

Abstract
Patients diagnosed with Pancreatic Ductal Adenocarcinoma (PDAC) frequently present with advanced metastatic disease and exhibit a poor response to therapy, resulting in poor outcomes. The tumor microenvironment cytokine Oncostatin-M (OSM) initiates PDAC plasticity, inducing the reprogramming to a stem-like/mesenchymal state, which enhances metastasis and therapy resistance. Using a panel of PDAC cells driven through epithelial-mesenchymal transition (EMT) by OSM or the transcription factors ZEB1 or SNAI1, we find that OSM uniquely induces tumor-initiation and gemcitabine resistance independently of its ability to induce a CD44HI/mesenchymal phenotype. In contrast, while ZEB1 and SNAI1 induce a CD44HI/mesenchymal phenotype and migration comparable to OSM, they are unable to promote tumor-initiation or robust gemcitabine resistance. Transcriptomic analysis identified that OSM-mediated stemness requires MAPK activation and sustained, feed-forward transcription of OSMR. MEK and ERK inhibitors prevented OSM-driven transcription of select target genes and stem-like/mesenchymal reprogramming, resulting in reduced tumor growth and re-sensitization to gemcitabine. We propose that the unique properties of OSMR, which hyperactivates MAPK signaling when compared to other IL-6 family receptors, make it an attractive therapeutic target, and that disrupting the OSM-OSMR-MAPK feed-forward loop may be a novel way to therapeutically target the stem-like behaviors common to aggressive PDAC.
Introduction
Pancreatic ductal adenocarcinoma (PDAC), which makes up 90% of clinically diagnosed pancreatic cancers, is the fourth leading cause of cancer-related deaths in both men and women (1, 2). The asymptomatic nature of PDAC increases the number of patients diagnosed with advanced, metastatic disease (3). The metastatic spread of PDAC is linked to therapy failure and remains an overarching clinical challenge for patients. Commonly, patients receive aggressive chemotherapy comprised of a cytidine analog gemcitabine, alone or in combination with either nab-paclitaxel, EGFR inhibitor (Erlotinib), or Capecitibine (4–6). However, chemotherapy extends progression-free survival only 6–12 months, and the 5-year survival rate remains less than 10%; clearly, there remains an urgent need for novel therapeutics to target PDAC (4).
Cancer cells with stem-like properties are considered the “roots” of aggressive tumors, including PDAC. Factors within the tumor microenvironment, which are secreted by cancer-associated immune cells (including macrophages and myeloid-derived suppressor cells) or fibroblasts, can profoundly influence cancer cells. Cytokines, chemokines and growth factors can cause cancer cells to transition from a more differentiated/epithelial state into a stem-like/mesenchymal state, which can be reversed upon removal of the stimulus, suggesting the adaptive response is plastic and reversible. In PDAC, cancer cells exploit developmental processes such as epithelial-mesenchymal transition (EMT) to induce a stem-like phenotype, which is linked with therapy failure, in addition to increasing migratory and invasive behaviors important for metastatic outgrowth (7, 8). Interestingly, in triple-negative breast cancer, single cell analysis determined that EMT in primary tumors proceeds through distinct, hybrid states, ranging from epithelial to mesenchymal (9). Tumor cells that express both mesenchymal markers (vimentin, N-cadherin, O-cadherin, and CD44) and epithelial markers (E-cadherin, CD24, epithelial cell adhesion molecule, cytokeratins) called epithelial-mesenchymal (EM) hybrid cells) harbor the greatest level of plasticity and are more efficient at intravasation, survival in circulation, extravasation, and metastatic outgrowth (9–12). EM circulating tumor cells (CTC) can be used to track a patient’s response to therapy, with increasing numbers of EM-CTC conferring increased risk of relapse (13). In addition, a sub-set of CTC responsible for initiating metastasis express high levels of the stem cell marker CD44 (14). Likewise, single-cell analysis identified predominantly de-differentiated, stem-like/mesenchymal cells expressing high levels of CD44 in micro-metastases, whereas macro-metastases are more differentiated/epithelial, more proliferative, and display greater similarity to the primary tumor of origin (15). Taken together, these findings suggest that cancer cell plasticity allows the initiation of metastatic dissemination and outgrowth at a secondary site, followed later by increased proliferation and differentiation. Thus, therapeutic strategies aiming to either (i) undermine a cancer cell’s ability to reprogram to an invasive, drug-tolerant, stem-like/mesenchymal state, or (ii) induce the differentiation of pre-existing stem-like/mesenchymal cells will help improve current treatments.
EMT inhibitors have demonstrated efficacy in preventing the acquisition of stem-like/mesenchymal phenotypes by targeting epigenetic regulators, glucose metabolism, or EMT transcription factors, like ZEB1 or SNAI1 (16). Additional studies have sought to define how cytokines and growth factors in the tumor microenvironment can positively or negatively influence the reprogramming responsible for enhancing metastasis and therapy failure. In particular, IL-6 family cytokines serve as prognostic markers, therapeutic targets, mediators of inflammation, and activators of signal transducer and activator of transcription 3 (STAT3), mitogen-activated protein kinase (MAPK), and phosphatidylinositol-3-kinase and protein kinase B (PI3K/Akt signaling) (17). Oncostatin-M (OSM) is a pleotropic IL-6 family member that is elevated in aggressive and therapy resistant cancers, such as PDAC, glioblastoma, non-small cell lung carcinoma, breast cancer, and ovarian cancer (18–21). OSM is a potent inducer of stem-like/mesenchymal reprogramming in PDAC marked by enhanced expression of CD44, ZEB1 and SNAI1, as well as the repression of epithelial markers E-cadherin and CD24 (22, 23). OSM signals through a heterodimer receptor complex comprised of g-protein-like receptor 130 (GP130) and either OSM-Receptor (OSMR) or LIF-Receptor (LIFR). Like other IL-6 family co-receptors, gp130 induces the phosphorylation of Janus kinases (JAKs) and STAT3, which can induce stem-like/mesenchymal phenotypes (24). Moreover, STAT3 activation induces the expression of OSM and/or OSMR, resulting in a feed-forward loop that further reinforces OSMR effector signaling (25, 26). In breast cancer, STAT3 cooperates with transforming growth factor beta (TGFβ) signaling effector mothers against decapentaplegic homolog 3 (SMAD3) to coordinate stem-like/mesenchymal reprogramming (27). Unfortunately, STAT inhibitors have thus far failed in the clinical setting because of non-specific, off-target effects arising from the fact that STAT family member proteins demonstrate high homology with each other (28). Moreover, JAK inhibitors, while capable of inhibiting STAT3 activation, also undermine anti-tumorigenic, type I interferon-mediated STAT1/2 activation, which may explain why in preclinical models of breast cancer, JAK inhibitors enhanced the formation of metastasis (29). However, OSMR binding antibodies may prove to be an effective strategy to block STAT3 activation, migration, and metastasis, as was recently reported in preclinical models of ovarian cancer (30).
Here, we demonstrate that OSM can simultaneously induce mesenchymal and stem-like behaviors, while other reprograming events, induced by the EMT transcription factors ZEB1 and SNAI1, do not bestow tumor initiating capacity. Moreover, OSM only confers therapy resistance if both STAT3 and ERK can be effectively activated. These findings suggest that mesenchymal and stem-like behaviors can be separable, independent programs. This may be in part, due to the uniqueness of OSMR, which can activate MAPK signaling differently than other IL6 family members, resulting in enhanced and sustained phosphorylation of both ERK and STAT3. OSM-OSMR feed-forward loop signaling further amplifies MAPK activation, tumor initiation, and gemcitabine resistance, which can be undermined by MEK and ERK inhibitors. Here, we show that MAPK activation is integral to the OSM-induced plasticity underlying the mesenchymal and stem-like programs responsible for tumor initiation and therapy failure.
Materials and Methods
Cell Culture
PDAC cell lines (HPAC, PANC1, Panc05.04, and Panc04.03) were obtained from ATCC and cultured in a humidified atmosphere containing 5% CO2 at 37°C. HPAC cell identity was validated by STR analysis, and monthly mycoplasma testing (#LT07–318; Lonza) confirmed all cell lines remained contaminant free. HPACs were cultured in DMEM/F12 (#10–092-CV; Corning) with 10% FCS (#S11150; Atlanta Biologicals), 0.005 mg/mL of human transferrin (#T2252; Sigma Aldrich), 10 ng/mL of human epidermal growth factor (#01–107; Millipore), 0.002 mg/mL of human insulin (#I9278; Sigma Aldrich), and 40 ng/mL of hydrocortisone (#H4001; Sigma Aldrich). PANC1’s were cultured in DMEM 1X (#10–013-CV; Corning) with 10% FCS (#S11150; Atlanta Biologicals). Panc05.04 were cultured in RPMI-1640 with 15% FBS (#MT35010AC22; Corning) and .2U/mL human insulin (#I9278; Sigma Aldrich). Panc04.03 were cultured in RPMI-1640 with 15% FCS (#MT35010AC22; Corning). Treatment of cells was performed as follows: 10 ng/mL human recombinant Oncostatin-M (OSM; #OSM01– 13; DAPCEL), 10 μM ruxolitinib (#INCB018424; Selleckchem),10 μM of U0126 (#S1102; Selleckchem), 10 μM Pimasertib (#AS-703026; Selleckchem), 10 μM PD184352 (#Cl-1040; Selleckchem), and 80 nM SCH772984 (#S7101; Selleckchem), unless otherwise stated in the figure legends. All short-term treatments were performed as denoted in the figure legend; all long-term treatments were given at each medium change unless denoted otherwise (~48 hours).
