

Abstract
Protein S-glutathionylation is emerging as a central oxidation that regulates redox signaling and biological processes linked to diseases. In recent years, the field of protein S-glutathionylation has expanded by developing biochemical tools for the identification and functional analyses of S-glutathionylation, investigating knockout mouse models, and developing and evaluating chemical inhibitors for enzymes involved in glutathionylation. This review will highlight recent studies of two enzymes, glutathione transferase omega 1 (GSTO1) and glutaredoxin 1 (Grx1), especially introducing their glutathionylation substrates associated with inflammation, cancer, and neurodegeneration and showcasing the advancement of their chemical inhibitors. Lastly, we will feature protein substrates and chemical inducers of LanC-like protein (LanCL), the first enzyme in protein C-glutathionylation.
Keywords: Cysteine, S-glutathionylation, C-glutathionylation, glutathione transferase omega, glutaredoxin, LanC-like protein
Graphical Abstract
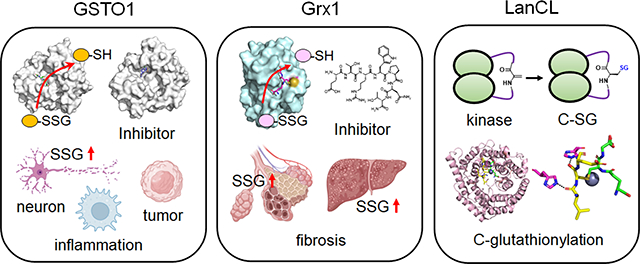
1. Introduction
Reactive oxygen species (ROS) intricately change the local or global redox state, thereby regulating signaling pathways (i.e., redox signaling) [1]. Due to a high intracellular glutathione (GSH) concentration, disulfide formation of protein cysteines with glutathione in response to ROS, termed protein S-glutathionylation (SSG), has been observed for decades [2,3]. Research in glutathionylation has been extensive in identifying oxidation-susceptible proteins and understanding the functional outcome of glutathionylation, where chemistry has been instrumental in developing various chemical tools that enable the identification of protein SSG with proteomics [4]. Previous studies demonstrated that SSG is involved in all major functional categories of biological processes, including metabolism (e.g., glycolysis [5]), signal transduction (e.g., G-protein, kinase, and phosphatase [6–8]), cytoskeletal organization (e.g., actin [9]), inflammation (e.g., IKK-β [10,11]), and transcription (e.g., p53 [12]). Biological studies also demonstrated that SSG regulates physiology (e.g., SERCA in calcium signaling [13], actin for cell migration [9], and titin in muscle contraction [14,15]) and contributes to pathologic disorders [16–19].
From a chemistry perspective, SSG can form via complex sulfur chemistry without enzymes: nucleophilic cysteine thiolate can react with glutathione disulfide (GSSG) via thiol exchange. Alternatively, protein cysteine can be oxidized to reactive intermediates, including sulfenic acid, which reacts with GSH forming SSG [20]. In contrast, intracellular GSH can reverse protein SSG to reduced cysteine (SH) in proteins while forming GSSG [20]. However, intracellularly, these chemical reactions are facilitated by redox enzymes that catalyze the formation and reduction of SSG (SSG enzyme hereafter) [20]. For example, glutathione transferase pi (GSTP) [21] increases SSG formation, whereas glutaredoxin (Grx) [22] and glutathione transferase omega (GSTO) [23] catalyze deglutathionylation (see a review for other enzymes [20]). Recently, new SSG biology with these enzymes was uncovered, especially combined with knockout (KO) mouse models. In addition, the first enzyme catalyzing protein C-glutathionylation (C-SG), namely LanC-like protein (LanCL), emerged. In parallel, the advancement of developing inhibitors for SSG enzymes unfolds therapeutic opportunities in diseases. This review will focus on the chemistry and biology of SSG enzymes, highlighting 1) emerging protein substrates of SSG enzymes (GSTO1 and Grx1) and their biological implications and 2) recent development of inhibitors for SSG enzymes (GSTO1 and Grx1). Lastly, we will introduce 3) emerging biological roles and chemical ligands of LanCL.
2. Substrates and inhibitors of enzymes in protein glutathionylation
2.1. Glutathione transferase omega 1 (GSTO1).
GSTO1 is a member of a broad family of glutathione transferases (GST) primarily involved in phase II detoxification via GSH conjugation [24]. In humans, GSTO1 and its isozyme GSTO2 were identified with a high sequence homology (64%) and a typical GST-fold structure [24]. However, unlike other GST classes, GSTO1/2 are unique for having catalytic cysteine (both C32) in the active site and catalyzing thioltransferase reaction (primarily deglutathionylation) similar to glutaredoxin (see a review for other GSTO reactions) [25,26]. In recent years, GSTO1 has stood out with evidence supporting its roles in positively regulating inflammation [27–30], contributing to cancer progression [31,32], and protecting from neurodegeneration [33,34], primarily via its deglutathionylation activity. This section will showcase selected examples of GSTO1 substrates for SSG while updating recent development of GSTO1 inhibitors.
Fused in sarcoma (FUS).
FUS was named after its fusion to a transcription factor (i.e., CHOP) in human myxoid liposarcomas, playing as an oncogene [35]. FUS translocates to the nucleus and regulates transcription, RNA splicing, and DNA repair response [36]. Interestingly, FUS genetic mutations are causatively linked to neurogenerative diseases, especially amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) [36,37]. FUS protein is intrinsically prone to form aggregation and pathological inclusion bodies [38]. Therefore, FUS overexpression or mutations increase its cytosolic accumulation and deposition, which causes cytosolic toxicity responsible for pathological degeneration [38].
Cha et al. reported that Drosophila expressing neuronal FUS shows FUS-induced neuronal toxicity (e.g., reducing locomotive activity and lifespan) and mitochondrial dysfunction (e.g., mitochondrial fragmentation and reduced complex I and III) [33,34]. Importantly, FUS-expressing flies dramatically reduced GstO2 protein (a Drosophila homolog of human GSTO1) (Figure 1a), whereas GstO2 expression prevented neurotoxicity and mitochondrial dysfunction, suggesting the neuroprotective role of GstO2. Subsequently, FUS was found glutathionylated at conserved cysteine 447 (C447) at the zinc finger domain (ZnF) (Figure 1a). FUS SSG enhanced its aggregation in the cytoplasm, whereas GstO2 expression prevented FUS SSG and aggregation (Figure 1a). Likewise, human GSTO1 WT expression in mammalian neuronal cells reduced FUS aggregation by deglutathionylation, decreasing cytoplasmic FUS localization and delaying neuronal toxicity. Lastly, human GSTO1 level was significantly reduced in patients carrying FUS mutations associated with ALS, supporting the roles of GSTO1 and SSG in ALS pathogenesis. These findings suggest that modulating GSTO1 expression may be a therapeutic direction to treat neurodegenerative diseases and ALS.
Figure 1.
