK1.4), immuno-staining was performed at 4°C for 30min. Cells were washed twice with FACS buffer (PBS, 2mM EDTA, 2% FBS, 0.09% Sodium Azide), prior to fixation and ran on an Attune NxT auto sampler (Life Technologies). FCS files for myeloid stains were analyzed with FlowJo 10.2 (Tree Star) and processed similarly as described in50.
Single-cell library preparation
Whole live cells were washed twice in 1x PBS + 0.04% BSA and immediately submitted to the Mayo Clinic Genome Analysis Core for Single Cell sorting. The cells were first counted and measured for viability using either the Vi-Cell XR Cell Viability Analyzer (Beckman-Coulter, Brea, CA) or a basic hemocytometer and light microscope. The barcoded Gel Beads were thawed from −80C and the cDNA master mix was prepared according to the manufacture’s instruction for Chromium Next GEM Single Cell 3’ Library and Gel Bead Kit (10x Genomics, Pleasanton, CA). A per sample concentration of 400,000 cells per milliliter was required for the standard targeted cell recovery of approximately 5000 cells. The cell suspension and master mix, thawed Gel Beads and partitioning oil were added to a Chromium Single Cell G chip. The filled chip was loaded into the Chromium Controller, where each sample was processed and the individual cells within the sample were partitioned into uniquely labeled GEMs (Gel Beads-In-Emulsion). The GEMs were collected from the chip and taken to the bench for reverse transcription, GEM dissolution, and cDNA clean-up. The resulting cDNA contained a pool of uniquely barcoded molecules. A portion of the cleaned and measured pooled cDNA continued to library construction, where standard Illumina sequencing primers and a 10x Genomics unique i7 Sample index were added to each cDNA pool. All cDNA pools and resulting libraries were measured using Qubit High Sensitivity assays (Thermo Fisher Scientific, Waltham, MA) and Agilent Bioanalyzer High Sensitivity chips (Agilent, Santa Clara, CA). Libraries are sequenced at between 40,000 and 50,000 fragment reads per cell following Illumina’s standard protocol using the Illumina cBot and HiSeq 3000/4000 PE Cluster Kit (Illumina, San Diego, CA). The flow cells are sequenced as 100 X 2 paired end reads on an Illumina HiSeq 4000 HD using HiSeq 3000/4000 sequencing kit and HCS v3.4.0.38 collection software. Base-calling is performed using Illumina’s RTA version 2.7.7.
Single cell RNA-seq analysis
Sequencing output was demultiplexed and aligned to the mouse reference genome GR. Cm38 using 10x Genomics Cell Ranger 3.0.2. Alignment output from Cell Ranger in the form of gene-barcode matrix were analyzed using R package Seurat 3.2.351–53. Genes were filtered to remove those expressing in less than 3 cells. Cells were also filtered so that only those with at least 500 expressed genes, at least 1,000 unique molecular identifiers (UMIs) and mitochondrial gene content less than 50% UMI. Cells with UMI count larger than the average UMI of all cells plus 3 times the standard deviation of UMI were also removed. Raw counts were log-transformed and multiplied by a factor of 1000. A subset of 2000 genes with high variance across all the cells were selected as variable features. The dataset was then centered and scaled after regressing out the number of UMI of each cell. Principal component analysis (PCA) was performed on the previously identified 2000 most variable genes and an elbow plot showing number of PCs vs. cumulative total variance explained was generated to decide the optimal number of top principal components (PCs) to use for downstream analysis, which was 10 in this study. The Uniform Manifold Approximation and Projection (UMAP) method was used to project the cells represented by selected number of PCs into two-dimensional scatter plots for visualization. A graph-based clustering approach was used to group cells into clusters. First, a k-nearest neighbor (KNN) graph was constructed based on the selected top PCs using euclidean distance. Pairwise edge weights of cells were refined based on the shared overlap in their local neighborhoods (Jaccard similarity). This step was achieved by calling function FindNeighbors in the Seurat package. A modularity optimization process using the Louvain algorithm was applied to iteratively group cells together, with the goal of optimizing the standard modularity function. This is implemented in the function FindClusters in the Seurat package. A resolution of 0.6 was used to control the granularity of the clusters. Cluster marker genes were determined using function FindAllMarkers for each cluster and filtered by Bonferroni-corrected p-value < 0.05 and log2 fold change > 0.25 and expressing in at least 25% of cells in the cluster. Cluster marker genes were compared to published cell type markers to identify cell types.
