

Abstract
Renal cell carcinoma (RCC) of variant histology comprises approximately 20% of kidney cancer diagnoses, yet the optimal therapy for these patients and the factors that impact immunotherapy response remain largely unknown. To better understand the determinants of immunotherapy response in this population, we characterized blood- and tissue-based immune markers for patients with variant histology RCC, or any RCC histology with sarcomatoid differentiation, enrolled in a phase II clinical trial of atezolizumab and bevacizumab. Baseline circulating (plasma) inflammatory cytokines were highly correlated with one another, forming an “inflammatory module” that was increased in International Metastatic RCC Database Consortium (IMDC)-poor risk patients and was associated with worse progression-free survival (PFS) (p=0.028). At baseline, an elevated circulating VEGF-A level was associated with a lack of response (p=0.03) and worse PFS (p=0.021). However, a larger increase in on-treatment levels of circulating VEGF-A was associated with clinical benefit (p=0.01) and improved overall survival (p=0.0058). Among peripheral immune cell populations, an on-treatment decrease in circulating PD-L1+ T cells was associated with improved outcomes, with a reduction in CD4+PD-L1+ (HR:0.62[0.49–0.91], p=0.016) and CD8+PD-L1+ T cells (HR:0.59[0.39–0.87], p=0.009) correlated with improved PFS. Within the tumor itself, a higher percentage of terminally exhausted (PD-1+ and either TIM-3+ or LAG-3+) CD8+ T cells was associated with worse PFS (p=0.028). Overall, these findings support the value of tumor and blood-based immune assessments in determining therapeutic benefit for patients with RCC receiving atezolizumab plus bevacizumab and provide a foundation for future biomarker studies for patients with variant histology RCC receiving immunotherapy-based combinations.
Keywords: metastatic renal cell carcinoma, non-clear cell RCC, sarcomatoid, immune checkpoint inhibitors, vascular endothelial growth factor inhibitor
Introduction
The most common form of renal cell carcinoma (RCC) is clear cell RCC (ccRCC), which accounts for 75–80% of the approximately 75,000 cases diagnosed each year in the United States [1]. Variant histology RCC (commonly referred to as non–clear cell), which includes subtypes with distinct biological immunopathologic features (e.g., chromophobe, papillary, translocation) and is associated with adverse outcomes, represents 20–25% of RCC [2]. Additionally, any RCC histology may be associated with sarcomatoid differentiation, which typically confers a more aggressive clinical behavior and worse outcomes [3]. Although immune checkpoint inhibition (ICI) has shifted the treatment paradigm of RCC, most large clinical trials have included only patients with ccRCC [4]–[6]. Thus, there is an unmet need to understand the clinical and biological features that determine the therapeutic response to ICIs in patients with variant histology RCC or RCC with sarcomatoid features [7]–[11].
The combination of atezolizumab, a monoclonal antibody specific for programmed death-ligand 1 (PD-L1), and bevacizumab, a monoclonal antibody specific for vascular endothelial growth factor A (VEGF-A), has clinical activity in patients with ccRCC, particularly in specific molecular subtypes [12], [13]. In addition, among patients with variant histology RCC or RCC with sarcomatoid differentiation, atezolizumab plus bevacizumab had an acceptable safety profile and clear clinical activity, particularly in tumors positive for PD-L1 expression or sarcomatoid differentiation [14]. Moving forward, it will be critical to determine the biological factors that determine therapeutic response, with the goal of informing treatment selection and sequencing.
Although the International metastatic RCC Database Consortium (IMDC) prognostic model continues to perform well in variant histology RCC, the circulating and intratumoral factors that determine response to ICI in these tumor types remain largely unknown [15]. Herein, we performed a blood- and tissue-based biomarker analysis of a multicenter phase II clinical trial of atezolizumab and bevacizumab for patients with metastatic RCC (mRCC) of variant histology or any histology with sarcomatoid features [14]. In this phase II clinical trial of atezolizumab and bevacizumab that included 60 patients with variant histology mRCC or any mRCC histology with ≥20% sarcomatoid differentiation, the objective response rate was 33% in the overall study population, 50% in patients with ccRCC with sarcomatoid differentiation and 26% in patients with variant histology RCC [14].In this study, we defined baseline circulating inflammatory (IL-1, IL-6, IL-13, MCP-1, MIP-1β) and angiogenic soluble (VEGF-A) factors associated with clinical risk and therapeutic outcome. We also sought to assess the association between the presence of intratumoral terminally exhausted CD8+ T cells and clinical outcomes. This study provides insights into the biological factors that impact response to ICI and anti-angiogenic agents in variant histology RCC and supports future validation studies of circulating and intratumoral biomarkers in these diseases.
Methods
Study design and clinical endpoints
This study utilized patient data and biospecimens (blood [N=60] and tumor [N=38]) from a prospective phase-II clinical trial of the anti–PD-L1 atezolizumab and the anti–VEGF-A bevacizumab in patients with mRCC with variant histology and/or sarcomatoid differentiation (NCT02724878) [14]. This study was conducted in accordance with the principles of the Declaration of Helsinki. Written informed consent was obtained from all participants prior to their participation in the study. Blood samples were collected at baseline, prior to therapy (C1D1: cycle one day one), and on-treatment, following two cycles of therapy (C3D1: cycle three day one). Plasma was assayed for the levels of soluble factors (e.g., cytokines and growth factors), and peripheral blood mononuclear cells (PBMCs) were immunophenotyped using flow cytometry. For Dana-Farber/Harvard Cancer Center (DF/HCC) participating sites, 30 mL of blood were collected as per the Laboratory Manual and couriered within 6 hours of collection to The Center of Immunooncology Laboratory. For non-DF/HCC participating sites, 30 mL of blood were collected and stored as per the Laboratory Manual and batch shipped at the end of study to The Center of Immunooncology Laboratory. In addition, formalin-fixed, paraffin-embedded (FFPE) archival or pre-treatment study tumor biopsies were analyzed by immunohistochemistry and immunofluorescence for enrolled subjects with available biospecimen. Archived tumor tissue was collected pre-study as per the Laboratory Manual and stored at ambient temperature until the end of study at which time the specimens were shipped to the Signoretti Laboratory. Fresh tumor biopsy was conducted pre-study as per the Laboratory Manual. Frozen specimens were stored at −80°C and specimens fixed in formalin were stored at 4°C until shipping. At the end of study, the tumor specimens were shipped to the Signoretti Laboratory.
Baseline demographics, clinical characteristics, and treatment outcomes, including progression-free survival (PFS), overall survival (OS), and clinical benefit (CB) [16], were collected and analyzed from the trial database. PFS was defined as the time from treatment initiation to radiographic progression or death from any cause or censored at the date of last disease assessment. OS was calculated from treatment initiation to the date of death from any cause or censored at the date of last follow-up. Objective response was assessed by the clinical investigator using RECIST version 1.1 every six weeks during the first 24 weeks and then every 12 weeks after treatment initiation until radiographic progression occurred, intolerable toxicity, or withdrawal of consent. CB was defined [16] by the sum of objective responses (complete or partial) and stable disease (SD) where patients had tumor shrinkage and PFS ≥6 months. Patients with progressive disease (PD) and PFS <3 months were classified as having no clinical benefit (NCB). All other patients were classified as having intermediate clinical benefit (ICB) [17]. This study was approved by each participating institution’s institutional review board.
Cytokines
The Luminex fluorescent bead array platform (Bio-Plex MAGPIX) was used to quantify multiple soluble biomarkers simultaneously in human plasma at baseline and on-treatment (Bio-Rad, Inc.). The samples were analyzed in duplicates, and the fluorescent values were extrapolated to a standard curve for quantification. The following markers were analyzed: EGF, IFNg, IFNa2, TNFa, TNFb, IL-1b, IL-1a, IL-1RA, IL-2, sIL-2R, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12, IL-13, IL-15, IL-17, MIP-1a, MIP-1β, MCP-1, MCP-3, CXCL6, CXCL5, IP-10, GROb, VEGF-A, FGF2, Ang-1, Ang-2, Eotaxin, TGFa, G-CSF, Flt-3L, Fractalkine, and sCD40L and sPDL-1 (R&D systems).
Flow cytometry
For flow cytometry analysis, PBMCs were thawed in a 37°C water bath and transferred to a 15 mL conical tube containing warm DMEM (GIBCO, 11965–092). Cells were pelleted at 1,800 RPM at 4°C (Sorvall Legend XTR Centrifuge, Thermofisher) for 5 minutes and washed with Phosphate Buffered Saline (PBS, Gibco). To discriminate between living and dead cells, PBMCs were incubated with Near Infrared Fixable Viability Dye (NIR, Biolegend; Invitrogen™ LIVE/DEAD™ Fixable Near-IR Dead Cell Stain Kit, for 633 or 635 nm excitation; L10119) in PBS for 15 minutes on ice and then washed once with 1 ml PBS per manufacturer protocol. PBMCs were next incubated on ice with Fc block (Human TruStain FcX Biolegend, 422302) in FACS buffer (PBS/2.5% FBS) for 20 minutes, washed once, and divided into single tubes (approximately 500,000 cells per tube). Next, cells were incubated on ice with the antibodies listed in Supplementary Table S1 in PBS/2.5% FBS. Cells were washed twice with FACS buffer and kept in the dark on ice until flow cytometry analysis (no more than 30 minutes after the last wash). Data was acquired using a Fortessa analyzer (BD Biosciences), and FCS files were analyzed using FlowJo software 10.7.1 (Treestar,Inc.). The gating strategy that was adopted is presented in Supplementary Figs.S1–S3.
Immunohistochemistry (IHC) and multiplex immunofluorescence (MIF) on tumor tissue
Tumor tissue samples (N=38) were collected before the initiation of therapy and analyzed using IHC and MIF.
MIF
The assay was performed on a Bond RX Autostainer (Leica Biosystems) using the Opal multiplex IHC system (PerkinElmer/Akoya Biosciences Cat# NEL871001KT) as described in detail previously [18]. A previously optimized panel of antibodies specific for CD8 (Agilent, M710301–2, RRID:AB_2075537), PD-1 (clone EH33, a gift from Dr. Gordan Freeman, Dana-Farber Cancer Institute), TIM3 (R&D Systems, AF2365, RRID:AB_355235) and LAG3 (LifeSpan Biosciences, LS‑B2237–50, RRID:AB_1650048) was used (Supplementary Table S2). In brief, whole slide multispectral images were acquired at 10x magnification using Vectra 3.0 (PerkinElmer/Akoya Biosciences). Next, they were visually inspected using Phenochart 1.0.9 (PerkinElmer/Akoya Biosciences, RRID:SCR_019160) to select at least five intra-tumoral CD8+ T cell–enriched regions of interest (ROIs) per case where each ROI had an area=669 μm x 500 μm. The number of hotspot ROIs was determined so that a minimum of 100 CD8+ T cells would be analyzed. ROIs were scanned at 20x magnification, imported into InForm v2.2.0 (PerkinElmer, RRID:SCR_019161) and deconvoluted using a multispectral library built with single stain slides. Image analysis was performed using HALO v2.1.1637.18 (Indica Labs, RRID:SCR_018350) and the Indica Labs High-Plex FL v2.0 module. A unique algorithm was created for each case, and its accuracy was validated through visual inspection by two pathologists (T. Denize and M. Ficial) with extensive experience in image analysis.