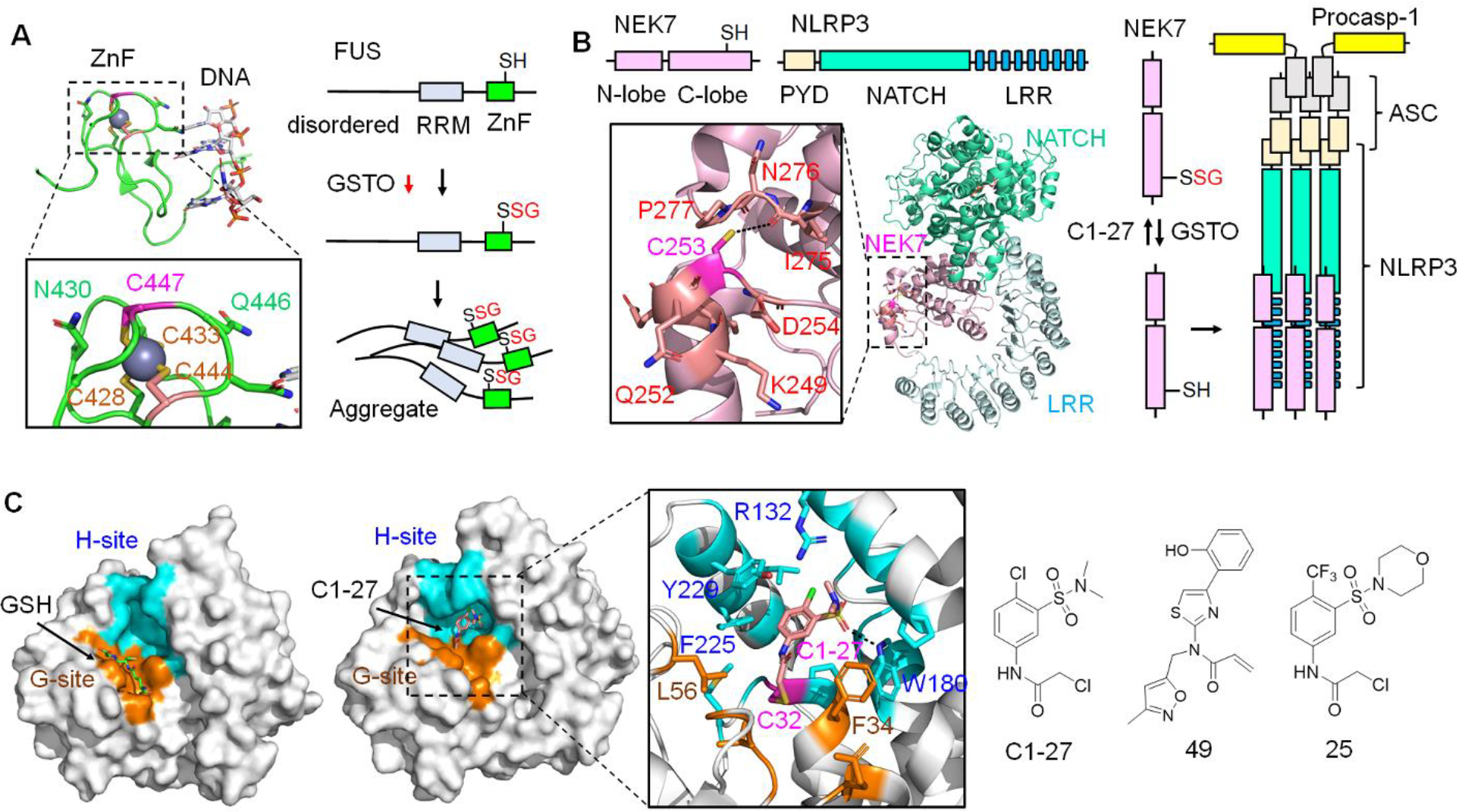
GSTO1 substrates and Inhibitors. A. GSTO1 regulates FUS SSG associated with amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). FUS is susceptible to SSG at C447 (bound to zinc) in the zinc finger domain (ZnF, PDB: 6G99). FUS SSG increases its aggregation, causing toxicity in motor neurons linked to neurodegeneration. GSTO1 reduces FUS SSG and protects from FUS-induced toxicity. B. GSTO1 activates NLRP3 inflammasome via NEK7 deglutathionylation. NEK7 was found glutathionylated at C253 (kinase C-lobe, PDB: 6NPY) in macrophages. GSTO1 binds to NEK7, causing its deglutathionylation. NEK7 deglutathionylation enables its interaction with NLRP3, inducing NLRP3 inflammasome complex formation with ASC and pro-caspase-1. The NLRP3 inflammasome activates caspase-1, which activates inflammatory cytokines (IL-1β and IL-18). C1–27 inhibits GSTO1, thus increasing NEK7 SSG and reducing NLRP3 inflammasome-mediated IL-1β release. C. GSTO1 inhibitors. GSTO1 has reactive cysteine (C32) with a GSH binding site (G-site, orange) and a hydrophobic site (H-site, cyan) (PDB: 1EEM). C1–27 inhibits GSTO1 by primarily binding at the H-site (cyan) while covalently conjugated to C32 (PDB: 4YQM). Dotted lines indicate a distance less than 4 Å. C1–27 was used as a lead compound to develop GSTO1 inhibitors, acrylamide-containing compound 49 with the highest potency (IC50 = 0.22 nM) and α-chloroacetamide derivative 25 with improved microsomal stability (half-life = 1.4 h).
NIMA-related kinase 7 (NEK7).
NEK7 is one of the mammalian NIMA-related kinases implicated in mitosis [39]. Recently, NEK7 was found to be an essential activator of NLRP3 inflammasome [40], a multiprotein complex that plays a central role in the innate immune system and inflammatory diseases [41]. NLRP3 inflammasome activation involves two steps [41]: pathogen or damage-associated molecular patterns, including lipopolysaccharide (LPS), increase expressions of NLRP3 and cytokines (pro-IL-1β and pro-IL-18) (step 1, priming). Next, various stimuli, including extracellular ATP and nigericin, activate and lead to NLRP3 inflammasome assembly, where NEK7-NLRP3 binding induces NLRP3 oligomerization with ASC and pro-caspase 1 (step 2, activation) (Figure 1b). Eventually, activated caspase-1 cleaves and increases pro-inflammatory IL-1β and IL-18.
Board and O’Neill’s group reported that macrophages in GSTO1 KO mice show impaired NLRP3 inflammasome activation in response to LPS/ATP [27]. In addition, GSTO1 inhibitor (C1–27) or siRNA caused similar outcomes, decreasing inflammatory cytokines. Biochemically, NEK7 was found to be glutathionylated at cysteine 253 (C253) (Figure 1b). LPS/ATP stimulation increased the association of GSTO1 with NEK7, causing NEK7 deglutathionylation (Figure 1b). In agreement, bone marrow-derived macrophage (BMDM) with GSTO1 KO versus WT showed higher NEK7 SSG, supporting GSTO1 in reversing NEK SSG. Lastly, compared to WT, cells with NEK7 C253A showed higher IL-1β release in response to nigericin, suggesting that NEK7 without SSG (similar to C253A) enhances NLRP3 inflammasome assembly and activation (Figure 1b). In an independent study, ASC SSG was also observed in macrophages where GSTO1-induced ASC deglutathionylation was necessary for NLRP3 inflammasome assembly and activation [29], suggesting several layers of SSG regulation in NLRP3 inflammasome. In addition to NLRP3, GSTO1 was previously shown to regulate TLR4 and Nf-kB inflammatory pathways, supporting the pro-inflammatory role of GSTO1 [30].
Interferon genes and tissue factor.
Compared to inflammation, GSTO1’s role in cancer is less understood. The Neamati group surveyed GSTO1 expression in 9,718 patients (34 tumor types), supporting GSTO1 upregulation in selected cancers (e.g., glioma, renal carcinoma, melanoma) correlated with decreased patient survival [31]. To validate experimentally, three cancer cell lines (A172, HCT116, U87-MG) with GSTO1 KO were produced. Xenograft studies showed suppressed tumor growth of GSTO1 KO cell lines versus WT, albeit dependent on individual cell types. Interestingly, GSTO1 depletion in HCT116 impeded three-dimensional (3D) tumor growth but not its proliferation in two-dimensional (2D) culture, suggesting that GSTO1 may regulate the tumor microenvironment. Transcriptional profile and proteomic analyses identified that GSTO1 KO cell lines enrich interferon (IFN) genes. In addition, GSTO KO significantly correlated with the downregulated tissue factor (TF) gene (known as factor III or F3), a transmembrane glycoprotein in the clotting cascade. Notably, IFN genes play a significant role in tumor cells by inhibiting proliferation, inducing apoptosis, and stimulating antitumor immunity [42]. Likewise, TF expression in tumor cells facilitates hematogenous metastasis and angiogenesis [43]. Although direct GSTO1 substrates for SSG were not determined, this report demonstrates that GSTO1 KO upregulates IFN genes and downregulates TF gene impeding cancer progression, suggesting the therapeutic potential of GSTO1 in cancers. It is worth indicating that GSTO1 also exerts its roles in cancers without its deglutathionylation activity. For example, GSTO1 was shown to mediate breast cancer stem cell enrichment potentially via protein-protein interaction [44,45].
Chemical Inhibitors.
GSTO1 upregulation in many tumors and its pro-inflammatory roles spurred the development of GSTO1 inhibitors. The active site of GSTO1, like a GST fold, retains a GSH-binding site (G-site) and a hydrophobic pocket (H-site) (Figure 1c) [24]. Notably, catalytic C32 in GSTO1 is one of the highly nucleophilic cysteines based on its reactivity with many electrophiles [46]. Therefore, various electrophilic scaffolds, including α-chloroacetamide [47,48], acrylamide [49], fluorosulfate [50], and cyclopropene [51], were utilized to develop GSTO1 inhibitors (see a review for inhibitors) [46].
C1–27.
