

Mining data derived from high throughput DNA or RNA sequencing approaches, including metagenomics, has led to the discovery of a multitude of uncultivated virus genome sequences1–12. These sequences improve our knowledge of the representation of the global virosphere and fuel the expansion and refinement of virus taxonomy. Inclusion of these newly discovered viral sequences into high-quality reference databases is a bottleneck to virology. For formal taxonomic classification, International Committee on Taxonomy of Viruses (ICTV) guidelines stipulate that genome sequences have to be available from a public database. However, the correct use of nomenclature and inclusion of standardized metadata fields is equally as important as the availability of the sequence data to enable the use and reuse of the data by the global research community. Here, we present standards and recommendations for the submission of virus genome sequence data to public databases for the purpose of taxonomic classification. These represent a conceptual and practical extension to the Minimum Information about an Uncultivated Virus Genome (MIUViG) standards that include standards on reporting the virus origin, genome quality, genome annotation, taxonomic classification, biogeographic distribution and host prediction13. Aspects of these standards have been reiterated in a recently published consensus view stating that viruses inferred from metagenomic sequences require strict quality control before they can be used for taxonomic assignments14. The guidelines presented here focus on the MIUViG standards on genome quality and expand on naming of sequences and submission to public databases.
ICTV coordinates the classification of viruses into 15 taxonomic ranks from species up to realm15–17 (Figure 1). It is important to note that the ICTV is not responsible for the classification of viruses below the rank of species, such as strains, variants, isolates, lineages, genotypes, or serotypes within individual species, which are instead generally classified by community consensus over time or by non-ICTV expert groups18,19. At the species rank, the ICTV requires that the complete genome sequence of a representative member or “exemplar virus” (isolated or identified by [meta]genomic sequencing) is available as an annotated sequence record in one of the International Nucleotide Sequence Database Collaboration (INSDC) member databases20. Practically, this means that the annotated genome sequence of any exemplar virus should be submitted to GenBank (National Center for Biotechnology Information [NCBI]), the European Nucleotide Archive (ENA), or the DNA Data Bank of Japan (DDBJ)21,22. This choice was guided by the long-term proven reliability, global accessibility, and visibility of INSDC databases. Due to this requirement, at least one fully sequenced virus genome per ICTV-ratified species is now readily available to the global research community and can be used as a reference in comparative genomics analyses.
