

Abstract
BACKGROUND:
Hereditary Hemorrhagic Telangiectasia (HHT) is a vascular disorder characterized by arteriovenous malformations (AVMs) and blood vessel enlargements. However, there are no effective drug therapies to combat AVM formation in HHT patients. Here, we aimed to address whether elevated levels of angiopoietin-2 (ANG2) in the endothelium is a conserved feature in mouse models of the three major forms of HHT that could be neutralized to treat brain AVMs and associated vascular defects. In addition, we sought to identify the angiogenic molecular signature linked to HHT.
METHODS:
Cerebrovascular defects, including AVMs and increased vessel calibers, were characterized in mouse models of the three common forms of HHT using transcriptomic and dye injection labeling methods.
RESULTS:
Comparative RNA-seq analyses of isolated brain endothelial cells revealed a common, but unique pro-angiogenic transcriptional program associated with HHT. This included a consistent upregulation in cerebrovascular expression of ANG2 and downregulation of its receptor Tyr kinase with Ig and EGF homology domains (TIE2/TEK) in HHT mice compared to controls. Furthermore, in vitro experiments revealed TEK signaling activity was hampered in an HHT setting. Pharmacological blockade of ANG2 improved brain vascular pathologies in all HHT models, albeit to varying degrees. Transcriptomic profiling further indicated that ANG2 inhibition normalized the brain vasculature by impacting a subset of genes involved in angiogenesis and cell migration processes.
CONCLUSIONS:
Elevation of ANG2 in the brain vasculature is a shared trait among the mouse models of the common forms of HHT. Inhibition of ANG2 activity can significantly limit or prevent brain AVM formation and/or blood vessel enlargement in HHT mice. Thus, ANG2-targeted therapies may represent a compelling approach to treat AVMs and vascular pathologies related to all forms of HHT.
Keywords: angiopoietin, Tie2, Tek, arteriovenous malformations, brain, endothelial, angiogenesis, HHT, cell migration
Graphical Abstract

Introduction
Hereditary Hemorrhagic Telangiectasia (HHT) is an autosomal dominant vascular disorder that affects approximately 1 in 5,000 people worldwide 1,2. HHT is caused by heterozygous loss-of-function gene variants in members of the Transforming Growth Factor beta (TGFβ) signaling pathway, including activin receptor-like kinase 1 (ACVRL1/ALK1), endoglin (ENG), mothers against decapentaplegic homolog 4 (SMAD4), or bone morphogenetic protein 9 (BMP9/GDF2) 1–9. These pathogenic variants primarily affect the endothelial cells (ECs) and account for the different types of HHT (ENG – HHT1; ALK1 – HHT2; SMAD4 – Juvenile Polyposis/HHT; BMP9 – HHT-like), with most patients harboring mutations in ALK1 and ENG (>85%) 3,10. HHT patients commonly develop mucosa and/or skin telangiectasias (abnormally enlarged small vessels) and visceral arteriovenous malformations (AVMs). AVMs are fragile, enlarged capillaries that form shunts between arteries and veins 4,11 and regularly develop in major organs, such as the brain, liver, and lung 2. AVMs are prone to rupturing and can lead to stroke or lethal hemorrhaging. Patients also experience severe and frequent nose bleeds (epistaxis), anemia and cardiac complications, further adding to the debilitating nature of HHT.
Despite knowing the HHT causative genes, effective drug treatments for the symptoms of this disease are limited, though this area continues to be actively investigated at the research and clinical levels12,13. Subsequently, invasive surgeries, such as cauterization or embolization, are the typical methods used to treat telangiectasias and AVMs respectively in patients 10,14–16. However, the recurring and progressive nature of HHT further complicates the use of these procedures as routine treatments given the inherent risks, especially in certain tissues such as the brain. In this regard, anti-angiogenic drugs have been tested in the clinic, as HHT pathogenesis is thought to be driven by an abnormal, reactivated angiogenic process. Several studies using bevacizumab, an anti-vascular endothelial growth factor A (VEGF-A) based drug, have shown promising results in treating patients with recurring epistaxis and high output heart failure 17,18. However, VEGF signaling remains a highly complex process, and drugs targeting VEGF-A can elicit serious side-effects and not all patients respond to treatment. Therefore, given the severity of this disease and a lack of understanding the precise pro-angiogenic nature of HHT, there remains a need to identify additional drug therapies, including anti-angiogenic drugs, that could benefit HHT patients.
Increased efforts to uncover potential therapeutic targets for HHT has been a recent focus in several studies using animal models of HHT, including testing the efficacy of inhibiting angiopoietin-2 (ANG2) 19, phosphatidyl inositol 3-kinase (PI3K) 20–22, mammalian target of rapamycin (mTOR) 23,24, tyrosine kinase receptors 22,24, integrin-yes-associated protein (YAP)/taffazzin (TAZ) 25 and VEGF 26, and activating ALK1 23,27. The effectiveness of these drugs has mostly been tested in the murine retina due to ease of accessibility, a 2-dimensional like vascular structure that forms via a natural angiogenic process in the early postnatal days, and consistent vascular phenotypes in mouse models of HHT that include the AVMs 28–33. However, retinal vascular problems are not a common occurrence in HHT patients. Considering the mounting evidence showing organ-specific prevalence of vascular pathologies associated with the various types of HHT 34,35, there is an urgent need to determine the therapeutic effects for clinically relevant organs, like the brain.
Approximately 10–20% of HHT patients possess brain AVMs (bAVMs), which are thought to be primarily congenital. HHT-associated bAVMs can lead to seizure, headache and spontaneous or recurrent intracranial hemorrhage, causing significant morbidity and mortality 36,37. Homozygous loss-of-function HHT mouse models have been a key resource in our current understanding of bAVM development, in part, due to the consistent formation of these lesions in vivo. Furthermore, the use of these models has been boosted by a recent study indicating that telangiectasia formation may arise via focal bi-allelic loss of ALK1 and ENG 38, though the causal nature of loss of heterozygosity in these malformations is unclear. It should also be noted that bi-allelic loss of HHT genes have not been reported in AVMs39, and homozygous mouse models of HHT, in general, exhibit more widespread vascular defects compared to HHT patients. In terms of animal models, bAVMs have been reported individually in Alk1, Eng and Smad4 mouse models of HHT 21,40–44, but have not been extensively characterized in a comparative manner. In fact, few comparative studies between all three models have been described 42 yet could provide critical context as these animal models appear to develop both shared and unique vascular phenotypes.
Our previous work showed that SMAD4, a central transcription factor in TGF-β signaling pathway, directly suppressed expression of angiopoietin-2 (Ang2), a pro-angiogenic ligand, in endothelial cells (ECs). Upon loss of endothelial Smad4, elevated ANG2 was associated with retinal AVM formation and other HHT associated defects, such as increased blood vessel diameters. Importantly, these retinal vascular phenotypes could be prevented and reversed by inhibiting ANG2 in a Smad4 retinal-based HHT mouse model 19. Since ALK1 and ENG co-receptors in the TGFβ signaling pathway are located upstream of SMAD4 3, we hypothesized that ANG2 inhibition could be used as an anti-angiogenic therapeutic strategy to normalize HHT related brain vascular defects in the most common forms of HHT. Here, we utilized Alk1, Eng and Smad4 HHT mouse models, which account for over 90% of HHT patient-type pathogenic variants, to examine and characterize HHT-associated peripheral cerebrovascular defects. In addition, we observed elevated levels of antagonistic ANG2, and decreased expression of the angiopoietin receptor, Tyr kinase with Ig and EGF homology domains (TIE2/TEK), in the brain ECs of all HHT mouse models. Consistent with these results, in vitro analyses revealed a substantial reduction in the ANG-TEK vascular signaling pathway, indicating its potential role in HHT. Furthermore, comparative RNA-seq analyses revealed a common, but unique pro-angiogenic transcriptional program in brain ECs associated with HHT. Significantly, inhibition of ANG2 utilizing a monoclonal blocking antibody exhibited vascular benefits on all HHT mutant brains, but in different aspects and levels. Lastly, we provided evidence that ANG2 inhibition mainly functioned through the interference of angiogenesis and cell migration mediated processes.
Methods
The data that support the findings of this study, and those which are not publicly available, are available from the corresponding author upon reasonable request.
Mice and Treatments
All in vivo experiments were conducted in accordance with Tulane University’s Institutional Animal Care and Use Committee policy. Smad4f/f 45, Alk1f/f 46, and Engf/f 31,47 mice were crossed with Cdh5-CreERT2 48 mice to generate endothelial cell specific, tamoxifen inducible knockout mice (Smad4iECKO, Alk1iECKO, and EngiECKO). To induce gene deletion, tamoxifen (Sigma, T5648–5G) was orally fed to neonatal pups at designed postnatal days (P) based on previous studies29,31,32. 300 μg of tamoxifen was delivered orally at P1, P2, and P3 (in total 900 μg) for Smad4 and Eng mouse models, while 100 μg of tamoxifen was given at P4 for the Alk1 mouse model. 30 μg of anti-ANG2 monoclonal blocking antibody (Roche, LC10) or IgG control were administered intraperitoneally (i.p.) into P2 and/or P4 pups 19. For survival studies, Ang2 loxP mice (Ang2f/f49,50) were used to incorporate an Ang2 floxed allele into the Smad4-HHT background and subsequently produce Ang2f/WT;Smad4f/f control and Ang2iECKO/WT;Smad4iECKO mutant mice. The sex of the pups was not distinguished in these studies, as HHT affects males and females equally.
Blue Latex Dye Perfusion
After anesthetizing P6/P7 pups on ice, abdominal and thoracic cavities were opened to expose the hearts and 400 ml of blue latex dye (Connecticut Valley Biological Supply Co., BR80B) was injected into left ventricle using a 30-gauge, 1 ml syringe. Latex perfused brains were fixed in 4% paraformaldehyde (PFA) overnight, followed by washing with PBS. After removing the skulls, brains were removed and washed in PBS with 0.01% sodium azide (NaN3) and stored at 4°C. Brains were positioned in PBS and imaged using a Leica M205 FA stereomicroscope. Analysis was done using Fiji software.
Brain Endothelial Cell Isolation
Whole brain tissue was digested using a Neural Tissue Dissociation Kit (P) (Milteni, 130–092-628) following the manufacture instructions. Brain endothelial cells (ECs) were purified using dynabeads (Invitrogen 11035) coated with platelet endothelial cell adhesion molecule (PECAM) antibody (BD, 553370), followed by immediate RNA or protein extraction. RNA was extracted using Thermo Fisher GeneJET RNA Purification Kit (Thermo, K0732). Protein was collected using RIPA buffer (Thermo, 89900) supplemented with 100x protease inhibitor cocktail (Thermo, 78430) and 10x phosphatase inhibitor (Roche, 04906845001). We note that we routinely assess the purity of the brain EC isolation in comparison to the non-EC population via qPCR and western blot analyses.
RNA Sequencing and Analysis
As previously detailed 19, total RNA quality was determined using Bioanalyzer RNA 6000 Nano Assay Kit (Agilent, 5067–1511). 0.5 μg RNA per sample was used for RNA library construction with a TruSeq RNA Library Prep Kit v2 (Illumina, RS-122–2001); independent biological samples from HHTf/f and HHTiECKO mice were used to create individual RNA-Seq libraries (three or five RNA-Seq libraries were constructed for each genotype or treatment group as noted in the respective graphs). The quality and quantity of mRNA library was determined by Qubit dsDNA High Sensitivity Assay Kit (Thermo, Q32851) and Bioanalyzer DNA1000 assay kit (Agilent, 5067–1505), respectively. The libraries were sequenced using the NextSeq 500/550 High Output Kit v2.5 (Illumina, 20024907) on a NextSeq 550 System (Illumina, SY-415–1002). Obtained data was analyzed using Illumina Basespace tools, including RNA-seq Alignment (version 2.0.1) and RNA-seq Differential Expression (version 1.0.1), which utilizes DESeq2. Gene expression was normalized in DESeq2 using median of ratios (counts divided by sample-specific size factors determined by median ratio of gene counts relative to geometric mean per gene). Venn diagrams were generated using InteractiVenn 51. Gene Ontology (GO) analysis was performed on Web Gestalt 52. After organizing original gene counts data, heatmaps were generated using Heatmapper 53; each column in the heatmap represents an independent biological sample for the corresponding genotypes and treatments noted. RNA-Seq data were deposited in the National Center for Biotechnology Information’s Gene Expression Omnibus (GSE197105):
To review GEO accession GSE197105: Go to https://nam11.safelinks.protection.outlook.com/?url=https%3A%2F%2Fwww.ncbi.nlm.nih.gov%2Fgeo%2Fquery%2Facc.cgi%3Facc%3DGSE197105&data=04%7C01%7Cxzhou15%40tulane.edu%7Cbf261b37cba74cf21bde08d9f622f474%7C9de9818325d94b139fc34de5489c1f3b%7C0%7C0%7C637811452256607517%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C3000&sdata=%2FKYT48YZsWXCTuNE126%2BhMTyZAOlSYO0eAMGE0vqdtE%3D&reserved=0.
Quantitative Polymerase Chain Reaction (qPCR)
Total RNA concentration was quantified using a Nanodrop (Thermo). 300 ng RNA was transcribed into cDNA using iScript Reverse Transcription Supermix kit (Biorad, 1708840). qPCR was run using PerfeCTa SYBR Green SuperMix (Quantabio, 95071) on CFX96 system (Biorad). Three technical replicates were included for each sample and one average was plot in the final figure. ΔΔCt method was used to determine relative gene expression. The used primers were listed below: ornithine decarboxylase (Odc) Fwd 5′-ACCGTGCTGTGAGTGTTTCC-3′; Odc Rev 5′-TGTGGCAGTCAAACTCGTCC-3′; Smad4 Fwd 5′-GCCCCATCCTGGACATTACT-3′; Smad4 Rev 5′-CCCGAAGGATCCACATAGCC-3′; Alk1 Fwd 5′-GGGCCTTTTGATGCTGTCG-3′; Alk1 Rev 5′-TGGCAGAATGGTCTCTTGCAG-3′; Eng Fwd 5′- TTCTCACACACGTGGCCC-3′; Eng Rev 5′- CCGATGCTGTGGTTGGTACT-3′; Ang2 Fwd 5′-TGACAGCCACGGTCAACAAC-3′; Ang2 Rev 5′-ACGGATAGCAACCGAGCTCTT-3′; Tek Fwd: 5′-GTAAA CAAGAGCGAGTGGACC-3′; Tek Rev: 5′-CCATGGCGCCTTCTA CTACT-3′ 19.
