

Abstract
There is a paucity of data on the transfer of ketamine from maternal blood into human milk. Quantification of ketamine in human milk provides information about the potential exposure of the infant to ketamine and its metabolites from the mother during lactation. A highly specific, reproducible, and sensitive UPLC-MS/MS based analytical method was developed and validated for the quantitation of ketamine and its metabolites (norketamine and dehydronorketamine) in human milk. Samples were subjected to a simple protein precipitation and ketamine-d4 and norketamine-d4 were used as internal standards. Separation of the analytes was achieved using an Acquity UPLC equipped with BEH RP18 1.7μm, 2.1×100 mm column. Mass spectrometric analysis of the analyte ions was carried out using electrospray with negative ionization and multiple reaction monitoring mode. The assay was linear over a concentration range of 1–100 ng/mL for ketamine and norketamine, and 0.1–10 ng/mL for dehydronorketamine. Acceptable intra-day and inter-day accuracy and precision were observed for all the analytes. High recovery of the analytes and minimal matrix effect were observed. Stability of analytes was confirmed at the tested conditions. This assay was successfully used to measure analytes in human milk samples collected from lactating women enrolled in a clinical research study. This is the first validated method that simultaneously quantified ketamine and its metabolites in human milk.
Keywords: Ketamine, Lactation, UPLC-MS/MS, Human milk
Graphical abstract

Introduction
Intravenous ketamine is well-recognized as an adjuvant analgesic and has been shown to result in better outcomes and decreased opioid consumption in the non-pregnant surgical population [1]. However, use of ketamine is not recommended as an analgesic adjuvant in pregnant and lactating population due to lack of safety data [2]. An important concern while administering ketamine in this population is the potential exposure of the drug and metabolite to the newborn during breastfeeding. Previous studies that explored the analgesic effect of ketamine in women undergoing cesarian section have not investigated the potential for drug exposure to the infant during lactation [3–7]. There is lack of data regarding the safety of ketamine as an adjuvant for post-cesarean analgesia in pregnant and lactating populations. Additionally, ketamine is increasingly being used for managing postpartum disorders such as depression despite limited clinical evidence [8]. Therefore, it is necessary to assess and establish the safety of ketamine use during breastfeeding. To quantify the secretion of ketamine and its metabolites into human milk, we developed a sensitive analytical method that quantifies the drug and its metabolites in the human milk collected from lactating women participating in a clinical research study.
Ketamine undergoes extensive metabolism in the liver that is mediated by several cytochrome P450 isoforms, with CYP3A4 as the primary enzyme responsible for N-demethylation of ketamine leading to the generation of its primary metabolite, norketamine, which undergoes further metabolism to generate the hydroxy norketamine and dehydronorketamine [9, 10]. Recent preclinical and clinical studies reviewed pharmacological activity of ketamine and its metabolites, and the evidence supporting the relevance of ketamine and its metabolites to the clinical effects suggested that ketamine metabolites may have broader clinical relevance, with hydroxynorketamine metabolites showing antidepressant activity in preclinical models [11, 12]. An assay method that can simultaneously measure ketamine and its metabolites is critical for any exposure response study.
Limited information is available about the secretion of ketamine and its metabolites into breast milk. To the best of our knowledge, there is only one manuscript in the published literature so far which reported simultaneous measurement of ketamine and its metabolites in human milk collected from four lactating women and they used protein precipitation and solid phase extraction followed by LC-MS/MS analysis, but did not report details on the sample processing and assay validation [13]. Wolfson et al. reported minimal transfer of ketamine and its metabolites into human milk and a rapid decline in drug concentrations in milk at 12 hours after IM dose [13]. However, there is no information available about the safety and transfer of ketamine into human milk during lactation after intravenous (bolus or infusion) administration [14].
Herein, we report the development and validation of a robust and sensitive UPLC-MS/MS assay with a quick and simple sample processing for determining ketamine, and two of its metabolites (norketamine and dehydronorketamine) in human milk. The developed assay was simple, sensitive, and reproducible and was successfully applied to the measurement of ketamine and its metabolites in human milk samples collected from lactating women enrolled in a clinical research study.
Materials and Methods
Chemicals and reagents
Standard stock solutions for +/− ketamine hydrochloride, +/− norketamine hydrochloride, dehydronorketamine hydrochloride, ketamine-d4 hydrochloride and +/− norketamine-d4 hydrochloride was purchased from Cerilliant corporation, Round Rock, TX. LC-MS grade solvents were obtained from Fischer Scientific (Fair Lawn, NJ, USA). Blank human milk was procured from Mid-Atlantic mother’s milk bank (Pittsburgh, PA).
