

ABSTRACT
Background
Wilson's disease (WD) is a rare genetic condition characterized by a copper overload in organs secondary to mutation in ATP7B gene. Lifelong decoppering treatments are the keystone of the treatment but must be regularly adapted to obtain a correct copper balance and could lead to copper deficiency (CD).
Objectives
Study the characteristics of CD in WD patients.
Methods
CD cases from our cohort of 338 WD patients have been investigated. CD was defined by the association of serum copper, exchangeable copper and urinary copper excretion assays less than two standard deviations from the mean with cytopenia and/or neurological damage of spinal cord origin. A systematic review of literature about cases of CD in WD patient was performed in PubMed database according to PRISMA guidelines.
Results
Three WD patients were diagnosed with CD in our cohort. Review of the literature found 17 other patients. Most of the patients had anemia and neutropenia associated with neurological symptoms (especially progressive posterior cord syndrome). All the patients were treated with Zinc salts and the symptoms occurred more than a decade after the initiation of treatment. The adaptation of the treatment allowed a correction of the cytopenia but only a partial improvement of the neurological symptoms.
Conclusions
WD patients can develop CD after many years of zinc therapy. Anemia and neutropenia are red flags that should evoke CD.
Keywords: anemia, copper deficiency, myeloneuropathy, Wilson's disease, zinc salts
Wilson's disease (WD) is a rare genetic condition, with fewer than 1000 patients in France. 1 It is characterized by a copper overload in liver, brain and other organs secondary to homozygous or compound heterozygous variants in ATP7B gene, which encodes a transmembrane copper‐transporting ATPase. Lifelong decoppering treatments including chelators and zinc salts, which reverse copper overload by different mechanisms, are the keystone of the treatment but have to be regularly adapted to obtain a correct copper balance. Chelators (D‐penicillamine and Trientine salts) mobilize tissue copper stores, increasing the urinary copper excretion and allow normalization of body copper balance. 2 Tetrathiomolybdate forms, in digestive tractus, a tripartite complex with copper and protein preventing the absorption of complexed copper. In blood, it complexes copper with albumin, making the copper unavailable for cellular uptake. 3 This treatment is currently under phase III evaluation. Zinc salt reduces the intestinal absorption of copper by increasing the production of metallothionein, a protein that can bind various metal ions but with a stronger affinity, both in enterocytes, reducing metal intestinal absorption into portal circulation, and in hepatocytes, reducing the damaging effects of free liver copper. 2 , 3 , 4 , 5
If their doses are not adapted during the maintenance phase of the disease, chronic treatments could lead to copper deficiency (CD) which is characterized by progressive cytopenia associated with low urinary copper excretion, then neurological symptoms, in particular posterior column syndrome (PCS). Description of symptomatic CD in WD patients are scarce and only a few clinical cases have been published. 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16
The objectives of this study are (1) to report the prevalence, the clinical, biological, radiological features and the evolution of WD patients with symptomatic CD from the cohort of patients followed in our Wilson's Disease Reference Center (Rothschild Foundation Hospital, Paris, France) and (2) to perform a review of the literature to identify other cases of WD patients with symptomatic CD.
Materials and Methods
Original Cases
WD patients from the Wilson's Disease Reference Center in Rothschild Foundation Hospital (Paris, France) and presenting with CD were studied. Copper deficiency was defined by the association of serum copper, exchangeable copper and urinary copper excretion assays less than two standard deviations from the mean with cytopenia and/or neurological damage of spinal cord origin. All patients gave their informed consent for the genetic analysis and for anonymous study of their data (consent for research) before their inclusion in the French WD Registry. This study was approved by the Institutional Review Board (IRB00003888, IORG0003254, FWA00005831) of the French Institute of medical research and Health (N°19–550). Data were collected prospectively from our cohort of patients followed bi‐annually from 2005 to today, the analysis was done retrospectively from these data. Collected data included (1) WD information at diagnosis (age, sex, ATP7B mutations, initial phenotype, hepatic score (from 0 to 6), 17 , 18 UWDRS score, ophthalmological score 17 and brain Magnetic resonance imaging (MRI) data as well as WD treatment (drug, dose, duration of treatment), (2) information concerning the CD: first symptoms, age at CD diagnosis, clinical data, spinal and brain MRI, ENMG study, biological data and (3) evolution of CD at last follow‐up.
Review of the Literature
We systematically searched the PubMed database for publications (in English or French) on case reports of CD in WD patients without any timeframe. The following search terms (without any PubMed search filters) were used: [WILSON DISEASE] AND [COPPER DEFICIENCY]. Publications on adult and/or pediatric patients were included. Relevant articles were selected by considering the title, the abstract, and the full text. Previous reviews of the literature and related articles were included. Articles not related to WD and CD or with incomplete data were excluded. The results of the systematic review were reported in accordance with the Preferred Reporting Items for Systematic Reviews and Meta‐Analyses guidelines (http://www.prisma-statement.org).
Results
Three WD patients (3 males, 39, 78 and 57 years old respectively for patient 1, 2, and 3) were diagnosed with CD, representing 0.9% of the 338 WD of the cohort (Tables 1 and 2).
TABLE 1.
