

Summary
Glycosylation of eukaryotic virus particles is common and influences their uptake, trafficking, and immune recognition. In contrast, glycosylation of bacteriophage particles has not been reported; phage virions typically do not enter the cytoplasm upon infection, and do not generally inhabit eukaryotic systems. We show here that several genomically distinct phages of Mycobacteria are modified with glycans attached to the C-terminus of capsid and tail tube protein subunits. These O-linked glycans influence antibody production and recognition, shielding viral particles from antibody binding and reducing production of neutralizing antibodies. Glycosylation is mediated by phage-encoded glycosyltransferases and genomic analysis suggests that they are relatively common among mycobacteriophages. Putative glycosyltransferases are also encoded by some Gordonia and Streptomyces phages, but there is little evidence of glycosylation among the broader phage population. The immune response to glycosylated phage virions in mice suggests that glycosylation may be an advantageous property for phage therapy of Mycobacterium infections.
Graphical Abstract
eTOC:
Freeman et al. describe mycobacteriophages with glycosylated capsid and/or tail tube protein subunits. This modification is mediated by phage-encoded glycosyltransferases and there is evidence for glycosylation throughout the mycobacteriophage population. Glycans alter the immune processing of phages and the binding affinity of antibodies to intact phages.
Introduction
Many viruses of eukaryotes have glycan modifications on the viral particles that play important roles in infection and host interactions.1 Glycosylation can be important for receptor recognition as illustrated by binding of SARS-CoV-2 to its ACE2 receptor,2 but also influences intracellular trafficking3 and protection against host restriction factors.4 Viral glycosylation is most commonly N-linked, can be quite extensive, and is usually mediated by host-encoded glycosyltransferases.1 Virion glycans can sterically obscure protein epitopes from antibody neutralization, providing a protective ‘glycan shield’.5
Double-stranded DNA (dsDNA) tailed bacteriophages – viruses infecting bacterial hosts – act fundamentally differently from viruses of eukaryotes in that the proteinaceous phage body remains outside of the cell during infection, with the dsDNA genome exiting the capsid, traveling through the tail tube, and across the cytoplasmic membrane. Glycosylation of phage virions has not been reported, although it is of potential interest with a growing exploration of the therapeutic use of phages,6,7 and the possibility that glycan shielding could be exploited to minimize antibody recognition.
Mycobacteriophages are viruses of Mycobacteria, a genus containing a number of important human and animal pathogens including M. tuberculosis, M. abscessus, and M. avium. Genomic sequences of over 2,200 individual phages isolated on Mycobacterium smegmatis mc2155 provide a high-resolution view of the genomic diversity of phages of a single host strain.8,9 These can be sorted into 38 distinct groups that share minimal nucleotide sequence similarity (31 clusters and seven ‘singletons’), and there is great sequence diversity within each group.10 Mycobacterium genomes commonly harbor integrated prophages, which are similarly diverse but are not closely related to the M. smegmatis phages.11–13
A small genomically-defined subset of mycobacteriophages have broad host ranges and infect strains of M. tuberculosis and M. abscessus as well as M. smegmatis.14–16 These have been used therapeutically for 20 compassionate use cases in patients with Mycobacterium infections, with eleven having favorable clinical or microbiological outcomes.15,17–19 Intravenous administration is commonly used for phage therapies6 and patient antibody responses to the phages are common.17 Although these can correlate with poor clinical outcomes,20 antibody responses that neutralize phages in vitro do not necessarily prevent favorable outcomes.17 Overall, the role of immune responses in phage therapy remains unclear.
Here, we show that several mycobacteriophages have extensive O-linked glycosylation of the virions mediated by phage-encoded glycosyltransferases. Inactivating a glycosyltransferase gene from phage Che8 removes all glycosylation of both the capsid and tail tube subunits although the virions are fully viable and stable. Virion glycans alter antibody binding and the production of phage-neutralizing antibodies in mice.
Results
Identification of glycosylated mycobacteriophage virions
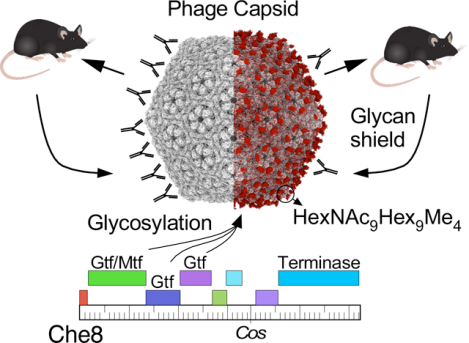
Bioinformatic analysis of mycobacteriophage genomes identified potential glycosyltransferase genes in phage Che8.21 To explore whether Che8 virions are a potential target for this glycosyltransferase, we purified phage particles from Che8 and several other mycobacteriophages and separated the virion proteins by SDS-PAGE (Fig. 1). For most of these phages, Coomassie staining showed one or two abundant proteins consistent in molecular weight with the predicted capsid and tail tube subunits, as well as several other less abundant virion proteins (Fig. 1A). These proteins were also visualized using a glycoprotein stain (Fig. 1B). Glycosylated proteins were detected in phages Che8, Corndog, and Myrna, but not the other phages. For all three glycosylated phages, the glyco-staining aligns with the most abundant virion proteins. Furthermore, SDS-PAGE migration of these bands is somewhat slower than the predicted molecular weights of major capsid and/or tail tube subunits, suggesting that they are modified (Fig. 1A). For Che8, the glycosylation candidates are the major capsid (gp6) and tail tube (gp11) subunits which are similarly sized (29.1 and 29.9 kDa, respectively) and likely co-migrate. For Corndog the corresponding tail tube subunit (gp49) and major capsid (gp41) proteins are 29.9 kDa and 43 kDa, respectively, although monomeric capsid subunit is not observed because of wholesale covalent crosslinking,22 with only some very high molecular weight proteins (likely pentameric and hexameric incompletely crosslinked capsomers) observed at the top of the gel (Fig. 1A). The glycostaining suggests that both tail tube and cross-linked capsid subunits could be glycosylated (Fig. 1B). It is less clear which Myrna virion proteins are glycosylated, although the capsid subunit (gp99, 37.5 kDa) does not appear to be a candidate based on the protein migration pattern in SDS-PAGE (Fig. 1A). Glyco-staining of Myrna revealed three discrete protein bands, labeled 1, 2, and 3 in Fig. 1B.
Figure 1. Mycobacteriophages with glycosylated virions.
A and B. SDS-PAGE analysis of mycobacteriophage virions. Replicate gels were stained with either Coomassie Blue (A) or glycostain (B). Molecular weight markers (M), a glycostain positive control (C), and phages as labeled are shown. The major glyco-stained bands in Che8 and Corndog and the three Myrna glyco-stained bands labeled 1, 2, and 3 were excised for analysis by MS/MS. C. Genome segments of phages Che8, Corndog, and Myrna encoding glycosyltransferase genes. Genes are shown as colored boxes above the genome ruler and putative functions are indicated. D. Glycopeptide CID-MS/MS spectra from the tail tube subunit of Corndog (gp49, top), the capsid subunit from Che8 (gp6, middle) and a minor capsid subunit of Myrna (gp98, bottom) from band 1; the precursor ions were m/z 1516.63+, m/z 1202.83+ and m/z 1351.45+, respectively (underlined in Fig. S1). Glycan oxonium ions (m/z 163.1, 204.1, 325.2, 366.1, etc.) are present in the lower half of the MS/MS spectra. Partial methylation of hexoses in Corndog and Che8 is evident by the presence of the oxonium ions at m/z 177.1, 339.2 and 380.2. Fragment ions corresponding to the peptide (Pep) and the peptide linked to HexNAc (Pep+HexNAc) are identified in all the MS/MS spectra. Ions marked with an asterisk in the Corndog spectrum are derived from a co-eluting peptide and are un-related to the glycopeptide under investigation. Details of the MS analyses are in Supplementary Data Set 1. HexNAc, N-Acetylhexosamine, Hex, unmodified hexose, Me, methyl.
Phages Che8, Corndog, and Myrna encode glycosyltransferases
Che8 and Corndog both have siphoviral morphologies, although Che8 has an isometric capsid and Corndog has a prolate capsid;23,24 Myrna has a myoviral morphology.25 Genomic analysis reveals that all three phages carry genes coding for glycosyltransferases (Fig. 1C). In Che8 these are located at the extreme 3’ end of the virion genome, closely linked to the virion structural genes when the cos ends are joined. Numerous families of glycosyltransferases have been described26 and Che8 gp110 has a glycosyltransferase family_25 motif, gp109 is an unrelated glycosyltransferase, and gp108 has both glycosyltransferase family_2 and methyltransferase family_24 domains (Fig. 1C). Corndog gp37 and gp38 are distant homologues of Che8 gp109 and gp110 (56% and 47% identity, respectively), and gp35 has a methyltransferase family_24 domain; Corndog gp36 is a predicted polypeptide Nacetylgalactosaminyltransferase family_12 protein. These genes are located within the virion structural gene operon situated between the portal and capsid subunit genes (Fig. 1C). The virion structural genes are not well defined in Myrna, but Myrna gp238 is a glycosyltransferase related to Che8 gp110 and Corndog gp38, and Myrna gp234 is a glycosyltransferase Family_GT2 protein.
O-glycosylation of phage virion proteins
The major glycol-stained bands identified in phage Che8, Corndog, and Myrna (Fig. 1B) were excised, trypsin digested, and analyzed by mass spectrometry (Fig. 1D, Extended Dataset 1). The most abundant protein species detected were the capsid and tail tube subunits in Che8 (gp6 and gp11, respectively), the tail tube subunit in Corndog (gp49), and both the major and minor capsid subunits of Myrna27 (gp99 and gp98, respectively). Less abundant co-migrating proteins were also present (Table S1). Further inspection of the MS spectra detected a series of weak, multiply charged ions in each of the tryptic digests (Fig. S1), and targeted MS/MS analysis revealed these to be families of related glycopeptides from the major capsid subunit of Che8 (gp6) and the tail tube subunit of Corndog (gp49). In Myrna, all three proteins appear to be the minor capsid subunit, gp98,27 modified by glycans of varying length; band 1 contains the longest glycan chains and its mass spectrum is shown in Fig. 1D. Within and between glycopeptide families, the glycans vary in the number of monosaccharides and/or methylations present for all three phages investigated, the glycan was observed on the C-terminal peptide of the protein. Data are consistent with the glycan being attached to the C-terminal tryptic peptide of the protein, presumably O-linked, to the C-terminal serine residue. Glycopeptides of the Che8 tail tube subunit (gp11) were not identified, likely because the large (~3500 Da) predicted C-terminal peptide (also ending in serine) escaped detection due to its size. However, subsequent chymotrypsin digestion confirmed that the Che8 tail tube subunit is similarly glycosylated.
Further examination of the MS/MS spectra (Fig. 1D) revealed different glycosylation patterns in the three phages. The patterns suggest that Corndog’s glycans are long chains of partially methylated hexose sugars (Hex) linked to the C-terminal serine via a single N-acetylated hexosamine residue (HexNAc); one of the more abundant species is peptide-HexNAc1Hex16Me4 (Fig. 1D). Che8’s glycans are composed of equal numbers of HexNAc and partially methylated Hex residues, linked to the peptide via HexNAc; the most abundant species are peptide-HexNAc9Hex9Me3 and peptide-HexNAC9Hex9Me4 (Fig. 1D). Myrna’s glycans also contain equal numbers of HexNAc and Hex residues but without methylation and are linked via HexNAc to the peptide; the most abundant is peptide–HexNAc13Hex13 (Fig. 1D). The oxonium ions in the MS/MS spectra suggest that the monosaccharides are differently arranged in Che8 and Myrna. For example, there is evidence for chains of linked HexNAc residues (i.e. HexNAc4) in Che8 but not Myrna. Interestingly, there is also evidence for Hex linked to HexNAc in both strains but not for chains of Hex residues. We note that specific isomers of the monosaccharides cannot be determined by mass alone and so it was not possible to determine which isomers of hexose or N-acetylated hexosamine are present.
Che8 gp110 is required for virion glycosylation
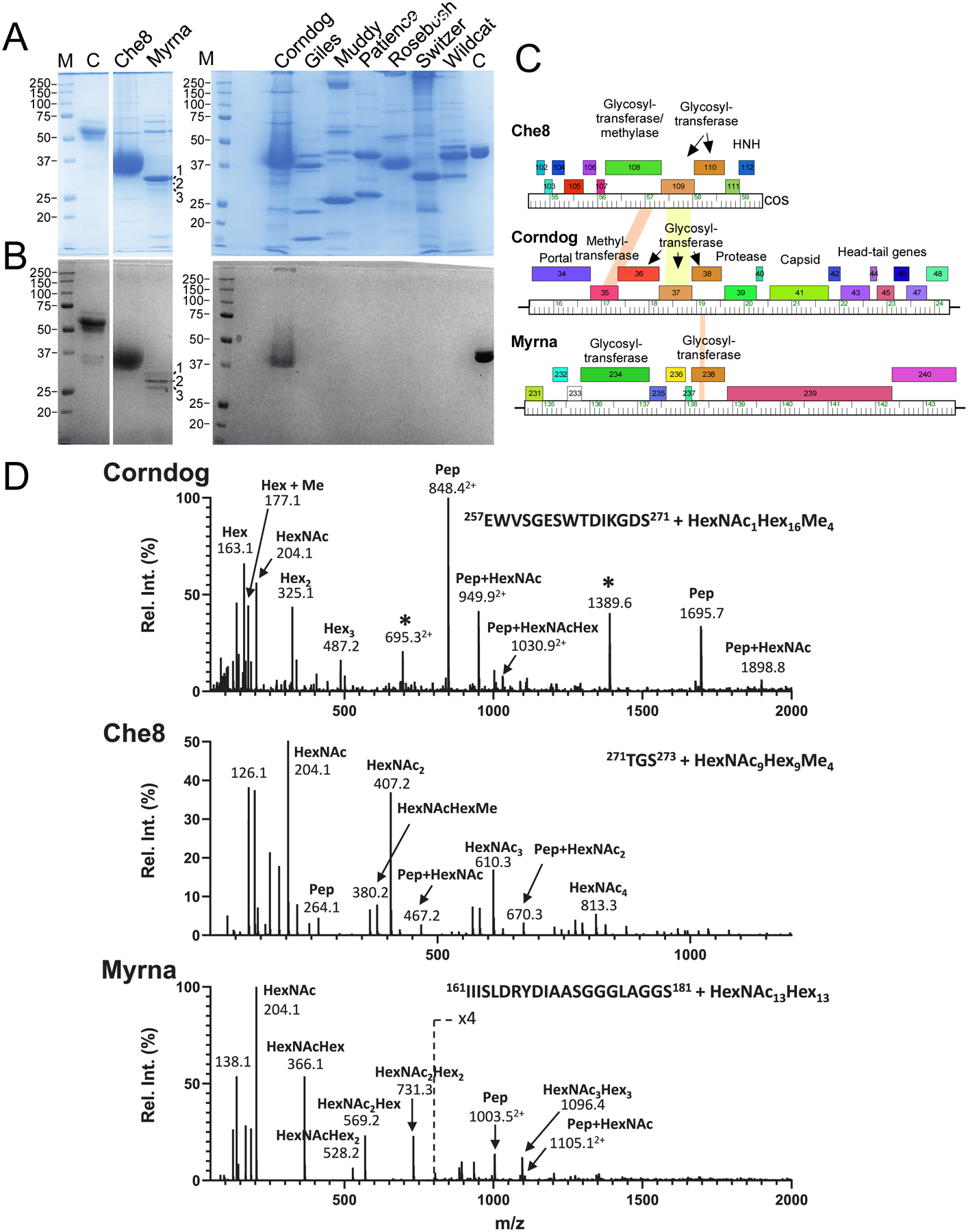
Although the three glycosylated phages shown in Figure 1 code for several different glycosyltransferases, they all encode homologues of Che8 gp110, which is thus a candidate for catalyzing glycan addition. To make a Che8 mutant defective in glycosylation, we first constructed a strain expressing a sgRNA targeting gene 110 together with an active Cas9 enzyme (Fig. 2A) and demonstrated that expression of this ribonucleoprotein complex reduces the plaquing efficiency of Che8 by about 100-fold (Fig. 2B). Screening of CRISPR-escape survivors identified a mutant (designated Che8Δ110-1) with a 7 bp deletion within gene 110, introducing a frameshift and premature translation termination (Fig. 2A). We note that an additional deletion removes genes 85-94, although these are all of unknown function except for an MPME1 transposon28 and are poorly conserved in Subcluster F1 phages.