IHC
Tumor sections had been previously stained with a validated antibody against PD-L1 (05.9A11 mouse monoclonal antibody, 1:125, 13 μg/mL [a gift from Dr. Gordon Freeman, and commercially available through Cell Signaling Technology, Danvers, MA]) with BOND RX autostainer (Leica Biosystems, Buffalo Grove, IL) using the BOND Polymer Refine Detection Kit (DS9800; Leica Biosystems) and BOND Polymer Refine Red Detection Kit (DS9390; Leica Biosystems), as previously described [14]. The PD-L1 score was calculated by dividing the number of PD-L1+ tumor cells by the total number of tumor cells. A cutoff of 1% was used to determine PD-L1 positivity.
Statistical analysis
To characterize the immune landscape of patients with mRCC with variant histology and/or sarcomatoid differentiation, the relationships between biomarker variables were analyzed using Pearson correlations and hierarchical clustering using Euclidean distance. All biomarker variables were Z-score normalized. We calculated an inflammatory module, the systemic inflammation score (SIS), which corresponds to the average score of the five highly correlated inflammatory cytokines: MIP-1β, IL-1, MCP-1, IL-6, and IL-13. To evaluate the association of SIS with IMDC risk groups and CB groups, pairwise Wilcoxon signed-rank tests were applied. The relationships of SIS with PFS and OS were assessed by log-rank tests and univariate Cox regression models. PFS and OS were estimated using Kaplan–Meier methodology. Biomarkers were dichotomized based on the median for all measures and the 25th percentile for SIS. Values were consistent across duplicates. To account for the different proportions of cells obtained across gating strategies, we calculated an average score for each cell type. All tests were two-sided with a significance threshold of 0.05 and R (version 4.1.1) was used to carry out the above analyses. We conducted all our analyses in the entire cohort and in different subsets according to sarcomatoid status and histology.
Data availability
The data generated in this study are available in the in this article and its Supplementary Data files or upon request from the corresponding author.
Results
We determined circulating plasma-derived cytokine signatures and peripheral immune-cell profiles of all 60 patients at baseline and after two cycles of therapy to explore determinants of response to treatment and survival outcomes. Additionally, we utilized IHC and MIF on baseline FFPE tumor biospecimens of 38 patients to identify intratumoral T-cell phenotypes associated with response to therapy (Fig.1; Supplementary Table S3).
Figure 1. Study design of the clinical trial and correlative analysis evaluating the combination of atezolizumab and bevacizumab in variant histology RCC and any RCC with sarcomatoid features.

Abbreviations: BP: Blood pressure, C1D1: Day 1 of Cycle 1 of treatment, C3D1: Day 1 of Cycle 3 of treatment, ccRCC: clear-cell renal cell carcinoma, ECOG-PS: Eastern Cooperative Oncology Group Performance Status, FFPE: Formalin-Fixed Paraffin-Embedded, ORR: overall response rate, RCC: renal cell carcinoma, RECIST: Response Evaluation Criteria in Solid Tumors.
Circulating inflammatory module is associated with therapeutic resistance and worse clinical outcomes.
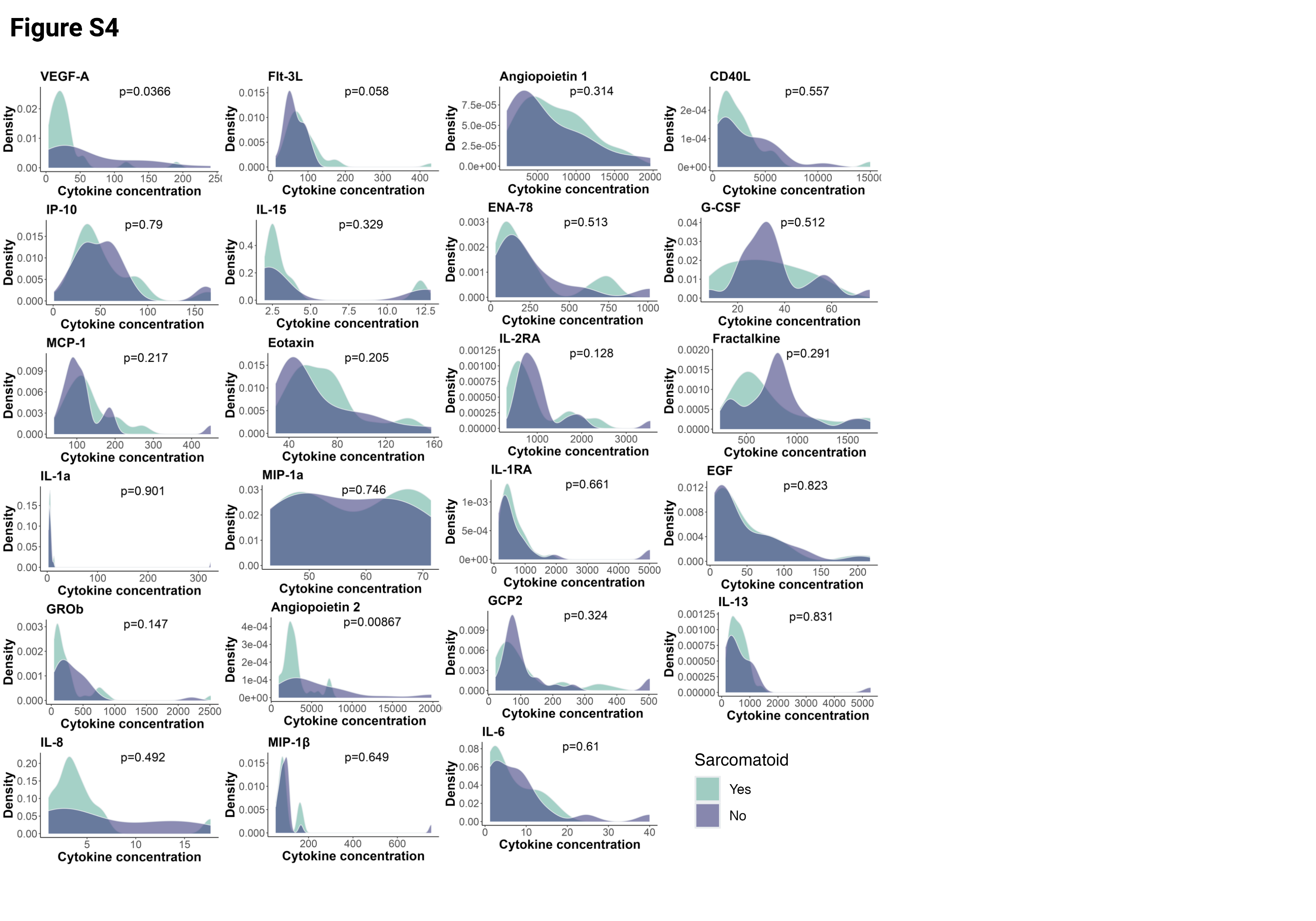
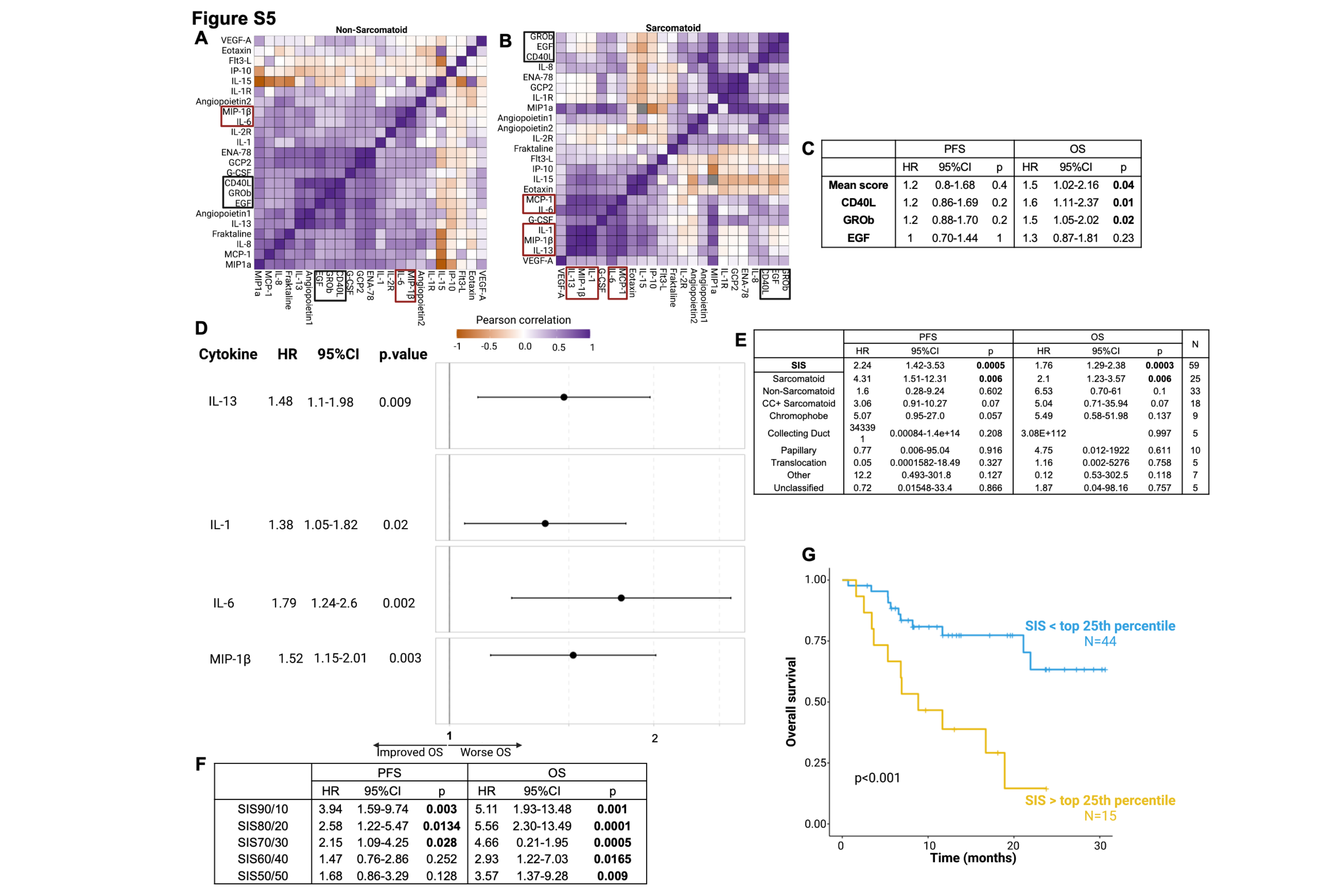
To identify plasma cytokine and soluble factor signatures associated with response to therapy, we investigated the distribution of each cytokine in the whole cohort and in sarcomatoid status–defined subgroups (Supplementary Fig.S4). Apart from two angiogenic cytokines (VEGF-A and Angiopoietin 2), which were more abundant in patients with non-sarcomatoid tumors, circulating cytokines had similar distributions between sarcomatoid and non-sarcomatoid subgroups. We performed pairwise correlations between all quantified plasma proteins at baseline. Hierarchical clustering of this correlation matrix revealed an “inflammatory module” consisting of highly correlated levels of five critical inflammatory cytokines (IL-1, IL-6, IL-13, MIP-1β, and MCP-1; Fig.2A). Inflammatory proteins also strongly correlated in the sarcomatoid (IL-1, IL-6, MCP-1, IL-13, MIP-1β; Supplementary Fig.S5A) and non-sarcomatoid (MIP-1β and IL-6; Supplementary Fig.S5B) subgroups. CD40L, GROb, and EGF also appeared to be highly correlated in the overall cohort (Fig.2A), and in the sarcomatoid (Supplementary Fig.S5A) and non-sarcomatoid (Supplementary Fig.S5B) subgroups. However, we observed no clear association between CD40L, GROb, and EGF (individually or as a module) with PFS (Supplementary Fig.S5C). By contrast, when we individually examined the five inflammatory cytokines (IL-1, IL-6, IL-13, MIP-1β, and MCP-1), elevated levels of four of these five cytokines corresponded with lower PFS (Fig.2B). Cox regression analyses uncovered negative associations between PFS and plasma levels of IL-1 (HR=1.71,p=0.004), IL-6 (HR=1.70,p= 0.012), IL-13 (HR=1.64,p=0.014), and MIP-1β (HR=2.05,p=0.007). Additionally, higher expression levels of IL-1 (HR=1.38,p=0.02), MIP-1β (HR=1.52,p=0.003), IL-6 (HR=1.79,p=0.002), and IL-13 (HR=1.48,p=0.009) were individually associated with worse OS (Supplementary Fig.S5D). We subsequently calculated a systemic inflammation score (SIS) based on the mean level of these five correlated inflammatory cytokines. Elevated SIS was associated with worse PFS (Cox regression: HR=2.24,p<0.001; Fig.2C) and OS (HR=1.76,p=0.003 Fig.2C). The same trend appeared in the sarcomatoid (HR=4.31,p=0.006) and non-sarcomatoid (HR=1.6,p=0.602) subgroups as well as in CC+sarcomatoid (HR=3.06,p=0.07), chromophobe (HR=5.07,p=0.057), collecting duct (HR=343391,p=0.208), and other (HR=12.2,p=0.127) histologies (Supplementary Fig.S5E). Similar results were found for OS in these subgroups as well as in translocation and unclassified histologies (Supplementary Fig.S5E). In the overall population, the SIS (continuous variable) was associated with worse PFS and OS. When dichotomized into “high” and “low” groups, higher SIS was associated with worse survival at numerous thresholds (Fig.2C; Supplementary Fig.S5F). Patients with higher SIS had improved PFS (log-rank-test, p=0.028; Fig.2D) and OS (log-rank-test,p=0.036; Supplementary Fig.S5G). Even after controlling for the IMDC score in a multivariate analysis, we observed a trend of association between high SIS and worse PFS (HR=1.43,p=0.099) and OS (HR=1.30,p=0.093).
