

Abstract
Aging is associated with cognitive decline and is the main risk factor for a myriad of conditions including neurodegeneration and stroke. Concomitant with aging is the progressive accumulation of misfolded proteins and loss of proteostasis. Accumulation of misfolded proteins in the endoplasmic reticulum (ER) leads to ER stress and activation of the unfolded protein response (UPR). The UPR is mediated, in part, by the eukaryotic initiation factor 2α (eIF2α) kinase protein kinase R-like ER kinase (PERK). Phosphorylation of eIF2α reduces protein translation as an adaptive mechanism but this also opposes synaptic plasticity. PERK, and other eIF2α kinases, have been widely studied in neurons where they modulate both cognitive function and response to injury. The impact of astrocytic PERK signaling in cognitive processes was previously unknown. To examine this, we deleted PERK from astrocytes (AstroPERKKO) and examined the impact on cognitive functions in middle-aged and old mice of both sexes. Additionally, we tested the outcome following experimental stroke using the transient middle cerebral artery occlusion (MCAO) model. Tests of short-term and long-term learning and memory as well as of cognitive flexibility in middle-aged and old mice revealed that astrocytic PERK does not regulate these processes. Following MCAO, AstroPERKKO had increased morbidity and mortality. Collectively, our data demonstrate that astrocytic PERK has limited impact on cognitive function and has a more prominent role in the response to neural injury.
Keywords: Astrocytes, protein kinase R-like ER kinase (PERK), unfolded protein response, learning and memory, stroke, aging, MCAO, glia
Graphical Abstract:
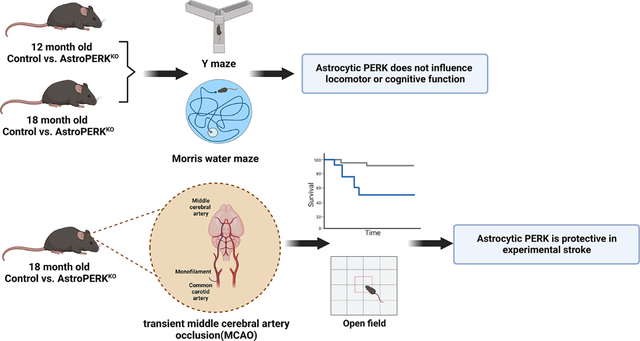
Selective deletion of PERK from astrocytes does not impact cognitive function but worsens outcome from experimental stroke. Created with BioRender.com
Introduction
Aging is associated with cognitive decline (1) and is the main risk factor for neurodegeneration and stroke (2,3). With age, cells accumulate damage, mutations, and gradually decline in the maintenance of protein homeostasis (proteostasis) (4). The loss of proteostasis is associated with age-related cognitive decline (5,6) and neurodegeneration (6–8). The mechanisms ensuring proteostasis are numerous and involve molecular chaperones, transcription factors, and organelle-specific sensors that monitor protein disturbances (7).
The endoplasmic reticulum (ER) is a vital organelle within this proteostasis network (9). The ER is essential for synthesis, folding, and processing of secretory and transmembrane proteins (10). Diverse pathological insults lead to the accumulation of misfolded proteins within the ER, which subsequently results in ER stress (11). To restore homeostasis, cells initiate the evolutionarily conserved unfolded protein response (UPR) (12). The UPR is orchestrated by three trans-ER membrane proteins, PKR(protein kinase R)-like ER kinase (PERK), inositol requiring enzyme (IRE) 1, and activating transcription factor (ATF) 6, each of which mediate distinct downstream signaling pathways with substantial crosstalk among them (13). The activation of the UPR ultimately allows cells to cope with increased protein folding load (12,14). However, chronic UPR activation in response to unresolved ER stress can lead to aberrant non-resolving inflammation and results in cell death (12,15). The UPR is partly orchestrated by the trans-ER membrane Ser/Thr protein kinase PERK which, after sensing misfolded protein accumulation in the ER lumen, is activated by dimerization and autophosphorylation (16,17). Activated PERK phosphorylates the eukaryotic initiation factor 2α (eIF2α), which in turn leads to global attenuation of protein translation while simultaneously increasing expression of selected proteins including ER resident molecular chaperons in order to restore proteostasis (12,16,18). This ER monitoring network is perturbed during aging (6). ER resident chaperones and UPR signaling molecules in CNS have been reported to decrease in old rodents compared to young (19) and the responsiveness of IRE1 is also diminished (5), suggesting their reduced function may contribute to age-related disease. PERK has been widely studied in neurodegeneration and injury. Neural injury, such as ischemic stroke, leads to suppression of protein synthesis through PERK-dependent phosphorylation of eIF2α at serine 51 (20–26). Recently it was shown that PERK deletion from neurons worsens middle cerebral artery occlusion (MCAO)-induced infarct volume and behavioral deficits, indicating a functional role in outcome following injury (27). Collectively, many studies have demonstrated that PERK signaling has a functional role in neural injury and disease. These same pathways are also important in learning and memory (28). In neurons, eIF2α has emerged as a convergence point of proteostasis and cognition (29).
Memory formation requires synaptic plasticity, which is the activity dependent changes of neuronal synaptic transmission (30). In addition, new protein translation at the preexisting synapses is particularly necessary for memory formation (28,31). As such, the phosphorylation of eIF2α impedes memory formation (28,32). Consistent with this, ER stress and PERK activation have been causally implicated in memory impairment in rodents (33–38). Moreover, targeting the PERK pathway by either genetic or pharmacological approaches has been reported to ameliorate neuronal loss and memory impairment in various neurodegenerative and neural injury models (33,35,39–43). Importantly, physiological phosphorylation of eIF2α allows cognitive flexibility and disruption of P-eIF2α or the downstream transcription factor ATF4 impairs this process (44,45). Currently, it is unknown if the PERK-dependent regulation of cognitive function is exclusive to neurons or if glial cells, such as astrocytes, also influence this process.
Astrocytes are a major glial cell type, which orchestrate diverse critical functions in the central nervous system such as formation and function of synapses, providing trophic support to neurons, development of white and gray matter, blood flow regulation, driving neuroinflammation and promoting CNS repair after neurological insult (46,47). Astrocytes regulate synaptic transmission by forming the “tripartite synapse” (46). Moreover, astrocytes by virtue of producing various neuroactive compounds, can modulate synaptic plasticity (48) which makes them an ideal candidate for the regulation of complex brain functions such as learning and memory (49). Indeed, previous studies have shown that astrocytes are critical in learning and memory (49–53).
The functional role of astrocytic PERK in the context of learning and memory has not been studied previously. Here, we deleted PERK from astrocytes and examined the impact on cognitive function and cerebral ischemia in aged mice. Our data showed that PERK deletion from astrocytes does not worsen age-related cognitive decline but does result in greater morbidity and mortality following experimental stroke.
Materials and Methods
Animals
PERKfl/fl (“Control”: Jackson Laboratories) (RRID:IMSR_JAX:023066) (54) and astrocyte selective Cre-recombinase expressing (GFAP-Cre, line 77.6) (RRID:IMSR_JAX:024098) (55) mice were purchased, subsequently housed and bred under the care of the Office of Lab Animal Resources at West Virginia University. All animal studies were conducted with approval from the WVU institutional animal care and use committee and in accordance with the guide for the Care and Use of Laboratory Animals. Female GFAP-Cre mice were crossed with male PERKfl/fl mice and progeny F1 heterozygous females with GFAP-Cre were backcrossed with male PERKfl/fl mice to obtain the astrocytic PERK knockout (“AstroPERKKO”; GFAP-Cre, PERKfl/fl) mice. The Cre allele was always maintained as hemizygous. Mice were on a 12/12 hour light/dark cycle with food and water ad libitum. Tail biopsies were taken from 17–21 days old animals and genomic DNA was isolated using Wizard SV genomic DNA purification system (Promega). Genotyping was done by standard polymerase chain reaction (PCR) using platinum II hot start green PCR master mix (Invitrogen). Subsequently, genotype was determined by agarose gel electrophoresis. The genotyping primers (Integrated DNA Technologies) are used to detect the presence of Cre-recombinase allele (forward primer: GCT AAC CAT GTT CAT GCC TTC; reverse primer: AGG CAA ATT TTG GTG TAC GG) and PERK floxed allele (forward primer: TTG CAC TCT GGC TTT CAC TC; reverse primer: AGG AGG AAG GTG GAA TTT GG).
Open field test
All behavioral tests were conducted by an experimenter blinded to the genotypes of the mice. General locomotor activity was measured using an open field apparatus that was an acrylic rectangular box without a lid [dimensions: 40.64 cm (length) × 40.64 cm (width) × 38.1 cm (height); San Diego Instruments PAS-Open Field System PAS004634] containing photocells. Data were collected by photo beam disruption due to animal movement (16 photo beams in both x and y axis directions for locomotor activity and 16 elevated photo beams in z axis for rearing activity) and analyzed by PAS system software. The center area was defined as an inner section 7.62 cm away from each of the walls. Ambulation, rearing activity, center and peripheral activity measured by counting the number of photo beam interruptions using PAS-Open field software. Each mouse was placed at the center of the apparatus and were allowed to move freely within. Data were collected for 30 minutes.
Y-maze test
Spontaneous alternation Y-maze test was performed to assess short-term spatial working memory as described previously (56) with minor modifications. The Y shaped maze made of grey polycarbonate [dimensions: 38 cm (length) × 8 cm (width) × 10 cm (height)] was surrounded by black colored maze cues on the walls of the room. Each mouse was placed in one arm of the maze and allowed to roam freely inside the maze for 8.5 minutes. All trials were video recorded and analyzed by ANY-maze video tracking software (Stoelting) (RRID:SCR_014289). Entry to an arm was recorded only when all of the animal’s limbs were within that particular arm. Percent spontaneous alternation was calculated as follows: the number of triads containing entries into three arms/maximum possible alternations (total number of arms entered-2) X 100.
Morris water maze test
Morris water maze test was performed as described previously (57) with minor modifications in the protocol. A circular pool (1.07 m in diameter) was filled with water and non-toxic paint was used to make the water opaque. A circular platform (diameter: 10 cm) was submerged below the water surface. The location of the hidden platform was constant throughout the training phase. The water maze was surrounded by black colored maze cues on the walls of the room. Briefly, mice were trained for seven consecutive days [4 trials/day/mouse from each direction (north, south, east, west) in a random order] to find hidden platform in a circular water pool. Maximum duration for each trial was 60 s and the trial ended when a mouse found the hidden platform successfully. Once mice found the platform, they were allowed to sit on the platform for 15 s and then returned to a heated drying cage. Mice that did not find the platform within the trial period were guided to the platform, and then were allowed to sit there for 15 s before returning to the cage. The probe trial was administered at the 7th day right after the last training session. During the probe trial, the platform was removed from the maze and mice were allowed to swim for 60 s after which they were returned to the heated cage. To assess cognitive flexibility, mice underwent reversal test where mice were retrained for two consecutive days as described above to learn a new platform location. At the end of last training session on day 2, another probe trial (reversal probe trial) was administered as described before. Each trial was video recorded and analyzed by ANY-maze video tracking software (Stoelting) (RRID:SCR_014289).
Primary astrocyte culture
Primary astrocytes were prepared from postnatal day 0–1 control (PERKfl/fl) and AstroPERKKO (GFAP-Cre, PERKfl/fl) mice as previously described (58). In brief, pups were euthanized by decapitation, and the brains were collected into cold media. Meninges, cerebellum and olfactory bulbs were removed and cerebra were collected. Tissue was disrupted by trituration and filtered through a 100 μm cell strainer. Cells were centrifuged at 300 × g for 5 min, resuspended in fresh astrocyte medium (Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS) (Corning), 16 mM 2-[4-(2-hydroxyethyl)piperazin-1-yl] ethanesulfonic acid (HEPES) (Gibco), 1X non-essential amino acids (Corning), 2 mM L-glutamine (Sigma-Aldrich), 100 units/ml penicillin (Gibco), 100 μg/ ml streptomycin (Gibco), and 50 μg/ml gentamicin (Sigma-Aldrich), and transferred onto T-75 tissue culture flasks. The cultures were maintained for approximately 12–14 days at 37 °C in humidified 5% CO2/95% air atmosphere. One half of the media was changed every 3–4 days. Astrocytes were separated from microglia by shaking at 200 rpm for 2 hours. Subsequently, media in the flask was removed and astrocytes were incubated with trypsin (Gibco) for 5 min at 37°C. Cells were collected in media and centrifuged for 5 min at 300 g. Thereafter, astrocytes were seeded in 6 well plates and 48–72 hours later cells were collected for protein.
Western Blotting
Primary astrocytes were washed twice with 1X Dulbecco’s phosphate buffered saline (DPBS) (Gibco) and lysed with immunoprecipitation (IP) lysis buffer (20 mM Tris [pH 7.5], 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 0.5% NP-40, and 1X phosphatase/protease inhibitor cocktail (Thermo scientific). Fifteen-month-old (mean age = 462.4 days) control and AstroPERKKO mice were anesthetized using isoflurane (Vet one). Following cessation of breathing, mice were transcardially perfused with 30 ml ice cold 1X DPBS (Gibco). Brains were removed and cortex and hippocampus fractions were isolated. Tissues were disrupted by trituration and filtered through a 100 μm cell strainer. Cells were centrifuged at 300 × g for 5 min, resuspended in IP lysis buffer. The lysates were subsequently centrifuged for 15 min at 17000 × g at 4 °C and clear supernatants were taken for further analysis. Protein concentrations were determined using the bicinchonicic acid assay (BCA) assay (Thermo scientific). Equal amounts of protein from each sample were solubilized in Laemmli sample buffer [2% Sodium Dodecyl Sulfate (SDS)] and heated for 5 min at 95 °C. Proteins were separated by 8% or 10% SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (Bio-Rad). The membranes were blocked in 5% milk in wash buffer (20 mM Tris base, 137 mM NaCl and 0.05% Tween-20 (TBST)) followed by an overnight incubation at 4°C with primary antibody diluted in 5% bovine serum albumin (BSA) (Fisher) or milk, according to the manufacturer’s recommendation. Primary antibodies were diluted as follows; PERK 1:2000 (Cell signaling, 3192S), P-eIF2α 1:2000 (Cell signaling, 3398S), eIF2α 1:3000 (Cell signaling, 5324S), GLAST 1:2000 (Abcam, ab416), GFAP 1:3000 (Cell signaling, 12389S), GAPDH 1:12000 (Sigma-Aldrich, MAB374). Antibody used are listed in table 1. Membranes were washed for 1 h with frequent changes to TBST. Membranes were incubated with horseradish peroxidase-conjugated goat anti-rabbit or goat anti-mouse (1:3000 dilution, Jackson Immuno Research) secondary antibody diluted in 5% milk in TBST for 1 h at room temperature. Membranes were again washed for 1 h and TBST was changed frequently. Proteins were detected by enhanced chemiluminescence (Thermo-scientific). Immunoblots were imaged on an ChemiDoc Touch imaging system (Bio-Rad) and quantified using Image Lab software (Bio-Rad) (RRID:SCR_014210).
