

Abstract
Data mining involves the computational analysis of a plethora of publicly available datasets to generate new hypotheses that can be further validated by experiments for the improved understanding of the pathogenesis of neurodegenerative diseases. Although the number of sequencing datasets is on the rise, microarray analysis conducted on diverse biological samples represent a large collection of datasets with multiple web-based programs that enable efficient and convenient data analysis. In this review, we first discuss the selection of biological samples associated with neurological disorders, and the possibility of a combination of datasets, from various types of samples, to conduct an integrated analysis in order to achieve a holistic understanding of the alterations in the examined biological system. We then summarize key approaches and studies that have made use of the data mining of microarray datasets to obtain insights into translational neuroscience applications, including biomarker discovery, therapeutic development, and the elucidation of the pathogenic mechanisms of neurodegenerative diseases. We further discuss the gap to be bridged between microarray and sequencing studies to improve the utilization and combination of different types of datasets, together with experimental validation, for more comprehensive analyses. We conclude by providing future perspectives on integrating multi-omics, to advance precision phenotyping and personalized medicine for neurodegenerative diseases.
Keywords: microarray analysis, biological samples, messenger RNA (mRNA), microRNA (miRNA), circular RNA (circRNA), long non-coding RNA (lncRNA), multi-omics integration, translational neuroscience, biomarker discovery, therapeutic development
1. Introduction
Over the past few decades, methods for quantifying the transcriptome have developed and expanded from microarray gene expression and quantitative polymerase chain reaction [1,2] to bulk RNA-seq and single-cell or single-nucleus RNA sequencing (sc/snRNA-seq) [3]. RNA-seq techniques have been at the forefront of studies aimed at understanding the heterogeneity of neurological diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), and multiple sclerosis (MS) [3,4]. It also has the unique ability of being able to detect novel sequences and splice variants [3,5]. However, RNA-seq methods are generally more labor intensive in data analysis and not as cost efficient in terms of data storage, and they may possess transcript length bias, which is currently mediated by long-read sequencing [5]. Although microarray gene expression analysis is limited to transcripts that are already established for the model organism being analyzed, it is able to detect highly varied genes [6]. Despite the technical differences, results from microarray and RNA-seq analyses have been shown to be highly consistent with each other [7]. In the context of data mining, microarray analysis is still widely adopted due to its low cost, high efficiency, limited bias [8], greater statistical power [9], and vast number of public neuroscience datasets available for data mining [3,10].
Data mining enables the utilization and comparison of deposited datasets containing high-dimensional features to efficiently acquire information related to translational neuroscience, which can be used to generate new hypotheses and may be validated experimentally [11]. When mining for transcriptomics data, it is important to take into account the type of RNA data and biological samples used in the analysis. For example, to probe for the pathogenic mechanisms of neurodegenerative diseases, the RNA profiles of post-mortem brain tissues from patients will provide insights into the specific alterations of the biological pathways that might play key roles in disease pathogenesis. On the other hand, regarding biomarker discovery, alterations in the RNA signatures from biological samples that can be obtained non-invasively, such as blood, could be used. Changes in the RNA profiles of cerebrospinal fluid (CSF) are sometimes utilized for biomarker discovery, although it is worth noting that CSF extraction can be invasive. Hence, there is a pressing need for the further investigation and establishment of blood biomarkers of neurodegenerative diseases [12,13]. Finally, drug discovery in translational neuroscience requires the testing of therapeutics in physiologically relevant models. Induced pluripotent stem cells (iPSCs) derived from patients are important models that can be used to test how therapeutics alter RNA profiles, protein expression, and cellular functions.
In this review, we will discuss the various biological samples that are commonly used for transcriptomic analysis of neurological diseases, with a specific focus on their limitations and advantages. We then discuss the pipeline for mining of microarray gene expression data, including the identification of datasets, quality control checks, statistical analysis, and functional annotations of genes. We further summarize the various types of RNA samples, including protein coding messenger RNA (mRNA) and non-coding RNA such as microRNA (miRNA), circular RNA (circRNA), and long non-coding RNA (lncRNA), that have been studied using microarray analysis for translational neuroscience applications. Furthermore, we discuss the gaps to be bridged between microarray and RNA-seq techniques and highlight how these two methods are complementary to each other. We conclude by providing future perspectives for the advancement of multi-omics integration, precision phenotyping, and personalized medicine for neurodegenerative diseases.
2. Biological Samples for Microarray Analysis
The selection of biological samples for data mining forms the core basis of both bioinformatics and experimental analyses, and it determines the outcomes and conclusions of studies. In neuroscience, these biological samples mostly consist of brain tissues, CSF, peripheral blood, as well as human stem cells. Samples collected from healthy controls or patients with neurodegenerative diseases are subjected to an array of quantitative and qualitative biological measurements, such as microarray characterizations and image analysis. The results of these measurements are interpreted to understand the brain functionality throughout various stages of disease [14,15]. It is important to classify biological samples based on basic demographic information, as well as genotypes (e.g., patients containing pathogenic mutations), phenotypes (e.g., observable characteristics that arise from the diseases), and clinical outcomes (e.g., patient-derived characteristics such as the Braak stage) of the subjects [16]. Another consideration lies in the ease of obtaining the biological samples for analysis, including the experimental procedures involved and whether it is invasive. Furthermore, the selection of biological samples should also be determined based on the applications of the studies, whether it is for biomarker identification, drug discovery, or the elucidation of disease mechanisms.
2.1. Brain Tissues
The use of human post-mortem brain tissues provides direct observations concerning the pathology and disease state when the patient is deceased. However, the inability to obtain brain tissue samples from living patients over time, and only during the last stage of life, creates bias and cannot be used to assess the initial stages of the disease. It is also difficult to elucidate the course of disease progression that determines clinical outcomes and disease phenotypes associated with the patients [17,18]. Furthermore, the main issues with using tissue samples for analysis lie in tissue heterogeneity, including diversity and variability, as well as low reproducibility across patient subjects [19,20]. Heterogeneity associated with neurodegeneration is further supported with a machine-learning technique that analyzes imaging datasets to reveal data-driven disease phenotypes, temporal progression, and trajectories that are distinct across patients [21]. Due to these abnormalities, a combined analysis of multiple datasets, containing different batches of samples and large number of patients, would provide a more accurate analysis. The increased utilization of data mining of brain tissue-associated microarray datasets holds promise for biomarker discovery and enhanced understanding of disease mechanisms in neurological disorders, leading to improved prognosis [22,23].
2.2. CSF and Peripheral Blood
To profile living patients, CSF and peripheral blood are often used as biological samples due to their extractability and diagnostic applications [24,25], and they are less likely to be affected by heterogeneity [19]. Unlike peripheral blood with expansive applications, the primary application of CSF is for the detection and diagnosis of neurological diseases [26]. Among several other candidates, established CSF biomarkers such as β-amyloid and tau can be used for the early diagnosis of AD [27], whereas α-synuclein and neurofilament light chains have been shown to aid the diagnosis of PD and MS, respectively [28,29,30,31]. On the other hand, establishing blood biomarkers has been a highly sought after strategy due to their extremely low invasiveness, low cost, and accessibility [32,33,34]. Currently, work is being conducted in order to increase the precision of measurements and to increase the corroboration between blood biomarkers and established CSF biomarkers [35]. This suggests that blood biomarkers may be used in clinical practice for diagnosing neurodegenerative diseases in the near future. However, for blood biomarkers to be fully implemented and consistent with observations from other biological samples, such as brain tissues, more correlation studies need to be conducted and new analysis methods need to be developed to take into account the variability between samples and individual patients [35,36]. Multiple studies utilizing peripheral blood have been focusing on examining miRNA expression levels due to their biomarker-quality characteristics [37,38,39]. More specifically, miRNAs are small non-coding RNAs that are being utilized for analyzing dysregulated genes due to their abundance, tissue specificity, and stability [40]. Currently, CSF and peripheral blood are often used in combination for disease evaluation, with the disease state being confirmed by established CSF biomarkers, and differentially expressed genes (DEGs) are isolated from peripheral blood to help reinforce the credibility of the proposed blood biomarkers [26,41]. With the need to provide treatment for asymptomatic patients with neurodegenerative diseases, such as AD, as well as to screen for risk in large numbers of young individuals, the development of biomarkers is shifting from a focus on CSF to peripheral blood, due to the ease of extractability and decreased invasiveness [42].
2.3. Human Stem Cells
The use of human stem cells is on the rise due to the high applicability of these cells in understanding disease mechanisms, as well as in regenerative therapy [43]. Stem cell therapy offers the ability to regenerate neural tissue and ameliorate the effects of neurodegeneration [44,45]. In addition, human iPSCs from patient fibroblasts can be differentiated to derive a vast source of central nervous system (CNS) cell types and contribute to a generation of multicellular organoids [46]. Other advantages associated with using stem cells include their ability to proliferate while maintaining developmental potential, the ease of modifying their genes, and the direct modeling of human biology without species-specific confounding factors [47]. Compendium-based big data approaches have been proposed to characterize the identity of each differentiated cell type from stem cells, which holds the key to understand the molecular events associated with the cell type and its biological applications [48,49,50]. For example, it has been shown that Aβ secreted from early-onset familial AD iPSC-derived neurons, was highly responsive to γ-secretase inhibitors and modulators, indicating their potential use for the identification and validation of candidate drugs [51]. The use of stem cells in model systems has also led to an increase in the utilization of iPSCs for drug screening and in vitro drug analysis [52]. These studies illustrate the potential of using datasets obtained from human stem cells for bioinformatic analysis to provide extensive insights into biomarker discovery and therapeutic development. Lastly, stem cells play a key role in regenerative medicine, and it is important to understand the mechanisms that regulate regeneration across different species and in different tissues. Recently, a Regeneration Roadmap database has been constructed which contains a comprehensive and systematic collection of gene expression and omics data associated with regenerative biology, and it can facilitate data mining studies [53].
In addition to the abovementioned biological samples, bioinformatic analysis also utilizes other samples including plasma, urine, feces, gut microbiome, mucus, saliva, and sputum to study metabolic changes of metabolites [54,55,56,57,58,59]. One study has shown that gut microbiome samples can be isolated and quantified using sequencing to study MS [60]. Additionally, gut microbiome alterations have been shown to modulate CNS autoimmunity in animal studies [60,61]. In vitro cell lines, as well as in vivo models, including transgenic and knock-in mice, have also been used to validate human data acquired from mining datasets associated with neurodegenerative diseases [62]. To this end, the array of biological samples that can be used for bioinformatics analysis is vast. A combination of datasets from various types of samples could be utilized for integrated analysis to understand the biological changes in localized regions (e.g., brain tissues) or in circulation (e.g., CSF and/or blood). It may also be used to understand the correlation and association within physiological systems in response to treatments, drug responses, and disease progression. Therefore, it is important to pinpoint the research question of interest to ensure the usage of appropriate datasets for computational analysis, and so that they fit into the correct biological context for meaningful interpretation. In many instances, the utilization of multiple datasets of various biological samples, in conjunction with experimental validation, is necessary to confirm findings.
