

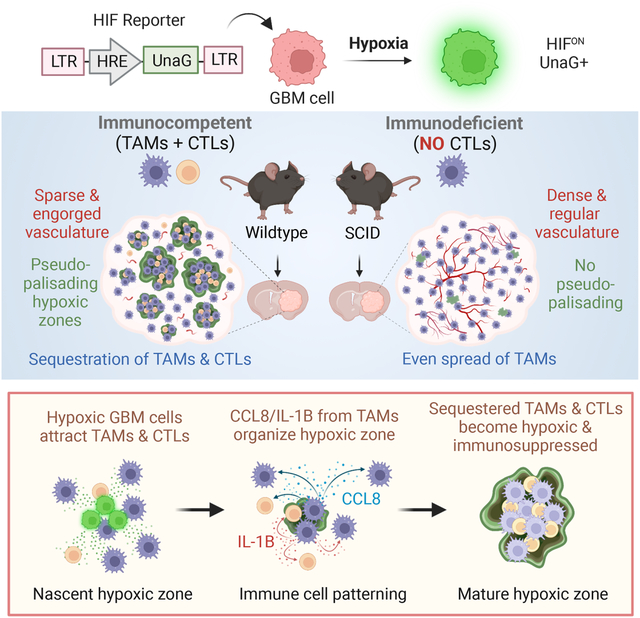
SUMMARY
Glioblastoma (GBM), a highly lethal brain cancer, is notorious for immunosuppression, but the mechanisms remain unclear. Here, we documented a temporospatial patterning of tumor-associated myeloid cells (TAMs) corresponding to vascular changes during GBM progression. As tumor vessels transitioned from the initial dense regular network to later scant and engorged vasculature, TAMs shifted away from perivascular regions and trafficked to vascular-poor areas. This process was heavily influenced by the immunocompetence state of the host. Utilizing a sensitive fluorescent UnaG reporter to track tumor hypoxia coupled with single-cell transcriptomics, we revealed that hypoxic niches attracted and sequestered TAMs and cytotoxic T lymphocytes (CTLs), where they were reprogrammed towards an immunosuppressive state. Mechanistically, we identified chemokine CCL8 and cytokine IL-1β as two hypoxic niche factors critical for TAM trafficking and co-evolution of hypoxic zones into pseudopalisading patterns. Therefore, perturbation of TAM patterning in hypoxic zones may improve tumor control.
Keywords: GBM, immune landscape, tumor-associated microglia/macrophages, TAM, cytotoxic T lymphocytes (CTLs), immunosuppression, tumor hypoxia, tumor vasculature, CCL8, IL-1β
Graphical Abstract
eTOC blurb/In brief
Glioblastoma is notorious for immunosuppression, but the mechanisms are unclear. Sattiraju et al. report that hypoxic zones in GBM attract and sequester tumor-associated myeloid cells and cytotoxic T cells, where they are reprogrammed into an immunosuppressive state. This process is influenced by the immunocompetence state of the host and involves CCL8 and IL-1B as niche factors in hypoxic zones.
INTRODUCTION
Glioblastoma (GBM), the most common primary brain cancer, remains deadly. A major contributing factor is its immunologically cold status1, which poses hurdles for immunotherapy2,3. Hence, insights into the governing factors of GBM immunosuppression are needed.
Tumor-associated myeloid cells (TAMs) constitute up to 50% of cells in GBM4,5, and they support GBM expansion, angiogenesis, and T cell suppression6. Current strategies to target TAMs by either reducing their number or reversing their pro-tumorigenic state are promising in animal models of GBM7,8, but no survival benefit has been achieved for GBM patients9,10. Thus, mapping the spatial organization of TAMs and dissecting contextual cues may facilitate strategies to disrupt TAM reprogramming.
To delineate the link between TAM patterning and metabolic state of tumor microenvironment (TME), a faithful reporter is crucial to reveal hypoxic niches. Current approaches such as pimonidazole11, EF512 or PET imaging13,14 are limited by low cellular resolution, incomplete tissue penetration, and no direct link to hypoxia-inducible factor (HIF), a master transcription factor for cellular adaptation to low oxygen15. Genetic HIF reporters using standard fluorescent proteins such as GFP are hampered by the requirement of oxygen for fluorophore maturation. We thus employed UnaG, a fluorescent protein that does not require oxidation to become fluorescent16. Indeed, UnaG reporter under the control of HIF response elements (HRE) can faithfully track HIF activity in hypoxic cells17.
Here, we transplanted murine GBM cells engineered with a lentiviral HRE-UnaG reporter in a fully immunocompetent background. We charted a temporospatial TAM patterning corresponding to vascular changes, the emergence of hypoxic zones, and subsequent maturation of pseudopalisades. We complemented the spatial information with single cell transcriptomics to reveal signaling communication between GBM cells and immune cells in hypoxic niches, featuring immune cell recruitment and immunosuppression. Validation in human GBM patient samples showed that TAM patterning correlated with glioma grade, hypoxic burden, and mesenchymal subtype. Mechanistically, we demonstrated that immune cell trafficking and maturation of hypoxia zones involved CCL8 and IL-1β. Our findings indicated that targeting TAM patterning in GBM by perturbing hypoxic niches may improve tumor control.
RESULTS
Host immune status influences temporospatial patterning of TAMs during GBM progression
To characterize the spatial organization of TAMs in a fully immunocompetent microenvironment, we first studied GL261, a murine orthotopic GBM transplant model in syngeneic C57BL/6 wildtype (B6-WT) background (Figure 1A). Four weeks post-transplant, immunofluorescence (IF) for myeloid marker IBA1 or macrophage antigen F4/80 revealed a highly heterogenous TAM distribution (Figure 1A). This also applied to TAMs expressing phagocytic marker CD68 or immunosuppression marker CD206 (mannose receptor 1 encoded by Mrc1) (Figure 1B). The spatial pattern was also observed for monocyte-derived macrophage (MDMs) labeled by integrin α4 (CD49d)18, which mostly resided in GBM interior, in contrast to microglia (TMEM119+) which accumulated largely at tumor periphery (Figure 1C).
Figure 1. Temporospatial patterning of TAMs parallels vascular changes during GBM progression and is influenced by host immunocompetence status.
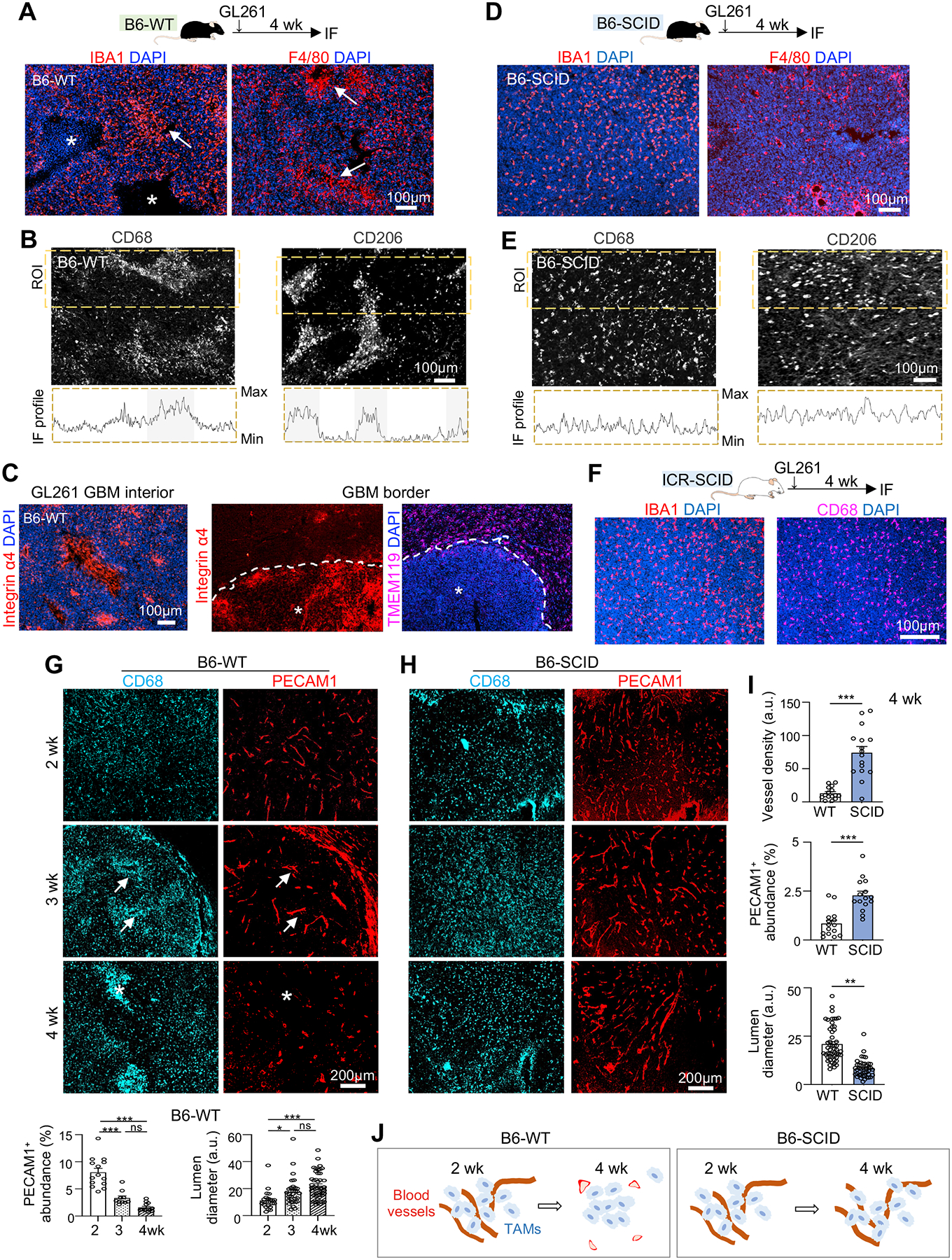
(A) Top, experimental scheme of intracranial transplant of GL261 GBM cells into C57BL/6 wild-type (B6-WT) hosts and IF analysis 4 weeks later. Bottom, representative IF images from n=3 mice show spatial patterning of IBA1+ and F4/80+ TAMs. Asterisks, necrotic cores; arrows, TAM aggregates.
(B) IF images and profile plots of IF intensities of CD68 or CD206 within region of interest (ROI, dashed boxes).
(C) IF images of MDM (integrin α4+) or microglia (TMEM119+) in GBM interior (asterisks) or GBM border (dashed line) at 4 weeks post-transplant in B6-WT host.
(D) Top, experimental scheme of GL261 transplanted into C57BL/6-SCID (B6-SCID) hosts and IF analysis 4 weeks later. Bottom, representative IF images from n=3 mice show distribution of TAMs (IBA1+ or F4/80+).
(E) IF images and profile plots of IF intensities of CD68+ and CD206+ TAMs within ROI in B6-SCID host.
(F) Representative IF images show distribution of IBA1+ and CD68+ TAMs in GL261 established in ICR-SCID host (n=3 mice).
(G-I) IF images and quantifications of vascular changes and temporospatial transition of CD68+ TAMs in distinct zones of GL261 GBM established in B6-WT host (G) or B6-SCID host (H). Quantifications in bottom of (G): PECAM1 abundance, n=10 tumor areas; lumen diameter, n=30 tumor areas, from n=3 mice per timepoint. One-way ANOVA; a.u., arbitrary units. Quantification of vascular comparison (I): n=15 randomly chosen tumor areas from n=5 mice per group; unpaired t-test. *P<0.05; **P<0.01; ***P<0.001; ns, not significant.
(J) Diagrams depicting TAM spatial patterning in parallel to vascular changes in dependence of host immune status.
As immunodeficient mice are commonly used for patient-derived xenotransplant (PDX) models, we wondered whether the host immune status might affect TAM patterning. We thus transplanted GL261 cells intracranially into C57BL/6-SCID (B6-SCID) mice that lack functional T and B lymphocytes because of impaired VDJ recombination. We observed a rather uniform distribution of TAMs expressing IBA1, F4/80, CD68, or CD206 (Figures 1D–1E). These findings were further confirmed in ICR-SCID hosts, a common outbred strain of SCID mice (Figure 1F). Hence, the host immunocompetence status exerts a strong influence on the geographic patterning of TAMs.
TAM patterning parallels vascular changes during GBM progression
The spatial patterning of TAMs in B6-WT hosts was highly dynamic during GL261 expansion: at early stage (2 weeks post-transplant), CD68+ TAMs were evenly distributed, but by 3–4 weeks they became congregated in distinct zones (Figure 1G). In contrast, CD68+ TAMs in B6-SCID hosts remained evenly spread throughout GBM progression (Figure 1H).
As blood-borne MDMs constituted the majority of TAMs in GBM interior, we wondered if TAM patterning might reflect vascular rearrangement during GBM expansion. Indeed, we found a drastic difference of tumor vasculature (PECAM1+) in B6-WT versus B6-SCID hosts, paralleled by distinct TAM organization (Figures 1G–1I). In B6-WT hosts, tumor vessels progressively transformed from the initial dense and regular network to later sparse, engorged, and tortuous vasculature; in parallel, CD68+ TAMs shifted from initial perivascular location at 3 weeks (reflecting transvascular influx of MDMs into GBM) to later vascular-poor areas at 4 weeks. In contrast, in B6-SCID hosts, with blood vessels remaining dense and regularly patterned, CD68+ TAMs remained evenly distributed. Hence, the spatial organization of TAMs coincided with progressive vascular alterations during GBM expansion, and this process was influenced by host immune status (Figure 1J).
HRE-UnaG reporter reveals the emergence of hypoxic tumor zones
As vascular aberrancy would lead to hypoxia, we next asked if TAM patterning might be associated with the emergence of hypoxic zones in GBM. We thus engineered GL261 cells with a lentiviral HRE-UnaG reporter, with UnaG fused to a PEST degron for fast turnover (Figure 2A). We first confirmed in culture a robust induction of UnaG upon exposure to hypoxia (1% O2) and a rapid decline upon return to normoxia (Figure 2B).
Figure 2. TAM trafficking coincides with the emergence of hypoxic zones and development of pseudopalisades in GBM.
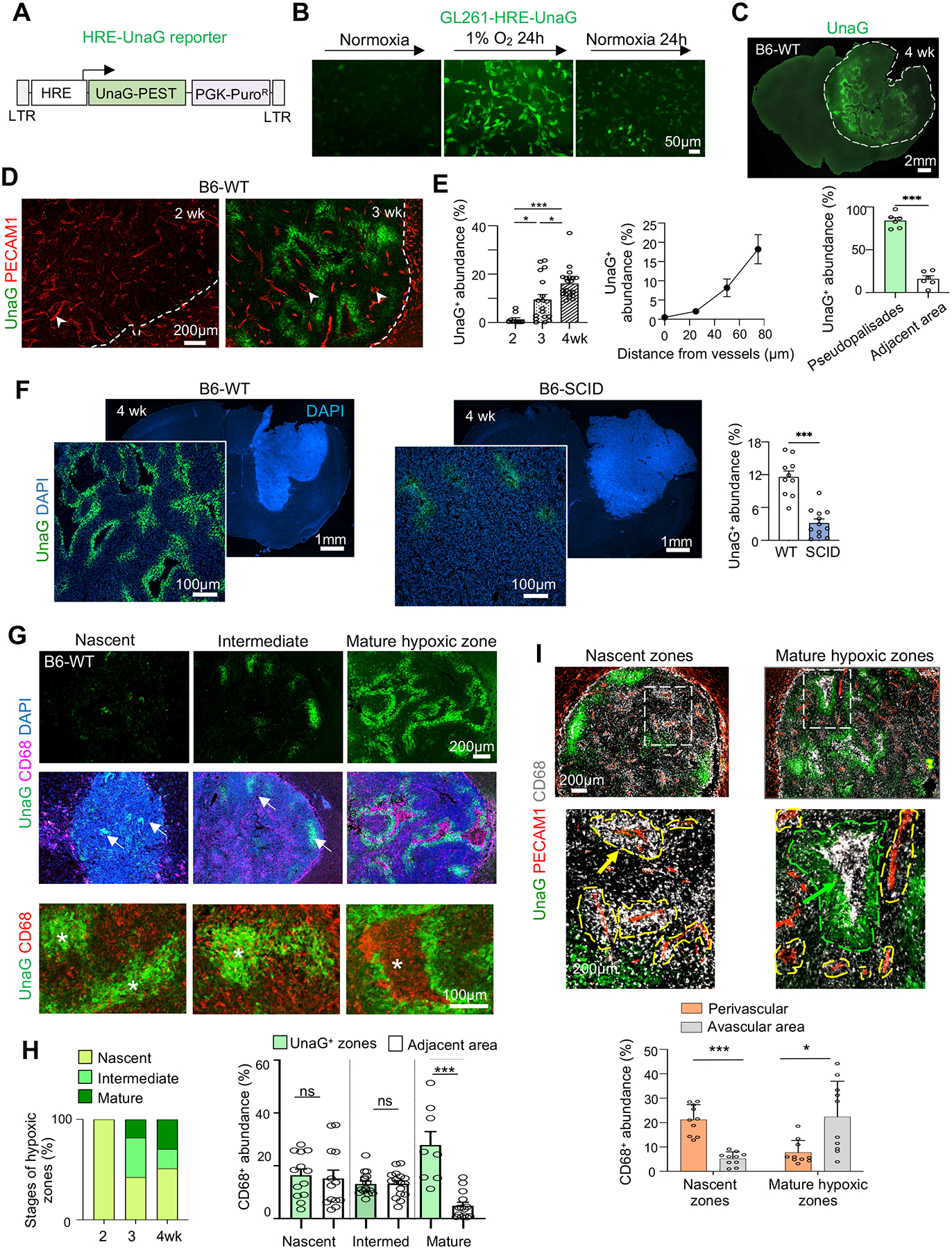
(A) Diagram of lentiviral HRE-UnaG reporter.