Figure 1:
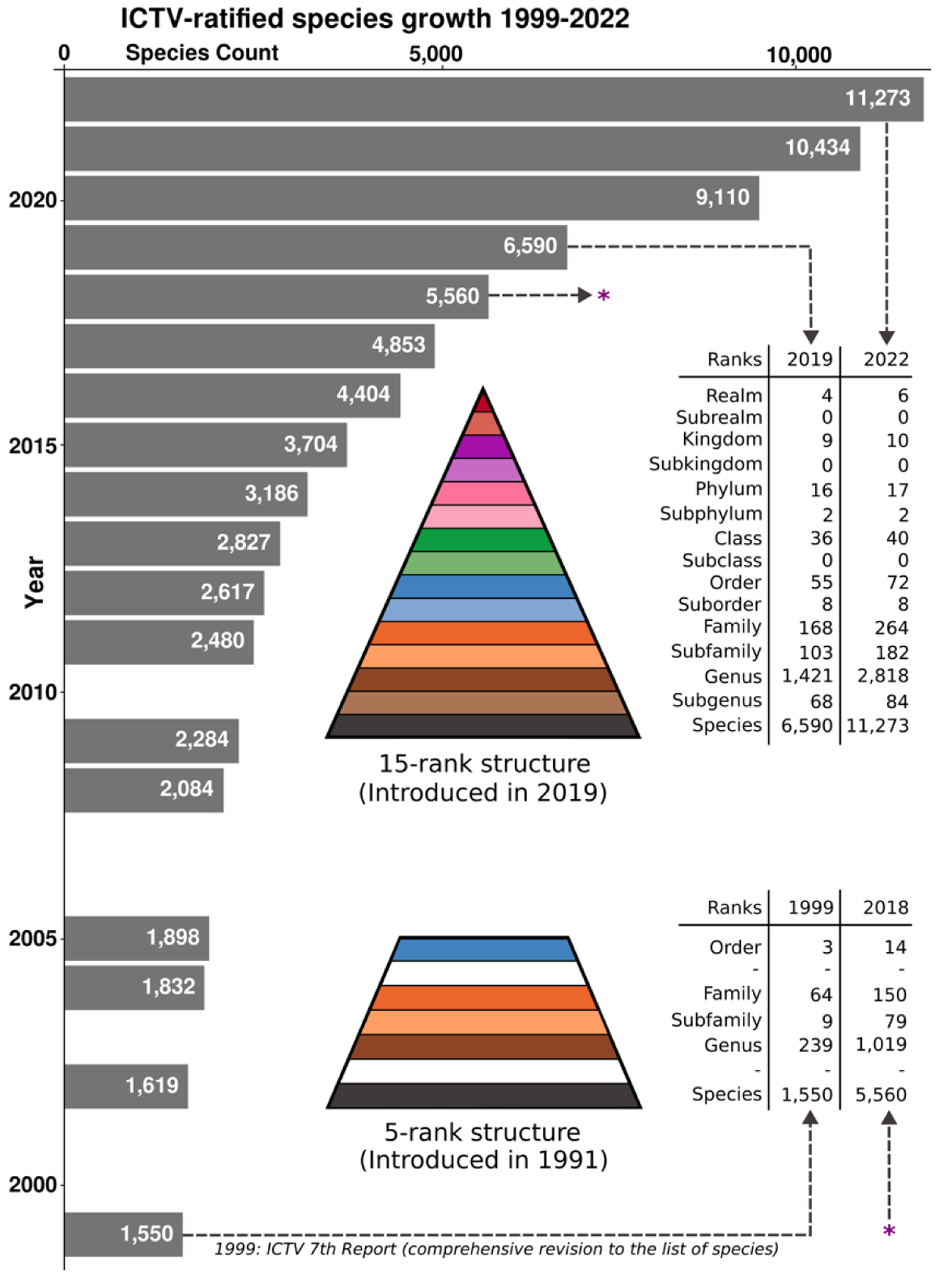
Growth in ICTV-ratified species numbers since the 7th ICTV Report in 1999. The report in 1999 was based on a five-rank structure that was introduced in 1991. The 15-rank taxonomic structure that comprised new ranks such as class, phylum, kingdom, and realm, was introduced in 2019. This figure illustrates the ongoing increase in the number of assigned taxa and the framework that allows classification of UViGs.
We note that many complete, coding-complete, and incomplete virus genome sequences and genomic fragments are available in public repositories other than INSDC (e.g., IMG/VR12, BV-BRC23, RAST24, iVirus25 or GISAID26), whereas other databases such as the Sequence Read Archive (SRA) and Whole Genome Shotgun (WGS) contain unassembled sequencing reads and unannotated or draft genomes, respectively (example guidance from NCBI: https://www.ncbi.nlm.nih.gov/sra/docs/submit/ and https://www.ncbi.nlm.nih.gov/genbank/wgs/). Such repositories provide a resource for data mining of virus genome sequences if these genomes are further assembled and annotated27,28. By mandating the deposition of annotated sequences into the INSDC databases, ICTV limits the scattering of exemplar genome sequences across databases and promotes the accessibility of the taxonomically-classified exemplar viruses. Furthermore, the close links between the ICTV and INSDC through NCBI enables better database organization and updating because taxonomy identifiers are persistent and the identifiers are updated routinely with each new ICTV taxonomy release.
A virus genome sequence may be submitted to INSDC databases using the dedicated portals of NCBI (BankIt or table2asn), ENA (Webin), or DDBJ (Nucleotide Sequence Submission System [NSSS]), choosing the submission route for individual complete genomes, or through batch submission. If the virus genome sequence was assembled from datasets that were generated by the submitter, submission follows the same protocols as submission of a virus isolate genome. The sequencing reads should be deposited in the SRA database with the metadata linked through BioProject and BioSample29, which contain biological data related to individual initiatives (projects) and descriptions of biological source materials (samples) respectively. Metadata in these databases are provided in structured ontologies including the Biological Sample Ontology, the Environment Ontology30, and the Disease Ontology. Although the availability of raw data cannot be enforced and no mandatory requirements currently exist from the ICTV, submitting such data is a best practice that will be useful for future work, including virus discovery and population genetics studies.