Figure 2. A plasma-derived systemic “inflammation module” is associated with worse clinical outcomes.

Blood samples from 60 patients were analyzed using a custom multiplex immunoassay to quantify circulating cytokine levels at baseline.
A) Unsupervised hierarchical clustering of Pearson correlations between cytokine levels at baseline demonstrating that specific circulating inflammatory cytokines are highly correlated.
B) Forest plots for PFS hazard ratios by level of circulating inflammatory cytokines from univariate Cox regression model (N=60).
C) Hazard ratios for PFS and OS by level of SIS as a continuous variable, from univariate Cox regression model (N=60).
D) Kaplan-Meier curves showing that a higher SIS is associated with worse progression-free survival (log-rank-test; N=60).
E) Boxplots demonstrating that the SIS is highest in non-responding patients (NCB) (pairwise Wilcoxon tests; N=56).
F) Boxplots showing that the SIS is higher among patients in the poor IMDC risk group (pairwise Wilcoxon tests; N=60).
The threshold for statistical significance in all the analyses was set at a p-value of 0.05.
Boxplots: Median (solid line within the box): the line inside the box represents the median, which is the central value in the dataset. Box (rectangle): the box represents the interquartile range (IQR), which spans from the 25th percentile (lower edge of the box) to the 75th percentile (upper edge of the box). Whiskers (vertical lines extending from the box): the whiskers depict the range of the data excluding outliers, they extend to 1.5 times the IQR. Abbreviations: CB: clinical benefit, CI: confidence interval, HR: hazard ratio, ICB: intermediate clinical benefit, IMDC: International Metastatic RCC Database Consortium, NCB: no clinical benefit, OS: overall survival, PFS: progression-free survival, SIS: systemic inflammation score.
In panel D, time is since initiation of therapy.
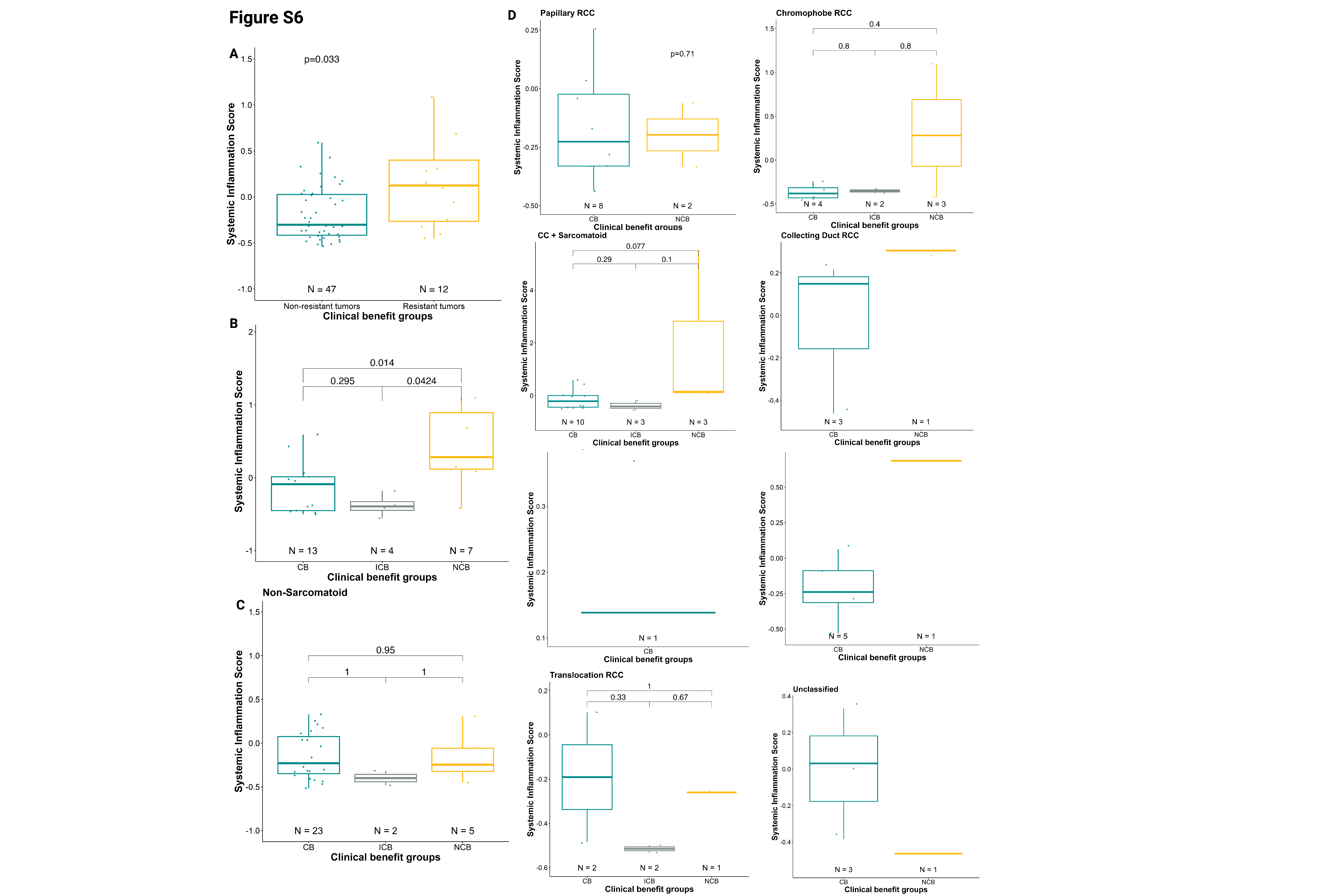
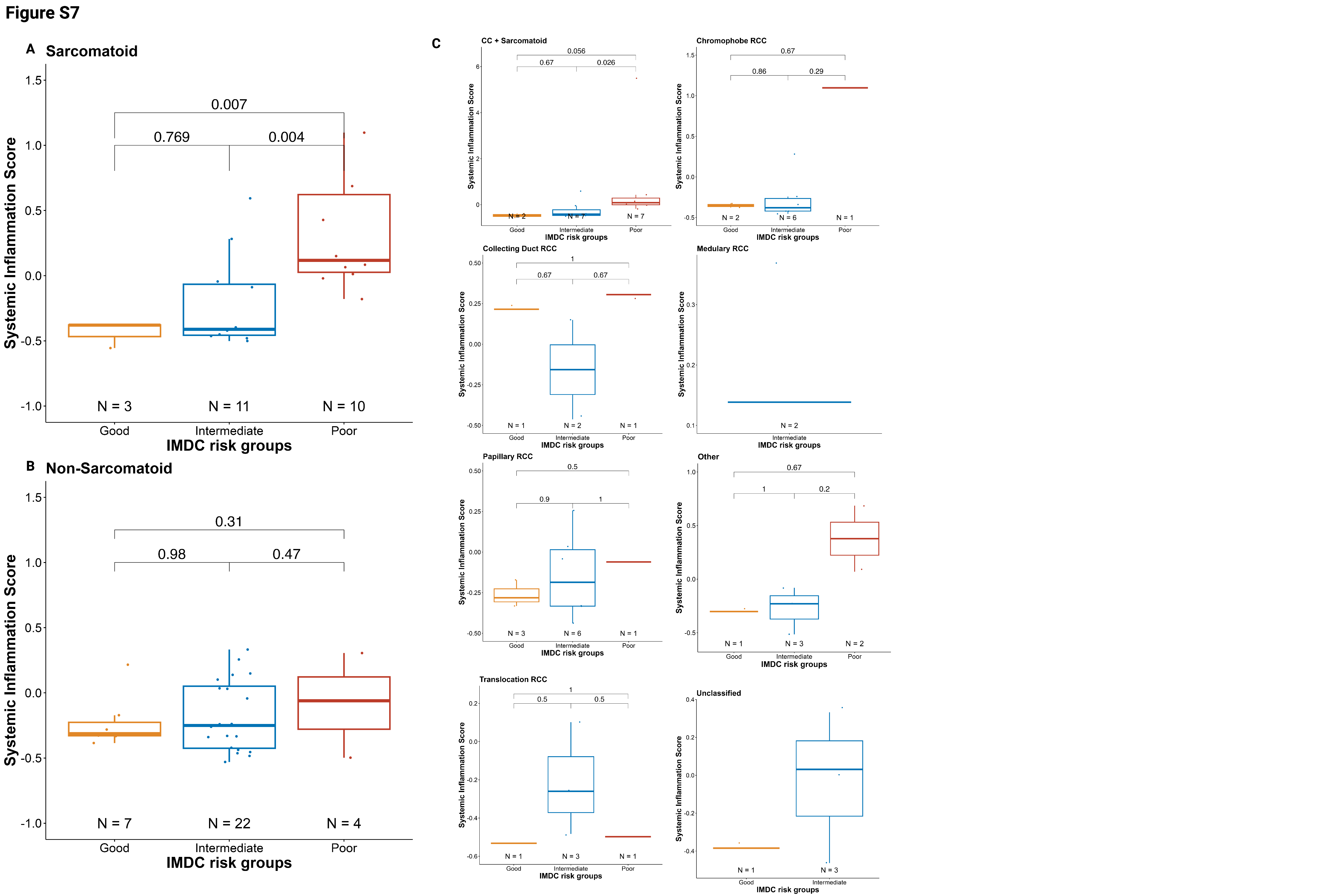
We observed that an elevated SIS score was also associated with a lack of clinical benefit (Wilcoxon test, NCB vs. CB/ICB, p=0.033, Supplementary Fig.S6A). When comparing individual subgroups, we found that an elevated SIS score was associated with a lack of CB as compared with intermediate ICB (Wilcoxon test, NCB vs. ICB, p=0.044; Fig.2E). A similar (non-significant) trend was observed for CB vs NCB (Wilcoxon test,p=0.058; Fig.2E).In subgroup analyses, higher SIS was associated with resistance to therapy in patients with sarcomatoid differentiation and increased baseline (Supplementary Fig.S6B); however, those without sarcomatoid component did not show any association between SIS and clinical response to treatment (Supplementary Fig.S6C). The same trends were consistent across histologies (Supplementary Fig.S6D). SIS also differed between IMDC risk groups, with the poor-risk group having significantly higher inflammation scores than the intermediate-risk (Wilcoxon test, p=0.03) and favorable-risk (Wilcoxon test, p=0.01; Fig.2F) groups. In a subgroup analysis by sarcomatoid versus non-sarcomatoid tumors, the same pattern was observed, with patients in the IMDC poor-risk group exhibiting higher SIS (Supplementary Fig.S7A–B). This result was consistent across different histologies (Supplementary Fig.S7C).
