

Abstract
In head and neck squamous cell carcinoma (HNSCC), a significant proportion of tumors have inactivating mutations in the histone methyltransferase NSD1. In these tumors, NSD1 inactivation is a driver of T cell exclusion from the tumor microenvironment (TME). A better understanding of the NSD1-mediated mechanism regulating infiltration of T cells into the TME could help identify approaches to overcome immununosuppression. Here, we demonstrated that NSD1 inactivation results in lower levels of H3K36 di-methylation and higher levels of H3K27 tri-methylation, the latter being a known repressive histone mark enriched on the promoters of key T cell chemokines CXCL9 and CXCL10. HNSCC with NSD1 mutations had lower levels of these chemokines and lacked responses to PD-1 immune checkpoint blockade. Inhibition of KDM2A, the primary lysine demethylase that is selective for H3K36, reversed the altered histone marks induced by NSD1 loss and restored T cell infiltration into the TME. Importantly, KDM2A suppression decreased growth of NSD1-deficient tumors in immunocompetent, but not in immunodeficient, mice. Together, these data indicate that KDM2A is an immunotherapeutic target for overcoming immune exclusion in HNSCC.
Keywords: HNSCC, NSD1, CXCL9, CXCL10, KDM2A, H3K36, H3K27, immune-cold phenotype, immunotherapy
INTRODUCTION
Head and neck squamous cell carcinoma (HNSCC), the 6th most common malignancy worldwide, is a devastating malignancy of the mucosal lining of the upper aerodigestive tract. The aggressive nature of the cancer, as well as the treatment itself, have significant impact on critical functions like speech, breathing, and swallowing. Despite improved multi-modal treatment strategies, patients with advanced HNSCC still suffer from a low survival rate (1).
Recently, immunotherapy approaches with checkpoint blocking antibodies, such as pembrolizumab and nivolumab, have shown impressive responses in recurrent/metastatic HNSCC, though only in a minority of patients (2,3). In general, the overall response rate to immune checkpoint blockade is limited, and in HNSCC, it is <20% (4–6). Numerous strategies to enhance endogenous and synthetic immune-mediated rejection of tumors are under intense investigation; however, all face significant challenges pending better understanding of the interface between tumor cells and the immune system within the tumor microenvironment (7). A particular challenge for immune checkpoint blockade approaches has been the development of ways to overcome tumor microenvironments that lack T cell infiltration – what is sometimes called an immune-cold phenotype (8,9).
Previous analyses of multiomics data from The Cancer Genome Atlas (TCGA) revealed a distinct immune-cold subset of HNSCC tumors with a particularly low level of tumor associated T cells (10). This human papilloma virus (HPV)-negative subset of HNSCC was also unique in that 57% of these tumors had inactivating mutations in the histone methyl transferase NSD1 (nuclear receptor binding SET domain protein 1). Further, all HNSCC tumors in the TCGA with NSD1 inactivating mutations have multiomics profiles placing them in the immune-cold subset (11,12). These data, in addition to recent mouse studies showing that NSD1 inactivation results in an immune-cold phenotype (13) support a role for NSD1 in modulating T cell infiltration in HNSCC. Indeed, we previously showed that inhibition of NSD1 expression confers the exclusion of T cells from the tumor microenvironment in vivo (10); however, the mechanism underlying this is not understood.
NSD1 is a SET-domain containing histone methyltransferase that catalyzes di-methylation of histone 3 specifically at lysine 36 (H3K36me2). Synthesis of the key chromatin silencing modification tri-methylation of H3K27 (H3K27me3) by the Polycomb repressive complex (PRC2) is directly antagonized by H3K36 di- and tri-methylation (14–17). Indeed, recent studies in murine embryonic stem cells demonstrated that reduction in global levels of H3K36me2 due to NSD1 depletion increased H3K27me3 across the epigenome (15). Because H3K27me3 is enriched on the repressed promoters of the key T cell recruiting chemokines CXCL9 an CXCL10 (18), here, we test the hypothesis that loss of NSD1 function – and the subsequent effects on H3K36 and H3K27 methylation states – impacts CXCL9 and CXCL10 expression in the tumor microenvironment. Importantly, our studies demonstrate the potential of targeting the opposing H3K36me2-specific lysine demethylase, KDM2A, as a therapeutic strategy to reverse the effects of NSD1 inactivation in HNSCC and enhance T cell infiltration.
MATERIALS and METHODS
Cell lines
The FaDu human HNSCC cell line was obtained from ATCC; early passage cells were used. The MOC1 cell line was provided by Dr. Ravindra Uppaluri, who developed the cell line from murine oral squamous cell carcinomas induced by topical 7,12-dimethylbenz(a) anthracene (DMBA) administration; early passage cells were used. Cells were cultured in complete DMEM/F12 medium containing 10% fetal bovine serum (FBS), 1% Non-Essential Amino Acid (NEAA) and 1% penicillin and streptomycin. Cells were maintained at 37 deg C in a humidified atmosphere containing 5% CO2. The 293GP cell line (RRID:CVCL_E072) was a kind gift from Dr. Crystal Mackall (Department of Pediatrics - Hematology & Oncology, Stanford University). The Lenti-X 293T cell line was purchased from Takara Bio USA, Inc (RRID:CVCL_0063). Cells were cultured in complete DMEM medium containing 10% FBS, 1% penicillin and streptomycin. Cells were maintained at 37 deg C in a humidified atmosphere containing 5% CO2.
T cells
Our use of human donor blood was approved by the Institutional Review Board at Stanford University. Leukoreduction system (LRS) chambers were obtained from the Stanford Blood Center. T cells were enriched using the RosetteSep™ Human T Cell Enrichment Cocktail (STEMCELL Technologies) followed by centrifugation on Ficoll-Paque™ Premium (GE Healthcare). T cells were cultured in complete RPMI/1640 medium containing 10% FBS, 1% penicillin and streptomycin, 100 U/mL IL-2 and 1% HEPES at 37 deg C in a humidified atmosphere containing 5% CO2. T cells were activated by anti-CD3 and anti-CD28 antibody (Biolegend) according to the manufacturer’s instructions 3 days before use. Specific information about the antibodies is in Supp. Table 1. Plates were pre-coated with 50 uL of 10 ug/mL anti-CD3 and anti-CD28 antibody in each microwell of the 96-well plate at 4 deg C overnight. The wells were washed, and 200 uL of 1–2×106/mL T cells were added to each well. The cells were incubated at 37 deg C in a humidified atmosphere containing 5% CO2 for 3 days before use.
Lentiviral production and transduction
The Lenti-X 293T cell line was used to package and produce the lentiviral particles. Briefly, the cells were transfected with the packaging plasmid pCMV-dR8.2 (RRID:Addgene_8455), the envelope plasmid pCMV-VSV-G (RRID:Addgene_8454), and the transfer plasmids containing the shRNA (Supp. Table 2) or the transgene, using Lipofectamine® 2000 (ThermoFisher) according to the manufacturer’s instructions. Supernatants were collected 48 to 72 hours post-transfection and used to transduce the target cells in 6-well plates at a concentration of 3×105 cells per well with 8 ug/mL polybrene. The medium was changed 24 hours later.
