

Abstract
The nucleus accumbens (NAc) is a key brain region involved in reward processing and is linked to multiple neuropsychiatric conditions such as substance use disorder, depression, and chronic pain. Recent studies have begun to investigate persistent changes in NAc gene expression at a single-cell resolution, however, our understanding of the cellular heterogeneity of the NAc epigenomic landscape remains limited. In this study, we utilize single-nucleus assay for transposase-accessible chromatin using high-throughput sequencing (snATAC-seq) to map cell-type-specific differences in chromatin accessibility in the NAc. Our findings not only reveal the transcription factors and putative gene regulatory elements that may contribute to these cell-type-specific epigenomic differences but also provide a valuable resource for future studies investigating epigenomic changes that occur in neuropsychiatric disorders.
Keywords: Gene regulation, enhancers, epigenomics, nucleus accumbens
Introduction
The nucleus accumbens (NAc) is an important brain reward region involved in processing reinforcing and aversive stimuli (de Jong et al., 2019; de Jong et al., 2022; Nestler and Luscher, 2019). The NAc is thus a central substrate for a number of neuropsychiatric conditions involved in disordering reward behavior, including substance use disorder, depression, and chronic pain (Nestler and Waxman, 2020; Russo and Nestler, 2013; Tsankova et al., 2007). Most NAc neurons are medium spiny neurons (MSNs) that are broadly divided by their preferential expression of dopamine D1 receptor or D2 receptor, and which exert opposing effects on motivated behavior (Gerfen and Surmeier, 2011; Kupchik et al., 2015; Lobo et al., 2010). Recent single-cell transcriptomic studies have largely supported the primary transcriptional division between D1 and D2 receptor expressing MSNs, but they have also revealed greater transcriptional heterogeneity than had been previously appreciated (Avey et al., 2018; Chen et al., 2021; He et al., 2021; Savell et al., 2020; Stanley et al., 2020; Tran et al., 2021; Zhao et al., 2022). The exact number of D1 and D2 MSN subtypes described depends on the study, analytical approach, and whether spatial features are included in subtype determinations (Avey et al., 2018; Chen et al., 2021; He et al., 2021; Savell et al., 2020; Stanley et al., 2020; Tran et al., 2021). For example, Chen et al. identified two primary D1 MSN subpopulations (hereafter referred to as D1_Alpha and D1_Beta) and one primary D2 MSN subpopulation when clustering based on variable genes across all cell types and 8 D1 MSN and 8 D2 MSN subpopulations when subclustering based only on variable genes across D1/D2 MSNs.
While the functional role of transcriptionally-defined D1 and D2 MSN subpopulations remains an area of ongoing research, the distinct behavioral and physiological roles D1 and D2 MSNs in NAc play in encoding reward has been explored using pharmacological and genetic approaches (e.g. D1 or D2 MSN-specific knock-in mice expressing Cre recombinase) (Bock et al., 2013; Calipari et al., 2016; Gerfen and Surmeier, 2011; Kravitz et al., 2012; Kupchik et al., 2015; MacAskill et al., 2012; Saunders et al., 2015; Yawata et al., 2012) that have yielded important insight into functional and molecular changes that occur within these distinct NAc cell types in health and disease (Chandra et al., 2015; Heiman et al., 2008; Khibnik et al., 2016; Kronman et al., 2019; Lee et al., 2006; Lobo et al., 2010). Given the role of epigenomic regulation in neuropsychiatric disease (Nestler and Luscher, 2019; Renthal and Nestler, 2008; Tsankova et al., 2007), several studies have profiled cell-type-specific gene expression/translation and/or bulk epigenomic profiling of sorted NAc MSN subtypes (Avey et al., 2018; Chen et al., 2021; Gallegos et al., 2022; He et al., 2021; Heiman et al., 2008; Khibnik et al., 2016; Kronman et al., 2019; Renthal et al., 2009; Rizzardi et al., 2019; Robison and Nestler, 2011; Savell et al., 2020; Scherma et al., 2020; Stanley et al., 2020; Tran et al., 2021; Xu and Heller, 2018; Zhao et al., 2022). However, these studies were not able to characterize the full diversity of NAc MSN subtypes and often require the use of transgenic mouse lines. Improved understanding of gene regulatory mechanisms in distinct NAc cell types may provide both new avenues for treatment as well as new tools for gaining genetic access to distinct NAc cell types across a range of species. Here, we present single-nucleus epigenomic profiling of mouse NAc and describe the analytical groundwork for identifying gene regulatory elements that may mediate cell-type-specific gene expression in the NAc, information that could be useful for designing novel cell-type-specific and cell-state-specific AAVs.
Methods
Mice
8-week old C57BL6/J male mice were obtained from the Jackson Laboratory (strain #000664) and housed on a 12 hr light-dark cycle with access to food and water ad libitum. All experiments were conducted after mice were habituated our facility for > 2 weeks, during the light cycle, not previously handled by the investigator, and according to institutional animal care and safety guidelines at Brigham and Women’s Hospital.
Nuclei isolation from mouse NAc
Isolation of NAc nuclei was performed according to the ‘non-gradient’ nuclear isolation protocol we described previously (Yang et al., 2022) with minor modifications. Briefly, bilateral NAc were dissected from 1 mm coronal sections using 14g tissue biopsy punches (centered on 1.6 AP, 0.9 ML, −4.4 DV) and frozen on dry ice (Renthal et al., 2007). Each sample included NAc pooled from two mice. Nuclei from two biological replicates were isolated separately; they were thawed in homogenization buffer (73 mM NaCl, 0.5 mM CaCl2, 10 mM MgCl2,5 mM Tris-HCl pH 7.5, 1% BSA, and 0.1 U/μl RNase inhibitor (Promega)) and transferred to a total of 5mL of homogenization buffer in a Dounce homogenizer for 10 strokes with the tight pestle. Tween 20 (0.01%), NP40 (0.01%), and Digitonin (0.001%) was then added and 5 additional strokes with the tight pestle were performed. The homogenate was then passed through a 40μM filter and diluted 1:1 with homogenization buffer without detergent. Nuclei were then centrifugated at 500g for 10min at 4C and resuspended in 1X PBS, 0.04% BSA, and 0.1 U/μL RNAse inhibitor and quantified using a hemocytometer.
snATAC-seq of NAc nuclei
Approximately 10,000 nuclei in total from two biological replicate were added to a 10X Genomics ATAC-seq chip. Biological replicates were processed independently according to the manufacturer’s instructions. Library fragment distribution was confirmed using an Agilent Tapestation and libraries were sequenced on an Illumina Nextseq 550 using a 75 cycle high output kit. After sequencing, Illumina BCL files were mapped the mouse reference genome (GRCm38) using the Cellranger ATAC v1.1.0 pipeline using default settings.
Pre-processing, clustering, and annotation of snATAC-seq data
Data pre-processing, clustering, and annotation of snATAC-seq data was similar to that described previously (Yang et al., 2022). Briefly, accessible peaks and the peak-cell matrix for each sample was obtained from the Cellranger ATAC v1.1.0 output using default settings. An aggregated fragments file was produced by running cellranger-atac aggr on default settings. Nuclei with > 3000 fragments overlapping with peaks were included for downstream analysis. Average number of reads per nucleus in nuclei which passed the filter is 15,025, which resulted in a 10,347 median fragments per nuclei (Figure S1A–B). To annotate snATAC-seq data, a pseudo gene expression matrix was generated by counting the fragments overlapping within 2000 bp upstream of TSS of each cell-type-specific gene identified from NAc scRNA-seq data (Chen et al., 2021) using the GeneActivity() function in Signac (Stuart et al., 2019). This pseudo gene expression matrix was then used to anchor epigenomically profiled nuclei to the annotated scRNA-seq dataset using FindTransferAnchors(reduction = “cca”) using 2500 variable features from the scRNA-seq dataset which were also present as variable features in the snATAC object of the “gene activity” assay and TransferData() in Seurat. Nuclei profiled by snATAC-seq with anchoring prediction score < 0.6 were excluded from downstream analyses (Figure S1C). Cell types with < 35 nuclei remaining after filtering were also excluded from downstream analysis, which resulted in the removal of endothelial/mural cells, interneurons, neural stem/neuroblast cells and macrophages. Quality of integration of samples was assessed by integrative Local Inverse Sampson’s Index (iLISI) scoring (Korsunsky et al., 2019) across both samples. All scores were generated using the R package scPOP package by running lisi() with default parameters (Figure S1D). Distribution of cell types per sample was also even across both samples (Figure S1E).
