

Abstract
Over the last several decades, there has been continued interest in developing novel therapeutic approaches targeting protein lysine methyltransferases (PKMTs). Along with PKMT inhibitors, targeted protein degradation (TPD) has emerged as a promising strategy to attenuate aberrant PKMT activity. Particularly, proteolysis targeting chimeras (PROTACs) effectively eliminate PKMTs of interest, suppressing all enzymatic and non-enzymatic functions. PROTACs and other TPD approaches add new depth to PKMT research and novel therapeutics discovery. This review focuses on recent advances in PKMT degrader and inhibitor development over the last several years.
Introduction
Epigenetic regulation plays a crucial role in human diseases such as cancer, inspiring a generation of novel therapeutic interventions that target critical epigenetic factors [1–7]. Protein lysine methyltransferases (PKMTs) are among epigenetic regulators implicated in oncogenesis, with mutations or aberrant regulation promoting cancer initiation, progression, and metastasis [4, 7–9]. PKMTs transfer methyl groups from the cofactor S-5′-adenosyl-L-methionine (SAM) to catalyze the mono-, di- and tri-methylation of lysine residues, resulting in active or repressive gene transcription [7, 8]. PKMTs of particular interest that have been implicated in cancers such as breast [10] and leukemia [11] include the Polycomb repressive complex (PRC2) [12, 13], G9a/GLP [14, 15], ASH1L [16, 17], the NSD protein family [18], WDR5 [19, 20], and SETD2 [21]. Many small molecule inhibitors targeting these PKMTs have been published and previously reviewed [2, 9, 22, 23].
In addition to catalytic inhibitors, novel PKMT-targeting technologies have been developed in recent years such as targeted protein degradation (TPD) [24]. One popular TPD approach is proteolysis targeting chimeras (PROTAC) (Figure 1) [24–28]. PROTAC degraders are heterobifunctional molecules consisting of a protein of interest (POI) binder attached to an E3-ligase ligand via a linker. The linker provides close proximity between the POI and an E3-ligase, such as VHL or CRBN, to induce polyubiquitination and subsequent 26S proteasomal degradation, effectively eliminating the POI and suppressing all its catalytic and non-catalytic activity (Figure 1) [24–28].
Figure 1.
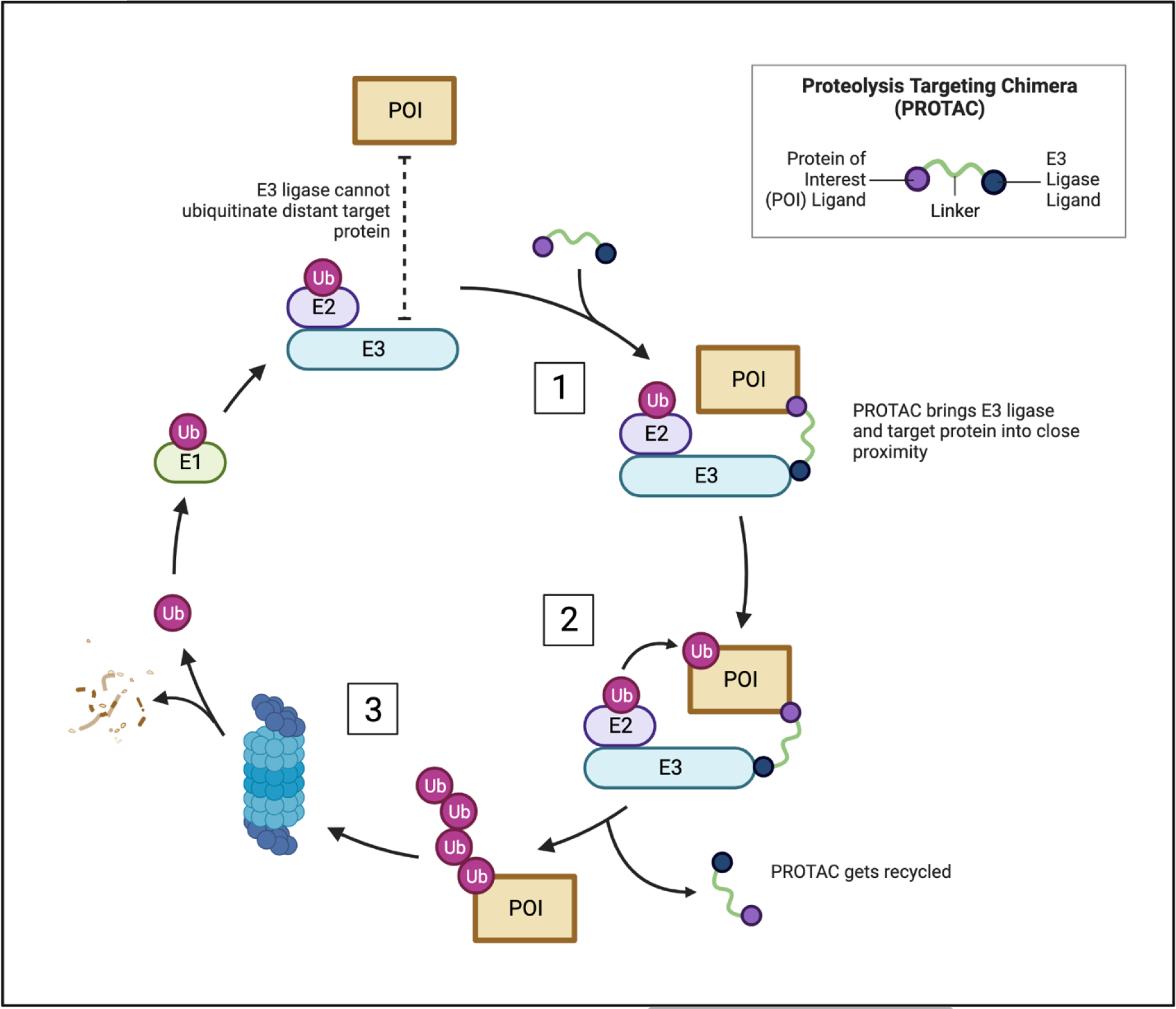
Mode of action of PROTACs (Made using Biorender (licensed)). 1. PROTAC recruits the E3 ligase complex to the POI by binding both the POI and E3 ligase. 2. The E3 ligase complex induces ubiquitination of the POI. 3. The POI gets polyubiquitinated and subsequently degraded via the 26S proteasome. The PROTAC gets recycled, and the process is repeated.