Characteristics of the three Wilson's disease patients with copper deficiency at Wilson's disease diagnosis
| Patients | 1 | 2 | 3 |
|---|---|---|---|
| Gender | Male | Male | Male |
| At Wilson's disease diagnosis | |||
| Age (years) | 34 | 58 | 23 |
| Mutation 1 | His1069Gln‐exon14 | Phe714Leu‐exon8 | Thr977Met‐ex13 |
| Mutation 2 | Pro1098Arg‐exon15 | deletion‐exon 4 | Ile1148Thr‐ex16 |
| Clinical form of WD | Hepatic + KFR | Hepatic and neurologic | Presymptomatic |
| UWDRS | 0 | 3 (writer cramp) | 0 |
| Hepatic score (0–6) | 5 (compensated cirrhosis, platelets 70,000/mm4) | 3 (mild cirrhosis, no cytopenia) | 0 |
| Ophthalmologic score (0–2) | 2 | 0 | 0 |
| Brain MRI | Normal | Normal | Normal |
| Treatments | zinc acetate (150 mg/day) | D‐Penicillamine 900 mg/d during 6 years then zinc acetate 150 mg/d | D‐Penicillamine 900 mg/d during thirteen years, then Trientine 2HCL 600 mg/d + zinc sulfate 1200 mg/d |
TABLE 2.
Characteristics of the three Wilson's disease patients at the diagnosis of copper deficiency and evolution
| Patients | 1 | 2 | 3 |
|---|---|---|---|
| At copper deficiency diagnosis | |||
| Age (years) | 39 | 78 | 57 |
| Weight (kg) | 80 | 54 | 64 |
| Delay since WD diagnosis/first treatment/ (years) | 5.8 | 20 | 34 |
| Current treatment | Zinc acetate (150 mg/day) | Zinc acetate (150 mg/day) | Trientine 2HCL (600 mg/day) + zinc sulphate (1200 mg/day) |
| Duration of WD current treatment (years) | 5.8 | 14 | 21 |
| First clinical symptoms of CD | None | Progressive gait instability over 6 months and LL paresthesia | Progressive LL paresthesia, then LL distal motor weakness and neuropathic pain |
| Delay between first anomalies and CD diagnosis (months) | 6 | 24 | 6 |
| Clinical exam | Normal | Posterior cord syndrome | Posterior cord syndrome |
| Spinal MRI | Normal | Normal | Normal |
| Brain MRI | Normal | Normal | Normal |
| ENMG | Not done | Moderate axonal sensitive neuropathy of LL | Moderate axonal sensory and motor polyneuropathy of LL |
| Hemogram | Pancytopenia | Bicytopenia | Pancytopenia |
| Hemoglobin (N = 120–160 g/L) | 66 | 99 | 110 |
| Platelets (N = 150,000–400,000/mm4) | 35,000 | 437,000 | 140,000 |
| Leukocytes (N = 4000–10,000/mm4) | 3000 | 5300 (but 300 neutrophiles) | 3800 |
| Serum copper (N = 12.7–22.2 μmol/l) | 0.27 | 0.34 | 0.48 |
| Ceruloplasmin (N = 0.2–0.5 g/l) | 0.09 | 0.02 | 0.02 |
| Exchangeable copper (N = 0.62–1.15 μmol/l) | 0.08 | 0.1 | 0.08 |
| REC (%) | 29.6 | 29.4 | 16.7 |
| Urine copper excretion UCE * | 0.22 | 0.34 | 0.1 |
| Zinc blood level (N = 12.5–18 μmol/l) | 30 | 29.1 | 23.7 |
| Urine zinc excretion (N = 4–13 μmol/l) | 89 | 98 | 53.7 |
| Serum Iron (N = 12–28 μmol/l) | 4.9 | 5.5 | 7.3 |
| TSF (N = 1.7–2.7 g/L) | 4 | 3.6 | 3.12 |
| IBC (N = 15–50%) | 8 | 6 | 9 |
| B12 vitamin level | Normal | Normal | Normal |
| Modification of WD treatment | Yes, dose of zinc acetate decreased to 50 mg/d | Yes, treatment stopped | Yes, Trientine stopped and zinc sulphate decreased to 200 mg/d |
| Evolution at last follow‐up | |||
| Delay since CD diagnosis (years) | 10 | 8 | 5 |
| Neurological examination | Normal | Gait instability persists but walking distance >500 m intermittent paresthesia | Gait instability and slight LL weakness persist no more pain |
| Hemogram | Leucopenia improved, anemia disappeared, thrombopenia persisted | Normalization | Normalization |
| Serum copper (N = 12.7–22.2 μmol/l) | 3.49 | 2.14 | 1.89 |
| Ceruloplasmin (N = 0.2–0.5 g/l) | 0.11 | 0.04 | 0.02 |
| Exchangeable copper (N = 0.62–1.15 μmol/l) | 0.33 | 0.61 | 0.64 |
| Urine copper excretion UCE* | 1.07 | 1.27 | 0.78 |
| Serum Iron (N = 12–28 μmol/l) | 24.2 | 14.1 | 13 |
Abbreviations: CD, Copper Deficiency; IBC, iron binding capacity; LL, lower limbs; REC, relative exchangeable copper; TSF, transferrin.
Objectives of UCE under treatment (maintenance phase): 1.5 μmol/l for zinc salt; 3–8 μmol/l for chelators.
