

Abstract
The high-risk subtype human papillomaviruses (hrHPVs) infect and oncogenically transform basal epidermal stem cells associated with the development of squamous-cell epithelial cancers. The viral E6 oncoprotein destabilizes the p53 tumor suppressor, inhibits p53 K120-acetylation by the Tat-interacting protein of 60 kilodaltons (TIP60, or Kat5), and prevents p53-dependent apoptosis. Intriguingly, the p53 gene is infrequently mutated in HPV+ cervical cancer clinical isolates which suggests a possible paradoxical role for this gatekeeper in viral carcinogenesis. Here, we demonstrate that E6 activates the TP53-induced glycolysis and apoptosis regulator (TIGAR) and protects cells against oncogene-induced oxidative genotoxicity. The E6 oncoprotein induces a Warburg-like stress response and activates PI3K/PI5P4K/AKT-signaling that phosphorylates the TIGAR on serine residues and induces its hypoxia-independent mitochondrial targeting in hrHPV-transformed cells. Primary HPV+ cervical cancer tissues contain high levels of TIGAR, p53, and c-Myc and our xenograft studies have further shown that lentiviral-siRNA-knockdown of TIGAR expression inhibits hrHPV-induced tumorigenesis in vivo. These findings suggest the modulation of p53 pro-survival signals and the antioxidant functions of TIGAR could have key ancillary roles during HPV carcinogenesis.
Keywords: Human papillomavirus, E6 oncoprotein, p53, TIGAR, c-Myc, ROS, mitochondria, hypoxia
1. Introduction
There are myriad mechanisms by which oncogenic viruses cooperate with and deregulate host cellular growth/proliferative-signaling pathways to cause human cancers. The human papillomaviruses (HPVs) are members of the Papovaviridae family and are small spherical, non-enveloped dsDNA viruses that infect basal epithelial stem cells associated with the development of benign localized skin lesions (common or plantar warts; papillomas). However, many of the alpha-genus, high-risk HPV subtypes (hrHPVs; e.g., HPV16, HPV18, HPV31, HPV33, and HPV45) are sexually transmitted and can infect urogenital and oropharyngeal mucosal squamous epithelia and have been etiologically linked to uterine cervical dysplasia and a number of HPV+ epithelial malignancies, including cervical, vaginal, penile, and anogenital carcinomas, as well as certain subsets of head-and-neck cancers (Mix et al., 2021; Graham, 2017b; The Cancer Genome Atlas Network, 2015; Castellsagué et al., 2016). The viral genome is maintained and replicates as non-integrated circular episomes within the nuclei of HPV-infected cells. The switch from latency to vegetative genomic replication for the production of infectious particles is intricately linked to the keratinocyte differentiation program and is mediated by a viral regulatory nucleotide sequence, known as the long control region (LCR), and the early proteins: E1, E2, E4, E5, E6, and E7 (Münger et al., 2004; Graham, 2017a; Longworth and Laimins, 2004; Jones and Münger, 1997; Genther et al., 2003; Wang et al., 2009). Whereas the E1 and E2 proteins are responsible for episomal replication in HPV-infected basal epithelial cells, the E5, E6, and E7 proteins regulate vegetative genomic replication within infected suprabasal keratinocytes and differentiated spinous epithelial cells by reprogramming these cells to express S-phase-promoting components, such as Myc, Piwil2, and cyclin/cyclin-dependent kinases, which promote nucleotide biogenesis and DNA-replication (Genther et al., 2003; Wang et al., 2009; Feng et al., 2016; Vande Pol and Klingelhutz, 2013; Roman and Munger, 2013; Malanchi et al., 2002; Martin et al., 1998).
The viral E6 and E7 oncoproteins deregulate the cell-cycle by interfering with the functions of the tumor suppressor proteins, p53 and retinoblastoma (Rb; Vande Pol and Klingelhutz, 2013; Roman and Munger, 2013; Scheffner et al., 1993; Huibregtse et al., 1991; Münger et al., 1989; Boyer et al., 1996). The amino terminus of E7 binds to the pocket protein Rb and induces its ubiquitin-dependent degradation and dissociates Rb-E2F inhibitory complexes to promote the G1/S transition in infected differentiated keratinocytes (Münger et al., 1989; Boyer et al., 1996). The hrHPV E7 oncoprotein also degrades the protein tyrosine phosphatase PTPN14, independent of its ability to inactivate Rb, which inhibits keratinocyte differentiation and could contribute to cellular immortalization and viral carcinogenesis (Hatterschide et al., 2019). Although the p53 gene is infrequently mutated in primary HPV+ cervical cancer clinical isolates (Schultheis et al., 2021; Lee et al., 1994), the E6 oncoprotein ubiquitinates and reduces the expression of p53 through interactions with the E6-associated protein (E6AP) and inhibits acetylation of the p53 protein on lysine residue K120 and prevents p53-dependent cellular apoptosis by destabilizing the Tat-interacting protein of 60 KiloDaltons (TIP60, or Kat5) through interactions with the E3 ubiquitin ligase, EDD1/UBR5 (Scheffner et al., 1993; Huibregtse et al., 1991; Martinez-Zapien et al., 2016; Jha et al., 2010; Subbaiah et al., 2016). The hrHPV E6/E7 oncoproteins also cooperate with cellular protooncogenes and augment the expression and transactivation functions of the basic domain/leucine zipper (bZIP) transcription factor, Myc. Strickland and Vande Pol, 2016 have demonstrated that the E7 protein enhances the cap-independent translation of c-Myc by increasing the utilization of an internal ribosome entry site (IRES) within c-myc mRNA transcripts. Further, the E6 oncoprotein directly interacts with Myc and recruits E6/Myc/Max complexes to E-box enhancer elements within the human telomerase reverse transcriptase (htert) gene promoter associated with Myc-dependent transactivation and keratinocyteimmortalization (Zhang et al., 2017; McMurray and McCance, 2003; Liu et al., 2008). As mounting evidence suggests the remaining p53 protein present in HPV-transformed epithelial cells is transcriptionally active, and p53 is a downstream target of the Myc protooncogene (Butz et al., 1995; Banuelos et al., 2003; Kimple et al., 2013; Reisman et al., 1993; Roy et al., 1994; Zindy et al., 1998), it remains to be fully determined how E6 cooperates with Myc to induce the G1/S transition in HPV-infected keratinocytes, while overcoming the oxidative stress and p53-dependent genotoxicity associated with the unscheduled activation of Myc pro-replicative signals (Zindy et al., 1998; Vafa et al., 2002; Hermeking and Eick, 1994; Chen et al., 2013; Gorrini et al., 2007).
The TP53-induced glycolysis and apoptosis regulator (TIGAR) is a 2,6-bisfructose-phosphatase that localizes to the outer membranes of mitochondria under conditions of hypoxia, glucose-deprivation, or ischemic injury and contributes to mitochondrial quality control and prevents ischemia-induced mitophagy and the accumulation of damaging reactive oxygen species (ROS) by increasing the intracellular levels of NADPH and reduced glutathione (Bensaad et al., 2006; Cheung et al., 2012; Cheung et al., 2013; Li et al., 2014; Zhou et al., 2016; Bensaad et al., 2009). The mitochondrial targeting of TIGAR occurs through a mechanistic process that requires activation of hypoxia-inducible factor-1 alpha (HIF-1α) and molecular interactions with hexokinase-2 (HK2; Cheung et al., 2012). The TIGAR protein also increases the production of ribose-5-phosphate through glucose catabolism by the pentose-phosphate pathway which could promote nucleotide biosynthesis in proliferating cells (Bensaad et al., 2006). Several studies have implicated the overexpression of TIGAR as a key determinant of therapy-responsiveness and indicator of aggressive disease progression and poor clinical outcomes for many types of cancers, including gliomas, colorectal, esophageal, nasopharyngeal, and renal cell carcinomas, gastric cancers, multiple myeloma, chronic lymphocytic leukemia, acute myeloid leukemia, adult T-cell leukemia, lymphoblastic leukemia, and non-Hodgkin’s lymphoma (Cheung et al., 2013; Tang and He, 2019; Maurer et al., 2019; Chu et al., 2020; Wong et al., 2015; Wang et al., 2020; Liu et al., 2019; Yin et al., 2014; Hong et al., 2016; Qian et al., 2016; Romeo et al., 2018; Hutchison et al., 2018). The TIGAR has also been shown to contribute to the epithelial-to-mesenchymal transition (EMT), tumor cell migration/invasiveness, and metastasis related to Met-signaling in non-small-cell lung cancers (Shen et al., 2018). Here, we demonstrate that hrHPV E6 oncoproteins activate HIF-1α/HIF-2α and cellular kinases which phosphorylate the TIGAR protein on serine residues to induce its hypoxia-independent mitochondrial targeting and prevent c-Myc-induced oxidative DNA-damage, associated with enhanced oncogenic cellular transformation in vitro and HPV tumorigenesis in vivo. Hypoxia has been reported to transcriptionally inhibit the expression of the viral E6/E7 oncoproteins in HPV-infected cells through the phosphatidylinositol-3-kinase (PI3K)/AKT/mechanistic target of rapamycin (mTOR) complex 2 (mTORC2) pathway (Bossler et al., 2019) and, therefore, our studies allude to a functionally distinct role for E6-induced HIF-1α/HIF-2α pseudohypoxic signaling for the activation of mitochondrial TIGAR. These findings shed new light on the molecular mechanisms by which hrHPVs cooperate with cellular growth/proliferative signals to reprogram differentiated cells and suggest that the E6 oncoprotein augments p53 pro-survival functions by inducing a pseudohypoxic stress response to counter the metabolic cytotoxicity and replicative stress caused by aberrant protooncogene activation which could contribute to viral carcinogenesis. These studies further provide the first evidence that phospho-signaling regulates the mitochondrial localization and antioxidant functions of TIGAR which could lead to new therapeutic strategies to target this key pro-oncogenic factor in human cancers.
1. Materials and methods
1.1. Cell culture
All cells used for these studies were grown at 37°C under 5% CO2 in a humidified incubator. The HPV18+ Hela cervical adenocarcinoma cell-line was cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; No. 30–2002; ATCC, Manassas, VA), supplemented with 10% fetal bovine serum (FBS; Biowest, Riverside, MO), 100 U/ml penicillin and 100 Pg/ml streptomycin sulfate (Invitrogen, Waltham, MA). The HPV18+ C-4 I, HPV18/45+ MS751, and HPV16+ SiHa cervical carcinoma cell-lines were cultured in Eagle’s Minimum Essential Medium (EMEM; ATCC No. 30–2003), supplemented with 10% FBS and antibiotics as described. The HPV16+ Ca Ski cervical carcinoma cell-line was cultured in Roswell Park Memorial Institute-1640 (RPMI-1640) Medium (ATCC No. 30–2001), supplemented with 10% FBS and antibiotics. The HFL1 immortalized human fibroblast cell-line was cultured in F-12K Medium (ATCC No. 30–2004), supplemented with 10% FBS and antibiotics; and the HT-1080 fibrosarcoma and 293 HEK cell-lines were cultured in EMEM, supplemented with 10% FBS and antibiotics as described. The HCT116 p53-null (CRISPR KO) carcinoma cell-line (No. HD 104–001; Horizon Discovery Biosciences, Cambridge, UK) was grown in McCoy’s 5A Medium (ATCC No. 30–2007), supplemented with 10% FBS and antibiotics; and the 293FT cell-line was cultured in EMEM, supplemented with 4% D-(+)-glucose, 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin sulfate, and 500 μg/ml Geneticin (G418; ATCC No. 30–2305). Primary epidermal keratinocytes, human neonatal foreskin (HEKn; ATCC No. PCS-200–010), were cultured in Dermal Cell Basal Medium (ATCC No. PCS-200–030), supplemented with Keratinocyte Growth Kit (ATCC No. PCS-200–040) components (0.4% Extract-P, 0.5 ng/ml recombinant human TGF-α, 6 mM L-glutamine, 100 ng/ml hydrocortisone hemisuccinate, 5 μg/ml recombinant human insulin, 1.0 μM epinephrine, and 5 μg/ml apo-transferrin), 10 U/ml penicillin, 10 μg/ml streptomycin, 0.025 μg/ml amphotericin B, and 33 μM phenol red (ATCC No. PCS-999–001). Primary dermal fibroblasts, human neonatal foreskin (HDFn; ATCC No. PCS-201–010), were cultured at 37°C under 5% CO2 in Iscove’s Modified Dulbecco’s Medium (IMDM; ATCC No. 30–2005), supplemented with 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin sulfate. All plasmid DNA transfections were performed using Lipofectamine LTX with Plus reagent (ThermoFisher Scientific, Rockford, IL) as per the manufacturer’s recommended protocol. The source information for the cell-lines, chemicals and reagents, lentiviruses, and DNA plasmids used in this study is provided in the Supplemental information (Table S1).
1.2. Colony-formation assays
In vitro colony-formation experiments to assess oncogenic anchorage-independent cell growth were performed as follows: 2 × 105 primary human dermal fibroblasts (HDFn) were seeded on 12-well tissue-culture plates and cotransfected with various expression constructs for c-Myc (FLAG) and HPV16 E6 (HA) or HPV18 E6 (HA), or a CβS vector as negative control. After 24 hrs, certain samples were also transduced with lentiviral-siRNA-tigar or an empty lentiviral vector. The samples were then trypsinized and re-seeded on a 12-well plate coated with 0.5% base agar + IMDM, supplemented with 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin sulfate, and mixed with 0.3% top agar + IMDM, supplemented 10% FBS and antibiotics. After 3 weeks, the transformed colonies were stained with a solution of 0.005% crystal violet in 10% ethanol/dH2O, rinsed and destained with dH2O, and visualized and quantified by counting using an inverted microscope. Three independent experiments were performed and the data are representative of the mean ± SD.
1.3. Lentiviral vectors
The lentiviral pLenti-siRNA-tigar vector was generated by annealing and TA-cloning the oligodeoxynucleotide primers: 5’-ATATACCAGCAGCTGCTGCA-3’ and 5’-GCAGCAGCTGCTGGTATATA-3’ (Biosynthesis, Lewisville, TX), into the spectinomycin-resistant GATEWAY pCR8/GW/TOPO entry vector (Invitrogen) which was transformed into chemically competent TOP10 E. coli bacteria (Invitrogen). The orientation of the dsDNA insert was verified by PCR and sequencing using the M13 forward and reverse primers. Clonase recombination reactions were then performed with the pLenti6.2/V5-DEST destination plasmid (Invitrogen) and the subcloned products were transformed into One Shot Stbl3 E. coli cells (Invitrogen) to generate pLenti-siRNA-tigar. The selected recombinant clones were screened by restriction endonuclease digestion with Afl II and Xho I, or Eco RI alone, and then cotransfected together with ViraPower Lentiviral Packaging Mix (Invitrogen) plasmids (i.e., pLP1, pLP2, and pLP/VSVG) into 293FT cells using Lipofectamine 2000 reagent (Invitrogen). The transfected cultures were monitored by microscopy for syncytia formation and, after 96 hrs, the supernatants were collected and clarified by centrifugation at 260 x g at 4°C for 10 min and passed through a 0.22 μm polyethersulfone filter. The pLenti-siRNA-tigar lentivirus contains a CMV promoter which drives the expression of a siRNA specifically targeted against tigar mRNA transcripts, and the ability of this vector to inhibit TIGAR protein expression was demonstrated by transducing HeLa cells and then performing denaturing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting using an Anti-TIGAR primary antibody.
1.4. Immunoprecipitations and immunoblotting
The expression of the TIGAR, c-Myc, p53, H-Ras, HIF-1α, HIF-2α, and HPV16/18 E6 proteins was analyzed in HPV-transformed epithelial cell-lines and cotransfected cells by SDS-PAGE and immunoblotting and quantified by densitometry. For immunoblotting, the cultured cells were harvested by scraping and then washed 2x in PBS, pH 7.4, and centrifuged at 260 x g for 7 min at 4°C. The cell pellets were resuspended in 100 Pl of Lysis Buffer (Promega, Madison, WI); and the cells were lysed by repeated freeze-thawing over dry-ice and shearing using a 27-gauge tuberculin syringe. The samples were clarified by brief centrifugation at 10,000 x g for 2 min and then mixed with 2x Laemmli Sample Buffer with 2-mercaptoethanol (BioRad Laboratories, Hercules, CA), heated at 95°C for 5 min, and resolved by denaturing SDS-PAGE using a 4% Tris-glycine polyacrylamide stacking gel and either 12.5% or 15% running gel. The electrophoresed proteins were transferred onto a Protran BA83 0.2 3μm nitrocellulose membrane (Whatman, Maidstone, UK) using a model TE 77 PWR semi-dry blotting unit (Amersham Biosciences, Little Chalfont, UK). The membranes were then incubated in Blocking buffer (3% w/v bovine serum albumin and 0.5% v/v Tween-20 in PBS, pH 7.4) with gentle agitation for 1 hr at room temperature, and incubated with various primary antibodies (diluted 1:1000 or 1:2000 in Blotto buffer: 50 mM Tris-HCl, pH 8.0, 0.2 mM CaCl2, 80 mM NaCl, 0.2% v/v IGEPAL-CA630, 0.02% w/v sodium azide, and 5% w/v nonfat dry milk) for 2 hrs on an orbital shaker at room temperature. The membranes were washed 2x by incubating with Blotto buffer with gentle agitation for 10 min, followed by incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies (diluted 1:500; Jackson Immunoresearch Laboratories, West Grove, PA) for 1.5 hrs with agitation at room temperature. The membranes were subsequently washed 2x with Blotto buffer and once with TMN buffer (100 mM NaCl, 5 mM MgCl2, 100 mM Tris-HCl, pH 9.5) for 10 min each, and then analyzed by chemiluminescence detection with Pierce ECL Western Blot Reagent (ThermoFisher Scientific) and a ChemiDoc Touch imaging system (BioRad Laboratories). The protein bands were quantified by densitometry using Image Lab 6.0.1 software (BioRad Laboratories) and the mean levels of TIGAR, c-Myc, H-Ras, and p53 were normalized relative to Actin protein expression with statistical analysis and standard deviations shown for three experimental replicates.