Table 1:
List of antibodies used in the study
| Antibody name | Structure of the immunogen against which the animal was immunized | The manufacturer, Catalog or lot number, RRID, Species it was raised in, Monoclonal or polyclonal | Concentration used |
|---|---|---|---|
| PERK | A synthetic peptide corresponding to residues surrounding Val248 of human PERK protein | (Cell Signaling Technology Cat# 3192, RRID:AB_2095847) Rabbit Monoclonal |
1:2000 |
| P-eIF2α | A synthetic phosphopeptide corresponding to residues surrounding Ser51 of human eIF2α | Cell signaling 3398S RRID:AB_2096481 Rabbit Monoclonal |
1:2000 |
| Total eIF2α | A purified recombinant protein fragment representing sequence in the central region of human eIF2α | (Cell Signaling Technology Cat# 5324, RRID:AB_10692650) Rabbit Monoclonal |
1:3000 |
| GFAP | A synthetic peptide corresponding to residues surrounding Asp395 of human GFAP protein. | (Cell Signaling Technology Cat# 12389, RRID:AB_2631098) Rabbit Monoclonal |
1:3000 |
| GLAST | Synthetic peptide corresponding to Rat EAAT1 (C terminal). Corresponding to 20 residues from the C-terminal | Abcam ab416 RRID:AB_304334 Rabbit Polyclonal |
1:2000 |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase from rabbit muscle | (Millipore Cat# MAB374, RRID:AB_2107445) Mouse Monoclonal |
1:12000 |
Reverse transcription (RT)-PCR
RNA was isolated using 1 ml of TRIzol (Life technologies) according to the manufacturer’s protocol. RNA was quantified using a NanoDrop system (Thermo scientific). For cDNA synthesis, 1 μg of RNA was mixed with oligo dT primer and incubated at 70°C for 5 min followed immediately by 5 minutes on ice. A mix containing reaction buffer (Promega), moloney murine leukemia virus reverse transcriptase (Promega), deoxynucleotide triphosphates (dNTP) (Promega), and ribonuclease inhibitor (Promega) was added and incubated at 42°C for 1 hour. The reaction was terminated by incubation at 95°C for 5 minutes. Subsequently, cDNA was used as input DNA for PCR (using platinum II hot start green PCR master mix (Invitrogen)) to detect the presence of Cre-recombinase (forward primer: GCT AAC CAT GTT CAT GCC TTC; reverse primer: AGG CAA ATT TTG GTG TAC GG). Cre presence was confirmed by running PCR products on agarose gel.
Retro-orbital injection of adeno-associated virus (AAV)
Pre-made AAV viral stock was purchased from Addgene. pAAV-FLEX-tdTomato was a gift from Edward Boyden (Addgene viral prep # 28306-PHPeB) (RRID:Addgene_28306). Mice were anesthetized briefly with isoflurane. Gentle pressure was applied to the skin dorsal and ventral to the eye so that the eye partially protrudes. The needle (27 gauge) of the syringe was placed in the medial canthus to access the retro-orbital sinus. An injection volume of 150 μl containing 5 × 1011 viral genomes (vg) was used. Mice were analyzed 3 – 4 weeks post-injection.
Microscopy
Following euthanasia, mice were transcardially perfused with 30 ml of cold DPBS (Gibco) followed by 30 ml of cold 4% paraformaldehyde (PFA). Brains were removed and post-fixed in 4% PFA overnight. Brains were then washed with PBS and immersed in 20% then 30% sucrose, incubating at 4° until the tissue sinks at each step. Brains were then frozen and sectioned at 25 μm on a cryostat (Leica). Sections were placed on glass slides and mounted using ProLong glass antifade mountant with NucBlue (ThermoFisher). Slides were then imaged on a slide scanner (Olympus) and subsequently imaged on a Nikon A1R confocal microscope.
For H&E staining, coronal sections of formalin-fixed paraffin embedded brain were used. Slides were evaluated by a pathologist blinded to the experimental groups. Microscopic changes were graded as to severity utilizing the International Harmonization of Nomenclature and Diagnostic (INHAND) standards. Histopathology was conducted by iHisto inc.
Immunostaining
Following TTC staining, tissue was fixed in 10% formalin overnight then paraffin embedded. Immunostaining was conducted in a blinded fashion by iHisto inc. Tissues were sectioned, dewaxed, and stained using anti-CHOP (ThermoFisher, MA1-250) and anti GFAP (Cell Signaling, #3670). Immunostaining was enhanced using Tyramide Signal Amplification. Slides were imaged using a slide scanner at 20x magnification. Quantification was conducted by selecting semi-random fields based on GFAP staining in the deep cortex and the caudate putamen, using the corpus callosum as a reference landmark. Cells were quantified from the ipsilateral and contralateral hemispheres from three animals of each genotype with a minimum of 300 cells per hemisphere were counted. Total cell number, based on DAPI staining, was determined in an automated fashion using Photoshop. GFAP and CHOP positive cells were then counted manually from the same field. To assess astrocyte number, branching degree, and Feret diameter, images were analyzed with Cell Profiler V4 (www.cellprofiler.org, RRID: nif-0000-00280) (59). The mean measurements from each animal were used for statistical assessment.
Middle cerebral artery occlusion (MCAO)
MCAO surgeries were conducted as described previously (60), with minor modifications. Briefly, mice were anesthetized with isoflurane and an incision in the skin made to visualize the proximal common carotid artery. A small incision was made in the vessel and then a sterile monofilament (occluder) threaded into the internal carotid artery to a point that prevents blood flow into the right middle cerebral artery. The occluder remained in place for 60 min and then was removed. Surgeries were performed by a researcher blinded to the genotypes. Neuroscores were assessed based on the following scale: 0 – no deficit, 1 – forelimb weakness and/or turning to one side when suspended by tail, 2 – circling, 3 – unable to bear weight or falling to the affected side, 4 – lack of spontaneous movement or barrel rolling (61,62).
Statistical analysis
All statistical analyses were performed using GraphPad Prism (version 9) (RRID:SCR_002798) software. Unpaired two-tailed t test or Mann-Whitney test was used to compare between two groups. Ordinary two-way ANOVA test was used to compare between repeatedly measured datasets. Sidak’s multiple comparison test was used for post-hoc analysis. Ordinary two-way ANOVA test was performed to determine interactions between genotype and sex, genotype and age as well as age and sex. Tukey’s multiple comparison test was used for post-hoc analysis. Three-way ANOVA was performed to examine interactions between genotype, age and sex. Tukey’s multiple comparison test was performed for post-hoc analysis. Statistical significance is defined as p < 0.05. Data are represented as Mean ± SD on all bar charts. For the violin plots, dark spaced line on the top, middle and bottom of a violin represents 75th Percentile, median and 25th percentile of the dataset respectively. Statistical tests are listed in table 2.
TABLE 2.
Summary of statistics for each figure.
| Figure | Test parameter | Who vs Who | N | Is data distribution normal? (Shapiro-Wilk test) | Test name | P value | F (DFn, DFd) | Post-Hoc analysis |
|---|---|---|---|---|---|---|---|---|
| 1E | Cortex PERK/GAPDH Western blot densitometry |
Control vs AstroPERKKO | Control= 5 (M= 4; F= 1) AstroPERKKO= 5 (M= 3; F=2) |
No | Mann-Whitney test | 0.9524 | N/A | N/A |
| 1E | Cortex P-eIF2α/ Total eIF2α Western blot densitometry |
Control vs AstroPERKKO | Control= 5 (M= 4; F= 1) AstroPERKKO= 5 (M= 3; F=2) |
Yes | Unpaired two-tailed t test | 0.3917 | N/A | N/A |
| 1E | Cortex GFAP/GAPDH Western blot densitometry |
Control vs AstroPERKKO | Control= 5 (M= 4; F= 1) AstroPERKKO= 5 (M= 3; F=2) |
Yes | Unpaired two-tailed t test | 0.1446 | N/A | N/A |
| 1E | Cortex GLAST/GAPDH Western blot densitometry |
Control vs AstroPERKKO | Control= 5 (M= 4; F= 1) AstroPERKKO= 5 (M= 3; F=2) |
Yes | Unpaired two-tailed t test | 0.9723 | N/A | N/A |
| 1G | Hippocampus PERK/GAPDH Western blot densitometry |
Control vs AstroPERKKO | Control= 5 (M= 4; F= 1) AstroPERKKO= 5 (M= 3; F=2) |
Yes | Unpaired two-tailed t test | 0.8589 | N/A | N/A |
| 1G | Hippocampus P-eIF2α/ Total eIF2α Western blot densitometry |
Control vs AstroPERKKO | Control= 5 (M= 4; F= 1) AstroPERKKO= 5 (M= 3; F=2) |
Yes | Unpaired two-tailed t test | 0.6128 | N/A | N/A |
| 1G | Hippocampus GFAP/GAPDH Western blot densitometry | Control vs AstroPERKKO | Control= 5 (M= 4; F= 1) AstroPERKKO= 5 (M= 3; F=2) |
Yes | Unpaired two-tailed t test | 0.8941 | N/A | N/A |
| 1G | Hippocampus GLAST/GAPDH Western blot densitometry |
Control vs AstroPERKKO | Control= 5 (M= 4; F= 1) AstroPERKKO= 5 (M= 3; F=2) |
Yes | Unpaired two-tailed t test | 0.4582 | N/A | N/A |
| 2A | Total Ambulation (open field test) | Middle-aged (12 months) Control vs AstroPERKKO |
Control= 17 (M= 7; F= 10) AstroPERKKO= 17 (M= 8; F=9) |
No | Mann-Whitney test | 0.1309 | N/A | N/A |
| 2B | Total Rearing (open field test) | Middle-aged (12 months) Control vs AstroPERKKO |
Control= 17 (M= 7; F= 10) AstroPERKKO= 17 (M= 8; F=9) |
No | Mann-Whitney test | 0.3986 | N/A | N/A |
| 2C | % Center Tendency (open field test) | Middle-aged (12 months) Control vs AstroPERKKO |
Control= 17 (M= 7; F= 10) AstroPERKKO= 17 (M= 8; F=9) |
Yes | Unpaired two-tailed t test | 0.0944 | N/A | N/A |
| 2D | Total Ambulation (open field test) | Old (18 months) Control vs AstroPERKKO |
Control= 8 (M= 3; F= 5) AstroPERKKO= 9 (M= 5; F=4) |
No | Mann-Whitney test | 0.4234 | N/A | N/A |
| 2E | Total Rearing (open field test) | Old (18 months) Control vs AstroPERKKO |
Control= 8 (M= 3; F= 5) AstroPERKKO= 9 (M= 5; F=4) |
Yes | Unpaired two-tailed t test | 0.6264 | N/A | N/A |
| 2F | % Center Tendency (open field test) | Old (18 months) Control vs AstroPERKKO |
Control= 8 (M= 3; F= 5) AstroPERKKO= 9 (M= 5; F=4) |
Yes | Unpaired two-tailed t test | 0.5894 | N/A | N/A |
| 2G | No Of Arm Entries (Y Maze test) | Middle-aged (12 months) Control vs AstroPERKKO |
Control= 17 (M= 7; F= 10) AstroPERKKO= 17 (M= 8; F=9) |
Yes | Unpaired two-tailed t test | 0.9079 | N/A | N/A |
| 2H | % Spontaneous Alternation (Y maze test) | Middle-aged (12 months) Control vs AstroPERKKO |
Control= 17 (M= 7; F= 10) AstroPERKKO= 17 (M= 8; F=9) |
Yes | Unpaired two-tailed t test | 0.0999 | N/A | N/A |
| 2I | No Of Arm Entries (Y Maze test) | Old (18 months) Control vs AstroPERKKO |
Control= 8 (M= 3; F= 5) AstroPERKKO= 9 (M= 5; F=4) |
Yes | Unpaired two-tailed t test | 0.8176 | N/A | N/A |
| 2J | % Spontaneous Alternation (Y maze test) | Old (18 months) Control vs AstroPERKKO |
Control= 8 (M= 3; F= 5) AstroPERKKO= 9 (M= 5; F=4) |
Yes | Unpaired two-tailed t test | 0.2216 | N/A | N/A |
| 3A | Latency To Platform Entry (Morris water maze, Training phase) | Middle-aged (12 months) Control vs AstroPERKKO |
Control= 15 (M= 6; F= 9) AstroPERKKO= 14 (M= 5; F=9) |
N/A | Ordinary two-way ANOVA | 0.3633 | F (1, 167) = 0.8310 | Sidak’s multiple comparisons test |
| 3B | % Time In Quadrants (Morris water maze, Probe Trial) | Middle-aged (12 months) Control vs AstroPERKKO |
Control= 15 (M= 6; F= 9) AstroPERKKO= 14 (M= 5; F=9) |
N/A | Ordinary two-way ANOVA | 0.9943 | F (1,108) = 5.129e-005 | Sidak’s multiple comparisons test |
| 3C | Time Spent In Platform Area (Morris water maze, Probe Trial) | Middle-aged (12 months) Control vs AstroPERKKO |
Control= 15 (M= 6; F= 9) AstroPERKKO= 14 (M= 5; F=9) |