(B) Representative images of UnaG expression in GL261 cells (stably transduced with lenti-HRE-UnaG) when exposed to 1% O2 or normoxia. n=3 cultures.
(C) Fluorescent image and quantification of UnaG+ cells in pseudopalisading or adjacent areas of GL261-HRE-UnaG GBM established in B6-WT host at 4 wk post-transplant. n=6 tumor areas from 4 mice.
(D) Representative IF images from n=3 mice show emergence of UnaG+ tumor cells between 2–3 weeks post-transplant in vascular poor areas. Arrowheads point to tumor vasculatures with engorged lumen.
(E) Left, quantification of UnaG+ areas during GBM progression. n=10 tumor areas from 3 mice per timepoint. Right, quantification of UnaG abundance in relation to the distance from blood vessels. n=11 tumor fields for each distance.
(F) Representative IF images from n=3 mice show an overview of GBM burden (DAPI at low magnification) and hypoxic zones (UnaG at high magnification) in B6-SCID vs. B6-WT hosts. Quantification: n= 10 tumor areas from 3 mice per group.
(G, H) Top two rows, IF images show transition of hypoxic zones (UnaG+) from nascent (arrows) to mature pseudopalisades and corresponding spatial patterns of CD68+ TAMs during GL261 progression. Bottom row, high magnification IF images show cell-cell sorting of CD68+ TAMs and UnaG+ tumor cells in hypoxia areas (asterisks). Quantification: n=10 tumor areas from 3 mice for each group.
(I) IF images and quantification show transition of CD68+ TAM at perivascular location in nascent zones to vascular poor area in mature hypoxic zones of GL261 GBM. Enlarged images of boxed areas are shown below. n= 13 tumor areas from 3 mice per group.
Unpaired t-test (C, F, H), One-way ANOVA (E, I), *P<0.05, ***P<0.001, ns, not significant.
See also Figure S1.
We then transplanted GL261-HRE-UnaG cells into B6-WT hosts and detected the emergence of UnaG+ (HIFON) cells at 2–3 weeks post-transplant in areas distant from blood vessels, which progressively expanded into pseudopalisading patterns, coinciding with vascular alterations (Figures 2C–2E).
UnaG+ cells highly expressed glucose transporter 1 (GLUT1), a direct HIF target19 (Figure S1A). We also administered pimonidazole (Pimo), a compound that becomes covalently linked to cellular proteins at low oxygen tension11. As compared to Pimo, HRE-UnaG labeled more tumor cells in wider areas, especially in nascent hypoxic zones devoid of Pimo labeling (Figure S1B). In more mature hypoxic zones with distinctive pseudopalisading patterns, UnaG and Pimo signals largely overlapped (Figure S1B). HRE-UnaG thus enabled us to detect nascent hypoxic zones with physiological activation of HIF, in line with its capability to report HIF activity at 5% O2 tension and below17,20, whereas Pimo mostly labels cells below 1% O2 tension21.
TAM patterning parallels hypoxic zone emergence and maturation of pseudopalisades
Given the marked differences of tumor vasculature and TAM patterning in B6-WT vs. B6-SCID hosts, we next compared UnaG+ populations, which were highly abundant in B6-WT hosts, but scant in B6-SCID hosts despite advanced tumor sizes (Figure 2F).
The sensitive HRE-UnaG reporter enabled a clear delineation of the spatial relationship of nascent hypoxic zones and the surrounding immune landscape during GBM progression. The spatial transition of CD68+ TAMs (from initial accumulation around the blood vessels to later confinement in vascular-poor areas) coincided with expansion/maturation of hypoxic zones and pseudopalisades, involving active recruitment, cell-cell sorting, and corralling of CD68+ TAMs by a rim of UnaG+ tumor cells in pseudopalisading pattern (Figures 2G–2I).
By contrast, in B6-SCID hosts, small clusters of UnaG+ tumor cells were found scattered throughout GBM tissues at 2–3 weeks post-transplant (likely reflecting fast tumor expansion outpacing vascular development), but by 4 weeks, they did not mature into large hypoxic zones (Figure S1C), in line with stable tumor vasculature.
TAM patterning in association with hypoxic zones was also observed in two additional mouse GBM models in immunocompetent background: RCAS GBM, an autochthonous GBM model induced with the RCAS avian virus system expressing PDGFB/shTrp5322, and CT2A (a syngeneic murine GBM transplant model in C57BL/6 host)23 (Figures S1D–S1E).
Spatial patterning of TAMs in GBM patients correlates with glioma grade and GBM subtype
To examine whether TAM spatial patterning applies to human GBM, we examined a tissue microarray from 71 glioma patients. Immunostaining for GLUT1 (a HIF target, also independently expressed in vascular cells) demonstrated a positive correlation of hypoxic burden with glioma grade (Figures 3A–3B; Table S1A). Moreover, accumulation of CD68+ TAMs in GLUT1+ regions was also in proportion to hypoxic burden (Figures 3C and S1F).
Figure 3. Spatial patterning of TAMs corresponds to hypoxic zones in GBM patient specimens.
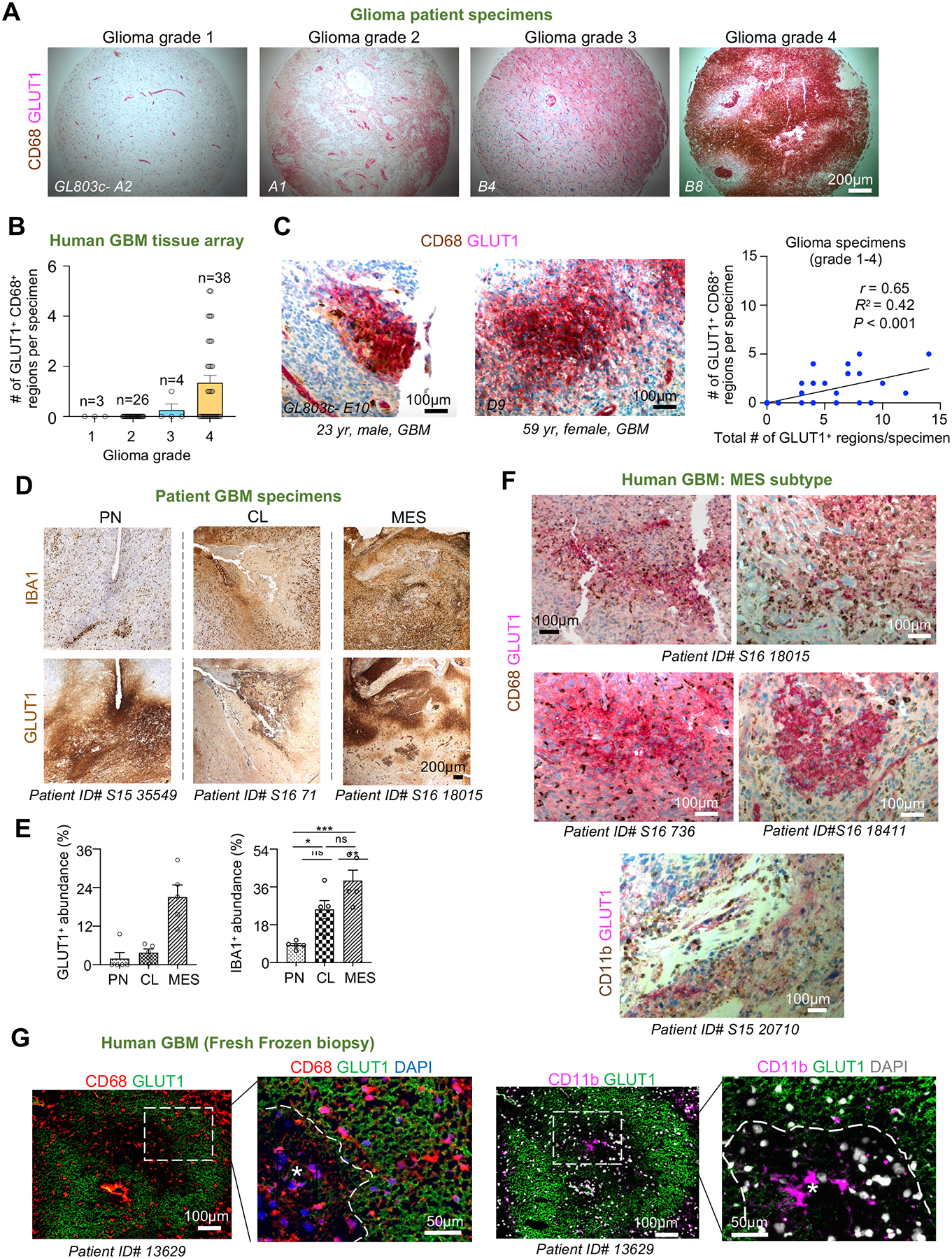
(A) IHC on tissue microarray of glioma biopsy specimens of different grade (n=71 patients, US Biomax, no. GL803c) co-stained for GLUT1 (pink) and CD68 (brown). Hematoxylin for nuclear counterstaining.
(B) Quantification of the relative abundance of hypoxic zones containing CD68+ TAMs in relation to glioma grades (n= 71 patient samples).
(C) High magnification IHC images of GBM patient biopsy specimens co-stained for GLUT1 and CD68. Pearson correlation of hypoxic burden (GLUT1+) in relation to the abundance of hypoxic zones harboring TAMs (n= 71 patient samples).
(D, E) IHC images and quantifications show the abundance of GLUT1+ and IBA1+ areas in GBM subtypes. Hematoxylin for nuclear counterstaining. Specimen IDs are denoted below each image. n=5 randomly selected zones for each subtype; one-way ANOVA; *P<0.05, **P<0.01, ***P<0.001, ns, not significant.
(F) Tissue sections of human GBMs of MES subtype co-stained for GLUT1 and CD68. Hematoxylin for nuclear counterstaining.
(G) IF images of sections from fresh frozen human GBM samples show distribution of CD68+ or CD11b+ TAMs in relation to hypoxic zones (asterisks) outlined by GLUT1.
See also Figure S1.
The mesenchymal (MES) transcriptional subtype of GBM is known for worse survival and increased TAM compartment compared to proneural (PN) and classical (CL) subtypes24,25. Immunostaining showed that the MES subtype contained more prominent GLUT1+ pseudopalisades and higher abundance of IBA1+ TAMs (Figures 3D–3E; Table S1B)26. Co-labeling for GLUT1 and CD68 in MES human GBM specimens verified a close spatial association of hypoxia and TAMs (Figure 3F), a finding further corroborated by IF staining on fresh frozen biopsies showing confinement of CD68+ or CD11b+ TAMs within GLUT1+ pseudopalisades (Figure 3G).
Targeting hypoxic niches disrupts TAM organization and improves tumor control
To further link hypoxic zones with TAM patterning, we investigated how targeting hypoxic niches may impact the tumor immune landscape. At two weeks post-transplant, B6-WT mice bearing GL261-HRE-UnaG GBM were treated with radiation therapy (RT), evofosfamide (Evo), a chemotherapy pro-drug activated under hypoxia27, or both (RT+Evo) for 2 weeks. Evo or RT alone reduced tumor burden, but the combined modality achieved even better tumor reduction (Figures 4A–4B).
Figure 4. Targeting hypoxic niches disrupts spatial patterning of TAMs.
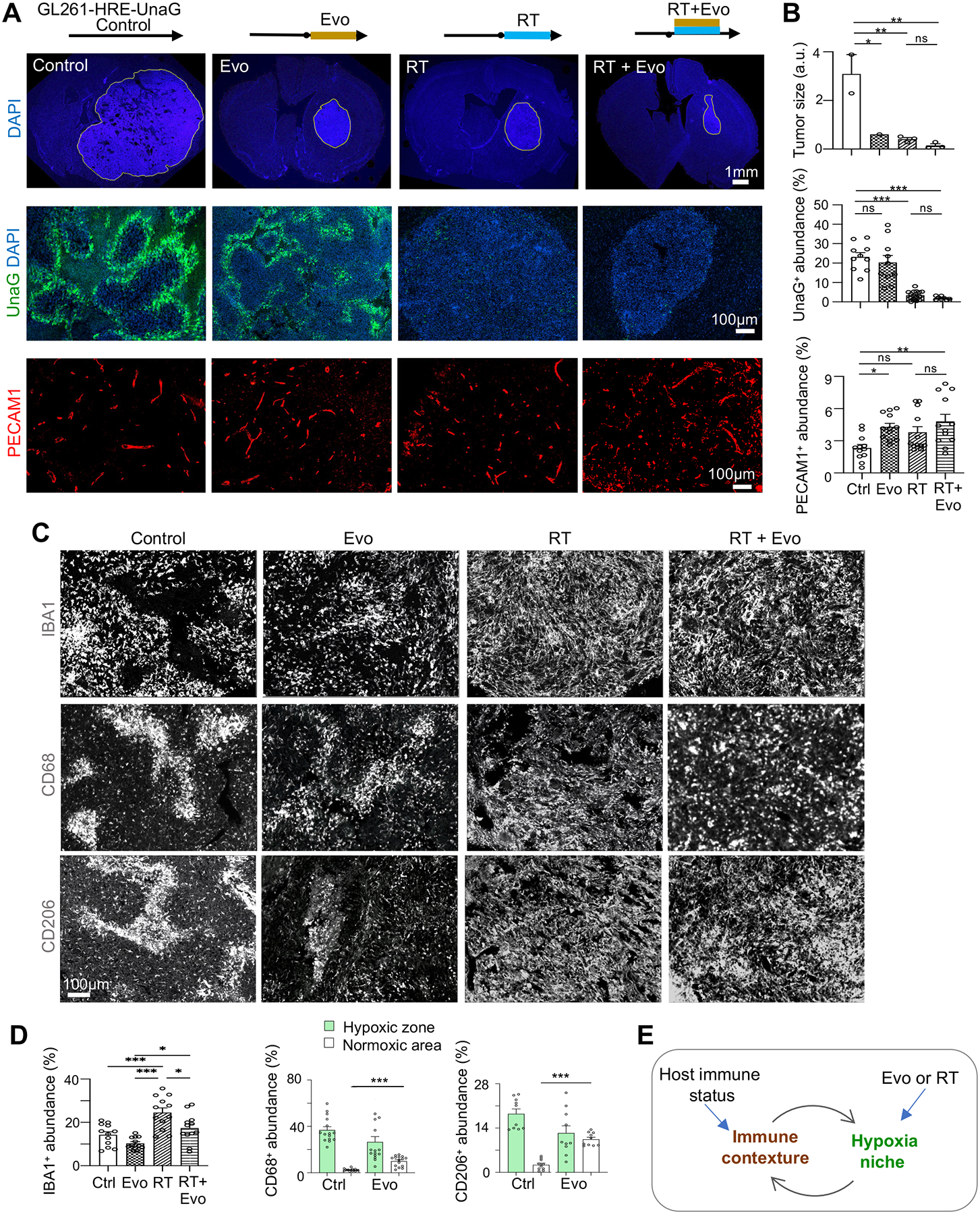
(A, B) Two weeks after transplantation, B6-WT mice bearing GL261 GBM were treated for 2 weeks with evofosfamide (Evo), radiation treatment (RT), or both. IF images (A) and quantifications (B) show tumor area, hypoxic zones (UnaG+), and tumor vasculature (PECAM1+) after treatment. n=3 mice for tumor size; n=10 for UnaG, and n=12 for PECAM1 abundance in randomly selected tumor areas from 3 mice per cohort.
