

Abstract
Purpose
A subset of common variable immunodeficiency (CVID) patients either presents with or develops autoimmune and lymphoproliferative complications, such as granulomatous lymphocytic interstitial lung disease (GLILD), a major cause of morbidity and mortality in CVID. While a myriad of phenotypic lymphocyte derangements has been associated with and described in GLILD, defects in T and B cell antigen receptor (TCR/BCR) signaling in CVID and CVID with GLILD (CVID/GLILD) remain undefined, hindering discovery of biomarkers for disease monitoring, prognostic prediction, and personalized medicine approaches.
Methods
To identify perturbations of immune cell subsets and TCR/BCR signal transduction, we applied mass cytometry analysis to peripheral blood mononuclear cells (PBMCs) from healthy control participants (HC), CVID, and CVID/GLILD patients.
Results
Patients with CVID, regardless of GLILD status, had increased frequency of HLADR+CD4+ T cells, CD57+CD8+ T cells, and CD21lo B cells when compared to healthy controls. Within these cellular populations in CVID/GLILD patients only, engagement of T or B cell antigen receptors resulted in discordant downstream signaling responses compared to CVID. In CVID/GLILD patients, CD21lo B cells showed perturbed BCR-mediated phospholipase C gamma and extracellular signal-regulated kinase activation, while HLADR+CD4+ T cells and CD57+CD8+ T cells displayed disrupted TCR-mediated activation of kinases most proximal to the receptor.
Conclusion
Both CVID and CVID/GLILD patients demonstrate an activated T and B cell phenotype compared to HC. However, only CVID/GLILD patients exhibit altered TCR/BCR signaling in the activated lymphocyte subsets. These findings contribute to our understanding of the mechanisms of immune dysregulation in CVID with GLILD.
Keywords: Common variable immunodeficiency, Granulomatous lymphocytic interstitial lung disease, Inborn errors of immunity, Mass cytometry, T and B cell antigen receptor-mediated signaling
Introduction
Common variable immunodeficiency (CVID) comprises a clinically and immunologically heterogenous group of inborn errors of immunity (IEI) characterized by hypogammaglobulinemia, failure to mount specific antibody responses to vaccination, and susceptibility to recurrent infections [1]. Patients have a profound reduction in serum IgG, with decreased serum IgM and/or IgA, that is at least two standard deviations below the age-corrected mean values [2]. Estimated to affect 1:25,000 to 1:100,000 individuals of European descent, CVID is the most prevalent group of symptomatic IEI in older children and adults [3–5]. Remarkably, up to 60–70% of CVID patients display one or more non-infectious complications (simultaneously or sequentially over their disease course) that significantly increase their morbidity and mortality. These complications include (i) autoimmunity (cytopenias, thyroiditis, rheumatoid arthritis, etc.); (ii) lymphoma (majority non-Hodgkin’s) and other malignancy; (iii) enteropathy; (iv) chronic liver disease due to granulomas, nodular hyperplasia, primary sclerosing/biliary cholangitis; (v) chronic pulmonary disease such as lymphoid interstitial pneumonia (LIP), granulomatous and lymphocytic interstitial lung disease (GLILD); and (vi) other nonmalignant lymphoid hyperplasia [6–11].
The risk of death is estimated at 11 times higher in CVID patients with noninfectious complications compared to those without [12, 13]. Increased mortality risk is closely associated with several organ-specific pathologies, such as lung impairment (hazard ratio, HR = 2.06), liver disease (HR = 2.48), gastrointestinal disease (HR = 2.78), and lymphoma (HR = 2.44) [12]. Importantly, while the use of immunoglobulin replacement and prophylactic antimicrobials has greatly reduced infectious complications, it has not prevented or ameliorated the noninfectious complications. These noninfectious complications are thought to derive from dysregulated innate and adaptive cellular activation responses due to underlying defects that affect maintenance of tolerance [14, 15], which are not addressed by normalization of immunoglobulin levels.
Less than 30% of CVID patients have an identifiable underlying monogenic defect, with each of the monogenic subtypes being very rare [16–20]. Monogenic defects in CVID fall broadly into two categories—those that affect almost exclusively B cell development, differentiation, activation, and/or survival, such as cluster of differentiation 19, 20, 21, or 27, and those that control/affect crosstalk between B and T cells, affecting immune tolerance and antimicrobial defense, hence resulting in immune dysregulation, such as nuclear factor kappa-B, subunit 1 (NFKB1) [21] or phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit delta (PIK3CD). In the latter, autoimmune/inflammatory/neoplastic complications commonly co-exist (or dominate) with infectious susceptibility [10].
Certain immune phenotypic abnormalities have been associated with specific autoimmune/lymphoproliferative complications in CVID patients, such as the expansion of CD21lo B cells (> 10%) with splenomegaly [8] and autoimmune cytopenias [22], expansion of transitional B cells (> 9%) with lymphadenopathy [16], significant decrease of isotype switched B cells as an independent risk factor for granulomas, autoimmunity, and splenomegaly [23], and decreased interferon (IFN)γ mRNA expression with intestinal disease [24]. These associations, however, are not absolute, cannot be generalized, and have not been shown to predict either the development of specific noninfectious complications over the disease course, or treatment response to pathway-specific immunomodulation.
While a myriad of immunological derangements has been associated with and described in GLILD, the immunopathogenesis of GLILD in CVID patients remains elusive. GLILD occurs in approximately 25% of CVID patients [25–27]. Untreated, GLILD leads to progressive pulmonary fibrosis and is associated with increased mortality [26, 28]. Small case studies of GLILD involving immunohistochemical analysis performed on lung biopsies have shown lymphocytic infiltrates with the presence of T cells and variable findings of B cell follicles within the infiltrates [26, 29, 30]. Maglione et al. found increased serum levels of B cell activating factor (BAFF) in CVID patients with a progressive course of ILD compared to stable GLILD, suggesting that a BAFF-induced resistance to apoptosis drives pulmonary B cell hyperplasia [30, 31]. In a study by Fraz et al., GLILD patients’ sera, compared to CVID patients’ with other noninfectious complications, were found to have higher levels of soluble CD25 and T cell immunoglobulin and mucin domain-3 (TIM-3). Combined with the Th1 response signature cytokines TNF and IFN-γ, these findings suggest that activated T cells play an important role in GLILD pathogenesis [32]. A combination of rituximab and azathioprine improved radiographic abnormalities and pulmonary functional tests (PFTs) in patients with CVID and GLILD [28, 33]. Additionally, induction therapy with high-dose corticosteroids has also been shown to improve high resolution CT scans and PFT with long-term remission in 42% of patients [34].
Numerous antigen receptor signaling defects have been identified in CVID patients, including defective zeta-chain-associated protein kinase 70 (ZAP70) recruitment [35], impaired extracellular signal-regulated kinase (ERK) signaling in CD21lo B cells [36], reduced calcium mobilization [37–40], lower phosphorylation of protein kinase B (AKT) and ribosomal protein S6 (S6) [41], and reduced canonical NFκB activation [21]. The consequences of these signaling defects may include the disruption of central and peripheral tolerance, potentially culminating in autoimmunity. To elucidate disease-associated defects in CVID and CVID with GLILD specifically, we applied mass cytometry analysis to peripheral blood mononuclear cells (PBMCs) from CVID patients with (CVID/GLILD) and without GLILD (CVID) to identify perturbations of immune cell subsets and T and B cell antigen receptor (TCR/BCR) signaling.
Methods
Study Approval
All human donors were consented under human subjects’ research protocol, approved by the Institutional Review Board of the Research Compliance Office at Medical College of Wisconsin. Under these practices, the study was in line with the ethical principles outlined in the Declaration of Helsinki.
Blood Processing and Stimulation
Whole blood was collected in heparinized vacutainers for isolation of PBMCs using Ficoll-Paque Plus™. PBMCs were stimulated via the addition of biotinylated anti-CD3 (BioLegend 300,404, clone UCHT1, final [10 μg/mL]), biotinylated anti-CD28 (BioLegend 302,904, clone CD29.2, final [10 μg/mL]), F(ab’)2 anti-IgM (Southern Biotech 2022–01, final [20 μg/mL]), F(ab’)2 anti-IgG (Southern Biotech 6000–01, final [20 μg/mL]), and streptavidin (Invitrogen 21,125, final [35 ng/mL]). Following 2- and 20-min stimulation, cells were fixed with paraformaldehyde and stored at −80C for mass cytometry analyses.
Mass-Tag Barcoding and Antibody Staining
Mass-tag cell barcoding of stimulated and fixed samples, followed by antibody staining and permeabilization was performed as previously described [42]. Antibody panels can be found in Sup Table 1.
PhenoGraph Analyses
R studio version 1.1.383 was downloaded from R website (http://www.r-project.org/). Cytofkit version 3.4 was downloaded from Bioconductor. Manually gated single cells (191Ir+ 193Ir+), CD19+, CD4+, or CD8+ events were processed using PhenoGraph. Clusters were based on expression of the 26 surface markers in the antibody panel (Sup Table 1). Additional settings include (1) merge method: ceil (5000 max events), (2) transformation: cytofAsinh, (3) cluster method: RphenoGraph, (4) visualization method: tSNE, and (5) cellular progression: NULL. Cellular cluster maps were displayed within the “Shiny” feature of the R package. Output.csv files, such as “cluster median data” and “cluster cell percentage”, produced by the PhenoGraph analysis were used to determine cluster phenotype, relative abundance, and statistical significance between groups.
Statistical Analyses of Cellular Phenotyping
We compared cellular frequencies and median protein expression of manually gated cell populations and PhenoGraph output.csv files to determine significant difference between the 3 patient groups. We used the nonparametric Kruskal–Wallis followed by post hoc pair-wise comparisons using Dunn’s multiple comparison test on GraphPad Prism (version 8). A p < 0.05 was regarded as significant.