Higher pretreatment VEGF-A concentrations are associated with worse clinical outcomes.
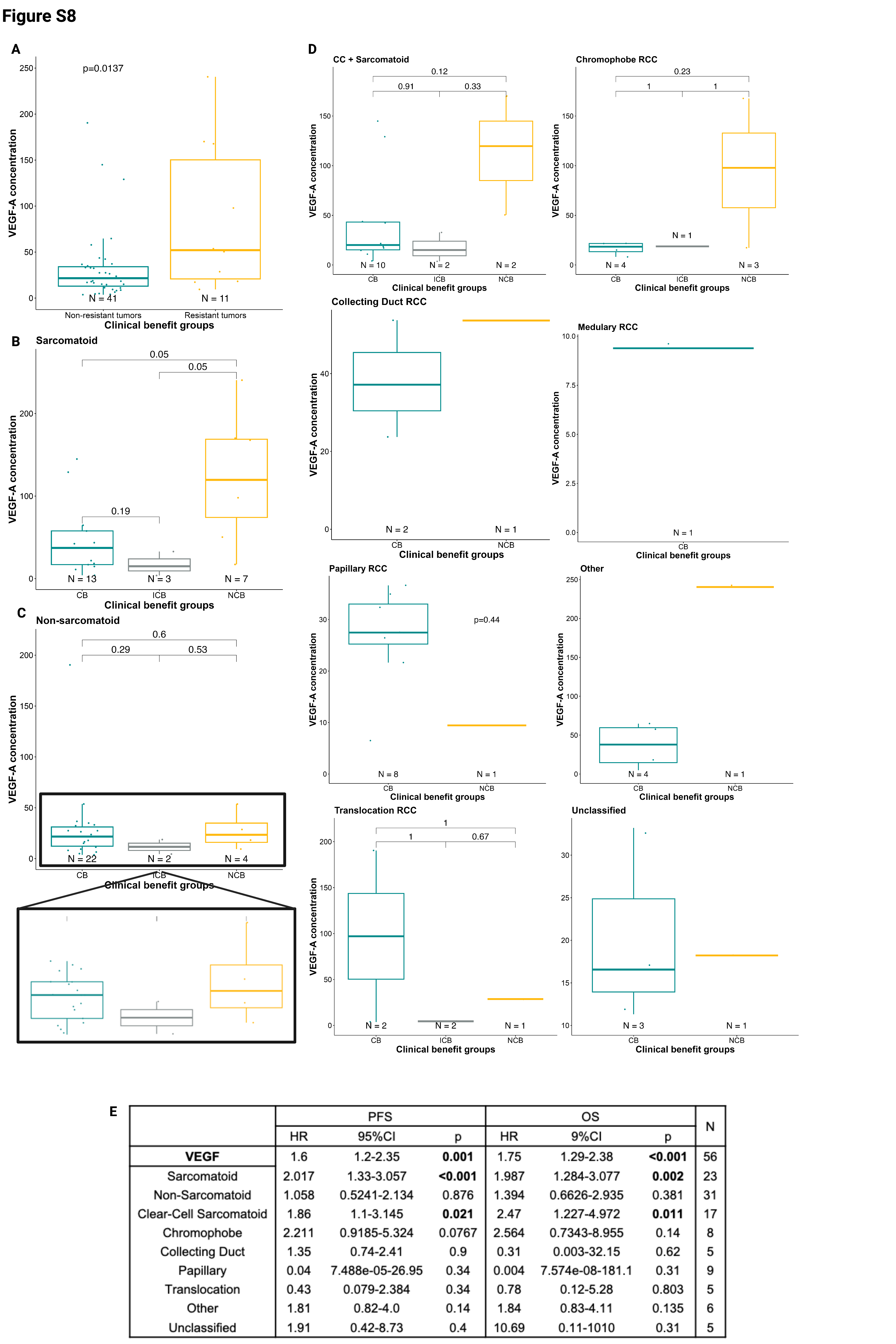
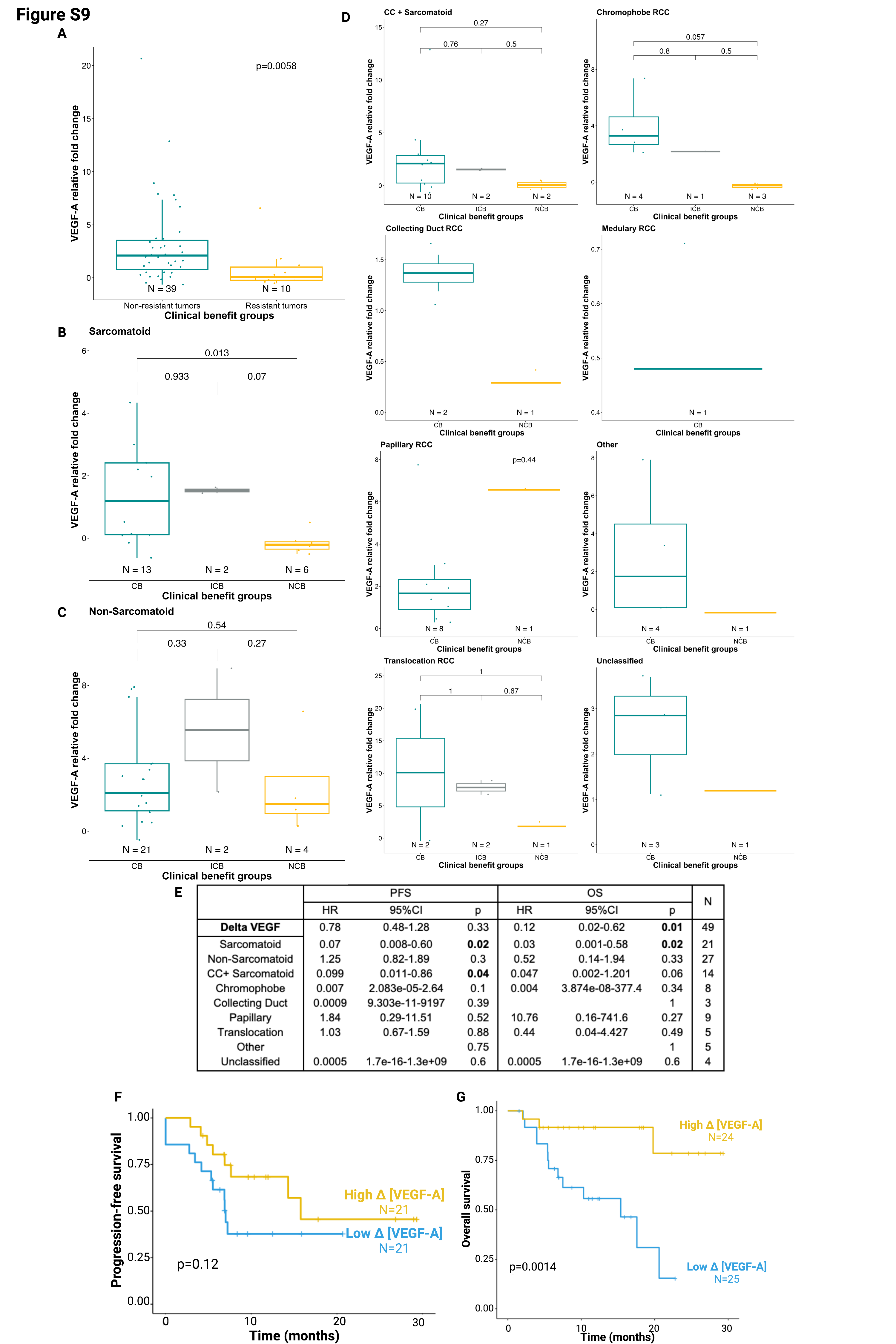
VEGF-A levels at baseline were higher in patients with therapy-resistant disease (NCB) compared to patients with responsive disease (CB; Wilcoxon test, p=0.027) or intermediate benefit (ICB; Wilcoxon test, p=0.01; Fig.3A). More broadly, VEGF-A levels were higher in resistant disease (NCB) compared to non-resistant disease (CB+ICB; Wilcoxon test, p=0.0137; Supplementary Fig.S8A). In the sarcomatoid subgroup, patients with higher baseline VEGF-A levels exhibited resistance to treatment when compared with patients who benefited from atezolizumab+bevacizumab (p=0.04; Supplementary Fig.S8B). A similar (non-significant) trend was observed in the non-sarcomatoid subset (p=0.6; Supplementary Fig.S8C). The association of baseline VEGF-A levels and CB in individual histologies were overall concordant Supplementary (Fig.S8D). Higher baseline VEGF-A levels (as a continuous variable, and dichotomized at the median) were associated with lower PFS (Cox regression, HR=2.017, p=0.001; Supplementary Fig.S8E; log-rank-test, p=0.021; Fig.3B) and OS (Cox regression, HR=1.75,p<0.001; Supplementary Fig.S8E; log-rank-test,p=0.0031; Fig.3C). In addition, a higher baseline VEGF-A level tended to be associated with shorter PFS independently of sarcomatoid status (sarcomatoid: HR=2.02,p<0.001; non-sarcomatoid: HR=1.06,p=0.876) and in all histologies except papillary and translocation RCC (Supplementary Fig.S8E).
Figure 3. Association of baseline and early changes in VEGF-A concentrations with clinical outcomes.

Paired blood samples from 60 patients (at baseline and on-treatment) were analyzed using a multiplex immunoassay to quantify circulating VEGF-A levels at baseline and on treatment.
(A-C) Patients with higher baseline VEGF-A expression have worse clinical outcomes with atezolizumab plus bevacizumab, with (A) higher rate of therapy resistance (NCB; pairwise Wilcoxon test; N=52), and shorter (B) PFS and (C) OS (log-rank-tests; N=56).