Preparation of standards
The primary stock solutions for ketamine hydrochloride (1 mg/mL) and norketamine hydrochloride (1 mg/mL), ketamine-d4 hydrochloride (100 μg/mL) and norketamine-d4 hydrochloride (100 μg/mL) were prepared in methanol, and the stock solution for dehydronorketamine hydrochloride (100 μg/mL) was prepared in acetonitrile. The working stocks for standards and quality control (QC) samples were prepared by dilution of primary stock solutions with methanol. A working stock solution was prepared by mixing primary stock solutions to obtain a mixture containing 1 μg/mL of ketamine and norketamine, and 0.1 μg/mL dehydronorketamine. A working solution for internal standard mix containing 5 ng/mL of each internal standard was prepared in acetonitrile. Blank human milk was homogenized to reconstitute the supernatant lipid and was spiked with working solution mixture to a known concentration to obtain calibration standards and QC samples. The calibration standards were prepared by serial dilutions to generate a concentration range for calibration curve of 1–100 ng/mL for ketamine and norketamine (1, 2.5, 5, 10, 25, 50, 75, and 100 ng/mL), and 0.1–10 ng/mL for dehydronorketamine (0.1, 0.25, 0.5, 1, 2.5, 5, 7.5, and 10 ng/mL). Four QCs were prepared at concentrations of 2, 20, 40 and 80 ng/mL (and respective 10-fold lower concentrations for dehydronorketamine) spread across the standard curve range, by spiking separate working solutions of analytes into blank human milk. The working standards, spiked milk standards and QC samples were all stored at −80°C. The working internal standard solution was stored at 4°C.
Sample preparation
An aliquot (50 μL) of calibration standards, QCs, or samples was mixed with 450 μL of acetonitrile containing 5 ng/mL internal standard mixture. The contents were vortex mixed for 30 sec and centrifuged at 15,000 rpm for 12 mins at 8°C. The supernatant (450 μL) was transferred to a clean glass tube and dried under air stream. The dried residue was reconstituted in 100 μL reconstitution mixture containing the 4 parts solvent A (95% 2mM ammonium acetate + 5% acetonitrile + 0.1 % formic acid) and 1 part solvent B (95% acetonitrile + 5% water + 0.1% formic acid) of the mobile phase. The reconstituted solution was centrifuged at 15,000 rpm for 12 mins at 8°C, and the supernatant was transferred to waters WISP maximum recovery vials, and 3 μL was injected onto the column and analyzed using UPLC-MS/MS system.
Chromatographic and mass spectrometric conditions
Waters UPLC System equipped with Acquity UPLC BEH RP18 1.7 μm, 2.1 × 100 mm column, combined with a Waters guard column (289002078) was used for separation of analytes. The LC method consisted of a gradient mobile phase of solvent A and solvent B. The solvent gradient started at 100% solvent A which was held for 0.5 min and then changed to 80% solvent A and 20% solvent B by 1 min following a linear gradient. The solvent composition then changed to 65% solvent A and 35% solvent B over the next 3 min, and then immediately to 30% solvent A and 70% solvent B over 0.1 min, and the composition was maintained for 1 min. At 5.2 min, column was then re-equilibrated to initial solvent condition of 100% solvent A. The total run time was 7 mins with a flow rate of 0.3 mL/min. Auto-sampler and column were maintained at 12°C and 50°C respectively.
Analytes were detected using Multiple Reaction Monitoring (MRM) on Waters Xevo TQ-S Mass Spectrometer (Waters Corporation, Milford, MA, USA) in positive electrospray ionization (+ESI) mode. The settings for this instrument were as follows: capillary voltage 1.5kV, de-solvation temperature 500°C, cone gas flow 150 L/h, de-solvation gas flow 1000 L/h; argon pressure 0.15 mL/min, and nitrogen pressure 7 bar. The extracted ions following MRM transitions monitored were m/z 238.2 to 125.1 for ketamine, m/z 224.2 to 125.1 for norketamine, m/z 222.16 to 142.0 for dehydronorketamine, m/z 228.2 to 129.1 for norketamine-d4 (IS), and m/z 242.2 to 129.0 for ketamine-d4 (IS). The cone and collision energy for ketamine, its metabolite, and internal standards are presented in Table 1. The LC and mass spectrometer were controlled, and data collected by Masslynx® software version 4.2 (Waters Corporation, Milford, MA).
Table 1.