Results
We applied mass cytometry analysis to PBMCs from age-matched healthy individuals (HC, n = 24), patients with CVID (n = 12), and patients with both CVID and GLILD (n = 23) (Table 1). GLILD was diagnosed based on lung biopsy. Both CVID and CVID/GLILD had significantly lower IgG levels compared to HC, but the mean IgG levels were not significantly different between CVID and CVID/GLILD. Neither patient population had lymphopenia nor decreased CD4+, CD8+, and CD56+ cells compared to HC at the time of study. Compared to CVID patients, CVID/GLILD patients demonstrated decreased CD19+ B cells but similar CD4+ and CD8+ T cell counts (Table 1). None of the patients were being treated with systemic corticosteroids, azathioprine, or rituximab at the time they were studied. Whole exome sequencing of CVID/GLILD patients identified previously published pathogenic variants in XIAP, NFKB2, KMT2D, and IKZF1 (Sup Table 2). To determine immune cell subset and TCR/BCR signaling differences, we designed a mass cytometry antibody panel for evaluation of 26 phenotypic cell surface and 13 functional phospho-epitope markers that evaluate proximal (close to cellular membrane, TCR/BCR-specific) and distal (pathways common to TCR/BCR and signaling by other receptors) signaling events (Sup Fig. 1, Sup Table 1). PBMCs were stimulated and data were analyzed using both supervised manual gating (Sup Fig. 2) and PhenoGraph, an unsupervised clustering algorithm [43, 44]. Compared to CVID and HC, CVID/GLILD patients’ CD19+ B cells, isotype switched, and mature naïve B cells were significantly decreased while CD4+ and CD8+ T cells were not (absolute counts Sup Fig. 3).
Table 1.
Patient cohort characteristics
| Characteristics | Patients with CVID | Patients with GLILD | p value |
|---|---|---|---|
|
| |||
| No. of patients | 12 | 23 | |
| Sex (Female %) | 58% | 75% | |
| Age (range) | 45 (17–63) | 41 (22–67) | NS |
| Plasma IgM, mg/dL (range) | 19 (5–87) | 17 (4–66) | NS |
| Plasma IgA, mg/dL (range) | 25 (0–177) | 15 (0–5) | |
| Plasma IgG, mg/dL (range) | 203 (33–477) | 217 (7–832) | NS |
| Absolute lymphocyte count (cells/mm3) | 1109 (368–1569) | 1288 (330–6909) | NS |
| CD4+ T cells (Abs) | 512 (110–1101) | 545 (1203–1324) | NS |
| CD8+ T cells (Abs) | 397 (59–1217) | 537 (64–4836) | NS |
| CD19+ B cells (Abs) | 98 (0–246) | 65 (0–553) | 0.03 |
| CD56+ NK cells (Abs) | 85 (31–221) | 113 (3–384) | NS |
| 1/12 (8%) | 13/23 (61%) | ||
| Lymphadenopathy | |||
| Splenomegaly | 6/12 (50%) | 20/23 (87%) | |
| Enteropathy | 0/12 (0%) | 1/23 (4%) | |
| Autoimmune cymiddleenia | 1/12 (8%) | 6/23 (26%) | |
CVID and CVID/GLILD Patients Demonstrate Increased Frequency of CD21lo B Cells
We used the PhenoGraph algorithm [43, 44] to visualize cellular populations from each individual in our study. PhenoGraph relies on networks of recorded events and the connections between them to cluster events into phenotypic categories. We applied PhenoGraph to the unstimulated patient samples to generate a cellular cluster map of baseline major leukocyte populations (Fig. 1a, b). Cellular populations derived from traditional hierarchical gating strategies rely on priori knowledge of surface markers to define specific lymphocyte populations. Unsupervised clustering via PhenoGraph is achieved through the simultaneous evaluation of surface marker expression (Sup Table 1). As a result, we do not expect these methodologies to yield identical numbers of subpopulations [43, 45–47]. We assessed differential expression of surface markers on PhenoGraph-derived subpopulations and defined populations accordingly. CVID/GLILD patients demonstrated an overall reduced frequency and absolute counts of CD19+ B lymphocytes compared to HC and CVID, while no differences were seen in CD4+ T cell and CD8+ T cell frequencies and absolute counts among PBMCs (Fig. 1c, Sup Fig. 3).
Fig. 1.
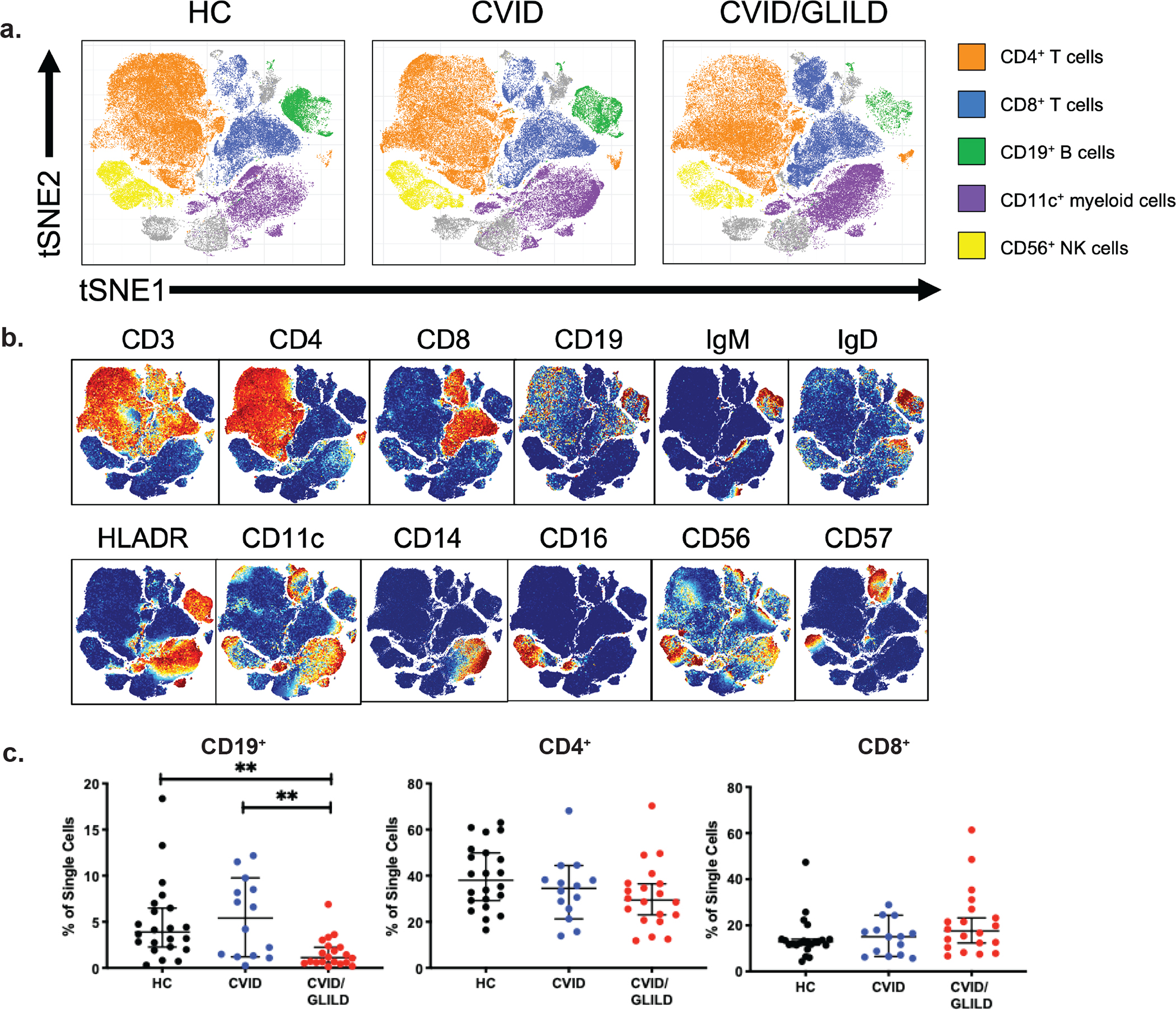
Major leukocyte populations revealed through PhenoGraph. Unsupervised PhenoGraph cluster map of single cells from HC, CVID, and CVID/GLILD groups (a). Stratification of single cell events based on surface marker expression (b). Percentages of lymphocyte populations identified through PhenoGraph analysis (c). Error bars represent median and 95% CI. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Not significant unless stated by asterisk in figure. Kruskal–Wallis accounting for multiple comparisons was used for statistical significance
Memory B cells can be identified based on co-expression of CD19 and CD27, a transmembrane protein expressed by both peripheral T cells [48] and memory B cells [49]. As previously reported [50], no difference was observed in the median frequency of CD27+CD19+ B cells across the three groups (HC vs. CVID vs. CVID/GLILD) (Fig. 2a). CVID/GLILD patients showed a significant increase in CD21lo B cell frequency (CD21loIgM+IgD+) (Fig. 2b) along with a decrease in mature naïve B cell (CD27−IgM+IgD+) percentages relative to CVID and HC (Fig. 2c). The percentages of immature naïve B cells (CD27−IgM+IgD−), IgM memory B cells ( CD27+IgM+IgD−), and pre-isotype switched B cells ( CD27+IgM+IgD+) of CVID/GLILD patients, were also decreased relative to HC, while atypical memory B cells ( CD27−IgM−IgD−) were elevated in CVID/GLILD patients compared to both CVID patients and HC (Fig. 2d–g). No frequency differences were observed in anergic B cells ( CD27−IgM−IgD+) (Fig. 2h). C-delta class switched B cells have undergone Cμ-to-Cδ class switch recombination at the genetic level and may have increased autoreactivity [51, 52]. We found no difference in frequency of these cells in our cohort ( CD27+IgM−IgD+) (Fig. 2i). We observed a reduction in the percentage of CXCR5+ B cells in both CVID and CVID/GLILD patients relative to HC, suggesting defective B cell trafficking to peripheral lymphoid organs (Fig. 2j). Lower CXCR5 expression on CD21lo B cells from CVID and CVID/GLILD was also observed (Fig. 2k).
Fig. 2.