Western blot analysis, and quantitative real-time RT-PCR
Western blots were conducted using whole-cell protein extracts and enhanced by either chemiluminescence or fluorescence as annotated in the figure legends and as described previously (31). Primary antibodies used were Pan-Actin (#MCA-5J11; Encore), E-cadherin (#3195; Cell Signaling Technology), SNAI1 (#3879; Cell Signaling Technology), phosphorylated STAT3Y705 (#9145; Cell Signaling Technology), STAT3 (#9139; Cell Signaling Technology), phosphorylated ERK1/2 (#4370; Cell Signaling Technology), ERK (#9102; Cell Signaling Technology), phosphorylated MEK1/2 (#2338; Cell Signaling Technology), OSMR (sc-271695; SantaCruz Biotechnology, ab232684; Abcam), ZEB1 (#3396; Cell Signaling Technology), P-Cadherin (CDH3; #2189; Cell Signaling Technology), EPCAM (#36746;Cell Signaling Technology), Occludin (#91131;Cell Signaling Technology), GAPDH (#97166;Cell Signaling Technology). Secondary antibodies used were HRP-linked anti-mouse (#7076; Cell Signaling Technology) and HRP-linked anti-rabbit (#7074; Cell Signaling Technology) for chemiluminescence blots, while Dylight800 anti-Rabbit (5151S; ThermoFisher) and Dylight680 anti-Mouse (5470S; Thermofisher) were used for Fluorescent Blots. For quantitative real-time RT-PCR (qPCR), total RNA was isolated as described previously (31). RNA (1–2 μg) was reverse transcribed by iScript cDNA Synthesis Kit (#170–8891; Bio-Rad). Gene expression was identified using iQ SYBR Green Supermix (#170–8880; Bio-Rad) and a CFX96 thermocycler (Bio-Rad). All samples were normalized to GAPDH or ACTIN expression, as indicated, and error bars represent ± SEM for a representative experiment performed in triplicate or quadruplicate. A two-tailed unpaired Student t test was performed to determine significance (*, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001). Primer sequences were as follows: OSMR forward: 5′-TCCCAATACCACAAGCACAG-3′; OSMR reverse: 5′-GCAAGTTCCTGAGAGTATCCTG-3′; SNAI1 forward: 5′-GGAAGCCTAACTACAGCGAG-3′; SNAI1 reverse: 5′-CAGAGTCCCAGATGAGCATTG-3′; ZEB1 forward: 5′-ACCCTTGAAAGTGATCCAGC-3′; ZEB1 reverse: 5′-CATTCCATTTTCTGTCTTCCGC-3′; GAPDH forward: 5′-TGCACCACCAACTGCTTAGC-3′; GAPDH reverse: 5′-GGCATGGACTGTGGTCATGAG-3′; CDH1 forward: 5′-CCCAATACATCTCCCTTCACAG-3′; CDH1 reverse: 5′-CCACCTCTAAGGCCATCTTTG-3′; ACTIN forward: 5’ CAGCCATGTACGTTGCTATCCAGG-3’; ACTIN reverse: AGGTCCAGACGCAGGATGGCATG-3’; SOCS3 forward: 5’CAAGCACAAGAAGCCAAC-3’; SOCS3 reverse: 5’- TTCCCTCCAACACATTCC-3’ CD44s forward: 5’-AGCAGCGGCTCCTCCAGTGA-3’; CD44s reverse: 5’-CCCACTGGGGTGGAATGTGTCT-3’.
Flow cytometry, migration, and growth assays
For flow cytometry and FACS, cells (~0.5–2 × 106) were stained with anti-human CD44 APC (clone BJ18; #338806; BioLegend) and anti-human CD24 PE (clone ML5; #311106; BioLegend). Cells were then either sorted using the FACS-ARIA or analyzed using a BD LSRII and FACSDiva software. Migration assays were performed using the IncuCyte ZOOM or Sx5 imaging system (Essen BioScience) as indicated in the figure legends. Briefly, cells (1000 cells/well) were suspended in their base media (DMEM/F-12, RPMI, DMEM) containing 0.5% FBS and seeded onto 96-well ClearView-Chemotaxis plates with 8-mm pores. HPAC cells were stained with live cell NIR Nuclight Dye (Sartorius; #4804) before the plates were incubated and imaged over the indicated time points. Cells migrating to the bottom chamber across the pores were imaged and quantified. Gemcitabine outgrowth assays were performed using the live cell IncuCyte ZOOM or Sx5 imaging system (Essen BioScience) after cells were stained with Incucyte Nuclight Rapid Red Dye, which marks live Cells (#4717; Sartorius) and plated at 2000 cells/well in 96-well flat-bottom plates (#3599; Corning), prior to treatment with gemcitabine (#G6423–50Mg; Sigma Aldrich) at the dose indicated in the figure legends. After gemcitabine addition, cells were imaged at regular intervals and quantified with Incucyte software.
Limiting Dilution Assay
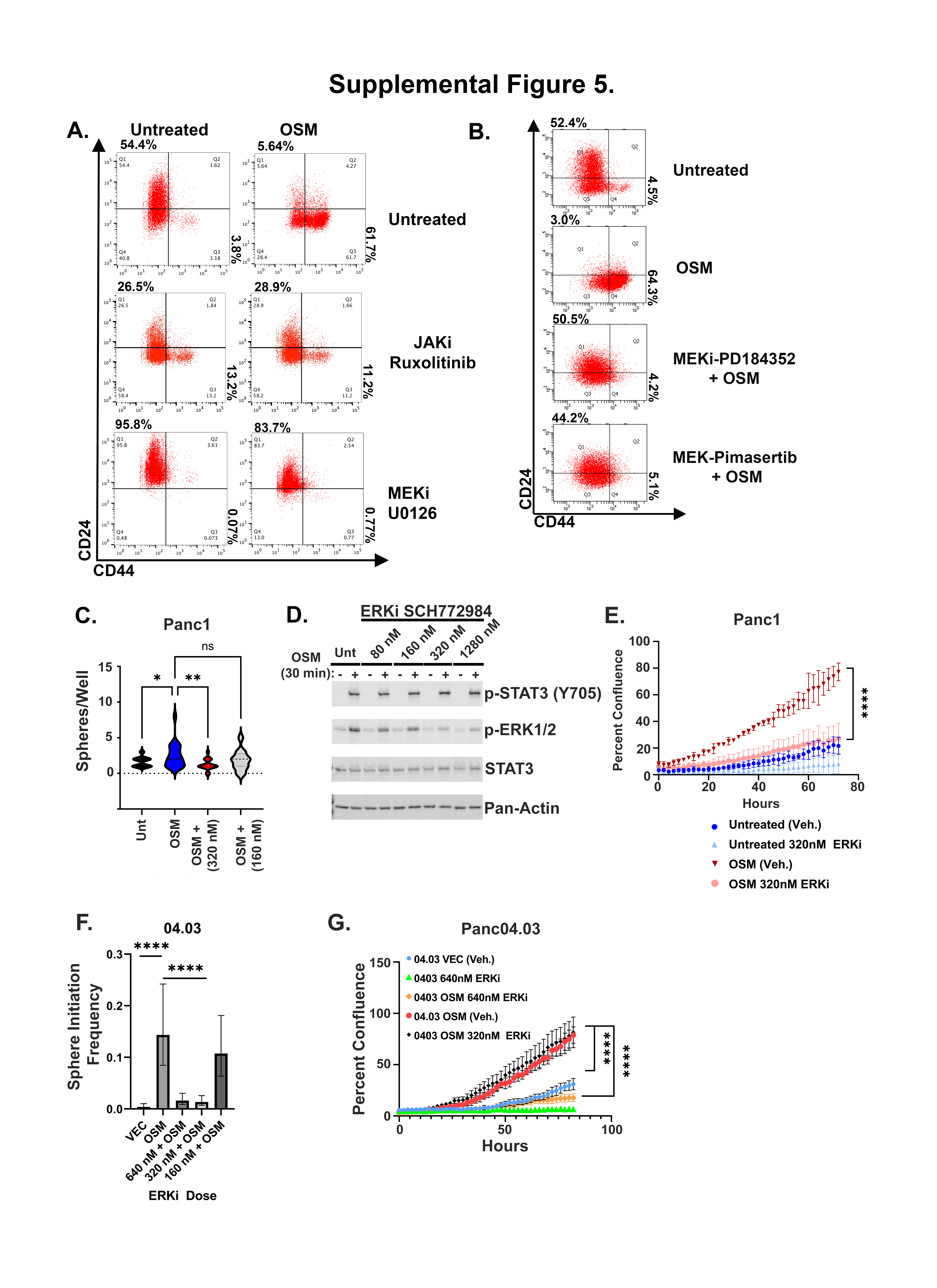
Panc1, Panc04.03, and Panc05.04 were live-sorted at limiting dilutions (1, 5, 10, 25, and 100 cells/well) into 96-well corning Ultra-Low attachment, polystyrene, flat bottom, plates (#3474; Corning-Costar). As specified in the figure legends, cells were fed with media or media supplemented with ERKi (50 uL/well) every 4 days over the course of 14 days and monitored for sphere initiation. Panc04.03 and Panc05.04 stem cell frequency for was calculated using an Extreme Limiting Dilution Assay (ELDA) as described previously (32). After ERKi treatment, Panc1 spheroid number was quantified using the Fiji platform (https://hpc.nih.gov/apps/Fiji.html). Fiji performs a series of image-analysis steps, which result in the creation of selection around areas of the image consisting of pixels, which can be translated into multiple parameters, including the area of the signal. Background noise was removed during the analysis. Data on the number of spheres identified by Fiji and total and average spheroids area from the 10, 25, and 100 cell groups were collected and plotted in Prism.