(D- F) Relative increase in VEGF-A concentration on-treatment is associated with improved (D) response (pairwise Wilcoxon test; N=49), (E) PFS, and (F) OS (log-rank-test; N=49).
In panels B, C, E, F time is since initiation of therapy.
High Δ[VEGF-A] corresponds to a higher relative increase in VEGF concentration, low Δ [VEGF-A] is a lower relative increase or a decrease in VEGF concentration. The threshold for statistical significance in all the analyses was set at a p-value of 0.05.
Boxplots: Median (solid line within the box): the line inside the box represents the median, which is the central value in the dataset. Box (rectangle): the box represents the interquartile range (IQR), which spans from the 25th percentile (lower edge of the box) to the 75th percentile (upper edge of the box). Whiskers (vertical lines extending from the box): the whiskers depict the range of the data excluding outliers, they extend to 1.5 times the IQR.
Abbreviations: Δ [VEGF-A]: ([VEGF-A (C3D1) – [VEGF-A (C1D1)])/ – [VEGF-A (C1D1), C1D1: Day 1 of Cycle 1 of treatment, C3D1: Day 1 of Cycle 3 of treatment, VEGF-A: vascular endothelial growth factor A.
We defined the dynamic evolution of circulating VEGF-A as Δ [VEGF-A]=([VEGF-A(C3D1) – [VEGF-A(C1D1)])/ VEGF-A(C1D1), and dichotomized it using the median. In contrast to the pattern observed prior to treatment, those patients who had a greater increase in plasma VEGF-A (high Δ[VEGF-A]) on-treatment (C3D1) compared to baseline (C1D1), had overall improved clinical outcomes than patients with more modest increases or decreases in VEGF-A (low Δ[VEGF-A]) (Fig.3D–F). Patients with therapy-resistant disease (NCB) had decreases or only modest increases in plasma VEGF-A as compared to patients who responded to treatment (CB; Wilcoxon test,p=0.012), or to those with ICB (Wilcoxon test,p=0.013; Fig.3D), and more broadly to those with non-resistant disease (CB+ICB; p=0.0058; Supplementary Fig.S9A) in the overall cohort. The same trend was observed in different subgroups according to sarcomatoid status or individual histology (Supplementary Fig.S9B–D). Patients with larger increases in plasma VEGF-A levels during therapy also tended to have improved PFS (Cox regression, HR=0.78,p=0.33; Supplementary Fig.S9E; log-rank-test, p=0.059; Fig.3E [Kaplan-Meier curve starting at C1D1]; p=0.12; Supplementary Fig.S9F [Kaplan-Meier curve starting at C3D1]) and OS (Cox regression, HR=0.12,p=0.01; Supplementary Fig.S9E; log-rank-test, p=0.0058; Fig.3F [Kaplan-Meier curve starting at C1D1]; p=0.0014; Supplementary Fig.S9G [Kaplan-Meier curve starting at C3D1]).
Large decreases in circulating CD8+PD-L1+ T cells are associated with worse PFS.
To better understand how the circulating immune landscape impacts response to therapy in variant histology RCC, we used known lineage markers to analyze the baseline peripheral blood samples by high-dimensional flow cytometry (Fig.4). Across all samples, we found no large modules of correlated immune cell types at baseline (Pearson correlations, Fig.4A). We did, however, find areas of moderate correlation among CD4+PD-1+ and CD4+ T-cell effector memory populations (TEM, r=0.64,p<0.0001; and TEMRA, r=0.75,p<0.0001). Additionally, CD4+PD-L1+ T-cell levels correlated with CD8+PD-L1+ T-cell levels (r=0.71,p<0.0001; Fig.4A).
Figure 4. Dynamic changes in PD-L1 expressing circulating T cells are associated with treatment-related outcomes.

Paired blood samples from 60 patients (at baseline and on-treatment) were analyzed using a flow cytometry to quantify circulating immune cells at baseline and on treatment.
A) Unsupervised hierarchical clustering of Pearson correlations between baseline immune cell levels.
B) Boxplots demonstrating the association of the change of CD8+ PD-L1+ cell levels (from C1D1 to C3D1) with CB (pairwise Wilcoxon tests; N=50).
C) Forest plots for PFS hazard ratios in patients treated with atezolizumab+bevacizumab by ratio of on-treatment to baseline circulating CD4+ and CD8+ cells expressing PD-L1 (Cox univariate regression; N=50).
D) Kaplan-Meier curves showing that lower decrease (C3D1/C1D1 ratio ≤ median ratio) in CD8+ PD-L1+ cells on treatment is associated to improved PFS (log-rank-test; N=50).
In panel D, time is since initiation of therapy. The threshold for statistical significance in all the analyses was set at a p-value of 0.05.
Boxplots: Median (solid line within the box): the line inside the box represents the median, which is the central value in the dataset. Box (rectangle): the box represents the interquartile range (IQR), which spans from the 25th percentile (lower edge of the box) to the 75th percentile (upper edge of the box). Whiskers (vertical lines extending from the box): the whiskers depict the range of the data excluding outliers, they extend to 1.5 times the IQR.
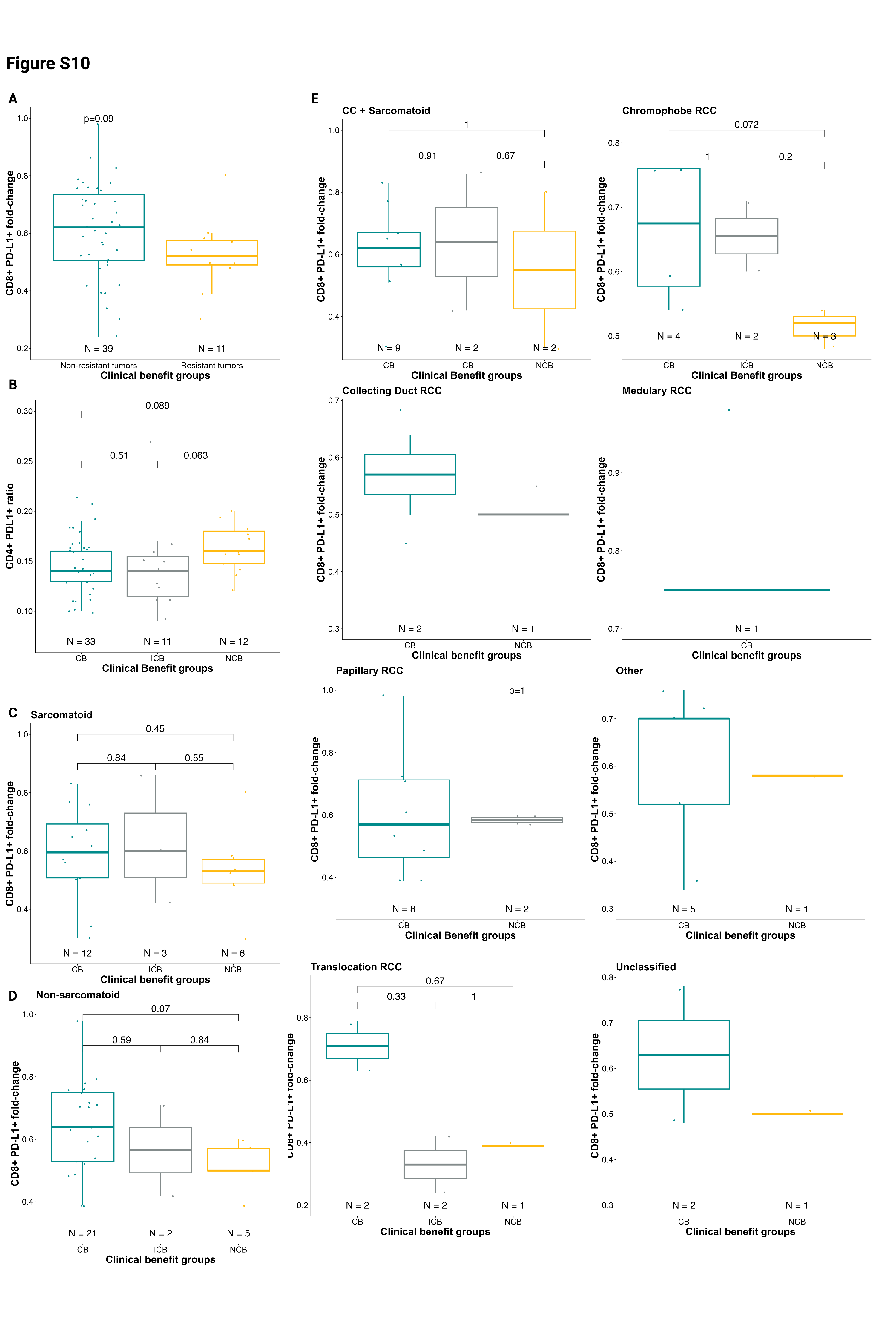
To further understand the importance of PD-L1+ T cells, we investigated their relative changes at C3D1 compared to C1D1. The proportion of CD8+PD-L1+ T cells decreased with treatment in all patients (Fig.4B). However, patients with therapeutic response (CB) had a smaller decrease in the proportion of CD8+PD-L1+ T cells than patients with therapeutic resistance (NCB) with atezolizumab plus bevacizumab treatment (p=0.06; Fig.4B). The larger decrease in CD8+PD-L1+ T cells appeared to specifically be associated with resistance (Wilcoxon test, NCB vs. CB+ICB, p=0.09; Supplementary Fig.S10A). However, a similar trend was not clearly observed with the relative change in CD4+ PD-L1+ T cells on-treatment (p=0.22; Supplementary Fig.S10B). Furthermore, we did not find any associations between baseline PD-L1+ T cells and outcomes in different subsets (Supplementary Fig.S10C–E).
