

Abstract
Background and Aims:
TGFβ plays pleiotropic roles in pancreatic cancer including promoting metastasis, attenuating CD8 T cell activation, and enhancing myofibroblast differentiation and deposition of extracellular matrix. However, single-agent TGFβ inhibition has shown limited efficacy against pancreatic cancer in mice or humans.
Methods:
We evaluated the TGFβ blocking antibody NIS793 in combination with either gemcitabine/n(ab)-paclitaxel or FOLFIRINOX chemotherapy in orthotopic pancreatic cancer models. Single-cell RNA-seq and immunofluorescence were used to evaluate changes in tumor cell state and the tumor microenvironment.
Results:
Blockade of TGFβ with chemotherapy reduced tumor burden in poorly immunogenic pancreatic cancer, without affecting the metastatic rate of cancer cells. Efficacy of combination therapy was not dependent on CD8 T cells, as response to TGFβ blockade was preserved in CD8-depleted or RAG2−/− mice. TGFβ blockade decreased total αSMA+ fibroblasts but had minimal effect on fibroblast heterogeneity. Bulk RNA-seq on tumor cells sorted ex vivo revealed that tumor cells treated with TGFβ blockade adopted a classical lineage consistent with enhanced chemosensitivity, and immunofluorescence for cleaved caspase 3 confirmed that TGFβ blockade increased chemotherapy-induced cell death in vivo.
Conclusions:
TGFβ regulates pancreatic cancer cell plasticity between classical and basal cell states. TGFβ blockade in orthotropic models of pancreatic cancer enhances sensitivity to chemotherapy by promoting a classical malignant cell state. This study provides scientific rationale for evaluation of NIS793 with either FOLFIRINOX or gemcitabine/n(ab)paclitaxel chemotherapy backbone in the clinical setting and supports the concept of manipulating cancer cell plasticity to increase the efficacy of combination therapy regimens.
Keywords: TGFβ, pancreatic cancer, tumor microenvironment, immunotherapy, chemotherapy
Pancreatic ductal adenocarcinoma (PDAC) is the third leading cause of cancer death, with a 5-year survival rate of 11%1. PDAC is characterized by dense desmoplasia, resulting in a hypoxic, nutrient-poor immunosuppressive tumor microenvironment2. Response to combination chemotherapy is limited, and immunotherapy has been largely unsuccessful to date2.
The cytokine TGFβ plays an important regulatory role in cellular homeostasis through nuclear translocation of SMAD family members, and inactivation of its signaling can lead to tumorigenesis. In early-stage PDAC, blockade of TGFβ signaling in epithelial cells leads to rapid outgrowth and disease progression3, 4. However, loss of SMAD4, a frequent event in PDAC, renders these tumors insensitive to the growth inhibitory effects of TGFβ5, 6. Mouse models with genetic deficiencies in TGFβ signaling in KrasG12D-expressing acinar cells develop earlier and more aggressive tumors4, 5, 7, 8. Although these mice have lower rates of metastasis than littermates with intact TGFβ signaling, their more aggressive primary tumors lead to shorter overall survival5. Nevertheless, the reduction in metastasis supports a role for TGFβ in promoting epithelial to mesenchymal transitions in mice. In human PDAC organoids, exposure to TGFβ promotes a mesenchymal or basal-like state9.
TGFβ also acts on stromal cells. LRRC15+ disease-associated fibroblasts are dependent on TGFβ signaling as mice selectively lacking TGFβR in fibroblasts fail to develop the LRRC15+ CAF subset10. In the context of metastasis, myeloid cells responding to TGFβ in the primary tumor can facilitate tumor cell metastasis11. TGFβ signaling also plays a role in T cell function. TGFβ promotes Foxp3-expressing regulatory CD4 T cells which protect against tumor-promoting inflammation in early stages of disease but inhibit CD8 T cell function in established tumors12–14. TGFβ signaling in CD8 T cells attenuates their proliferation, cytokine production, and ability to express CXCR3 for entry into tumors15, 16. In models of colorectal and other fibrotic cancers, TGFβ blockade alters the stromal compartment to allow for influx of activated CD8 T cells and, in combination with PD-1 or PD-L1 blockade, immune-mediated tumor regression17, 18. Thus, the stromal and tumor cell state modulating properties of TGFβ blockade may synergize in appropriately designed combination strategies.
Although genetic depletion of TGFβ signaling in mice promotes more aggressive oncogenesis4–8, blockade of TGFβ in established cancer may be beneficial due to the pleiotropic effects of this cytokine on multiple cell types19. TGFβ inhibition using multiple strategies in mouse pancreatic cancer models appears promising20, 21. Ongoing attempts to block TGFβ in PDAC have included small molecule inhibitors of TGFβR122, antisense oligonucleotides23, and bispecific fusion proteins consisting of the TGFβRII extracellular domain and several different partners including antiEGFR, PD-L1 blockade or IL-1524, 25. The most advanced agent that has completed clinical trials in pancreatic cancer is galunisertib, a small molecule inhibitor of TGFβR1. In first-line locally advanced and metastatic PDAC, the combination of galunisertib with gemcitabine increased median survival by 1.8 months compared to gemcitabine alone26. However, a phase I trial of galunisertib and PD-L1 blockade in second-line metastatic PDAC showed no benefit over historical controls27. As a monotherapy, TGFβ inhibition is unlikely to be effective, and understanding the definitive mechanism of action of TGFβ in PDAC is critical to developing rational combination therapies.
The monoclonal antibody NIS793 blocks TGFβ1/2 ligand, to a lesser extent TGFβ3, and is structurally distinct from TGFβ trap fusion proteins. In mouse models of breast and colorectal cancer, NIS793 repolarized fibroblasts to an interferon-licensed CAF population and increased CD8 T cell infiltration into the tumor interior28. Here we evaluated antibody blockade of TGFβ in several mouse models of orthotopic and metastatic PDAC. Although we recapitulate findings of decreased αSMA+ fibroblasts and increased T cell infiltration into tumors, neither of these effects are required for disease control. The primary effect of TGFβ blockade in mouse PDAC models is to polarize tumor cells toward the classical lineage, thereby enhancing sensitivity to combination chemotherapies. We demonstrate the efficacy of TGFβ blockade combined with either gemcitabine/n(ab)paclitaxel or FOLFIRINOX, the two most frequently used regimens in patients with PDAC. These data support clinical evaluation of NIS793 in combination with gemcitabine/n(ab)paclitaxel or FOLFIRINOX.
Material and Methods
Cell lines
KPC.1 cells derived from a LSL-KrasG12D;p53+/floxed, Pdx-cre mouse were a gift from Dr. Anirban Maitra (MD Anderson). KPCY cell line 6694c2 was a gift from Dr. Ben Stanger (University of Pennsylvania)29.
Mice
All animal protocols were approved by the Dana-Farber Cancer Institute Committee on Animal Care (protocol #14–019, 14–037, 10–055) and are in compliance with the NIH/NCI ethical guidelines for tumor-bearing animals. The following mouse strains were purchased from Jackson labs: C57BL/6 (000664), TCRα−/− (002116), β2m−/− (002087), RAG2−/− (008449).
In vivo treatments
NIS793 and human IgG2c isotype control antibodies were provided by Novartis Pharmaceuticals and dosed i.p. at 200 μg/mouse every two days as indicated. In some experiments, mice received depleting antibodies (150μg/mouse) on the day of tumor inoculation and then every 3 days until sacrifice. Antibodies were purchased from Bioxcell: αCD4[GK1.5], αCD8[2.43]. Chemotherapies were obtained from the Dana-Farber Cancer Institute Pharmacy and were administered as per the dosing schedules indicated in each relevant figure. Gemcitabine was administered IP; all other chemotherapies were administered IV by tail vein injection.
Statistical analysis
Graphpad Prism software was used to analyze data. Significance between two groups was determined using a two-sided Mann-Whitney test, without assuming Gaussian distribution. Significance among more than two groups was determined using ANOVA with multiple hypothesis testing. Individual data points shown in each graph are independent biological replicates. For some figures, tumor weights were normalized to the average of the vehicle group within each experiment and data from multiple experiments were combined and presented as “normalized tumor weights”.
Results
TGFβ blockade does not affect rate of metastasis in a mouse model of neoadjuvant chemotherapy.
To test the role of TGFβ blockade (αTGFβ, TGFβ blocking antibody NIS793) in metastasis formation, we generated a metastatic mouse PDAC model from the poorly immunogenic pancreatic cancer cell line 6694c229. 6694c2 parental cells were orthotopically inoculated into mice and allowed to spontaneously metastasize to the lung. Metastases were isolated ex vivo and passaged to generate the 6694c2-met cell line (Supplemental Figure 1). To confirm that TGFβ signaling can be effectively blocked by αTGFβ, we cultured 6694c2-met cells with TGFβ ligand or TGFβ blocking antibody and analyzed phosphorylated SMAD2 by immunoblot (Figure 1A). 6694c2-met cells showed intact TGFβ signaling that can be activated by TGFβ ligand and blocked by αTGFβ.
Figure 1: TGFβ blockade does not affect metastasis formation in vivo.