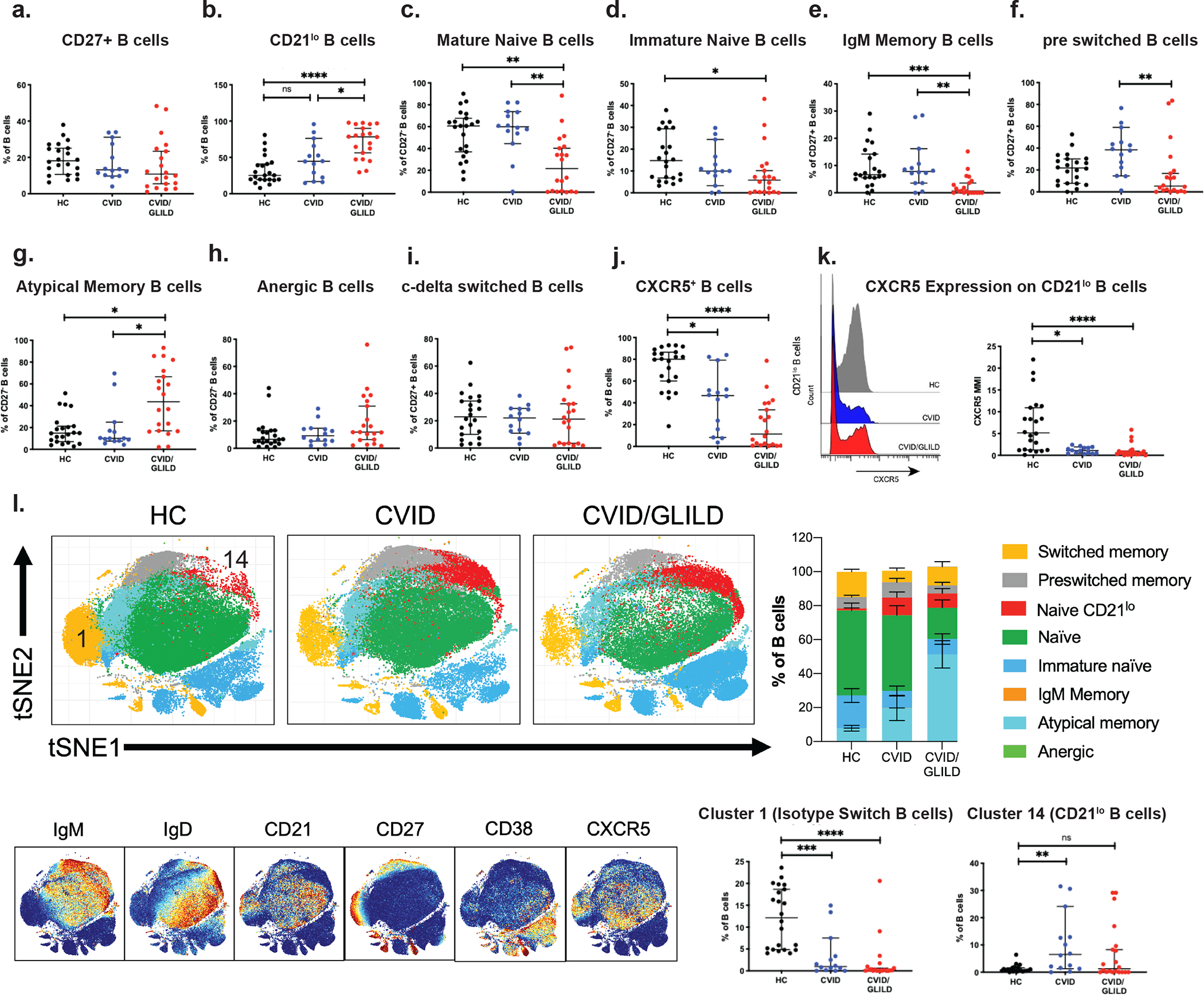
CVID and CVID/GLILD patients demonstrate increased frequency of CD21lo B cells. B cell subpopulation percentages derived from manual gating of mass cytometry data for CD27+ B cells (a), CD21lo B cells (b), mature naïve B cells (c), immature naïve B cells (d), IgM memory B cells (e), pre switched B cells (f), atypical memory B cells (g), anergic B cells (h), c-delta switched B cells (i), and CXCR5+ B cells (j). CXCR5 median intensity on CD21lo B cells (k). Unsupervised PhenoGraph clustering of CD19+ cells (l) and surface marker expression (m). Cluster frequencies corresponding to isotype switched B cells (cluster 1) and CD21lo B cells (cluster 14) (n). Error bars represent median and 95% CI. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Not significant unless stated by asterisk in figure. Kruskal–Wallis accounting for multiple comparisons was used for statistical significance
PhenoGraph clustering of CD19+ cells was used to delineate B cell subsets (Fig. 2l). The resulting cluster map resolved various B cell subpopulations defined by the expression level of B cell-specific phenotypic markers (Fig. 2m, Sup Fig. 4). Both the CVID and CVID/GLILD groups showed a higher proportion of CD21lo B cells (Fig. 2n). Further phenotyping of this CD21lo B cell cluster classifies it as CD19+IgM+IgD+CD45RA+CD27lo. Increases in CD21lo B cells among CD19+ B cells have been associated with various autoimmune manifestations in CVID [53, 54], consistent with the increased frequency of CD21lo B cells in CVID/GLILD patients (compared to both HC and CVID).
CVID and CVID/GLILD Patients Show Increased Frequency of Activated HLADR+CD4+ T Cells
We found no differences in CD4+ T cell frequency among PBMCs across HC, CVID, and CVID/GLILD (Fig. 1c). We measured expression of CD45RA and CD27 on T cells to facilitate assignment to the following T cell subsets[55–57]: naïve (CD45RA+CD27+), effector memory (CD45RA−CD27−, central memory (CD45RA−CD27+), or TEMRA ( CD45RA+CD27−). Examination of these subpopulations revealed altered proportions in CVID/GLILD patients compared to CVID and HC. Specifically, we observed decreased frequency of naïve CD4+ T cells (CD27+CD45RA+), and increased frequencies of CD4+ central memory T cells (CD27+CD45RA−) and CD4+ effector memory T cells (CD27−CD45RA−) (Fig. 3a–d). CVID patients’ T cells were previously reported to have increased expression of programmed cell death protein 1 (PD-1) [58]. In the context of CVID with immune dysregulation, expression of surface molecules that regulate adaptive immune responses, such as inducible T cell costimulator (ICOS) and CTLA4, were also elevated [59]. Both the CVID and CVID/GLILD groups displayed elevated frequency of I COS+CD4+ and PD-1+CD4+ T cells compared to HC (Fig. 3e, f). While not significant, CTLA4+CD4+ T cell frequencies from both CVID and CVID/GLILD patient groups trended higher compared against HC (Fig. 3g). Higher frequencies of CD4+ and CD8+ T cells that express human leukocyte antigen-DR isotype (HLADR) and CD38 are also evidence of enhanced immune activation in CVID [22, 60–62]. In agreement with previous findings, we found an increase in the frequency of activated HLADR+CD4+ T cells in both CVID and CVID/GLILD (Fig. 3h), with elevated expression of HLADR in CVID patients (Fig. 3i). We observed no differences in frequency of CD38+CD4+ T cells (Fig. 3j), but expression of CD38 on CD38+CD4+ T cells was elevated in both CVID and CVID/GLILD patients (Fig. 3k). Frequency of HLADR+CD38+ T cells in both the CVID and the CVID/GLILD patient groups was also elevated (Fig. 3l). These observations are consistent with PhenoGraph clustering of CD4+ T cells, where clusters of HLADR+CD4+ T cells are increased in CVID and CVID/GLILD patients (Fig. 3m–o, Sup Fig. 5).
Fig. 3.
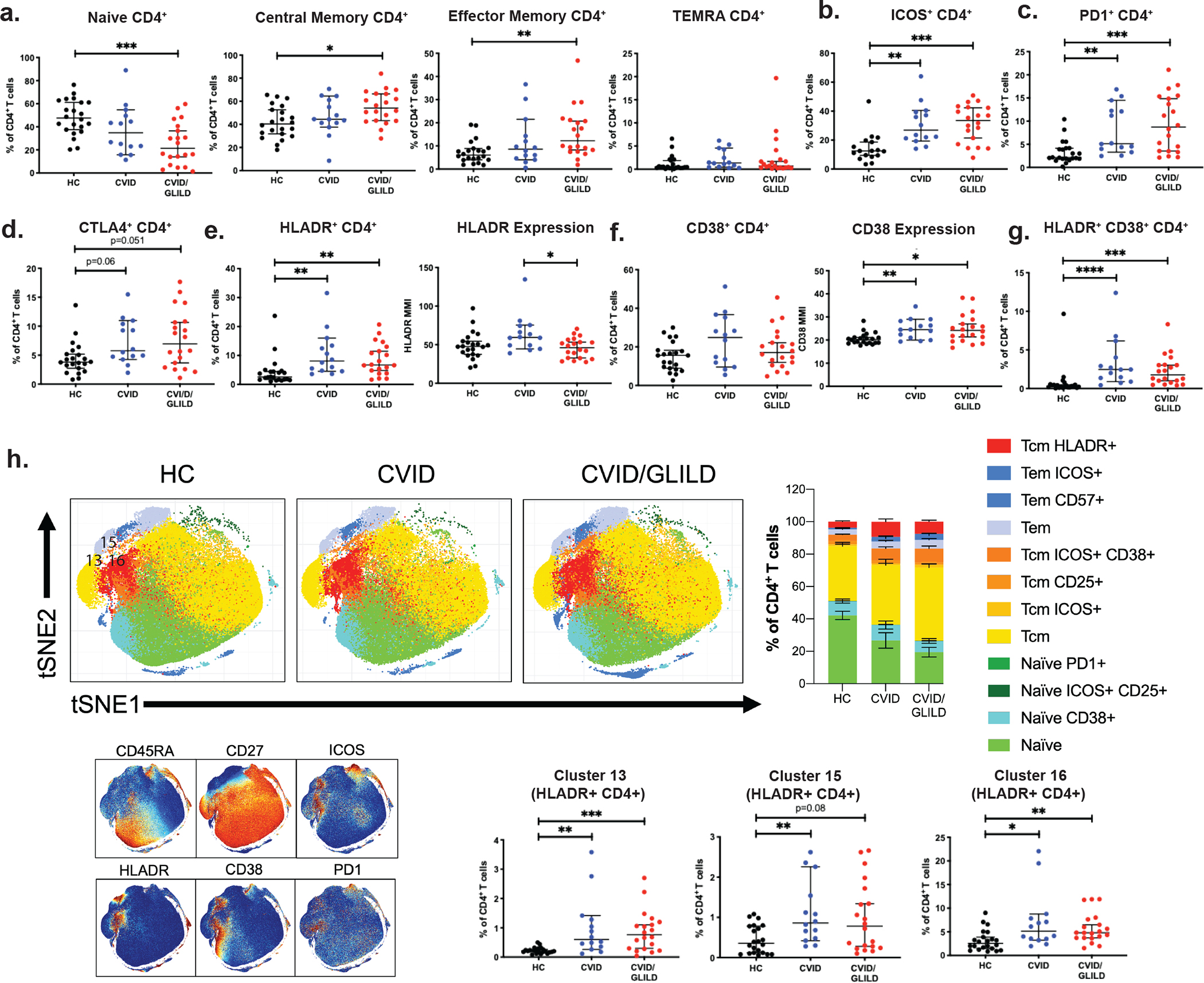
CVID and CVID/GLILD patients show increased frequency of activated HLADR+CD4+ T cells. Percentages of CD4 T cell populations derived from manual gating of mass cytometry data for naïve CD4+ (a), central memory CD4+ (b), effector memory CD4+ (c), TEMRA CD4+ (d), ICOS+CD4+ (e), PD-1+CD4+ (f), CTLA4+CD4+ (g). HLADR+CD4+ T cell percentage (h) and median intensity of HLADR surface expression on HLADR+CD4+ (i). CD38 +CD4+ T cell percentage (j) and median intensity of CD38 surface expression on CD38+CD4+ (k). Percentage of HLADR+CD38+CD4+ (l). Unsupervised PhenoGraph-derived clustering of CD4+ single cell events (m), cluster map surface marker expression (n) and cluster frequencies corresponding to HLADR+CD4+ T cells (clusters 13, 15, and 16) (o). Error bars represent median and 95% CI. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Not significant unless stated by asterisk in figure. Kruskal–Wallis accounting for multiple comparisons was used for statistical significance
CVID and CVID/GLILD Patients Exhibit Increased Frequency of Exhausted CD57+CD8+ Central Memory T Cells
Examination of major CD8+ T cell subpopulation frequency in our cohort revealed altered proportions of naïve and T cell effector memory CD8+ T cells in CVID/GLILD patients. Relative to HC and CVID, CVID/GLILD patients showed decreased frequency of naïve CD8+ T cells (Fig. 4a). No differences in central memory or effector memory CD8 + T cells were observed (Fig. 4b, c). Concurrently, the TEMRA (CD27−CD45RA+) CD8+ T cells percentage in CVID/GLILD patients was increased compared to HC and CVID (Fig. 4d). ICOS+CD8+ T cell frequency was elevated only in the CVID group (Fig. 4e). No differences in PD-1+CD8+ T cell frequency was observed (Fig. 4f). We observed increased frequency of activated HLADR+CD8+ and CD57+CD8+ T cells among PBMC in CVID and CVID/GLILD compared to HC (Fig. 4g–o, Sup Fig. 6).