Tissue Collection and Processing
For brain collections, P6/P7 pups were anesthetized on ice, abdominal and thoracic cavities were opened to expose the hearts and followed by systemic perfusion with 10 ml ice-cold PBS and 10 ml ice-cold PFA. Brains were fixed in 4% PFA for 24 hours. After removing the skulls, brains were removed and stored in PBS with 0.01% NaN3 at 4°C. Brain sections were obtained by embedding in 4% agarose in PBS and sectioning coronally at 50 μm on a vibrating microtome (Leica VT1000S). All brain slices were collected serially and stored in 0.01% NaN3 at 4°C. In situ hybridization was performed within 4 weeks, while immunofluorescent staining was performed within 6 months after collection. Retinas were collected as previously detailed 54. First, eyeballs were removed and fixed in 4% PFA for 1 hour at 4°C. After washing eyeballs in PBS, retinas were dissected out, followed by immediate immunofluorescent staining protocol or serial dehydrating to store in 75% ethanol at −20°C. For in situ hybridization, retinas were re-fixed in 4% PFA overnight at 4°C before being serial dehydrated into 75% ethanol and stored at −20°C.
In situ hybridization
Brain slices were rinsed with PBS, mounted on glass slides and air dried at 4°C overnight. Samples were permeabilized using 10 μg/ml proteinase K for 10 min at RT, rinsed with PBS quickly, refixed in 4% PFA for 5 min at RT. After rinsing with PBS, samples were incubated in appropriate digoxigenin-labeled probe/hybridization mixture overnight at 65°C. Samples were washed in a series of SSC, followed by incubation with the Anti-digoxigenin-AP Fab fragments antibody (Roche, 11093274910) in blocking reagent (Roche, 11096176001). Samples were then washed with a maleic acid buffer solution, placed in BM purple (Roche, 11442074001) for colorimetric reaction, and stopped by fixation in 4% PFA for 1 hour at RT. After washing in PBS, brain slices were mounted by Permount (Fisher chemical, SP15–100). For retinas, in situ hybridization experiments were performed as previously described 30. After color metric reaction, retinas were bleached to remove remaining tissue pigmentation and mounted on slides using the ProLong™ Diamond Antifade Mountant (Fisher scientific, P36961). All images were taken by Leica M205 FA stereomicroscope. Anti-sense RNA in situ hybridization probes were generated as previously reported: Ang2 and Tek 19; Apln 29.
Immunofluorescent Staining
Brain slices were rinsed with PBS, mounted on glass slides and air dried at 4°C overnight. Samples were permeabilized in 1% Triton X in PBS for 30min, followed by blocking in CAS-block (Thermo, 88120) for 1 hour at RT. Brain slices were then incubated in primary antibodies diluted in CAS-block at 4°C overnight, followed by secondary antibodies for 2–3 hours at RT. Whole mount retinas were permeabilized in 1% Triton X in PBS for 30 min, followed by blocking in CAS-block (Thermo, 88120) for 30min at RT. Retinas were then incubated in primary antibodies diluted in CAS-block at 4°C overnight, followed by secondary antibodies for 4 hours at RT. Brain slices and retinas were mounted using the ProLong™ Diamond Antifade Mountant (Fisher scientific, P36961). Antibodies: IB4 (Thermo, I21411, 1:200), FOXO1 (Cell signaling, 2880, 1:100), ESM1 (R&D, AF1999, 1:100).
Whole Brain Lysate Collection
Isolated brains were kept on ice while proceeding to immediate protein extraction or snap-frozen on dry ice and stored in −80°C for protein extraction later. Tissue Extraction Reagent I (Invitrogen, FNN0071) supplemented with 100x protease inhibitor cocktail (Thermo, 78430) and 10x phosphatase inhibitor (Roche, 04906845001) was pre-chilled on ice. Each brain was homogenized manually in 800 μl of ice-cold buffer with a grinder pestle on ice. The mixture of minced brains and buffer was incubated while rotating at 4°C for 10 min. Supernatant was collected and aliquoted after centrifuging the mixture for 20 min at 13,000g at 4°C.
Western Blot
Protein concentrations of iBECs, HUVECs, and whole brain lysates were determined using the Pierce™ Coomassie (Bradford) protein assay kit (Thermo, 23200). Laemmli buffer (Biorad, 1610737) supplemented with β-mercaptoethanol (Biorad, 1610710) was added to the protein samples, followed by boiling at 95°C for 8 min. Protein lysate was separated on 4–20% Mini-PROTEAN TGX Precast gels (Biorad, 4568094) and blotted onto 0.2 μm PVDF (Biorad, 1704156) or nitrocellulose (Thermo, 88018) membranes. Membranes were blocked for 30 min at RT with blocking buffer consisting of 5% BSA in TBST (0.1% Tween 20 in TBS) and incubated at 4°C overnight in primary antibodies diluted in the blocking buffer. Membranes were washed in TBST and incubated with appropriate secondary antibodies (LI-COR, IRDye, 1:5000; or Southern Biotech HRP-conjugated anti-mouse and anti-rabbit, 1:5000) for 1 hour at room temperature. Membranes were imaged using a LI-COR Odyssey imaging system or regular X-ray films for ECL. Bands were quantified using Image Studio or ImageJ softwares. The primary antibodies used were as follows: AKT (Cell Signaling Technology, #9272), pS473-AKT (Cell Signaling Technology, #4060), ANG2 (Santa Cruz Biotechnology, sc-74403, 1:250), ID1 (Biocheck, BCH-1/195–14-50, 1:1000), TEK (R&D, AF762, 1:500), pY992-TEK (R&D, AF2720, 1:500), ALK1 (Abcam, ab263902, 1:1000), SMAD4 (Abcam, ab40759, 1:500), β-ACTIN (Cell Signaling Technology, 3700, 1:5000).
HUVECs and Stimulation Conditions
HUVECs were isolated from umbilical veins obtained from anonymous donors and subcultured in 5% fetal bovine serum (FBS)-containing EC growth medium (ScienCell), as described before55. Cells were treated as described in the Figure’s legend with ALK1-Fc (R&D, 370-AL-100) and rANG1 (R&D, 923-AN-025). 5–20 μg of protein cell extracts were analyzed by western blot as described above. For the secretion assays, HUVECs were treated as described in the Figure 4 legend, media was then replaced with 0.05% serum medium for 3 hours and then collected for ELISA quantification.
Figure 4. Disrupted ALK1 signaling leads to increased production of ANG2 and a reduction of ANG-TEK signaling in vitro.
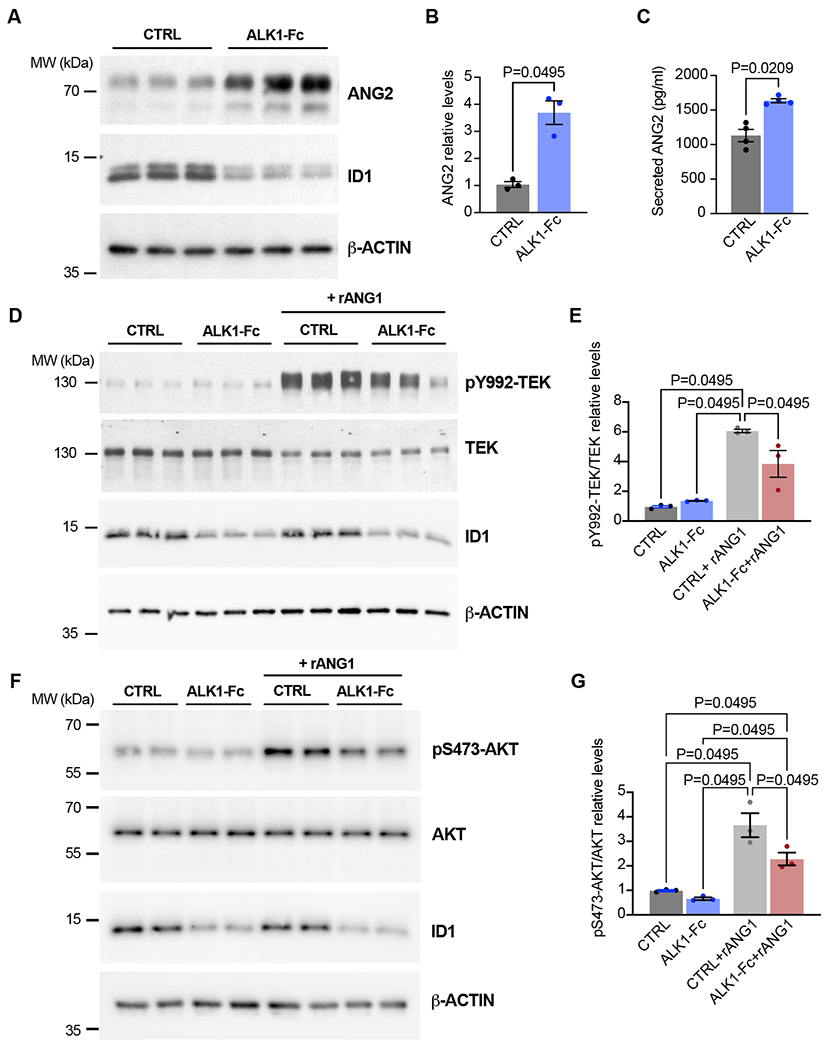
(A-G) Human umbilical vein ECs (HUVECs) were treated or not for 24 hours with ALK1-Fc (1 ug/ml) in complete medium (conditioned for 1–2 days) in the absence or presence of rANG1 (300 ng/ml) for 24 hours. Cell extracts were analyzed by western blot using antibodies directed against the indicated proteins (A,B, D-G) and ANG2 levels from cultured media were analyzed by ELISA (C). β-ACTIN served as a protein loading control. Shown are representative western blots of 2–3 independent experiments. Densitometric analyses and quantification of ANG2 (B), pY992-TEK/TEK (E), and pS473-AKT/AKT (G) relative levels are shown. Colorimetric analyses and quantification of ANG2 levels are shown (C). n = 3–4 independent measurements in B, C, E, and GA-G. All values are mean ± SEM. Non-parametric, Wilcoxon rank sum test.
Enzyme-linked Immunosorbent Assay (ELISA)
Whole brain lysate concentration was determined using Qubit Protein Assay Kit (Thermo, Q33211). Preliminary experiments were conducted to determine the appropriate amount of protein loaded, ensuring the targeted protein concentrations would fall near the middle of the linear range in the standard curve. Equal amount of total protein for all samples was utilized for detecting ANG2 and TEK protein levels. For the measurements of secreted ANG2 from HUVECs, conditioned media, prepared as described above, were spun down at 2000xg for 10 min to remove particulates. Media were then assayed according to the manufacturer’s instructions (R&D Systems™ Human Angiopoietin-2 Quantikine ELISA Kit, #DANG20). Mouse Angiopoietin 2 ELISA Kit (Abcam, ab171335) and mouse Tie-2 Quantikine ELISA Kit (LSBio, LS-F5441) were used according to the manufacturers’ instructions. Duplicates were included for each sample as suggested and one average was plotted in the final figure.
Statistical Analysis
All statistical analyses and graphs were performed and generated using GraphPad Prism. Bar graphs with individual values and mean± standard deviation, shown as error bars, were presented for all quantified data. Sample size (n) indicated the number of independent biological samples. Non-parametric, Wilcoxon rank sum test were used for statistical analysis via SAS 9.4 software. A p-value < 0.05 was considered statistically significant.
Results
Loss of endothelial Smad4, Alk1, or Eng leads to shared and unique peripheral cerebrovascular defects and cerebral AVM formation.
In an effort to characterize the cerebrovascular defects among the common forms of HHT, we generated inducible endothelial cell-specific knockout mice (iECKO) for Alk1, Eng and Smad4 by crossing floxed HHT-allelic mice (Alk1f/f, Engf/f and Smad4f/f) with a common Cdh5-CreERT2 driver (Alk1f/f;Cdh5-CreERT2, Engf/f;Cdh5-CreERT2 and Smad4f/f;Cdh5-CreERT2: referred throughout as Alk1iECKO, EngiECKO and Smad4iECKO respectively), as previously reported 19,21,22,29,31,32,56. To induce gene deletion, 300 μg of tamoxifen was fed orally for 3 consecutive days beginning at postnatal day 1 (P1) for Eng and Smad4 HHT mouse models, and brains were collected at P7 (Fig 1A), when retinal AVMs and other vascular defects have been reported to be robust 19,29,31,56. Previous experiments have shown that Alk1iECKO pups begin to perish 48 hours post-tamoxifen treatment 21,32. To account for this limiting factor, we employed a previously described tamoxifen-induction strategy that would allow vascular phenotypes to develop within a constrained timeframe yet would be comparable to the analyzation time points of the Eng and Smad4 HHT models 19,21,29,31,32. Accordingly, to produce the Alk1 HHT mouse model, 100 μg of tamoxifen was administered at P4 and brain samples were collected at P6 (Fig 1A). Littermate Alk1f/f, Engf/f and Smad4f/f pups treated with tamoxifen served as corresponding controls.
Figure 1. Cerebrovascular defects in HHT mouse models.