Among many inhibitors, recent development and evaluation of C1–27 and its derivatives are notable (Figure 1c) [47]. Based on an α-chloroacetamide scaffold for cysteine reactivity, the Neamati group searched and screened a library of α-chloroacetamide-containing compounds, identifying C1–27 as a potent GSTO1 inhibitor (IC50 of 31 nM) that forms covalent conjugation with C32 (Figure 1c) [47]. Because of covalent inhibition, a C1–27 derivative with BODIPY was developed and used in proteome labeling, demonstrating that C1–27 is selective (up to 1 μM concentration) toward GSTO1 but with weak affinity or reactivity to protein disulfide isomerase (PDI). Co-crystal structural analysis showed that C1–27 primarily binds to H-site but with additional interactions with G-site (Figure 1c). Subsequently, evaluation in a panel of cancer cell lines (e.g., HT29 and HCT116) demonstrated that C1–27 inhibits cancer cell proliferation (GI50 of 1.2–4.3 μM) and the clonogenic survival of HCT116 (C1–27 in 0.2–3 μM). Additionally, C1–27 was tested for in vivo efficacy with colon cancer cell line-derived xenograft, where C1–27 (25–45 mg/kg) showed significant inhibition of tumor growth without toxicity or weight loss at its high concentration (45 mg/kg). C1–27 also showed modest inhibition of colorectal cancer patient-derived xenograft (KRAS-mutant CRM-13–180 PDX), demonstrating its anti-cancer activity.
The Board group recently used C1–27 to investigate the role of GSTO1 in NLRP3 inflammasome activation (Figure 1b) [27]. C1–27 inhibited IL-1β release in BMDM and peripheral blood mononuclear cells (PBMC) in response to LPS/ATP without affecting IkB degradation or TNFα production, supporting its selective modulation of NLRP3 complex than TLR4 signaling or other inflammasomes. Moreover, C1–27 reduced NLRP3 activation (i.e., IL-1β release) in mice with colitis and decreased the clinical score in mice with experimental autoimmune encephalomyelitis (EAE), supporting its in vivo efficacy.
49.
Although C1–27 shows potent GSTO1 inhibition, it was noted that C1–27 appears to have many off-targets, especially at high concentrations, likely due to α-chloroacetamide reactivity, thus retaining cytotoxicity even with GSTO1 knockdown [47]. Therefore, the Neamati group sought to replace α-chloroacetamide in C1–27 with acrylamide [49], which retains cysteine selectivity and is observed in many FDA-approved drugs [52]. However, co-crystal structural analyses indicated that simply installing acrylamide in C1–27 in place of α-chloroacetamide changes its binding orientation, thus impairing the binding potency. Therefore, through co-crystal structural analysis and synthesis, the group came to develop compound 49 (Figure 1c), which showed remarkable inhibition potency (IC50 = 0.22 nM) [49]. Despite its high potency, it is insightful to learn that highly potent compounds in vitro, including 49, did not display significant cytotoxicity (IC50 > 30 μM for 49) in HCT116 and HT29, an observation also seen with KT53 [53]. Despite such observation, 49 may be effective in 3D tumor models versus 2D cultures, as shown by GSTO1 KO cancer cell lines [31]. Alternatively, 49 would be an excellent candidate for GSTO1 inhibition in inflammatory diseases.
25.
The Board and Baell group independently developed selective GSTO1 inhibitors derived from C1–27, especially with drug-likeness analysis [48]. It was noted that C1–27 metabolic stability is low (half-life 2–7 min in human or mouse microsome). Pharmacokinetics (PK) analysis of C1–27 indicated its rapid absorption (maximum plasma concentration achieved at 5 min post-dose) and fast elimination (half-life < 15 min), suggesting its PK improvement is necessary. Through synthesis and evaluation, compound 25 (Figure 1c) was developed with high inhibitory potency for GSTO1 (IC50 = 0.25 μM; kinact = 0.85 min−1; KI = 0.61 μM) comparable to C1–27 (IC50 = 0.13 μM; kinact = 0.78 min−1; KI = 0.44 μM). Impressively, 25 showed significant inhibition of IL-1β release in BMDM (IL-1β = 3.4% relative to untreated at 5 μM) than C1–27 (IL-1β = ca. 65% at 5 μM). In addition, 25 showed improved PK with its higher concentration in plasma (half-life = 1.4 h) and slower elimination than C1–27, which is encouraging but still shows a limited in-vivo half-life. Nevertheless, both compounds would be instrumental for analyzing GSTO1 inhibition in inflammatory diseases and cancers.
2.2. Glutaredoxin 1 (Grx1)
Grx1, a mammalian glutaredoxin member, is a ubiquitous oxidoreductase with a redox-active CXXC motif [54]. Extensive mechanistic studies demonstrated Grx1 selectivity for glutathionylated cysteines or proteins as substrates, primarily via an encounter catalytic mechanism [55], while retaining two GSH bindings sites [56]. Therefore, Grx1 presumably has broad substrates for deglutathionylation and stands out as a central player of SSG, especially associated with inflammation [57–60] and fibrosis [61–63]. In this section, we highlight recent studies revealing the role of Grx1 and SSG in fibrosis while showcasing Grx1 inhibitors.
SMAD family member 3 (SMAD3).
SMAD3 is a member of the SMAD family that plays a central role in TGF-β signaling [64]. In the canonical TGF-β signaling (Figure 2a), TGF-β binds to TGF-β receptor I and II (TGFBR1 and TGFBR2) complex, which recruits and phosphorylates SMAD2 and SMAD3. The phosphorylated SMAD2 and SMAD3 gain interaction with SMAD4. The trimer increases its translocation to the nucleus for transcriptional activation of genes associated with diverse biological processes, including fibrosis [64].
Figure 2.
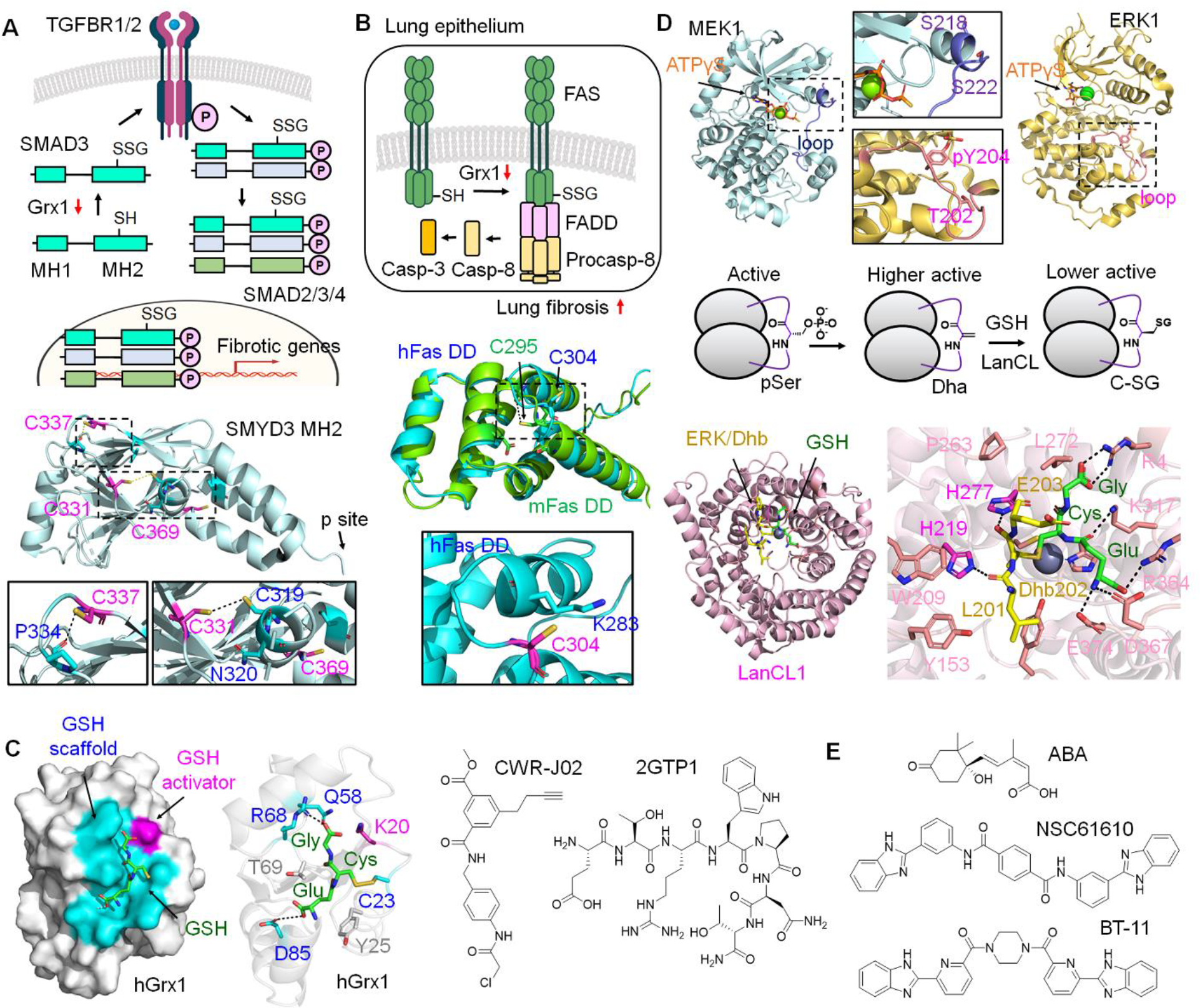
Grx1 and LanCL substrates and chemical modulators. A. Grx1 reduces SMAD3 SSG and decreases TGFβ-induced liver fibrosis. Liver fibrosis decreases Grx1 expression, which increases SMAD3 SSG. Glutathionylated SMAD3 increases its interaction with TGFBR1, enhancing SMAD3 phosphorylation. Phosphorylated SMAD3 forms a trimer made of SMAD2/3/4, which translocates to the nucleus and induces fibrotic gene expression. SMAD3 forms SSG potentially at C331, C337, and C369 in the MH2 domain (PDB: 1MJS) responsible for protein interactions (e.g., TGFBR1). C331 is within 4.0 Å from C319, which may form disulfide. B. Grx1 reduces lung fibrosis that correlates with Fas SSG elevation. Fas forms SSG at C295 (murine Fas) at its cytoplasmic death domain (DD). C295 in mouse Fas (mFas, green, UniProt alpha-fold structure) is equivalent to C304 in human Fas (hFas, blue, PDB: 1DDF). Fas SSG enhances Fas complex formation with FADD and pro-caspase-8, which activates caspase-3 and induces lung epithelium apoptosis responsible for stimulating lung fibrosis. C. Grx1 inhibitors. Grx1 has two GSH binding sites, a GSH scaffold site (cyan) and a GSH activator site (including K19, pink), with catalytic C23 that forms disulfide with GSH during deglutathionylation (PDB: 1B4Q). α-chloroacetamide-containing Grx1 inhibitor, CWR-J02, was developed that covalently inhibits Grx1. 