

Abstract
Technical advancements over the past 2 decades have enabled the measurement of the panoply of molecules of cells and tissues including transcriptomes, epigenomes, metabolomes, and proteomes at an unprecedented resolution. Unbiased profiling of these molecular landscapes in the context of aging can reveal important details about mechanisms underlying age-related functional decline and age-related diseases. However, the high-throughput nature of these experiments creates unique analytical and design demands for robustness and reproducibility. In addition, ‘omic’ experiments are generally onerous, making it crucial to effectively design them to eliminate as many spurious sources of variation as possible, as well as account for any biological or technical parameter that may influence such measures. In this manuscript, we provide general guidelines on best practices in the design and analysis of ‘omic’ experiments in aging research, from experimental design to data analysis and considerations for long-term reproducibility and validations of such studies.
Introduction
With the democratization of high-throughput ‘omics’ experiments, it is now easier than ever to get genome-wide information about the molecular pathways governing how cells, tissues and organisms age. Indeed, over the past decades, we have gained the ability to generate robust high-throughput measures describing the molecular landscapes of biological systems at multiple levels (e.g. genome, epigenome, transcriptome, metabolome, proteome). Such high-level analyses enable unbiased analyses of how these systems respond to complex biological contexts (e.g. aging), and enable researchers to generate new hypotheses about the molecular networks that regulate such processes. Their high-throughput nature creates unique analytical and design demands. For example, the ideal number of replicates in low-throughput experiments can be statistically determined using power analyses or simulations, whereas every molecule in an ‘omic’ experiment will have its own effect size and variance in response to biological cues, which cannot be reasonably determined a priori. In addition, because of the cost involved, it is critical to effectively design such experiments to account for biological or technical parameters that may influence measures of cellular ‘omes’ with aging. This is important not. only to improve reproducibility and interpretability of experiments, but also to determine how general the conclusions that can be derived from an ‘omic’ experiment are to explain aging (i.e. only relevant to one sex/strain, or multiple).
Although the most mature and robust ‘omics’ technologies usually interrogate populations of cells (i.e. bulk ‘omics’), recent advances now allow such investigation to occur at the level of single cells or single nuclei. Single cell profiling technology theoretically has the unique power to detect underlying cell population heterogeneity. Indeed, single cell ‘omics’ can be very useful in the identification of the cellular composition of a population or tissue of interest, although their power is more limited for identification of, for example, differentially expressed genes in a cell type of interest, due to sampling bias, relatively high technical noise and an illusion of large numbers (e.g. dropouts1, high false discovery2). Thus, special considerations should be taken when designing and analyzing such experiments in aging research and beyond.
Aging research has rightfully embraced the “omics” revolution, which has already enabled many important discoveries about the aging process. Unbiased high-throughput experiments in aging animals has already provided important insights about aging: discovery of widespread immune activation across tissues3, 4, 5, development biomarkers and clocks to quantify aging using machine-learning approaches6, 7, and identification of determinants of species longevity8, 9, 10. Due to laborious and time consuming nature of aging experiments, effective design, generation and analysis of “omics” data is especially critical in aging research. General guidelines for “omics” studies have been discussed elsewhere11, 12, 13, 14, 15, 16, 17. In this manuscript, we discuss specific guidelines for best practices in the design and analysis of ‘omic’ experiments in aging research, focusing on biological, technical and analytical considerations, and validation of such “omics” results, with a more particular focus on sequencing-based omics technologies.
1. Biological considerations for ‘omic’ studies of aging
For any successful aging omics experiment, many key steps must occur very early in the design and sample collection phase. Since any mistake at this stage cannot be compensated for in the downstream analysis, it is crucial to consider sources of biological variation (e.g. sex, reproductive status, circadian variation, genetic background, microbiome, etc.) and how they interact with the studied process (e.g. biological aging). Indeed, uncontrolled influence of such sources of biological variation will lead to confounded designs from which no robust interpretable results can be derived. Like any other aging experiment using animals, aging “omics” should adhere to general guidelines for animal use in aging reseach18 and general reporting guidelines for animal research (ARRIVE)19. In addition, we discuss here important sources of biological variation that may be relevant to account for in the design of a robust “omics” experiment for the study of aging (Figure 1; Table 1).
Figure 1: Considerations for the use of “omics” in aging research.
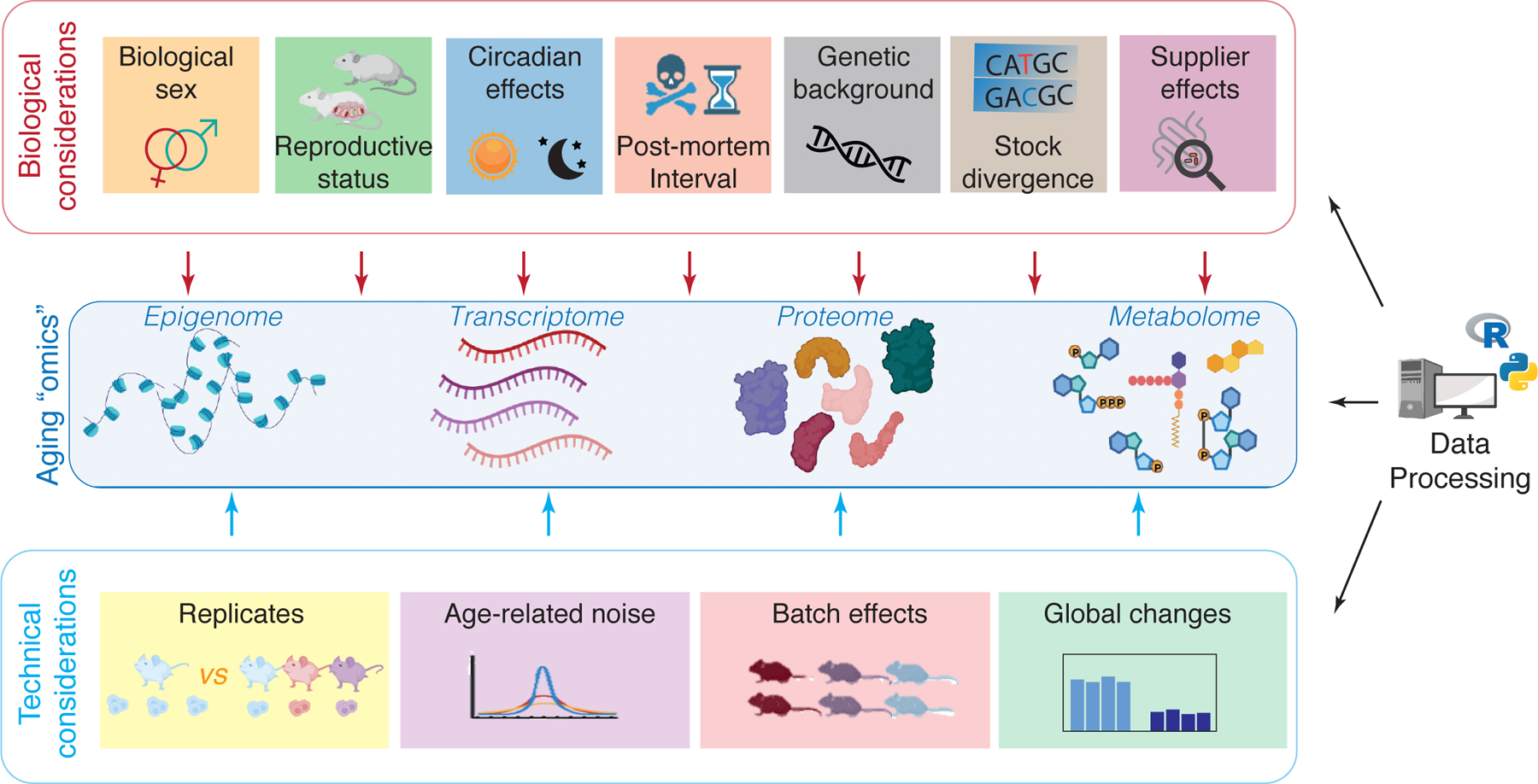
In addition to general considerations for “omics” experiments, aging “omics” requires additional care in experimental design and data processing due to unique challenges. Important biological variables can interact with aging signatures and should be controlled for carefully (e.g. sex). In addition, due to timescales involved in aging research, there are also important technical issues to take into account (e.g. batch effects). Both biological considerations and technical considerations should be taken into account during data processing to extract features of aging at the “omic” level. Some elements created with BioRender.com.