Figure 2. Che8 110 is required for glycosylation.
A and B. A sgRNA-expressing plasmid was designed to target Che8 gene 110 encoding a putative N-acetyl-galactosyl transferase. When sgRNA and Cas9 expression is induced (+ATc) the efficiency of plaquing of Che8 is reduced approximately two orders of magnitude (panel B). Candidate CRISPR-escape mutants were isolated and one was shown to contain a seven base pair deletion (underlined) within the sequence targeted by the sgRNA (blue type). The reading frame shift produces a truncated variant of gp110 (amino acids in red type). C. SDS-PAGE of Che8 and Che8Δ110-1 virions, stained with either Coomassie Blue or Glycostain as indicated. Seven bands are resolved in the 37 kDa size range in wild type Che8, all of which are glycosylated. In Che8Δ110-1 these collapse to a single band containing both the major capsid (29.04 kDa) and tail tube subunits (29.78 kDa), both of which are unglycosylated. D. An expanded view of the glycosylated Che8 proteins, and a schematic of the arrangements of sugars in each band. HexNAc, N-Acetylhexosamine, Hex, unmodified hexose, Me, methyl. Further mass spectrometry analysis is shown in Figure S1 and in Supplementary Data Set 2. E. Viability of wild type and mutant Che8 particles with titers starting at either 1011 or 1012 pfu/mL after prolonged storage at room temperature or 4 °C. Averages of two or three technical replicates and standard error bars are shown. F. Adsorption of wild type and mutant Che8 to M. smegmatis. G. Fecundity of wild type and mutant Che8 as determined by the number of infectious particles in individual plaques. Six technical replicates are shown as individual datapoints on top of a box plot indicating the mean (central line) +/− one standard error (box boundaries).
Separation of mutant virion proteins by SDS-PAGE showed loss of Coomassie stained protein in the 34–40 kDa size range, and appearance of an abundant protein band at ~ 30 kDa (Fig. 2C), which is not recognized by the glycostain (Fig. 2C). Che8 gp110 is thus likely required for addition of all glycans to Che8 virion proteins. Although minor virion proteins are less abundant, we observed no other differences between wild type and Che8Δ110-1 particles, suggesting that the capsid and tail tube subunits are the only glycosylated proteins in Che8 virions (Fig. 2C). We compared the mutant and wild type particles for stability, adsorption, and fecundity, and observed no evident differences (Fig. 2E – G). Virion glycosylation thus does not appear to play role in phage structural integrity, phage binding to its host, or viral reproduction.
Higher resolution SDS-PAGE separates seven resolvable Che8 protein species in the 34–40 kDa size range, all of which are glycosylated and with the smallest being 3–5 kDa larger than the unglycosylated major protein band visualized in the Che8Δ110-1 lane (Fig. 2C). The seven glycoprotein bands were separately excised together with the major unglycosylated band from Che8Δ110-1, digested with chymotrypsin and analyzed by nanoLC-MS/MS (Fig. 2D, Extended Data Set 2). The unglycosylated Che8Δ110-1 protein band primarily contains the capsid and the tail tube subunits as expected (Table S2). The capsid subunit is the most abundant protein in each of the seven wild type bands; the tail tube protein is also present but at an apparent lower abundance (Table S2). The C-terminal peptide of the capsid protein was observed glycosylated in all seven bands, and the glycopeptides in the successive bands differed by the mass of a HexNAc-Hex pair (365 Da; Fig. 2D). Using targeted HCD-MS/MS analysis, we also confirmed that glycans are attached to the C-terminal chymotryptic peptide of the tail tube subunit and that they are of similar composition to those of the capsid subunit (Fig. S1). These data together with the initial ms analysis (Fig. 1) are all consistent with O-linked glycosylation of the C-terminal serine, although linkage through other residues cannot be excluded.
Structure of Che8 virions with C-terminal glycosylation of capsid subunits.
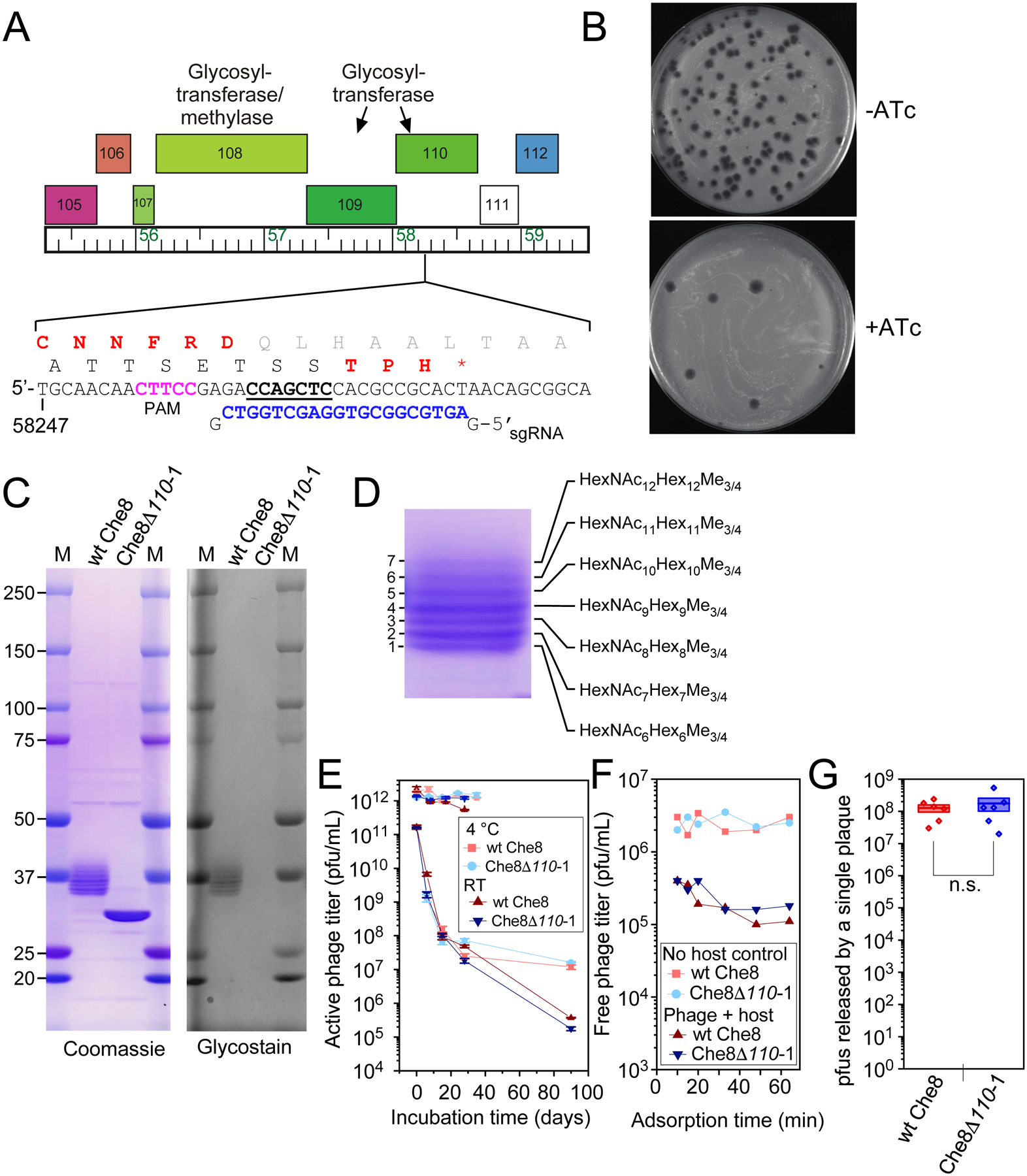
We have recently described a high resolution Cryo-EM structure of the Che8 capsid, which has T=9 symmetry and a HK97-type protein fold.24 We have also determined the cryo-EM structure of the Che80Δ110-1 mutant capsid which possesses a structure nearly identical to the wild type virion (Fig. 3). The only difference between the two maps was observed at the C-terminal end of the major capsid protein (serine 273, Fig. 3A). In the wild-type map, density can be seen above the serine (Fig. 3B, C), which is not observed in the mutant map (Fig. 3D, right). Modeling showed that the observed density can accommodate a short glycan molecule of one to two sugars (Fig. 3C, right panel). Further contouring of the map suggests there is additional density extending beyond that shown in Figure 4C, although the density is poorly resolved at lower contour levels. Presumably, the longer glycans identified by mass spectrometry are highly flexible and insufficiently ordered to yield coherent density beyond 1–2 sugars. In the Che8Δ110-1 capsid, no density was detected above the C-terminal serine, even at very high contour levels (Fig. 3D, right).
Figure 3. Cryo-EM structures of Che8 and Che8 Δ110-1 capsids.
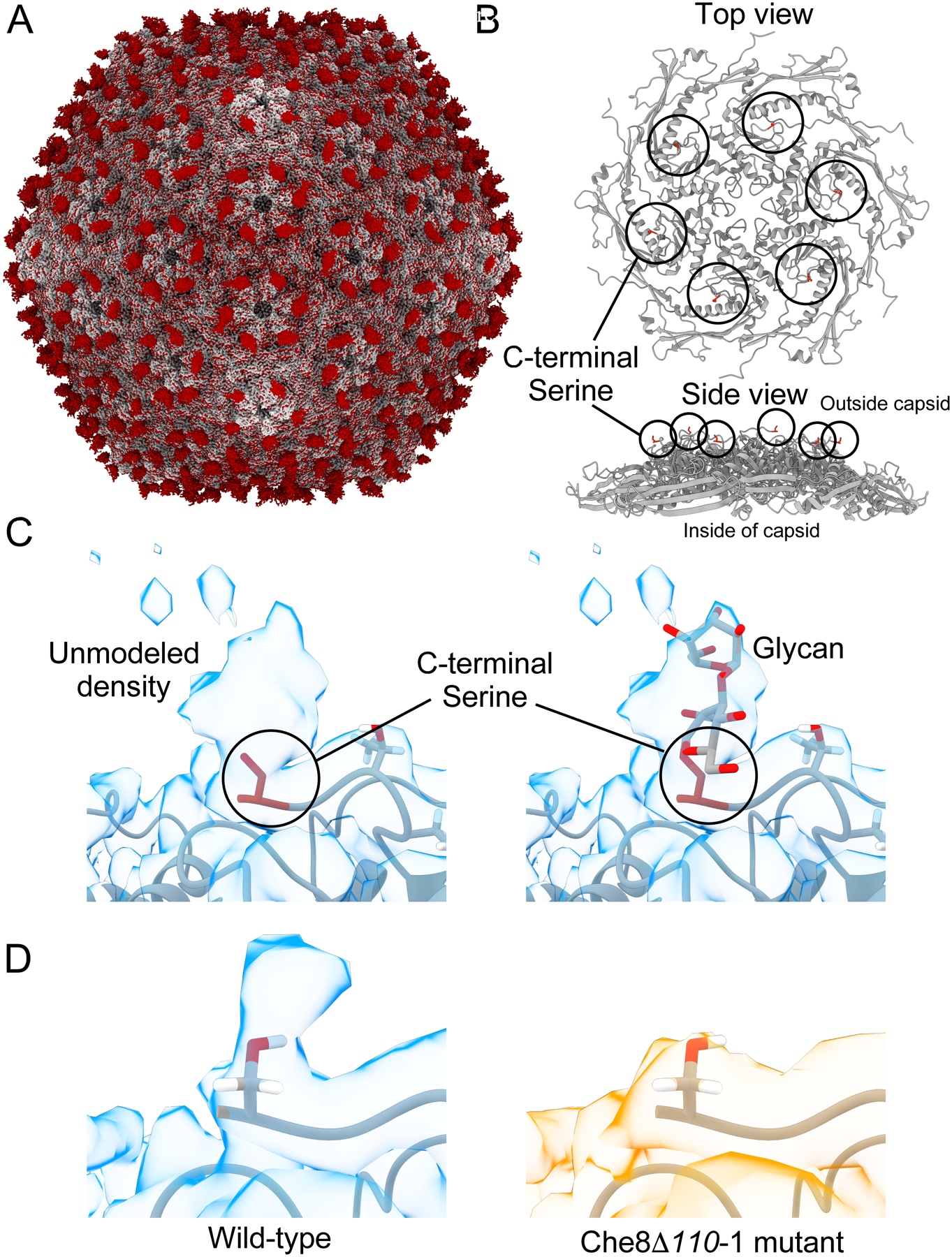
A. The non-glycosylated Che8 Δ110-1 capsid (gray) aligned with a difference map of the Che8 and Che8 Δ110-1 capsid densities (red). Excess red density at each capsomere is capsid glycosylation. B. The model of a single hexamer of the major capsid protein from the Che8 wild type (PDB:8E16) highlighting the C-terminal serine 273 in red. C. The wild type Che8 major capsid protein model (PDB:8E16) fitted into the cryo-EM map (EMD-27824) contoured at level 2. An O-glycosylated alpha-D-galactopyranose-(1–3)-2-acetamido-s-desoxy-beta-D-glucopyranose glycan has been modeled into the density on the right for illustration purposes. D. The wild type capsid compared to the Che8 Δ110-1 mutant at the putative glycosylation site. Both maps have been contoured at the same level.
Figure 4. Glycosylation shields wild-type Che8 from antibody recognition.