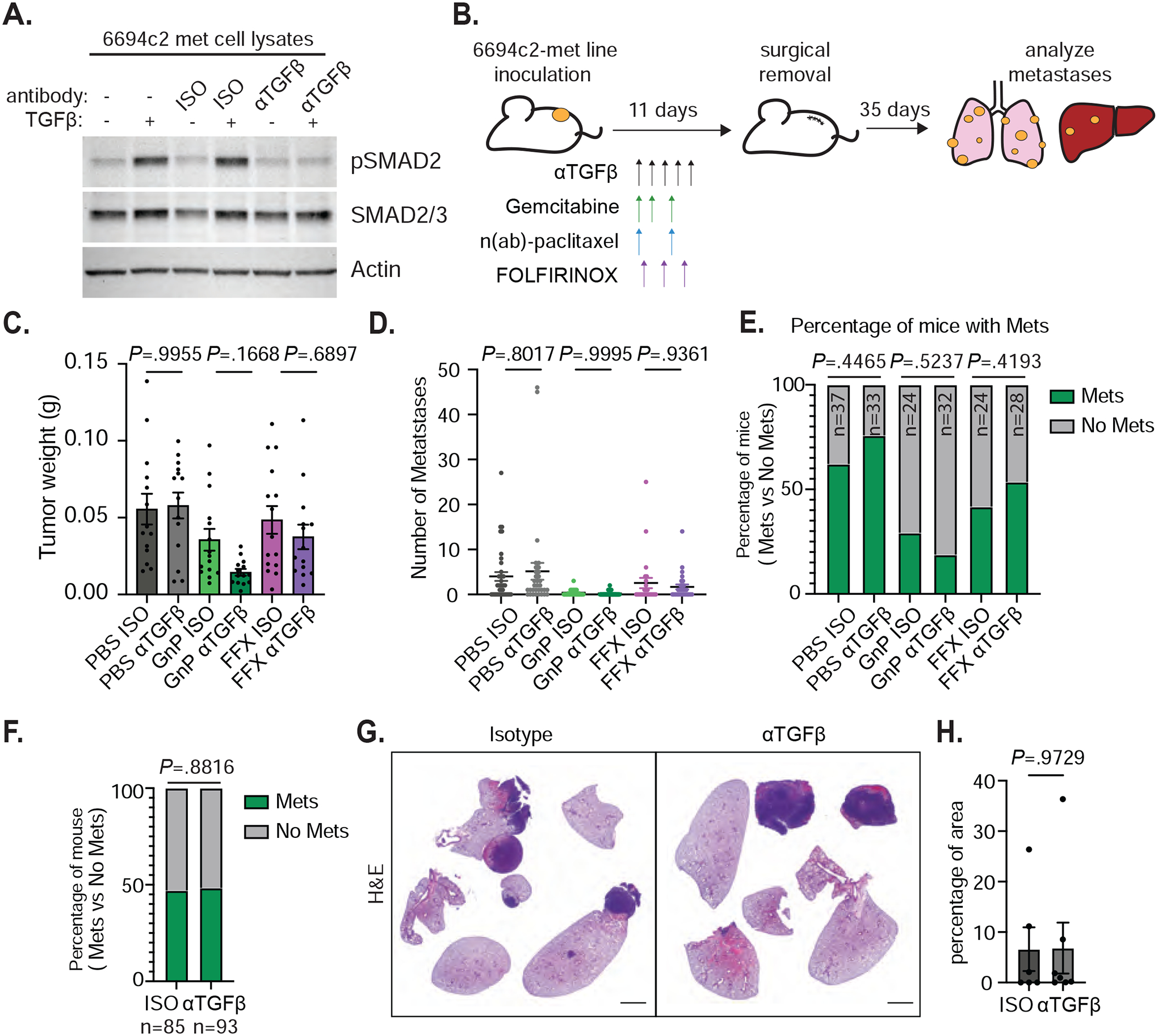
A) 6694c2-met cells were cultured with 5ng/ml TGFβ, and/or 20ug/ml TGFβ blocking or isotype control antibody for 24h. Protein lysates were collected and analyzed by immunoblot. B) Diagram of experimental metastasis model. 180,000 6694c2-met cells were implanted subcutaneously in the lower back of C57BL/6 mice. Mice were treated with αTGFβ or isotype control i.p. every 2 days starting on day 2 for 5 doses. Gemcitabine was given on days 2, 4 and 7; nab-paclitaxel was dosed on days 2 and 7. FOLFIRINOX (5mg/kg oxaliplatin, 50mg/kg irinotecan, 75mg/kg leucovorin, and 75mg/kg 5-FU) was dosed on day 3. Primary tumors grew for 11 days before surgical removal. Metastases in the lung, liver and lymph node were analyzed at day 46. C) Primary tumor weight upon surgical removal. Data are representative of 3 independent experiments. D) Total metastases visually counted in different treatment groups. E) The percentage of mice containing metastases. F) The percentage of mice containing metastases from all isotype-treated or all αTGFβ-treated groups pooled from 3 independent experiments. G) Representative H&E image of metastases in the lungs from isotype and αTGFβ single agent treated mice. H) Quantification was performed on the H&E image for the percentage of area of metastases in the lungs from isotype and αTGFβ single agent treated mice. Scale bars = 2000px. Error bars are SEM throughout. Mann-Whitney t-test was used for comparison between two groups.
Patients with resectable PDAC are commonly treated with neoadjuvant chemotherapy followed by surgical resection, but most patients experience disease progression post-surgery2. To model this experience in mice, 6694c2-met cells were inoculated subcutaneously and grew for 11 days prior to surgical removal. Mice were treated with αTGFβ and/or chemotherapies before surgery. We included the two most common chemotherapy regimens for PDAC, FOLFIRINOX (5-fluorouracil, leucovorin, irinotecan, and oxaliplatin) and GnP (gemcitabine plus n(ab)-paclitaxel). Macro- and micro-metastasis rates were counted by direct visualization and microscopic imaging with H&E staining at day 46 after initial inoculation (Figure 1B).
TGFβ blockade alone did not affect primary tumor weight as measured at surgical removal (Figure 1C). When combined with GnP, and to a lesser extent FOLFIRINOX, αTGFβ reduced the primary tumor burden compared to chemotherapy alone (Figure 1C). GnP reduced the rate of metastasis, consistent with decreased primary tumor burden. Addition of TGFβ blockade affected neither the metastasis rate to the lung, lymph nodes and the liver, nor the percentage of mice with metastasis across any of the treatment backbones (Figure 1D–E, Supplemental Figure 1A–C). To further define the key factors that affect rate of tumor metastasis in our mouse model, we applied a zeroinflated Poisson regression model, which showed that primary tumor weight strongly predicts the number of metastases. TGFβ blockade does not affect metastasis in any of the tested conditions (Figure 1F and Supplemental Figure 1D–E). H&E staining further revealed that αTGFβ does not affect the area of micro- and macro-metastasis in the lung (Figure 1G–H).
In spontaneous models of pancreatic cancer, genetic loss of the genes encoding SMAD4 or TGFbR2 lead to more aggressive primary tumors4, 5, 7, 8. Paradoxically, conditional activation of TGFβ signaling in adult acinar cells expressing oncogenic KRAS leads to acinar ductal metaplasia (ADM) and more rapid oncogenesis30. Deletion of TGFbR2 in mutant KRAS acinar cells in the context of pancreatitis also leads to increased ADM31, suggesting that the anti-proliferative effects of TGFβ are important for restoration of homeostasis after inflammation, but that constitutive TGFβ signaling in the absence of inflammation can lead to spontaneous, pathologic ADM. Here we wondered whether pharmacologic blockade of TGFβ in KPC mice (LSL-KRASG12D/+;LSLTrp53R172H/+; Ptf1aCre/+) with ultrasound-confirmed tumors would lead to increased acinar cell proliferation or ADM formation in the tumor-adjacent normal regions of the pancreas, similar to that observed in the genetically deficient models. We did not observe major differences in Ki67+ epithelial cells, αSMA+ fibroblasts or ADM formation in KPC mice treated with αTGFβ or αTGFβ + chemotherapy (Supplemental Figure 2). This could be reflective of the shorter time course or partial blockade of TGFβ achieved by the antibody as compared to genetic loss of SMAD4 or TGFβR2.
Combination of TGFβ blockade with GnP is effective in poorly immunogenic orthotopic 6694c2 tumors.
To determine the effect of TGFβ blockade on primary tumor progression, we implanted 6694c2 parental cells orthotopically in the pancreas. 6694c2 cells have intact TGFβ signaling and can respond to TGFβ ligand and TGFβ blockade (Figure 2A). TGFβ blockade in other cancer types reshapes the tumor microenvironment by reducing cancer associated fibroblasts (CAFs)28. We compared the percentage of myofibroblasts in orthotopic 6694c2 tumors treated with isotype control or αTGFβ by immunofluorescence staining of alpha-smooth muscle actin (αSMA) (Figure 2B–C). Consistent with previous findings, αTGFβ monotherapy reduced αSMA+ cells in the tumor, suggesting successful delivery of the antibody to the tumor microenvironment28. Although full-size antibodies may have difficulty penetrating dense tumors32, we were able to detect the human IgG2c heavy chain of αTGFβ by ELISA in the tumors of treated mice at levels comparable to those achieved by direct mixing of αTGFβ with a tumor cell-matrigel inoculum (Supplemental Figure 3A). The αTGFβ antibody persisted at higher concentrations in tumors compared to isotype control, presumably due to on-target retention. Although orthotopic tumors were more easily reached by the antibody, we also observed high levels of αTGFβ drug in densely fibrotic tumors arising spontaneously in KPC mice (Supplemental Figure 3B–C).
Figure 2: Anti-TGFβ GnP combination treatment is highly effective in poorly immunogenic mouse tumors.