In this review, we will cover the most recently reported PKMT-targeting degraders and inhibitors developed over the past three years (Table 1).
Table 1.
List of degraders and inhibitors of PKMTs reported in last several years.
| Methylation Mark | Target Protein | Compound Name | Mode of Action | Tumor Type (Cell Lines) | In Vivo |
|---|---|---|---|---|---|
| H3K27me3 | EED | UNC6582 | PROTAC (VHL) | DLBCL (DB, Pfieffer) Other: HeLa |
|
| EED | PROTAC 1 | PROTAC (VHL) | DLBCL (Karpas422) | ||
| EZH2 | MS1943 | Hydrophobic tag | TNBC (MDA-MB-468, BT549, HCC70, MDA-MB-231, HCC1187, HCC70, HCC1954) DLBCL (Karpas422, SUDHL8) Other: PNT2, MCF7, K562, A549, HCC827, MCF10A |
Y | |
| EZH2 | MS8815 | PROTAC (VHL) | TNBC (MDA-MB-453, BT549, SUM159, MDA-MB-468, 515a*) | Y | |
| EZH2 | YM281 | PROTAC (VHL) | Lymphoma (SU-DHL-2, SU-DHL-4, SU-DHL-6) Other: 22Rv1 |
Y | |
| EZH2 | U3i | PROTAC (VHL) | TNBC (MDA-MB-231, MDA-MB-468, BT549) Leukemia (MV4;11, HL-60) Other: MCF7, MCF-10A, LO2, HK-2 |
||
| EZH2 (dual w/ cMyc) |
MS177 | PROTAC (CRBN) | MLL-r AML (EOL-1, MV4;11, MOLM-13, RS4;11) Other: HeLa, MM1.S, Kelly, HSPC, K562 |
Y | |
| EZH2 | E7 | PROTAC (CRBN) | NSCLC (A549, H1299) TNBC (MDA-MB-468) DLBCL (Pfeiffer, WSU-DLCL-2) Prostate (LNCaP, DU 145) Ovarian (SKOV3, A2780) |
||
| H3K36me2 | ASH1L | AS-99 | Inhibitor | Leukemia (MV4;11, MOLM13, KOPN8, K562, SET2) Other: CD34+, CD45+ |
Y |
| NSD1 | BT5 | Covalent Inhibitor | Leukemia (K562, SET2, MOLM13) Other: HEK293T, HM2, NUP98-NSD1, CD34+ |
||
| NSD2 | UNC6934 | Inhibitor | MM (KMS-11, MM1.S, UTMC2) Other: U2OS, HCT116, HEK293, HT1080, MCF7, RS4;11 |
||
| NSD2 (dual w/ IKZF1/3) | MS159 | PROTAC (CRBN) | MM (KMS11, H929) Other: 293FT |
Y | |
| NSD3 | MS9715 | PROTAC (VHL) | MLL-r AML (EOL-1, MOLM13, RS4;11) Other: MM1.S, 293FT, K562 |
||
| NSD3 | Compound 8 | PROTAC (VHL) | NSCLC (A549, NCI-H1703, H520) | Y | |
| H3K36me3 | SETD2 | EPZ-719 | Reversible Inhibitor |
MM (KMS34, KMS11) Other: A549 |
Y |
| H3K4me2 | WDR5 | Compound 8g | PROTAC (VHL) | Leukemia (MV4;11, HL-60) | |
| WDR5 | MS67 | PROTAC (VHL) |
MLL-r AML (MV4;11, EOL-1, RS4;11, THP-1, MOLM13, KOPN8) PDAC (MIA PaCa-2, HPAF-II, Panc 10.05, BxPC-3) Other: 239FT, HL-60, MCF7, NCI-H2009, PC3, SK-ES-1 |
Y | |
| WDR5 (dual w/ IKZF1/3) | MS40 | PROTAC (CRBN) |
MLL-r AML (MV4;11, RS4;11, EOL-1, KOPN8, MOLM13, THP-1) Breast (BT549, SUM149, SUM159, MCF7, T-47D, MDA-MB-231, HCC1806, WHIM12) Other: K562 |
Y | |
| H3K9me2 | G9a/GLP | MS8511 | Covalent Inhibitor | MDA-MB-231 K562 |
Y = in vivo data included
Degraders of PRC2
Numerous small molecule EZH2 and EED catalytic inhibitors have been published over the years [2, 22, 23], including the FDA-approved tazemetostat [29]. More recently, select inhibitors have been utilized in the discovery of novel EZH2- and EED-targeting protein degraders. These compounds effectively degrade their target protein to actively suppress the canonical and non-canonical oncogenic functions of PRC2 core components.