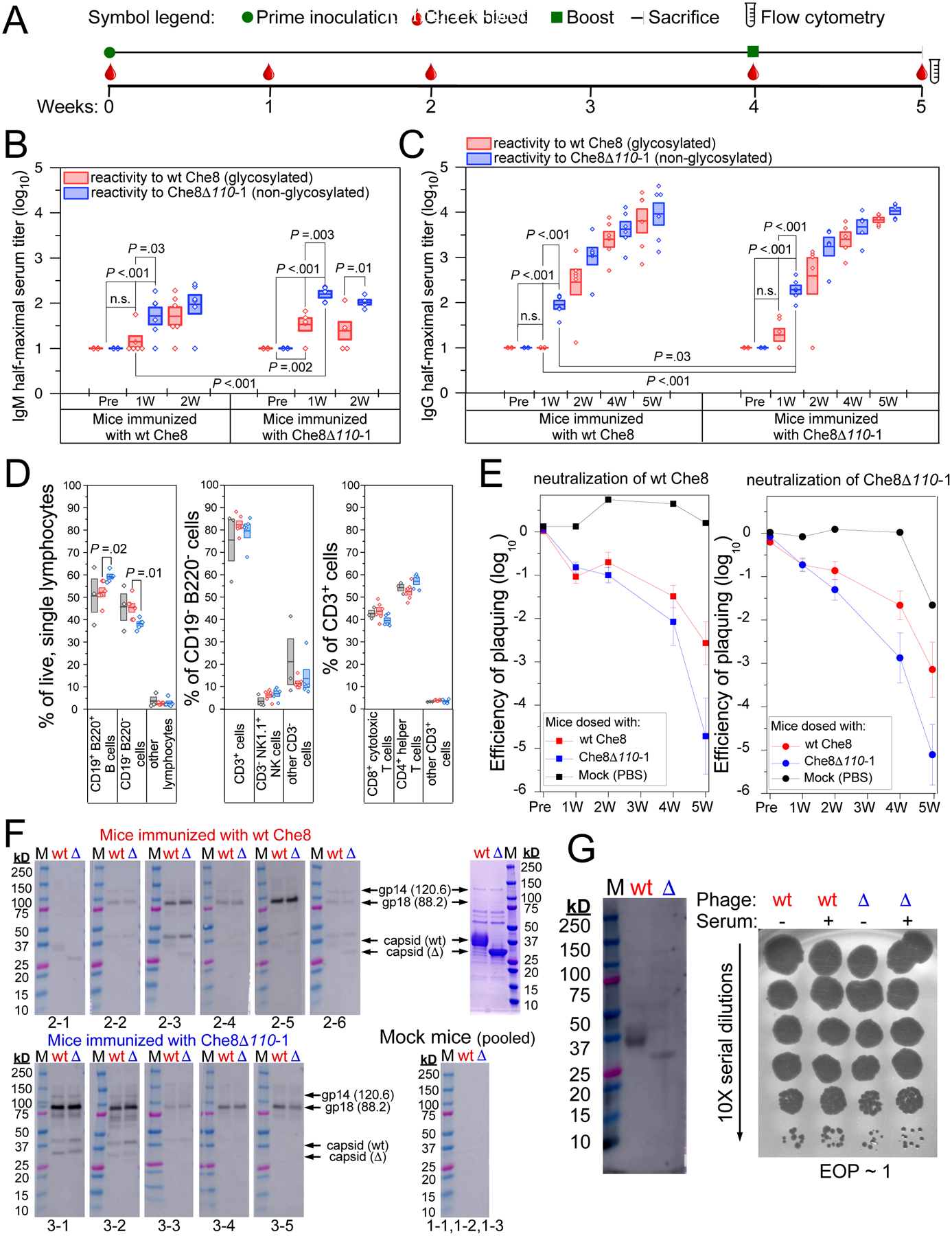
A. Timeline of the C57BL/6J mouse study. Mice were inoculated with PBS, wt Che8, or Che8 Δ110-1 (Table S3) and serum collected via submandibular bleed as indicated. A second dose was administered at four weeks, after serum collection, and at five weeks mice were sacrificed and terminal sera and spleens were harvested. B and C. ELISA were used to quantify IgM (B) and IgG (C) titers in sera from mice inoculated with wild type Che8 (left panels) and Che8Δ110-1 (right panels) at the indicated timepoints. Box plots show half-maximal titers for individual mice with a line showing mean values and the boxes representing the mean value +/− 1 standard error. Two sample t-tests were performed between relevant pairs and P values are shown where differences are significant. Antibody recognition of wild type Che8 and the Che8Δ110-1 mutant are shown in red and blue, respectively. D. Multicolor flow cytometry was gated as shown in Figure S4. Shown are proportions of CD19− B220− vs CD19+ B220+ B cells in the live, single lymphocyte population; of CD3+ vs CD3− cells in the CD19− B220− population; and of CD4+ and CD8+ T cell subtypes in the CD3+ population. E. Phage neutralization by serum was determined by incubating phages with sera and measuring reductions in phage titers from the starting input (109 pfu/ml). Each panel presents the average +/− standard error neutralization of Che8 (left) and Che8Δ110-1 (right) by sera from mock–, Che8– or Che8Δ110-1–inoculated mice shown in black, red, and blue, respectively. F. Western blots showing IgG reactivity to virion proteins of Che8 (wt) and Che8Δ110-1 (Δ) in sera collected from individual mice (as shown below each blot) five weeks after inoculation; molecular weight makers, M. Upper and lower panels use sera from Che8-inoculated and Che8Δ-inoculated mice, respectively. Serum from mock-inoculated mice were pooled. Panel at the upper right shows Coomassie stained proteins used in the Western blots. Putative protein identities are indicated. G. Western blot (left) using normal human serum showing some reactivity to virion proteins (left panel), but no phage neutralization (right panel).
Virion glycans shield Che8 from antibody recognition
To examine antibody responses to Che8, highly purified wild type and Che8Δ110-1 virions were inoculated into C57BL/6J mice (Table S3), serum was collected weekly (Fig. 4A), and IgM and IgG titers specific to Che8 and Che8Δ110-1 were determined (Fig. 4B, C, S2). IgM responses were measured one and two weeks after inoculation, and mice inoculated with either wild type or mutant virions showed phage-specific reactivity with higher affinity binding to mutant (unglycosylated) particles than to wild type Che8 (Fig. 4B). This difference was greatest at the one-week time point, and the overall reactivity was similar at week-two except for increased recognition of wild type Che8 in the mice inoculated with the same phage (Fig. 4B). IgG titers increased across a five-week time span, and at early time points we similarly observed significantly higher antibody titers recognizing mutant particles relative to wild type virions (Fig. 4C). This difference in antibody specificity is greatest one week after inoculation, when IgG antibodies binding to the glycosylated particles are undetectable (in sera from mice inoculated with wild type virions) or low abundant (mice inoculated with mutant virions). IgG titers increased at later time points and the greater reactivity to unglycosylated virions continues, although the differences are less prominent at later times (Fig. 4C). IgG antibody titers did not differ greatly depending on which phage was used to inoculate the mice. These data suggest that Che8 glycosylation shields the virus from antibody binding in vitro (Fig. 4C).
We also harvested splenocytes from mice five weeks after inoculation (one week after restimulation with a second dose of phage) and characterized the cellular populations. Among the populations of live, single lymphocytes, there was a small difference in the proportions of CD19+ B220+ B cells and CD19− B220− cells, with mice inoculated with unglycosylated virions enriched with B cells. The CD19− B220− populations were further gated into CD3+ and CD3− cell types, with no evident differences between groups of mice. Examination of T cell subsets within the CD3+ population reveals small, but not statistically significant, differences: a small enrichment of CD4+ helper T cells in mice inoculated with Che8Δ110-1 is balanced by a corresponding decrease in CD8+ cytotoxic T cells in that population (Fig. 4D, S4). Taken together, these differences, while modest, are consistent with greater immunogenicity and antigenicity of the Che8Δ110-1 mutant relative to glycan-shielded wild type Che8.
A second set of mice were also inoculated, and immune responses monitored similarly (Figs. S4 – S6). The responses were similar to the first experiment, with mice inoculated with the unglycosylated virions having reduced IgM and IgG recognition of glycosylated virions at early time points; however, this difference was less evident in mice inoculated with wild type Che8 (Fig. S5, S6). Flow cytometry of the mice splenocytes showed similar patterns to the first experiment, although the magnitudes of the differences are somewhat greater (Fig. S4 and Fig. S5). The differences in the two experiments may result from differences in genetic backgrounds of the mice, different ages of the mice at the time of initial inoculation, or from different timings of the booster dose (this second study restimulated mice at Week 7.5, versus Week 4 in the primary mouse study presented in Fig. 4) (Table S3).
Che8 virion glycosylation alters production of neutralizing antibodies
We also tested mice sera for their ability to neutralize Che8 infection in vitro. Neutralization was mild at early times and increased up to five weeks (Fig. 4E, S3). Interestingly, neutralization of both wild type and mutant phage particles was substantially greater with serum from mice inoculated with the Che8Δ110-1 mutant than with wild type glycosylated Che8 particles (Fig. 4E). This was somewhat unexpected as neutralization is anticipated to result from antibody binding and blocking of receptor binding proteins at the tail tip, and not antibody recognition of either the capsid or the tail tube. Virion glycosylation thus not only shields the particles from in vitro antibody binding to the capsid and tail but may also influence how the particles are processed by the human immune system such as to alter which virion proteins are presented as antigens and trigger antibody production. Greater neutralization of virions by serum from mice inoculated with unglycosylated Che8Δ110-1 was also observed in the second experiment but the difference was more modest (Fig. S5, S7).
To explore the specificity of antibody recognition, we analyzed antibody binding to virion proteins at five weeks post-inoculation using Western blots (Fig. 4F). The reaction patterns differ substantially in different mice, but most mice show their strongest reactions to a minor virion constituent ~85 kDa that is barely visible by Coomassie staining and is shown by mass spectrometry (Extended Data Set 3) to be gp18, a predicted ~88 kDa minor tail protein component of the tail tip (gp18 is the second largest Che8 virion protein, with the tape measure protein being 120 kDa, and the next largest being the 67 kDa minor tail protein gp20). In general, recognition of the denatured capsid and tail tube proteins in the Westerns is relatively weak, although can be observed in some mice – especially those inoculated with wild type Che8. There are no obvious and systematic differences between recognition of the wild type and mutant particles in any serum. We note that recognition of the denatured proteins by Westerns is biased toward linear epitopes and may differ substantially from recognition of conformational epitopes on native virions seen by ELISA assays. Mock-infected mice showed no reactions to Che8 proteins, but some recognition of capsid and/or tail tube proteins was observed in pooled normal human serum, although this is not neutralizing (Fig. 4G).
Because antibody responses increase considerably with time, we examined the recognition of virion proteins by Western blot analysis of two mice from each phage inoculation at the weekly time points, each representing different antibody recognition by ELISA and neutralization (Fig. 5). For all four mice, the antibody titers are too weak at weeks 1 and 2 to detect antibody binding but binding to the putative minor tail proteins (especially gp18) is seen in most mice at weeks 4 and 5 (Fig. 5); the exception is mouse 2–1 serum, which has recognition of the capsid and/or tail tube protein, but not the minor tail proteins. This mouse serum also shows a relatively poor response by ELISA and no neutralization at any time point (Fig. 5). Overall, these observations are consistent with the interpretation that antibody binding to proteins at the tail tip (gp18) are associated with neutralization. However, we note that although antibodies strongly recognize intact highly purified virions, they have much weaker binding to denatured virion proteins in the Western analyses. A notable example is mouse 2–4 (Table S3), which has among the strongest ELISA response at four weeks, with only weak binding by Western blotting and mild neutralization. At five weeks, the ELISA IgG signal is somewhat greater, but the binding to minor tail proteins is much stronger by Western blotting, and neutralization is considerable (Fig. 5).
Figure 5. Antibody production in individual mice.
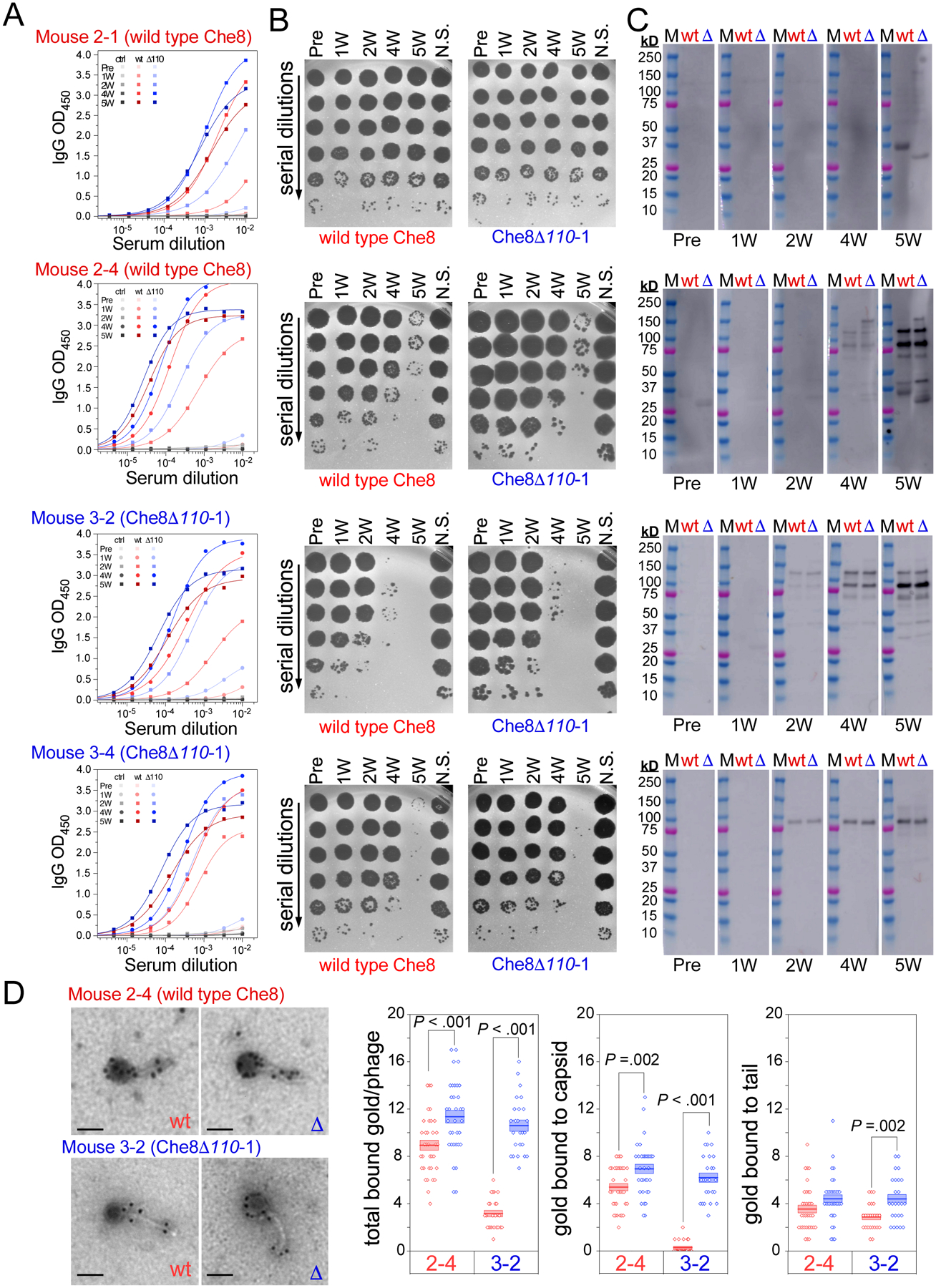
The sera from four individual mice at each timepoint were used in A. ELISAs, B. neutralization assays, and C. Western blots. Mice 2–1 and 2–4 were immunized with wild type Che8, while mice 3–2 and 3–4 were immunized with Che8 Δ110-1, as indicated above each dataset (see also Table S3). Logistic fits of the ELISA curves yield half-maximal serum titers of antibodies that bind to both wild type Che8 (red) and Che8 Δ110-1 (blue), as well as uncoated wells (black). Neutralization assays were performed against both phages, as indicated above the plaque plate images. Western blots probe the reactivity of the mouse sera to both wild type Che8 (wt, red) and Che8 Δ110-1 (Δ blue) and were exposed for 2 minutes 59.4 seconds for sera from mice immunized with wild type Che8 while Western blots using sera from Mouse 3–2 and Mouse 3–4 (dosed with Che8 Δ110) were exposed for 18.4 seconds. D. Immunogold staining of wild type and mutant Che8 particles. Sera from mice 2–4 and 3–2 collected at week 2 were tested and the numbers of gold particles per virion (total) or binding to either capsid or tail are plotted (three rightmost panels). Datapoints are shown for at least 24 virions imaged for each and represented as box plots with lines showing mean values and boundaries of +/− one standard error. Representative micrographs are shown at the left; scale bar corresponds to 100 nm.