Fig. 4.
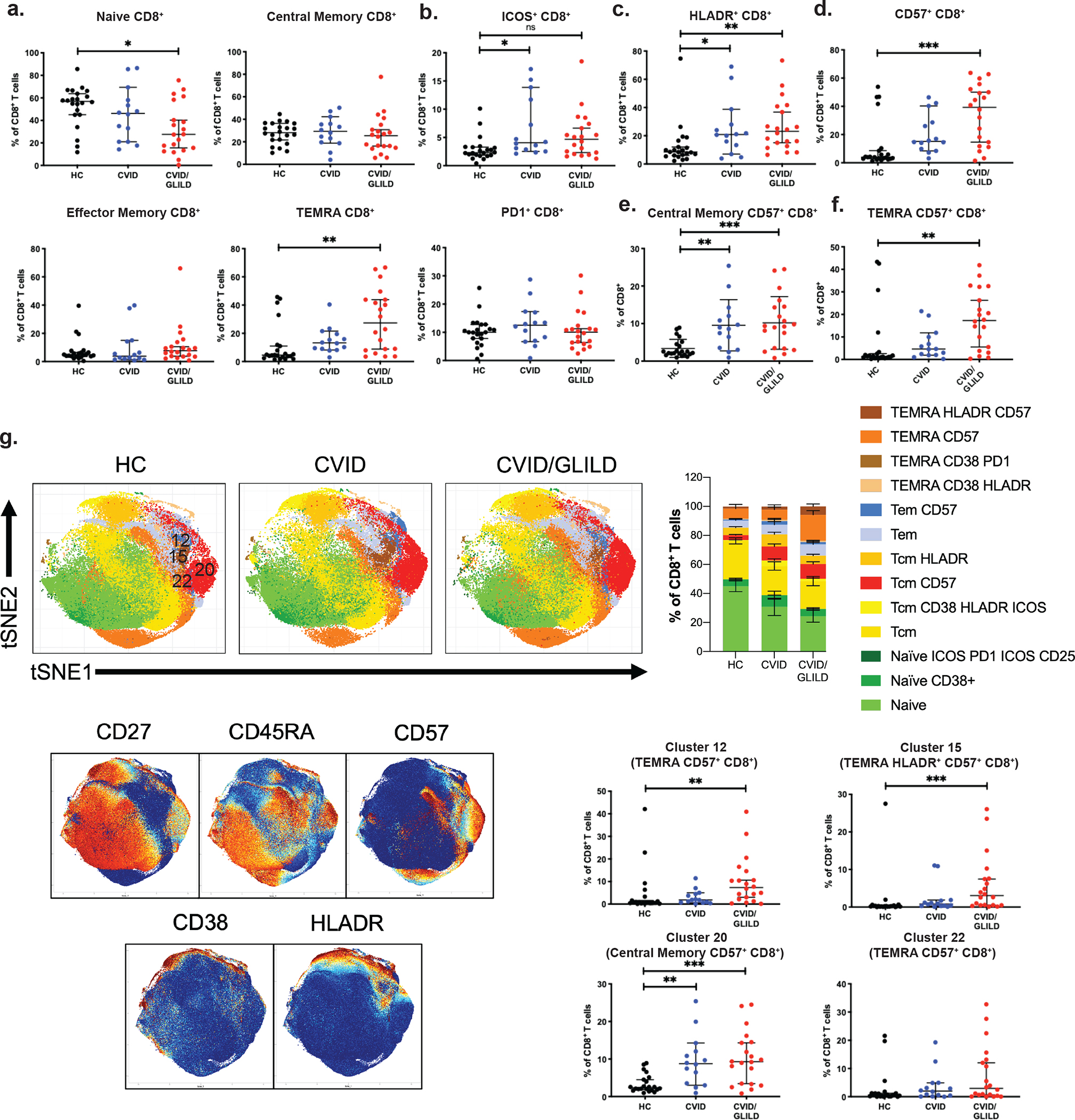
CVID/GLILD patients exhibit increased frequency of exhausted CD57+ CD8+ T cells. Percentages of CD8 T cell populations derived from manual gating of mass cytometry data for naïve CD8+ (a), central memory CD8+ (b), effector memory CD8 + (c), TEMRA CD8+ (d), ICOS+CD8+ (e), PD-1+CD8+ (f), HLADR+CD8+ (g), CD57 +CD8+ (h), naïve CD57+CD8+ (i), central memory CD57+CD8+ (j), effector memory CD57+CD8+ (k), and TEMRA CD57+CD8+ (l). Unsupervised PhenoGraph clustering of CD8+ single cell events (m), cluster map surface marker expression (n), and cluster frequencies corresponding to CD57+CD8+ TEMRA (cluster 12 and cluster 22), HLADR+CD57+CD8+ TEMRA (cluster 15), and CD57+CD8+ central memory (cluster 20) (o). Error bars represent median and 95% CI. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Not significant unless stated by asterisk in figure. Kruskal–Wallis accounting for multiple comparisons was used for statistical significance
CVID/GLILD Patients’ CD21lo B Cells Demonstrate Uncoordinated ERK Activation
We stimulated patients’ PBMCs and measured TCR/BCR-mediated signal transduction. In CD21lo B cells, canonical indicators of BCR signaling were seen (Fig. 5a). For each intracellular marker, a signaling response index as shown by differences between the 2-min stimulated and unstimulated (T2-T0) were calculated (Fig. 5b). Detection of co-expressed genes, where changes in expression of two or more genes were correlated with each other, have been used to generate gene networks involved in various disease contexts [63–66]. We adopted a similar strategy to assess the differential co-induction/phosphorylation of signaling proteins in HC, CVID, and CVID/GLILD. To do this, we examined the correlation of the response index (median T2-T0 differences) in signaling markers to measure the correlation between proximal (TCR/BCR-specific) and distal (TCR/BCR and signling by other receptors) cellular signaling response. This correlation was corroborated via examination of the T2-T0 changes in the PhenoGraph-resolved populations of naïve CD19+ B cells, naïve CD4+ T cells, and naïve CD8+ T cells from healthy individuals, which demonstrated positive correlations between markers along the various signaling pathways (Sup Fig. 7).
Fig. 5.