(C, D) IF images (C) and quantifications (D) show abundance and distribution of TAMs expressing IBA1, CD68, or CD206 after treatments. n=10–15 randomly selected images from 3 mice per treatment condition.
(E) Diagram depicting interaction of immune response and tumor hypoxia.
One-way ANOVA (B, D); *P<0.05, **P<0.01, ***P<0.001, ns, not significant.
Regarding tumor hypoxia, RT or combined treatment drastically reduced the UnaG + population, whereas Evo treatment alone resulted in a trend of reduction of the UnaG+ population (Figures 4A–4B). Assessment of tumor vasculature revealed that Evo or combined treatment led to a slightly higher vascular density, while RT did not reduce vascular density (Figures 4A–4B). These findings signify a high radiosensitivity of hypoxic GBM cells and ongoing neoangiogenesis, contrary to the model that RT would increase tumor hypoxia due to radiosensitivity of blood vessels28.
We next examined the impact of RT or Evo on the abundance and, importantly, spatial patterning of TAMs. RT or combined treatment increased infiltration of IBA1+ TAMs with uniform distribution (Figures 4C–4D), coinciding with eradication of UnaG+ cells and disappearance of pseudopalisades/hypoxic zones after RT. Likewise, after Evo treatment, UnaG+ pseudopalisading patterns were less defined, as were the spatial patterns of TAMs expressing IBA1, CD68, or CD206. Altogether, tumor hypoxia and TAM patterning influenced one another to co-evolve into pseudodopalisading patterns (Figure 4E).
scRNA-seq reveals immune signaling by hypoxic GBM cells
To explore the ligand-receptor signaling between GBM cells and immune cells in hypoxic vs. non-hypoxic niches, we carried out single cell RNA-sequencing (scRNA-seq) on GL261 HRE-UnaG GBM at 4 weeks post-transplant. The 9 major cell types included GBM cells (76.5%), TAMs (macrophages (11%), microglia (4%)), other immune cells (T cells (4.7%), dendritic cells (1.9%), B cells (0.68%), and mast cells (0.38%)), endothelial cells (0.6%), and a small population of astrocytes (0.3%) (Figures 5A and S2A–S2B).
Figure 5. Single cell RNA-seq reveals distinct in vivo GBM hypoxia gene signature and the presence of a hypoxic subpopulation of immune cells.
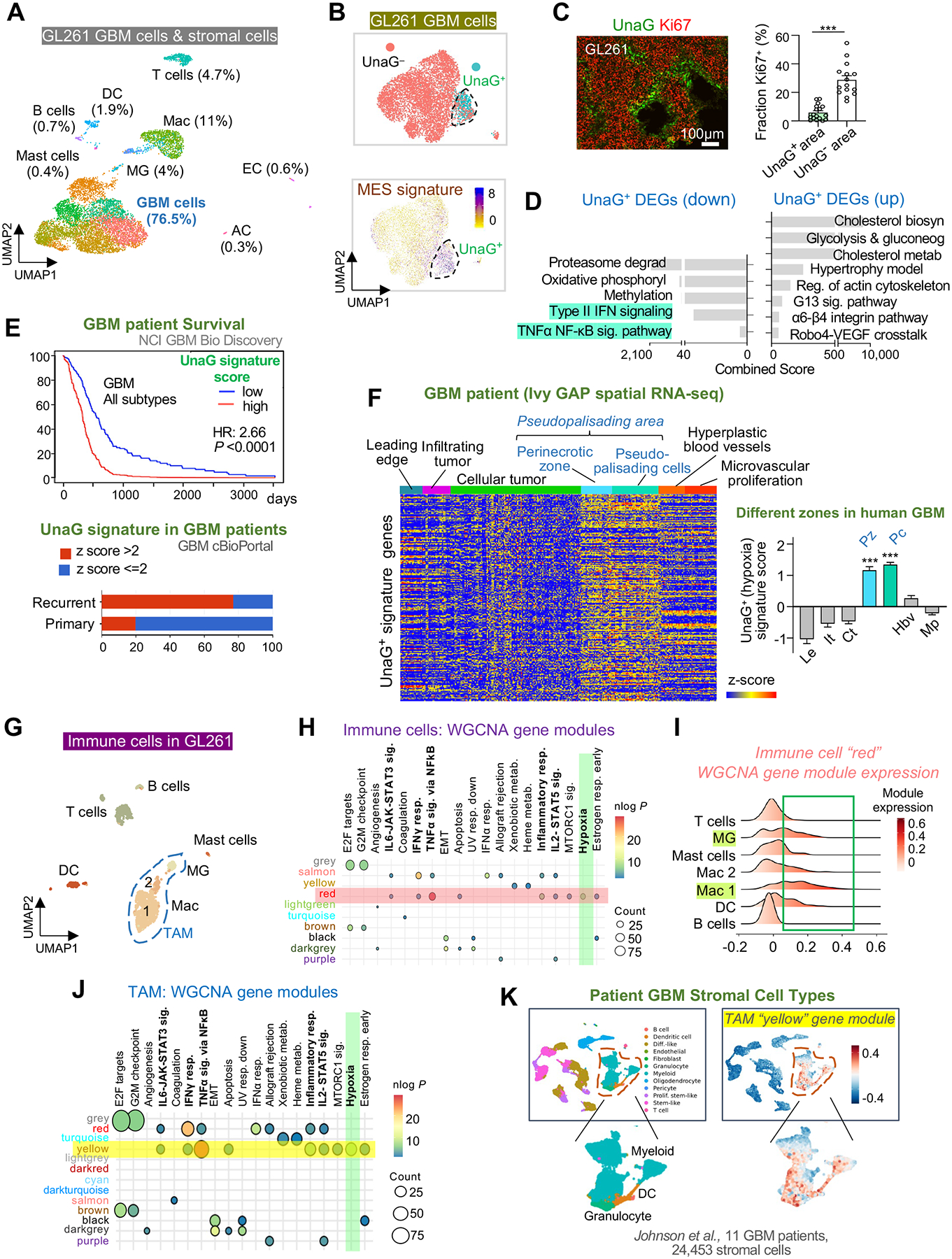
(A) UMAP plot of cell types identified by scRNA-seq of GL261 tumor in B6-WT host at 4 weeks post-transplant.
(B) UMAP plot of GL261 GBM cells shows UnaG+ and UnaG− cell clusters and expression scores for MES2 gene signature (Neftel et al., 2019).
(C) IF image and quantification show fraction of Ki67+ cells in UnaG+ and UnaG− areas. n=15 tumor areas from 3 mice per group; unpaired t-test; ***P<0.001.
(D) ENRICHR pathway enrichment analysis of up- and downregulated DEGs in UnaG+ vs. UnaG− GBM cells (WikiPathways 2019 gene sets).
(E) Top, survival curve of human GBM patients stratified into high or low expressors of UnaG+ GBM gene signature based on TCGA GBM database (NCI BioDiscovery portal). Bottom, representation of UnaG+ gene signature in primary vs. recurrent human GBM (cBioPortal).
(F) Heatmap and bar graph show relative enrichment of UnaG+ gene signature in different zones of human GBM based on Ivy GBM Atlas Project (Ivy GAP) database. One-way ANOVA; ***P<0.001.
(G) UMAP of immune cells in GL261 GBM.
(H) Transcriptome of immune cells analyzed by weighted gene correlated network analysis (WGCNA). Co-expressed gene modules (rows with color names) were assessed for enrichment of hallmark gene sets (columns). Highlighted row and column denote enrichment of module “red” for “Hypoxia” gene set (green).
(I) Expression scores for WGCNA gene module “red” in different immune cells of GL261 GBM.
(J) Transcriptome of TAMs analyzed by WGCNA. Selected co-expressed gene modules (rows) were assessed for enrichment of hallmark gene sets (columns). Highlighted row and column denote enrichment of gene module “yellow” for “Hypoxia” gene set (green).
(K) Mapping of hypoxic TAM gene module “yellow” onto human GBM patient scRNA-seq dataset (Johnson et al., 2021) shows enrichment scores in myeloid cells, as well as granulocytes and dendritic cells (DC).
See also Figures S2–S4.
The HRE-UnaG reporter allowed us to distinguish HIFON and HIFOFF cells based on UnaG mRNA reads. Indeed, 13.6% of GBM cells were HIFON (UnaG+), forming a separate cluster from UnaG− cells and displaying a mesenchymal shift in gene expression (Figures 5B and S2C–S2D). This echoed a recent human GBM scRNA-seq study showing an association of MES2 cell state with hypoxia29. UnaG+ GBM cells also appeared relatively quiescent, indicated by downregulation of cell cycle genes (Rgcc and Cdc20) and reduced proliferative index (6% Ki67+ vs. 30% in UnaG− cells) (Figures 5C and S2E). This resonated with our recent study linking GBM quiescence with HIF signaling30.
Further confirming faithful reporting of HIF activity by HRE-UnaG, the top upregulated differentially expressed genes (DEGs) in UnaG+ GBM cells included many canonical HIF target genes for metabolic adaptation (e.g., Aldoa, Ldha, Slc2a1 (encoding GLUT1)), angiogenesis (Vegfa), autophagy (Bnip3), apoptosis (Eno1), and notably, macrophage chemotaxis (Mif)), and cytokines (Cxcl10, Cxcl9) (Figure S2E; Table S2A). UMAP feature plots highlighted high expression of Mif and Lgals3 in UnaG+ GBM cells (Figure S2F), both known to regulate macrophage trafficking and immune suppression31–34. MIF also promotes the synthesis of secondary inflammatory cytokines, particularly IL-1β in macrophages35, lymphocytes36, and neutrophils37.
Pathway enrichment analyses by Enrichr, Ingenuity Pathway Analysis (IPA), and Gene Set Enrichment Analysis (GSEA)38–40 further revealed that aside from metabolism and angiogenesis, gene signatures of UnaG+ GBM cells exhibited negative enrichment for Type II interferon (IFN) signaling, TNFα and NF-κB pathway, Neuroinflammation signaling, and IFNα and IFNγ responses (Figures 5D and S2G–S2H; Table S2B), while upstream regulators included cytokines such as IL1B (Figures S2I). Furthermore, the gene ontology (GO) ‘Positive Regulation of Immune System Process’ was downregulated in UnaG+ GBM cells; conversely, the GO ‘Negative Regulation of Immune System Process’ was predominantly upregulated in UnaG+ tumor cells (Table S3). In aggregate, transcriptomic data implicated engagement of immune signaling by UnaG+ GBM cells favoring immunosuppression.
We next compared our in vivo GBM HIFON gene signature (top 200 upregulated DEGs in UnaG+ GBM cells) with the “Hallmark Hypoxia” gene set from the MSigDB database (200 genes compiled from multiple hypoxia studies, including many in vitro studies)41. This revealed 58 overlapping genes (related to glycolysis, Robo4-VEGF signaling, oxidative stress), but also 142 unique genes captured by the HRE-UnaG reporter concerning cholesterol metabolism, actin cytoskeleton, peroxisome, PPAR (regulator of lipid metabolism and inflammation), and notably chemokine signaling (Figure S3A; Table S4), confirming the distinct characteristics of the GBM HIFON gene signature in vivo.
UnaG+ gene signature is more represented in recurrent GBM and predicts worse outcome
To assess the prognostic value of our in vivo UnaG+ gene signature, we mined the TCGA human GBM data resource. We found that GBM patients in the high expressor group for UnaG+ signature had shorter survival than the low expressor group, and this applied to all three GBM transcriptional subtypes (Figures 5E and S3B–C, Table S5A). Consistently, the MES subtype (more malignant than PN or CL) displayed the highest enrichment for UnaG+ gene signature (Figure S3D). Moreover, the UnaG+ gene signature was also more represented in recurrent than primary GBMs, underscoring the importance of a HIFON gene program in GBM relapse (Figure 5E).
As a further validation of the clinical relevance of our UnaG+ gene signature, we analyzed the human Ivy Glioblastoma Atlas Project (Ivy GAP) database, which contains gene expression profiles from distinct anatomical zones, including perinecrotic zone (Pz), pseudopalisading cells (Pc), cellular tumor, leading edge, and microvascular proliferation area42. Aligned with above findings, Ivy GAP analysis revealed that the UnaG+ signature was enriched in the Pz and Pc zones (Figure 5F; Table S5B). A survey of Ivy GAP mRNA in situ hybridization data showed higher transcription of top UnaG+ DEGs in the Pz of human GBM in pseudopalisading patterns (Figure S3E).
UnaG+ cells were not uniform, as a second round of clustering revealed 4 subpopulations, each with distinct functional specializations, with subcluster (sc)-a concerning cell cycle regulation, apoptotic signaling, and neural crest differentiation, sc-b angiogenesis, sc-c unfolded protein response (UPR) and focal-adhesion, and sc-d IFN signaling (Figures S3F–S3G). Resonating with immune signaling by UnaG+ cells, various UnaG+ subclusters were also enriched for immune pathways, e.g., cytoplasmic retention of NF-κB for sc-a, macrophage chemotaxis for sc-b, and type I and II IFN signaling for sc-d. Of note, NF-κB is a transcription factor controlling numerous pro-inflammatory cytokines43; thus cytoplasmic retention of NF-κB signified immune repression44, again linking GBM hypoxia and immunosuppression. Comparative IPA similarly revealed that sc-b UnaG+ cells featured IL6, VEGF and CXCR4 signaling, while sc-d featured IFN, neuroinflammation, and necroptosis pathways (Figure S3H). Consistently, IL1B, IFN, and interferon regulator factors (IRFs) were predicted as upstream regulators for sc-b and sc-d gene programs (Figure S3I).
Hypoxic TAMs express immunotolerance markers
We further examined our scRNA-seq data of immune cells, which comprised two main macrophage clusters, one microglia cluster, a sizable population of T cells, followed by dendritic cells, mast cells, and B cells (Figure 5G). Weighted Gene Co-expression Network Analysis (WGCNA) revealed 10 major co-expressed gene modules in the immune cells, with the one (denoted as “red”) enriched for hypoxia signaling and many immune pathways, and it was predominantly expressed in a subpopulation of microglia and macrophages, with a small representation in dendritic cells and T cells (Figures 5H–5I).
We next focused on TAMs (microglia and macrophages), where WGCNA similarly revealed a distinct co-expressed gene module (“yellow”) enriched for hypoxia signaling and immune pathways, e.g., IL1 signal transduction, with top co-expressed genes including Il1b, Ccrl2, and Cxcr4 (Figures 5J and S4A–S4B). We validated in two human GBM scRNA-seq datasets that the “yellow” hypoxic TAM gene module was specifically represented in a subpopulation of tumor-associated macrophages, granulocytes, and dendritic cells (Figures 5K and S4C).
We also compared TAMs to homeostatic microglia in normal brain45. The DEGs in TAMs featured type II IFN signaling and immune cell chemotaxis (Figures S4D–S4E). Thus, unbiased single cell transcriptomic analysis revealed the presence of a subpopulation of immune cells displaying hypoxia gene signatures.
Hypoxic GBM niches drive sequestration and immunosuppression of TAMs and cytotoxic T cells
To further understand TAM diversity, we conducted unsupervised clustering, which identified eight TAM subclusters (sc) in GL261, each with distinct functional specifications, e.g., proinflammatory response (sc-1, −2), proliferation (sc-5, −8), metabolic pathways (sc-5), T cell activation (sc-6), angiogenesis (sc-3, −7), while microglia (sc-4) formed a cluster distinct from macrophages (Figures 6A and S4F–S4G; Table S6A). We also surveyed macrophage subclusters for perivascular macrophages, a subpopulation characterized by the markers Lyve1 and Cd16346, but did not find overt expression among TAMs.
Figure 6. Entrapped TAMs in hypoxic zones express immunosuppressive markers.

(A) UMAP plot of TAM subclusters in GL261.
(B) Violin plots of marker gene expression in TAM subcluster 3 (hypoxic) vs. other subclusters combined.
(C) Heatmap showing DEGs in hypoxic TAMs, with top upregulated DEGs labeled.
(D, E) Representative IF images from GL261 transplants (n=3) show expression of CD68 or CD206 in relation to hypoxia markers GLUT1 or Pimo.
(F) IF images show expression of immunotolerance markers Arginase-1 (Arg1) and CD206 in sequestered TAMs in hypoxic zones (asterisks). Quantification: n=10 randomly selected tumor areas from 3 mice per condition.
(G) Schematic of TAM patterning in hypoxic zones of GBM.
(H) IF image and quantifications of CD8+ CTLs or FOXP3+ Tregs in relation to hypoxic cores (asterisks) outlined by UnaG+ cells (dashed lines) of GL261 at 4 wks post-transplant. n=15 tumor areas from 3 mice per group.
(I) Multiplex 5-color IF imaging from n=3 tumor sections show localization of CTLs (CD8a+) and TAMs (CD68+) in relation to hypoxic zones (UnaG+) or blood vessels (PECAM1+).