Retroviral production and transduction
The 293GP cell line was used to produce the retroviral particles for CAR transduction. The 293GP cells were transfected with the envelope plasmid RD114 and the plasmids encoding CD19 CAR (a kind gift from Dr. Crystal Mackall)(19), using Lipofectamine® 2000 according to the manufacturer’s instructions. The supernatants were collected 48 to 72 hours post-transfection and then stored at −80 deg C. Retroviral supernatants were diluted 1:1 in T cell culture media and applied to non-treated 6-well plates (Corning) coated with retronectin (Takara). Plates were centrifuged at 3200 rpm for 2–3 hours at 32 deg C. Vector-containing supernatants were removed, and activated T cells (1×106 cells/well) were added to the plates. The plates were centrifuged at 1000 g for 45 min at 32 deg C and then incubated at 37 deg C.
Mice
B6.129S7-Rag1tm1Mom/J mice (Rag1 KO mice) (RRID:IMSR_JAX:002216) and C57BL/6J (B6 mice) (RRID:IMSR_JAX:000664) were obtained from Jackson Laboratory. The mice were housed in laminar flow cabinets under specific pathogen-free conditions and fed ad libitum. All procedures were performed in accordance with protocols approved by an Institutional Animal Care and Use Committee at Stanford University. For in vivo experiments with murine HNSCC cell lines, 2×106 cells were injected subcutaneously in the flanks of the Rag1 KO mice or wild type B6 mice. Tumor volume (mm3) was determined by caliper measurements performed every two to three days and calculated by using the following formula: volume=length×width2×0.5. The tumors were harvested and processed for TIL analysis.
Tumor dissociation and tumor-infiltrating lymphocyte analysis
Tumor tissue was digested as follows. The tissue was minced, transferred into gentleMACS™ C-tubes with digestion solution (DMEM-F12+1%FBS, 1% Pen strip, 25mM HEPES and 10% collagenase/Hyaluronidase), and dissociated. The dissociated tissue was incubated while rotating at 37 deg C for 1 hour. After incubation, the suspension was filtered through a 40 μM filter. If the cell pellet was heavily contaminated with red blood cells, a brief ACK lysis was performed. The cells were washed and stained with antibodies to CD3 and CD45 (Biolegend). See Supp. Table 1. Data were acquired on a BD LSRFortessa or BD FACSAria II. Events collected were analyzed using FlowJo Version 10.5.0 software (RRID:SCR_008520).
Immunofluorescence (IF) staining and microscopy of tumor tissue
OCT-embedded tissue sample sections (3–5 μm) were dried for 30 minutes at room temperature and then in ice-cold acetone for 5–10 min. Washed sections were then permeabilized in 0.1% Triton X-100, blocked in 5% BSA for 60 min at room temperature. The slides were then stained with anti-CD3 antibody (Biolegend), anti-CXCL9 antibody (Invitrogen), or anti-CXCL10 antibody (Bioss) at 4 deg C overnight. Specific information about the antibodies is in Supp. Table 1. Subsequently, the slides were incubated at room temperature for 1 hour with a secondary antibody (Invitrogen), followed by DAPI staining for 20 minutes. The samples were mounted on glass slides and analyzed by LSM700 confocal microscopy (Zeiss).
Quantitative RT-PCR
RNA was extracted with the RNeasy Mini Kit (Qiagen), and cDNA synthesis was performed using Maxima First Strand cDNA Synthesis Kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. The relative gene expression was analyzed using the Luminaris Color Probe High ROX qPCR Master Mix (Thermo). Relative gene expression was obtained by the ΔΔCt method (20) after normalization to HPRT1. Data is available in Supp. Data 1.
Western blot
Histone protein was extracted with the Histone Extraction Kit (Abcam), according to the manufacturer’s protocol. The protein samples were denatured and separated by Novex™ WedgeWell™ 4–12% Tris-Glycine Mini Gels (Thermo Fisher Scientific) and transferred onto 0.2 μm PVDF Pre-cut Blotting Membranes (Thermo Fisher Scientific). After blocking with 5% non-fat milk at room temperature for 2 hours, the membranes were incubated with primary antibodies to H3K36me2 or H3K27me3 or total H3 at 4 deg C overnight. Antibody details are in Supp. Table 1. Subsequently, the membranes were incubated with secondary antibodies for 1 hour at room temperature. Finally, protein signals were detected by Pierce™ ECL Western Blotting Substrate (Thermo Fisher Scientific) and visualized after exposure to the Hyperfilm™ ECL™ (GE Healthcare). The relative optical density ratio was calculated with the Image J software (RRID:SCR_003070) by comparison to H3. Original Western blot gel images and Image J data are provided in Supp. Data 2 and Supp. Data 3.
Spheroid T cell infiltration assay
CD19 (truncated) expressing FaDu cells (7×103) were resuspended in 1 mL of liquefied Matrigel on ice. Aliquots of 30 μL of resuspended cells in Matrigel were plated as droplets in the center of wells of a 24-well plate. Plates were incubated at 37 deg C in 5% CO2 for 15 minutes to solidify Matrigel domes. Domes were overlaid with 1 mL of EN medium and incubated at 37 deg C in a 5% CO2 humidified incubator. EN medium consisted of DMEM/F12 medium containing 10 mM Nicotinomide, 1 mM N-acethylcysteine, 1X B-27™ Supplement, 1X Antibiotic-Antimycoti, 50 ng/mL EGF, and 100 ng/mL Noggin. Culture medium was changed twice a week. After 2 weeks, 2×105 CD19 CAR T cells were added to each well. For the KDM2A inhibitor treated group, 1.5 μM daminozide was added to the medium the day before adding the T cells. After 3 days of co-culture, the spheroid Matrigel domes were harvested and analyzed by microscopy. Domes were fixed with 4% paraformaldehyde for 30 minutes at room temperature. The domes were washed with PBS three times, and the organoids were then incubated in 0.5% Triton X-100 + 5% BSA in PBS for 2 hours at room temperature. The permeabilized spheroids were then incubated with antibodies at 4 deg C overnight, washed, and then incubated with DAPI for 20 mins. The stained samples were mounted on glass slides and analyzed by LSM700 confocal microscopy (Zeiss). Cell counts are available in Supp. Data 4.
Enzyme-linked immunosorbent assay
CXCL9 and CXCL10 protein levels in the conditioned culture medium were measured by enzyme-linked immunosorbent assay (ELISA), using the Human CXCL9 (MIG) Mini ABTS ELISA Development Kit (Peprotech) and the Human CXCL10 (IP-10) ELISA MAX™(BioLegend), following the manufacturer’s instructions. Briefly, plates were pre-coated with anti-CXCL9 or anti-CXCL10 antibody. Standards and cell culture supernatants were diluted in sample diluent buffer and incubated for 2 hours at room temperature. Detection antibodies were diluted in antibody diluent buffer and incubated for 2 hours at room temperature. After washing steps, HRP-conjugate was diluted and incubated for 30 minutes at room temperature. After washing steps, ABTS liquid substrate was added to each well, and absorbance was measured at a wavelength of 405 nm with correction wavelength of 650 nm using a microplate (ELISA) reader (SpectraMax M3, Molecular Devices). An eight-point standard curve was used to calculate the concentration (pg/mL) of CXCL9 and CXCL10 in the samples.