Diagnosis of WD was made at the age of 34, 58, and 23 years old for patient 1, 2, and 3 respectively. All patients were compound heterozygous with different mutations. Patient 1 had a compensated cirrhosis with thrombocytopenia (70,000/mm3, N > 150,000/mm3) and a Kayser‐Fleischer Ring, patient 2 had a mild cirrhosis (without cytopenia) associated with an isolated writer cramp and patient 3 was initially asymptomatic, diagnosed on familial screening but developed neurological symptoms secondary to bad adherence to treatment (but no leucothrombocytopenia).
CD appeared 17.4 ± 14 [min 5.8‐max 33] years after initiation of WD treatment and was characterized by extremely low levels of serum copper, exchangeable copper and urinary copper excretion (respectively 0.36 ± 0.11; 0.22 ± 0.12; 0.09 ± 0.01 μmol/L) while serum and urinary zinc values were high (respectively 27.6 ± 3.4; 80.2 ± 23.4 μmol/L, Fig. 1). All patients showed an iron deficiency: low iron blood level (5.9 ± 1.25 μmol/L), elevated transferrin blood level (3.57 ± 0.44 g/L) and low iron‐binding capacity (7.67 ± 1.53%) (see Table 2 for normal values).
FIG. 1.
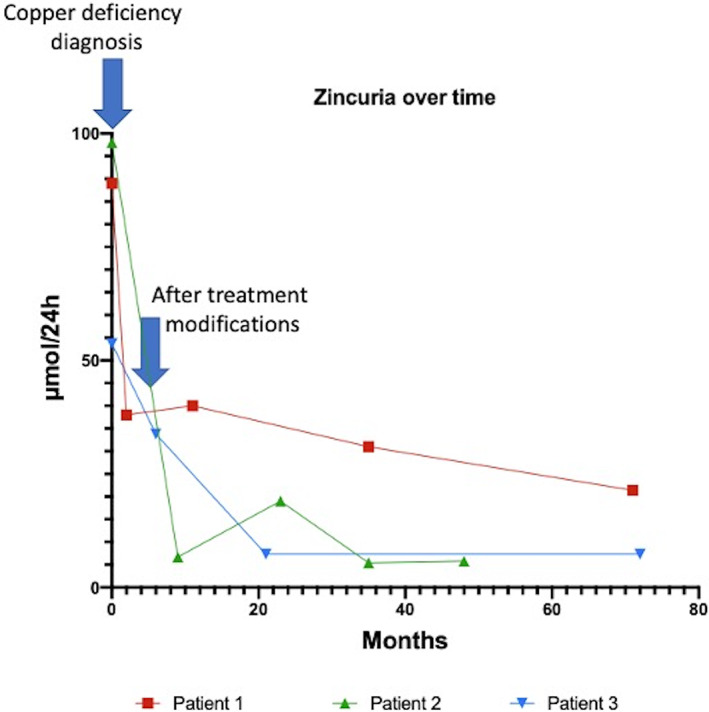
Zincuria values over time.
Apart from the copper disturbances, patient 1 worsened his initial thrombocytopenia and developed anemia and neutropenia. In patient 2 and 3, first symptom was a progressive sensory ataxic gait disorder associated with lower limbs paresthesia. Spinal cord MRI was normal in all, but sensory axonal electrophysiological pattern was displayed in the two patients with neuropathic symptoms of the lower limbs. Patient 2 also had an anemia with neutropenia while patient 3 had an anemia and a leuconeutropenia. The mean time to diagnose CD was 12 ± 10.4 [min 6‐max 24] months.
Two patients (patient 1 and 2) were on zinc acetate 150 mg/d and one patient (patient 3) on zinc sulphate 1200 mg/d associated with Trientine 2HCL 600 mg/d. No cause of acquired CD was found (malabsorption, digestive troubles etc.).
In patient 1, zinc acetate was decreased to 50 mg/day. In patient 2, zinc acetate was stopped, and an intravenous iron supplementation was performed. In patient 3, zinc sulphate was lowered to 400 then 200 mg/day and Trientine 2HCL was stopped. At 6‐ and 15 months’ follow‐up, neurological patients were subjectively better but biological data were unchanged. After a mean 8 ± 3 years follow‐up, anemia recovered in all patients. Neurological improvement continued in the two patients but without electrophysiological neither clinical normalization. Exchangeable copper values were still low but urinary copper excretion started to increase.
Literature Review
Our PubMed search revealed 419 publications, of which 404 were excluded (Fig. 2). A total of 15 publications (concerning 17 patients with WD and symptomatic CD) were reviewed (Table 3). Sex ratio was 1.43 (10 women/7 men) with a median age at diagnosis of WD of 16.5 years and median age at diagnosis of symptomatic CD of 37 years. Initial clinical form of WD was neurologic in 10 patients, hepatic in three, asymptomatic in two and not found in two. All patients received zinc, three of them were also treated by D‐Penicillamine and two by Trientine 2HCl. Symptoms of CD started on average 16 years after the diagnosis of WD. Six patients had cytopenia without neurological symptoms, nine had both cytopenia and neurological involvement and two had neurological symptoms but without available blood count. Concerning the cytopenia, anemia (12 patients) and leuconeutropenia (12 patients also) were the most frequent; pancytopenia was relatively rare (two patients only). After adaptation of the WD treatment, most of the patients have a complete normalization of the cytopenia and incomplete improvement of the neurological symptoms. Unfortunately, the delay between therapeutics adjustment and the evolution was not indicated in most publications.
FIG. 2.