The Cone and Collision Energy settings in UPLC/MS-MS for analytes and internal standards
| Parent m/z | Daughter m/z | Dwell (sec) | Cone Energy (V) | Collision Energy (V) | |
|---|---|---|---|---|---|
| Ketamine | 238.2 | 125.1 | 0.044 | 60 | 26 |
| Norketamine | 224.2 | 125.1 | 0.044 | 16 | 22 |
| Dehydronorketamine | 221.16 | 142.0 | 0.044 | 22 | 22 |
| Ketamine-d4 (IS) | 242.2 | 129.0 | 0.044 | 28 | 24 |
| Norketamine-d4 (IS) | 228.2 | 129.1 | 0.044 | 22 | 22 |
Assay validation
Standard curve linearity
Calibration curves were constructed over several days to assess assay linearity and reproducibility. The response for each analyte was assessed as ratio of peak area of observed mass to charge transitions for parent ion and product ions of analyte and to the peak area of the m/z of the respective internal standard (ketamine-d4 for ketamine and norketamine-d4 for both norketamine and dehydronorketamine). The response of the analyte on Y-axis was plotted against the theoretical concentration on X-axis for each analyte separately, and a regression line was drawn with a weighting of 1/x, The slope, intercept, and correlation coefficient were calculated for each analyte. A deviation of ≤15% as compared to the theoretical concentration was accepted for the standard and QC samples. The lower limit of quantification (LLOQ) was set to be the lowest concentration of the analyte giving a response fivefold higher than the blank response. A deviation of ≤20% from the theoretical value was considered acceptable.
Accuracy and precision
The accuracy and precision of the assay were determined using the analytes QC samples spread across the standard curve range. Triplicate QC samples were run on the same day for calculating the variability of the response within the same day (intraday variation). A set of QC samples were also analyzed over three different days to calculate the inter-day variation (). Accuracy was calculated as % difference of the measured concentration from the theoretical value. The precision was determined by calculating the coefficient of variation using the formula:
Recovery and matrix effect
The recovery of the analytes after extraction was determined by comparing the peak areas of analytes from human milk spiked with known amounts of ketamine, its metabolites, and internal standards prior to extraction, with the extracted milk samples to which the analytes were added at the same nominal concentrations just before injection. Recoveries of ketamine, norketamine, and dehydronorketamine were estimated at 2, 20, 40, or 80 ng/ml (0.2, 2, 4 and 8 ng/ml for dehydronorketamine). A single concentration of internal standards (5 ng/ml each IS) was used to determine their recovery. Results were expressed as .
Matrix effect was investigated by comparing the peak areas of analyte standards spiked in blank human milk following extraction with non-extracted standard analyte solutions (in methanol) at the same nominal concentration. The difference from 100% recovery was attributed to matrix effect also termed as ion suppression. Four QC concentrations for analytes and a fixed concentration of internal standards (5 ng/ml each IS) were analyzed. Results were expressed in terms of .
Stability
The stability of ketamine, norketamine and dehydronorketamine in human milk was evaluated in triplicates at each QC level under three different conditions. The milk samples spiked with known concentrations of analytes were stored for 24 hours at room temperature (benchtop), 24 hours at 4°C, or frozen at −80°C and then thawed at room temperature for 3 cycles before analysis. Long term stability of the samples was assessed by analyzing the QC samples stored at −80°C for 12 months. In all cases, the measured analyte concentrations in stability QC samples were compared against freshly prepared standard curve and QCs.
Assay application
Breast milk samples were collected from postpartum lactating women participating in a clinical research study approved by the University of Pittsburgh IRB (STUDY18120046) and a written informed consent was obtained from all participants before performing any study related procedures. The studies were performed approximately 6 months after delivery. Participants received a sub-anesthetic ketamine infusion at 0.1 mg/kg/hour for 12 hours (not to exceed 8 mg/hr) dose. Each participant was asked to collect the entire expressed milk by pumping at seven different time points covering a 20-hour period after the start of the infusion (time points: 2, 4, 6, 12, 14, 16, and 20 h). Human milk samples aliquots were used for estimating the concentration of ketamine and its metabolites. If the concentration of analytes in the samples were beyond the standard curve range, the samples were diluted with blank milk and reanalyzed.
Results
Mass spectral analysis
Methanolic solutions of five analytes, ketamine, norketamine, dehydronorketamine, ketamine-d4, and norketamine-d4, were individually infused into the mass spectrometer in the + ESI mode and the mass-to-charge transition ratios (m/z ratio) from parent molecule to product ion were optimized. Table 1 shows the m/z transition ratios for all the 5 compounds. The MS instrument settings were optimized to achieve the maximum intensity.
Separation and relative retention time
The retention times for ketamine and ketamine-d4, norketamine and norketamine-d4 and dehydronorketamine were 2.90 min, 2.87 min, and 2.72 min, respectively. The total run time for each sample was 7 mins. The gradient elution was necessary to achieve an optimal separation of analytes with sharp and symmetrical peaks. Representative chromatograms for blank milk samples, and milk samples spiked with analytes at LLOQ, and 12 h milk sample collected from a patient are shown in Figure 1 (right panel).
Figure 1:

Representative chromatograms for ketamine, norketamine, dehydronorketamine, ketamine-d4 and norketamine-d4 in blank human milk (left panel), blank human milk spiked with analytes at LLOQ concentrations (middle panel) and chromatograms from human milk sample collected from subject 6 at 12h after initiating ketamine infusion (right panel).