Table 1:
Biological considerations in aging “omics”
| Biological variable | Biological impact | Remedy | References |
|---|---|---|---|
| Biological sex | ‐ X-linked and Y-linked gene expression ‐ Sex hormone signaling |
‐ Balanced representation of biological sex across groups | 20, 21, 24 |
| Reproductive status | ‐ Sex hormone signaling ‐ Immune remodeling |
‐ Matching reproductive status across groups ‐ Eliminate routine use of retired breeding |
25, 26, 27 |
| Circadian effects | ‐ Circadian gene regulation | ‐ Staggered collection approach | 28, 29 |
| Post-mortem interval | ‐ Gene program activation post-mortem | ‐ Staggered collection approach | 30 |
| Genetic background | ‐ Changes in basal gene regulation ‐ Impacts responses to longevity intervention |
‐ Only compare animals with the same genetic backgrounds | 34, 35, 51 |
| Genetic divergence of inbred animal stocks | ‐ Possible changes in gene regulation | ‐ Only compare animals from the same genetic stock | 37, 38 |
| “Supplier” effects and microbiome | ‐ Microbiome composition impacted by housing ‐ Systemic changes linked to microbiome changes |
‐ Only compare animals from the same supplier/animal facility | 40, 41 |
Biological sex.
Accumulating evidence has shown that biological processes in general, and aging in particular, are very sex-dimorphic even outside of reproductive function20. Typically, sex-differences can derive from differential gonadal hormone prevalence (i.e. estrogens vs. androgens) or differences in sex chromosome complement (i.e. XX vs. XY in mammals)21. Of specific relevance to omics studies, gene expression and chromatin structure show marked differences throughout life22, 23, even when focusing only on autosomes. In addition, different age-related ‘omic’ trajectories can be observed depending on biological sex24. For these reasons, aging omics studies should ideally include both female and male samples in sufficient numbers (see below) to detect any potential sex differences in aging trajectories, especially if studies aim at deriving generalizable conclusions for the field. In the case that inclusion of both sexes is not feasible (e.g. cost reasons), which sex was used should be explicitly and prominently mentioned, and this choice should be discussed as a caveat. Further, it is never appropriate to: (i) not describe the sex of the samples used, or (ii) use animals of different sexes in different groups (e.g. males only for the young group and females only for the old group). In addition, it is also generally discouraged to derive samples pooled from animals of mixed sex, unless there is reasonable biological justification, as there is a concern that effects could average out and provide a misleading picture existing in neither sex separately.
Reproductive status.
In addition to their role in reproduction, sex hormones influence somatic tissues by signaling through broadly expressed dedicated receptors, such as estrogen receptors (e.g. Erα, Erβ) or androgen receptor (i.e. Ar). Thus, different reproductive states are not neutral with respect to somatic biological processes, including at the “omic” level. Indeed, gene expression and chromatin accessibility can be directly regulated by cyclic estrogen fluctuations during the murine estrus cycle, for example in the brain25. Pregnancy itself is accompanied by major hormonal changes and has been shown to impact gene expression in human immune cells26, although how long these changes persist after parturition is unclear. Further, mating itself (even in the absence of fertilization or pregnancy) alters the rate of aging in female mice27. Thus, the long-standing practice of using retired breeder mice as a source of cost-effective aged animals may have unforeseen and uncontrolled impact on omics data. Thus, we recommend prioritizing the use of unmated animals unless the impact of breeding and/or co-housing on aging ‘omic’ phenotypes is a desired outcome variable.
Circadian effects.
In animals, the circadian system controls daily biological rhythms, with both central regulation and cell autonomous circuits, whose dysregulation is thought to play an important role during aging28. Circadian biology is mediated in part by large changes in transcriptional regulation that lead to tissue adaptation throughout the day, with the daily regulation of thousands of genes29. This impact of circadian rhythms on genomic regulation has several consequences to avoid confounds in omics experiments. First, one should make sure to stagger sample collection across considered groups so that there is no circadian pattern to the collection of each group (e.g. in an experiment comparing young and old animals, alternate sampling of old and young animals). Second, the approximate time of day at which animals are euthanized for sample collection should be reported, so that data can be appropriately contextualized in future studies.
Post-mortem interval.
Related to circadian impacts, the post-mortem interval has been found to yield reproducible transcriptional changes in human30 and mouse31 tissues. In addition, disease status32 and storage length33 can also affect gene expression in post-mortem tissues, especially relevant to human samples (see below). Since this phenomenon is likely to exist across species and tissues, post-mortem interval should be accounted for in ‘omic’ experimental designs. Thus, we recommend using staggered collection approaches, so as to help mitigate the impact of post-mortem transcriptional remodeling and to avoid confounding the impact of age groups with that of post-mortem transcriptional remodeling.
Genetic background.
The impact of genetics on gene expression patterns in humans has become better appreciated with large scale efforts like GTex34.Because genetic factors can regulate organismal aging in model organisms, and because response to pro-longevity interventions (including at the ‘omic’ level) may be influenced by genetic background (e.g. calorie restriction in mice35, 36), accounting for genetic variability is key for statistical power in ‘omics’ studies. Since genetic variation may decrease the signal-to-noise ratio of such experiments, note that profiling genetically diverse samples may require higher sample numbers to achieve adequate sensitivity (see below).
Genetic divergence of inbred animal stocks.