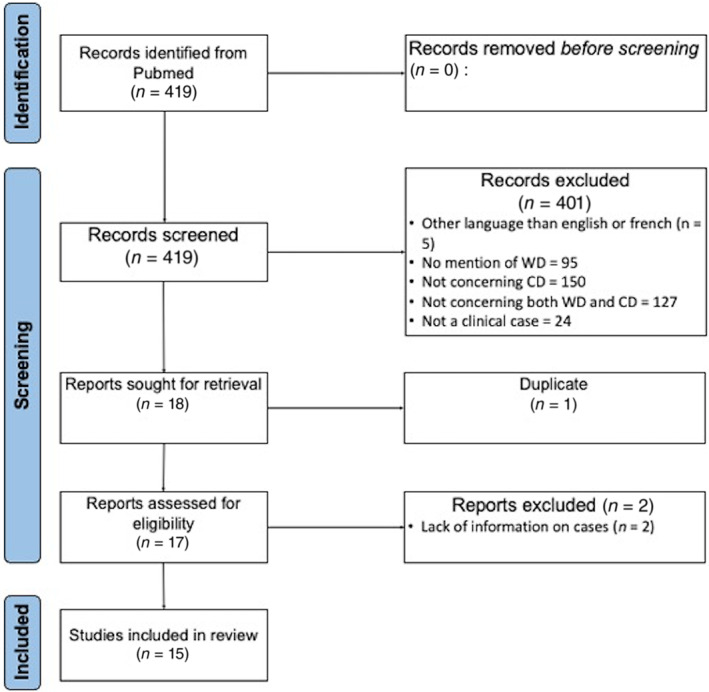
Flow chart of the literature review.
TABLE 3.
Review of the literature: Wilson's disease patients with copper deficiency
| References | Sex | Age at diagnosis of WD (years) | Clinical form of WD | Age at diagnosis of CD (years) | WD treatment when the CD is diagnosed | Symptoms | Resolution |
|---|---|---|---|---|---|---|---|
| Van Den Hamer and Hogenrad (1989) 6 | Male | ND | ND | 56 | Zinc sulphate (1200 mg/d) | Anemia | Total resolution after parenteral copper administration |
| Neutropenia | |||||||
| Cu = 0.78 μmol/l | |||||||
| UCE = ND | |||||||
| Narayan et al (2006) 7 | Male | 9 | Neurologic | 13 | Zinc sulphate (280 mg/d) + D‐Penicillamine (750 mg/d) | Anemia | ND |
| Cu = 2.5 μmol/L | |||||||
| UCE = ND | |||||||
| CNS demyelination | |||||||
| Foubert‐Samier et al (2009) 8 | Male | 15 | Neurologic | 43 | Zinc acetate (400 mg/d) + Trientine 2HCl (900 mg/d) | Anemia | Improvement in cytopenia without improvement in peripheral neuropathy after withdrawn of zinc acetate |
| Neutropenia | |||||||
| Cu = 0.5 μmol/L | |||||||
| UCE = 1.7 μmol/L | |||||||
| Axonal sensory‐motor peripheral neuropathy | |||||||
| Horvath et al (2010) 9 | Male | 25 | Neurologic | 41 | Zinc sulphate (1100 mg/d) | Anemia | Improvement in cytopenia without improvement in peripheral neuropathy after withdrawn of zinc acetate |
| Leucopenia | |||||||
| Cu = 0.5 μmol/L | |||||||
| UCE = 0.54 μmol/L | |||||||
| Axonal sensory‐motor peripheral neuropathy | |||||||
| Benbir et al (2010) 10 | Male | 16 | Neurologic | 21 | Zinc acetate (100 mg/d) + D‐Penicillamine (1200 mg/d) | Cu = 0.34 μmol/L | Total resolution after withdrawn of WD therapeutics |
| UCE = ND | |||||||
| Partial seizures | |||||||
| (No blood count available) | |||||||
| Cortese et al (2011) 11 | Female | 27 | Neurologic | 51 | Zinc sulphate (1200 mg/d) | Anemia | Improvement in cytopenia without improvement in peripheral neuropathy |
| Neutropenia | |||||||
| Cu = 0.78 μmol/L | |||||||
| UCE = 0.31 μmol/L | |||||||
| Sensory‐motor peripheral neuropathy | |||||||
| Da Silva Jr et al (2011) 12 | Female | 29 | Neurologic | 44 | Zinc acetate (450 mg/d) | Macrocytosis without anemia | Stabilization of clinical status |
| Leukopenia | |||||||
| Thrombocytopenia | |||||||
| Cu = 0.47 μmol/L | |||||||
| UCE = 0.11 μmol/L | |||||||
| Myeloneuropathy | |||||||
| Lozano Herrero et al (2012) 13 | Female | 18 | Hepatic | 56 | Zinc acetate (150 mg/d) | Anemia | Minimal improvement |
| Neutropenia | |||||||
| Cu = 0.47 μmol/L | |||||||
| UCE = indétectable | |||||||
| Subacute combined degeneration | |||||||
| Teodoro et al (2013) 14 | Male | 20 | Neurologic | 36 | Zinc sulphate (330 mg/d) + Trientine 2HCl (500 mg/d) | Anemia | Partial regression |
| Cu = 2.09 μmol/L | |||||||
| UCE = 0.63 μmol/L | |||||||
| Posterior dorsal cord myelopathy | |||||||
| Dziezyc et al (2014) 15 | Female | 19 | Presymptomatic | 37 | Zinc sulphate (180 mg/d) | Neutropenia | Clinical and biological improvement |
| Cu <0.78 μmol/L | |||||||
| UCE = 0.17 μmol/L | |||||||
| Posterior dorsal cord | without total resolution | ||||||
| myelopathy | |||||||
| Female | 16 | Hepatic | 41 | Zinc sulphate (180 mg/d) | Neutropenia | Total resolution after decreasing | |
| Cu <0.78 μmol/L | |||||||
| UCE = 0.09 μmol/L | the treatment | ||||||
| Female | 12 | Presymptomatic | 18 | Zinc sulphate (180 mg/d) | Pancytopenia | Total resolution after withdrawn of the treatment | |
| Cu = 1.10 μmol/L | |||||||
| UCE = 0.18 μmol/L | |||||||
| Rau et al (2014) 19 | Male | 14 | ND | 16 | Zinc sulphate | Cu = 4.87 μmol/L | Total resolution after withdrawn of the treatment |
| UCE = 0.50 μmol/L | |||||||
| Anemia | |||||||
| Neutropenia | |||||||
| Mohamed et al (2018) 20 | Female | 13 | Neurologic | 26 | Zinc sulphate (600 mg/d) | Pancytopenia | Total resolution after withdrawn of the treatment |
| Cu <0.1 μmol/L | |||||||
| UCE = ND μmol/L | |||||||
| Cai et al (2019) 21 | Female | 7 | Hepatic | 11 | Zinc gluconate (240 mg/d) | Anemia | Total resolution after withdrawn of the treatment |
| Neutropenia | |||||||
| Cu = 0.88 μmol/L | |||||||
| UCE = 0.47 μmol/L | |||||||
| Abnormal gait | |||||||
| Wu et al (2020) 16 | Female | 17 | Neurologic | 18 | Zinc sulphate (225 mg/d) | Cu = ND | Total resolution after copper supplementation |
| UCE = significantly diminished | |||||||
| Subacute combined degeneration of the spinal cord | |||||||