Linearity
The calibration curves of the three analytes showed excellent linearity over a concentration range of 1–100 ng/mL for ketamine and norketamine and 0.1–10 ng/mL for dehydronorketamine. The mean correlation coefficients for analytes (ketamine, norketamine and dehydronorketamine) standard curves were , and ; Figure 2), respectively. All the calibration standards including the LLOQ showed an acceptable deviation (±15%).
Figure 2.
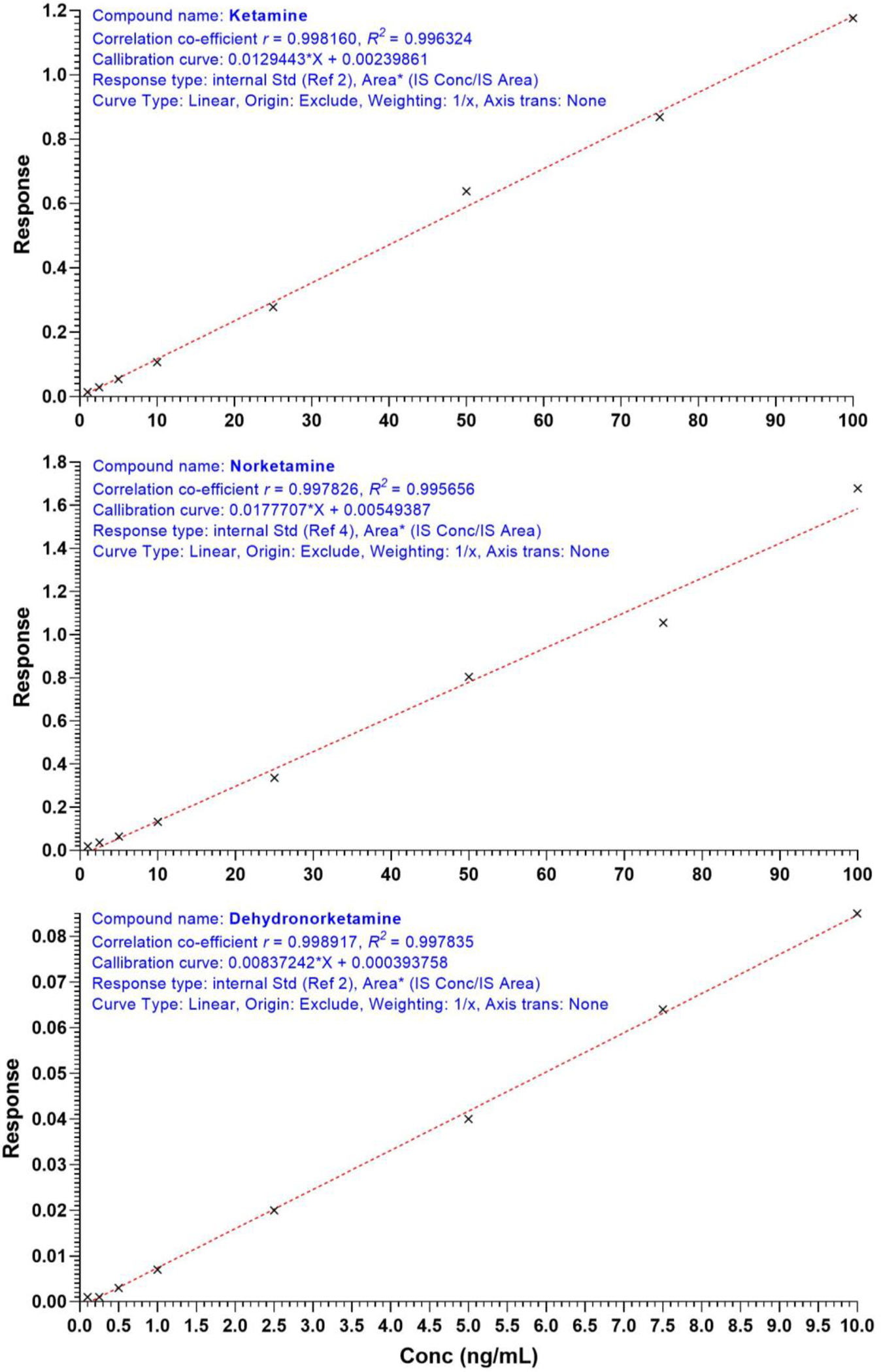
Representative standard curves for ketamine (top), norketamine (middle) and Dehydronorketamine respectively.
Precision and accuracy
The intra-day and inter-day accuracy and precision was assessed using the QC samples, and the mean of measured concentrations, %CV and bias for the QC samples is presented in Table 2. All the measured concentrations were within −5.7% to 6.5% of the theoretical concentrations at intra-day and inter-day and were therefore considered acceptable. Also, the CV ranged from 0.33% to 13.4% for all analytes at all QC levels indicating assay reproducibility.