No | Mann-Whitney test | 0.1546 | N/A | N/A |
| 3D | Latency To Platform Entry (Morris water maze, Reversal training phase) | Middle-aged (12 months) Control vs AstroPERKKO |
Control= 15 (M= 6; F= 9) AstroPERKKO= 14 (M= 5; F=9) |
N/A | Ordinary two-way ANOVA | 0.1940 | F (1, 54) = 1.729 | Sidak’s multiple comparisons test |
| 3E | % Time In Quadrants (Morris water maze, Reversal Probe Trial) | Middle-aged (12 months) Control vs AstroPERKKO |
Control= 15 (M= 6; F= 9) AstroPERKKO= 14 (M= 5; F=9) |
N/A | Ordinary two-way ANOVA | 0.9965 | F (1,108) = 1.976e-005 | Sidak’s multiple comparisons test |
| 3F | Time Spent In Platform Area (Morris water maze, Reversal Probe Trial) | Middle-aged (12 months) Control vs AstroPERKKO |
Control= 15 (M= 6; F= 9) AstroPERKKO= 14 (M= 5; F=9) |
No | Mann-Whitney test | 0.1088 | N/A | N/A |
| 4A | Latency To Platform Entry (Morris water maze, Training phase) | Old (18 months) Control vs AstroPERKKO |
Control= 8 (M= 3; F= 5) AstroPERKKO= 9 (M= 5; F=4) |
N/A | Ordinary two-way ANOVA | 0.0685 | F (1,105) = 3.388 | Sidak’s multiple comparisons test |
| 4B | % Time In Quadrants (Morris water maze, Probe Trial) | Old (18 months) Control vs AstroPERKKO |
Control= 8 (M= 3; F= 5) AstroPERKKO= 9 (M= 5; F=4) |
N/A | Ordinary two-way ANOVA | 0.9884 | F (1, 60) = 0.0002127 | Sidak’s multiple comparisons test |
| 4C | Time Spent In Platform Area (Morris water maze, Probe Trial) | Old (18 months) Control vs AstroPERKKO |
Control= 8 (M= 3; F= 5) AstroPERKKO= 9 (M= 5; F=4) |
Yes | Unpaired two-tailed t test | 0.0845 | N/A | N/A |
| 4D | Latency To Platform Entry (Morris water maze, Reversal training phase) | Old (18 months) Control vs AstroPERKKO |
Control= 8 (M= 3; F= 5) AstroPERKKO= 9 (M= 5; F=4) |
N/A | Ordinary two-way ANOVA | 0.7721 | F (1, 30) = 0.08542 | Sidak’s multiple comparisons test |
| 4E | % Time In Quadrants (Morris water maze, Reversal Probe Trial) | Old (18 months) Control vs AstroPERKKO |
Control= 8 (M= 3; F= 5) AstroPERKKO= 9 (M= 5; F=4) |
N/A | Ordinary two-way ANOVA | 0.6896 | F (1, 60) = 0.1610 | Sidak’s multiple comparisons test |
| 4F | Time Spent In Platform Area (Morris water maze, Reversal Probe Trial) | Old (18 months) Control vs AstroPERKKO |
Control= 8 (M= 3; F= 5) AstroPERKKO= 9 (M= 5; F=4) |
Yes | Unpaired two-tailed t test | 0.5011 | N/A | N/A |
| 5A | Neuroscore (24 h post MCAO) | Old (18 months) Control vs AstroPERKKO |
Control= 11 (M= 6; F= 5) AstroPERKKO= 13 (M= 9; F=4) |
Yes | Unpaired two-tailed t test | 0.0007 | N/A | N/A |
| 5B | Total Ambulation (open field test post MCAO) | Old (18 months) Control vs AstroPERKKO |
Control= 10 (M= 5; F= 5) AstroPERKKO= 7 (M= 5; F=2) |
Yes | Unpaired two-tailed t test | 0.0476 | N/A | N/A |
| 5C | Total Rearing (open field test post MCAO) | Old (18 months) Control vs AstroPERKKO |
Control= 10 (M= 5; F= 5) AstroPERKKO= 7 (M= 5; F=2) |
Yes | Unpaired two-tailed t test | 0.0397 | N/A | N/A |
| 5D | % Survival (post MCAO) | Old (18 months) Control vs AstroPERKKO |
N/A | N/A | Log-rank (Mantel-Cox) test | 0.0475 | N/A | N/A |
| 6B | % of CHOP + cells in GFAP + cells Immunofluorescence | Old (18 months) Control vs AstroPERKKO |
Control= 3 AstroPERKKO= 3 |
N/A | Ordinary two-way ANOVA | 0.0033 | F (1, 8) = 17.01 | Tukey’s multiple comparisons test |
| Contralateral hemisphere vs Ipsilateral hemisphere | <0.0001 | F (1, 8) = 61.87 | ||||||
| 6B | % of CHOP positive cells in GFAP- cells (post MCAO) Immunofluorescence | Old (18 months) Control vs AstroPERKKO |
Control= 3 AstroPERKKO= 3 |
N/A | Ordinary two-way ANOVA | 0.7250 | F (1, 8) = 0.1328 | Tukey’s multiple comparisons test |
| Contralateral hemisphere vs Ipsilateral hemisphere | <0.0001 | F (1, 8) = 68.89 | ||||||
| 7A | Astrocytes per field (post MCAO) Immunofluorescence | Old (18 months) Control vs AstroPERKKO |
Control= 3 AstroPERKKO= 3 |
N/A | Ordinary two-way ANOVA | 0.7133 | F (1, 8) = 0.1450 | Tukey’s multiple comparisons test |
| Contralateral hemisphere vs Ipsilateral hemisphere | 0.6106 | F (1, 8) = 0.2807 | ||||||
| 7B | Branching Degree (post MCAO) Immunofluorescence | Old (18 months) Control vs AstroPERKKO |
Control= 3 AstroPERKKO= 3 |
N/A | Ordinary two-way ANOVA | 0.8165 | F (1, 8) = 0.05753 | Tukey’s multiple comparisons test |
| Contralateral hemisphere vs Ipsilateral hemisphere | 0.6920 | F (1, 8) = 0.1688 | ||||||
| 7D | Feret Diameter (post MCAO) Immunofluorescence | Old (18 months) Control vs AstroPERKKO |
Control= 3 AstroPERKKO= 3 |
N/A | Ordinary two-way ANOVA | 0.9889 | F (1, 8) = 0.0002043 | Tukey’s multiple comparisons test |
| Contralateral hemisphere vs Ipsilateral hemisphere | 0.8592 | F (1, 8) = 0.03354 | ||||||
| Supplementary 1A | % Time In Target Quadrant (Probe Trial Morris Water Maze) | Middle-aged (12 months) Control vs Old (18 months) |
Middle aged =29 (Control: M= 6, F= 9; AstroPERKKO: M= 5, F=9) Old =17 (Control: M= 3, F= 5; AstroPERKKO: M= 5; F=4) |
No | Mann-Whitney test | 0.0099 | N/A | N/A |
| Supplementary 1B | Time Spent In Platform Area (Morris water maze, Probe Trial) | Middle-aged (12 months) Control vs Old (18 months |
Middle aged =29 (Control: M= 6, F= 9; AstroPERKKO: M= 5, F=9) Old =17 (Control: M= 3, F= 5; AstroPERKKO: M= 5; F=4) |
Yes | Unpaired two-tailed t test | 0.0065 | N/A | N/A |
| Supplementary 1C | No Of Platform Line Crossing (Morris water maze, Probe Trial) | Middle-aged (12 months) Control vs Old (18 months |
Middle aged =29 (Control: M= 6, F= 9; AstroPERKKO: M= 5, F=9) Old =17 (Control: M= 3, F= 5; AstroPERKKO: M= 5; F=4) |
Yes | Unpaired two-tailed t test | 0.0453 | N/A | N/A |
| Supplementary 1D | % Time In Target Quadrant (Reversal Probe Trial Morris Water Maze) | Middle-aged (12 months) Control vs Old (18 months |
Middle aged =29 (Control: M= 6, F= 9; AstroPERKKO: M= 5, F=9) Old =17 (Control: M= 3, F= 5; AstroPERKKO: M= 5; F=4) |
Yes | Unpaired two-tailed t test | 0.2123 | N/A | N/A |
| Supplementary 1E | Time Spent In Platform Area (Morris water maze, Reversal Probe Trial) | Middle-aged (12 months) Control vs Old (18 months |
Middle aged =29 (Control: M= 6, F= 9; AstroPERKKO: M= 5, F=9) Old =17 (Control: M= 3, F= 5; AstroPERKKO: M= 5; F=4) |
No | Mann-Whitney test | 0.7478 | N/A | N/A |
| Supplementary 1F | No Of Platform Line Crossing (Morris water maze, Reversal Probe Trial) | Middle-aged (12 months) Control vs Old (18 months |
Middle aged =29 (Control: M= 6, F= 9; AstroPERKKO: M= 5, F=9) Old =17 (Control: M= 3, F= 5; AstroPERKKO: M= 5; F=4) |
No | Mann-Whitney test | 0.8508 | N/A | N/A |
Results
Deletion of PERK in astrocytes.
To examine the impact of astrocytic PERK in learning and memory, we generated conditional knockout mice by crossing PERKfl/fl with GFAP-Cre (AstroPERKKO). To validate the astrocyte-selective Cre, we retro-orbitally injected both control (PERKfl/fl) and AstroPERKKO mice with adeno-associated virus (PHP.eB serotype) carrying a flex-switch controlled, Cre-inducible tdTomato (63). As shown in Figures 1A and 1B, Cre expressing mice show widespread tdTomato labeling of astrocytes, as determined by the complex and bushy cell morphology. No tdTomato expression was observed in Cre negative animals. To confirm PERK knockout in astrocytes, we isolated and cultured primary astrocytes from the control and AstroPERKKO mice. As shown in Figure 1C, PERK is markedly reduced in Cre expressing astrocytes. The levels of PERK, phospho-eIF2α (P- eIF2α), the astrocytic glutamate transporter GLAST, and GFAP were immunoblotted from the cortex and hippocampus. As shown in Figure 1D–1G, PERK expression was not reduced in the cortex and hippocampus fraction of AstroPERKKO animals, suggesting PERK expression is low in astrocytes, which is consistent with previously reported single cell sequencing data (64). Furthermore, expression of P-eIF2α also remained unchanged between the genotypes in both brain regions. GFAP and GLAST expression were also similar in both genotypes. Taken together, these data demonstrate that PERK is effectively deleted from GFAP-expressing astrocytes without causing obvious alterations in cellular phenotype.
Figure 1: Deletion of PERK from GFAP expressing astrocytes.
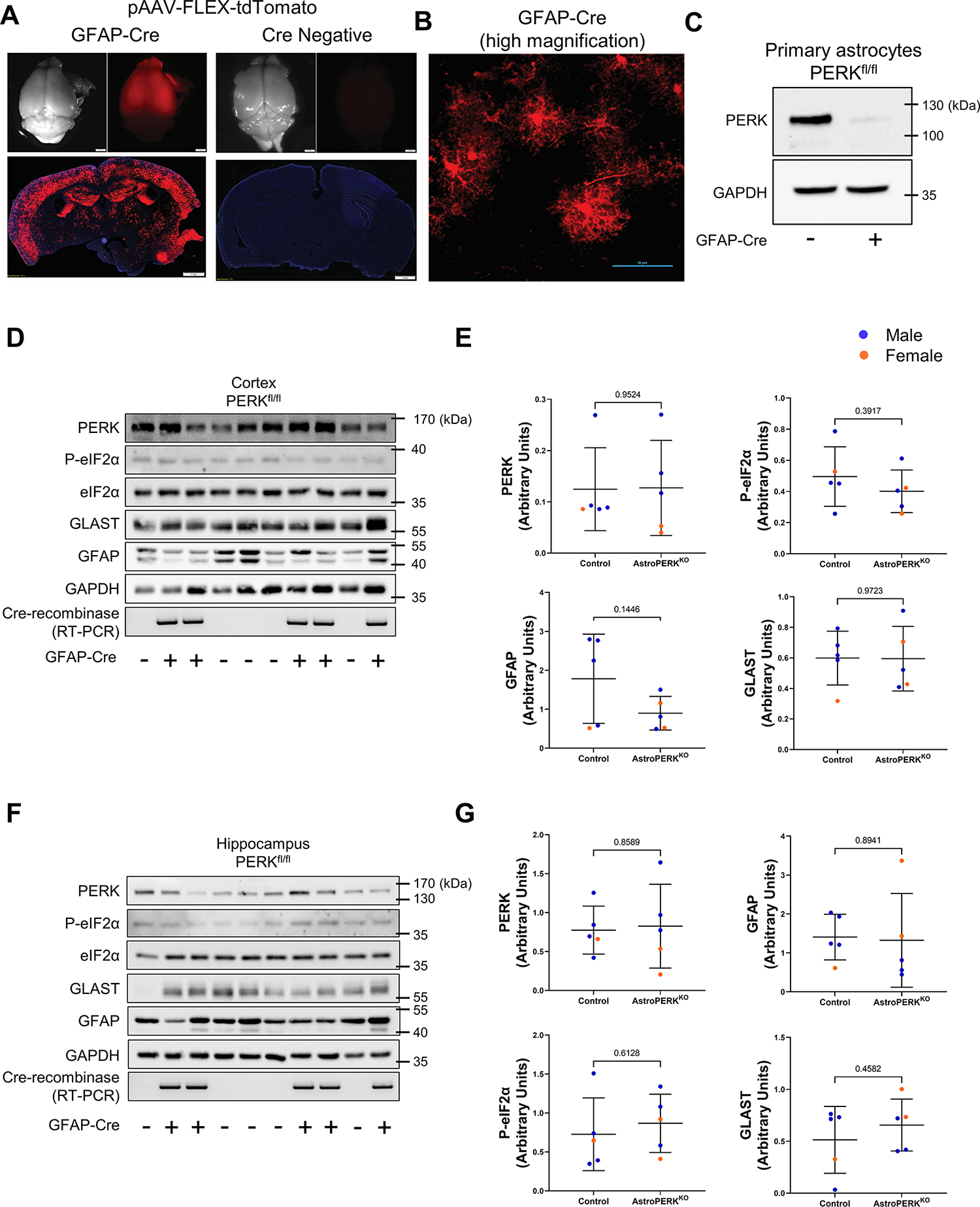
(A) AstroPERKKO (GFAP-Cre, PERKfl/fl) mice (left) and Cre negative PERKfl/fl (right) mice were retro-orbitally injected with 5 × 1011 viral genomes of the Cre-inducible pAAV-FLEX-tdTomato (PHP.eB serotype). Three weeks post injection, tdTomato expression was evaluated as an indicator of Cre activity. Scale bar = 1 mm. (B) Higher magnification showing tdTomato expressing cells display astrocyte morphology. Scale bar = 50 μm. (C) Primary astrocytes were isolated from PERKfl/fl mice without or with GFAP-Cre followed by immunoblotting for PERK and GAPDH. Cortex (D-E) and hippocampus (F-G) were isolated from 15-month-old control (PERKfl/fl) and AstroPERKKO mice. Levels of PERK, P-eIF2α, eIF2α, GLAST, GFAP and GAPDH are measured by immunoblotting and quantified from the cortex (D-E) and hippocampus (F-G). Cre expression is confirmed by RT-PCR. Data are represented as means ± SD. Each point represents an individual animal (blue dot = male, orange dot = female). Numerical p values are shown.
Astrocytic PERK ablation does not affect general locomotion.
To assess the behavioral effects of astrocytic PERK deletion, we first tested general locomotor activity using open field test in age and sex matched middle-aged (12 months; Mean age: 349.9 days) (Figure 2A–C) and old (18 months; Mean age: 527.2 days) (Figure 2D–F) mice (Table 3). We first analyzed the dataset to detect sex specific effects and found none. Therefore, we combined males and females together for further downstream analysis. We found no difference in total ambulation (Figure 2A) and total rearing (Figure 2B) between control and AstroPERKKO mice for middle-aged as well as old (Figure 2D, 2E) mice. Rodents typically show distinct aversions to large, brightly lit, open and unknown environments and typically reside near the periphery of the open field chamber (65). Consistent with this behavior, both control and AstroPERKKO mice mostly occupied the periphery and showed no difference between genotypes in the center activity for middle-aged (Figure 2C) and old (Figure 2F) mice, respectively. Additionally, we did not observe any age specific effect on total ambulation, total rearing and center activity (Table 3) in our animals. Overall, these data indicate that PERK deletion from GFAP+ astrocytes does not affect general locomotor functions.
Figure 2: PERK deletion in astrocytes does not alter general locomotion and short-term working memory.
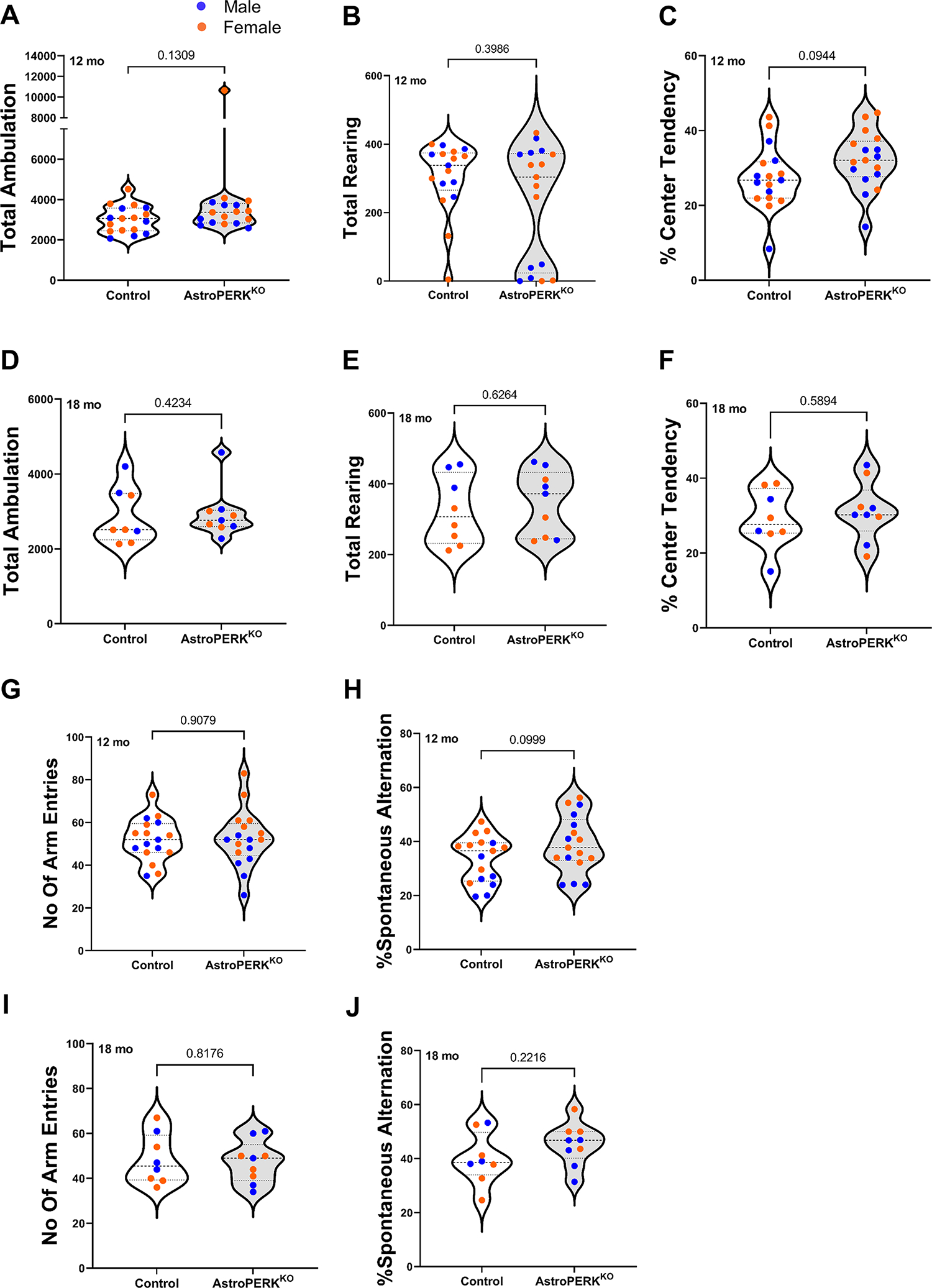
(A-C) Middle-aged (12 months) control, AstroPERKKO (n=17 for each genotype) and (D-E) old (18 months) control (n=8), AstroPERKKO (n=9) mice underwent open field test. Total ambulation, total rearing and percent center activity between two genotypes for middle-aged (A, B, C) and old (D, E, F) mice are plotted respectively. (G-H) Middle-aged and (I-J) old control and AstroPERKKO mice underwent Y-maze spontaneous alternation test. Total number of arm entries and spontaneous alternation rate are plotted for (G-H) middle-aged and (I-J) old mice respectively. Each point represents an individual animal (blue dot = male, orange dot = female). Numerical p values are shown.