The phosphorylation of the TIGAR protein was analyzed by performing immunoprecipitations with various monoclonal phospho-specific antibodies (i.e., Anti-Phospho-Serine, Anti-Phospho-Threonine, or Anti-Phospho-Tyrosine; Millipore-Sigma, St. Louis, MO) and Protein-G agarose beads (BioRad Laboratories). As a control for the phospho-specific antibodies, in certain experiments the HPV16+ SiHa cell extracts were treated for 1 hr at 37°C with a recombinant Serine/threonine-protein phosphatase (2,000 U; Abcam, Cambridge, MA) prior to immunoprecipitation. Also, the effects of overexpressing TIP60 upon the acetylation of the p53 protein on lysine residue K120 in the HPV18-transformed cervical carcinoma cell-lines, HeLa and C-4 I, were determined by immunoprecipitations using a monoclonal Anti-K120-Acetyl-p53 antibody (Abcam). The cells were harvested by scraping, pelleted by centrifugation at 260 x g for 7 min at 4°C, and then resuspended in RIPA buffer (0.15 M NaCl, 50 mM Tris-HCl, pH 7.4, 0.5% sodium deoxycholate, 0.5% Nonidet P-40, 0.1% SDS) containing 50 ng/ml of each of the protease inhibitors: pepstatin, leupeptin, chymostatin, bestatin, and antipain-dihydrochloride (Roche Applied Science, Indianapolis, IN). The cells were lysed by repeated freeze-thawing and syringe shearing using a 27-gauge tuberculin syringe, and then clarified by centrifugation at 10,000 x g for 2 min at 4°C. The extracts were pre-cleared by incubating the samples with 20 μl of a 50% slurry of Protein-G agarose in PBS, pH 7.4, for 5 min at 4°C with gentle agitation. The samples were centrifuged at 5,000 x g for 5 min at 4°C and the supernatants were transferred to new 1.5 ml microtubes. Immunoprecipitations were performed by adding 4 μl of either Anti-Phospho-Serine, Anti-Phospho-Threonine, Anti-Phospho-Tyrosine (Millipore-Sigma), or an Anti-K120-Acetyl-p53 (10E5; Abcam) antibody and then incubating the samples overnight at 4°C on a roller shaker. Forty microliters of a 50% slurry of Protein-G agarose beads were then added and the samples were incubated for 2 hrs at 4°C with gentle agitation. Following incubation, the agarose beads were pelleted by centrifugation at 5,000 x g for 5 min at 4°C, washed 2x by resuspension in 250 μl of ice-cold PBS, pH 7.4, with incubation for 5 min at 4°C on a roller shaker, followed by centrifugation, and finally the beads were resuspended in 30 μl of 2x Laemmli Sample Buffer with 2-mercaptoethanol. The precipitated products were eluted by heating the samples at 95°C for 5 min and then briefly centrifuged before loading them onto denaturing SDS-PAGE gels for subsequent analysis by immunoblotting and chemiluminescence-detection. The amounts of precipitated serine-phosphorylated TIGAR or K120-acetylated p53 were quantified by densitometry using Image Lab 6.0.1 software and normalized relative to the input levels of these proteins with statistical analysis and standard deviations provided for three replicates. The source information for all antibodies used throughout this study is in the Supplemental information (Table S1).
1.5. Isolation of mitochondria
To determine if phosphorylation effects the subcellular trafficking and mitochondrial targeting of the TIGAR protein, cytoplasmic and mitochondrial fractions were prepared from HPV18-transformed cervical carcinoma cell-lines (HeLa and MS751) and cotransfected HT-1080 cells containing TIGAR (FLAG) and the HPV16/18 E6 (HA) oncoproteins or an empty CβS vector as negative control. The cells were cultured in 100 mm2 poly-L-lysine-coated dishes (Corning, Glendale, AZ) and then harvested by scraping and pelleted by centrifugation at 260 x g for 10 min at 4°C. Mitochondria were isolated using a Qproteome Mitochondria Isolation Kit (Qiagen, Germantown, MD) per the manufacturer’s suggested protocol. The cells were lysed by sonication over an ice-bath using a Misonix S-4000 instrument (Cole-Parmer, Vernon Hills, IL) equipped with a microtip probe set at 70% amplitude. The lysates were then clarified by centrifugation at 1,200 x g for 10 min at 4°C and the supernatants were transferred to new 1.5 ml microtubes. The supernatants were centrifuged again at 6,400 x g for 10 min at 4°C and the microsomal fractions were transferred to separate 1.5 ml microtubes. The pellets containing isolated mitochondria were resuspended in 1 ml of ice-cold Mitochondrial Storage Buffer (Qiagen) and washed by centrifugation at 6,400 x g for 20 min at 4°C. Finally, the mitochondria were resuspended in 100 μl of Mitochondrial Storage Buffer and subsequently analyzed by performing immunoprecipitations using monoclonal Anti-Phospho-Serine and Anti-Phospho-Tyrosine antibodies (Millipore-Sigma) and Protein-G agarose beads (BioRad Laboratories), followed by SDS-PAGE, Western blotting and densitometry to quantify the relative ratios of serine-phosphorylated and tyrosine-phosphorylated (i.e., as a negative control) TIGAR present in the mitochondrial and cytoplasmic fractions. The Mit/Cyt Phospho-Ser Indices were calculated by dividing the ratio of mitochondrial phospho-Ser:phospho-Tyr by the ratio of cytoplasmic phospho-Ser:phosho-Tyr with the mean values, statistical analysis, and standard deviations shown for three experimental replicates. The purity of the cytoplasmic and mitochondrial input fractions was assessed by SDS-PAGE and immunoblotting to detect IκB-α as a cytoplasmic marker and the outer membrane protein TOM20 as a mitochondrial marker.
1.6. Measuring ROS and mitochondrial membrane depolarization
The effects of inhibiting TIGAR expression upon the accumulation of damaging ROS and mitochondrial membrane polarity were determined by transducing HPV18-transformed cervical carcinoma cell-lines (HeLa, MS751, and C-4 I) and cotransfected HT-1080 cells, containing c-Myc, the HPV16/18 E6 oncoproteins, or an empty CβS vector, with lentiviral-siRNA-tigar or the pLenti vector as a negative control. For certain experiments, HT-1080 cells were transfected with expression constructs for either TIP60 or the dominant-negative p53-R175H DNA-binding mutant. The samples were stained using a fluorescent chemical ROS probe, CellROX-Deep Red (Invitrogen), or JC-1 (Invitrogen) to detect changes in mitochondrial membrane polarity and then analyzed by fluorescence-confocal microscopy. The chemical uncoupler, carbonyl cyanide 3-chlorophenylhydrazone (CCCP, 50 μM; Millipore-Sigma), was included as a positive control for ROS and mitochondrial membrane depolarization. The relative percentages of ROS (CellROX-Deep Red)-positive cells per field were quantified by counting triplicate visual fields at 200x magnification. A differential interference contrast (DIC) filter was used to visualize all the cells. The ratios of damaged/depolarized mitochondria (i.e., JC-1 green fluorescence) to normal polarized mitochondria (i.e., JC-1 red fluorescence) were determined by measuring the relative fluorescence intensities for the green and red signals across a randomly positioned, standard 5 μm2 area using Carl Zeiss ZEN OS imaging software (Carl Zeiss Microscopy, Jena, Germany). Twenty individual cells were analyzed for each data point.
1.7. Quantification of apoptosis and DNA-damage
To assess whether the lentiviral siRNA-inhibition of TIGAR expression causes oxidative genotoxicity and programmed cell-death, the relative percentages of apoptotic cells were quantified by washing the samples 2x with PBS, pH 7.4, and then staining them with Annexin V-FITC and a propidium iodide (PI) solution (10 mg/ml; BD Biosciences, Franklin Lakes, NJ) for subsequent analysis by fluorescence-confocal microscopy. The chemical uncoupler CCCP was included as a positive control for apoptosis. The percentages of Annexin V-FITC and/or PI-positive cells within each field was determined by counting triplicate visual fields at 200x magnification. A DIC filter was used to visualize all the cells. To determine whether HPV+ cells are generally more resistant to oxidative stress-induced cytotoxicity, HPV18+ HeLa cells and 293 HEK (HPV-neg) cells were treated with hydrogen peroxide (H2O2, 2 μM) for 2 hrs and then the samples were stained with Annexin V-FITC and PI and the relative percentages of apoptotic cells were quantified by confocal microscopy and counting triplicate visual fields at 20x magnification. Alternatively, the samples were stained using a 594 nm fluorescent Click-IT Plus Terminal deoxynucleotidyl dUTP nick end-labeling (TUNEL) kit (Invitrogen) to detect ssDNA breaks and the relative numbers of TUNEL-positive foci per cell were quantified by confocal microscopy. Certain samples were permeabilized and treated with deoxyribonuclease I (DNAse I; Qiagen) as a positive TUNEL control. Cell nuclei were stained using Hoechst 33342. Twenty individual cells were analyzed for each data point.
To determine the effects of inhibiting TIGAR expression upon oncogene-induced DNA-damage, HPV18-transformed cervical carcinoma cells (HeLa, MS751, and C-4 I) or HPV-negative HFL1 fibroblasts, and cotransfected HT-1080 cells containing c-Myc and the HPV16/18 E6 oncoproteins or an empty CβS vector were transduced with either lentiviral-siRNA-tigar or the pLenti vector as a negative control. For certain experiments, HT-1080 cells were transfected with expression constructs for TIP60 or the dominant-negative p53-R175H mutant. The samples were then immunostained using a rabbit polyclonal Anti-Phospho-γH2A.X (Ser 139) primary antibody (Santa Cruz Biotechnology, Dallas, TX) and a rhodamine red (TRITC)-conjugated fluorescent secondary antibody (Jackson Immunoresearch Laboratories) and the relative numbers of phospho-γH2A.X foci per cell were quantified by immunofluorescence-confocal microscopy. Cell nuclei were visualized by 4’6-diamidino-2-phenyl-indole (DAPI; Invitrogen)-staining. Twenty individual cells (or fifteen in Fig. 4C) were analyzed for each data point.
1.8. In vivo xenograft model of HPV tumorigenesis
In vivo xenograft studies of HPV tumorigenesis were performed as follows: immunodeficient NIH III-nude mice (Crl:NIH-Lystbg-J Foxn1nu Btkxid; Charles River Laboratories, Shrewsbury, MA), all between 6–12 months of age, were anesthetized by inhalation of 3–5% isoflurane/O2 and then subcutaneously engrafted over each hind flank with 1 × 106 HPV18+ HeLa adenocarcinoma cells that were transduced with lentiviral-siRNA-tigar or an empty pLenti vector as negative control, and suspended in 250 μl of the vehicle (i.e., sterile Ca2+/Mg2+-free PBS, pH 7.4) using a 27-gauge tuberculin syringe (n-value = 12 animals per sample group). Another sample group was injected with the Vehicle alone for comparison. The experimental animals were housed and maintained in a sterile individually ventilated cage (IVC) rack system and monitored daily over a period of 8 weeks for the development/growth of primary tumor masses at the injection sites as well as any signs of pain, morbidity, or other changes to their overall health status. After 8 weeks, the experimental animals were humanely sacrificed in accordance with 2020 AVMA guidelines using inhalation of 30% CO2 to induce rapid loss of consciousness, followed by 70% CO2 to cause death, with cervical dislocation as a secondary physical method to ensure expiration. Necropsies were performed to harvest the HPV18+ HeLa xenograft tumor tissues, which were weighed using a Mettler Toledo XP26 microanalytical balance (ThermoFisher Scientific) and measured using a digital caliper. The volumes of the primary tumor masses were determined using the calculation: (D × d2)/2, where ‘D’ equals the longest diameter and ‘d’ equals the shortest diameter. The tumor tissues were fixed in a 10% neutral-buffered formalin solution (3.75% formaldehyde, 33.3 mM sodium phosphate monobasic, 45.8 mM sodium phosphate dibasic, in distilled deionized water, pH 6.8), dehydrated by incubation at room temperature with gentle agitation in 50x volume of 70% ethanol for 1 hr, 95% ethanol for 1.5 hrs, 100% ethanol for 1.5 hrs, and xylene (Millipore-Sigma) for 1 hr, and then embedded in Histoplast IM paraffin (ThermoFisher Scientific) and stored frozen at −80°C until ready for use. The HPV+ xenograft tumor tissues were sectioned using a Microm HM360 rotary microtome (Carl Zeiss Microscopy), mounted on glass microscope slides, deparaffinized, and subsequently analyzed by immunofluorescence-confocal microscopy to detect the expression of TIGAR and the c-Myc oncoprotein within the engrafted HeLa tumor (i.e., Anti-human Ki67-positive) tissues. At least three tissue sections were analyzed for each sample. The in vivo tumorigenesis studies were carried out in accordance with protocol A16–001-HARR which was approved by the Southern Methodist University-Institutional Animal Care & Use Committee.
1.9. Analysis of primary HPV16+ cervical cancer clinical samples
To determine if the expression of p53-regulated pro-survival signals coincides with oncogene dysregulation associated with viral carcinogenesis, the expression of the p53, TIGAR, c-Myc, and H-Ras proteins in primary HPV-infected cervical cancer clinical specimens was analyzed by immunofluorescence-confocal microscopy. De-identified HPV16+ cervical carcinoma tissue samples (from 9 different patients) were provided as formalin-fixed paraffin-embedded (FFPE) sections under approved Institutional Review Board protocols: 2016-HARR-HARR (Southern Methodist University) and CHS No. 22431 (University of Hawaii) in accordance with Declaration of Helsinki Principles, by the Pathology Shared Resource of the University of Hawaii Cancer Center to support these studies. These samples were immunostained to detect and quantify the expression of TIGAR, c-Myc, p53, and H-Ras within tumor cells that were positive for the HPV16 E6 oncoprotein. Immortalized human HFL1 fibroblasts (HPV-negative) were also immunostained for comparison as an antibody control. The tissue sections were deparaffinized by treating the samples 2x with xylene for 3 min at room temperature, followed by a 1:1 solution of xylene/ethanol for 3 min, 2x with 100% ethanol for 3 min, 95% ethanol for 3 min, 70% ethanol for 3 min, 50% ethanol for 3 min, and then rinsing 2x with distilled deionized water. The microscope slides with the mounted sections were incubated in fixative solution (2% formaldehyde and 0.2% glutaraldehyde in PBS, pH 7.4) for 20 min at room temperature, washed 2x with PBS, and incubated in Blocking buffer for 1 hr with gentle agitation. After the buffer was removed, the tissue samples were immunostained using primary antibodies (diluted 1:1000 or 1:2000 in BLOTTO buffer) that recognize TIGAR, c-Myc, p53, H-Ras, or the HPV16 E6 oncoprotein (see Table S1) for 2 hrs on an orbital shaker at room temperature. The slides were then washed 2x with BLOTTO buffer for 10 min, followed by incubation with various fluorescence-conjugated secondary antibodies (diluted 1:500 in BLOTTO) for 1 hr with gentle agitation. DAPI was included for the visualization of cell nuclei. The samples were washed 2x with BLOTTO buffer and 1x with PBS for 10 min, fluorescence mounting medium (Kirkegaard & Perry Laboratories, Gaithersburg, MD) and glass coverslips were added, and the tissue sections were subsequently analyzed by immunofluorescence-confocal microscopy. The relative numbers of HPV16 E6-positive cells which contained TIGAR, c-Myc, and p53 protein expression were quantified by counting triplicate visual fields at 200x magnification.
1.10. Confocal microscopy
Immunofluorescence and direct confocal microscopy analyses were performed on a Zeiss LSM800 instrument with an Airyscan detector and stage CO2 incubator (Carl Zeiss Microscopy). All images were taken using either a Plan-Apochromat 20x/0.8 objective or Plan-Apochromat 40x/1.3 oil immersion objective lens and Zeiss ZEN OS software, with eight averages for each. The colocalization between the Anti-TIGAR and MitoTracker Orange fluorescent signals was measured across a standard 5 μm2 area using the ZEN Colocalization tool and Pearson correlation coefficient. The expression of GFP in transfected primary human keratinocytes and dermal fibroblasts was visualized using a Nikon Eclipse TE2000-U inverted microscope and D-Eclipse C1 confocal system (Nikon Instruments, Melville, NY) with a 488 nm Ar laser, differential interference contrast (DIC) filter, and Plan Apo 20x/0.75 objective lens.
1.11. Statistical analysis
The statistical significance of experimental data sets was determined using unpaired two-tailed Student’s t-tests (alpha = 0.05) and calculated P-values using the Shapiro-Wilk normality test and GraphPad Prism 8.0 software (GraphPad Software, San Diego, CA), with the P-values defined as: 0.1234 (ns), 0.0332 (*), 0.0021 (**), 0.0002 (***), < 0.0001 (****). The numbers of animals to include in each sample group to ensure robust statistically-significant results were determined by power analyses using NCSS PASS 2020 sample-size statistical software (NCSS Statistical Software LLC, Kaysville, UT); and these calculations allow for a 20% dropout rate due to non-experimental causes. All experiments were performed at least three times (N-value ≥ 3), except for the in vivo xenograft tumorigenesis studies using NIH III-nude mice with 12 animals per sample group.