Macrophage experiments
In vitro experiments were done on cells isolated from healthy 3-month-old WT and ATTAC mice. Peritoneal cells were isolated via peritoneal lavage as described previously6. Isolated cells were seeded in 10cm dish and after 2 hours, non-attached lymphocytes were washed away. Adherent cells constituting macrophages were detached by short trypsinization plus scraping. Macrophages were counted and reseeded in chambered slides (20,000 cells/well). After ~24 hours, cells were treated with vehicle (0.1% ethanol) or AP20187 (500 μM stock in ethanol, B/B homodimerizer, Takara #635059) at the following concentrations: 0 nM, 10 nM, 100 nM and 500 nM. Additionally, macrophages were treated with liposomal clodronate at 1 ug/mL (Clodrosome #SKU CLD-8909) as a positive control for macrophage cell death29,54. Cells were monitored and manually counted with a hemocytometer after 48 hours. To assess cell death, TUNEL staining was performed after 48 hours of drug treatment following the manufacture’s protocol (Roche, In Situ Cell Death Detection Kit, Fluorescein #11684795910). Bone marrow and bone marrow cells were isolated as described previously55. Isolated cells were cultured in DMEM supplemented with 10% FBS, gentamycin, penicillin and streptomycin and differentiated using 20 ng/mL M-CSF (R&D systems #416-ML-010/CF). Cells were cultured for seven days with medium changes every 2–3 days. At day 7, BMDMs were detached by short trypsinization plus scraping, counted and reseeded at 20,000 cells/well (chambered slide) for TUNEL staining and 100,000 cells/well (24-well plate) for manual cell counts. After 24 hours, BMDMs were subjected to the same drug treatments and analyses as peritoneal macrophages. In vivo experiments were performed in ATTAC;Kras mice. 6-week-old mice were treated with liposomal clodronate (Clodrosome #SKU-CLD-8909) twice a week for two weeks at a dose of 12 μl/g.
In vitro clodronate treatment of senescent cells
Mouse embryonic fibroblasts were obtained from embryonic day E13.5 C57BL/6 embryos according to standard techniques. Human lung fibroblasts, IMR-90 cells, were purchased from ATCC (#CCL-186). Cell culture, induction of cellular senescence via 10 Gy γ-radiation (137Caesium source) and MTS assays were performed as previously described56. Briefly, cycling, non-senescent cells (1,000 cells/well) and irradiation-induced senescent cells (2,000 cells/well) were seeded in 96-well plates. Each MEF line or IMR-90 replicate was seeded in technical duplicates. The next day, medium was changed to medium containing liposomal clodronate (Clodrosome #SKU CLD-8909) at 0.1 to 50 ug/ ml. After 48 hours, cells were imaged and subjected to MTS assays to determine cell survival (CellTiter 96 AQueous MTS Reagent (Promega, G1112) with phenazine methosulfate (Sigma, P9625)). Technical duplicates for each cell line were averaged and values were compared to untreated control cells.
Quantification and statistical analysis
GraphPad Prism 9 (version 9.3.1) was used for all statistical analyses. Scatter plots with bars represent the mean with SEM. Statistical significance was determined by ordinary one-way ANOVA with Tukey’s multiple comparison test or a Student’s two-tailed unpaired t-test. For consistency in these comparisons, the following denotes significance in all figures: *P<0.05, **P<0.01, ***P<0.001. We note that no power calculations were used. No samples were excluded. Investigators were blinded to allocation during experiments and outcome assessment, except for special instances in which blinding was not possible.
Supplementary Material
Highlights.