UNC6582 [30] and PROTAC 1 [31] (Table 1 & Figure 2A) are two recently published EED-targeting PROTAC degraders. The VHL-recruiting degraders feature different selective EED-binders and potently degrade PRC2 core components in a proteasome-, concentration- and time-dependent manner. Both UNC6582 and PROTAC 1 display potent anti-proliferative activity in DLBCL cell lines containing EZH2 gain-of-function mutations and offer effective chemical probes to further explore EED and PRC2 biology [30, 31]. In addition to the EED-targeting PROTAC degraders, six EZH2-targeting protein degraders have been reported. Among them is the hydrophobic tag (HyT) degrader, MS1943 (Table 1 & Figure 2A) [32]. Unique in its mode of action among the other degraders, HyTs most likely mimic protein misfolding or unfolding and trigger 26S proteasomal degradation by linking a hydrophobic, bulky moiety to a POI binder. MS1943 phenocopies the effect of EZH2 KO/KD and selectively kills EZH2-sensitive TNBC cell lines while EZH2 inhibitors do not. Moreover, the PK/PD profile in xenograft mice demonstrates its in vivo efficacy and ability to suppress tumor growth while being well tolerated. As expected for HyT degraders, MS1943 activity is mediated by the unfolded protein response (UPR) pathway. MS1943 treatment led to prolonged ER stress and sustained UPR activation, ultimately increasing apoptosis in EZH2-sensitive TNBC cells and altering several PRC2 target gene protein levels, including those in the Wnt/b-Catenin signaling pathway [32].
Figure 2.
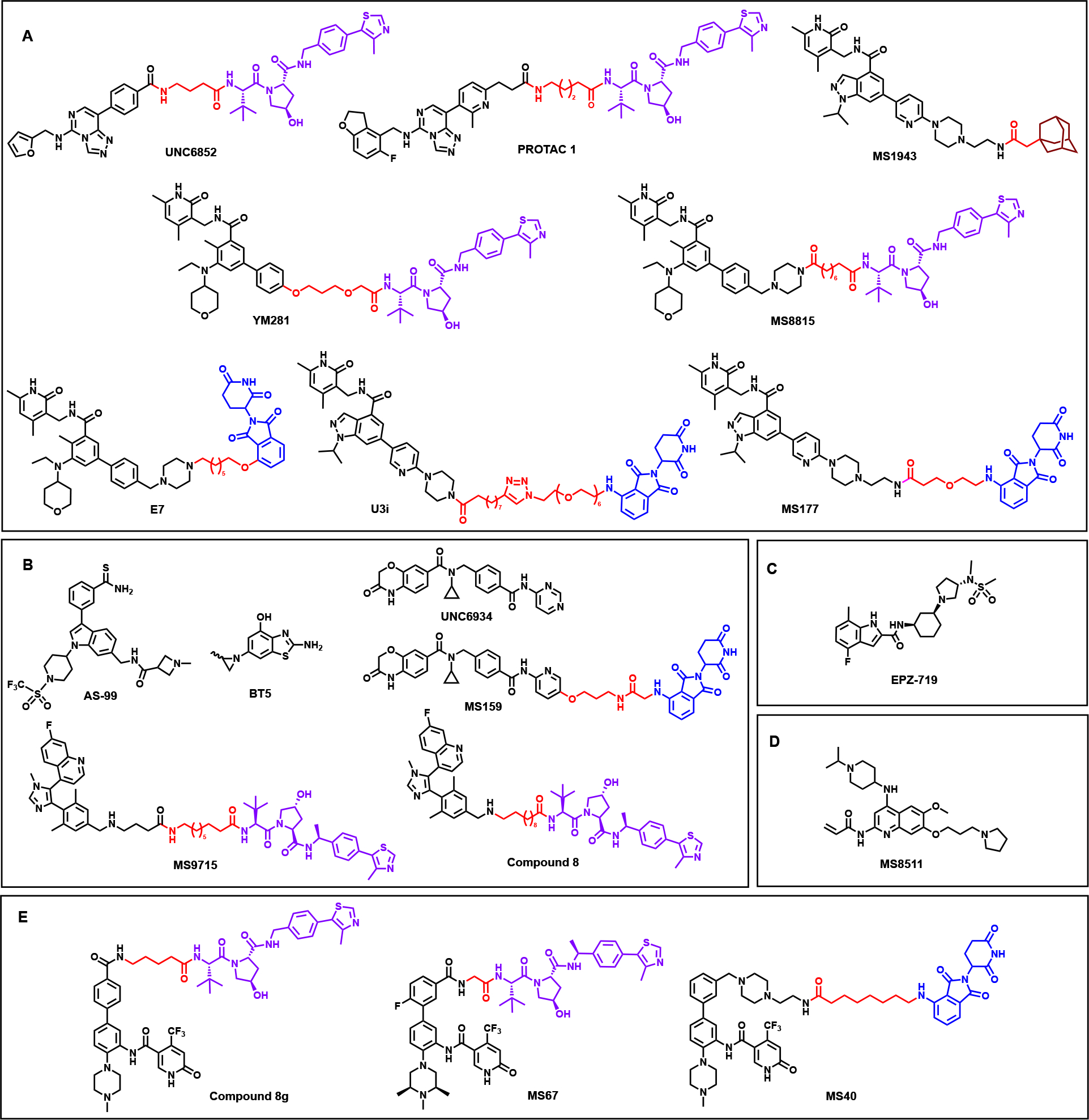
Chemical structures of recently discovered PKMT-targeting degraders and inhibitors. For the PROTAC degraders the POI binding ligands are shown in black, the E3 ligase recruiting ligands indicated in red (CRBN-recruiting ligands) and purple (VHL-recruiting ligands), and the connecting linkers are shown in red. (Made using ChemDraw (licensed)).