To visualize antibody binding to intact virions, sera collected from two mice at week 2 were used to immunogold stain phage particles (Fig. 5D). Prominent staining of capsids and tails was observed, consistent with robust antibody titers seen by ELISA at this timepoint, even though capsid and tail tube recognition by Western is weak. Quantification showed greater staining of unglycosylated virions relative to wild type Che8, with the greatest differences in serum from mouse 3–2 inoculated with unglycosylated phage.
Glycosylation of other actinobacteriophages
A bioinformatic survey of other actinobacteriophages revealed many others that possess potential glycosyltransferases, some encoding for a single glycosyltransferase (Gtf) similar to Myrna, and others encoding for two or more Gtfs similar to Che8 and Corndog (Table 1). A subset of these phages also code for capsid and tail tube proteins that both have C-terminal serine residues and so are strong candidates for also having similarly glycosylated virions. They span 18 distinct genomic types (clusters/subclusters/singletons, Table 1). Some of these phages also have putative methyltranferase (Mtf) genes closely linked to the glycosyltransferase genes, suggesting the possibility of methylated glycans as in Che8 and Corndog (Table 1). These phages infect Mycobacterium, Gordonia, and Streptomyces host bacteria.
Table 1.
Actinobacteriophages encoding glycosyltransferases
| Cluster1 | Example2 | Capsid3 | Tail4 | gtf 5 | mtf 6 | Host | Virion glyco7 |
|---|---|---|---|---|---|---|---|
| AA (2) | Phrappuccino | 54 (S)8 | None | 142, 143, 154, | 145 | Mycobacterium | Pred |
| C (174) | Myrna (C1) | 98 (S)8 | None | 234 | ND | Mycobacterium | Exp |
| F (213)9 | Che8 (F1) | 6 (S) | 11 (S) | 108, 109, 110, | 108 | Mycobacterium | Exp |
| I (7)9 | Sbash (I2) | 6 (S) | 12 (S) | 87, 88 | ND | Mycobacterium | Pred |
| J (39) | Omega | 15 (A) | 31 (S) | 16, 23 | 17 | Mycobacterium | Exp |
| K4 (18) | Fionnbharth | 14 (S) | 19 (V) | 6 | 95 | Mycobacterium | Exp |
| O (23) | Corndog | 41 (S) | 49 (S) | 37, 38 | 35 | Mycobacterium | Exp |
| S (18) | Marvin | 38 (S) | 43 (S) | 81, 83 | 80 | Mycobacterium | Pred |
| X (2) | Nebkiss | 5 (S) | 10 (S) | 166 | 167 | Mycobacterium | Exp |
| Singleton | MooMoo | 5 (S) | 11 (S) | 94, 96 | ND | Mycobacterium | Pred |
| Singleton | Sparky | 6 (S) | 11 (S) | 88, 89 | ND | Mycobacterium | Pred |
| FC (4) | Mimi | 161 (G) | ND | 253, 256, 257 | ND | Arthrobacter | unk |
| AT (4) | Kitkat | 25 (A) | 31 (R) | 29, 93 | 100 | Arthrobacter | unk |
| CQ (14) | BrutonGaster (CQ2) | 23 (S) | 30 (S) | 105 | ND | Gordonia | Pred |
| CZ (78) | Adora (CZ4) | 6 (A) | 12 (P) | 30 | ND | Gordonia | unk |
| DD (2) | Yvonnetastic | 33 (S) | 39 (S) | 24, 25, 138 | ND | Gordonia | Pred |
| DK (2) | GodonK | 76 (S) | 78 (S) | 81, 82 | ND | Gordonia | Pred |
| DP (6) | Ziko | 42 (S) | 48 (S) | 123, 142 | ND | Gordonia | Pred |
| DS (6) | Forza | 87 (S) | 89 (S) | 93, 94 | ND | Gordonia | Pred |
| DU (7) | Kudefre (DU1) | 16 (A) | 21 (S) | 123, 125, 126 | ND | Gordonia | Pred |
| EA (163) | TeddyBear | 6 (P) | 14 (A) | 47 | ND | Microbacterium | unk |
| EC (37) | Antares | 27 (A) | ND | 85 | ND | Microbacterium | unk |
| GC (4) | Dewdrop | 60 (P) | ND | 153, 154, 155, 156 | 158 | Microbacterium | unk |
| GD (5) | PauloDiaboli | 88 (N) | 90 (I) | 179, 180 | ND | Microbacterium | unk |
| CB (3) | Grayson | 79 (D) | 83 (T) | 62 | ND | Rhodococcus | unk |
| CE (2) | Trina | 72 (D) | 78 (T) | 56 | ND | Rhodococcus | unk |
| CB (3) | Grayson | 79 (D) | 83 (T) | 62 | ND | Rhodococcus | unk |
| BE (49) | Angela (BE1) | 47 (A) | 53 (A) | 39 | ND | Streptomyces | unk |
| BK (24) | Belfort (BK1) | 29 (A) | 37 (V) | 20, 21 | ND | Streptomyces | unk |
| BM (6) | Kela | 25 (S) | 52 (S) | 47, 48 | ND | Streptomyces | Pred |
| BN (8) | Dryad | 16 (S) | 22 (S) | 34 | 36 | Streptomyces | Pred |
| BO (3) | Wheeheim | 18 (I) | ND | 26 | ND | Streptomyces | unk |
| BR (2) | Zuko | 11 (A) | 19 (T) | 4, 5 | ND | Streptomyces | unk |
| Singleton | TurkishDelight | 17 (S) | 21 (S) | 20, 22 | ND | Streptomyces | Pred |
| Singleton | Kromp | 7 (N) | 14 (A) | 79 | ND | Streptomyces | unk |
| Singleton | Cantare | 14 (A) | 21 (A) | 95 | ND | Brevibacterium | unk |
| Singleton | LuckyBarnes | 13 (A) | 19 (G) | 39, 40 | ND | Brevibacterium | unk |
| Singleton | TJE1 | 22 (A) | 36 (K) | 6 | 8 | Tetrasphaera | unk |
| Singleton | phiAsp2 | 14 (P), 15 (P) | 16 (S) | 33, 35 | 34 | Actinoplanes | unk |
Cluster designation is shown with the total number of cluster/subcluster members in parentheses; most if not all members are simple to the example shown in column 2, although the numbers and types of gtf genes may differ. The subcluster is shown (e.g. K4), if only that subcluster has gtf genes.
An example genome is listed with its subcluster shown in parentheses where the cluster contains subclusters.
The gene coding for the major capsid subunit(s) is shown with the C-terminal amino residue shown in parentheses.
The gene coding for the tail tube subunit is shown with the C-terminal amino residue shown in parentheses. For some phages, the tail tube subunit has not been determined (ND).
gtf genes encoding putative glycosyltransferase proteins are shown.
genes encoding putative methyltransferases or Mtf domains are shown. ND, not determined.
Virion glycosylation determined experimentally (Exp), predicted (Pred), or unknown (Unk)
Genes encode capsid decoration proteins which are glycosylated or likely glycosylated.
All subclusters likely have glycosylated virions, but there are variations in gtf and mtf genes/domains among phage members.
Some of the glycosylated phage clusters are relatively large such as mycobacteriophage Cluster F which has over 200 members (Table 1) and is divided into five subclusters. All of the phages within all five F subclusters code for glycosyltransferases near the right genome end, but there are at least four distinct configurations (Fig. 6A). Three of these include genes related to Che8 110 coding for a Gtf, and all contain at least one additional Gtf albeit from three different sequence families. Three configurations code for an Mtf (e.g. phages Cornie and Ariel) or a combined Gft-Mtf protein (e.g. Che8 108), and two (e.g. DeadP and Akhila) code for a closely-linked serine/threonine kinase (Fig. 6A). A similar pattern is seen with the 39 Cluster J phages (Table 1), with five different organizations of gtf genes, positioned between the capsid and tail genes (Fig. 6B). One gtf gene (e.g. Ariel 21) is common to all five organizations, and all phages have at least one and as many as three additional gtf genes (Fig. 6B). All also code for a Mtf or a protein with both Gtf and Mtf domains (e.g. Squint 17); three of the organizations code for serine-threonine kinases (Fig. 6B). Of the five Cluster J organization types, one resembles the Cluster F phages (represented by DeadP, Fig. 6A, B), with BAKA genes 12 (S/T kinase), 13 (Gtf), and 14 (Gtf) related to DeadP genes 101, 102, and 103; the others have more distantly related gtf and mtf genes than in Cluster F phages. These patterns illustrate the considerable diversity of the systems, and the marked variations within genomes that are otherwise quite closely related. This genetic diversity is likely to be reflected in the composition and structure of the glycan modifications expressed by the different actinobacteriophages.
Figure 6. Glycosylation in other actinobacteriophages.
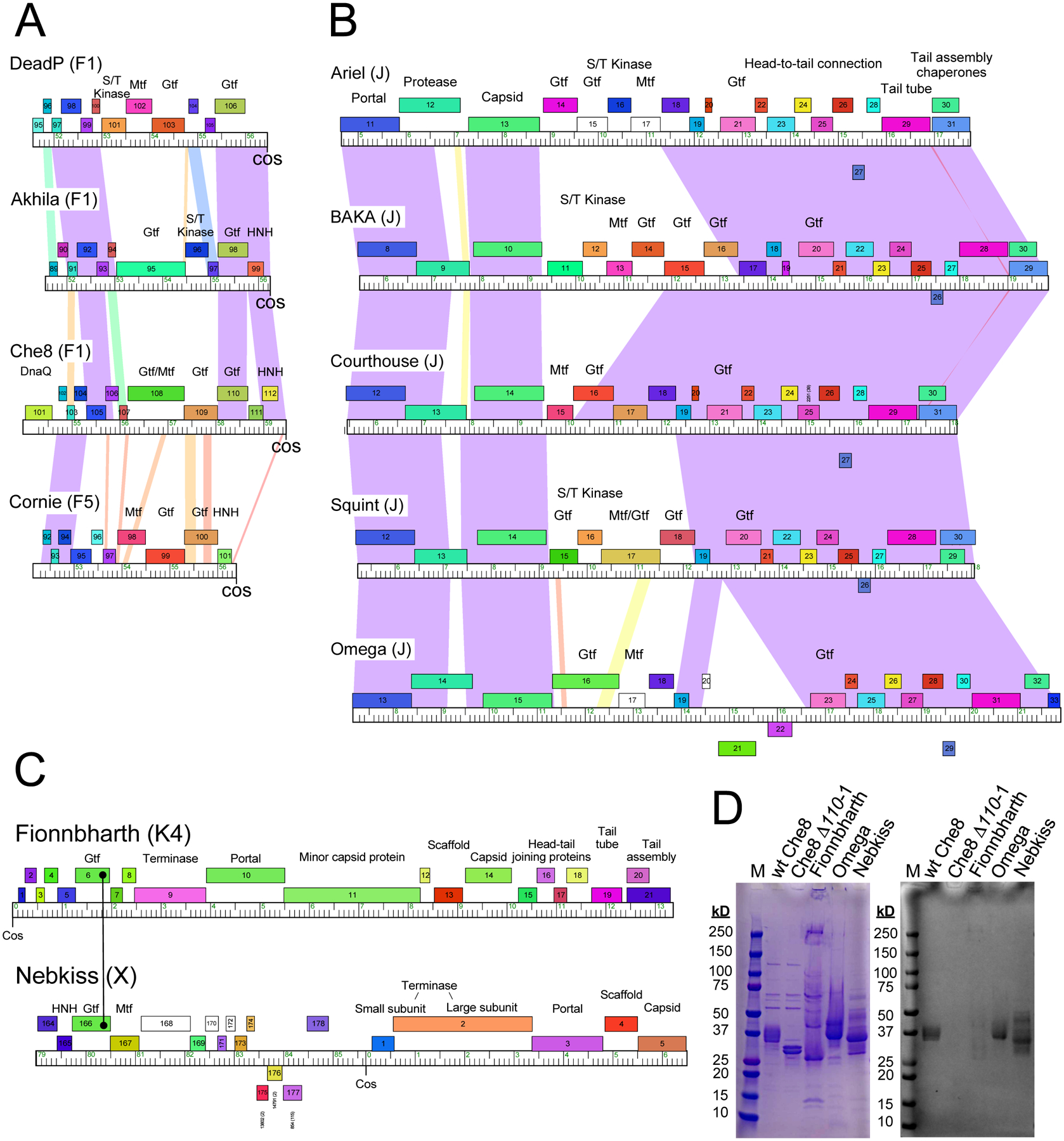
A. Cluster F phages coding for glycosyltransferases. Cluster F is a large group (216 individual phage members) of mycobacteriophages organized into five subclusters (F1 – F5). All of the phages code for glycosyltransferases, arranged into four different organizations, represented by phages DeadP, Akhila, Che8, and Cornie (subcluster designations are shown in parentheses). The organization represented by Akhila is the most common and is in 85% of Cluster F phages; the Che8, DeadP, and Cornie organizations are less common and are present in 20, 3 and 1 Cluster F phages, respectively. Segments from the extreme right ends of the genome are shown with genes shown as colored boxes; all genes shown are transcribed rightwards and are colored according to their phamily assignments (i.e. sequence-related genes are similarly colored)42. Shading between genomes reflects BLASTN pairwise similarity and is spectrum colored with violet being the most similar and red the least similar above a threshold E value of 10−4. Putative functions are noted above the genes: Mtf, methyltransferase; Gtf, glycosyltransferase; HNH, homing endonuclease; S/T kinase, serine-threonine kinase). B. Cluster J phages coding for glycosyltransferases. Genomes are represented as in panel A, with genes above and below each genome transcribed rightwards and leftwards, respectively. C. Organizations of phages Fionnbharth and Nebkiss from Clusters/Subclusters K4 and X, respectively. Maps are annotated as in panels A and B. For Nebkiss, the extreme left and right genome ends have been joined at cos, as they are in replicating genomes. Fionnbharth gp6 and Nebkiss gp166 are related Gtf’s containing Stealth CR2 domains. D. Evidence of glycosylation of Fionnbharth, Omega and Nebkiss virions. Phage proteins were separated by SDS-PAGE and stained by either Coomassie (left) or Glycostain (right), as in Figure 1.