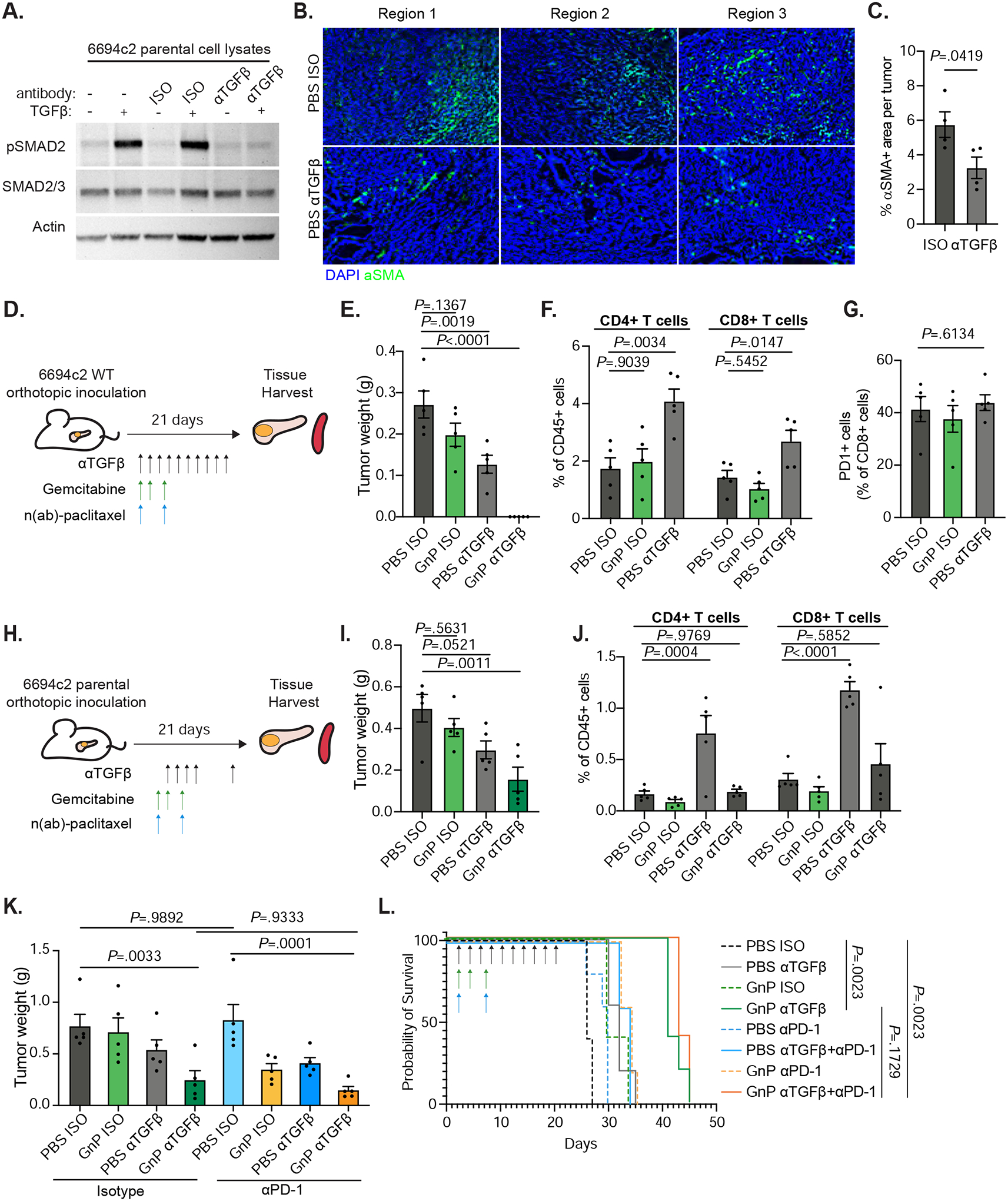
A) 6694c2 parental cells were cultured with 5ng/ml TGFβ, and/or 20ug/ml TGFβ blocking or isotype control antibody for 24h. Protein lysates were collected and analyzed by immunoblot. B) 6694c2 tumors were inoculated into C57BL/6 mice, treated with TGFβ blocking antibody or isotype control and harvested at the midstage of growth on day 15 and frozen in OCT. Representative images from different regions of tumors stained with an antibody against αSMA and DAPI nuclear counterstain are shown. Scale bars = 100μm. C) Quantification of αSMA+ area per tumor. D) Diagram of experimental protocol for orthotopic 6694c2 mouse model for Figure 2E–G. 50,000 6694c2 parental cells were inoculated orthotopically into the pancreas of C57BL/6 mice. E). Tumor weights were measured on day 21. F) Tumor infiltrating CD4 and CD8 T cells were analyzed by flow cytometry of day 21 tumors. G) Percentage of PD1+ tumor infiltrating CD8 T cells. H) Diagram of experimental protocol Figure 2I–J. TGFβ blockade or isotype were dosed i.p. on day 4, day 6, day 8, day 10 and day 20; gemcitabine was given i.p. on day 4, day 7 and day 10; nab-paclitaxel was dosed i.v. on day 4 and day 10. I) Tumor weights were measured on day 21. J) Tumor infiltrating CD4 T cells and CD8 T cells were analyzed by flow cytometry. K-L) 6694c2 tumors were inoculated into mice, treated with TGFβ blockade (every 2 days), PD-1 blockade (twice a week) or corresponding isotype controls until day 20, along with gemcitabine and nab-paclitaxel treatments. K) Tumor weights were measured on day 21; L) Survival of a separate cohort was monitored. N=5 per group. Error bars are SEM throughout.
We treated mice with αTGFβ and/or GnP and showed that, whereas αTGFβ monotherapy induced a slight reduction in tumor weight compared to isotype control, the mice treated with αTGFβ GnP were tumor-free (Figure 2D–E). TGFβ blockade alone induced a significant increase in intratumoral CD4 and CD8 T cells, consistent with reports that TGFβ restricts accumulation of T cells in tumors17, 18 (Figure 2F, Supplemental Figure 4A). Notably, the percentage of PD-1+ CD8 T cells did not change upon TGFβ blockade (Figure 2G). We adjusted the dosing strategy of αTGFβ and GnP to start later at day 4, when tumors are established and approximately 50mm3 in volume (Figure 2H). The αTGFβ GnP combination again reduced tumor weight compared to αTGFβ monotherapy but did not increase intratumoral CD4 and CD8 T cells (Figure 2I–J).
6694c2 pancreatic tumors were previously reported to be T cell low and unresponsive to checkpoint blockade including PD-1 blockade, similar to most patients with PDAC29, 32–34. We confirmed that 6694c2 orthotopic tumors are not responsive to PD-1 blockade (Figure 2K). Addition of PD-1 blockade (αPD-1) to TGFβ blockade, with or without chemotherapy, did not significantly affect tumor weight (Figure 2K). Anti-TGFβ GnP treatment extended median survival of 6694c2 orthotopic tumor-bearing mice from 26 days to 41 days, whereas addition of PD-1 blockade to this regimen provided no significant difference in survival (Figure 2L). Using immunohistochemistry, we found a significant increase in intratumoral CD3+ cells and intratumoral PD1+ cells in αTGFβ and aPD-1 combination treated mice, consistent with previous findings in a mouse mammary carcinoma model17. Interestingly, the addition of aPD-1 to αTGFβ GnP does not reliably increase T cell infiltrates, suggesting that the PD-(L)1 axis may not have a central role in αTGFβ GnP combination therapy treated tumors (Supplemental Figure 4B–C).
Single-cell RNA-seq analysis reveals increased T cell proliferation with combination TGFβ blockade and gemcitabine/nab-paclitaxel
To understand how TGFβ blockade, especially with GnP, affects the tumor microenvironment, we performed single-cell transcriptomic analysis. 6694c2-zsGreen cells were inoculated orthotopically into C57BL/6 mice. Tumors were excised and dissociated at day 14 post-inoculation, allowing us to capture and evaluate changes in the tumor microenvironment at the mid-stage of tumor growth. Single-cell suspensions were stained with antibodies against CD45, followed by fluorescence-activated cell sorting (FACS) with a generous gate allowing all the cells except zsGreen+ tumor cells to be captured (Figure 3A). Droplet-based single-cell transcriptional profiling was performed on 17,463 cells from 4 different treatment groups. The sequenced cells were visualized on a Uniform Manifold Approximation and Projection (UMAP) plot where twodimensional distances approximate global differences in gene expression profile (Figure 3B). Unsupervised clustering identified 20 clusters of tumor-infiltrating cells that were labeled based on their expression of canonical cell marker genes (Figure 3C and Supplemental Figure 5). By analyzing cell representation across different treatment groups, we found that the αTGFβ GnP combination treated group had increased DC-SIGN+ DCs (Cd209a, H2-Ab1) and proliferating T cells (Top2a, Mki67, Cd3e). Combination treated tumors also had decreased frequencies of Lrg1high granulocytes and TNF/IL23a high granulocytes compared to either αTGFβ or GnP monotherapies or untreated groups, suggesting that αTGFβ GnP combination therapy reduces the immunosuppressive tumor microenvironment (Figure 3D–E).
Figure 3. Anti-TGFβ GnP combination treatment reverses the immunosuppressive environment, yet is not dependent on T cells.
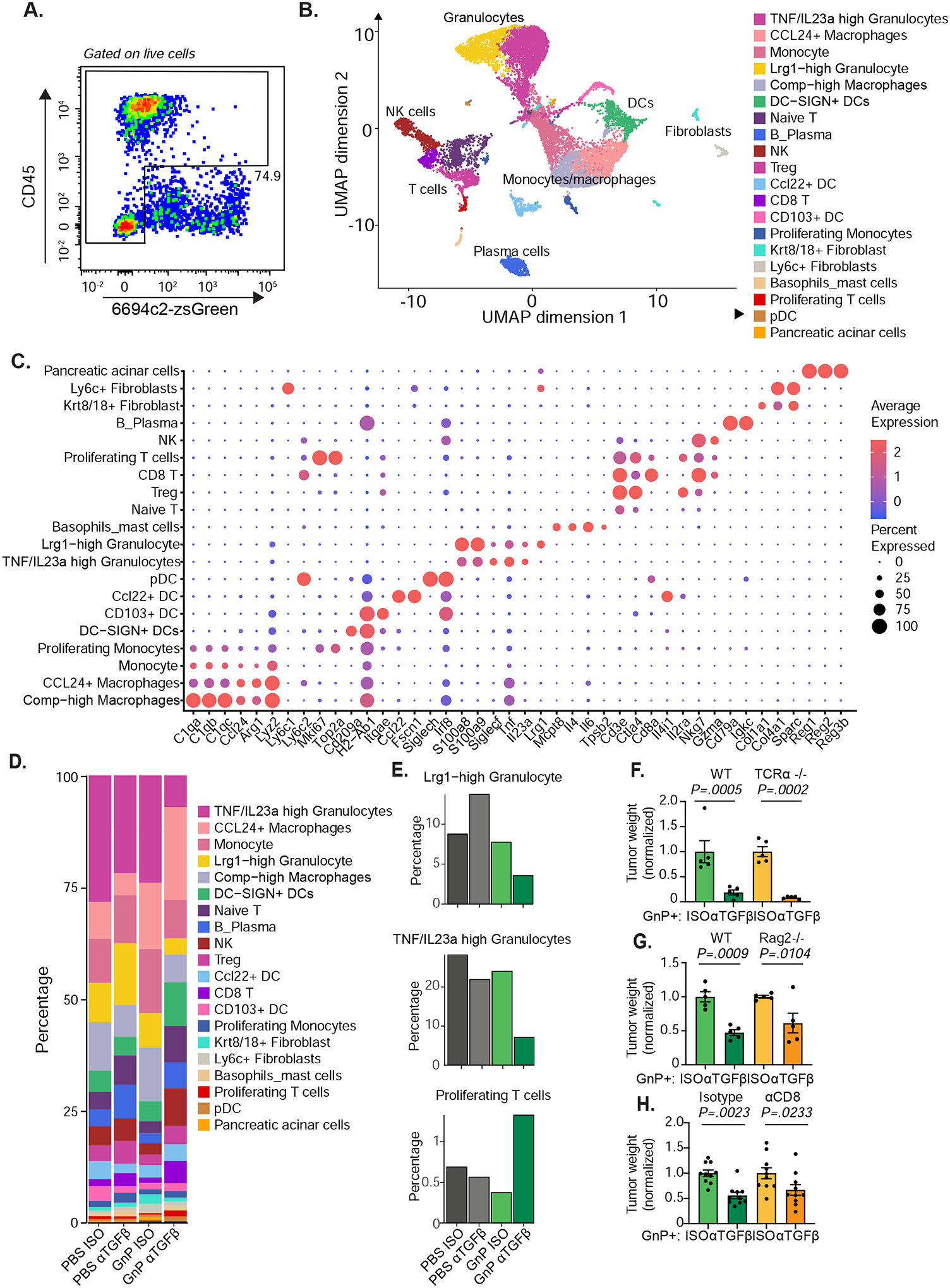
A) 6694c2-zsGreen cells were inoculated orthotopically into C57BL/6 mice, treated with αTGFβ or Isotype +/− GnP as illustrated in Figure 2D. Tumors were harvested on day 15, digested, and live single cells except zsGreen+ tumor cells were sorted by FACS for single-cell transcriptional profiling. n = 5 tumors pooled per group. Representative plot and gating strategy are shown. B) UMAP plot of single-cell RNAseq data. C) Bubble plot outlining the expression of canonical lineage cell markers used for cell cluster annotation. D) Cluster representation per sample. E) Fractional representation of indicated clusters across different treatment groups. F-G) 6694c2 cells were inoculated orthotopically in C57BL/6 or TCRα−/− (F), or RAG2−/− (G) mice. Mice were treated with αTGFβ or Isotype +/− GnP as illustrated in Figure 2D. Tumors were harvested at 21 days after inoculation and weighed. H) 6694c2 cells were inoculated orthotopically in C57BL/6 treated with αTGFβ or Isotype +/− GnP as illustrated in Figure 2D. Mice are also treated with depleting antibodies against to CD4 and CD8 (150μg each antibody per mouse) starting day 0, every 3 days. Control mice received isotype treatment. Tumors were harvested at day 21. Error bars are SEM throughout.