The recently reported EZH2 PROTAC degraders include two VHL-recruiting compounds, YM281 [33], and MS8815 [34], and three CRBN-recruiting compounds, E7 [35], U3i [36], and MS177 [37], a dual EZH2-cMyc degrader (Table 1 & Figure 2A). All degraders operate via the ubiquitin-proteasome system (UPS) to efficiently degrade PRC2 components and display superior anti-tumor activity compared to their parent EZH2 catalytic inhibitors. YM281 displays superior cell killing activity in DLBCL and lymphoma cell lines, including patient cells, and reduces tumor growth in xenograft mouse models while the inhibitor does not. Moreover, YM281-induced EZH2 degradation led to improved cell cycle arrest and increased apoptosis, again outperforming its parent inhibitor [33]. These results indicate that enzymatic inhibition alone may not be enough to attenuate lymphoma cells in vivo, thus highlighting a potential advantage of EZH2 PROTAC degraders. The other VHL-recruiting degrader, MS8815, displayed slightly better growth inhibition and EZH2 degradation than YM281 in various TNBC cell lines [34]. The CRBN-recruiting dual EZH2-cMyc degrader, MS177, aided in a study that uncovered a novel non-canonical role of EZH2 in MLL-rearranged leukemias. The study found that EZH2 binds to cMyc via a hidden transactivation domain that directly interacts with cMyc and coactivators, establishing a PRC2-independent role in gene activation crucial for MLL-r AML growth. MS177 potently degrades multiple PRC2 components while also ubiquitinating and depleting cMyc protein levels, ultimately suppressing both cMyc and EZH2 oncogenic activity. Dual degradation of EZH2 and cMyc by MS177 displayed stronger anti-cancer effects in various cell lines and patient-derived models than enzymatic inhibition of EZH2 alone or treatment with the EED degrader, UNC6852. MS177 also performed well in vivo with a notable PK/PD profile, suppressing oncogenesis in xenograft and PDX models of multiple cell types and enhancing overall survival [37]. All these EZH2 degraders offer capable chemical tools to further investigate PRC2 biology and suggest that pharmacological degradation of EZH2 over pharmacological inhibition is a very promising therapeutic strategy for treating various EZH2-dependent cancers.
Inhibitors and degraders of H3K36 PKMTs
PKMTs that regulate H3K36 methylation, including ASH1L, SETD2 and the NSD family, share a unique feature in their catalytic SET domain: a regulatory autoinhibitory loop that blocks access to the substrate binding site [38–40]. While the autoinhibitory loop previously posed a challenge for drug design, the following recently published small molecule compounds overcome the structural difficulties and utilize alternative approaches, offering a blueprint for future compound development.
AS-99 (Table 1 & Figure 2B) [41] is the first-in-class ASH1L SET domain small molecule inhibitor. AS-99 was discovered through fragment-based screening, crystal structures of ASH1L in complex with earlier fragment hits, and medicinal chemistry optimization via structure-based design. AS-99 contains a thioamide group, allowing for key interactions like the chalcogen bond between the group’s sulfur and a backbone carbonyl. The thioamide group is essential for potent binding, as its replacement with an amide resulted in ~100-fold reduced activity. Structural studies show that the inhibitors bind adjacent to the autoinhibitory loop, possibly stabilizing the region and allosterically blocking ASH1L enzymatic activity. AS-99 is highly selective for ASH1L, blocks cell proliferation, induces apoptosis, downregulates MLL fusion target genes in MLL leukemia cells, and reduces tumor burden in MLL leukemia mouse models. In addition to discovering AS-99, the group validated that the ASH1L SET domain is required for transformation of MLL fusion proteins and leukemogenesis [41]. AS-99 is a selective and effective inhibitor useful for exploring ASH1L biology and offers insight into designing autoinhibitory loop-containing SET domain inhibitors.
Due to the aforementioned challenges, potent, selective, and well-characterized NSD family inhibitors are still lacking [42, 43]. A recent study reported a covalent NSD1 SET domain inhibitor, BT5 (Table 1 & Figure 2B) [44]. A crystal structure of the NSD1 SET domain with an earlier covalent hit showed a conformational change in the loop that led to the formation of a channel-like site available for small molecule binding. Based on the crystal structure, BT5 was developed using an aziridine moiety as a covalent warhead to target the C2026 buried in the hydrophobic autoinhibitory loop. BT5 potently inhibits NSD1, engages the SET domain in cells, and reduces H3K36me2 levels. Additionally, BT5 inhibited growth of NUP98-NSD1-transformed cells and demonstrated on-target activity through downregulated HoxA genes. Though BT5 exhibited enhanced selectivity for NSD1, inhibition of NSD2 and NSD3 at higher concentrations was also observed as the C2026 residue is conserved in all three NSD SET domains. While further optimization is needed to attain full potential as a chemical probe, BT5 provides groundwork for future development of NSD SET domain inhibitors. In addition to BT5, three PROTAC degraders targeting NSD2 and NSD3 have recently been published. MS159 (Table 1 & Figure 2B) [45] is a CRBN-recruiting degrader based on the selective NSD2-PWWP1 antagonist UNC6934 [46]. UNC6934 is incapable of inhibiting NSD2 catalytic activity and ineffective in suppressing multiple myeloma (MM) cell growth, rendering the PROTAC approach advantageous for MM treatment. MS159 is selective for NSD2, effectively degrades NSD2 in a proteasome-dependent manner, and inhibits MM cell growth. Additionally, MS159 degrades CRBN neo-substrates IKZF1 and IKZF3, validated onco-targets of MM, but not GSTP1, which plays a critical role in most cells and can lead to potential toxicities when degraded. MS159 is bioavailable in mice and largely phenocopies NSD2 KD in MM cell lines, making it a valuable chemical tool for exploring the role of NSD2 and IKZF1/3 in vivo. MS159 also highlights the potential of NSD2-IZKF1/3 dual degradation as a novel therapeutic approach for treating MM [45]. The recently published VHL-recruiting NSD3 degraders are MS9715 [47] and Compound 8 [48] (Table 1 & Figure 2B). Both degraders utilize the NSD3 antagonist BI-9321 [49] that binds to the PWWP1-NSD3 domain. MS9715 effectively degrades both the long and short isoforms of NSD3 in MLL-r AML and MM cells. The degrader is highly selective for NSD3, depletes NSD3S in a concentration-, time-, and proteasome-dependent manner, and effectively suppresses NSD3-related gene expression. MS9715 displayed superior antiproliferative effects and increased apoptosis in hematological cancer cells compared to BI-9321. Moreover, MS9715 treatment decreased cMyc protein levels and is positively correlated with the suppression of cMyc-related genes. These results taken together resemble the effects of NSD3 KO. The degradation of NSD3 and cMyc by MS9715 demonstrates the benefits of co-suppressing these oncogenic networks as a strategy for treating NSD3-dependent cancers. Compound 8 degraded NSD3 in A549 xenograft models, however, tumor regression data was not reported [48].