Inbred animals have been broadly used in aging research, since their lack of genetic diversity may limit non relevant noise in data and makes the analysis of the role of specific genes more tractable (e.g. through knock-out experiments). For instance, labs use mice from C57BL/6 substrains, worms from the N2 strain, African turquoise killifish from the GRZ strain, etc. However, due to reproductive isolation and genetic drift (since mutation rates are not null), such inbred stocks tend to significantly diverge with time37, 38. For mice, since they can be sourced from different vendors (e.g. Jackson labs, Charles River), this has led to the emergence of vendor-specific substrains (e.g. C57BL/6J vs. N), that have fixed dozens of genetic variants leading to many phenotypic differences with relevance to aging37. Vendors like Jackson labs try to minimize the impact of genetic drift on their animals by regularly rederiving animals from frozen embryos. For animal colonies maintained by labs and not a central provider (e.g. C. elegans, killifish), reproductive isolation is expected to lead to genetic drift as each lab maintains their own animal colony, with potential impact on aging phenotypes38. Unfortunately, it is not always possible to source all animals over multi-year studies from the same vendor (e.g. mice), or to control for mutation accumulation over repeated generations of breeding in a lab-maintained colony (e.g. worms, killifish). Thus, in the context of omics studies, it is recommended to ensure that all samples to be compared are derived from the same substrains (and/or within a similar timeframe), or, if unavoidable, that all groups to be profiled contain representation from all different isolates.
“Supplier” effects and the microbiome.
Housing conditions across different animal facilities can vary widely (e.g. chow composition, temperature, ambient noise, etc.). Indeed, even with dedicated standardization efforts, the NIA intervention testing program has reported differences in basal longevity and response to interventions in genetically controlled UM-HET3 mice39. A potential driver for such differences is the microbiome, an emerging factor in aging, longevity and age-related diseases40. Importantly, microbiome composition can vary widely across mouse suppliers41. In controlled microbial transplant experiments, compositional differences in healthy microbiome can significantly influence metabolism, behavior, cognition, blood cell counts41 – and, although this has not been assayed yet, there is no reason to believe that there would not be concomitant ‘omics’ changes as well. Thus, similar as above, it is recommended to make sure that all samples to be compared are derived from animals obtained from the same vendor and within a reasonable timeframe, or, if unavoidable, that all groups contain animals from all sources (i.e. young animals cannot be compared to old animals if sourced from a different supplier, even if they come from the same genetic stock).
Pathological findings and censorship.
There is much debate on whether studying aging should include only “healthy” aging examples, or whether the spectrum of “unhealthy” aging should also be represented. Significant numbers of laboratory mice past middle-age will present with some age-related pathology (neoplastic or non-neoplastic)42. Although this has not necessarily been studied quite as closely, it is expected to occur in other commonly used models in aging research as well. Although the choice to only include macroscopically “healthy” animals may make sense for some study designs, we believe that this decision should be made explicit, and criteria for animal censorship should be transparent. Such reporting is crucial so that the scientific community can interpret a dataset as representative of “normal” vs. “healthy” aging.
Choice of age groups in animal models.
Choosing age groups for “omics” profiling will have large impact on the results. The simplest design for aging “omics” experiments using model organisms will include a “young” group compared to an “old” group. In general, young animals should be past sexual maturity, ideally when reproductive processes are stabilized18. In contrast, old animals ideally correspond to advanced ages before population crash to avoid the so called “survivorship bias”18. To note, ages representative of young vs. old states will differ as a function of species, but also of system of study (e.g. reproductive vs. somatic aging occur on different time scales in mammalian females). Inclusion of additional intermediate time points may reveal non-linear dynamic regulation of molecular networks, and reveal differential age-related dynamics in abundance changes of transcripts, proteins or metabolites (e.g. transcripts or small RNAs43, proteins44). To note, although informative, adding additional time points may not always be feasible based on animal availability and costs. In addition, multi-time point designs will also require more complex analytical tools. Thus, we encourage the community to consider the inclusion of ages across the lifespan when feasible.
Considerations for human samples.
The considerations discussed above are also broadly applicable to human ‘omics’ datasets (e.g. the need to account for sex/gender, reproductive status, circadian effects, etc.). However, unlike most animal models, humans have greater genetic diversity and heterogeneity. Indeed, human ‘omic’ data can be influenced by behavioral factors (e.g. dietary preferences45, etc.) or even the socio-economic status46 of individuals. Because these factors are not always easy to account for using biobanked samples or even during study recruitment, this makes aging ‘omics’ studies using human samples uniquely challenging to identify consistent age-related patterns. If human samples are obtained from biobanked post-mortem tissues, sample integrity and quality must be insured by evaluating the purity, quantity, concentration and integrity of the samples. In addition, it is key to account for post-mortem interval when selecting samples from biobanks to account for its effect on ‘omics’ profiles30 (see above). Importantly, humans are generally more genetically heterogenous than animal models, with human genetic diversity having been shown to impact gene expression34, chromatin binding47, chromatin accessibility48, DNA methylation49 or proteome50 landscapes. Importantly, detecting significant transcriptional changes with age in the GTEx RNA-seq data required accounting for genetic variability using the top 3 principle components of the genotype of donors51, highlighting the need to account for genetic variation in human ‘omic’ studies. Greater genetic diversity often leads to significant variation in gene expression and regulation between individuals52. Therefore, extra care is needed in experimental design using human samples to account for factors, including population diversity, ethnicity, gender/sex, socioeconomic status and comorbidities. Because of these unique challenges, specialized analytical tools may be needed to account for unique patterns of genetic or environmentally-driven heterogeneity across human samples53, 54 to gain meaningful insights from ‘omics’ aging studies. Finally, it is critical to ensure that appropriate ethical clearance and informed consent has been obtained for the collection and use of the human tissues/samples, and that any data generated is handled, stored and shared in accordance with relevant data privacy regulations55, 56.
2. Technical considerations for ‘omic’ studies of aging
Although the design and analysis of aging ‘omic’ studies shares many technical considerations with that of other biological contexts, there are also important considerations that will be more specifically relevant in the context of aging studies. Here, we discuss and highlight both more general and more specific technical considerations for successful, reproducible, and robust analysis of ‘omic’ changes with aging (Figure 1).
Defining biological vs. technical replicates.
Biological replicates are central in any ‘omics’ experiments to assess the reproducibly of observed changes and identify features that are consistently different between young vs. old animals. Biological replicates are samples that are taken from different animals (or different pools of animals) within a population. Samples derived from the same individual (e.g. coming from any technical variation in sample processing, derived from separate fragments of the same tissue, etc.) constitute technical replicates. When pooling multiple replicates to increase the quantity of input in a bulk ‘omics’ experiment (which may be needed in some instances e.g. if sample quantities are inadequate to produce enough material from a single animal), a pooled sample is still considered as a single biological replicate, and multiple ‘pooled’ samples should be included in the design. While technical replicates may be important to identify non-biological sources of experimental variability, as a rule of thumb, a minimum of three biological replicates should be included for each group in bulk omics experiments (e.g. RNA-seq), to minimize the odds of an outlier sample disproportionally driving results. The reliability of differential expression analysis improves with increasing numbers of biological replicates, especially for genes with lower fold change57, 58, 59. Use of lower numbers of replicates does not increase the false positive rate (i.e. features incorrectly labelled to be differentially expressed), but reduces power to detect truly differentially regulated genes57, 58, 59. Thus, based on benchmarking studies, ‘omic’ experiments with 4–6 biological replicates are likely to capture key salient changes between the conditions of interest (i.e. 60–90% of changes detected with larger replicate number57, 58, 59), although they may miss more subtle changes (e.g. higher rates of false negatives in low sample conditions). To note, based on sample availability and/or budget constraints, smaller number of replicates per time point may be considered for aging time-series experiments given that there is sufficient depth in time-series sampling60. In general, emerging approaches for statistical power analysis may be used to identify the replicates and sample size for differential expression studies, provided that some adequate pilot ‘omic’ data exists61, 62. However, an important caveat to consider for any power calculation in ‘omics’ is that each gene, feature, or region has its own effect size (i.e. fold change in expression or accessibility) and variance (i.e. level variability between samples), and thus its own power threshold. If required due to higher sample variability, it is possible to add new samples in separate batches in follow up experiments after a pilot, as long as all biological groups are represented across subsequent batches (see below). However, it is important to note that adding new samples after a pilot experiment in ‘omics’ will lead to issues related to batch effects (see below), so using a reasonable number of replicates (4–6 per group) a priori should be preferred to maximize biological signal, and avoid the need for batch correction.