2. Results
2.1. High-risk HPV E6 proteins induce hypoxia-independent mitochondrial targeting of the TIGAR
The expression of TIGAR is positively regulated by p53 and the myc protooncogene and protects proliferating cells from toxic metabolic byproducts, such as damaging ROS, by increasing the levels of NADPH and reduced glutathione (Bensaad et al., 2006; Bensaad et al., 2009; Romeo et al., 2018; Cheung et al., 2016). The TIGAR protein is predominantly cytoplasmic, although a significant fraction has been shown to relocate to the outer membranes of mitochondria under conditions of hypoxic stress or ischemic injury –which requires HIF-1α activation and molecular interactions with HK2 (Cheung et al., 2012; Li et al., 2014). Interestingly, the HPV E6 oncoprotein has been shown to induce HIF-1α protein expression by preventing the degradation of HIF-1 by the von Hippel Lindau (VHL) ubiquitin ligase (Guo et al., 2014) and to enhance the expression of HK2 in HeLa cells (Hoppe-Seyler et al., 2017). We therefore investigated whether HPV E6 might induce a pseudohypoxic stress response, associated with the activation of HIF-1α and HIF-2α, and promote the mitochondrial targeting of TIGAR –which could help protect HPV-infected cells against c-Myc-induced oxidative damage. Indeed, these experiments revealed that the HPV16/18 E6 oncoproteins markedly induced the expression of both HIF-1α and HIF-2α in transfected primary human keratinocytes that were cultured under normal oxygen levels (19.95% O2; Figs. 1A and 1B). Our studies have further revealed that HIF-1α and HIF-2α are upregulated in HPV18+ (HeLa and C-4 I) and HPV16+ (SiHa) cervical carcinoma cell-lines, as compared to cultured primary keratinocytes (supplementary Fig. S1A). Also, ectopic expression of the HPV16/18 E6 (HA-tagged) oncoproteins induced the expression of HIF-1α and HIF-2α as determined by immunoblotting, compared to an empty vector control in transfected HT-1080 cells (Fig. S1B). Whereas HIF-1α is stabilized in response to reduced oxygen levels (i.e., hypoxia), the HIF-2α protein is frequently upregulated in tumor cells under normoxic conditions which represents a phenomenon known as “pseudohypoxia” (Pietras et al., 2010). We next examined the expression of TIGAR, p53, c-Myc, and the viral E6 oncoprotein in HPV18 and HPV16-transformed cervical carcinoma cell-lines, as compared to HPV-negative cells (i.e., human HFL1 fibroblasts or primary keratinocytes). These studies revealed that TIGAR is upregulated in the hrHPV+ cell-lines and correlated with oncogenic c-Myc dysregulation and detectable p53 protein levels (Figs. 1C and 1D). We then tested whether ectopic expression of the E6 oncoprotein might augment TIGAR expression and found that the hemagglutinin (HA)-tagged HPV16/18 E6 oncoproteins moderately increased (~5-fold) the levels of TIGAR in transfected HT-1080 cells as compared to an empty vector control (Fig. 1E). The expression of Myc and p53 was not significantly altered (Fig. 1E). As the HPV16/18 E6 oncoproteins were found to induce pseudohypoxic signaling associated with the activation of HIF-1α/HIF-2α, we investigated whether these viral proteins could induce mitochondrial targeting of the TIGAR in transfected cells. These experiments surprisingly revealed that the hrHPV E6 oncoproteins induced hypoxia-independent mitochondrial targeting of the TIGAR protein as compared to an empty vector control (Fig. 1F), suggesting the E6 oncoprotein could cooperate with cellular protooncogenes by activating the pro-survival functions of TIGAR in HPV-infected differentiated cells.
Fig. 1.
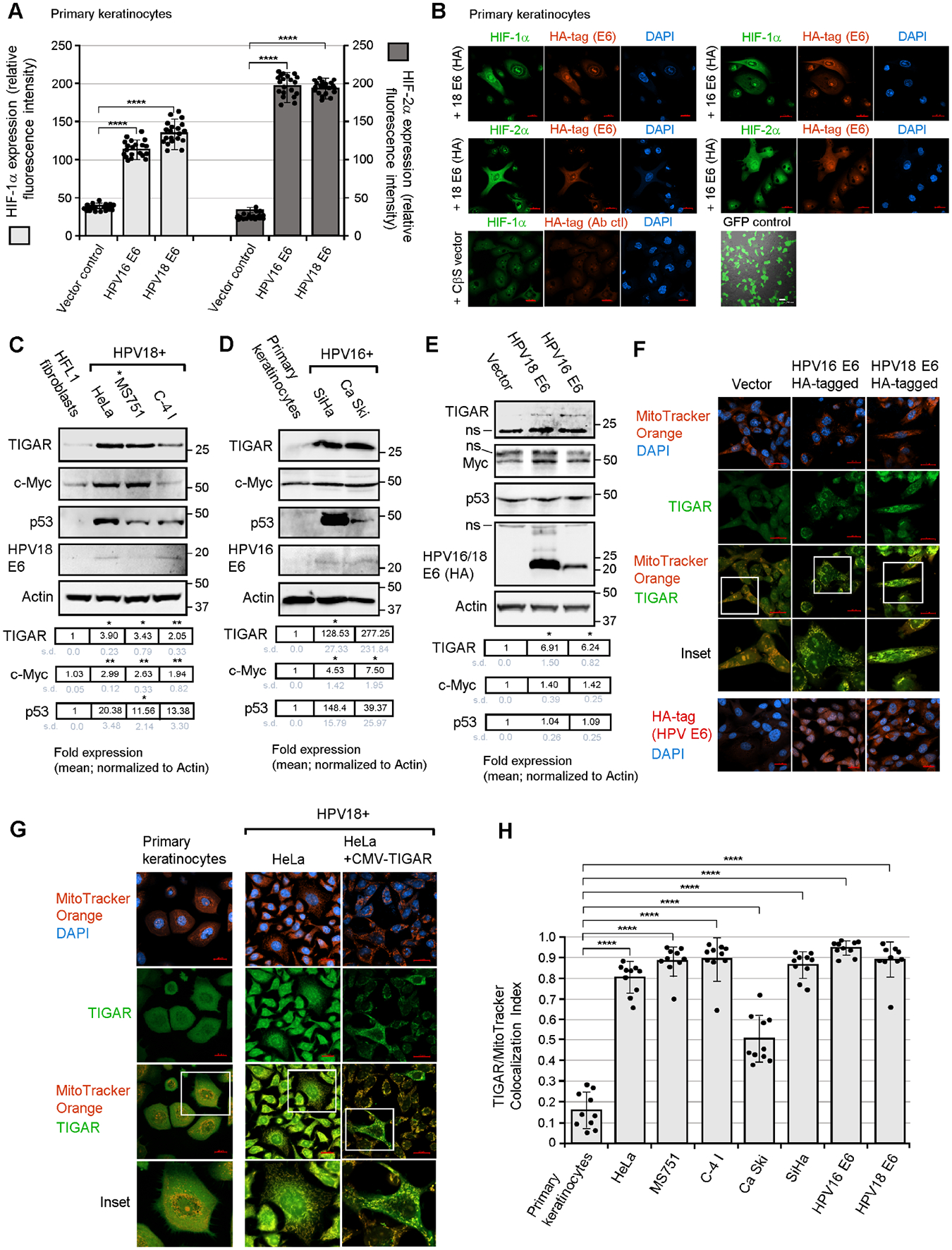
The hrHPV E6 oncoproteins induce hypoxia-independent mitochondrial targeting of TIGAR in HPV+ cervical carcinoma cells. (A and B) The hrHPV E6 oncoproteins activate the hypoxia-inducible factors, HIF-1α and HIF-2α, in transfected cells cultured under normoxic conditions. Primary human keratinocytes were transfected with expression constructs for HPV16/18 E6 (HA-tagged) or an empty CβS vector as negative control, and immunofluorescence-confocal microscopy was performed to visualize and quantify the expression of HIF-1α or HIF-2α (green signal) within the HPV16/18 E6 (HA)-positive (red signal) cells as shown in B. A green fluorescent protein (N3-pEGFP) transfection control is also shown. The relative fluorescence intensities of the HIF-1α and HIF-2α-specific signals were measured relative to the vector control (graph in A). (C) The HPV18-transformed cervical carcinoma cell-lines, HeLa, MS751, and C-4 I, were analyzed for the expression of TIGAR, c-Myc, p53, and HPV18 E6 by immunoblotting using the indicated primary antibodies (*MS751 cells also contain HPV45 sequences). Human HFL1 fibroblasts were included as a negative control. The increased levels of TIGAR in the HPV18+ cell-lines correlated with expression of the c-Myc oncoprotein and p53. (D) The expression of TIGAR, c-Myc, and p53 was also analyzed in the HPV16-transformed cell-lines, SiHa and Ca Ski. The HPV16 E6 oncoprotein was detected by immunoblotting. Primary keratinocytes were included as a negative control. “ns” denotes a nonspecific band. (E) HT-1080 cells were transfected with HPV18 E6 or HPV16 E6 (HA-tagged) expression constructs and the induction of TIGAR was analyzed by immunoblotting and quantified relative to Actin protein levels. (F) To determine if the HPV16/18 E6 (HA)-tagged) oncoproteins could induce hypoxia-independent mitochondrial targeting of the TIGAR protein, the cells were transfected and then labeled with MitoTracker Orange and immunofluorescence-confocal microscopy was performed using an Anti-TIGAR primary antibody (green signal). The E6-expressing cells exhibited punctate colocalization of the TIGAR-specific signal with the MitoTracker Orange probe, as compared to the vector control which showed diffuse cytoplasmic TIGAR immunostaining (see insets). (G and H) The hypoxia-independent mitochondrial targeting of the TIGAR protein (green signal) in HPV18+ (HeLa, MS751, and C-4 I) and HPV16+ (SiHa and Ca Ski) cervical carcinoma cells was visualized by labeling the cells with MitoTracker Orange (red signal) and then performing immunofluorescence-confocal microscopy. Primary keratinocytes are shown for comparison. An enlarged inset area is depicted in the lower panels. As a positive control for antibody-staining, HeLa cells were transfected with a CMV-TIGAR (FLAG-tagged) expression construct and these cells exhibited punctate TIGAR immunostaining that colocalized with the MitoTracker Orange probe. Scale bar, 20 μm. The colocalization between TIGAR and the MitoTracker Orange signal was quantified using the ZEN OS colocalization module in H (micrographs for the SiHa, CaSki, MS751, and C-4 I cell-lines are provided in Fig. S2A). Data is mean ± SD. N-value = 3. The asterisks denote statistical significance as determined using unpaired two-tailed Student’s t-tests (****P<0.0001).
We next examined the subcellular localization of TIGAR in HPV18 (HeLa, MS751, and C-4 I) and HPV16 (Ca Ski and SiHa)-transformed cervical carcinoma cell-lines using immunofluorescence-confocal microscopy. As shown in Fig. 1G and supplementary Fig. S2A, the TIGAR protein was significantly targeted to mitochondria in the HPV16/18-transformed cell-lines under normoxic conditions as compared to primary keratinocytes or HFL1 fibroblasts which exhibited a diffuse, primarily cytoplasmic distribution of TIGAR (micrographs for the HPV16-transformed cell-lines, SiHa and Ca Ski, the HPV18-transformed cell-lines, MS751 and C-4 I, and HFL1 fibroblasts are provided in Fig. S2A). As a control for antibody-staining, we examined the localization of a FLAG epitope-tagged TIGAR protein in transfected HPV18+ HeLa cells. These studies demonstrated that FLAG-tagged TIGAR exhibited a punctate mitochondria-like immunostaining pattern and colocalized with the MitoTracker Orange probe in a hypoxia-independent manner in hrHPV-transformed cells (Fig. 1G, right panels). The colocalization between the TIGAR and MitoTracker Orange-specific fluorescent signals in HPV18 and HPV16-transformed cervical carcinoma cell-lines and transfected HT-1080 cells expressing the HA-tagged HPV16/18 E6 oncoproteins was quantified across a randomly positioned 5 μm2 area using the ZEN OS colocalization module and is graphically represented in Fig. 1H. To further validate these findings, we demonstrated that the HPV18 E6 oncoprotein induced hypoxia-independent mitochondrial targeting of FLAG-tagged TIGAR in cotransfected HT-1080 cells, in contrast to cells expressing the TIGAR (FLAG) protein and an empty vector control (Fig. S2B; the expression of the FLAG-TIGAR protein was demonstrated by immunoblotting as shown in Fig. S2C).
2.2. The hrHPV E6 oncoproteins activate cellular kinases and induce serine-phosphorylation of the TIGAR associated with its mitochondrial targeting
The hrHPV E6 oncoproteins activate cellular signaling pathways –in particular, the Met-receptor which is a tyrosine kinase receptor that binds to hepatocyte growth factor/scatter factor (HGF/SF) and has been shown to promote TIGAR-induced cellular invasiveness/metastasis in certain cancers (Shen et al., 2018; Qian et al., 2016; Scott et al., 2018). Moreover, the inhibition of c-Met kinase activity was found to downregulate TIGAR associated with the depletion of NADPH in nasopharyngeal carcinoma cells (Lui et al., 2011). Intracellular signaling by HGF/SF-Met also coordinately activates the small GTPase, Ras, which stimulates its downstream effectors, PI3K and the Raf1 mitogen-activated protein kinase (MAPK; Hartmann et al., 1994). Beyond the ubiquitination of TIGAR by the E3 ligase, tripartite motif-31 (TRIM31), which promotes its degradation under conditions of ischemic injury (Zeng et al., 2021), there exists a general knowledge gap regarding other posttranslational modifications that could potentially modulate the functions of TIGAR. We thus investigated whether phosphorylation contributes to the E6-induced hypoxia-independent mitochondrial targeting of TIGAR. The results shown in Fig. 2A demonstrate that the chemical kinase inhibitors, wortmannin (a broad-specificity PI3K-inhibitor) and SB203580 (a p38MAPK-inhibitor), but not staurosporine (an inhibitor of protein kinase C and other calcium-dependent kinases) or a dimethyl sulfoxide (DMSO) solvent control, significantly blocked the mitochondrial localization of TIGAR in treated HPV18+ HeLa cells (representative micrographs are provided in Fig. S3A). We next performed immunoprecipitations using various phospho-specific monoclonal antibodies and discovered the TIGAR protein is strongly phosphorylated on serine residues in HeLa cells, which was not altered by treatment with staurosporine (Fig. 2B). These data were similarly reproduced in HPV16+ SiHa cells treated with the chemical kinase-inhibitors, staurosporine, wortmannin, and SB203580, followed by immunoprecipitation with the Anti-Phospho-Serine or Anti-Phospho-Threonine monoclonal antibodies and immunoblotting to detect the phosphorylated TIGAR protein (supplementary Fig. S4). A phospho-specific antibody control was included by treating the HPV16+ SiHa extract with a recombinant Serine/threonine-protein phosphatase prior to immunoprecipitation (Fig. S4). Consistent with our confocal imaging data, the chemical inhibitors wortmannin and SB203580 prevented the phosphorylation of TIGAR on serine residues in treated HPV18+ HeLa cells and HPV16+ SiHa cells (Figs. 2B and S4). We then tested whether constitutively-active mutants of PI3K, or the closely-related phosphatidylinositol-5-phosphate 4-kinase-beta (PI5P4Kβ; Emerling et al., 2013), or AKT could induce mitochondrial targeting of a FLAG-tagged TIGAR protein in cotransfected HT-1080 cells (the expression of the mutant kinases and FLAG-tagged TIGAR is shown in Fig. S3B). These studies demonstrated that the kinase mutants, PI3K, PI5P4K, and AKT, induced mitochondrial targeting of the TIGAR (FLAG) in these cells, as compared to an empty vector control which exhibited diffuse cytoplasmic TIGAR immunostaining (Fig. 2C). As PI3K and AKT are downstream effectors of Ras-signaling, and others have reported that the H-Ras oncoprotein is dysregulated in aggressive hrHPV+ cervical cancers associated with metastasis and high incidences of recurrence (Landro et al., 2008), we examined the expression of H-Ras in HPV18-transformed cervical carcinoma cell-lines. These results are provided in Fig. S3C and demonstrate that H-Ras is elevated in these cells as compared to an HPV-negative control.
Fig. 2.