7-mer peptide Grx1 inhibitor (ETRWPNT, named 2GTP1) was developed that reversibly inhibits Grx1. D. LanCL-induced C-glutathionylation (C-SG) in MEK1 and ERK1. pSer and pThr in the activation loops of MEK1 (PDB: 3EQD) and ERK1 (PDB: 2ZOQ) are susceptible to forming Dha/Dhb, which increases kinase activity. LanCL catalyzes C-SG formation between GSH and Dha/Dhb, which reduces kinase activity, thus preventing kinase hyperactivation. The co-crystal structure shows LanCL (light pink) and GSH (green) conjugated to Dhb-containing peptide (GFL-Dhb-EY, yellow) derived from the activation loop (GFL-T202-EY) in ERK1 (PDB: 8CZK). His219 and His277 (pink) play a role in stabilizing an enolate intermediate, while the zinc atom (gray sphere) activates GSH nucleophilicity. Dotted lines indicate a distance less than 4 Å. E. LanCL ligands. Hormone abscisic acid (ABA) exerts anti-inflammatory and anti-diabetic roles via binding to LanCL2 (and likely LanCL1). Two compounds (NSC61610 and BT-11) were developed that bind to LanCL2, showing their anti-inflammatory activities.
Xi and Xie et al. reported that liver fibrosis correlates with low expression of Grx1 and high SSG, whereas pirfenidone, an anti-fibrotic drug, inhibited liver fibrosis via Grx1 elevation [62]. Grx1 KO mice increased sensitivity to liver fibrosis and showed high activation of hepatic stellate cells (HSC) that produce profibrogenic cytokines. Conversely, Grx1 WT overexpression, but not inactive Grx1 C23S, to mice or HSC inhibited liver fibrotic progression and HSC activation. Biochemically, Grx1 overexpression in HSC reduced SMAD3-TGFBR1 interaction, SMAD3 phosphorylation, and SMAD3 nuclear localization in response to TGF-β. Subsequent analysis demonstrated that SMAD3 is a Grx1 substrate and susceptible to forming SSG at potentially C332, C338, and C370 in its MH2 domain (Figure 2a), which is responsible for interacting with protein partners, including TGFBR1/2 [64]. Accordingly, a SMAD3 cysteine mutant (SMAD3 C332A/C338A/C370A) failed to induce basal or TGF-β-stimulated gene expression [62], suggesting SMAD3 SSG increases TGF-β-stimulated SMAD3 phosphorylation and fibrogenic gene expression (Figure 2a). This observation reports the significant role of Grx1 and SSG in the progression of liver fibrosis and suggests the therapeutic potential of Grx1 overexpression in fibrotic pathogenesis.
Fas.
Fas is a cell-death receptor belonging to the tumor necrosis factor receptor (TNFR) superfamily with a conserved death domain (DD) in its cytoplasmic tail [65]. Fas activation by Fas ligand stimulates signaling complex formation comprised of Fas, Fas-associated DD (FADD), and pro-caspase-8, resulting in activation of caspase-8 and -3 and subsequent apoptosis (Figure 2b) [65]. Previously, Fas SSG was observed at its cysteine 295 (murine Fas C295) (Figure 2b) as a consequence of Grx1 degradation, promoting Fas aggregation and signaling complex formation, and propagating apoptosis [66].
Recently, the Janssen-Heininger group reported that Grx1 expression was significantly decreased in lung tissues of individuals with idiopathic pulmonary fibrosis (IPF) in accordance with Fas SSG elevation (Figure 2b) [61]. Similarly, mice lacking Grx1 were more susceptible to pulmonary fibrosis induced by TGF-β or bleomycin while displaying Fas SSG elevation and caspase-3 activation. Notably, direct administration of recombinant Grx1 WT, not C23S, into the mouse airway resulted in Grx1 elevation in alveolar regions while reducing IPF progression. Therefore, this report supports that Fas SSG may enhance lung epithelial cell apoptosis, increasing lung fibrogenesis, which suggests the anti-fibrotic effect of Grx1. Similarly, Fas SSG elevation and Grx1 ablation were also observed in hepatic injury with ethanol exposure [67].
Chemical Inhibitors.
The central role of Grx1 in SSG and redox biology, along with its pro-inflammatory role, drove the development of Grx1 inhibitors. However, a lack of deep substrate binding pockets in the Grx1 active site (Figure 2c), combined with its encounter mechanism that indicates no significant substrate binding interaction during catalysis [55], challenges developing selective inhibitors with high binding affinity. Therefore, available inhibitors mainly depend on their reactivity with redox-active C23 in Grx1 [68,69], thus difficult to achieve high selectivity considering many nucleophilic cysteines in the proteome. In this section, recent examples of Grx1 inhibitors are introduced.
CWR-J02.
Screening >500 thiol-reactive compounds in Grx1 enzyme assay identified a chloroacetamide-derived covalent inhibitor, named CWR-J02 (Figure 2c), with modest potency (IC50 = 32 μM, KI = 40 μM, kinact = 0.5 min−1 for Grx assay), which conjugates with C23 [68]. Molecular dynamics suggested potential reversible interactions of CWR-J02 with Grx1 before covalent inactivation. CWR-J02 (32 μM) was then shown to inhibit Grx1 activity and LPS-induced inflammatory cytokines (TNF-α, IL-6, and IL-1β) in BV2 microglial cells. However, the anti-inflammatory effect of CWR-J02 was also observed even after Grx1 knockdown (siRNA), suggesting CWR-J02 exerts its effects via other targets in addition to Grx1. For example, the chloroacetamide in CWR-J02 may inhibit GSTO1 by its cysteine reactivity.
Peptide 2GTP1.
Although non-covalent Grx1 inhibition without cysteine conjugation was considered impractical [68], our group developed a peptide-based non-covalent Grx1 inhibitor devoid of an electrophilic group [70]. Phage library with linear 7-mer peptides was screened against Grx1 or Trx1, evolving peptide binders of Grx1 and Trx1, which resulted in finding one 7-mer peptide (ETRWPNT, named 2GTP1, Figure 2c) that binds to both Grx1 (KD = 1.2 μM) and Trx1 (KD = 2.5 μM). 2GTP1 showed its selective binding to Grx1 and Trx1 over Grx3. Interestingly, 2GTP1-Grx1 interaction was preserved upon incubating GSH (1–5 mM), whereas 2GTP1 did not retain its binding with oxidized Grx1, supporting 2GTP1’s binding to reduced Grx1. The enzyme assays confirmed that 2GTP1 inhibits in vitro Grx1 deglutathionylation activity (IC50 = 11 μM) and Trx1 disulfide reduction activity, albeit only ca. 50% Trx1 inhibition even at high concentrations, suggesting more selective inhibition of Grx1 than Trx1. Interestingly, TAT-derived 2GTP1 (TAT-2GTP1) did not induce ROS elevation nor endoplasmic reticulum (ER) stress, as opposed to electrophilic compounds, suggesting that it may not induce or change global SSG in the proteome. However, 2GTP1 is unique for its non-covalent interaction with Grx1, thus suggesting its potential use in developing selective Grx1 inhibitors.