Table 2.
The Intra-day and Inter-day precision and accuracy of analytes quantification in human milk
| Added concentration (ng/mL) | Intra-day (N = 3) | Inter-day (N = 9) | |||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Mean ± SD | CV (%) | Bias (%) | Mean ± SD | CV (%) | Bias (%) | ||
|
| |||||||
| Ketamine | 2.0 | 2.1 ± 0.02 | 0.7 | 6.3 | 2.1 ± 0.1 | 6.4 | 3.6 |
| 20 | 21.3 ± 0.3 | 1.3 | 6.5 | 19.5 ± 2.3 | 11.8 | −2.6 | |
| 40 | 41.3 ± 0.3 | 0.8 | 3.1 | 39.1 ± 1.9 | 4.9 | −2.2 | |
| 80 | 75.5 ± 1.7 | 2.3 | −5.7 | 75.8 ± 2.8 | 3.7 | −5.2 | |
|
| |||||||
| Norketamine | 2.0 | 2.09 ± 0.02 | 1.0 | 4.5 | 2.07 ± 0.1 | 5.5 | 3.4 |
| 20 | 20.6 ± 0.1 | 0.3 | 3.1 | 19.2 ± 2.6 | 13.4 | −3.8 | |
| 40 | 42.0 ± 0.3 | 0.7 | 5.0 | 39.4 ± 2.6 | 6.7 | −1.6 | |
| 80 | 78.3 ± 0.4 | 0.5 | −2.2 | 77.5 ± 2.5 | 3.2 | −3.2 | |
|
| |||||||
| Dehydronorketamine | 0.2 | 0.21 ± 0.01 | 4.8 | 5.0 | 0.21 ± 0.01 | 6.1 | 6.1 |
| 2.0 | 2.01 ± 0.03 | 1.5 | 0.7 | 1.96 ± 0.1 | 5.1 | −2.2 | |
| 4.0 | 4.01 ± 0.04 | 1.1 | 0.3 | 3.8 ± 0.2 | 6.1 | −5.1 | |
| 8.0 | 7.79 ± 0.1 | 1.2 | −2.7 | 7.78 ± 0.2 | 2.6 | −2.7 | |
Recovery and matrix effect
High analyte recovery was obtained at all QC levels and ranged between 94.8% and 116.6%. Minimal matrix effect was observed with responses ranging 95.2% to 122.6% for ketamine and norketamine and 105% to 111.2% for dehydronorketamine using QC samples. The recovery of internal standards for all analytes ranged between 100.5% to 108.3% and minimal matrix effect was observed for internal standards with responses ranging 97.6% to 115.7%. Recovery and matrix effect data are summarized in Table 3.
Table 3.
Summary of assay validation in human milk
| Added concentration (ng/mL) | % Recovery (N = 3) | Matrix Effect (%) (N = 3) | 24 hours at RT (N = 3) | 24 hours at 4°C (N = 3) | Freeze-thaw (N = 3) | 12 months at (−80°C) (N=3) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||
| Mean ± SD | CV (%) | Mean ± SD | CV (%) | CV (%) | Bias (%) | CV (%) | Bias (%) | CV (%) | Bias (%) | CV (%) | Bias (%) | ||
|
| |||||||||||||
| Ketamine | 2.0 | 99.1 ± 1.5 | 1.5 | 1.9 ± 2.4 | 2.4 | 1.4 | 2.1 | 1.6 | 2.7 | 1.3 | 3.0 | 8.5 | 13.6 |
| 20 | 94.8 ± 1.2 | 1.2 | −4.6 ± 1.4 | 1.5 | 0.8 | 3.5 | 1.6 | −0.3 | 1.6 | −2.3 | 7.0 | 9.7 | |
| 40 | 95.7 ± 1.1 | 1.2 | −4.8 ± 1.2 | 1.3 | 1.3 | 8.2 | 1.9 | 2.2 | 2.2 | 8.8 | 4.4 | 11.5 | |
| 80 | 98.0 ± 0.4 | 0.4 | −2.08 ± 0.6 | 0.6 | 2.4 | 4.5 | 1.5 | −0.8 | 2.3 | 5.8 | 4.3 | 3.2 | |
|
| |||||||||||||
| Norketamine | 2.0 | 116.6 ± 1.2 | 1.1 | 22.6 ± 1.2 | 0.9 | 0.4 | 3.7 | 1.4 | 3.8 | 1.0 | 3.8 | 1.3 | 8.9 |
| 20 | 116.0 ± 1.7 | 1.4 | 18.9 ± 1.3 | 1.1 | 0.7 | 2.8 | 0.6 | −0.7 | 0.5 | −3.5 | 1.6 | 3.4 | |
| 40 | 115.8 ± 0.8 | 0.7 | 14.8 ± 1.5 | 1.3 | 0.5 | 9.4 | 0.3 | 2.3 | 0.8 | 8.0 | 1.1 | 6.2 | |
| 80 | 106.6 ± 0.4 | 0.3 | 6.6 ± 0.7 | 0.7 | 0.5 | 6.0 | 0.7 | −0.2 | 0.7 | 5.8 | 1.0 | 0.2 | |
|
| |||||||||||||
| Dehydronorketamine | 0.2 | 105.0 ± 2.4 | 2.3 | 11.2 ± 3.1 | 2.8 | 2.4 | 2.9 | 4.4 | 0 | 0 | 4.4 | 0.8 | 14.4 |
| 2.0 | 103.4 ± 2.2 | 2.1 | 6.1 ± 2.4 | 2.2 | 2.1 | 6.8 | 2.7 | −6.1 | 1.3 | −1.5 | 2.6 | −0.8 | |
| 4.0 | 104.9 ± 1.6 | 1.5 | 5.03 ± 2.0 | 1.9 | 0.9 | 8.3 | 0.6 | −5.7 | 1.9 | 7.8 | 1.8 | 1.9 | |
| 8.0 | 106.9 ± 0.8 | 0.7 | 8.2 ± 2.2 | 2.1 | 0.7 | 3.3 | 0.7 | −9.0 | 0.5 | 4.5 | 0.5 | −4.1 | |
|
| |||||||||||||