Senescent cells accelerate spontaneous KrasLA1-driven lung tumorigenesis
Senescent alveolar macrophages accumulate in aged and tumorous tissue
Surprisingly, knocking out p16INK4a or p21Cip1 delays neoplasia in KrasLA1 mice
Senescent alveolar macrophages counteract cytotoxic T cell accumulation
Acknowledgments
We would like to thank A. Aziz for his valuable input during the initial design of the experiments; G. Nelson, T. Thao, and R. Fierro Velasco for genotyping and animal support; T. McNeely and M. Schaefer for input on the design of the RNA-ISH experiment; H. Okuyama for guidance on T cell characterization, the Mayo Clinic Genome Analysis Core for RT-qPCR instrumentation and assistance with the single cell RNA-seq experiment; and B. Childs, J. van Deursen, T. Bussian, P. Ng, and J. Elisseeff for helpful feedback on the manuscript. This work was supported by the National Institutes of Health (R01AG053229 to D.J.B., R01AG068076 to D.J.B., T32AG049672 to I.S., R01AG056318 to H.L., R01AG061796 to H.L., P50CA136393 to H.L., R03OD34496 to H.L., CA240323 to H.L.), the Glenn Foundation for Medical Research (D.J.B. and H.L.), Mayo Clinic Center for Biomedical Discovery (H.L.), Mayo Clinic Center for Individualized Medicine (H.L.), Mayo Clinic Cancer Center (P30CA015083 to H.L.), Mayo Clinic Department of Artificial Intelligence and informatics (H.L.), David F. and Margaret T. Grohne Cancer Immunology and Immunotherapy Program (H.L.), Eric and Wendy Schmidt Fund for AI Research and Innovation (H.L.), and the Mayo Clinic Graduate School of Biomedical Sciences (stipends supporting L.I.P. and S.I.G.). This research was conducted while I.S. was a Glenn Foundation for Medical Research Postdoctoral Fellow.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
L.I.P., I.S., S.I.G., and D.J.B. have a potential financial interest related to this research. They are co-inventors on patents held by Mayo Clinic and D.J.B. is a co-inventor on patent applications licensed to or filed by Unity Biotechnology, a company developing senolytic strategies for treatment of age-related disorders, including small molecules that selectively eliminate senescent cells. Research in the Baker laboratory has been reviewed by the Mayo Clinic Conflict of Interest Review Board and is being conducted in compliance with Mayo Clinic Conflict of Interest policies. The other authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
References
- 1.Serrano M, Lin AW, McCurrach ME, Beach D, and Lowe SW (1997). Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602. [DOI] [PubMed] [Google Scholar]
- 2.van Deursen JM (2014). The role of senescent cells in ageing. Nature 509, 439–446. 10.1038/nature13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, et al. (2011). Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479, 547–551. 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- 4.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, and Lowe SW (2007). Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656–660. 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prieto LI, Graves SI, and Baker DJ (2020). Insights from In Vivo Studies of Cellular Senescence. Cells 9. 10.3390/cells9040954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sturmlechner I, Zhang C, Sine CC, van Deursen EJ, Jeganathan KB, Hamada N, Grasic J, Friedman D, Stutchman JT, Can I, et al. (2021). p21 produces a bioactive secretome that places stressed cells under immunosurveillance. Science 374, eabb3420. 10.1126/science.abb3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tasdemir N, Banito A, Roe JS, Alonso-Curbelo D, Camiolo M, Tschaharganeh DF, Huang CH, Aksoy O, Bolden JE, Chen CC, et al. (2016). BRD4 Connects Enhancer Remodeling to Senescence Immune Surveillance. Cancer Discov 6, 612–629. 10.1158/2159-8290.CD-16-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krtolica A, Parrinello S, Lockett S, Desprez PY, and Campisi J (2001). Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A 98, 12072–12077. 10.1073/pnas.211053698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu D, and Hornsby PJ (2007). Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res 67, 3117–3126. 10.1158/0008-5472.CAN-06-3452. [DOI] [PubMed] [Google Scholar]
- 10.Wang L, Lankhorst L, and Bernards R (2022). Exploiting senescence for the treatment of cancer. Nat Rev Cancer 10.1038/s41568-022-00450-9. [DOI] [PubMed] [Google Scholar]