TCGA and cBioPortal
The cBioPortal (https://www.cbioportal.org) (RRID:SCR_014555) was used to analyze the HPV-negative Head and Neck Squamous Cell Carcinoma (TCGA, PanCancer Atlas) dataset (21,22).
CXCL9 and CXCL10 in human HNSCC datasets
RNAseq data from a neoadjuvant pembrolizumab HNSCC cohort (23) was examined for CXCL9 and CXCL10 transcripts. The scRNA-Seq dataset, which was generated using Smart-Seq2 technology as part of a study reported by (24), was accessed from Gene Expression Omnibus (RRID:SCR_005012) (Accession number: GSE103322) as a preprocessed series matrix file. The dataset was subsequently analyzed using Seurat (25): The series matrix was loaded into a Seurat object and was filtered to exclude poor quality or dying cells by removing cells with a mitochondrial genome fraction of 0.4 or greater as well as those with a unique feature count of fewer than 200 genes. Potential doublets were excluded by removing cells with a unique feature count of greater than 4000. Integration was then performed by splitting the dataset into separate Seurat objects, with each object containing all the cells that derived from one sample. Gene expression counts for each cell were normalized using regularized negative binomial regression, and variable genes (N=2000) were found for each sample using the ‘vst’ method. Samples were then integrated into a single gene expression object by finding integration anchors using the ‘FindIntegrationAnchors’ and ‘IntegrateData’ commands. The combined genes were then scaled and centered using linear models. This integration approach resulted in clustering of cells by cell type and cell cycle stage rather than by sample. Cell type labels that were previously assigned by Puram et al. were accessed from the GEO meta-data. Expression of CXCL9 and CXCL10 was analyzed within samples that had been profiled using whole exome sequencing, such that the NSD1 mutation status was known. NSD1 somatic mutation calls were accessed from the Puram et al report. For each gene (CXCL9 and CXCL10), Fisher’s extract test was used to test for differences the number of malignant cells that expressed the gene (i.e., had a normalized count value greater than zero) between NSD1 mutated and NSD1 wild type HNSCC samples. For primary and metastatic tumors independently, comparisons were made between malignant cells of the NSD1 HNSCC versus malignant cells of each NSD1 wild-type HNSCC separately.
ChIP-seq analysis
The H3K36me2 and H3K27me3 ChIP-seq data were downloaded from GSE149670 (26). ChIP-seq clean reads were aligned to the human genome (GRCh38) using Bowtie2 (RRID:SCR_016368) (27). Then, aligned reads were filtered to remove PCR duplicates and reads from chromosomes 1–21, X and Y are retained. ChIP-seq signal profiles were converted to BigWig tracks using deeptools (28), then visualized by Integrated Genome Viewer (IGV).
Statistical analysis
Data were expressed as mean ± standard error of mean (SEM) and analyzed by t-test or one way ANOVA. Statistical analysis was performed with GraphPad Prism software (RRID:SCR_002798). Values of p<0.05 were considered to be statistically significant.
Data availability
Data generated in this study are available within the article and its supplementary data files and are available upon request from the corresponding author. Publicly available data generated by others and analyzed in this study were obtained from the database of Genotypes and Phenotypes (dbGaP) at phs001623.v2.p1 and the Gene Expression Omnibus (GEO) at GSE103322; GSE149670; and GSE131967.
RESULTS
Inactivation of NSD1 results in downregulated expression of CXCL9 and CXCL10 in HNSCC
Analysis of data from a multicenter, phase II, window-of-opportunity trial (NCT02296684) (23), in which neoadjuvant pembrolizumab was administered to patients with locally advanced, HPV-unrelated HNSCC two to three weeks prior to definitive surgical resection, revealed a significant correlation between pathologic tissue response and expression of the key T cell-recruiting chemokines CXCL9 and CXCL10 (Fig. 1A, Supp. Fig. 1). None of the tumors with low CXCL9 or CXCL10 expression exhibited response to pembrolizumab. Two of the patients in the study had tumors with inactivating mutations in NSD1. Both were non-responders and were among those with the lowest expression of CXCL9 and CXCL10. Interestingly, histologic examination of the NSD1-mutant tumors demonstrated a microenvironment that was devoid of T cells at baseline (Fig. 1B). Notably, the T cells appeared to be excluded from the tumor microenvironment and confined to the stromal compartment between the tumor cell nests.
Figure 1: NSD1 inactivation downregulates the expression of CXCL9 and CXCL10 in HNSCC.
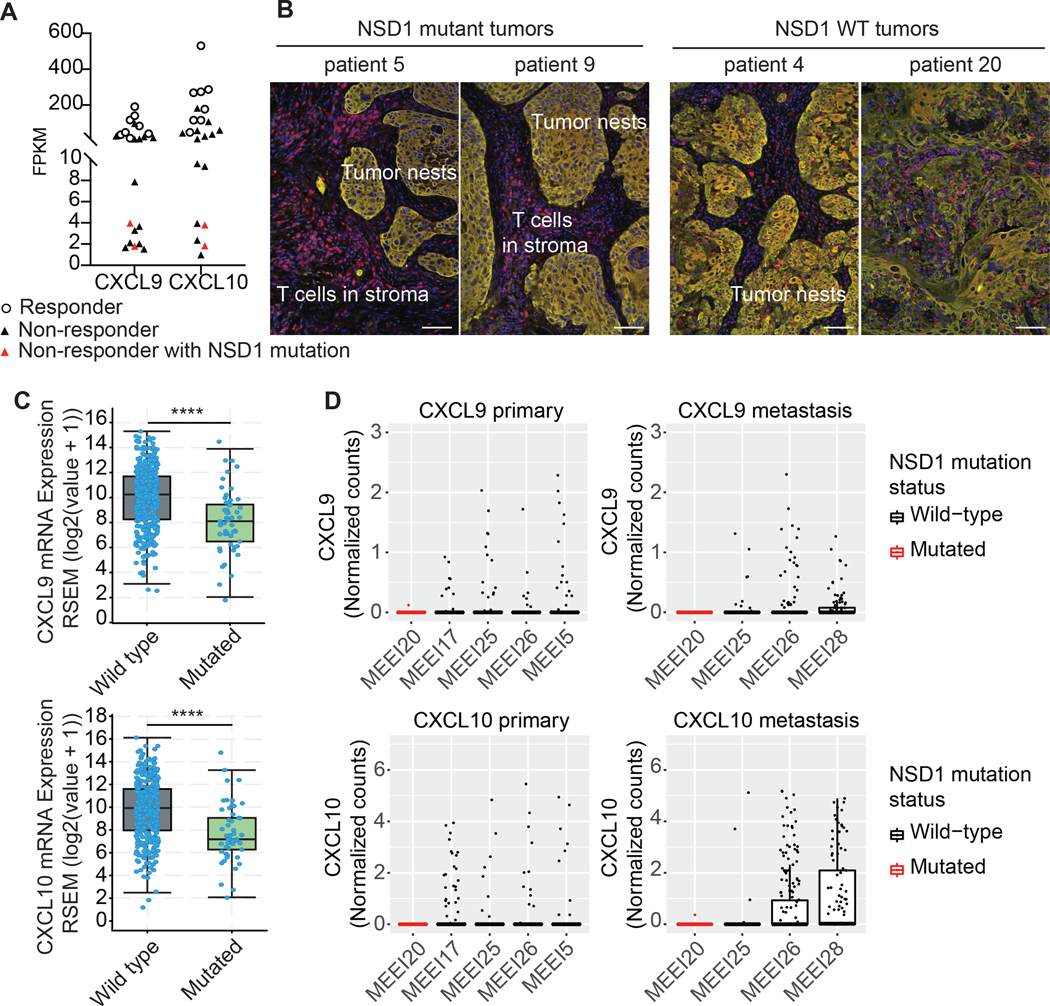
RNA expression and genomic sequencing data from a multicenter, phase II, window-of-opportunity trial (NCT02296684) (23) was examined for CXCL9 and CXCL10 expression and NSD1 mutations. In this trial, neoadjuvant pembrolizumab was administered to patients with locally advanced, resectable, HPV-unrelated HNSCC two to three weeks prior to definitive surgical resection. (A) Comparison of CXCL9 and CXCL10 baseline expression to pathologic tumor response (pTR), as defined in (23), to the anti-PD-1 antibody pembrolizumab. 〇 represent responders. ▲ represent non-responders with wild-type NSD1. ▲ represent non-responders with mutated NSD1. (B) Tumor histology from the two patients in the trial that had tumors with NSD1 mutations. Histology of NSD wild-type tumors is also shown. Red = CD3 staining of T cells. Yellow = pancytokeratin staining of the tumor cells. Blue = DAPI. White scale bars represent ~100 μm. (C) Comparison of the CXCL9 and CXCL10 expression in NSD1 wild-type and mutated HPV-negative HNSCC tumors in the Cancer Genome Atlas. ****p<0.0001. (D) Expression of CXCL9 and CXCL10 in malignant cells of NSD1 wild-type and mutated primary and metastatic HNSCCs. Box plots show expression (Normalized counts) of CXCL9 and CXCL10 in malignant single cells (points) of HNSCC samples, including primary and metastatic (lymph node metastasis) tumors. Black and red boxes and points represent NSD1 wild-type and NSD1 mutated HNSCCs, respectively. Single cell RNA-Sequencing data was accessed from a previous study by Puram et al. (24).