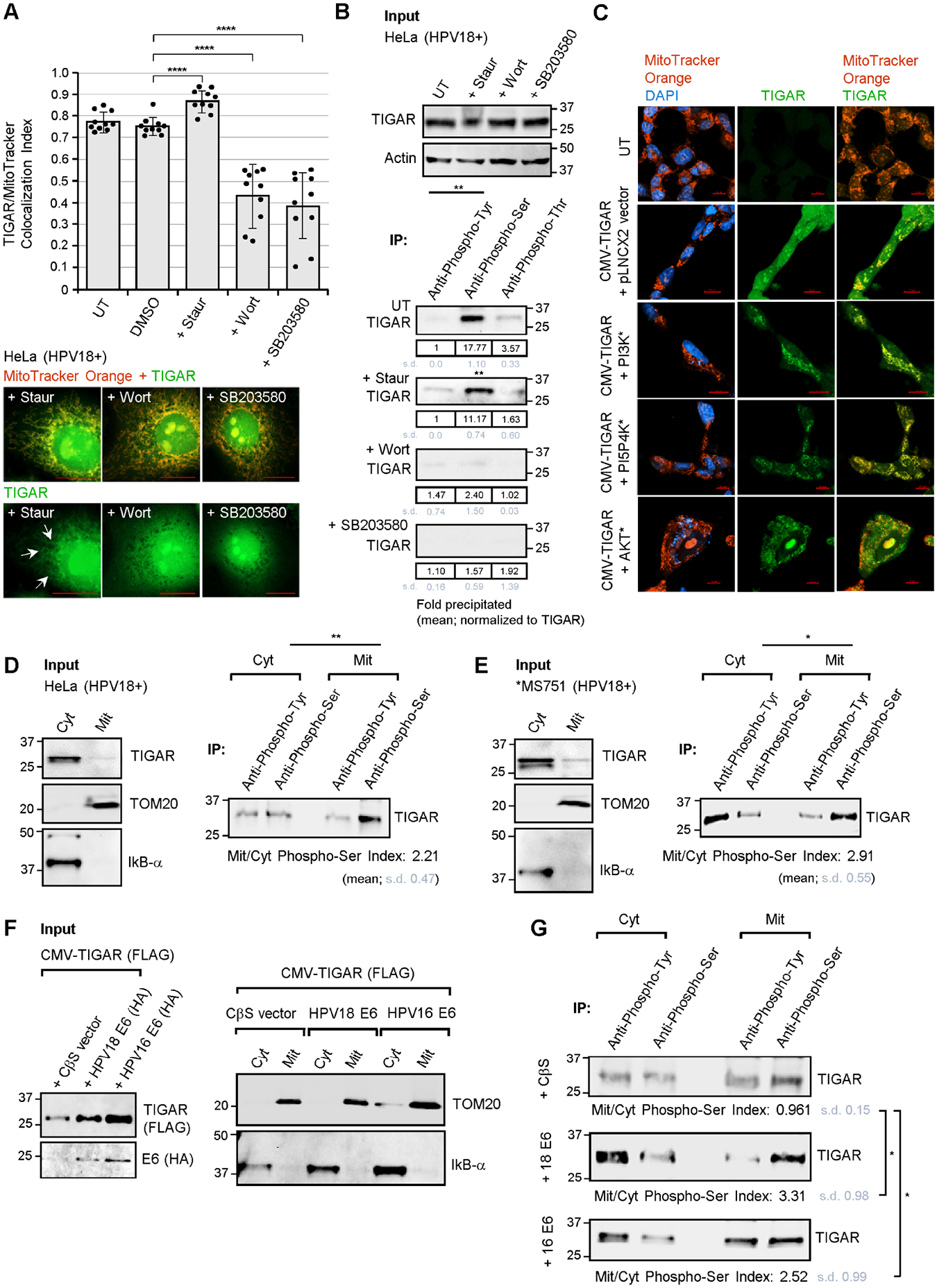
The viral E6 oncoprotein activates cellular kinases and induces serine-phosphorylation of the TIGAR protein associated with its mitochondrial localization. (A) The mitochondrial targeting of TIGAR in HPV18+ HeLa cervical carcinoma cells that were treated with various chemical kinase inhibitors (i.e., staurosporine, wortmannin, or SB203580) was assessed by measuring the colocalization between TIGAR and the MitoTracker Orange label by immunofluorescence-confocal microscopy using the ZEN OS colocalization module. Representative micrographs depict colocalization between the TIGAR protein (green signal; see arrows) and MitoTracker Orange-labeled structures (red signal) in staurosporine-treated cells, as compared to the kinase inhibitors wortmannin and SB203580 (additional micrographs are provided in Fig. S3A). (B) The effects of the chemical kinase inhibitors, staurosporine, wortmannin, and SB203580, upon the phosphorylation of TIGAR were determined by immunoprecipitating the TIGAR protein from HeLa cell extracts using monoclonal Anti-Phospho-Tyrosine, Anti-Phospho-Serine, and Anti-Phospho-Threonine antibodies (Millipore-Sigma) and Protein G-agarose beads and then immunoblotting with a rabbit polyclonal Anti-TIGAR antibody (Millipore-Sigma, ab2). The serine-phosphorylation of TIGAR was inhibited by the chemical PI3K-inhibitor, wortmannin, and the p38MAPK-inhibitor, SB203580, but not by staurosporine. (C) To determine whether constitutively-active mutants of PI3K, or the closely related PI5P4K, and AKT could effect mitochondrial targeting of the TIGAR protein, HT-1080 cells were cotransfected with CMV-TIGAR (FLAG) and expression constructs for the various kinase mutants or an empty pLNCX2 vector as negative control. The expression of the constitutively-active kinase mutants and TIGAR-FLAG was detected by immunoblotting as shown in Fig. S3B. The cells were labeled with MitoTracker Orange (red signal) and immunofluorescence-confocal microscopy was performed to visualize mitochondrial localization of the TIGAR protein (green signal). DAPI nuclear-staining (blue signal) was included for reference in the merged images. Scale bar, 20 μm. (D and E) To confirm the role of serine-phosphorylation in the mitochondrial targeting of TIGAR, cytoplasmic and mitochondrial extracts were prepared from HPV18-transformed HeLa (in D) and MS751 cells (in E; *MS751 cells also contain HPV45 sequences) and immunoprecipitations were performed using Anti-Phospho-Serine and Anti-Phospho-Tyrosine antibodies and Protein G-agarose beads. The relative input levels of TIGAR are shown in the left panels. The precipitated phosphorylated TIGAR protein was detected by immunoblotting and the Mitochondrial/Cytoplasmic Phospho-Serine Index was calculated as described in the Materials and methods. (F and G) To determine if the HPV E6 oncoprotein induces serine-phosphorylation of the TIGAR protein associated with its mitochondrial targeting, HT-1080 cells were cotransfected with CMV-TIGAR (FLAG-tagged) and expression constructs for HPV16/18 E6 (HA-tagged) or an empty CβS vector as negative control. The input levels of TIGAR (FLAG) and the HPV16/18 E6 (HA) proteins were assessed by immunoblotting (in F). Cytoplasmic and mitochondrial fractions were prepared and immunoprecipitations were performed as described. The precipitated phosphorylated TIGAR protein was detected by immunoblotting and quantified using densitometry (in G). Data is mean ± SD. N-value = 3. The asterisks denote statistical significance as determined using unpaired two-tailed Student’s t-tests ((*P<0.0332, **P<0.0021, ****P<0.0001).
To determine if the E6-induced serine-phosphorylation of the TIGAR protein correlates with its translocation to mitochondria, we isolated mitochondrial and cytoplasmic fractions from the HPV18-transformed cell-lines, HeLa and MS751, and then performed immunoprecipitations using various phospho-specific antibodies. The purity of the input fractions was confirmed by immunoblotting to detect the inhibitor of kappa B-alpha (IκB-α; a cytoplasmic marker) and TOM20 (a mitochondrial marker; Figs. 2D and 2E, Input panels). As noted by others, the majority of the TIGAR protein was localized in the cytoplasm with a minor fraction targeted to mitochondrial membranes (Figs. 2D and 2E, Input panels). Surprisingly, the TIGAR protein immunoprecipitated from mitochondrial fractions was strongly phosphorylated on serine-residues in both HPV18+ cell-lines (Figs. 2D and 2E, IP panels). Our microscopy data in Figs. 1F and 1H, and supplementary Figs. S2B and S2C demonstrated that the ectopically expressed hrHPV E6 oncoproteins induce hypoxia-independent mitochondrial targeting of the TIGAR. We therefore isolated mitochondrial and cytoplasmic fractions from HT-1080 cells that were cotransfected with expression constructs for FLAG-tagged TIGAR and either HPV16/18 E6 (HA) or an empty vector as negative control. TOM20 and IκB-α served as markers for the mitochondrial and cytoplasmic fractions, respectively (Fig. 2F). Immunoprecipitations were performed using phospho-specific antibodies and these experiments demonstrated that the mitochondrial TIGAR (FLAG) protein, consistently, was strongly phosphorylated on serine residues in cells expressing the hrHPV E6 oncoproteins as compared to an empty vector control (Fig. 2G). We would note that tyrosine-phosphorylation of the TIGAR protein was also observed in some experiments; however, this modification was not consistently associated with the subcellular trafficking and mitochondrial targeting of TIGAR (i.e., compare the mean Cyt/Mit Anti-Phospho-Tyr signals in Figs. 2D and the lower panel of Fig. 2G). Furthermore, the tyrosine-phosphorylation signal was much weaker than the serine-phosphorylation of TIGAR in the IP results for untreated HeLa cells in Fig. 2B. Based upon these findings, we conclude that E6 activates cellular signaling components (possibly through HGF/SF-Met and/or Ras) and induces the serine-phosphorylation and hypoxia-independent mitochondrial targeting of TIGAR which could protect proliferating HPV-infected cells against metabolic oxidative stress and ROS-induced cytotoxicity.
2.3. The hrHPV E6 proteins cooperate with the cellular c-Myc oncoprotein in a TIGAR-dependent manner
The E6 and E7 proteins coordinately activate the expression of S-phase-promoting genes to drive vegetative replication of the HPV episomal dsDNA genome in differentiated keratinocytes during the infection cycle. The E7 oncoprotein has been shown to increase the expression of c-Myc by enhancing its cap-independent translation through an IRES which is independent of the ability of E7 to degrade Rb (Strickland and Vande Pol, 2016). The E6 protein also functionally cooperates with c-Myc and induces cellular immortalization through c-Myc-dependent transactivation of E-box enhancer elements within the htert promoter (McMurray and McCance, 2003; Liu et al., 2008; Peta et al., 2018). Interestingly, the p53 gene is a downstream target of Myc and aberrant oncogenic expression of c-Myc can result in increased ROS production, oxidative genomic DNA-damage and p53-dependent cellular apoptosis (Reisman et al., 1993; Zindy et al., 1998; Vafa et al., 2002; Hermeking and Eick, 1994). We therefore investigated whether the hypoxia-independent activation of mitochondrial TIGAR is required for cooperation between the hrHPV E6 and c-Myc oncoproteins. Our results in Fig. 3A demonstrate that lentiviral small-interfering RNA (siRNA)-knockdown of TIGAR expression resulted in the accumulation of damaging ROS in transduced HPV18-transformed cervical cancer cell-lines (HeLa, MS751, and C-4 I), as compared to HPV18+ cells transduced with an empty lentiviral vector or HPV-negative (HFL1) cells. Representative micrographs are provided in Fig. 3A (lower panels). Next, to determine if the HPV16/18 E6 proteins could inhibit the production of ROS in cells which overexpress the c-Myc oncoprotein, HT-1080 cells were cotransfected with expression constructs for HPV16/18 E6 and/or c-Myc. The samples were then transduced with lentiviral-siRNA-tigar to inhibit endogenous TIGAR expression, or an empty pLenti vector as negative control. As shown in Fig. 3B, these studies revealed that the E6 oncoproteins cooperate with c-Myc and prevented the oxidative stress and accumulation of ROS induced by aberrant c-Myc overexpression. The transduction of c-Myc/hrHPV E6-expressing cells with lentiviral-siRNA-tigar countered the ability of E6 to protect against c-Myc-induced ROS, as compared to cells transduced with an empty lentiviral vector (Fig. 3B). We further tested whether the induction of TIGAR’s antioxidant functions by hrHPV E6 oncoproteins could prevent c-Myc-induced mitochondrial membrane depolarization by staining the cotransfected cells with the fluorescent probe, JC-1 (Invitrogen), which emits red fluorescence when it aggregates on polarized mitochondrial membranes, but releases a cytoplasmic green fluorescent signal associated with damaged/depolarized mitochondria. As shown in Fig. 3C, oncogenic c-Myc expression resulted in a higher green-to-red signal ratio, indicative of increased mitochondrial membrane depolarization, that was countered by ectopic co-expression of the HPV16/18 E6 proteins (see representative micrographs). The transduction of c-Myc/hrHPV E6-expressing cells with lentiviral-siRNA-tigar interfered with the ability of E6 to prevent damage to mitochondrial membranes caused by c-Myc overexpression, as compared to an empty lentiviral vector control (Fig. 3C). The siRNA-knockdown of TIGAR similarly resulted in increased mitochondrial membrane depolarization in transduced HPV18-transformed cervical cancer cell-lines, compared to HPV18+ cells transduced with an empty lentiviral vector or HPV-negative cells (Fig. 1D). The inhibition of TIGAR expression in transduced HeLa cells by lentiviral-siRNA-tigar was demonstrated by immunoblotting and quantified by densitometry and is shown in Fig. 3E; also, the expression of the FLAG-tagged c-Myc protein in transfected cells is shown in Fig. 1F. Consistent with a general protective role of TIGAR’s antioxidant functions in HPV+ cells, our studies further demonstrated that HPV18-transformed HeLa cells are more resistant to oxidative stress-induced cytotoxicity than HPV-negative 293 HEK cells as determined by treating the cultures with hydrogen peroxide, followed by the quantitation of cellular apoptosis by Annexin V-FITC/PI-staining and confocal microscopy analysis (Figs. S5A and S5B).
Fig. 3.
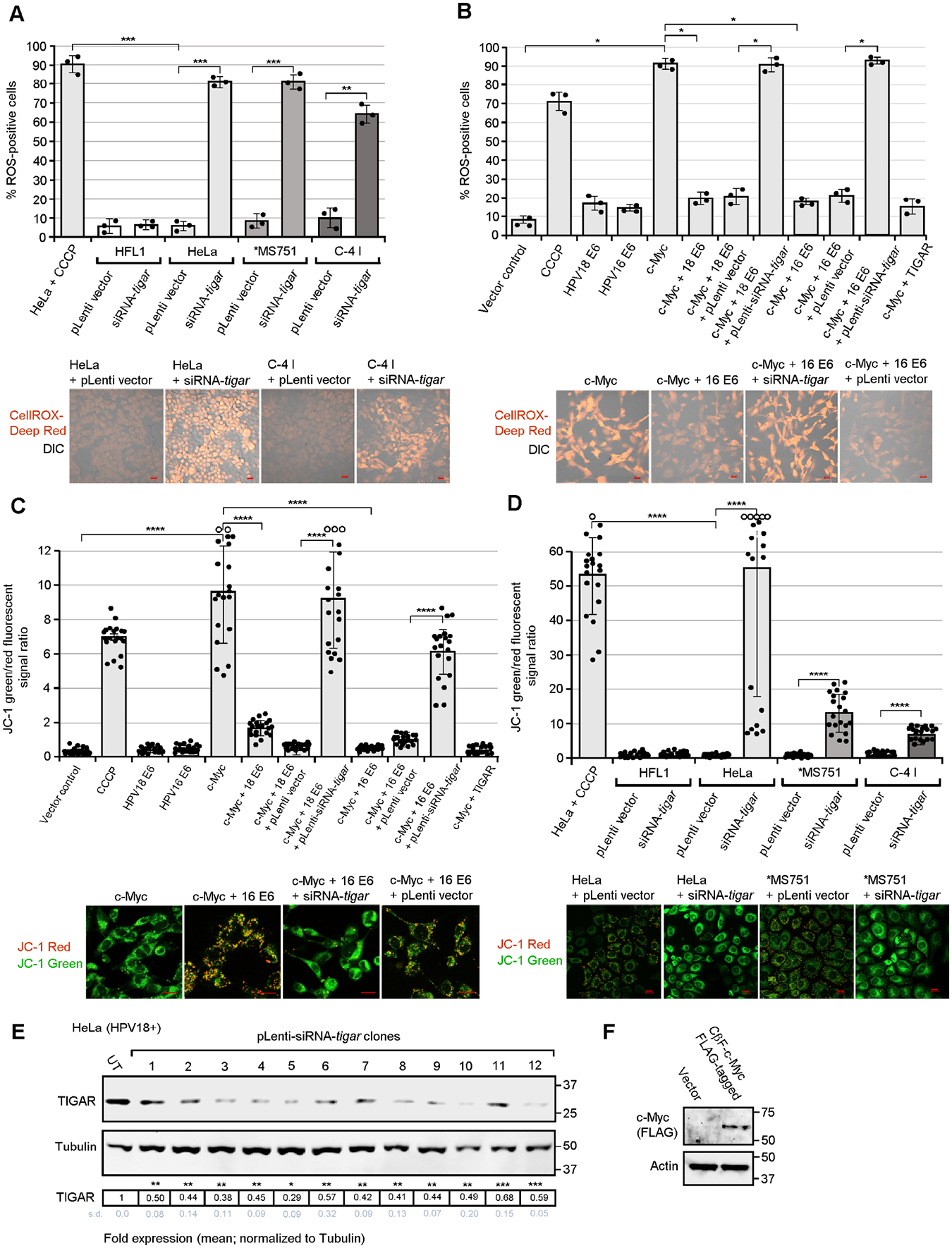
The hrHPV E6 oncoproteins cooperate with c-Myc and inhibit oncogene-induced oxidative stress and mitochondrial membrane depolarization in HPV-transformed cervical carcinoma cells through the activation of TIGAR. (A) To determine the effects of inhibiting TIGAR upon the accumulation of damaging ROS, the HPV18+ cell-lines, HeLa, MS751, and C-4 I were transduced with a lentiviral siRNA-tigar expression vector to knockdown endogenous TIGAR protein levels or an empty lentiviral vector (pLenti) as negative control. The transduced cells were labeled with a fluorescent ROS chemical probe, CellROX-Deep Red (Invitrogen), and the relative percentages of ROS-positive cells were quantified by confocal microscopy. HeLa cells treated with the chemical uncoupler CCCP (50 μM; Millipore-Sigma) were included as a positive control. Human HFL1 fibroblasts (HPV-negative) are shown for comparison. Representative micrographs are provided in the lower panels. (B) The ability of hrHPV E6 oncoproteins to cooperate with c-Myc and prevent the accumulation of oncogene-induced ROS was assessed by cotransfecting HT-1080 cells with a CβF-c-Myc (FLAG-tagged) construct and either an HPV16/18 E6 expression plasmid or empty CβS vector as negative control. To determine if the protective effects of E6 were dependent upon TIGAR’s antioxidant functions, certain samples were transduced with an empty pLenti vector or lentiviral-siRNA-tigar to inhibit TIGAR expression. (C and D) The effects of the viral E6 oncoprotein and TIGAR functions upon c-Myc-induced mitochondrial membrane depolarization were determined by transducing HT-1080 cells, cotransfected with expression constructs for c-Myc and/or the HPV16/18 E6 oncoproteins (in C), or the HPV18+ cell-lines: HeLa, MS751, and C-4 I (in D), with lentiviral-siRNA-tigar or an empty pLenti vector as negative control. The samples were then stained with the fluorescent chemical probe JC-1 (Invitrogen) and confocal microscopy was performed to quantify the ratios of the relative fluorescence intensities for the JC-1 red (indicative of polarized mitochondrial membranes) and green (which indicates depolarization) signals using Carl Zeiss Microscopy ZEN OS imaging software. The open circles indicate data out-of-range. Graphed data in A-D is mean ± SD. Representative micrographs are shown in the lower panels. Scale bar, 20 Pm. (E) HPV18-transformed HeLa cells were transduced with various lentiviral-siRNA-tigar clones and the effective knockdown of TIGAR protein expression was assessed by immunoblotting and densitometry quantitation relative to Tubulin levels. UT, untransduced cells. (F) The FLAG-tagged c-Myc oncoprotein was also detected in transfected cells by immunoblotting. N-value = 3. The asterisks denote statistical significance as determined using unpaired two-tailed Student’s t-tests (*P<0.0332, **P<0.0021, ***P<0.0002, ****P<0.0001).