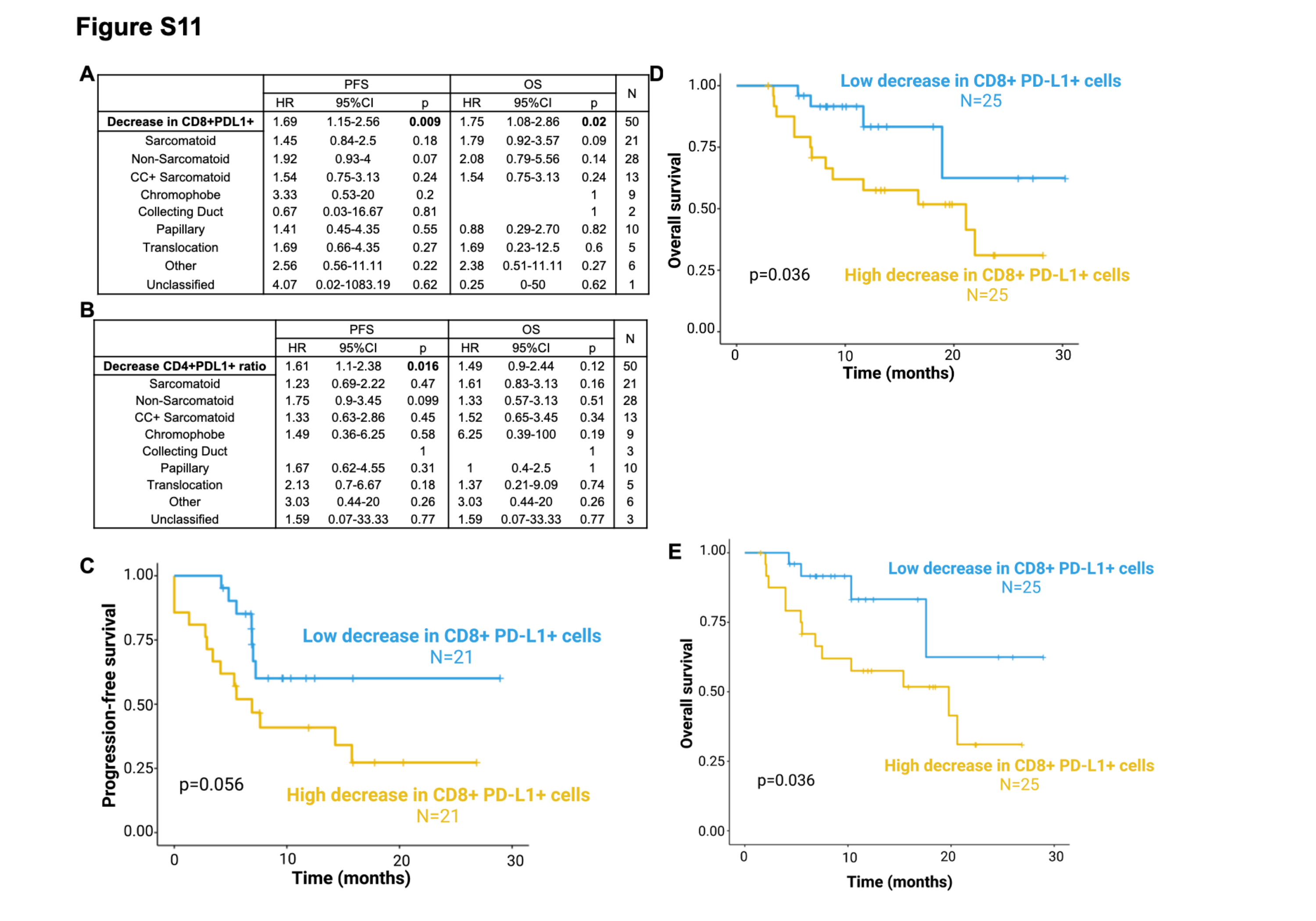
Differences in the relative changes in PD-L1+ T cells on-treatment were also associated with altered PFS and OS. A larger decrease of PD-L1+ T cells on-treatment was associated with worse PFS for both CD4+PD-L1+ T cells (HR=1.61, p=0.016; Fig.4C) and CD8+PD-L1+ T cells (HR=1.69, p=0.009; Fig.4C). The same trend was observed for OS for both CD4+PD-L1+ T cells (HR=1.49, p=0.12) and CD8+PD-L1+ T cells (HR=1.75, p=0.025; Supplementary Fig.S11A, B). For patients with higher (>median) on-treatment decrease in CD8+PD-L1+ T cells, the median PFS was 5.5 months and median OS was 21.3 months. In contrast, among patients with lower (≤median) on-treatment decreases in CD8+PD-L1+ T cells, the median PFS and median OS were not reached (PFS, p=0.02; Fig.4D [Kaplan-Meier curve starting at C1D1]; p=0.056; Supplementary Fig.S11C [Kaplan-Meier curve starting at C3D1]; OS, p=0.036; Supplementary Fig.S11D–E). Among both sarcomatoid/non-sarcomatoid groups and different histologies, a higher decrease in PD-L1+ circulating T cells remained associated with worse PFS (Supplementary Fig.S11A–B).
Higher percentages of terminally exhausted CD8+ T cells are associated with worse PFS.
Since the tumor microenvironment also plays a crucial role in determining the response ICI, we assessed PD-L1 expression on tumor cells by IHC. Additionally, we performed detailed spatial phenotyping of T cells, investigating the expression of markers of antigen experience (PD-1) and terminal exhaustion (TIM-3 and LAG-3) on CD8+ tumor-infiltrating T cells using MIF.
The average PD-L1 expression was 19.6% of tumor cells across sarcomatoid tumors and 27.2% across non-sarcomatoid tumors (Fig.5A–B). There were no differences in PD-L1 expression between sarcomatoid and non-sarcomatoid tumor samples (Wilcoxon test,p=0.77). There were no associations between the various histologies and their tumor cell PD-L1 expression.
Figure 5. Evaluation of antigen-experienced and terminal exhaustion T-cell markers in nccRCC.

Tissue samples from 38 patients were collected before initiation of therapy and stained (immunohistochemistry and multiplex immunofluorescence) to characterize PD-L1 status of the tumor and the intratumoral immune microenvironment.
A-B) Distribution of tumor samples according to T cell markers by (A) sarcomatoid status and (B) histology (N=11).
C) Kaplan-Meier curves showing that patients with a lower percentage of terminally exhausted CD8 T cells had improved PFS (log-rank-test; N=38).
In panel C, time is since initiation of therapy.
The percentage of terminally exhausted CD8+ cells is calculated relatively to all CD8+ cells. Low corresponds to percentages that are inferior to the median while high corresponds to percentages that are higher than the median. The threshold for statistical significance in all the analyses was set at a p-value of 0.05.
There was no clear association between RCC histology and the proportion of antigen-experienced (PD-1+ but TIM-3– and LAG-3–) or terminally exhausted (PD-1+ with TIM-3+ or LAG-3+) CD8+ T cells, although the analysis was restricted by a limited number of available tumor samples. There was no significant difference in the density of terminally exhausted CD8+ T cells between clear-cell sarcomatoid (mean=151.2cell/mm2), non-clear cell sarcomatoid (mean=20.3cell/mm2) and non-sarcomatoid (mean=76.5cell/mm2) tumors (p>0.5, Fig.5A). Additionally, no significant differences were observed in the density of antigen-experienced CD8+ T cells between clear-cell sarcomatoid (mean=23.7cell/mm2), non-clear cell sarcomatoid (mean=114.1cell/mm2) and non-sarcomatoid (mean=97.4cell/mm2) tumors (p>0.5, Fig.5A). Of note, the two tumor samples with papillary histology had a relatively low density of terminally exhausted CD8+ T cells (3.6, 7.2cell/mm2, Fig.5B).
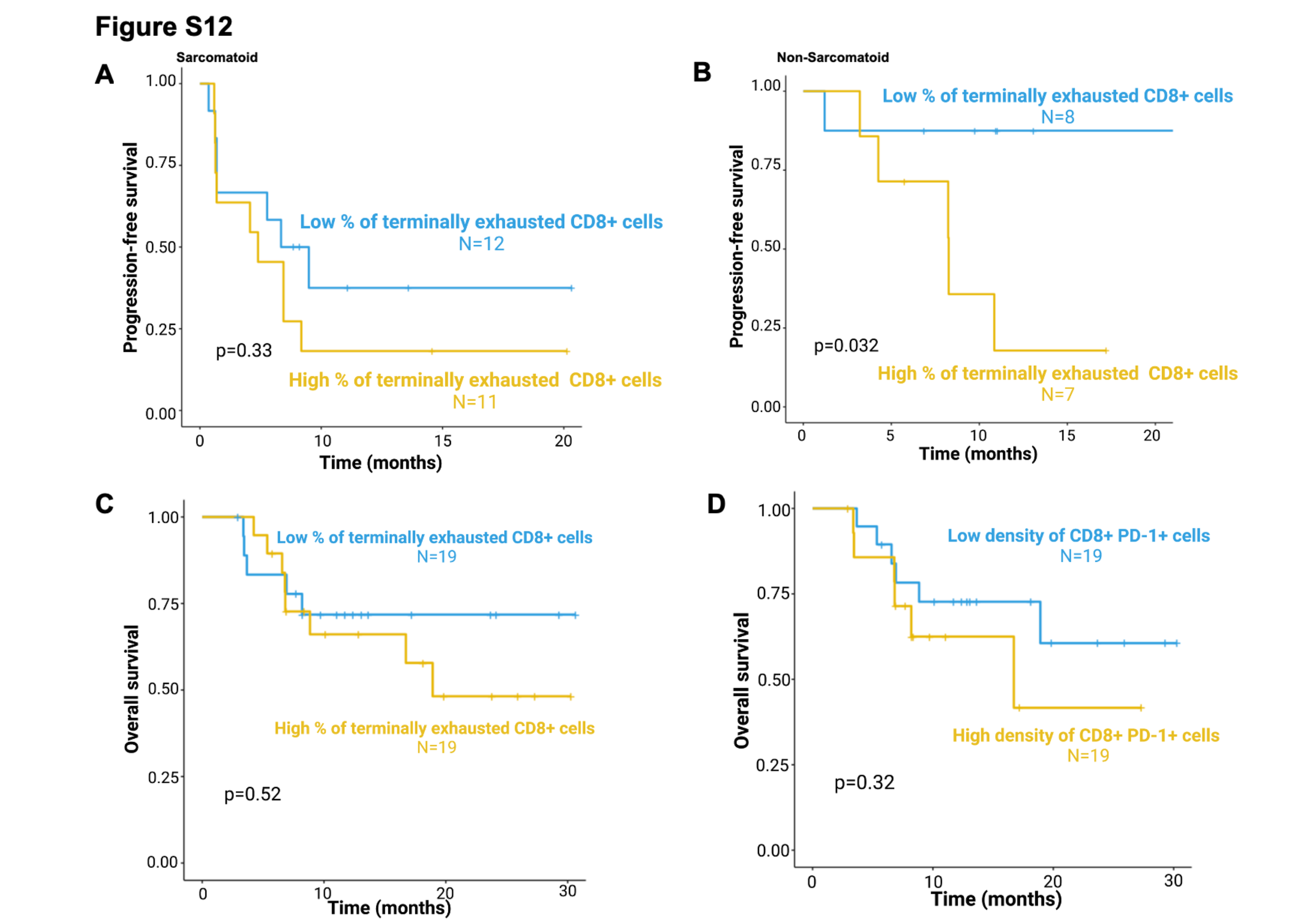
Across all histologies, there was a clear association between a higher percentage of intratumoral terminally exhausted CD8+ T cells and worse PFS. Patients with high (>median) percentages of terminally exhausted CD8+ T cells had a median PFS of only 6.9 months, while those with low percentages of terminally exhausted CD8+ T cells did not reach the median PFS (log-rank-test, p=0.028, Fig.5C). A higher percentage of intratumoral terminally exhausted CD8+ T cells was associated with worse outcomes in patients with nccRCC without sarcomatoid component (p=0.032, Supplementary Fig.S12B), and a similar (non-significant) effect was seen in sarcomatoid tumors (p=0.33, Supplementary Fig.S12A). However, there was no significant association between the percentage of terminally exhausted CD8+ T cells (log-rank-test, p=0.52, Supplementary Fig.S12C) or CD8+PD-1+ T cells (Supplementary Fig.S12D) and OS.