Replicates and age-related stochasticity.
Specific to aging biology, aging is thought to harbor a strong stochastic component and different animals may age along different trajectories with distinct molecular changes63. For example, aging has been associated with increased gene expression and epigenetic noise, with higher inter-individual variability observed in old animals64, 65, 66. To note, a recent meta-analysis suggests that age-related transcriptional noise may be driven by changes in cell composition of tissues67. Thus, it is possible that additional biological replicates may be required when including older animals to detect robust aging signatures despite increased variability. Specific benchmarking studies including large number of biological replicates in young vs. old samples will have to be conducted to determine whether age-related stochasticity warrants increased numbers of replicates. However, one should be careful with proactively using increased numbers of replicates for old vs. young animals, as sample number imbalance itself can greatly impact the results of differential gene expression analyses68. The importance of not over-concluding from relative lack of changes in low sample studies related to aging cannot be overstate. Indeed, although early studies of aging proteomics failed to detect robust age-related changes69, later more powered studies were able to detect robust differences44. Higher numbers of replicates are particularly important for studies involving human subjects with high heterogeneity and diversity among individuals.
Replication in single-cell aging ‘omics’.
Similar to bulk ‘omics’, biological replicates in single cell experiments should be derived from cells coming from distinct individuals. Because single-cell experiments are considerably more expensive than bulk ‘omic’ experiments, there are economic constraints to consider when performing multiple sets of independent experiments coming from different animals. This may be especially the case in the context of aging where many rare cell types may no longer be sufficiently abundant in old animals (e.g. neural stem cells70). A key strategy to save cost without compromising quality can be to multiplex biological replicates at earlier stages of library preparation (while allowing sample-level cell tracking71, 72, 73), with downstream computational de-multiplexing to assess biological variability. However, sample mixing after oligo-antibody labeling is only possible where high-quality antibodies can reasonably label all cells in a specific sample (i.e. pan-immune CD45 antigen71), and if the labeling technique does not lead to sample degradation74. For fragile or ultra-low input samples, pooling cells/nuclei from replicate animals in equal starting amounts before library generation can be a solution to sample biological variation, although replicate libraries should still be considered for statistical robustness.
Minimizing and/or accounting for batch effects.
Batch effects will occur when technical variation between sets of processed samples overshadows biological variation, in such a way that biological variation can no longer accurately be measured75. Batch effects are a notorious issue in ‘omic’ data, and often require additional computational pre-processing steps to correct75, 76, 77. To note, batch effects in ‘omics’ can occur at multiple steps, such as (i) original sample collection/storage, (ii) target enrichment (e.g. chromatin immunoprecipitation step, RNA isolation step, etc.), (iii) library construction step, or (iv) sequencing step (e.g. sequencing of replicate libraries on different lanes), and each batch layer can have compounded effects. A unique challenge in ‘omics’ studies with aging animals is that the timescale of experiments can span from days to years for longer-lived species (e.g. mice, humans). This feature can make it impractical to collect all biological samples in a very tight timeframe, which requires staggered study enrollment. Since correcting for batch effects may erase true biological signatures78, whenever possible, aging ‘omic’ experiments should be designed to avoid batch effects. However, when batches are unavoidable (e.g. studies involving human samples or longitudinal collection), a proactive study design should enable downstream correction by ensuring that all (or most) biological groups are represented in each set of samples. In that case, computational algorithms can be used to correct ‘omic’ measurements before downstream analyses are performed both for bulk experiments (e.g. SVA77, RUV79) or single-cell experiments (e.g. Harmony, LIGER80).
Accounting for global changes in aging ‘omics’.
Algorithms for differential abundance analysis (e.g. DEseq281) usually assume that most features are unchanged. However, when global changes in transcription, chromatin accessibility or levels of a chromatin mark occur, these assumptions are violated82, 83, 84. In this case, inclusion of exogenous molecules of the same type as those assayed can be included as “spike-in” controls (i.e. synthetic RNAs82, chromatin from a different species83, 84, etc.). Spike-ins can then be used to perform normalization before downstream analyses are carried out79. To note, in the absence of global changes in ‘omic’ landscapes, use of spike-ins will increase technical noise85. However, if they are unnecessary, spike-in molecules can be safely omitted from downstream analyses4, 86. Thus, if global changes are expected in response to aging, inclusion of spike-ins should be considered when possible.
Impact of tissue composition.
The cellular make-up of tissue can change with aging87. A common change stems from infiltration of immune cells in aged tissues88, 89, which may drive general inflammation- and immune-related gene signatures upregulated with aging. Thus, if cell composition shifts are a concern in a tissue, it may be useful to pair bulk ‘omic’ analyses with analyses of cell type composition in this tissue at corresponding ages. Alternatively, transcriptional profiles of pure cell types that can be found in the cognate tissue, derived from atlas-type single-cell RNA-seq efforts or FACS-sorted cells (e.g. tabula muris, human cell atlas) may be used to perform transcriptomic deconvolution and detect shifts in cell composition (e.g. CIBERSORT, DeconRNAseq)90. Information on underlying cell composition may then be used as modelling covariates to prioritize the identification of cell-autonomous ‘omic’ changes in bulk tissue aging datasets.
Sample preparation methods.
For sequencing-based omics, selection strategies and library construction methods can broadly impact results. First, the selection strategies chosen for the assay should be carefully considered for each ‘omic’ layer: RNA species included in RNA-seq (i.e. polyA selection vs. ribo-depletion), specificity of the antibody used for ChIP-seq, sequencing of controls libraries (e.g. input or IgG immunoprecipitation), etc. In addition, for any sequencing-based methods, it is recommended to keep the number of PCR cycles as low as possible to limit the impact of bottlenecking or GC-biases. When this is difficult due to low input material, such as single cell RNA-seq, UMI-based profiling can help mitigate the impact of such biases. In the context of single-cell profiling, there are additional technical considerations related to sample preparation, compounded by considerations already outlined for bulk assays. Indeed, comparison of single-nucleus RNA sequencing to single-cell RNA-seq on human brain tissue revealed that genes related to microglia activation may be depleted in nuclei samples compared to whole cells91. Thus, it is crucial to use a uniform sample preparation method so that such preparation-related differences do not impact the results of comparisons across age groups. Importantly, since single-cell/nuclei RNA profiling can be very sensitive to ambient RNA in the preparation (derived from dead/dying cells or debris generated during cell/nuclei isolation), it may be important to estimate sample-wise ambient RNA levels and correct them (i.e. SoupX92)., especially if aging could differentially impact cell viability. Finally, it is important to note that the process of cell dissociation for the purpose of single-cell profiling can artefactually induce stress signalling pathways. This issue is well documented in muscle tissue, where it is now recommended to fix the tissue prior to dissociation to single-cell profiling93, 94, although it is likely relevant to other solid tissues requiring dissociation. Such technical issues are more likely to impact profiling of molecules with fast turnover (e.g. mRNA, phosphorylation events) than longer-lived molecules (e.g. proteins). However, the potential impact of dissociation should be taken into account when designing single-cell profiling “omics” experiments.