If a genome sequence was assembled from a public dataset, submission to an INSDC database should be done as a Third Party Annotation (TPA), a protocol that was initiated for cases where the original data does not belong to the submitter (see http://www.insdc.org/tpa.html for details and Tisza and Buck (2021)7 for an example). Even when the original dataset is in the public domain, we recommend that – whenever possible – the submitter of a newly (re-) assembled or (re-) annotated genome sequence contacts the original data depositor(s) to communicate that the data are being reused.
Practical aspects of submission to INSDC databases, with GenBank as an example, are briefly discussed here and published as a detailed standalone guide in Supplementary File 1. Practical guidelines for batch submission of Uncultivated Virus Genome (UViG) sequences are provided in Supplemental File 2.
Genome completeness and sequence quality:
To be considered valid for taxonomic classification, genome sequences should be properly assembled. Assembled genome sequences should be checked for terminal redundancy or other evidence of genome termini31, contigs should be checked for chimerism by evaluating the distribution of mapped reads and read pairs, and partially mapped or unmapped reads remaining in the dataset should be assessed and interpreted. The deposited genomes of exemplar viruses should at least be coding-complete, meaning that all open reading frames (ORFs) in the viral genome are fully sequenced32, whereas genomic non-coding terminal regions or repeat sequences may be incomplete. Incomplete genome sequences or fragments can still be used to provide context for taxonomic classification, but a coding-complete genome sequence is always required to establish a new taxon. More detailed comments and recommendations on genome sequence completeness can be found in Supplementary File 1, sections 1&3.
UViG sequence submission and naming:
GenBank requires every sequence record to have a species-rank taxonomic assignment in the <ORGANISM> field. A problem arises when a sequence belongs to a species that was not previously established. In such cases, a species-rank node is created and named according to the format “<lowest fitting taxon> sp.”, in which the <lowest fitting taxon> consists of the formal ICTV name of the lowest ranking taxon that can be confidently assigned according to the demarcation criteria and “sp.” for “species” indicates a novel species that has not yet been taxonomically established and named (Figure 2). Examples are “Sapovirus sp.”, “Herelleviridae sp.”, and “Cressdnaviricota sp.”. There is currently no ICTV-approved method to automatically assign a virus query sequence to its lowest fitting taxon because demarcation criteria for assigning sequences to taxa vary widely and should be cross-referenced with taxonomy proposals. Viral ecologists have defined operational clustering of viral sequences into viral operational taxonomic units (vOTUs) based on universal sequence similarity cutoffs13, but ICTV-ratified taxa go beyond such preliminary clusters by ensuring some robustness and providing additional information about the members of a taxon. In the GenBank record, metagenomic sequences should be given the /metagenomic, /metagenome_source=“…” and /environmental_sample source qualifiers. If further study shows that some or all the sequences in a metagenomic set have been misclassified, submitters may request an update (https://www.ncbi.nlm.nih.gov/genbank/update/) and GenBank will rename and reclassify the sequences, e.g., from “Siphoviridae sp.” to “Vequintavirinae sp.”. GenBank may also update the organism name in the record, e.g., from “Sapovirus sp.” to “Herelleviridae sp.” without submitter’s approval if ICTV sequence analysis indicates that a virus containing an “sp.” label has been misfiled.