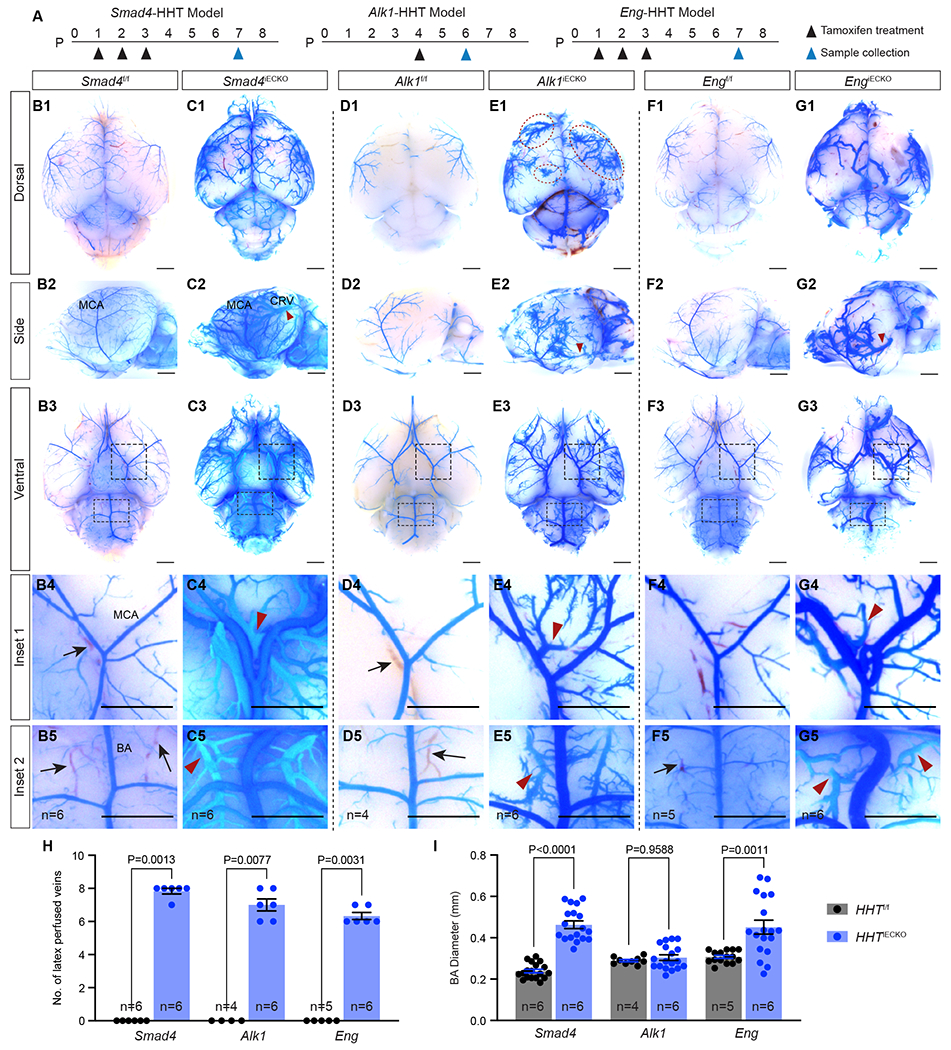
(A) Schematic of tamoxifen treatment and sample collection time points for postnatal (P) endothelial cell deletion of Smad4, Alk1 or Eng in each HHT mouse model. (B-G) Representative bright field images of blue latex dye perfused HHTf/f control and HHTiECKO mutant brains. B-G; 1–3 show dorsal, lateral, and ventral views. B-G; 4–5 are insets showing areas near the middle cerebral artery (MCA) and basilar artery (BA) from ventral views in B-G; 3. Red arrowheads point to blue latex dye perfused veins, which are absent in control littermates. Blood filled veins lacking dye (black arrows) are readily observed in control brains but not in HHTiECKO mice. Red dotted circles, bushy-like vascular structures. All scale bars are 3 mm. (H) Quantification of the number (No.) of latex perfused veins, including the veins on both sides of the MCA, the two veins parallel to the BA, caudal rhinal vein (CRV) and rostral rhinal vein on both sides in the lateral views (8 veins in total). (I) Quantification of BA diameter. n = the biological sample number. All values are mean ± SEM. Non-parametric, Wilcoxon rank sum test.
In order to visualize the brain vasculature of the HHT models, blue latex dye perfusion experiments were performed. Typically, blue latex dye injected into the left ventricle travels throughout the arterial system but is unable to pass through the capillaries and subsequent venous system due to the size of the latex dye particles 21,42,56. This arterial patterning of the dye was evident, and similar, in the peripheral brain vasculature between all control HHT mice (Fig 1B,D,F). Conversely, dye-filled blood vessels of all three HHT models (collectively referred to as HHTiECKO) were substantially different compared to controls, exhibiting both common and unique cerebral vascular defects (Fig 1C,E,G). First, as a proxy for AVMs, we screened for the presence of dye-filled veins on the surface of the brain; enlarged capillary vessels that constitute AVMs allow blue latex dye to populate the venous vessels. While dye-filled veins were absent in controls, we consistently found a significant number of latex dye perfused veins in HHTiECKO brains (Fig 1B–H, red arrowheads), indicating the presence of cerebral AVMs. In particular, dye perfused veins were evident adjacent to the middle cerebral (MCA) and basilar arteries (BA) located on the ventral brain surface (Fig 1C4–5, E4–5, G4–5). Moreover, lateral views of HHTiECKO brains showed consistent AVM-like structures whereby a large artery and vein, surrounded by intertwining branches from both vessels, appeared to come in close contact and overlap (Fig 1C2, E2, G2). In addition, the overall caliber of the arterial diameters was substantially greater. We found that the BA was significantly enlarged in Smad4iECKO and EngiECKO brains compared to floxed controls, but not in Alk1iECKO brains (Fig 1B5–G5,I). These results are consistent with previous work showing blood vessel enlargement in various vascular beds of Eng and Smad4 HHT mouse models, but not being an overt phenotype observed in Alk1 deficient mice 19,21,29,32,42,56. Incidentally, this suggested that enlarged vessel diameters could be a secondary consequence of increased blood flow, which may not have fully advanced in Alk1 HHT mice due to their short life span.
In terms of unique features, Smad4iECKO brains regularly had a greater abundance of dye-filled vessels, which also covered more of the brain compared to the Alk1 and Eng mutant models (Fig 1C). In Alk1iECKO brains, we noticed distinct bushy-like vessels that were often associated with veins and seldom seen in the brains of EngiECKO and Smad4iECKO pups (Fig 1E1, red dotted circles). These seemingly hyperplastic structures resembled the strong hypervascular phenotypes displayed in the retina of Alk1iECKO neonate pups, which were also notably more pronounced when compared to the Eng and Smad4 retinal HHT models 19,21,29,32,42,56. Although EngiECKO brains displayed notably enlarged vessels and AVM-like structures, we consistently observed fewer dye-filled vessels overall when compared to the other models, especially on the dorsal side of the brains suggesting a lower overall penetrance of the dye (Fig 1G). All together, these results showed that structures resembling AVMs reliably developed in Alk1iECKO, EngiECKO and Smad4iECKO mice and are therefore useful models to study peripheral brain vasculature pathologies associated with HHT. Moreover, HHT model-specific vascular abnormalities, such as enlarged vessels in EngiECKO and Smad4iECKO mice and bushy-like vessels in the Alk1iECKO mice, were present and could be used as distinguishable characteristics.
Transcriptional profiles of brain ECs revealed a unique angiogenic program associated with all three HHT models.
To gain comprehensive insight into the transcriptional changes associated with HHT, we performed RNA-sequencing (RNA-seq) on isolated brain ECs (iBECs) for all three HHT mouse models and corresponding controls. We found 5509, 5381, and 2663 differential expressed genes (DEGs) in Smad4iECKO, Alk1iECKO, and EngiECKO iBECs, respectively (Fig 2A). A comparison of these data sets showed that 1,232 DEGs overlapped between the HHTiECKO samples (Fig 2A) with 688 upregulated and 441 downregulated DEGs shared among the different models (Sup Fig 1A,C). Next, to identify those common DEGs that could be directly regulated at the SMAD4-transcriptional intersection of the HHT pathway, we incorporated SMAD4 chromatin immunoprecipitation sequencing (ChIP-seq) data obtained from a mouse EC line (Ms1) stimulated with BMP9 and 10 that we previously published 19. Integration of SMAD4 ChIP-seq data revealed 193 common DEGs (Fig 2B), which included 118 upregulated and 49 downregulated DEGs (Sup Fig 1B,D). In addition, Gene Ontology (GO) analyses of the RNA-seq data sets revealed biological processes that were impacted by the loss of Alk1, Eng or Smad4 (Fig 2C–E). Interestingly, of the top ten represented GO terms, angiogenesis was the only one shared amongst the three groups. Furthermore, GO analysis of the 193 shared DEGs (Fig 2B), which included the SMAD4 ChIP-seq data, indicated angiogenesis as one of the top significant GO terms (Fig 2F, Sup Fig 1F). The GO results are noteworthy because HHT is thought to arise via an abnormal angiogenesis process. However, the angiogenic signature of HHT is still unclear, even though it likely holds important contextual clues for understanding the pathogenic process of this disease. With this gap of knowledge in mind, heatmaps using the gene list from the angiogenesis GO category in Fig 2F were generated for each HHT model (Fig 2G). Among the 23 angiogenic genes, 14 were consistently upregulated in HHTiECKO iBECs, while 5 were consistently downregulated. Moreover, these angiogenesis related genes were located in a strikingly similar pattern in scatter plots of the RNA-seq data when compared between the HHTiECKO models (Fig 2H–J). Among the upregulated genes, a number have well established pro-angiogenic roles in angiogenesis, including Ang2, apelin (Apln), collagen type IV alpha 1 chain (Col4a1) and endothelial cell specific molecule 1 (Esm1) 33,57–63. Similarly, reduced Tek expression is expected to lead to an active, angiogenic vascular network 60, while loss of Sox17 results in loss of arterial identity, acquisition of venous characteristics and hypersprouting 64. Taken together, these comprehensive transcriptional studies uncovered commonalities in gene expression changes within HHTiECKO iBECs and implicated angiogenesis as an important process in brain related HHT pathogenesis. Our results also indicated a more refined and unique pro-angiogenic profile linked to HHT, as defined by the common angiogenic genes identified.
Figure 2. Transcriptional profiles of brain ECs isolated from HHT models.
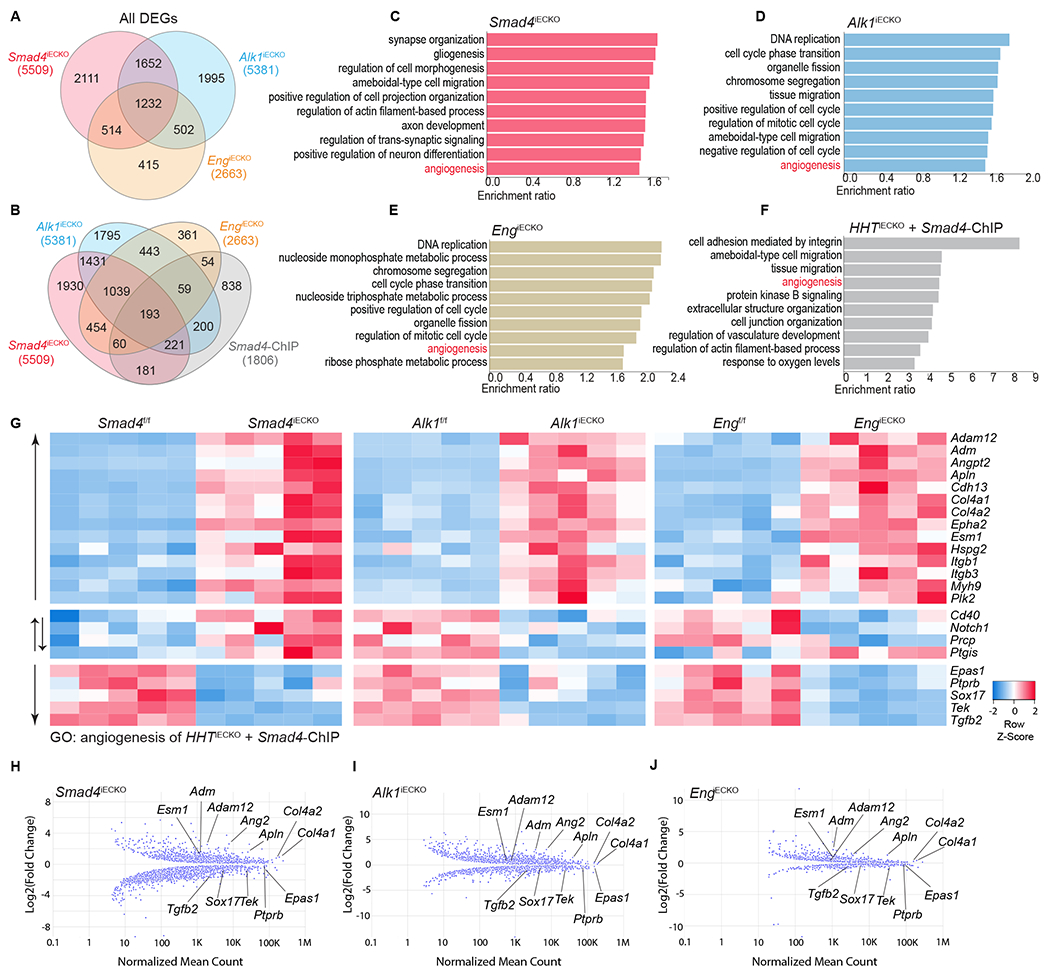
(A) Venn diagram showing the overlap of differential expressed genes (DEGs) identified from RNA-sequencing experiments utilizing iBECs from all three HHT models. (B) Venn diagram showing overlap between DEGs and Smad4 ChIP-seq data, previously obtained in BMP9/10 stimulated Ms1 cells 19. (C-E) Top 10 gene ontology (GO) terms in each HHTiECKO DEGs data set; false discovery rate (FDR)<0.05 applies to all GO terms. (F) Top 10 GO terms among the 193 overlapping genes from all three HHTiECKO DEGs and Smad4 ChIP seq data sets; FDR<0.05 applies to all GO terms. (G) Heatmap depicting the DEGs belonging to angiogenesis GO term in F. n = 5 (each column represents an independent biological sample) (H-J) Graphical dot plot of Log2 fold changes in DEGs obtained from HHTiECKO iBECs, as compared to respective HHTf/f controls. Multiple representative angiogenic genes from G are highlighted and show a similar pattern between the different HHT models.
Dysregulation in expression of ANG2 and TEK signaling components is conserved in HHTiECKO brains
The ANG-TEK signaling pathway is intimately involved in controlling vascular related processes, including vascular permeability, inflammation, and pathological angiogenic responses 60. ANG2 is expressed during vascular remodeling and classically acts as an antagonist of ANG1 for the TEK receptor in a context-dependent manner. We previously showed that SMAD4 transcriptionally repressed the critical pro-angiogenic factor Ang2 in ECs and that ANG2 was robustly upregulated in the absence of SMAD4, while TEK showed diminished expression overall. By inhibiting ANG2, we successfully rescued AVMs and normalized the vasculature in Smad4iECKO retinas 19. In the context of understanding whether similar approaches could directly affect the overall effectiveness of targeting this pathway therapeutically in the brain, it was crucial to verify that upregulated ANG2 and downregulated TEK were a consistent occurrence in the cerebral vascular networks of all three HHT mouse models.