| (No blood count available) | |||||||
| Ueda et al (2022) 22 | Female | 20 | Neurologic | 57 | Zinc acetate (150 mg/d) + D‐penicillamine (1000 mg/d) | Anemia | Improvement in cytopenia with partial improvement in myeloneuropathy after withdrawn of zinc acetate and D‐penicillamine |
| Cu = 1.73 μmol/L | |||||||
| UCE = 1.16 μmol/L | |||||||
| Posterior dorsal cord myelopathy |
Abbreviations: CD, copper deficiency; CNS, central nervous system; ND, no data; WD, Wilson's disease.
Discussion
Copper deficiency in WD patients exists although it remains extremely rare. The pre‐existing hepatic or neurological form of WD is not predictable of the risk of CD under treatment. In the same way, and despite the description of only male patients in our cohort, the CD could touch any gender. There are no published data on the correlation between CD phenotype and mutations in WD. Our patients carry different mutations suggesting that the causative mutation is not involved in the phenotype of CD. Symptomatic CD occurred more than a decade and half after the initiation of WD treatment, a delay that could be related to the time requiring eliminating the excess of copper before developing a deficiency.
In our cohort, like in the literature review, CD led to cytopenia, especially anemia and leuconeutropenia, and neurological disorders. Indeed, copper is a cofactor in several oxidative enzymes vital to the function of hematopoietic, vascular, skeletal tissue and nervous system. These enzymes are involved in electron‐transporting proteins and in antioxidant metabolism. The ferroxidases (hephaestin and ceruloplasmin) and the antioxidant cytochrome C oxidase are particularly implicated in normal hematopoiesis. Hephaestin is required to oxidize Fe2+ in Fe3+ for binding to transferrin ensuring its transport to the bone marrow and a normal hematopoiesis. Ceruloplasmin is a transport protein delivering copper from the liver to peripheral tissue. As hephaestin, it also serves as a ferroxidase in converting ferrous iron to ferric iron. Cytochrome C oxidase is required in the mitochondria for reducing the ferric iron into ferrous iron, required for the incorporation of iron into the protoporphyrin structure for hemoglobin synthesis. 23 CD therefore leads to a decrease activity in these enzymes and so impaired iron absorption, transport of iron across the intestinal cells, conversion of ferrous iron into ferric iron and vice‐versa. Moreover, the lack of antioxidant enzyme Cytochrome C could lead to a possibly red blood cell membrane defect and so reduced their lifespan. Leukopenia/neutropenia could be secondary to an impaired maturation and an increased destruction of myeloid precursors within the bone marrow. CD impact also the self‐renewal of CD34+ hematopoietic progenitors’ cells. 23 , 24 , 25 Thrombocytopenia seems to be less frequent. 25 Nevertheless, cytopenia could also be linked to the hypersplenism secondary to cirrhosis. Two of our patients had cirrhosis and cytopenia, but it is highly probable that the cytopenia was linked to an associated symptomatic copper deficiency. Indeed, patient 1 had very profound thrombocytopenia and developed secondarily anemia, which is unusual in hypersplenism; patient 2 had a mild cirrhosis without initial cytopenia, and developed anemia without thrombocytopenia during the CD diagnosis. Moreover, cytopenia is a rare side‐effect of trientine and D‐penicillamine. Mechanism of cytopenia induced by chelators are not clear but could be an immune cytopenia 26 or a bone marrow toxicity. 27
Concerning the neurological disorders, the most common presentation seems to be a PCS (Fig. 3). This disorder could evolve to a clinical pattern mimicking the subacute combined degeneration secondary to vitamin B12 deficiency. 24 , 25 , 28 Sensory ataxic gait seems to be the first symptom of PCS. 7 Others neurological symptoms include optic neuropathy, central nervous system demyelination, brainstem involvement, myeloneuropathy with spastic paraparesis or tetraparesis, myelo‐optico‐neuropathy, motor neuron disease, sensory‐motor peripheral neuropathy and small fiber involvement have been described. 11 , 25 , 28 Optic neuropathy typically has an indolent course. It presents with unilateral symptoms and can lead to total cecity if the CD is not corrected. However, if supplementation prevent further progression of the disease, it has no effect on visual recovery. 25
FIG. 3.