- 11.Sieben CJ, Sturmlechner I, van de Sluis B, and van Deursen JM (2018). Two-Step Senescence-Focused Cancer Therapies. Trends Cell Biol 28, 723–737. 10.1016/j.tcb.2018.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu JY, Souroullas GP, Diekman BO, Krishnamurthy J, Hall BM, Sorrentino JA, Parker JS, Sessions GA, Gudkov AV, and Sharpless NE (2019). Cells exhibiting strong p16 (INK4a) promoter activation in vivo display features of senescence. Proc Natl Acad Sci U S A 116, 2603–2611. 10.1073/pnas.1818313116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burd CE, Sorrentino JA, Clark KS, Darr DB, Krishnamurthy J, Deal AM, Bardeesy N, Castrillon DH, Beach DH, and Sharpless NE (2013). Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell 152, 340–351. 10.1016/j.cell.2012.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, and van Deursen JM (2011). Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236. 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang B, Wang L, Gasek NS, Zhou Y, Kim T, Guo C, Jellison ER, Haynes L, Yadav S, Tchkonia T, et al. (2021). An inducible p21-Cre mouse model to monitor and manipulate p21-highly-expressing senescent cells in vivo. Nat Aging 1, 962–973. 10.1038/s43587-021-00107-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, et al. (2016). Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530, 184–189. 10.1038/nature16932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, and Jacks T (2001). Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 410, 1111–1116. 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 18.Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, Benguria A, Zaballos A, Flores JM, Barbacid M, et al. (2005). Tumour biology: senescence in premalignant tumours. Nature 436, 642. 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- 19.Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, et al. (2008). ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 68, 3421–3428. 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 20.Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, and Baker DJ (2018). Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 562, 578–582. 10.1038/s41586-018-0543-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, and van Deursen JM (2016). Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science (New York, N.Y.) 354, 472–477. 10.1126/science.aaf6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Faber AC, Farago AF, Costa C, Dastur A, Gomez-Caraballo M, Robbins R, Wagner BL, Rideout WM 3rd, Jakubik CT, Ham J, et al. (2015). Assessment of ABT-263 activity across a cancer cell line collection leads to a potent combination therapy for small-cell lung cancer. Proc Natl Acad Sci U S A 112, E1288–1296. 10.1073/pnas.1411848112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mantovani A, Allavena P, Sica A, and Balkwill F (2008). Cancer-related inflammation. Nature 454, 436–444. 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 24.Coussens LM, Zitvogel L, and Palucka AK (2013). Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science (New York, N.Y.) 339, 286–291. 10.1126/science.1232227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hanahan D (2022). Hallmarks of Cancer: New Dimensions. Cancer Discov 12, 31–46. 10.1158/2159-8290.CD-21-1059. [DOI] [PubMed] [Google Scholar]
- 26.Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, Rydkina E, Vujcic S, Balan K, Gitlin II, et al. (2017). p16(Ink4a) and senescence-associated beta-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging (Albany NY) 9, 1867–1884. 10.18632/aging.101268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, Rydkina E, Vujcic S, Balan K, Gitlin I, et al. (2016). Aging of mice is associated with p16(Ink4a)- and beta-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging (Albany NY) 8, 1294–1315. 10.18632/aging.100991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prattichizzo F, Bonafe M, Olivieri F, and Franceschi C (2016). Senescence associated macrophages and “macroph-aging”: are they pieces of the same puzzle? Aging (Albany NY) 8, 3159–3160. 10.18632/aging.101133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeisberger SM, Odermatt B, Marty C, Zehnder-Fjällman AHM, Ballmer-Hofer K, and Schwendener RA (2006). Clodronate-liposome-mediated depletion of tumour-associated macrophages: a new and highly effective antiangiogenic therapy approach. British Journal of Cancer 95, 272–281. 10.1038/sj.bjc.6603240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borriello L, Coste A, Traub B, Sharma VP, Karagiannis GS, Lin Y, Wang Y, Ye X, Duran CL, Chen X, et al. (2022). Primary tumor associated macrophages activate programs of invasion and dormancy in disseminating tumor cells. Nat Commun 13, 626. 10.1038/s41467-022-28076-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, et al. (2014). Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20. 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mantovani A, Marchesi F, Malesci A, Laghi L, and Allavena P (2017). Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol 14, 399–416. 10.1038/nrclinonc.2016.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dong B, Wu C, Huang L, and Qi Y (2021). Macrophage-Related SPP1 as a Potential Biomarker for Early Lymph Node Metastasis in Lung Adenocarcinoma. Front Cell Dev Biol 9, 739358. 10.3389/fcell.2021.739358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu L, Wang X, Li X, Wu X, Tang M, and Wang X (2018). Upregulation of IGF1 by tumor-associated macrophages promotes the proliferation and migration of epithelial ovarian cancer cells. Oncol Rep 39, 818–826. 10.3892/or.2017.6148. [DOI] [PubMed] [Google Scholar]