Table 3: Open field test.
Middle-aged (12 months) and old (18 months) control and AstroPERKKO mice underwent open field test. Composite data from the tests are depicted in the table. Data are represented as Mean ± SEM. Abbreviations: M= Male, F= Female, AstroPERKKO= Astrocyte specific PERK knockout mice
| Age | 12 months | 18 months | ||||||
|---|---|---|---|---|---|---|---|---|
| Genotype | Control | AstroPERKKO | Control | AstroPERKKO | ||||
| Sex | M | F | M | F | M | F | M | F |
| N | 7 | 10 | 8 | 9 | 3 | 5 | 5 | 4 |
| Total beam break | 3510 ± 229.2 | 3794.8 ± 206.31 | 3718.5 ± 187.94 | 4818.22 ± 718.7 | 4161.33 ± 400.03 | 3173.4 ± 221.29 | 3811 ± 365.62 | 3468.25 ± 87.03 |
| Total ambulation | 2821.43 ± 223.9 | 3168.7 ± 204.32 | 3172.25 ± 171.59 | 4207.67 ± 770.41 | 3392.67 ± 409.09 | 2550.4 ± 209.87 | 3056.8 ± 358.39 | 2785.75 ± 86.64 |
| Total rearing | 330.14 ± 20.23 | 286.7 ± 38.31 | 205 ± 64.27 | 257 ± 48.61 | 430.33 ± 16.98 | 260.8 ± 19.13 | 384 ± 35.5 | 300.75 ± 34.56 |
| % Center activity | 25.99 ± 3.13 | 28.31 ± 2.49 | 28.12 ± 2.29 | 35.64 ± 2.12 | 25.14 ± 4.55 | 31.43 ± 2.63 | 31.59 ± 3.08 | 30.6 ± 3.97 |
Astrocytic PERK deletion does not alter spatial working memory.
Genetic ablation of PERK in neurons impairs short-term spatial working memory (36). We hypothesized that long-term deletion of PERK from astrocytes would diminish support for neurons and impair memory. To test this, we performed Y-maze spontaneous alternation test to examine short-term working memory in control and AstroPERKKO mice. As before, there were no sex specific effects and therefore we combined males and females for subsequent analysis. Our data demonstrated that there were no differences in total arm entries (Figure 2G, 2I) or spontaneous alternation rate (Figure 2H, 2J) between control and AstroPERKKO mice of both age groups. Typically, young mice exhibit an alternation rate of approximately 70% and alternation rates decline progressively as animals age (66). In accordance with previous studies, both middle-aged (12 months) and old (18 months) mice displayed an average spontaneous alternation rate of below 50% suggesting age-related impairment that is not worsened by PERK deletion. However, there was no age specific effect in the Y-maze performance (Table 4). Taken together, these data suggest that astrocytic PERK does not regulate short-term spatial working memory.
Table 4: Y-maze test.
Middle-aged (12 months) and old (18 months) control and AstroPERKKO mice underwent Y-maze spontaneous alternation test. Composite data from the tests are depicted in the table. Data are represented as Mean ± SEM. Abbreviations: M= Male, F= Female, AstroPERKKO= Astrocyte specific PERK knockout mice
| Age | 12 months | 18 months | ||||||
|---|---|---|---|---|---|---|---|---|
| Genotype | Control | AstroPERKKO | Control | AstroPERKKO | ||||
| Sex | M | F | M | F | M | F | M | F |
| N | 7 | 10 | 8 | 9 | 3 | 5 | 5 | 4 |
| Number of arm entries | 50.71 ± 3.12 | 52.7 ± 3.32 | 43.88 ± 3.21 | 59.89 ± 3.67 | 50.67 ± 4.28 | 47.20 ± 3.23 | 48.20 ± 5.02 | 46.25 ± 1.95 |
| % Spontaneous alternation | 27.23 ± 2.57 | 37.93 ± 2.01 | 37.12 ± 4.07 | 40.88 ± 2.79 | 43.47 ± 4.03 | 37.79 ± 4.15 | 41.10 ± 2.67 | 50.48 ± 2.62 |
Astrocytic PERK ablation does not alter long-term memory and cognitive flexibility in middle-aged mice.
PERK inhibition by genetic and pharmacological means improves memory consolidation (37) and hippocampal-dependent learning and memory (67) in rodents. Moreover, PERK inhibition has been reported to improve spatial memory following traumatic brain injury (35) and in an Alzheimer’s disease model (33). Therefore, we examined whether the AstroPERKKO mice have differences in long-term spatial memory at middle-age (12 months). As before, there were no sex specific effects and therefore we combined males and females for subsequent analysis. In the training phase, mice learned the location of the hidden platform, as shown by a decrease in the latency to platform entry (p < 0.0001; ordinary two-way ANOVA). There were no significant differences in the learning ability between control and AstroPERKKO mice (Figure 3A). After the last training session, we conducted probe trial (Figure 3B–C). Both the control and AstroPERKKO mice spent significantly more time in the target quadrant (TQ) (two-way ANOVA, Sidak’s multiple comparison test) that contained the platform during the training phase compared to other quadrants (Figure 3B). There were no significant differences in time spent in target quadrant (Figure 3B) and time spent in the platform area (Figure 3C) between control and AstroPERKKO mice. Both control and AstroPERKKO mice employed a similar strategy to find the platform during the probe trial, which was quantified by the parameter path efficiency to first platform entry (Table 5).
Figure 3: Astrocytic PERK ablation does not alter long-term memory and cognitive flexibility in middle-aged mice.
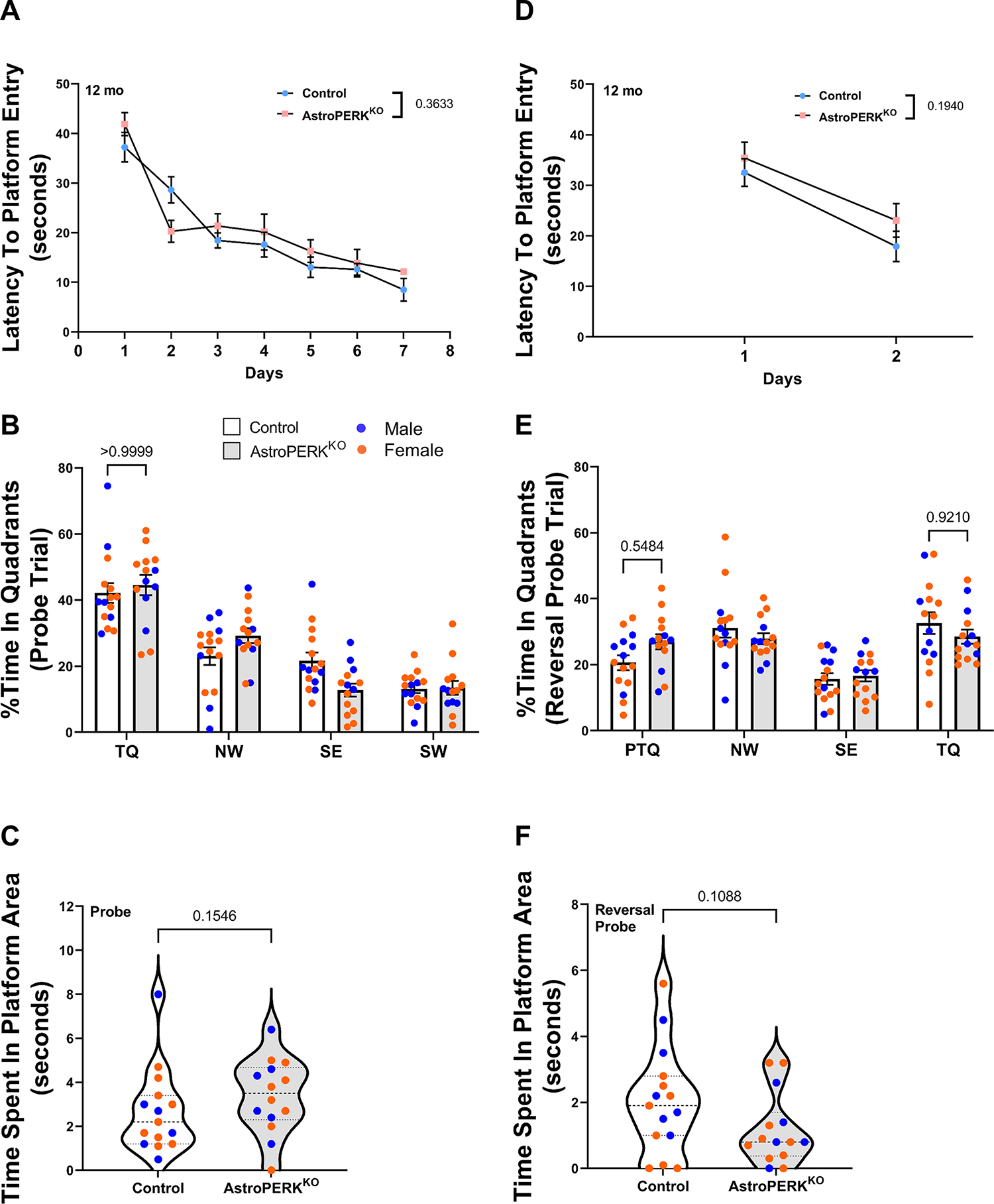
(A-C) Middle-aged (12 months) control and AstroPERKKO (n=17 for each genotype) mice were tested in the Morris water maze. Mice were trained to learn the hidden platform location for seven consecutive days. (A) Average latency (time) to find platform during the training phase is plotted for each genotype. Probe trials were administered right after the last training session. During probe trials, (B) percent time spent in individual quadrants and (C) time spent in platform area are plotted for each genotype. (D-F) The next day, the platform location was changed, and mice were retrained to learn the new platform position for two consecutive days (reversal training phase). (D) Average latency to find the platform during reversal training phase are plotted for each genotype. Reversal probe trials were conducted immediately after the last training session. (E) Percent time spent in individual quadrants and (F) time spent in platform are plotted for each genotype. Each point represents an individual animal (blue dot = male, orange dot = female). Numerical p values are shown. Abbreviations: TQ= target quadrant, PTQ= Target quadrant in previous probe trial, SE= southeast quadrant, NW= northwest quadrant, SW= southwest quadrant).
Table 5: Morris water maze test (Probe trial).
Middle-aged (12 months) and old (18 months) control and AstroPERKKO mice underwent morris water maze test. Composite data from the probe trials are depicted in the table. Data are represented as Mean ± SEM. Abbreviations: M= Male, F= Female, AstroPERKKO= Astrocyte specific PERK knockout mice, TQ= Target quadrant, NE= Northeast quadrant, NW= Northwest quadrant, SE= Southeast quadrant, SW= Southwest quadrant
| Age | 12 months | 18 months | ||||||
|---|---|---|---|---|---|---|---|---|
| Genotype | Control | AstroPERKKO | Control | AstroPERKKO | ||||
| Sex | M | F | M | F | M | F | M | F |
| N | 6 | 9 | 5 | 9 | 3 | 5 | 5 | 4 |
| Total distance travelled (decimeter) | 10.66 ± 0.68 | 11.72 ± 0.34 | 11.19 ± 0.38 | 11.90 ± 0.45 | 11.26 ± 0.73 | 10.99 ± 0.74 | 12.63 ± 0.56 | 11.62 ± 0.95 |
| Latency to first platform entry (seconds) | 14.98 ± 7.98 | 12.50 ± 1.80 | 11.76 ± 1.62 | 10.34 ± 1.45 | 24.67 ± 12.10 | 21.98 ± 5.39 | 8.78 ± 2.94 | 13.6 ± 3.92 |
| Path efficiency to first platform entry | 0.53 ± 0.10 | 0.43 ± 0.06 | 0.41 ± 0.07 | 0.51 ± 0.09 | 0.31 ± 0.11 | 0.28 ± 0.06 | 0.55 ± 0.09 | 0.48 ± 0.11 |
| Number of platform line crossing | 3.67 ± 0.81 | 4.11 ± 0.55 | 5.20 ± 0.59 | 5.11 ± 0.94 | 3 ± 0.94 | 1.8 ± 0.33 | 5 ± 0.8 | 2.75 ± 0.65 |
| Time spent in platform area (seconds) | 2.85 ± 1.00 | 2.56 ± 0.42 | 3.04 ± 0.56 | 3.57 ± 0.59 | 1.77 ± 0.48 | 0.82 ± 0.21 | 2.60 ± 0.48 | 1.38 ± 0.19 |
| Percent time spent in target (NE) quadrant | 45.98 ± 6.21 | 39.76 ± 2.19 | 42.00 ± 2.80 | 45.98 ± 4.24 | 40.78 ± 4.72 | 28.57 ± 2.66 | 37.97 ± 4.02 | 32.71 ± 3.71 |
| Percent time spent in NW quadrant | 22.17 ± 5.51 | 23.63 ± 2.16 | 28.43 ± 4.16 | 29.69 ± 2.40 | 28.50 ± 1.65 | 22.70 ± 2.95 | 26.57 ± 2.66 | 24.63 ± 3.67 |
| Percent time spent in SE quadrant | 21.61 ± 4.34 | 21.74 ± 2.66 | 19.07 ± 2.30 | 9.26 ± 1.79 | 14.83 ± 3.12 | 26.80 ± 1.8 | 18.13 ± 2.28 | 21.58 ± 1.73 |
| Percent time spent in SW quadrant | 10.56 ± 1.70 | 14.83 ± 1.47 | 10.53 ± 0.87 | 15.07 ± 2.90 | 15.94 ± 3.04 | 21.93 ± 0.87 | 17.30 ± 2.54 | 20.96 ± 1.03 |
Genetic ablation of PERK from forebrain neurons leads to impairment of cognitive flexibility (36,44). To examine the effect of astrocytic PERK ablation in cognitive flexibility, we conducted reversal probe trial. During the reversal training phase, control and AstroPERKKO mice learned the new platform location (p<0.0001; ordinary two-way ANOVA) equally well (Figure 3D). In the reversal probe trial, control mice spent significantly more time in new target quadrant than the previous target quadrant (PTQ; quadrant which contained platform during the probe trial) (p = 0.0087, control TQ vs control PTQ, two-way ANOVA with Tukey’s multiple comparison test) (Figure 3E). However, AstroPERKKO mice spent comparable amount of time in both target quadrant and previous target quadrant (p = 0.9998, AstroPERKKO TQ vs AstroPERKKO PTQ, two-way ANOVA with Tukey’s multiple comparison test) (Figure 3E) which indicates that AstroPERKKO mice were less efficient in learning the new platform location compared to control. However, there was no significant difference between AstroPERKKO and control mice in the time spent in the target quadrant or the previous target quadrant (Figure 3E). Additionally, control and AstroPERKKO mice spent comparable amount of time in the platform area (Figure 3F). Overall, our results demonstrate that astrocytic PERK ablation does not alter long-term spatial memory but may suggest a subtle difference in cognitive flexibility in middle-aged mice.