To validate our RNA-seq results, we conducted qPCR analyses on RNA extracted from iBECs. As anticipated, elevated Ang2 and reduced Tek transcript levels were confirmed upon endothelial deficiency of Smad4, Alk1, or Eng (Fig 3A–C). Furthermore, ELISA methods on whole brain lysates revealed that ANG2 and TEK protein levels were significantly altered in a manner comparable to the RNA quantifications in Smad4iECKO and Alk1iECKO mice, while EngiECKO mutants exhibited a trend in TEK reduction (Fig 3D–I). To visualize spatiotemporal expression of Ang2 transcripts, we performed in situ hybridization on brain slices from Smad4iECKO and EngiECKO P7 mice, Alk1iECKO P6 mice and equivalent stage-matched controls. In floxed-control brains, we found that Ang2 transcripts were primarily expressed in regions of the thalamus, with low level of diffuse expression in other areas of the brain, but not in an arrangement typical of the pervading blood vessels (Fig 3J–L). However, Ang2 mRNA was strongly elevated and ectopically expressed in a vascular-like pattern, especially in the cortex and hippocampus regions of HHTiECKO brains (Fig 3M–O). Ang2 transcripts were also retained in the thalamus region of the HHT models, similar to controls brains. As an additional verification to our RNA-seq data (Fig 2G), examination of Apln mRNA via in situ hybridization showed that transcripts are markedly induced throughout the vasculature of HHTiECKO brains compared to HHT controls, which exhibited lower levels of Apln expression throughout the cerebral blood vessels (Sup Fig 2A). Our prior work utilizing P7 Smad4iECKO retinas revealed that Ang2 transcripts, which are normally confined to the active tip cells of the growing vasculature, were considerably elevated and expressed in both the tip and trailing stalk cells at the vascular front 19. Likewise, in P6 Alk1iECKO and P7 EngiECKO retinas, we observed a noticeable increase of Ang2 RNA transcripts in both the tip and stalk cells (Sup Fig 2B). We also found that the pro-angiogenic factors, Apln (mRNA level) and ESM1 (protein level), which are largely expressed in the vascular front ECs of the retina, appeared to be upregulated and their expression expanded into the non-tip cell vasculature of all HHTiECKO retinas (Sup Fig 2C–D, white arrows). In summary, our expression analyses demonstrated that elevations in ANG2 and reductions in TEK levels are a conserved feature in the brain ECs of all three HHT models. Furthermore, upregulated and ectopic expression of typical endothelial tip cell markers implicated an abnormal, pro-angiogenic environment within the HHT vasculature.
Figure 3. Elevation in expression of ANG2, and reduction of TEK, is consistent across HHT mutant brains.
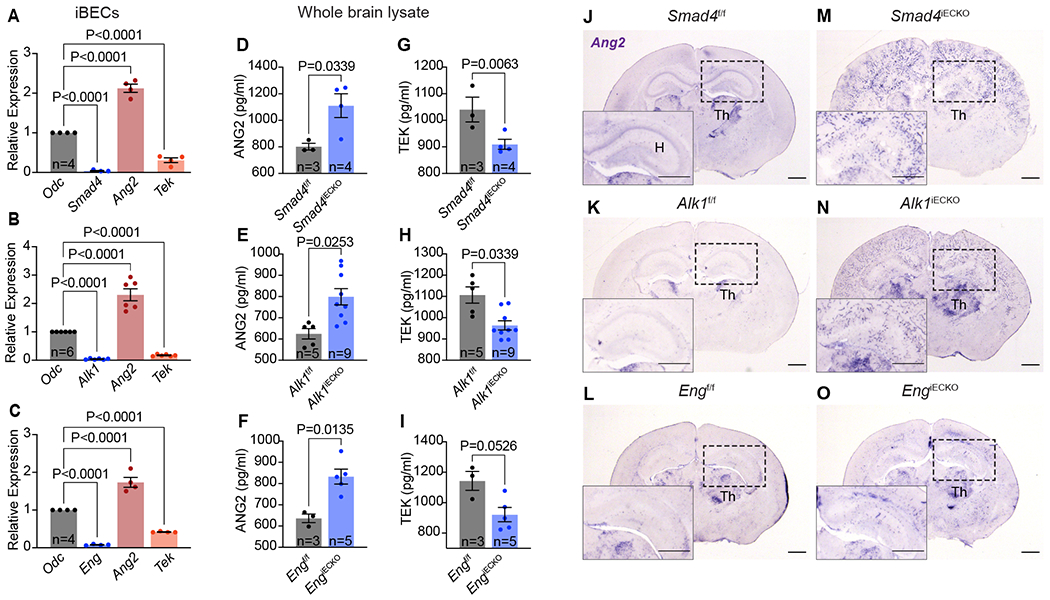
(A-C) qPCR analyses for the specified genes using iBECs from each HHT model (A, Smad4iECKO; B, Alk1iECKO; C, EngiECKO). ornithine decarboxylase (Odc) served as a housekeeping reference mRNA. (D-I) ELISA quantifications of ANG2 and TEK protein concentrations in whole brain lysates of HHTf/f and HHTiECKO mice. (J-O) In situ hybridization analyses of Ang2 mRNA in HHTf/f and HHTiECKO brain slices. Insets show a region of the hippocampus (H). Notice retained expression of Ang2 transcripts in a region of the thalamus (TH), while Ang2 mRNA is detected robustly and ectopically in blood vessels of HHTiECKO brains only. All scale bars, 3 mm. n = biological sample number. All values are mean ± SEM. Non-parametric, Wilcoxon rank sum test.
Loss of Alk1 leads to a reduction in ANG-TEK signaling in vitro.
ANG2 acts as a context-dependent modulator of TEK activity during vessel remodeling 65. Concordant data have now shown that ANG2 is overexpressed in pro-angiogenic pathological settings and that it acts as a TEK antagonist 60,66. Based on our collective data, we expected this same relationship to exist in a HHT background, but this has remained unclear. To address TEK signaling activities in a similar context in vitro, we first confirmed in human umbilical vein ECs (HUVECs) that ALK1 inhibition, via treatment with ALK1-Fc (a soluble form of ALK1 that binds to BMP9 and 10), led to a significant and robust increase in ANG2 protein levels (Fig 4A,B), as we have previously observed 23,33. Likewise, inhibitor of DNA binding 1 (ID1), a direct downstream target of SMAD4, was reduced in the presence of ALK1-Fc, as previously reported 23,24,33; RNA-seq analyses on the iBECs also showed that Id1 was significantly reduced in all three models of HHT (data not shown). In addition, ANG2 was detected at significantly higher levels in cultured media from HUVEC treated with ALK1-Fc compared to those without, demonstrating that ANG2 secretion is also elevated in a reduced ALK1 signaling background (Fig 4C). Knowing that ANG2 elevation is expected to impact TEK activity, we next evaluated if ALK1 inhibition could interfere with TEK signaling capacity. TEK phosphorylation (pTEK), upon binding to its agonist ligand ANG1, propagates signaling to activate various downstream effectors, such as the TEK-(PI3K) target, protein kinase B (AKT) 67. Therefore, recombinant human ANG1 was used to elicit TEK phosphorylation. Indeed, ANG1 treated HUVECs exhibited a robust increase in pTEK (Fig 4D,E). However, pTEK was appreciably diminished when HUVECs were treated with ALK1-Fc (Fig 4D,E) suggesting that the robust increases in ANG2 levels, via ALK1 inhibition, were able to interfere with ANG1-mediated TEK activation. As an additional control of TEK activity, we found that ANG1 treatment was able to substantially increase the phosphorylation status of AKT (pAKT) in control HUVECs (Fig4 F,G). However, treatment with ALK1-Fc resulted in a notable reduction in pAKT further signifying an overall loss of TEK signaling. Thus, TEK activity, in the context of an HHT background, is significantly downregulated in vitro .
Pharmacological inhibition of ANG2 improved HHTiECKO cerebrovascular defects.
The conserved elevations of ANG2 in ECs of the HHTiECKO models suggested that ANG2-targeted therapies could have a positive impact on vascular pathologies associated with all three major types of HHT. To test this approach, we administered the HHT and matching control mice with an efficacious ANG2 monoclonal blocking antibody (referred to as LC10; Roche) 19,68,69 or negative IgG control (Roche) via intraperitoneal injection (i.p.). Using the same tamoxifen-induction parameters described for each model in Figure 1, 30μg of LC10 or IgG were injected at various time points (Fig 5A) to determine if blocking ANG2 function could prevent the peripheral cerebrovascular phenotypes initially characterized (Fig 1). Blue latex dye perfusion experiments were conducted to visualize the brain peripheral vasculature.
Figure 5. ANG2 inhibition improves HHT associated cerebrovascular defects.
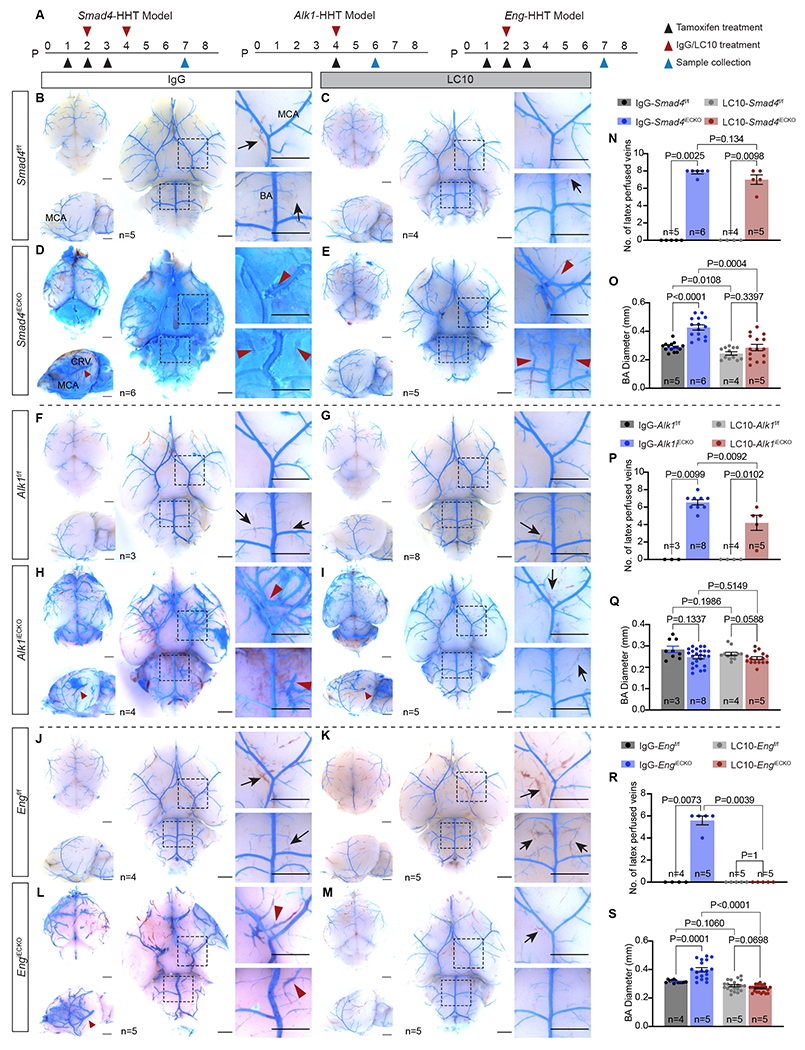
(A) Schematic of tamoxifen, IgG and LC10 treatment plans and sample collection times for each HHT model. P, postnatal day. (B-M) Representative bright field images of blue latex dye perfused brains. Red arrowheads, dye-filled veins; black arrows, blood-filled veins; MCA, middle cerebral artery; BA, basilar artery; CRV, caudal rhinal vein. All scale bars are 3mm. (N-S) Quantification of the number (No.) of latex perfused veins (8 veins in total were assessed) and basilar artery diameters. N = biological sample number. All values are mean ± SEM. Non-parametric, Wilcoxon rank sum test.
In the Smad4-HHT model experiments, we observed no noticeable differences in the overall arterial patterning or morphology of Smad4f/f mice treated with IgG control or LC10 (Fig 5B–C) and there were no dye-filled veins (Fig 5B–C,N) or changes in BA diameter (Fig 5B–C,O). Thus, it appeared that the LC10 treatments alone did not have a negative effect on the neonate peripheral brain vasculature. However, Smad4iECKO IgG treated pups displayed severe vascular defects (Fig 5D), similar to the phenotypes observed in untreated Smad4iECKO brains (Fig 1B). Most noticeably, significant amounts of latex dye frequently leaked out of the blood vessels (4 of 5 brains exhibited dye leakage) and covered large areas of the brain indicating weak vessel integrity. Due to the leakage, in several instances, we had to carefully remove areas of the dye so that we could assess the presence of dye-filled veins and measure BA diameters. In both cases, we discovered a significant number of latex-filled veins (Fig 5D,N) and increased BA calibers (Fig 5D,O). We note that dye leakage also occurred in Smad4iECKO pups in the initial characterization studies at a slightly reduced rate (3 of 6 brains). Furthermore, Smad4iECKO experimental animals received two doses of ANG2 inhibitor or IgG (Fig 5A), as preliminary experiments indicated a more positive response required an additional dose of LC10 (data not shown). Utilizing two doses, LC10 treated Smad4iECKO pups displayed a vasculature more comparable to the IgG and LC10 treated Smad4f/f brains (Fig 5E). In addition, those LC10-Smad4iECKO brains that exhibited dye leakage were less severe in appearance with noticeably less amount of leaked dye compared to those that received IgG. On the other hand, we still observed a similar number of dye-filled veins compared to IgG-Smad4iECKO mice (Fig 5D–E,N) suggesting AVMs were still present. However, the lateral AVM-like structures were not as prominent and completely absent in 1 of the 5 Smad4iECKO brains (Fig 5E). It also appeared that the vein diameters in the LC10 treated Smad4iECKO brains were much smaller than those treated with IgG, but this was not readily quantifiable because of the dye leakage throughout the brains of IgG-Smad4iECKO mice. However, if we compared the vein diameters of these LC10 treated Smad4iECKO mice and Smad4iECKO brains from our initial characterizations (Fig 1), vein calibers were significantly reduced in the presence of LC10 (Sup Fig 3). In terms of BA diameter, LC10 treatments successfully prevented arterial diameter increases, as they were similar in size to both IgG and LC10 treated Smad4f/f pups (Fig 5B–E,O). Overall, these results demonstrated that functional inhibition of ANG2 can promote a distinctly improved brain vasculature in a Smad4-HHT background by reducing latex dye leakage and diameters in the venous network, while also completely limiting arterial vessel enlargements. We note that vessel diameter readouts, such as the BA measurements, were used as an alternative and additional criterion to assess phenotypes associated with HHT. AVMs and telangiectasias are characterized, in part, by vessel enlargement; however, our blue latex dye methodology did not allow us to compare capillary beds/vessels versus AVMs in control and HHTiECKO mice, respectively (latex dye does not enter the capillary system in control flox mice).