2.3. LanC-like protein 1 and 2 (LanCL 1 and 2)
Eukaryotic LanCLs are orthologs of bacterial LanC enzymes that catalyze thioether formation in lanthipeptide biosynthesis [71]. However, such bacterial enzyme activity is not conserved in eukaryotic LanCL [72], and the biochemistry of LanCL has been a mystery for many years. Nonetheless, LanCL interaction with GSH was noted early [73], suggesting LanCL may be involved in redox biology. Recent findings report that LanCL catalyzes C-glutathionylation (C-SG). This section will present LanCL substrates for C-SG and chemical ligands.
MEK and ERK.
It has been observed that phosphoserine (pSer) and phosphothreonine (pThr) in long-lived proteins in the eye and other tissues spontaneously undergo β-elimination of phosphate, resulting in 2,3-dehydroalanine (Dha) or 2,3-didehydrobutyrine (Dhb), respectively (Figure 2d) [74]. Dha and Dhb can react and form a thioether bond with intracellular GSH via the Michael addition, called C-SG (Figure 2d) [75]. Alternatively, pathogenic bacteria retain pThr lyases (e.g., SpvC, OspF, VirA, and HopA1), which catalyze the formation of Dhb in phosphorylated kinases, likely disrupting kinase signaling during pathogenesis [76,77]. Recently, the van der Donk and Davis group reported that LanCL catalyzes C-SG between GSH and Dha/Dhb in kinases (Figure 2d) [78]. LanCL1/2 were found to interact physically with many kinases, including MEK1/2 and ERK1/2. To investigate LanCL biochemistry in vitro, recombinant MEK1 and ERK1 with Dha or Dhb at the sites of pSer and pThr in activation loops were prepared (Figure 2d). LanCL1/2 increased the rate of C-SG formation in Dha/Dhb-containing MEK1 and ERK1. Functionally, MEK1 with Dha in the activation loop displayed higher activity, whereas MEK1 with C-SG reduced its kinase activity versus its Dha form (Figure 2d), suggesting that LanCL prevents MEK1 hyperactivation via forming C-SG. Structural and biochemical analyses support that LanCL binds first with GSH (KD = 9 μM) in the active site, where a zinc ion increases GSH nucleophilicity, and His277 and His219 play a role in stabilizing and interacting with an enolate intermediate (Figure 2d) [79]. Notably, the active site recognizes a β-turn posed by dehydroamino acid in substrates without significant sequence preference [79], suggesting a broad range of LanCL1 substrates for C-SG.
Chemical Inducers.
Besides LanCL’s role in C-SG, LanCL2 has been known as a target for a terpenoid hormone, abscisic acid (ABA) (Figure 2e), for many years [80]. Studies demonstrated that LanCL2 (and likely LanCL1) is necessary for ABA’s actions in anti-inflammation (e.g., activating PPARγ) and anti-diabetes (e.g., insulin-releasing activity) [80,81]. In addition, irrespective of ABA, LanCL is also implicated in pro-survival (i.e., stimulating Akt via mTORC2) [82] and anti-apoptosis (i.e., suppressing JNK) [83]. Thus, these analyses stimulated the development of LanCL2 chemical ligands.
BT-11.
The Bassaganya-Riera group used virtual screening with LanCL2, identifying the compound NSC61610 (Figure 2e), which induced PPARγ genes for anti-inflammatory function in immune cells and mice with inflammatory bowel diseases (IBD) [84]. Next, due to the limited solubility of NSC61610, its derivatives with drug-likeness were sought, resulting in developing BT-11 (Figure 2e) [85]. BT-11 directly binds to LanCL2 (KD = 7.7 μM). BT-11 suppressed TNF-α-stimulated inflammation in splenocytes expressing LanCL2 versus its KO. BT-11 also showed protective efficacy in several IBD models of mice expressing LanCL2 versus KO [85]. Further studies demonstrated BT-11’s anti-inflammatory effects via stabilizing CD4+ T cell subsets, namely lamina propria regulatory T cells (Tregs), and suppressing inflammatory cytokines in PBMC isolated from Crohn’s disease patients [86]. BT-11 is orally active and did not cause clinical signs of toxicity in rats and dogs [87]. Phase I clinical trial (healthy volunteers, n=7) was positive without serious adverse events or dose-limiting toxicities up to 100 mg/kg [88]. Interestingly, BT-11 is considered a LanCL2 activator probably because it increases LanCL2 expression level [86], although its exact mechanism of action is unclear. Therefore, it is not known whether the anti-inflammatory effects of LanCL2 or BT-11 are related to glutathione, C-SG, or redox biology. However, because of its direct binding affinity to LanCL2 (and likely LanCL1 due to their high structural similarity), BT-11 could be a useful chemical tool to investigate LanCL1/2 in the research of C-SG.
Conclusion
Protein SSG is a central protein oxidation regulating physiology and contributing to diseases [4]. The field of SSG has broadened with the development of proteomic approaches [89–91] for identifying glutathionylated cysteines (n > 2,000 in databases [92]) and functional analyses of SSG in individual proteins. Impressively, recent biological studies of SSG enzymes (i.e., Grx1 and GSTO1) unveiled significant roles of SSG in regulating inflammation, fibrosis, and neurodegeneration. In parallel, chemistry has enabled significant progress in developing selective SSG enzyme inhibitors while demonstrating their positive therapeutic potential. Although not discussed here, it deserves to point out important SSG biology associated with other SSG enzymes (e.g., Grx2 [93,94] and GSTP [95,96]). Lastly, LanCL and C-SG open up new research directions in the field of SSG. Future studies will continue investigating SSG and its regulatory enzymes while developing their therapeutic inhibitors.
Highlights.
GSTO1 and Grx1 are central players of protein S-glutathionylation
Protein S-glutathionylation substrates of GSTO1 and Grx1 regulate inflammation, fibrosis, cancer, or neurodegeneration.
Chemical inhibitors of GSTO1 and Grx1 were developed and investigated in inflammatory diseases and cancers.
The first regulatory enzyme, LanCL, for C-glutathionylation was uncovered.
Acknowledgments
This work was supported by the National Institute of Health (NIH) [R01 HL131740 (Y.-H.A) and R01 GM143214 (Y.-H.A)].