3. RNA Based Microarray Gene Expression Analysis
AD and PD are the most common neurodegenerative diseases in the world, and they are characterized by progressive neuron loss [63,64]. On the other hand, MS is a prevalent neuroinflammatory and neuroimmunological disorder characterized by the loss of myelination in the CNS. It also has a neurodegenerative component in the progressive phase which currently does not have effective treatments [65]. Pathogenic mechanisms of neuroinflammation and neurodegeneration includes the dysregulation of biological processes, such as altered signaling pathways [66,67,68], as well as mutant protein production and toxic protein aggregation [69,70,71]. While targeting aberrant signaling pathways and toxic protein aggregates represent important therapeutic strategies [72,73,74], gene-level interventions can also be useful for treatment of neurodegenerative diseases. This is especially when preventing the expression of toxic gain-of-function genes does not detrimentally affect homeostatic cellular processes [75]. Therefore, it is vitally important to understand the changes in RNA expression of different biological samples in diseased states (Figure 1A), and to understand how alterations in RNA levels could have potential therapeutic efficacy. In this section, we will discuss the different types of RNA that are quantified using microarray (Figure 1B) and current microarray-based data mining studies that are associated with neurodegenerative diseases (Figure 1C), which can provide more insights into translational neuroscience applications (Figure 1D).
Figure 1.

Data mining of different types of RNA in various biological samples for translational neuroscience applications. (A) Various types of biological samples, including post-mortem brain tissues, CSF, peripheral blood, and human stem cells. (B) Different types of coding (mRNA) and non-coding RNA (miRNA, circRNA, and lncRNA) obtained from biological samples. (C) Data mining of microarray datasets associated with neurodegenerative diseases. Different genes detected by the microarray analysis are illustrated by different colors. (D) Translational neuroscience applications including drug discovery, the elucidation of disease mechanisms, and biomarker identification. The figure was created using BioRender.
3.1. Pipeline for the Data Mining of Microarray Datasets
The microarray method has been one of the most commonly used methods of transcriptomic analysis. It is used for identifying protein-encoding transcripts or non-coding RNAs that are differentially expressed in diseased states (as compared with healthy controls) by quantifying various RNA expression levels [76]. There are multiple databases archiving microarray datasets [77,78,79,80], with the Gene Expression Omnibus (GEO) database being the predominant repository [79]. The GEO database has a built-in tool, GEO2R, which is a graphical user interface that can be used to compare two or more groups of samples to identify the DEGs with statistical significance [79]. When isolating DEGs from the data mining of transcriptomic datasets, using a web-based tool such as GEO2R, or a command line-based analysis method using R scripts (Figure 2A), it is important to take into account the necessary quality control steps (Figure 2B) and statistical methods (Figure 2C) for the analysis. For quality control, it may be necessary to normalize the raw data [81], to perform a quality control assessment of the alignment, and to account for any contaminating species [82]. Feature selection may be required to remove genes that serve no biological purpose due to consistent expression across all samples [83,84], depending on the distribution of expression values of the samples used. For statistical analysis, using GEO2R as an example, it utilizes the R studio limma package for a differential analysis, where empirical Bayes moderated t-statistics and associated P-values, together with fold change values, are produced and used to evaluate the significance and extent of gene expression changes between diseased samples and healthy controls [85]. GEO2R also provides several graph plotting functions, such as the production of volcano and box plots, as well as uniform manifold approximation and projection (UMAP), that provides a further understanding of the expression level of genes and their expression changes under diseased conditions. Venn diagram analysis can also be performed to obtain overlapping DEGs across different datasets concerning similar disease conditions, in order to increase the stringency when determining significant genes with expression changes.
Figure 2.

Pipeline for the data mining of microarray gene expression. (A) Searching for suitable microarray datasets to be analyzed using web-based tools or command line scripts. Different genes identified by microarray analysis are represented by different colors. (B) Pre-processing of datasets via normalization, quality control, and feature selection. (C) Statistical tests to obtain DEGs with corresponding P-values (significance) and LogFC (fold-change). Downregulated genes are illustrated in red and upregulated genes are illustrated in purple. (D) Enrichment analysis and visualization using pathway analysis, gene set enrichment analysis (GSEA), and network analysis to provide a biological interpretation of the DEGs. Different pathways or functional annotations of the DEGs are illustrated by different colors. The figure was created using BioRender.
After obtaining the DEGs, there are multiple web-based and application-based programs that are available to elucidate the functional annotations of the DEGs and to provide insight into their role in disease mechanisms (Figure 2D). Tools aimed at conducting pathways analysis and identifying functionally enriched biological processes include the Database for Annotation, Visualization, and Integrated Discovery (DAVID) [86], Ingenuity Pathway Analysis (IPA) [87], Gene Set Enrichment Analysis (GSEA) [88], Centrality-based Pathway enrichment (CePa) [89], Signaling Pathway Impact Analysis (SPIA) [90], FunRich [91], and ExpressAnalyst [92]. Such tools are used to identify specific genes that are involved in biological processes that may pertain to disease pathogenesis, and to investigate how those biological processes are dysregulated under disease conditions compared with control conditions. Although the isolated DEGs are the inputs for most analysis tools, it is important to note that GSEA not only takes into account the DEGs and their expression values, but all transcriptomic expression values from each sample, for every gene in the raw data. Using an entire gene set, as opposed to specifically isolated DEGs, enables a more holistic analysis of the dysregulation of genes in diseased states. Programs and tools aimed at providing information on network visualization and specific protein–protein interactions include STRING [93], Cytoscape [94], and NetworkAnalyst [95]. These programs not only enable the isolation of nodes involved in functionally enriched biological pathways, but they also allow the identification of hub genes and assess the extent to which interactions and connectivity occur.
3.2. Microarray Analysis of Coding RNA (mRNA)
Microarray analysis has been used to assemble data that are representative of the mRNA expression levels of tens of thousands of genes in different neurodegenerative diseases. It can also identify DEGs in these disease states, which can be further explored to establish therapeutics or biomarkers. Here, we summarize results from data mining studies to elucidate the changes in gene expression in various sample types under different neurological conditions (Table 1).
Table 1.
Summary of upregulated and downregulated genes identified from a microarray analysis of mRNA obtained from brain tissues and blood samples in AD and PD, as well as from CSF in MS. Arrows represent the direction of changes of the gene levels.
| AD | Brain tissue |
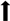 HDAC1, WWTR1, ITGB1, PDGFRB, PLOD1, MAP4K4, NFKBIA, TYROBP, GSN, TIMP1 HDAC1, WWTR1, ITGB1, PDGFRB, PLOD1, MAP4K4, NFKBIA, TYROBP, GSN, TIMP1
|
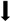 SIRT3, RAB7A, BDNF, VLDLR, APLP2 SIRT3, RAB7A, BDNF, VLDLR, APLP2
| ||
| Blood |
VCAM1, TYK2, TCIRG1, PPP3CB, SNCB, SACS, GSN, TIMP1
|
|
|
CTSD, RPL11, SNCA, FKBP1B, BDNF, VLDLR, APLP2
| ||
| PD | Brain tissue |
SRRM2
|
|
MAPK8, CDC42, NDUFS1, COX4I1, SDHC
| ||
| Blood |
LILRB3, CSF3R, SRRM2
|
|
|
ICAM1
| ||
| MS | CSF |
NLRP3, LILRB2, C1QB, CD86, C1QA, CSF1R, IL1B, TLR2
|
In AD, a vast number of data mining studies have been conducted to examine altered gene expression in brain tissues (upregulated: HDAC1 [96], WWTR1 [97], ITGB1 [98], PDGFRB [99], PLOD1 [99], MAP4K4 [99], NFKBIA [99,100]; downregulated: SIRT3 [101], BDNF [97], RAB7A [98]) and in peripheral blood (upregulated: VCAM1 [102], TYK2 [103], TCIRG1 [103], PPP3CB [103], SNCB [103], SACS [103]; downregulated: CTSD [102], RPL11 [104], SNCA [103], FKBP1B [103]). Although there is a limited number of studies analyzing CSF samples in microarray analysis, a meta-analysis has reported that NRGN is upregulated in the CSF samples of AD patients [105]. Another data mining study utilizing the GEO datasets of human AD brain tissues found that TYROBP is a key regulator of pathogen phagocytosis in microglia, and it is upregulated in late-onset AD [106]. Importantly, they further validated their results using a mouse model expressing TYROBP in microglia, and they revealed gene expression changes that significantly overlapped with the TYROBP network in the human brain [106]. In terms of biomarker discovery, a data mining study found five potential biomarker genes of AD. More specifically, GSN, BDNF, TIMP1, VLDLR, and APLP2 were validated both in bioinformatic analysis using AD GEO datasets of human brain tissues, and in an experimental validation using peripheral blood from AD patients [107].
In PD data mining studies, LILRB3 and CSF3R have been shown to be upregulated [108], and ICAM1 was shown to be downregulated in whole blood analysis [109]. Additionally, MAPK8, CDC42, NDUFS1, COX4I1, and SDHC have been shown to be significantly downregulated in PD brain tissues [110]. Another study revealed a significant upregulation of the RNA splicing factor, serine/arginine repetitive matrix 2 (SRRM2), in PD through the computational analysis of GEO datasets containing human brain tissues, cells, and whole blood. This was validated experimentally, and the analysis showed a significant upregulation of the upstream exons of SRRM2 [111]. In MS, a data mining study analyzing CSF found NLRP3, LILRB2, C1QB, CD86, C1QA, CSF1R, IL1B, and TLR2 to be downregulated in MS [112], many of which have been supported and validated experimentally in other studies [113,114,115]. Another study found that similar genes, involved in inflammation or immune responses, existed in MS and COVID-19 patients [116]. Additionally, through network analysis, they found that genes IL1B, P2RX7, IFNB1, TNF, and CASP1 enhanced the network connectivity between the combined gene sets of MS and COVID-19, which is associated with NOD-like receptor signaling [116].
Multiple neurodegenerative studies have been utilizing both microarray and RNA-seq analyses simultaneously, and they have isolated DEGs common to both methods of expression quantification [117,118,119]. For example, a systemic biological approach has also been adopted to integrate RNA-seq datasets for COVID-19 and microarray datasets for AD. This was conducted in order to examine and identify the common transcriptional alterations between COVID-19 and AD patients [119]. This study identified 26 hub genes that could be potential biomarkers and therapeutic targets for COVID-19 patients with AD comorbidities. Another PD study made use of both microarray and RNA-seq datasets to identify 12 significant genes that are commonly dysregulated between the blood and brain, including C10orf32, CCDC82, COL5A2, COQ7, GPNMB, HSD17B1, KANSL1, NCKIPSD, PM20D1, SP1, FRRS1L, and IL1R2 [117]. This study demonstrates that both disease processes and systemic disease factors may affect brain and blood cells in a similar manner. The correlation and corroboration between blood and brain transcriptomic data are further exemplified in a recent study in PD [120]. Together, these findings identify molecular signatures in PD patients’ brain and blood for potential pathophysiologic and prognostic importance, and these findings may potentially be applicable to other diseases, including AD and MS.
3.3. Microarray Analysis of Non-Coding RNA (miRNA, circRNA, and lncRNA)
In addition to studying mRNA, several non-coding RNAs, including miRNA, circRNA, and lncRNA, are isolated from peripheral blood for diagnosis or mechanistic analysis [121]. With non-coding RNAs making up most of the human genome, they are becoming increasingly sought after in neurodegenerative studies due to their role in neural cell specification during development, and in higher cognitive processes such as memory and plasticity [122]. Similar to mRNA, non-coding RNAs can also exhibit cell type specific expression levels to shape the cellular expression landscape and reinforce cellular identity. Importantly, non-coding RNAs are capable of modulating gene expression at the post-transcriptional level, binding to protein factors, controlling epigenetic mechanisms, and playing key roles in regulating many biological processes [123,124].