Figure 2:
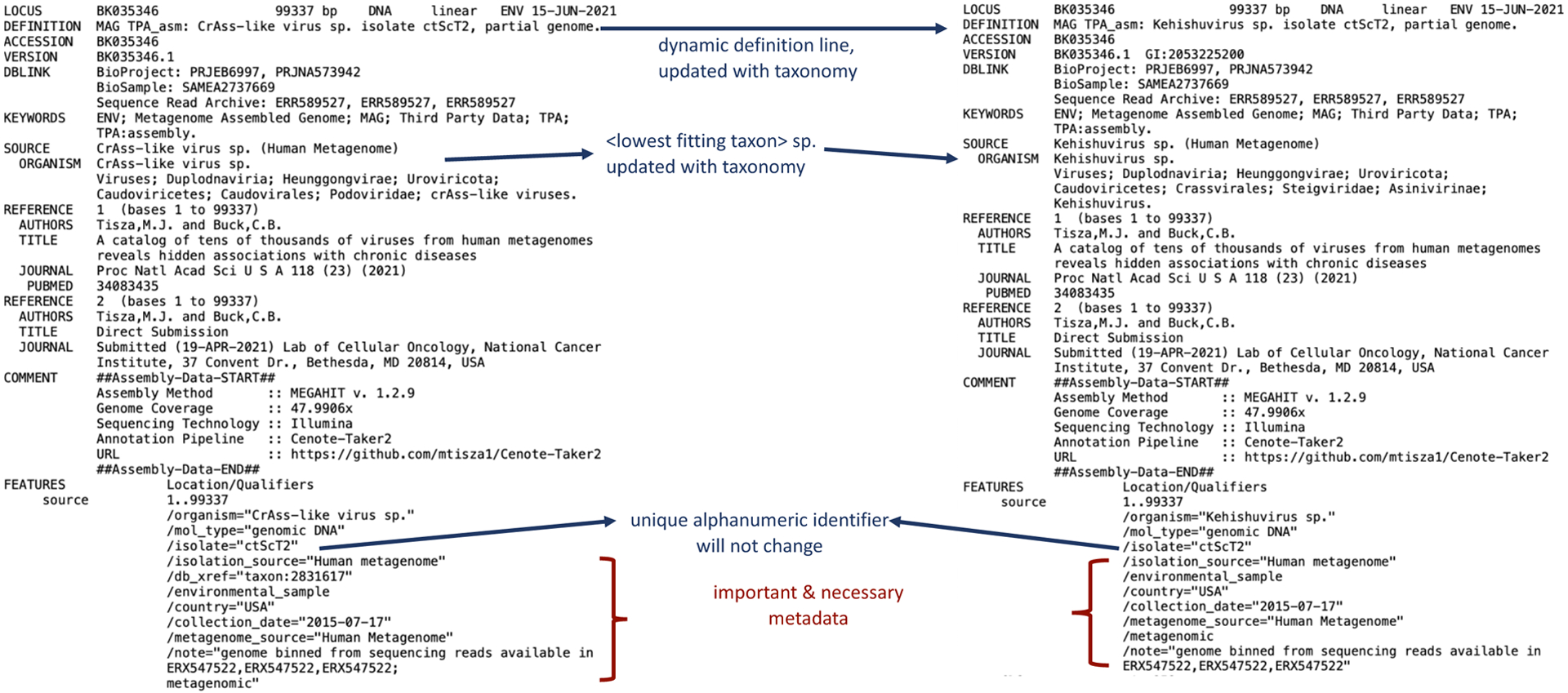
GenBank example of record BK035346. Left: as submitted with the taxonomy at the time of submission; Right: updated GenBank record after a later update to the International Committee on Taxonomy of Viruses (ICTV) taxonomy. The ORGANISM name was updated from CrAss-like virus sp. to Kehishuvirus sp. now showing the new taxonomic lineage information. The DEFINITION line was updated according to the ORGANISM change.
Using the GenBank record format as a model (Figure 2), we recommend the following:
<DEFINITION>: This field is automatically populated from the features in the record using a combination of <ORGANISM> and <ISOLATE> name.
<ORGANISM>: For UViGs, enter the “<lowest fitting taxon> sp.”. For an isolate, enter the virus name.
<ISOLATE>: Enter a unique name/code to describe this specific virus genome sequence. Ensure that this field is unique and is unlikely to be used in another study. Do not use taxonomy information in this field, because virus taxonomy is dynamic. As viruses are reclassified, taxonomy information in the <ORGANISM> field will automatically update, but isolate and genome designations are stable over time and hence should not be at odds with taxonomic names. For example, a novel virus <ISOLATE> should not be called “novel flavivirus 5”, as it may turn out not to be a flavivirus in the current or future classification.
Most databases can, at present, only accommodate the 26 letters of the Medieval Latin alphabet (i.e., ISO basic), ten numbers, and a few special characters, such as hyphens, underscores, and forward slashes. If an official virus name contains Greek letters, special characters or diacritics (e.g., Đakrông virus), feel free to enter them but be aware that most databases will convert them to the standard Latin-script letters (e.g., Dakrong virus), or may even produce an error; the correct spelling in publications should remain Đakrông virus. Underscores and hyphens may be used; forward slashes are typically included in IDs for virus pathogens with formatting requirements, such as members of Filoviridae19, Caliciviridae, and influenza A/B/C/D viruses.