3. Analytical considerations for reproducible omics in aging and beyond
Due to variations in proposed algorithms to analyze various ‘omic’ experiments, as well as updates to algorithms, it is crucial to establish important rules for long-term reproducibility of ‘omic’ experiments. Although these are not exclusive to the aging field, some recommendations to promote reproducible ‘omic’ analyses are provided below.
Assessing data quality.
An important factor to enhance reproducibility in aging genomics experiments is to perform proper quality controls on the raw data to ensure data quality. The precise quality control metrics will depend on the underlying data. For example, for sequencing-based data it is important to ensure that the quality of raw reads is good (e.g. Phred > 20), that the average alignment rate to the reference genome is reasonable (>70%), and that alignment rate and sequencing depth are comparable across samples. For epigenomic datasets (e.g. ATAC-seq, ChIP-seq), additional quality control metrics should be considered (e.g. enrichment at transcription start sites, enrichment for specific binding motifs). Guidelines for quality assessment for quantitative proteomics95, 96 and metabolomics16 have been discussed elsewhere, and will apply to such datasets in the context of aging as well97. After raw data processing, dimensionality reduction techniques (e.g. PCA, MDS) can help visualize global similarities and relationships across samples, and can be powerful tools in detecting unwanted batch effects and (un-)expected patterns (e.g. separation as a function of age/sex).
Meta-data and covariate inclusion.
Because both biological and technical factors can influence ‘omic’ data collection, it is important to systematically include metadata in computational analyses, in addition to age groups (e.g. sex, genetic backgrounds, batch, library kit, RNA quality, etc.) (Table 2). This can help detect uncorrected batch effects and technical variation but should also be considered to enhance researcher’s ability to detect true aging effects. If warranted, appropriate batch correction methods can be applied before proceeding to final data analysis. Evaluation of biological covariates (e.g. sex, genetic background, vendor, etc.) is also crucial at this stage. Indeed, non-linear interactions of such covariates with age can create analytical issues when using algorithms based on generalized linear models (e.g. DESeq2 Wald-Test81). In the case of non-linear interactions, alternative statistical tests should be used (e.g. DESeq2 Likelihood Ratio Test81).
Table 2:
Metadata collection for aging “omics”
| Variable | Sample-level recording | |
|---|---|---|
| Required | Optional | |
| Biological sex | ‐ Biological sex | ‐ Sex hormone levels |
| Reproductive status | ‐ Mating status | ‐ Estropause/Menopause ‐ Estrus cycle phase (rodents) |
| Circadian effects | ‐ Timeframe of euthanasia | ‐ Day/light cycle of animal facility |
| Feeding and metabolism | ‐ Chow ‐ Weight |
‐ Fasting period |
| Genetics | ‐ Strain and supplier | |
| Pathology | ‐ Gross pathological findings | ‐ Pictures at euthanasia |
| Batch processing | ‐ Sample set processing | ‐ Euthanasia date ‐ Processing date |
| Collection scheme | ‐ Synchronous or asynchronous for age groups | ‐ Relatedness (if non inbred) |
| Replicates | ‐ Number of animals per sample ‐ Labeling of technical replicates (if any) |
‐ Additional per animal information if animals were pooled |
| Processing pipeline | ‐ Software versions ‐ Genome build ‐ Code and processing scripts |
‐ Intermediate processing files |
Data filtering and result reporting.
Although specific pipelines to analyze aging ‘omic’ datasets should be selected to best fit specific experimental design, it is important to appropriately estimate false discovery rates [FDR] in analyses. For instance, a common practice has been to use log fold-change thresholding on differential gene expression analysis results after applying a significance threshold to reduce the number of significant genes to consider. However, when applied a posteriori to standard differential analysis methods (e.g. default DESeq2, edgeR), such post-hoc filtering leads to poor FDR control98. If gene expression change amplitude is crucial to analyses, tests specifically designed to determine differential expression with respect to a specific threshold should be used98, or at least the null hypothesis should be amended to reflect this choice (as is possible in more recent versions of DESeq2). Alternatively, if researchers only wish to shorten the list of potentially interesting genes, test stringency can be increased by decreasing the FDR significance threshold (e.g. from 0.05 to 0.01). Finally, common pipelines should include filtering steps for regions known to yield technical artefacts in specific ‘omic’ types, e.g. ENCODE blacklist regions for epigenomic analysis99 or mitochondrial genome for chromatin accessibility anaysis100.
4. Reproducibility and validation in omics studies
Experimental validation of aging ‘omics’ studies.
There has been much debate, even beyond the aging field, about the need to “validate” the results of high-throughput omics studies (e.g. RNA-seq, ChIP-seq, proteomics) with low throughput methods (e.g. qPCR, Western Blot/ELISA). Historically, this practice stemmed from technical limitations of the first ‘omics’ method (i.e. cDNA microarrays): because microarray technology is based on hybridization of fluorescent cDNA to probes immobilized on a glass slide, followed by imaging, the technique suffers from poor sensitivity and accuracy in the low (i.e. below or equal to background fluorescence) and high expression (i.e. signal saturation) ranges101. Thus, the validation of top targets derived from ‘omics’ experiments on microarray platforms (e.g. cDNA, ChIP-on-chip) by qPCR (e.g. RT-qPCR, ChIP-qPCR) was widely adopted as a way to limit false positives that plagued the early days of microarray experiments.
In contrast, most of the technological limitations of microarray-based methods do not apply to sequencing-based methods, which demonstrate both exceptional sensitivity and specificity (i.e. robust detection of lowly and highly expressed genes)102. By force of habit, many believe that qPCR-based validation of sequencing experiments (e.g. RNA-seq) should be required for robustness. However, to be meaningful, external validation of an ‘omics’ dataset should only be done with a superior method (i.e. one with less technical shortcomings), or on independent samples/molecules. Unfortunately, as a specific example, RT-qPCR suffers from at least one major bias that makes it more noisy and less reliable than RNA-seq: reliance on normalization to “housekeeping” genes (e.g. Actb, Gapdh, Hprt, etc.), with the assumption that their expression is invariant across samples. However, such housekeeping genes can be very variable across conditions103. In addition, since only a handful of genes may feasibly be “validated” this way, sampling bias is such that it may not represent the reproducibility of the whole experiment. Thus, in general, RNA-seq or similar experiment types should not require qPCR validation103, 104, 105. A potential exception to this rule is if differences being are either very small or with large sample-to-sample variation in RNA-seq, and may thus be unduly influenced by unwanted technical noise. However, if qPCR is performed (e.g. to include independent samples in a different strain, or in a different ethnicity for human samples), the choice of normalizing amplicons should be informed by the ‘omics’ dataset itself to avoid introducing noise due to variable housekeeping gene expression. Similar considerations exist in the context for the validation of high-throughput quantitative proteomics by low-throughput Western Blot experiments, as standard housekeeping controls also vary at the protein level between conditions, leading to experts qualifying such validation as unnecessary and invalid106, 107.