Differential chromatin accessibility analysis
Cell-type-specific snATAC-seq peaks were identified using the FindAllMarkers() function in Seurat (Stuart et al., 2021), comparing nuclei from one cell type to all other NAc cell types. Cell-type-specific snATAC-seq peaks are reported for each subtype if Log2FC > 0.1 and adj. p-value < 0.05. Peaks were annotated to closest genes using ChIPseeker 3.16. (Table S1)
Gene Regulatory Network analysis
We used SCENIC+ (González-Blas et al., 2022) for gene regulatory network (GRN) discovery using default parameters except quality control and cell type annotation was as describe above instead of in SCENIC+. Co-accessible groups of genomic regions were identified in each NAc cell type by topic modeling using pycisTopic (Bravo Gonzalez-Blas et al., 2019) with default parameters. First, a cisTopic object was created and the function evaluate_models() was used to select the optimal number of topics (32). Three types of region sets (otsu-binarized topic regions, top 3000 regions per topic, or cisTopic-called differentially accessible regions) were used for motif enrichment analysis using pycisTarget with default parameters. Motif enrichment results, the cisTopic object, and the scRNA AnnData object (raw counts matrix used) were passed as inputs to create a SCENIC+ object (create_SCENICPLUS_object()) and run SCENIC+ (run_scenicplus()). The list of mouse transcription factors was retrieved from https://resources.aertslab.org/cistarget/tf_lists/allTFs_mm.txt. A pseudo-multiome object containing SCENIC ‘metacells’ was generated by SCENIC+ using default parameters. Correlational outputs (TF-gene, region-gene) and importance scores produced by SCENIC+ (Table S2) were used as inputs to generate t-SNE coordinates for the aforementioned ‘metacells’. generate_pseudobulks() was used to generate pseudobulk cells containing filtered region- or gene-based eRegulon AUCs and gene expression. These pseudobulk cells were then used to create scatterplots of TF expression and eRegulon AUC (prune_plot()). All steps were performed under a development version of SCENIC+ and run on Python 3.7.4. Pandas 1.3.5 was used for all data frame analyses.
Transcription factor motif enrichment analysis
FindMotifs in Signac (Stuart et al., 2021) was used to identify overrepresented TF consensus binding motifs from the JASPAR2020 database in the differentially accessible peaks of each cell type (adj. p-value < 0.05). The background set was randomly sampled from 50,000 GC content and length-matched peaks from the all cell types using the MatchRegionStats() function in Signac (Table S3).
Putative gene regulatory element identification
To determine which snATAC-seq peaks may act as gene regulatory elements and the genes they may regulate, we computed the Pearson correlation between the counts of transposase-sensitive chromatin within a peak and the scRNA-seq expression of each gene on the same chromosome in the same cell type scRNA-seq (Chen et al., 2021). Distance between each snATAC-seq peak and gene was calculated between the center of the peak and the transcription start site of each gene. We then required putative cell-type-specific gene regulatory elements to themselves be cell-type-specific snATAC-seq peaks (Log2FC > 0.1, adj. p-value < 0.05, comparing accessibility in one NAc cell type to all others), have a Pearson’s r > 0.5 with an in cis cell-type-specific gene (Log2FC > 0.5 adj. p-value < 0.05, comparing gene expression in one NAc cell type to all others) in the same cell type, and be located within 200 kb of that gene. These peaks were then intersected with eRegulon regions from SCENIC+ (see Table S4).
Cell type association with UK Biobank GWAS
MAGMA_Celltyping R package (version 1.0.0) was used to calculate cell-type-specific enrichment p values (Skene et al., 2018). First, GWAS summary statistics were obtained from UK biobank: Ever addicted to any substance or behaviour (UK Biobank field 20401; 9,375 cases), Ongoing addiction to alcohol (field 20415; 1,536 cases), Tobacco smoking: Smokes on most or all days (field 22506; 2,870 cases), Recurrent depressive disorder (field F33; 768 cases), Bipolar affective disorder (field F31; 1,196 cases), Schizophrenia (field F20; 575 cases), Pain type(s) experienced in last month: Back pain (field 6159_4; 145,904 cases), Pain type(s) experienced in last month: Hip pain (field 6159_6; 64,827 cases), and Pain type(s) experienced in last month: Headache (field 6159_1; 112,303 cases). All the SNPs were mapped to a gene-level signature using “GRCh37” 10 kb upstream/1 kb downstream of each gene and gene-level p values were calculated. Second, the raw counts table is normalized to 10,000 using NormalizeData from Seurat and the gene expression specificity is ranked into 40 quantiles for each cell type. Finally, the cell type association analysis was calculated using linear mode. The cell type association p-value is corrected using Benjamini-Hochberg. To minimize false-positive associations, disease-celltype associations with FDR > 0.1 are not reported unless their p values are smaller than those at a narrower gene level window (1kbupstream/1kbdownstream).
Data visualization and statistical analyses
Plots and most statistical comparisons were performed in R version 4.2.2. ggplot2 and Integrative Genomics Viewer was used to visualize most data. Seaborn 0.11.2 and matplotlib 3.5.3 were also used to generate the plots for SCENIC+ analysis.
Results
snATAC-seq of mouse NAc
From two biological replicates, we generated a snATAC-seq dataset of 3,449 NAc nuclei after quality controls (see Methods) with an average of 11,643 transposase-sensitive fragments per nucleus (Figure S1A–B). These snATAC-seq fragments formed 96,177 peaks when aggregated across all NAc nuclei. The peaks most frequently mapped to intragenic regions (50.7%, not overlapping with promoter). The remainder of the fragments mapped to gene distal regions (26.9% within 200 kb of TSS, not overlapping with gene body or promoter), promoter regions (16.4%, ≤ 1,000 bp upstream or ≤ 100 bp downstream of TSS), intergenic (3%, >200 kb from any gene) and UTR (3%) regions (Figure 1A).
Figure 1.