| Ketamine-d4 (IS) | 5.0 | 100.5 ± 2.9 | 2.9 | 2.4 ± 3.1 | 3.2 | - | - | - | - | - | - | - | - |
|
| |||||||||||||
| Norketamine-d4 (IS) | 5.0 | 108.3 ± 8.3 | 7.7 | 15.7 ± 6.9 | 5.9 | - | - | - | - | - | - | - | - |
Storage stability
Analytes were stable at all the conditions tested, Table 3. The measured concentrations of all analytes deviated by only ± 15% of the nominal concentrations at all QC levels for 24 hours at room temperature, 24 hours at 4°C, after three freeze-thaw cycles and after 12 months at −80°C. Also, the CV% of the tested concentrations were less than 9% at all stability conditions.
Application to clinical sample analysis
The newly developed and validated assay methodology was successfully used to measure the concentrations of ketamine, norketamine and dehydronorketamine in human milk samples collected from a participant (randomly selected; # 6)) in the clinical study. Figure 1 right panel shows representative chromatograms of the analytes in breast milk sample collected from participant 6 at 12 hours after initiating of ketamine infusion (0.1 mg/kg/h for 12 hours). The breast milk concentrations vs time profile for all analytes observed in participant 6 in the clinical study are presented in Figure 3.
Figure 3:

Human milk concentrations (ng/mL) versus time (h) profile for ketamine, norketamine and dehydronorketamine during and post 12-hour 0.1 mg/kg/hr IV infusion in subject 6. X-axis represents time (h), left y-axis represents human milk concentrations of ketamine (ng/mL) and norketamine (ng/mL) and right y-axis represents human milk concentrations of dehydronorketamine (ng/mL).
Discussion
A recent review summarized chromatographic methods for quantifying ketamine and its metabolites in biological samples [15]. The majority of the presented methods were developed and validated in plasma and urine or in non-traditional matrices such as hair and nail [15]. Nojavan et al reported a novel method utilizing Electro Membrane Extraction (EME) for separation and HPLC-UV detection of analytes such as ketamine in human milk, plasma as well as in waste water [16, 17]. Sellers et al quantified ketamine in plasma and milk from Holstein cows using liquid-liquid extraction and HPLC-UV for separation and detection of the analytes [18]. Lopez-Garcia et al utilized protein precipitation followed by LC-MS/MS analysis for simultaneous determination of 40 psychoactive drugs including ketamine in human milk, cow milk, and milk matrices like fresh, powdered, whole and skimmed milk [19]. The infant exposure to ketamine and metabolites was recently predicted utilizing a LC-MS/MS assay that was not completely validated [13].
In the current study we developed and validated an analytical methodology for quantitation of ketamine and two of its metabolites norketamine and dehydronorketamine in human milk using LC-MS/MS. Deuterated compounds were used as the internal standards. Analytes were effectively separated using an analytical column and gradient methodology. Gradient elution resulted in a shorter run-time and ensured that all compounds of interest were eluted during a single run. Initially, all five pure compounds of interest were infused and scanned using the mass spectrometer using positive and negative mode to determine optimum response. ESI using positive mode was the most sensitive and was chosen for further analysis.