For example, miRNA plays a key role in the post-transcriptional gene regulation of mRNA expression [125]. Tissue specific miRNAs are becoming increasingly pursued for biomarker discovery due to their non-invasive extraction, accuracy, reproducibility, and predictability [126,127]. A data mining study that made use of blood datasets from MS patients found that the upregulation of miRNA hsa-miR-328-3p [128], hsa-miR-20a-5p [128], and miR-196 [129] occurred, as did the downregulation of miR-9 [129]. In AD, the dysregulation of miRNA has been identified in peripheral blood (upregulated: hsa-miR-186-5p [130]; downregulated: hsa-miR-125a-3p, hsa-miR-22-3p, hsa-miR-24-3p, hsa-miR-6131, and hsa-miR-125b-1-3p [131]) and in brain tissues (downregulated: hsa-miR-29c [132] and hsa-miR-26b-5p [133]). Another study found that the downregulation of miR-425 was implicated in AD pathogenesis, and a miR-425 deficient transgenic mouse was used to validate the results [134]. The loss of miR-425 in mice induced neuroinflammation and neuronal loss, it exacerbated cognitive impairment, and increased amyloid precursor protein amyloidogenic processing [134]. Interestingly, another study combined GEO datasets from AD tissues and experimental validation using blood samples from AD patients, and they found differential results with regard to hsa-miR-185-5p being upregulated in AD brain tissues, while being downregulated in AD blood samples [135].
With an integrative analysis using both microarray and RNA-seq datasets, seven miRNAs that interact with the eight DEGs were identified in early and late mild cognitive impairment patients [136]. Another study that focused on PD identified changes in miRNA expression in the PD patient’s blood leukocytes when compared with control patients. These changes were identified using RNA-seq techniques, microarray analysis, as well as data mining of GEO microarray data [137]. This study found 16 miRNAs that were differentially expressed in PD patients, and a specific interest in transcription factor FOXP1, which they found was implicated in a miRNA-mediated feedback loop that controlled the survival of midbrain dopaminergic neurons. In addition to these findings, it is worth noting the significance of miRNAs and other RNAs in other molecular mechanisms associated with brain diseases, such as vascular dysfunction [138] and retinopathies [139,140], which can deepen our understanding of disease mechanisms, and it may potentially serve as prognostic indicators for neurodegenerative diseases.
In addition to miRNA, circRNA has been suggested to have multiple functions, such as regulating transcription in the nucleus, binding to protein factors, acting as a miRNA “sponge” to compete for miRNA pairing with other RNAs, and being more stable in tissues compared with linear RNAs [141]. Furthermore, circRNA expression is enriched in the brain, aiding in its likelihood to be used in studies associated with neurodegenerative diseases [141]. A bioinformatics analysis using GEO datasets has found circRNAs originating from the following AD pathology-linked genes, DOCK1, NTRK2, DLG1, KIF1B, TRAPPC9, and APC, which are altered in AD [142]. A different study focusing on PD found that miRNA-7 (miR-7), which is bound to by circular the RNA sponge for miR-7 (ciRS-7) [143], is mostly expressed in neurons. Moreover, it represses α-synuclein protein, which ultimately protects against oxidative stress [143]. Although there are limited studies on role of circRNA in MS, an amyotrophic lateral sclerosis (ALS) study performing microarray analysis on the peripheral blood of ALS patients found that circRNAs hsa_circ_0000567 and hsa_circ_0023919 were downregulated, and hsa_circ_0063411 and hsa_circ_0088036 were upregulated [144]. These genes are involved in muscle differentiation in mice, clathrin-mediated endocytosis at neuromuscular junctions, Ago-mediated gene silencing, and there is speculation that they are involved in immune responses.
In addition, lncRNAs are non-coding RNA molecules that are more than 200 nucleotides in length [145], and their function is essential for many biological processes, including epigenetic regulation, cell signal transduction, immune response, and cell proliferation and differentiation. Moreover, their abnormal expression can result in a variety of neurodegenerative diseases [146]. By analyzing GEO datasets, a study has found that lncRNA-XIST was downregulated in the whole blood of PD patients [147]. Many neurodegenerative studies have focused on the specific upregulation of the nuclear paraspeckle assembly transcript 1 (NEAT1) under diseased conditions. An AD study found that the lncRNAs LOC100507557 (downregulated), LOC101929787 (upregulated), NEAT1 (upregulated), and JAZF1-AS1 (downregulated) were differentially expressed, and they were found to be dysregulated in five distinct anatomical regions of the brain [148]. With the 15-fold upregulation of NEAT1 in the entorhinal cortex, it is the highest upregulated lncRNA in all the analyzed brain regions and has the potential to serve as a biomarker of AD. Using an integrative analysis consisting of microarray, RNA-seq, and genome-wide association study (GWAS) datasets, a study identified five key lncRNAs associated with AD risk, and they were involved in the regulation of the immune system [149].
3.4. Bridging Gaps between Microarray and RNA-Seq Analysis
The experimental aspects of microarray and RNA-seq are similar in terms of how RNA is converted into cDNA. It is followed by signal quantification, although the technical details may provide slightly different information. In microarray analysis, cDNA is fluorescently labelled and hybridized with a complementary strand of a known gene, and the fluorescence release directly corresponds with the level of genetic expression of the specific gene in the biological sample [150]. In sequencing studies, gene expression levels are quantified by counts in RNA-seq, which is equivalent to the number of reads mapped on each gene. It is worth noting that newer forms of RNA-seq can directly sequence individual RNA strands with a method known as nanopore direct RNA-seq (DRS). Nanopore DRS allows for the sequencing of single RNA strands, including nucleotide modifications (e.g., methylation, 5′ end capping, 3′ polyadenylation) and all exons, and it has been used to sequence both coding and non-coding RNAs [151]. In sc/snRNA-seq, unique molecular identifiers (UMIs) are further acquired to provide cell-type specific information of the gene expression [152]. The counts may be varied depending on the covariates of the gene or samples such as library size and gene length. As discussed earlier, RNA-seq can detect splice variants and novel sequences, whereas RNA microarray is limited to established transcripts for the model organism being analyzed, although this difference may not affect studies that do not require this detailed level of information. On the other hand, sc/snRNA-seq techniques are of great interest because they not only provide the average expression level for an ensemble of cells, such as in the typical microarray and RNA-seq analyses [153], but also the ability to quantify gene expression levels in specific cell types [153,154,155,156]. Although single cell microarray analyses have previously been reported [152,157], the resolution and the heterogeneity that can be resolved might not be comparable to current sc/snRNA-seq, and they are dependent on the samples that can be obtained.
In addition to the experimental aspects, there have been major gaps in standardizing data analysis pipelines to process different raw data obtained by the different methods of RNA profiling. Although the data analysis for microarray datasets is more straightforward, as it is directly quantified at the expression level, analyses for RNA-seq and sc/snRNA-seq are more complex, with less standardized protocols, a need for more data storage, and a knowledge of coding [158]. With the increasing availability of publicly accessible transcriptomic datasets, many web-based and application-based tools are being created to aid in the analysis of such high-content data. The GEO2R [159] and Bioinformatics Array Research Tool (BART) [160] are web-based programs that are capable of carrying out statistical DEG analysis on deposited GEO microarray datasets. Web-based tools have also emerged to facilitate RNA-seq analysis such as BEAVR [161], RNAlysis [162], RNAdetector [163], OneStopRNAseq [164], and Integrative Differential Expression Analysis for Multiple EXperiments (IDEAMEX) [165]. They consist of graphical user interfaces to assist in conducting DEG statistical analyses, and to assist with the visualization of the results with RNA-seq data. Furthermore, recent studies have also provided simplified and practical guides [166,167], as well as streamlined scRNA-seq data analysis, such as ScAmpi [168]. Additionally, there are tools that enable the analysis and visualization of sc/snRNA-seq data, including Automated Single-cell Analysis Pipeline (ASAP) [169], and the CanceR Single Cell ExpressioN Toolkit (CReSCENT) [170]. We have included a summary table of the data mining tools and programs used to analyze microarray, RNA-seq, and sc/snRNA-seq datasets (Table 2).
Table 2.
Summary of data mining tools and programs that can be used analyze microarray, RNA-seq, and sc/snRNA-seq datasets.
| Data Mining Tools/Programs | Datasets Analyzed | References |
|---|---|---|
| GEO2R | Microarray data | [159] |
| BART | Microarray data | [160] |
| BEAVR | RNA-seq data | [161] |
| RNAlysis | RNA-seq data | [162] |
| RNAdetector | RNA-seq data | [163] |
| OneStopRNAseq | RNA-seq data | [164] |
| IDEAMEX | RNA-seq data | [165] |
| ScAmpi | ScRNA-seq data | [168] |
| ASAP | Sc/snRNA-seq data | [169] |
| CReSCENT | Sc/snRNA-seq data | [170] |
An approach to compare and reconcile microarray and RNA-seq analysis methods, specifically in terms of data mining, is check for the similarities between the specific DEGs identified, or to conduct enrichment analysis of all DEGs to see if there are similar pathways and networks obtained from the respective methods. To quantify the similarities between the data obtained from microarray and RNA-seq methods, studies have been carried out to examine the correlation between the expression intensity values. One study specifically used microarray and RNA-seq analysis to quantify the mRNA expression in human brains, which was collected from the Allen Human Brain Atlas. They found consistent, reproducible measurements between the two methods, with a high correlation between expression values (R = 0.78) [171]. They also showed that RNA-seq scaling factors can be applied to improve the sensitivity of microarrays to detect DEGs. Another study examining the lncRNA expression levels in iPSC-derived neurons illustrated a high correlation (R = 0.64) between the expression values of microarray and RNA-seq analysis [172]. These studies suggest that both are suitable methods used for high-throughput gene expression analysis. It is important to note that there is a possibility for false positives, particularly when using a small sample size [173,174]. Hence, the use of multiple datasets, rigorous analysis methods, and stringent statistical parameters is critical to reduce false positives.
3.5. Experimental Validation to Advance Therapeutic Development and Biomarker Identification
The current emergence of the big data era and the outburst of transcriptomic datasets from studies with different experimental conditions, biological samples, disease models, developmental states, and responses to treatments, indicate the need for an accurate and reliable data mining process. This presents the need for a thorough interpretation of the outcomes from data mining and their relevance to true biological observations. To test the effect of the DEGs obtained from data mining, we can either knockout or overexpress the key protein of interest, or we can administer modulators of certain signaling pathways to observe how the cellular systems respond to these alterations. It is also important to check whether any of these treatments are toxic to the cells. Typically, when alterations in protein expression or function correlate with disease progression, it indicates that the protein plays a major role in the disease mechanism. High-throughput screening of small molecules or antisense oligonucleotides that can modulate protein function would lead to a therapeutic discovery. Generally, biomarkers are established based on certain key proteins that can be detected in CSF or blood to provide a prognosis of the disease pathogenesis.
Studies have been conducted to quantify the variation and discrepancies between microarray and RNA-seq data [150,174,175], and programs have been created for their integration [176,177]. With evidence showing the corroboration of results between two methods of data collection [7,150], data mining of microarray datasets remain useful for generating novel hypotheses, validating existing RNA-seq data, or integrating novel RNA-seq analysis. Transcriptomics datasets can be extremely high dimensional and may contain tens of thousands of genes, whereas experimental datasets may only contain tens of genes [178]. Furthermore, different analysis criteria adopted in data mining, such as the biological samples used, the number and combination of datasets analyzed, and the cut-off parameters selected, may cause inconsistent results between studies. Hence, there is a definite need to experimentally validate the results obtained from data mining to ensure their accuracy and usefulness.