To determine if the activation of mitochondrial TIGAR by the E6 oncoprotein could prevent c-Myc-induced oxidative genomic DNA-damage, we performed 594 nm fluorescent terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL) analyses on HT-1080 cells transfected with expression constructs for c-Myc and the HPV16/18 E6 proteins that were also transduced with lentiviral-siRNA-tigar or an empty lentiviral vector. The results in Fig. 4A demonstrate that oncogenic c-Myc resulted in increased numbers of TUNEL-positive intracellular foci that were markedly reduced in the presence of the HPV16/18 E6 oncoproteins (see representative micrographs). This protective effect was countered by transducing c-Myc/hrHPV E6-expressing cells with lentiviral-siRNA-tigar, as compared to an empty lentiviral vector (Fig. 4A). We then tested the effects of inhibiting TIGAR upon recruitment of the serine-139 phosphorylated histone variant, γH2A.X, to sites of damaged chromatin associated with c-Myc-induced oxidative DNA-damage by performing immunofluorescence-confocal microscopy using an Anti-Phospho-γH2A.X primary antibody (see Table S1). The data in Fig. 4B demonstrate that c-Myc overexpression resulted in DNA-damage, as evidenced by increased numbers of phospho-γH2A.X foci (red fluorescent nuclear structures in the micrographs), that was countered by ectopic expression of the hrHPV E6 proteins. The lentiviral-siRNA-tigar knockdown of TIGAR blocked the ability of the viral E6 oncoproteins to prevent c-Myc-induced DNA-damage in transduced cells, compared to the empty lentiviral vector control (Fig. 4B). The siRNA-inhibition of TIGAR also resulted in increased genomic DNA-damage and phospho-γH2A.X foci in the transduced HPV18-transformed cervical cancer cell-lines, in contrast to the empty vector or HPV-negative cells (Fig. 4C; see circled nuclear foci in the enlarged inset).
Fig. 4.
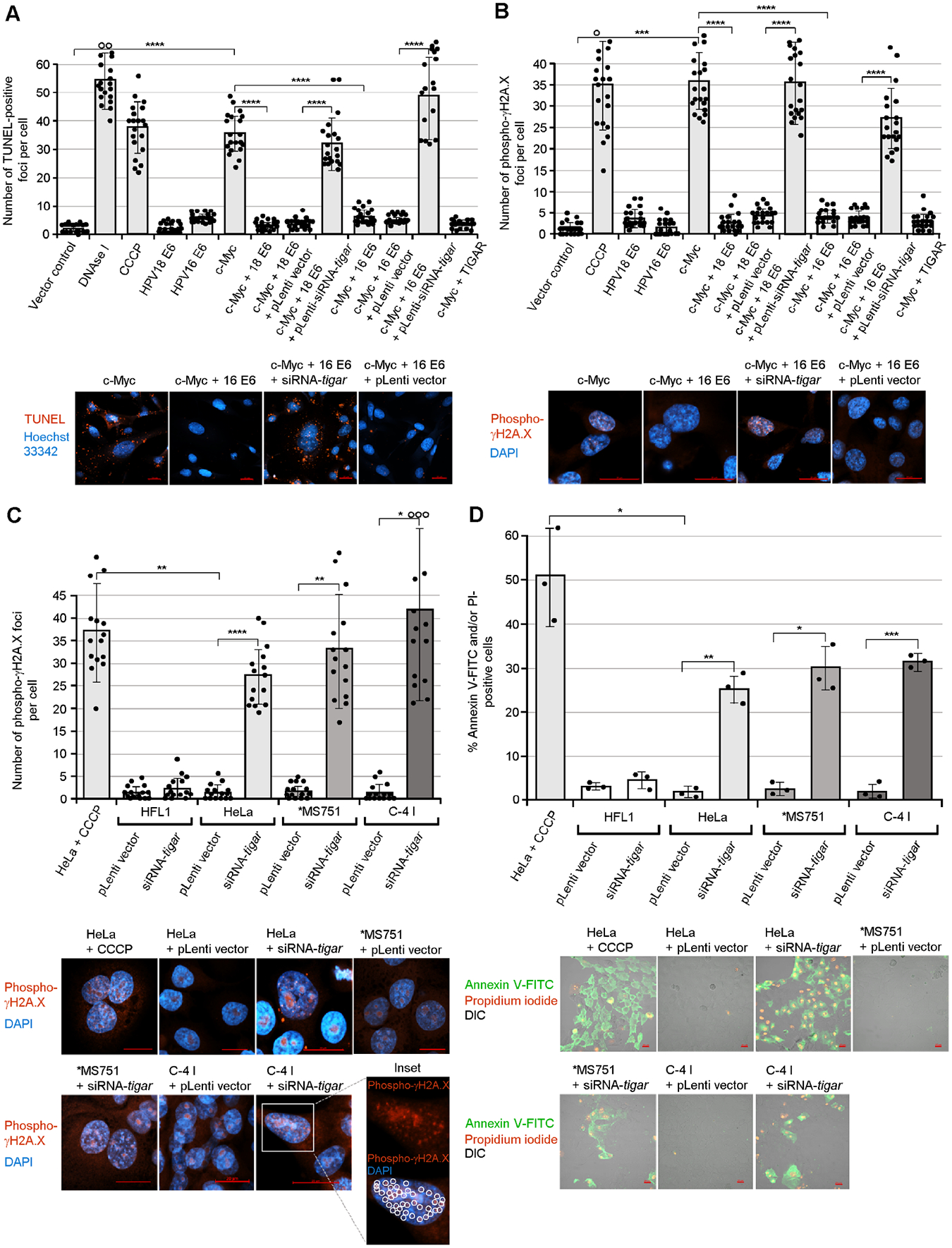
The viral E6 oncoproteins inhibit oncogene-induced genotoxicity and cellular apoptosis in a TIGAR-dependent manner. (A) HT-1080 cells were cotransfected with expression constructs for c-Myc and/or HPV16/18 E6 and then transduced with either lentiviral-siRNA-tigar or an empty pLenti vector as negative control. The samples were stained using a 594 nm fluorescent Click-iT Plus TUNEL kit (Invitrogen) and confocal microscopy was performed to quantify the relative numbers of TUNEL-positive foci per cell (red signal in the micrographs). Hoechst 33342 nuclear-staining (blue signal) is shown for reference. (B) To determine if the induction of TIGAR by hrHPV E6 oncoproteins protects c-Myc-expressing cells against oxidative DNA-damage, HT-1080 cells were transfected and transduced as described and then immunostained using a rabbit polyclonal Anti-Phospho-γH2A.X primary antibody and Rhodamine red-X-conjugated Anti-Rabbit secondary antibody to detect recruitment of this phosphorylated histone variant to sites of DNA-damage. Immunofluorescence-confocal microscopy was performed to quantify the relative numbers of phospho-γH2A.X foci per cell (red signal in the micrographs). (C) The accumulation of DNA-damage and nuclear phospho-γH2A.X foci in the HPV18+ cervical cancer cell-lines, HeLa, MS751, and C-4 I, that were transduced with either lentiviral-siRNA-tigar or an empty pLenti vector as negative control was detected and quantified using immunofluorescence-confocal microscopy as in B. The open circles indicate data out-of-range. Representative micrographs are provided in the lower panels. The red punctate phospho-γH2A.X foci are indicated by circles in the enlarged inset. (D) The requirement for TIGAR functions to protect against oncogene-induced apoptosis was confirmed by transducing HPV18-transformed cervical carcinoma cell-lines (HeLa, MS751, and C-4 I) with lentiviral-siRNA-tigar or an empty pLenti vector and then staining the cells with Annexin V-FITC and propidium iodide (PI; BD-Pharmingen). HeLa cells treated with CCCP were included as a positive apoptosis control; and transduced (HPV-negative) HFL1 cells are shown for comparison. The relative percentages of apoptotic (Annexin V-FITC and/or PI-positive) cells per field were quantified using confocal microscopy and counting triplicate fields at 200x magnification. The data in A-D is mean ± SD. N-value = 3. The asterisks denote statistical significance as determined using unpaired two-tailed Student’s t-tests (*P<0.0332, **P<0.0021, ***P<0.0002, ****P<0.0001). Representative micrographs are shown in the lower panels. Scale bar, 20 μm.
We next investigated whether the hypoxia-independent activation of mitochondrial TIGAR by E6 could prevent oncogene-induced apoptosis. Our results in Fig. 4D demonstrate that lentiviral-siRNA-tigar inhibition of TIGAR expression resulted in increased apoptosis in the HPV18-transformed cell-lines (HeLa, MS751, and C-4 I) compared to HPV18+ cells transduced with an empty lentiviral vector or HPV-negative cells, as determined by staining the samples with Annexin V-fluorescein isothiocyanate (Annexin V-FITC) and propidium iodide (PI) followed by confocal microscopy analysis. Moreover, as shown in supplementary Figs. S6A and S6B, the oncogenic overexpression of c-Myc resulted in significant apoptosis and an increased number of Annexin V-FITC and/or PI-positive cells that were countered by the hrHPV E6 oncoproteins. However, the inhibition of TIGAR functions by transducing c-Myc/hrHPV E6-expressing cells with lentiviral-siRNA-tigar interfered with the ability of E6 to protect cells against c-Myc-induced programmed cell death (Figs. S6A and S6B). Additional representative micrographs, demonstrating that the ability of the HPV18 E6 oncoprotein to protect against c-Myc-induced oxidative damage and genotoxicity is dependent upon TIGAR functions, are provided in supplementary Figs. S7A–D.
2.4. The activation of TIGAR by hrHPV E6 oncoproteins is dependent upon wild-type p53 functions and the inhibition of p53-K120-acetylation by TIP60
The HPV E6 oncoprotein destabilizes the acetyltransferase TIP60 through interactions with the ubiquitin ligase EDD1/UBR5 and inhibits TIP60-mediated p53-K120-acetylation and p53-dependent apoptosis (Jha et al., 2010; Subbaiah et al., 2016). We therefore investigated the effects of ectopic TIP60 overexpression upon p53-K120-acetylation and TIGAR protein expression in the HPV18-transformed cell-lines, C-4 I and HeLa. The data in Figs. 5A and 5B demonstrate that the overexpression of TIP60 markedly reduced the expression of TIGAR in these cells and correlated with increased p53-K120-acetylation, as determined by immunoprecipitations using a monoclonal Anti-K120-Acetyl-p53 antibody (the expression of HA-tagged TIP60 and p53 in the transfected HPV18+ cell-lines was visualized by immunofluorescence-microscopy as shown in Fig. S8C; and the TIP60 (HA) protein was also detected by immunoblotting as shown in Fig. S8B). To determine if the induction and mitochondrial targeting of TIGAR by the hrHPV E6 and c-Myc oncoproteins are dependent upon wild-type p53 functions, a p53-null carcinoma cell-line, HCT116 (which contains a homozygous CRISPR knockout of p53), was transfected with various expression constructs for HPV16/18 E6, c-Myc, wild-type p53, or a transcriptionally-inactive p53-R175H DNA-binding mutant (Hermeking et al., 1997) and the relative expression and subcellular localization of TIGAR were quantified by immunofluorescence-confocal microscopy. These results demonstrated that the induction of TIGAR expression by hrHPV E6/c-Myc requires p53 functions, since the co-expression of wild-type p53 resulted in higher TIGAR protein levels, in contrast to the DNA-binding mutant p53-R175H (Figs. 5C and 5D). We also observed that the TIGAR protein in HPV18 E6/c-Myc/wild-type p53-expressing cells was targeted to punctate mitochondria-like structures, as compared to HPV18 E6/c-Myc/p53-R175H-expressing cells or the ectopic TIGAR protein which exhibited a diffuse/cytoplasmic subcellular distribution (Fig. 5D, inset panels). The expression of the wild-type p53 protein in HPV18 E6/c-Myc-expressing HCT116 p53-null cells was confirmed by immunoblotting (Fig. S8A).
Fig. 5.
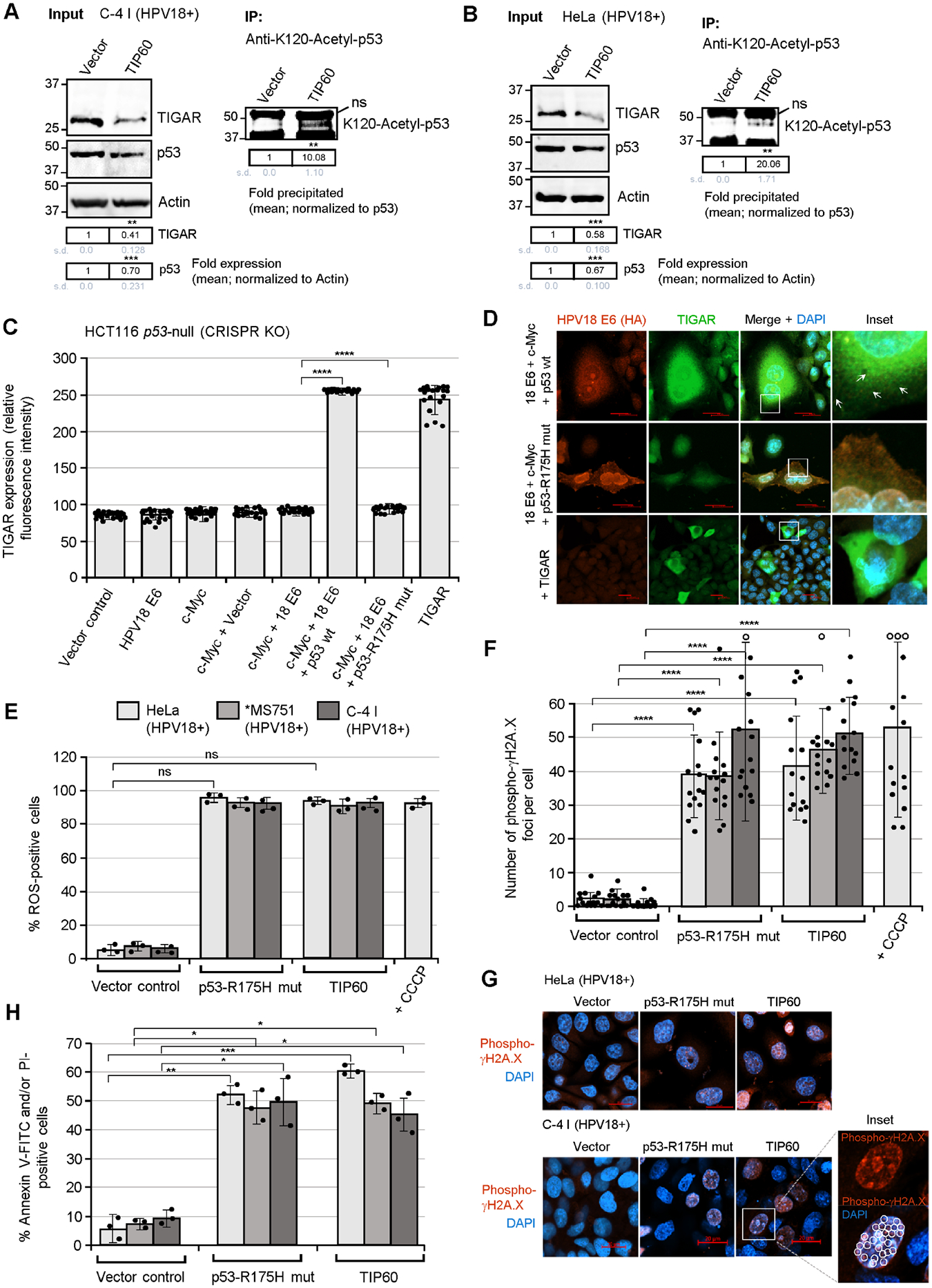
The activation of TIGAR by the E6 oncoprotein is dependent upon p53 functions and the inhibition of p53 lysine K120-acetylation in hrHPV-transformed cells. (A and B) To determine if the inhibition of TIP60-mediated lysine K120-acetylation of p53 is required for the expression of TIGAR in hrHPV-transformed cells, the HPV18+ C-4 I (in A) and HeLa (in B) cell-lines were transfected with either an expression construct for wild-type TIP60 (HA-tagged) or an empty CβS vector as negative control. The overexpression of TIP60 (HA) was confirmed by immunofluorescence-microscopy and immunoblotting and is shown in Figs. S8B and S8C. The input levels of TIGAR and p53 were quantified and normalized relative to Actin expression. The K120-acetylated p53 protein was immunoprecipitated using a monoclonal Anti-K120-Acetyl-p53 antibody (Abcam) and Protein G-agarose quantified with normalization for the relative input levels of p53 (right panels). (C and D) To determine whether p53 is required for the expression and mitochondrial targeting of TIGAR by the viral E6 oncoproteins, the p53-null human carcinoma cell-line, HCT116 p53−/− (which contains a homozygous CRISPR-knockout of the p53 gene; Horizon Discovery), was transfected with various expression constructs for c-Myc, HPV18 E6 (HA), wild-type p53, or a dominant-negative p53-R175H DNA-binding mutant, and immunofluorescence-confocal microscopy was performed to quantify and visualize the TIGAR protein in punctate mitochondrial structures (see arrows in D). The relative fluorescence intensity of the TIGAR-specific signal was measured using ZEN OS imaging software and graphed in C. The expression of wild-type p53 in the HCT116 p53-null cells cotransfected with expression constructs for HPV18 E6, c-Myc, and p53 was detected by immunoblotting and is shown in Fig. S8A. (E) The effects of the dominant-negative p53-R175H mutant and overexpression of TIP60 upon the accumulation of damaging ROS in hrHPV-transformed cells were assessed by transfecting the HPV18+ cell-lines, HeLa, MS751, and C-4 I, with CMV-p53-R175H, pOZ-TIP60 (HA), or an empty CβS vector control and then staining the cells with a fluorescent chemical ROS probe, CellROX-Deep Red, followed by confocal microscopy analysis. (F and G) The HPV18+ cell-lines, HeLa, MS751, and C-4 I, were transfected as described in E and then immunostained using an Anti-Phospho-γH2A.X primary antibody to visualize the recruitment of this phosphorylated histone variant to sites of DNA-damage. HeLa cells treated with CCCP were included as a positive control. Immunofluorescence-confocal microscopy was performed to quantify the relative numbers of phospho-JH2A.X foci (red signal in G) per cell. The open circles indicate data out-of-range. DAPI nuclear-staining (blue signal in G) is provided for reference. An enlarged inset area shows several phospho-γH2A.X-positive punctate structures (circles) within the nucleus of a C-4 I cell transfected with the pOZ-TIP60 expression construct. Scale bar, 20 μm. (H) The requirement for p53 functions and the effects of TIP60 overexpression upon cellular apoptosis in hrHPV-transformed cells were determined by transfecting HPV18+ cell-lines (HeLa, MS751, and C-4 I) with CMV-p53-R175H, pOZ-TIP60 (HA), or an empty CβS vector and then staining the cells with Annexin V-FITC and PI (BD-Pharmingen). A DIC filter was used to visualize all cells in the merged images. Data in C, E, F, and H is mean ± SD. N-value = 3. The asterisks denote statistical significance as determined using unpaired two-tailed Student’s t-tests (*P<0.0332, **P<0.0021, ***P<0.0002, ****P<0.0001, not significant (ns) 0.1234).