(J, K) IF images of fresh frozen human GBM sections stained for immune cell markers for TAMs or T cells and GLUT1 (asterisks) in hypoxic or adjacent non-hypoxic zones. DAPI for nuclear staining. Enlarged images of boxed area in (J) are shown on right.
Unpaired t-test (F, H); *P<0.01, ***P<0.001.
See also Figures S5–S7.
Notably, gene signatures of sc-3 macrophages featured hypoxia pathways and immunotolerance/anti-inflammatory signaling such as IL10, IL4 (Figure S4G). Indeed, sc-3 TAMs displayed enrichment for the “Hallmark Hypoxia” gene set (Figure S4H) and they highly expressed hypoxia marker genes Vegfa and Slc2a1, phagocytic marker Cd68, and immunotolerance genes Mrc1 (CD206) and Arg 1 (Arginase-1), while Aif1 (encoding IBA1) was ubiquitously expressed in TAM subclusters (Figure 6B). A pseudotime analysis implicated a differentiation path from Ccr2+ monocytes (sc-2) to sc-3 TAMs (Figure S4I). We further compared the transcriptomes of sc-3 TAMs with the rest of TAMs, which revealed Ccl8 and immunotolerance markers Mrc1 and Arg1 as among the top DEGs in hypoxic TAMs (Figure 6C; Table S6B).
Both IPA and GSEA further confirmed that sc-3 macrophages were positively enriched for HIF1α signaling and immunotolerance/anti-inflammatory pathways (e.g., IL10, IL4, PD-1/PD-L1), but negatively enriched for inflammatory pathways (e.g., IFNα and IFNγ response, Macrophage Classical Signaling Pathway, and IL2 signaling and Production) (Figures S4J–S4K). Predicted upstream regulators for sc-3 macrophages included MIF, IL1B, IL4, IL10, as well as TGFB and PDCD1 (immune-inhibitory receptor PD-1)47 (Figure S4L).
To complement the transcriptomic data revealing a distinct hypoxic subpopulation of TAMs (sc-3), we performed co-IF for spatial information, which revealed that entrapped TAMs (CD68+ or CD206+) were co-labeled by GLUT1 or Pimo, respectively (Figures 6D–6E). The entrapped TAMs also expressed ARG1 and were surrounded by UnaG+ GBM cells in pseudopalisading patterns (Figure 6F). Similar findings were also observed in RCAS and CT2A GBM models (see Figure S1). These data converged on a distinct hypoxic and immunotolerant TAM subpopulation that was sequestered by UnaG+ pseudopalisades (Figure 6G).
We next analyzed the transcriptomes of T cells (expressing Cd3e (CD3+)), which could be partitioned into 4 main subclusters: CD8 cytotoxic T lymphocytes (CTLs), Tregs, natural killer (NK) cells, and CD4 T helper cells (Figures S5A–S5C). CD8+ CTLs contained three subclusters: CTL-a Effector T cells (46%), CTL-b Tissue-resident Effector Memory T cells (30%), and CTL-c Naive T cells (24%) (Figures S5D–S5E; Table S6C). Notably, CTL-a cells featured Hypoxia and Exhausted T cell gene signatures and expressed T cell exhaustion marker Havcr2 (Figures S5F–S5G), consistent with earlier findings that CTLs infiltrating GBM become mostly exhausted48,49. Concordantly, CTL-a marker genes also showed negative enrichment for Inflammatory Response and IFN Signaling, but positive enrichment for immune checkpoint blockade (PD-1L expression and PD-1 checkpoint pathway) (Figures S5H–S5I).
Additional IF staining demonstrated sequestration of CD8a+ CTLs in UnaG+ zones, whereas Tregs (FOXP3+) largely congregated in UnaG− areas (Figure 6H). This also largely held true for RCAS and CT2A GBMs (Figures S5J–S5K). Hence, transcriptomic and histological studies indicated that a subpopulation of T cells (showing signs of hypoxia and immune exhaustion) was entrapped in pseudopalisading hypoxic zones, further linking tumor hypoxia to immunosuppression.
We conducted 5-color multiplex imaging, which confirmed congregation of CD68+ TAMs and CD8a+ CTLs inside of UnaG+ pseudopalisades in vascular-poor areas of GL261 (Figures 6I and S5L). We also conducted co-IF on fresh frozen human GBM biopsies, which similarly showed co-aggregation of CD11b+ TAMs and CD8a+ CTLs in hypoxic zones, whereas FOXP3+ Tregs accumulated in non-hypoxia areas (Figures 6J–6K; Table S1B).
Hypoxic GBM cells and immune cells display distinct ligand-receptor signaling communication
We next conducted CellPhoneDB50 analyses, which revealed a rich repertoire of distinct ligand-receptor signaling between various cell populations in GL261 GBM (Figures S6A–S6F). Specifically, comparison of the communication between hypoxic or non-hypoxic GBM cells and hypoxic TAMs revealed that the hypoxic pairs (i.e., UnaG+ GBM cells ↔ sc-3 TAMs) mainly communicated regarding phagocytosis of apoptotic cells (LGALS3–MERTK, C3–Integrin αMβ2, ICAM1–Integrin αXβ2), angiogenesis (VEGFA–FLT1, C3–C3AR1), as well as immune suppression and inhibition of TNF signaling (TNFRSF1B–GRN) (Figure S6A). In addition, communication of non-hypoxic GBM cells and hypoxic TAMs included SPP1–CD44 and SPP1–PTGER4 signaling, which mediates recruitment and co-stimulation of immune cells (Figure S6A).
Likewise, the hypoxic GBM cells ↔ non-hypoxic TAMs communication (presumably before becoming entrapped in UnaG+ pseudopalisades) also mainly concerned inhibition of TNF signaling (TNFRSF1B–GRN) and immune suppression (AXL–GAS6, C3–C3AR1), in addition to phagocytosis and macrophage migration using ICAM1-integrin ligand-receptor pair (Figure S6B).
The communication among tumor cells, e.g., hypoxic GBM cells ↔ non-hypoxic GBM cells mainly concerned angiogenesis, cholesterol metabolism, proliferation, and invasion; whereas the communication of non-hypoxic GBM cells ↔ non-hypoxic GBM cells included adhesion, stemness, and EMT (Figure S6C).
The communication of hypoxic GBM cells ↔ T cells featured T cell recruitment using ICAM1 and COL6A2 as ligands, whereas non-hypoxic GBM cells ↔ T cells communication utilized SPP1 and CXCL10 as ligands that regulate T cell activity (Figure S6D).
The communication between immune cells, e.g., hypoxic TAMs ↔ T cells featured T cell recruitment (FN1–integrin α4β1, FN1–integrin α4β7, CXCL10–CXCR3, CXCL10–DPP4) and immune co-stimulation (TNFSF9-TNFRSF9), whereas the communication of non-hypoxic TAMs ↔ T cells featured control of T cell activity or T cell recruitment (Figure S6E).
Next, we compared the signaling of hypoxic GBM cells with different T cell subpopulations. The communication of hypoxic GBM cells ↔ CTLs mainly concerned migration and adhesion of CTLs (e.g., Collagens–Integrin α1β1 complex), while hypoxic GBM cells ↔ Tregs interactions concerned homing and activation of Tregs and promotion of Treg function (Figure S6F).
We further examined recent human GBM spatial transcriptomics data51 (Table S1C), focusing on localization of gene signatures and ligand-receptor pairs in hypoxic zones. We validated spatial patterning of macrophages and T cells in hypoxic areas of human GBM (Figure S7A, Data S1). Additionally, we found that CD68+ spots and CD8A+ spots in hypoxic zones displayed hypoxia/immunotolerance gene signatures (Figures S7A–S7B, Data S1). We also confirmed co-localization of tissue spots expressing ligand-receptor pairs (e.g., FN1-Integrin αVβ1, CCL8-CCR1/CCR5 and IL1B-IL1R1) in hypoxic zones and in immune spots expressing CD68 or various T cell markers (Figure S7C). This also applied to signaling between hypoxic GBM cells ↔ hypoxic macrophages (e.g., MIF-CD74, LGALS3-MERTK) (Data S1), or between hypoxic GBM cells ↔ T cells (Data S1).
Progressive spatial confinement of TAMs in hypoxic zones involves CCL8 and IL-1β
Lastly, we explored the signaling mechanisms underlying TAM trafficking and sequestration in pseudopalisading areas and co-evolution of hypoxic zones. We focused on cytokines CCL8 and IL-1β, as Ccl8 was identified as a top upregulated DEG in the hypoxic TAMs (sc-3) (see Figure 6C), while IL-1β, a major cytokine of inflammatory responses and cytotoxicity52, was one of the top co-expressed genes in the “yellow” hypoxic TAM gene module and also predicted to be an upstream regulator of both sc-3 (hypoxic) macrophages and UnaG+ tumor cells. Indeed, scRNA-seq demonstrated a specific expression of Ccl8 and Il1b in TAMs, but not tumor cells (Figure 7A), with Ccl8 highly expressed in hypoxic TAMs (sc-3), while Il1b appeared more ubiquitously expressed in all TAMs. CCL8 receptors Ccr1 and Ccr5 were also highly expressed in TAMs (Figure 7A).
Figure 7. Progressive spatial confinement of TAMs in hypoxic zones involves CCL8 and IL-1β.
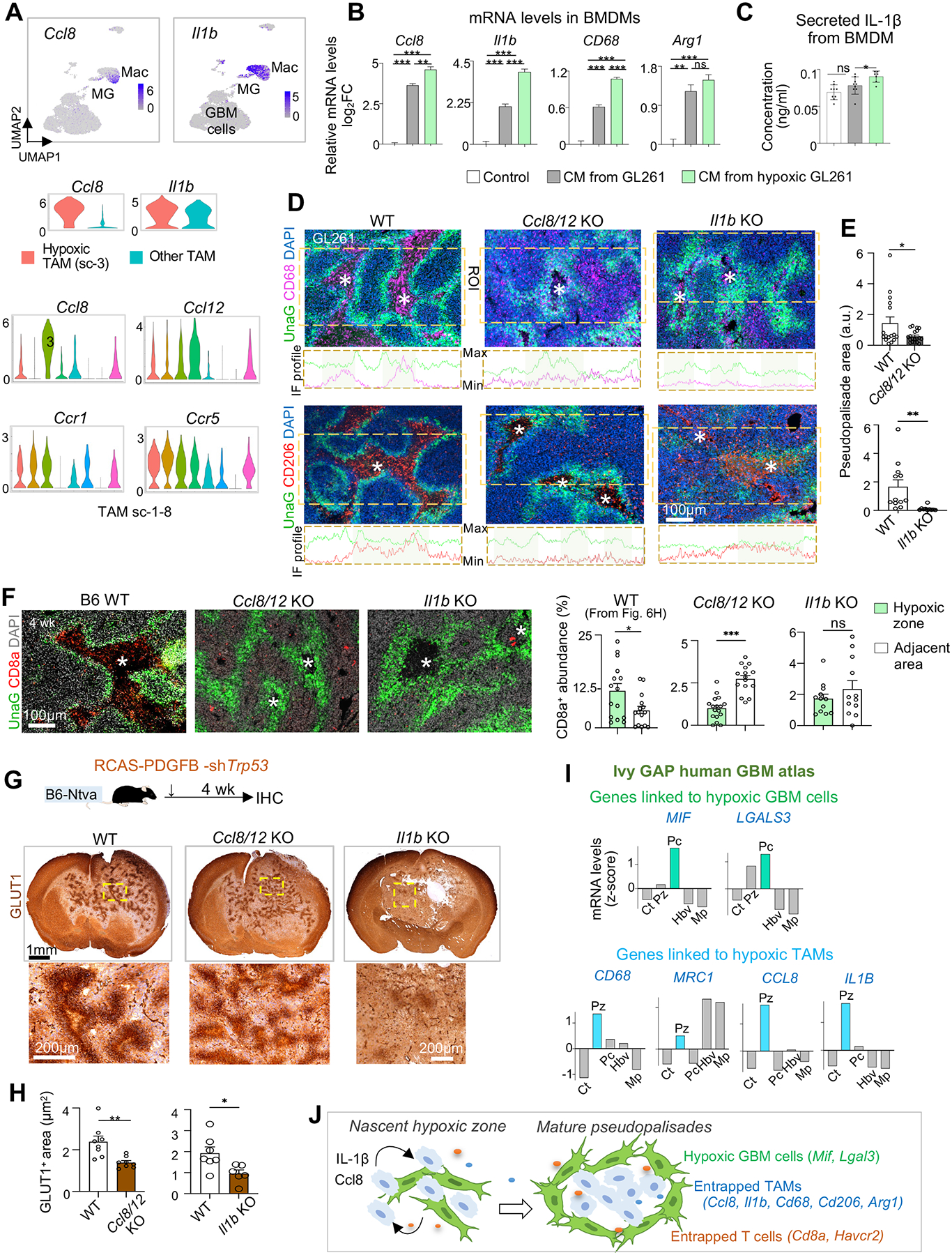
(A) Top, UMAP feature plots show the expression of Ccl8 and Il1b in tumor-associated macrophages (Mac) as compared to other cells in GL261 GBM. Middle, violin plots show the expression of Ccl8 and Il1b in sc-3 (hypoxic) TAMs vs. other TAMs combined. Bottom, violin plots show the expression of the indicated genes in TAM subclusters.
(B) qRT-PCR results of gene expression in cultured BMDMs after 24 hrs exposure to conditioned media (CM) of GL261 cells grown in normal or hypoxic (1% O2) condition. n=3 wells per condition.
(C) ELISA of secreted IL-1β from BMDMs when exposed to control or CM. n=3 wells per condition.
(D) Representative IF images from n=3 mice and profile plots of IF intensities for CD68 or CD206 in region of interest (ROIs, dashed boxes) in GL261 transplanted in WT or KO hosts.
(E) Quantifications of abundance of pseudopalisading areas (UnaG+) in GL261 transplanted in different hosts. n=12–16 areas from 3 mice per condition. a.u., arbitrary units.
(F) IF images and quantifications from n=3 mice show changes in CD8a+ CTL trafficking into hypoxic zones of GL261 transplanted in KO mice compared to control hosts. Asterisks denote hypoxic cores outlined by UnaG+ pseudopalisades.
(G, H) Experimental scheme of mouse RCAS GBM model in immunocompetent B6 background (G, top). Representative IHC images for GLUT1 (G, bottom) of brains bearing RCAS GBMs generated in WT or different KO mice. Quantifications (H) show the extent of GLUT1+ pseudopalisading areas in RCAS GBMs. n=6–8 hypoxic zones per genotype from n=3 mice.
(I) Expression of hypoxia niche genes associated with either UnaG+ tumor cells (top) or entrapped TAMs (bottom) in pseudopalisading cells (Pc) or perinecrotic zones (Pz) relative to other areas based on Ivy GAP human GBM data.
(J) Model of hypoxia-induced TAM and CTL trafficking, cell-cell sorting, gene reprogramming into immunosuppression, and maturation of pseudopalisades.
Unpaired t-test (E, F, H); One-way ANOVA (B, C); *P< 0.05, **P< 0.01, ***P<0.001; ns, not significant.
See also Figure S7.
We first tested whether hypoxic GBM cells send cues to TAMs to induce Ccl8 and Il1b expression. To this end, we exposed primary bone marrow-derived macrophages (BMDMs) to conditioned media (CM) from GL261 GBM cells, which led to a marked upregulation of Ccl8 and Il1b, while CM from hypoxic GL261 cells exerted an even stronger effect (Figure 7B). This also held true for Cd68 and Arg1 (Figure 7B). As IL-1β is secreted after proteolytic cleavage of pro-IL-1β22, ELISA confirmed that CM from hypoxic GL261 cells induced IL-1β secretion from BMDMs (Figure 7C).
To assess the functional significance of Ccl8 and Il1b induction in hypoxic niches for TAM patterning, we transplanted GL261-HRE-UnaG into Ccl8/12 or Il1b knockout (KO) mice22,53. Of note, the Ccl8/12 KO affects both Ccl8 and Ccl12 due to their close genomic proximity; however, Ccl12 was not specifically expressed in hypoxic TAMs (Figure 7A, Data S1), and it is absent from human genome, implying a lesser role for hypoxia-regulated TAM patterning. We found that while UnaG+ GBM cells were abundantly detected in both control and Ccl8/12 or Il1b KO hosts, the hypoxic zones outlined by UnaG+ pseudopalisades were smaller in the KO hosts, corresponding to the less developed spatial patterning of CD68+ or CD206+ TAMs (Figures 7D–7E). Moreover, CD8a+ CTLs accumulated mostly in non-hypoxic areas in both Ccl8/12 KO or Il1b KO hosts, in contrast to WT hosts where they congregated more in UnaG+ hypoxic zones (Figure 7F).