4. Summary and Future Perspectives
A vast number of studies related to neurodegenerative diseases have made use of microarray datasets for the data mining of transcriptomics data. This is mainly due to the expansiveness and diversity of deposited microarray datasets, as well as the ease of processing due to many accessible web-based analysis tools and established computational pipelines. Additionally, it is important to note the ability of microarray datasets to quantify the expression of non-coding RNA species, including miRNA, circRNA, and lncRNA. With the advancements in RNA sequencing technologies, microarray analysis has become less utilized, although it remains important to recognize the ability of microarray analysis as a resource to validate RNA-seq results and vice versa, and it may also be used as a basis for hypothesis testing and generation. It is important to note that data mining may subject to technological and biological biases as well as systematic errors that can impact downstream analyses [179]. A good strategy would be to combine the data mining of both microarray and RNA-seq datasets to increase the stringency and the accuracy of the DEGs identified.
The future of omics analysis lies at the interface of multi-omics integration, where genomics, transcriptomics, proteomics, metabolomics, lipidomics, as well as spatial omics can be utilized simultaneously [180]. One of the main challenges of integrative approaches concerns increased dimensionality due to the increased complexity of the omics datasets associated with the biological systems. An integrative analysis, such as independent biological integration or unsupervised machine learning, will enable the reconstruction of biological systems, with a holistic understanding of gene and protein regulation at different omic levels for translational applications [180,181]. Multi-omics data integration would provide a more sophisticated and accurate analysis for early disease detection (e.g., lysosomal dysfunction [182,183]), as well as increase precision phenotyping and personalized medicine [184,185,186,187]. It is also important to take into account pharmacogenomics to understand that individuals will respond differently to different medicines based on many biological and environmental factors. Exploring different omic datasets through established pipelines of multi-omics integration will unlock a broad range of opportunities for translational applications, including elucidation of disease mechanisms, biomarker discovery, and therapeutic development.
Acknowledgments
The authors thank the funding sources for supporting this work.
Author Contributions
Conceptualization, C.H.L.; methodology, L.M.O., B.A.O., J.Z. and C.H.L.; data curation, L.M.O. and B.A.O.; writing—original draft preparation, L.M.O. and C.H.L.; validation, C.H.L. and J.Z.; formal analysis, C.H.L. and J.Z.; investigation, L.M.O., B.A.O., J.Z. and C.H.L.; visualization, L.M.O. and C.H.L.; writing—review and editing, L.M.O., B.A.O., J.Z. and C.H.L.; supervision, C.H.L.; project administration, C.H.L.; funding acquisition, C.H.L. and J.Z. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
C.H.L. is supported by a Lee Kong Chian School of Medicine Dean’s Postdoctoral Fellowship (021207-00001) from Nanyang Technological University (NTU) Singapore and a Mistletoe Research Fellowship (022522-00001) from the Momental Foundation USA. J.Z. is supported by a Presidential Postdoctoral Fellowship (021229-00001) from NTU Singapore.
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Negi A., Shukla A., Jaiswar A., Shrinet J., Jasrotia R.S. In: Chapter 6-Applications and Challenges of Microarray and RNA-Sequencing. Singh D.B., Pathak R.K.B.T.-B., editors. Academic Press; Cambridge, MA, USA: 2022. pp. 91–103. [Google Scholar]
- 2.Costa C., Giménez-Capitán A., Karachaliou N., Rosell R. Comprehensive Molecular Screening: From the RT-PCR to the RNA-Seq. Transl. lung cancer Res. 2013;2:87–91. doi: 10.3978/j.issn.2218-6751.2013.02.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kukurba K.R., Montgomery S.B. RNA Sequencing and Analysis. Cold Spring Harb. Protoc. 2015;2015:951–969. doi: 10.1101/pdb.top084970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu A.R., Neff N.F., Kalisky T., Dalerba P., Treutlein B., Rothenberg M.E., Mburu F.M., Mantalas G.L., Sim S., Clarke M.F., et al. Quantitative Assessment of Single-Cell RNA-Sequencing Methods. Nat. Methods. 2014;11:41–46. doi: 10.1038/nmeth.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mantione K.J., Kream R.M., Kuzelova H., Ptacek R., Raboch J., Samuel J.M., Stefano G.B. Comparing Bioinformatic Gene Expression Profiling Methods: Microarray and RNA-Seq. Med. Sci. Monit. Basic Res. 2014;20:138–142. doi: 10.12659/MSMBR.892101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cui X., Churchill G.A. Statistical Tests for Differential Expression in CDNA Microarray Experiments. Genome Biol. 2003;4:210. doi: 10.1186/gb-2003-4-4-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kogenaru S., Yan Q., Guo Y., Wang N. RNA-Seq and Microarray Complement Each Other in Transcriptome Profiling. BMC Genomics. 2012;13:629. doi: 10.1186/1471-2164-13-629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oshlack A., Wakefield M.J. Transcript Length Bias in RNA-Seq Data Confounds Systems Biology. Biol. Direct. 2009;4:14. doi: 10.1186/1745-6150-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tarca A.L., Romero R., Draghici S. Analysis of Microarray Experiments of Gene Expression Profiling. Am. J. Obstet. Gynecol. 2006;195:373–388. doi: 10.1016/j.ajog.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harbola A., Negi D., Manchanda M., Kesharwani R.K. In: Chapter 27-Bioinformatics and Biological Data Mining. Singh D.B., Pathak R.K.B.T.-B., editors. Academic Press; Cambridge, MA, USA: 2022. pp. 457–471. [Google Scholar]
- 11.Wu W.-T., Li Y.-J., Feng A.-Z., Li L., Huang T., Xu A.-D., Lyu J. Data Mining in Clinical Big Data: The Frequently Used Databases, Steps, and Methodological Models. Mil. Med. Res. 2021;8:44. doi: 10.1186/s40779-021-00338-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hadar A., Gurwitz D. Peripheral Transcriptomic Biomarkers for Early Detection of Sporadic Alzheimer Disease? Dialogues Clin. Neurosci. 2018;20:293–300. doi: 10.31887/DCNS.2018.20.4/dgurwitz. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lake J., Storm C.S., Makarious M.B., Bandres-Ciga S. Genetic and Transcriptomic Biomarkers in Neurodegenerative Diseases: Current Situation and the Road Ahead. Cells. 2021;10 doi: 10.3390/cells10051030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He S., Dou L., Li X., Zhang Y. Review of Bioinformatics in Azheimer’s Disease Research. Comput. Biol. Med. 2022;143:105269. doi: 10.1016/j.compbiomed.2022.105269. [DOI] [PubMed] [Google Scholar]
- 15.Paananen J. Bioinformatics in the Identification of Novel Targets and Pathways in Neurodegenerative Diseases. Curr. Genet. Med. Rep. 2017;5:15–21. doi: 10.1007/s40142-017-0115-8. [DOI] [Google Scholar]
- 16.Koh E.J., Kim S.H., Hwang S.Y. Sample Management: A Primary Critical Starting Point for Successful Omics Studies. Mol. Cell. Toxicol. 2022;18:141–148. doi: 10.1007/s13273-021-00213-x. [DOI] [Google Scholar]
- 17.Clement C., Hill J.M., Dua P., Culicchia F., Lukiw W.J. Analysis of RNA from Alzheimer’s Disease Post-Mortem Brain Tissues. Mol. Neurobiol. 2016;53:1322–1328. doi: 10.1007/s12035-015-9105-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stan A.D., Ghose S., Gao X.M., Roberts R.C., Lewis-Amezcua K., Hatanpaa K.J., Tamminga C.A. Human Postmortem Tissue: What Quality Markers Matter? Brain Res. 2006;1123:1–11. doi: 10.1016/j.brainres.2006.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sturm G., List M., Zhang J.D. Tissue Heterogeneity Is Prevalent in Gene Expression Studies. NAR Genomics Bioinforma. 2021;3:lqab077. doi: 10.1093/nargab/lqab077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu M., Fang K., Wang W., Lin W., Guo L., Wang J. Identification of Key Genes and Pathways for Alzheimer’s Disease via Combined Analysis of Genome-Wide Expression Profiling in the Hippocampus. Biophys. Reports. 2019;5:98–109. doi: 10.1007/s41048-019-0086-2. [DOI] [Google Scholar]
- 21.Young A.L., Marinescu R.V., Oxtoby N.P., Bocchetta M., Yong K., Firth N.C., Cash D.M., Thomas D.L., Dick K.M., Cardoso J., et al. Uncovering the Heterogeneity and Temporal Complexity of Neurodegenerative Diseases with Subtype and Stage Inference. Nat. Commun. 2018;9:4273. doi: 10.1038/s41467-018-05892-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yin X., Wu Q., Hao Z., Chen L. Identification of Novel Prognostic Targets in Glioblastoma Using Bioinformatics Analysis. Biomed. Eng. Online. 2022;21:26. doi: 10.1186/s12938-022-00995-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang L., Zeng W., Sun H., Huang F., Yang C., Cai X., Lu Y., Zeng J., Yang K. Bioinformatical Analysis of Gene Expression Omnibus Database Associates TAF7/CCNB1, TAF7/CCNA2, and GTF2E2/CDC20 Pathways with Glioblastoma Development and Prognosis. World Neurosurg. 2020;138:e492–e514. doi: 10.1016/j.wneu.2020.02.159. [DOI] [PubMed] [Google Scholar]
- 24.Cai C., Langfelder P., Fuller T.F., Oldham M.C., Luo R., van den Berg L.H., Ophoff R.A., Horvath S. Is Human Blood a Good Surrogate for Brain Tissue in Transcriptional Studies? BMC Genomics. 2010;11:589. doi: 10.1186/1471-2164-11-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McEwen A.E., Leary S.E.S., Lockwood C.M. Beyond the Blood: CSF-Derived CfDNA for Diagnosis and Characterization of CNS Tumors. Front. Cell Dev. Biol. 2020;8:45. doi: 10.3389/fcell.2020.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robey T.T., Panegyres P.K. Cerebrospinal Fluid Biomarkers in Neurodegenerative Disorders. Future Neurol. 2019;14:FNL6. doi: 10.2217/fnl-2018-0029. [DOI] [Google Scholar]
- 27.Niemantsverdriet E., Valckx S., Bjerke M., Engelborghs S. Alzheimer’s Disease CSF Biomarkers: Clinical Indications and Rational Use. Acta Neurol. Belg. 2017;117:591–602. doi: 10.1007/s13760-017-0816-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katayama T., Sawada J., Takahashi K., Yahara O. Cerebrospinal Fluid Biomarkers in Parkinson’s Disease: A Critical Overview of the Literature and Meta-Analyses. Brain Sci. 2020;10:466. doi: 10.3390/brainsci10070466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parnetti L., Gaetani L., Eusebi P., Paciotti S., Hansson O., El-Agnaf O., Mollenhauer B., Blennow K., Calabresi P. CSF and Blood Biomarkers for Parkinson’s Disease. Lancet Neurol. 2019;18:573–586. doi: 10.1016/S1474-4422(19)30024-9. [DOI] [PubMed] [Google Scholar]