Critical UViG metadata including assembly methods and sequence quality descriptors can be added as structured comments based on the Minimum Information about any (x) Sequence (MIxS) and MIUViG checklists. The most important MIUViG fields are listed in Table 1.
Do not use a “complete genome” tag for the virus isolate/genome name unless it has been experimentally verified as complete (including termini determination by, for instance, rapid amplification of complementary DNA [cDNA] ends [RACE]). Currently, the only alternative to “complete genome” in GenBank is “partial genome”, which should be used in case of UViGs. To specify the genome completeness, we suggest using the categories from the MIUViG checklist as structured comments, with information about the prediction method provided in the genome metadata (Table 1, Supplementary File 1).
Table 1:
Information to provide when submitting UViG sequences to INSDC databases.
| Information to provide | Where to add | Description | Suggested syntaxa |
|---|---|---|---|
| organism | Submission portal + MIUViG checklist structured comment | UViG: lowest ranking taxon that can be confidently assigned according to ICTV demarcation criteria. Isolated virus: virus name. |
[ <“lowest fitting taxon” sp.> | virus name ] |
| isolate | Submission portal + MIUViG checklist structured comment | Unique name or code for this sequence. Do not use taxonomic information here. | <Unique identifier> |
| Source of UViG | MIUViG checklist structured comment | Type of sample used for UViG assembly | [ metagenome (not viral targeted) | viral fraction metagenome (virome) | sequence-targeted metagenome | metatranscriptome (not viral targeted) | viral fraction RNA metagenome (RNA virome) | sequence-targeted RNA metagenome | microbial single amplified genome (SAG) | viral single amplified genome (vSAG) | isolate microbial genome | other ] |
| Assembly software | MIUViG checklist structured comment | Tool(s) used for assembly and optionally binning. Include version and parameters. | {software};{version};{parameters} |
| Assembly quality | MIUViG checklist structured comment | Assembly quality in categories as per the MIUViG criteria. Finished: Single, validated, contiguous sequence per replicon without gaps or ambiguities, with extensive manual review and annotation. High-quality draft genome: One or multiple fragments, totalling ≥ 90% of the expected genome or replicon sequence or predicted complete. Genome fragment(s): One or multiple fragments, totalling < 90% of the expected genome or replicon sequence, or for which no genome length could be estimated. |
[ Finished genome | High-quality draft genome | Genome fragment(s) ] |
| Completeness score | MIUViG checklist structured comment | (Optional) Estimated completeness of the UViG in percentage. | {quality};{percentage} |
| Completeness approach | MIUViG checklist structured comment | (Optional) Approach used to estimate completeness, such as identification of terminal repeats or presence of all CDS | {text} |
| Virus identification software | MIUViG checklist structured comment | Tool(s) used for identification of sequence as virus. Include versions and parameters. | {software};{version};{parameters} |
| Predicted genome type | MIUViG checklist structured comment | Type of genome predicted for the UViG. | [ DNA | dsDNA | ssDNA | RNA | dsRNA | ssRNA | ssRNA (+) | ssRNA (−) | mixed | uncharacterized ] |
entries between []: choose one of the listed descriptors; entries between <>: fill in the UViG or virus information for this record; entries between {}: enter data for your methods used.
Providing appropriate metadata:
In INSDC databases, general sequence metadata such as the origin and source of isolation are stored as source modifiers (see more detailed description in Supplementary File 1, section 4). Using the principles of findability, accessibility, interoperability, and reusability (FAIR) for data stewardship33, all metadata fields should be provided as structured ontology terms (e.g., The Environment Ontology30, see also Supplementary File 1). The minimum recommended source modifiers to be used are <ISOLATION SOURCE>, <COLLECTION DATE>, and <COUNTRY>, with <SEGMENT> reserved for viruses with segmented genomes. Additional information specific to UViGs should be provided by submitting a MIUViG sequence13 metadata checklist34,35 for each UViG sequence and connecting the resulting BioSample package to the UViG genome sequence record by linking the BioSample ID to the GenBank submission. The definition, format, and expected values for each field in the MIUViG sequence checklist are available on the Genomic Standards Consortium (GSC) website. We refer to the GenBank Nucleotide record OP880254 as an example of how to implement the MIUViG standards (https://www.ncbi.nlm.nih.gov/nuccore/OP880254.1).