Mouse xenografts
All procedures were performed in compliance with the Case Western Reserve University Institutional Animal Care and Use Committee (IACUC). Athymic female nu/nu mice (6–8 weeks old) were purchased from the Athymic Animal and Xenograft Core of Case Western Reserve University (Cleveland, OH) and from The Jackson Laboratory (Ban Harbor, ME). Mice were anesthetized with 3% Isoflurane in 2l/min oxygen for any procedure requiring sedation. Tumor cells were resuspended in 50 μL of a 1:1 Matrigel:culture media solution and subcutaneously injected into both flanks. Tumor growth was monitored via Bioluminescence imaging (BLI). BLI was performed using the IVIS Spectrum In Vivo Imaging System (Perkin Elmer). Pre- anesthetized mice were injected Intraperitoneally (IP) with 150 μL Luciferin (GoldBio, 15 mg/mL in sterile water) 5 minutes prior to imaging. Mice were then transferred into the IVIS Spectrum analyzer where they were kept under Isoflurane anesthesia throughout the procedure. In vivo imaging was performed bi-weekly for 3 weeks. Tumor volume was measured by caliper and calculated using the formula: {[long side * (short side)2]/2}. Mice were sacrificed by CO2 asphyxiation followed by cervical dislocation once tumors reached 500 mm3 and/or animals showed signs of terminal illness. Size and weight of resected tumors were recorded. Intraperitoneal injections of SCH772984 (#S7101; Selleckchem (90 mg/kg in HP-β-CD (2-Hydroxylpropyl-beta-cyclodextrin) – PubChem) were performed daily after tumor size reached 50–100 mm3. Tumor volume was measured using caliper every other day. At the end point, mice were sacrificed by CO2 asphyxiation followed by cervical dislocation; tumors were then removed, measured by caliper and weighted.
RNA-Sequencing/Analysis
RNA sequencing was performed through the Genomics Core (Case Western Reserve School of Medicine, Cleveland, OH, USA). 20 uL of 50 ng/uL of RNA was prepped and sequenced single end (1×50bp) on an Illumina HiSeq platform. 30–50M reads was generated per sample and assessed for quality and trimmed for adapter sequences using TrimGalore! v0.4.2 (Babraham Bioinformatics), a wrapper script for FastQC and cutadapt. Reads that passed quality control were then aligned to human reference genome (mm10) using the STAR aligner v2.5.1. The alignment for the sequences were guided using the GENCODE annotation for mm10. Aligned reads were quantified and analyzed for differential expression using Cufflinks v2.2.1, a RNASeq analysis package which reports the fragments per kilobase of exon per million fragments mapped (FPKM) for each gene. Differential analysis report was generated using Cuffdiff. Differential genes were identified using a significance cutoff of q-value < 0.05. Further pathway analysis was done using iPathwayGuide (AdvaitaBio). GSEA was performed on CD24HI-VEC and CD44HI-VEC RNA-seq data sets with MSigDB gene set h.all.v7.0.symbol.gmt [Hallmarks] using publically available Gene Set Enrichment Software.
Kaplan Meier Curves
Kaplan Meier Curves were plotted using Kaplan Meier Plotter, a meta-analysis tool that can assess the correlation between expression of 30 000 genes (using mRNA, miRNA, or protein) and survival in 25 000 samples from 21 different tumor types from Gene Expression Omnibus, European Genome-Phenome Archive, and The Cancer Genome Atlas databases (33). All Kaplan-Meier Plots were graphed using the median as the cut off value for high and low expression.
Viral constructs and virus production
VEC, OSM, SNAI1 (Addgene Plasmid #23347), and ZEB1 (Addgene Plasmid #42100) expression plasmids were packaged into pLenti-Dest-Puro (w118–1) (Addgene #17452) or pLenti-Dest-Neo (705–1) (Addgene Plasmid #17392), as described previously and sequence verified before being used to infect target cells (34). Lenti-CRISPRV2 constructs with the following small-guide sequences were packaged and used to target OSMR, (GenScript_ pLenti-CRISPRV2 SgS 5’- CACCGCGCGATAGCGCGAATATATTGTTT-3’, Sg1 5’- ATTCTACGCGTCAGAGTTTG-3’, Sg2 5’- CCACAACCTTCCTTATCATC-3’, Sg3 5’- ATCATACTGTGACCTTATTC-3’, Sg4 5’-TGAGGACTTACCAGAGTGAA-3’), SMAD3 (GenScript_ pLenti-CRISPRV2 SgS 5’-CACCGCGCGATAGCGCGAATATATTGTTT-3’, Sg1 5’-CCGATCGTGAAGCGCCTGC-3’, Sg2 5’-TTCACGATCGGGGGAGTGAA-3’, Sg3 5’- AACGTGGAAAGGCGCAGCTC-3’), and STAT3 (GenScript_pLenti-CRISPRV2 SgS 5’-CACCGCGCGATAGCGCGAATATATTGTTT-3’, Sg3 5’- ACTGCTGGTCAATCTCTCCC-3’,.
Statistical Analyses
Statistical analysis was performed using GraphPad Prism version 8.0 software (GraphPad Software, San Diego, CA, USA). All data are presented as the mean ± SEM for quantitative-PCR and mean ± SD for limiting dilution, gemcitabine outgrowth, and migration assays. Differences between two groups were compared by two-tailed Student’s t tests or Mann Whitney test. Differences among the means of three groups were analyzed by ordinary one-way analysis of variance (ANOVA), while four groups were analyzed by two-way ANOVA followed by a multiple comparisons test. A p value less than 0.05 was considered to indicate statistical significance (*p < 0.05, **p < 0.01, ***p < 0.001, ****p<0.0001).
Data Availability
All RNA-sequencing data was generated by Case Western Reserve University Genetics and Genomics Core facility and is available upon request from corresponding author.
Results
OSM induces tumor-initiation independently of its ability to induce a CD44HI/mesenchymal phenotype.
OSM induces epithelial PDAC cells to acquire a CD44HI/mesenchymal phenotype that is associated with cancer stem-like behaviors, which include tumor-initiating capacity, and enhanced migratory and invasive potential (22). Importantly however, we show that a CD44HI/mesenchymal phenotype is itself not sufficient to induce key stem-like behaviors, including tumor initiation or gemcitabine resistance (Fig. 1). Using HPAC cells, we sorted and expanded the small fraction of spontaneous mesenchymal CD24LO/CD44HI cells (which we refer to as CD44HI) from the parental population (which is largely epithelial and CD24HI/CD44LO; Fig. 1A). Epithelial CD24HI/CD44LO cells were also sorted, which we refer to as CD24HI (Fig. 1A). Western blots of epithelial and mesenchymal markers demonstrate loss of E- and P- cadherin (CDH1 and CDH3), EPCAM, and Occludin in the CD44HI population and transcription of both ZEB1 and SNAI1 with OSM (Fig. 1B and S1A).