Astrocytic PERK ablation does not alter long-term memory and cognitive flexibility in old mice.
We next examined long-term memory and cognitive flexibility in old (18 months) mice to determine if astrocytic PERK deletion would alter age-related cognitive impairment. As before, there were no sex specific effects and therefore we combined males and females for subsequent analysis. During the training phase, both the control and AstroPERKKO mice learned the platform location. (Figure 4A) (p = 0.0002, ordinary two-way ANOVA). Consistent with the middle-aged mice, there were no significant differences in time spent in target quadrant (Figure 4B) or platform area (Figure 4C) between control and AstroPERKKO mice during the probe trial. Both control and AstroPERKKO mice employed a similar strategy to find the platform during the probe trial, which was quantified by the parameter path efficiency to first platform entry (Table 6). Overall, our data suggest that like the middle-aged cohort, PERK deletion from astrocytes does not affect long-term spatial memory in old mice.
Figure 4: Astrocytic PERK deletion does not alter long-term memory and cognitive flexibility in old mice.
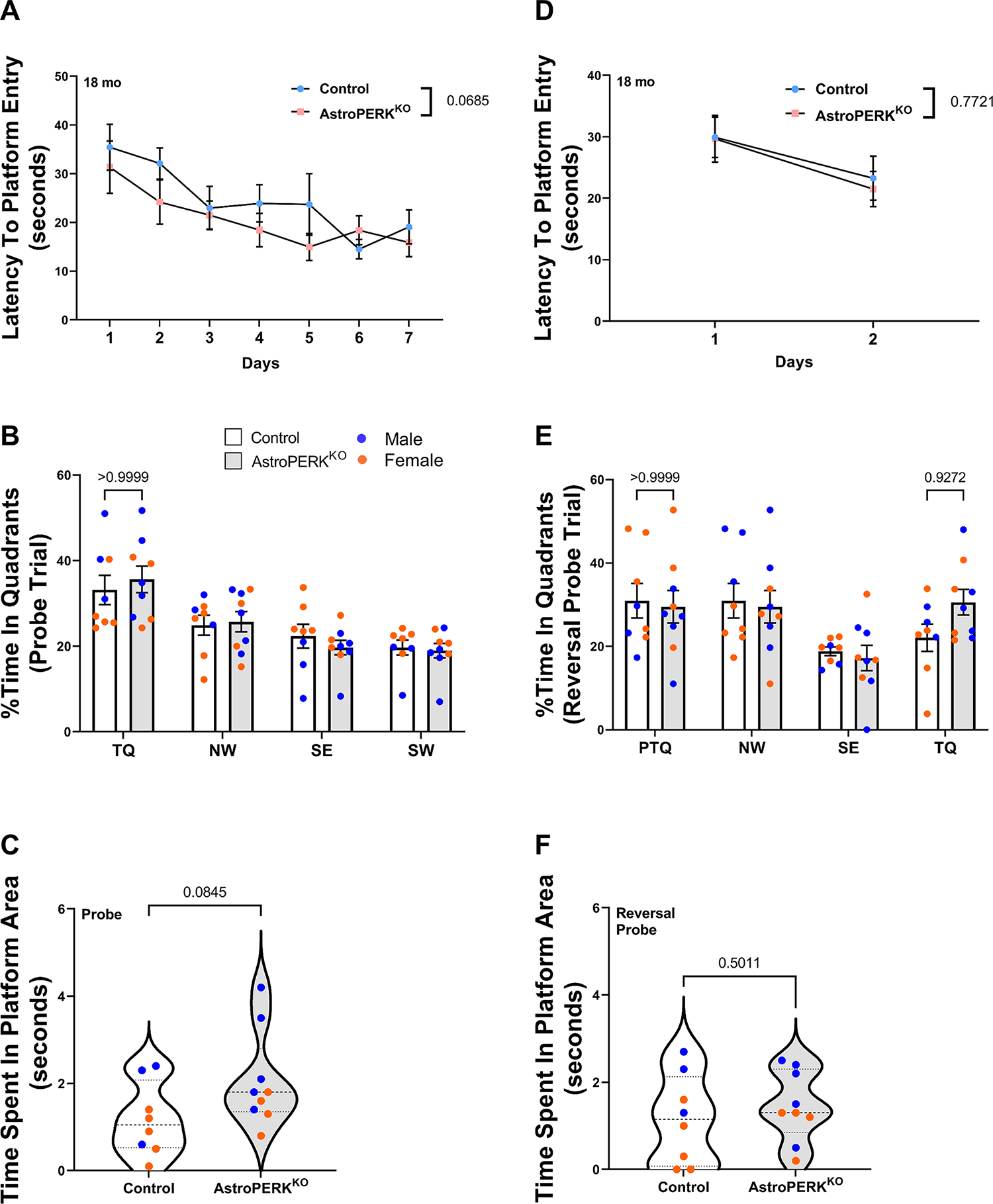
(A-C) Old (18 months) control (n=8), and AstroPERKKO (n=9), mice were tested in the Morris water maze. Mice were trained to learn the hidden platform location for seven consecutive days. (A) Average latency to find platform during training phase is plotted for each genotype. Probe trials were administered right after the last training session. During the probe trials, (B) percent time spent in individual quadrants and (C) time spent in platform are plotted for each genotype. (D-F) The next day, the platform location was changed, and mice were retrained to learn the new platform position for two consecutive days (reversal training phase). (D) Average latency to find the platform during reversal training phase are plotted for each genotype. Reversal probe trials were conducted immediately after the completion of training. (E) Percent time spent in individual quadrants and (F) time spent in the platform area are plotted for each genotype. Each point represents an individual animal (blue dot = male, orange dot = female). Numerical p values are shown. (Abbreviations: TQ= target quadrant, PTQ= Target quadrant in previous probe trial, SE= southeast quadrant, NW= northwest quadrant, SW= southwest quadrant).
Table 6: Morris water maze test (Reversal probe trial).
Middle-aged (12 months) and old (18 months) control and AstroPERKKO mice underwent morris water maze test. Composite data from the reversal probe trials are depicted in the table. Data are represented as Mean ± SEM. Abbreviations: M= Male, F= Female, AstroPERKKO= Astrocyte specific PERK knockout mice, TQ= Target quadrant, PTQ= Previous target quadrant, NE= Northeast quadrant, NW= Northwest quadrant, SE= Southeast quadrant, SW= Southwest quadrant.
| Age | 12 months | 18 months | ||||||
|---|---|---|---|---|---|---|---|---|
| Genotype | Control | AstroPERKKO | Control | AstroPERKKO | ||||
| Sex | M | F | M | F | M | F | M | F |
| N | 6 | 9 | 5 | 9 | 3 | 5 | 5 | 4 |
| Total distance travelled (decimeter) | 12.28 ± 0.71 | 12.26 ± 0.53 | 14.00 ± 1.09 | 11.87 ± 0.50 | 13.23 ± 0.79 | 11.29 ± 0.92 | 12.74 ± 0.89 | 11.03 ± 0.67 |
| Latency to first platform entry (seconds) | 11.55 ± 2.51 | 15.30 ± 2.22 | 8.70 ± 2.80 | 22.26 ± 4.57 | 14.37 ± 1.38 | 28.27 ± 7.76 | 12.90 ± 3.7 | 18.43 ± 3.72 |
| Path efficiency to first platform entry | 0.44 ± 0.11 | 0.33 ± 0.06 | 0.55 ± 0.10 | 0.28 ± 0.06 | 0.33 ± 0.07 | 0.28 ± 0.11 | 0.41 ± 0.09 | 0.31 ± 0.05 |
| Number of platform line crossing | 3.67 ± 0.38 | 3.33 ± 0.99 | 2.40 ± 0.61 | 2.00 ± 0.54 | 4 ± 0.47 | 1.2 ± 0.52 | 3.2 ± 0.66 | 2.25 ± 0.41 |
| Time spent in platform area (seconds) | 2.40 ± 0.50 | 1.79 ± 0.57 | 1.12 ± 0.39 | 1.20 ± 0.38 | 2.10 ± 0.34 | 0.58 ± 0.28 | 1.82 ± 0.33 | 1.00 ± 0.23 |
| Percent time spent in NE (PT) quadrant | 23.44 ± 2.42 | 18.72 ± 3.06 | 22.67 ± 2.97 | 29.33 ± 2.73 | 31.39 ± 1.46 | 26.33 ± 1.38 | 23.20 ± 3.25 | 22.17 ± 4.69 |
| Percent time spent in NW quadrant | 25.36 ± 3.53 | 34.96 ± 3.53 | 24.83 ± 2.14 | 29.56 ± 1.96 | 23.39 ± 2.91 | 35.47 ± 4.93 | 24.93 ± 3.39 | 35.17 ± 6.08 |
| Percent time spent in SE quadrant | 18.39 ± 2.96 | 13.83 ± 1.80 | 21.00 ± 1.68 | 14.20 ± 1.90 | 19.50 ± 1.38 | 18.40 ± 1.3 | 20.70 ± 3.15 | 12.83 ± 4.23 |
| Percent time spent in target (Sw) quadrant | 32.72 ± 4.31 | 32.50 ± 4.50 | 31.47 ± 3.13 | 26.87 ± 2.57 | 25.83 ± 1.73 | 19.83 ± 4.48 | 31.17 ± 4.15 | 29.75 ± 3.92 |
To examine the cognitive flexibility in old mice, reversal probe trial was conducted. During the reversal training phase, both control and AstroPERKKO mice learned the platform location equally well (Figure 4D) (p = 0.0376; ordinary two-way ANOVA). During the reversal probe trial, both control and AstroPERKKO mice spent a comparable amount of time in the target quadrant and the previous target quadrant, indicating they did not effectively extinguish the previously learned location (Figure 4E). Additionally, both genotypes spent a similar amount of time in the platform area (Figure 4F).
We next analyzed the data using age as the only independent variable. Expectedly, middle-aged mice performed significantly better in water maze test compared to old mice (Supplementary Figure 1A–C). During the probe trial, middle-aged mice spent significantly more time in target quadrant and platform area as well as performed more platform line crossings (Supplementary Figure 1A–C) which indicates superior long-term memory. Taken together, our data demonstrate that middle-aged mice have superior long-term memory compared to old mice. However, during the reversal probe trial, both middle-aged and old mice spent comparable amount of time in target quadrant and platform area (Supplementary Figure 1D–F). Additionally, there was no significant difference in the number of platform line crossing between the middle-aged and old mice (Supplementary Figure 1F). Our data are consistent with previous reports that described similar age-related cognitive decline in rodents (68,69). Collectively, these data suggest that PERK deletion from astrocytes does not worsen age-related cognitive impairment.
Deletion of PERK from astrocytes worsens experimental stroke outcome in aged mice.
Our initial hypothesis that long-term PERK deletion from astrocytes would impair cognitive function was not supported by the data. Therefore, we next tested if PERK deletion would affect outcome following an ER stress-inducing injury. For this, we used the experimental ischemic stroke model of middle cerebral artery occlusion (MCAO). This model has been reliably shown to induce ER stress and PERK activation (24,25,27,70–72). Additionally, PERK deletion from neurons resulted in greater functional impairment following MCAO (27). Using the same cohort of mice as in Figure 4 (plus 3 – 4 test-naïve mice per genotype to ensure prior behavioral testing would not affect outcome) these mice underwent 60 min transient MCAO. Mice were assessed at 24 h post-MCAO and impairment scored. The AstroPERKKO mice were significantly more impaired 24 h after MCAO (Figure 5A). The mice were further assessed at 72 h post-MCAO by general locomotor activity. Again, the AstroPERKKO showed more impairment with less activity in the open field and less rearing activity (Figures 5B and 5C). Consistent with worse outcome, AstroPERKKO had significantly higher mortality following MCAO (Figure 5D). To confirm ER stress and PERK activation in astrocytes following MCAO, tissue sections were stained for CHOP and GFAP. As shown in Figure 6A, control animals had robust CHOP staining in the ipsilateral side in both GFAP positive astrocytes and GFAP negative cells. The AstroPERKKO mice also had CHOP expression in the ipsilateral side but was largely restricted to GFAP negative cells. The percentage of CHOP positive astrocytes following MCAO was selectively and significantly reduced in the AstroPERKKO animals (Figure 6B). These data confirm PERK activation in astrocytes following MCAO. The infarct caused by the MCAO was variable and did not correlate with functional outcome (Supplemental Figure 2). The apparent lack of difference in infarction between the genotypes is likely due to significantly more of the AstroPERKKO dying before collection for TTC staining preventing detection of potentially larger infarcts in the knockout animals. Histopathological evaluation from animals that survived to 72 h showed neuronal necrosis, edema, and neuropil vacuolation, as expected. No pathology was noted in the contralateral hemispheres of the control or AstroPERKKO mice, indicating that PERK deletion does not affect normal CNS development or architecture (Supplemental Figure 3). Collectively, these data show that life-long deletion of PERK from astrocytes leads to worse outcome and increased risk of death from experimental stroke.
Figure 5: Astrocytic PERK ablation worsens experimental stroke outcome in old mice.
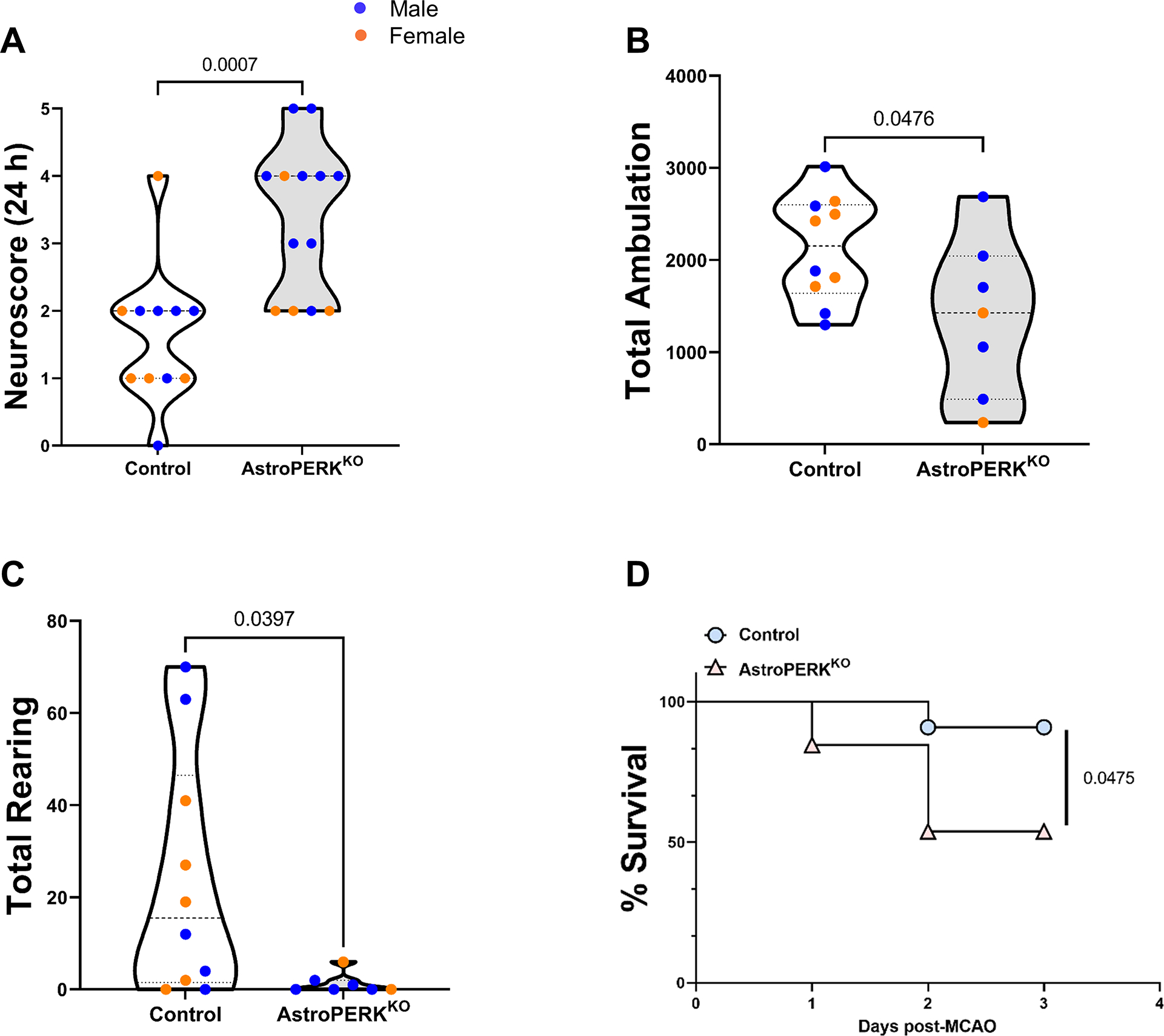
Old (18 months; mean age= 550.1 days) control (n = 11) and AstroPERKKO (n = 13) mice underwent MCAO. (A) Neuroscore was determined 24 hours after the surgery and are plotted for each genotype. (B-C) Seventy-two hours after the surgery, mice underwent open field test. (B) Total ambulation and (C) total rearing activity are plotted for each genotype. (D) Percent survival until 72 hours after the surgery are plotted for each genotype. Each point represents an individual animal (blue dot = male, orange dot = female). Numerical p values are shown.