SETD2 is the only known PKMT that catalyzes H3K36 tri-methylation [50, 51]. While previous SETD2 inhibitors were not suitable for in vivo studies [40], the recently published inhibitor EPZ-719 (Table 1 & Figure 2C) displays improved ADME properties that can be used for in vivo studies [52]. Structure-aided medicinal chemistry optimization determined that the aminopyrrolidine scaffold of EPZ-719 contributed to its superior in vitro and in vivo activity. A crystal structure confirmed binding of EPZ-719 with SETD2. The reversible inhibitor also displays potent antiproliferative effects in MM cell lines. Together, the in vitro and in vivo activity of EPZ-719 suggests that the inhibitor is ready for testing in xenograft models. Accordingly, in vivo studies are reportedly underway. EPZ-719 is a valuable chemical probe to study SETD2 biology both in vitro and in vivo.
Inhibitor of H3K9me2 PKMTs
Recently, MS8511 (Table 1 & Figure 2D) was the first covalent G9a/GLP inhibitor reported [53]. Based on co-crystal structures, MS8511 was synthesized by exploiting cysteine residues found in the inhibitor binding sites of G9a and GLP (Cys1098 and Cys1186 respectively). MS8511 is selective for G9a/GLP and reduces H3K9me2 in K562 and MDA-MB-231 cells more potently than the widely used G9a/GLP chemical probe UNC0642 [54]. Additionally, binding affinity studies suggest that MS8511 might have a slight preference for G9a over GLP, warranting further investigation as the shared 80% sequence identity in their conserved SET domains poses a challenge for developing paralog-specific chemical probes. Overall, MS8511 is a potent, G9a/GLP-selective, and cell-active covalent chemical probe that can be used to study G9a/GLP biology in vitro and aid in future compound development [53].
Degraders of WDR5
As part of the MLL/KMT2A lysine methyltransferase complex, WDR5 overexpression is implicated in oncogenesis [19, 20]. Previous PPI inhibitors that interrupt complex formation have weak anti-tumor activity, most likely because they do not address non-catalytic WDR5 oncogenic activity [55]. As such, recently published WDR5 PROTAC degraders offer an alternative strategy that targets both catalytic and non-catalytic oncogenic activity. The recently reported first-in-class VHL-recruiting WDR5 degrader, compound 8g, selectively induces robust WDR5 degradation but does not potently inhibit MV4;11 cell growth [56] (Table 1 & Figure 2E). Another VHL-recruiting WDR5 degrader, MS67 (Table 1 & Figure 2E), was developed via structure-based design with a high-resolution crystal structure of the WDR5-degrader-E3 ligase ternary complex [57]. The ternary complex structure shows extensive new interactions between the WDR5, and VHL induced by PROTAC MS67 (Figure 3, violet residues, PBD ID: 7JTP). This study displays the advantage of obtaining ternary structures for the design of more potent and selective PROTAC degraders. MS67 is selective for WDR5, potently degrades WDR5 in a proteasome-dependent manner, and displays superior in vitro and in vivo anti-tumor activity compared to WDR5 PPI inhibitors The degrader effectively suppresses transcription of WDR5-regulated genes, depletes the MLL complex, displays improved cell cycle arrest and apoptosis, and phenocopies WDR5 KO in certain cell lines. MS67 treatment led to potent, effective, and selective degradation of WDR5 in a panel of MLL-r AML and PDAC cell lines, patient cells, and xenograft models. Overall, MS67 displays superior in vitro and in vivo anti-tumor activity, a promising PK/PD profile, and is well tolerated in mice, highlighting its potential for future development as a therapeutic agent [57]. The CRBN-recruiting degrader MS40 (Table 1 & Figure 2E) selectively degrades WDR5 in addition to CRBN neo-substrates IKZF1 and IKZF3 [58]. The degrader significantly decreases H3K4me2, represses MLL complex function, and downregulates target genes of WDR5 and IKZF1/3, as seen in WDR5 KD. MS40 has superior anti-tumor activity than the PPI inhibitor OICR-9429 [59] in MLL-r AML, breast cancer, and primary AML cell lines, as well as promising and well-tolerated in vivo activity in an AML PDX model. The improved antitumor activity of MS40 is likely due to dual targeting of WDR5 and IKZF1/3, which have distinct separate oncogenic functions. Coupled with favorable in vivo PK/PD and lack of toxicity, MS40 has potential as a therapeutic agent for WDR5- and IKZF1/3-dependent cancers [58]. Both MS40 and MS67 display improved anti-cancer activity compared to inhibitors, suggesting that TPD is an effective alternative approach for targeting proteins with both catalytic and non-catalytic functions like WDR5.
Figure 3.