Based upon these findings, we next sought to determine whether wild-type p53 functions and the inhibition of TIP60-mediated p53-K120-acetylation are needed to protect hrHPV-infected cells against oncogene-induced oxidative damage and cytotoxicity. For these studies, the HPV18+ cervical cancer cell-lines, HeLa, MS751, and C-4I, were transfected with expression constructs for the dominant-negative p53 DNA-binding mutant, p53-R175H, or wild-type TIP60, or an empty vector as negative control. These results demonstrate that the inhibition of endogenous p53 functions by the p53-R175H mutant, or overexpression of TIP60, caused the accumulation of damaging ROS (Fig. 5E) and greater numbers of phospho-γH2A.X foci in HPV18-transformed cells as compared to the empty vector control (Figs. 5E–G). We also found that the p53-R175H mutant or overexpression of the acetyltransferase TIP60 resulted in increased apoptosis in the HPV18-transformed cell-lines, as determined by staining the samples with Annexin V-FITC and PI, followed by confocal microscopy analysis (Fig. 5H).
2.5. TIGAR expression is essential for HPV-induced tumorigenesis and correlates with oncogenic c-Myc dysregulation in HPV16+ cervical cancer clinical isolates
The hrHPV E6 oncoproteins have been shown to cooperate with c-Myc and deregulate cellular growth/proliferation in vitro (McMurray and McCance, 2003; Liu et al., 2008; Peta et al., 2018). We therefore tested if the viral E6 oncoproteins and c-Myc could induce the anchorage-independent growth of cotransfected primary human dermal (neonatal foreskin) fibroblasts in soft agar colony-formation assays and whether the oncogenic cooperation between hrHPV E6/c-Myc is dependent upon TIGAR functions. The data in Fig. 6A demonstrate that the co-expression of either HPV18E6/c-Myc or HPV16E6/c-Myc resulted in significant colony-formation and increased numbers of anchorage-independent spheroids which were inhibited by transducing these cells with lentiviral-siRNA-tigar, as compared to an empty lentiviral vector control. The representative micrographs in the lower panels show the relative numbers of colonies formed in HPV16 E6/c-Myc transfected cells and the HPV16 E6/c-Myc/siRNA-tigar or HPV16 E6/c-Myc/pLenti vector transduced cells (Fig. 6A). These functional studies demonstrate that the cooperation between the hrHPV E6 oncoproteins and c-Myc is dependent upon the pro-survival activity of TIGAR.
Fig. 6.
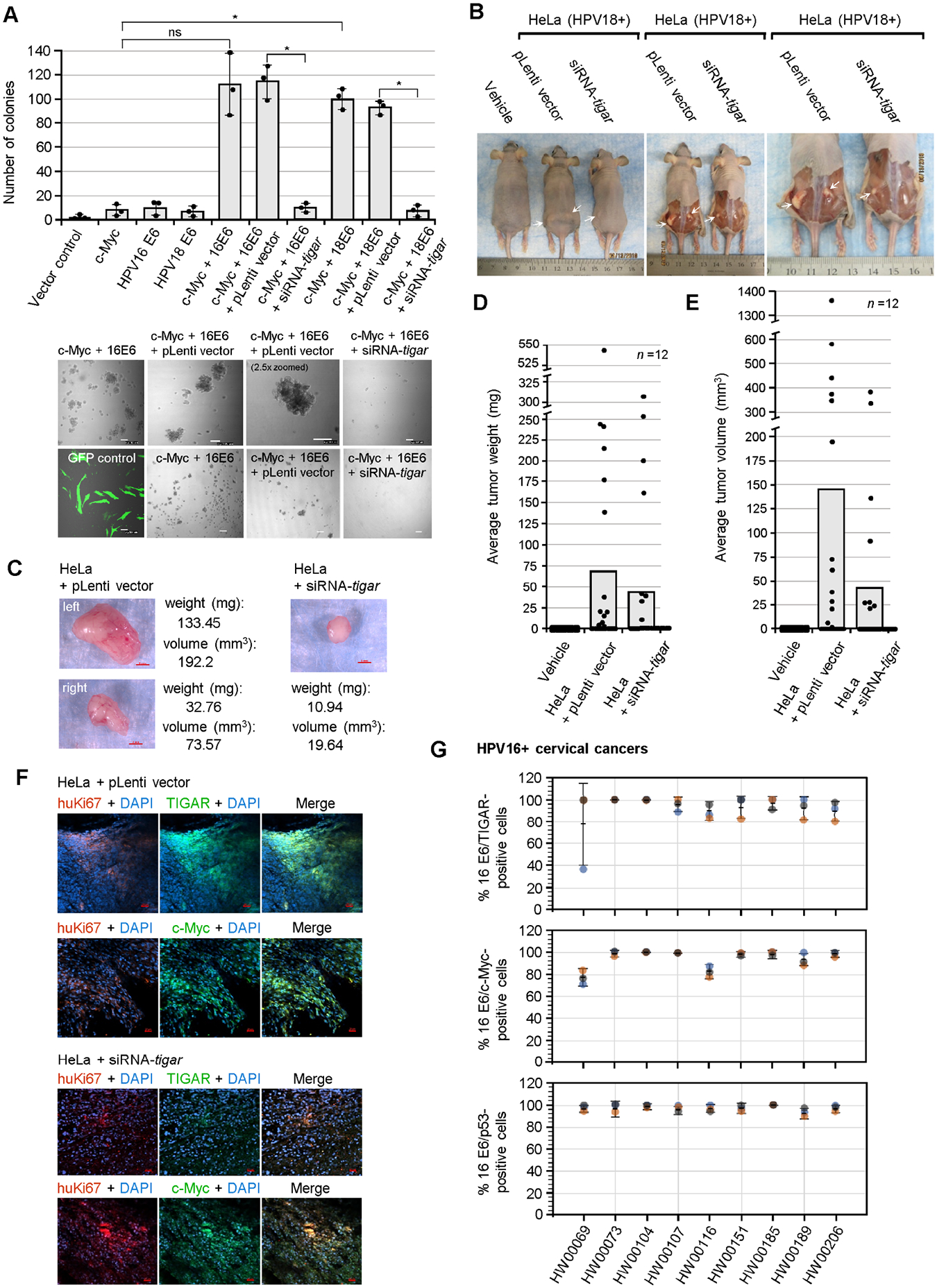
TIGAR is required for the oncogenic cooperation between E6/c-Myc and hrHPV-induced tumorigenesis in vivo. (A) The role of TIGAR in oncogenic cellular transformation by the viral E6 oncoprotein and c-Myc was assessed by cotransfecting primary human dermal fibroblasts (neonatal foreskin, HDFn; ATCC) with various expression constructs for HPV16/18 E6, c-Myc, or an empty CβS vector and then transducing the cells with lentiviral-siRNA-tigar or the pLenti vector as a negative control. The transfection efficiency was evaluated using a N3-pEGFP plasmid and performing fluorescence-microscopy with a DIC filter overlay to visualize GFP expression (bottom left). Soft agar colony assays were performed over a period of 3 weeks and the transformed anchorage-independent spheroids were quantified by counting. Representative micrographs of the transformed colonies are shown in the lower panels. Scale bar, 100 μm. Data is mean ± SD. N-value = 3. The asterisks denote statistical significance as determined using unpaired two-tailed Student’s t-tests (*P<0.0332, not significant (ns) 0.1234). (B) To determine if TIGAR functions are required for hrHPV-induced tumorigenesis in vivo, athymic NIH III-nude mice were subcutaneously engrafted over each hind flank with HPV18+ HeLa cells that were transduced with lentiviral-siRNA-tigar to inhibit TIGAR expression, or an empty lentiviral (pLenti) vector as negative control. Alternatively, a group of animals was injected with the vehicle alone (i.e., sterile Ca2+/Mg2+-free PBS, pH 7.4) for comparison (n-value = 12). After 8 weeks, the experimental animals were humanely sacrificed and necropsies were performed to analyze the growth of the primary xenograft tumors (a Kaplan-Meier plot of tumor development, as well as graphs of the relative percentages of animals that developed tumors or exhibited bilateral tumor growth are provided in Figs. S9A–C). The arrows in B indicate the significantly larger tumor masses present in the HeLa/pLenti vector animals as compared to the HeLa/lentiviral-siRNA-tigar animals. (C) The harvested tumor masses were weighed and their volumes were determined by measuring with a digital caliper. Representative images are provided. Scale bar, 2 mm. (D) The tumors derived from HeLa/lentiviral-siRNA-tigar engrafted animals exhibited a reduced average weight as compared to the HeLa/pLenti vector animals. (E) The tumor masses from the HeLa/lentiviralsiRNA-tigar animals also exhibited a smaller average volume than the HeLa/pLenti vector sample group. (F) The expression of TIGAR and c-Myc (green signals) in the xenograft tumor tissues from engrafted HeLa/pLenti vector and HeLa/siRNA-tigar experimental animals was analyzed by immunofluorescence-confocal microscopy. The engrafted HeLa tumor cells were visualized by co-staining the samples with an Anti-human-Ki67 (huKi67) primary antibody (red signal). DAPI nuclear-staining (blue signal) is included in the merged images for reference. Scale bar, 20 μm. (G) The TIGAR, c-Myc, and p53 proteins are detectably expressed in the E6-positive tumor cells of primary HPV16+ cervical cancer patient isolates. Clinically biopsied HPV16+ cervical cancer tissue specimens were provided by the Pathology Shared Resource of the University Hawaii Cancer Center and the relative percentages of HPV16 E6/TIGAR-positive, HPV16 E6/c-Myc-positive, and HPV16 E6/p53-positive cells per field were quantified by immunofluorescence-confocal microscopy and counting triplicate visual fields at 200x magnification (representative micrographs for the HPV16+ clinical samples as well as the HPV-negative HFL1 antibody control are provided in Figs. S10A–B). The error bars are mean ±SD, n-value = 3.
Next, to determine if the antioxidant effector TIGAR contributes to HPV-induced tumorigenesis and neoplastic disease progression in vivo, immunodeficient NIH III-nude mice (n-value = 12) were anesthetized and subcutaneously engrafted over each hind quarter with HPV18+ HeLa adenocarcinoma cells that were transduced with lentiviralsi-RNA-tigar or an empty lentiviral vector as negative control. For comparison, another sample group was injected with the Vehicle alone. The experimental animals were closely monitored over a period of 8 weeks for the development of tumors and other signs of disease progression (e.g., aggressive tumor growth, lethargy, or difficulty with ambulation). As shown in Fig. 6B, the siRNA-inhibition of TIGAR resulted in markedly reduced tumor growth and the development of fewer masses (see arrows), in contrast to the HeLa/pLenti vector-engrafted animals. A Kaplan-Meier plot of tumor development in the HeLa/lentiviral-siRNA-tigar and HeLa/pLenti vector animals, as compared to the Vehicle alone, is provided in Fig. S9A (the percentages of animals positive for tumor growth and that developed bilateral tumors are shown in Figs. S9B and S9C). The tumors from the HeLa/lentiviral-siRNA-tigar animals were notably smaller and exhibited poor vascularization compared to the larger tumors in the HeLa/pLenti vector animals (Figs. 6C–E). Consistent with these findings, the xenograft tumor tissues (i.e., human Ki67-positive) from HeLa/pLenti vector animals contained high levels of the pro-angiogenic markers, HIF-1α and vascular endothelial growth factor (VEGF), in addition to p53 and the H-Ras oncoprotein (Fig. S9D). These tumor tissues also exhibited significant TIGAR protein expression that correlated with c-Myc dysregulation, in contrast to the smaller tumors in HeLa/lentiviral-siRNA-tigar experimental animals (Fig. 6F).
We next analyzed the expression of TIGAR, c-Myc, and p53 in the E6-positive tumor cells of primary HPV16+ cervical cancer clinical isolates (from 9 different patients) obtained from the University of Hawaii Cancer Center (see Table S1). The formalin-fixed tumor tissue sections were deparaffinized and used for immunofluorescence-confocal microscopy experiments to quantify the relative percentages of HPV16 E6/p53, E6/c-Myc, and E6/TIGAR-positive cells in each visual field at 200x magnification (representative micrographs as well as data for the HPV-negative HFL1 antibody-staining control are provided in Fig. S7A). All HPV16+ cervical cancer tissues contained detectable expression of TIGAR, c-Myc, and p53 as compared to the HPV-negative HFL1 control cells (Figs. 6G and S10A). Moreover, these clinical samples also expressed the H-Ras oncoprotein, consistent with the deregulation of Ras effector-signaling (PI3K and MAPK) in hrHPV-infected cells (Fig. S10B). These findings collectively suggest that E6-induced pseudohypoxic signaling and the activation of mitochondrial TIGAR contribute to the cooperation between hrHPVs and cellular oncogenes during viral carcinogenesis (Fig. 7).
Fig. 7.
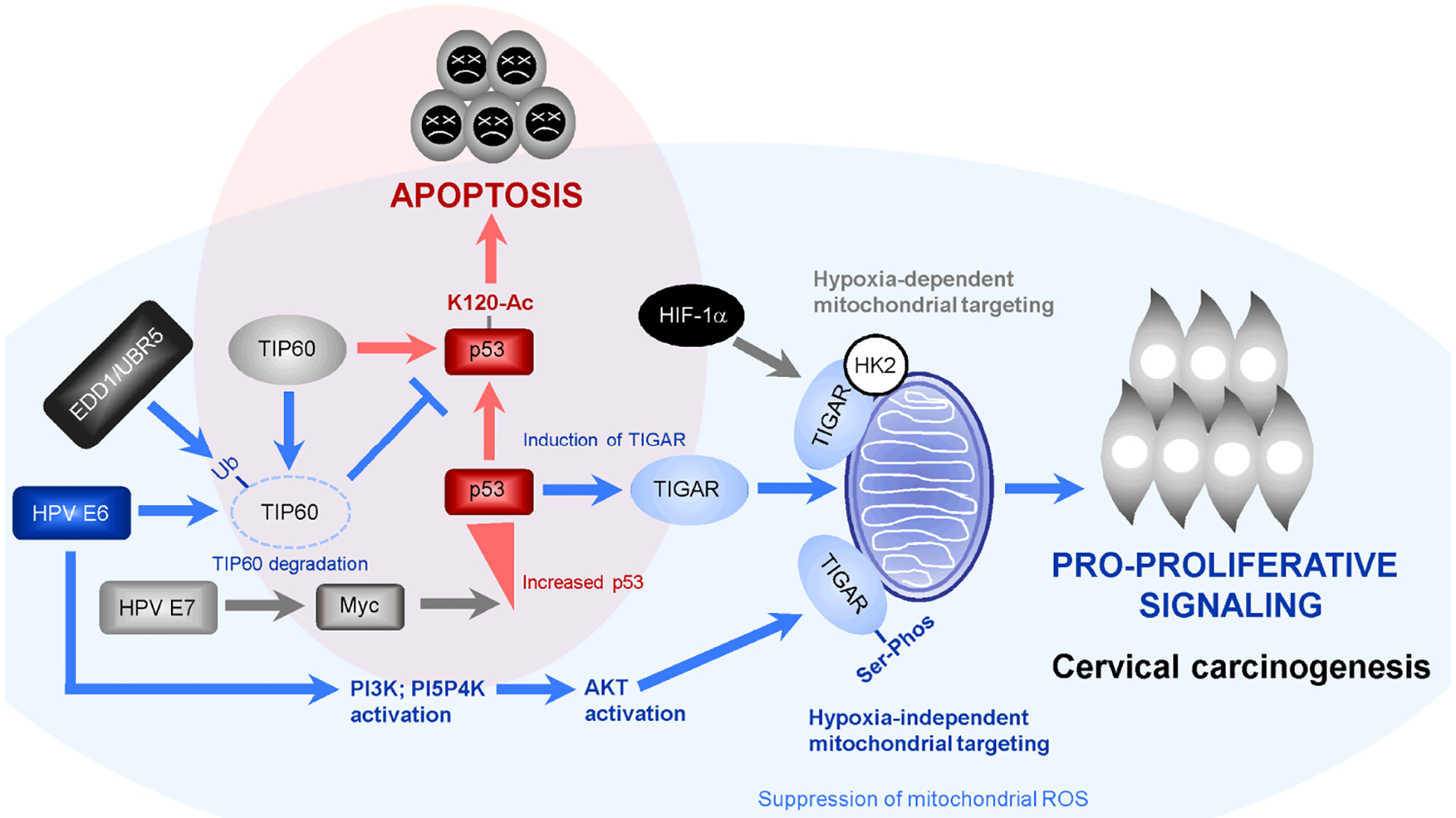
Model of the roles of p53-regulated pro-survival signals and hypoxia-independent mitochondrial targeting of TIGAR in the cooperation between hrHPV E6 and cellular oncogenes during viral carcinogenesis. The HPV E6/E7 oncoproteins have been shown to activate c-Myc-dependent transcription and cooperate with c-Myc to induce cellular proliferation and immortalization (Strickland and Vande Pol, 2016; Zhang et al., 2017; McMurray and McCance, 2003). However, aberrant oncogenic c-Myc expression can cause DNA-damage, activate p53, and induce p53-K120-acetylation and p53-dependent cellular apoptosis (shaded in red; Vafa et al., 2002; Hermeking and Eick, 1994; Romeo et al., 2018). The HPV E6 oncoprotein destabilizes the p53-cofactor TIP60 through recruitment of the E3 ubiquitin-ligase, EDD1/UBR5, and inhibits p53-K120-acetylation and p53-dependent apoptosis (Jha et al., 2010; Subbaiah et al., 2016). Whereas the TIGAR protein has been shown to target mitochondrial membranes and protect cells against the accumulation of damaging ROS under hypoxic conditions –dependent upon HIF-1α activation and interactions with hexokinase-2 (HK2) (Bensaad et al., 2006; Cheung et al., 2012), our studies demonstrate that the hrHPV E6 oncoproteins induce a pseudohypoxic response and activate PI3K/PI5P4K/AKT-signaling to promote the serine-phosphorylation and mitochondrial targeting of TIGAR which counters the oxidative stress/cytotoxicity associated with oncogenic proproliferative signaling during HPV-associated cervical carcinogenesis (shaded in blue).