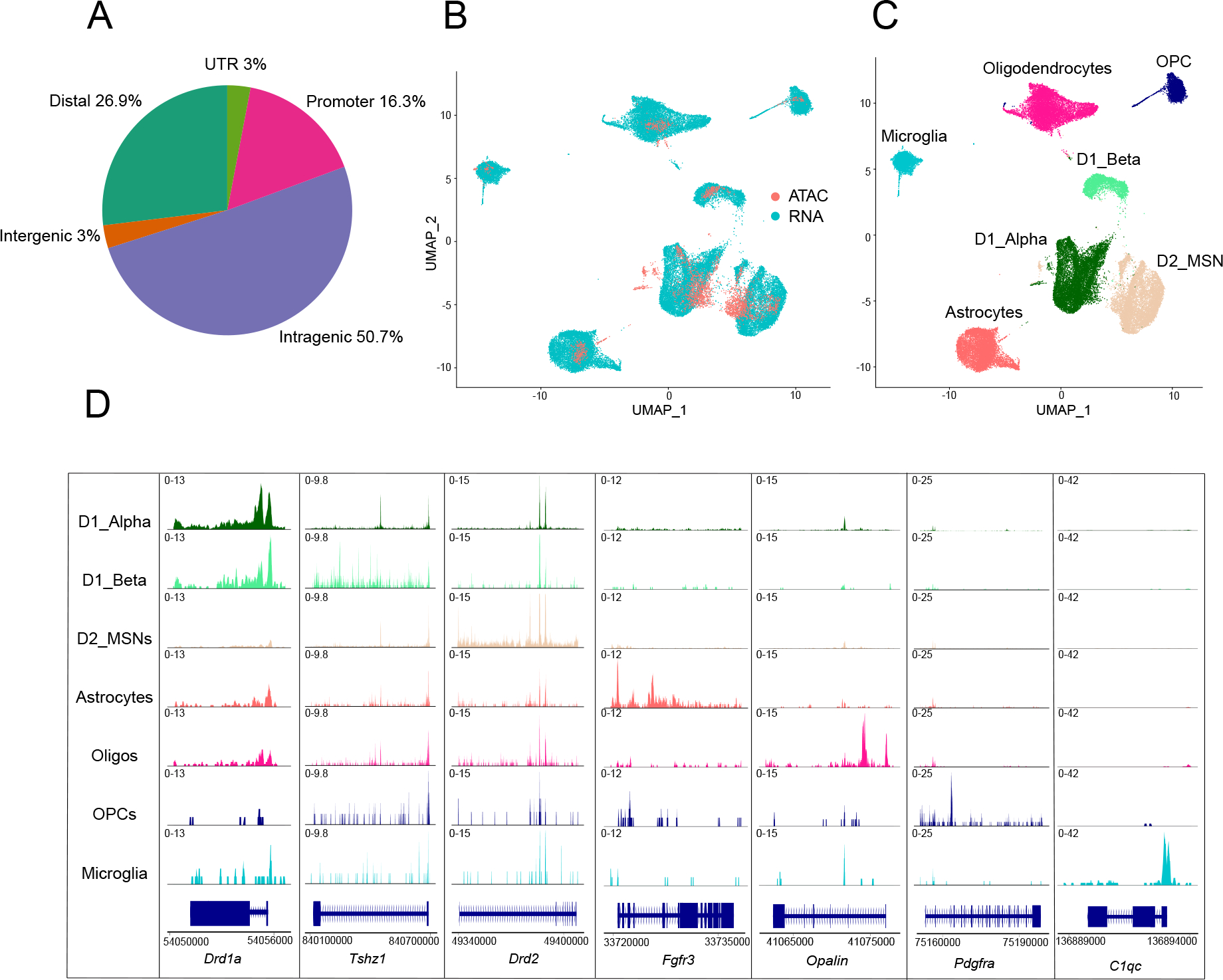
A: Proportion of peaks mapping to promoter regions (−1000bp or +100bp from the TSS of gene), intragenic regions (within gene body excluding promoter and UTR regions), distal regions (<200kb upstream or downstream of TSS, excluding promoters), intergenic regions (>200kb upstream or downstream of TSS) and UTR regions.
B-C: UMAP plots of 3,449 mouse NAc nuclei profiled by snATAC-seq anchored to 41,433 NAc nuclei profiled by scRNA-seq. Nuclei are colored by profiling method (B) or cell-type classification (C). MSN = medium spiny neurons; Oligos = oligodendrocytes; OPC = oligo precursor cells.
D: Coverage plots for selected cell-type-specific marker genes for each NAc cell type (rows). Chromatin accessibility is displayed at cell-type-specific genes (columns), which is displayed as the average frequency of sequenced DNA fragments per cell for each cell type; y axis is scaled for each gene (column).
We next assigned cell types to each snATAC-seq nucleus by multi-omic label transfer (anchoring) (Stuart et al., 2019) from published NAc single-cell RNA-seq data (Chen et al., 2021) (see Methods). This label-transfer algorithm allowed us to reliably assign known cell types to epigenomically defined NAc nuclei (Figure 1B–C) (see Methods) and yielded cell-type percentages of 35.9% D1_Alpha MSNs, 6.4% D1_Beta MSNs, 39.2% D2 MSNs, as well as 6.9% oligodendrocytes, 1.8% oligodendrocyte precursor cells (OPC), 7.9% astrocytes and 1.9% microglia. A small number of NAc interneurons and endothelial cells were also sequenced but not included in downstream analyses because of limited statistical power (see Methods). The anchoring and distribution of cell types was similar in both biological replicates (Figure S1C–E). We found broad consistency in cell-type-specific accessibility within cell-type marker genes; for example, compared to all NAc cell types, the accessibility of Drd1a is preferentially enriched in neurons annotated as D1 MSNs, and the accessibility of Drd2 is preferentially enriched in neurons annotated as D2 MSNs. Similarly, non-neuronal cell type marker genes such as Fgfr3 in astrocytes, C1qc in microglia, Opalin in oligodendrocytes and Pdgfra in OPCs are also preferentially accessible in their respective cell types compared to all other NAc cell types (Figure 1D).
Cell-type-specific chromatin accessibility and transcription factor activity in NAc
We next asked which genomic regions are selectively accessible in distinct NAc cell types. To do this, we performed differential accessibility analysis of our snATAC-seq data and identified 9,887 genomic regions that are preferentially accessible across 7 distinct NAc cell types (Log2FC > 0.1, adj. p-val < 0.05, comparing accessibility within one NAc cell type to that of all others) (Figure 2A, Table S1). These cell-type-specific epigenomic data are valuable both for identifying cell-type-specific actions of distinct TFs as well predicting putative gene regulatory elements (e.g. enhancers). To identify such regulatory events in the NAc, we used SCENIC+ (González-Blas et al., 2022) for identifying, at single-cell resolution, cell-type-specific gene regulatory networks (GRNs) associated with transcription factors. For each transcription factor (TF) for which a SCENIC+ motif annotation was available, we identified sets of genomic regions to which each TF is predicted to bind and their putative target genes (collectively termed an enhancer-driven ‘Regulon’, or ‘eRegulon). We identified 176 unique activator TFs targeting a total of 15,356 unique genomic regions and 9,049 unique genes (Table S2). Clustering of nuclei by the components of eRegulon activity reveals cell-type-specificity of GRNs that is consistent with their established cell type annotation (Figure 2B), indicating that SCENIC+ is faithfully capturing the regulatory logic involved in cell-type-specific gene expression patterns. For example, the regulon activity of ISL1 is highest in D1_Alpha MSNs and positively correlates with Isl1 expression (Figure 2C). We also found that the regulon activity of DLX5 and MEIS2 are highest in D1_Beta and D2 MSNs, respectively, both of which also correlated with the expression of the respected TFs (Figure 2C). Indeed, most of the TFs whose consensus DNA binding motifs are enriched in distinct NAc cell types compared to all other NAc cell types can function as activators of downstream cell-type-specific regulons (Figure 2C). Consistent with our SCENIC+ analyses, compared to all NAc cell types, we also observed significant enrichment of ISL1 consensus DNA binding motifs in differentially accessible regions of D1_Alpha MSNs, DLX5 motifs in D1_Beta MSNs, and MEIS2 motifs in D2 MSNs (Figure S2, Table S3). Moreover, the regulons described here likely contribute to known functions of each TF in their respective cell type, such as ISL1 in establishing D1 MSN transcriptional identity (Lu et al., 2014), and DLX5 and MEIS2 in promoting D1 and D2 MSN development (Lindtner et al., 2019; Su et al., 2022). Together, these insights point to the utility of GRN models in understanding the activity of TFs in the mouse NAc.
Figure 2.