Interestingly, we also note that prophages are abundant in some mycobacterial strains including M. abscessus,12,29 and a subset of these encode glycosyltransferases, including those grouped in Clusters MabA and MabR (Table 1). Both code for capsid and tail tube subunits which both have C-terminal serine residues, and the gtf/mtf genes are closely linked to the virion structural genes. Prophages in both Clusters MabA and MabR have been shown to be intact and can be spontaneously induced and lytically propagated on a suitable M. abscessus host strain.16 We predict that these also have glycosylated virions.
Although the Cluster J phages, which includes Omega, have gtf and mtf genes within their structural gene operons, only the tail tube subunit has a C-terminal serine; the capsid subunit does not (Table 1). Phages Fionnbharth and Nebkiss30 (in Clusters/Subclusters K4 and X, respectively) both code for Gtf proteins that contain Stealth CR2 domains, distinct from the Gtfs in the other phages characterized here (Fig. 6C). This is the sole predicted Gtf in both phages, although both also code for an Mtf (Fig. 6C). Only the capsid subunit of Fionnbharth (gp14) has a C-terminal serine, whereas in Nebkiss both capsid and tail tube subunits have C-terminal serine residues (Fig. 6C, Table 1). We note that in about one half of the genome types that encode glycosyltransferases (Table 1), neither the capsid nor tail tube subunits have the C-terminal serine residues. Though a C-terminal serine is conserved in all the phage proteins that have so far been experimentally shown to be glycosylated, it is not yet known whether this residue is obligatory for glycosylation or if glycosylation can occur at other solvent-exposed amino acid residues (Table 1).
To test whether some of these other actinobacteriophages that encode Gtf proteins are glycosylated, we separated virion proteins by SDS-PAGE and stained with the glycostain (Fig. 6D). We observed a single glycostain-positive band of phage Omega (Cluster J) containing both the major capsid protein (gp15, 51.6 kDa) and the tail tube subunit (gp31, 35.9 kDa), as determined by mass spectrometry (Fig. 6, Extended Data Set 3). It is likely that one or both are glycosylated. For Nebkiss (Cluster X), three glycostaining bands were observed, each of which contains both capsid (gp5, 34.7 kDa) and tail tube (gp10, 21.0 kDa) subunits, both of which have C-terminal serine residues (Table 1; Fig. 6, Extended Data Set 3). It is plausible that both the capsid and tail tube subunits have O-linked glycan attached to their C-terminus. We observed only weak staining of Fionnbharth proteins, and subunit identification is less clear. A weakly staining high molecular weight band may correspond to interlinked and crosslinked capsid proteins (gp14, 32.2 kDa; Fig. 6D) as indicated by mass spectrometry (Extended Data Set 3). There are additional weakly glycostaining bands in the 25–50 kDa region and mass spectrometry shows these to contain mixtures of several virion proteins, including portal (gp10, 58.4 kDa), and capsid subunits, both of which have C-terminal serine residues (Extended Data Set 3). Further analysis is required to confirm if the proteins identified in the glyco-stained bands are glycosylated and if so, the nature and the location of the glycan(s).
A search of the RefSeq collection of ~4,500 phage genomes in GenBank shows that relatively few encode one or more predicted Gtfs (<1.5%). Only 14 non-actinobacteriophage genomes code for two or more Gtfs and are the best candidates for potential virion modification (Table S4). Nine of these infect marine hosts (Synechococcus, Prochlorococcus, Flavobacterium), and with the exception of Bacillus phage BCP8–2, all infect gram-negative bacteria. With the exception of Enterobacteria phage SfI (38.3 kbp), all have large genomes (>140 kbp) and six are jumbo phages with genomes over 200 kb (Table S4). The glycosyltransferases of some these phages are predicted to be involved in cell wall glycosylation,31 and none of the phages have annotated capsid or tail tube subunits with C-terminal serine residues. Phage glycosylation may be relatively rare among the phage population at large, even though it appears to be prevalent among mycobacteriophages.
Discussion
Here, we have shown that the virions of several mycobacteriophages are O-glycosylated on their capsid and/or tail tube subunits. Mass spectrometry characterization of the capsids of Che8, Myrna and Corndog gave data consistent with O-linkage of large glycans to the C-terminal serine residues. Initial analysis of the phages Omega, Nebkiss and Fionnbharth, which also express putative glycosyltransferases, suggests that capsid and/or tail tube subunit proteins may also be glycosylated in these phages. A bioinformatic survey reveals that the majority of actinobacteriophages encoding glycosyltransferase genes contain at least one major structural protein with a C-terminal serine residue (Table 1), suggesting a possible conserved mechanism of phage virion glycosylation. Glycan chemical composition is different in Che8, Corndog, and Myrna, and in Che8 and Corndog some sugars are methylated. The O-glycan chains are quite long in all three phages and extend to at least twelve hexose and twelve HexNAc units in Che8, one HexNAc and 17 hexose units in Corndog, and 13 Hex and 13 HexNAc units in Myrna. The sugars proximal to the peptide on the capsid subunits of phage Che8 were visualized by cryoEM. All glycan modification was lost when Che8 gene 110 encoding a glycosyltransferase was inactivated.
Although most of the glycan chains in Che8 are likely disordered and not visualized by cryoEM, they provide extensive coating of the phage virions, with consequences for recognition of the underlying protein chains by external ligands including antibodies. Inoculation of wild type glycosylated phage Che8 and the unglycosylated mutant into mice results in phage-specific antibody titers that are generally similar, but both IgM and IgG antibodies preferentially recognize the unglycosylated phage particles at early times after mouse inoculation. However, serum from mice inoculated with glycosylated virions is less effective at phage neutralization in vitro, especially at later times, presumably reflecting differences in how the two phages are processed by the adaptive immune response. We note that our analysis of B and T cell responses to glycosylated and unglycosylated phages was limited by a lack of defined MHC-I and MHC-II restricted epitopes for these phages, thus limiting our analysis to bulk cellular responses.
Glycans can provide a shield against neutralizing antibodies5, but also affect antigen processing to induce antibodies and effector T cells. Glycosylated peptides represent a minority of defined T cell epitopes for HIV32, hepatitis C virus33, and SARS-CoV-2.34 Moreover, evidence suggests that glycosylation interferes with CD4+ and CD8+ T cell epitope recognition.35 Thus, unglycosylated mutant phages may result in enhanced epitope presentation by MHC-I and MHC-II molecules, driving more robust antibody and T cell responses.
The six phages shown to be glycosylated all contain at least one and often multiple genes coding for glycosyltransferases, and some code for methyltransferases; these sets of genes are typically closely linked to genes coding for their target virion structural proteins (Figs 1, 6). Several other mycobacteriophages share these genomic organizations and are likely also glycosylated, although the variation and diversity of the numbers and types of glycosyltransferase genes suggests that these are actively evolving. A few phages of Gordonia and Streptomyces may also be glycosylated, but glycosylation does not appear to be a feature of bacteriophages in general, at least among those for which genomic sequences are available. It is possible that some phages have alternative systems for glycosylation.
The role of glycosylation in these phages is unclear. However, several Mycobacterium species can thrive in intracellular environments, a property that may reflect environmental interactions with amoebae.36–38 The three phages characterized here, Corndog, Che8, and Myrna, do not infect the intracellular pathogen M. tuberculosis or the opportunistic pathogens M. avium or M. abscessus,14,16,39 although we note that Fionnbharth can infect M. tuberculosis.14 In general, it seems unlikely that these phages encounter bacterial hosts in mammalian systems, but in environmental situations their hosts could be engulfed by amoebae, thus limiting access to the phage. Indeed, bacterial survival of amoebae ingestion could have evolved as a phage defense mechanism. Virion glycosylation of the phages may represent a counter measure that facilitates uptake of phage particles by amoebae and plausibly trafficking to bacterial intracellular locations. Glycosylation could also facilitate association with mucoidal surfaces and thus promote access to its host, as proposed for Ig-like domains on phage particles.40
Phage glycosylation has implications for the therapeutic use of phages, especially for control of Mycobacterium infections. Although such therapeutic use has shown promise7,10,41 antibody responses arise when the phage are administered intravenously over prolonged periods, and could interfere with phage efficacy against Mycobacterium infection. Phage neutralization in vitro is relatively common by patient sera17 although does not necessarily correlate with clinical outcomes. Our observations here suggest that use of glycosylated phage particles may have some clinical advantages due to the weaker anti-phage reactivity generated in early responses, and diminished neutralization especially at later times. Although none of the phages shown here have yet been used therapeutically, suitable genetic modifications of phages that have useful host tropisms, and/or growth of phages in recombinant strains could combine the potential advantages of glycosylation with host range and lytic activity.
Limitations of the study
The immune responses reported here were observed in mice and immune responses in human patients may differ substantially. It thus remains unclear as to whether phage glycosylation will offer any substantial therapeutic benefit, and clinical trials comparing glycosylated and unglycosylated phages will be required to address any therapeutic potential. The detailed mechanisms of glycosylation warrant further exploration to determine the roles of each phage encoded glycosyltransferase and methyltransferase, and any possible role of host enzymes. It remains unclear when and where during phage virion assembly the process of glycosylation occurs. The specific monosaccharide isomers composing the glycans still need to be determined as well as their arrangement and linkages. This initial study suggests that glycosylation may be widespread among mycobacteriophages but is likely highly diverse. An exploration of glycosylation in a wider variety of phages, including non-mycobacteriophages without obvious glycosyltransferases, is warranted.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Graham Hatfull (gfh@pitt.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE43 partner repository and are publicly available as of the date of publication. The Che8 Δ110-1 cryo-EM map was deposited at the EMBD and the Che8 Δ110-1 cryo-EM raw data was deposited at EMPIAR; these are publicly available as of the date of publication. Accession numbers for all standardized datasets are listed in the key resources table. Original western blot images and the four Extended Data Set files are available on Mendeley at: doi: 10.17632/49hmcxgjnr.2
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat Anti-Mouse IgG H&L (HRP) preadsorbed | Abcam | ab97040, RRID: AB_10698223 |
| Goat Anti-Mouse IgM mu chain (HRP) preadsorbed | Abcam | ab98679, RRID: AB_10696527 |
| Goat Anti-Human IgG Fc (HRP) preadsorbed | Abcam | ab98624, RRID: AB_10673832 |
| Normal Human Serum | ThermoFisher | 31876, RRID: AB_2532169 |
| Goat anti-Mouse IgG H&L (10nm gold) | Abcam | ab39619, RRID: AB_954440 |
| ACK Lysis Buffer | Gibco | A10492–01 |
| Live/Dead Violet Stain | Invitrogen | L34955 |
| αCD16/32 Fc block | Tonbo Biosciences | 70–0161-M001 |
| CD3e-BUV395 | BD Biosciences | BD 565992, RRID: AB_2739443 |
| CD4-AF700 | BioLegend | BioLegend 100536, RRID: AB_493701 |
| CD8a-PE-Cy7 | BD Biosciences | BD 552877, RRID: AB_394506 |
| CD19-BV785 | BioLegend | BioLegend 115543, RRID: AB_11218994 |
| B220-APC | BioLegend | BioLegend 103212, RRID: AB_312997 |
| NK1.1-BV421 | BD Biosciences | BD 562921, RRID: AB_2728688 |
| CD44-APC-Cy7 | BD Biosciences | BD 560568, RRID: AB_1727481), |
| CD62L-BUV563 | BD Biosciences | BD 741230, RRID: AB_2870784 |
| BD Brilliant Stain Buffer | BD Biosciences | BD 566349 |
| Foxp3 Fix/Perm | ThermoFisher | 00-5523-00 |
| Normal rat serum | Invitrogen | 10710C, RRID: AB_2532985 |
| Foxp3 isotype control antibody | Invitrogen | 45-4321-80, RRID: AB_906259 |
| Foxp3-PerCPCy5.5 antibody | Invitrogen | 45-5773-82, RRID: AB_914351 |
| Bacterial and virus strains | ||
| Mycobacterium smegmatis mc2155 | Snapper et al., 1990 | NC_008596 |
| Mycobacteriophage Che8 | Pedulla et al., 2003 | AY129330.1 |
| Mycobacteriophage Che8 Δ110-1 | This paper | N/A |
| Mycobacteriophage Myrna | Hatfull et al., 2010 | EU826466 |
| Mycobacteriophage Corndog | Pedulla et al., 2003 | AY129335 |
| Mycobacteriophage Fionnbharth | Pope et al., 2015 | JN831653 |
| Mycobacteriophage Omega | Pedulla et al., 2003 | AY129338 |
| Mycobacteriophage Nebkiss | Pope et al., 2015 | MK016501 |
| Biological samples | ||
| Mouse sera | This paper | N/A |
| Mouse spleens | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| 3,3′,5,5′-Tetramethylbenzidine (TMB) Liquid Substrate System for ELISA | Sigma Aldrich | Cat# T0440–1 |
| Carbonate-bicarbonate buffer capsules | Sigma Aldrich | Cat. # C3041 |
| Sulfuric acid | EMD Millipore | CAS: 7664-93-9 |
| ECL substrate | https://www.thermofisher.com/order/catalog/product/34580 | |
| Imperial protein stain | Fisher Scientific | Cat# 4615 |
| Sequencing grade modified trypsin | Promega | Cat#V5113 |
| Chymotrypsin | Sigma | Cat#C3142 |
| Critical commercial assays | ||
| Deposited data | ||
| Mass spectrometry proteomics data | ProteomeXchange Consortium via PRIDE | PXD041690 and 10.6019/PXD041690 |
| Mycobacteriophage Che8 Δ110-1 cryo-EM capsid map | EMDB | EMD-28761 |
| Mycobacteriophage Che8 Δ110-1 raw cryo-EM data | EMPIAR | EMPIAR-11285 |
| Supplemental Data Set 1 | Mendeley | 10.17632/49hmcxgjnr.2 |
| Supplemental Data Set 2 | Mendeley | 10.17632/49hmcxgjnr.2 |
| Supplemental Data Set 3 | Mendeley | 10.17632/49hmcxgjnr.2 |
| Supplemental Data Set 4 | Mendeley | 10.17632/49hmcxgjnr.2 |
| Western blot raw images | Mendeley | 10.17632/49hmcxgjnr.2 |
| Experimental models: Cell lines | ||
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | Jackson Laboratory | 000664, RRID: JAX:000664 |
| IFNβ-EYFP reporter mice (C57BL/6J background) | Jackson Laboratory | 10818, RRID: JAX:010818 |
| Oligonucleotides | ||
| Forward primer for sgRNA plasmid construction: GGGAGAGTGCGGCGTGGAGCTGGTC | This paper | N/A |
| Reverse primer for sgRNA plasmid construction: AAACGACCAGCTCCACGCCGCACTC | This paper | N/A |
| Recombinant DNA | ||
| Plasmid pIRL53 | Rock et al., 2017 | |
| Software and algorithms | ||
| OriginLab | Version 10.05(2023b) | |
| FlowJo | Version v10.8.1 | |
| Gen5 Microplate Reader and Imager Software | BioTek | TS Installation Version 2.06.10 |
| Mascot Server version 2.7.0 | Matrix Science | https://www.matrixscience.com/ |
| Other | ||
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
In vivo animal studies
This study utilized healthy adult C57BL/6J (Jackson Laboratory strain 000664, RRID: JAX:000664) and IFNβ-EYFP reporter mice, which are on the C57BL/6J genetic background (Jackson Laboratory strain 10818, RRID: JAX:010818). Mice were randomly assigned to experimental groups as reported in Table S3 and were not involved in any previous procedures. The sex and age of each mouse, individual animal designators used in the text and figures, and inoculation details are also reported in Table S3; in general, we used both male and female mice with starting ages ranging from 6 to 11 weeks old. We did not use sufficient mice to determine if there are sex differences, which we identify as a limitation of the study.