To investigate further the relationship between NSD1 mutations and CXCL9/CXCL10 expression, we examined previously reported human HNSCC datasets. In The Cancer Genome Atlas (TCGA), HNSCC tumors with NSD1 mutations had significantly lower expression of CXCL9/CXCL10 (Fig. 1C). In fact, of all T cell related chemokines/cytokines (29) that had differences in expression, CXCL9 and CXCL10 had the most significantly altered expression levels in an NSD1 mutant background (Supp. Fig. 2) and are well known to have an important role in the recruitment of T cells into the tumor microenvironment. Similarly, analysis of CXCL9 and CXCL10 expression in a previously published HNSCC single cell RNA-sequencing (scRNA-seq) dataset (24) also revealed essentially absent expression of these chemokines in the context of NSD1 mutations (Fig. 1D). This dataset included scRNA-seq profiles for a primary and a metastatic (lymph node metastasis) tumor of an HPV-negative patient that had a somatic point mutation (nonsense) in NSD1 (detected by whole exome sequencing), as well as primary (N=4) and metastatic (N=3) tumors of NSD1 wild-type HNSCCs (i.e., tumors in which NSD1 mutations were not detected). This revealed that CXCL9 and CXCL10 were expressed (i.e., detected) within a subset of malignant cells of each primary or metastatic NSD1 wild-type HNSCC, but were undetected or detected in only one malignant cell of the NSD1 mutated primary and metastatic HNSCCs. Indeed, for both primary and metastatic HNSCCs, the frequency of cells with detectable CXCL9 or CXCL10 expression was significantly lower within malignant cells of the NSD1 mutated tumor compared with malignant cells of each NSD1 wild-type counterpart. Finally, because NSD1-mediated catalysis of H3K36 di-methylation directly antagonizes H3K27 tri-methylation by PRC2 (14–17), we hypothesized that NSD1 inactivation would correlate with enrichment of H3K27me3 on CXCL9 and CXCL10 loci, a known repressive mark for these genes (18), and indeed, analysis of published ChIP-seq data from HNSCC cell lines (26) revealed an increase in H3K27me3 and a reduction in H3K36me2 on CXCL9 and CXCL10 loci when NSD1 was knocked out (Supp. Fig. 3). Further, of known H3K27me3-regulated genes (30), CXCL9 and CXCL10 were among those with the most significantly altered expression in the NSD1 mutant subset of HNSCC tumors in the TCGA (Supp. Fig. 4). Together, these data indicate a downstream role of NSD1 inactivation on CXCL9 and CXCL10 expression and point to disruption of the NSD1-CXCL9/CXCL10 relationship being associated with resistance to immune checkpoint blockade.
The effect of NSD1 inactivation on CXCL9 and CXCL10 expression can be reversed by inactivation of KDM2A
We next investigated the effects of NSD1 inactivation on H3K36 and H3K27 methylation and chemokine expression. Lentiviral transduction of NSD1 shRNA in human and mouse HNSCC cells resulted in a decrease in H3K36me2 and an increase in H3K27me3, as well as a concomitant decrease in CXCL9 and CXCL10 expression (Fig. 2A-F), in line with what we observed in the human HNSCC datasets (Fig. 1).
Figure 2: KDM2A inhibition restores CXCL9 and CXCL10 expression in the absence of NSD1.

Quantitative RT-PCR (qRT-PCR) analysis of NSD1 expression after inhibition of NSD1 expression by shRNA transduction in human (FaDu) (A) and mouse (MOC1) (B) HNSCC cell lines. Representative Western blots of H3K36me2 and H3K27me3 levels in FaDu (C) and MOC1 (D) cells after inhibition of NSD1 expression by shRNA transduction. Quantification of mRNA expression by qRT-PCR of CXCL9 and CXCL10 in FaDu (E) and MOC1 (F) cells transduced to express NSD1 shRNA. Data shown are representative of experiments repeated at least three times. Error bars represent standard error of the mean (SEM), ** p <0.01, *** p ≤0.001. (G) Reciprocal relationship of H3K36me2 and H3K27me3. NSD1 catalyzes the di-methylation of H3K36 (H3K36me2). KDM2A is a lysine demethylase with specificity for H3K36me2. Tri-methylation of H3K27 (H3K27me3) is directly antagonized by H3K36me2 (and H3K36me3). (H) KDM2A mRNA expression (assessed by qRT-PCR and normalized to HPRT1) in FaDu and MOC1 cell lines, transduced to express shRNA targeting NSD1 with and without shRNA targeting KDM2A. H3K36me2 and H3K27me3 levels were assessed by Western blot analysis of FaDu (I) and MOC1 (J) cells, transduced to express shRNA targeting NSD1 with and without shRNA targeting KDM2A. All experiments were repeated at least three times. Error bars represent standard error of the mean (SEM), *p<0.05, ** p <0.01, *** p ≤0.001. CXCL9 and CXCL10 mRNA expression levels were assessed by qRT-PCR and normalized to expression of HPRT1 in FaDu (K) and MOC1 (L) cell lines, transduced to express shRNA targeting NSD1 with and without shRNA targeting KDM2A. Data shown are representative of experiments repeated at least three times. Error bars represent standard error of the mean (SEM), * p <0.05, *** p ≤0.001.