Figure 1: OSM induces tumor-initiation independently of its ability to induce a CD44HI/mesenchymal phenotype.
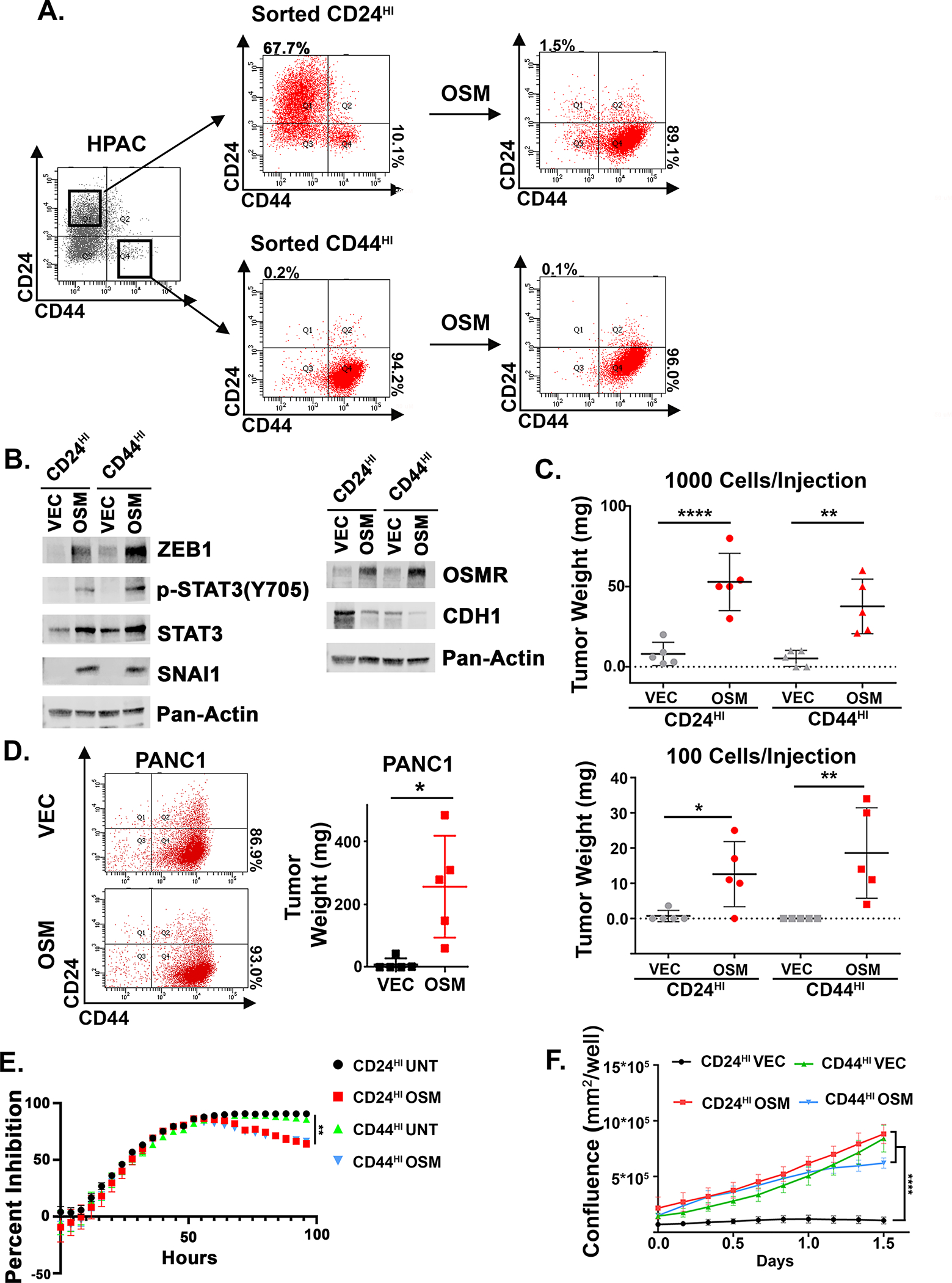
A, B HPAC cells were sorted using FACS for CD24HI/CD44LO (referred to as CD24HI) and CD24LO/CD44HI (referred to as CD44HI). CD24HI and CD44HI cells were infected with lentiviruses encoding OSM (or control, VEC) and assessed by flow cytometry for CD24 and CD44, and Western blot as indicated. C HPAC derivatives were subcutaneously injected into the flanks of nude mice at the indicated cell numbers and tumor development was monitored over 1 month. Data were calculated using a Mann-Whitney test and shown as mean ± S.D. *P<0.05, **P<0.01, ****P<0.0001. D PANC1 cells were infected with lentiviruses encoding OSM (or control, VEC). Cells were assessed by flow cytometry and 1 million cells were injected subcutaneously into mice and monitored over 1 month. Data are shown as a mean ± S.D., and statistical significance was determined by t-test where *P<0.05. E CD24HI and CD44HI HPAC cells were pre-treated with recombinant OSM (10 ng/mL) for 48 hours before 16 nM of gemcitabine or vehicle was added; cell numbers were continuously monitored and quantified from several random fields within each well for an additional 96 hours using an Incucyte imager. Data are plotted as cell number in gemcitabine at a specific timepoint divided by cell number of the corresponding untreated control at the same timepoint. Statistical significance was determined by two-way ANOVA where **P<0.01. F Transwell migration of HPAC derivatives was quantified from several random fields within each well for an additional 96 hours using an Incucyte imager and are shown as mean ± S.D. Statistical significance was determined by two-way ANOVA where ****P<0.0001.
The sorted CD24HI and CD44HI populations were each infected with a lentivirus encoding OSM expression (or a control; VEC) and assessed by Western blot and flow cytometry, before testing tumor-initiating capacity. As expected, CD24HI cells expressing OSM (CD24HI-OSM) converted to a CD44HI/mesenchymal state with elevated phospho-STAT3, ZEB1, SNAI1, and OSMR expression, and OSM induced tumor formation with as few as 100 cells were injected (Fig. 1B, C). Likewise, the sorted, spontaneous CD44HI/mesenchymal cells expressing OSM (CD44HI-OSM), formed tumors equally efficiently (Fig. 1C). However, neither control CD24HI-VEC or spontaneous CD44HI-VEC formed tumors at the time points tested (Fig. 1C). Next, we assessed the impact of OSM in three additional K-Ras-mutant PDAC lines, Panc1, Panc 05.04, and Panc 04.03, following expression of OSM. In Panc 05.04, OSM induced CD44 expression, tumorsphere initiation, and migration (S1E, I, J, L). In Panc1 and Panc 04.03 cells, OSM enhanced tumor initiating capacity, tumorsphere formation, and migration, but did not increase CD44 expression (Fig. 1D, S1B, F, E and S5E, G). Taken together, our finding across multiple cell models suggest that a CD44HI/mesenchymal state is not sufficient for tumor-initiating capacity and that OSM is inducing tumor initiation and migration above and beyond its ability to induce CD44 expression.
Next, the growth and survival of CD24HI and CD44HI HPAC populations was assessed following treatment with gemcitabine, as increased resistance to chemotherapy is another hallmark of stem-like/mesenchymal cells. Sorted CD24HI and CD44HI cells were pre-treated with recombinant OSM (or left untreated) for 48 hours, followed by the addition of gemcitabine and assessment of cell growth over an additional 96 hours; gemcitabine-treated cells were normalized to their untreated population, and percent growth inhibition was plotted. Initially, all populations were strongly and equally inhibited 48 hours after gemcitabine addition. However, from 48 hours onward, both the OSM-treated CD24HI and CD44HI populations began recovering, with ~35 and 40% outgrowth, respectively (Fig. 1E). In contrast, the untreated CD24HI and CD44HI populations remained strongly inhibited. OSM also provided a modest resistance in Panc 05.04, but was unable to protect Panc1 or Panc 04.03 (S1C, H, K). Together, this further indicates that a CD44HI/mesenchymal state alone or exposure to OSM may not be sufficient for enhancing survival following chemotherapy and suggests that tumor-initiation and gemcitabine resistance can be governed by independent programs.
OSMR is essential for OSM-mediated stem-like/mesenchymal conversion and is correlated with poor prognosis.