Discussion:
This correlative analysis of blood and tumor specimens from the phase II clinical trial of atezolizumab+bevacizumab in variant histology RCC revealed both circulating and intratumoral determinants of clinical outcomes with ICI and antiangiogenic agents. Analysis of soluble factors in plasma revealed a highly coordinated “inflammatory module” of circulating immunomodulatory cytokines (IL-1, IL-6, IL-13, and MIP-1β) associated with adverse clinical outcomes. Beyond static baseline measurements, dynamic changes in circulating angiogenic factors (e.g., VEGF-A) and peripheral immune populations (e.g., CD8+PD-L1+ T cells) also impacted clinical outcomes. Finally, within the tumor, a high percentage of terminally exhausted CD8+ T cells in the tumor microenvironment were associated with worse PFS. These results provide novel insights into the biology of variant histology RCC or ccRCC with sarcomatoid features in the immunotherapy era and a reference framework for future biomarker studies. Many of these findings build on prior observations in ccRCC and other tumor types, and it is notable that the heterogeneous underlying biology of different RCC histologies and sarcomatoid differentiation does represent a limitation. Nevertheless, our main findings linking systemic inflammatory cytokines, baseline and dynamic changes in VEGF-A, dynamic changes in circulating T-cell populations, and intratumoral terminally exhausted CD8+ T cells with altered clinical outcomes have not been described in variant/sarcomatoid RCC histologies.
We observed that increased baseline levels of circulating proinflammatory cytokines are correlated with poorer clinical outcomes, which is consistent with previous observations [19]–[23]. The IMDC risk score for metastatic RCC, which has been validated as a reliable predictor of OS in patients with variant histology or “non-clear cell” RCC [15], includes several systemic inflammatory correlates including neutrophilia, anemia, and thrombocytosis, as negative prognostic factors [24]. It is notable that SIS discriminates between response and resistance even in the sarcomatoid group, which is typically characterized by a highly inflammatory state. In the IMmotion150 trial, which enrolled patients with clear-cell mRCC (including tumors with a sarcomatoid component), an elevated myeloid inflammation signature within the tumor was associated with shorter PFS in patients receiving atezolizumab-containing regimens [19]. Further, anti-IL-1β therapy also remodeled the myeloid compartment and delayed tumor outgrowth in the RENCA RCC model [20], suggesting a potential therapeutic approach. In fact, in the CANTOS trial that was designed to test directly the inflammatory hypothesis of atherothrombosis, patients who received canakinumab, an anti-IL-1, showed lower cancer mortality [21], [22]. Additionally, baseline circulating IL-6 has been implicated as a potential negative predictive biomarker of response to ICI in NSCLC, and in vitro experiments indicate that IL-6 can upregulate PD-L1 via JAK1/Stat3 signaling [23]. Other circulating inflammatory cytokines, such as IL-8, have also be shown to have a negative predictive value for clinical outcomes in patients treated with ICI [25]. Further investigations of the immunosuppressive role of systemic inflammation in RCC with variant histology, as well as corresponding circulating biomarkers, is warranted.
Our observation that increased baseline VEGF-A carries a worse prognosis is established in the literature [26], [27], although the implications of this finding for therapy selection are unclear. For instance, Hegde et al. analyzed several clinical trials evaluating bevacizumab for metastatic colorectal cancer, NSCLC, and mRCC. They found that increased baseline plasma VEGF-A levels conferred a worse OS prognosis; however, VEGF-A appeared to be a prognostic, but not a predictive biomarker [28]. On the other hand, a meta-analysis of clinical trials investigating bevacizumab for breast cancer found that bevacizumab improved PFS for patients with high (>median) but not low (≤median) VEGF-A levels [29].
Our finding that greater increases in VEGF-A on-treatment are associated with a better prognosis also has uncertain implications. Among patients with clear cell or papillary RCC treated with adjuvant sunitinib or sorafenib after nephrectomy, exposure to these VEGFR inhibitors were previously found to result in host-mediated compensatory increase in circulating VEGF-A [30]. This suggests that increases in VEGF-A are associated with target engagement by VEGF pathway inhibitors and may explain our observed association between increased VEGF-A on therapy and improved clinical outcomes. A review of biomarkers of response to bevacizumab by Lambrechts et al. found that changes in circulating VEGF-A levels during treatment have been inconsistent across five trials for different cancer types (not including RCC) [31]. Furthermore, one study that included patients with platinum-resistant recurrent ovarian cancer treated with bevacizumab and gemcitabine showed that rapid on-treatment decrease of VEGF-A levels is associated with worse response, clinical benefit rates, PFS and OS [32]. The significant differences in PFS and OS associated with on-treatment VEGF-A changes are striking and should be explored in future biomarker studies.
Upon treatment with atezolizumab (plus bevacizumab) peripheral CD8+PD-L1+ populations universally decreased after one treatment cycle. Smaller decreases in the CD8+PD-L1+ population after treatment were associated with CB and longer PFS. Previous studies in melanoma and non-small cell lung cancer have demonstrated that increased CD8+PD-L1+ cells are associated with an immunosuppressed tumor microenvironment and worse clinical outcomes, and have resulted in mixed outcomes in response to immunotherapy [33], [34]. In gastric and gastro-esophageal junction adenocarcinoma, there was a correlation of PD-L1 expression on tumor and immune cells (i.e., CD8+ T cells) with worse PFS and OS, but only that of tumor cells reached statistical significance [35]. In cervical cancer, the presence of PD-L1-expressing tumor-infiltrating lymphocytes (TILs) predicted poor prognosis [35]. However, to our knowledge, no studies have been conducted to evaluate the effect of CD8+PD-L1+ T cells on the tumor microenvironment in variant RCC or ccRCC with sarcomatoid features. Further work is required to understand why a decrease in CD8+PD-L1+ T cells was associated with improved clinical outcomes when baseline CD8+PD-L1+ T cells are otherwise correlated with worse clinical outcomes in other tumor types, and whether CD8+PD-L1+ T cells could be a predictive biomarker for response to immunotherapy in variant histology (and clear cell) RCC.
While we did not detect any clear correlations in tumor PD-L1 expression and tumor histology, we recognize that identifying such patterns may be limited by the small number of tumors analyzed. Sarcomatoid RCC has been characterized by high PD-L1 tumoral expression compared to non-sarcomatoid RCC [36]. In agreement with other studies in ccRCC [37], [38], increased terminally exhausted intratumoral CD8+ T cells were associated with worse survival outcomes. To our knowledge, this is the first study to describe antigen-experienced and terminally exhausted CD8+ T cells in variant histology RCC. It would have also been interesting to dissect the distribution of the IMmotion151 molecular clusters [13] across this nccRCC cohort.
In conclusion, when examining the immune landscape of patients with RCC of variant histology treated with atezolizumab plus bevacizumab, both the peripheral (circulating) and intratumor immune topography, were associated with treatment-related outcomes. Furthermore, our data demonstrate that a baseline inflammatory state is associated with worse outcomes, whereas a lower baseline VEGF-A concentration and higher increase in VEGF-A levels on treatment correlated with CB and improved survival. In addition, marked decreases in circulating CD8+PD-L1+ T cells had a negative prognostic value. Finally, patients with high levels of intratumoral terminally exhausted CD8+ T cells exhibited shorter PFS. In aggregate, these findings support the further investigation of both peripheral and intratumoral immune biomarkers as part of an integrative approach in mRCC with variant histology or sarcomatoid differentiation.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Synopsis.
The authors show high levels of a defined set of plasma inflammatory cytokines and intratumoral terminally exhausted CD8+ T cells correlate with worse immunotherapy outcomes for patients with variant histology RCC, providing a foundation for future biomarker studies.
Acknowledgements:
S. Signoretti and T. K. Choueiri are supported in part by the Dana-Farber/Harvard Cancer Center Kidney SPORE (2P50CA101942-16) and Program 5P30CA006516-56. T. K. Choueiri is supported in part by the the Kohlberg Chair at Harvard Medical School and the Trust Family, Michael Brigham, Pan Mass Challenge and Loker Pinard Funds for Kidney Cancer Research at DFCI. Roche/Genentech provided support to this investigator-initiated study (NCT02724878).
Funding Information:
S. Signoretti and T. K. Choueiri are supported in part by the Dana-Farber/Harvard Cancer Center Kidney SPORE (2P50CA101942-16) and Program 5P30CA006516-56. T. K. Choueiri is supported in part by the the Kohlberg Chair at Harvard Medical School and the Trust Family, Michael Brigham, Pan Mass Challenge and Loker Pinard Funds for Kidney Cancer Research at DFCI. Roche/Genentech provided support to this investigator-initiated study (NCT02724878).
Author’s Disclosures:
Z. Bakouny reports honoraria from UpToDate and research support from BMS and Genentech/imCORE. W. Xie reports a consultant role at Convergent Therapeutics, Inc. R. Flippot reports honoraria for consulting at Astellas, Pfizer, MSD, BMS, Ipsen, Bayer, Janssen. X. Wei reports institutional research support from BMS and a position on the advisory board of Novartis. LC. Harshman reports previous employment at Dana-Farber Cancer Institute and grants to institution from Bayer, Sotio, Bristol-Myers Squib, Merck, Takeda, Dendreon/Valient, Jannsen, Medivation/Astellas, Genentech, Pfizer, and Endocyte (Novartis), Advisory or consulting services for Bayer, Genentech, Pfizer, Medivation/Astellas, Corvus, Merck, Exelixis, Novartis, Advanced Accelerator Applications, Jounce, Bristol-Myers Squibb, EMD Serono, Michael J Hennessy Associates (Healthcare Communications Company and several brands such as OncLive and PER), ASIM CME, and Ology Medical Education during the conduct of the study, as well as current employment (including stock options) at Surface Oncology and travel support form Bayer and Genentech outside the submitted work. U.N. Vaishampayan reports receiving research support from BMS and Merck, honoraria for consulting from Exelixis, Sanofi, AAA, Aveo, Bayer, Gilead, Pfizer, Merck and Alkermes. DF. McDermott reports honoraria from BMS, Pfizer, Merck, Alkermes, Inc., EMD Serono, Eli Lilly and Company, Iovance, Eisai Inc., Werewolf Therapeutics, Calithera Biosciences, Synthekine, Inc., Johnson & Johnson, Aveo for consulting and research support from BMS, Merck, Genentech, Pfizer, Exelixis, X4 Pharma, Alkermes, Inc., Checkmate Pharmaceuticals, CRISPR Therapeutics for cancer research. W. Xu reports advisory board fees from Exelixis and Jazz Pharmaceuticals, and continuing medical education honoraria from MedNet and Physicians’ Education Resource. E.M. Van Allen is a consultant for ango Therapeutics, Genome Medical, Genomic Life, Enara Bio, Manifold Bio, Monte Rosa, Novartis Institute for Biomedical Research, Riva Therapeutics, Serinus Bio; received research support from Novartis, Bristol-Myers Squibb and Sanofi; and is an equity holder of Tango Therapeutics, Genome Medical, Genomic Life, Syapse, Enara Bio, Manifold Bio, Microsoft, Monte Rosa, Riva Therapeutics, Serinus Bio; serves on the editorial boards of JCO Precision Oncology, Science Advances; he reports that institutional patents filed on chromatin mutations and immunotherapy response, and methods for clinical interpretation; intermittent legal consulting on patents for Foaley & Hoag. BA. McGregor reports receiving consulting fees from Astella, Bayer, Bristol Myers Squibb, Calithera, Dendreon, Exelixis, Ipsen, Pfizer, Seattle Genetics and institutional fees from Bristol Myers Squibb, Calithera, Exelixis, Pfizer, Seattle Genetics, Aveo. S.Signoretti reports receiving commercial research grants from Bristol-Myers Squibb, AstraZeneca, Exelixis and Novartis; is a consultant/advisory board member for Merck, AstraZeneca, Bristol-Myers Squibb, CRISPR Therapeutics AG, AACR, and NCI; receives royalties from Biogenex; and mentored several non-US citizens on research projects with potential funding (in part) from non-US sources/Foreign Components. Dr. Choueiri reports institutional and personal, paid and/or unpaid support for research, advisory boards, consultancy, and honoraria from: AstraZeneca, Aravive, Aveo, Bayer, Bristol Myers-Squibb, Calithera, Circle Pharma, Eisai, EMD Serono, Exelixis, GlaxoSmithKline, IQVA, Infinity, Ipsen, Jansen, Kanaph, Lilly, Merck, Nikang, Nuscan, Novartis, Pfizer, Roche, Sanofi/Aventis, Surface Oncology, Takeda, Tempest, Up-To-Date, CME events (Peerview, OncLive, MJH and others), outside the submitted work. Institutional patents filed on molecular alterations and immunotherapy response/toxicity, and ctDNA. Equity: Tempest, Pionyr, Osel, Precede Bio. CureResponse. Committees: NCCN, GU Steering Committee, ASCO/ESMO, ACCRU, KidneyCan. Medical writing and editorial assistance support may have been funded by Communications companies in part. No speaker’s bureau. Mentored several non-US citizens on research projects with potential funding (in part) from non-US sources/Foreign Components. The institution (Dana-Farber Cancer Institute) may have received additional independent funding of drug companies or/and royalties potentially involved in research around the subject matter. T. K. Choueiri is supported in part by the Dana-Farber/Harvard Cancer Center Kidney SPORE (2P50CA101942–16) and Program 5P30CA006516–56, the Kohlberg Chair at Harvard Medical School and the Trust Family, Michael Brigham, Pan Mass Challenge, Hinda and Arthur Marcus Fund and Loker Pinard Funds for Kidney Cancer Research at DFCI. R.R. McKay reports consulting or advisory Role at Janssen, Novartis, Tempus, Exelixis, Pfizer, Bristol Myers Squibb, Astellas Medivation, Dendreon, Bayer, Sanofi, Merck, Vividion Therapeutics, Calithera Biosciences, AstraZeneca, Myovant Sciences, Caris Life Sciences, Sorrento Therapeutics, AVEO and research funding from Pfizer (Inst), Bayer (Inst), Tempus (Inst). D.A.Braun. reports nonfinancial support from Bristol Myers Squibb, honoraria from LM Education/Exchange Services, advisory board fees from Exelixis and AVEO, personal fees from Charles River Associates, Schlesinger Associates, Imprint Science, Insight Strategy, Trinity Group, Cancer Expert Now, Adnovate Strategies, MDedge, CancerNetwork, Catenion, OncLive, Cello Health BioConsulting, PWW Consulting, Haymarket Medical Network, Aptitude Health, ASCO Post/Harborside, Targeted Oncology, AbbVie, and research support from Exelixis and AstraZeneca, outside of the submitted work.