The JHDM1/KDM2 and JHDM3/JMJD2/KDM4 family comprise the two lysine demethylase families with activity on methylated H3K36. Notably, the JHDM1/KDM2 family, which consists of two human members, KDM2A and KDM2B, is selective for lower methylation states of H3K36 (31). In HNSCC tumors, KDM2A is the most highly expressed H3K36-active lysine demethylase gene (Supp. Fig. 5A-B). We independently observed the relatively high expression of KDM2A in our cell lines (Supp. Fig. 5C-D) and thus, hypothesized that in the context of NSD1 inactivation, the targeted inhibition of KDM2A expression might be able to restore methylation of H3K36 (via other secondary methyl transferases other than NSD1). Because tri-methylation of H3K27 by PRC2 is directly antagonized by H3K36 tri-methylation (14–17), we also hypothesized that inhibition of KDM2A would indirectly lead to decreased levels of H3K27me3 (Fig. 2G).
As previously observed, inhibited expression of NSD1 resulted in decreased H3K36me2 levels and increased H3K27me3 levels (Fig. 2I-J). Strikingly, the concurrent knockdown of KDM2A in the NSD1 knockdown cells was sufficient to restore (and even exceed) H3K36me2 levels compared to the parental controls. Similarly, the levels of H3K27me3 levels were reduced closer to baseline control levels with the concomitant KDM2A knockdown. Thus, KDM2A targeting was sufficient to reverse the histone methylation effects of NSD1 inhibition in these cells.
These effects on H3K36 and H3K27 methylation states by KDM2A inhibition also had significant impact on the expression of CXCL9 and CXCL10. KDM2A knockdown resulted in the increased expression of CXCL9 and CXCL10 that was equal to or greater than the expression seen in the parental cells (Fig. 2K-L) – indicating that the indirect effects of targeting KDM2A on the methylation state of H3K27 has physiologic relevance.
Inhibition of KDM2A enhances T cell infiltration into the tumor microenvironment of HNSCC
As mentioned, NSD1 inactivating mutations in HNSCC have a strong correlation with an immune cell-deficient tumor microenvironment (10). Given our observations of NSD1 inactivation on CXCL9 and CXCL10 expression (Fig. 1–2), we investigated the role of these chemokines on T cell recruitment in the context of altered NSD1 and KDM2A activity. To do this, we utilized in vitro cultured 3-dimensional tumor spheroids grown from HNSCC cell lines that could be manipulated to express chemokines and/or shRNA (Fig. 3A-B). Co-culture of these spheroids with human donor-derived T cells enabled the assessment of T cell infiltration into the tumor microenvironment. Because these were not tumor antigen-specific T cells, we transduced the T cells to express a CD19-specific chimeric antigen receptor (CD19-CAR) (19) and the tumor cells to express a truncated surface CD19 molecule that lacked the intracellular portion (32). In this manner, the CD19-CAR provided T cell specificity for the tumor cells, and we were able to use this system to assess how tumor cell-intrinsic mechanisms influence T cell infiltration.
Figure 3. KDM2A inhibition reverses the immune cold phenotype induced by NSD1 inactivation and induces T cell infiltration into the tumor microenvironment.

(A) Overview and illustration of experiment workflow. Tumor spheroids were established from HNSCC tumor cells transfected to express truncated CD19 on the cell surface and grown in submerged Matrigel. The spheroids were co-cultured with CD19-CAR-T cells for three days and then isolated for analysis by confocal microscopy. (B) Assessment by qRT-PCR of CXCL9 and CXCL10 expression after CXCL9 or CXCL10 overexpression (OE) in FaDu cells transduced to express shRNA targeting NSD1 (NSD1-sh). (C) Representative images of tumor spheroids after co-cultured with CD19-CAR-T cells for 3 days. Ctrl = vector control; NSD1-sh = shRNA targeting NSD1; CXCL9-OE = overexpression construct for CXCL9; CXCL10-OE = overexpression construct for CXCL10; KDM2A-sh = shRNA targeting KDM2A. Scale bar=50um. (D) Representative images of tumor spheroids stained with anti-CD3 antibody (red) and DAPI (blue). Scale bar=50 um. (E) Quantification of CD3+ cells per 100 cells in the spheroids. (F) Representative images of FaDu tumor spheroids expressing control or NSD1 targeted shRNA with or without the KDM2A inhibitor (daminozide) treatment. The spheroids were stained with anti-CD3 antibody (red) and DAPI (blue). Scale bar=50 um. (G) Quantification of CD3+ cells per 100 cells in the spheroids. Data shown are representative of experiments repeated at least three times. Error bars represent standard error of the mean (SEM). * p <0.05, ** p <0.01, *** p ≤0.001.
The CD19-expressing tumor cells were transduced to express shRNA targeting either NSD1 or KDM2A and/or transduced to express either CXCL9 or CXCL10 prior to forming tumor spheroids. The expression of the chemokines was confirmed by qRT-PCR (Fig. 3B) and ELISA (Supp. Fig. 6). The spheroids grew in a similar fashion compared to each other and compared to control transduced cells; however, the spheroids expressing the shRNA targeting NSD1 had a distinctly sharper border and had fewer T cells aggregating near them when in co-culture (Fig. 3C). To assess the ability of the T cells to infiltrate the microenvironment of the spheroids, we analyzed co-cultured spheroids by confocal immunofluorescence microscopy using labeled anti-CD3 antibody (Fig. 3D). The number of T cells in each spheroid was quantified relative to the number of total cells (Fig. 3E). We observed significantly fewer infiltrating T cells in the NSD1 knockdown spheroids compared to control spheroids. Notably, forced expression of CXCL9 or CXCL10 by the tumor cells was sufficient to restore the number of infiltrating T cells in the context of NSD1 knockdown. This forced expression of CXCL9 and CXCL10 was supraphysiologic, and thus, other mechanisms may be involved in T cell exclusion in NSD1-mutant tumors besides the suppressed expression of CXCL9/10. Nevertheless, inhibition of KDM2A expression restored T cell infiltration in the NSD1 knockdown tumor spheroids (Fig. 3D-E), consistent with the ability of KDM2A inhibition to rescue CXCL9 and CXCL10 expression in NSD1 knockdown tumor cells (Fig. 2K-L). An orthogonal approach of using a KDM2A small molecule inhibitor (daminozide) also increased the number of infiltrating T cells into tumor spheroids, similar to what was observed with the shRNA inhibition of KDM2A expression (Fig. 3F-G). Thus, targeting KDM2A in NSD1-inactivated tumors can rescue T cell infiltration into the microenvironment.