Figure 6: Cerebral ischemia drives PERK-dependent CHOP expression in astrocytes.
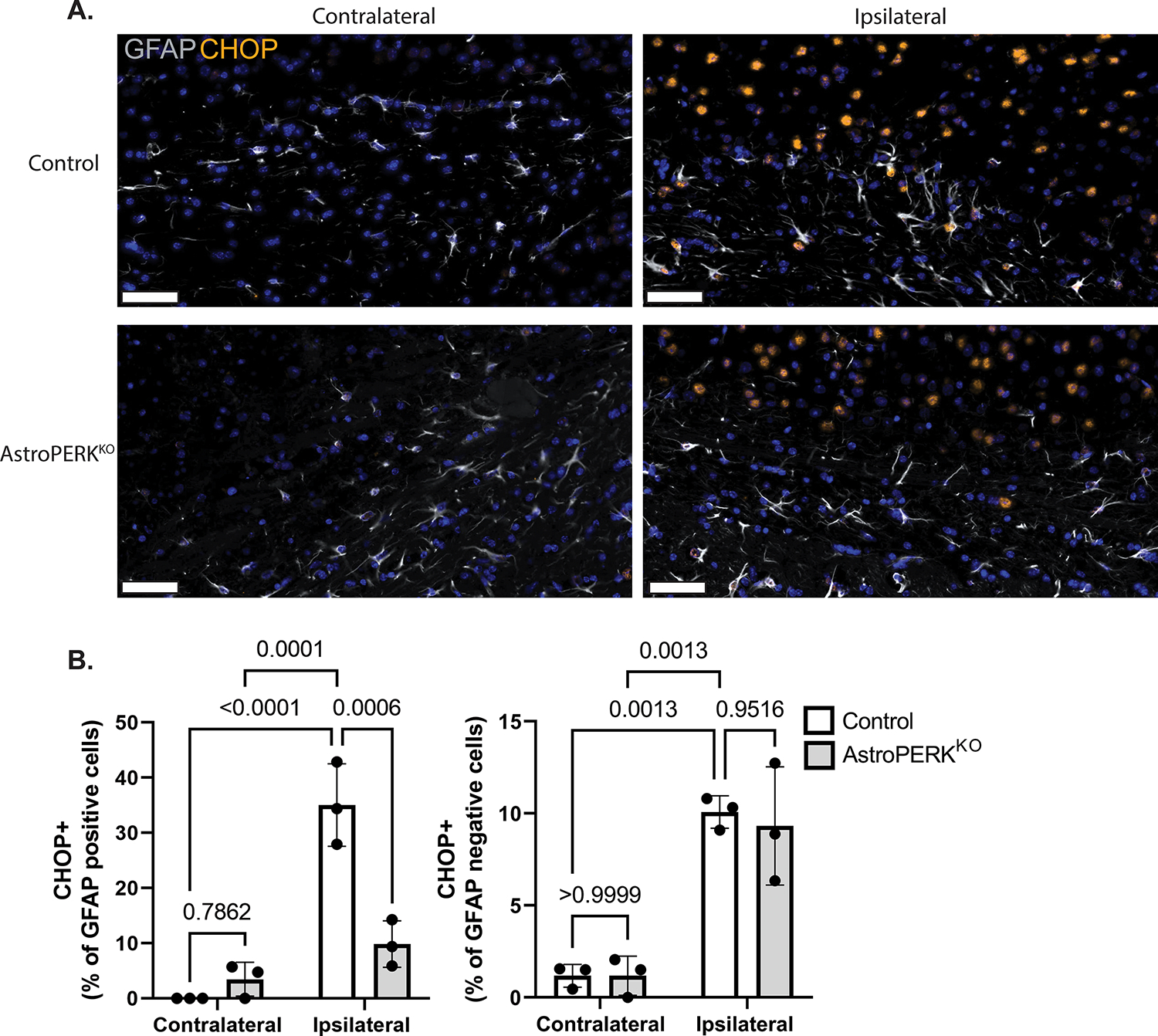
Tissue from a subset of animals shown in figure 5 were used for immunostaining. (A) Representative images from contralateral and ipsilateral hemispheres stained from GFAP (white) and CHOP (orange). Scale bar = 50 μm (B) Quantification of CHOP positive astrocytes (GFAP positive) and GFAP negative cells. Data are represented as means ± SD. Each point represents an individual animal. Numerical p values are shown.
We next examined if PERK deletion impacts astrocyte numbers or morphology in the uninjured or injured tissue. The number of astrocytes was similar between control and AstroPERKKO (Figure 7A). Morphology was examined based on GFAP staining. The branching degree (number of terminal branches / branches originating from the soma) was quantified and showed that PERK deletion did not impact the complexity of the cytoskeleton (Figure 7B & 7C). Additionally, we assessed astrocyte size based on Feret’s diameter of GFAP staining and found that PERK deletion did not significantly impact size (Figure 7D & 7E). These data show that PERK does not regulate astrocyte number or morphology following ischemic injury. The data from the uninjured contralateral tissue also suggests that PERK deletion does not impact astrocyte development nor lead to astrocyte loss or overt morphological defects in the healthy CNS.
Figure 7: PERK does not regulate astrocyte number or morphology.
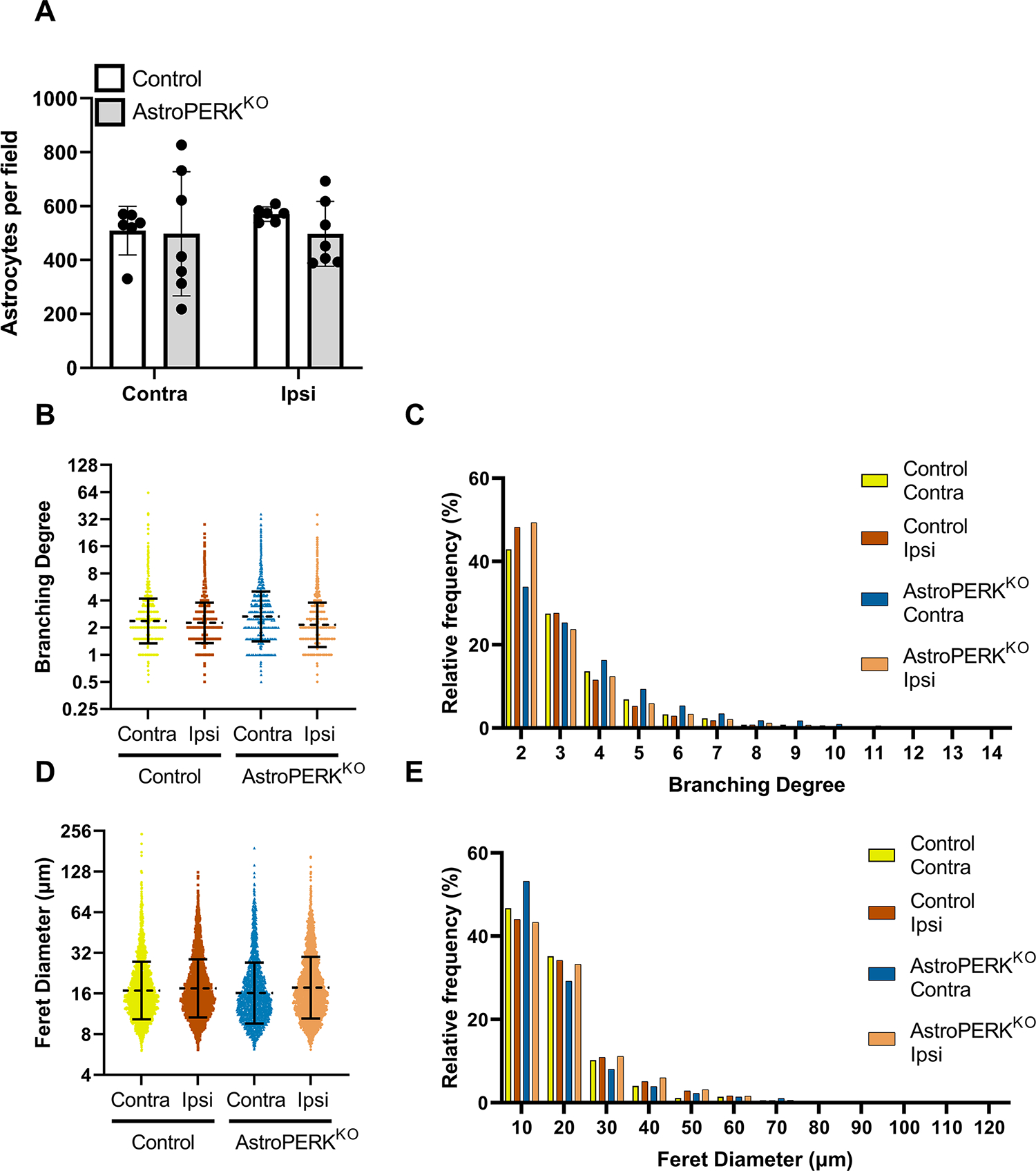
Tissue from a subset of animals shown in figure 5 were used for immunostaining. (A) GFAP+ astrocytes were quantified from 2 – 3 fields per animal from 3 mice of each genotype. Each point represents a microscopy field. (B) Branching degree (number of terminal branches / branches originating from the soma) was quantified and each data point represents an individual astrocyte. Dashed line and error bars represent geometric mean and SD. (C) Relative distribution of cells based on branching degree (graph is truncated when frequency reached < 0.2%). (D) Feret diameter was quantified, and each data point represents an individual astrocyte. Dashed line and error bars represent geometric mean and SD. (E) Relative distribution of cells based on Feret diameter (graph is truncated when frequency reached < 0.2%).
Discussion
The relationship between protein synthesis and long-term memory has been known for more than 50 years as protein synthesis inhibitors were found to impair long-term memory in mammals (73). Phosphorylation of the α subunit of eukaryotic initiation factor 2 (eIF2α) has been described as a focal point for the control of translation initiation (74,75). This phosphorylation occurs at serine 51 and is mediated in a stimulus-dependent fashion by four kinases: PERK, protein kinase R (PKR), general control nonderepressible (GCN) 2, and heme-regulated inhibitor (HRI) (76). Further studies have established a role of eIF2α phosphorylation in regulation of learning and long-term spatial memory in rodents (32,77,78). As a result, eIF2α kinases, including PERK, are central regulators of cognitive function (29). Physiologically, the phosphorylation of eIF2α serves as a rheostat that properly limits memory formation, allowing for behavioral flexibility (45). However, in various neurodegenerative diseases, chronic PERK activation and subsequent eIF2α phosphorylation in CNS is observed (79). As a result, translation of key synaptic proteins is suppressed and may contribute to cognitive decline (42,80–82). These previous studies have clearly defined PERK signaling in control of protein translation as a regulator of cognitive function. However, previous studies have focused on examining the contribution of neuronal PERK signaling in cognitive function. Therefore, an important question remains: does PERK signaling in glia also contribute to learning and memory?
To begin to answer this question, we deleted PERK in astrocytes. From our immunofluorescent and histological analyses, we can infer that PERK deletion from astrocytes did not impact the gross morphology and structure of the CNS or the normal development and survival of astrocytes. We hypothesized that loss of PERK, which is a central regulator of proteostasis, would in the long-term diminish astrocyte support of neurons and manifest as cognitive impairment. Aging is associated with progressive proteostasis impairment in various model organisms as well as in humans (83–85). Therefore, we subjected middle-aged (12 months) and old (18 months) cohorts of control and AstroPERKKO mice to a battery of behavioral tests to assess cognitive function. We found that PERK deletion from astrocytes did not impair general locomotor function, which is consistent with findings from global pharmacological inhibition of PERK (86). Moreover, astrocytic PERK ablation did not affect learning and memory in either age cohort. This may suggest that the effects of PERK signaling are cell-intrinsic to neurons. However, other glial cells in the CNS such as microglia and oligodendrocytes are also known to modulate learning and memory (87,88). Whether PERK signaling in microglia and oligodendrocytes can affect memory function still remains an open question. Consistent with previous reports (89,90), we observed age related decline in long-term memory. However, astrocytic PERK deletion did not exacerbate this cognitive decline.
Protein translation is also impacted by disease and neural injury. It has been known for several decades that transient cerebral ischemia, in both rodents and non-human primates, leads to suppression of protein synthesis in the affected region (20,21). These findings have been consistently reproduced by others that have shown that transient cerebral ischemia increases P-eIF2α in a PERK-dependent fashion leading to suppression of protein synthesis (22–26). Targeting PERK by small molecule inhibition was shown to attenuate MCAO-induced infarct volume (70). In contrast, recent work has shown that deletion of PERK in neurons is detrimental following MCAO (27). Our data demonstrate that deletion of PERK from astrocytes also leads to worse outcome from experimental stroke. Together, these studies show that PERK signaling in both neurons and astrocytes is beneficial following experimental stroke. In this study, MCAO was conducted on mice that were over 18 months old. This is highly relevant as stroke primarily effects elderly individuals (91). Importantly, PERK was deleted for the life of the animal. Considering that, like neurons, astrocytes are long-lived cells (46) it is possible that disruption of PERK signaling sensitizes astrocytes to ischemic damage, but that short-term inhibition or deletion could be protective. Inducible deletion studies to allow temporal control of PERK deletion from astrocytes are underway to test this.
Another potential mechanism by which PERK deletion from astrocytes could be detrimental is through an impaired antioxidant response. The nuclear factor erythroid-2-related factor 2 (Nrf2) is a central regulator of the antioxidant response through driving expression of detoxifying enzymes and by promoting glutathione production (92). Nrf2 is activated following cerebral ischemia and has a protective role (93). Additionally, Nrf2 is activated in a PERK-dependent fashion during ER stress and the Nrf2 pathway has been shown to be protective in astrocytes (94–96). As such, deletion of PERK may impair Nrf2 activation in astrocytes following MCAO. Astrocytes have been shown to protect neurons through a variety of mechanisms including providing antioxidant defense (97). This raises the possibility that PERK deletion may impair the astrocyte-mediated protection of neurons thus contributing to a worse outcome following MCAO.
UPR mediators, particularly PERK, have garnered attention recently as their activation has been reported in various rodent models of neurodegenerative disorders as well as in human patients suffering from these ailments (79). Moreover, studies have also demonstrated that targeting eIF2α phosphorylation can be a potential therapeutic approach in prion disease and frontotemporal dementia (41,42,80,81,98). While there is great potential for modulation of PERK signaling as a therapeutic, there may be differential outcomes depending on the pathological condition. Our study, as well as others, have shown that targeting PERK can have detrimental consequences on CNS injury or disease (27,99–102). However, targeting PERK in memory dysfunction remains promising. CNS activation of the UPR has been reported to significantly impair long-term spatial memory in multiple studies (34,103,104). In line with this, Sidrauski et al. demonstrated that reversing translational inhibition by administration of the small molecule ISRIB (Integrated Stress Response Inhibitor) enhanced long-term spatial memory (38). Additionally, inhibition or knockdown of PERK boosted long-term memory and behavioral plasticity (37,67). Forebrain specific deletion of PERK using Camk2a-cre ameliorates memory decline in a model of Alzheimer’s disease (33). In line with this finding, in vivo administration of soluble PERK inhibitor (GSK2656157) reverses traumatic brain injury (TBI) induced memory decline and anxiety-like behavior (35,86). Post stroke cognitive impairment is common (105) and is potentially another condition that could benefit from pharmacological enhancement of translation initiation.