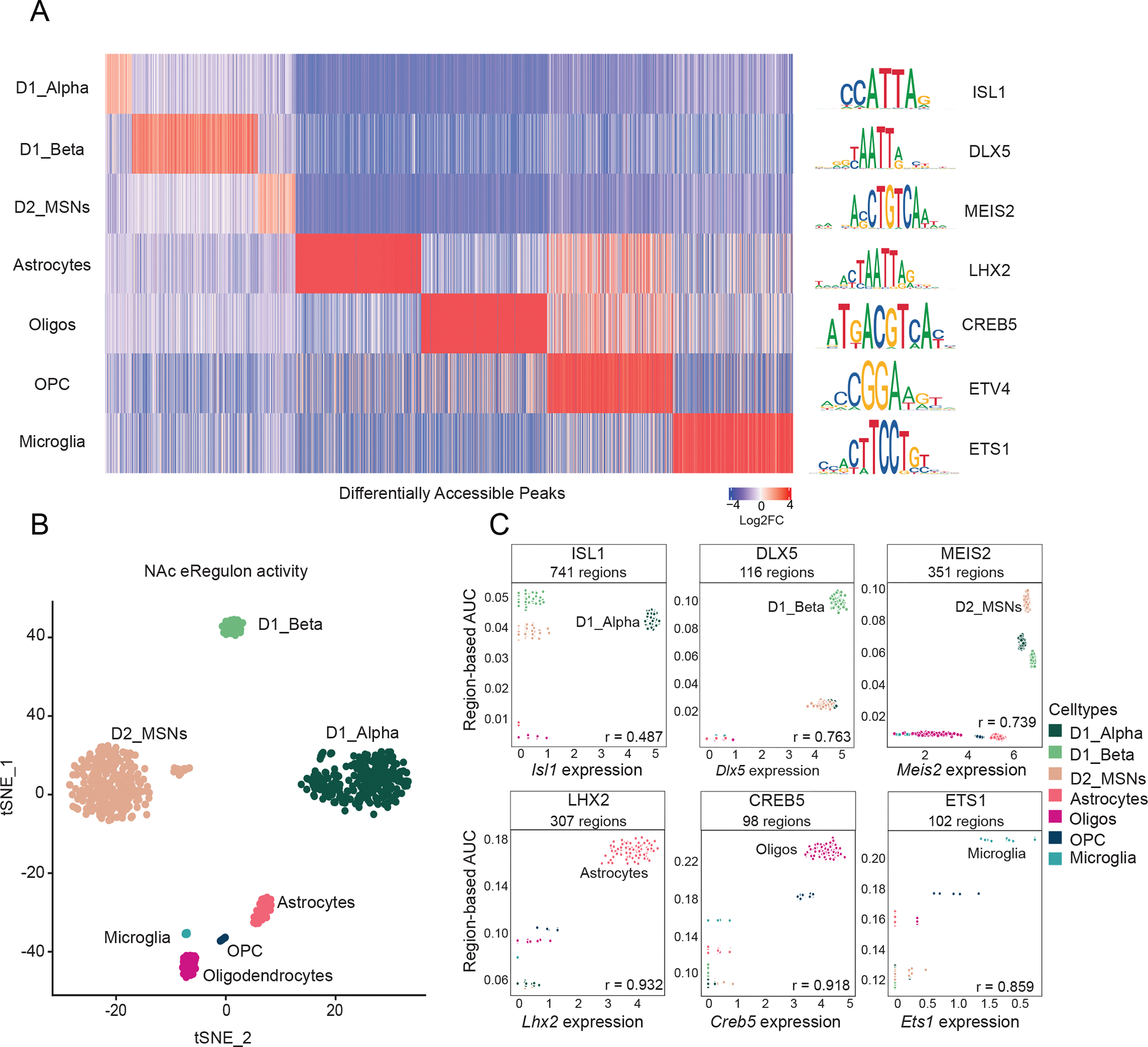
A: Left) Differential chromatin accessibility of 2,740 cell-type-specific peaks (log2FC > 0.1, FDR < 0.05 in one NAc cell type compared to all other NAc cell types, top 500 peaks shown for each cell type) (see Table S1). Heatmap displays log2FC for each peak (columns) in the respective NAc cell type (rows). (Right) The transcription factor (TF) DNA binding motifs that are most significantly enriched within each cell type’s differentially accessible peaks compared to randomly selected peaks (Log2FC > 0.1, FDR < 0.05, see Table S2 for full set).
B: tSNE dimensionality reduction plot of 688 NAc pseudo-multiome cells based on gene and region eRegulon activity as computed by SCENIC+ (see methods).
C: Scatter plots of region-based eRegulon activity (AUC) (y-axis) in each NAc pseudobulk cell (see SCENIC+ methods) for TF motifs that are significantly enriched in distinct NAc cell types compared to all other NAc cell types and the normalized expression of the respective TF in each nucleus (x-axis). All Pearson’s r values are positive and are significant (p-value < 0.01).
Putative gene regulatory elements contributing to MSN heterogeneity
Improved understanding of cell-type-specific gene regulatory mechanisms both yields new mechanistic insight into the factors that determine cell identity and provides new ways to design reporter mice and viral vectors for obtaining targeted genetic access to distinct cell types. To prioritize the peaks of chromatin accessibility that are most likely to function as cell-type-specific gene enhancers, we used two complementary approaches. First, we recovered the putative enhancers regions identified genome-wide for each NAc cell type using TF-region-gene triplets calculated above by SCENIC+ (Table S2). Second, we calculated the correlation between each cell-type-specific peak of chromatin accessibility and the expression of cell-type-specific genes in each cell type. We found 320,530 positively correlated (Pearson’s r > 0.5) cell-type-specific peak-gene pairs that are distributed at a median distance of 1,784 kb upstream of their target gene center. We then intersected the genomic regions identified by both approaches to generate a working map of the distal gene regulatory landscape for distinct NAc cell types (Table S4). The potential utility of these data can be seen at loci for the cell-type-defining dopamine receptor genes, Drd1a and Drd2) (Figure 3A–C). For the Drd1a gene, we observed putative enhancer that is predicted to be activated by ISL1 at chr13:54030344–54031396, ~25 kb upstream the TSS. Closer inspection of the Drd1a locus also identified regions if differential chromatin accessibility 1.1 kb downstream and 3 kb downstream of the TSS that are correlated with Drd1a expression may also function in a regulatory capacity but were overlooked by upstream peak calling (Figure 3A). For the Drd2 gene, we observed a putative enhancer that is predicted to be activated by MEIS2 at chr9:49409367–49409797, ~46kb downstream of the TSS. Inspection of Drd2 locus also identified D2 MSN-specific regions of chromatin accessibility 19.5 kb upstream to 71.2 kb downstream of the Drd2 TSS that is correlated with Drd2 expression and were overlooked by our peak calling pipeline (Figure 3B). We also observed an interesting inverse relationship between Isl1 expression and a regulon’s activity that contains the p_e2 Drd2 enhancer, which suggests that in D1 MSNs, ISL1 may contribute to both Drd1 activation and Drd2 repression (Figure S3). These and other putative enhancers described here (Table S4) may also be of particular interest for driving cell-type-specific gene expression from viral vectors, though additional functional testing is needed. Together, these findings provide new insight into the gene regulatory dynamics of distinct NAc cell types and are of particular interest for developing new tools for improving genetic access to distinct MSN subtypes.
Figure 3.