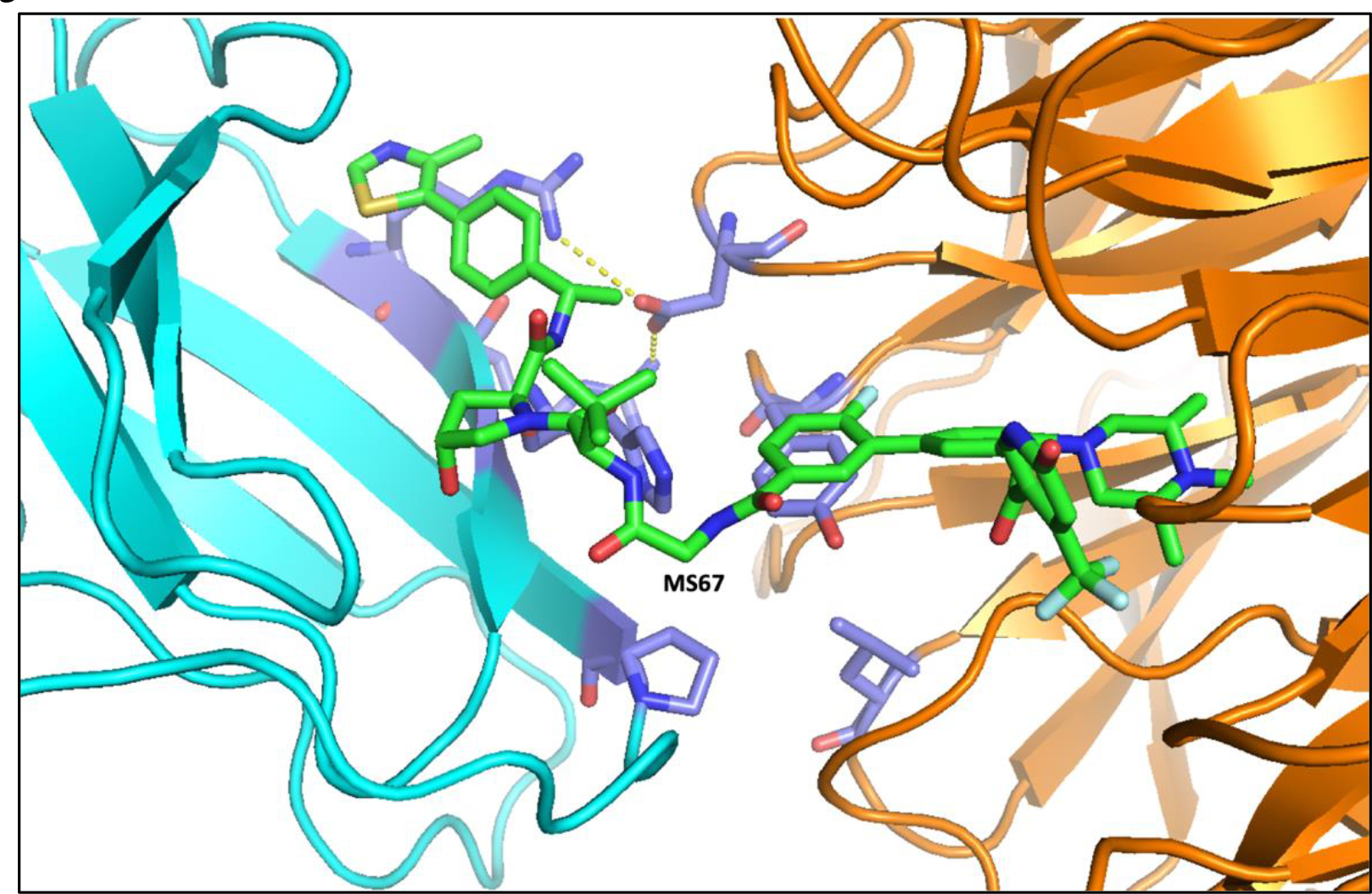
Crystal structure of the VHL (cyan)-MS67(green)-WDR5 (orange) ternary complex. Violet residues indicate new protein-protein interactions induced by MS67. Hydrogen bon interactions were indicated by yellow dotted lines. PBD ID: 7JTP.
Conclusion
Over the past few years, there have been many advancements in PKMT-targeting small molecule compounds. These novel compounds overcame previous challenges, including inhibitor resistance and poor in vivo activity. As laid out in this review, effective PKMT-targeting degraders with improved anti-cancer activity have been developed as novel, promising therapeutics, including those targeting EED (UNC6582, EED-PROTAC-1) [30, 31], EZH2 (MS1943, MS8815, YM281, U3i, E7, MS177) [32–37], NSD2 (MS159) [45], NSD3 (MS9715) [47], and WDR5 (MS40, MS67) [57, 58]. The novel inhibitors for ASH1L (AS-99) [41], NSD1 (BT5) [44], SETD2 (EPZ-719) [52], and G9a/GLP (MS8511) [53] presented here overcame structural challenges and have improved properties that allow for in vivo studies. Overall, these compounds provide effective chemical tools to investigate PKMT biology, offer potential anti-cancer therapeutic approaches to expand on, and lay the groundwork for future development of even more effective PKMT-targeting agents.
Acknowledgements:
J.J. acknowledges the support by the grants R01CA218600, R01CA230854, R01CA260666, R01CA268384, and R01CA268519 from the National Cancer Institute (NCI) at the U.S. National Institutes of Health (NIH), and an endowed professorship by the Icahn School of Medicine at Mount Sinai. J.V. acknowledges the support by the grant R01CA218600S1 from the NCI and the Training Grant in Cancer Biology (T32CA078207) at the Icahn School of Medicine at Mount Sinai from the U.S. NIH.