Each analyte was checked for four m/z combinations and the most sensitive m/z combination for each analyte was selected for identification and quantitation of analyte. Different combinations of m/z for ketamine (238.2 to 125.1, 238.2 to 163.1, 238.2 to 179.2, and 238.2 to 207.1), norketamine (224.2 to 67.2, 224.2 to 125.1, 224.2 to 153.4, and 224.2 to 165.5) and dehydronorketamine (222.2 to 115.1, 222.2 to 142.0, 222.2 to 152.1, and 222.2 to 170.1) were evaluated. The final m/z transitions selected for the assay are presented in Table.1. The selected m/z’s for ketamine (238.2 to 125.1), norketamine (224.2 to 125.1) and dehydronorketamine (222.2 to 142.0) were 60%, 10-fold and 3-fold more sensitive respectively, than the response observed for next m/z combination. Additionally, a small peak observed in the dehydronorketamine channel at the retention time for ketamine was generated from ketamine and the extra peak did not interfere with dehydronorketamine analysis. Moreover, the response obtained for m/z combination (222.2 to 142.0) with ketamine was 400-fold lower than m/z combination selected for ketamine.
We anticipated high content of proteins and fat in the milk and initially explored liquid-liquid extraction with a mixture of dichloromethane and isopropyl alcohol. Liquid-liquid extraction has been previously used for extraction of ketamine from cowmilk [20]. We used human milk and adopted the reported method to optimize extraction process, but the method showed inconsistent recovery for the QC samples and was time-consuming and thus, not practical for analysis of large number of samples collected during a clinical study. Hence, we chose to use a much simpler sample processing approach using protein precipitation with acetonitrile, which resulted in high recovery of all analytes.
The newly developed method is robust, accurate, precise, sensitive, and reproducible. The inter-day and intra-day coefficients were less than 15% and % bias was observed to be less than 5% at all levels of QC’s. Recovery was close to 100% for the three analytes measured and for internal standards. The only other published method for simultaneous quantification of ketamine and its metabolites utilized solid phase extraction which is time-consuming, expensive and requires large volume of sample (~200 μL) [13], whereas our methodology uses much lower sample volume and is sensitive in measuring close to 1.5 pg on the column.
To test the stability of the analytes, the conditions were selected based on the storage conditions encountered during the analysis of the samples. Human milk samples are usually stored at −80°C immediately after collection and may be subjected to freeze thaw cycles depending on the objectives of the study. The QC samples in breast milk were subjected to 3 freeze-thaw cycles, stored at 4°C, and at room temperature for 24 hours, then analyzed to assess the effect of these conditions on stability of ketamine and its metabolites. Additionally, long term stability of the analytes at sample storage conditions was tested by storing the QCs at −80°C for 12 months. There was no significant effect of temperature conditions on analytes stability and the % bias was within 15%. Overall, these observations demonstrate negligible loss of ketamine and its metabolites under various conditions that simulate the real-life situations.
This assay was utilized for analysis of human milk samples obtained from postpartum women enrolled in a clinical research study assessing the safety of ketamine as an adjuvant anesthetic during cesarean section. The concentrations of ketamine and its metabolites were successfully measured in breast milk samples using this assay, indicating the suitability of the assay for quantifying clinically relevant concentrations of analytes in human milk. As depicted in Figure 3, ketamine was detected at higher levels in human milk as compared to its metabolites, which could be related in part to the lower lipophilicity of the measured metabolites as compared to the parent drug [9, 10]. Ketamine is metabolized by dealkylation to produce norketamine which is further oxidized into dehydronorketamine [21]. Our observation supports the previous findings of Wolfson et al who reported comparable levels of ketamine and norketamine and low exposure of dehydronorketamine in breast milk after intramuscular administration of 0.5 mg/kg and 1 mg/kg ketamine to four postpartum women [13].
Conclusion
A highly specific, sensitive, and reproducible UPLC-MS/MS based assay using a simple, and robust approach for extraction using protein precipitation for simultaneous extraction and measurement of ketamine, norketamine and dehydronorketamine concentrations in human milk was successfully developed and validated. The assay utilized deuterated ketamine and norketamine as internal standards. Our is the first validated assay methodology for quantification of ketamine and its metabolites in human milk. The developed method is easy to perform and extremely practical for analysis of large number of clinical samples.
Highlights.
A simple LC-MS/MS method for quantification of ketamine and its metabolites.
Assay employed simple protein precipitation & deuterated internal standards.
Validated in human milk with high recovery, sensitivity, and reproducibility.
Successfully applied to human milk samples collected in a clinical study.
ACKNOWLEDGMENTS
This project was supported by NIH K12HD043441, the UPMC Department of Anesthesiology & Perioperative Medicine, and the CTSI Virginia Kaufmann Pain Research Challenge 2018.
We thank Prerna Dodeja, MS for assistance in designing the graphical abstract.