Features:
Sequence annotations, such as ORFs, introns, encoded proteins, and regulatory elements, are stored as features. Feature annotations should be provided for all UViG sequences that are to be used as exemplar genomes to represent new species. At a minimum, the coding sequences should be specified, including functional annotations based on homology searches, phylogenetic analysis, and conserved protein domains, which should be labelled “putative” until experimentally validated.
The availability of complete and consistently annotated records is crucial for the use and reuse of virus sequences and advancing the virology research field. We aim to assist and support the virology community in its expanding use of (meta-) genomic data and the associated taxonomic efforts by promoting the use of this set of standards. While our recommendations are primarily aimed at viruses inferred from metagenome data (UViGs), they are universally applicable to all viruses. Our capacity to generate sequences still outpaces our ability to classify them, so submitting new virus data according to these outlined guidelines will greatly facilitate their findability, accessibility, and reusability as ICTV strives to build a robust virus taxonomy.
Supplementary Material
Acknowledgements
The authors thank Anya Crane (Integrated Research Facility at Fort Detrick/National Institute of Allergy and Infectious Diseases/National Institutes of Health, Fort Detrick, Frederick, MD, USA) for critically editing the manuscript. The views and conclusions contained in this document are those of the authors and should not be interpreted as necessarily representing the official policies, either expressed or implied, of the U.S. Department of Health and Human Services or of the institutions and companies affiliated with the authors, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
E.M.A. gratefully acknowledges the support of the Biotechnology and Biological Sciences Research Council (BBSRC); this research was funded by the BBSRC Institute Strategic Program Gut Microbes and Health BB/R012490/1 and its constituent project(s) BBS/E/F/000PR10353 and BBS/E/F/000PR10356. The work conducted by the U.S. Department of Energy Joint Genome Institute (S.R.) was supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. Work by J.R.B. and I.K.M. was supported by the National Center for Biotechnology Information of the National Library of Medicine (NLM), National Institutes of Health. This work was supported in part through Laulima Government Solutions, LLC, prime contract with the U.S. National Institute of Allergy and Infectious Diseases (NIAID) under Contract No. HHSN272201800013C. J.H.K. performed this work as an employee of Tunnell Government Services (TGS), a subcontractor of Laulima Government Solutions, LLC, under Contract No. HHSN272201800013C. MBS was supported by the US National Science Foundation Award #1759874. R.A.E. was supported by the National Institute of Diabetes And Digestive and Kidney Diseases of the National Institutes of Health under Award Number RC2DK116713 and by the Australian Research Council under Award Number DP220102915. C.L. was supported by a Postdoctoral Mandate from KU Leuven (PDMt2/21/038) and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy - EXC 2051 - Project-ID 390713860. E.J.L. was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number U24AI162625. P.S. was supported by a Wellcome Trust Biomedical Resource grant (WT108418AIA). S.S. acknowledges support from the Mississippi Agricultural and Forestry Experiment Station (MAFES), USDA-ARS project 58-6066-9-033 and the National Institute of Food and Agriculture, U.S. Department of Agriculture, Hatch Project, under Accession Number 1021494. B.E.D. was supported by the European Research Council (ERC) Consolidator Grant 865694: DiversiPHI, the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy - EXC 2051 - Project-ID 390713860, the Alexander von Humboldt Foundation in the context of an Alexander von Humboldt-Professorship founded by German Federal Ministry of Education and Research, and the European Union’s Horizon 2020 research and innovation program, under the Marie Skłodowska-Curie Actions Innovative Training Networks grant agreement no. 955974 (VIROINF).
Footnotes
The authors do not declare any conflicts of interest.