Conceptually, one can also approach validation of omics studies on another level – are the predictions derived from the high-throughput dataset concordant with other measures? For instance, we believe that by cross-referencing biological measures across large datasets, multi-omic analyses are a good way to validate findings from one omics dataset (e.g. bulk vs. single cell RNA-seq, comparison with ATAC-seq, etc.). Although not necessary, the degree of validation would be increased if agreement across omics methods is derived from independent sample sets. Importantly, we recommend focusing on agreement at the level of enriched pathways/gene sets rather than specific genes, which will be more robust to exceptions. In addition, since omics is often hypothesis-generating, validation of functional predictions derived from the datasets in functional assays (e.g. differentiation potential of a stem cell, ability to clear protein aggregates, etc.) is also a good form of validation for an omics dataset. Finally, although not necessary, the validation of a few key genes at the protein level by western blot or ELISA would constitute another low-throughput option if validated/specific antibodies exist. However, we also note the usefulness of resource-type omics datasets, that can then be mined by the community for hypothesis generation (e.g. Tabula Muris Senis)87. In these cases, external validation may not be tractable at scale, but extensive quality-checking and meta-data recording should be done to make the datasets robust and reusable. Thus, different criteria of required validation should be considered if the dataset aims at hypothesis-generation vs. hypothesis-validation.
Reproducibility in aging omics research: sharing code and data.
Similar to ‘bench’ protocols, data processing and algorithmic choices are key to reproduce ‘omic’ results. However, since the format of methods sections cannot accommodate every coding detail, it is crucial for long-term reproducibility that all scripts used to analyze data always be deposited in free repositories (e.g. GitHub) or provided as supplemental zip files when corresponding manuscripts are evaluated. In addition, since different versions of the same tools or databases will provide different results, it is important that version numbers for all package/software and databases be provided for analytical reproducibility. Alternatively, the pipelines can also be packaged in ready-to-use formats, such as Conda environments or Docker images.
Most journals, like Nature Aging, now require deposition of the raw ‘omic’ data to public repositories (e.g. SRA, GEO, ProteomeXChange). Going further, we suggest that sharing intermediate processed data (e.g. count tables, peak files, etc.), although not mandatory, will help improve reproducibility and useability of ‘omics’ datasets (e.g. as part of a GitHub repository or as a supplemental archive). In contrast, for single-cell datasets, the final annotated object (i.e. including cell type assignments, meta-data, etc.) must be shared for long-term useability and reproducibility. This can be done readily by uploading relevant files on FigShare or Zenodo (i.e. text files of counts and metadata, RData object, etc.), or on dedicated single-cell browsing platforms (e.g. Broad Single cell portal; https://singlecell.broadinstitute.org/single_cell).
Conclusions
Our goal was to bring attention to best practices in designing and analyzing ‘omic’ studies in aging research. Many considerations that we highlight are generally applicable to ‘omic’ studies in general, but we also discuss unique challenges related to aging research. In general, emerging evidence shows that many biological variables and technical choices can impact biological measures in sometimes unpredictable ways. Thus, we highlight specific sources of biological variation relevant to aging, which should be taken into account when designing aging ‘omic’ experiments (e.g. sex, reproductive status, genetic background, microbiome, circadian effects, etc.). Since aging experiments can span years, we also discuss the importance of accounting for technical variation and batch effects for aging ‘omics’.
Some challenges in the use of ‘omics’ specifically in aging research remain open. For instance, since ‘omics’ experiments on older animals can show increased inter- and intra-sample variability 108, 109, addressing this inherent variability not as a mere confound, but potentially as a biologically relevant outcome, may provide important biological insights. Indeed, at least for single-cell modalities, algorithms are being developed to identify differentially variable genes110. Finally, ‘omic’ layers may become less correlated or decoupled with aging44, 111, 112, 113, 114, creating unique challenges for multi-omic integration in the context of aging. In particular, decoupling between mRNA and protein levels has been observed broadly in post-mitotic tissues (e.g. brain 112, 113, heart 44, kidney111) or quiescent cells (e.g. muscle stem cells114). Proteins likely to accumulate in old tissues (e.g. extra-cellular matrix components, aggregation-prone proteins) may also be more susceptible to decorrelation than other proteins. Thus, robust and reproducible aging ‘omics’ should help the field better understand the systems biology of aging and age-related decline.
Acknowledgements
Some figure elements were created with BioRender.com. We thank Mr. Juan Bravo and Dr. Bryan Teefy for their insights on the manuscript. We apologize to those whose work was not cited due to space constraints.
P.P.S is supported by University of California, San Francisco and the UCSF Bakar Aging Research Institute. B.A.B. is supported by NIGMS R35 GM142395, NIA R01 AG076433, Simons Collaboration on Plasticity in the Aging Brain grant SF811217, Pew Biomedical Scholar Award #00034120, and the Kathleen Gilmore Biology of Aging research award.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Qiu P Embracing the dropouts in single-cell RNA-seq analysis. Nat Commun 11, 1169 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Squair JW et al. Confronting false discoveries in single-cell differential expression. Nat Commun 12, 5692 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schaum N et al. Ageing hallmarks exhibit organ-specific temporal signatures. Nature 583, 596–602 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benayoun BA et al. Remodeling of epigenome and transcriptome landscapes with aging in mice reveals widespread induction of inflammatory responses. Genome Res 29, 697–709 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dulken BW et al. Single-cell analysis reveals T cell infiltration in old neurogenic niches. Nature 571, 205–210 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horvath S & Raj K DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet 19, 371–384 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Rutledge J, Oh H & Wyss-Coray T Measuring biological age using omics data. Nat Rev Genet 23, 715–727 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang Z et al. Longitudinal comparative transcriptomics reveals unique mechanisms underlying extended healthspan in bats. Nat Ecol Evol 3, 1110–1120 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Ma S et al. Comparative transcriptomics across 14 Drosophila species reveals signatures of longevity. Aging Cell 17, e12740 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma S et al. Cell culture-based profiling across mammals reveals DNA repair and metabolism as determinants of species longevity. Elife 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Landt SG et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res 22, 1813–1831 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conesa A et al. A survey of best practices for RNA-seq data analysis. Genome Biol 17, 13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luecken MD & Theis FJ Current best practices in single-cell RNA-seq analysis: a tutorial. Mol Syst Biol 15, e8746 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan F, Powell DR, Curtis DJ & Wong NC From reads to insight: a hitchhiker’s guide to ATAC-seq data analysis. Genome Biol 21, 22 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baek S & Lee I Single-cell ATAC sequencing analysis: From data preprocessing to hypothesis generation. Comput Struct Biotechnol J 18, 1429–1439 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alseekh S et al. Mass spectrometry-based metabolomics: a guide for annotation, quantification and best reporting practices. Nat Methods 18, 747–756 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakayasu ES et al. Tutorial: best practices and considerations for mass-spectrometry-based protein biomarker discovery and validation. Nat Protoc 16, 3737–3760 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller RA & Nadon NL Principles of animal use for gerontological research. J Gerontol A Biol Sci Med Sci 55, B117–123 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Percie du Sert N et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biol 18, e3000410 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Austad SN Sex differences in health and aging: a dialog between the brain and gonad? Geroscience 41, 267–273 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Y, Kim M, Paye S & Benayoun BA Sex as a Biological Variable in Nutrition Research: From Human Studies to Animal Models. Annu Rev Nutr (2022). [DOI] [PMC free article] [PubMed]