OSM can activate OSMR- or LIFR-containing receptor complexes to engage downstream signaling. Direct comparison of OSMR and LIFR expression in normal versus cancerous pancreas tissues identified that OSMR is significantly elevated in cancerous tissue and correlated with significantly lower patient survival (Fig. 2A–B). In contrast, LIFR expression was not elevated in PDAC and was actually correlated with improved patient survival (Fig. 2A, S2A). Moreover, IL6ST (encoding the OSMR co-receptor GP130) was not correlated with patient survival (Fig. 2B), indicating that OSMR expression may be a determining factor in patient outcomes. In addition to LIFR, elevated expression of other IL6 family receptors (IL6R, CNTFR, and IL11RA) correlate with an improved patient survival; elevated expression of IL31RA was the only IL-6 family receptor correlated with lower patient survival in PDAC (S2). To confirm the key role of OSMR in OSM-mediated stem-like/mesenchymal reprogramming, we utilized CRISPR/Cas9 to knock-out OSMR in HPAC cells. Four small-guide RNAs constructs targeting OSMR (sgOR1–4) were used, as well as a scrambled control (sgS), with SgOR1 and SgOR3 displaying the most effective knock-out of full-length OSMR (Fig. 2C). Selected populations were only partially inhibited from acquiring a CD44HI/mesenchymal phenotype (S3A); therefore, individual clones were selected from the SgS, SgOR1, and SgOR3 populations and re-examined for loss of OSMR function (S3B-D). Complete OSMR KO clones were identified from both the SgOR1 and SgOR3 populations, SgOR1–2 and SgOR3–1, respectively, with clones being unresponsive to recombinant OSM or lentiviral-expressed OSM, as evidenced by the inability to phosphorylate STAT3, transcriptionally induce SNAI1, SOCS3, and ZEB1, and convert to a CD44HI/mesenchymal phenotype (Fig. 2D–F, S3B–D). Furthermore, while control SgS clones displayed dramatic increases in OSMR protein in response to OSM, due to a positive feed-forward activation of OSMR expression, the SgOR1 and SgOR3 clones showed no increase in OSMR (Fig. 2D). Finally, the CD24HI cells were sorted from the SgS, SgOR1, and SgOR3 clones, and infected with lentiviruses encoding OSM (or a vector control) and CD24/CD44 expression was monitored over the course of 3 weeks. Importantly, not only did OSMR KO prevent the conversion of cells to a CD44HI/mesenchymal state, but it also prevented the spontaneous development of CD44HI/mesenchymal cells, further supporting the importance of OSMR in the conversion to a CD44HI/mesenchymal state (Fig. 2F).
Figure 2: OSMR is essential for OSM-mediated stem-like/mesenchymal conversion and is correlated with poor prognosis.
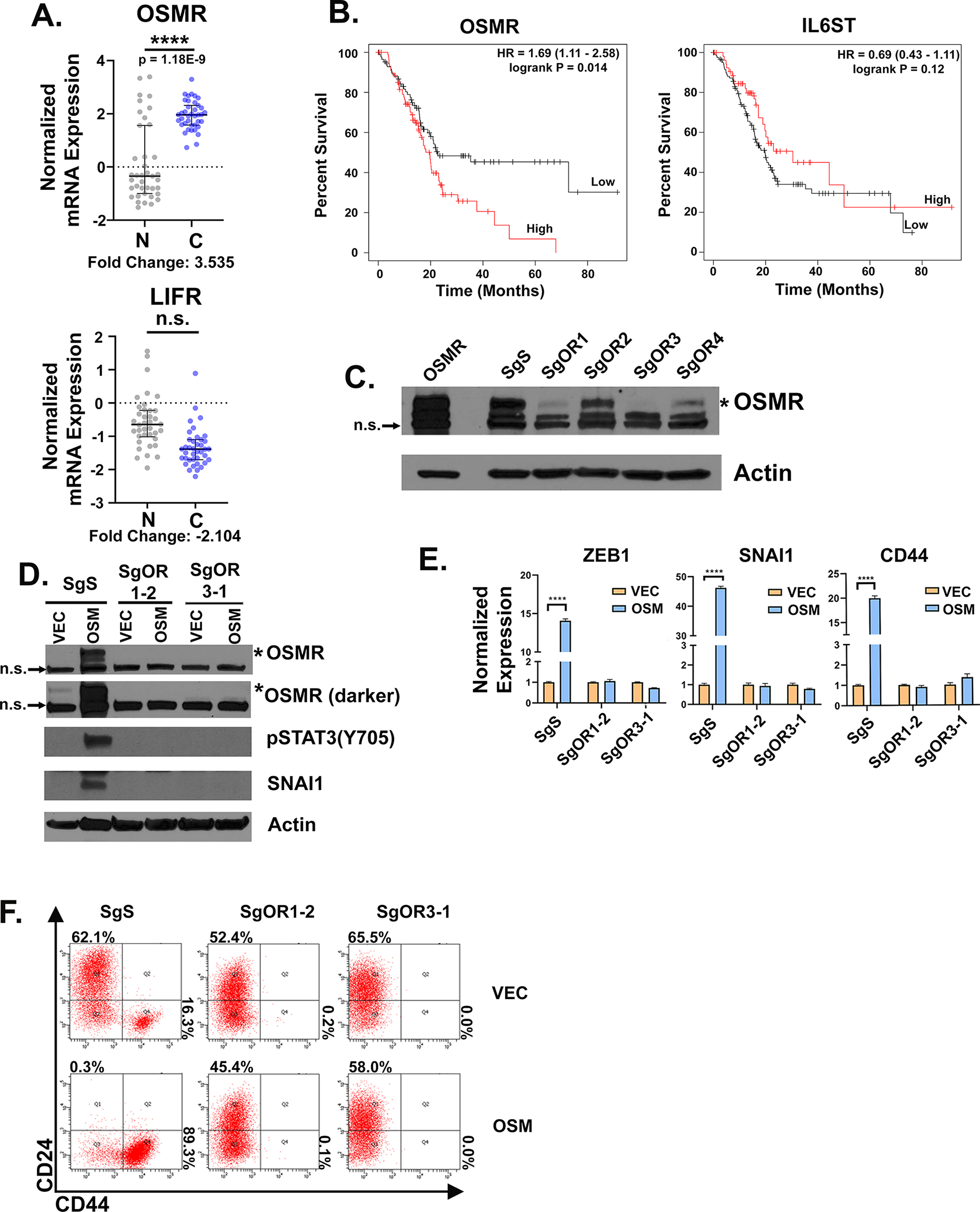
A RNA expression of LIFR and OSMR from patient non-cancerous (N) versus cancerous (C) pancreatic tissues from the Oncomine data mining platform (Badea Pancreas dataset, from 39 normal and 39 PDAC patients; (68)). B Kaplan-Meier plot of patients with pancreatic cancer that express high or low levels of OSMR or IL6ST using the median as the cutoff value for high and low expression (33). Statistical significance was determined by KM-plotter where **P<0.01. C HPAC cells were infected with Lenti-CRISPRV2s targeting OSMR (SgOR1-SgOR4; or a control scrambled guide, SgS). Western blot of selected populations confirms SgOR1 and SgOR3 most efficiently knock-out OSMR (indicated by the asterisk*); the lower two bands are non-specific or “n.s.”. D-F Individual clones selected from the SgS, SgOR1, and SgOR3 populations were infected with lentiviruses encoding OSM (or a control lentivirus, VEC) and assessed by Western blot D, quantitative PCR E, or flow cytometry F, for the indicated genes and proteins. D Data are shown as mean ± S.E.M, and statistical significance was determined by two-way ANOVA where ****P<0.0001.
Sustained OSMR feed-forward signaling is required for therapy resistance
The OSM-OSMR feed-forward loop facilitates enhanced responsiveness to OSM and amplifies signaling leading to stem-like/mesenchymal reprogramming. Here, we wanted to assess how breaking the OSM-OSMR feed-forward loop would inhibit and potentially reverse OSM-driven stem-like/mesenchymal behaviors. HPACs were treated with recombinant OSM for 10 days to convert cells to a CD44HI state, then OSM was either removed for 1, 3, or 10 days or maintained (rOSM). Upon OSM removal, there was a stark and rapid reduction in OSM target genes, including OSMR, as well as SOCS3 and SNAI1 within 1 day of OSM removal (Fig. 3A). In a second experiment, HPACs were treated with recombinant OSM for 2 weeks and then the CD44HI cells were sorted, and OSM was either removed for 7 days or maintained (continued OSM; Fig. 3B). Importantly, removal of OSM from the sorted CD44HI cells did not reduce CD44 expression (Fig. 3B). The stable and sustained expression of CD44 following OSM removal contrasted with the stark reduction in phospho-STAT3, SNAI1, and OSMR, which were reduced by 75–95% after 7 days of OSM removal (Fig. 3B–D). Similar to CD44 expression, both ZEB1 mRNA and protein expression remained elevated following OSM removal (Fig. 3C, D). These findings suggest that once expressed, ZEB1 and CD44 may be difficult to repress despite a robust disruption of the OSM/OSMR feed-forward loop and other OSMR effectors. As shown earlier, the presence of OSM signaling and not the CD44HI/mesenchymal state created cells with enhanced tumor-initiating capacity and resistance to gemcitabine (Fig. 1). Likewise, the removal of OSM reduced cell outgrowth in the presence of gemcitabine, despite the maintenance of the CD44HI/mesenchymal phenotype (Fig. 3E). Together, our data suggest that sustained OSM/OSMR signaling is a key-determinant of tumor-initiating capacity and gemcitabine resistance, and that these OSM/OSMR-induced phenotypes are not solely due to the cells being in a CD44HI/mesenchymal state.