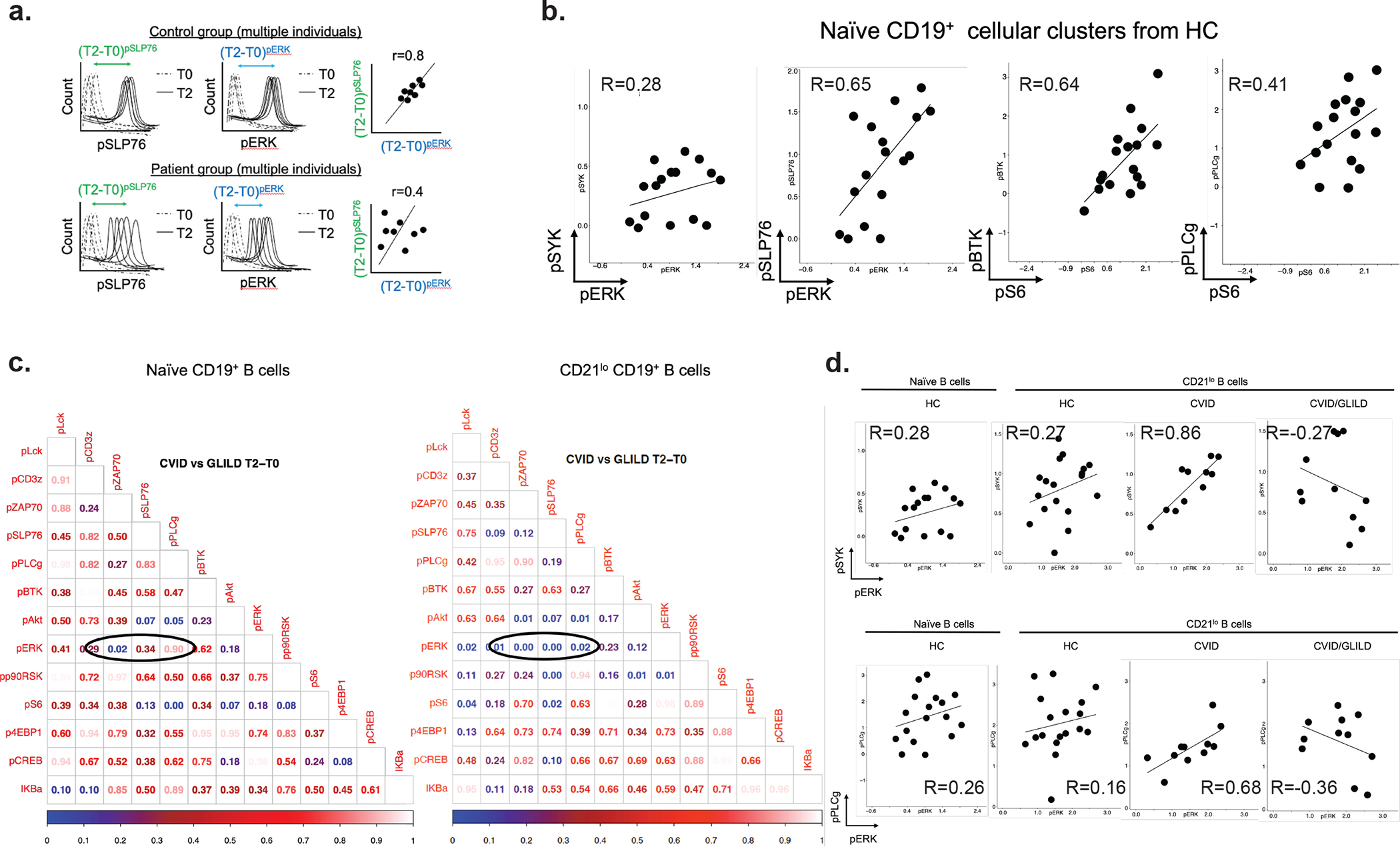
CVID/GLILD CD21lo B cells demonstrate discordant BCR signaling events. BCR stimulation responses of pSYK, pPLCγ, and pERK within PhenoGraph-derived CD21lo B cell cluster (a). Diagrammatic illustration of correlation of stimulation responses between pairs of signaling proteins (b). Tabulated p value matrices of differential pairwise signaling protein correlations between CVID and CVID/GLILD groups in PhenoGraph-d erived naïve B cells and CD21lo B cells (c). Correlation scatterplots of differentially correlated signaling protein relationships between CD21lo B cells from CVID and CVID/GLILD (d, e) Error bars represent median and 95% CI. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Not significant unless stated by asterisk in figure. Kruskal–Wallis accounting for multiple comparisons was used for statistical significance
We explored antigen receptor-mediated signaling responses in PhenoGraph-derived populations that were elevated in CVID and CVID/GLILD: CD21lo B cells, HLADR+CD4+ T cells, and CD57+CD8+ T cells. These activated subpopulations were compared against their naïve counterparts (Sup Fig. 7). Unlike naïve CD4+ T cells from CVID/GLILD, the naïve B and CD8+ T cells did not demonstrate the TCR/BCR signaling abnormalities as described below. Among CD21lo B cells, we examined the correlation of the proximal TCR/BCR signaling response (T2-T0 differences) of each signaling marker against every other in a pairwise fashion (i.e., shifts in spleen tyrosine kinase (pSYK) vs. shifts in pERK). Positive correlation coefficient represents synchronized/coordinated activation of the two signaling proteins. We then compared these signaling protein correlations within CD21lo B cells between two patient groups (i.e., shifts in pSYK vs. shifts in pERK for CVID vs. shifts in pSYK vs. shifts in pERK for CVID/GLILD) and tabulated the statistical significance of the correlation differences (Fig. 5c). Significant p values (p < 0.05) indicated that the correlations of response index (T2-T0) for one signaling protein vs. the other (X vs. Y axes) were significantly different in the two patient groups being compared (CVID vs. CVID/GLILD). However, the same comparison and analyses was not statistically significant in the naïve B cell compartment (Fig. 5c).
In CD21lo B cells from HC and CVID patients, we found ERK phosphorylation to be correlated with phosphorylation of both SYK and phospholipase C gamma (PLCγ) (at T2-T0) (Fig. 5d, e). CD21lo B cells from CVID/GLILD patients, however, demonstrated a heterogenous, inverted correlation between ERK activity and early BCR signaling events. For a subset of CVID/GLILD patients, activation of early BCR signaling events did not result in ERK activation. Conversely, a separate subset of CVID/GLILD patients demonstrated ERK activation despite decreased proximal BCR signaling activity, suggesting that downstream cellular processes may be decoupled from antigen receptor stimulation. These data indicate that while both CVID and CVID/GLILD patients demonstrate increased frequency of activated/exhausted CD21lo B cells, only those from CVID/GLILD patients exhibit altered BCR downstream signaling, which likely leads to tolerance breakdown and lymphocytic infiltration in the lungs and GLILD complication.
CVID/GLILD Patients’ HLADR+CD4+ T Cells Demonstrate Asynchronous PI3K/AKT and ERK Signaling
We explored TCR-mediated intracellular signaling relationships in HLADR+CD4+ T cell to determine whether signaling profiles are unique in the CVID/GLILD patients, and thus provide insight into disease etiology. Comparison of the T2-T0 median differences within HLADR+CD4+ T cells between CVID and CVID/GLILD patients revealed altered signaling relationships for T cell surface glycoprotein CD3 zeta chain (CD3ζ) with the following signaling proteins: AKT, ERK, and S6 (Fig. 6a, b). HLADR+CD4+ T cells from CVID patients demonstrated proportional stimulation responses for CD3ζ activation against AKT and ERK, indicating concordantly responsive PI3K/AKT and ERK activation downstream of CD3ζ phosphorylation (Fig. 6c–e). However, HLADR+CD4+ T cells from CVID/GLILD patients showed heterogeneity and disproportionality in these signaling relationships. We report HLADR+CD4+ T cells from CVID/GLILD patients possess attenuated PI3K/AKT and ERK activity despite phosphorylation of CD3ζ. A subset of these patients demonstrated decreased CD3ζ activation, yet are still able to activate PI3K/AKT, ERK, and S6, which suggests that cellular processes that proceed from TCR signal initiation may be decoupled. This dysregulated TCR downstream signaling likely contributes to breakdown of tolerance in CVID/GLILD and development of non-infectious complication.
Fig. 6.
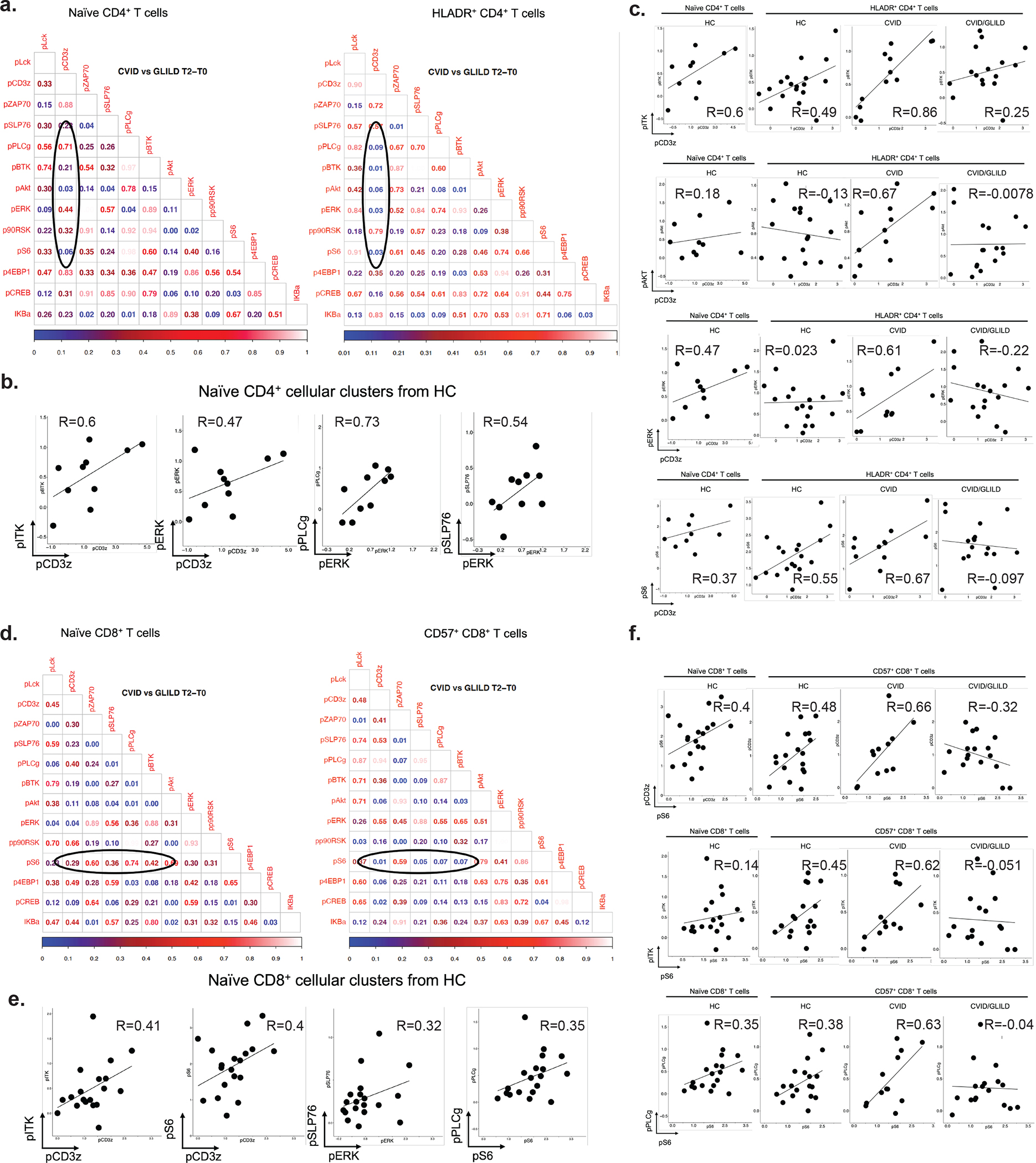
CVID/GLILD HLADR +CD4+ T cells demonstrate discordant TCR signaling events. p Value matrices of differential pairwise signaling protein correlations between CVID and CVID/GLILD groups in naïve CD4+ and HLADR+CD4+ T cells (a). TCR stimulation responses of pCD3ζ, pAkt, pERK, and pS6 within PhenoGraph-derived HLADR+CD4+ T cell cluster (b). Correlation scatterplots of differentially correlated signaling protein relationships within HLADR+CD4+ T cells (c–e). Error bars represent median and 95% CI. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Not significant unless stated by asterisk in figure. Kruskal–Wallis accounting for multiple comparisons was used for statistical significance
CVID/GLILD Patients’ CD57+CD8+ T Cells Demonstrate PLCγ Activation That Is Uncoupled from S6 Phosphorylation
Examination of the correlation between signaling responses (T2-T0) of different signaling markers against each other within CD57+CD8+ T cells from CVID/GLILD patients, compared to those from CVID patients, demonstrated altered pS6 signaling relationships (Fig. 7a, b). Specifically, CD57+CD8+ T cells from CVID patients displayed proportionate activation of pS6 relative to phosphorylation of CD3ζ and PLCγ. In CVID patients, engagement of the TCR resulted in proportional activation of proximal TCR signaling proteins and S6 phosphorylation. In contrast, pS6 activation in CD57+ CD8 + T cells from CVID/GLILD patients was disproportionate to proximal TCR signaling and PLCγ activation (Fig. 7c, d). While not abrogated, the activation of the proximal TCR signaling proteins did not correlate with downstream S6 phosphorylation. In summary, these CD57+CD8+ T cells with an exhausted/senescent phenotype exhibit diminished proximal TCR signaling but equivalent downstream S6 phosphorylation in CVID/GLILD patients only. This phenomenon of blunted TCR stimulation with preservation or even enhancement of downstream kinase activation is also observed in monogenic etiologies of CVID where autoimmune/inflammatory disorders are pervasive, suggesting that disordinate TCR proximal and distal signaling response is a common finding in primary immune regulatory disorders.