To further corroborate these findings, we also examined the effect of Ccl8/12 or Il1b ablation in RCAS GBM22. One month after the induction of GBM by RCAS-PDGFB/shTrp53 virus, we examined the development of hypoxic zones (Figure 7G). Similar to the GL261 model, the RCAS GBMs established in Ccl8/12 KO or Il1b KO mice harbored smaller hypoxic zones (outlined by GLUT1+ cells) and less developed pseudopalisades than controls (Figures 7G–7H). Of note, in the RCAS GBM model, Ccl8 or Il1b were absent from both tumor and non-tumor cells, whereas in the GL261 model, gene deletions only occurred in stromal cells, which may account for the more pronounced phenotype of spatial perturbance of TAMs and hypoxic zones in the RCAS GBM model.
Echoing the human GBM spatial transcriptomics data shown in Fig. S7D–S7F, re-examination of the human GBM Ivy GAP database confirmed high expression of CCL8, IL1B, CD68, and MRC1 in hypoxic areas (Pz), while MIF and LGALS3 (two top DEGs in UnaG+ GBM cells) were highly expressed by pseudopalisading cells (Pc) (Figure 7I). In addition, TCGA data showed that these hypoxia-associated genes and niche factors as well as CCL8 receptors CCR1 and CCR5 are expressed at higher levels in GBM of MES subtype and largely predict poor outcome (Data S1). In aggregate, our data supported a mutual interaction – hypoxic GBM cells release cues to induce CCL8 and IL-1β in TAMs, which in turn promote trafficking/sequestration of TAMs and CTLs in hypoxic zones and the maturation of pseudopalisades (Figure 7J).
DISCUSSION
Our study highlighted hypoxic zones as a major driver of TAM patterning and immunosuppression during GBM progression. We also unveiled a reciprocal influence between host immune response and tumor hypoxia, with adaptive immunity exacerbating vascular abnormalities and thus tumor hypoxia, whereas hypoxic niches attracted and sequestered TAMs, leading to immunosuppression.
Even though GBM is well known for immunosuppression and tumor hypoxia predicts poor prognosis54, the underlying mechanisms linking the two are unclear. Our studies with fully immunocompetent GBM models revealed temporospatial pattering of TAMs coinciding with vascular alterations and emergence of hypoxic niches. We showed that in the GBM interior, majority of TAMs were blood-borne MDMs, whereas microglia largely resided in GBM periphery. Notably, at early stages, MDM aggregated around blood vessels that were dense and regular; later, as vasculature became sparse, tortuous, and engorged, TAMs shifted from perivascular regions to poorly vascularized areas. Our scRNA-seq and IF analyses converged on the presence of a subpopulation of TAMs and CTLs that were hypoxic and immunosuppressed. Hence, two potential mechanisms may account for hypoxia-induced immunosuppression: i) hypoxic zones attract and sequester immune cells, and ii) entrapped immune cells experience hypoxia and become immunosuppressed. The development of pseudopalisading structures in GBM facilitates inflammatory containment, by confining cytotoxic immune cells and necrotic tumor cells (which are immunogenic) inside hypoxic cores and by limiting communication of antigen-presenting cells to effector immune cells.
It is assumed that tumor hypoxia arises from rapid tumor expansion outstripping blood supply; in contrast, our data unveiled adaptive immunity as a driving force of vascular aberrancy and thereby tumor hypoxia. In GBM established in immunocompetent hosts, while the initial tumor vasculatures appeared regular and dense, they became progressively engorged, tortuous, and sparse. In contrast, in SCID hosts lacking functional T and B cells, although GBM tumors reached advanced sizes, tumor vasculatures remained regular and dense, with only scattered hypoxic microregions that never transformed into pseudopalisades. Hence, immunity-driven vascular aberrancy is a determining force in shaping tumor hypoxia and TAM patterning, leading to immunosuppression.
The distinct in vivo HIFON GBM gene signature captured by the HRE-UnaG reporter and scRNA-seq indicated that hypoxic GBM cells were not only engaged in metabolic adaptation, stress response, but also immune signaling. In addition, the UnaG+ GBM gene signature indicated quiescence and a mesenchymal shift, both linked to malignant potency30,55; it is also more represented in recurrent GBM, in MES GBM subtype, and prognostic for poor survival for GBM patients. scRNA-seq analyses also revealed distinct ligand-receptor communications between tumor cells and immune cells in hypoxic niches, featuring not only angiogenesis, phagocytosis, but also immune cell recruitment, immune checkpoints, and immunosuppression.
The clinical relevance of our study is further supported by the observation of TAM spatial organization in patient GBM samples, correlated with hypoxic burden, glioma grade and MES subtype. Both mouse and human GBM transcriptomic analyses identified a co-expressed gene module in TAMs enriched in hypoxia and immune pathways, including IL1. Moreover, hypoxic niche genes associated with hypoxic GBM cells (MIF, LGALS3) or hypoxic TAMs (CCL8, IL1B, CD68, MRC1) were expressed higher in perinecrotic zones or pseudopalisading cells of GBM patients, particularly for the MES subtype that is linked to elevated malignancy and high immune response24. Our results thus substantiated the recent report of tumor and immune cell interactions driving transition to MES-like states in GBM patients56.
From a therapeutic point of view, targeting the hypoxic niche may synergize with conventional chemoradiation treatment, not only by reducing the hypoxic tumor population (which is therapy resistant, as chemodrugs target mainly proliferative cells and irradiation requires oxygenation to be effective), but also by attenuating immunosuppression. Current clinical trials with Evo for recurrent GBM have so far yielded only limited positive results57, thus combinatorial strategies may be better equipped to bring out therapeutic benefits of perturbing hypoxic niches in GBM. Indeed, we found in GL261 model that combining evofosfamide (Evo) and irradiation eradicated hypoxic niches, abolished TAM sequestration, and achieved better tumor control. Attenuating CCL8, IL1B, or MIF signaling could be another promising strategies, and neutralizing antibodies are being tested for autoimmune disorders and solid cancers (e.g., anti-CCL858–60; anti-IL1B: Gevokizumab61, Canakinumab62; anti-MIF: Imalumab63, 4-iodo-6-phenylpyrimidine64, Milatuzumab65). Our results also raised the awareness that while boosting anti-cancer immunity is desirable, it may lead to a cascade of interconnected events – cell toxicity, vascular aberrancy, higher hypoxic burden, and immunosuppression –, thus dampening the efficacy of immunotherapy.
In summary, the mutual communication between tumor cells and immune cells in the hypoxic niche plays a determining role in sculpting the immune landscape, which may limit inflammatory spread but also induce a tolerogenic/immunosuppressive microenvironment. The understanding of the reciprocal influence of immune response and tumor hypoxia will have important clinical ramifications for prognosis and advancing immunotherapy for GBM patients.
Limitations of the study
Our study leveraged 10X Visium transcriptomic data to validate cell type specific interactions of immune cells at hypoxic niches in human GBM, with the caveat that the transcriptomic data was not on single cell level (Visium spots can comprise about 10 cells). Future investigations with single-cell resolution will be needed to further dissect cell-type specific signaling at the hypoxic niche.
Our comparison of immunodeficient SCID vs. immunocompetent WT hosts (receiving identical GL261 GBM transplants) highlighted phenotypic differences of stromal players in shaping tumor hypoxia and TAM patterning. This sets the stage for future studies to address outstanding questions: what would be the phenotypes after B and/or T cell depletion, which subpopulation of T and/or B cells is mainly responsible, are MDMs differentially primed in B6 WT vs. B6 SCID upon entry into the brain and within GBM? Alternative hypotheses (e.g., neutrophil infiltration, pericyte activation) also await further testing. Of note, GL261 is a relatively immunogenic GBM model; it is thus worthwhile to expand investigations in other less immunogenic GBM models.
Our study has not fully revealed the regulatory mechanisms of Ccl8 and Il1b induction in hypoxic TAMs, which occurs most likely as result of reciprocal interactions with hypoxic tumor cells. Several factors such as IL1B, IL4, IL6, IL10, MIF, TGFβ, and VEGF were predicted by IPA upstream regulator analysis, and these factors could shape the transcriptional responses of TAMs or neighboring GBM cells in hypoxic niches. Indeed, MIF and VEGF were top DEGs in UnaG+ tumor cells, thus likely to be present in conditioned media of hypoxic GL261 cells. Functional analyses and time course studies will be needed to reveal further details of these interactions. Similar scenarios of communication have been described for the cancer stem cell (CSC) population of GBM and infiltrating immune cells66. Phenotypic alignment of hypoxic GBM cells and CSCs awaits future studies.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Hongyan Zou (hongyan.zou@mssm.edu).
Materials availability
Plasmids generated in this study have been deposited at Addgene or are available upon request from the lead contact.
Data and code availability
scRNA-seq data has been deposited in NCBI Gene Expression Omnibus database and is publicly available from the date of publication. Accession number is listed in the Key Resources Table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Arg1 (M-20) | Santa Cruz Biotechnology | Cat#sc-18355; RRID: AB_2058957 |
| Anti-mouse CD68 (FA-11) | Bio-Rad | Cat# MCA1957GA; RRID: AB_324217 |
| Anti-CD8a (53–6.7) | Thermo Fisher Scientific | Cat# 14-0081-82; RRID: AB_467087 |
| Anti-mouse MMR/ CD206 | R and D Systems | Cat# AF2535; RRID: AB_2063012 |
| Anti-mouse CD16/32 (2.4G2) | BD Biosciences | Cat# 553142; RRID: AB_394657 |
| Anti-FOXP3 (FJK-16s) | Thermo Fisher Scientific | Cat# 14-5773-82; RRID: AB_467576 |
| Anti-GLUT1 (SA0377) | Thermo Fisher Scientific | Cat# MA5-31960; RRID: AB_2809254 |
| Anti-Iba1 | FUJIFILM Wako Shibayagi | Cat# 019-19741; RRID: AB_839504 |
| Anti-mouse CD49d / integrin alpha4 (9C10 (MFR4.B)) | BioLegend | Cat# 103701; RRID: AB_313042 |
| Anti-Ki67 | Abcam | Cat# ab15580; RRID: AB_443209 |
| Anti-cMyc tag | Novus | Cat# NB600-335; RRID: AB_10002720 |
| Anti-CD31/ PECAM1 | Abcam | Cat# ab28364; RRID: AB_726362 |
| Anti-PU.1 (E.388.3) | Thermo Fisher Scientific | Cat# MA5-15064; RRID: AB_10986949 |
| Anti-Pimonidazole Dylight 594 (4.3.11.3) | Hydroxyprobe | Cat# HP7-100Kit |
| Bacterial and virus strains | ||
| Stbl3 E. coli | Thermo Fisher Scientific | C737303 |
| Biological samples | ||
| Primary bone marrow derived macrophages (mouse) | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Pimonidazole | Hydroxyprobe | Cat# HP-100mg |
| Evofosfamide/ TH-302 | AdooQ Bioscience | Cat# A11144 |
| Recombinant murine M-CSF | Peprotech | Cat# 315-02 |
| Percoll | GE Healthcare | Cat# 17089101 |
| BrainBits Hibernate E Low Fluorescence | Fisher Scientific | Cat# NC0285514 |
| DNAse I | Worthington | Cat# LS006344 |
| RBC lysis buffer | BioLegend | Cat# 420301 |
| Critical commercial assays | ||
| Mouse IL-1 beta/IL-1F2 DuoSet ELISA | R and D Systems | Cat# DY401 |
| DuoSet ELISA Ancillary Reagent Kit 2 | R and D Systems | Cat# DY008 |
| RNeasy mini kit | Qiagen | Cat# 74104 |
| SuperScript III First Strand Synthesis System | Thermo Fisher Scientific | Cat# 18080051 |
| Neural Tissue Dissociation Kit (P) | Miltenyi Biotec | Cat# 130-092-628 |
| Deposited data | ||
| Analyzed scRNA-seq data | This paper | Gene Expression Omnibus, GEO: GSE179077 |
| Experimental models: Cell lines | ||
| GL261 (murine GBM cell line) | National Cancer Institute | NCI-DTP Cat# Glioma 261; RRID: CVCL_Y003 |
| GL261-HRE-UnaG | This paper | N/A |
| CT2A | Millipore | Millipore Cat# SCC194, RRID: CVCL_ZJ44 |
| HEK293T | ATCC | CRL-3216; RRID: CVCL_0063 |
| Experimental models: Organisms/strains | ||
| C57BL/6J | The Jackson Laboratory | Strain# 000664; RRID: IMSR_JAX:000664 |
| C57BL/6J SCID; B6.Cg-Prkdcscid/SzJ | The Jackson Laboratory | Strain# 001913; RRID: IMSR_JAX:001913 |
| ICR-SCID; IcrTac:ICR-Prkdcscid | Taconic Biosciences | Model# ICRSC-M; RRID: IMSR_TAC:icrsc |
| N-tva-Ccl8/12 KO | Sabina Islam | N/A |
| N-tva-Il1b KO | Dmitry Shayakhmetov | N/A |
| Oligonucleotides | ||
| Mouse Arg-1 forward: 5’-GTGGCTTTAACCTTGGCTTG-3’ | This paper | N/A |
| Mouse Arg-1 reverse: 5’-CTGTCTGCTTTGCTGTGATG-3’ | This paper | N/A |
| Mouse Ccl8 forward: 5’-ACAATATCCAGTGCCCCATG-3’ | This paper | N/A |
| Mouse Ccl8 reverse: 5’-TGAAGGTTCAAGGCTGCAG-3’ | This paper | N/A |
| Mouse CD68 forward: 5’-ACTTCGGGCCATGTTTCTCT-3’ | This paper | N/A |
| Mouse CD68 reverse: 5’-GCTGGTAGGTTGATTGTCGT-3’ | This paper | N/A |
| Mouse Il1b forward: 5’-CCAAGCAACGACAAAATACC-3’ | This paper | N/A |
| Mouse Il1b reverse: 5’-GTTGAAGACAAACCGTTTTTCC-3’ | This paper | N/A |
| Mouse Gapdh forward: 5’-ACTGCCACCCAGAAGACTGT-3’ | This paper | N/A |
| Mouse Gapdh reverse: 5’-GATGCAGGGATGATGTTCT-3’ | This paper | N/A |
| Recombinant DNA | ||
| pcDNA-dUnaG | Dr. Friedemann Kiefer | N/A |
| pENTR/D-TOPO | Thermo Fisher Scientific | Cat#: K240020 |
| pLenti X1 Puro DEST | Addgene | Plasmid# 17297; RRID: Addgene_17297 |
| pMD2.G | Addgene | Plasmid# 12259; RRID: Addgene_12259 |
| psPAX2 | Addgene | Plasmid# 12260; RRID: Addgene_12260 |
| pLenti-HRE-dUnaG | This paper | Addgene #124372 |
| Software and algorithms | ||
| Microsoft Office | Microsoft | RRID: SCR_016137 |
| Adobe Photoshop | Adobe | RRID: SCR_014199 |
| Fiji Is Just ImageJ (FIJI) | Public domain, National Institutes of Health | https://imagej.net/software/fiji/; RRID: SCR_003070 |
| Prism 9 | GraphPad | RRID: SCR_002798 |
| SnapGene | Dotmatics | RRID: SCR_015052 |
| Zen Blue | Zeiss | RRID: SCR_013672 |
| Cell Ranger | 10x Genomics | RRID: SCR_017344 |
| Seurat R package | Rahul Satija Lab | https://satijalab.org/seurat/index.html; RRID: SCR_007322 |
| SCRABBLE R package | Kai Tan Lab | https://github.com/tanlabcode/SCRABBLE |
| Enrichr | Avi Ma’ayan Lab | https://maayanlab.cloud/Enrichr/; RRID: SCR_001575 |
| GBM Bio Discovery Portal | NCI | https://gbm-biodp.nci.nih.gov |
| cBioPortal for Cancer Genomics | MSKCC | https://docs.cbioportal.org/about-us/; RRID: SCR_014555 |
| Ivy Glioblastoma Atlas Project | Allen Institute for Brain Science | https://glioblastoma.alleninstitute.org; RRID: SCR_005044 |
| high dimensional WGCNA R package | Vivek Swarup Lab | https://smorabit.github.io/hdWGCNA/index.html |
| Leica Application Suite X | Leica Microsystems | RRID: SCR_013673 |
| Ingenuity Pathway Analysis | Qiagen Digital Insights | RRID: SCR_008653 |
| Gene Set Enrichment Analysis | Broad Institute | RRID: SCR_003199 |
| The Cancer Genome Atlas | National Institutes of Health | https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga; RRID: SCR_003193 |
| Monocle3 | Cole Trapnell Lab | https://cole-trapnell-lab.github.io/monocle3/; RRID: SCR_018685 |
| CellPhoneDB 4.0 | Roser Vento-Tormo Lab | https://github.com/ventolab/CellphoneDB; RRID: SCR_017054 |
| GlioVis | Public domain, Massimo Squatrito Lab | http://gliovis.bioinfo.cnio.es |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Mice
C57BL/6J wild-type mice and C57BL/6J-SCID mice were obtained from The Jackson Laboratory. ICR-SCID mice were purchased from Taconic Biosciences. Ccl8/12 KO mice53 were gifted by Dr. Sabina Islam and Il1b KO mice67 were gifted by Dr. Dmitry Shayakhmetov, and were both bred in our colony on C57BL/6J genetic background. Mice of both sexes in the age range of 8–16 weeks were used for experiments. All animals were housed in a climate-controlled, pathogen-free facility with access to food and water ad libitum under a 12-hour light/dark cycle. All animal procedures were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) of Icahn School of Medicine at Mount Sinai.