T cells are not required for the efficacy of TGFβ blockade in either poorly immunogenic or highly immunogenic pancreatic cancer models.
To determine if T cells are required for efficacy of αTGFβ GnP combination therapy, 6694c2 parental cells were implanted orthotopically into mice lacking αβ T cells (TCRα−/−), or mice lacking mature T cells or B cells (RAG2−/−). TCRα−/− mice retained the therapeutic effect of combination treatment (Figure 3F). The additional benefit of TGFβ blockade compared to chemotherapy alone was retained in RAG2−/− compared to wildtype mice, indicating that T cells are not required. Similarly, mice treated with a depleting antibody to CD8 T cells were still able to respond to αTGFβ GnP treatment (Figure 3G–H). All together, we demonstrate that TGFβ blockade along with gemcitabine and nab-paclitaxel can reduce the immunosuppressive tumor microenvironment to some extent, but that T cells are not necessary for the efficacy of αTGFβ GnP combination therapy in this poorly immunogenic orthotopic 6694c2 pancreatic cancer model.
One possible explanation for the surprising lack of a requirement for T cells in the response of 6694c2 tumors to TGFβ+GnP therapy could be that these poorly immunogenic tumors lack a sufficient endogenous T cell response33. We therefore expanded our mouse models to include KPC.1, a T cell-high immunogenic mouse model of pancreatic cancer. KPC.1 tumors respond to combination checkpoint blockade and express model neoantigens derived from minor histocompatibility antigens when implanted into recipient C57BL/6 mice35, 36; these tumors are more reflective of the rare high TMB subset of human PDAC. TGFβ blockade in a breast cancer mouse model induces the relocalization of T cells from an excluded phenotype to the center of the tumor17, 28. In control orthotopic KPC.1 tumors treated with GnP alone, T cells clustered predominantly around the edges of the tumor mass, but upon treatment with αTGFβ and GnP, we observed increased T cells in the tumor interior, consistent with previous reports in breast and colorectal cancer models (Figure 4A). Combination therapy significantly reduced primary tumor weight of KPC.1 orthotopic tumors whereas αTGFβ or GnP monotherapies had no effect (Figure 4B). Antibody depletion of CD4 and CD8 T cells caused an increase in the primary tumor weight, confirming that the endogenous T cell response controls growth of this highly immunogenic model (Figure 4C). To our surprise, αTGFβ GnP combination treatment was still effective even in the absence of CD4 and CD8 T cells, which conclusively demonstrates that the efficacy of αTGFβ GnP combination therapy is independent of T cells.
Figure 4. TGFβ blockade significantly increases tumor infiltrating T cells in highly immunogenic pancreatic cancer.
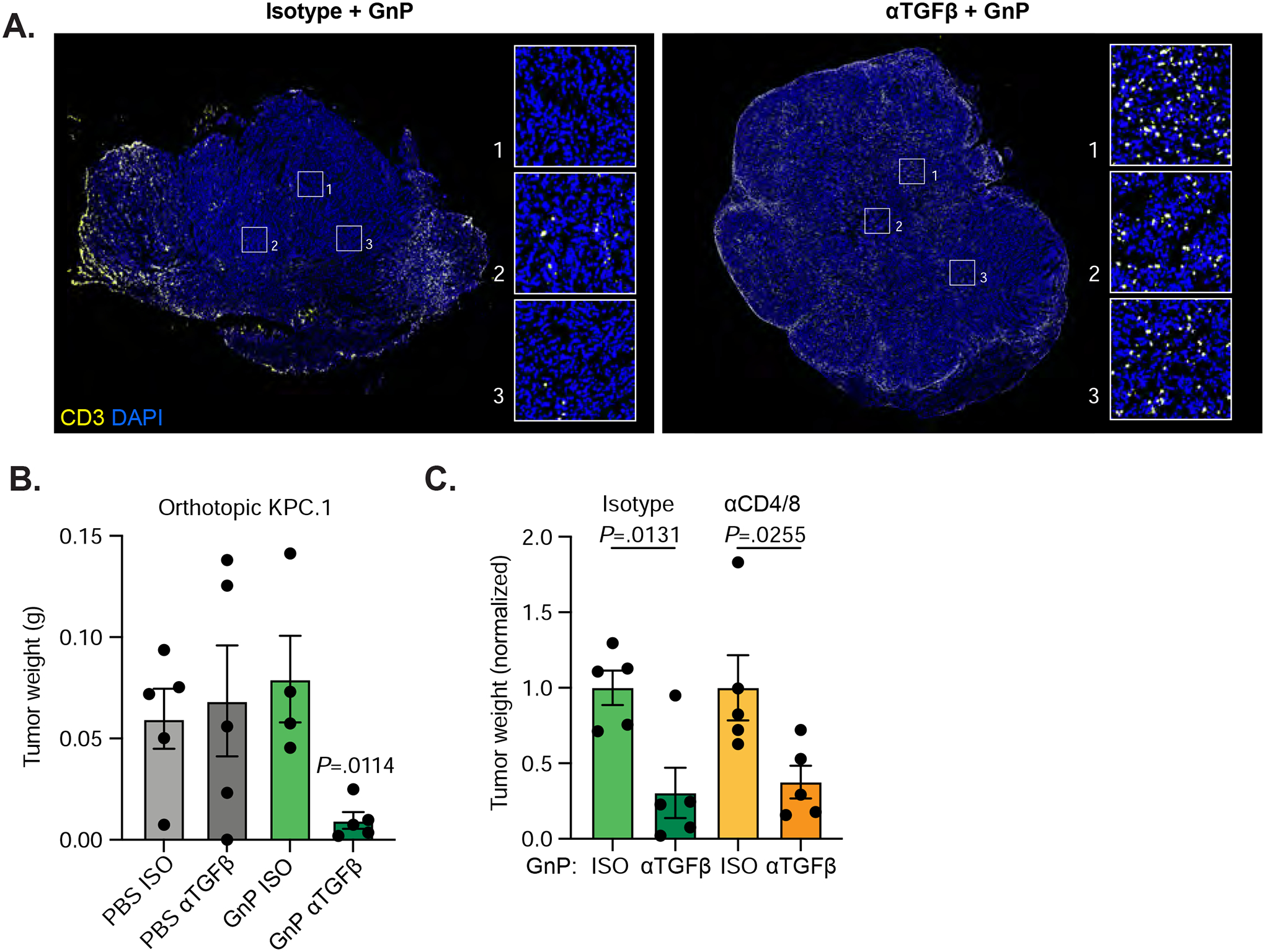
A) KPC.1 cells were inoculated orthotopically into C57BL/6 mice treated with αTGFβ or isotype +/− GnP as in Figure 2D. Tumors were harvested on day 18. Representative images from tumors treated with isotype+GnP or αTGFβ+GnP and stained with anti-CD3. Scale bars = 1000μm. B) Tumors weights at harvest. n = 5 per group. C) C57BL/6 mice inoculated orthotopically with KPC.1 cells were treated with αTGFβ or Isotype +/− GnP as in Figure 2D. Mice were also treated with depleting antibodies against CD4 and CD8 (150μg each antibody per mouse) or isotype control antibodies every 3 days starting at day 0. Tumors were harvested at day 21.
TGFβ blockade is effective in combination with FOLFIRINOX
FOLFIRINOX combination chemotherapy is frequently used as first-line standard of care for pancreatic cancer but is difficult to dose in mice due to sensitivity of mice to 5-fluorouracil37. We first dosed mice with a FOLFIRINOX regimen containing 5mg/kg oxaliplatin, 50mg/kg irinotecan, 75mg/kg leucovorin, and 75mg/kg 5-FU (FFXhigh) which resulted in reduced tumor burden but significant loss in body weight leading to euthanasia (Supplemental Figure 6A–B). We then tested different dosing regimens of FOLFIRINOX to find a dosing strategy that minimized toxicity while retaining efficacy. We found that dosing mice at days 3, 6, and 9 with 5mg/kg oxaliplatin, 50mg/kg irinotecan, 75mg/kg leucovorin, and 15mg/kg 5-FU (FFX) significantly decreased tumor burden without significant weight loss (Figure 5A–C). In patients with pancreatic cancer, FOLFIRINOX treatment induces neutropenia. In mice, we found that both neutrophils and monocytes are significantly depleted in spleens of FFXhigh treated but not GnP treated mice, further supporting that this chemotherapy regimen models the human treatments (Figure 5D). To evaluate whether αTGFβ can further reduce tumor burden when combined with FOLFIRINOX, we inoculated 6694c2 cells orthotopically into C57BL/6 mice and treated the mice with FOLFIRINOX or GnP with or without TGFβ blockade (Figure 5A). For both chemotherapy regimens, addition of TGFβ blockade significantly reduced tumor burden (Figure 5C).