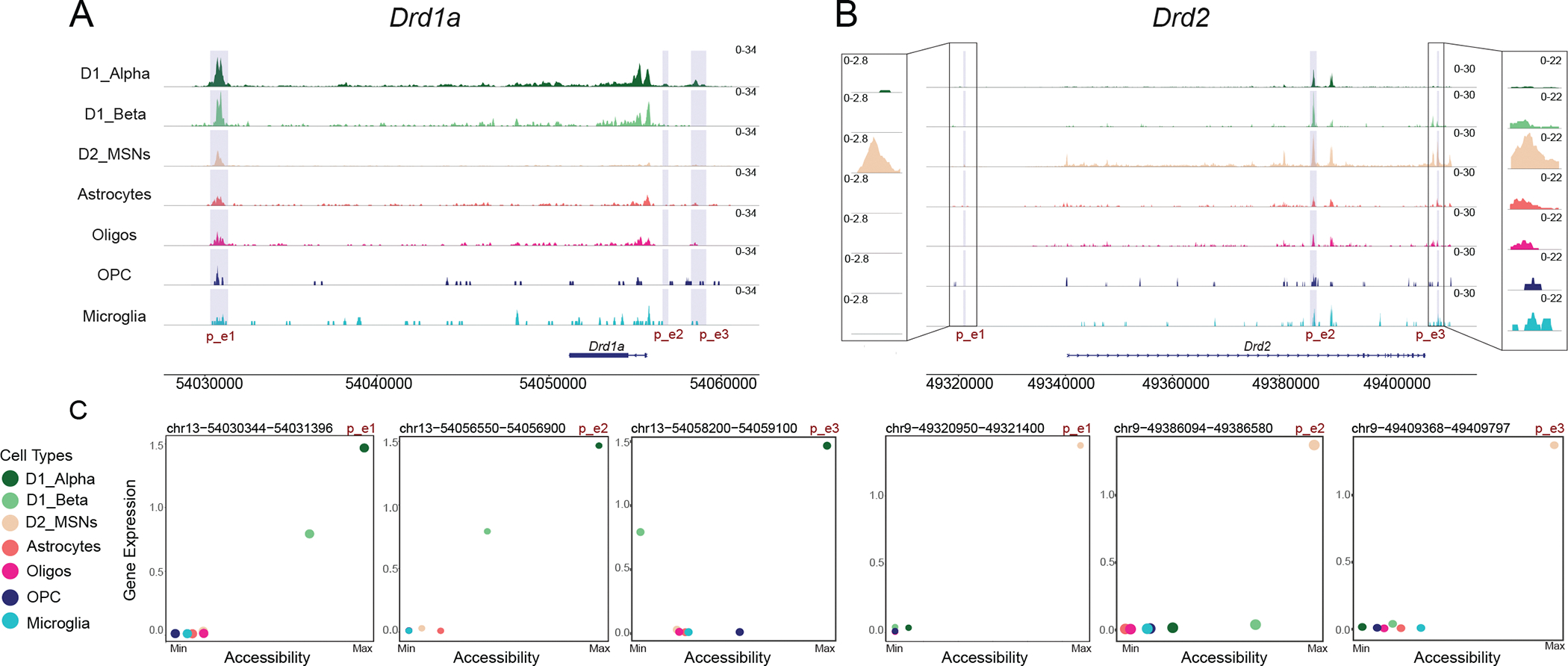
A-B: Chromatin accessibility is displayed at the genomic loci of (A) Drd1a and (B) Drd2. At each genomic locus, chromatin accessibility is displayed as the average fraction of transposase-sensitive fragments per nucleus at that region. Accessibility at each locus (y axis) is scaled to the max value across all cell types (column). Highlighted regions denote putative enhancer (p_e) regions that are examined in C. Insets in B allow for better visualization of differential chromatin accessibility of p_e1 and p_e3.
C: Scatterplot of chromatin accessibility (calculated as mean counts of snATAC-seq fragments present within highlighted p_e region across cells of each cell type) and mean expression of the respective cell-type-specific gene each cell type (chromatin accessibility is normalized to their max values). Colors indicate cell types.
Cell-type-specific expression of genes associated with neuropsychiatric disease
In addition to providing important insight into the gene regulatory mechanisms that establish cell identity within the NAc, cell-type-specific gene expression data has been particularly powerful when combined with human genetics for understanding the potential role of these cell types across a range of human disease (Cusanovich et al., 2018; Skene et al., 2018). We thus turned to UK Biobank genome-wide association studies (GWAS) that describe genomic variation associated with a range of neuropsychiatric conditions in which the NAc has been previously implicated (e.g. depression, substance use disorders, chronic pain), and asked whether genetic variation associated with these conditions disproportionally affects gene expression patterns in certain NAc cell types. To do this, we used MAGMA Single Cell (Skene et al., 2018), which first links genomic variants associated with a given disease to genes, and then calculates whether these associated genes are significantly enriched in distinct NAc cell types compared to all other NAc cell types (see Methods). While we did not observe any significant associations between distinct NAc cell-type-specific gene expression patterns and GWAS of substance use disorders, including tobacco use, alcohol use, or to any substance or behavior, we did observe interesting trends suggesting D1 and D2 MSNs may be enriched for genes that are associated with multiple pain conditions, depression, and a weak association with alcohol use (Figure 4A–B). The small number of statistically significant GWAS associations in the UK biobank for several neuropsychiatric conditions and the use of mouse data likely contributes to weak cell type enrichment patterns for some conditions. Indeed, stronger associations between cell-type-specific gene expression and neuropsychiatric GWAS loci has been reported using a larger schizophrenia GWAS and gene expression from human NAc (Tran et al., 2021). Overall, these studies must be interpreted in the context of their limitations, as there are only a small number of statistically significant genomic variants reported for most neuropsychiatric conditions in the UK Biobank (Figure 4A). Nevertheless, this analytical framework provides a starting point for mechanistically exploring the role of genetic variation in neuropsychiatric disease in the context of specific NAc cell types.
Figure 4.