GBM cell lines
The murine high-grade glioma cell line GL261 (KrasG12T, Trp53G153C, Pten−/−, c-myc and Egfr amplification)68–70 was obtained from the repository of the National Cancer Institute. CT2A cells23 were provided by Dr. Roger Abounader (University of Virgina). Glioma cells were cultured in DMEM media with Glutamax (Gibco) supplemented with 10% FBS (Thermo Fisher Scientific) and 1% Penicillin-Streptomycin antibiotics (Gibco). For long term storage, cells were cryopreserved in medium containing 10% DMSO. Cells were passaged after thawing for at least two passages before use in experiments. For in vitro hypoxia studies, GL261 cells were seeded into 6 well plates and placed for 24 hr in a hypoxia chamber (C-Chamber; Biospherix) set to 1% oxygen. HEK293T cells were obtained from American Type Culture Collection (ATCC).
RCAS glioma
A genetically modified mouse model using the RCAS/t-va system was used to generate murine GBM, as previously described22. Briefly, DF1 avian fibroblast cells (ATCC) were grown at 39°C, expanded to passage 4 and transfected with RCAS-PDGFB-HA or RCAS-shp53-RFP using a Fugene 6 Transfection kit (Roche, 11814443001). Cells were cultured with DMEM media (Gibco, 11995–065) supplemented with 1% L-glutamine, 1% penicillin/streptomycin, and 10% FBS (ATCC). Transfected DF1 cells were used for injections before reaching passage 25. DF-1 cells (4×104) were stereotactically delivered with a Hamilton syringe equipped with a 30-gauge needle for tumor generation. The injection site was the frontal striatum with the coordinates AP −1.7 mm and right −0.5 mm from bregma; depth −2.0 mm from dural surface. Mice were continually monitored for signs of tumor burden and were sacrificed upon observation of endpoint symptoms including head tilt, lethargy, seizures, and excessive weight loss.
Human participants
All human studies were performed on de-identified archived tissue specimens from GBM patients. A tissue microarray slide with Formalin-Fixed Paraffin-Embedded (FFPE) specimens from 71 glioma patients (collected before commencement of standard of care) was obtained from US Biomax, Inc. The following FFPE specimens of grade 4 gliomas were obtained from Emory University: GBM patient sample S15 35549, from a 57 year old male, diagnosed with recurrent grade 4 glioma; patient had undergone radiation therapy and GBM transcriptional subtype was determined as Proneural upon Nanostring RNA-seq. GBM patient sample S16 71 was obtained from a 56 year old female, diagnosed with a primary grade 4 glioma; patient had undergone treatment with radiation therapy, temozolomide, lomustine and bevacizumab and GBM subtype was determined as Classical. GBM patient samples S16 18015, S15 20710, S16 18411 and S16 736 were obtained from a 67 year old male, 73 year old male, 70 year old female, and a 56 year old female respectively. GBM subtype was determined as Mesenchymal. Patient S16 18015 had undergone treatment with temozolomide26. Fresh frozen GBM patient samples (tissue ID 13629) were obtained at The Mount Sinai Hospital biorepository from a 70 year old male, who was diagnosed with a primary malignant grade 4 glioma.
METHOD DETAILS
Generation of lentiviral HIF reporter
The plasmid dUnaG was obtained from the Kiefer laboratory17 and used as template to amplify an HRE-dUnaG PCR fragment, which was inserted into the Gateway entry vector pENTR/D-TOPO (Invitrogen) and then transferred by Gateway LR reaction (Invitrogen) into the destination plasmid pLenti X1 Puro DEST (Addgene #17297)71. The final pLenti-HRE-dUnaG plasmid (deposited as Addgene #124372) carries an array of five hypoxia-response elements (HRE) and a minimal CMV promoter to drive the expression of UnaG fused to a PEST degron and C-terminal myc tag, and in addition a puromycin resistance gene driven by a constitutive phosphoglycerate kinase (PGK) promoter. Of note, the myc tag was used in some immunofluorescence experiments to stain for UnaG with an anti-myc antibody to extended stability of fluorescence for storage of sections.
Lentiviral particles were produced by transfection of HEK293T cells with the pLenti plasmid, envelope plasmid pMD2.G, and packaging plasmid psPAX2 (Addgene #12259 and #12260; deposited by Didier Trono, EPFL Lausanne). Media supernatants were collected 2–3 days after transfection and viral particles were concentrated by ultracentrifugation.
Transduction of GBM cells
GL261 cells were transduced with a mixture of HRE-UnaG lentiviral particles and polybrene (5 mg/ml). Transduced cells were selected with 1 μg/ml puromycin, starting 48 hr after transduction. Early passages of GL261-HRE-UnaG cells were frozen 1 wk after puromycin selection, and subsequent passages were cultured in media with 0.5 μg/ml puromycin to prevent silencing of lentiviral vectors.
Intracranial transplantation of tumor cells
Mice were anaesthetized in an induction chamber with a 2.5% isoflurane/oxygen mixture and secured to a stereotaxic apparatus (Stoelting). Anesthesia was maintained with a 1.5% isoflurane/oxygen mixture, which was delivered via a nose-cone. A lubricant ophthalmic ointment (Artificial Tears, Akron) was applied. A cranial hole was drilled through a scalp incision 2.0 mm lateral and 0.5 mm posterior to Bregma. GBM cells (105 cells) suspended in 5 μl PBS were then injected through the hole at a depth of 3.2 mm using a 10 μl gas tight syringe (Hamilton) and a Nanomite programmable syringe pump (Harvard Apparatus) with a constant infusion rate of 1 μl/min to prevent backflow. After injection, syringe was incrementally raised using the stereotaxic apparatus over a period of 5 min. Scalp incision was sealed using a tissue adhesive (Vetbond, 3M).
Immunofluorescence and immunohistochemistry
Mice carrying intracranial GBM transplants or de novo tumors induced by RCAS/t-va system were intracardially perfused with PBS followed by 4% PFA/PBS. Brains were harvested and fixed overnight in 4% PFA/PBS followed by two successive overnight incubations in 12.5% and 25% sucrose/PBS. Brains were then embedded in O.C.T compound (Fisher Scientific), frozen on dry ice. Cryosections of 20 μm thickness were cut using a cryostat (Leica) and collected in PBS and stored as floating sections at 4°C.
For immunofluorescence, floating sections were blocked for 1 hr (blocking buffer: PBS with 5% donkey serum and 0.3% Triton X-100), then incubated overnight with primary antibodies in antibody dilution buffer (PBS with 1% BSA and 0.3% Triton X-100), followed by staining with Alexa-labeled secondary antibodies (Jackson ImmunoResearch) for 2 hr, and counterstaining with DAPI (Invitrogen). Sections were washed in PBS and mounted with Fluoromount G (Southern Biotech).
For immunohistochemistry of microtome sections from patient GBM specimens and from mouse brains carrying RCAS induced tumors, sections from FFPE tissue blocks were processed at the Mount Sinai Pathology core using a Ventana system (Roche).
Antibodies are listed in the Key Resources Table.
Single-cell RNA sequencing
Animals were euthanized, brain tissue containing the main tumor bulk was dissected on ice, and a tissue piece of approximately 3 mm edge length was diced with scalpel blades and dissociated into single cell suspension using papain digestion (Miltenyi Neural Tissue Dissociation Kit (P) 130-092-628).
The cell suspension was pelleted and resuspended in 7 ml of HBSS (without Ca/Mg), mixed with 1.2 ml of fetal bovine serum (FBS) and 3.6 ml of 100% Percoll (GE Healthcare). The Percoll cell suspension was overlaid with 1 ml of 10% FBS in DMEM and spun at 800g for 15 min, and pellet was collected in a new 15 ml tube and resuspended in 0.5 ml of FACS buffer ((Hibernate-E low fluorescence (BrainBits) with 0.2% BSA and 20 μg/ml DNase (Worthington)). Red blood cells (RBCs) were lysed by incubating cells with RBC lysis buffer (BioLegend) for 15 min at room temperature; cells were washed and resuspended in FACS buffer. The final cell suspension was submitted for single cell sequencing with the 10X Genomics system at the Mount Sinai Genomics core facility (using ~10,000 viable cells from the sample).
Bioinformatic analysis
Clustering analysis of scRNA-seq data was performed with the Seurat software package on the R platform72. We calculated signature score for GBM cell state with the scrabble software package (jlaffy.github.io/scrabble/), using the gene list of the MES2 signature29.
Gene set enrichment analysis (GSEA) of gene lists ranked by expression changes was performed with the GSEA platform39,73. We used the Enrichr website for the pathway analysis for differentially expressed genes (cut-off: P < 0.05; fold-change > 2-fold)38.
The GBM biodiscovery portal (http://gbm-biodp.nci.nih.gov; accessed 02/2021) was used to match UnaG+ gene expression signature with patient survival74. cBioPortal (https://www.cbioportal.org; accessed 02/2021) was used to match occurrence in recurrent vs. primary GBM (TCGA-GBM Firehose legacy dataset, mRNA expression (mRNA expression z-scores relative to all samples (log RNA Seq V2 RSEM))75,76. Pseudotime analysis was performed with Monocle 3 software package on R platform77. The Ivy GAP database, containing expression data from 122 micro-dissected anatomical domains of 10 GBM patients, was used for GBM anatomical transcriptional analysis (glioblastoma.alleninstitute.org)42.
Raw fastq files were aligned to mouse genome reference mm10 using CellRanger v5.0.0 (10X Genomics). CellRanger-filtered count matrix was processed and analyzed using R package Seurat v4.0.5. Normalization was performed using NormalizeData function with normalization.method = ‘LogNormalize’. Dimensionality reduction was computed on the top 2,000 variable features using FindVariableFeatures, ScaleData and RunPCA functions. UMAPs were generated using the top 15 PCs. Identification of modules of co-expressed genes was carried out using Weighted Gene Co-expression Network Analysis (WGCNA) using the R package hdWGCNA v0.1.1.9006 (https://github.com/smorabit/hdWGCNA). Metacells of 25 cells (k=25, overlap=10) were generated with MetacellsByGroups function following by identification of co-expression modules using ConstructNetwork function with following parameters: softPower=6, deepSplit=1, mergeCutHeight = 0.25. Only the top 10,000 variable genes were considered. Enrichment analysis on the WGCNA modules was carried out with clusterProfiler v.4.2.0 using the GSEA MSigDB HALLMARK gene set annotation for mouse genes (downloaded at https://bioinf.wehi.edu.au/MSigDB/ on 09/08/2022). Cell-cell interaction analysis was carried out with CellPhoneDB v478.
A 10X Visium spatial transcriptomic dataset of 17 GBM patient samples51 was downloaded from Datadryad (https://datadryad.org/stash/dataset/doi:10.5061/dryad.h70rxwdmj) and analyzed using the Seurat R package. All GBM patient samples were scored for Hallmark Hypoxia gene signature. Patient samples UKF265, UKF255 and UKF248 were selected for our analyses as they showed the most hypoxic burden of all samples. Spots corresponding to hypoxic zones were selected from UMAPs and presence of immune cells in spatially correlated spots was deduced by analyzing expression of marker genes.
Pimonidazole staining
Mice bearing intracranial GL261 tumors were intraperitoneally injected with 60 mg/kg pimonidazole (Hydroxyprobe), which was diluted in 0.9% saline. To stain for pimonidazole labeled cells, brain cryosections were blocked overnight and incubated the with mouse Dylight-549 anti-pimonidazole antibody (clone 4.3.11.3; Hydroxyprobe). Sections were mounted with Fluoromount-G (SouthernBiotech) and images were acquired by fluorescence microscopy with a Zeiss Axio microscope.
Irradiation and evofosfamide treatment
Mice bearing GBM were randomly divided into four cohorts (Control, XRT only, Evo only, XRT+Evo). Treatments were administered two weeks after intracranial transplantations to allow for tumor establishment. For XRT, mice were irradiated in a X-Rad 320 irradiator (Precision X-Ray), twice every week with a 2.5 Gy dose for two weeks (5 Gy total dose/week). A lead shield was placed over the body of the mice to only expose heads to radiation. Mice in the Evo cohort were administered with 50 mg/kg i.p. injection of Evo (AdooQ Bioscience) every day for two weeks. Mice in the XRT+Evo treatment cohort were injected daily with Evo (50 mg/kg) for two weeks and were concurrently treated with 2.5 Gy radiation twice every week. Mice in all cohorts were weighed daily and monitored for ambulatory, feeding, and grooming activities, and animals meeting humane endpoints were euthanized.
qRT-PCR analysis of bone marrow derived macrophages treated with tumor conditioned media
The isolation of bone marrow-derived macrophages followed previously described protocols79. In brief, mice were sacrificed by cervical dislocation and bone marrow was extracted from tibia and femur by flushing out with DMEM media containing 10% FBS with a 25-gauge needle attached to a 10 ml syringe. The cell suspension was passed through a 70 μm strainer and centrifuged at 300 g for 5 min at room temperature. The cell pellet was resuspended with DMEM media containing 10% FBS and MCSF (25 ng/ml; Peprotech). The media was changed every two days for 7 days to obtain pure differentiated BMDMs.
To produce conditioned media, 300,000 cells of murine GBM cell lines GL26170 were plated in 10 cm culture dish in DMEM media containing 10% FBS. After one day, media was changed to fresh media and the cells were incubated for another 2 days. To obtain media supernatant conditioned by hypoxic GBM cells, dishes were placed in a 1% oxygen hypoxic chamber (Biospherix). The conditioned media was collected after 2 days and the supernatant was centrifuged at 300 g for 5 min to remove cell debris.
BMDM cultures were treated with supernatant for 48 hours. For qRT-PCR analysis, RNA of BMDMs was extracted using the RNeasy mini kit (QIAGEN) and cDNA was synthesized using SuperScript III First Strand Synthesis System (Invitrogen). Quantitative PCR was performed using PerfeCTa SYBR Green FastMix Rox (Quanta Biosciences) in the ABI 7900HT qPCR system (Applied Biosystems). Primers are listed in the Key Resources Table. Gapdh was used as the housekeeping gene for normalization.
ELISA of bone marrow derived macrophages treated with tumor conditioned media
ELISA was performed using the Mouse IL-1 beta/IL-1F2 DuoSet ELISA kit (R&D Systems DY401-05), using additional reagents from the DuoSet ELISA ancillary reagent kit 2 (R&D Systems DY008). Briefly, a 96-well microplate was coated with IL-1β capture antibody, then 100 μl of samples or standards was added to each well, followed by streptavidin-HRP, and color reaction. The plate was then immediately read at 450 nm using a microplate reader. Microsoft Excel and GraphPad Prism 9 software were then used to generate a standard curve to calculate values for samples.
QUANTIFICATION AND STATISTICAL ANALYSIS
Image quantifications
Image data were quantified with Fiji Is Just ImageJ (FIJI) package of Image J80. To quantify abundance of marker or UnaG reporter expression, in each selection of region of interest (ROI), percentage of ROI area positive for fluorescent or immunosignals was automatically counted after applying a threshold mask followed by morphological segmentation using MorphoLib J Plugin81. For vessel density, in each selected region of interest (ROI), PECAM1+ vessel sections were manually counted, and numbers were normalized to area size of ROI. To quantify signal abundance per unit area, images were spatially calibrated and total number of signal positive areas were divided by area of ROI. The ‘plot profile’ function of Image J was used to generate profiles of relative immunofluorescence in a chosen ROI (this function sums up the pixel values of vertical lines in a ROI and plots them as profile).
Statistical analysis
Statistical analyses were performed using GraphPad Prism 9 software. Bar graphs represent means and error bars represent SEM. One-way ANOVA with Tukey’s post-hoc correction (for three or more experimental groups) and t test were performed to assess if experimental groups were significantly different from each other. P < 0.05 was considered to be statistically significant (*). P < 0.01, **; P < 0.001, ***.