Figure 5. Anti-TGFβ FOLFIRINOX combination treatment effectively reduces tumor burden in poorly immunogenic mouse tumors.
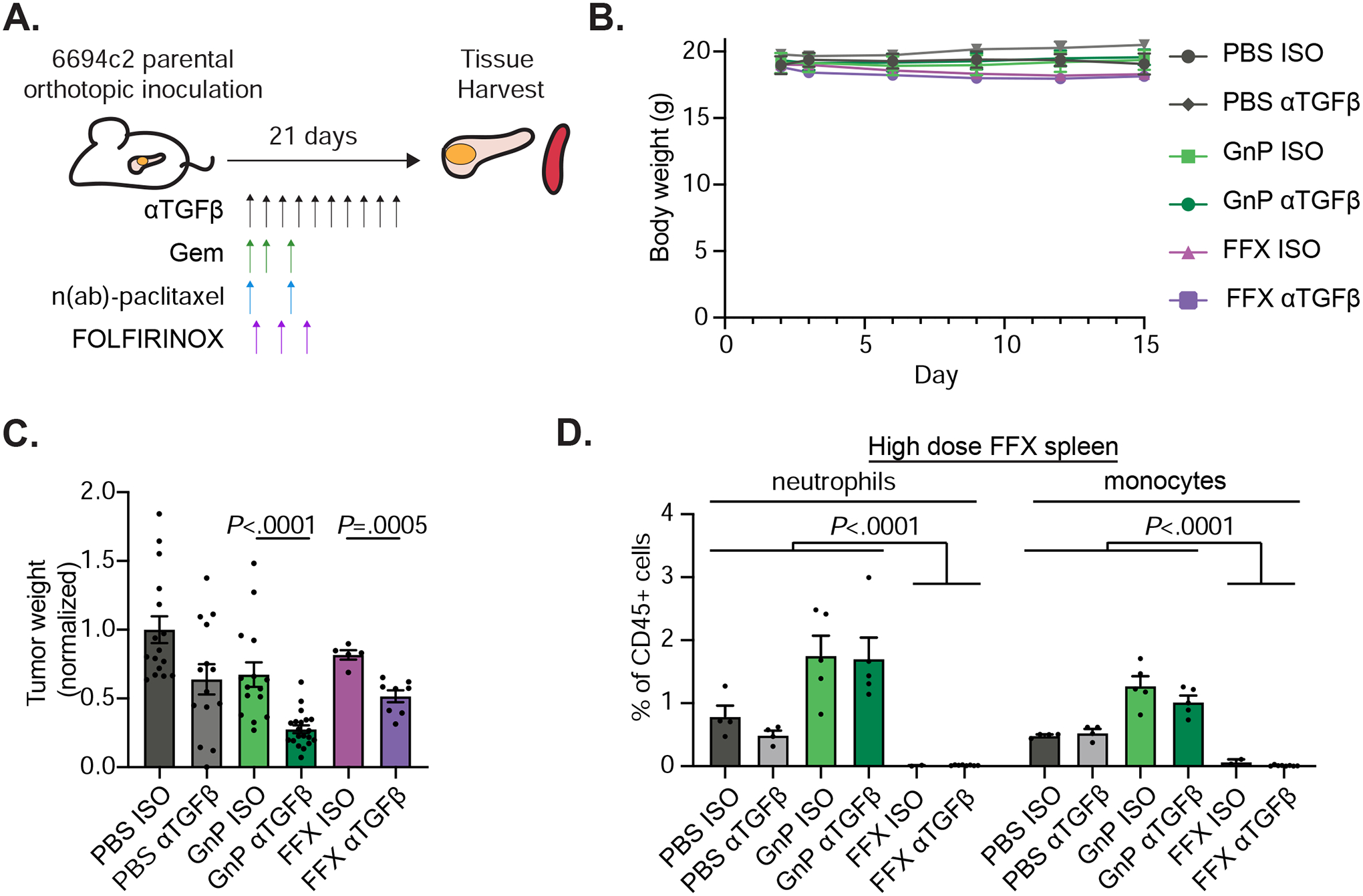
A) Diagram of experimental protocol for orthotopic 6694c2 mouse model with different treatments. TGFβ blocking antibody and GnP were given as in Figure 2D. FOLFIRINOX (5mg/kg oxaliplatin, 75mg/kg leucovorin, 50mg/kg irinotecan, and 15mg/kg 5-FU) were dosed i.v. at days 3, 6, and 9. B) Mouse body weight was measured. C) Tumor weights at day 15. Results are combined from three independent experiments. D) Mice inoculated with 6694c2 cells were treated with TGFβ blocking antibody or isotype control, +/−GnP, or +/− high dose FOLFIRINOX (5mg/kg oxaliplatin, 75mg/kg leucovorin, 50mg/kg irinotecan, and 75mg/kg 5-FU; dosed i.v. at day 3 and day 6). Spleens were harvested 15 days after inoculation. Spleen neutrophils (CD11b+, Gr-1high, SSChigh) and monocytes (CD11b+, Gr-1mid, SSCmid) were analyzed by flow cytometry.
FOLFIRINOX induces a distinct population of cancer associated fibroblasts.
Pancreatic CAFs are heterogeneous with distinct phenotypes and functional traits38. To better understand if fibroblast heterogeneity in the tumor microenvironment is affected by different chemotherapies and TGFβ blockade, we applied single-cell transcriptomic analysis to stromaenriched samples from 6694c2 orthotopic pancreatic cancer. 6694c2 cells were orthotopically implanted into C57BL/6 mice and mid-stage tumors harvested at day 14. For each treatment group, single-cell suspensions were generated from the digested tumors and were enriched for stromal cells using antibodies against CD31 and CD90, followed by positive selection with magnetic beads and single-cell transcriptional analysis. Different components of the tumor microenvironment were clustered based on expression of canonical cell marker genes (Figure 6A and Supplemental Figure 5). Fibroblasts were further sub-clustered to identify heterogenous populations corresponding to myofibroblasts (myCAFs), inflammatory fibroblasts, MHC class II+ fibroblasts, proliferating fibroblasts, and a novel population identified by expression of CXCL5 and other chemokines (Figure 6B). This novel CAF/CXCL5 population appeared predominantly in the αTGFβ and FOLFIRINOX (αTGFβ FFX) combination treated tumors (Figure 6C and Supplemental Figure 5). By performing RNA velocity analysis, which can infer the near-future states of cells from their unspliced mRNA, we found that both the BCAT1-high myCAF and the CAF/CXCL5 clusters are derived from cycling fibroblasts (Supplemental Figure 6B). Due to low cell recovery, we were unable to obtain single-cell transcriptional data from FOLFIRINOX treated tumors. To determine whether the novel CAF/CXCL5 population was induced by FOLFIRNOX versus the combination therapy, we used the orthogonal strategy of immunofluorescence staining for the protein marker PTX3 to identify CAF/CXCL5 cells and PDGFRαβ to identify total fibroblasts when quantifying fibroblast heterogeneity. We found that the CAF/CXCL5 cluster is present in both FFX and αTGFβ FFX treated groups, but not in any other treatment groups, indicating this CAF/CXCL5 cluster is induced by FOLRININOX treatment rather than being unique to αTGFβ FFX combination therapy (Figure 6D). No obvious changes in CAF subset distribution were observed in GnP, αTGFβ, or GnP αTGFβ groups compared to the control group from the single-cell transcriptomic analysis. TGFβ blockade did not consistently affect fibroblast subset distribution, making CAF heterogeneity unlikely to explain why TGFβ blockade effectively reduced tumor burden in combination with chemotherapy.
Figure 6. FOLFIRINOX treated mice have a distinct population of cancer associated fibroblasts.