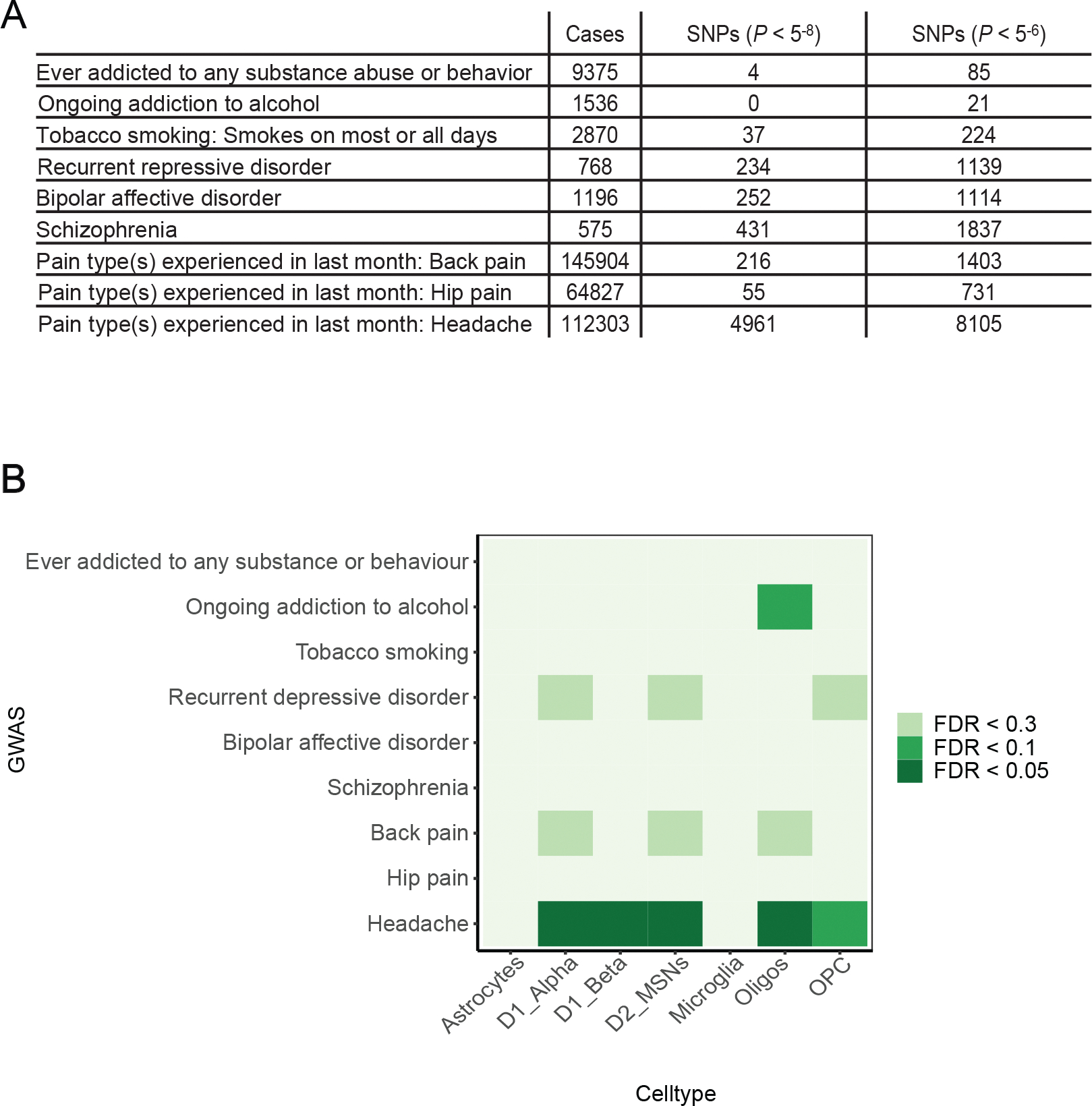
A: Table of UK Biobank neuropsychiatric GWAS included in our analyses. The number of cases and associated SNPs (P < 5−8 and P < 5−6) are shown.
B: Heatmap displays the cell type enrichment of genes associated with each UK Biobank neuropsychiatric GWAS. Cell type enrichment is determined by the expression of genes implicated by each GWAS in one cell NAc cell type compared to all other NAc cell types (FDR is displayed, see methods).
Discussion:
In this study, we used snATAC-seq to profile accessible chromatin regions in distinct NAc cell types. Our analyses of these data provide new insight into the gene regulatory mechanisms underlying cell-type-specific gene expression in NAc as well as an analytical framework for extending these studies to animal models of neuropsychiatric disease and tissues from human donors who suffered from these conditions.
Given their central role in mediating reward behavior and relative abundance in our dataset, our analyses largely focused on distinct subtypes of MSNs. We observed distinct epigenomic profiles between D1 and D2 MSNs subtypes, identified the cell-type-specific regions of differential chromatin accessibility that are likely to function as gene regulatory elements, and described the TFs whose consensus binding motifs are enriched within distinct NAc cell types. We did not include the comparatively rare D1/2-negative NAc interneuron population in our analyses, some of which have been recently epigenomically profiled (Gallegos et al., 2022), as they were too sparse in our data to make meaningful genome-wide comparisons. Another limitation of the study is that we only profiled male mice. As sex differences are important modifiers of neuropsychiatric disease risk, it will be important for future studies to explore sex differences in the epigenomic and transcriptional landscape of distinct NAc cell types.
Some of the TFs whose consensus DNA binding sequences we find enriched in distinct NAc neuronal subtypes compared to all NAc cell types have already been reported to play an important role in striatal development and function. For example, ISL1 is known to function in striatal neuron development to establish D1 transcriptional identity both by upregulating D1-specific genes and repressing D2-specific genes (Ehrman et al., 2013; Lu et al., 2014), findings which are consistent with our analyses of Isl1 expression and regulon activity in D1 MSNs (Figure S3). The example of ISL1 illustrates some face validity to our analyses and suggest that future studies into the cell-type-specific TFs and regulons (Table S2) implicated by our epigenomic data may reveal new insight into the gene regulatory mechanisms underlying NAc development, celltype identity, and function.
As many cell-type-specific regions of chromatin accessibility are likely reflections of rather than drivers of cell-type-specific gene expression programs, we bioinformatically prioritized the genomic regions which are most likely to function as cell-type-specific gene regulatory elements genome-wide (Table S4). For example, we described putative enhancers for Drd1a and Drd2 whose chromatin accessibility is cell-type-specific, correlated with their respective gene’s expression, and often part of SCENIC+ TF-region-gene triplets/eRegulons. Future functional studies are required to test to what degree these elements promote their respective target gene expression in vivo.
In addition to providing new insight into the gene regulatory mechanisms underlying NAc cell-type-specific gene expression patterns, the epigenomic data presented here also lays the groundwork for future applications. For example, the cell-type-specific distal gene regulatory elements described here can be engineered into viral vectors to drive viral gene expression in distinct NAc cell types. These distal regulatory elements are often shorter and offer greater cell-type-specificity compared to the proximal promoter sequence of known cell-type-specific genes. Indeed, efforts in this regard have been successful for targeting multiple CNS interneuron subtypes (Hrvatin et al., 2019; Mich et al., 2021; Vormstein-Schneider et al., 2020) and may be able to further improve the specificity of currently available D1 and D2 MSN-specific AAVs (Ferguson et al., 2011).
While our study aimed to understand the gene regulatory mechanisms that mediate cell-type-specific gene expression differences of NAc cell types in naïve mice, similar efforts in animal models of neuropsychiatric disease and human donors that suffer from them are likely to be fruitful. Indeed, the discovery of cell state-specific gene regulatory elements could enable genetic access to disordered cells and networks across a range of neuropsychiatric conditions such as substance use disorder, depression, or chronic pain.
Conclusion
Our study provides a deeper understanding of the regulatory processes that control gene expression in distinct NAc cell types. This information may be useful for understanding NAc function in states of health and for developing tools to access these cell types in states of neuropsychiatric disease.
Supplementary Material
A: NAc snATAC-seq metrics. Top row displays box plots of number of fragments per nucleus (log10 transformed) in each sample and the bottom row displays the number of fragments per peak (log10 transformed) in each sample. Boxes indicate quartiles and whiskers are 1.5-times the interquartile range (Q1-Q3). The median is a black line inside each box. The distribution is aggregated across all samples and displayed on the horizontal histogram.
B: Distribution of NAc snATAC-seq fragment lengths in each sample.
C: UMAP plots of 3,449 mouse NAc nuclei profiled by snATAC-seq anchored to NAc nuclei profiled by scRNA-seq as in Figure 1B–C. Only nuclei profiled by snATAC-seq are displayed and are colored by sample.
D: Violin plot of cells with iLISI integration scores (see methods) in NAc snATAC-seq samples.
E: Stacked barplot showing distribution of cell types in NAc snATAC-seq samples.
Scatterplots of average gene expression of TF-mRNA and TF-motif fold enrichment (Z-score normalized) found from Signac in each mouse NAc cell type compared to all other NAc cell types.
Scatter plots of region-based eRegulon activity (AUC) (y-axis) in each NAc pseudobulk cell (see SCENIC+ methods) for ISL1 motif that is significantly enriched in D1_Alpha and D2 MSNs but with negative correlation to D2 MSNs (left) and positive correlation to D1_Alpha MSNs (right) compared to all other NAc cell types and the normalized expression of the respective TF in each nucleus (x-axis). All Pearson’s r values are positive and are significant (p-value < 0.01).