3. Discussion
The MYST-family acetyltransferase TIP60 (Kat5) is a transcriptional cofactor for both c-Myc and p53 (Patel et al., 2004; Frank et al., 2003; Awasthi et al., 2005; Tang et al., 2006; Sykes et al., 2006; Kurash et al., 2008); and the p53 gene is a downstream target of c-Myc (through the activation of E-box elements within the p53 promoter and protein stabilization by p14ARF) which has been shown to mediate oncogene-induced DNA-damage, genomic instability and cellular apoptosis (Reisman et al., 1993; Roy et al., 1994; Zindy et al., 1998; Vafa et al., 2002; Hermeking and Eick, 1994; Chen et al., 2013; Gorrini et al., 2007; Chen et al., 2010). Intriguingly, the E6 oncoproteins of hrHPVs cooperate with c-Myc and destabilize TIP60 through interactions with the E3 ubiquitin ligase, EDD1/UBR5, and thereby inhibit p53-K120-acetylation and p53-dependent apoptosis (Jha et al., 2010; Subbaiah et al., 2016; Strickland and Vande Pol, 2016; McMurray and McCance, 2003; Liu et al., 2008). Although the viral E6 oncoprotein is known to ubiquitinate and constitutively destabilize the p53 tumor suppressor through molecular interactions with E6AP (Scheffner et al., 1993; Huibregtse et al., 1991; Martinez-Zapien et al., 2016), the p53 protein is nevertheless detectably expressed in HPV16 and HPV18-transformed epithelial cell-lines and HPV16+ cervical cancer clinical isolates (Figs. 1C, 1D, and 6G); and others have shown that the remaining p53 present within hrHPV-transformed cells is transcriptionally active (Butz et al., 1995; Banuelos et al., 2003; Kimple et al., 2013). These findings, together with the fact that p53 mutations are infrequently observed in HPV+ clinical samples (Schultheis et al., 2021; Lee et al., 1994), prompted us to investigate whether the hrHPVs might paradoxically exploit the functions of certain p53-regulated pro-survival genes –independent of the E6-E6AP-p53 axis, to protect HPV-infected cells against oncogene-induced oxidative stress, genomic DNA-damage, and cytotoxicity associated with viral carcinogenesis.
Our results in the present study have demonstrated that the TIGAR protein is highly expressed in hrHPV-transformed cervical cancer cell-lines and correlates with oncogenic c-Myc and p53 expression (Figs. 1C and 1D). We were surprised to discover that the viral E6 oncoprotein induces hypoxia-independent mitochondrial targeting of the TIGAR protein in transfected cells and HPV16/18-transformed cervical carcinoma cell-lines (Figs. 1E–H, S2A–C). The HPV16/18 E6 oncoproteins induced the expression of HIF-1α as well as the HIF-2α subunit in transfected keratinocytes cultured under normal oxygen (normoxic) conditions (Figs. 1A and 1B), suggesting the hrHPVs may induce a Warburg-like pseudohypoxic stress response to activate p53-regulated pro-survival signals and the mitochondrial targeting of TIGAR to help protect proliferating infected cells against metabolic byproducts (e.g., ROS) that could cause oxidative damage and apoptosis (Bensaad et al., 2006; Cheung et al., 2012; Bensaad et al., 2009). The HPV+ cervical carcinoma cell-lines: HeLa, C-4 I, and SiHa, also exhibited upregulation of HIF-1α/HIF-2α as compared to cultured primary keratinocytes (Fig. S1A). Bossler et al., 2019 have reported that hypoxic conditions activate AKT-signaling and transcriptionally repress the expression of the viral E6/E7 oncoproteins through the upstream modulators PI3K and mTORC2 which, by contrast, alludes to a functional distinction with E6-induced HIF-1α/HIF-2α pseudohypoxia and the activation of PI3K/AKT that phosphorylates TIGAR and induces its mitochondrial targeting in HPV-infected cells under normoxic conditions. While previous studies by others have shown that hrHPV E6 oncoproteins induce the expression of HIF-1α and HK2 (Guo et al., 2014; Hoppe-Seyler et al., 2017), it remains to be determined whether these factors contribute to the E6-induced hypoxia-independent mitochondrial targeting of TIGAR (Fig. 7). Interestingly, Blancher et al., 2001 have demonstrated the expression of HIF-1α, but not HIF-2α, in breast cancer cells cultured under hypoxic conditions was associated with Ras-signaling and dependent upon PI3K-activation. Moreover, the PI3K/AKT pathway is constitutively deregulated in many HPV+ cervical cancers through 3q26.3 chromosomal alterations affecting the p110α catalytic subunit of class IA PI3K (PIK3CA) locus and contributes to HPV-induced cellular transformation and oncogenesis (Bertelsen et al., 2006; Henken et al., 2011). The Ras effector, p38MAPK, is also essential for HPV genomic replication in differentiated keratinocytes (Satsuka et al., 2015). Our results in Figs. 2A, 2B, and S3A demonstrate that the chemical kinase inhibitors, wortmannin and SB203580, inhibited serine-phosphorylation of the TIGAR protein and prevented its subcellular trafficking and mitochondrial targeting in treated HPV18+ HeLa cells. We have also shown that wortmannin and SB203580, but not staurosporine, inhibited the phosphorylation of the TIGAR protein in HPV16+ SiHa cells (Fig. S4). Consistent with these data, the constitutively-active mutants of PI3K, PI5P4K, and AKT induced the mitochondrial localization of FLAG-tagged TIGAR in cotransfected HT-1080 cells which suggests the activation of cellular kinases and phosphorylation of TIGAR are important for its mitochondrial targeting under normoxic conditions (Fig. 2C and S3B). Immunoprecipitation experiments, using various phospho-specific antibodies and isolated mitochondrial or cytoplasmic fractions from HPV16/18 E6-expressing cells and HPV18-transformed cervical carcinoma cells (HeLa and MS751), further demonstrated that the TIGAR protein in mitochondria was specifically phosphorylated on serine residues (Figs. 2D–G). This work provides the first evidence that phosphorylation modulates the antioxidant functions and translocation of TIGAR to mitochondrial membranes. However, the specific amino acid residue(s) within the TIGAR protein that is phosphorylated by E6 remains to be biochemically identified and these studies are ongoing. Although the primary sequence of the TIGAR protein doesn’t appear to contain a consensus AKT peptide recognition sequence, there are potential MAPK D-motif target sites for phosphorylation at aa residues 203–209 (RSLFDYF) and 214–224 (KCSLPATLSRS) and an S/P site which spans residues 157–163 (SPSNCL), and these may be targeted through site-directed mutagenesis experiments. The hrHPV E6 oncoproteins are known to activate the Met-receptor tyrosine kinase (Qian et al., 2016; Scott et al., 2018), and Shen et al., 2018 have shown that oncogenic c-Met signaling mediates the TIGAR-induced EMT and cell invasiveness/metastasis in non-small-cell lung cancers. It is therefore possible that E6 could activate cellular kinases and induce serine-phosphorylation of the TIGAR through the HGF/SF-Met receptor signaling pathway, with concomitant activation of the Ras GTPase and its downstream effectors, PI3K/AKT and Raf1/MAPK (Qian et al., 2016; Scott et al., 2018).
The aberrant oncogenic expression of c-Myc can lead to the increased production of damaging metabolic byproducts (e.g., ROS) that cause oxidative stress, genomic instability, and cytotoxicity in proliferating cells (Zindy et al., 1998; Vafa et al., 2002; Hermeking and Eick, 1994). By inhibiting endogenous TIGAR expression with a lentiviral-siRNA-tigar vector, we demonstrated that TIGAR is essential for the ability of HPV16/18 E6 to suppress the accumulation of oncogene-induced ROS in transduced cells containing E6 and c-Myc (Figs. 3B, 3E, and 3F). These studies have further shown that the oncogenic cooperation between hrHPV E6 oncoproteins and c-Myc and the ability of E6/c-Myc to induce colony-formation in vitro are dependent upon TIGAR functions (Fig. 6A). The siRNA-knockdown of TIGAR markedly inhibited tumor development and growth in an in vivo xenograft model of HPV-induced adenocarcinoma using NIH III-nude mice that were subcutaneously engrafted with HeLa cells transduced with lentiviral-siRNA-tigar or an empty lentiviral (pLenti) vector as negative control (Figs. 6B–F, S9A–C). The tumors that formed in the HeLa/siRNA-tigar experimental animals were comparatively smaller and were poorly vascularized and contained fewer blood vessels than the HeLa/pLenti vector tumors (Figs. 6B–E). Consistent with these results, the angiogenic markers HIF-1α and VEGF were expressed in the huKi67-positive cells of the HeLa/pLenti vector tumor tissues (Fig. S9D). The physiological relevance and impact of these findings were underscored by the expression of TIGAR and wild-type p53 that correlated with oncogenic c-Myc and H-Ras dysregulation in the E6-positive tumor cells of HPV16+ cervical cancer clinical isolates (Fig. 6G, S10A, and S10B; see Table S1 for source information). The TCGA-cervical cancers database has shown that the p53 protein and p53 tetrameric complexes are present and many p53-regulated genes, including tigar, are activated in HPV16/18+ tumors (The Cancer Genome Atlas Network, 2017). In particular, although TIGAR was detectable in most tumor types (i.e., adenocarcinomas, keratin-high squamous-cell carcinomas, and keratin-low basal-cell carcinomas), TIGAR was most upregulated in adenocarcinomas and the less-differentiated basal-cell carcinomas and correlated with c-Myc dysregulation. Many HPV+ cervical cancers also exhibited HIF-1α activation and a hypoxic gene expression profile (The Cancer Genome Atlas Network, 2017) in agreement with our present findings which provide a mechanistic link between E6-mediated pseudohypoxia, PI3K/PI5P4K/AKT-signaling, and the activation of mitochondrial TIGAR functions in HPV+ tumor cells.
4. Conclusions
These aggregate data demonstrate that hrHPVs modulate the expression of certain p53-regulated pro-survival genes by inhibiting TIP60-mediated p53-K120-acetylation (Jha et al., 2010; Subbaiah et al., 2016), and induce a pseudohypoxic stress response to promote the serine-phosphorylation and mitochondrial targeting of TIGAR which could help protect HPV-infected cells against oncogene-induced oxidative stress and genotoxicity associated with the development and progression of hrHPV+ epithelial cancers (Fig. 7). Targeting the cellular kinases that phosphorylate the TIGAR protein and modulate its subcellular trafficking and antioxidant functions could lead to new therapeutic strategies to inhibit the pro-oncogenic activity of TIGAR in malignant tumors.
Supplementary Material
Highlights.
HPV E6 oncoproteins induce a pseudohypoxic stress response mediated by HIF-1α/2α.
E6 activates cellular kinases that phosphorylate the TIGAR protein on Ser residues.
The viral E6 protein induces hypoxia-independent mitochondrial targeting of TIGAR.
siRNA-inhibition of TIGAR expression inhibits HPV xenograft tumorigenesis in vivo.
• HPV+ cervical cancer clinical samples contain high levels of TIGAR, p53, and c-Myc.
Acknowledgements
We kindly thank K. Vousden for providing the pcDNA3.1-TIGAR (FLAG) expression construct, Y. Nakatani for the pOZ-TIP60 expression vector, and B. Vogelstein for the pCEP-p53 (wt) and pCEP-p53-R175H plasmids. Other members of the Harrod lab: M. Saberi, N. Adams, and J. Savage, are thanked for their helpful technical assistance. We also thank L. Banks for generously providing the HPV16/18 E6 (HA) expression constructs and the Pathology Shared Resource of the University of Hawaii Cancer Center for the primary HPV16+ cervical cancer clinical samples to support these studies.
Funding information
This work was supported by National Cancer Institute/National Institutes of Health grants 1R15CA202265-01A1 and 1R15CA267892-01A1 and an SMU Dean’s Research Council grant to RH. LY was supported by an SMU Moody School of Graduate and Advanced Studies Dissertation Fellowship, and TB was supported by an SMU Dean’s Dissertation Fellowship and an American Society for Microbiology-Graduate Student Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CRediT authorship contribution statement
Lacin Yapindi: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Data curation, Visualization, Writing – review & editing. Tetiana Bowley: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Data curation, Writing – review & editing. Nick Kurtaneck: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Data curation. Rachel Bergeson: Formal analysis, Investigation. Kylie James: Formal analysis, Investigation. Jillian Wilbourne: Formal analysis, Investigation. Carolyn Harrod: Methodology, Formal analysis, Investigation. Brenda Hernandez: Formal analysis, Resources. Brooke Emerling: Formal analysis, Resources. Courtney Yates: Formal analysis, Resources. Robert Harrod: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Data curation, Visualization, Writing – original draft, Writing – review & editing, Supervision, Project administration, Funding acquisition.
Declaration of competing interest
The authors declare that they have no conflicts of interest with the contents of this article.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Supplemental information
This article contains supplemental information.
Data availability
All data are contained within the article.