Footnotes
Conflicts of interest:
J. V. declares no conflicts of interest. H.Ü.K is a consultant for EpiCypher Inc. J.J. is a cofounder and equity shareholder in Cullgen, Inc., a scientific cofounder and scientific advisory board member of Onsero Therapeutics, Inc., and a consultant for Cullgen, Inc., EpiCypher, Inc., Accent Therapeutics, Inc, and Tavotek Biotherapeutics, Inc. The Jin laboratory received research funds from Celgene Corporation, Levo Therapeutics, Inc., Cullgen, Inc. and Cullinan Oncology, Inc.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
* of special interest
** of outstanding interest
- 1.Bernstein BE, Meissner A, and Lander ES, The mammalian epigenome. Cell, 2007. 128(4): p. 669–81. [DOI] [PubMed] [Google Scholar]
- 2.Kaniskan HÜ and Jin J, Recent progress in developing selective inhibitors of protein methyltransferases. Curr. Opin. Chem. Biol, 2017. 39: p. 100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arrowsmith CH, et al. , Epigenetic protein families: a new frontier for drug discovery. Nat. Rev. Drug Discov, 2012. 11(5): p. 384–400. [DOI] [PubMed] [Google Scholar]
- 4.Dawson MA and Kouzarides T, Cancer epigenetics: from mechanism to therapy. Cell, 2012. 150(1): p. 12–27. [DOI] [PubMed] [Google Scholar]
- 5.Helin K and Dhanak D, Chromatin proteins and modifications as drug targets. Nature, 2013. 502(7472): p. 480–8. [DOI] [PubMed] [Google Scholar]
- 6.Schapira M, Chemical Inhibition of Protein Methyltransferases. Cell Chem. Biol, 2016. 23(9): p. 1067–1076. [DOI] [PubMed] [Google Scholar]
- 7.Bannister AJ and Kouzarides T, Regulation of chromatin by histone modifications. Cell Res, 2011. 21(3): p. 381–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martin C and Zhang Y, The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol, 2005. 6(11): p. 838–49. [DOI] [PubMed] [Google Scholar]
- 9.Zagni C, Chiacchio U, and Rescifina A, Histone methyltransferase inhibitors: novel epigenetic agents for cancer treatment. Curr. Med. Chem, 2013. 20(2): p. 167–85. [DOI] [PubMed] [Google Scholar]
- 10.Garcia-Martinez L, et al. , Epigenetic mechanisms in breast cancer therapy and resistance. Nat. Commun, 2021. 12(1): p. 1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu H, et al. , Genetic and Epigenetic Targeting Therapy for Pediatric Acute Lymphoblastic Leukemia. Cells, 2021. 10(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Margueron R and Reinberg D, The Polycomb complex PRC2 and its mark in life. Nature, 2011. 469(7330): p. 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim KH and Roberts CW, Targeting EZH2 in cancer. Nat. Med, 2016. 22(2): p. 128–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tu WB, et al. , MYC interacts with the G9a histone methyltransferase to drive transcriptional repression and tumorigenesis. Cancer cell, 2018. 34(4): p. 579–595. e8. [DOI] [PubMed] [Google Scholar]
- 15.Sun T, et al. , G9a Promotes Invasion and Metastasis of Non–Small Cell Lung Cancer through Enhancing Focal Adhesion Kinase Activation via NF-κB Signaling PathwayG9a Promotes Invasion and Metastasis in NSCLCs through FAK. Mol. Cancer Res, 2021. 19(3): p. 429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu L, et al. , ASH1L Links Histone H3 Lysine 36 Dimethylation to MLL Leukemia. Cancer Discov, 2016. 6(7): p. 770–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones M, et al. , Ash1l controls quiescence and self-renewal potential in hematopoietic stem cells. J. Clin. Invest, 2015. 125(5): p. 2007–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bennett RL, et al. , The Role of Nuclear Receptor-Binding SET Domain Family Histone Lysine Methyltransferases in Cancer. Cold Spring Harb Perspect Med, 2017. 7(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan X, et al. , PI3K/AKT-mediated upregulation of WDR5 promotes colorectal cancer metastasis by directly targeting ZNF407. Cell Death Dis., 2017. 8(3): p. e2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen X, et al. , Upregulated WDR5 promotes proliferation, self-renewal and chemoresistance in bladder cancer via mediating H3K4 trimethylation. Sci. Rep, 2015. 5: p. 8293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duns G, et al. , Histone methyltransferase gene SETD2 is a novel tumor suppressor gene in clear cell renal cell carcinoma. Cancer Res, 2010. 70(11): p. 4287–91. [DOI] [PubMed] [Google Scholar]
- 22.Kaniskan HÜ, Konze KD, and Jin J, Selective inhibitors of protein methyltransferases. J. Med. Chem, 2015. 58(4): p. 1596–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaniskan HÜ and Jin J, Chemical probes of histone lysine methyltransferases. ACS Chem. Biol, 2015. 10(1): p. 40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao L, et al. , Targeted protein degradation: mechanisms, strategies and application. Signal Transduct. Target Ther, 2022. 7(1): p. 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dale B, et al. , Advancing targeted protein degradation for cancer therapy. Nat. Rev. Cancer, 2021. 21(10): p. 638–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cromm PM and Crews CM, Targeted Protein Degradation: from Chemical Biology to Drug Discovery. Cell Chem.Biol, 2017. 24(9): p. 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27*. Vogelmann A, et al. , Proteolysis targeting chimeras (PROTACs) for epigenetics research. Curr. Opin. Chem. Biol, 2020. 57: p. 8–16. Comprehensive review of PROTAC degraders as tools for advancing epigenetic research.
- 28.Webb T, Craigon C, and Ciulli A, Targeting epigenetic modulators using PROTAC degraders: Current status and future perspective. Bioorg. Med. Chem. Lett, 2022. 63: p. 128653. [DOI] [PubMed] [Google Scholar]
- 29.Mullard A, FDA approves an inhibitor of a novel ‘epigenetic writer’. Nat. Rev. Drug Discov, 2020. 19: p. 156. [DOI] [PubMed] [Google Scholar]
- 30.Potjewyd F, et al. , Degradation of Polycomb Repressive Complex 2 with an EED-Targeted Bivalent Chemical Degrader. Cell Chem. Biol, 2020. 27(1): p. 47–56 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsu JH, et al. , EED-Targeted PROTACs Degrade EED, EZH2, and SUZ12 in the PRC2 Complex. Cell Chem. Biol, 2020. 27(1): p. 41–46 e17. [DOI] [PubMed] [Google Scholar]
- 32**. Ma A, et al. , Discovery of a first-in-class EZH2 selective degrader. Nat Chem Biol, 2020. 16(2): p. 214–222. First EZH2-targeted protein degrader published and one of the first hydrophobic tag-based degrader compounds reported.