Fig. 7.
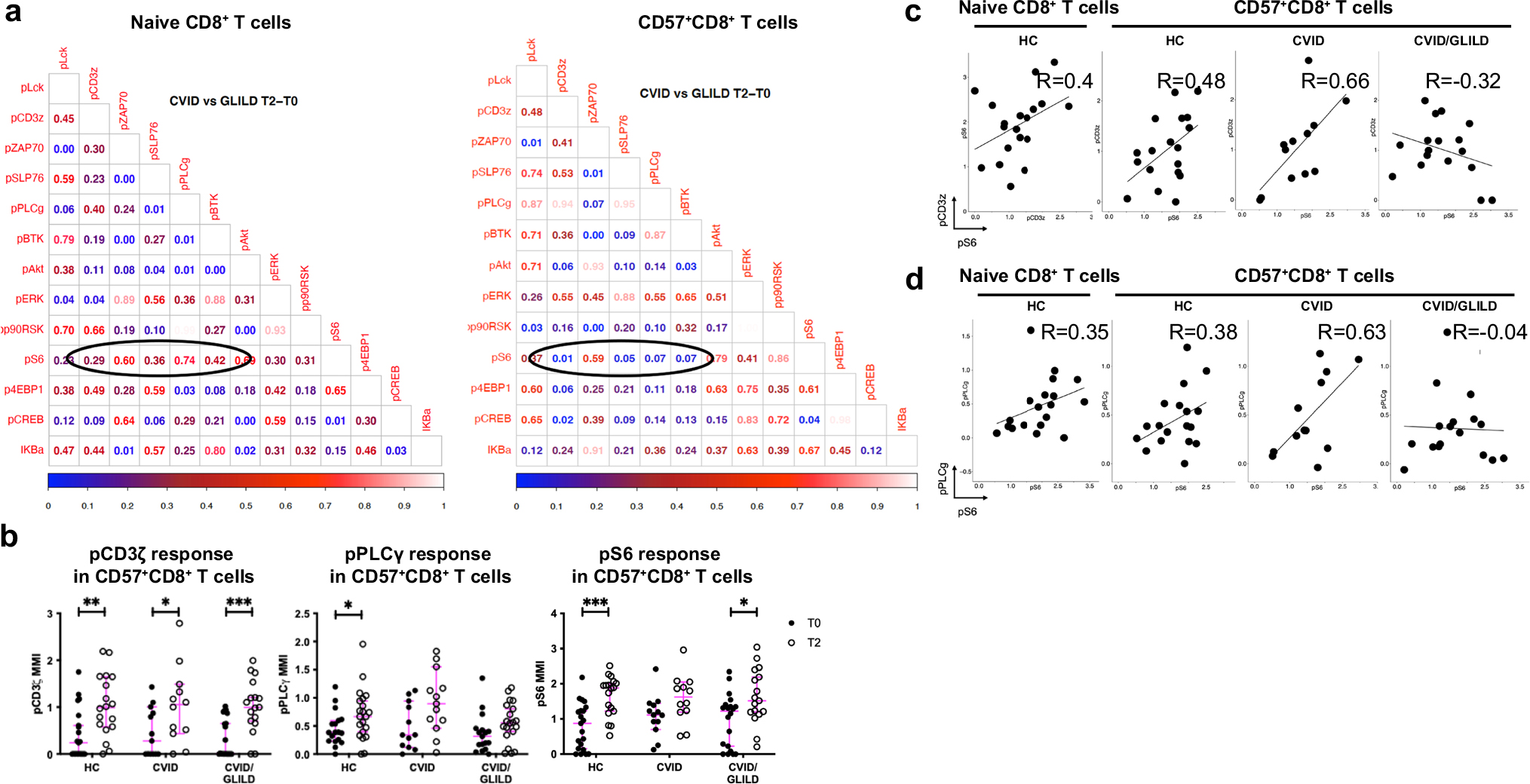
CVID/GLILD CD57+CD8+ T cells demonstrate discordant TCR signaling events. p Value matrices of differential pairwise signaling protein correlations between CVID and CVID/GLILD groups in naïve CD8+ and CD57+CD8+ T cells (a). TCR stimulation responses of pCD3ζ, pPLCγ, and pS6 within PhenoGraph-derived CD57+CD8+ T cell cluster (b). Correlation scatterplots of differentially correlated signaling protein relationships within CD57+CD8+ T cells from CVID and CVID/GLILD (c, d). Error bars represent median and 95% CI. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Not significant unless stated by asterisk in figure. Kruskal–Wallis accounting for multiple comparisons was used for statistical significance
Discussion
Respiratory failure is the leading cause of death among CVID patients [12]. Our poor understanding of the immunopathogenesis of GLILD constitutes an obstacle to both successful personalized medicine and consensus regarding treatment options. Here, we applied mass cytometry to the study of HC, CVID, and CVID/GLILD patients’ PBMCs to 1) define the phenotypes of T, B, NK, and myeloid immune cell subsets, and 2) evaluate T and B cell antigen receptor signal transduction (Sup Fig. 1). These analyses revealed that T and B cell compartments of CVID and CVID/GLILD patients display increased activated cells, such as increased frequency of CD21lo B cells, HLADR+CD4+ T cells, and CD57+CD8+ T cells (Figs. 2, 3, and 4). Each of these findings has been recognized independently in previous literature—supporting different subclassifications in CVID. Importantly, in CVID/GLILD patients only, TCR/BCR signaling in these activated T and B cells is altered, suggesting that while increased frequencies of activated lymphocyte subsets may be a common finding in CVID and CVID/GLILD, the downstream cellular function is only disturbed in CVID/GLILD. These data support the use of TCR/BCR immunomodulation in GLILD specifically. These data presented provide a cohesive picture of the phenotypic and functional (TCR/BCR signal transduction readouts) differences among HC vs. CVID vs. CVID/GLILD. They establish a foundation upon which to define specific abnormalities in signaling pathways in CVID/GLILD patients.
Similar to previous reports [67], we found that CD21lo B cells demonstrate decreased expression of CXCR5 suggesting increased tissue-homing nature of this B cell subpopulation in CVID and CVID/GLILD patients (Fig. 2j). However, unlike previous reports, but consistent with the core phenotype of B cell activation/exhaustion, we demonstrated defects in BCR signal transduction in this population in CVID/GLILD patients only (Fig. 5). These findings suggest that exhaustion of these cells is the cause (or effect) of abnormal lymphocyte receptor signaling processes. Exhaustion may be due to inability to clear microbial antigens leading to chronic antigen-induced B cell activation, and/or aberrant responses to self-antigen as seen in autoimmune disorders where CD21lo B cell frequency is increased [54, 67, 68]. Additionally, several CVID genetic etiologies associated with defects in TCR/BCR signaling, e.g., PIK3CD, NFκB1, or IKZF1, have been shown to present with increased CD21lo B cell frequency, and CD4+ and CD8+ T cell activation. However, detailed evaluation of TCR/BCR signaling has not been addressed in the context of these genetic defects.
With respect to T cell abnormalities, decreased CD4+ naïve T cell numbers and CD8+ T cell exhaustion have been previously described in CVID. Cytokine profiles, T cell proliferation outcomes, and expression of high levels of activation and memory markers consistent with persistent T cell activation and exhaustion have all been demonstrated in CVID[69–72]. Our findings of T cell immunophenotyping are consistent with previous literature, as we detected increased frequency and absolute numbers of HLADR+CD4+ T cells and CD57+CD8+ T cells in CVID/GLILD patients (Figs. 3 and 4, Sup Fig. 3). Additionally, we demonstrated altered TCR downstream signal transduction in these T cell subpopulations compared to HC and CVID. These findings are consistent with chronic stimulation cell antigenic stimulation in CVID/GLILD (Figs. 6 and 7). Chronic TCR stimulation may underlie the T cell phenotypic abnormalities seen. Remarkably, TCR signal transduction in CD8+ naïve T cell subpopulations remain normal and similar between CVID/GLILD and HC, suggesting that the activated/exhausted T cell phenotype is tied to TCR signal transduction defects.
B and T cell immunophenotyping has long been pursued to subclassify CVID, such as the previously published classification of CVID based on CD21lo B cell percentage, and its prediction of types of autoimmune/inflammatory complications. However, findings have not been applied to clinical practice because these immunological/clinical phenotypic correlations have not been consistently demonstrated across (i) heterogeneous CVID cohorts, with diverse clinical phenotypes and from multiple institutions; (ii) multiple distinct clinical flow cytometry panels/definitions (i.e., inconsistent B cell phenotype flow cytometry panels used by different immune diagnostic labs, and hence divergent gating and B cell subpopulation definitions); and (iii) clinical practitioner standard of care laboratory assessments (i.e., nonstandardized clinical laboratory evaluation).