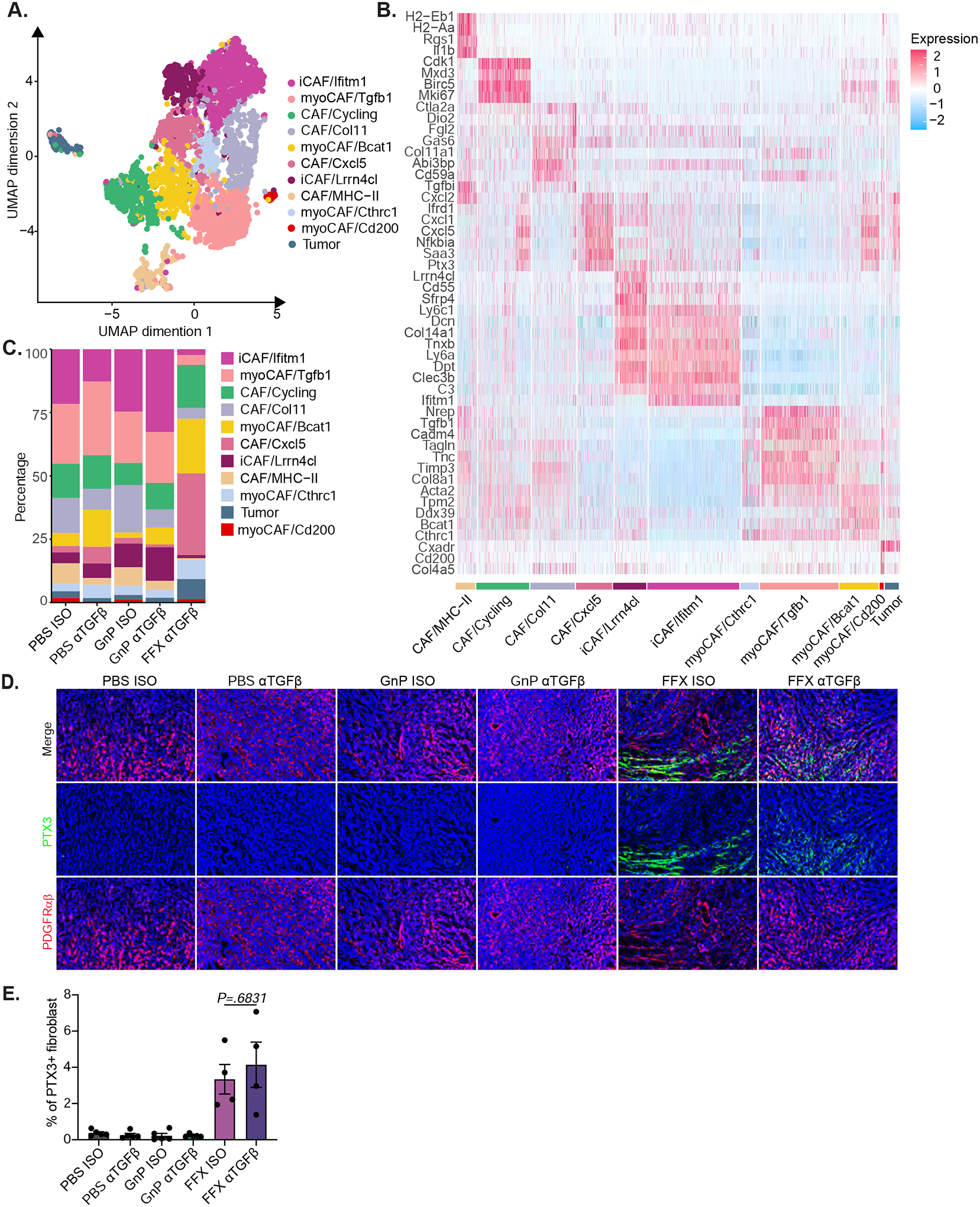
A) 6694c2-zsGreen cells were inoculated orthotopically into C57BL/6 mice treated with αTGFβ or Isotype, +/− GnP as illustrated in Figure 2D, or +/− FOLFIRINOX (5mg/kg oxaliplatin, 75mg/kg leucovorin, 50mg/kg irinotecan, and 75mg/kg 5-FU; dosed by i.v. at day 3 and day 6). Tumors were digested and enriched for fibroblasts using CD31 and CD90 positive selection with magnetic beads followed by single-cell transcriptional analysis. n ≥ 5 tumors pooled per group. Combined UMAP plot of single-cell RNAseq data for fibroblast populations. B) Expression heatmap of representative genes differently expressed. C) Cluster representation per sample. D) C57BL/6 mice inoculated orthotopically with 6694c2 WT cells were treated with αTGFβ or isotype control, +/− GnP as illustrated in Figure 2D, or +/− FOLFIRINOX (5mg/kg oxaliplatin, 75mg/kg leucovorin, 50mg/kg irinotecan, and 15mg/kg 5-FU; dosed i.v. at day 3, day 6 and day 9). Tumors were cryopreserved at day 15, sectioned and stained with DAPI and antibodies against PDGFRαβ and PTX3. Representative images are shown. Scale bars = 100μm. E) Quantification of PTX3+ PDGFRαβ+ area per tumor.
TGFβ blockade enhances tumor cell susceptibility to chemotherapy by promoting a basal-like to classical cell state transition.
Work from our group and others has demonstrated that pancreatic cancer cells can be classified based on transcriptional status into basal-like and classical states, which correspond to differential prognoses and sensitivity to therapies9, 39, 40. TGFβ ligand treated pancreatic cancer organoids exhibited a pronounced shift from classical to basal-like state, which displayed broadly decreased sensitivity to chemotherapies9. Therefore, we investigated whether blockade of TGFβ might have the opposite effect and polarize cells toward a more classical phenotype. 6694c2 tumor cells analyzed by single-cell RNA-seq demonstrated a greater expression of the classical state upon αTGFβ treatment in both PBS and GnP treated groups (Supplemental Figure 7A–C). To further investigate intrinsic changes in tumor cells, we performed bulk transcriptional profiling on FACS sorted tumor cells (CD45−zsGreen+) from mid-stage orthotopic 6694c2-zsGreen tumors (Figure 7A). Principal component analysis (PCA) highlighted the significant degree of divergence between individual samples, showing that replicates within each treatment group are similar, with αTGFβ treatment shifting samples to the right along the axis of greatest variation (PC1) (Figure 7B). Importantly, αTGFβ treatment shifted the tumor cells to a more classical state in all settings, with or without the presence of chemotherapies (Figure 7C). Genes associated with the classical state are upregulated in αTGFβ treated tumor cells alone or in combination with FOLFIRINOX or GnP (Figure 7D). To determine whether this shift to a classical state corresponded to increased sensitivity to chemotherapy in vivo, we used immunofluorescence staining for cleaved caspase 3 as a marker of apoptotic cell death in mid-stage orthotopic 6694c2 tumors from mice that underwent different treatments. The αTGFβ monotherapy, GnP treatment and FFX treatment alone caused minor increases in the percentage of cleaved caspase 3 per tumor area. In contrast, both αTGFβ GnP and αTGFβ FFX combination therapies showed significantly more apoptotic cells in the tumor than the respective chemotherapy-only regimens (Figure 7E–F). Overall, these findings indicate that TGFβ blockade mediated more profound chemotherapy-induced tumor cell death by switching tumor cell transcriptional states to increasingly classical expression.
Figure 7. TGFβ blockade enhances tumor cell susceptibility to chemotherapy by polarizing tumor cells to a classical state.
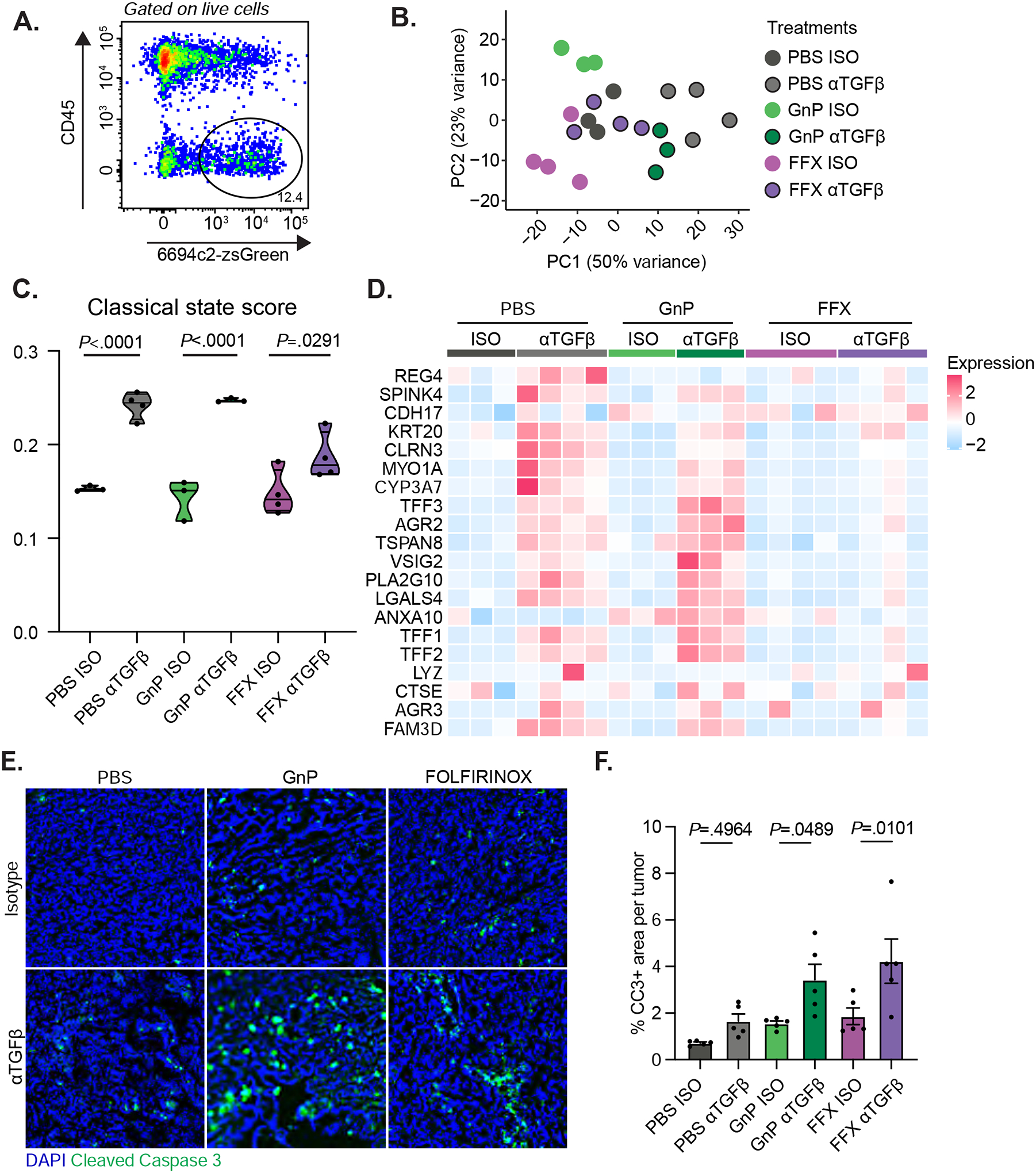
A) 6694c2-zsGreen cells were inoculated orthotopically into C57BL/6 mice, treated with αTGFβ or Isotype, +/− GnP as illustrated in Figure 2D, or +/− FOLFIRINOX (5mg/kg oxaliplatin, 75mg/kg leucovorin, 50mg/kg irinotecan, and 15mg/kg 5-FU; dosed by i.v. at day 3, day 6 and day 9) as in Figure 5A. Tumors were harvested on day 15, digested, and stained with antibody against CD45. Live zsGreen+CD45− cells were sorted by FACS for limited input bulk transcriptional profiling. B) PCA plot with each dot representing an individual sample from different treatment groups. C) Violin plots depicting the classical state score across different treatment groups. D) Expression heatmap of genes associated with the classical state across different treatment groups. E) 6694c2 WT cells were inoculated orthotopically into C57BL/6 mice; treated with αTGFβ or isotype every 2 days starting at day 2 post-inoculation; +/− gemcitabine treatment on days 3, 6, and 9, paclitaxel treatment on days 3 and 9; +/− FOLFIRINOX treatment (5mg/kg oxaliplatin, 75mg/kg leucovorin, 50mg/kg irinotecan, and 15mg/kg 5-Fu) on days 3, 6, and 9. Tumors were harvested at day 12 and stained with DAPI and anti-cleaved caspase 3. Representative images are shown. Scale = 100μm. F) Percentage area of cleaved caspase 3 per tumor. 10 random tumor regions were imaged and quantified per tumor excluding the edge of the tumors or the necrotic centers. Each dot represents the average CC3+ area of the 10 random tumor regions from the same tumor sample. N=5 tumors per group.