All mice were bred and maintained in specific pathogen free conditions in accordance with the Institutional Animal Care and Use Committee of University of Pittsburgh and the Guide for the Care and Use of Laboratory Animals (NIH publication no. 85–23. Revised 1985). Mice were handled according to protocols approved by the University of Pittsburgh Subcommittee on Animal Care (IACUC protocol 21028897).
Bacterial strains
This study utilized M. smegmatis mc2 155, colonies of which were grown on Middlebrook 7H10 agar plates and cultured in Middlebrook 7H9 supplemented with ADC at 37 °C.44
METHOD DETAILS
Bioinformatics
Phage genome analyses were performed using two databases made using Phamerator42 and MMseqs245 (“Actinobacteriophage_2422” and “Actino_Draftv501”) containing completely sequenced Actinobacteriophage genomes.
Construction of Che8Δ110-1
Che8Δ110-1 was constructed by isolating escape mutants of CRISPR-mediated selection against phages with the wild type Che8 gene 110 sequence. pIRL53 is a plasmid containing the active Cas9 gene from Streptococcus thermophilus under the control of a tet promoter and was a kind gift from Dr. Jeremy Rock.46 The 8 kb plasmid vector was linearized using BsmBI and gel purified. For the sgRNA targeting sequence, complementary primers for Che8 110 were designed to match a portion of the gene 110 upstream of a PAM site. Primers used were 5’-GGGAGAGTGCGGCGTGGAGCTGGTC and 5’-AAACGACCAGCTCCACGCCGCACTC. These were annealed together in a thermocycler, and the annealed primers were then inserted into the pIRL53 linearized vector using T4 ligase. Ligation reactions were transformed into chemically competent DH10B and plated on LB plus kanamycin selective media. Surviving colonies were grown overnight in liquid LB with kanamycin, and miniprepped; plasmids were sequenced by Genewiz to verify construction. Cas9 plasmids containing complementary sequence to Che8 110 were transformed into electrocompetent M. smegmatis mc2155 and plated on 7H10 plus kanamycin selective media. Surviving colonies were grown to saturation in liquid 7H9 with kanamycin and this culture was mixed with phage and plated with top agar onto bacterial lawns with and without inducing agent anhydro tetracycline (ATc). The induced plates strongly select against the wild type phage, and an escape mutant was isolated, purified and propagated. Illumina whole genome sequencing was performed, and genome analysis confirmed inactivation of gene 110 through a 7-base deletion (coordinates 58266–58272 in the wild type Che8 genome) which results in a frameshift and introduction of an early stop codon. Sequencing also showed an off-target deletion of wild type Che8 coordinates 49730 – 52095 in the Che8 Δ110-1 mutant.
Phage preparation
Isolation and CsCl banding of mycobacteriophages in this study was described previously.21 Briefly, 30 large Petri plates of 7H10 medium plus dextrose, carbenicillin, and cycloheximide were pour-plated with host bacteria and sufficient phages to generate a webbed surface; these plates were flooded with phage buffer (10 mM Tris, pH 7.5, 10 mM MgSO4, 68 mM NaCl) and incubated at 4 °C overnight. The lysate was harvested and NaCl to 0.5M and 10% PEG 8000 were added, and allowed to precipitate overnight at 4°C. The PEG pellet was harvested by centrifugation and resuspended by rocking in phage buffer overnight at 4 °C. CsCl was added at a ratio of 0.75 g per 1 mL of phage solution. The resuspended phages were transferred to heatseal tubes and ultracentrifuged at 40,000 rpm for 16 hours in a fixed angle rotor. Visible phage bands (~0.5 to 1 mL) were pulled from the tube using a syringe and transferred to Slide-a-Lyzer cassettes (MWCO 10,000 Da); these were dialyzed against two 1 L changes of phage buffer in room temperature prior to further analysis.
Phages used in mouse studies were prepared similarly, except phages were pelleted by centrifugation at 100,000 x g for one hour instead of PEG precipitated. These samples were dialyzed extensively against PBS++ (PBS, Sigma Aldrich P3813, supplemented with 1mM CaCl2 and 1mM MgCl2).
SDS-PAGE and glycoprotein staining
For large format gels (as in Fig. 2), 2 × 1011 pfu of phage were pelleted by centrifugation at 13,000 rpm for 30 minutes. Pellets were resuspended in 75 μl of 20 mM dithiothreitol (DTT), then 2 μl of 0.5M Ethylenediaminetetraacetic acid (EDTA, pH 8) was added and the samples were incubated at 75°C for 2 minutes to release the DNA. To digest the DNA, 4 μL of 1M MgSO4 and 2 μL of 1 mg mL−1 DNase I were added to the samples before incubation at 30°C for 20 minutes. Proteins were denatured by adding 28 μL of 4x SDS loading dye (200 mM Tris-HCl pH 6.8, 8% SDS, 40% glycerol, 4% β-mercaptoethanol, 50 mM EDTA, 0.08% bromophenol blue) and boiling at 95°C for 2.5 minutes. From these samples, 10 μl was loaded into each well of large format 6–15% SDS PAGE gradient gel (~2 × 1011 pfu per well) and the proteins were separated by electrophoresis at 120 V for 20 hours. Part of the gel was excised and stained with Imperial protein stain (ThermoFisher 24615) while the other part was stained with Glycoprotein stain (ThermoFisher 24562). For small format gels (as in Fig. 1), similar phage preparations were separated on a 12% SDS polyacrylamide gel at 100 V until the dye front ran to the bottom of the gel. Gels were stained with either Bio-Safe Coomassie Stain (Bio-Rad 1610786) or Glycoprotein stain.
In-gel tryptic digestion and nanoLC-MS/MS analysis on Q-Tof hybrid mass spectrometer.
Protein bands from the Coomassie-stained mini-gel corresponding to the Glycostain-reactive bands were excised, placed in clean Eppendorf tubes and destained with 100 mM ammonium bicarbonate, 30% acetonitrile. The gel bands were dehydrated with acetonitrile, reduced with 10 mM DTT in 50 mM ammonium bicarbonate at 56°C for 1 hour then alkylated with 55 mM iodoacetamide in 50 mM ammonium bicarbonate at room temperature in the dark for 1 hour. The gel bands were dehydrated again with acetonitrile then ~100 ng of trypsin (Promega) in 50 mM ammonium bicarbonate was added. Sufficient 50 mM ammonium bicarbonate to cover the gel bands was added and the tubes were incubated overnight at 37° C.
The digests were analyzed using a MClass nanoHPLC (Waters) coupled to a Premier Q-Tof or a Synapt G2S1 Q-Tof hybrid mass spectrometer (Waters). The samples were injected onto a C18 PepMap100 5 μm trap (Thermo Scientific) coupled to a nanoACQUITY BEH 1.7 μm 100 μm × 100 mm C18 column (Waters) and separated using a 60-minute gradient. Mobile phase A was 0.1% formic acid in ddH2O, and mobile phase B was 0.1% formic acid in acetonitrile. Peptides were eluted using a linear gradient of 0.2% to 40% mobile phase B over 44 min, 40% to 95% mobile phase B over 4 min. The flow rate was 0.5 μL/min. MS spectra were acquired from m/z 200 to 2000 in positive electrospray ionization mode at 10 K and 30 K resolution on the Premier and Synapt Q-Tof’s respectively. Peptide and glycopeptide MS/MS spectra were acquired in Data Dependent Acquisition (DDA) mode. Mascot™ was used to search the MS/MS spectra against a Custom database containing all annotated protein sequences from the Myrna, Corndog and Che8 genomes. Glycopeptide MS and MS/MS spectra were examined manually.
In-gel chymotryptic digestion and nanoLC-MS/MS on Orbitrap Eclipse Tribrid mass spectrometer
The glycostain-reactive gel bands from the Che8 SDS-PAGE gel were excised, destained and dehydrated as described above but not reduced and alkylated, then treated with chymotrypsin (200 ng) in 50 mM ammonium bicarbonate and incubated at 25°C overnight. The resulting digests were analyzed on an Orbitrap Eclipse Tribrid mass spectrometer equipped with an electrospray ionization source (Thermo Scientific) connected to an UltiMate 3000 nano-LC system (Thermo Scientific). The same reverse phase column setup described above was used to resolve the peptides and glycopeptides prior to MS analysis. Mobile phase A was 0.1% formic acid in ddH2O, and mobile phase B was 0.1% formic acid in acetonitrile. Peptides were eluted using a linear gradient of 0.2% to 37% mobile phase B over 45 min, 37% to 95% mobile phase B over 3 min. The flow rate was 0.5 μL/min. MS spectra were acquired in the Eclipse from m/z 200 – 2000 or 350 – 2000, depending on the analysis, and in positive electrospray ionization mode at 120 K resolution. The most intense ions (threshold = 1e6, dynamic exclusion = 25 sec) were selected in the quadrupole for HCD-MS/MS (isolation window = 1.6 m/z) and a fixed HCD activation energy was selected based on peptide m/z and charge state using a decision tree. Fragment ion spectra were acquired in the Orbitrap at 15 K resolution in profile mode. Cycle time was 1 second. The MS and MS/MS spectra were analyzed in the same manner as described above.
Adsorption Assay
M. smegmatis mc2155 was grown to mid-log phase (OD600 = 0.6) in 7H9/ADC/CaCl2 and inoculated with phage to an MOI of 0.01. Each reaction contained 20 mL of bacterial culture and 20 μL of phage lysate. Separate negative control reactions were set up by adding 20μL of phage buffer + 1 mM CaCl2 to 20 mL of sterile media. Immediately upon addition of phage/buffer the flasks were started shaking at 150 rpm at 37 °C. At various timepoints 1 mL samples were removed from the flasks and centrifuged at 11,000 x g to pellet bacteria and adsorbed phage. A sample of the supernatant was taken and serially diluted before spotting 10 μL spots onto prepared lawns of M. smegmatis mc2155. Plaques were counted by hand after 24 hours of incubation at 37 °C and titers of non-adsorbed phage were calculated and plotted.
Fecundity Assay
Agar plugs containing entire well-isolated plaques were excised from bacterial lawns using the large end of a 1000 μL pipet tip. These were squeezed into 1 mL of phage buffer supplemented with 1 mM CaCl2 and the sample was vortexed to break up the agar plug and dissolve the bacterial lawn layer containing the plaque. This sample was centrifuged at 17,000 × g for 1 minute to pellet the bacterial and agar, and a portion of the supernatant was removed and serially diluted. Full-plate plaque assays were performed with 10 μL of the 10−5 dilution for each sample. Plaques were counted by hand after 24 hours of incubation at 37 °C and the number of phages released into the initial 1 mL volume were calculated and plotted. These experiments were performed with six technical replicates.
Stability Assay
Phages at 1011 and 1012 pfu/mL in PBS++ were transferred to screw-cap tubes and incubated at 4 °C or room temperature (~20 °C). At various timepoints the samples were serially diluted and quantified by full-plate plaque assays with 10 μL samples of the dilutions. Plaques were counted by hand after 24 hours of incubation at 37 °C and titers were calculated and plotted. These experiments included two or three technical replicates and data are plotted as mean +/− standard error.
Cryo-Electron Microscopy
To prepare grids, 5 μL of mutant (Che8 Δ110-1) phage particles (~10 mg/mL) were added to Au-flat 2/2 (2 μm hole, 2 μm space) microscopy grid (Protochips, Morrisville, NC, USA) with a Vitrobot Mk IV (Thermo Fisher Scientific, Waltham, Massachusetts, USA). Grids were blotted for 5 s with a force of 5 before being plunged into liquid ethane.
Data were collected on a 300 keV Titan Krios (Thermo Fisher Scientific, Waltham, Massachusetts, USA) at the Pacific Northwest Center for Cryo-EM with a K3 detector (Gatan, Pleasanton, CA, USA). Supporting Table 1 provides the collection parameters for each phage. The cryoSPARC47 software was used for phage capsid reconstructions using the standard workflow (import, motion correction, CTF estimation, blob picker, extraction, 2D classification, homogenous refinement) using the default settings. Particles were extracted using an 800 pixel box size. Homogenous refinement was carried out with “optimize-per-particle defocus”, “optimize per-group CTF params” and Ewald sphere correction using the default parameters. The wild-type Che8 major capsid protein model (PDB: 8E16) was fitted into the mutant capsid map using ChimeraX version 1.448 and the “Fit in Map” command.48 The model fitted the map with no changes needed. Figure 3 was created using ChimeraX and contouring both the wild-type map (EMD-27824) and the mutant map to the same level.
Mouse studies
Phages or PBS++ (PBS supplemented with 1 mM MgSO4 and 1 mM CaCl2) were administered to mice intraperitoneally in a 100 μL volume. The total protein concentration of phages was measured with Bradford assay and adjusted to 10 μg/mL such that each 100 μL dose delivered 1 μg. Serum was collected by submandibular bleeding at indicated times after initial inoculation (Fig. 4 and S5). Mice were humanely euthanized at the terminal timepoint and spleens and terminal blood were harvested.
ELISA
ELISAs were performed as previously described.49 Briefly, highly purified bacteriophage samples were diluted to 225 ng/mL (as measured by Bradford assay, corresponds to ~ 5 × 109 pfu/mL) in carbonate-bicarbonate coating buffer (Sigma C3041) and 100 μl was used to coat the wells of EIA microplates (Corning CLS3590) at 4° C overnight. The coating solution was removed and the plates were washed with 200 μl of PBST (10 mM phosphate, 138 mM NaCl, 2.7 mM KCl, pH 7.4 + 0.05% Tween 20), then blocked with 300 μL of PBST + 3% milk overnight at 4° C. The blocking buffer was removed and 100 μl of serum serial dilutions were added to the wells such that each individual serum is assayed at eight dilutions starting at 1:100 and increasing in dilution power by factors of 3. After an overnight incubation with serum dilutions at 4° C, sera were aspirated, and the plates were washed five times with 200 μL of PBST before the addition of secondary antibody. Secondary antibodies were diluted 1:10,000 into PBST and include Goat Anti-Mouse IgG H&L (HRP) preadsorbed (Abcam ab97040, RRID: AB_10698223) and Goat Anti-Mouse IgM mu chain (HRP) preadsorbed (Abcam ab98679, RRID: AB_10696527). After one hour of incubation at room temperature the secondary antibody was removed and the plates were washed three times with PBST and two times with PBS, then developed by adding 100 μL of TMB substrate (Sigma T0440) and, after 6–8 minutes of incubation, 100 μL of 2N H2SO4. The plates were immediately quantified in a plate reader by measuring the optical density at 450 (signal) and 570 (background). The background-subtracted data were plotted as a function of serum dilution on a semi-log plot and fit with a fixed-baseline logistic curve using OriginLab. Logistic fits were compared for each experimental dataset and its corresponding uncoated well control; datasets with signals < 4X the signal from the corresponding uncoated control well were deemed below the limit of detection and their half-maximal titer values were set to half of the limit of detection (2log10/2 = 1log10) to allow statistical analysis. For samples with signals > 4X the background, half-maximal serum dilutions were obtained from the logistic fit. Half-maximal serum titers presented in Fig. 4 are calculated as log10(1/half-maximal serum dilution).