- 30.Yang J., Hamade M., Wu Q., Wang Q., Axtell R., Giri S., Mao-Draayer Y. Current and Future Biomarkers in Multiple Sclerosis. Int. J. Mol. Sci. 2022;23:5877. doi: 10.3390/ijms23115877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deisenhammer F., Zetterberg H., Fitzner B., Zettl U.K. The Cerebrospinal Fluid in Multiple Sclerosis. Front. Immunol. 2019;10:726. doi: 10.3389/fimmu.2019.00726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mankhong S., Kim S., Lee S., Kwak H.B., Park D.H., Joa K.L., Kang J.H. Development of Alzheimer’s Disease Biomarkers: From CSF-to Blood-Based Biomarkers. Biomedicines. 2022;10:850. doi: 10.3390/biomedicines10040850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.D’Ambrosio A., Pontecorvo S., Colasanti T., Zamboni S., Francia A., Margutti P. Peripheral Blood Biomarkers in Multiple Sclerosis. Autoimmun. Rev. 2015;14:1097–1110. doi: 10.1016/j.autrev.2015.07.014. [DOI] [PubMed] [Google Scholar]
- 34.Feng L., Li J., Zhang R. Current Research Status of Blood Biomarkers in Alzheimer’s Disease: Diagnosis and Prognosis. Ageing Res. Rev. 2021;72:101492. doi: 10.1016/j.arr.2021.101492. [DOI] [PubMed] [Google Scholar]
- 35.Park S.A., Jang Y.J., Kim M.K., Lee S.M., Moon S.Y. Promising Blood Biomarkers for Clinical Use in Alzheimer’s Disease: A Focused Update. J. Clin. Neurol. 2022;18:401–409. doi: 10.3988/jcn.2022.18.4.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Obrocki P., Khatun A., Ness D., Senkevich K., Hanrieder J., Capraro F., Mattsson N., Andreasson U., Portelius E., Ashton N.J., et al. Perspectives in Fluid Biomarkers in Neurodegeneration from the 2019 Biomarkers in Neurodegenerative Diseases Course—A Joint PhD Student Course at University College London and University of Gothenburg. Alzheimers. Res. Ther. 2020;12:20. doi: 10.1186/s13195-020-00586-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roser A.E., Gomes L.C., Schünemann J., Maass F., Lingor P. Circulating MiRNAs as Diagnostic Biomarkers for Parkinson’s Disease. Front. Neurosci. 2018;12:625. doi: 10.3389/fnins.2018.00625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Swarbrick S., Wragg N., Ghosh S., Stolzing A. Systematic Review of MiRNA as Biomarkers in Alzheimer’s Disease. Mol. Neurobiol. 2019;56:6156–6167. doi: 10.1007/s12035-019-1500-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Søndergaard H.B., Hesse D., Krakauer M., Sørensen P.S., Sellebjerg F. Differential MicroRNA Expression in Blood in Multiple Sclerosis. Mult. Scler. J. 2013;19:1849–1857. doi: 10.1177/1352458513490542. [DOI] [PubMed] [Google Scholar]
- 40.Wang Z., Lu Y., Han J. Wang Peripheral Blood MicroRNAs: A Novel Tool for Diagnosing Disease? Intractable Rare Dis. Res. 2012;1:98–102. doi: 10.5582/irdr.2012.v1.3.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zou K., Abdullah M., Michikawa M. Current Biomarkers for Alzheimer’s Disease: From CSF to Blood. J. Pers. Med. 2020;10:85. doi: 10.3390/jpm10030085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Janeiro M.H., Ardanaz C.G., Sola-Sevilla N., Dong J., Cortés-Erice M., Solas M., Puerta E., Ramírez M.J. Biomarkers in Alzheimer’s Disease. Adv. Lab. Med. / Av. en Med. Lab. 2021;2:27–37. doi: 10.1515/almed-2020-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zakrzewski W., Dobrzyński M., Szymonowicz M., Rybak Z. Stem Cells: Past, Present, and Future. Stem Cell Res. Ther. 2019;10:68. doi: 10.1186/s13287-019-1165-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hung C.W., Liou Y.J., Lu S.W., Tseng L.M., Kao C.L., Chen S.J., Chiou S.H., Chang C.J. Stem Cell-Based Neuroprotective and Neurorestorative Strategies. Int. J. Mol. Sci. 2010;11:2039–2055. doi: 10.3390/ijms11052039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoang D.M., Pham P.T., Bach T.Q., Ngo A.T.L., Nguyen Q.T., Phan T.T.K., Nguyen G.H., Le P.T.T., Hoang V.T., Forsyth N.R., et al. Stem Cell-Based Therapy for Human Diseases. Signal Transduct. Target. Ther. 2022;7:272. doi: 10.1038/s41392-022-01134-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim J., Koo B.-K., Knoblich J.A. Human Organoids: Model Systems for Human Biology and Medicine. Nat. Rev. Mol. Cell Biol. 2020;21:571–584. doi: 10.1038/s41580-020-0259-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kabir M.H., O’Connor M.D. Stems Cells, Big Data and Compendium-Based Analyses for Identifying Cell Types, Signalling Pathways and Gene Regulatory Networks. Biophys. Rev. 2019;11:41–50. doi: 10.1007/s12551-018-0486-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Müller G.A., Tarasov K.V., Gundry R.L., Boheler K.R. Human ESC/IPSC-Based “omics” and Bioinformatics for Translational Research. Drug Discov. Today Dis. Model. 2012;9:e161–e170. doi: 10.1016/j.ddmod.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Novak G., Kyriakis D., Grzyb K., Bernini M., Rodius S., Dittmar G., Finkbeiner S., Skupin A. Single-Cell Transcriptomics of Human IPSC Differentiation Dynamics Reveal a Core Molecular Network of Parkinson’s Disease. Commun. Biol. 2022;5:49. doi: 10.1038/s42003-021-02973-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Billing A.M., Dib S.S., Bhagwat A.M., da Silva I.T., Drummond R.D., Hayat S., Al-Mismar R., Ben-Hamidane H., Goswami N., Engholm-Keller K., et al. A Systems-Level Characterization of the Differentiation of Human Embryonic Stem Cells into Mesenchymal Stem Cells. Mol. Cell. Proteomics. 2019;18:1950–1966. doi: 10.1074/mcp.RA119.001356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yagi T., Ito D., Okada Y., Akamatsu W., Nihei Y., Yoshizaki T., Yamanaka S., Okano H., Suzuki N. Modeling Familial Alzheimer’s Disease with Induced Pluripotent Stem Cells. Hum. Mol. Genet. 2011;20:4530–4539. doi: 10.1093/hmg/ddr394. [DOI] [PubMed] [Google Scholar]
- 52.Pandey S., Jirásko M., Lochman J., Chvátal A., Chottova Dvorakova M., Kučera R. IPSCs in Neurodegenerative Disorders: A Unique Platform for Clinical Research and Personalized Medicine. J. Pers. Med. 2022;12:1485. doi: 10.3390/jpm12091485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kang W., Jin T., Zhang T., Ma S., Yan H., Liu Z., Ji Z., Cai Y., Wang S., Song M., et al. Regeneration Roadmap: Database Resources for Regenerative Biology. Nucleic Acids Res. 2022;50:D1085–D1090. doi: 10.1093/nar/gkab870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hyvärinen E., Savolainen M., Mikkonen J.J.W., Kullaa A.M. Salivary Metabolomics for Diagnosis and Monitoring Diseases: Challenges and Possibilities. Metabolites. 2021;11:587. doi: 10.3390/metabo11090587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Minale G., Saesong T., Temkitthawon P., Waranuch N., Nuengchamnong N., Chootip K., Kamkaew N., Kongbangkerd T., Engsuwan J., Ingkaninan K. Characterization of Metabolites in Plasma, Urine and Feces of Healthy Participants after Taking Brahmi Essence for Twelve Weeks Using Lc-Esi-Qtof-Ms Metabolomic Approach. Molecules. 2021;26:2944. doi: 10.3390/molecules26102944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim Y., Koh I.S., Rho M. Deciphering the Human Microbiome Using Next-Generation Sequencing Data and Bioinformatics Approaches. Methods. 2015;79:52–59. doi: 10.1016/j.ymeth.2014.10.022. [DOI] [PubMed] [Google Scholar]
- 57.Yao F., Hong X., Li S., Zhang Y., Zhao Q., Du W., Wang Y., Ni J. Urine-Based Biomarkers for Alzheimer’s Disease Identified Through Coupling Computational and Experimental Methods. J. Alzheimers. Dis. 2018;65:421–431. doi: 10.3233/JAD-180261. [DOI] [PubMed] [Google Scholar]
- 58.Gomez J.L., Chen A., Diaz M.P., Zirn N., Gupta A., Britto C., Sauler M., Yan X., Stewart E., Santerian K., et al. A Network of Sputum Micrornas Is Associated with Neutrophilic Airway Inflammation in Asthma. Am. J. Respir. Crit. Care Med. 2020;202:51–64. doi: 10.1164/rccm.201912-2360OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shao L., Liao J., Qian J., Chen W., Fan X. MetaGeneBank: A Standardized Database to Study Deep Sequenced Metagenomic Data from Human Fecal Specimen. BMC Microbiol. 2021;21:263. doi: 10.1186/s12866-021-02321-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jangi S., Gandhi R., Cox L.M., Li N., Von Glehn F., Yan R., Patel B., Mazzola M.A., Liu S., Glanz B.L., et al. Alterations of the Human Gut Microbiome in Multiple Sclerosis. Nat. Commun. 2016;7:12015. doi: 10.1038/ncomms12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang Y., Kasper L.H. The Role of Microbiome in Central Nervous System Disorders. Brain. Behav. Immun. 2014;38:1–12. doi: 10.1016/j.bbi.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lipponen A., Natunen T., Hujo M., Ciszek R., Hämäläinen E., Tohka J., Hiltunen M., Paananen J., Poulsen D., Kansanen E., et al. In Vitro and In Vivo Pipeline for Validation of Disease-Modifying Effects of Systems Biology-Derived Network Treatments for Traumatic Brain Injury—Lessons Learned. Int. J. Mol. Sci. 2019;20:5395. doi: 10.3390/ijms20215395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Collaborators G.B.D. 2017 U.S.N.D. Burden of Neurological Disorders Across the US From 1990-2017: A Global Burden of Disease Study. JAMA Neurol. 2021;78:165–176. doi: 10.1001/jamaneurol.2020.4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gan L., Cookson M.R., Petrucelli L., La Spada A.R. Converging Pathways in Neurodegeneration, from Genetics to Mechanisms. Nat. Neurosci. 2018;21:1300–1309. doi: 10.1038/s41593-018-0237-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cree B.A.C., Arnold D.L., Chataway J., Chitnis T., Fox R.J., Ramajo A.P., Murphy N., Lassmann H. Secondary Progressive Multiple Sclerosis. Neurology. 2021;97:378 LP–388. doi: 10.1212/WNL.0000000000012323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thakur S., Dhapola R., Sarma P., Medhi B., Reddy D.H. Neuroinflammation in Alzheimer’s Disease: Current Progress in Molecular Signaling and Therapeutics. Inflammation. 2023;46:1–17. doi: 10.1007/s10753-022-01721-1. [DOI] [PubMed] [Google Scholar]
- 67.Dong-Chen X., Yong C., Yang X., Chen-Yu S., Li-Hua P. Signaling Pathways in Parkinson’s Disease: Molecular Mechanisms and Therapeutic Interventions. Signal Transduct. Target. Ther. 2023;8:73. doi: 10.1038/s41392-023-01353-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wilson D.M., 3rd, Cookson M.R., Van Den Bosch L., Zetterberg H., Holtzman D.M., Dewachter I. Hallmarks of Neurodegenerative Diseases. Cell. 2023;186:693–714. doi: 10.1016/j.cell.2022.12.032. [DOI] [PubMed] [Google Scholar]
- 69.Lo C.H., Sachs J.N. The Role of Wild-Type Tau in Alzheimer’s Disease and Related Tauopathies. J. life Sci. 2020;2:1–17. doi: 10.36069/JoLS/20201201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lo C.H. Heterogeneous Tau Oligomers as Molecular Targets for for Alzheimer’s Disease and Related Tauopathies. Biophysica. 2022;2:440–451. doi: 10.3390/biophysica2040039. [DOI] [Google Scholar]
- 71.Lo C.H. Recent Advances in Cellular Biosensor Technology to Investigate Tau Oligomerization. Bioeng. Transl. Med. 2021;6:e10231. doi: 10.1002/btm2.10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lo C.H., Lim C.K.W., Ding Z., Wickramasinghe S.P., Braun A.R., Ashe K.H., Rhoades E., Thomas D.D., Sachs J.N. Targeting the Ensemble of Heterogeneous Tau Oligomers in Cells: A Novel Small Molecule Screening Platform for Tauopathies. Alzheimer’s Dement. 2019;15:1489–1502. doi: 10.1016/j.jalz.2019.06.4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McAlary L., Plotkin S.S., Cashman N.R. Emerging Developments in Targeting Proteotoxicity in Neurodegenerative Diseases. CNS Drugs. 2019;33:883–904. doi: 10.1007/s40263-019-00657-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dong Y., Dekens D.W., De Deyn P.P., Naudé P.J.W., Eisel U.L.M. Targeting of Tumor Necrosis Factor Alpha Receptors as a Therapeutic Strategy for Neurodegenerative Disorders. Antibodies. 2015;4:369–408. doi: 10.3390/antib4040369. [DOI] [Google Scholar]