KDM2A inhibition restores T cell infiltration into immune cell-depleted NSD1-inactivated tumors in vivo
As mentioned, inhibition of NSD1 and/or KDM2A expression did not have any apparent effect on tumor spheroid growth in vitro (Fig. 3C). We also confirmed that expression of shRNA targeting NSD1 and KDM2A did not affect tumor cell proliferation kinetics in vitro (Supp. Fig. 7). Similarly, inhibition of NSD1 and KDM2A in a C57BL/6 murine oral cancer model (MOC1) did not have any effect on tumor growth kinetics in vivo when assessed in syngeneic C57BL/6 Rag1-deficient (Rag1 KO) mice, which lack T and B cells (Fig. 4A; Supp. Fig. 8). However, interestingly, when assessed in immunocompetent wild-type (WT) C57BL/6 mice, NSD1 inhibition or inactivation in MOC1 cells resulted in significantly greater tumor volumes and fewer infiltrating T cells compared to control MOC1 cells (Fig. 4B-C; Supp Fig. 8). The concomitant inhibition of KDM2A expression in the MOC1-NSD1 shRNA cells inhibited the tumor volumes back to levels of the control MOC1 cells in WT mice but not in Rag1 KO mice (Fig. 4D-E; Supp Fig. 8), indicating that tumor inhibition was immune mediated. Consistent with our tumor spheroid data (Fig. 3D-G), we found that there were very few CD45+ CD3+ T cells in the MOC1 tumors expressing shRNA targeting NSD1 in WT mice, as assessed by flow cytometry of suspended cells from dissociated tumors and by immunofluorescence of tissue section (Fig. 4C and 4F). Further, there was little to no expression of CXCL9 and CXCL10 seen in these tumors (Fig. 4F). However, the concomitant expression of shRNA targeting KDM2A was sufficient to restore both the chemokine expression and CD3+ T cell infiltration into the tumor microenvironment of NSD1 knockdown MOC1 cells grown in WT mice (Fig. 4F, Supp. Fig. 9). Thus, the inhibition of KDM2A provides a powerful strategy for improving T cell infiltration into HNSCC tumors devoid of immune cells and offers a potential immunotherapeutic approach to enhance responses to immune checkpoint blockade.
Figure 4. KDM2A inhibition induces T cell infiltration of the tumor microenvironment in vivo.
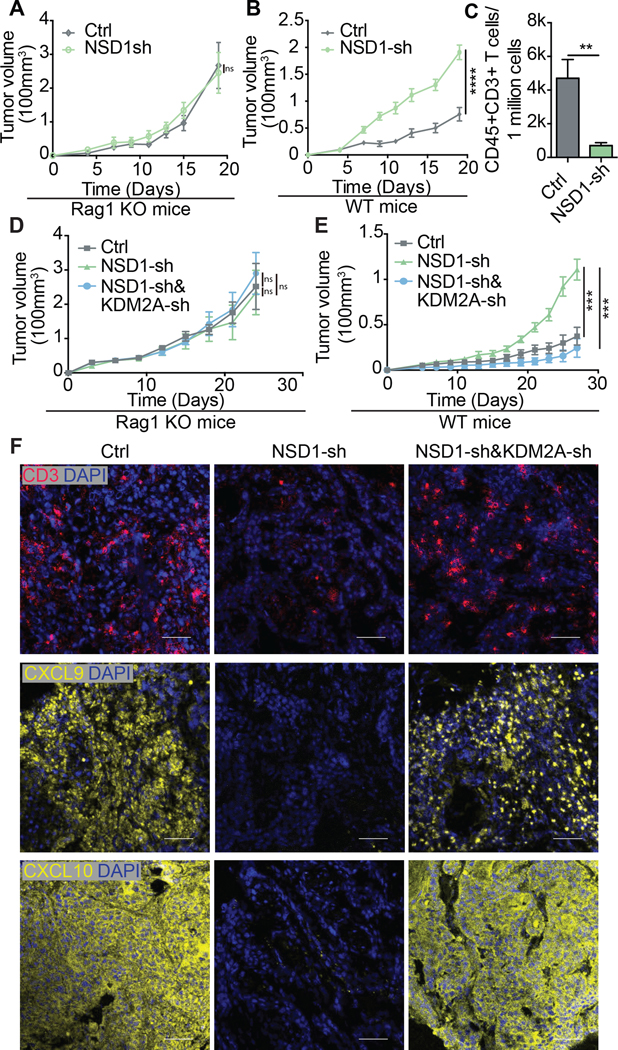
Growth curves of MOC1 control (vector control) and NSD1 knockdown (NSD1-sh) tumors in syngeneic Rag1–/– (Rag1 KO) mice (n=6 for each group) (A) and wild-type mice (n=8 per group) (B). (C) Tumor infiltrating T cells (CD45+CD3+) were quantified by flow cytometry of dissociated tumors. Growth curves of MOC1 control, NSD1 knockdown (NSD1-sh) and NSD1 and KDM2A double-knockdown (NSD1-sh & KDM2A-sh) cells in Rag1 KO mice (n=6 for each group) (D) and wild-type mice (n=5 for each group) (E). (F) Representative immunofluorescence images of CD3, CXCL9, and CXCL10 expression (red) in tumors formed from MOC1 cells transduced with vector control (Ctrl), NSD1 shRNA (NSD1-sh), or both NSD1 shRNA and KDM2A shRNA (NSD1-sh&KDM2A-sh). Scale bar=50 um. Blue stain=DAPI. Error bars represent standard error of the mean (SEM). *** p ≤0.001. ns = not significant.
DISCUSSION
Successful anti-tumor immune responses following PD-1/PD-L1 checkpoint blockade require recruitment and clonal proliferation of tumor-specific T cells present in the tumor microenvironment (TME). Tumor expression of the interferon (IFN) γ-inducible chemokines CXCL9 (also known as monokine induced by gamma interferon, or MIG) and CXCL10 (also known as interferon γ-induced protein 10, or IP-10) is correlated with the presence of tumor infiltrating CD8+ T cells and the activation of Th1 type immunity within the TME (33). As such, these chemokines are considered critical for robust responses to immune checkpoint inhibitors (e.g. anti-PD-1 and anti-CTLA-4 antibodies) and have been associated with improved patient outcomes (34).