- 33.Tu Y, et al. , Design, Synthesis, and Evaluation of VHL-Based EZH2 Degraders to Enhance Therapeutic Activity against Lymphoma. J. Med. Chem, 2021. 64(14): p. 10167–10184. [DOI] [PubMed] [Google Scholar]
- 34.Dale B, et al. , Targeting Triple-Negative Breast Cancer by a Novel Proteolysis Targeting Chimera Degrader of Enhancer of Zeste Homolog 2. ACS Pharmacol. Transl. Sci, 2022. 5(7): p. 491–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Z, et al. , Design and Synthesis of EZH2-Based PROTACs to Degrade the PRC2 Complex for Targeting the Noncatalytic Activity of EZH2. J. Med. Chem, 2021. 64(5): p. 2829–2848. [DOI] [PubMed] [Google Scholar]
- 36.Wang C, et al. , Discovery of precision targeting EZH2 degraders for triple-negative breast cancer. Eur. J. Med. Chem, 2022. 238: p. 114462. [DOI] [PubMed] [Google Scholar]
- 37**. Wang J, et al. , EZH2 noncanonically binds cMyc and p300 through a cryptic transactivation domain to mediate gene activation and promote oncogenesis. Nat. Cell Biol, 2022. 24(3): p. 384–399. This study uncovered a novel non-canonical oncogenic function of EZH2, and includes the first dual EZH2-cMyc PROTAC degrader published.
- 38.Rogawski DS, et al. , Two Loops Undergoing Concerted Dynamics Regulate the Activity of the ASH1L Histone Methyltransferase. Biochemistry, 2015. 54(35): p. 5401–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qiao Q, et al. , The structure of NSD1 reveals an autoregulatory mechanism underlying histone H3K36 methylation. J. Biol. Chem, 2011. 286(10): p. 8361–8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zheng W, et al. , Sinefungin derivatives as inhibitors and structure probes of protein lysine methyltransferase SETD2. J. Am. Chem. Soc, 2012. 134(43): p. 18004–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41**. Rogawski DS, et al. , Discovery of first-in-class inhibitors of ASH1L histone methyltransferase with anti-leukemic activity. Nat. Commun, 2021. 12(1): p. 2792. This inhibitor overcame the structural challenge of the SET domain autoinhibitory loop and can serve as a basis for future compound developement.
- 42.Shen Y, et al. , Identification of LEM-14 inhibitor of the oncoprotein NSD2. Biochem. Biophys. Res. Commun, 2019. 508(1): p. 102–108. [DOI] [PubMed] [Google Scholar]
- 43.Coussens NP, et al. , High-throughput screening with nucleosome substrate identifies small-molecule inhibitors of the human histone lysine methyltransferase NSD2. J. Biol. Chem, 2018. 293(35): p. 13750–13765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang H, et al. , Covalent inhibition of NSD1 histone methyltransferase. Nat. Chem. Biol, 2020. 16(12): p. 1403–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45*. Meng F, et al. , Discovery of a first-in-class degrader for nuclear receptor binding SET domain protein 2 (NSD2) and Ikaros/Aiolos. J. Med. Chem, 2022. 65(15): p. 10611–10625. First report of a dual NSD2 and IKZF1/3 PROTAC degrader that displays superior anticancer activity due to dual targeting.
- 46.Dilworth D, et al. , A chemical probe targeting the PWWP domain alters NSD2 nucleolar localization. Nat. Chem. Biol, 2022. 18(1): p. 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47*. Xu C, et al. , A NSD3-targeted PROTAC suppresses NSD3 and cMyc oncogenic nodes in cancer cells. Cell Chem. Biol, 2022. 29(3): p. 386–397 e9. A NSD3 PROTAC degrader that alters the downstream target genes of both NSD3 and cMyc.
- 48.Sun Y, et al. , Discovery of a potent and selective proteolysis targeting chimera (PROTAC) degrader of NSD3 histone methyltransferase. Eur. J. Med. Chem, 2022: p. 114528. [DOI] [PubMed] [Google Scholar]
- 49.Bottcher J, et al. , Fragment-based discovery of a chemical probe for the PWWP1 domain of NSD3. Nat. Chem. Biol, 2019. 15(8): p. 822–829. [DOI] [PubMed] [Google Scholar]
- 50.Sun XJ, et al. , Identification and characterization of a novel human histone H3 lysine 36-specific methyltransferase. J. Biol. Chem, 2005. 280(42): p. 35261–71. [DOI] [PubMed] [Google Scholar]
- 51.Edmunds JW, Mahadevan LC, and Clayton AL, Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J, 2008. 27(2): p. 406–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lampe JW, et al. , Discovery of a First-in-Class Inhibitor of the Histone Methyltransferase SETD2 Suitable for Preclinical Studies. ACS Med. Chem. Lett, 2021. 12(10): p. 1539–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Park K-S, et al. , Discovery of the First-in-Class G9a/GLP Covalent Inhibitors. J. Med. Chem, 2022. 65(15): p. 10506–10522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu F, et al. , Discovery of an in vivo chemical probe of the lysine methyltransferases G9a and GLP. J. Med. Chem, 2013. 56(21): p. 8931–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Senisterra G, et al. , Small-molecule inhibition of MLL activity by disruption of its interaction with WDR5. Biochem. J, 2013. 449(1): p. 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dölle A, et al. , Design, Synthesis, and Evaluation of WD-Repeat-Containing Protein 5 (WDR5) Degraders. J. Med. Chem, 2021. 64(15): p. 10682–10710. [DOI] [PubMed] [Google Scholar]
- 57*. Yu X, et al. , A selective WDR5 degrader inhibits acute myeloid leukemia in patient-derived mouse models. Sci. Transl. Med, 2021. 13(613): p. eabj1578. Two high-resolution crystal structures of the WDR5-PROTAC-E3 ligase complex and an effective WDR5 PROTAC degrader.
- 58.Li D, et al. , Discovery of a dual WDR5 and Ikaros PROTAC degrader as an anti-cancer therapeutic. Oncogene, 2022. 41(24): p. 3328–3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grebien F, et al. , Pharmacological targeting of the Wdr5-MLL interaction in C/EBPα N-terminal leukemia. Nat. Chem. Biol, 2015. 11(8): p. 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]