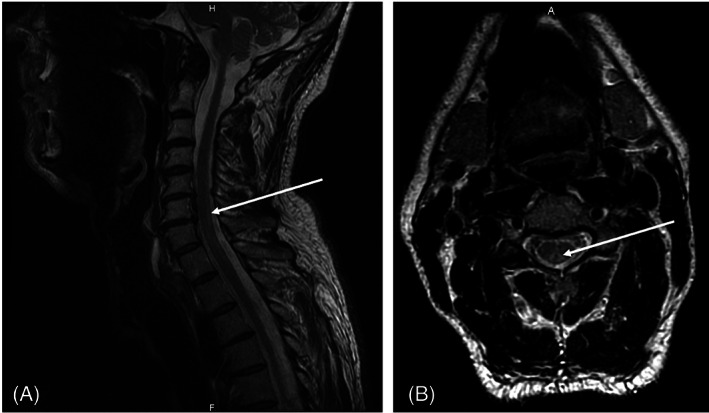
Sagittal (A) and axial (B) MRI section of a patient with copper deficiency showing an hypersignal in FLAIR sequence corresponding to a posterior column syndrome.
Spinal cord MRI usually shows a T2‐weighted sequences hypersignal, most often involving cervical and/or thoracic cord. MRI can be normal despite neurological presentation of PCS. 25 , 28 Brain MRI can be abnormal with widespread periventricular white matter lesion which could be due to vascular leukoencephalopathy or demyelination lesions. 28 , 29 In fact, animal models of CD suggest that spinal cord damage is secondary to demyelination, Wallerian degeneration and gliosis. 28 , 30 Copper is involved in the methylation cycle so CD could lead to a failure in myelin maintenance leading to demyelination lesions. 28 , 31 Cerebral spinal fluid examination is usually normal. 28
One of the patients in our literature review presented with recurrent partial seizures. Seizures related to hypocupremia are not well described in the literature, but copper is known to be a component of key metalloenzymes that have a critical role in the structure and function of the nervous system. Nevertheless, in Menkes disease, a X‐linked genetic disorder of intracellular copper transport, epileptic seizures are well‐described. Also, copper replacement therapy has shown to be effective in seizure control in one patient with Menkes disease. In the case of the WD patient described in our literature review, the cessation of the Zinc therapy led to a discontinuation of the epileptic seizures (while anti‐epileptic treatments were not effective) supporting the role of CD in the partial seizures.
Another manifestation described in the literature but not observed in our patients is a decrease in immune functions. Indeed, the involvement of copper in the immune system was suspected in the early 1980's when copper‐deficient animals showed increase susceptibility to Salmonella or Candida albicans infections. The role of copper in immune function is still unclear but CD seems to reduce the production of interleukin by T‐lymphocytes. 24 , 32 Copper could be implicated in destruction of microorganisms through the generation of hydroxyl radicals and in the intracellular trafficking to phagosomal compartment in macrophages threw the ATP7A. 24 , 33 , 34
All the patients, in the literature and in our cohort, were treated with zinc salt. Cases of CD, in general population, secondary to chronic absorption of oral zinc, in particular with the use of zinc‐enriched dental cream, have been reported in the literature. 28 , 35 , 36 , 37 Interestingly, CD symptoms have not yet been reported in patients treated only with chelator agents despite the superior decoppering potential of these agents compared to zinc salt. This observation could suggest that zinc overload could contribute to the symptoms observed in our patients. 12 However, the patient reported by Teodoro et al had a normal zinc blood level arguing against this hypothesis. 14 But the zinc blood level could not reflect the real zinc overload. Thus, a level of zincuria >2000 μg/24 h (>30 μmol/24 h) is often retained to assess a good adherence to treatment. 38 In our cases, all the patients had a zincuria much higher than 50 μmol/l, confirming the zinc overload (Fig. 2). After treatment withdrawn or modification, zincuria decreased below 2000 μg/24 h (30 μmol/24 h). Another hypothesis is the chelator leads to an elimination of the excess of copper in organs but does not influence the copper balance unlike to zinc salts. CD could be secondary to several other causes 25 (Table 4).
TABLE 4.