A subset of HNSCC with mutations in NSD1 has an immune-cold TME, and we previously demonstrated that inactivation of this H3K36-specific histone methyltransferase can induce a TME phenotype that is resistant to T cell infiltration (10). Here, we report that the expression of CXCL9 and CXCL10 is repressed in the context of NSD1 inactivation and that targeting of KDM2A, a lysine demethylase specific for H3K36me2, leads to restored expression of these chemokines and subsequent recruitment of immune cells into the TME. The repression of CXCL9 and CXCL10 is likely due to the increased levels of H3K27me3 observed when NSD1 is inactivated. These data are consistent with previous reports that tri-methylation of H3K27 by PRC2 is directly antagonized by H3K36me2 and -me3 (14–17). In the setting of NSD1 inactivation, it is not clear which methyl transferase is responsible for di-methylation of H3K36 following KDM2A inhibition; however, NSD2 has been noted to physiologically oppose KDM2A on H3K36me2 levels (35), and SMYD2, another histone methyltransferase with specificity for catalyzing H3K36 di-methylation (36), has been shown to be overexpressed in HNSCC (37) and thus, may be active in this context.
The ability to inhibit H3K27 tri-methylation indirectly by targeting KDM2A has potential implications for cancer therapeutic approaches. EZH2 overexpression and H3K27me3 dysregulation are observed in many cancers, and EZH2’s catalytic (methyltransferase) and noncatalytic roles have been shown to contribute to tumor development and progression (38,39). In HNSCC, H3K27 tri-methylation resulting from EZH2 overexpression is negatively correlated with MHC I expression on the tumor cells and response to PD-1 checkpoint blockade (40). This and other preclinical immune studies have led to the proposal of combining EZH2 inhibition with immunotherapy (39,41). Over the years, significant effort has been directed toward the development of approaches to target EZH2, and several small molecule inhibitors of EZH2 are now in early clinical trials(39) However, EZH2 is also critical for CD8+ effector T cell survival and antitumor activity (42) and is important for Th1 and Th2 differentiation from naïve CD4 T cells (43,44). Thus, some concern has been raised regarding the possibility that T cell dysfunction may be observed with global EZH2 inhibition, and thus, there is rationale to explore alternative ways to reverse the repressive effects of H3K27me3 on key promoter regions. In the NSD1-inhibited MOC1 mouse model, we observed that EZH2 inhibition does reduce tumor growth in WT mice but not to the extent that KDM2A inhibition appeared to achieve (Supp. Fig. 10). Although it was difficult to observed synergistic effects of dual inhibition of EZH2 and KDM2A, with or without PD-1 checkpoint blockade (Supp. Fig. 10), there is evidence that NSD1 inactivation results in resistance to EZH2 inhibition and that KDM2A inhibition can overcome this resistance (45). Thus, these strategies need to be studied further in the context of immune response. Here, our study demonstrates that inhibition of KDM2A induces the increased methylation of H3K36 and decreased methylation of H3K27, resulting in both the increased expression of CXCL9/CXCL10 and the increased infiltration of T cells into the TME. Because our chemokine overexpression rescue experiments resulted in supraphysiologic levels of CXCL9/CXCL10 expression (Fig. 3B), it is possible that the influence of NSD1 inactivation on T cell exclusion from the TME is the result of other mechanisms in addition to the repression of CXCL9 and CXCL10 expression. Nevertheless, the co-inactivation of KDM2A was able to reverse these effects of NSD1 inactivation on T cell infiltration, and thus, these data introduce KDM2A as a novel target for immunotherapy.
Since response rates to immune checkpoint blockade correlate with the extent of immune cell infiltration in the TME, KDM2A inhibition may enhance clinical response to anti-PD-1 therapy and warrants further investigation.
Supplementary Material
SIGNIFICANCE STATEMENT.
The altered epigenetic landscape of NSD1-deficient tumors confers sensitivity to inhibition of the histone modifying enzyme KDM2A as an immunotherapeutic strategy to stimulate T cell infiltration and suppress tumor growth.
ACKNOWLEDGEMENTS
Our studies were supported by the Stanford Cancer Institute Tissue Bank for procurement of tumor samples and blood, the Lokey Stem Cell Research Building Flow Cytometry core facility for flow cytometric analysis, and the Stanford Blood Center for healthy donor blood. This work was supported by funding from the NIH (R35DE030054) to J.B.S.
Footnotes
COMPETING INTEREST STATEMENT:
No interests to declare.
REFERENCES
- 1.Johnson DE, Burtness B, Leemans CR, Lui VWY, Bauman JE, Grandis JR. Head and neck squamous cell carcinoma. Nat Rev Dis Primers [Internet]. 2020;6:92. Available from: 10.1038/s41572-020-00224-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zolkind P, Uppaluri R. Checkpoint immunotherapy in head and neck cancers. Cancer and Metastasis Reviews [Internet]. Springer US; 2017. [cited 2018 Dec 29];36:475–89. Available from: 10.1007/s10555-017-9694-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vaddepally RK, Kharel P, Pandey R, Garje R, Chandra AB. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers (Basel) [Internet]. MDPI; 2020;12:738. Available from: https://pubmed.ncbi.nlm.nih.gov/32245016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferris RL, Blumenschein GJ, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med. 2016;375:1856–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burtness B, Harrington KJ, Greil R, Soulières D, Tahara M, de Castro GJ, et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet. England; 2019;394:1915–28. [DOI] [PubMed] [Google Scholar]
- 6.Cohen EEW, Bell RB, Bifulco CB, Burtness B, Gillison ML, Harrington KJ, et al. The Society for Immunotherapy of Cancer consensus statement on immunotherapy for the treatment of squamous cell carcinoma of the head and neck (HNSCC). J Immunother Cancer. 2019;7:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oliva M, Spreafico A, Taberna M, Alemany L, Coburn B, Mesia R, et al. Immune biomarkers of response to immune-checkpoint inhibitors in head and neck squamous cell carcinoma. Ann Oncol [Internet]. Oxford University Press; 2019;30:57–67. Available from: https://pubmed.ncbi.nlm.nih.gov/30462163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, et al. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front Immunol. 2019;10:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daud AI, Loo K, Pauli ML, Sanchez-Rodriguez R, Sandoval PM, Taravati K, et al. Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J Clin Invest [Internet]. American Society for Clinical Investigation; 2016. [cited 2018 Mar 17];126:3447–52. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27525433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brennan K, Shin JH, Tay JK, Prunello M, Gentles AJ, Sunwoo JB, et al. NSD1 inactivation defines an immune cold, DNA hypomethylated subtype in squamous cell carcinoma. Sci Rep [Internet]. 2017. [cited 2018 Dec 30];7:17064. Available from: http://www.nature.com/articles/s41598-017-17298-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pan C, Izreig S, Yarbrough WG, Issaeva N. NSD1 mutations by HPV status in head and neck cancer: differences in survival and response to DNA-damaging agents. Cancers Head Neck [Internet]. 2019;4:3. Available from: 10.1186/s41199-019-0042-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brennan K, Koenig JL, Gentles AJ, Sunwoo JB, Gevaert O. Identification of an atypical etiological head and neck squamous carcinoma subtype featuring the CpG island methylator phenotype. EBioMedicine [Internet]. 2017/03/01. Elsevier; 2017;17:223–36. Available from: https://pubmed.ncbi.nlm.nih.gov/28314692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Y, Goldberg EM, Chen X, Xu X, McGuire JT, Leuzzi G, et al. Histone methylation antagonism drives tumor immune evasion in squamous cell carcinomas. Mol Cell [Internet]. Elsevier; 2022;82:3901–3918.e7. Available from: 10.1016/j.molcel.2022.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaffe JD, Wang Y, Chan HM, Zhang J, Huether R, Kryukov GV, et al. Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat Genet. 2013;45:1386–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Streubel G, Watson A, Jammula SG, Scelfo A, Fitzpatrick DJ, Oliviero G, et al. The H3K36me2 Methyltransferase Nsd1 Demarcates PRC2-Mediated H3K27me2 and H3K27me3 Domains in Embryonic Stem Cells. Mol Cell. United States; 2018;70:371–379.e5. [DOI] [PubMed] [Google Scholar]