In regard to the Alk1-HHT experiments, the vascular parameters in both IgG and LC10 treated Alk1f/f mice were indistinguishable and appeared normal (Fig 5F–G,P). In contrast, Alk1iECKO pups given IgG exhibited an overall defective vasculature highlighted by venous vessels filled with latex dye (Fig 5H,P) and bushy-like growth on many vessels (Fig 5H), similar to untreated Alk1iECKO brains (Fig 1E). In contrast, the cerebrovasculature of Alk1iECKO mice treated with LC10 more closely resembled that of the Alk1f/f mice (Fig 5I). Significantly fewer dye perfused veins were observed compared to IgG treated Alk1iECKO mice signifying a reduced occurrence of brain AVMs (Fig 5H–I,P). Intriguingly, ANG2 inhibition had little to no effect on the formation of the hypervascular, bushy-like vessel structures, as they were also observed in LC10-Alk1iECKO brains (Fig 5I). Given that BA diameters remained normal sizes in Alk1iECKO brains (Fig 1E,I), it was not surprising that the BA diameters were similar among all experimental conditions (Fig 5F–I,Q). Thus, in the context of the Alk1-HHT model, administration of LC10 appeared to reduce overall AVM severity, as assessed by a substantial reduction in the number of dye-filled veins and allowed the blood vessel network to retain an overall morphology more characteristic of those observed in the various control brains.
Administration of LC10 had the most profound effects on the Eng-HHT model. As with the other models, treatment of Engf/f pups with IgG or LC10 had no noticeable impacts on the brain peripheral vasculature (Fig 1F and 5J–K), while IgG-EngiECKO brains displayed severe vascular phenotypes, including AVM-like structures, dye perfused veins, and greatly increased arterial calibers (Fig 5L). Strikingly, LC10 treatment was able to completely prevent these cerebrovascular defects in EngiECKO brains with one dose (Fig 5M). We did not find any latex dye-perfused veins, and the BA diameters in LC10 treated EngiECKO pups remained the same size as Engf/f mice treated with IgG or LC10 (Fig 5J–K,M,R,S). In addition, no AVM-like structures were found on the lateral sides of LC10-EngiECKO brains that were easily distinguished in the IgG treated EngiECKO brains (Fig 5L,M). Therefore, based on the criteria we used to analyze the brain vasculature, we concluded that inhibition of ANG2 completely blocked the formation of peripheral vascular defects associated with loss of Eng, which implicated a potentially strong beneficial impact on ENG-HHT pathologies.
A genetic link between the HHT and ANG-TEK signaling pathways
Several lines of evidence strongly supported the notion that the HHT and ANG-TEK signaling pathways are genetically linked. Our prior work showed that SMAD4 transcriptionally repressed Ang2 and that loss of Smad4 in the Smad4-HHT mouse model led to a robust increase in its expression 19. That work, along with these studies, also demonstrated that the various HHT backgrounds share similar changes in expression of ANG2 and TEK and that ANG2 inhibition substantially improved their HHT vascular pathologies (Fig 3,4) 19. To further explore this genetic connection, we performed survival studies on mice whereby a floxed Ang2 allele was incorporated into the Smad4-HHT model. Based upon our studies, we expected that inactivation of the Ang2 gene would prolong the survival of Smad4iECKO mice. We chose to perform these experiments in the Smad4-HHT background because of SMAD4’s direct transcriptional link to Ang2 and the severe vascular phenotypes exhibited in Smad4iECKO mice. In this survival study, Smad4f/f and Ang2f/wt;Smad4f/f (wt, wild type) mice treated with the same tamoxifen regimen (Fig 1A, Sup Fig 4A) served as controls. Accordingly, all these mice survived well into adulthood (Sup Fig 4B). In contrast, Smad4iECKO mice died between P8 and P10 (Sup Fig 4B), as previously reported 29,56. However, simultaneous inactivation of one allele of Ang2 in Ang2f/wt;Smad4f/f;Cdh5-CreERT2 mice (referred to as Ang2iECKO/wt;Smad4iECKO) was sufficient to increase survival rates, as these mice died between P8 to P18 with 50% of these mice perishing after P10 (Sup Fig 4B). Thus, genetic loss of Ang2 significantly extended the survival period for Smad4iECKO mice, further supporting a genetic link between the HHT and ANG-TEK pathways and the systemic benefits of blocking ANG2 function.
ANG2 inhibition corrected a gene expression signature in Smad4iECKO mice that implicated angiogenesis and cell migration processes.
To explore the transcriptional effects of ANG2 inhibition, we performed RNA-seq on iBECs from IgG and LC10 treated Smad4-HHT animal models and controls, similar to experiments in Fig 5A–E. First, we compared IgG treated Smad4f/f and Smad4iECKO iBECs to obtain a set of DEGs representing a base line of mis-regulated genes with endothelial Smad4 deficiency; 2636 DEGs were identified and referred to as DEGs set 1 (Fig 6A–B). Next, we compared IgG and LC10 treated Smad4iECKO iBECs to generate a list of genes that were influenced by blocking ANG2 activity in a Smad4 mutant background. This resulted in DEGs set 2, which was comprised of 317 genes (Fig 6A–B). By comparing the two DEG data sets, it was revealed that 189 genes were shared between the different experimental conditions analyzed (Fig 6B). In addition, GO analyses were conducted on each of the DEG sets 1 and 2, and on the 189 overlapping genes between DEG sets 1 and 2 (Sup Fig 5A–B and Fig 6C). In focusing on the genes shared between sets 1 and 2, we found that the top GO terms centered around 2 main biological processes: angiogenesis (FDR=3.8117e-8) and cell migration (FDR=3.6000e-11) (Fig 6C). This was also evident when we generated Directed Acyclic Graphs (DAG) utilizing the GO terms (Fig 6D and Sup Fig 5C,D). DAGs present GO terms as nodes and link those that are connected to a common biological process. The upper terms are more general (for example: locomotion and localization of cell), while the lower connected nodes show more process specificity (for example: cell migration). Together, GO and DAG analyses pointed to angiogenesis and cell migration as being the processes most impacted in Smad4 mutants upon functional inhibition of ANG2.
Figure 6. Functional inhibition of ANG2 corrects a gene signature in Smad4iECKO iBECs.
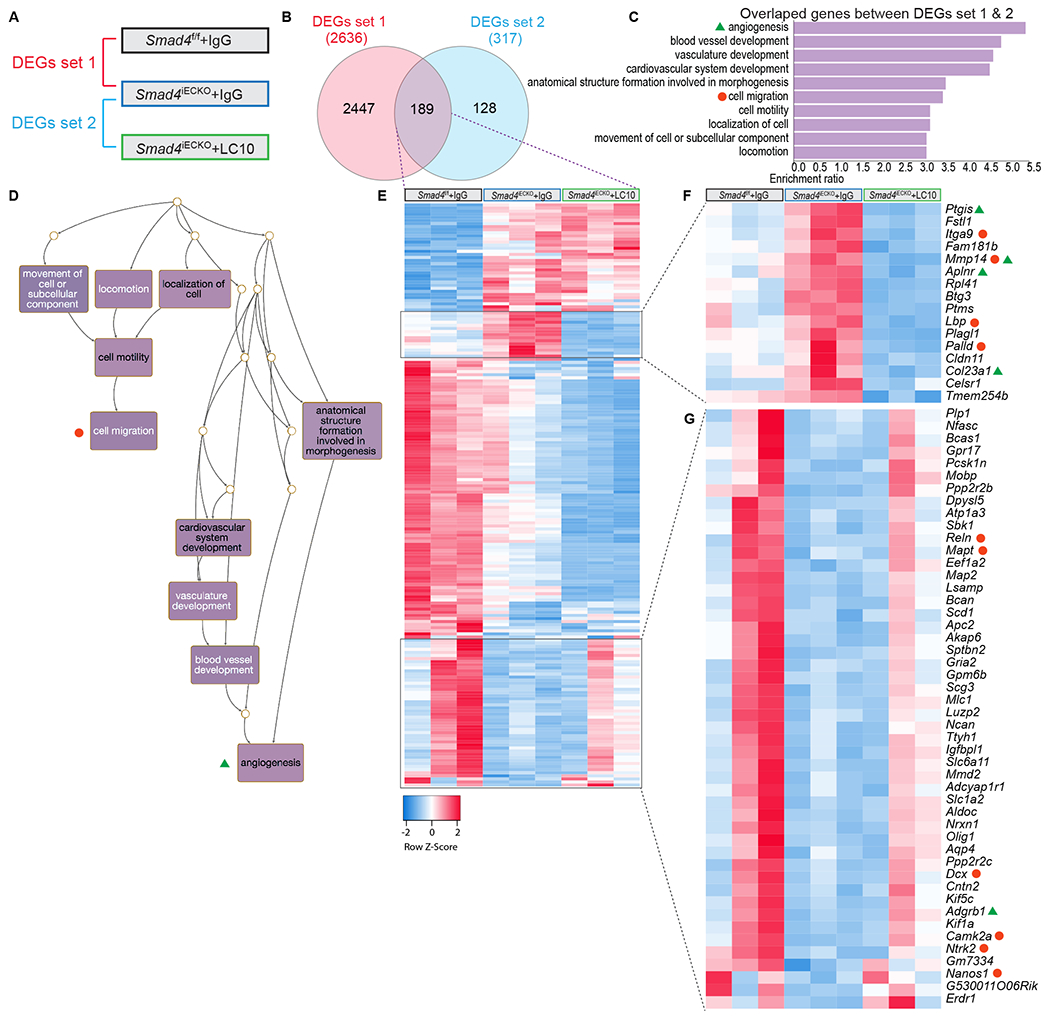
(A) Diagram depicting the comparison strategy to generate differentially expressed genes (DEGs) data sets from IgG and LC10 treated Smad4f/f and Smad4iECKO iBECs. (B) Venn diagram showing overlap between DEGs set 1 and 2. (C) Top 10 gene ontology (GO) terms among the overlapping genes between DEGs set 1 and 2. False discovery rate (FDR)<0.05 applies to all GO terms; angiogenesis (FDR=3.8117e-8) and cell migration (FDR=3.6000e-11). (D) Directed acyclic graph (DAG) for the top 10 GO terms in C underscores processes involved in angiogenesis and cell migration. (E) Heatmap depicting the 189 overlapping genes between DEGs set 1 and 2 in B. n = 3 (each column represents an independent biological sample) (F-G) Enlarged heatmaps of two gene clusters in E indicated a transcriptional correction of Smad4 mutant ECs in the presence of LC10. Genes labeled with green triangles and orange circles belonged to angiogenesis and cell migration GO terms, respectively.
To take a deeper look at the transcriptional signatures, we next generated a heatmap of the 189 common genes between DEGs set 1 and 2 (Fig 6E). We identified two gene clusters in which the expression profiles in LC10 treated Smad4iECKO iBECs more strongly resembled those observed in IgG-Smad4f/f mice (Fig 6F–G), as compared to IgG-Smad4iECKO iBECs (Fig 6E). These clusters were important because they represented a transcriptional signature of those genes potentially responsible for retaining a more normal peripheral brain vasculature in LC10 treated Smad4iECKO pups (Fig 5B–E,O). In particular, one gene cluster consisting of 15 genes showed a strong response to ANG2 inhibition (Fig 6F). This set of genes was highly elevated in Smad4iECKO iBECs, but in the presence of LC10, their expression was more comparable to IgG-Smad4f/f control levels and often even further reduced (Fig 6F). Moreover, when assessing what GO terms these genes belonged to, we found that Ptgis, Mmp14, Aplnr, and Col23a1 were categorized in the angiogenesis biological process, while Itga9, Mmp14, Lbp, and Palld were grouped into cell migration (Fig 6F). Additionally, several genes in the second gene cluster were linked to angiogenesis and cell migration processes (Fig 6G). Thus, our transcriptional studies demonstrated that ANG2 blockade via LC10 partially corrected a cerebral EC gene signature in Smad4iECKO mice that predominately involved angiogenesis and cell migration processes.
Discussion
There is a critical need to identify biological targets and therapeutic remedies for HHT patients, as a cure remains elusive, and no drugs have proven to be effective against AVMs. The findings of this study suggest that approaches targeting ANG2 may hold value for treating vascular pathologies associated with the various forms of HHT. The first suggestion of this was observed in our expression analyses demonstrating that robust and ectopic expression of ANG2 in the brain endothelium is shared among all three mouse models of HHT (Figs 2 and 3). In addition, we and other groups have detected increased levels of vascular Ang2 in different HHT mouse models. In a BMP9/10 immunoblocking mouse model of HHT, Ang2 mRNA was shown to be highly elevated in retinal ECs 33, while ANG2 protein was observed to be elevated in the lungs of Alk1+/− heterozygous mutant mice 70. Comparable results were observed in vitro where, similar to Figure 4, ANG2 was demonstrated to be increased in cultured ECs with blocked BMP9/10 signaling 23,33. Given that ANG2 is an extracellular ligand, it would also be expected that in an HHT setting ANG2 is secreted outside of the cell in excess. Accordingly, we found that loss of HHT signaling in HUVECs, via ALK1-Fc, resulted in increased secretion of ANG2 into the media (Fig 4C). Thus, multiple studies substantiate local increases of endothelial ANG2 expression in HHT animal models and in vitro backgrounds.
However, despite these observations, seemingly contradictory results have been observed in HHT patients and some animal models. Circulating plasma levels of ANG2 have been shown to be significantly decreased in ALK1-HHT2 patients, though ENG-HHT1 patients showed no statistical changes 71. Recently, combined measurements of circulating ANG2 from patients with the three major forms of HHT, although not statistically significant, trended towards a decrease in ANG2 levels 72. Furthermore, cultured blood outgrowth ECs from ENG-HHT1 and ALK1-HHT2 patients and several tissues from Eng+/− heterozygous mutant mice exhibited substantial decreases in ANG2 expression 70,73. Taking these discrepancies into consideration, several explanations could account for these observed variances. ANG2 is produced and secreted from ECs and acts via an autocrine mechanism, often in an antagonistic manner towards ANG1-mediated activation of TEK 74,75. It is possible that the upregulated levels of ANG2 are further participating in the autocrine loop and subsequently captured within the local HHT endothelial microenvironment, which is independent from systemic circulating levels of ANG2. Additionally, aside from Ang2 overexpression in the brain endothelium and potentially other vascular beds, it is also detected in various tissues and cell types, including the cerebral cortex, thalamus, smooth muscle cells, adipocytes, and fibroblasts (Figure J–O; proteinatlas.org). It is likely that different cell types exhibit regulations in ANG2 expression that differ from ECs and therefore, their contributions to the systemic circulation of ANG2 may surpass that of the endothelium.