Amplitude of proximal (i.e., ZAP70) and distal (i.e., ERK or S6) TCR signal transduction events and their correlation may most accurately reflect alterations that result in T cell activation/exhaustion, and thus may constitute a surrogate for autoimmune/inflammatory complications in CVID. We conclude that proximal and distal antigen receptor-mediated signaling is uncoordinated in CD21lo B cells, HLADR+CD4+ T cells, and CD57+CD8+ T cells from CVID/GLILD patients, suggesting that CVID patients with GLILD are more likely to suffer from an underlying genetic defect affecting TCR/BCR signal transduction than those without (Figs. 6 and 7, Sup Fig. 7). Hence, TCR/BCR signaling immunomodulators may be an effective therapy for CVID/GLILD patients. We propose that discordance of these signaling events can be used to (i) distinguish CVID/GLILD patients from all CVID patients, and (ii) raise suspicion (and support further workup) of an underlying genetic defect. Validation is required in larger and more heterogeneous cohorts of CVID patients with autoimmune/inflammatory complications, and such studies should be pursued in the future to better define the underlying immunopathogenesis and delineate prognostic measures of immune dysregulation in CVID.
Conclusions
Both CVID and CVID/GLILD patients demonstrate increased frequencies of activated/exhausted T and B cell subsets (HLADR+CD4+ T cells, CD57+CD8 T cells, and CD21lo B cells). However, CVID/GLILD patients are unique in demonstrating discordant TCR/BCR signaling in these activated cells. Altogether, these findings suggest that in CVID/GLILD, (i) an underlying genetic defect(s) is responsible for abnormal TCR/BCR signaling and breakdown of tolerance, and (ii) the use of TCR/BCR signaling immunomodulation as an effective therapy.
Supplementary Material
Acknowledgements
We would like to thank the University of Colorado Cancer Center Flow Cytometry Shared Resource for the mass cytometry services. We also thank the study participants and their families. We would like to thank John Cambier for his intellectual input and helpful comments.
Funding
VGL has received funding from NIH T32 AI07405 and NSF Graduate Research Fellowship DGE-1938058. TG and DG are funded by the Grohne-Stepp Endowed Chair in Cancer Research from the University of Colorado Cancer Center. This work is funded by the Jeffrey Modell Foundation Translational Research Award and NIH K23AR070897.
Footnotes
Conflict of Interest JMR and JWV receive personal paid fees from Up to Date.
Ethics Approval All human donors were consented under human subjects’ research protocol, approved by the Institutional Review Board of the Research Compliance Office at Medical College of Wisconsin.
Consent to Participate All patients provided informed consent to participate in this study.
Consent for Publication The authors consent to publish the content of this original report.
Supplementary Information The online version contains supplementary material available at https://doi.org/10.1007/s10875-023-01485-9.
Data Availability
Mass cytometry data is available at the following link: https://flowrepository.org/id/RvFrTO9BWuHjW6cmXfzpPE9jXEzo8KXlbpxapGEJ4R8Go78ElSPFMJfNZGCSuhgX.
References
- 1.Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol. 1999;92:28–34. [DOI] [PubMed] [Google Scholar]
- 2.Bonilla FA, Khan DA, Ballas ZK, Chinen J, Frank MM, Hsu JT, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. J Allergy Clin Immunol. 2014;136(5):1186–1205.e78. [DOI] [PubMed] [Google Scholar]
- 3.Selenius JS, Martelius T, Pikkarainen S, Siitonen S, Mattila E, Pietikainen R, et al. Unexpectedly high prevalence of common variable immunodeficiency in Finland. Front Immunol. 2017;8:1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Westh L, Mogensen TH, Dalgaard LS, Bernth Jensen JM, Katzenstein T, Hansen ABE, et al. Identification and characterization of a nationwide danish adult common variable immunodeficiency cohort. Scand J Immunol. 2017;85(6):450–61. [DOI] [PubMed] [Google Scholar]
- 5.Gathmann B, Grimbacher B, Beaute J, Dudoit Y, Mahlaoui N, Fischer A, et al. The European internet-based patient and research database for primary immunodeficiencies: results 2006–2008. Clin Exp Immunol. 2009;157 Suppl 1:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112(2):277–86. [DOI] [PubMed] [Google Scholar]
- 7.Quinti I, Soresina A, Spadaro G, Martino S, Donnanno S, Agostini C, et al. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol. 2007;27(3):308–16. [DOI] [PubMed] [Google Scholar]
- 8.Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. 2008;111(1):77–85 [DOI] [PubMed] [Google Scholar]
- 9.Hampson FA, Chandra A, Screaton NJ, Condliffe A, Kumararatne DS, Exley AR, et al. Respiratory disease in common variable immunodeficiency and other primary immunodeficiency disorders. Clin Radiol. 2012;67:587–95. [DOI] [PubMed] [Google Scholar]
- 10.Ho HE, Cunningham-Rundles C. Non-infectious complications of common variable immunodeficiency: updated clinical spectrum, sequelae, and insights to pathogenesis. Front Immunol. 2020;7:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cabañero-Navalon MD, Garcia-Bustos V, Nuñez-Beltran M, Císcar Fernández P, Mateu L, Solanich X, et al. Current clinical spectrum of common variable immunodeficiency in Spain: the multicentric nationwide GTEM-SEMI-CVID registry. Front Immunol. 2022;28:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. 2012;119(7):1650–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fischer A, Provot J, Jais JP, Alcais A, Mahlaoui N, Adoue D, et al. Autoimmune and inflammatory manifestations occur frequently in patients with primary immunodeficiencies. J Allergy Clin Immunol. 2017;140(5):1388–1393.e8. [DOI] [PubMed] [Google Scholar]
- 14.Berbers RM, Drylewicz J, Ellerbroek PM, van Montfrans JM, Dalm V, van Hagen PM, et al. Targeted proteomics reveals inflammatory pathways that classify immune dysregulation in common variable immunodeficiency. J Clin Immunol. 2021;41(2):362–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hultberg J, Ernerudh J, Larsson M, Nilsdotter-Augustinsson Å, Nyström S. Plasma protein profiling reflects TH1-driven immune dysregulation in common variable immunodeficiency. J Allergy Clin Immunol. 2020;146(2):417–28. [DOI] [PubMed] [Google Scholar]
- 16.Bonilla FA, Barlan I, Chapel H, Costa-Carvalho BT, Cunningham-Rundles C, de la Morena MT, et al. International Consensus Document (ICON): common variable immunodeficiency disorders. J Allergy Clin Immunol Pract. 2016;4(1):38–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Schouwenburg PA, Davenport EE, Kienzler AK, Marwah I, Wright B, Lucas M, et al. Application of whole genome and RNA sequencing to investigate the genomic landscape of common variable immunodeficiency disorders. Clin Immunol. 2015;160(2):301–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bogaert DJA, Dullaers M, Lambrecht BN, Vermaelen KY, De Baere E, Haerynck F. Genes associated with common variable immunodeficiency: one diagnosis to rule them all? J Med Genet. 2016;53(9):575–90. [DOI] [PubMed] [Google Scholar]
- 19.Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human inborn errors of immunity: 2022 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2022;42(7):1473–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edwards ESJ, Bosco JJ, Ojaimi S, O’Hehir RE, van Zelm MC. Beyond monogenetic rare variants: tackling the low rate of genetic diagnoses in predominantly antibody deficiency. Cell Mol Immunol. 2021;18(3):588–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fliegauf M, Bryant VL, Frede N, Slade C, Woon ST, Lehnert K, et al. Haploinsufficiency of the NF-κB1 Subunit p50 in common variable immunodeficiency. Am J Hum Genet. 2015;97(3):389–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mouillot G, Carmagnat M, Gérard L, Garnier JL, Fieschi C, Vince N, et al. B-cell and T-cell phenotypes in CVID patients correlate with the clinical phenotype of the disease. J Clin Immunol. 2010;30(5):746–55. [DOI] [PubMed] [Google Scholar]
- 23.Sánchez-Ramón S, Radigan L, Yu JE, Bard S, Cunningham-Rundles C. Memory B cells in common variable immunodeficiency: clinical associations and sex differences. Clin Immunol. 2008;128(3):314–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agarwal S, Smereka P, Harpaz N, Cunningham-Rundles C, Mayer L. Characterization of immunologic defects in patients with common variable immunodeficiency (CVID) with intestinal disease. Inflamm Bowel Dis. 2011;17(1):251–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ardeniz Ö, Cunningham-Rundles C. Granulomatous disease in common variable immunodeficiency. Clin Immunol. 2009;133(2):198–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rao N, Mackinnon AC, Routes JM. Granulomatous and lymphocytic interstitial lung disease: a spectrum of pulmonary histopathologic lesions in common variable immunodeficiency–histologic and immunohistochemical analyses of 16 cases. Hum Pathol. 2015;46(9):1306–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bates CA, Ellison MC, Lynch DA, Cool CD, Brown KK, Routes JM. Granulomatous-lymphocytic lung disease shortens survival in common variable immunodeficiency. J Allergy Clin Immunol. 2004;114(2):415–21. [DOI] [PubMed] [Google Scholar]
- 28.Verbsky JW, Hintermeyer MK, Simpson PM, Feng M, Barbeau J, Rao N, et al. Rituximab and antimetabolite treatment of granulomatous and lymphocytic interstitial lung disease in common variable immunodeficiency. J Allergy Clin Immunol. 2021;147(2):704–712.e17. [DOI] [PubMed] [Google Scholar]
- 29.Patel S, Anzilotti C, Lucas M, Moore N, Chapel H. Interstitial lung disease in patients with common variable immunodeficiency disorders: several different pathologies? Clin Exp Immunol. 2019;198(2):212–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maglione PJ, Overbey JR, Cunningham-Rundles C. Progression of CVID interstitial lung disease accompanies distinct pulmonary and laboratory findings. J Allergy Clin Immunol Pr. 2015;3(6):941–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maglione PJ, Gyimesi G, Cols M, Radigan L, Ko HM, Weinberger T, et al. BAFF-driven B cell hyperplasia underlies lung disease in common variable immunodeficiency. JCI Insight. 2019;4(5):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fraz MSA, Michelsen AE, Moe N, Aalokken TM, Macpherson ME, Nordoy I, et al. Raised serum markers of T cell activation and exhaustion in granulomatous-lymphocytic interstitial lung disease in common variable immunodeficiency. J Clin Immunol. 2022;42(7):1553–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chase NM, Verbsky JW, Hintermeyer MK, Waukau JK, Tomita-Mitchell A, Casper JT, et al. Use of combination chemotherapy for treatment of granulomatous and lymphocytic interstitial lung disease (GLILD) in patients with common variable immunodeficiency (CVID). J Clin Immunol. 2013;33(1):30–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smits B, Goldacker S, Seneviratne S, Malphettes M, Longhurst H, Mohamed OE, et al. The efficacy and safety of systemic corticosteroids as first line treatment for granulomatous lymphocytic interstitial lung disease. J Allergy Clin Immunol. 2022. Online ahead of print. [DOI] [PubMed] [Google Scholar]