References
- [1].“Kidney and Renal Pelvis Cancer — Cancer Stat Facts.” [Online]. Available: https://seer.cancer.gov/statfacts/html/kidrp.html. [Accessed: 13-Oct-2022]. [Google Scholar]
- [2].Lopez-Beltran A, Scarpelli M, Montironi R, and Kirkali Z, “2004 WHO Classification of the Renal Tumors of the Adults,” Eur. Urol, vol. 49, no. 5, pp. 798–805, May 2006. [DOI] [PubMed] [Google Scholar]
- [3].Blum KA et al. , “Sarcomatoid renal cell carcinoma: biology, natural history and management,” Nat. Rev. Urol. 2020 1712, vol. 17, no. 12, pp. 659–678, Oct. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Motzer RJ et al. , “Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma,” N. Engl. J. Med, vol. 378, no. 14, pp. 1277–1290, Apr. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Rini BI et al. , “Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma,” N. Engl. J. Med, vol. 380, no. 12, pp. 1116–1127, Mar. 2019. [DOI] [PubMed] [Google Scholar]
- [6].Rini BI et al. , “Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): a multicentre, open-label, phase 3, randomised controlled trial,” Lancet, vol. 393, no. 10189, pp. 2404–2415, Jun. 2019. [DOI] [PubMed] [Google Scholar]
- [7].TK C.and RJ M, “Systemic Therapy for Metastatic Renal-Cell Carcinoma,” N. Engl. J. Med, vol. 376, no. 4, pp. 152–162, Jun. 2017. [DOI] [PubMed] [Google Scholar]
- [8].Van EM Allen and Choueiri TK, “Dissecting the immunogenomic biology of cancer for biomarker development,” Nat. Rev. Clin. Oncol, vol. 18, no. 3, pp. 133–134, Mar. 2021. [DOI] [PubMed] [Google Scholar]
- [9].Choueiri TK et al. , “A Focus on Translational Research.” [Google Scholar]
- [10].Ravi P, Bakouny Z, Schmidt A, and Choueiri TK, “Novel Therapeutic Approaches and the Evolution of Drug Development in Advanced Kidney Cancer,” Cancer J., vol. 26, no. 5, pp. 464–470, Sep. 2020. [DOI] [PubMed] [Google Scholar]
- [11].Braun DA et al. , “Beyond conventional immune-checkpoint inhibition — novel immunotherapies for renal cell carcinoma,” Nat. Rev. Clin. Oncol, vol. 18, no. 4, pp. 199–214, Jan. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].McDermott DF et al. , “Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma.” [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Motzer RJ et al. , “Molecular Subsets in Renal Cancer Determine Outcome to Checkpoint and Angiogenesis Blockade,” Cancer Cell, vol. 38, no. 6, pp. 803–817.e4, Dec. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].McGregor BA et al. , “Results of a multicenter phase II study of atezolizumab and bevacizumab for patients with metastatic renal cell carcinoma with variant histology and/or sarcomatoid features,” J. Clin. Oncol, vol. 38, no. 1, pp. 63–70, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kroeger N.et al. , “Metastatic non–clear cell renal cell carcinoma treated with targeted therapy agents: Characterization of survival outcome and application of the International mRCC Database Consortium criteria,” Cancer, vol. 119, no. 16, pp. 2999–3006, Aug. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Braun DA et al. , “Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma,” Nat. Med, vol. 26, no. 6, pp. 909–918, Jun. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Miao D.et al. , “Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma,” Science, vol. 359, no. 6377, p. 801, Feb. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].“6-plex IF Protocol on the BOND RX (Leica Biosystems).” [Online]. Available: https://www.protocols.io/view/6-plex-if-protocol-on-the-bond-rx-leica-biosystems-j8nlkexm1l5r/v1. [Accessed: 16-Aug-2022].
- [19].McDermott DF et al. , “Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma,” Nat. Med, vol. 24, no. 6, pp. 749–757, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Aggen DH et al. , “Blocking IL1 beta promotes tumor regression and remodeling of the myeloid compartment in a renal cell carcinoma model: multidimensional analyses,” Clin. Cancer Res, vol. 27, no. 2, pp. 608–621, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ridker PM et al. , “Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease,” N. Engl. J. Med, vol. 377, no. 12, pp. 1119–1131, 2017. [DOI] [PubMed] [Google Scholar]
- [22].Ridker PM et al. , “Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial,” Lancet, vol. 390, no. 10105, pp. 1833–1842, Oct. 2017. [DOI] [PubMed] [Google Scholar]
- [23].Liu C.et al. , “Systematic analysis of IL-6 as a predictive biomarker and desensitizer of immunotherapy responses in patients with non-small cell lung cancer,” 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Heng DYC et al. , “Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor-targeted agents: Results from a large, multicenter study,” J. Clin. Oncol, vol. 27, no. 34, pp. 5794–5799, Dec. 2009. [DOI] [PubMed] [Google Scholar]
- [25].Bakouny Z.and Choueiri TK, “IL-8 and cancer prognosis on immunotherapy Interleukin-8, produced by intratumoral and circulating myeloid cells, correlates with an immunosuppressive myeloid-enriched tumor microenvironment and adverse cancer prognosis,” Nat. Med. [DOI] [PubMed] [Google Scholar]
- [26].Lacin S.and Yalcin S, “The Prognostic Value of Circulating VEGF-A Level in Patients With Hepatocellular Cancer,” Technol. Cancer Res. Treat, vol. 19, Nov. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Li S, Wang L, Meng Y, Chang Y, Xu J, and Zhang Q, “Increased levels of LAPTM4B, VEGF and survivin are correlated with tumor progression and poor prognosis in breast cancer patients,” Oncotarget, vol. 8, no. 25, pp. 41282–41293, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hegde PS et al. , “Predictive impact of circulating vascular endothelial growth factor in four phase III trials evaluating bevacizumab,” Clin. Cancer Res, vol. 19, no. 4, pp. 929–937, Feb. 2013. [DOI] [PubMed] [Google Scholar]
- [29].Vieira dos Santos L, Rocha Cruz M, de Lima Lopes G, and Paulo J.da Silveira Nogueira Lima, “VEGF-A levels in bevacizumab-treated breast cancer patients: a systematic review and meta-analysis.” [DOI] [PubMed] [Google Scholar]
- [30].Xu W.et al. , “Angiogenic factor and cytokine analysis among patients treated with adjuvant VEGFR TKIs in resected renal cell carcinoma,” Clin. Cancer Res, vol. 25, no. 20, pp. 6098–6106, Oct. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lambrechts D, Lenz HJ, De Haas S, Carmeliet P, and Scherer SJ, “Markers of response for the antiangiogenic agent bevacizumab,” J. Clin. Oncol, vol. 31, no. 9, pp. 1219–1230, Mar. 2013. [DOI] [PubMed] [Google Scholar]
- [32].Soyama H.et al. , “Rapid decrease in serum VEGF-A levels may be a worse prognostic biomarker for patients with platinum-resistant recurrent ovarian cancer treated with bevacizumab and gemcitabine,” Cancer Chemother. Pharmacol, vol. 85, no. 5, pp. 941–947, May 2020. [DOI] [PubMed] [Google Scholar]
- [33].Brochez L, Meireson A, Chevolet I, Sundahl N, Ost P, and Kruse V, “Challenging PD-L1 expressing cytotoxic T cells as a predictor for response to immunotherapy in melanoma.” [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhang L.et al. , “Massive PD-L1 and CD8 double positive TILs characterize an immunosuppressive microenvironment with high mutational burden in lung cancer,” J Immunother Cancer, vol. 9, p. 2356–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Thompson ED et al. , “Patterns of PD-L1 expression and CD8 T cell infiltration in gastric adenocarcinomas and associated immune stroma,” Gut, vol. 66, no. 5, pp. 794–801, Jan. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bakouny Z.et al. , “Integrative molecular characterization of sarcomatoid and rhabdoid renal cell carcinoma,” Nat. Commun. 2021 121, vol. 12, no. 1, pp. 1–14, Feb. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ficial M.et al. , “Expression of T-Cell exhaustion molecules and human endogenous retroviruses as predictive biomarkers for response to nivolumab in metastatic clear cell renal cell carcinoma,” Clin. Cancer Res, vol. 27, no. 5, pp. 1371–1380, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Pignon J-C et al. , “Precision Medicine and Imaging irRECIST for the Evaluation of Candidate Biomarkers of Response to Nivolumab in Metastatic Clear Cell Renal Cell Carcinoma: Analysis of a Phase II Prospective Clinical Trial.” [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available in the in this article and its Supplementary Data files or upon request from the corresponding author.