Discussion
Many cell types in the tumor microenvironment express receptors for TGFβ, yet we were unable to demonstrate a role for T cells or fibroblasts in the efficacy of TGFβ blockade in murine PDAC models. Instead, a major effect of TGFβ blockade in mouse pancreatic tumors is to polarize malignant cells to a more classical expression state. This mechanism of action supports combination of TGFβ blockade with chemotherapy as a possible therapeutic avenue, as classicalpredominant tumors account for the majority of PDAC. Moreover, classical tumors display increased sensitivity to chemotherapy both in vitro and in clinical trials and confer an overall improved prognosis9, 40, 41. We confirm in mice that blockade of TGFβ increases sensitivity to either gemcitabine/n(ab)paclitaxel or FOLFIRINOX despite highly divergent effects of these two chemotherapy regimens on immune and stromal cells in the microenvironment.
Single-cell studies in PDAC have revealed tremendous cell state heterogeneity within malignant, immune, and fibroblast cell populations within the PDAC tumor microenvironment9, 42–44. Tumor cell plasticity allows for cell state changes, and classical and basal-like transcriptional subtypes exist on a continuum, with most PDAC tumors harboring a mixture of classical and basal-like cell types9, 42–44. The COMPASS trial evaluated patients with metastatic PDAC assigned to either gemcitabine/n(ab)paclitaxel or FOLFIRINOX paired with genetic, histologic and molecular profiling45. In this study, heterogeneity was the main predictor of poor prognosis, although stromalrich tumors were associated with worse response to chemotherapy, and chemotherapy treatment tended to switch tumors from immune-infiltrated to stroma-rich subtypes46. Tumor cell state transitions from classical to basal have also been observed with chemotherapy and have been proposed as a major pathway for acquired resistance47.
Multiple groups have shown that the classical state and decreased heterogeneity in PDAC are associated with both better prognosis and better response to therapy39, 41, 45, 47–50. However, therapeutic manipulation of tumor cell state for clinical benefit has been more difficult to achieve. Emerging evidence suggests that TGFβ superfamily members may regulate tumor cell state. Whereas TGFβ polarizes tumor cells to a basal-like state, the BMP inhibitor GREM1 has the opposite effect and pushes cells toward an epithelial or classical lineage51. The high expression of TGFβ superfamily members in the PDAC tumor microenvironment suggests that a network of paracrine signaling coordinated by TGFβ and BMP ligands maintains tumor cell heterogeneity. Therapeutic manipulation of these soluble ligands and/or their receptors may be a readily achievable means of classical polarization in PDAC.
Multiple distinct subtypes of CAFs have been described in PDAC, including myofibroblasts (myCAFs) inflammatory fibroblasts (iCAFs) and a minor population of MHC II+ fibroblasts38. MyCAFs express αSMA, deposit extracellular matrix, have elevated TGFβ signaling and tend to surround tumor cells. In contrast, iCAFs express Ly6C and IL-6 in response to JAK/STAT signaling and are spatially distanced from tumor cells. TGFβ and IL-1 have been proposed to regulate the ratio of myCAFs to iCAFs in PDAC model systems52; therefore, we hypothesized that blockade of TGFβ would reduce the ratio of myCAFs:iCAFs and potentially generate IFN-licensed CAFs as previously observed in a breast cancer model using the same αTGFβ agent28. However, single-cell analysis and immunofluorescence showed a decrease in total fibroblasts, including αSMA+ CAFs, but no consistent effect on fibroblast subtype composition. We observed a novel population of PTX3+ fibroblasts induced by FOLFIRINOX and marked by high expression of myeloid cell recruiting chemokines. This chemotherapy-induced CAF population could potentially explain the intense recruitment of macrophages observed in resected PDAC tumors from patients who received neoadjuvant FOLFIRINOX and requires further study53.
TGFβ is an immunosuppressive cytokine that can induce Treg differentiation and suppress CD8 T cell effector function and memory formation19. TGFβ produced by macrophages and stromal cells contributes to resolution of inflammation during wound healing and likely plays a similar role in cancer. We were surprised to find that TGFβ blockade was equally effective at controlling orthotopic PDAC in wild type and T cell deficient mouse models. This apparent lack of a role for T cells was observed in both poorly immunogenic 6694c2 tumors and in highly immunogenic KPC.1 tumors which are heavily infiltrated by CD8 T cells and respond to checkpoint blockade immunotherapy. Nevertheless, TGFβ blockade relocalized T cells from the tumor exterior to the tumor interior, which could be a first step toward achieving a T cell-directed response in PDAC. We did not observe an added benefit of TGFβ blockade with PD-1 blockade, although other T cell targeted therapies could be more effective.
Our proposed mechanism of action involves tumor cell intrinsic signaling. One limitation of our current study is that we evaluated effects in mouse models, all of which have intact TGFβ signaling pathways. Given that genetic or functional loss of SMAD4 is common in PDAC54, it will be important to test whether SMAD4 status affects clinical response to TGFβ blockade. NIS793 in combination with gemcitabine/n(ab)paclitaxel is currently being tested in phase II and phase III trials of first line metastatic PDAC (NCT04390763, NCT04935359), and based on the results shown here, trials in combination with FOLFIRINOX have now started in both resectable and first line metastatic PDAC (NCT05546411, NCT05417386). Although tumor cell intrinsic effects were dominant in mouse models, it is possible that TGFβ signaling in fibroblasts, T cells, or macrophages may be more critically important in human PDAC. Even if T cells are not involved in the mechanism of action of combination chemotherapy and TGFβ blockade in humans, T cell directed therapies may be beneficial in future combination trials. T cells in human PDAC express the co-inhibitory receptor TIGIT at higher levels than PD-1, and therapies directed at activation of dendritic cells, such as agonistic anti-CD40 or innate immune adjuvants, may help increase the repertoire of tumor-specific T cells55, 56. Comprehensive profiling of on-treatment samples from these clinical trials and subgroup analysis of SMAD4 mutant tumors will help identify the definitive role of TGFβ in human PDAC.
Supplementary Material
Acknowledgements:
SKD, LQ, MTH, AJA, and SR were funded by the Hale Center for Pancreatic Cancer Research. SKD and AJA were funded by NIH U01 CA224146-01 and 1 U01 CA274276-01. SKD was funded by the Ludwig Center at Harvard, the Dana-Farber Cancer Institute/Novartis Program in Drug Discovery and NIH 1R01AI158488-01. LQ was funded by a SITC-Bristol-Myers Squibb Postdoctoral Cancer Immunotherapy Translational Fellowship. MTH was funded by a Baruj Benacerraf Fellowship. JIC was funded by a Kyoto Prize Scholarship and a Certus Scholarship. PL was funded by NIH T32CA207021. V C-R was funded by a fellowship from the Pancreatic Cancer Action Network and the Francois Wallace Monahan Fund in loving memory of Michael Insel. MD was funded by R01CA177684, National Institutes of Health Mentored Clinical Scientist Development Award 1K08DK114563-01, the Fariborz Maseeh Award for Innovative Medical Education, and The Peter and Ann Lambertus Family Foundation. We thank Jason Pyrdol and the DFCI Center for Cancer Immunology Research for 10x sample processing. We also thank Kun Huang and the DFCI Molecular Imaging Core for assistance with imaging and the Harvard Medical School Rodent Histopathology Core and Brigham and Women’s Hospital Pathology Core for assistance with histology.