Causes of copper deficiency (adapted from Myint et al)
| Causes | Diseases |
|---|---|
| Drug‐related | Excessive zinc ingestion (from dental paste, excessive oral supplementation or in Wilson's disease treatment) or iron supplements |
| Decreased/inadequate intake | Anorexic patients, malnutrition, Kwashiorkor, vegetarian, parenteral or enteral nutrition without adequate copper supplements |
| Increase copper loss or inadequate absorption | Inflammatory bowel disease (Crohn's disease, ulcerative colitis), celiac disease, bypass surgery, gastrectomy, prolonged diarrhea, protein‐losing enteropathy, tropical and non‐tropical spure |
| Increase demand | Pregnant women, lactating women, premature infants, children |
| Hereditary | Menkes disease |
Finally, in all the cases cytopenia seems to resolve after withdrawn of WD treatment while neurological symptoms regressed little or not at all. However, one of these cases (reported by Wu et al) had a rapid apparition of CD symptoms and total resolution of neurological symptoms after copper supplementation. 16 This observation suggests that early detection of zinc‐induced CD, and stringent follow‐up, is important to prevent iatrogenic effect of zinc in WD patients. Early recognition and copper replacement could permit a complete resolution of neurological symptoms. 16 , 39
Conclusion
The appearance of cytopenia and/or a progressive PCS in a WD patient on long‐term zinc therapy should raise the possibility of CD. Early recognition of CD, requiring a rigorous follow‐up, is important to prevent the occurrence of hematological and/or neurological symptoms. Reduction of treatment usually allows resolution of cytopenia but only partial resolution of neurological symptoms.
Author Roles
(1) Research project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing of the first draft, B. Review and Critique.
K.C.: 1C, 2C, 3A.
M.A.O.: 1B, 3B.
N.D.O.: 1B, 2A, 2B.
A.P.: 1A, 1B, 2C, 3B.
Disclosures
Ethical Compliance Statement: All patients gave their informed consent for the genetic analysis and for anonymous study of their data (consent for research) before their inclusion in the French WD Registry. This study was approved by the Institutional Review Board (IRB00003888, IORG0003254, FWA00005831) of the French Institute of medical research and Health (N°19–550). We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest: None of the authors declare any financial disclosures or conflict of interest.
Financial Disclosures for the Previous 12 Months: None of the authors declare any financial disclosures for the previous 12 months.
References
- 1. Poujois A, Woimant F, Samson S, Chaine P, Girardot‐Tinant N, Tuppin P. Characteristics and prevalence of Wilson's disease: a 2013 observational population‐based study in France. Clin Res Hepatol Gastroenterol 2018;42(1):57–63. [DOI] [PubMed] [Google Scholar]
- 2. Aggarwal A, Bhatt M. Advances in treatment of Wilson disease. Tremor Other Hyperkinet Mov 2018;8:525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brewer GJ. Zinc acetate for the treatment of Wilson's disease. Expert Opin Pharmacother 2001;2(9):1473–1477. [DOI] [PubMed] [Google Scholar]
- 4. Hedera P. Clinical management of Wilson disease. Ann Transl Med 2019;7(S2):S66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Roberts EA, Schilsky ML. Diagnosis and treatment of Wilson disease: an update. Hepatology 2008;47(6):2089–2111. [DOI] [PubMed] [Google Scholar]
- 6. Van Den Hamer CJA, Hoogenraad TU. Copper deficiency in Wilson's disease. Lancet 1989;334(8660):442. [DOI] [PubMed] [Google Scholar]
- 7. Narayan S, Kaveer N. CNS demyelination due to hypocupremia in Wilson's disease from overzealous treatment. Neurol India 2006;54(1):110. [DOI] [PubMed] [Google Scholar]
- 8. Foubert‐Samier A, Kazadi A, Rouanet M, Vital A, Lagueny A, Tison FO, Meissner W. Axonal sensory motor neuropathy in copper‐deficient Wilson's disease: neuropathy in WD. Muscle Nerve 2009;40(2):294–296. [DOI] [PubMed] [Google Scholar]
- 9. Horvath J, Beris P, Giostra E, Martin P‐Y, Burkhard PR. Zinc‐induced copper deficiency in Wilson disease. J Neurol Neurosurg Psychiatry 2010;81(12):1410–1411. [DOI] [PubMed] [Google Scholar]
- 10. Benbir G, Gunduz A, Ertan S, Ozkara C. Partial status epilepticus induced by hypocupremia in a patient with Wilson's disease. Seizure 2010;19(9):602–604. [DOI] [PubMed] [Google Scholar]
- 11. Cortese A, Zangaglia R, Lozza A, Piccolo G, Pacchetti C. Copper deficiency in Wilson's disease: peripheral neuropathy and myelodysplastic syndrome complicating zinc treatment: letters to the editor. Mov Disord 2011;26(7):1361–1362. [DOI] [PubMed] [Google Scholar]
- 12. da Silva‐Junior FP, Machado AAC, Lucato LT, Cancado ELR, Barbosa ER. Copper deficiency myeloneuropathy in a patient with Wilson disease. Neurology 2011;76(19):1673–1674. [DOI] [PubMed] [Google Scholar]
- 13. Lozano Herrero J, Muñoz Bertrán E, Ortega González I, Gómez Espín R, López Espín MI. Myelopathy secondary to copper deficiency as a complication of treatment of Wilson's disease. Gastroenterol Hepatol 2012;35(10):704–707. [DOI] [PubMed] [Google Scholar]