Supplementary Material
Table S1. Patient information for human glioma samples, related to Figure 3, Figure 6 and Figure S7. A) Patient information for glioma tissue microarray. B) Patient information for fresh frozen and subtype classified GBM samples. C) Patient information for samples analyzed by 10X Visium (Ravi et al., Cancer Cell 2022).
Table S2. Top DEGs and ranked gene expression changes in UnaG+ vs. UnaG− GL261 GBM cells, related to Figure S2. A) Top DEGs (up- and down-regulated) in UnaG+ vs. UnaG− GL261 GBM cells. B) Ranked gene expression changes in UnaG+ vs. UnaG− GL261 GBM cells.
Table S3. List of genes of the GO terms Positive regulation/Negative regulation of Immune System Process, related to Figure S2.
Table S4. Comparison of Hallmark hypoxia gene set and UnaG+ gene set, related to Figure S3.
Table S5. DEGs of subcluster 5 (UnaG+) GL261 GBM cells related to Figure 5 and Figure S3. A) DEGs in subcluster 5 (UnaG+) GL261 GBM cells, cut-off: log2FC >0.25, and P < 0.05. B) Genes listed in Ivy GAP database that match with UnaG+ GBM gene signature.
Table S6. Top upregulated genes in each of the TAM subclusters and DEGs in hypoxic TAMs and CTLs, related to Figure 5, Figure 6 and Figure S4. A) Top upregulated genes in each of the TAM subclusters. B) DEGs in hypoxic TAMs (vs. all other TAMs), cut-off: log2FC > 0.25, P<0.05. C) DEGs in hypoxic CTLs (vs. all other CTLs), cut-off: log2FC > 0.25, P<0.05.
Highlights.
Host immune status influences tumor vasculature and hypoxic zone formation in GBM
Spatial patterning of TAM and CTL parallels hypoxic zone maturation to pseudopalisades
Sequestered TAM and CTL in hypoxic zones are reprogrammed towards immunosuppression
TAM/CTL organization involves CCL8 and IL-1B as niche factors in hypoxic zones
ACKNOWLEDGEMENTS
We thank Friedemann Kiefer (Max-Planck Institute for Molecular Biomedicine, Münster, Germany) for sharing HRE-UnaG plasmids, Kristin Beaumont (Icahn School of Medicine at Mount Sinai) for advising on single-cell RNA sequencing experiments, and Sanjana Shroff (Mount Sinai Genomics Core Facility) for performing quality control and library preparations for single-cell RNA sequencing. We also thank Raymund Yong for providing patient GBM specimens (Mount Sinai Biorepository and Pathology Core) and Ruben Fernandez-Rodriguez (Histology Core Lab) for help with IHC of patient specimens; and Glenn Doherty and Nikolaos Tzavaras (Mount Sinai Microscopy Core Facility) for assisting in imaging. We thank Dalia Halawani and all members of Zou and Friedel laboratories for their valuable suggestions. Graphical abstract was created with BioRender.com. This work was supported by the National Institutes of Health/National Institute of Neurological Disorders and Stroke grants R01NS107462 (to HZ and RHF), R21NS125700 (to HZ and RHF), R01NS092735 (to RHF), F31NS124259 (to VM), and R01NS100864 (to DH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- 1.Pombo Antunes AR, Scheyltjens I, Duerinck J, Neyns B, Movahedi K, and Van Ginderachter JA (2020). Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. eLife 9. 10.7554/eLife.52176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Touat M, Li YY, Boynton AN, Spurr LF, Iorgulescu JB, Bohrson CL, Cortes-Ciriano I, Birzu C, Geduldig JE, Pelton K, et al. (2020). Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 580, 517–523. 10.1038/s41586-020-2209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brahm CG, van Linde ME, Enting RH, Schuur M, Otten RHJ, Heymans MW, Verheul HMW, and Walenkamp AME (2020). The Current Status of Immune Checkpoint Inhibitors in Neuro-Oncology: A Systematic Review. Cancers (Basel) 12. 10.3390/cancers12030586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wei J, Chen P, Gupta P, Ott M, Zamler D, Kassab C, Bhat KP, Curran MA, de Groot JF, and Heimberger AB (2020). Immune biology of glioma-associated macrophages and microglia: functional and therapeutic implications. Neuro Oncol 22, 180–194. 10.1093/neuonc/noz212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klemm F, Maas RR, Bowman RL, Kornete M, Soukup K, Nassiri S, Brouland JP, Iacobuzio-Donahue CA, Brennan C, Tabar V, et al. (2020). Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 181, 1643–1660.e1617. 10.1016/j.cell.2020.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andersen BM, Faust Akl C, Wheeler MA, Chiocca EA, Reardon DA, and Quintana FJ (2021). Glial and myeloid heterogeneity in the brain tumour microenvironment. Nat Rev Cancer 21, 786–802. 10.1038/s41568-021-00397-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, et al. (2013). CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 19, 1264–1272. 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akkari L, Bowman RL, Tessier J, Klemm F, Handgraaf SM, de Groot M, Quail DF, Tillard L, Gadiot J, Huse JT, et al. (2020). Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF-1R inhibition as a strategy to overcome resistance. Science Translational Medicine 12, eaaw7843. 10.1126/scitranslmed.aaw7843. [DOI] [PubMed] [Google Scholar]
- 9.Butowski N, Colman H, De Groot JF, Omuro AM, Nayak L, Wen PY, Cloughesy TF, Marimuthu A, Haidar S, Perry A, et al. (2016). Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro Oncol 18, 557–564. 10.1093/neuonc/nov245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bejarano L, Jordāo MJC, and Joyce JA (2021). Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov 11, 933–959. 10.1158/2159-8290.CD-20-1808. [DOI] [PubMed] [Google Scholar]
- 11.Varia MA, Calkins-Adams DP, Rinker LH, Kennedy AS, Novotny DB, Fowler WC, and Raleigh JA (1998). Pimonidazole: a novel hypoxia marker for complementary study of tumor hypoxia and cell proliferation in cervical carcinoma. Gynecol Oncol 71, 270–277. 10.1006/gyno.1998.5163. [DOI] [PubMed] [Google Scholar]
- 12.Koch CJ, Evans SM, and Lord EM (1995). Oxygen dependence of cellular uptake of EF5 [2-(2-nitro-1H-imidazol-1-yl)-N-(2,2,3,3,3-pentafluoropropyl)a cet amide] : analysis of drug adducts by fluorescent antibodies vs bound radioactivity. Br J Cancer 72, 869–874. 10.1038/bjc.1995.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stokes AM, Hart CP, and Quarles CC (2016). Hypoxia Imaging With PET Correlates With Antitumor Activity of the Hypoxia-Activated Prodrug Evofosfamide (TH-302) in Rodent Glioma Models. Tomography (Ann Arbor, Mich.) 2, 229–237. 10.18383/j.tom.2016.00259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verhoeven J, Bolcaen J, De Meulenaere V, Kersemans K, Descamps B, Donche S, Van den Broecke C, Boterberg T, Kalala JP, Deblaere K, et al. (2019). Technical feasibility of [Radiat Oncol 14, 89. 10.1186/s13014-019-1290-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weidemann A, and Johnson RS (2008). Biology of HIF-1alpha. Cell Death Differ 15, 621–627. 10.1038/cdd.2008.12. [DOI] [PubMed] [Google Scholar]
- 16.Kumagai A, Ando R, Miyatake H, Greimel P, Kobayashi T, Hirabayashi Y, Shimogori T, and Miyawaki A (2013). A bilirubin-inducible fluorescent protein from eel muscle. Cell 153, 1602–1611. 10.1016/j.cell.2013.05.038. [DOI] [PubMed] [Google Scholar]
- 17.Erapaneedi R, Belousov VV, Schäfers M, and Kiefer F (2016). A novel family of fluorescent hypoxia sensors reveal strong heterogeneity in tumor hypoxia at the cellular level. EMBO J 35, 102–113. 10.15252/embj.201592775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, Dhara S, Simpson K, Gardner EE, Iacobuzio-Donahue CA, et al. (2016). Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep 17, 2445–2459. 10.1016/j.celrep.2016.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harris AL (2002). Hypoxia--a key regulatory factor in tumour growth. Nat Rev Cancer 2, 38–47. 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 20.Schmitz C, Pepelanova I, Seliktar D, Potekhina E, Belousov VV, Scheper T, and Lavrentieva A (2020). Live reporting for hypoxia: Hypoxia sensor-modified mesenchymal stem cells as in vitro reporters. Biotechnol Bioeng 117, 3265–3276. 10.1002/bit.27503. [DOI] [PubMed] [Google Scholar]
- 21.Young RJ, and Möller A (2010). Immunohistochemical detection of tumour hypoxia. Methods Mol Biol 611, 151–159. 10.1007/978-1-60327-345-9_12. [DOI] [PubMed] [Google Scholar]
- 22.Herting CJ, Chen Z, Maximov V, Duffy A, Szulzewsky F, Shayakhmetov DM, and Hambardzumyan D (2019). Tumour-associated macrophage-derived interleukin-1 mediates glioblastoma-associated cerebral oedema. Brain 142, 3834–3851. 10.1093/brain/awz331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seyfried TN, el-Abbadi M, and Roy ML (1992). Ganglioside distribution in murine neural tumors. Mol Chem Neuropathol 17, 147–167. 10.1007/BF03159989. [DOI] [PubMed] [Google Scholar]
- 24.Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, deCarvalho AC, Lyu S, Li P, Li Y, et al. (2017). Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 32, 42–56.e46. 10.1016/j.ccell.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. (2010). Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110. S1535–6108(09)00432–2 [pii] 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaffes I, Szulzewsky F, Chen Z, Herting CJ, Gabanic B, Velazquez Vega JE, Shelton J, Switchenko JM, Ross JL, McSwain LF, et al. (2019). Human Mesenchymal glioblastomas are characterized by an increased immune cell presence compared to Proneural and Classical tumors. Oncoimmunology 8, e1655360. 10.1080/2162402X.2019.1655360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brenner A, Zuniga R, Sun JD, Floyd J, Hart CP, Kroll S, Fichtel L, Cavazos D, Caflisch L, Gruslova A, et al. (2018). Hypoxia-activated evofosfamide for treatment of recurrent bevacizumab-refractory glioblastoma: a phase I surgical study. Neuro Oncol 20, 1231–1239. 10.1093/neuonc/noy015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barker HE, Paget JTE, Khan AA, and Harrington KJ (2015). The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nature Reviews Cancer 15, 409–425. 10.1038/nrc3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, Richman AR, Silverbush D, Shaw ML, Hebert CM, et al. (2019). An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 178, 835–849.e821. 10.1016/j.cell.2019.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tejero R, Huang Y, Katsyv I, Kluge M, Lin JY, Tome-Garcia J, Daviaud N, Wang Y, Zhang B, Tsankova NM, et al. (2019). Gene signatures of quiescent glioblastoma cells reveal mesenchymal shift and interactions with niche microenvironment. EBioMedicine 42, 252–269. 10.1016/j.ebiom.2019.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mittelbronn M, Platten M, Zeiner P, Dombrowski Y, Frank B, Zachskorn C, Harter PN, Weller M, and Wischhusen J (2011). Macrophage migration inhibitory factor (MIF) expression in human malignant gliomas contributes to immune escape and tumour progression. Acta Neuropathologica 122, 353. 10.1007/s00401-011-0858-3. [DOI] [PubMed] [Google Scholar]
- 32.Bach JP, Deuster O, Balzer-Geldsetzer M, Meyer B, Dodel R, and Bacher M (2009). The role of macrophage inhibitory factor in tumorigenesis and central nervous system tumors. Cancer 115, 2031–2040. 10.1002/cncr.24245. [DOI] [PubMed] [Google Scholar]
- 33.Kouo T, Huang L, Pucsek AB, Cao M, Solt S, Armstrong T, and Jaffee E (2015). Galectin-3 Shapes Antitumor Immune Responses by Suppressing CD8+ T Cells via LAG-3 and Inhibiting Expansion of Plasmacytoid Dendritic Cells. Cancer Immunology Research 3, 412–423. 10.1158/2326-6066.Cir-14-0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dumic J, Dabelic S, and Flögel M (2006). Galectin-3: An open-ended story. Biochimica et Biophysica Acta (BBA) - General Subjects 1760, 616–635. 10.1016/j.bbagen.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 35.Mitchell RA, Liao H, Chesney J, Fingerle-Rowson G, Baugh J, David J, and Bucala R (2002). Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: regulatory role in the innate immune response. Proc Natl Acad Sci U S A 99, 345–350. 10.1073/pnas.012511599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stojanovic I, Cvjeticanin T, Lazaroski S, Stosic-Grujicic S, and Miljkovic D (2009). Macrophage migration inhibitory factor stimulates interleukin-17 expression and production in lymph node cells. Immunology 126, 74–83. 10.1111/j.1365-2567.2008.02879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Galvao I, Dias AC, Tavares LD, Rodrigues IP, Queiroz-Junior CM, Costa VV, Reis AC, Ribeiro Oliveira RD, Louzada-Junior P, Souza DG, et al. (2016). Macrophage migration inhibitory factor drives neutrophil accumulation by facilitating IL-1beta production in a murine model of acute gout. J Leukoc Biol 99, 1035–1043. 10.1189/jlb.3MA0915-418R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, Lachmann A, Wojciechowicz ML, Kropiwnicki E, Jagodnik KM, et al. (2021). Gene Set Knowledge Discovery with Enrichr. Curr Protoc 1, e90. 10.1002/cpz1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krämer A, Green J, Pollard J, and Tugendreich S (2014). Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530. 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Puchalski RB, Shah N, Miller J, Dalley R, Nomura SR, Yoon JG, Smith KA, Lankerovich M, Bertagnolli D, Bickley K, et al. (2018). An anatomic transcriptional atlas of human glioblastoma. Science 360, 660–663. 10.1126/science.aaf2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oeckinghaus A, and Ghosh S (2009). The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol 1, a000034. 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lawrence T (2009). The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol 1, a001651. 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zeisel A, Hochgerner H, Lönnerberg P, Johnsson A, Memic F, van der Zwan J, Häring M, Braun E, Borm LE, La Manno G, et al. (2018). Molecular Architecture of the Mouse Nervous System. Cell 174, 999–1014.e1022. 10.1016/j.cell.2018.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Faraco G, Park L, Anrather J, and Iadecola C (2017). Brain perivascular macrophages: characterization and functional roles in health and disease. J Mol Med (Berl) 95, 1143–1152. 10.1007/s00109-017-1573-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strauss L, Mahmoud MAA, Weaver JD, Tijaro-Ovalle NM, Christofides A, Wang Q, Pal R, Yuan M, Asara J, Patsoukis N, and Boussiotis VA (2020). Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci Immunol 5. 10.1126/sciimmunol.aay1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Broekman ML, Maas SLN, Abels ER, Mempel TR, Krichevsky AM, and Breakefield XO (2018). Multidimensional communication in the microenvirons of glioblastoma. Nat Rev Neurol 14, 482–495. 10.1038/s41582-018-0025-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Watson MJ, Vignali PDA, Mullett SJ, Overacre-Delgoffe AE, Peralta RM, Grebinoski S, Menk AV, Rittenhouse NL, DePeaux K, Whetstone RD, et al. (2021). Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature 591, 645–651. 10.1038/s41586-020-03045-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garcia-Alonso L, Lorenzi V, Mazzeo CI, Alves-Lopes JP, Roberts K, Sancho-Serra C, Engelbert J, Mareckova M, Gruhn WH, Botting RA, et al. (2022). Single-cell roadmap of human gonadal development. Nature 607, 540–547. 10.1038/s41586-022-04918-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ravi VM, Will P, Kueckelhaus J, Sun N, Joseph K, Salie H, Vollmer L, Kuliesiute U, von Ehr J, Benotmane JK, et al. (2022). Spatially resolved multi-omics deciphers bidirectional tumor-host interdependence in glioblastoma. Cancer Cell 40, 639–655 e613. 10.1016/j.ccell.2022.05.009. [DOI] [PubMed] [Google Scholar]