The various studies referenced above also indicated differences in ANG2 expression depending on the type of HHT and tissue/organ assessed. For instance, levels of ANG2 skewed higher in brain and lung AVMs but were reduced in liver AVMs when combined HHT patient data was analyzed, though this was not statistically significant or separated between the types of HHT 72. When comparing ANG2 levels in heterozygous HHT models, pulmonary ANG2 was found to be unchanged in Eng+/− mice but increased in the lungs of Alk1+/− mice compared to corresponding controls 70. These studies are relevant because incidences of tissue/organ specific AVMs differ between the various types of HHT 3. It is tempting to speculate that variations in ANG2 expression may underlie these differences in prevalence. Moreover, it is unclear whether ANG2 could be involved in the formation of AVMs or telangiectasias in multiple organs and tissues. Based on our results and others, we suggest an inductive role for ANG2 in both brain and retinal AVMs. Incidentally, we have also observed robust increases of Ang2 mRNA in the lung ECs of Smad4-HHT mice (data not shown). Therefore, an assessment of the ANG2 levels in various organs and tissues of HHT patients and animal models (also anti-ANG2 treatments) could provide further clues linking ANG2 to tissue-specific AVM formation and HHT pathologies.
In terms of animal models, homozygous loss-of-function HHT mouse models, such as those used in this study, have played an important role in improving our molecular understanding of this disease. However, there are also limitations to these models. For instance, while consistent and robust formation of AVMs and other vascular pathologies has been an extremely useful resource, the acute manner of these phenotypes and short lifespan of these mice are somewhat at odds with the developing and progressive nature observed in HHT patients. Unfortunately, few mammalian models effectively capture the disease progression observed in humans 76. Therefore, additional HHT animal models that better mimic the conditions faced in medicine are needed. An emphasis on creating new HHT animal models with prolonged phenotypes—perhaps mouse models expressing HHT patient-based variants or organ-specific vascular deletion—would offer the ability to better assess the usefulness of various therapeutic measures on AVM regression and progression and related HHT vascular deficiencies.
In addition to our data implicating the ANG2-TEK pathway in HHT, our work is also consistent with other studies reporting that ANG2 acts in an antagonistic fashion to limit TEK receptor signaling in various pathological settings 66. Importantly, this same relationship was identified in human sporadic brain AVMs where elevated and decreased levels of ANG2 and TEK were observed, respectively 77,78. Recent investigations utilizing single cell RNA-seq methods have shown that Ang2 expression is dramatically increased in the ECs of various human cerebrovascular malformations, such as idiopathic brain AVMs, cavernomas, hemangioblastomas, meningiomas, and lung cancer brain metastasis 79,80. Plus, ANG2 elevations were detected in gain-of-function G protein subunit alpha q (GNAQ-R183Q) variant driven capillary malformations (CM) in Sturge-Weber syndrome brains 81. Therefore, increasing amounts of data indicate that ANG2 may be a pathogenic driver of multiple vascular malformations and pathologies and support the notion that ANG2 neutralizing strategies may have beneficial impacts on a broader scale.
In support of this idea, inhibition of ANG2 function using an anti-ANG2 monoclonal blocking antibody (LC10) improved the peripheral brain vasculature in mouse models of the three main forms of HHT, though at varying degrees (Fig 5). This included the ability to prevent blood vessel enlargements that are common in both Smad4 and Eng HHT animal models and characteristic of AVMs and telangiectasias in HHT patients. In addition, the formation of AVM-like structures and presence of dye-filled veins was substantially reduced in LC10 treated Alk1-HHT mice, while these malformations and dye perfused vessels were completely absent in Eng-HHT mice (Fig 5). We also previously showed that neutralization of ANG2 resulted in the prevention and alleviation of retinal AVMs and increased vessel diameters19. Furthermore, pharmacological blockade of ANG2 in a Pdcd10 mouse model of cerebral cavernous malformation (CCM) has been demonstrated to inhibit CCM lesion formation 82. Recently, a similar outcome was observed in a human xenograft model of CM; GNAQ-R183Q expressing ECs, which develop into CM-like enlarged blood vessels when implanted in mice, formed vessels with calibers similar to control ECs in the absence of Ang2 81. Taken together, these animal studies highlight the potential for ANG2 inhibitory-based therapeutics on human vascular diseases. Accordingly, several ANG2 blocking antibodies, in combination with VEGF-A inhibitors, are currently being tested in clinical trials to combat solid tumor formation, wet age-related macular degeneration (wAMD) and diabetic macular edema (DME) 66. Perhaps the results from these studies will offer new evidence and support for applying similar strategies to HHT patients.
Our studies also revealed a distinct but shared pro-angiogenic signature associated with the different HHT mouse models. HHT pathogenesis has been thought to be driven by abnormal angiogenic events, however, the molecular identity of this process has not been fully characterized. We found that in addition to elevated levels of ANG2, noted pro-angiogenic factors Apln and Esm1 were also part of this signature and highly upregulated in the brain and retinal ECs of all HHT mouse models (Fig 2; Sup Fig 2). These findings are similar to single cell expression studies in human brain ECs 80; analyses indicated that in contrast to human adult brain ECs, fetal and malformed brain ECs exhibit an active “angiogenic capillary” phenotype as defined by the induction of several pro-angiogenic markers, including Ang2, Apln and Esm1. In this context, multi-pronged approaches aimed at inhibiting these specific pro-angiogenic factors may have positive effects on AVMs and other vascular malformations. In further support of this strategy, it was recently demonstrated that pharmacological antagonism of APLN in a recombination signal-binding protein for immuno-globulin Jκ regions (Rbpj) mediated mouse model of brain AVMs led to improved blood vessel morphology and reduced arteriovenous connection diameters 83. In our data, we observed that the Apelin receptor, Apj, was downregulated in Smad4-HHT brain ECs treated with LC10 (Fig 6F) further indicating a possible role for APLN in HHT pathogenesis. Thus, there is the potential that anti-ANG2 therapeutics could be enhanced to treat HHT pathologies by simultaneously inhibiting the function of APLN and/or ESM1.
Additionally, our transcriptomic studies in the Smad4-HHT mouse model strongly implicated cell migration as an additional and crucial activity involved in HHT vascular phenotypes (Fig 6). Indeed, several prior studies observed defective flow-induced EC migration in both zebrafish and mouse models of Alk1-HHT 25,84. Intriguingly, one of these studies linked defects in cell migration and polarity to integrins. Specifically, robust overexpression of several integrins, including integrin β1 (ITGB1), integrin α5 (ITGA5) and integrin αv (ITGAV) was observed in the retinal blood vessels of Alk1 deficient mice, while integrin signaling in conjunction with the VEGF receptor 2 (Vegfr2) was increased in HUVECs 25. Also, an integrin inhibitor was shown to reduce AVM formation in the retina and rescue cell polarity defects, suggesting EC migration was impacted during treatment. Similarly, we found that both Itgb1 and Itgb5 were markedly upregulated in the brain ECs of all three HHT models (Fig 2) further supporting the idea that overactive integrin-mediated cell migration is involved in the HHT disease process. Future investigations into a possible mechanistic connection between HHT, ANG2 and integrin signaling are of particular interest because in context-dependent situations, ANG2 can bind to and promote integrin signaling in ECs 85–87.
In summary, this study expands our current understanding of the connection between ANG2-TEK signaling and the HHT pathway. We revealed that elevated levels of ANG2 are associated with mouse models of the three most common forms of HHT (Alk1, Eng and Smad4) and that ANG2 neutralization mitigates cerebrovascular defects, in varying degrees, associated with these models. Furthermore, comprehensive transcriptomic analyses identified a common but distinctive molecular signature that heavily implicated pro-angiogenic and cell migration processes in HHT pathogenesis and the preventative effects of ANG2 inhibition. Collectively, our results, along with studies in the HHT field and newly developing works characterizing human malformed brain ECs, provide accumulating evidence that pro-angiogenic ANG2 activity is involved in the development of vascular malformations. Thus, anti-ANG2 based therapeutics present a promising avenue for treating vascular diseases.
Supplementary Material
Supplemental Figure 1. Transcriptional profiles of cerebral ECs associated with HHT. (A-D) Venn diagrams showing the overlap of upregulated or downregulated differential expressed genes (DEGs) detected in iBECs from three HHT models with or without Smad4 ChIP-seq data, previously obtained in BMP9/10 stimulated Ms1 cells. (E) Top 10 gene ontology (GO) terms in the common genes between HHTiECKO upregulated DEGs data sets. (F) Top 10 GO terms in the 118 overlapping genes of HHTiECKO upregulated DEGs and Smad4-ChIP-seq data sets. Notice that only angiogenesis has a false discovery rate (FDR)<0.05 (blue bars); grey bars indicate FDR>0.05. (G) Top 10 GO terms in the common genes between HHTiECKO downregulated DEGs data sets. (H) Top 10 GO terms in the 49 overlapping genes between HHTiECKO downregulated DEGs and Smad4-ChIP-seq data sets.
Supplemental Figure 2. Increased and ectopic expression of pro-angiogenic genes in HHTiECKO tissues. (A) In situ hybridization analyses of Apln mRNA in HHTf/f and HHTiECKO brain slices. (B) In situ hybridization analyses of Ang2 mRNA in HHTf/f and HHTiECKO retinas. (C) In situ hybridization analyses of Apln mRNA in HHTf/f and HHTiECKO retinas. (D) Immunofluorescent antibody staining for ESM1 in HHTf/f and HHTiECKO retinas. White arrows, ESM1 expressed in retinal stalk cells. n, biological sample number; IB4, isolectin IB4. Scale bars are 3 mm in A, 0.3 mm in B-C, 50 μm in D.
Supplemental Figure 3. ANG2 inhibition prevents vein dilation in Smad4iECKO mice. (A) Quantification of vein diameter in Smad4iECKO and LC10 treated Smad4iECKO blue latex dye perfused brains. The veins parallel to the basilar artery were analyzed. n = indicated biological sample number. All values are mean ± SEM. Non-parametric, Wilcoxon rank sum test.
Supplemental Figure 4. Genetic loss of Ang2 substantially prolongs the survival of Smad4iECKO mice. (A) Tamoxifen treatment plan for survival studies. P, postnatal days. (B) Survival curves for Smad4f/f, Smad4iECKO, Ang2f/wt;Smad4f/f, Ang2iECKO/wt;Smad4iECKO pups. n = indicated biological sample number. Non-parametric, Wilcoxon rank sum test was performed in A. Log-rank (Mantel-Cox) test was performed in C.
Supplemental Figure 5. Transcriptional effects on Smad4iECKO iBECs subjected to ANG2 blockade. (A,B) Top 10 gene ontology (GO) terms in DEGs set 1 and 2. False discovery rate (FDR)< 0.05 applies to all GO terms. (C,D) Directed acyclic graph (DAG) for the top 10 GO terms in A and B, respectively.
Highlights:
Cerebrovascular defects, including arteriovenous malformation (AVM) and blood vessel enlargement, were characterized in animal models of the three most common forms of HHT (ALK1, ENG and SMAD4).
Angiopoietin-2 (ANG2), a pathophysiological pro-angiogenic factor, is robustly and ectopically elevated in the brain endothelial cells (ECs) of HHT mouse models.
Pharmacological neutralization of ANG2 in HHT mice limited or prevented brain AVM formation and blocked blood vessel enlargement in vivo.
Transcriptomic analyses revealed a unique angiogenic profile shared among the brain ECs of HHT mice; ANG2 inhibition in Smad4-HHT mice partially impeded gene expression changes associated with angiogenesis and cell migration.
Acknowledgements
We would like to thank Jone Garai and Jovanny Zabaleta at Louisiana State University Health Sciences Center for their continued assistance with RNA-seq studies. The Alk1 conditional floxed mice were kindly provided by Paul Oh at the Barrow Neurological Institute through the assistance of Joshua Wythe at Baylor College of Medicine. We are grateful to Hua Su for transferring the Eng conditional floxed mice from UC San Francisco in coordination with Helen Arthur. We would also like to express appreciation to Susan Quaggin at Northwestern University Feinberg School of Medicine for providing the Ang2 conditional floxed mice, which were developed from the following resource: O’Brien Kidney Center Award P30DK114857. We thank Prodyot K. Chatterjee (The Feinstein Institutes) for their technical assistance with HUVEC cultures and Dr. Christine N. Metz (The Feinstein Institutes) for generously providing us with HUVECs. Biographical abstract created with BioRender.com.
Sources of Funding
XZ is supported by an American Heart Association predoctoral fellowship (20PRE35120237). SMM is supported by NIH grants R01 HL163196 and R01 HL139713, and Department of Defense Investigator-imitated Research Award PR160198. PM is supported by NIH grants R01HL139778 and R01HL150040, and Department of Defense grant PR161205.
Nonstandard Abbreviations and Acronyms
- ACVRL1/ALK1
activin receptor-like kinase 1
- AKT
protein kinase B
- ANG1
angiopoietin-1
- ANG2
angiopoietin-2
- APLN
apelin
- AVM
arteriovenous malformation
- BA
basilar artery
- bAVM
brain arteriovenous malformation
- BMP9/GDF2
bone morphogenetic protein 9
- ChIP-seq
chromatin immunoprecipitation sequencing
- DAG
directed acyclic graph
- DEG
differential expressed genes
- ENG
endoglin
- ESM1
endothelial cell specific molecule 1
- f
floxed loxPs
- GO
gene ontology
- HHT
Hereditary Hemorrhagic Telangiectasia
- HUVEC
human umbilical vein endothelial cell
- iBECs
isolated brain endothelial cells
- ID1
inhibitor of DNA binding 1
- iECKO
inducible endothelial cell knockout
- IgG
immunoglobin g
- LC10
anti-ANG2 monoclonal blocking antibody
- MCA
middle cerebral artery
- mTOR
mammalian target of rapamycin
- P
postnatal day
- PI3K
phosphatidyl inositol 3-kinase
- RNA-seq
RNA sequencing
- SMAD4
mothers against decapentaplegic homolog 4
- TAZ
taffazzin
- TIE2/TEK
Tyr kinase with Ig and EGF homology domains
- TGFβ
transforming growth factor beta
- VEGF-A
vascular endothelial growth factor A
- wt
wildtype
- YAP
yes-associated protein
Footnotes
Conflict of interest: CU and CK declare employment, stock ownership and patents with Roche. The other authors have declared that no conflict of interest exists.
Disclosure
Christoph Ullmer and Christian Klein declare employment, stock ownership and patents with Roche.