- 35.Boncristiano M, Majolini MB, D’Elios MM, Pacini S, Valensin S, Ulivieri C, et al. Defective recruitment and activation of ZAP70 in common variable immunodeficiency patients with T cell defects. Eur J Immunol. 2000;30:2632–8. [DOI] [PubMed] [Google Scholar]
- 36.Visentini M, Marrapodi R, Conti V, Mitrevski M, Camponeschi A, Lazzeri C, et al. Dysregulated extracellular signal-regulated kinase signaling associated with impaired B-cell receptor endocytosis in patients with common variable immunodeficiency. J Allergy Clin Immunol. 2014;134(2):401–10. [DOI] [PubMed] [Google Scholar]
- 37.Van De Ven AAJM, Compeer EB, Bloem AC, Van De Corput L, Van Gijn M, Van Montfrans JM, et al. Defective calcium signaling and disrupted CD20-B-cell receptor dissociation in patients with common variable immunodeficiency disorders. J Allergy Clin Immunol. 2012;129(3). [DOI] [PubMed] [Google Scholar]
- 38.Foerster C, Voelxen N, Rakhmanov M, Keller B, Gutenberger S, Goldacker S, et al. B cell receptor-mediated calcium signaling is impaired in B lymphocytes of type Ia patients with common variable immunodeficiency. J Immunol. 2010;184(12):7305–13. [DOI] [PubMed] [Google Scholar]
- 39.Fischer MB, Hauber I, Eggenbauer H, Thon V, Vogel E, Schaffer E, et al. A defect in the early phase of T-cell receptor-mediated T-cell activation in patients with common variable immunodeficiency. Blood. 1994;84(12):4234–41. [PubMed] [Google Scholar]
- 40.Paccani SR, Boncristiano M, Patrussi L, Ulivieri C, Wack A, Valensin S, et al. Defective Vav expression and impaired F-actin reorganization in a subset of patients with common variable immunodeficiency characterized by T-cell defects. Blood. 2005;106(2):626–34. [DOI] [PubMed] [Google Scholar]
- 41.del Pino-Molina L, Torres Canizales JM, Rodríguez-Pena R, López-Granados E. Evaluation of B-cell intracellular signaling by monitoring the PI3K-Akt axis in patients with common variable immunodeficiency and activated phosphoinositide 3-kinase delta syndrome. Cytom Part B - Clin Cytom. 2021;100(4):460–6. [DOI] [PubMed] [Google Scholar]
- 42.Zunder ER, Finck R, Behbehani GK, Amir EAD, Krishnaswamy S, Gonzalez VD, et al. Palladium-based mass tag cell barcoding with a doublet-filtering scheme and single-cell deconvolution algorithm. Nat Protoc. 2015;10(2):316–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Levine JH, Simonds EF, Bendall SC, Davis KL, Amir EAD, Tadmor MD, et al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell. 2015;162(1):184–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kimball AK, Oko LM, Bullock BL, Nemenoff RA, van Dyk LF, Clambey ET. A beginner’s guide to analyzing and visualizing mass cytometry data. J Immunol. 2018;200(1):3–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Gorman WE, Kong DS, Balboni IM, Rudra P, Bolen CR, Ghosh D, et al. Mass cytometry identifies a distinct monocyte cytokine signature shared by clinically heterogeneous pediatric SLE patients. J Autoimmun. 2017;81:74–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Galbraith MD, Kinning KT, Sullivan KD, Araya P, Smith KP, Granrath RE, et al. Specialized interferon action in COVID-19. PNAS. 2022;119(11):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Galbraith MD, Kinning KT, Sullivan KD, Baxter R, Araya P, Jordan KR, et al. Seroconversion stages COVID19 into distinct pathophysiological states. Elife. 2021;1:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hintzen RQ, Lens SM, Beckmann MP, Goodwin RG, Lynch D, Van Lier RAW. Characterization of the human CD27 ligand, a novel member of the TNF gene family. J Immunol. 1994;152(4):1762–73. [PubMed] [Google Scholar]
- 49.Agematsu K, Hokibara S, Nagumo H, Komiyama A. CD27: A memory B-cell marker. Immunol Today. 2000;21(5):204–6. [DOI] [PubMed] [Google Scholar]
- 50.Brouet JC, Chedeville A, Fermand JP, Royer B. Study of the B cell memory compartment in common variable immunodeficiency. Eur J Immunol. 2000;30(9):2516–20. [DOI] [PubMed] [Google Scholar]
- 51.Yasui H, Akahori Y, Hirano M, Yamada K, Kurosawa Y. Class switch from mu to delta is mediated by homologous recombination between alpha-mu and sigma -mu sequences in human immunoglobulin gene loci. Eur J Immunol. 1989;19:1399–403. [DOI] [PubMed] [Google Scholar]
- 52.Koelsch K, Zheng NY, Zhang Q, Duty A, Helms C, Mathias MD, et al. Mature B cells class switched to IgD are autoreactive in healthy individuals. J Clin Invest. 2007;117(6):1558–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.López-Herrera G, Segura-Méndez NH, O’Farril-Romanillos P, Nuñez-Nuñez ME, Zarate-Hernández MC, Mogica-Martínez D, et al. Low percentages of regulatory T cells in common variable immunodeficiency (CVID) patients with autoimmune diseases and its association with increased numbers of CD4+CD45RO+ T and CD21low B cells. Allergol Immunopathol (Madr). 2019;47(5):457–66. [DOI] [PubMed] [Google Scholar]
- 54.Warnatz K, Wehr C, Drager R, Schmidt S, Eibel H, Schlesier M, et al. Expansion of CD19(hi)CD21(lo/neg) B cells in common variable immunodeficiency (CVID) patients with autoimmune cytopenia. Immunobiology. 2002;206(5):502–13. [DOI] [PubMed] [Google Scholar]
- 55.van der Burg M, Kalina T, Perez-Andres M, Vlkova M, Lopez-Granados E, Blanco E, et al. The EuroFlow PID orientation tube for flow cytometric diagnostic screening of primary immunodeficiencies of the lymphoid system. Front Immunol. 2019;10:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tussey L, Speller S, Gallimore A, Vessey R. Functionally distinct CD8+ memory T cell subsets in persistent EBV infection are differentiated by migratory receptor expression. Eur J Immunol. 2000;30(7):1823–9. [DOI] [PubMed] [Google Scholar]
- 57.Appay V, Dunbar PR, Callan M, Klenerman P, Gillespie GMA, Papagno L, et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med. 2002;8(4):379–85. [DOI] [PubMed] [Google Scholar]
- 58.Le CC, Bengsch B, Khanna C, Trofa M, Ohtani T, Nolan BE, et al. Common variable immunodeficiency-associated endotoxemia promotes early commitment to the T follicular lineage. J Allergy Clin Immunol. 2019;144(6):1660–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berbers RM, van der Wal MM, van Montfrans JM, Ellerbroek PM, Dalm VASH, van Hagen PM, et al. Chronically activated T-cells retain their inflammatory properties in common variable immunodeficiency. J Clin Immunol. 2021;41(7):1621–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Paquin-Proulx D, Sandberg JK. Persistent immune activation in CVID and the role of IVIg in its suppression. Front Immunol. 2014;5:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carbone J, Sarmiento E, Micheloud D, Rodríguez-Molina J, Fernández-Cruz E. Elevated levels of activated CD4 T cells in common variable immunodeficiency: association with clinical findings. Allergol Immunopathol (Madr). 2006;34(4):131–5. [DOI] [PubMed] [Google Scholar]
- 62.Giovannetti A, Pierdominici M, Mazzetta F, Marziali M, Renzi C, Mileo AM, et al. Unravelling the complexity of t cell abnormalities in common variable immunodeficiency. J Immunol. 2007;178(6):3932–43. [DOI] [PubMed] [Google Scholar]
- 63.Kayano M, Shiga M, Mamitsuka H. Detecting differentially coexpressed genesfrom labeled expression data: a brief review. IEEE/ACM Trans Comput Biol Bioinforma. 2014;11(1):154–67. [DOI] [PubMed] [Google Scholar]
- 64.Amar D, Safer H, Shamir R. Dissection of regulatory networks that are altered in disease via differential co-expression. PLoS Comput Biol. 2013;9(3):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Choi JK, Yu U, Yoo OJ, Kim S. Differential coexpression analysis using microarray data and its application to human cancer. Bioinformatics. 2005;21(24):4348–55. [DOI] [PubMed] [Google Scholar]
- 66.Bhattacharyya M, Bandyopadhyay S. Studying the differential co-expression of microRNAs reveals significant role of white matter in early Alzheimer’s progression. Mol Biosyst. 2013;9(3):457–66. [DOI] [PubMed] [Google Scholar]
- 67.Rakhmanov M, Keller B, Gutenberger S, Foerster C, Hoenig M, Driessen G, et al. Circulating CD21low B cells in common variable immunodeficiency resemble tissue homing, innate-like B cells. PNAS. 2009;106(32):13451–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Freudenhammer M, Voll RE, Binder SC, Keller B, Warnatz K. Naive- and memory-like CD21 low B cell subsets share core phenotypic and signaling characteristics in systemic autoimmune disorders. J Immunol. 2020;205(8):2016–25. [DOI] [PubMed] [Google Scholar]
- 69.Boileau J, Mouillot G, Gérard L, Carmagnat M, Rabian C, Oksenhendler E, et al. Autoimmunity in common variable immunodeficiency: correlation with lymphocyte phenotype in the French DEFI study. J Autoimmun. 2011;36(1):25–32. [DOI] [PubMed] [Google Scholar]
- 70.Lanio N, Sarmiento E, Gallego A, Carbone J. Immunophenotypic profile of T cells in common variable immunodeficiency: is there an association with different clinical findings? Allergol Immunopathol (Madr). 2009;37(1):14–20. [DOI] [PubMed] [Google Scholar]
- 71.Vlková M, Thon V, Šárfyová M, Bláha L, Svobodník A, Lokaj J, et al. Age dependency and mutual relations in T and B lymphocyte abnormalities in common variable immunodeficiency patients. Clin Exp Immunol. 2006;143(2):373–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Viallard JF, Blanco P, André M, Etienne G, Liferman F, Neau D, et al. CD8+HLA-DR+ T lymphocytes are increased in common variable immunodeficiency patients with impaired memory B-cell differentiation. Clin Immunol. 2006;119(1):51–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mass cytometry data is available at the following link: https://flowrepository.org/id/RvFrTO9BWuHjW6cmXfzpPE9jXEzo8KXlbpxapGEJ4R8Go78ElSPFMJfNZGCSuhgX.