References
- Awasthi S, Sharma A, Wong K, Zhang J, Matlock EF, Rogers L, Motloch P, Takemoto S, Taguchi H, Cole MD, Lüscher B, Dittrich O, Tagami H, Nakatani Y, McGee M, Girard A-M, Gaughan L, Robson CN, Monnat RJ Jr., Harrod R, 2005. A human T-cell lymphotropic virus type 1 enhancer of Myc transforming potential stabilizes Myc-TIP60 transcriptional interactions. Mol. Cell. Biol 25, 6178–6198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banuelos A, Reyes E, Ocadiz R, Alvarez E, Moreno M, Monroy A, Gariglio P, 2003. Neocarzinostatin induces an effective p53-dependent response in human papillomavirus-positive cervical cancer cells. J. Pharmacol. Exp. Ther 306, 671–680. [DOI] [PubMed] [Google Scholar]
- Bensaad K, Cheung EC, Vousden KH, 2009. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 28, 3015–3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaad K, Tsuruta A, Selak MA, Calvo Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH, 2006. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126, 107–120. [DOI] [PubMed] [Google Scholar]
- Bertelsen B, Steine SJ, Sandvei R, Molven A, Laerum OD, 2006. Molecular analysis of the PI3K-AKT pathway in uterine cervical neoplasia: frequent PIK3CA amplification and AKT phosphorylation. Int. J. Cancer 118, 1877–1883. [DOI] [PubMed] [Google Scholar]
- Blancher C, Moore JW, Robertson N, Harris AL, 2001. Effects of ras and von Hippel-Lindau (VHL) gene mutations on hypoxia-inducible factor (HIF)-1alpha, HIF-2alpha, and vascular endothelial growth factor expression and their regulation by the phosphatidylinositol 3’-kinase/Akt signaling pathway. Cancer Res. 61, 7349–7355. [PubMed] [Google Scholar]
- Bossler F, Kuhn BJ, Günther T, Kraemer SJ, Khalkar P, Adrian S, Lohrey C, Holzer A, Shimobayashi M, Dürst M, Mayer A, Rösl F, Grundhoff A, Krijgsveld J, Hoppe-Seyler K, Hoppe-Seyler F, 2019. Repression of human papillomavirus oncogene expression under hypoxia is mediated by PI3K/mTORC2/AKT signaling. mBio 10, e02323–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer SN, Wazer DE, Band V, 1996. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 56, 4620–4624. [PubMed] [Google Scholar]
- Butz K, Shahabeddin L, Geisen C, Spitkovsky D, Ullmann A, Hoppe-Seyler F, 1995. Functional p53 protein in human papillomavirus-positive cancer cells. Oncogene 10, 927–936. [PubMed] [Google Scholar]
- Castellsagué X, Alemany L, Quer M, Halec G, Quirós B, Tous S, et al. , 2016. HPV involvement in head and neck cancers: comprehensive assessment of biomarkers in 3680 patients. J. Natl. Cancer Inst 108, djv403. [DOI] [PubMed] [Google Scholar]
- Chen D, Kon N, Zhong J, Zhang P, Yu L, Gu W, 2013. Differential effects on ARF stability by normal versus oncogenic levels of c-Myc expression. Mol. Cell 51, 46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Shan J, Zhu W-G, Qin J, Gu W, 2010. Transcription-independent ARF regulation in oncogenic stress-mediated p53 responses. Nature 464, 624–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung EC, Athineos D, Lee P, Ridgway RA, Lambie W, Nixon C, Strathdee D, Blyth K, Sansom OJ, Vousden KH, 2013. TIGAR is required for efficient intestinal regeneration and tumorigenesis. Dev. Cell 25, 463–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung EC, Lee P, Ceteci F, Nixon C, Blyth K, Sansom OJ, Vousden KH, 2016. Opposing effects of TIGAR- and RAC1-derived ROS on Wnt-driven proliferation in the mouse intestine. Genes Dev. 30, 52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung EC, Ludwig RL, Vousden KH, 2012. Mitochondrial localization of TIGAR under hypoxia stimulates HK2 and lowers ROS and cell death. Proc. Natl. Acad. Sci., U.S.A 109, 20491–20496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J, Niu X, Chang J, Shao M, Peng L, Xi Y, Lin A, Wang C, Cui Q, Luo Y, Fan W, Chen Y, Sun Y, Guo W, Tan W, Lin D, Wu C, 2020. Metabolic remodeling by TIGAR overexpression is a therapeutic target in esophageal squamous-cell carcinoma. Theranostics 10, 3488–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerling BM, Hurov JB, Poulogiannis G, Tsukazawa KS, Choo-Wing R, Wulf GM, Bell EL, Shim H-S, Lamia KA, Rameh LE, Bellinger G, Sasaki AT, Asara JM, Yuan X, Bullock A, Denicola GM, Song J, Brown V, Signoretti S, Cantley LC, 2013. Depletion of a putatively druggable class of phosphatidylinositol kinases inhibits growth of p53-null tumors. Cell 155, 844–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng D, Yan K, Zhou Y, Liang H, Liang J, Zhao W, Dong Z, Ling B, 2016. Piwil2 is reactivated by HPV oncoproteins and initiates cell reprogramming via epigenetic regulation during cervical cancer tumorigenesis. Oncotarget 7, 64575–64588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank SR, Parisi T, Taubert S, Fernandez P, Fuchs M, Chan H-M, Livingston DM, Amati B, 2003. MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO Rep. 4, 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genther SM, Sterling S, Duensing S, Münger K, Sattler C, Lambert PF, 2003. Quantitative role of the human papillomavirus type 16 E5 gene during the productive stage of the viral life cycle. J. Virol 77, 2832–2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrini C, Squatrito M, Luise C, Syed N, Perna D, Wark L, Martinato F, Sardella D, Verrecchia A, Bennett S, Confalonieri S, Cesaroni M, Marchesi F, Gasco M, Scanziani E, Capra M, Mai S, Nuciforo P, Crook T, Lough J, Amati B, 2007. Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature 448, 1063–1067. [DOI] [PubMed] [Google Scholar]
- Graham SV, 2017a. Keratinocyte differentiation-dependent human papillomavirus gene regulation. Viruses 9, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham SV, 2017b. The human papillomavirus replication cycle, and its links to cancer progression: a comprehensive review. Clin. Sci. (Lond.) 131, 2201–2221. [DOI] [PubMed] [Google Scholar]
- Guo Y, Meng X, Ma J, Zheng Y, Wang Q, Wang Y, Shang H, 2014. Human papillomavirus 16 E6 contributes HIF-1α induced Warburg effect by attenuating the VHL-HIF-1α interaction. Int. J. Mol. Sci 15, 7974–7986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann G, Weidner KM, Schwartz H, Birchmeier W, 1994. The motility signal of scatter factor/hepatocyte growth factor mediated through the receptor tyrosine kinase met requires intracellular action of Ras. J. Biol. Chem 269, 21936–21939. [PubMed] [Google Scholar]
- Hatterschide J, Bohidar AE, Grace M, Nulton TJ, Kim HW, Windle B, Morgan IM, Munger K, White EA, 2019. PTPN14 degradation by high-risk human papillomavirus E7 limits keratinocyte differentiation and contributes to HPV-mediated oncogenesis. Proc. Natl. Acad. Sci., U.S.A 116, 7033–7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henken FE, Banerjee NS, Snijders PJF, Meijer CJLM, Arce JDC, Rösl F, Broker TR, Chow LT, Steenbergen RDM, 2011. PIK3CA-mediated PI3-kinase signalling is essential for HPV-induced transformation in vitro. Mol. Cancer 10, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermeking H, Eick D,1994. Mediation of c-Myc-induced apoptosis by p53. Science 265, 2091–2093. [DOI] [PubMed] [Google Scholar]
- Hermeking H, Lengauer C, Polyak K, He TC, Zhang L, Thiagalingam S, Kinzler KW, Vogelstein B, 1997. 14–3-3sigma is a p53-regulated inhibitor of G2/M progression. Mol. Cell 1, 3–11. [DOI] [PubMed] [Google Scholar]
- Hong M, Xia Y, Zhu Y, Zhao H-H, Zhu H, Xie Y, Fan L, Wang L, Miao K-R, Yu H, Miao Y-Q, Wu W, Zhu H-Y, Chen Y-Y, Xu W, Qian S-X, Li J-Y, 2016. TP53-induced glycolysis and apoptosis regulator protects from spontaneous apoptosis and predicts poor prognosis in chronic lymphocytic leukemia. Leuk. Res 50, 72–77. [DOI] [PubMed] [Google Scholar]
- Hoppe-Seyler K, Honegger A, Bossler F, Sponagel J, Bulkescher J, Lohrey C, Hoppe-Seyler F, 2017. Viral E6/E7 oncogene and cellular hexokinase 2 expression in HPV-positive cancer cell lines. Oncotarget 8, 106342–106351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huibregtse JM, Scheffner M, Howley PM, 1991. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 10, 4129–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison T, Malu A, Yapindi L, Bergeson R, Peck K, Romeo M, Harrod C, Pope J, Smitherman L, Gwinn W, Ratner L, Yates C, Harrod R, 2018. The TP53-induced glycolysis and apoptosis regulator mediates cooperation between HTLV-1 p30II and the retroviral oncoproteins Tax and HBZ and is highly expressed in an in vivo xenograft model of HTLV-1-induced lymphoma. Virology 520, 39–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha S, Vande Pol S, Banerjee NS, Dutta AB, Chow LT, Dutta A, 2010. Destabilization of TIP60 by human papillomavirus E6 results in attenuation of TIP60-dependent transcriptional regulation and apoptotic pathway. Mol. Cell 38, 700–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DL, Münger K, 1997. Analysis of the p53-mediated G1 growth arrest pathway in cells expressing the human papillomavirus type 16 E7 oncoprotein. J. Virol 71, 2905–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimple RJ, Smith MA, Blitzer GC, Torres AD, Martin JA, Yang RZ, Peet CR, Lorenz LD, Nickel KP, Klingelhutz AJ, Lambert PF, Harari PM, 2013. Enhanced radiation sensitivity in HPV-positive head and neck cancer. Cancer Res. 73, 4791–4800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurash JK, Lei H, Shen Q, Marston WL, Granda BW, Fan H, Wall D, Li E, Gaudet F, 2008. Methylation of p53 by Set7/9 mediates p53 acetylation and activity in vivo. Mol. Cell 29, 392–400. [DOI] [PubMed] [Google Scholar]
- Landro ME, Dalbert D, Picconi MA, Cúneo N, González J, Vornetti S, Bazán G, Mural J, Basiletti J, Teyssié AR, Alonio LV, 2008. Human papillomavirus and mutated H-ras oncogene in cervical carcinomas and pathological negative pelvic lymph nodes: a retrospective follow-up. J. Med. Virol 80, 694–701. [DOI] [PubMed] [Google Scholar]
- Lee J-H, Kang Y-S, Koh J-W, Park S-Y, Kim B-G, Lee E-D, Lee K-H, Park K-B, Seo Y-L, 1994. P53 gene mutation is rare in human cervical carcinomas with positive HPV sequences. Int. J. Gynecol. Cancer 4, 371–378. [DOI] [PubMed] [Google Scholar]
- Li M, Sun M, Cao L, Gu J-H, Ge J, Chen J, Han R, Qin Y-Y, Zhou Z-P, Ding Y, Qin Z-H, 2014. A TIGAR-regulated metabolic pathway is critical for protection of brain ischemia. J. Neurosci 34, 7458–7471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Dakic A, Chen R, Disbrow GL, Zhang Y, Dai Y, Schlegel R, 2008. Cell-restricted immortalization by human papillomavirus correlates with telomerase activation and engagement of the hTERT promoter by Myc. J. Virol 82, 11568–11576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Wu Y, Zhang Y, Yuan M, Li X, Gao J, Zhang S, Xing C, Qin H, Zhao H, Zhao Z, 2019. TIGAR promotes tumorigenesis and protects tumor cells from oxidative and metabolic stresses in gastric cancer. Front. Oncol 9, 1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longworth MS, Laimins LA, 2004. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol. Mol. Biol. Rev 68, 362–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui VWY, Wong EYL, Ho K, Ng PKS, Lau CPY, Tsui SKW, Tsang C-M, Tsao S-W, Cheng SH, Ng MHL, Ng YK, Lam EKY, Hong B, Lo KW, Mok TSK, Chan ATC, Mills GB, 2011. Inhibition of c-Met downregulates TIGAR expression and reduces NADPH production leading to cell death. Oncogene 30, 1127–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malanchi I, Caldeira S, Krützfeldt M, Giarre M, Alunni-Fabbroni M, Tommasino M, 2002. Identification of a novel activity of human papillomavirus type 16 E6 protein in deregulating the G1/S transition. Oncogene 21, 5665–5672. [DOI] [PubMed] [Google Scholar]
- Martin LG, Demers GW, Gallaway DA, 1998. Disruption of the G1/S transition in human papillomavirus type 16 E7-expressing human cells is associated with altered regulation of cyclin E. J. Virol 72, 975–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Zapien D, Ruiz FX, Poirson J, Mitschler A, Ramirez J, Forster A, Cousido-Siah A, Masson M, Vande Pol S, Podjarny A, Travé G, Zanier K, 2016. Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature 529, 541–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer GD, Heller S, Wanka C, Rieger J, Steinbach JP, 2019. Knockdown of the TP53-induced glycolysis and apoptosis regulator (TIGAR) sensitizes glioma cells to hypoxia, irradiation and temozolomide. Int. J. Mol. Sci 20, 1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray HR, McCance DJ, 2003. Human papillomavirus type 16 E6 activates TERT gene transcription through induction of c-Myc and release of USF-mediated repression. J. Virol 77, 9852–9861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mix JM, Saraiya M, Thompson TD, Querec TD, Greek A, Tucker TC, Peters ES, Lynch CF, Hernandez BY, Copeland G, Goodman MT, Unger ER, 2021. Prevalence of human papillomavirus genotypes in high-grade cervical precancer and invasive cervical cancer from cancer registries before and after vaccine introduction in the United States. Cancer 127, 3614–3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münger K, Baldwin A, Edwards KM, Hayakawa H, Nguyen CL, Owens M, Grace M, Huh K, 2004. Mechanisms of human papillomavirus-induced oncogenesis. J. Virol 78, 11451–11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münger K, Werness BA, Dyson N, Phelps WC, Harlow E, Howley PM, 1989. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 8, 4099–4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JH, Du Y, Ard PG, Phillips C, Carella B, Chen C-J, Rakowski C, Chatterjee C, Lieberman PM, Lane WS, Blobel GA, McMahon SB, 2004. The c-Myc oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol. Cell. Biol 24, 10826–10834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peta E, Sinigaglia A, Masi G, Di Camillo B, Grassi A, Trevisan M, Messa L, Loregian A, Manfrin E, Brunelli M, Martignoni G, Palù G, Barzon L, 2018. HPV16 E6 and E7 upregulate the histone lysine demethylase KDM2B through the c-Myc/miR-146a-5p axis. Oncogene 37, 1654–1668. [DOI] [PubMed] [Google Scholar]
- Pietras A, Johnsson AS, Pahlman S, 2010. The HIF-2α-driven pseudo-hypoxic phenotype in tumor aggressiveness, differentiation, and vascularization. Curr. Top. Microbiol. Immunol 345, 1–20. [DOI] [PubMed] [Google Scholar]
- Qian G, Wang D, Magliocca KR, Hu Z, Nannapaneni S, Kim S, Chen Z, Sun S-Y, Shin DM, Saba NF, Chen ZG, 2016. Human papillomavirus oncoprotein E6 upregulates c-Met through p53 downregulation. Eur. J. Cancer 65, 21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian S, Li J, Hong M, Zhu Y, Zhao H, Xie Y, Huang J, Lian Y, Li Y, Wang S, Mao J, Chen Y, 2016. TIGAR cooperated with glycolysis to inhibit the apoptosis of leukemia cells and associated with poor prognosis in patients with cytogenetically normal acute myeloid leukemia. J. Hematol. Oncol 9, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisman D, Elkind NB, Roy B, Beamon J, Rotter V, 1993. c-Myc trans-activates the p53 promoter through a required downstream CACGTG motif. Cell Growth Differ. 4, 57–65. [PubMed] [Google Scholar]
- Roman A, Munger K, 2013. The papillomavirus E7 proteins. Virology 445, 138–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo M, Hutchison T, Malu A, White A, Kim J, Gardner R, Smith K, Nelson K, Bergeson R, McKee R, Harrod C, Ratner L, Lüscher B, Martinez E, Harrod R, 2018. The human T-cell leukemia virus type-1 p30II protein activates p53 and induces the TIGAR and suppresses oncogene-induced oxidative stress during viral carcinogenesis. Virology 518, 103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy B, Beamon J, Balint E, Reisman D, 1994. Transactivation of the human p53 tumor suppressor gene by c-Myc/Max contributes to elevated mutant p53 expression in some tumors. Mol. Cell. Biol 14, 7805–7815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satsuka A, Mehta K, Laimins L, 2015. p38MAPK and MK2 pathways are important for the differentiation-dependent human papillomavirus life cycle. J. Virol 89, 1919–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffner M, Huibregtse JM, Vierstra RD, Howley PM, 1993. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75, 495–505. [DOI] [PubMed] [Google Scholar]
- Schultheis AM, de Bruijn I, Selenica P, Macedo GS, da Silva EM, Piscuoglio S, Jungbluth AA, Park KJ, Klimstra DS, Wardelmann E, Hartmann W, Gerharz CD, von Petersdorff M, Buettner R, Reis-Filho JS, Weigelt B, 2021. Genomic characterization of small cell carcinomas of the uterine cervix. Mol. Oncol 16, 833–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott ML, Coleman DT, Kelly KC, Carroll JL, Woodby B, Songock WK, Cardelli JA, Bodily JM, 2018. Human papillomavirus type-16 E5-mediated upregulation of Met in human keratinocytes. Virology 519, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M, Zhao X, Zhao L, Shi L, An S, Huang G, Liu J, 2018. Met is involved in TIGAR-regulated metastasis of non-small-cell lung cancer. Mol. Cancer 17, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland SW, Vande Pol S, 2016. The human papillomavirus 16 E7 oncoprotein attenuates AKT signaling to promote ribosomal entry site-dependent translation and expression of c-Myc. J. Virol 90, 5611–5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbaiah VK, Zhang Y, Rajagopalan D, Abdullah LN, Yeo-The NSL, Tomaić V, Banks L, Myers MP, Chow EK, Jha S, 2016. E3 ligase EDD1/UBR5 is utilized by the HPV E6 oncogene to destabilize tumor suppressor TIP60. Oncogene 35, 2062–2074. [DOI] [PubMed] [Google Scholar]
- Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, McMahon SB, 2006. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol. Cell 24, 841–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Luo J, Zhang W, Gu W, 2006. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol. Cell 24, 827–839. [DOI] [PubMed] [Google Scholar]
- Tang Z, He Z, 2019. TIGAR promotes growth, survival and metastasis through oxidation resistance and AKT activation in glioblastoma. Oncol. Lett 18, 2509–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Network, 2015. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517, 576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Network, 2017. Integrated genomic and molecular characterization of cervical cancer. Nature 543, 378–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafa O, Wade M, Kern S, Beeche M, Pandita TK, Hampton GM, Wahl GM, 2002. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol. Cell 9, 1031–1044. [DOI] [PubMed] [Google Scholar]
- Vande Pol SB, Klingelhutz AJ, 2013. Papillomavirus E6 oncoproteins. Virology 445, 115–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HS, Duffy AA, Broker TH, Chow LT, 2009. Robust production and passaging of infectious HPV in squamous epithelium of primary human keratinocytes. Genes Dev. 23, 181–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li R, Chen R, Huang G, Zhou X, Liu J, 2020. Prognostic values of TIGAR expression and 18F-FDG PET/CT in clear cell renal cell carcinoma. J. Cancer 11, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong EYL, Wong SCC, Chan CML, Lam EKY, Ho LY, Lau CPY, Au TCC, Chan AKC, Tsang CM, Tsao SW, Lui VWY, Chan ATC, 2015. TP53-induced glycolysis and apoptosis regulator promotes proliferation and invasiveness of nasopharyngeal carcinoma cells. Oncol. Lett 9, 569–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L, Kufe T, Avigan D, Kufe D, 2014. Targeting MUC1-C is synergistic with bortezomib in downregulating TIGAR and inducing ROS-mediated myeloma cell death. Blood 123, 2997–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng S, Zhao Z, Zheng S, Wu M, Song X, Li Y, Zheng Y, Liu B, Chen L, Gao C, Liu H, 2021. The E3 ubiquitin ligase TRIM31 is involved in cerebral ischemic injury by promoting degradation of TIGAR. Redox Biol. 45, 102058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Dakic A, Chen R, Dai Y, Schlegel R, Liu X, 2017. Direct HPV E6/Myc interactions induce histone modifications, Pol II phosphorylation, and hTERT promoter activation. Oncotarget 8, 96323–96339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J-H, Zhang T-T, Song D-D, Xia Y-F, Qin Z-H, Sheng R, 2016. TIGAR contributes to ischemic tolerance induced by cerebral preconditioning through scavenging of reactive oxygen species and inhibition of apoptosis. Sci. Rep 6, 27096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF, 1998. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 12, 2424–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the article.