- 75.Ghosh R., Tabrizi S.J. Gene Suppression Approaches to Neurodegeneration. Alzheimer’s Res. Ther. 2017;9:82. doi: 10.1186/s13195-017-0307-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sealfon S.C., Chu T.T. Methods in Molecular Biology. Volume 671. Humana Press; Totowa, NJ, USA: 2011. RNA and DNA Microarrays; pp. 3–34. [DOI] [PubMed] [Google Scholar]
- 77.Kodama Y., Mashima J., Kosuge T., Ogasawara O. DDBJ Update: The Genomic Expression Archive (GEA) for Functional Genomics Data. Nucleic Acids Res. 2019;47:D69–D73. doi: 10.1093/nar/gky1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bono H. All of Gene Expression (AOE): An Integrated Index for Public Gene Expression Databases. PLoS ONE. 2020;15:e0227076. doi: 10.1371/journal.pone.0227076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M., et al. NCBI GEO: Archive for Functional Genomics Data Sets-Update. Nucleic Acids Res. 2013;41:D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Alonso-Betanzos A., Bolón-Canedo V., Morán-Fernández L., Sánchez-Maroño N. A Review of Microarray Datasets: Where to Find Them and Specific Characteristics. Methods Mol. Biol. 2019;1986:65–85. doi: 10.1007/978-1-4939-9442-7_4. [DOI] [PubMed] [Google Scholar]
- 81.Park T., Yi S.-G., Kang S.-H., Lee S., Lee Y.-S., Simon R. Evaluation of Normalization Methods for Microarray Data. BMC Bioinformatics. 2003;4:33. doi: 10.1186/1471-2105-4-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhou Q., Su X., Jing G., Chen S., Ning K. RNA-QC-Chain: Comprehensive and Fast Quality Control for RNA-Seq Data. BMC Genomics. 2018;19:144. doi: 10.1186/s12864-018-4503-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li Z., Xie W., Liu T. Efficient Feature Selection and Classification for Microarray Data. PLoS ONE. 2018;13:e0202167. doi: 10.1371/journal.pone.0202167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Townes F.W., Hicks S.C., Aryee M.J., Irizarry R.A. Feature Selection and Dimension Reduction for Single-Cell RNA-Seq Based on a Multinomial Model. Genome Biol. 2019;20:295. doi: 10.1186/s13059-019-1861-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dennis G., Sherman B.T., Hosack D.A., Yang J., Gao W., Lane H.C., Lempicki R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:R60. doi: 10.1186/gb-2003-4-9-r60. [DOI] [PubMed] [Google Scholar]
- 87.Krämer A., Green J., Pollard J.J., Tugendreich S. Causal Analysis Approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30:523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gu Z., Liu J., Cao K., Zhang J., Wang J. Centrality-Based Pathway Enrichment: A Systematic Approach for Finding Significant Pathways Dominated by Key Genes. BMC Syst. Biol. 2012;6:56. doi: 10.1186/1752-0509-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.SPIA: Signaling Pathway Impact Analysis (SPIA) Using Combined Evidence of Pathway Over-Representation and Unusual Signaling Perturbations. [(accessed on 21 July 2023)]. Available online: https://rdrr.io/bioc/SPIA/
- 91.Pathan M., Keerthikumar S., Ang C.-S., Gangoda L., Quek C.Y.J., Williamson N.A., Mouradov D., Sieber O.M., Simpson R.J., Salim A., et al. FunRich: An Open Access Standalone Functional Enrichment and Interaction Network Analysis Tool. Proteomics. 2015;15:2597–2601. doi: 10.1002/pmic.201400515. [DOI] [PubMed] [Google Scholar]
- 92.Liu P., Ewald J., Pang Z., Legrand E., Jeon Y.S., Sangiovanni J., Hacariz O., Zhou G., Head J.A., Basu N., et al. ExpressAnalyst: A Unified Platform for RNA-Sequencing Analysis in Non-Model Species. Nat. Commun. 2023;14:2995. doi: 10.1038/s41467-023-38785-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Szklarczyk D., Gable A.L., Lyon D., Junge A., Wyder S., Huerta-Cepas J., Simonovic M., Doncheva N.T., Morris J.H., Bork P., et al. STRING V11: Protein-Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019;47:D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Stark C., Breitkreutz B.-J., Reguly T., Boucher L., Breitkreutz A., Tyers M. BioGRID: A General Repository for Interaction Datasets. Nucleic Acids Res. 2006;34:D535-9. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhou G., Soufan O., Ewald J., Hancock R.E.W., Basu N., Xia J. NetworkAnalyst 3.0: A Visual Analytics Platform for Comprehensive Gene Expression Profiling and Meta-Analysis. Nucleic Acids Res. 2019;47:W234–W241. doi: 10.1093/nar/gkz240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lv L., Zhang D., Hua P., Yang S. The Glial-Specific Hypermethylated 3′ Untranslated Region of Histone Deacetylase 1 May Modulates Several Signal Pathways in Alzheimer’s Disease. Life Sci. 2021;265:118760. doi: 10.1016/j.lfs.2020.118760. [DOI] [PubMed] [Google Scholar]
- 97.Yu W.W., Yu W.W., Yang Y., Lü Y. Exploring the Key Genes and Identification of Potential Diagnosis Biomarkers in Alzheimer’s Disease Using Bioinformatics Analysis. Front. Aging Neurosci. 2021;13:602781. doi: 10.3389/fnagi.2021.602781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ma G., Liu M., Du K., Zhong X., Gong S., Jiao L., Wei M. Differential Expression of MRNAs in the Brain Tissues of Patients with Alzheimer’s Disease Based on GEO Expression Profile and Its Clinical Significance. Biomed Res. Int. 2019;2019:1–9. doi: 10.1155/2019/8179145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang Q., Li J., Weng L. Identification and Validation of Aging-Related Genes in Alzheimer’s Disease. Front. Neurosci. 2022;16:905722. doi: 10.3389/fnins.2022.905722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang H.H., Zhang Y., Zheng C., Yang S., Chen X., Wang H.H., Gao S. A 3-Gene-Based Diagnostic Signature in Alzheimer’s Disease. Eur. Neurol. 2022;85:6–13. doi: 10.1159/000518727. [DOI] [PubMed] [Google Scholar]
- 101.Song S., Li B., Jia Z., Guo L. Sirtuin 3 MRNA Expression Is Downregulated in the Brain Tissues of Alzheimer’s Disease Patients: A Bioinformatic and Data Mining Approach. Med. Sci. Monit. 2020;26:e923547. doi: 10.12659/MSM.923547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pang X., Zhao Y., Wang J., Zhou Q., Xu L., Kang D., Liu A.L., Du G.H. The Bioinformatic Analysis of the Dysregulated Genes and MicroRNAs in Entorhinal Cortex, Hippocampus, and Blood for Alzheimer’s Disease. Biomed Res. Int. 2017;2017:1–16. doi: 10.1155/2017/9084507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li X., Wang H., Long J., Pan G., He T., Anichtchik O., Belshaw R., Albani D., Edison P., Green E.K., et al. Systematic Analysis and Biomarker Study for Alzheimer’s Disease. Sci. Rep. 2018;8:17394. doi: 10.1038/s41598-018-35789-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Qin H., Hu C., Zhao X., Tian M., Zhu B. Usefulness of Candidate MRNAs and MiRNAs as Biomarkers for Mild Cognitive Impairment and Alzheimer’s Disease. Int. J. Neurosci. 2021;133:89–102. doi: 10.1080/00207454.2021.1886098. [DOI] [PubMed] [Google Scholar]
- 105.Liu W., Lin H., He X., Chen L.L., Dai Y., Jia W., Xue X., Tao J., Chen L.L. Neurogranin as a Cognitive Biomarker in Cerebrospinal Fluid and Blood Exosomes for Alzheimer’s Disease and Mild Cognitive Impairment. Transl. Psychiatry. 2020;10:125. doi: 10.1038/s41398-020-0801-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang B., Gaiteri C., Bodea L.G., Wang Z., McElwee J., Podtelezhnikov A.A., Zhang C., Xie T., Tran L., Dobrin R., et al. Integrated Systems Approach Identifies Genetic Nodes and Networks in Late-Onset Alzheimer’s Disease. Cell. 2013;153:707–720. doi: 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yao F., Zhang K., Zhang Y., Guo Y., Li A., Xiao S., Liu Q., Shen L., Ni J. Identification of Blood Biomarkers for Alzheimer’s Disease through Computational Prediction and Experimental Validation. Front. Neurol. 2019;9:1158. doi: 10.3389/fneur.2018.01158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bao J., Chang W., Zhao Y. Diagnosis and Drug Prediction of Parkinson’s Disease Based on Immune-Related Genes. J. Mol. Neurosci. 2022;72:1809–1819. doi: 10.1007/s12031-022-02043-5. [DOI] [PubMed] [Google Scholar]
- 109.Tan C., Liu X., Chen J. Microarray Analysis of the Molecular Mechanism Involved in Parkinson’s Disease. Parkinsons. Dis. 2018;2018:1–12. doi: 10.1155/2018/1590465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chi J., Xie Q., Jia J., Liu X., Sun J., Deng Y., Yi L. Integrated Analysis and Identification of Novel Biomarkers in Parkinson’s Disease. Front. Aging Neurosci. 2018;10:178. doi: 10.3389/fnagi.2018.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shehadeh L.A., Yu K., Wang L., Guevara A., Singer C., Vance J., Papapetropoulos S. SRRM2, a Potential Blood Biomarker Revealing High Alternative Splicing in Parkinson’s Disease. PLoS ONE. 2010;5:e9104. doi: 10.1371/journal.pone.0009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li Z., Liu Y., Jia A., Cui Y., Feng J. Cerebrospinal Fluid Cells Immune Landscape in Multiple Sclerosis. J. Transl. Med. 2021;19:125. doi: 10.1186/s12967-021-02804-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hagan N., Kane J.L., Grover D., Woodworth L., Madore C., Saleh J., Sancho J., Liu J., Li Y., Proto J., et al. CSF1R Signaling Is a Regulator of Pathogenesis in Progressive MS. Cell Death Dis. 2020;11:904. doi: 10.1038/s41419-020-03084-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Olcum M., Tastan B., Kiser C., Genc S., Genc K. Microglial NLRP3 Inflammasome Activation in Multiple Sclerosis. Adv. Protein Chem. Struct. Biol. 2020;119:247–308. doi: 10.1016/bs.apcsb.2019.08.007. [DOI] [PubMed] [Google Scholar]
- 115.Van Wageningen T.A., Gerrits E., Brouwer N., Breve J.J.P., Geurts J.J.G., Eggen B.J.L., Erik Boddeke H.W.G.M., Van Dam A.M. Distinct Gene Expression in Demyelinated White and Grey Matter Areas of Patients with Multiple Sclerosis. Brain Commun. 2022;4:fcac005. doi: 10.1093/braincomms/fcac005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Qiu D., Zhang D., Yu Z., Jiang Y., Zhu D. Bioinformatics Approach Reveals the Critical Role of the NOD-like Receptor Signaling Pathway in COVID-19-Associated Multiple Sclerosis Syndrome. J. Neural Transm. 2022;129:1031–1038. doi: 10.1007/s00702-022-02518-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Moni M.A., Rana H.K., Islam M.B., Ahmed M.B., Xu H., Hasan M.A.M., Lei Y., Quinn J.M.W. A Computational Approach to Identify Blood Cell-Expressed Parkinson’s Disease Biomarkers That Are Coordinately Expressed in Brain Tissue. Comput. Biol. Med. 2019;113:103385. doi: 10.1016/j.compbiomed.2019.103385. [DOI] [PubMed] [Google Scholar]