Figure 3: The OSM-OSMR feed-forward loop is required for SNAI1 expression and therapy resistance.
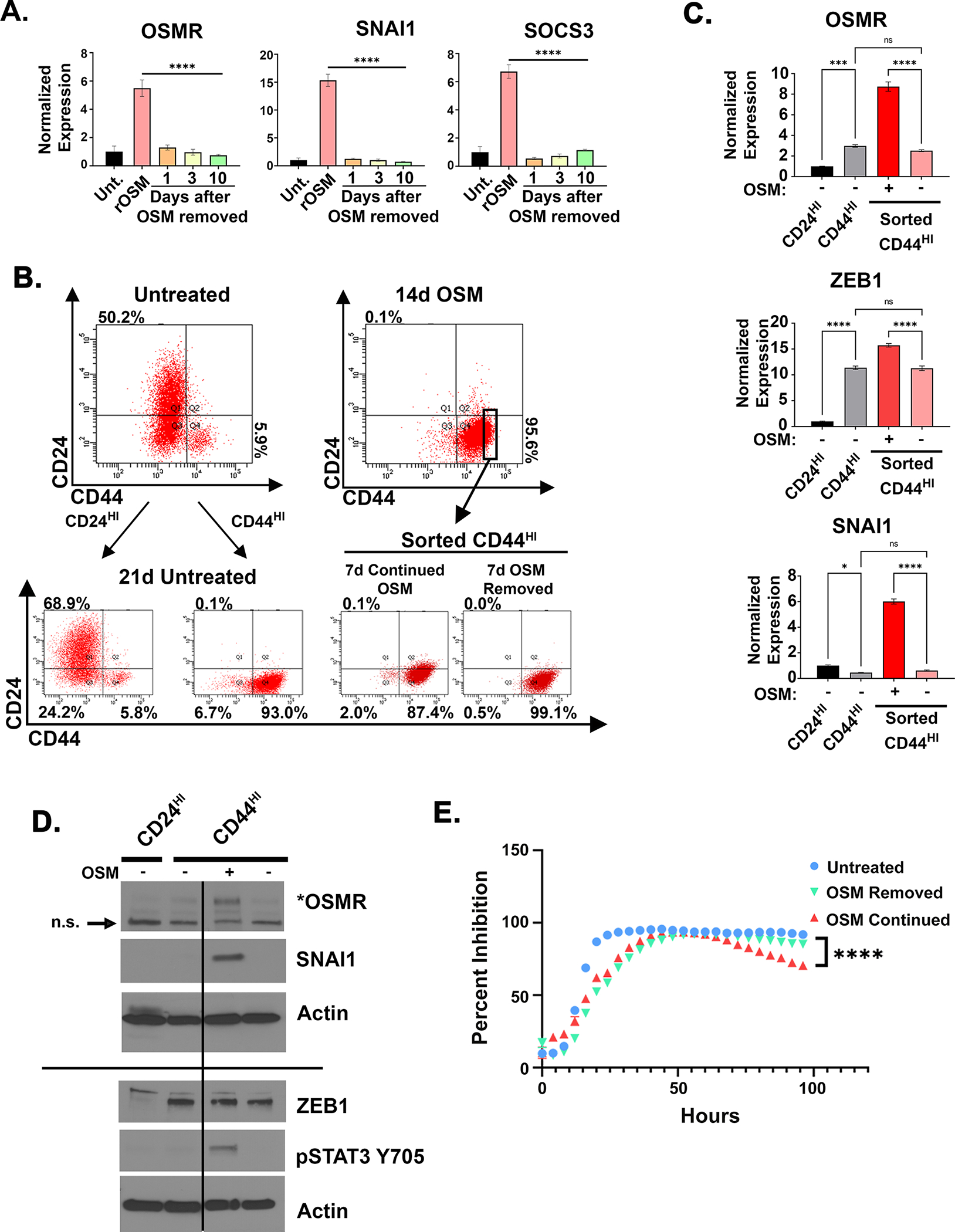
A HPAC cells were treated for 10 days with recombinant OSM (10 ng/mL) (rOSM). After 10 days, OSM was either maintained or removed for 1, 3, or 10 days, followed by quantitative PCR for the indicated genes. B-D HPAC cells were treated with OSM for 14 days, before being sorted for CD24HI (untreated controls only) or CD44HI, as indicated. B Following the sort, OSM was either maintained or removed for 7 days and the cells assessed by flow cytometry for CD24 and CD44 expression. C quantitative PCR with cells from B. Data are shown as mean ± S.E.M, and statistical significance was determined by ordinary one-way ANOVA with ns = not significant, *P<0.05, ***P<0.001, ****P<0.0001. D Western blot for the indicated genes and proteins. E Cell growth in 16 nM of gemcitabine with continued recombinant OSM treatment or removal for 96 hours. Data are plotted as cell number ± S.D. in gemcitabine at a specific timepoint divided by cell number of the corresponding untreated control at the same timepoint. Statistical significance was determined by two-way ANOVA where ****P<0.0001.
OSM-induced tumor initiation is not recapitulated by EMT transcription factors.
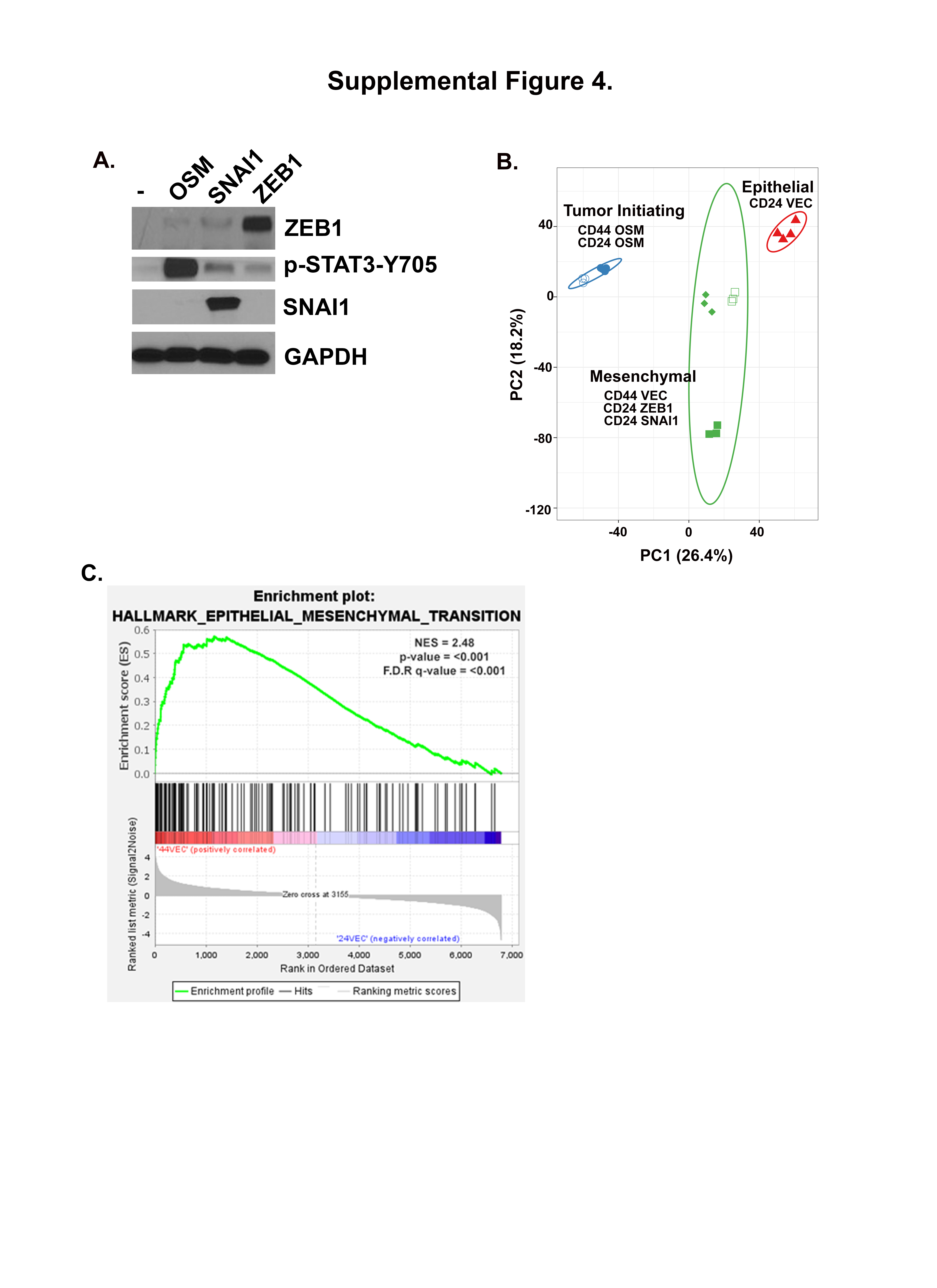
The EMT transcription factors SNAI1 and ZEB1 have been implicated in promoting stem-like reprogramming in addition to EMT (35, 36). To better understand the role of OSM-induced SNAI1 and ZEB1 in the stem-like/mesenchymal reprogramming reported here, we compared HPACs converted to a CD44HI/mesenchymal state by the expression of OSM, SNAI1, or ZEB1 (S4A). Importantly, despite consistent conversion to a CD44HI/mesenchymal cells state (Fig. 4A), and comparable increases in migratory behavior by OSM, SNAI1, and ZEB1 (Fig. 4B), only OSM was capable of enhancing tumor-initiating capacity when injected into immunocompromised mice (Fig. 4C). Interestingly, both SNAI1- and ZEB1-expression induced modest resistance to gemcitabine when compared to OSM (Fig. 4D).