Genetic or pharmacological manipulation of proteostasis signaling pathways, particularly the unfolded protein response (UPR), can affect learning and memory deficits due to aging and neurological disorders (5,33,35). However, previous studies have focused on translational regulation in neurons and the contributions of these signaling events in non-neuronal cells in the context of cognitive function have not been well studied. Here, we have provided initial evidence that PERK signaling in astrocytes is more likely playing a role in the response to neural injury and has limited effects on cognitive function in normal unstressed condition. This is consistent with previous work showing that astrocytic PERK promotes neuroinflammation and neurodegeneration (106–108). However, much remains unknown. Based on previous single cell sequencing data, PERK is lowly expressed in astrocytes compared to other cells in the CNS (64). Therefore, modulation of other eIF2α kinases in astrocytes may reveal a role in cognition. Similarly, other UPR mediators in astrocytes may have a role in cognition. IRE1α signaling in neurons has been implicated in long-term memory where genetic deletion of IRE1 and/or the downstream effector transcription factor XBP1 resulted in memory impairment (5,109). Additionally, we deleted PERK in GFAP-expressing astrocytes, thus the role of PERK in non-GFAP astrocytes cannot be excluded. Astrocytic PERK is likely to influence cognitive function in the context of neurological diseases through non-cell-autonomous effects on neurons, as previous work has shown that PERK signaling in astrocytes drives neurodegeneration in a mouse model of prion disease (108). If astrocytic PERK has a similar role in other neurological diseases remains to be determined. Future studies will likely show that, under pathological conditions, PERK signal in glial cells is multifaceted and can influence cognitive, inflammatory, and reparative processes.
Supplementary Material
Supplementary Figure 1: Age alters long-term memory. (A-C) During the probe trials, (A) percent time spent in target quadrant, (B) time spent in platform area and (C) number of platform line crossings are plotted for the middle-aged and old mice respectively. (D-F) During the reversal probe trials, (D) percent time in target quadrant, (E) time spent in platform are and (F) number of platform line crossings are plotted for both age groups, respectively. Each point represents an individual animal (blue dot = male, orange dot = female). Numerical p values are shown.
Supplementary Figure 2: MCAO-induced infarction. At 72 h post-MCAO, the surviving animals shown in Figure 5 were euthanized and brains were stained with TTC. Images from each animal are shown. The space between the dashes on the top and right of the images is 1 mm.
Supplementary Figure 3: Histopathology. Representative H&E images from cortex, striatum, and hippocampal CA1 region of control and AstroPERKKO mice 72 h post-MCAO. Contralateral and ipsilateral sections are shown. Inset indicates location of enlarged image.
Significance Statement.
Neuronal PERK signaling is a vital regulator of cognitive function. In this study, we have examined the contribution of astrocytic PERK signaling in learning and memory. Our results indicate that astrocytic PERK does not influence short-term or long-term memory in aged mice. The results support the notion that PERK-dependent regulation of learning and memory is likely cell intrinsic to neurons. Additionally, we have also demonstrated that astrocytic PERK signaling is critical to limiting detrimental outcome and death from experimental stroke.
Acknowledgements
We would like to thank Dr. Elizabeth Chiurazzi and Ms. Terri Poling for helping in choosing appropriate behavioral test and analyses. We also thank Dr. Wen Zheng and Dr. Edwin Wan for assistance with cryosectioning, and Dr. Amanda Ammer for imaging. This work was supported by grants from the NIH (R01NS099304) (Stroke COBRE GM109098) (WVU CTSI U54GM104942). The WVU imaging facility is supported, in part, by grants from the NIH (U54GM104942 and P20GM103434). The WVU experimental stroke and behavioral core facilities are also supported by the NIH (GM109098).
Abbreviations:
- ATF6
Activating transcription factor 6
- ATF4
activating transcription factor 4
- AstroPERKKO
astrocyte specific PERK knockout
- CHOP
C/EBP homologous protein
- CNS
central nervous system
- Cre
cyclization recombinase
- ER
endoplasmic reticulum
- eIF2α
eukaryotic initiation factor 2α
- GCN2
general control nonderepressible 2
- GFAP
glial fibrillary acidic protein
- GLAST
glutamate-aspartate transporter
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HRI
heme-regulated inhibitor
- IRE1
inositol requiring enzyme 1
- ISRIB
integrated Stress Response Inhibitor
- MCAO
middle cerebral artery occlusion
- MWM
morris water maze
- NE
northeast quadrant
- NW
northwest quadrant
- Nrf2
nuclear factor erythroid 2–related factor 2
- P-eIF2α
phospho-eIF2α
- PTQ
previous target quadrant
- PERK
protein kinase R-like ER kinase
- PKR
protein kinase R
- SE
southeast quadrant
- SW
southwest quadrant
- TQ
target quadrant
- UPR
unfolded protein response
- XBP1
X-box binding protein 1
Footnotes
Conflict of interest
The authors declare they have no competing interests.
Data availability statement
Data are available upon request.
Reference
- 1.Deary IJ, Corley J, Gow AJ, Harris SE, Houlihan LM, Marioni RE, Penke L, Rafnsson SB, and Starr JM (2009) Age-associated cognitive decline. British Medical Bulletin 92, 135–152 [DOI] [PubMed] [Google Scholar]
- 2.(2019) Global, regional, and national burden of neurological disorders, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 18, 459–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wyss-Coray T (2016) Ageing, neurodegeneration and brain rejuvenation. Nature 539, 180–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.López-Otín C, Blasco MA, Partridge L, Serrano M, and Kroemer G (2013) The Hallmarks of Aging. Cell 153, 1194–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cabral-Miranda F, Tamburini G, Martinez G, Medinas D, Gerakis Y, Miedema T, Duran-Aniotz C, Ardiles AO, Gonzalez C, Sabusap C, Bermedo-Garcia F, Adamson S, Vitangcol K, Huerta H, Zhang X, Nakamura T, Sardi SP, Lipton SA, Kenedy BK, Cárdenas JC, Palacios AG, Plate L, Henriquez JP, and Hetz C (2020) Control of mammalian brain aging by the unfolded protein response (UPR). bioRxiv, 2020.2004.2013.039172 [Google Scholar]
- 6.Martínez G, Duran-Aniotz C, Cabral-Miranda F, Vivar JP, and Hetz C (2017) Endoplasmic reticulum proteostasis impairment in aging. Aging Cell 16, 615–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Labbadia J, and Morimoto RI (2015) The Biology of Proteostasis in Aging and Disease. Annual Review of Biochemistry 84, 435–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hetz C, and Mollereau B (2014) Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nature Reviews Neuroscience 15, 233–249 [DOI] [PubMed] [Google Scholar]
- 9.Braakman I, and Bulleid NJ (2011) Protein Folding and Modification in the Mammalian Endoplasmic Reticulum. Annual Review of Biochemistry 80, 71–99 [DOI] [PubMed] [Google Scholar]
- 10.Schwarz DS, and Blower MD (2016) The endoplasmic reticulum: structure, function and response to cellular signaling. Cellular and Molecular Life Sciences 73, 79–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sprenkle NT, Sims SG, Sánchez CL, and Meares GP (2017) Endoplasmic reticulum stress and inflammation in the central nervous system. Mol Neurodegener 12, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walter P, and Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 [DOI] [PubMed] [Google Scholar]
- 13.Brewer JW (2014) Regulatory crosstalk within the mammalian unfolded protein response. Cell Mol Life Sci 71, 1067–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ron D, and Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8, 519–529 [DOI] [PubMed] [Google Scholar]
- 15.Zhang K, and Kaufman RJ (2008) From endoplasmic-reticulum stress to the inflammatory response. Nature 454, 455–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harding HP, Zhang Y, and Ron D (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274 [DOI] [PubMed] [Google Scholar]
- 17.Liu CY, Schröder M, and Kaufman RJ (2000) Ligand-independent dimerization activates the stress response kinases IRE1 and PERK in the lumen of the endoplasmic reticulum. J Biol Chem 275, 24881–24885 [DOI] [PubMed] [Google Scholar]
- 18.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, and Ron D (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11, 619–633 [DOI] [PubMed] [Google Scholar]
- 19.Paz Gavilán M, Vela J, Castaño A, Ramos B, del Río JC, Vitorica J, and Ruano D (2006) Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol Aging 27, 973–982 [DOI] [PubMed] [Google Scholar]
- 20.Cooper HK, Zalewska T, Kawakami S, Hossmann KA, and Kleihues P (1977) The effect of ischaemia and recirculation on protein synthesis in the rat brain. J Neurochem 28, 929–934 [DOI] [PubMed] [Google Scholar]
- 21.Kleihues P, Hossmann KA, Pegg AE, Kobayashi K, and Zimmermann V (1975) Resuscitation of the monkey brain after one hour complete ischemia. III. Indications of metabolic recovery. Brain Res 95, 61–73 [DOI] [PubMed] [Google Scholar]
- 22.Mengesdorf T, Proud CG, Mies G, and Paschen W (2002) Mechanisms underlying suppression of protein synthesis induced by transient focal cerebral ischemia in mouse brain. Experimental neurology 177, 538–546 [DOI] [PubMed] [Google Scholar]
- 23.Althausen S, Mengesdorf T, Mies G, Oláh L, Nairn AC, Proud CG, and Paschen W (2001) Changes in the phosphorylation of initiation factor eIF-2alpha, elongation factor eEF-2 and p70 S6 kinase after transient focal cerebral ischaemia in mice. J Neurochem 78, 779–787 [DOI] [PubMed] [Google Scholar]
- 24.Sanderson TH, Deogracias MP, Nangia KK, Wang J, Krause GS, and Kumar R (2010) PKR-like endoplasmic reticulum kinase (PERK) activation following brain ischemia is independent of unfolded nascent proteins. Neuroscience 169, 1307–1314 [DOI] [PubMed] [Google Scholar]
- 25.Owen CR, Kumar R, Zhang P, McGrath BC, Cavener DR, and Krause GS (2005) PERK is responsible for the increased phosphorylation of eIF2alpha and the severe inhibition of protein synthesis after transient global brain ischemia. J Neurochem 94, 1235–1242 [DOI] [PubMed] [Google Scholar]
- 26.Kumar R, Azam S, Sullivan JM, Owen C, Cavener DR, Zhang P, Ron D, Harding HP, Chen JJ, Han A, White BC, Krause GS, and DeGracia DJ (2001) Brain ischemia and reperfusion activates the eukaryotic initiation factor 2alpha kinase, PERK. J Neurochem 77, 1418–1421 [DOI] [PubMed] [Google Scholar]
- 27.Wang YC, Li X, Shen Y, Lyu J, Sheng H, Paschen W, and Yang W (2020) PERK (Protein Kinase RNA-Like ER Kinase) Branch of the Unfolded Protein Response Confers Neuroprotection in Ischemic Stroke by Suppressing Protein Synthesis. Stroke; a journal of cerebral circulation 51, 1570–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Costa-Mattioli M, Sossin WS, Klann E, and Sonenberg N (2009) Translational control of long-lasting synaptic plasticity and memory. Neuron 61, 10–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trinh MA, and Klann E (2013) Translational control by eIF2α kinases in long-lasting synaptic plasticity and long-term memory. Neurobiol Learn Mem 105, 93–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Citri A, and Malenka RC (2008) Synaptic Plasticity: Multiple Forms, Functions, and Mechanisms. Neuropsychopharmacology 33, 18–41 [DOI] [PubMed] [Google Scholar]
- 31.Klann E, and Sweatt JD (2008) Altered protein synthesis is a trigger for long-term memory formation. Neurobiol Learn Mem 89, 247–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Costa-Mattioli M, Gobert D, Stern E, Gamache K, Colina R, Cuello C, Sossin W, Kaufman R, Pelletier J, Rosenblum K, Krnjević K, Lacaille JC, Nader K, and Sonenberg N (2007) eIF2alpha phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell 129, 195–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma T, Trinh MA, Wexler AJ, Bourbon C, Gatti E, Pierre P, Cavener DR, and Klann E (2013) Suppression of eIF2α kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat Neurosci 16, 1299–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin L, Cao J, Yang SS, Fu ZQ, Zeng P, Chu J, Ning LN, Zhang T, Shi Y, Tian Q, Zhou XW, and Wang JZ (2018) Endoplasmic reticulum stress induces spatial memory deficits by activating GSK-3. J Cell Mol Med 22, 3489–3502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sen T, Gupta R, Kaiser H, and Sen N (2017) Activation of PERK Elicits Memory Impairment through Inactivation of CREB and Downregulation of PSD95 After Traumatic Brain Injury. J Neurosci 37, 5900–5911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu S, Henninger K, McGrath BC, and Cavener DR (2016) PERK Regulates Working Memory and Protein Synthesis-Dependent Memory Flexibility. PLoS One 11, e0162766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ounallah-Saad H, Sharma V, Edry E, and Rosenblum K (2014) Genetic or pharmacological reduction of PERK enhances cortical-dependent taste learning. J Neurosci 34, 14624–14632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sidrauski C, Acosta-Alvear D, Khoutorsky A, Vedantham P, Hearn BR, Li H, Gamache K, Gallagher CM, Ang KK, Wilson C, Okreglak V, Ashkenazi A, Hann B, Nader K, Arkin MR, Renslo AR, Sonenberg N, and Walter P (2013) Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife 2, e00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chou A, Krukowski K, Jopson T, Zhu PJ, Costa-Mattioli M, Walter P, and Rosi S (2017) Inhibition of the integrated stress response reverses cognitive deficits after traumatic brain injury. Proc Natl Acad Sci U S A 114, E6420–e6426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Halliday M, Radford H, Sekine Y, Moreno J, Verity N, le Quesne J, Ortori CA, Barrett DA, Fromont C, Fischer PM, Harding HP, Ron D, and Mallucci GR (2015) Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell death & disease 6, e1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Radford H, Moreno JA, Verity N, Halliday M, and Mallucci GR (2015) PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol 130, 633–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, Ortori CA, Willis AE, Fischer PM, Barrett DA, and Mallucci GR (2013) Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med 5, 206ra138. [DOI] [PubMed] [Google Scholar]
- 43.Devi L, and Ohno M (2014) PERK mediates eIF2α phosphorylation responsible for BACE1 elevation, CREB dysfunction and neurodegeneration in a mouse model of Alzheimer’s disease. Neurobiology of aging 35, 2272–2281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trinh MA, Kaphzan H, Wek RC, Pierre P, Cavener DR, and Klann E (2012) Brain-specific disruption of the eIF2α kinase PERK decreases ATF4 expression and impairs behavioral flexibility. Cell Rep 1, 676–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oliveira MM, and Klann E (2022) eIF2-dependent translation initiation: Memory consolidation and disruption in Alzheimer’s disease. Semin Cell Dev Biol 125, 101–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sofroniew MV, and Vinters HV (2010) Astrocytes: biology and pathology. Acta Neuropathol 119, 7–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sofroniew MV (2009) Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci 32, 638–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ota Y, Zanetti AT, and Hallock RM (2013) The role of astrocytes in the regulation of synaptic plasticity and memory formation. Neural Plast 2013, 185463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adamsky A, and Goshen I (2018) Astrocytes in Memory Function: Pioneering Findings and Future Directions. Neuroscience 370, 14–26 [DOI] [PubMed] [Google Scholar]