- 22.Oliva M et al. The impact of sex on gene expression across human tissues. Science 369 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qu K et al. Individuality and variation of personal regulomes in primary human T cells. Cell Syst 1, 51–61 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu RJ et al. Multi-omic profiling of primary mouse neutrophils predicts a pattern of sex and age-related functional regulation. Nat Aging 1, 715–733 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jaric I, Rocks D, Greally JM, Suzuki M & Kundakovic M Chromatin organization in the female mouse brain fluctuates across the oestrous cycle. Nat Commun 10, 2851 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Knight AK et al. Characterization of gene expression changes over healthy term pregnancies. PLoS One 13, e0204228 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garratt M, Try H, Smiley KO, Grattan DR & Brooks RC Mating in the absence of fertilization promotes a growth-reproduction versus lifespan trade-off in female mice. Proc Natl Acad Sci U S A 117, 15748–15754 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Acosta-Rodriguez VA, Rijo-Ferreira F, Green CB & Takahashi JS Importance of circadian timing for aging and longevity. Nat Commun 12, 2862 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang R, Lahens NF, Ballance HI, Hughes ME & Hogenesch JB A circadian gene expression atlas in mammals: implications for biology and medicine. Proc Natl Acad Sci U S A 111, 16219–16224 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferreira PG et al. The effects of death and post-mortem cold ischemia on human tissue transcriptomes. Nat Commun 9, 490 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bonadio RS et al. Insights into how environment shapes post-mortem RNA transcription in mouse brain. Sci Rep 11, 13008 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Highet B, Parker R, Faull RLM, Curtis MA & Ryan B RNA Quality in Post-mortem Human Brain Tissue Is Affected by Alzheimer’s Disease. Front Mol Neurosci 14, 780352 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.White K et al. Effect of Postmortem Interval and Years in Storage on RNA Quality of Tissue at a Repository of the NIH NeuroBioBank. Biopreserv Biobank 16, 148–157 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Consortium GT et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liao CY, Rikke BA, Johnson TE, Diaz V & Nelson JF Genetic variation in the murine lifespan response to dietary restriction: from life extension to life shortening. Aging Cell 9, 92–95 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mitchell SJ et al. Effects of Sex, Strain, and Energy Intake on Hallmarks of Aging in Mice. Cell Metab 23, 1093–1112 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simon MM et al. A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol 14, R82 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Urban ND et al. Explaining inter-lab variance in C. elegans N2 lifespan: Making a case for standardized reporting to enhance reproducibility. Exp Gerontol 156, 111622 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harrison DE et al. Acarbose, 17-alpha-estradiol, and nordihydroguaiaretic acid extend mouse lifespan preferentially in males. Aging Cell 13, 273–282 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim M & Benayoun BA The microbiome: an emerging key player in aging and longevity. Transl Med Aging 4, 103–116 (2020). [PMC free article] [PubMed] [Google Scholar]
- 41.Ericsson AC et al. Supplier-origin mouse microbiomes significantly influence locomotor and anxiety-related behavior, body morphology, and metabolism. Commun Biol 4, 716 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pettan-Brewer C & Treuting PM Practical pathology of aging mice. Pathobiol Aging Age Relat Dis 1 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Teefy BB et al. Dynamic regulation of gonadal transposon control across the lifespan of the naturally short-lived African turquoise killifish. Genome Res 33, 141–153 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keele GR et al. Global and tissue-specific aging effects on murine proteomes. bioRxiv, 2022.2005.2017.492125 (2022). [DOI] [PMC free article] [PubMed]
- 45.Herrera-Marcos LV, Lou-Bonafonte JM, Arnal C, Navarro MA & Osada J Transcriptomics and the Mediterranean Diet: A Systematic Review. Nutrients 9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gaye A, Gibbons GH, Barry C, Quarells R & Davis SK Influence of socioeconomic status on the whole blood transcriptome in African Americans. PLoS One 12, e0187290 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ni Y, Hall AW, Battenhouse A & Iyer VR Simultaneous SNP identification and assessment of allele-specific bias from ChIP-seq data. BMC Genet 13, 46 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kumasaka N, Knights AJ & Gaffney DJ Fine-mapping cellular QTLs with RASQUAL and ATAC-seq. Nat Genet 48, 206–213 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kader F & Ghai M DNA methylation-based variation between human populations. Mol Genet Genomics 292, 5–35 (2017). [DOI] [PubMed] [Google Scholar]
- 50.Wu L et al. Variation and genetic control of protein abundance in humans. Nature 499, 79–82 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang X et al. Tissue-specific expression and regulation of sexually dimorphic genes in mice. Genome Res 16, 995–1004 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haraksingh RR & Snyder MP Impacts of variation in the human genome on gene regulation. J Mol Biol 425, 3970–3977 (2013). [DOI] [PubMed] [Google Scholar]
- 53.Stegle O, Parts L, Piipari M, Winn J & Durbin R Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat Protoc 7, 500–507 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goncalves ANA et al. Assessing the Impact of Sample Heterogeneity on Transcriptome Analysis of Human Diseases Using MDP Webtool. Front Genet 10, 971 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bonomi L, Huang Y & Ohno-Machado L Privacy challenges and research opportunities for genomic data sharing. Nat Genet 52, 646–654 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.O’Doherty KC et al. Toward better governance of human genomic data. Nat Genet 53, 2–8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu Y, Zhou J & White KP RNA-seq differential expression studies: more sequence or more replication? Bioinformatics 30, 301–304 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schurch NJ et al. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA 22, 839–851 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lamarre S et al. Optimization of an RNA-Seq Differential Gene Expression Analysis Depending on Biological Replicate Number and Library Size. Front Plant Sci 9, 108 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sefer E, Kleyman M & Bar-Joseph Z Tradeoffs between Dense and Replicate Sampling Strategies for High-Throughput Time Series Experiments. Cell Syst 3, 35–42 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schmid KT et al. scPower accelerates and optimizes the design of multi-sample single cell transcriptomic studies. Nat Commun 12, 6625 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Poplawski A & Binder H Feasibility of sample size calculation for RNA-seq studies. Brief Bioinform 19, 713–720 (2018). [DOI] [PubMed] [Google Scholar]