References
- 1.Kjeldsen AD, Vase P, Green A. Hereditary haemorrhagic telangiectasia: a population-based study of prevalence and mortality in Danish patients. Journal of internal medicine. 1999;245:31–39. [DOI] [PubMed] [Google Scholar]
- 2.Shovlin CL. Hereditary haemorrhagic telangiectasia: pathophysiology, diagnosis and treatment. Blood Rev. 2010;24:203–219. doi: 10.1016/j.blre.2010.07.001 [DOI] [PubMed] [Google Scholar]
- 3.McDonald J, Wooderchak-Donahue W, VanSant Webb C, Whitehead K, Stevenson DA, Bayrak-Toydemir P. Hereditary hemorrhagic telangiectasia: genetics and molecular diagnostics in a new era. Front Genet. 2015;6:1. doi: 10.3389/fgene.2015.00001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Govani FS, Shovlin CL. Hereditary haemorrhagic telangiectasia: a clinical and scientific review. European journal of human genetics : EJHG. 2009;17:860–871. doi: 10.1038/ejhg.2009.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bayrak-Toydemir P, McDonald J, Markewitz B, Lewin S, Miller F, Chou LS, Gedge F, Tang W, Coon H, Mao R. Genotype-phenotype correlation in hereditary hemorrhagic telangiectasia: mutations and manifestations. American journal of medical genetics Part A. 2006;140:463–470. doi: 10.1002/ajmg.a.31101 [DOI] [PubMed] [Google Scholar]
- 6.Gallione C, Aylsworth AS, Beis J, et al. Overlapping spectra of SMAD4 mutations in juvenile polyposis (JP) and JP-HHT syndrome. American journal of medical genetics Part A. 2010;152a:333–339. doi: 10.1002/ajmg.a.33206 [DOI] [PubMed] [Google Scholar]
- 7.Gallione CJ, Repetto GM, Legius E, Rustgi AK, Schelley SL, Tejpar S, Mitchell G, Drouin E, Westermann CJ, Marchuk DA. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet. 2004;363:852–859. doi: 10.1016/S0140-6736(04)15732-2 [DOI] [PubMed] [Google Scholar]
- 8.Gallione CJ, Richards JA, Letteboer TG, Rushlow D, Prigoda NL, Leedom TP, Ganguly A, Castells A, Ploos van Amstel JK, Westermann CJ, Pyeritz RE, Marchuk DA. SMAD4 mutations found in unselected HHT patients. J Med Genet. 2006;43:793–797. doi: 10.1136/jmg.2006.041517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996;13:189–195. doi: 10.1038/ng0696-189 [DOI] [PubMed] [Google Scholar]
- 10.Robert F, Desroches-Castan A, Bailly S, Dupuis-Girod S, Feige JJ. Future treatments for hereditary hemorrhagic telangiectasia. Orphanet J Rare Dis. 2020;15:4. doi: 10.1186/s13023-019-1281-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Botella LM, Albinana V, Ojeda-Fernandez L, Recio-Poveda L, Bernabeu C. Research on potential biomarkers in hereditary hemorrhagic telangiectasia. Frontiers in genetics. 2015;6:115. doi: 10.3389/fgene.2015.00115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faughnan ME, Mager JJ, Hetts SW, et al. Second International Guidelines for the Diagnosis and Management of Hereditary Hemorrhagic Telangiectasia. Ann Intern Med. 2020;173:989–1001. doi: 10.7326/m20-1443 [DOI] [PubMed] [Google Scholar]
- 13.Shovlin CL, Buscarini E, Sabbà C, et al. The European Rare Disease Network for HHT Frameworks for management of hereditary haemorrhagic telangiectasia in general and speciality care. Eur J Med Genet. 2022;65:104370. doi: 10.1016/j.ejmg.2021.104370 [DOI] [PubMed] [Google Scholar]
- 14.Garg N, Khunger M, Gupta A, Kumar N. Optimal management of hereditary hemorrhagic telangiectasia. Journal of blood medicine. 2014;5:191–206. doi: 10.2147/jbm.S45295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kühnel T, Wirsching K, Wohlgemuth W, Chavan A, Evert K, Vielsmeier V. Hereditary Hemorrhagic Telangiectasia. Otolaryngol Clin North Am. 2018;51:237–254. doi: 10.1016/j.otc.2017.09.017 [DOI] [PubMed] [Google Scholar]
- 16.McDonald J, Stevenson DA. Hereditary Hemorrhagic Telangiectasia. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Mirzaa GM, Amemiya A, eds. GeneReviews(®). Seattle (WA): University of Washington, Seattle Copyright © 1993-2022, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.; 1993. [PubMed] [Google Scholar]
- 17.Dupuis-Girod S, Ginon I, Saurin JC, et al. Bevacizumab in patients with hereditary hemorrhagic telangiectasia and severe hepatic vascular malformations and high cardiac output. JAMA. 2012;307:948–955. doi: 10.1001/jama.2012.250 [DOI] [PubMed] [Google Scholar]
- 18.Al-Samkari H, Kasthuri RS, Parambil JG, et al. An international, multicenter study of intravenous bevacizumab for bleeding in hereditary hemorrhagic telangiectasia: the InHIBIT-Bleed study. Haematologica. 2021;106:2161–2169. doi: 10.3324/haematol.2020.261859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crist AM, Zhou X, Garai J, Lee AR, Thoele J, Ullmer C, Klein C, Zabaleta J, Meadows SM. Angiopoietin-2 Inhibition Rescues Arteriovenous Malformation in a Smad4 Hereditary Hemorrhagic Telangiectasia Mouse Model. Circulation. 2019;139:2049–2063. doi: 10.1161/CIRCULATIONAHA.118.036952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alsina-Sanchis E, Garcia-Ibanez Y, Figueiredo AM, Riera-Domingo C, Figueras A, Matias-Guiu X, Casanovas O, Botella LM, Pujana MA, Riera-Mestre A, Graupera M, Vinals F. ALK1 Loss Results in Vascular Hyperplasia in Mice and Humans Through PI3K Activation. Arterioscler Thromb Vasc Biol. 2018;38:1216–1229. doi: 10.1161/ATVBAHA.118.310760 [DOI] [PubMed] [Google Scholar]
- 21.Ola R, Dubrac A, Han J, et al. PI3 kinase inhibition improves vascular malformations in mouse models of hereditary haemorrhagic telangiectasia. Nat Commun. 2016;7:13650. doi: 10.1038/ncomms13650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jin Y, Muhl L, Burmakin M, Wang Y, Duchez AC, Betsholtz C, Arthur HM, Jakobsson L. Endoglin prevents vascular malformation by regulating flow-induced cell migration and specification through VEGFR2 signalling. Nat Cell Biol. 2017;19:639–652. doi: 10.1038/ncb3534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruiz S, Chandakkar P, Zhao H, Papoin J, Chatterjee PK, Christen E, Metz CN, Blanc L, Campagne F, Marambaud P. Tacrolimus rescues the signaling and gene expression signature of endothelial ALK1 loss-of-function and improves HHT vascular pathology. Hum Mol Genet. 2017;26:4786–4798. doi: 10.1093/hmg/ddx358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ruiz S, Zhao H, Chandakkar P, et al. Correcting Smad1/5/8, mTOR, and VEGFR2 treats pathology in hereditary hemorrhagic telangiectasia models. J Clin Invest. 2020;130:942–957. doi: 10.1172/JCI127425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park H, Furtado J, Poulet M, Chung M, Yun S, Lee S, Sessa WC, Franco CA, Schwartz MA, Eichmann A. Defective Flow-Migration Coupling Causes Arteriovenous Malformations in Hereditary Hemorrhagic Telangiectasia. Circulation. 2021;144:805–822. doi: 10.1161/CIRCULATIONAHA.120.053047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han C, Choe SW, Kim YH, Acharya AP, Keselowsky BG, Sorg BS, Lee YJ, Oh SP. VEGF neutralization can prevent and normalize arteriovenous malformations in an animal model for hereditary hemorrhagic telangiectasia 2. Angiogenesis. 2014;17:823–830. doi: 10.1007/s10456-014-9436-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hwan Kim Y, Vu PN, Choe SW, Jeon CJ, Arthur HM, Vary CPH, Lee YJ, Oh SP. Overexpression of Activin Receptor-Like Kinase 1 in Endothelial Cells Suppresses Development of Arteriovenous Malformations in Mouse Models of Hereditary Hemorrhagic Telangiectasia. Circ Res. 2020;127:1122–1137. doi: 10.1161/CIRCRESAHA.119.316267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pitulescu ME, Schmidt I, Benedito R, Adams RH. Inducible gene targeting in the neonatal vasculature and analysis of retinal angiogenesis in mice. Nat Protoc. 2010;5:1518–1534. doi: 10.1038/nprot.2010.113 [DOI] [PubMed] [Google Scholar]
- 29.Crist AM, Lee AR, Patel NR, Westhoff DE, Meadows SM. Vascular deficiency of Smad4 causes arteriovenous malformations: a mouse model of Hereditary Hemorrhagic Telangiectasia. Angiogenesis. 2018;21:363–380. doi: 10.1007/s10456-018-9602-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crist AM, Young C, Meadows SM. Characterization of arteriovenous identity in the developing neonate mouse retina. Gene Expr Patterns. 2017;23–24:22–31. doi: 10.1016/j.gep.2017.01.002 [DOI] [PubMed] [Google Scholar]
- 31.Mahmoud M, Allinson KR, Zhai Z, Oakenfull R, Ghandi P, Adams RH, Fruttiger M, Arthur HM. Pathogenesis of arteriovenous malformations in the absence of endoglin. Circ Res. 2010;106:1425–1433. doi: 10.1161/CIRCRESAHA.109.211037 [DOI] [PubMed] [Google Scholar]
- 32.Tual-Chalot S, Mahmoud M, Allinson KR, Redgrave RE, Zhai Z, Oh SP, Fruttiger M, Arthur HM. Endothelial depletion of Acvrl1 in mice leads to arteriovenous malformations associated with reduced endoglin expression. PLoS One. 2014;9:e98646. doi: 10.1371/journal.pone.0098646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruiz S, Zhao H, Chandakkar P, Chatterjee PK, Papoin J, Blanc L, Metz CN, Campagne F, Marambaud P. A mouse model of hereditary hemorrhagic telangiectasia generated by transmammary-delivered immunoblocking of BMP9 and BMP10. Sci Rep. 2016;5:37366. doi: 10.1038/srep37366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rattner A, Williams J, Nathans J. Roles of HIFs and VEGF in angiogenesis in the retina and brain. J Clin Invest. 2019;129:3807–3820. doi: 10.1172/jci126655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Augustin HG, Koh GY. Organotypic vasculature: From descriptive heterogeneity to functional pathophysiology. Science. 2017;357. doi: 10.1126/science.aal2379 [DOI] [PubMed] [Google Scholar]
- 36.Kilian A, Latino GA, White AJ, et al. Genotype-Phenotype Correlations in Children with HHT. J Clin Med. 2020;9. doi: 10.3390/jcm9092714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gamboa NT, Joyce EJ, Eli I, Park MS, Taussky P, Schmidt RH, McDonald J, Whitehead KJ, Kalani MYS. Clinical presentation and treatment paradigms of brain arteriovenous malformations in patients with hereditary hemorrhagic telangiectasia. J Clin Neurosci. 2018;51:22–28. doi: 10.1016/j.jocn.2018.01.019 [DOI] [PubMed] [Google Scholar]
- 38.Snellings DA, Gallione CJ, Clark DS, Vozoris NT, Faughnan ME, Marchuk DA. Somatic Mutations in Vascular Malformations of Hereditary Hemorrhagic Telangiectasia Result in Bi-allelic Loss of ENG or ACVRL1. Am J Hum Genet. 2019;105:894–906. doi: 10.1016/j.ajhg.2019.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bourdeau A, Cymerman U, Paquet ME, Meschino W, McKinnon WC, Guttmacher AE, Becker L, Letarte M. Endoglin expression is reduced in normal vessels but still detectable in arteriovenous malformations of patients with hereditary hemorrhagic telangiectasia type 1. Am J Pathol. 2000;156:911–923. doi: 10.1016/s0002-9440(10)64960-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi EJ, Chen W, Jun K, Arthur HM, Young WL, Su H. Novel brain arteriovenous malformation mouse models for type 1 hereditary hemorrhagic telangiectasia. PLoS One. 2014;9:e88511. doi: 10.1371/journal.pone.0088511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park SO, Wankhede M, Lee YJ, Choi EJ, Fliess N, Choe SW, Oh SH, Walter G, Raizada MK, Sorg BS, Oh SP. Real-time imaging of de novo arteriovenous malformation in a mouse model of hereditary hemorrhagic telangiectasia. J Clin Invest. 2009;119:3487–3496. doi: 10.1172/JCI39482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim YH, Choe SW, Chae MY, Hong S, Oh SP. SMAD4 Deficiency Leads to Development of Arteriovenous Malformations in Neonatal and Adult Mice. J Am Heart Assoc. 2018;7:e009514. doi: 10.1161/jaha.118.009514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Choi EJ, Walker EJ, Shen F, Oh SP, Arthur HM, Young WL, Su H. Minimal homozygous endothelial deletion of Eng with VEGF stimulation is sufficient to cause cerebrovascular dysplasia in the adult mouse. Cerebrovasc Dis. 2012;33:540–547. doi: 10.1159/000337762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shaligram SS, Zhang R, Zhu W, et al. Bone Marrow-Derived Alk1 Mutant Endothelial Cells and Clonally Expanded Somatic Alk1 Mutant Endothelial Cells Contribute to the Development of Brain Arteriovenous Malformations in Mice. Transl Stroke Res. 2021. doi: 10.1007/s12975-021-00955-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang X, Li C, Herrera PL, Deng CX. Generation of Smad4/Dpc4 conditional knockout mice. Genesis. 2002;32:80–81. [DOI] [PubMed] [Google Scholar]
- 46.Park SO, Lee YJ, Seki T, Hong KH, Fliess N, Jiang Z, Park A, Wu X, Kaartinen V, Roman BL, Oh SP. ALK5- and TGFBR2-independent role of ALK1 in the pathogenesis of hereditary hemorrhagic telangiectasia type 2. Blood. 2008;111:633–642. doi: 10.1182/blood-2007-08-107359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Allinson KR, Carvalho RL, van den Brink S, Mummery CL, Arthur HM. Generation of a floxed allele of the mouse Endoglin gene. Genesis (New York, NY : 2000). 2007;45:391–395. doi: 10.1002/dvg.20284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Y, Nakayama M, Pitulescu ME, et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. 2010;465:483–486. doi: 10.1038/nature09002 [DOI] [PubMed] [Google Scholar]