Conflict of interest:
SKD received research funding for this project from Novartis Institute for Biomedical Research. VC and MP are employees of Novartis Institute for Biomedical Research. SKD received research funding unrelated to this project from Eli Lilly and Company, Genocea, and Bristol-Myers Squibb and is a founder, science advisory board member (SAB) and equity holder in Kojin. MD has research funding unrelated to this project from Eli Lilly; he has received consulting fees from Genentech, ORIC Pharmaceuticals, Partner Therapeutics, SQZ Biotech, AzurRx, Eli Lilly, Mallinckrodt Pharmaceuticals, Aditum, Foghorn Therapeutics, Palleon, and Moderna; and he is a member of the Scientific Advisory Board for Neoleukin Therapeutics, Veravas, and Cerberus Therapeutics. SR holds equity in Amgen. AJA has consulted for Anji Pharmaceuticals, Arrakis Therapeutics, AstraZeneca, Boehringer Ingelheim, Oncorus, Inc., Merck & Co., Inc., Mirati Therapeutics, Nimbus Therapeutics, Plexium, Revolution Medicines, Reactive Biosciences, Riva Therapeutics, Servier Pharmaceuticals, Syros Pharmaceuticals, Tknife Therapeutics. AJA holds equity in Riva Therapeutics. AJA has research funding from Bristol Myers Squibb, Deerfield, Inc., Eli Lilly, Mirati Therapeutics, Novartis, Novo Ventures, Revolution Medicines, and Syros Pharmaceuticals.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions:
Study concept and design: LQ, MTH, LRA, MP, VC, SKD
Acquisition of data: LQ, MTH, JIC, LK, AT, PL, SJW, EB, V C-R, GAU
Analysis and interpretation of data: LQ, MTH, LRA, JIC, LK, AT, PL, SJW, EB, V C-R, GAU, AY, MD, AJA, SR, MP, VC, SKD
Drafting of the manuscript: LQ and SKD
Critical revision of the manuscript for important intellectual content: all authors
Statistical analysis: LRA
Obtained funding: SKD
Technical, or material support: MP, VC
Study supervision: LQ, MTH, MP, VC, SKD
References
- 1.Rahib L, Smith BD, Aizenberg R, et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014;74:2913–21. [DOI] [PubMed] [Google Scholar]
- 2.Hosein AN, Dougan SK, Aguirre AJ, et al. Translational advances in pancreatic ductal adenocarcinoma therapy. Nat Cancer 2022;3:272–286. [DOI] [PubMed] [Google Scholar]
- 3.Principe DR, DeCant B, Mascarinas E, et al. TGFbeta Signaling in the Pancreatic Tumor Microenvironment Promotes Fibrosis and Immune Evasion to Facilitate Tumorigenesis. Cancer Res 2016;76:2525–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhong Y, Macgregor-Das A, Saunders T, et al. Mutant p53 Together with TGFbeta Signaling Influence Organ-Specific Hematogenous Colonization Patterns of Pancreatic Cancer. Clin Cancer Res 2017;23:1607–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whittle MC, Izeradjene K, Rani PG, et al. RUNX3 Controls a Metastatic Switch in Pancreatic Ductal Adenocarcinoma. Cell 2015;161:1345–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aguirre AJ, Nowak JA, Camarda ND, et al. Real-time Genomic Characterization of Advanced Pancreatic Cancer to Enable Precision Medicine. Cancer Discov 2018;8:1096–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bardeesy N, Cheng KH, Berger JH, et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev 2006;20:3130–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ijichi H, Chytil A, Gorska AE, et al. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev 2006;20:314760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raghavan S, Winter PS, Navia AW, et al. Microenvironment drives cell state, plasticity, and drug response in pancreatic cancer. Cell 2021;184:6119–6137 e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krishnamurty AT, Shyer JA, Thai M, et al. LRRC15(+) myofibroblasts dictate the stromal setpoint to suppress tumour immunity. Nature 2022;611:148–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pang Y, Gara SK, Achyut BR, et al. TGF-beta signaling in myeloid cells is required for tumor metastasis. Cancer Discov 2013;3:936–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Lazarus J, Steele NG, et al. Regulatory T-cell Depletion Alters the Tumor Microenvironment and Accelerates Pancreatic Carcinogenesis. Cancer Discov 2020;10:422–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen W, Jin W, Hardegen N, et al. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med 2003;198:1875–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sugiyarto G, Prossor D, Dadas O, et al. Protective low-avidity anti-tumour CD8+ T cells are selectively attenuated by regulatory T cells. Immunother Adv 2021;1:ltaa001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gunderson AJ, Yamazaki T, McCarty K, et al. TGFbeta suppresses CD8(+) T cell expression of CXCR3 and tumor trafficking. Nat Commun 2020;11:1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang L, Yu Z, Muranski P, et al. Inhibition of TGF-beta signaling in genetically engineered tumor antigen-reactive T cells significantly enhances tumor treatment efficacy. Gene Ther 2013;20:575–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mariathasan S, Turley SJ, Nickles D, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018;554:544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tauriello DVF, Palomo-Ponce S, Stork D, et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018;554:538–543. [DOI] [PubMed] [Google Scholar]
- 19.Batlle E, Massague J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019;50:924–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li D, Schaub N, Guerin TM, et al. T Cell-Mediated Antitumor Immunity Cooperatively Induced By TGFbetaR1 Antagonism and Gemcitabine Counteracts Reformation of the Stromal Barrier in Pancreatic Cancer. Mol Cancer Ther 2021;20:1926–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang H, Zhang Y, Gallegos V, et al. Targeting TGFbetaR2-mutant tumors exposes vulnerabilities to stromal TGFbeta blockade in pancreatic cancer. EMBO Mol Med 2019;11:e10515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pujala B, Ramachandran SA, Sonawane M, et al. Discovery of MDV6058 (PF06952229), a selective and potent TGFbetaR1 inhibitor: Design, synthesis and optimization. Bioorg Med Chem Lett 2022;75:128979. [DOI] [PubMed] [Google Scholar]
- 23.Schlingensiepen KH, Jaschinski F, Lang SA, et al. Transforming growth factorbeta 2 gene silencing with trabedersen (AP 12009) in pancreatic cancer. Cancer Sci 2011;102:1193–200. [DOI] [PubMed] [Google Scholar]
- 24.Liu B, Zhu X, Kong L, et al. Bifunctional TGF-beta trap/IL-15 protein complex elicits potent NK cell and CD8(+) T cell immunity against solid tumors. Mol Ther 2021;29:2949–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lan Y, Zhang D, Xu C, et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-beta. Sci Transl Med 2018;10. [DOI] [PubMed] [Google Scholar]
- 26.Melisi D, Garcia-Carbonero R, Macarulla T, et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br J Cancer 2018;119:1208–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Melisi D, Oh DY, Hollebecque A, et al. Safety and activity of the TGFbeta receptor I kinase inhibitor galunisertib plus the anti-PD-L1 antibody durvalumab in metastatic pancreatic cancer. J Immunother Cancer 2021;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grauel AL, Nguyen B, Ruddy D, et al. TGFbeta-blockade uncovers stromal plasticity in tumors by revealing the existence of a subset of interferon-licensed fibroblasts. Nat Commun 2020;11:6315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li J, Byrne KT, Yan F, et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 2018;49:178–193 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chuvin N, Vincent DF, Pommier RM, et al. Acinar-to-Ductal Metaplasia Induced by Transforming Growth Factor Beta Facilitates KRAS(G12D)-driven Pancreatic Tumorigenesis. Cell Mol Gastroenterol Hepatol 2017;4:263–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grabliauskaite K, Saponara E, Reding T, et al. Inactivation of TGFbeta receptor II signalling in pancreatic epithelial cells promotes acinar cell proliferation, acinarto-ductal metaplasia and fibrosis during pancreatitis. J Pathol 2016;238:434–45. [DOI] [PubMed] [Google Scholar]
- 32.Dougan M, Ingram JR, Jeong HJ, et al. Targeting Cytokine Therapy to the Pancreatic Tumor Microenvironment Using PD-L1-Specific VHHs. Cancer Immunol Res 2018;6:389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roehle K, Qiang L, Ventre KS, et al. cIAP1/2 antagonism eliminates MHC class I-negative tumors through T cell-dependent reprogramming of mononuclear phagocytes. Sci Transl Med 2021;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stump CT, Roehle K, Manjarrez Orduno N, et al. Radiation combines with immune checkpoint blockade to enhance T cell priming in a murine model of poorly immunogenic pancreatic cancer. Open Biol 2021;11:210245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crowley SJ, Bruck PT, Bhuiyan MA, et al. Neoleukin-2 enhances anti-tumour immunity downstream of peptide vaccination targeted by an anti-MHC class II VHH. Open Biol 2020;10:190235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lenehan PJ, Cirella A, Uchida AM, et al. Type 2 immunity is maintained during cancer-associated adipose tissue wasting. Immunother Adv 2021;1:ltab011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Delahoussaye AM, Abi Jaoude J, Green M, et al. Feasibility of administering human pancreatic cancer chemotherapy in a spontaneous pancreatic cancer mouse model. BMC Cancer 2022;22:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elyada E, Bolisetty M, Laise P, et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting CancerAssociated Fibroblasts. Cancer Discov 2019;9:1102–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moffitt RA, Marayati R, Flate EL, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet 2015;47:1168–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Kane GM, Grunwald BT, Jang GH, et al. GATA6 Expression Distinguishes Classical and Basal-like Subtypes in Advanced Pancreatic Cancer. Clin Cancer Res 2020;26:4901–4910. [DOI] [PubMed] [Google Scholar]
- 41.Collisson EA, Sadanandam A, Olson P, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med 2011;17:500–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bernard V, Semaan A, Huang J, et al. Single-Cell Transcriptomics of Pancreatic Cancer Precursors Demonstrates Epithelial and Microenvironmental Heterogeneity as an Early Event in Neoplastic Progression. Clin Cancer Res 2019;25:2194–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chan-Seng-Yue M, Kim JC, Wilson GW, et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat Genet 2020;52:231–240. [DOI] [PubMed] [Google Scholar]
- 44.Hwang WL, Jagadeesh KA, Guo JA, et al. Single-nucleus and spatial transcriptome profiling of pancreatic cancer identifies multicellular dynamics associated with neoadjuvant treatment. Nat Genet 2022;54:1178–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aung KL, Fischer SE, Denroche RE, et al. Genomics-Driven Precision Medicine for Advanced Pancreatic Cancer: Early Results from the COMPASS Trial. Clin Cancer Res 2018;24:1344–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grunwald BT, Devisme A, Andrieux G, et al. Spatially confined sub-tumor microenvironments in pancreatic cancer. Cell 2021;184:5577–5592 e18. [DOI] [PubMed] [Google Scholar]
- 47.Porter RL, Magnus NKC, Thapar V, et al. Epithelial to mesenchymal plasticity and differential response to therapies in pancreatic ductal adenocarcinoma. Proc Natl Acad Sci U S A 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Andres MP, Jackson RJ, Felipe I, et al. GATA4 and GATA6 loss-ofexpression is associated with extinction of the classical programme and poor outcome in pancreatic ductal adenocarcinoma. Gut 2022. [DOI] [PubMed] [Google Scholar]
- 49.Connor AA, Denroche RE, Jang GH, et al. Integration of Genomic and Transcriptional Features in Pancreatic Cancer Reveals Increased Cell Cycle Progression in Metastases. Cancer Cell 2019;35:267–282 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ligorio M, Sil S, Malagon-Lopez J, et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 2019;178:160–175 e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lan L, Evan T, Li H, et al. GREM1 is required to maintain cellular heterogeneity in pancreatic cancer. Nature 2022;607:163–168. [DOI] [PubMed] [Google Scholar]
- 52.Biffi G, Oni TE, Spielman B, et al. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov 2019;9:282–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dias Costa A, Vayrynen SA, Chawla A, et al. Neoadjuvant chemotherapy is associated with altered immune cell infiltration and an anti-tumorigenic microenvironment in resected pancreatic cancer. Clin Cancer Res 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang YH, Hu J, Chen F, et al. ID1 Mediates Escape from TGFbeta Tumor Suppression in Pancreatic Cancer. Cancer Discov 2020;10:142–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Freed-Pastor WA, Lambert LJ, Ely ZA, et al. The CD155/TIGIT axis promotes and maintains immune evasion in neoantigen-expressing pancreatic cancer. Cancer Cell 2021;39:1342–1360 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Steele NG, Carpenter ES, Kemp SB, et al. Multimodal Mapping of the Tumor and Peripheral Blood Immune Landscape in Human Pancreatic Cancer. Nat Cancer 2020;1:1097–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.