Footnotes
CONFLICT OF INTEREST STATEMENT
There are no conflicts of interest to disclose.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The study was approved by the University of Pittsburgh IRB (Protocol #18120046) and a written informed consent was obtained from all participants before performing any study related procedures.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Laskowski K, et al. , A systematic review of intravenous ketamine for postoperative analgesia. Canadian Journal of Anesthesia/Journal canadien d’anesthésie, 2011. 58(10): p. 911. [DOI] [PubMed] [Google Scholar]
- 2.Ketamine Hydrochloride injection. Par Pahrmaceutical, Inc. March/2022. [Google Scholar]
- 3.Adhikari P, et al. , Analgesic effects of intravenous ketamine after spinal anaesthesia for non-elective caesarean delivery: a randomised controlled trial. BMJ Open, 2021. 11(6): p. e044168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Behaeen K, et al. , Analgesic effect of low dose subcutaneous ketamine administration before and after cesarean section. Iran Red Crescent Med J, 2014. 16(3): p. e15506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Han SY, et al. , The Effect of Low-dose Ketamine on Post-caesarean Delivery Analgesia after Spinal Anesthesia. Korean J Pain, 2013. 26(3): p. 270–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Samuel H, Aweke S, and Tuni J, Effect of low-dose intravenous ketamine on postoperative pain following cesarean section under spinal anesthesia: A prospective cohort study, Ethiopia. Ann Med Surg (Lond), 2022. 77: p. 103570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zangouei A, et al. , Effect of Low-Dose Intravenous Ketamine on Prevention of Headache After Spinal Anesthesia in Patients Undergoing Elective Cesarean Section: A Double-Blind Clinical Trial Study. Anesth Pain Med, 2019. 9(6): p. e97249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen-Li D, et al. , Ketamine as potential treatment for postpartum depression: A narrative review. Ann Clin Psychiatry, 2022. 34(4): p. 264–274. [DOI] [PubMed] [Google Scholar]
- 9.Turfus SC, et al. , Use of human microsomes and deuterated substrates: an alternative approach for the identification of novel metabolites of ketamine by mass spectrometry. Drug Metab Dispos, 2009. 37(8): p. 1769–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hijazi Y and Boulieu R, Contribution of CYP3A4, CYP2B6, and CYP2C9 isoforms to N-demethylation of ketamine in human liver microsomes. Drug Metab Dispos, 2002. 30(7): p. 853–8. [DOI] [PubMed] [Google Scholar]
- 11.Zanos P, et al. , Ketamine and Ketamine Metabolite Pharmacology: Insights into Therapeutic Mechanisms. Pharmacol Rev, 2018. 70(3): p. 621–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang K, Yao Y, and Hashimoto K, Ketamine and its metabolites: Potential as novel treatments for depression. Neuropharmacology, 2023. 222: p. 109305. [DOI] [PubMed] [Google Scholar]
- 13.Wolfson P, et al. , The Pharmacokinetics of Ketamine in the Breast Milk of Lactating Women: Quantification of Ketamine and Metabolites. J Psychoactive Drugs, 2022: p. 1–5. [DOI] [PubMed] [Google Scholar]
- 14.Cobb B, et al. , Breastfeeding after Anesthesia: A Review for Anesthesia Providers Regarding the Transfer of Medications into Breast Milk. Transl Perioper Pain Med, 2015. 1(2): p. 1–7. [PMC free article] [PubMed] [Google Scholar]
- 15.Göktaş EF and Arıöz F, A review of chromatographic methods for ketamine and its metabolites norketamine and dehydronorketamine. Biomed Chromatogr, 2018. 32(1). [DOI] [PubMed] [Google Scholar]
- 16.Asadi S and Nojavan S, Two-step voltage dual electromembrane extraction: A new approach to simultaneous extraction of acidic and basic drugs. Anal Chim Acta, 2016. 923: p. 24–32. [DOI] [PubMed] [Google Scholar]
- 17.Tabani H, et al. , Introduction of agarose gel as a green membrane in electromembrane extraction: An efficient procedure for the extraction of basic drugs with a wide range of polarities. J Chromatogr A, 2017. 1497: p. 47–55. [DOI] [PubMed] [Google Scholar]
- 18.Sellers G, et al. , Pharmacokinetics of ketamine in plasma and milk of mature Holstein cows. J Vet Pharmacol Ther, 2010. 33(5): p. 480–4. [DOI] [PubMed] [Google Scholar]
- 19.Lopez-Garcia E, et al. , Simultaneous LC-MS/MS determination of 40 legal and illegal psychoactive drugs in breast and bovine milk. Food Chem, 2018. 245: p. 159–167. [DOI] [PubMed] [Google Scholar]
- 20.Rofael HZ and Abdel-Rahman MS, Development and validation of a high-performance liquid chromatography method for the determination of cocaine, its metabolites and ketamine. J Appl Toxicol, 2002. 22(2): p. 123–8. [DOI] [PubMed] [Google Scholar]
- 21.Dinis-Oliveira RJ, Metabolism and metabolomics of ketamine: a toxicological approach. Forensic Sci Res, 2017. 2(1): p. 2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]