Abbreviation
- Nf-kB
nuclear factor kappaB
- IkB
inhibitor of Nf-kB
- IKK-β
IkB kinase beta subunit
- SERCA
sarcoendoplasmic reticulum calcium ATPase
- CHOP
CCAAT/enhancer-binding protein homologous protein
- NIMA
never in mitosis gene a
- NLRP3
nucleotide-binding domain, leucine-rich-containing family, pyrin domain containing 3
- IL-1β
interleukin-1β
- IL-18
interleukin-18
- ASC
apoptosis-associated speck-like protein containing a CARD
- TLR4
toll-like receptor 4
- TNFα
tumor necrosis factor alpha
- SMAD
suppressor of mothers against decapentaplegic
- TGFβ
transforming growth factor beta
- Trx1
thioredoxin 1
- TAT
trans-activator of transcription
- MEK1
mitogen-activated protein kinase kinase
- ERK
extracellular signal-regulated kinase
- PPARγ
peroxisome proliferator-activated receptor-gamma
- Akt
Ak strain transforming or protein kinase B
- mTORC2
mammalian target of rapamycin complex 2
- JNK
Jun N-terminal kinase
Footnotes
Competing interests
Authors declare no competing interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Schieber M, Chandel NS: ROS function in redox signaling and oxidative stress. Curr Biol (2014) 24(10):R453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garel MC, Domenget C, Caburi-Martin J, Prehu C, Galacteros F, Beuzard Y: Covalent binding of glutathione to hemoglobin. I. Inhibition of hemoglobin S polymerization. J Biol Chem (1986) 261(31):14704–14709. [PubMed] [Google Scholar]
- 3.Craescu CT, Poyart C, Schaeffer C, Garel MC, Kister J, Beuzard Y: Covalent binding of glutathione to hemoglobin. II. Functional consequences and structural changes reflected in NMR spectra. J Biol Chem (1986) 261(31):14710–14716. [PubMed] [Google Scholar]
- 4.Kukulage DSK, Matarage Don NNJ, Ahn YH: Emerging chemistry and biology in protein glutathionylation. Curr Opin Chem Biol (2022) 71:102221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mailloux RJ: Protein S-glutathionylation reactions as a global inhibitor of cell metabolism for the desensitization of hydrogen peroxide signals. Redox Biol (2020) 32:101472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adachi T, Pimentel DR, Heibeck T, Hou X, Lee YJ, Jiang B, Ido Y, Cohen RA: S-glutathiolation of Ras mediates redox-sensitive signaling by angiotensin II in vascular smooth muscle cells. J Biol Chem (2004) 279(28):29857–29862. [DOI] [PubMed] [Google Scholar]
- 7.Yang Y, Dong X, Zheng S, Sun J, Ye J, Chen J, Fang Y, Zhao B, Yin Z, Cao P, Luo L: GSTpi regulates VE-cadherin stabilization through promoting S-glutathionylation of Src. Redox Biol (2020) 30:101416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abdelsaid MA, El-Remessy AB: S-glutathionylation of LMW-PTP regulates VEGF-mediated FAK activation and endothelial cell migration. J Cell Sci (2012) 125(20):4751–4760. [DOI] [PubMed] [Google Scholar]
- 9.Sakai J, Li J, Subramanian KK, Mondal S, Bajrami B, Hattori H, Jia Y, Dickinson BC, Zhong J, Ye K, Chang CJ et al. : Reactive oxygen species-induced actin glutathionylation controls actin dynamics in neutrophils. Immunity (2012) 37(6):1037–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reynaert NL, van der Vliet A, Guala AS, McGovern T, Hristova M, Pantano C, Heintz NH, Heim J, Ho YS, Matthews DE, Wouters EF et al. : Dynamic redox control of NF-kB through glutaredoxin-regulated S-glutathionylation of inhibitory kB kinase beta. Proc Natl Acad Sci USA (2006) 103(35):13086–13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Checconi P, Limongi D, Baldelli S, Ciriolo MR, Nencioni L, Palamara AT: Role of glutathionylation in infection and inflammation. Nutrients (2019) 11(8):1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Velu CS, Niture SK, Doneanu CE, Pattabiraman N, Srivenugopal KS: Human p53 is inhibited by glutathionylation of cysteines present in the proximal DNA-binding domain during oxidative stress. Biochemistry (2007) 46(26):7765–7780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schoneich C, Cohen RA: S-glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med (2004) 10(11):1200–1207. [DOI] [PubMed] [Google Scholar]
- 14.Alegre-Cebollada J, Kosuri P, Giganti D, Eckels E, Rivas-Pardo JA, Hamdani N, Warren CM, Solaro RJ, Linke WA, Fernandez JM: S-glutathionylation of cryptic cysteines enhances titin elasticity by blocking protein folding. Cell (2014) 156(6):1235–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loescher CM, Breitkreuz M, Li Y, Nickel A, Unger A, Dietl A, Schmidt A, Mohamed BA, Kotter S, Schmitt JP, Kruger M et al. : Regulation of titin-based cardiac stiffness by unfolded domain oxidation (UnDOx). Proc Natl Acad Sci USA (2020) 117(39):24545–24556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rashdan NA, Shrestha B, Pattillo CB: S-glutathionylation, friend or foe in cardiovascular health and disease. Redox Biol (2020) 37:101693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chia SB, Elko EA, Aboushousha R, Manuel AM, van de Wetering C, Druso JE, van der Velden J, Seward DJ, Anathy V, Irvin CG, Lam YW et al. : Dysregulation of the glutaredoxin/S-glutathionylation redox axis in lung diseases. Am J Physiol Cell Physiol (2020) 318(2):C304–C327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hayes JD, Dinkova-Kostova AT, Tew KD: Oxidative stress in cancer. Cancer Cell (2020) 38(2):167–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cha SJ, Kim H, Choi HJ, Lee S, Kim K: Protein glutathionylation in the pathogenesis of neurodegenerative diseases. Oxid Med Cell Longev (2017) 2017:2818565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gallogly MM, Mieyal JJ: Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr Opin Pharmacol (2007) 7(4):381–391. [DOI] [PubMed] [Google Scholar]
- 21.Tew KD, Manevich Y, Grek C, Xiong Y, Uys J, Townsend DM: The role of glutathione S-transferase P in signaling pathways and S-glutathionylation in cancer. Free Radic Biol Med (2011) 51(2):299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shelton MD, Chock PB, Mieyal JJ: Glutaredoxin: role in reversible protein S-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid Redox Signal (2005) 7(3–4):348–366. [DOI] [PubMed] [Google Scholar]
- 23.Menon D, Board PG: A role for glutathione transferase omega 1 (GSTO1–1) in the glutathionylation cycle. J Biol Chem (2013) 288(36):25769–25779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Board PG, Coggan M, Chelvanayagam G, Easteal S, Jermiin LS, Schulte GK, Danley DE, Hoth LR, Griffor MC, Kamath AV, Rosner MH et al. : Identification, characterization, and crystal structure of the omega class glutathione transferases. J Biol Chem (2000) 275(32):24798–24806. [DOI] [PubMed] [Google Scholar]
- 25.Whitbread AK, Masoumi A, Tetlow N, Schmuck E, Coggan M, Board PG: Characterization of the omega class of glutathione transferases. Methods Enzymol (2005) 401:78–99. [DOI] [PubMed] [Google Scholar]
- 26.Board PG, Menon D: Structure, function and disease relevance of omega-class glutathione transferases. Arch Toxicol (2016) 90(5):1049–1067. [DOI] [PubMed] [Google Scholar]
- 27. Hughes MM, Hooftman A, Angiari S, Tummala P, Zaslona Z, Runtsch MC, McGettrick AF, Sutton CE, Diskin C, Rooke M, Takahashi S et al. : Glutathione transferase omega-1 regulates NLRP3 inflammasome activation through NEK7 deglutathionylation. Cell Rep (2019) 29(1):151–161. •• This paper reports GSTO1 selectively regulating NLRP3 inflammasome via NEK7 glutathionylation and investigates the effects of C1–27 and GSTO1 KO in the inflammatiory mouse models
- 28.Hughes MM, McGettrick AF, O’Neill LAJ: Glutathione and glutathione transferase omega 1 as key posttranslational regulators in macrophages. Microbiol Spectr (2017) 5(1):MCHD-0044–2016. [DOI] [PubMed] [Google Scholar]
- 29.Li S, Wang L, Xu Z, Huang Y, Xue R, Yue T, Xu L, Gong F, Bai S, Wu Q, Liu J et al. : ASC deglutathionylation is a checkpoint for NLRP3 inflammasome activation. J Exp Med (2021) 218(9): e20202637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Menon D, Coll R, O’Neill LA, Board PG: GSTO1–1 modulates metabolism in macrophages activated through the LPS and TLR4 pathway. J Cell Sci (2015) 128(10):1982–1990. [DOI] [PubMed] [Google Scholar]
- 31. Xu Y, Bankhead A 3rd, Tian X, Tang J, Ljungman M, Neamati N: Deletion of glutathione S-transferase omega 1 activates type I interferon genes and downregulates tissue factor. Cancer Res (2020) 80(17):3692–3705. • This paper reports GSTO1 upregulation in cancer patients and describes biological pathway analysis of GSTO1 KO in three cancer cell lines and xenograft models, supporting the role of GSTO1 in cancer progression.
- 32.Wang K, Zhang FL, Jia W: Glutathione S-transferase omega 1 promotes the proliferation, migration and invasion, and inhibits the apoptosis of non-small cell lung cancer cells, via the JAK/STAT3 signaling pathway. Mol Med Rep (2021) 23(1):71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cha SJ, Lee S, Choi HJ, Han YJ, Jeon YM, Jo M, Lee S, Nahm M, Lim SM, Kim SH, Kim HJ et al. : Therapeutic modulation of GSTO activity rescues FUS-associated neurotoxicity via deglutathionylation in ALS disease models. Dev Cell (2022) 57(6):783–798. •• This paper reports the protective role of GSTO1 in ALS and neurodegeneration via FUS deglutathionylation in Drosophila and ALS-associated patient neurons and suggests the therapeutic potential of GSTO1 upregulation in ALS pathogenesis.