- 63.Gladyshev VN Aging: progressive decline in fitness due to the rising deleteriome adjusted by genetic, environmental, and stochastic processes. Aging Cell 15, 594–602 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hernando-Herraez I et al. Ageing affects DNA methylation drift and transcriptional cell-to-cell variability in mouse muscle stem cells. Nat Commun 10, 4361 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.White RR et al. Comprehensive transcriptional landscape of aging mouse liver. BMC Genomics 16, 899 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Angelidis I et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat Commun 10, 963 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ibañez-Solé O, Ascensión AM, Araúzo-Bravo MJ & Izeta A Lack of evidence for increased transcriptional noise in aged tissues. eLife 11, e80380 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang K, Li J & Gao H The impact of sample imbalance on identifying differentially expressed genes. BMC Bioinformatics 7 Suppl 4, S8 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Walther DM & Mann M Accurate quantification of more than 4000 mouse tissue proteins reveals minimal proteome changes during aging. Mol Cell Proteomics 10, M110 004523 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kalamakis G et al. Quiescence Modulates Stem Cell Maintenance and Regenerative Capacity in the Aging Brain. Cell 176, 1407–1419 e1414 (2019). [DOI] [PubMed] [Google Scholar]
- 71.Stoeckius M et al. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol 19, 224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mylka V et al. Comparative analysis of antibody- and lipid-based multiplexing methods for single-cell RNA-seq. Genome Biol 23, 55 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang Y et al. Sample-multiplexing approaches for single-cell sequencing. Cell Mol Life Sci 79, 466 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.10xGenomics. Which nuclei isolation protocols are supported for use with the 3’ CellPlex Kit for Cell Multiplexing? 2022. [cited 2023 2023–01–11]Available from: https://kb.10xgenomics.com/hc/en-us/articles/360061929592-Which-nuclei-isolation-protocols-are-supported-for-use-with-the-3-CellPlex-Kit-for-Cell-Multiplexing-
- 75.Nyamundanda G, Poudel P, Patil Y & Sadanandam A A Novel Statistical Method to Diagnose, Quantify and Correct Batch Effects in Genomic Studies. Sci Rep 7, 10849 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kujawa T, Marczyk M & Polanska J Influence of single-cell RNA sequencing data integration on the performance of differential gene expression analysis. Front Genet 13, 1009316 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Leek JT svaseq: removing batch effects and other unwanted noise from sequencing data. Nucleic Acids Res 42, e161 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jaffe AE et al. Erratum to: Practical impacts of genomic data “cleaning” on biological discovery using surrogate variable analysis. BMC Bioinformatics 17, 302 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Risso D, Ngai J, Speed TP & Dudoit S Normalization of RNA-seq data using factor analysis of control genes or samples. Nat Biotechnol 32, 896–902 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tran HTN et al. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol 21, 12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Loven J et al. Revisiting global gene expression analysis. Cell 151, 476–482 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stewart-Morgan KR, Reveron-Gomez N & Groth A Transcription Restart Establishes Chromatin Accessibility after DNA Replication. Mol Cell 75, 284–297 e286 (2019). [DOI] [PubMed] [Google Scholar]
- 84.Orlando DA et al. Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell Rep 9, 1163–1170 (2014). [DOI] [PubMed] [Google Scholar]
- 85.Baik B, Yoon S & Nam D Benchmarking RNA-seq differential expression analysis methods using spike-in and simulation data. PLoS One 15, e0232271 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen Y, Bravo JI, Son JM, Lee C & Benayoun BA Remodeling of the H3 nucleosomal landscape during mouse aging. Transl Med Aging 4, 22–31 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tabula Muris C A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 583, 590–595 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Singh P et al. Lymphoid neogenesis and immune infiltration in aged liver. Hepatology 47, 1680–1690 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nevalainen T, Autio A & Hurme M Composition of the infiltrating immune cells in the brain of healthy individuals: effect of aging. Immun Ageing 19, 45 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Avila Cobos F, Alquicira-Hernandez J, Powell JE, Mestdagh P & De Preter K Benchmarking of cell type deconvolution pipelines for transcriptomics data. Nat Commun 11, 5650 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Thrupp N et al. Single-Nucleus RNA-Seq Is Not Suitable for Detection of Microglial Activation Genes in Humans. Cell Rep 32, 108189 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Young MD & Behjati S SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. Gigascience 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Machado L et al. Tissue damage induces a conserved stress response that initiates quiescent muscle stem cell activation. Cell Stem Cell 28, 1125–1135 e1127 (2021). [DOI] [PubMed] [Google Scholar]
- 94.Machado L et al. In Situ Fixation Redefines Quiescence and Early Activation of Skeletal Muscle Stem Cells. Cell Rep 21, 1982–1993 (2017). [DOI] [PubMed] [Google Scholar]
- 95.Gatto L et al. Initial recommendations for performing, benchmarking and reporting single-cell proteomics experiments. Nat Methods 20, 375–386 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pappireddi N, Martin L & Wuhr M A Review on Quantitative Multiplexed Proteomics. Chembiochem 20, 1210–1224 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Contrepois K et al. Cross-Platform Comparison of Untargeted and Targeted Lipidomics Approaches on Aging Mouse Plasma. Sci Rep 8, 17747 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.McCarthy DJ & Smyth GK Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics 25, 765–771 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Amemiya HM, Kundaje A & Boyle AP The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci Rep 9, 9354 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Corces MR et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14, 959–962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Argyropoulos C, Daskalakis A, Nikiforidis GC & Sakellaropoulos GC Background adjustment of cDNA microarray images by Maximum Entropy distributions. J Biomed Inform 43, 496–509 (2010). [DOI] [PubMed] [Google Scholar]
- 102.Wang Z, Gerstein M & Snyder M RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10, 57–63 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Panina Y, Germond A, Masui S & Watanabe TM Validation of Common Housekeeping Genes as Reference for qPCR Gene Expression Analysis During iPS Reprogramming Process. Sci Rep 8, 8716 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hughes TR ‘Validation’ in genome-scale research. J Biol 8, 3 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Coenye T Do results obtained with RNA-sequencing require independent verification? Biofilm 3, 100043 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mehta D, Ahkami AH, Walley J, Xu SL & Uhrig RG The incongruity of validating quantitative proteomics using western blots. Nat Plants 8, 1320–1321 (2022). [DOI] [PubMed] [Google Scholar]
- 107.Aebersold R, Burlingame AL & Bradshaw RA Western blots versus selected reaction monitoring assays: time to turn the tables? Mol Cell Proteomics 12, 2381–2382 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Martinez-Jimenez CP et al. Aging increases cell-to-cell transcriptional variability upon immune stimulation. Science 355, 1433–1436 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mahmoudi S et al. Heterogeneity in old fibroblasts is linked to variability in reprogramming and wound healing. Nature 574, 553–558 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vallejos CA, Richardson S & Marioni JC Beyond comparisons of means: understanding changes in gene expression at the single-cell level. Genome Biol 17, 70 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Takemon Y et al. Proteomic and transcriptomic profiling reveal different aspects of aging in the kidney. Elife 10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ori A et al. Integrated Transcriptome and Proteome Analyses Reveal Organ-Specific Proteome Deterioration in Old Rats. Cell Syst 1, 224–237 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kelmer Sacramento E et al. Reduced proteasome activity in the aging brain results in ribosome stoichiometry loss and aggregation. Mol Syst Biol 16, e9596 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schuler SC et al. Extensive remodeling of the extracellular matrix during aging contributes to age-dependent impairments of muscle stem cell functionality. Cell Rep 35, 109223 (2021). [DOI] [PubMed] [Google Scholar]