- 35.Gocheva V, Wang HW, Gadea BB, Shree T, Hunter KE, Garfall AL, Berman T, and Joyce JA (2010). IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev 24, 241–255. 10.1101/gad.1874010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu B, Wu Y, Huang S, Zhang R, Son YM, Li C, Cheon IS, Gao X, Wang M, Chen Y, et al. (2021). Uncoupling of macrophage inflammation from self-renewal modulates host recovery from respiratory viral infection. Immunity 54, 1200–1218 e1209. 10.1016/j.immuni.2021.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kale A, Sharma A, Stolzing A, Desprez PY, and Campisi J (2020). Role of immune cells in the removal of deleterious senescent cells. Immun Ageing 17, 16. 10.1186/s12979-020-00187-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gasek NS, Kuchel GA, Kirkland JL, and Xu M (2021). Strategies for Targeting Senescent Cells in Human Disease. Nat Aging 1, 870–879. 10.1038/s43587-021-00121-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Q, Li A, Tian Y, Wu JD, Liu Y, Li T, Chen Y, Han X, and Wu K (2016). The CXCL8-CXCR1/2 pathways in cancer. Cytokine Growth Factor Rev 31, 61–71. 10.1016/j.cytogfr.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang F, Zhang S, Meng Q, Zhou F, Pan B, Liu F, and Yu Y (2021). CXCR1 correlates to poor outcomes of EGFR-TKI against advanced non-small cell lung cancer by activating chemokine and JAK/STAT pathway. Pulm Pharmacol Ther 67, 102001. 10.1016/j.pupt.2021.102001. [DOI] [PubMed] [Google Scholar]
- 41.Teijeira A, Garasa S, Gato M, Alfaro C, Migueliz I, Cirella A, de Andrea C, Ochoa MC, Otano I, Etxeberria I, et al. (2020). CXCR1 and CXCR2 Chemokine Receptor Agonists Produced by Tumors Induce Neutrophil Extracellular Traps that Interfere with Immune Cytotoxicity. Immunity 52, 856–871 e858. 10.1016/j.immuni.2020.03.001. [DOI] [PubMed] [Google Scholar]
- 42.Di Micco R, Krizhanovsky V, Baker D, and d’Adda di Fagagna F (2021). Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol 22, 75–95. 10.1038/s41580-020-00314-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baeuerle PA, and Gires O (2007). EpCAM (CD326) finding its role in cancer. Br J Cancer 96, 417–423. 10.1038/sj.bjc.6603494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saade M, Araujo de Souza G, Scavone C, and Kinoshita PF (2021). The Role of GPNMB in Inflammation. Front Immunol 12, 674739. 10.3389/fimmu.2021.674739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walsh PT, Smith LM, and O’Connor R (2002). Insulin-like growth factor-1 activates Akt and Jun N-terminal kinases (JNKs) in promoting the survival of T lymphocytes. Immunology 107, 461–471. 10.1046/j.1365-2567.2002.01525.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Casanova-Acebes M, Dalla E, Leader AM, LeBerichel J, Nikolic J, Morales BM, Brown M, Chang C, Troncoso L, Chen ST, et al. (2021). Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature 595, 578–584. 10.1038/s41586-021-03651-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, Wu EA, Horner JW, and DePinho RA (2001). Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 413, 86–91. 10.1038/35092592. [DOI] [PubMed] [Google Scholar]
- 48.Baker DJ, Perez-Terzic C, Jin F, Pitel KS, Niederlander NJ, Jeganathan K, Yamada S, Reyes S, Rowe L, Hiddinga HJ, et al. (2008). Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nature cell biology 10, 825–836. 10.1038/ncb1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baker DJ, Dawlaty MM, Wijshake T, Jeganathan KB, Malureanu L, van Ree JH, Crespo-Diaz R, Reyes S, Seaburg L, Shapiro V, et al. (2013). Increased expression of BubR1 protects against aneuploidy and cancer and extends healthy lifespan. Nature cell biology 15, 96–102. 10.1038/ncb2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goplen NP, Huang S, Zhu B, Cheon IS, Son YM, Wang Z, Li C, Dai Q, Jiang L, and Sun J (2019). Tissue-Resident Macrophages Limit Pulmonary CD8 Resident Memory T Cell Establishment. Front Immunol 10, 2332. 10.3389/fimmu.2019.02332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 33, 495–502. 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Borriello L, Coste A, Traub B, Sharma VP, Karagiannis GS, Lin Y, Wang Y, Ye X, Duran CL, Chen X, et al. (2022). Primary tumor associated macrophages activate programs of invasion and dormancy in disseminating tumor cells. Nature Communications 13, 626. 10.1038/s41467-022-28076-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Toda G, Yamauchi T, Kadowaki T, and Ueki K (2021). Preparation and culture of bone marrow-derived macrophages from mice for functional analysis. STAR Protoc 2, 100246. 10.1016/j.xpro.2020.100246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sturmlechner I, Sine CC, Jeganathan KB, Zhang C, Fierro Velasco RO, Baker DJ, Li H, and van Deursen JM (2022). Senescent cells limit p53 activity via multiple mechanisms to remain viable. Nat Commun 13, 3722. 10.1038/s41467-022-31239-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed data for single-cell RNA-seq have been deposited at Gene Expression Omnibus GSE201247.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.