- 34.Cha SJ, Yoon JH, Han YJ, Kim K: Knockdown of glutathione S-transferase leads to mislocalization and accumulation of cabeza, a drosophila homolog of FUS, in the brain. J Neurogenet (2022) 1–5. [DOI] [PubMed] [Google Scholar]
- 35.Crozat A, Aman P, Mandahl N, Ron D: Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature (1993) 363(6430):640–644. [DOI] [PubMed] [Google Scholar]
- 36.Dormann D, Haass C: Fused in sarcoma (FUS): an oncogene goes awry in neurodegeneration. Mol Cell Neurosci (2013) 56:475–486. [DOI] [PubMed] [Google Scholar]
- 37.Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J et al. : Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science (2009) 323(5918):1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun Z, Diaz Z, Fang X, Hart MP, Chesi A, Shorter J, Gitler AD: Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol (2011) 9(4):e1000614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun Z, Gong W, Zhang Y, Jia Z: Physiological and pathological roles of mammalian NEK7. Front Physiol (2020) 11:606996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He Y, Zeng MY, Yang D, Motro B, Nunez G: NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature (2016) 530(7590):354–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swanson KV, Deng M, Ting JP: The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 19(8):477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parker BS, Rautela J, Hertzog PJ: Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer (2016) 16(3):131–144. [DOI] [PubMed] [Google Scholar]
- 43.Kasthuri RS, Taubman MB, Mackman N: Role of tissue factor in cancer. J Clin Oncol (2009) 27(29):4834–4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu H, Chen I, Shimoda LA, Park Y, Zhang C, Tran L, Zhang H, Semenza GL: Chemotherapy-induced Ca(2+) release stimulates breast cancer stem cell enrichment. Cell Rep (2017) 18(8):1946–1957. [DOI] [PubMed] [Google Scholar]
- 45.Manupati K, Debnath S, Goswami K, Bhoj PS, Chandak HS, Bahekar SP, Das A: Glutathione S-transferase omega 1 inhibition activates JNK-mediated apoptotic response in breast cancer stem cells. FEBS J (2019) 286(11):2167–2192. [DOI] [PubMed] [Google Scholar]
- 46.Xie Y, Dahlin JL, Oakley AJ, Casarotto MG, Board PG, Baell JB: Reviewing hit discovery literature for difficult targets: glutathione transferase omega-1 as an example. J Med Chem (2018) 61(17):7448–7470. [DOI] [PubMed] [Google Scholar]
- 47. Ramkumar K, Samanta S, Kyani A, Yang S, Tamura S, Ziemke E, Stuckey JA, Li S, Chinnaswamy K, Otake H, Debnath B et al. : Mechanistic evaluation and transcriptional signature of a glutathione S-transferase omega 1 inhibitor. Nat Commun (2016) 7:13084. • This paper reports the development of a potent GSTO1 inhibitor, C1–27, with its evaluation in cancer cell line-derived xenograft models and the mechanistic pathway analysis upon GSTO1 inhibition.
- 48.Xie Y, Tummala P, Oakley AJ, Deora GS, Nakano Y, Rooke M, Cuellar ME, Strasser JM, Dahlin JL, Walters MA, Casarotto MG et al. : Development of benzenesulfonamide derivatives as potent glutathione transferase omega-1 inhibitors. J Med Chem (2020) 63(6):2894–2914. [DOI] [PubMed] [Google Scholar]
- 49. Dai W, Samanta S, Xue D, Petrunak EM, Stuckey JA, Han Y, Sun D, Wu Y, Neamati N: Structure-based design of N-(5-phenylthiazol-2-yl)acrylamides as novel and potent glutathione S-transferase omega 1 inhibitors. J Med Chem (2019) 62(6):3068–3087. • This paper describes the design and development of a highly potent GSTO inhibitor, 49, through elegant medicinal chemistry combined with co-crystal structural analysis and biological evaluation
- 50.Mortenson DE, Brighty GJ, Plate L, Bare G, Chen W, Li S, Wang H, Cravatt BF, Forli S, Powers ET, Sharpless KB et al. : “Inverse drug discovery” strategy to identify proteins that are targeted by latent electrophiles as exemplified by aryl fluorosulfates. J Am Chem Soc (2018) 140(1):200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wormer GJ, Hansen BK, Palmfeldt J, Poulsen TB: A cyclopropene electrophile that targets glutathione S-transferase omega-1 in cells. Angew Chem Int Ed Engl (2019) 58(34):11918–11922. [DOI] [PubMed] [Google Scholar]
- 52.Jackson PA, Widen JC, Harki DA, Brummond KM: Covalent modifiers: a chemical perspective on the reactivity of alpha, beta-unsaturated carbonyls with thiols via hetero-Michael addition reactions. J Med Chem (2017) 60(3):839–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsuboi K, Bachovchin DA, Speers AE, Spicer TP, Fernandez-Vega V, Hodder P, Rosen H, Cravatt BF: Potent and selective inhibitors of glutathione S-transferase omega 1 that impair cancer drug resistance. J Am Chem Soc (2011) 133(41):16605–16616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lillig CH, Berndt C, Holmgren A: Glutaredoxin systems. Biochim Biophys Acta (2008) 1780(11):1304–1317. [DOI] [PubMed] [Google Scholar]
- 55.Gallogly MM, Starke DW, Mieyal JJ: Mechanistic and kinetic details of catalysis of thiol-disulfide exchange by glutaredoxins and potential mechanisms of regulation. Antioxid Redox Signal (2009) 11(5):1059–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Begas P, Liedgens L, Moseler A, Meyer AJ, Deponte M: Glutaredoxin catalysis requires two distinct glutathione interaction sites. Nat Commun (2017) 8:14835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guo Y, Liu Y, Zhao S, Xu W, Li Y, Zhao P, Wang D, Cheng H, Ke Y, Zhang X: Oxidative stress-induced FABP5 S-glutathionylation protects against acute lung injury by suppressing inflammation in macrophages. Nat Commun (2021) 12(1):7094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luo Y, Mo D, Guo H, Ye C, Chen B, Zhu H, Deng C, Deng Q, Guo C, Qiu L: NF-kappaB inactivation attenuates the M1 macrophage polarization in experimental necrotizing enterocolitis by glutaredoxin-1 deficiency. Cell Biol Int (2022) 46(11):1886–1899. [DOI] [PubMed] [Google Scholar]
- 59.Ahn YJ, Wang L, Tavakoli S, Nguyen HN, Short JD, Asmis R: Glutaredoxin 1 controls monocyte reprogramming during nutrient stress and protects mice against obesity and atherosclerosis in a sex-specific manner. Nat Commun (2022) 13(1):790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Manuel AM, van de Wetering C, MacPherson M, Erickson C, Murray C, Aboushousha R, van der Velden J, Dixon AE, Poynter ME, Irvin CG, Taatjes DJ et al. : Dysregulation of pyruvate kinase M2 promotes inflammation in a mouse model of obese allergic asthma. Am J Respir Cell Mol Biol (2021) 64(6):709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Anathy V, Lahue KG, Chapman DG, Chia SB, Casey DT, Aboushousha R, van der Velden JLJ, Elko E, Hoffman SM, McMillan DH, Jones JT et al. : Reducing protein oxidation reverses lung fibrosis. Nat Med (2018) 24(8):1128–1135. •• This paper describes reduced Grx1 expression and Fas-SSG elevation in correlation with pulmonary fibrosis. The paper also reports the protective outcome of recombinant Grx1 administration to the lung airway against TGFβ-induced fibrosis.
- 62. Xi Y, Li Y, Xu P, Li S, Liu Z, Tung HC, Cai X, Wang J, Huang H, Wang M, Xu M et al. : The anti-fibrotic drug pirfenidone inhibits liver fibrosis by targeting the small oxidoreductase glutaredoxin-1. Sci Adv (2021) 7(36):eabg9241. •• This paper describes Grx1 downregulation and SMAD3 SSG elevation associated with liver fibrosis. The paper also describes Grx1 upregulation by anti-fibrotic pirfenidone and the role of SMAD3 SSG in enhancing TGFβ-induced liver fibrosis.
- 63.Tsukahara Y, Ferran B, Minetti ET, Chong BSH, Gower AC, Bachschmid MM, Matsui R: Administration of glutaredoxin-1 attenuates liver fibrosis caused by aging and non-alcoholic steatohepatitis. Antioxidants (2022) 11(5):867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Massague J: TGFbeta signalling in context. Nat Rev Mol Cell Biol (2012) 13(10):616–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Strasser A, Jost PJ, Nagata S: The many roles of FAS receptor signaling in the immune system. Immunity (2009) 30(2):180–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Anathy V, Aesif SW, Guala AS, Havermans M, Reynaert NL, Ho YS, Budd RC, Janssen-Heininger YM: Redox amplification of apoptosis by caspase-dependent cleavage of glutaredoxin 1 and S-glutathionylation of Fas. J Cell Biol (2009) 184(2):241–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sun X, Ye C, Deng Q, Chen J, Guo C: Contribution of glutaredoxin-1 to Fas S-glutathionylation and inflammation in ethanol-induced liver injury. Life Sci (2021) 264:118678. [DOI] [PubMed] [Google Scholar]
- 68.Gorelenkova Miller O, Cole KS, Emerson CC, Allimuthu D, Golczak M, Stewart PL, Weerapana E, Adams DJ, Mieyal JJ: Novel chloroacetamido compound CWR-J02 is an anti-inflammatory glutaredoxin-1 inhibitor. PLoS One (2017) 12(11):e0187991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Haffo L, Lu J, Bykov VJN, Martin SS, Ren X, Coppo L, Wiman KG, Holmgren A: Inhibition of the glutaredoxin and thioredoxin systems and ribonucleotide reductase by mutant p53-targeting compound APR-246. Sci Rep (2018) 8(1):12671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kekulandara DN, Nagi S, Seo H, Chow CS, Ahn YH: Redox-Inactive peptide disrupting Trx1-Ask1 interaction for selective activation of stress signaling. Biochemistry (2018) 57(5):772–780. • This paper describes the first non-covalent reversible peptide inhibitor of Grx1 developed via phage display.