- 50.Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, and Alberini CM (2011) Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 144, 810–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Han J, Kesner P, Metna-Laurent M, Duan T, Xu L, Georges F, Koehl M, Abrous DN, Mendizabal-Zubiaga J, Grandes P, Liu Q, Bai G, Wang W, Xiong L, Ren W, Marsicano G, and Zhang X (2012) Acute cannabinoids impair working memory through astroglial CB1 receptor modulation of hippocampal LTD. Cell 148, 1039–1050 [DOI] [PubMed] [Google Scholar]
- 52.Stehberg J, Moraga-Amaro R, Salazar C, Becerra A, Echeverría C, Orellana JA, Bultynck G, Ponsaerts R, Leybaert L, Simon F, Sáez JC, and Retamal MA (2012) Release of gliotransmitters through astroglial connexin 43 hemichannels is necessary for fear memory consolidation in the basolateral amygdala. Faseb j 26, 3649–3657 [DOI] [PubMed] [Google Scholar]
- 53.Ben Menachem-Zidon O, Avital A, Ben-Menahem Y, Goshen I, Kreisel T, Shmueli EM, Segal M, Ben Hur T, and Yirmiya R (2011) Astrocytes support hippocampal-dependent memory and long-term potentiation via interleukin-1 signaling. Brain Behav Immun 25, 1008–1016 [DOI] [PubMed] [Google Scholar]
- 54.Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, Gannon M, Ma K, McNaughton K, and Cavener DR (2002) The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol 22, 3864–3874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gregorian C, Nakashima J, Le Belle J, Ohab J, Kim R, Liu A, Smith KB, Groszer M, Garcia AD, Sofroniew MV, Carmichael ST, Kornblum HI, Liu X, and Wu H (2009) Pten deletion in adult neural stem/progenitor cells enhances constitutive neurogenesis. J Neurosci 29, 1874–1886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prieur EAK, and Jadavji NM (2019) Assessing Spatial Working Memory Using the Spontaneous Alternation Y-maze Test in Aged Male Mice. Bio Protoc 9, e3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vorhees CV, and Williams MT (2006) Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc 1, 848–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meares GP, Ma X, Qin H, and Benveniste EN (2012) Regulation of CCL20 expression in astrocytes by IL-6 and IL-17. Glia 60, 771–781 [DOI] [PubMed] [Google Scholar]
- 59.Stirling DR, Swain-Bowden MJ, Lucas AM, Carpenter AE, Cimini BA, and Goodman A (2021) CellProfiler 4: improvements in speed, utility and usability. BMC Bioinformatics 22, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hattori K, Lee H, Hurn PD, Crain BJ, Traystman RJ, and DeVries AC (2000) Cognitive deficits after focal cerebral ischemia in mice. Stroke; a journal of cerebral circulation 31, 1939–1944 [DOI] [PubMed] [Google Scholar]
- 61.Liu Q, Johnson EM, Lam RK, Wang Q, Bo Ye H, Wilson EN, Minhas PS, Liu L, Swarovski MS, Tran S, Wang J, Mehta SS, Yang X, Rabinowitz JD, Yang SS, Shamloo M, Mueller C, James ML, and Andreasson KI (2019) Peripheral TREM1 responses to brain and intestinal immunogens amplify stroke severity. Nat Immunol 20, 1023–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Longa EZ, Weinstein PR, Carlson S, and Cummins R (1989) Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke; a journal of cerebral circulation 20, 84–91 [DOI] [PubMed] [Google Scholar]
- 63.Chan KY, Jang MJ, Yoo BB, Greenbaum A, Ravi N, Wu WL, Sánchez-Guardado L, Lois C, Mazmanian SK, Deverman BE, and Gradinaru V (2017) Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat Neurosci 20, 1172–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, Goeva A, Nemesh J, Kamitaki N, Brumbaugh S, Kulp D, and McCarroll SA (2018) Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 174, 1015–1030.e1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Choleris E, Thomas AW, Kavaliers M, and Prato FS (2001) A detailed ethological analysis of the mouse open field test: effects of diazepam, chlordiazepoxide and an extremely low frequency pulsed magnetic field. Neurosci Biobehav Rev 25, 235–260 [DOI] [PubMed] [Google Scholar]
- 66.Li M, Su S, Cai W, Cao J, Miao X, Zang W, Gao S, Xu Y, Yang J, Tao YX, and Ai Y (2020) Differentially Expressed Genes in the Brain of Aging Mice With Cognitive Alteration and Depression- and Anxiety-Like Behaviors. Front Cell Dev Biol 8, 814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sharma V, Ounallah-Saad H, Chakraborty D, Hleihil M, Sood R, Barrera I, Edry E, Kolatt Chandran S, Ben Tabou de Leon S, Kaphzan H, and Rosenblum K (2018) Local Inhibition of PERK Enhances Memory and Reverses Age-Related Deterioration of Cognitive and Neuronal Properties. J Neurosci 38, 648–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kennard JA, and Woodruff-Pak DS (2011) Age sensitivity of behavioral tests and brain substrates of normal aging in mice. Frontiers in aging neuroscience 3, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.van Praag H, Shubert T, Zhao C, and Gage FH (2005) Exercise enhances learning and hippocampal neurogenesis in aged mice. J Neurosci 25, 8680–8685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li Y, Zhang Y, Fu H, Huang H, Lu Q, Qin H, Wu Y, Huang H, Mao G, Wei Z, and Liao P (2019) Hes1 Knockdown Exacerbates Ischemic Stroke Following tMCAO by Increasing ER Stress-Dependent Apoptosis via the PERK/eIF2α/ATF4/CHOP Signaling Pathway. Neurosci Bull [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Morimoto N, Oida Y, Shimazawa M, Miura M, Kudo T, Imaizumi K, and Hara H (2007) Involvement of endoplasmic reticulum stress after middle cerebral artery occlusion in mice. Neuroscience 147, 957–967 [DOI] [PubMed] [Google Scholar]
- 72.Rissanen A, Sivenius J, and Jolkkonen J (2006) Prolonged bihemispheric alterations in unfolded protein response related gene expression after experimental stroke. Brain Res 1087, 60–66 [DOI] [PubMed] [Google Scholar]
- 73.Glassman E (1969) The biochemistry of learning: an evaluation of the role of RNA and protein. Annu Rev Biochem 38, 605–646 [DOI] [PubMed] [Google Scholar]
- 74.Sonenberg N, Hershey JWB, and Mathews M (2000) Translational control of gene expression. [Google Scholar]
- 75.Krishnamoorthy T, Pavitt GD, Zhang F, Dever TE, and Hinnebusch AG (2001) Tight binding of the phosphorylated alpha subunit of initiation factor 2 (eIF2alpha) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Mol Cell Biol 21, 5018–5030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Costa-Mattioli M, and Walter P (2020) The integrated stress response: From mechanism to disease. Science 368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Costa-Mattioli M, Gobert D, Harding H, Herdy B, Azzi M, Bruno M, Bidinosti M, Ben Mamou C, Marcinkiewicz E, Yoshida M, Imataka H, Cuello AC, Seidah N, Sossin W, Lacaille JC, Ron D, Nader K, and Sonenberg N (2005) Translational control of hippocampal synaptic plasticity and memory by the eIF2alpha kinase GCN2. Nature 436, 1166–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Di Prisco GV, Huang W, Buffington SA, Hsu CC, Bonnen PE, Placzek AN, Sidrauski C, Krnjević K, Kaufman RJ, Walter P, and Costa-Mattioli M (2014) Translational control of mGluR-dependent long-term depression and object-place learning by eIF2α. Nat Neurosci 17, 1073–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Scheper W, and Hoozemans JJ (2015) The unfolded protein response in neurodegenerative diseases: a neuropathological perspective. Acta Neuropathol 130, 315–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, Halliday M, Morgan J, Dinsdale D, Ortori CA, Barrett DA, Tsaytler P, Bertolotti A, Willis AE, Bushell M, and Mallucci GR (2012) Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature 485, 507–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Halliday M, Radford H, Zents KAM, Molloy C, Moreno JA, Verity NC, Smith E, Ortori CA, Barrett DA, Bushell M, and Mallucci GR (2017) Repurposed drugs targeting eIF2α-P-mediated translational repression prevent neurodegeneration in mice. Brain 140, 1768–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Oliveira MM, Lourenco MV, Longo F, Kasica NP, Yang W, Ureta G, Ferreira DDP, Mendonça PHJ, Bernales S, Ma T, De Felice FG, Klann E, and Ferreira ST (2021) Correction of eIF2-dependent defects in brain protein synthesis, synaptic plasticity, and memory in mouse models of Alzheimer’s disease. Science Signaling 14, eabc5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Taylor RC (2016) Aging and the UPR(ER). Brain Res 1648, 588–593 [DOI] [PubMed] [Google Scholar]
- 84.Brehme M, Voisine C, Rolland T, Wachi S, Soper JH, Zhu Y, Orton K, Villella A, Garza D, Vidal M, Ge H, and Morimoto RI (2014) A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep 9, 1135–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hipp MS, Kasturi P, and Hartl FU (2019) The proteostasis network and its decline in ageing. Nature Reviews Molecular Cell Biology 20, 421–435 [DOI] [PubMed] [Google Scholar]
- 86.Sen T, Saha P, Gupta R, Foley LM, Jiang T, Abakumova OS, Hitchens TK, and Sen N (2020) Aberrant ER Stress Induced Neuronal-IFNβ Elicits White Matter Injury Due to Microglial Activation and T-Cell Infiltration after TBI. J Neurosci 40, 424–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Steadman PE, Xia F, Ahmed M, Mocle AJ, Penning ARA, Geraghty AC, Steenland HW, Monje M, Josselyn SA, and Frankland PW (2020) Disruption of Oligodendrogenesis Impairs Memory Consolidation in Adult Mice. Neuron 105, 150–164.e156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cornell J, Salinas S, Huang HY, and Zhou M (2022) Microglia regulation of synaptic plasticity and learning and memory. Neural Regen Res 17, 705–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shoji H, Takao K, Hattori S, and Miyakawa T (2016) Age-related changes in behavior in C57BL/6J mice from young adulthood to middle age. Molecular Brain 9, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shoji H, and Miyakawa T (2019) Age-related behavioral changes from young to old age in male mice of a C57BL/6J strain maintained under a genetic stability program. Neuropsychopharmacol Rep 39, 100–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Das SR, Delling FN, Djousse L, Elkind MSV, Ferguson JF, Fornage M, Jordan LC, Khan SS, Kissela BM, Knutson KL, Kwan TW, Lackland DT, Lewis TT, Lichtman JH, Longenecker CT, Loop MS, Lutsey PL, Martin SS, Matsushita K, Moran AE, Mussolino ME, O’Flaherty M, Pandey A, Perak AM, Rosamond WD, Roth GA, Sampson UKA, Satou GM, Schroeder EB, Shah SH, Spartano NL, Stokes A, Tirschwell DL, Tsao CW, Turakhia MP, VanWagner LB, Wilkins JT, Wong SS, Virani SS, Epidemiology A. H. A. C. o., Committee PS, and Subcommittee SS (2019) Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 139, e56–e528 [DOI] [PubMed] [Google Scholar]
- 92.Ma Q (2013) Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53, 401–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu L, Locascio LM, and Doré S (2019) Critical Role of Nrf2 in Experimental Ischemic Stroke. Frontiers in pharmacology 10, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gan L, Vargas MR, Johnson DA, and Johnson JA (2012) Astrocyte-specific overexpression of Nrf2 delays motor pathology and synuclein aggregation throughout the CNS in the alpha-synuclein mutant (A53T) mouse model. J Neurosci 32, 17775–17787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vargas MR, and Johnson JA (2009) The Nrf2-ARE cytoprotective pathway in astrocytes. Expert reviews in molecular medicine 11, e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, and Diehl JA (2003) Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol 23, 7198–7209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Baxter PS, and Hardingham GE (2016) Adaptive regulation of the brain’s antioxidant defences by neurons and astrocytes. Free Radic Biol Med 100, 147–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Halliday M, Radford H, Sekine Y, Moreno J, Verity N, le Quesne J, Ortori CA, Barrett DA, Fromont C, Fischer PM, Harding HP, Ron D, and Mallucci GR (2015) Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis 6, e1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lin W, Harding HP, Ron D, and Popko B (2005) Endoplasmic reticulum stress modulates the response of myelinating oligodendrocytes to the immune cytokine interferon-gamma. J Cell Biol 169, 603–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stone S, Yue Y, Stanojlovic M, Wu S, Karsenty G, and Lin W (2019) Neuron-specific PERK inactivation exacerbates neurodegeneration during experimental autoimmune encephalomyelitis. JCI insight 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Athanasiou D, Aguila M, Bellingham J, Kanuga N, Adamson P, and Cheetham ME (2017) The role of the ER stress-response protein PERK in rhodopsin retinitis pigmentosa. Human molecular genetics 26, 4896–4905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shacham T, Patel C, and Lederkremer GZ (2021) PERK Pathway and Neurodegenerative Disease: To Inhibit or to Activate? Biomolecules 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sun H, Yang Y, Shao H, Sun W, Gu M, Wang H, Jiang L, Qu L, Sun D, and Gao Y (2017) Sodium Arsenite-Induced Learning and Memory Impairment Is Associated with Endoplasmic Reticulum Stress-Mediated Apoptosis in Rat Hippocampus. Frontiers in Molecular Neuroscience 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chen G, Wei X, Xu X, Yu G, Yong Z, Su R, and Tao L (2021) Methamphetamine Inhibits Long-Term Memory Acquisition and Synaptic Plasticity by Evoking Endoplasmic Reticulum Stress. Frontiers in Neuroscience 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sun JH, Tan L, and Yu JT (2014) Post-stroke cognitive impairment: epidemiology, mechanisms and management. Annals of translational medicine 2, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Guthrie LN, Abiraman K, Plyler ES, Sprenkle NT, Gibson SA, McFarland BC, Rajbhandari R, Rowse AL, Benveniste EN, and Meares GP (2016) Attenuation of PKR-like ER Kinase (PERK) Signaling Selectively Controls Endoplasmic Reticulum Stress-induced Inflammation Without Compromising Immunological Responses. J Biol Chem 291, 15830–15840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Meares GP, Liu Y, Rajbhandari R, Qin H, Nozell SE, Mobley JA, Corbett JA, and Benveniste EN (2014) PERK-Dependent Activation of JAK1 and STAT3 Contributes to Endoplasmic Reticulum Stress-Induced Inflammation. Mol Cell Biol 34, 3911–3925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Smith HL, Freeman OJ, Butcher AJ, Holmqvist S, Humoud I, Schätzl T, Hughes DT, Verity NC, Swinden DP, Hayes J, de Weerd L, Rowitch DH, Franklin RJM, and Mallucci GR (2020) Astrocyte Unfolded Protein Response Induces a Specific Reactivity State that Causes Non-Cell-Autonomous Neuronal Degeneration. Neuron 105, 855–866.e855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Martínez G, Vidal René L., Mardones P, Serrano Felipe G., Ardiles Alvaro O., Wirth C, Valdés P, Thielen P, Schneider Bernard L., Kerr B, Valdés Jose L., Palacios Adrian G., Inestrosa Nibaldo C., Glimcher Laurie H., and Hetz C (2016) Regulation of Memory Formation by the Transcription Factor XBP1. Cell Reports 14, 1382–1394 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Age alters long-term memory. (A-C) During the probe trials, (A) percent time spent in target quadrant, (B) time spent in platform area and (C) number of platform line crossings are plotted for the middle-aged and old mice respectively. (D-F) During the reversal probe trials, (D) percent time in target quadrant, (E) time spent in platform are and (F) number of platform line crossings are plotted for both age groups, respectively. Each point represents an individual animal (blue dot = male, orange dot = female). Numerical p values are shown.
Supplementary Figure 2: MCAO-induced infarction. At 72 h post-MCAO, the surviving animals shown in Figure 5 were euthanized and brains were stained with TTC. Images from each animal are shown. The space between the dashes on the top and right of the images is 1 mm.
Supplementary Figure 3: Histopathology. Representative H&E images from cortex, striatum, and hippocampal CA1 region of control and AstroPERKKO mice 72 h post-MCAO. Contralateral and ipsilateral sections are shown. Inset indicates location of enlarged image.
Data Availability Statement
Data are available upon request.