References
- 1.Callanan J et al. Expansion of known ssRNA phage genomes: From tens to over a thousand. Sci. Adv 6, eaay5981 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gregory AC et al. The Gut Virome Database reveals age-dependent patterns of virome diversity in the human gut. Cell Host Microbe 28, 724–740.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zayed AA et al. Cryptic and abundant marine viruses at the evolutionary origins of Earth’s RNA virome. Science 376, 156–162 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hillary LS, Adriaenssens EM, Jones DL & McDonald JE RNA-viromics reveals diverse communities of soil RNA viruses with the potential to affect grassland ecosystems across multiple trophic levels. ISME Commun. 2, 34 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roux S, Krupovic M, Poulet A, Debroas D & Enault F Evolution and diversity of the Microviridae viral family through a collection of 81 new complete genomes assembled from virome reads. PLoS One 7, e40418 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krishnamurthy SR, Janowski AB, Zhao G, Barouch D & Wang D Hyperexpansion of RNA Bacteriophage Diversity. PLoS Biol. 14, e1002409 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tisza MJ & Buck CB A catalog of tens of thousands of viruses from human metagenomes reveals hidden associations with chronic diseases. Proc. Natl. Acad. Sci 118, e2023202118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gregory AC et al. Marine DNA viral macro- and microdiversity from pole to pole. Cell 177, 1109–1123.e14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Camarillo-Guerrero LF, Almeida A, Rangel-Pineros G, Finn RD & Lawley TD Massive expansion of human gut bacteriophage diversity. Cell 184, 1098–1109.e9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nayfach S et al. Metagenomic compendium of 189,680 DNA viruses from the human gut microbiome. Nat. Microbiol 6, 960–970 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Emerson JB et al. Host-linked soil viral ecology along a permafrost thaw gradient. Nat. Microbiol 3, 870–880 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roux S et al. IMG/VR v3: an integrated ecological and evolutionary framework for interrogating genomes of uncultivated viruses. Nucleic Acids Res. 49, D764–D775 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roux S et al. Minimum Information about an Uncultivated Virus Genome (MIUViG). Nat. Biotechnol 37, 29–37 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simmonds P et al. Four principles to establish a universal virus taxonomy. PLoS Biol. 21, e3001922 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koonin EV et al. Global organization and proposed megataxonomy of the virus world. Microbiol. Mol. Biol. Rev 84, e00061–19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gorbalenya AE et al. The new scope of virus taxonomy: partitioning the virosphere into 15 hierarchical ranks. Nat. Microbiol 5, 668–674 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simmonds P et al. Consensus statement: Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol 15, 161–168 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Rambaut A et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol 5, 1403–1407 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuhn JH et al. Virus nomenclature below the species level: A standardized nomenclature for natural variants of viruses assigned to the family Filoviridae. Arch. Virol 158, 301–311 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karsch-Mizrachi I, Nakamura Y & Cochrane G The International Nucleotide Sequence Database Collaboration. Nucleic Acids Res. 40, D33–D37 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salzberg SL Reminder to deposit DNA sequences. Nature 533, 179–179 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Blaxter M et al. Reminder to deposit DNA sequences. Science 352, 780–780 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Olson RD et al. Introducing the Bacterial and Viral Bioinformatics Resource Center (BV-BRC): a resource combining PATRIC, IRD and ViPR. Nucleic Acids Res. 51, D678–D689 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aziz RK et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9, 75 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bolduc B et al. iVirus 2.0: Cyberinfrastructure-supported tools and data to power DNA virus ecology. ISME Commun. 1, 77 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shu Y & McCauley J GISAID: Global initiative on sharing all influenza data – from vision to reality. Eurosurveillance 22, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mokili JL, Rohwer F & Dutilh BE Metagenomics and future perspectives in virus discovery. Curr. Opin. Virol 2, 63–77 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paez-Espino D et al. Uncovering Earth’s virome. Nature 536, 425–430 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Barrett T et al. BioProject and BioSample databases at NCBI: Facilitating capture and organization of metadata. Nucleic Acids Res. 40, 57–63 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buttigieg PL et al. The environment ontology in 2016: bridging domains with increased scope, semantic density, and interoperation. J. Biomed. Semantics 7, 57 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garneau JR et al. High-throughput identification of viral termini and packaging mechanisms in virome datasets using PhageTermVirome. Sci. Rep 11, 18319 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ladner JT et al. Standards for sequencing viral genomes in the era of high-throughput sequencing. MBio 5, e01360–14–e01360–14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilkinson MD et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 3, 160018 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.NCBI. BioSample Types and Attributes checklists. Available at: https://submit.ncbi.nlm.nih.gov/biosample/template/. (Accessed: 24th May 2023)
- 35.Genomics Standards Consortium MIUVIG checklist. Available at: https://genomicsstandardsconsortium.github.io/mixs/MIUVIG/. (Accessed: 24th May 2023)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.