- 71.van der Donk WA, Nair SK: Structure and mechanism of lanthipeptide biosynthetic enzymes. Curr Opin Struct Biol (2014) 29:58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.He C, Zeng M, Dutta D, Koh TH, Chen J, van der Donk WA: LanCL proteins are not involved in lanthionine synthesis in mammals. Sci Rep (2017) 7:40980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chung CH, Kurien BT, Mehta P, Mhatre M, Mou S, Pye QN, Stewart C, West M, Williamson KS, Post J, Liu L et al. : Identification of lanthionine synthase C-like protein-1 as a prominent glutathione binding protein expressed in the mammalian central nervous system. Biochemistry (2007) 46(11):3262–3269. [DOI] [PubMed] [Google Scholar]
- 74.Siodlak D: alpha, beta-Dehydroamino acids in naturally occurring peptides. Amino Acids (2015) 47(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Townsend DM, Lushchak VI, Cooper AJ: A comparison of reversible versus irreversible protein glutathionylation. Adv Cancer Res (2014) 122:177–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhu Y, Li H, Long C, Hu L, Xu H, Liu L, Chen S, Wang DC, Shao F: Structural insights into the enzymatic mechanism of the pathogenic MAPK phosphothreonine lyase. Mol Cell (2007) 28(5):899–913. [DOI] [PubMed] [Google Scholar]
- 77.Chambers KA, Abularrage NS, Scheck RA: Selectivity within a family of bacterial phosphothreonine lyases. Biochemistry (2018) 57(26):3790–3796. [DOI] [PubMed] [Google Scholar]
- 78. Lai KY, Galan SRG, Zeng Y, Zhou TH, He C, Raj R, Riedl J, Liu S, Chooi KP, Garg N, Zeng M et al. : LanCLs add glutathione to dehydroamino acids generated at phosphorylated sites in the proteome. Cell (2021) 184(10):2680–2695. •• This paper reports the first eukaryotic enzyme, LanCL, catalyzing C-glutathionylation between dehydroamino acids and GSH at the activation loop of kinases (ERK1 and MEK1) and the biological implication of LanCL1 and C-glutathionylation.
- 79. Ongpipattanakul C, Liu S, Luo Y, Nair SK, van der Donk WA: The mechanism of thia-Michael addition catalyzed by LanC enzymes. Proc Natl Acad Sci USA (2023) 120(3):e2217523120. • This paper reports co-crystal structures of LanCL1 with GSH and a dehydroamino acid-containing peptide substrate and the enzyme mechanism of LanCL1 in catalyzing C-glutathionylation.
- 80.Sturla L, Fresia C, Guida L, Bruzzone S, Scarfi S, Usai C, Fruscione F, Magnone M, Millo E, Basile G, Grozio A et al. : LANCL2 is necessary for abscisic acid binding and signaling in human granulocytes and in rat insulinoma cells. J Biol Chem (2009) 284(41):28045–28057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bassaganya-Riera J, Guri AJ, Lu P, Climent M, Carbo A, Sobral BW, Horne WT, Lewis SN, Bevan DR, Hontecillas R: Abscisic acid regulates inflammation via ligand-binding domain-independent activation of peroxisome proliferator-activated receptor gamma. J Biol Chem (2011) 286(4):2504–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zeng M, van der Donk WA, Chen J: Lanthionine synthetase C-like protein 2 (LanCL2) is a novel regulator of Akt. Mol Biol Cell (2014) 25(24):3954–3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang J, Xiao Q, Chen X, Tong S, Sun J, Lv R, Wang S, Gou Y, Tan L, Xu J, Fan C et al. : LanCL1 protects prostate cancer cells from oxidative stress via suppression of JNK pathway. Cell Death Dis (2018) 9(2):197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lu P, Hontecillas R, Horne WT, Carbo A, Viladomiu M, Pedragosa M, Bevan DR, Lewis SN, Bassaganya-Riera J: Computational modeling-based discovery of novel classes of anti-inflammatory drugs that target lanthionine synthetase C-like protein 2. PLoS One (2012) 7(4):e34643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Carbo A, Gandour RD, Hontecillas R, Philipson N, Uren A, Bassaganya-Riera J: An N,N-bis(benzimidazolylpicolinoyl)piperazine (BT-11): a novel lanthionine synthetase C-like 2-based therapeutic for inflammatory bowel disease. J Med Chem (2016) 59(22):10113–10126. [DOI] [PubMed] [Google Scholar]
- 86.Leber A, Hontecillas R, Zoccoli-Rodriguez V, Bassaganya-Riera J: Activation of LANCL2 by BT-11 ameliorates IBD by supporting regulatory T cell stability through immunometabolic mechanisms. Inflamm Bowel Dis (2018) 24(9):1978–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Leber A, Hontecillas R, Zoccoli-Rodriguez V, Ehrich M, Davis J, Chauhan J, Bassaganya-Riera J: Nonclinical toxicology and toxicokinetic profile of an oral lanthionine synthetase C-like 2 (LANCL2) agonist, BT-11. Int J Toxicol (2019) 38(2):96–109. [DOI] [PubMed] [Google Scholar]
- 88.Leber A, Hontecillas R, Zoccoli-Rodriguez V, Colombel JF, Chauhan J, Ehrich M, Farinola N, Bassaganya-Riera J: The safety, tolerability, and pharmacokinetics profile of BT-11, an oral, gut-restricted lanthionine synthetase C-like 2 agonist investigational new drug for inflammatory bowel disease: a randomized, double-blind, placebo-controlled phase I clinical trial. Inflamm Bowel Dis (2020) 26(4):643–652. [DOI] [PubMed] [Google Scholar]
- 89.VanHecke GC, Abeywardana MY, Ahn YH: Proteomic identification of protein glutathionylation in cardiomyocytes. J Proteome Res (2019) 18(4):1806–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.VanHecke GC, Yapa Abeywardana M, Huang B, Ahn YH: Isotopically labeled clickable glutathione to quantify protein S-glutathionylation. Chembiochem (2020) 21(6):853–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kramer PA, Duan J, Gaffrey MJ, Shukla AK, Wang L, Bammler TK, Qian WJ, Marcinek DJ: Fatiguing contractions increase protein S-glutathionylation occupancy in mouse skeletal muscle. Redox Biol (2018) 17:367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang P, Zhang Q, Li S, Cheng B, Xue H, Wei Z, Shao T, Liu ZX, Cheng H, Wang Z: iCysMod: an integrative database for protein cysteine modifications in eukaryotes. Brief Bioinform (2021) 22(5): bbaa400. [DOI] [PubMed] [Google Scholar]
- 93.Scalcon V, Folda A, Lupo MG, Tonolo F, Pei N, Battisti I, Ferri N, Arrigoni G, Bindoli A, Holmgren A, Coppo L et al. : Mitochondrial depletion of glutaredoxin 2 induces metabolic dysfunction-associated fatty liver disease in mice. Redox Biol (2022) 51:102277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li J, Tang X, Wen X, Ren X, Zhang H, Du Y, Lu J: Mitochondrial Glrx2 knockout augments acetaminophen-induced hepatotoxicity in mice. Antioxidants (2022) 11(9):1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.van de Wetering C, Manuel AM, Sharafi M, Aboushousha R, Qian X, Erickson C, MacPherson M, Chan G, Adcock IM, ZounematKermani N, Schleich F et al. : Glutathione-S-transferase P promotes glycolysis in asthma in association with oxidation of pyruvate kinase M2. Redox Biol (2021) 47:102160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jones JT, Qian X, van der Velden JL, Chia SB, McMillan DH, Flemer S, Hoffman SM, Lahue KG, Schneider RW, Nolin JD, Anathy V et al. : Glutathione S-transferase pi modulates NF-kB activation and pro-inflammatory responses in lung epithelial cells. Redox Biol (2016) 8:375–382. [DOI] [PMC free article] [PubMed] [Google Scholar]