Multicolor Flow Cytometry
One week after the second dose of phage, splenocytes were harvested by passing spleens through a 70 μm nylon cell strainer with RPMI/10% FBS (fetal bovine serum) and pelleted by centrifugation at 1500 rpm for 5 minutes. The supernatant was removed and red blood cells were lysed with 2 mL of ACK lysis buffer (Gibco A10492–01) for 1 minute. 10 mL RPMI/10% FBS was added after ACK Lysis and the remaining cells were again pelleted, the supernatant removed, and the cells transferred to a 96-well plate and re-pelleted. The cells were then washed with 200 μL PBS before adding 100 μl of Live/Dead Violet stain (Invitrogen L34955), diluted 1:1000 in PBS. After a 12-minute incubation in the dark, the cells were pelleted, washed with 200 μl PBS, and blocked with 100 μL of αCD16/32 Fc block (Tonbo Biosciences 70–0161-M001) diluted 1:100 in PBS/1% FBS. After a 10-minute incubation, the cells were pelleted and the supernatant removed, then 25 μL of surface antibody cocktail was added, followed by incubation at 4 °C in the dark for 40 minutes. The cocktail consisted of the following surface antibodies: CD3e-BUV395 (BD 565992, RRID: AB_2739443), CD4-AF700 (BioLegend 100536, RRID: AB_493701), CD8a-PE-Cy7 (BD 552877, RRID: AB_394506), CD19-BV785 (BioLegend 115543, RRID: AB_11218994), B220-APC (BioLegend 103212, RRID: AB_312997), NK1.1-BV421 (BD 562921, RRID: AB_2728688), CD44-APC-Cy7 (BD 560568, RRID: AB_1727481), and CD62L-BUV563 (BD 741230, RRID: AB_2870784). All surface antibodies were diluted 1:100 in BD Brilliant Stain Buffer BD Cat. #566349). After antibody incubation, cells were pelleted, supernatants removed, and the pellets washed with PBS/1%FBS. Cells were pelleted once more, then 200 μL of Foxp3 Fix/Perm (ThermoFisher 00-5523-00) was added and the cells were incubated at 4°C in the dark overnight. The next day the cells were pelleted at 1640 rpm for 3 minutes, supernatant removed, and washed once with 200 μL of Fix/Perm buffer, followed by addition of 25 μL of Fix/Perm buffer with normal rat serum (Invitrogen 10710C, RRID: AB_2532985). From these samples, 2 μL were removed and incubated at 4 °C for 1 hour with 25 μl of Fix/Perm buffer with rat serum and 2.5 μL of Foxp3 isotype control antibody (Invitrogen 45-4321-80, RRID: AB_906259). The remaining cells were incubated with 25 μL of the following ICS antibody cocktail: 250 μL Fix/Perm buffer + 25 μl Foxp3-PerCPCy5.5 antibody (Invitrogen 45-5773-82, RRID: AB_914351). After the incubation, cells were washed with 200 μL PBS/1%FBS, pelleted, and resuspend in 200 μL PBS/1%FBS. 100uL of BioLegend Precision Count beads (424902) were added and stained samples were run on Cytek® Aurora multispectral flow cytometer. Data were analyzed in FlowJo (v10.8.1) and cell population statistics were analyzed with OriginLab version 10.05(2023b).
Western Blots
Western blots were performed as previously described.49 Briefly, gel samples were prepared as described above but with a total of 2 × 1012 pfu of phage pelleted. From these samples, 10 μL was loaded into each well of precast 4–20% TGX gradient gels (Bio-Rad 4561096) and the proteins were separated by electrophoresis at 300 V for 20 minutes. Part of the gel was excised and used for staining (Imperial protein stain, ThermoFisher 24615) to confirm equal loading across wells and the rest was transferred to PVDF membrane (Bio-Rad 1620177) via wet transfer with Towbin buffer (25 mM Tris base, 192 mM glycine, 20% v/v MeOH) at 100V for 1 hour. Membranes were washed in TBS (25 mM Tris-HCl pH 7.5, 300 mM NaCl), blocked overnight at 4° C in TBS + 3% milk, and incubated with mouse serum diluted 1:200 in TBST + 3% milk (TBS with 0.05% Tween 20) overnight at 4° C. The blot shown in Fig. 4G was probed with normal human serum (ThermoFisher 31876, RRID: AB_2532169). After serum incubation, membranes were washed extensively in TBST before incubating for 45 minutes at room temperature with HRP-conjugated secondary antibody. For mouse sera, the secondary was Goat Anti-Mouse IgG H&L (HRP) preadsorbed (Abcam ab97040, RRID: AB_10698223), diluted 1:20,000 in TBST + 3% milk. For human serum, the secondary was Goat Anti-Human IgG Fc (HRP) preadsorbed (Abcam ab98624, RRID: AB_10673832), diluted 1:10,000 in TBST + 3% milk. After incubation with secondary the blots were washed six times in TBST and once in TBS, then incubated for 5 minutes with ECL substrate (SuperSignal WestPico PLUS Chemiluminescent Substrate, ThermoFisher 34579) and imaged with an Amersham Imager 600 (GE Healthcare).
Immunogold staining
TEM grids (Ted Pella 01814-F) were glow-discharged for 30 seconds then floated on 25 μL drops of wild type Che8 or Che8Δ110-1 at a titer of 1.0 × 1011 pfu/mL for 1 minute. Grids were then washed by floating on 25 μL drops of dH2O for 1 minute, then floated on 25 μL drops of blocking buffer (PBS + 0.05% Tween-20 + 0.3% BSA) for 15 minutes in a humidified chamber. After blocking, the grids were floated on 25 μL drops of sera from individual mice (diluted 1:200 in blocking buffer) for 1 hour in a humidified chamber before washing 5 times by floating on 25 μL drops of wash buffer (PBS + 0.05% Tween-20 + 0.03% BSA) for 3 minutes each time. Next, the grids were floated for 45 minutes on 25 μL drops of Goat anti-Mouse IgG H&L (10nm gold) gold-conjugated secondary antibody (Abcam ab39619, RRID: AB_954440), diluted 1:20 in blocking buffer. Finally, the grids were washed in 5 drops of wash buffer then 3 drops of phage buffer (10 mM Tris, pH 7.5, 10 mM MgSO4, 68 mM NaCl) for 3 minutes each wash before negative staining in 3 drops of 1% uranyl acetate. All grid preparation steps occurred at room temperature. Prepared grids were imaged at 36,000x using a TEM-FE-Morgani microscope on spot 2 and images were used to manually quantify the number of immunogold beads adhered to each virion.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed with OriginLab version 10.05(2023b). Statistical details including exact values of n (representing number of animals, number of biological replicates, or number of technical replicates as indicated) can be found in figure legends, in the method details subsection, and in Table S3. Statistical significance was assessed with unpaired (two-sample) t-tests and resulting P values are shown in figures for sample pairs that are significantly different.
Supplementary Material
Extended Data Set 1. Peptides identified by Mascot in the tryptic digests, related to the phage gel bands that reacted with glycostain, supporting Figure 1D. Some peptides were identified more than once but, for simplicity, only the top scoring hit is presented here. Peptides with scores less than 20 were considered unreliable and have been removed. Tabs include data for Corndog, Che8, and Myrna.
Extended Data Set 2. Peptides identified by Mascot in the chymotryptic digests, related to the Che8 WT and Che8Δ110-1 gel bands, supporting Figure 2D. Some peptides were identified more than once but, for simplicity, only the top scoring hit is presented here. Peptides with scores less than 20 were considered unreliable and have been removed. Tabs show data for each Che8 band shown in Figure 2 and the Che8Δ110-1 mutant.
Extended Data Set 3. Peptides identified by Mascot in the tryptic digests, related to the Che8 immunoreactive bands and the Omega, Fionnbharth, and Nebkiss glycostain-positive bands, supporting data shown in Figure 4F. Some peptides were identified more than once but, for simplicity, only the top scoring hit is presented here. Peptides with scores less than 20 were considered unreliable and have been removed.
Extended Data Set 4. Raw ELISA data, related to Figures 4, 5, S2, S5, and S6. Raw reads from the plate reader, background subtraction calculations, tabulated half-maximal and endpoint titers, and tabulated results from two-sample t-tests are included as separate tabs.
Highlights:
Some mycobacteriophages are glycosylated on capsid and/or tail tube subunits.
Glycosylation is mediated by phage-encoded glycosyltransferases.
The glycans are large, complex and vary significantly between phages.
Glycans alter antibody production and shield viral particles from antibody binding.
Acknowledgments
This work was supported by grants from National Institutes of Health grants GM131729 (GFH), AI085062 (JVW), GM008208 (OBP), and AI089443 (YZ), Howard Hughes Medical Institute Grant GT12053 (GFH), and by the Henry L. Hillman Foundation (JVW). A portion of this research was supported by NIH grant U24GM129547 and performed at the PNCC at OHSU and accessed through EMSL (grid.436923.9), a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research. We thank Theo Humphreys for help with cryoEM data collection, the Biostatistical Consulting Laboratory at the University of Pittsburgh Graduate School of Public Health for advice regarding statistical analyses, and Dan Russell for phage genome sequencing.
Declaration of interests
GFH received support from Janssen Inc through a Collaborative Research Agreement, which did not fund work in this report. JVW serves on the Scientific Advisory Board of Quidel and an Independent Data Monitoring Committee for GlaxoSmithKline, neither involved in the present work.
Inclusion and Diversity
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Feng T, Zhang J, Chen Z, Pan W, Chen Z, Yan Y, and Dai J (2022). Glycosylation of viral proteins: Implication in virus-host interaction and virulence. Virulence 13, 670–683. 10.1080/21505594.2022.2060464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhao X, Chen H, and Wang H (2021). Glycans of SARS-CoV-2 Spike Protein in Virus Infection and Antibody Production. Front Mol Biosci 8, 629873. 10.3389/fmolb.2021.629873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vigerust DJ, and Shepherd VL (2007). Virus glycosylation: role in virulence and immune interactions. Trends Microbiol 15, 211–218. 10.1016/j.tim.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chamontin C, Bossis G, Nisole S, Arhel NJ, and Maarifi G (2021). Regulation of Viral Restriction by Post-Translational Modifications. Viruses 13. 10.3390/v13112197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wagh K, Hahn BH, and Korber B (2020). Hitting the sweet spot: exploiting HIV-1 glycan shield for induction of broadly neutralizing antibodies. Curr Opin HIV AIDS 15, 267–274. 10.1097/COH.0000000000000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hatfull GF, Dedrick RM, and Schooley RT (2022). Phage Therapy for Antibiotic-Resistant Bacterial Infections. Annu Rev Med 73, 197–211. 10.1146/annurev-med-080219-122208. [DOI] [PubMed] [Google Scholar]
- 7.Strathdee SA, Hatfull GF, Mutalik VK, and Schooley RT (2023). Phage therapy: From biological mechanisms to future directions. Cell 186, 17–31. 10.1016/j.cell.2022.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hatfull GF (2018). Mycobacteriophages. Microbiol Spectr 6. 10.1128/microbiolspec.GPP3-0026-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hatfull GF (2020). Actinobacteriophages: Genomics, Dynamics, and Applications. Annu Rev Virol 7, 37–61. 10.1146/annurev-virology-122019-070009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hatfull GF (2022). Mycobacteriophages: From Petri dish to patient. PLoS Pathog 18, e1010602. 10.1371/journal.ppat.1010602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gauthier CH, Abad L, Venbakkam AK, Malnak J, Russell DA, and Hatfull GF (2022). DEPhT: a novel approach for efficient prophage discovery and precise extraction. Nucleic Acids Res. 10.1093/nar/gkac273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dedrick RM, Aull HG, Jacobs-Sera D, Garlena RA, Russell DA, Smith BE, Mahalingam V, Abad L, Gauthier CH, and Hatfull GF (2021). The Prophage and Plasmid Mobilome as a Likely Driver of Mycobacterium abscessus Diversity. mBio 12. 10.1128/mBio.03441-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glickman C, Kammlade SM, Hasan NA, Epperson LE, Davidson RM, and Strong M (2020). Characterization of integrated prophages within diverse species of clinical nontuberculous mycobacteria. Virol J 17, 124. 10.1186/s12985-020-01394-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guerrero-Bustamante CA, Dedrick RM, Garlena RA, Russell DA, and Hatfull GF (2021). Toward a Phage Cocktail for Tuberculosis: Susceptibility and Tuberculocidal Action of Mycobacteriophages against Diverse Mycobacterium tuberculosis Strains. mBio 12. 10.1128/mBio.00973-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dedrick R, Guerrero Bustamante C, Garlena RA, Russell DA, Ford K, Harris K, Gilmour KC, Soothill J, Jacobs-Sera D, Schooley RT, et al. (2019). Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nature Medicine 25, 730–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dedrick RM, Smith BE, Garlena RA, Russell DA, Aull HG, Mahalingam V, Divens AM, Guerrero-Bustamante CA, Zack KM, Abad L, et al. (2021). Mycobacterium abscessus Strain Morphotype Determines Phage Susceptibility, the Repertoire of Therapeutically Useful Phages, and Phage Resistance. mBio 12. 10.1128/mBio.03431-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dedrick RM, Smith BE, Cristinziano M, Freeman KG, Jacobs-Sera D, Belessis Y, Whitney Brown A, Cohen KA, Davidson RM, van Duin D, et al. (2022). Phage Therapy of Mycobacterium Infections: Compassionate-use of Phages in Twenty Patients with Drug-Resistant Mycobacterial Disease. Clin Infect Dis, ciac453. 10.1093/cid/ciac453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Little JS, Dedrick RM, Freeman KG, Cristinziano M, Smith BE, Benson CA, Jhaveri TA, Baden LR, Solomon DA, and Hatfull GF (2022). Bacteriophage treatment of disseminated cutaneous Mycobacterium chelonae infection. Nat Commun 13, 2313. 10.1038/s41467-022-29689-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nick JA, Dedrick RM, Gray AL, Vladar EK, Smith BE, Freeman KG, Malcolm KC, Epperson LE, Hasan NA, Hendrix J, et al. (2022). Host and pathogen response to bacteriophage engineered against Mycobacterium abscessus lung infection. Cell 185, 1860–1874 e1812. 10.1016/j.cell.2022.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dedrick RM, Freeman KG, Nguyen JA, Bahadirli-Talbott A, Smith BE, Wu AE, Ong AS, Lin CT, Ruppel LC, Parrish NM, et al. (2021). Potent antibody-mediated neutralization limits bacteriophage treatment of a pulmonary Mycobacterium abscessus infection. Nat Med 27, 1357–1361. 10.1038/s41591-021-01403-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pedulla ML, Ford ME, Houtz JM, Karthikeyan T, Wadsworth C, Lewis JA, Jacobs-Sera D, Falbo J, Gross J, Pannunzio NR, et al. (2003). Origins of highly mosaic mycobacteriophage genomes. Cell 113, 171–182. [DOI] [PubMed] [Google Scholar]