Cell-type-specific peaks of chromatin accessibility in NAc (Log2FC > 0.1, FDR < 0.05, each NAc cell type compared to all other NAc cell types)
Complete list of eRegulons produced by SCENIC+ including only eRegulons with positive region-to-gene and positive TF-gene correlation or positive region-to-gene correlation but negative TF-gene correlation.
Transcription factor consensus DNA binding motif enrichment
List of putative cell-type-specific gene regulatory elements in each NAc cell type.
Highlights.
Single-nucleus ATAC-seq of mouse nucleus accumbens
Description of cell-type-specific gene regulatory networks
Mapping of putative cell-type-specific gene enhancers
Distinct nucleus accumbens cell types are implicated in GWAS of neuropsychiatric disease
Acknowledgements
This work was supported by the National Institute of Drug Abuse DP1DA054343, the Burroughs Wellcome Fund, and the BWH Women’s Brain Initiative and Neurotechnology Studio.
Footnotes
Declaration of Competing Interests
The authors declare no competing interests.
Data Availability
Processed and raw data files along with metadata containing cell type annotations can be found on GEO (GSE232774).
Code for analyzing the raw data and generating the figures and supplementary tables can be found here: https://github.com/Renthal-Lab/nucleus_accumbens_atac.
References
- Avey D, Sankararaman S, Yim AKY, Barve R, Milbrandt J, and Mitra RD (2018). Single-Cell RNA-Seq Uncovers a Robust Transcriptional Response to Morphine by Glia. Cell Rep 24, 3619–3629 e3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock R, Shin JH, Kaplan AR, Dobi A, Markey E, Kramer PF, Gremel CM, Christensen CH, Adrover MF, and Alvarez VA (2013). Strengthening the accumbal indirect pathway promotes resilience to compulsive cocaine use. Nat Neurosci 16, 632–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo Gonzalez-Blas C, Minnoye L, Papasokrati D, Aibar S, Hulselmans G, Christiaens V, Davie K, Wouters J, and Aerts S (2019). cisTopic: cis-regulatory topic modeling on single-cell ATAC-seq data. Nat Methods 16, 397–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calipari ES, Bagot RC, Purushothaman I, Davidson TJ, Yorgason JT, Pena CJ, Walker DM, Pirpinias ST, Guise KG, Ramakrishnan C, et al. (2016). In vivo imaging identifies temporal signature of D1 and D2 medium spiny neurons in cocaine reward. Proc Natl Acad Sci U S A 113, 2726–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra R, Francis TC, Konkalmatt P, Amgalan A, Gancarz AM, Dietz DM, and Lobo MK (2015). Opposing role for Egr3 in nucleus accumbens cell subtypes in cocaine action. J Neurosci 35, 7927–7937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Blosser TR, Djekidel MN, Hao J, Bhattacherjee A, Chen W, Tuesta LM, Zhuang X, and Zhang Y (2021). Decoding molecular and cellular heterogeneity of mouse nucleus accumbens. Nat Neurosci 24, 1757–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusanovich DA, Hill AJ, Aghamirzaie D, Daza RM, Pliner HA, Berletch JB, Filippova GN, Huang X, Christiansen L, DeWitt WS, et al. (2018). A Single-Cell Atlas of In Vivo Mammalian Chromatin Accessibility. Cell 174, 1309–1324 e1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong JW, Afjei SA, Pollak Dorocic I, Peck JR, Liu C, Kim CK, Tian L, Deisseroth K, and Lammel S (2019). A Neural Circuit Mechanism for Encoding Aversive Stimuli in the Mesolimbic Dopamine System. Neuron 101, 133–151 e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong JW, Fraser KM, and Lammel S (2022). Mesoaccumbal Dopamine Heterogeneity: What Do Dopamine Firing and Release Have to Do with It? Annu Rev Neurosci 45, 109–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrman LA, Mu X, Waclaw RR, Yoshida Y, Vorhees CV, Klein WH, and Campbell K (2013). The LIM homeobox gene Isl1 is required for the correct development of the striatonigral pathway in the mouse. Proc Natl Acad Sci U S A 110, E4026–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson SM, Eskenazi D, Ishikawa M, Wanat MJ, Phillips PE, Dong Y, Roth BL, and Neumaier JF (2011). Transient neuronal inhibition reveals opposing roles of indirect and direct pathways in sensitization. Nat Neurosci 14, 22–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallegos DA, Minto M, Liu F, Hazlett MF, Aryana Yousefzadeh S, Bartelt LC, and West AE (2022). Cell-type specific transcriptional adaptations of nucleus accumbens interneurons to amphetamine. Mol Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, and Surmeier DJ (2011). Modulation of striatal projection systems by dopamine. Annu Rev Neurosci 34, 441–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Blas CB, De Winter S, Hulselmans G, Hecker N, Matetovici I, Christiaens V, Poovathingal S, Wouters J, Aibar S, and Aerts S (2022). SCENIC+: single-cell multiomic inference of enhancers and gene regulatory networks. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Kleyman M, Chen J, Alikaya A, Rothenhoefer KM, Ozturk BE, Wirthlin M, Bostan AC, Fish K, Byrne LC, et al. (2021). Transcriptional and anatomical diversity of medium spiny neurons in the primate striatum. Curr Biol 31, 5473–5486 e5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suarez-Farinas M, Schwarz C, Stephan DA, Surmeier DJ, et al. (2008). A translational profiling approach for the molecular characterization of CNS cell types. Cell 135, 738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrvatin S, Tzeng CP, Nagy MA, Stroud H, Koutsioumpa C, Wilcox OF, Assad EG, Green J, Harvey CD, Griffith EC, et al. (2019). A scalable platform for the development of cell-type-specific viral drivers. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khibnik LA, Beaumont M, Doyle M, Heshmati M, Slesinger PA, Nestler EJ, and Russo SJ (2016). Stress and Cocaine Trigger Divergent and Cell Type-Specific Regulation of Synaptic Transmission at Single Spines in Nucleus Accumbens. Biol Psychiatry 79, 898–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, and Raychaudhuri S (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravitz AV, Tye LD, and Kreitzer AC (2012). Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nat Neurosci 15, 816–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronman H, Richter F, Labonte B, Chandra R, Zhao S, Hoffman G, Lobo MK, Schadt EE, and Nestler EJ (2019). Biology and Bias in Cell Type-Specific RNAseq of Nucleus Accumbens Medium Spiny Neurons. Sci Rep 9, 8350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupchik YM, Brown RM, Heinsbroek JA, Lobo MK, Schwartz DJ, and Kalivas PW (2015). Coding the direct/indirect pathways by D1 and D2 receptors is not valid for accumbens projections. Nat Neurosci 18, 1230–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, Kim Y, Kim AM, Helmin K, Nairn AC, and Greengard P (2006). Cocaine-induced dendritic spine formation in D1 and D2 dopamine receptor-containing medium spiny neurons in nucleus accumbens. Proc Natl Acad Sci U S A 103, 3399–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindtner S, Catta-Preta R, Tian H, Su-Feher L, Price JD, Dickel DE, Greiner V, Silberberg SN, McKinsey GL, McManus MT, et al. (2019). Genomic Resolution of DLX-Orchestrated Transcriptional Circuits Driving Development of Forebrain GABAergic Neurons. Cell Rep 28, 2048–2063 e2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo MK, Covington HE 3rd, Chaudhury D, Friedman AK, Sun H, Damez-Werno D, Dietz DM, Zaman S, Koo JW, Kennedy PJ, et al. (2010). Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science 330, 385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu KM, Evans SM, Hirano S, and Liu FC (2014). Dual role for Islet-1 in promoting striatonigral and repressing striatopallidal genetic programs to specify striatonigral cell identity. Proc Natl Acad Sci U S A 111, E168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAskill AF, Little JP, Cassel JM, and Carter AG (2012). Subcellular connectivity underlies pathway-specific signaling in the nucleus accumbens. Nat Neurosci 15, 1624–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mich JK, Graybuck LT, Hess EE, Mahoney JT, Kojima Y, Ding Y, Somasundaram S, Miller JA, Kalmbach BE, Radaelli C, et al. (2021). Functional enhancer elements drive subclass-selective expression from mouse to primate neocortex. Cell Rep 34, 108754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, and Luscher C (2019). The Molecular Basis of Drug Addiction: Linking Epigenetic to Synaptic and Circuit Mechanisms. Neuron 102, 48–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, and Waxman SG (2020). Resilience to Stress and Resilience to Pain: Lessons from Molecular Neurobiology and Genetics. Trends Mol Med 26, 924–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Kumar A, Xiao G, Wilkinson M, Covington HE 3rd, Maze I, Sikder D, Robison AJ, LaPlant Q, Dietz DM, et al. (2009). Genome-wide analysis of chromatin regulation by cocaine reveals a role for sirtuins. Neuron 62, 335–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Maze I, Krishnan V, Covington HE 3rd, Xiao G, Kumar A, Russo SJ, Graham A, Tsankova N, Kippin TE, et al. (2007). Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron 56, 517–529. [DOI] [PubMed] [Google Scholar]
- Renthal W, and Nestler EJ (2008). Epigenetic mechanisms in drug addiction. Trends Mol Med 14, 341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzardi LF, Hickey PF, Rodriguez DiBlasi V, Tryggvadottir R, Callahan CM, Idrizi A, Hansen KD, and Feinberg AP (2019). Neuronal brain-region-specific DNA methylation and chromatin accessibility are associated with neuropsychiatric trait heritability. Nat Neurosci 22, 307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison AJ, and Nestler EJ (2011). Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci 12, 623–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo SJ, and Nestler EJ (2013). The brain reward circuitry in mood disorders. Nat Rev Neurosci 14, 609–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Oldenburg IA, Berezovskii VK, Johnson CA, Kingery ND, Elliott HL, Xie T, Gerfen CR, and Sabatini BL (2015). A direct GABAergic output from the basal ganglia to frontal cortex. Nature 521, 85–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savell KE, Tuscher JJ, Zipperly ME, Duke CG, Phillips RA 3rd, Bauman AJ, Thukral S, Sultan FA, Goska NA, Ianov L, et al. (2020). A dopamine-induced gene expression signature regulates neuronal function and cocaine response. Sci Adv 6, eaba4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherma M, Qvist JS, Asok A, Huang SC, Masia P, Deidda M, Wei YB, Soni RK, Fratta W, Fadda P, et al. (2020). Cannabinoid exposure in rat adolescence reprograms the initial behavioral, molecular, and epigenetic response to cocaine. Proc Natl Acad Sci U S A 117, 9991–10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene NG, Bryois J, Bakken TE, Breen G, Crowley JJ, Gaspar HA, Giusti-Rodriguez P, Hodge RD, Miller JA, Munoz-Manchado AB, et al. (2018). Genetic identification of brain cell types underlying schizophrenia. Nat Genet 50, 825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley G, Gokce O, Malenka RC, Sudhof TC, and Quake SR (2020). Continuous and Discrete Neuron Types of the Adult Murine Striatum. Neuron 105, 688–699 e688. [DOI] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Srivastava A, Madad S, Lareau CA, and Satija R (2021). Single-cell chromatin state analysis with Signac. Nat Methods 18, 1333–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z, Wang Z, Lindtner S, Yang L, Shang Z, Tian Y, Guo R, You Y, Zhou W, Rubenstein JL, et al. (2022). Dlx1/2-dependent expression of Meis2 promotes neuronal fate determination in the mammalian striatum. Development 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran MN, Maynard KR, Spangler A, Huuki LA, Montgomery KD, Sadashivaiah V, Tippani M, Barry BK, Hancock DB, Hicks SC, et al. (2021). Single-nucleus transcriptome analysis reveals cell-type-specific molecular signatures across reward circuitry in the human brain. Neuron 109, 3088–3103 e3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova N, Renthal W, Kumar A, and Nestler EJ (2007). Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci 8, 355–367. [DOI] [PubMed] [Google Scholar]
- Vormstein-Schneider D, Lin JD, Pelkey KA, Chittajallu R, Guo B, Arias-Garcia MA, Allaway K, Sakopoulos S, Schneider G, Stevenson O, et al. (2020). Viral manipulation of functionally distinct interneurons in mice, non-human primates and humans. Nat Neurosci 23, 1629–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu SJ, and Heller EA (2018). Single sample sequencing (S3EQ) of epigenome and transcriptome in nucleus accumbens. J Neurosci Methods 308, 62–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Xu M, Bhuiyan SA, Li J, Zhao J, Cohrs RJ, Susterich JT, Signorelli S, Green U, Stone JR, et al. (2022). Human and mouse trigeminal ganglia cell atlas implicates multiple cell types in migraine. Neuron 110, 1806–1821 e1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yawata S, Yamaguchi T, Danjo T, Hikida T, and Nakanishi S (2012). Pathway-specific control of reward learning and its flexibility via selective dopamine receptors in the nucleus accumbens. Proc Natl Acad Sci U S A 109, 12764–12769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ZD, Han X, Chen R, Liu Y, Bhattacherjee A, Chen W, and Zhang Y (2022). A molecularly defined D1 medium spiny neuron subtype negatively regulates cocaine addiction. Sci Adv 8, eabn3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A: NAc snATAC-seq metrics. Top row displays box plots of number of fragments per nucleus (log10 transformed) in each sample and the bottom row displays the number of fragments per peak (log10 transformed) in each sample. Boxes indicate quartiles and whiskers are 1.5-times the interquartile range (Q1-Q3). The median is a black line inside each box. The distribution is aggregated across all samples and displayed on the horizontal histogram.
B: Distribution of NAc snATAC-seq fragment lengths in each sample.
C: UMAP plots of 3,449 mouse NAc nuclei profiled by snATAC-seq anchored to NAc nuclei profiled by scRNA-seq as in Figure 1B–C. Only nuclei profiled by snATAC-seq are displayed and are colored by sample.
D: Violin plot of cells with iLISI integration scores (see methods) in NAc snATAC-seq samples.
E: Stacked barplot showing distribution of cell types in NAc snATAC-seq samples.
Scatterplots of average gene expression of TF-mRNA and TF-motif fold enrichment (Z-score normalized) found from Signac in each mouse NAc cell type compared to all other NAc cell types.
Scatter plots of region-based eRegulon activity (AUC) (y-axis) in each NAc pseudobulk cell (see SCENIC+ methods) for ISL1 motif that is significantly enriched in D1_Alpha and D2 MSNs but with negative correlation to D2 MSNs (left) and positive correlation to D1_Alpha MSNs (right) compared to all other NAc cell types and the normalized expression of the respective TF in each nucleus (x-axis). All Pearson’s r values are positive and are significant (p-value < 0.01).
Cell-type-specific peaks of chromatin accessibility in NAc (Log2FC > 0.1, FDR < 0.05, each NAc cell type compared to all other NAc cell types)
Complete list of eRegulons produced by SCENIC+ including only eRegulons with positive region-to-gene and positive TF-gene correlation or positive region-to-gene correlation but negative TF-gene correlation.
Transcription factor consensus DNA binding motif enrichment
List of putative cell-type-specific gene regulatory elements in each NAc cell type.
Data Availability Statement
Processed and raw data files along with metadata containing cell type annotations can be found on GEO (GSE232774).
Code for analyzing the raw data and generating the figures and supplementary tables can be found here: https://github.com/Renthal-Lab/nucleus_accumbens_atac.