- 14. Teodoro T, Neutel D, Lobo P, et al. Recovery after copper‐deficiency myeloneuropathy in Wilson's disease. J Neurol 2013;260(7):1917–1918. [DOI] [PubMed] [Google Scholar]
- 15. Dzieżyc K, Litwin T, Sobańska A, Członkowska A. Symptomatic copper deficiency in three Wilson's disease patients treated with zinc sulphate. Neurol Neurochir Pol 2014;48(3):214–218. [DOI] [PubMed] [Google Scholar]
- 16. Wu LM, Ekladious A, Wheeler L, Mohamad AA. Wilson disease: copper deficiency and iatrogenic neurological complications with zinc therapy. Intern Med J 2020;50(1):121–123. [DOI] [PubMed] [Google Scholar]
- 17. Poujois A, Trocello J‐M, Djebrani‐Oussedik N, et al. Exchangeable copper: a reflection of the neurological severity in Wilson's disease. Eur J Neurol 2017;24(1):154–160. [DOI] [PubMed] [Google Scholar]
- 18. Poujois A, Sobesky R, Meissner WG, et al. Liver transplantation as a rescue therapy for severe neurologic forms of Wilson disease. Neurology 2020;94(21):e2189–e2202. [DOI] [PubMed] [Google Scholar]
- 19. Rau AR, Usha M, Mallya P, Rau ATK. Cytopenia and bone marrow dysplasia in a case of Wilson's disease. Indian J Hematol Blood Transfus 2014;30(S1):433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mohamed M, Johnston A, Maclaine Cross A, Sharma A. Reversible pancytopenia caused by severe copper deficiency in a patient with Wilson disease. Med J Aust 2018;209(1):11–12. [DOI] [PubMed] [Google Scholar]
- 21. Cai S, Gong J‐Y, Yang J, Wang J‐S. Anemia following zinc treatment for Wilson's disease: a case report and literature review. BMC Gastroenterol 2019;19(1):120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ueda M, Katsuse K, Kakumoto T, et al. Copper deficiency in Wilson's disease with a normal zinc value. Intern Med. 2023;62(7):1073–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lazarchick J. Update on anemia and neutropenia in copper deficiency. Curr Opin Hematol 2012;19(1):58–60. [DOI] [PubMed] [Google Scholar]
- 24. Scheiber I, Dringen R, Mercer JFB. Copper: effects of deficiency and overload. In: Sigel A, Sigel H, Rko S, eds. Interrelations between essential metal ions and human diseases. Dordrecht: Springer Netherlands; 2013:359–387. [DOI] [PubMed] [Google Scholar]
- 25. Myint ZW, Oo TH, Thein KZ, Tun AM, Saeed H. Copper deficiency anemia: review article. Ann Hematol 2018;97(9):1527–1534. [DOI] [PubMed] [Google Scholar]
- 26. Chetri K et al. Wilson's disease in north‐East India: a case series. J Gastroenterol Hepatol 2019;34(Suppl. 3):517.30408229 [Google Scholar]
- 27. LiverTox: Clinical and Research Information on Drug‐Induced Liver Injury [Internet]. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases; 2012. [PubMed] [Google Scholar]
- 28. Poujois A, Djebrani‐Oussedik N, Ory‐Magne F, Woimant F. Neurological presentations revealing acquired copper deficiency: diagnosis features, aetiologies and evolution in seven patients: Neurological acquired copper deficiency. Intern Med J 2018;48(5):535–540. [DOI] [PubMed] [Google Scholar]
- 29. Prodan CI, Holland NR, Wisdom PJ, Burstein SA, Bottomley SS. CNS demyelination associated with copper deficiency and hyperzincemia. Neurology 2002;59(9):1453–1456. [DOI] [PubMed] [Google Scholar]
- 30. Tan N, Urich H. Menkes' disease and swayback. J Neurol Sci 1983;62(1–3):95–113. [DOI] [PubMed] [Google Scholar]
- 31. Wapnir RA. Copper absorption and bioavailability. Am J Clin Nutr 1998;67(5):1054S–1060S. [DOI] [PubMed] [Google Scholar]
- 32. Hopkins RG, Failla ML. Copper deficiency reduces Interleukin‐2 (IL‐2) production and IL‐2 mRNA in human T‐lymphocytes. J Nutr 1997;127(2):257–262. [DOI] [PubMed] [Google Scholar]
- 33. Hodgkinson V, Petris MJ. Copper homeostasis at the host‐pathogen Interface. J Biol Chem 2012;287(17):13549–13555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. White C, Lee J, Kambe T, Fritsche K, Petris MJ. A role for the ATP7A copper‐transporting ATPase in macrophage bactericidal activity. J Biol Chem 2009;284(49):33949–33956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jaiser SR, Winston GP. Copper deficiency myelopathy. J Neurol 2010;257(6):869–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Prasad AS. Hypocupremia induced by zinc therapy in adults. JAMA 1978;240(20):2166–2168. [PubMed] [Google Scholar]
- 37. Trocello J‐M, Hinfray S, Sanda N, et al. Une cause méconnue de myélopathie par carence en cuivre : l'utilisation de pâte adhésive dentaire. Rev Neurol 2011;167(6–7):537–540. [DOI] [PubMed] [Google Scholar]
- 38. Ranucci G, Di Dato F, Spagnuolo MI, Vajro P, Iorio R. Zinc monotherapy is effective in Wilson's disease patients with mild liver disease diagnosed in childhood: a retrospective study. Orphanet J Rare Dis 2014;9(1):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kumar N, Gross JB, Ahlskog JE. Copper deficiency myelopathy produces a clinical picture like subacute combined degeneration. Neurology 2004;63(1):33–39. [DOI] [PubMed] [Google Scholar]