- 118.Chiu I.M., Morimoto E.T.A., Goodarzi H., Liao J.T., O’Keeffe S., Phatnani H.P., Muratet M., Carroll M.C., Levy S., Tavazoie S., et al. A Neurodegeneration-Specific Gene-Expression Signature of Acutely Isolated Microglia from an Amyotrophic Lateral Sclerosis Mouse Model. Cell Rep. 2013;4:385–401. doi: 10.1016/j.celrep.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Premkumar T., Sajitha Lulu S. Molecular Crosstalk between COVID-19 and Alzheimer’s Disease Using Microarray and RNA-Seq Datasets: A System Biology Approach. Front. Med. 2023;10:1151046. doi: 10.3389/fmed.2023.1151046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Irmady K., Hale C.R., Qadri R., Fak J., Simelane S., Carroll T., Przedborski S., Darnell R.B. Blood Transcriptomic Signatures Associated with Molecular Changes in the Brain and Clinical Outcomes in Parkinson’s Disease. Nat. Commun. 2023;14:3956. doi: 10.1038/s41467-023-39652-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Salta E., De Strooper B. Noncoding RNAs in Neurodegeneration. Nat. Rev. Neurosci. 2017;18:627–640. doi: 10.1038/nrn.2017.90. [DOI] [PubMed] [Google Scholar]
- 122.Salta E., De Strooper B. Non-Coding RNAs with Essential Roles in Neurodegenerative Disorders. Lancet Neurol. 2012;11:189–200. doi: 10.1016/S1474-4422(11)70286-1. [DOI] [PubMed] [Google Scholar]
- 123.Latowska J., Grabowska A., Zarębska Ż., Kuczyński K., Kuczyńska B., Rolle K. Non-Coding RNAs in Brain Tumors, the Contribution of LncRNAs, CircRNAs, and SnoRNAs to Cancer Development—Their Diagnostic and Therapeutic Potential. Int. J. Mol. Sci. 2020;21:7001. doi: 10.3390/ijms21197001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang J., Yan S., Yang J., Lu H., Xu D., Wang Z. Non-Coding RNAs in Rheumatoid Arthritis: From Bench to Bedside. Front. Immunol. 2020;10:3129. doi: 10.3389/fimmu.2019.03129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wu J., Xiao J., Zhang Z., Wang X., Hu S., Yu J. Ribogenomics: The Science and Knowledge of RNA. Genomics, Proteomics Bioinforma. 2014;12:57–63. doi: 10.1016/j.gpb.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Condrat C.E., Thompson D.C., Barbu M.G., Bugnar O.L., Boboc A., Cretoiu D., Suciu N., Cretoiu S.M., Voinea S.C. MiRNAs as Biomarkers in Disease: Latest Findings Regarding Their Role in Diagnosis and Prognosis. Cells. 2020;9:276. doi: 10.3390/cells9020276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nagaraj S., Zoltowska K.M., Laskowska-Kaszub K., Wojda U. MicroRNA Diagnostic Panel for Alzheimer’s Disease and Epigenetic Trade-off between Neurodegeneration and Cancer. Ageing Res. Rev. 2019;49:125–143. doi: 10.1016/j.arr.2018.10.008. [DOI] [PubMed] [Google Scholar]
- 128.Yang Q., Pan W., Qian L. Identification of the MiRNA–MRNA Regulatory Network in Multiple Sclerosis. Neurol. Res. 2017;39:142–151. doi: 10.1080/01616412.2016.1250857. [DOI] [PubMed] [Google Scholar]
- 129.Ehya F., Tehrani H.A., Garshasbi M., Nabavi S.M. Identification of MiR-24 and MiR-137 as Novel Candidate Multiple Sclerosis MiRNA Biomarkers Using Multi-Staged Data Analysis Protocol. Mol. Biol. Res. Commun. 2017;6:127–140. doi: 10.22099/mbrc.2017.24861.1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Faruqui N.A., Prium D.H., Afrin Mowna S., Rahaman T.I., Dutta A.R., Farjana Akter M. Identification of Common Molecular Signatures Shared between Alzheimer’s and Parkinson’s Diseases and Therapeutic Agents Exploration: An Integrated Genomics Approach. bioRxiv. 2021 bioRxiv:2020.12.31.424962. [Google Scholar]
- 131.Lu L., Dai W.Z., Zhu X.C., Ma T. Analysis of Serum MiRNAs in Alzheimer’s Disease. Am. J. Alzheimers. Dis. Other Demen. 2021;36:153331752110217. doi: 10.1177/15333175211021712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Candido S., Lupo G., Pennisi M., Basile M.S., Anfuso C.D., Petralia M.C., Gattuso G., Vivarelli S., Spandidos D.A., Libra M., et al. The Analysis of MiRNA Expression Profiling Datasets Reveals Inverse MicroRNA Patterns in Glioblastoma and Alzheimer’s Disease. Oncol. Rep. 2019;42:911–922. doi: 10.3892/or.2019.7215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Li H., Zou L., Shi J., Han X. Bioinformatics Analysis of Differentially Expressed Genes and Identification of an MiRNA–MRNA Network Associated with Entorhinal Cortex and Hippocampus in Alzheimer’s Disease. Hereditas. 2021;158:25. doi: 10.1186/s41065-021-00190-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hu Y., Zhang Y., Ren R., Dammer E.B., Xie X., Chen S., Huang Q., Huang W., Zhang R., Chen H., et al. MicroRNA-425 Loss Mediates Amyloid Plaque Microenvironment Heterogeneity and Promotes Neurodegenerative Pathologies. Aging Cell. 2021;20:e13454. doi: 10.1111/acel.13454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sabaie H., Talebi M., Gharesouarn J., Asadi M.R., Jalaiei A., Arsang-Jang S., Hussen B.M., Taheri M., Jalili Khoshnoud R., Rezazadeh M. Identification and Analysis of BCAS4/Hsa-MiR-185-5p/SHISA7 Competing Endogenous RNA Axis in Late-Onset Alzheimer’s Disease Using Bioinformatic and Experimental Approaches. Front. Aging Neurosci. 2022;14:812169. doi: 10.3389/fnagi.2022.812169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Brito L.M., Ribeiro-dos-Santos Â., Vidal A.F., de Araújo G.S. Differential Expression and MiRNA–Gene Interactions in Early and Late Mild Cognitive Impairment. Biology. 2020;9:251. doi: 10.3390/biology9090251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Soreq L., Salomonis N., Bronstein M., Greenberg D., Israel Z., Bergman H., Soreq H. Small RNA Sequencing-Microarray Analyses in Parkinson Leukocytes Reveal Deep Brain Stimulation-Induced Splicing Changes That Classify Brain Region Transcriptomes. Front. Mol. Neurosci. 2013;6:10. doi: 10.3389/fnmol.2013.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Scimone C., Donato L., Alafaci C., Granata F., Rinaldi C., Longo M., D’Angelo R., Sidoti A. High-Throughput Sequencing to Detect Novel Likely Gene-Disrupting Variants in Pathogenesis of Sporadic Brain Arteriovenous Malformations. Front. Genet. 2020;11:146. doi: 10.3389/fgene.2020.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Donato L., Scimone C., Alibrandi S., Scalinci S.Z., Rinaldi C., D’Angelo R., Sidoti A. Epitranscriptome Analysis of Oxidative Stressed Retinal Epithelial Cells Depicted a Possible RNA Editing Landscape of Retinal Degeneration. Antioxidants. 2022;11:1967. doi: 10.3390/antiox11101967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Donato L., Alibrandi S., Scimone C., Rinaldi C., Dascola A., Calamuneri A., D’Angelo R., Sidoti A. The Impact of Modifier Genes on Cone-Rod Dystrophy Heterogeneity: An Explorative Familial Pilot Study and a Hypothesis on Neurotransmission Impairment. PLoS ONE. 2022;17:e0278857. doi: 10.1371/journal.pone.0278857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chen L., Wang C., Sun H., Wang J., Liang Y., Wang Y., Wong G. The Bioinformatics Toolbox for CircRNA Discovery and Analysis. Brief. Bioinform. 2021;22:1706–1728. doi: 10.1093/bib/bbaa001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cochran K.R., Veeraraghavan K., Kundu G., Mazan-Mamczarz K., Coletta C., Thambisetty M., Gorospe M., De S. Systematic Identification of Circrnas in Alzheimer’s Disease. Genes. 2021;12:1258. doi: 10.3390/genes12081258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Junn E., Lee K.-W., Jeong B.S., Chan T.W., Im J.-Y., Mouradian M.M. Repression of α-Synuclein Expression and Toxicity by MicroRNA-7. Proc. Natl. Acad. Sci. USA. 2009;106:13052–13057. doi: 10.1073/pnas.0906277106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Dolinar A., Koritnik B., Glavač D., Ravnik-Glavač M. Circular RNAs as Potential Blood Biomarkers in Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2019;56:8052–8062. doi: 10.1007/s12035-019-1627-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Li K., Wang Z. LncRNA NEAT1: Key Player in Neurodegenerative Diseases. Ageing Res. Rev. 2023;86:101878. doi: 10.1016/j.arr.2023.101878. [DOI] [PubMed] [Google Scholar]
- 146.Gong Y., Zhu W., Sun M., Shi L. Bioinformatics Analysis of Long Non-Coding RNA and Related Diseases: An Overview. Front. Genet. 2021;12:813873. doi: 10.3389/fgene.2021.813873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chi L.M., Wang L.P., Jiao D. Identification of Differentially Expressed Genes and Long Noncoding RNAs Associated with Parkinson’s Disease. Parkinsons. Dis. 2019;2019:1–7. doi: 10.1155/2019/6078251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wu J., Chen L., Zheng C., Xu S., Gao Y., Wang J. Co-Expression Network Analysis Revealing the Potential Regulatory Roles of LncRNAs in Alzheimer’s Disease. Interdiscip. Sci. Comput. Life Sci. 2019;11:645–654. doi: 10.1007/s12539-019-00319-w. [DOI] [PubMed] [Google Scholar]
- 149.Han Z., Xue W., Tao L., Zhu F. Identification of Key Long Non-Coding RNAs in the Pathology of Alzheimer’s Disease and Their Functions Based on Genome-Wide Associations Study, Microarray, and RNA-Seq Data. J. Alzheimers. Dis. 2019;68:339–355. doi: 10.3233/JAD-181051. [DOI] [PubMed] [Google Scholar]
- 150.van der Kloet F.M., Buurmans J., Jonker M.J., Smilde A.K., Westerhuis J.A. Increased Comparability between RNA-Seq and Microarray Data by Utilization of Gene Sets. PLOS Comput. Biol. 2020;16:e1008295. doi: 10.1371/journal.pcbi.1008295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Jain M., Abu-Shumays R., Olsen H.E., Akeson M. Advances in Nanopore Direct RNA Sequencing. Nat. Methods. 2022;19:1160–1164. doi: 10.1038/s41592-022-01633-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sena J.A., Galotto G., Devitt N.P., Connick M.C., Jacobi J.L., Umale P.E., Vidali L., Bell C.J. Unique Molecular Identifiers Reveal a Novel Sequencing Artefact with Implications for RNA-Seq Based Gene Expression Analysis. Sci. Rep. 2018;8:13121. doi: 10.1038/s41598-018-31064-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hwang B., Lee J.H., Bang D. Single-Cell RNA Sequencing Technologies and Bioinformatics Pipelines. Exp. Mol. Med. 2018;50:1–14. doi: 10.1038/s12276-018-0071-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Slovin S., Carissimo A., Panariello F., Grimaldi A., Bouché V., Gambardella G., Cacchiarelli D. Single-Cell RNA Sequencing Analysis: A Step-by-Step Overview. Methods Mol Biol. 2021;2284:343–365. doi: 10.1007/978-1-0716-1307-8_19. [DOI] [PubMed] [Google Scholar]
- 155.Svensson V., da Veiga Beltrame E., Pachter L. A Curated Database Reveals Trends in Single-Cell Transcriptomics. Database. 2020;2020:baaa073. doi: 10.1093/database/baaa073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ma S.-X., Lim S.B. Single-Cell RNA Sequencing in Parkinson’s Disease. Biomed. 2021;9:368. doi: 10.3390/biomedicines9040368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kamme F., Salunga R., Yu J., Tran D.-T., Zhu J., Luo L., Bittner A., Guo H.-Q., Miller N., Wan J., et al. Single-Cell Microarray Analysis in Hippocampus CA1: Demonstration and Validation of Cellular Heterogeneity. J. Neurosci. Off. J. Soc. Neurosci. 2003;23:3607–3615. doi: 10.1523/JNEUROSCI.23-09-03607.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Rao M.S., Van Vleet T.R., Ciurlionis R., Buck W.R., Mittelstadt S.W., Blomme E.A.G., Liguori M.J. Comparison of RNA-Seq and Microarray Gene Expression Platforms for the Toxicogenomic Evaluation of Liver from Short-Term Rat Toxicity Studies. Front. Genet. 2019;9:636. doi: 10.3389/fgene.2018.00636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.GEO2R. [(accessed on 21 July 2023)]; Available online: https://www.ncbi.nlm.nih.gov/geo/info/geo2r.html.