Figure 4: OSM-induced tumor initiation is not recapitulated by EMT transcription factors.
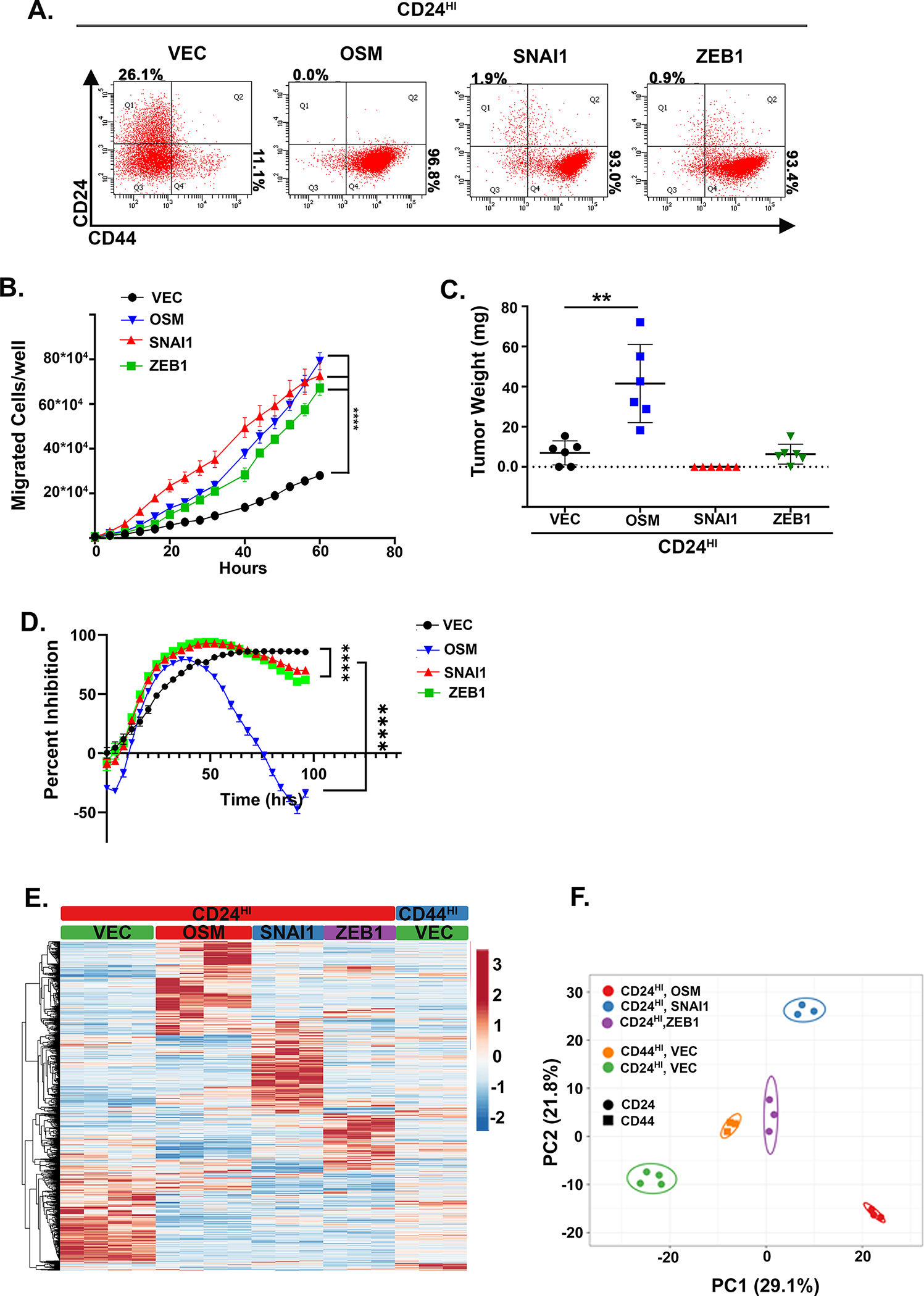
A CD24HI HPAC cells were infected with lentiviruses encoding OSM, ZEB1, or SNAI1 (or control, Vec). Following selection, cells were assessed by flow cytometry for CD24 and CD44. B Following, trans-well migration was assessed over 60 hours by Incucyte imager. Data is presented as mean ± S.D. with statistical significance determined by two-way ANOVA where ****P<0.0001. C tumor initiation after subcutaneous injection of 10,000 cells from A and monitoring for 1 month. Data were graphed as a mean ± S.D. and statistical significance calculated using a one-way ANOVA with multiple comparisons, **P<0.01. D Cell growth in 16 nM of gemcitabine was assessed over 96 hours. Data are plotted as cell number in gemcitabine at a specific timepoint divided by cell number of the corresponding untreated control at the same timepoint. Statistical significance was determined using two-way ANOVA, ****P<0.0001. E Heatmap with hierarchical clustering of expression lines from A with (n=2) for CD24HI-VEC and –OSM, -ZEB1, and -SNAI1 in a puromycin selection lentiviral plasmid, in addition to (n=2) CD24HI-VEC and –OSM and (n=3) CD44HI-VEC in a neomycin selection lentiviral plasmid. F PCA of cell lines from A, B showing clustering of a compressed representation of each group.
This result further supports our conclusion that the conversion to a CD44HI/mesenchymal state, which can occur spontaneously or following EMT transcription factor expression, is not sufficient to impart tumor initiation, and that OSM is simultaneously promoting EMT and tumor initiation. Importantly, the panel of cells generated here provide a unique opportunity to compare different cell states of interest using RNA-Seq. Analysis of the RNA-seq data identified a number of interesting observations. First, despite inducing a common CD24LO/CD44HI phenotype, OSM, ZEB1 and SNAI1 induced unique gene expression profiles, with a relative absence of a conserved EMT gene signature and considerable variation in gene expression among the CD44HI/mesenchymal populations (Fig. 4D). Additional GSEA of CD24HI-VEC and CD44HI-VEC demonstrates a significant enrichment in EMT-associated genes in CD44HI-VEC, consistent with the stark differences in morphology and EMT markers already noted (S4A & S1A). Principle component analysis confirmed that OSM-, ZEB1-, and SNAI1-induced gene expression patterns can be quite distinct and non-overlapping (Fig. 4E). Principal component analysis (PCA) combining CD24HI-VEC, CD24HI-OSM, CD44HI-VEC, and CD44HI-OSM, SNAI1, and ZEB1 groups confirms that OSM-induced tumor-forming cells share the most similarity and cluster separately from both epithelial and mesenchymal groups (S4B).
OSM tumor initiating capacity requires MAPK activation
To define an OSM-induced tumor-initiating gene signature, we compared the differentially expressed genes from the following groups: (i) CD24HI/epithelial cells that are not tumor-initiating (TI; CD24HI/epithelial-TILO, comprised of CD24HI-VEC cells); (ii) spontaneously generated CD44HI/mesenchymal cells that are not tumor-initiating (CD44HI/mesenchymal-TILO, comprised of CD44HI-VEC); and (iii) CD44HI/mesenchymal cells that are tumor-initiating (CD44HI/mesenchymal-TIHI, comprised of CD24HI and CD44HI cells expressing OSM). The CD24HI/epithelial-TILO and CD44HI/mesenchymal-TILO groups were subtracted from the CD44HI/mesenchymal-TIHI group, to specifically isolate an OSM-induced stem-like gene signature. Of 485 genes uniquely regulated by OSM, 207 were repressed and 278 were induced, with the OSM-induced TIHI groups sharing the most similarity by PCA (Fig. 5A, S4B). GO analysis of these gene sets identified the positive regulation of JAK/STAT and MAPK activity as well as stem-cell differentiation as specific to the OSM-induced CD44HI/mesenchymal-TIHI cells (Table 1). Interestingly, a TGFβ response in the OSM-induced CD44HI/mesenchymal-TICHI groups suggest a potential OSM-STAT3/SMAD3 contribution to stem-like reprogramming, as we have previously reported (27).