- 49.Kenig-Kozlovsky Y, Scott RP, Onay T, et al. Ascending Vasa Recta Are Angiopoietin/Tie2-Dependent Lymphatic-Like Vessels. J Am Soc Nephrol. 2018;29:1097–1107. doi: 10.1681/ASN.2017090962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Souma T, Thomson BR, Heinen S, et al. Context-dependent functions of angiopoietin 2 are determined by the endothelial phosphatase VEPTP. Proc Natl Acad Sci U S A. 2018;115:1298–1303. doi: 10.1073/pnas.1714446115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heberle H, Meirelles GV, da Silva FR, Telles GP, Minghim R. InteractiVenn: a web-based tool for the analysis of sets through Venn diagrams. BMC Bioinformatics. 2015;16:169. doi: 10.1186/s12859-015-0611-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Research. 2019;47:W199–W205. doi: 10.1093/nar/gkz401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Babicki S, Arndt D, Marcu A, Liang Y, Grant JR, Maciejewski A, Wishart DS. Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res. 2016;44:W147–153. doi: 10.1093/nar/gkw419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Crist A, Young C, Meadows SM. Characterization of Arteriovenous Identity in the Developing Neonate Mouse Retina. Gene Expression Patterns. 2017;23–24. doi: doi: 10.1016/j.gep.2017.01.002. [DOI] [PubMed] [Google Scholar]
- 55.Chatterjee PK, Al-Abed Y, Sherry B, Metz CN. Cholinergic agonists regulate JAK2/STAT3 signaling to suppress endothelial cell activation. Am J Physiol Cell Physiol. 2009;297:C1294–1306. doi: 10.1152/ajpcell.00160.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ola R, Kunzel SH, Zhang F, Genet G, Chakraborty R, Pibouin-Fragner L, Martin K, Sessa W, Dubrac A, Eichmann A. SMAD4 Prevents Flow Induced Arteriovenous Malformations by Inhibiting Casein Kinase 2. Circulation. 2018;138:2379–2394. doi: 10.1161/CIRCULATIONAHA.118.033842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rocha SF, Schiller M, Jing D, Li H, Butz S, Vestweber D, Biljes D, Drexler HC, Nieminen-Kelha M, Vajkoczy P, Adams S, Benedito R, Adams RH. Esm1 modulates endothelial tip cell behavior and vascular permeability by enhancing VEGF bioavailability. Circ Res. 2014;115:581–590. doi: 10.1161/CIRCRESAHA.115.304718 [DOI] [PubMed] [Google Scholar]
- 58.Kasai A, Shintani N, Kato H, Matsuda S, Gomi F, Haba R, Hashimoto H, Kakuda M, Tano Y, Baba A. Retardation of retinal vascular development in apelin-deficient mice. Arterioscler Thromb Vasc Biol. 2008;28:1717–1722. doi: 10.1161/ATVBAHA.108.163402 [DOI] [PubMed] [Google Scholar]
- 59.del Toro R, Prahst C, Mathivet T, Siegfried G, Kaminker JS, Larrivee B, Breant C, Duarte A, Takakura N, Fukamizu A, Penninger J, Eichmann A. Identification and functional analysis of endothelial tip cell-enriched genes. Blood. 2010;116:4025–4033. doi: 10.1182/blood-2010-02-270819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Augustin HG, Koh GY, Thurston G, Alitalo K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat Rev Mol Cell Biol. 2009;10:165–177. doi: 10.1038/nrm2639 [DOI] [PubMed] [Google Scholar]
- 61.Gale NW, Thurston G, Hackett SF, Renard R, Wang Q, McClain J, Martin C, Witte C, Witte MH, Jackson D, Suri C, Campochiaro PA, Wiegand SJ, Yancopoulos GD. Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter role is rescued by Angiopoietin-1. Dev Cell. 2002;3:411–423. [DOI] [PubMed] [Google Scholar]
- 62.Hackett SF, Ozaki H, Strauss RW, Wahlin K, Suri C, Maisonpierre P, Yancopoulos G, Campochiaro PA. Angiopoietin 2 Expression in the Retina: Upregulation During Physiologic and Pathologic Neovascularization. J Cell Physiol. 2000;184:275–284. [DOI] [PubMed] [Google Scholar]
- 63.Mundel TM, Kalluri R. Type IV collagen-derived angiogenesis inhibitors. Microvasc Res. 2007;74:85–89. doi: 10.1016/j.mvr.2007.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Corada M, Orsenigo F, Morini MF, Pitulescu ME, Bhat G, Nyqvist D, Breviario F, Conti V, Briot A, Iruela-Arispe ML, Adams RH, Dejana E. Sox17 is indispensable for acquisition and maintenance of arterial identity. Nat Commun. 2013;4:2609. doi: 10.1038/ncomms3609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim M, Allen B, Korhonen EA, Nitschke M, Yang HW, Baluk P, Saharinen P, Alitalo K, Daly C, Thurston G, McDonald DM. Opposing actions of angiopoietin-2 on Tie2 signaling and FOXO1 activation. J Clin Invest. 2016;126:3511–3525. doi: 10.1172/JCI84871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Saharinen P, Eklund L, Alitalo K. Therapeutic targeting of the angiopoietin-TIE pathway. Nat Rev Drug Discov. 2017;16:635–661. doi: 10.1038/nrd.2016.278 [DOI] [PubMed] [Google Scholar]
- 67.Akwii RG, Sajib MS, Zahra FT, Mikelis CM. Role of Angiopoietin-2 in Vascular Physiology and Pathophysiology. Cells. 2019;8. doi: 10.3390/cells8050471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ziegler T, Horstkotte J, Schwab C, et al. Angiopoietin 2 mediates microvascular and hemodynamic alterations in sepsis. J Clin Invest. 2013;123:3436–3445. doi: 10.1172/jci66549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Regula JT, Lundh von Leithner P, Foxton R, et al. Targeting key angiogenic pathways with a bispecific CrossMAb optimized for neovascular eye diseases. EMBO Mol Med. 2016;8:1265–1288. doi: 10.15252/emmm.201505889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ardelean DS, Jerkic M, Yin M, Peter M, Ngan B, Kerbel RS, Foster FS, Letarte M. Endoglin and activin receptor-like kinase 1 heterozygous mice have a distinct pulmonary and hepatic angiogenic profile and response to anti-VEGF treatment. Angiogenesis. 2014;17:129–146. doi: 10.1007/s10456-013-9383-4 [DOI] [PubMed] [Google Scholar]
- 71.Ojeda-Fernandez L, Barrios L, Rodriguez-Barbero A, Recio-Poveda L, Bernabeu C, Botella LM. Reduced plasma levels of Ang-2 and sEng as novel biomarkers in hereditary hemorrhagic telangiectasia (HHT). Clin Chim Acta. 2010;411:494–499. doi: 10.1016/j.cca.2009.12.023 [DOI] [PubMed] [Google Scholar]
- 72.Wetzel-Strong SE, Weinsheimer S, Nelson J, Pawlikowska L, Clark D, Starr MD, Liu Y, Kim H, Faughnan ME, Nixon AB, Marchuk DA. Pilot investigation of circulating angiogenic and inflammatory biomarkers associated with vascular malformations. Orphanet J Rare Dis. 2021;16:372. doi: 10.1186/s13023-021-02009-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fernandez LA, Garrido-Martin EM, Sanz-Rodriguez F, Pericacho M, Rodriguez-Barbero A, Eleno N, Lopez-Novoa JM, Duwell A, Vega MA, Bernabeu C, Botella LM. Gene expression fingerprinting for human hereditary hemorrhagic telangiectasia. Hum Mol Genet. 2007;16:1515–1533. doi: 10.1093/hmg/ddm069 [DOI] [PubMed] [Google Scholar]
- 74.Scharpfenecker M, Fiedler U, Reiss Y, Augustin HG. The Tie-2 ligand angiopoietin-2 destabilizes quiescent endothelium through an internal autocrine loop mechanism. J Cell Sci. 2005;118:771–780. doi: 10.1242/jcs.01653 [DOI] [PubMed] [Google Scholar]
- 75.Thomas M, Augustin HG. The role of the Angiopoietins in vascular morphogenesis. Angiogenesis. 2009;12:125–137. doi: 10.1007/s10456-009-9147-3 [DOI] [PubMed] [Google Scholar]
- 76.Tual-Chalot S, Garcia-Collado M, Redgrave RE, Singh E, Davison B, Park C, Lin H, Luli S, Jin Y, Wang Y, Lawrie A, Jakobsson L, Arthur HM. Loss of Endothelial Endoglin Promotes High-Output Heart Failure Through Peripheral Arteriovenous Shunting Driven by VEGF Signaling. Circ Res. 2020;126:243–257. doi: 10.1161/circresaha.119.315974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hashimoto T, Lam T, Boudreau NJ, Bollen AW, Lawton MT, Young WL. Abnormal balance in the angiopoietin-tie2 system in human brain arteriovenous malformations. Cir Res. 2001;89:111–113. [DOI] [PubMed] [Google Scholar]
- 78.Hashimoto T, Emala CW, Joshi S, Mesa-Tejada R, Quick CM, Feng L, Libow A, Marchuk DA, Young WL. Abnormal pattern of Tie-2 and vascular endothelial growth factor receptor expression in human cerebral arteriovenous malformations. Neurosurgery. 2000;47:910–918. [DOI] [PubMed] [Google Scholar]
- 79.Winkler EA, Kim CN, Ross JM, et al. A single-cell atlas of the normal and malformed human brain vasculature. Science. 2022:eabi7377. doi: 10.1126/science.abi7377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wälchli T, Ghobrial M, Schwab M, et al. Molecular atlas of the human brain vasculature at the single-cell level. bioRxiv. 2021:2021.2010.2018.464715. doi: 10.1101/2021.10.18.464715 [DOI] [Google Scholar]
- 81.Huang L, Bichsel C, Norris AL, Thorpe J, Pevsner J, Alexandrescu S, Pinto A, Zurakowski D, Kleiman RJ, Sahin M, Greene AK, Bischoff J. Endothelial GNAQ p.R183Q Increases ANGPT2 (Angiopoietin-2) and Drives Formation of Enlarged Blood Vessels. Arterioscler Thromb Vasc Biol. 2022;42:e27–e43. doi: 10.1161/atvbaha.121.316651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jenny Zhou H, Qin L, Zhang H, et al. Endothelial exocytosis of angiopoietin-2 resulting from CCM3 deficiency contributes to cerebral cavernous malformation. Nat Med. 2016;22:1033–1042. doi: 10.1038/nm.4169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Adhicary S, Fanelli K, Nakisli S, Ward B, Pearce I, Nielsen CM. Rbpj Deficiency Disrupts Vascular Remodeling via Abnormal Apelin and Cdc42 (Cell Division Cycle 42) Activity in Brain Arteriovenous Malformation. Stroke. 2023. doi: 10.1161/strokeaha.122.041853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rochon ER, Menon PG, Roman BL. Alk1 controls arterial endothelial cell migration in lumenized vessels. Development. 2016;143:2593–2602. doi: 10.1242/dev.135392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Felcht M, Luck R, Schering A, et al. Angiopoietin-2 differentially regulates angiogenesis through TIE2 and integrin signaling. J Clin Invest. 2012;122:1991–2005. doi: 10.1172/JCI58832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hakanpaa L, Sipila T, Leppanen VM, Gautam P, Nurmi H, Jacquemet G, Eklund L, Ivaska J, Alitalo K, Saharinen P. Endothelial destabilization by angiopoietin-2 via integrin β1 activation. Nat Commun. 2015;6:5962. doi: 10.1038/ncomms6962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mirando AC, Shen J, Silva RLE, Chu Z, Sass NC, Lorenc VE, Green JJ, Campochiaro PA, Popel AS, Pandey NB. A collagen IV-derived peptide disrupts α5β1 integrin and potentiates Ang2/Tie2 signaling. JCI Insight. 2019;4. doi: 10.1172/jci.insight.122043 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Transcriptional profiles of cerebral ECs associated with HHT. (A-D) Venn diagrams showing the overlap of upregulated or downregulated differential expressed genes (DEGs) detected in iBECs from three HHT models with or without Smad4 ChIP-seq data, previously obtained in BMP9/10 stimulated Ms1 cells. (E) Top 10 gene ontology (GO) terms in the common genes between HHTiECKO upregulated DEGs data sets. (F) Top 10 GO terms in the 118 overlapping genes of HHTiECKO upregulated DEGs and Smad4-ChIP-seq data sets. Notice that only angiogenesis has a false discovery rate (FDR)<0.05 (blue bars); grey bars indicate FDR>0.05. (G) Top 10 GO terms in the common genes between HHTiECKO downregulated DEGs data sets. (H) Top 10 GO terms in the 49 overlapping genes between HHTiECKO downregulated DEGs and Smad4-ChIP-seq data sets.
Supplemental Figure 2. Increased and ectopic expression of pro-angiogenic genes in HHTiECKO tissues. (A) In situ hybridization analyses of Apln mRNA in HHTf/f and HHTiECKO brain slices. (B) In situ hybridization analyses of Ang2 mRNA in HHTf/f and HHTiECKO retinas. (C) In situ hybridization analyses of Apln mRNA in HHTf/f and HHTiECKO retinas. (D) Immunofluorescent antibody staining for ESM1 in HHTf/f and HHTiECKO retinas. White arrows, ESM1 expressed in retinal stalk cells. n, biological sample number; IB4, isolectin IB4. Scale bars are 3 mm in A, 0.3 mm in B-C, 50 μm in D.
Supplemental Figure 3. ANG2 inhibition prevents vein dilation in Smad4iECKO mice. (A) Quantification of vein diameter in Smad4iECKO and LC10 treated Smad4iECKO blue latex dye perfused brains. The veins parallel to the basilar artery were analyzed. n = indicated biological sample number. All values are mean ± SEM. Non-parametric, Wilcoxon rank sum test.
Supplemental Figure 4. Genetic loss of Ang2 substantially prolongs the survival of Smad4iECKO mice. (A) Tamoxifen treatment plan for survival studies. P, postnatal days. (B) Survival curves for Smad4f/f, Smad4iECKO, Ang2f/wt;Smad4f/f, Ang2iECKO/wt;Smad4iECKO pups. n = indicated biological sample number. Non-parametric, Wilcoxon rank sum test was performed in A. Log-rank (Mantel-Cox) test was performed in C.
Supplemental Figure 5. Transcriptional effects on Smad4iECKO iBECs subjected to ANG2 blockade. (A,B) Top 10 gene ontology (GO) terms in DEGs set 1 and 2. False discovery rate (FDR)< 0.05 applies to all GO terms. (C,D) Directed acyclic graph (DAG) for the top 10 GO terms in A and B, respectively.