- 160.Amaral M.L., Erikson G.A., Shokhirev M.N. BART: Bioinformatics Array Research Tool. BMC Bioinformatics. 2018;19:296. doi: 10.1186/s12859-018-2308-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Perampalam P., Dick F.A. BEAVR: A Browser-Based Tool for the Exploration and Visualization of RNA-Seq Data. BMC Bioinformatics. 2020;21:221. doi: 10.1186/s12859-020-03549-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Teichman G., Cohen D., Ganon O., Dunsky N., Shani S., Gingold H., Rechavi O. RNAlysis: Analyze Your RNA Sequencing Data without Writing a Single Line of Code. BMC Biol. 2023;21:74. doi: 10.1186/s12915-023-01574-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.La Ferlita A., Alaimo S., Di Bella S., Martorana E., Laliotis G.I., Bertoni F., Cascione L., Tsichlis P.N., Ferro A., Bosotti R., et al. RNAdetector: A Free User-Friendly Stand-Alone and Cloud-Based System for RNA-Seq Data Analysis. BMC Bioinformatics. 2021;22:298. doi: 10.1186/s12859-021-04211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Li R., Hu K., Liu H., Green M.R., Zhu L.J. OneStopRNAseq: A Web Application for Comprehensive and Efficient Analyses of RNA-Seq Data. Genes. 2020;11:1165. doi: 10.3390/genes11101165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Jiménez-Jacinto V., Sanchez-Flores A., Vega-Alvarado L. Integrative Differential Expression Analysis for Multiple EXperiments (IDEAMEX): A Web Server Tool for Integrated RNA-Seq Data Analysis. Front. Genet. 2019;10:279. doi: 10.3389/fgene.2019.00279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Malhotra A., Das S., Rai S.N. Analysis of Single-Cell RNA-Sequencing Data: A Step-by-Step Guide. BioMedInformatics. 2022;2:43–61. doi: 10.3390/biomedinformatics2010003. [DOI] [Google Scholar]
- 167.He J., Lin L., Chen J. Practical Bioinformatics Pipelines for Single-Cell RNA-Seq Data Analysis. Biophys. Rep. 2022;8:158–169. doi: 10.52601/bpr.2022.210041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bertolini A., Prummer M., Tuncel M.A., Menzel U., Rosano-González M.L., Kuipers J., Stekhoven D.J., consortium T.P., Beerenwinkel N., Singer F. ScAmpi—A Versatile Pipeline for Single-Cell RNA-Seq Analysis from Basics to Clinics. PLOS Comput. Biol. 2022;18:e1010097. doi: 10.1371/journal.pcbi.1010097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Gardeux V., David F.P.A., Shajkofci A., Schwalie P.C., Deplancke B. ASAP: A Web-Based Platform for the Analysis and Interactive Visualization of Single-Cell RNA-Seq Data. Bioinformatics. 2017;33:3123–3125. doi: 10.1093/bioinformatics/btx337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mohanraj S., Díaz-Mejía J.J., Pham M.D., Elrick H., Husić M., Rashid S., Luo P., Bal P., Lu K., Patel S., et al. CReSCENT: CanceR Single Cell ExpressioN Toolkit. Nucleic Acids Res. 2020;48:W372–W379. doi: 10.1093/nar/gkaa437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Miller J.A., Menon V., Goldy J., Kaykas A., Lee C.-K., Smith K.A., Shen E.H., Phillips J.W., Lein E.S., Hawrylycz M.J. Improving Reliability and Absolute Quantification of Human Brain Microarray Data by Filtering and Scaling Probes Using RNA-Seq. BMC Genomics. 2014;15:154. doi: 10.1186/1471-2164-15-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lin M., Pedrosa E., Shah A., Hrabovsky A., Maqbool S., Zheng D., Lachman H.M. RNA-Seq of Human Neurons Derived from IPS Cells Reveals Candidate Long Non-Coding RNAs Involved in Neurogenesis and Neuropsychiatric Disorders. PLoS ONE. 2011;6:e23356. doi: 10.1371/journal.pone.0023356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Abedi M., Fatehi R., Moradzadeh K., Gheisari Y. Big Data to Knowledge: Common Pitfalls in Transcriptomics Data Analysis and Representation. RNA Biol. 2019;16:1531–1533. doi: 10.1080/15476286.2019.1652525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Li Y., Ge X., Peng F., Li W., Li J.J. Exaggerated False Positives by Popular Differential Expression Methods When Analyzing Human Population Samples. Genome Biol. 2022;23:79. doi: 10.1186/s13059-022-02648-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Eilertsen I.A., Moosavi S.H., Strømme J.M., Nesbakken A., Johannessen B., Lothe R.A., Sveen A. Technical Differences between Sequencing and Microarray Platforms Impact Transcriptomic Subtyping of Colorectal Cancer. Cancer Lett. 2020;469:246–255. doi: 10.1016/j.canlet.2019.10.040. [DOI] [PubMed] [Google Scholar]
- 176.Tang K., Ji X., Zhou M., Deng Z., Huang Y., Zheng G., Cao Z. Rank-in: Enabling Integrative Analysis across Microarray and RNA-Seq for Cancer. Nucleic Acids Res. 2021;49:e99. doi: 10.1093/nar/gkab554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Ge S.X., Son E.W., Yao R. IDEP: An Integrated Web Application for Differential Expression and Pathway Analysis of RNA-Seq Data. BMC Bioinformatics. 2018;19:534. doi: 10.1186/s12859-018-2486-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Can T. Introduction to Bioinformatics. Methods Mol. Biol. 2014;1107:51–71. doi: 10.1007/978-1-62703-748-8_4. [DOI] [PubMed] [Google Scholar]
- 179.Taub M.A., Corrada Bravo H., Irizarry R.A. Overcoming Bias and Systematic Errors in next Generation Sequencing Data. Genome Med. 2010;2:87. doi: 10.1186/gm208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.O’Connor L.M., O’Connor B.A., Lim S.B., Zeng J., Lo C.H. Integrative Multi-Omics and Systems Bioinformatics in Translational Neuroscience: A Data Mining Perspective. J. Pharm. Anal. 2023;13:836–850. doi: 10.1016/j.jpha.2023.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Subramanian I., Verma S., Kumar S., Jere A., Anamika K. Multi-Omics Data Integration, Interpretation, and Its Application. Bioinform. Biol. Insights. 2020;14:1177932219899051. doi: 10.1177/1177932219899051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Lo C.H., Zeng J. Defective Lysosomal Acidification: A New Prognostic Marker and Therapeutic Target for Neurodegenerative Diseases. Transl. Neurodegener. 2023;12:29. doi: 10.1186/s40035-023-00362-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Quick J.D., Silva C., Wong J.H., Lim K.L., Reynolds R., Barron A.M., Zeng J., Lo C.H. Lysosomal acidification dysfunction in microglia: An emerging pathogenic mechanism of neuroinflammation and neurodegeneration. J. Neuroinflammation. 2023;20:185. doi: 10.1186/s12974-023-02866-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Pitt D., Lo C.H., Gauthier S.A., Hickman R.A., Longbrake E., Airas L.M., Mao-Draayer Y., Riley C., De Jager P.L., Wesley S., et al. Toward Precision Phenotyping of Multiple Sclerosis. Neurol.-Neuroimmunol. Neuroinflammation. 2022;9:e200025. doi: 10.1212/NXI.0000000000200025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Lo C.H., Skarica M., Mansoor M., Bhandarkar S., Toro S., Pitt D. Astrocyte Heterogeneity in Multiple Sclerosis: Current Understanding and Technical Challenges. Front. Cell. Neurosci. 2021;15:726479. doi: 10.3389/fncel.2021.726479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Rossi S.L., Subramanian P., Bovenkamp D.E. The Future Is Precision Medicine-Guided Diagnoses, Preventions and Treatments for Neurodegenerative Diseases. Front. Aging Neurosci. 2023;15:1128619. doi: 10.3389/fnagi.2023.1128619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Hampel H., Gao P., Cummings J., Toschi N., Thompson P.M., Hu Y., Cho M., Vergallo A. The Foundation and Architecture of Precision Medicine in Neurology and Psychiatry. Trends Neurosci. 2023;46:176–198. doi: 10.1016/j.tins.2022.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.