- 16.Sengupta D, Zeng L, Li Y, Hausmann S, Ghosh D, Yuan G, et al. NSD2 dimethylation at H3K36 promotes lung adenocarcinoma pathogenesis. Mol Cell. 2021;81:4481–4492.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yuan G, Flores NM, Hausmann S, Lofgren SM, Kharchenko V, Angulo-Ibanez M, et al. Elevated NSD3 histone methylation activity drives squamous cell lung cancer. Nature. 2021;590:504–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagarsheth N, Peng D, Kryczek I, Wu K, Li W, Zhao E, et al. PRC2 Epigenetically Silences Th1-Type Chemokines to Suppress Effector T-Cell Trafficking in Colon Cancer. Cancer Res [Internet]. NIH Public Access; 2016. [cited 2018 Mar 17];76:275–82. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26567139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med [Internet]. 2015;21:581–90. Available from: 10.1038/nm.3838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Livak KJ, Schmittgen TD. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods [Internet]. 2001;25:402–8. Available from: https://www.sciencedirect.com/science/article/pii/S1046202301912629 [DOI] [PubMed] [Google Scholar]
- 21.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uppaluri R, Campbell KM, Egloff AM, Zolkind P, Skidmore ZL, Nussenbaum B, et al. Neoadjuvant and Adjuvant Pembrolizumab in Resectable Locally Advanced, Human Papillomavirus-Unrelated Head and Neck Cancer: A Multicenter, Phase II Trial. Clin Cancer Res. 2020;26:5140–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell. 2017;171:1611–1624.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farhangdoost N, Horth C, Hu B, Bareke E, Chen X, Li Y, et al. Chromatin dysregulation associated with NSD1 mutation in head and neck squamous cell carcinoma. Cell Rep. 2021;34:108769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods [Internet]. 2012;9:357–9. Available from: 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strazza M, Mor A. Consider the chemokines: a review of the interplay between chemokines and T cell subset function. Discov Med. United States; 2017;24:31–9. [PMC free article] [PubMed] [Google Scholar]
- 30.Cai Y, Zhang Y, Loh YP, Tng JQ, Lim MC, Cao Z, et al. H3K27me3-rich genomic regions can function as silencers to repress gene expression via chromatin interactions. Nat Commun. 2021;12:719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. England; 2006;439:811–6. [DOI] [PubMed] [Google Scholar]
- 32.Majzner RG, Rietberg SP, Sotillo E, Dong R, Vachharajani VT, Labanieh L, et al. Tuning the Antigen Density Requirement for CAR T-cell Activity. Cancer Discov. 2020;10:702–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dangaj D, Bruand M, Grimm AJ, Ronet C, Barras D, Duttagupta PA, et al. Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell [Internet]. 2019;35:885–900.e10. Available from: https://pubmed.ncbi.nlm.nih.gov/31185212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tokunaga R, Zhang W, Naseem M, Puccini A, Berger MD, Soni S, et al. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation - A target for novel cancer therapy. Cancer Treat Rev [Internet]. 2017/11/26. 2018;63:40–7. Available from: https://pubmed.ncbi.nlm.nih.gov/29207310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuan S, Natesan R, Sanchez-Rivera FJ, Li J, Bhanu NV, Yamazoe T, et al. Global Regulation of the Histone Mark H3K36me2 Underlies Epithelial Plasticity and Metastatic Progression. Cancer Discov [Internet]. 2020;10:854–71. Available from: 10.1158/2159-8290.CD-19-1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown MA, Sims RJ 3rd, Gottlieb PD, Tucker PW. Identification and characterization of Smyd2: a split SET/MYND domain-containing histone H3 lysine 36-specific methyltransferase that interacts with the Sin3 histone deacetylase complex. Mol Cancer. 2006;5:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohtomo-Oda R, Komatsu S, Mori T, Sekine S, Hirajima S, Yoshimoto S, et al. SMYD2 overexpression is associated with tumor cell proliferation and a worse outcome in human papillomavirus-unrelated nonmultiple head and neck carcinomas. Hum Pathol. United States; 2016;49:145–55. [DOI] [PubMed] [Google Scholar]
- 38.Ezponda T, Licht JD. Molecular pathways: deregulation of histone h3 lysine 27 methylation in cancer-different paths, same destination. Clin Cancer Res. 2014;20:5001–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eich M-L, Athar M, Ferguson JE 3rd, Varambally S. EZH2-Targeted Therapies in Cancer: Hype or a Reality. Cancer Res. 2020;80:5449–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou L, Mudianto T, Ma X, Riley R, Uppaluri R. Targeting EZH2 Enhances Antigen Presentation, Antitumor Immunity, and Circumvents Anti-PD-1 Resistance in Head and Neck Cancer. Clin Cancer Res. 2020;26:290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morel KL, Sheahan AV, Burkhart DL, Baca SC, Boufaied N, Liu Y, et al. EZH2 inhibition activates a dsRNA-STING-interferon stress axis that potentiates response to PD-1 checkpoint blockade in prostate cancer. Nat Cancer. 2021;2:444–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L, et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat Immunol [Internet]. NIH Public Access; 2016. [cited 2018 Mar 17];17:95–103. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26523864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tumes DJ, Onodera A, Suzuki A, Shinoda K, Endo Y, Iwamura C, et al. The polycomb protein Ezh2 regulates differentiation and plasticity of CD4(+) T helper type 1 and type 2 cells. Immunity. United States; 2013;39:819–32. [DOI] [PubMed] [Google Scholar]
- 44.Tong Q, He S, Xie F, Mochizuki K, Liu Y, Mochizuki I, et al. Ezh2 regulates transcriptional and posttranslational expression of T-bet and promotes Th1 cell responses mediating aplastic anemia in mice. J Immunol. 2014;192:5012–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Drosos Y, Myers JA, Xu B, Mathias KM, Beane EC, Radko-Juettner S, et al. NSD1 mediates antagonism between SWI/SNF and polycomb complexes and is required for transcriptional activation upon EZH2 inhibition. Mol Cell [Internet]. Elsevier; 2022;82:2472–2489.e8. Available from: 10.1016/j.molcel.2022.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data generated in this study are available within the article and its supplementary data files and are available upon request from the corresponding author. Publicly available data generated by others and analyzed in this study were obtained from the database of Genotypes and Phenotypes (dbGaP) at phs001623.v2.p1 and the Gene Expression Omnibus (GEO) at GSE103322; GSE149670; and GSE131967.