- 52.Gabay C, Lamacchia C, and Palmer G (2010). IL-1 pathways in inflammation and human diseases. Nat Rev Rheumatol 6, 232–241. 10.1038/nrrheum.2010.4. [DOI] [PubMed] [Google Scholar]
- 53.Tsou C-L, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M, and Charo IF (2007). Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. The Journal of Clinical Investigation 117, 902–909. 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barsoum IB, Koti M, Siemens DR, and Graham CH (2014). Mechanisms of hypoxia-mediated immune escape in cancer. Cancer Res 74, 7185–7190. 10.1158/0008-5472.CAN-14-2598. [DOI] [PubMed] [Google Scholar]
- 55.Bhat KPL, Balasubramaniyan V, Vaillant B, Ezhilarasan R, Hummelink K, Hollingsworth F, Wani K, Heathcock L, James JD, Goodman LD, et al. (2013). Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell 24, 331–346. 10.1016/j.ccr.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hara T, Chanoch-Myers R, Mathewson ND, Myskiw C, Atta L, Bussema L, Eichhorn SW, Greenwald AC, Kinker GS, Rodman C, et al. (2021). Interactions between cancer cells and immune cells drive transitions to mesenchymal-like states in glioblastoma. Cancer Cell 39, 779–792.e711. 10.1016/j.ccell.2021.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brenner AJ, Floyd J, Fichtel L, Michalek J, Kanakia KP, Huang S, Reardon D, Wen PY, and Lee EQ (2021). Phase 2 trial of hypoxia activated evofosfamide (TH302) for treatment of recurrent bevacizumab-refractory glioblastoma. Sci Rep 11, 2306. 10.1038/s41598-021-81841-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Naderi A, Farmaki E, Chavez B, Cai C, Kaza V, Zhang Y, Soltanmohammadi E, Daneshvar N, Chatzistamou I, and Kiaris H (2022). Beneficial effects of CCL8 inhibition at lipopolysaccharide-induced lung injury. iScience 25, 105520. 10.1016/j.isci.2022.105520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Honda F, Tsuboi H, Ono Y, Abe S, Takahashi H, Ito K, Yamada K, Kawano M, Kondo Y, Asano K, et al. (2021). Pathogenic roles and therapeutic potential of the CCL8-CCR8 axis in a murine model of IgG4-related sialadenitis. Arthritis Res Ther 23, 214. 10.1186/s13075-021-02597-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dangi A, Husain I, Jordan CZ, Yu S, Natesh N, Shen X, Kwun J, and Luo X (2022). Blocking CCL8-CCR8-Mediated Early Allograft Inflammation Improves Kidney Transplant Function. J Am Soc Nephrol 33, 1876–1890. 10.1681/ASN.2022020139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cavelti-Weder C, Babians-Brunner A, Keller C, Stahel MA, Kurz-Levin M, Zayed H, Solinger AM, Mandrup-Poulsen T, Dinarello CA, and Donath MY (2012). Effects of gevokizumab on glycemia and inflammatory markers in type 2 diabetes. Diabetes Care 35, 1654–1662. 10.2337/dc11-2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Howard C, Noe A, Skerjanec A, Holzhauer B, Wernsing M, Ligueros-Saylan M, and Thuren T (2014). Safety and tolerability of canakinumab, an IL-1beta inhibitor, in type 2 diabetes mellitus patients: a pooled analysis of three randomised double-blind studies. Cardiovasc Diabetol 13, 94. 10.1186/1475-2840-13-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mahalingam D, Patel MR, Sachdev JC, Hart LL, Halama N, Ramanathan RK, Sarantopoulos J, Volkel D, Youssef A, de Jong FA, and Tsimberidou AM (2020). Phase I study of imalumab (BAX69), a fully human recombinant antioxidized macrophage migration inhibitory factor antibody in advanced solid tumours. Br J Clin Pharmacol 86, 1836–1848. 10.1111/bcp.14289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rajasekaran D, Zierow S, Syed M, Bucala R, Bhandari V, and Lolis EJ (2014). Targeting distinct tautomerase sites of D-DT and MIF with a single molecule for inhibition of neutrophil lung recruitment. FASEB J 28, 4961–4971. 10.1096/fj.14-256636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stein R, Qu Z, Cardillo TM, Chen S, Rosario A, Horak ID, Hansen HJ, and Goldenberg DM (2004). Antiproliferative activity of a humanized anti-CD74 monoclonal antibody, hLL1, on B-cell malignancies. Blood 104, 3705–3711. 10.1182/blood-2004-03-0890. [DOI] [PubMed] [Google Scholar]
- 66.Bayik D, and Lathia JD (2021). Cancer stem cell-immune cell crosstalk in tumour progression. Nat Rev Cancer 21, 526–536. 10.1038/s41568-021-00366-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Horai R, Asano M, Sudo K, Kanuka H, Suzuki M, Nishihara M, Takahashi M, and Iwakura Y (1998). Production of mice deficient in genes for interleukin (IL)-1alpha, IL-1beta, IL-1alpha/beta, and IL-1 receptor antagonist shows that IL-1beta is crucial in turpentine-induced fever development and glucocorticoid secretion. J Exp Med 187, 1463–1475. 10.1084/jem.187.9.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Newcomb EW, and Zagzag D (2009). The Murine GL261 Glioma Experimental Model to Assess Novel Brain Tumor Treatments. In CNS Cancer, (Humana Press; ), pp. 227–241. 10.1007/978-1-60327-553-8_12. [DOI] [Google Scholar]
- 69.Oh T, Fakurnejad S, Sayegh ET, Clark AJ, Ivan ME, Sun MZ, Safaee M, Bloch O, James CD, and Parsa AT (2014). Immunocompetent murine models for the study of glioblastoma immunotherapy. Journal of Translational Medicine 12, 107. 10.1186/1479-5876-12-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Szatmári T, Lumniczky K, Désaknai S, Trajcevski S, Hídvégi EJ, Hamada H, and Sáfrány G (2006). Detailed characterization of the mouse glioma 261 tumor model for experimental glioblastoma therapy. Cancer Sci 97, 546–553. 10.1111/j.1349-7006.2006.00208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, and Kaufman PD (2009). A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One 4, e6529. 10.1371/journal.pone.0006529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e1821. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34, 267–273. 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 74.Celiku O, Johnson S, Zhao S, Camphausen K, and Shankavaram U (2014). Visualizing molecular profiles of glioblastoma with GBM-BioDP. PLoS One 9, e101239. 10.1371/journal.pone.0101239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1. 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404. 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, Zhang F, Mundlos S, Christiansen L, Steemers FJ, et al. (2019). The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502. 10.1038/s41586-019-0969-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Efremova M, Vento-Tormo M, Teichmann SA, and Vento-Tormo R (2020). CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat Protoc 15, 1484–1506. 10.1038/s41596-020-0292-x. [DOI] [PubMed] [Google Scholar]
- 79.Bailey JD, Shaw A, McNeill E, Nicol T, Diotallevi M, Chuaiphichai S, Patel J, Hale A, Channon KM, and Crabtree MJ (2020). Isolation and culture of murine bone marrow-derived macrophages for nitric oxide and redox biology. Nitric Oxide 100–101, 17–29. 10.1016/j.niox.2020.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Legland D, Arganda-Carreras I, and Andrey P (2016). MorphoLibJ: integrated library and plugins for mathematical morphology with ImageJ. Bioinformatics 32, 3532–3534. 10.1093/bioinformatics/btw413. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Patient information for human glioma samples, related to Figure 3, Figure 6 and Figure S7. A) Patient information for glioma tissue microarray. B) Patient information for fresh frozen and subtype classified GBM samples. C) Patient information for samples analyzed by 10X Visium (Ravi et al., Cancer Cell 2022).
Table S2. Top DEGs and ranked gene expression changes in UnaG+ vs. UnaG− GL261 GBM cells, related to Figure S2. A) Top DEGs (up- and down-regulated) in UnaG+ vs. UnaG− GL261 GBM cells. B) Ranked gene expression changes in UnaG+ vs. UnaG− GL261 GBM cells.
Table S3. List of genes of the GO terms Positive regulation/Negative regulation of Immune System Process, related to Figure S2.
Table S4. Comparison of Hallmark hypoxia gene set and UnaG+ gene set, related to Figure S3.
Table S5. DEGs of subcluster 5 (UnaG+) GL261 GBM cells related to Figure 5 and Figure S3. A) DEGs in subcluster 5 (UnaG+) GL261 GBM cells, cut-off: log2FC >0.25, and P < 0.05. B) Genes listed in Ivy GAP database that match with UnaG+ GBM gene signature.
Table S6. Top upregulated genes in each of the TAM subclusters and DEGs in hypoxic TAMs and CTLs, related to Figure 5, Figure 6 and Figure S4. A) Top upregulated genes in each of the TAM subclusters. B) DEGs in hypoxic TAMs (vs. all other TAMs), cut-off: log2FC > 0.25, P<0.05. C) DEGs in hypoxic CTLs (vs. all other CTLs), cut-off: log2FC > 0.25, P<0.05.
Data Availability Statement
scRNA-seq data has been deposited in NCBI Gene Expression Omnibus database and is publicly available from the date of publication. Accession number is listed in the Key Resources Table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Arg1 (M-20) | Santa Cruz Biotechnology | Cat#sc-18355; RRID: AB_2058957 |
| Anti-mouse CD68 (FA-11) | Bio-Rad | Cat# MCA1957GA; RRID: AB_324217 |
| Anti-CD8a (53–6.7) | Thermo Fisher Scientific | Cat# 14-0081-82; RRID: AB_467087 |
| Anti-mouse MMR/ CD206 | R and D Systems | Cat# AF2535; RRID: AB_2063012 |
| Anti-mouse CD16/32 (2.4G2) | BD Biosciences | Cat# 553142; RRID: AB_394657 |
| Anti-FOXP3 (FJK-16s) | Thermo Fisher Scientific | Cat# 14-5773-82; RRID: AB_467576 |
| Anti-GLUT1 (SA0377) | Thermo Fisher Scientific | Cat# MA5-31960; RRID: AB_2809254 |
| Anti-Iba1 | FUJIFILM Wako Shibayagi | Cat# 019-19741; RRID: AB_839504 |
| Anti-mouse CD49d / integrin alpha4 (9C10 (MFR4.B)) | BioLegend | Cat# 103701; RRID: AB_313042 |
| Anti-Ki67 | Abcam | Cat# ab15580; RRID: AB_443209 |
| Anti-cMyc tag | Novus | Cat# NB600-335; RRID: AB_10002720 |
| Anti-CD31/ PECAM1 | Abcam | Cat# ab28364; RRID: AB_726362 |
| Anti-PU.1 (E.388.3) | Thermo Fisher Scientific | Cat# MA5-15064; RRID: AB_10986949 |
| Anti-Pimonidazole Dylight 594 (4.3.11.3) | Hydroxyprobe | Cat# HP7-100Kit |
| Bacterial and virus strains | ||
| Stbl3 E. coli | Thermo Fisher Scientific | C737303 |
| Biological samples | ||
| Primary bone marrow derived macrophages (mouse) | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Pimonidazole | Hydroxyprobe | Cat# HP-100mg |
| Evofosfamide/ TH-302 | AdooQ Bioscience | Cat# A11144 |
| Recombinant murine M-CSF | Peprotech | Cat# 315-02 |
| Percoll | GE Healthcare | Cat# 17089101 |
| BrainBits Hibernate E Low Fluorescence | Fisher Scientific | Cat# NC0285514 |
| DNAse I | Worthington | Cat# LS006344 |
| RBC lysis buffer | BioLegend | Cat# 420301 |
| Critical commercial assays | ||
| Mouse IL-1 beta/IL-1F2 DuoSet ELISA | R and D Systems | Cat# DY401 |
| DuoSet ELISA Ancillary Reagent Kit 2 | R and D Systems | Cat# DY008 |
| RNeasy mini kit | Qiagen | Cat# 74104 |
| SuperScript III First Strand Synthesis System | Thermo Fisher Scientific | Cat# 18080051 |
| Neural Tissue Dissociation Kit (P) | Miltenyi Biotec | Cat# 130-092-628 |
| Deposited data | ||
| Analyzed scRNA-seq data | This paper | Gene Expression Omnibus, GEO: GSE179077 |
| Experimental models: Cell lines | ||
| GL261 (murine GBM cell line) | National Cancer Institute | NCI-DTP Cat# Glioma 261; RRID: CVCL_Y003 |
| GL261-HRE-UnaG | This paper | N/A |
| CT2A | Millipore | Millipore Cat# SCC194, RRID: CVCL_ZJ44 |
| HEK293T | ATCC | CRL-3216; RRID: CVCL_0063 |
| Experimental models: Organisms/strains | ||
| C57BL/6J | The Jackson Laboratory | Strain# 000664; RRID: IMSR_JAX:000664 |
| C57BL/6J SCID; B6.Cg-Prkdcscid/SzJ | The Jackson Laboratory | Strain# 001913; RRID: IMSR_JAX:001913 |
| ICR-SCID; IcrTac:ICR-Prkdcscid | Taconic Biosciences | Model# ICRSC-M; RRID: IMSR_TAC:icrsc |
| N-tva-Ccl8/12 KO | Sabina Islam | N/A |
| N-tva-Il1b KO | Dmitry Shayakhmetov | N/A |
| Oligonucleotides | ||
| Mouse Arg-1 forward: 5’-GTGGCTTTAACCTTGGCTTG-3’ | This paper | N/A |
| Mouse Arg-1 reverse: 5’-CTGTCTGCTTTGCTGTGATG-3’ | This paper | N/A |
| Mouse Ccl8 forward: 5’-ACAATATCCAGTGCCCCATG-3’ | This paper | N/A |
| Mouse Ccl8 reverse: 5’-TGAAGGTTCAAGGCTGCAG-3’ | This paper | N/A |
| Mouse CD68 forward: 5’-ACTTCGGGCCATGTTTCTCT-3’ | This paper | N/A |
| Mouse CD68 reverse: 5’-GCTGGTAGGTTGATTGTCGT-3’ | This paper | N/A |
| Mouse Il1b forward: 5’-CCAAGCAACGACAAAATACC-3’ | This paper | N/A |
| Mouse Il1b reverse: 5’-GTTGAAGACAAACCGTTTTTCC-3’ | This paper | N/A |
| Mouse Gapdh forward: 5’-ACTGCCACCCAGAAGACTGT-3’ | This paper | N/A |
| Mouse Gapdh reverse: 5’-GATGCAGGGATGATGTTCT-3’ | This paper | N/A |
| Recombinant DNA | ||
| pcDNA-dUnaG | Dr. Friedemann Kiefer | N/A |
| pENTR/D-TOPO | Thermo Fisher Scientific | Cat#: K240020 |
| pLenti X1 Puro DEST | Addgene | Plasmid# 17297; RRID: Addgene_17297 |
| pMD2.G | Addgene | Plasmid# 12259; RRID: Addgene_12259 |
| psPAX2 | Addgene | Plasmid# 12260; RRID: Addgene_12260 |
| pLenti-HRE-dUnaG | This paper | Addgene #124372 |
| Software and algorithms | ||
| Microsoft Office | Microsoft | RRID: SCR_016137 |
| Adobe Photoshop | Adobe | RRID: SCR_014199 |
| Fiji Is Just ImageJ (FIJI) | Public domain, National Institutes of Health | https://imagej.net/software/fiji/; RRID: SCR_003070 |
| Prism 9 | GraphPad | RRID: SCR_002798 |
| SnapGene | Dotmatics | RRID: SCR_015052 |
| Zen Blue | Zeiss | RRID: SCR_013672 |
| Cell Ranger | 10x Genomics | RRID: SCR_017344 |
| Seurat R package | Rahul Satija Lab | https://satijalab.org/seurat/index.html; RRID: SCR_007322 |
| SCRABBLE R package | Kai Tan Lab | https://github.com/tanlabcode/SCRABBLE |
| Enrichr | Avi Ma’ayan Lab | https://maayanlab.cloud/Enrichr/; RRID: SCR_001575 |
| GBM Bio Discovery Portal | NCI | https://gbm-biodp.nci.nih.gov |
| cBioPortal for Cancer Genomics | MSKCC | https://docs.cbioportal.org/about-us/; RRID: SCR_014555 |
| Ivy Glioblastoma Atlas Project | Allen Institute for Brain Science | https://glioblastoma.alleninstitute.org; RRID: SCR_005044 |
| high dimensional WGCNA R package | Vivek Swarup Lab | https://smorabit.github.io/hdWGCNA/index.html |
| Leica Application Suite X | Leica Microsystems | RRID: SCR_013673 |
| Ingenuity Pathway Analysis | Qiagen Digital Insights | RRID: SCR_008653 |
| Gene Set Enrichment Analysis | Broad Institute | RRID: SCR_003199 |
| The Cancer Genome Atlas | National Institutes of Health | https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga; RRID: SCR_003193 |
| Monocle3 | Cole Trapnell Lab | https://cole-trapnell-lab.github.io/monocle3/; RRID: SCR_018685 |
| CellPhoneDB 4.0 | Roser Vento-Tormo Lab | https://github.com/ventolab/CellphoneDB; RRID: SCR_017054 |
| GlioVis | Public domain, Massimo Squatrito Lab | http://gliovis.bioinfo.cnio.es |