- 22.Duda RL (1998). Protein chainmail: catenated protein in viral capsids. Cell 94, 55–60. [DOI] [PubMed] [Google Scholar]
- 23.Cresawn SG, Pope WH, Jacobs-Sera D, Bowman CA, Russell DA, Dedrick RM, Adair T, Anders KR, Ball S, Bollivar D, et al. (2015). Comparative genomics of Cluster O mycobacteriophages. PLoS One 10, e0118725. 10.1371/journal.pone.0118725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Podgorski JM, Freeman K, Gosselin S, Huet A, Conway JF, Bird M, Grecco J, Patel S, Jacobs-Sera D, Hatfull G, et al. (2023). A structural dendrogram of the actinobacteriophage major capsid proteins provides important structural insights into the evolution of capsid stability. Structure. 10.1016/j.str.2022.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hatfull GF, Jacobs-Sera D, Lawrence JG, Pope WH, Russell DA, Ko CC, Weber RJ, Patel MC, Germane KL, Edgar RH, et al. (2010). Comparative Genomic Analysis of 60 Mycobacteriophage Genomes: Genome Clustering, Gene Acquisition, and Gene Size. J Mol Biol 397, 119–143. 10.1016/j.jmb.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lairson LL, Henrissat B, Davies GJ, and Withers SG (2008). Glycosyltransferases: structures, functions, and mechanisms. Annu Rev Biochem 77, 521–555. 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]
- 27.Podgorski J, Calabrese J, Alexandrescu L, Jacobs-Sera D, Pope W, Hatfull G, and White S (2020). Structures of Three Actinobacteriophage Capsids: Roles of Symmetry and Accessory Proteins. Viruses 12. 10.3390/v12030294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sampson T, Broussard GW, Marinelli LJ, Jacobs-Sera D, Ray M, Ko CC, Russell D, Hendrix RW, and Hatfull GF (2009). Mycobacteriophages BPs, Angel and Halo: comparative genomics reveals a novel class of ultra-small mobile genetic elements. Microbiology 155, 2962–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cote J, Welch C, Kimble M, Archambault D, Ross JC, Orellana H, Amero K, Bourett C, Daigle A, Hutchison KW, and Molloy SD (2022). Characterization of the cluster MabR prophages of Mycobacterium abscessus and Mycobacterium chelonae. G3 (Bethesda) 12. 10.1093/g3journal/jkac188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pope WH, Bowman CA, Russell DA, Jacobs-Sera D, Asai DJ, Cresawn SG, Jacobs WR, Hendrix RW, Lawrence JG, Hatfull GF, et al. (2015). Whole genome comparison of a large collection of mycobacteriophages reveals a continuum of phage genetic diversity. Elife 4, e06416. 10.7554/eLife.06416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoetzinger M, Nilsson E, Arabi R, Osbeck CMG, Pontiller B, Hutinet G, Bayfield OW, Traving S, Kisand V, Lundin D, et al. (2021). Dynamics of Baltic Sea phages driven by environmental changes. Environ Microbiol 23, 4576–4594. 10.1111/1462-2920.15651. [DOI] [PubMed] [Google Scholar]
- 32.Olvera A, Cedeno S, Llano A, Mothe B, Sanchez J, Arsequell G, and Brander C (2020). Does Antigen Glycosylation Impact the HIV-Specific T Cell Immunity? Front Immunol 11, 573928. 10.3389/fimmu.2020.573928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim Y, Vaughan K, Greenbaum J, Peters B, Law M, and Sette A (2012). A meta-analysis of the existing knowledge of immunoreactivity against hepatitis C virus (HCV). PLoS One 7, e38028. 10.1371/journal.pone.0038028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parker R, Partridge T, Wormald C, Kawahara R, Stalls V, Aggelakopoulou M, Parker J, Powell Doherty R, Ariosa Morejon Y, Lee E, et al. (2021). Mapping the SARS-CoV-2 spike glycoprotein-derived peptidome presented by HLA class II on dendritic cells. Cell Rep 35, 109179. 10.1016/j.celrep.2021.109179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szabo TG, Palotai R, Antal P, Tokatly I, Tothfalusi L, Lund O, Nagy G, Falus A, and Buzas EI (2009). Critical role of glycosylation in determining the length and structure of T cell epitopes. Immunome Res 5, 4. 10.1186/1745-7580-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shamaei M, and Mirsaeidi M (2021). Nontuberculous Mycobacteria, Macrophages, and Host Innate Immune Response. Infect Immun 89, e0081220. 10.1128/IAI.00812-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rankine-Wilson LI, Shapira T, Sao Emani C, and Av-Gay Y (2021). From infection niche to therapeutic target: the intracellular lifestyle of Mycobacterium tuberculosis. Microbiology (Reading) 167. 10.1099/mic.0.001041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cardenal-Munoz E, Barisch C, Lefrancois LH, Lopez-Jimenez AT, and Soldati T (2017). When Dicty Met Myco, a (Not So) Romantic Story about One Amoeba and Its Intracellular Pathogen. Front Cell Infect Microbiol 7, 529. 10.3389/fcimb.2017.00529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jacobs-Sera D, Marinelli LJ, Bowman C, Broussard GW, Guerrero Bustamante C, Boyle MM, Petrova ZO, Dedrick RM, Pope WH, Science Education Alliance Phage Hunters Advancing, G., et al. (2012). On the nature of mycobacteriophage diversity and host preference. Virology 434, 187–201. 10.1016/j.virol.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barr JJ, Auro R, Furlan M, Whiteson KL, Erb ML, Pogliano J, Stotland A, Wolkowicz R, Cutting AS, Doran KS, et al. (2013). Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc Natl Acad Sci U S A 110, 10771–10776. 10.1073/pnas.1305923110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hatfull GF (2022). Phage Therapy for Nontuberculous Mycobacteria: Challenges and Opportunities. Pulm Ther. 10.1007/s41030-022-00210-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cresawn SG, Bogel M, Day N, Jacobs-Sera D, Hendrix RW, and Hatfull GF (2011). Phamerator: a bioinformatic tool for comparative bacteriophage genomics. BMC Bioinformatics 12, 395. 10.1186/1471-2105-12-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perez-Riverol Y, Bai J, Bandla C, Garcia-Seisdedos D, Hewapathirana S, Kamatchinathan S, Kundu DJ, Prakash A, Frericks-Zipper A, Eisenacher M, et al. (2022). The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res 50, D543–D552. 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Snapper SB, Melton RE, Mustafa S, Kieser T, and Jacobs WR Jr. (1990). Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol Microbiol 4, 1911–1919. [DOI] [PubMed] [Google Scholar]
- 45.Gauthier CH, Cresawn SG, and Hatfull GF (2022). PhaMMseqs: A new pipeline for constructing phage gene phamilies using MMseqs2. G3 (Bethesda). 10.1093/g3journal/jkac233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rock JM, Hopkins FF, Chavez A, Diallo M, Chase MR, Gerrick ER, Pritchard JR, Church GM, Rubin EJ, Sassetti CM, et al. (2017). Programmable transcriptional repression in mycobacteria using an orthogonal CRISPR interference platform. Nat Microbiol 2, 16274. 10.1038/nmicrobiol.2016.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296. 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- 48.Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, and Ferrin TE (2018). UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci 27, 14–25. 10.1002/pro.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Freeman KG, Wetzel KS, Zhang Y, Zack KM, Jacobs-Sera D, Walters SM, Barbeau DJ, McElroy AK, Williams JV, and Hatfull GF (2021). A Mycobacteriophage-Based Vaccine Platform: SARS-CoV-2 Antigen Expression and Display. Microorganisms 9. 10.3390/microorganisms9122414. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Extended Data Set 1. Peptides identified by Mascot in the tryptic digests, related to the phage gel bands that reacted with glycostain, supporting Figure 1D. Some peptides were identified more than once but, for simplicity, only the top scoring hit is presented here. Peptides with scores less than 20 were considered unreliable and have been removed. Tabs include data for Corndog, Che8, and Myrna.
Extended Data Set 2. Peptides identified by Mascot in the chymotryptic digests, related to the Che8 WT and Che8Δ110-1 gel bands, supporting Figure 2D. Some peptides were identified more than once but, for simplicity, only the top scoring hit is presented here. Peptides with scores less than 20 were considered unreliable and have been removed. Tabs show data for each Che8 band shown in Figure 2 and the Che8Δ110-1 mutant.
Extended Data Set 3. Peptides identified by Mascot in the tryptic digests, related to the Che8 immunoreactive bands and the Omega, Fionnbharth, and Nebkiss glycostain-positive bands, supporting data shown in Figure 4F. Some peptides were identified more than once but, for simplicity, only the top scoring hit is presented here. Peptides with scores less than 20 were considered unreliable and have been removed.
Extended Data Set 4. Raw ELISA data, related to Figures 4, 5, S2, S5, and S6. Raw reads from the plate reader, background subtraction calculations, tabulated half-maximal and endpoint titers, and tabulated results from two-sample t-tests are included as separate tabs.
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE43 partner repository and are publicly available as of the date of publication. The Che8 Δ110-1 cryo-EM map was deposited at the EMBD and the Che8 Δ110-1 cryo-EM raw data was deposited at EMPIAR; these are publicly available as of the date of publication. Accession numbers for all standardized datasets are listed in the key resources table. Original western blot images and the four Extended Data Set files are available on Mendeley at: doi: 10.17632/49hmcxgjnr.2
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat Anti-Mouse IgG H&L (HRP) preadsorbed | Abcam | ab97040, RRID: AB_10698223 |
| Goat Anti-Mouse IgM mu chain (HRP) preadsorbed | Abcam | ab98679, RRID: AB_10696527 |
| Goat Anti-Human IgG Fc (HRP) preadsorbed | Abcam | ab98624, RRID: AB_10673832 |
| Normal Human Serum | ThermoFisher | 31876, RRID: AB_2532169 |
| Goat anti-Mouse IgG H&L (10nm gold) | Abcam | ab39619, RRID: AB_954440 |
| ACK Lysis Buffer | Gibco | A10492–01 |
| Live/Dead Violet Stain | Invitrogen | L34955 |
| αCD16/32 Fc block | Tonbo Biosciences | 70–0161-M001 |
| CD3e-BUV395 | BD Biosciences | BD 565992, RRID: AB_2739443 |
| CD4-AF700 | BioLegend | BioLegend 100536, RRID: AB_493701 |
| CD8a-PE-Cy7 | BD Biosciences | BD 552877, RRID: AB_394506 |
| CD19-BV785 | BioLegend | BioLegend 115543, RRID: AB_11218994 |
| B220-APC | BioLegend | BioLegend 103212, RRID: AB_312997 |
| NK1.1-BV421 | BD Biosciences | BD 562921, RRID: AB_2728688 |
| CD44-APC-Cy7 | BD Biosciences | BD 560568, RRID: AB_1727481), |
| CD62L-BUV563 | BD Biosciences | BD 741230, RRID: AB_2870784 |
| BD Brilliant Stain Buffer | BD Biosciences | BD 566349 |
| Foxp3 Fix/Perm | ThermoFisher | 00-5523-00 |
| Normal rat serum | Invitrogen | 10710C, RRID: AB_2532985 |
| Foxp3 isotype control antibody | Invitrogen | 45-4321-80, RRID: AB_906259 |
| Foxp3-PerCPCy5.5 antibody | Invitrogen | 45-5773-82, RRID: AB_914351 |
| Bacterial and virus strains | ||
| Mycobacterium smegmatis mc2155 | Snapper et al., 1990 | NC_008596 |
| Mycobacteriophage Che8 | Pedulla et al., 2003 | AY129330.1 |
| Mycobacteriophage Che8 Δ110-1 | This paper | N/A |
| Mycobacteriophage Myrna | Hatfull et al., 2010 | EU826466 |
| Mycobacteriophage Corndog | Pedulla et al., 2003 | AY129335 |
| Mycobacteriophage Fionnbharth | Pope et al., 2015 | JN831653 |
| Mycobacteriophage Omega | Pedulla et al., 2003 | AY129338 |
| Mycobacteriophage Nebkiss | Pope et al., 2015 | MK016501 |
| Biological samples | ||
| Mouse sera | This paper | N/A |
| Mouse spleens | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| 3,3′,5,5′-Tetramethylbenzidine (TMB) Liquid Substrate System for ELISA | Sigma Aldrich | Cat# T0440–1 |
| Carbonate-bicarbonate buffer capsules | Sigma Aldrich | Cat. # C3041 |
| Sulfuric acid | EMD Millipore | CAS: 7664-93-9 |
| ECL substrate | https://www.thermofisher.com/order/catalog/product/34580 | |
| Imperial protein stain | Fisher Scientific | Cat# 4615 |
| Sequencing grade modified trypsin | Promega | Cat#V5113 |
| Chymotrypsin | Sigma | Cat#C3142 |
| Critical commercial assays | ||
| Deposited data | ||
| Mass spectrometry proteomics data | ProteomeXchange Consortium via PRIDE | PXD041690 and 10.6019/PXD041690 |
| Mycobacteriophage Che8 Δ110-1 cryo-EM capsid map | EMDB | EMD-28761 |
| Mycobacteriophage Che8 Δ110-1 raw cryo-EM data | EMPIAR | EMPIAR-11285 |
| Supplemental Data Set 1 | Mendeley | 10.17632/49hmcxgjnr.2 |
| Supplemental Data Set 2 | Mendeley | 10.17632/49hmcxgjnr.2 |
| Supplemental Data Set 3 | Mendeley | 10.17632/49hmcxgjnr.2 |
| Supplemental Data Set 4 | Mendeley | 10.17632/49hmcxgjnr.2 |
| Western blot raw images | Mendeley | 10.17632/49hmcxgjnr.2 |
| Experimental models: Cell lines | ||
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | Jackson Laboratory | 000664, RRID: JAX:000664 |
| IFNβ-EYFP reporter mice (C57BL/6J background) | Jackson Laboratory | 10818, RRID: JAX:010818 |
| Oligonucleotides | ||
| Forward primer for sgRNA plasmid construction: GGGAGAGTGCGGCGTGGAGCTGGTC | This paper | N/A |
| Reverse primer for sgRNA plasmid construction: AAACGACCAGCTCCACGCCGCACTC | This paper | N/A |
| Recombinant DNA | ||
| Plasmid pIRL53 | Rock et al., 2017 | |
| Software and algorithms | ||
| OriginLab | Version 10.05(2023b) | |
| FlowJo | Version v10.8.1 | |
| Gen5 Microplate Reader and Imager Software | BioTek | TS Installation Version 2.06.10 |
| Mascot Server version 2.7.0 | Matrix Science | https://www.matrixscience.com/ |
| Other | ||