Figure 5: OSM tumor initiating capacity requires MAPK activation.
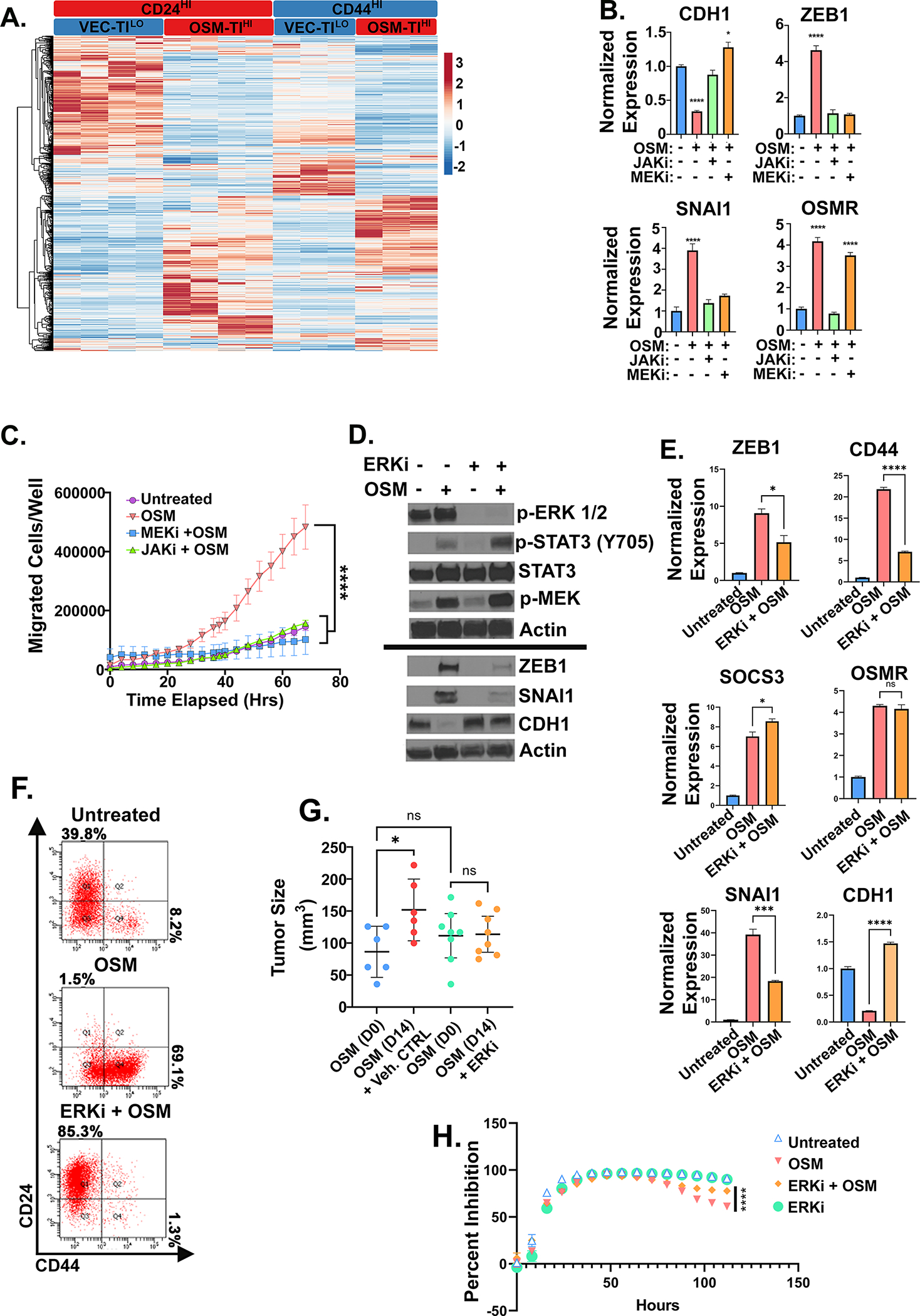
A Heatmap of RNA-seq analysis of HPAC derivatives expressing OSM (or control, VEC) used for defining the unique, stem-like program underlying OSM-induced tumor-initiation and gemcitabine resistance described in Table 1 with (n=2) for CD24HI-VEC and -OSM in a puromycin selection lentiviral plasmid, in addition to (n=2) CD24HI-VEC and –OSM and CD44HI-VEC and -OSM in a neomycin selection lentiviral plasmid. B Quantitative PCR of HPAC cells treated with non-cytotoxic/non-cytostatic doses of MEKi (U0126) or JAKi (Ruxolitinib) with or without OSM for 7 days with media changes every 48 hours. C Transwell migration of HPACs with MEKi (U0126) and JAKi, (Ruxolitinib) with or without recombinant OSM. Data is shown as mean ± S.D., and statistical significance was determined by two-way ANOVA and multiple comparisons test where ****P<0.0001. D Western blot, E quantitative PCR, and F flow cytometry of HPACs treated with rOSM with or without ERKi (SCH772984) for 5 days. E Statistical significance of quantitative PCR was determined by one-way ANOVA ns= not significant, *P<0.05, ***P<0.001, ****P<0.0001. G Tumor size measured by caliper after 1 week of tumor growth followed by 14 days of ERK inhibitor treatment. Statistical significance was determined by one-way ANOVA, where *P<0.01. H HPAC cells pre-treated with OSM with or without ERKi for 48 hours before the addition of 16 nM of gemcitabine. Data are plotted as cell number in gemcitabine at a specific timepoint divided by cell number of the corresponding untreated control at the same timepoint. Statistical significance was determined by two-way ANOVA where ****P<0.0001.
Table 1.
OSM-Tumor Initiating Gene Signature
| GO Terms | CD24HI/EP-TILO vs. CD44HI/MES-TILO | CD24HI/EP-TILO and CD44HI/MES-TILO vs. CD44HI/MES-TIHI | CD44HI/MES-TIHI | ||
|---|---|---|---|---|---|
| CD24-VEC vs CD44-VEC | CD24-VEC vs CD24-OSM | CD44-VEC vs. CD44-OSM | CD44-VEC vs. CD24-OSM | CD44-OSM vs CD24-OSM | |
| Epithelial to Mesenchymal Transition | 0.01 | 0.007 | 0.124 | 0.242 | 0.408 |
| Positive Regulation of epithelial to mesenchymal transition | 3.44E-04 | 3.17E-04 | 0.073 | 0.222 | 0.157 |
| Positive Regulation of MAPK activity | 0.206 | 5.57E-04 | 0.01 | 0.002 | 0.314 |
| Receptor signaling pathway via JAK/STAT3 | 0.316 | 0.01 | 0.01 | 3.85E-04 | 0.418 |
| Positive regulation of receptor signaling pathway via JAK/STAT3 | 0.144 | 1.76E-04 | 3.39E-04 | 2.68E-04 | 0.142 |
| Stem cell differentiation | 0.513 | 0.018 | 0.009 | 0.016 | 0.505 |
| Response to TGFB | 0.19 | 7.89E-06 | 0.004 | 0.018 | 0.866 |
| Positive regulation of vascular endothelial growth factor production | 0.152 | 0.002 | 4.53E-05 | 0.005 | 0.216 |
| Regulation of interleukin-1 beta production | 0.125 | 2.22E-05 | 1.04E-05 | 7.85E-05 | 0.332 |
Among the IL-6 family receptors, OSMR has unique characteristics, distinct signaling, and often more pronounced effects compared to other IL-6 family receptor complexes (37). OSMR uniquely activates a stem-like/mesenchymal phenotype in HPAC cells when compared to IL-6; the enhanced OSMR activity correlates with increased MAPK signaling due to the unique recruitment of SHC to OSMR, which recruits Grb2-SOS independently of the common GP130 co-receptor (37). Here, we evaluated the importance of MAPK signaling downstream of OSMR activation by using non-cytotoxic and non-cytostatic doses of MEK and ERK inhibitors. First, we assessed the role of MEK1/2 and STAT3 in inducing a CD44HI/mesenchymal phenotype, using MEK inhibitors, U0126, PD184352, and Pimasertib, or a JAK inhibitor, Ruxolitinib (Fig. 5, S5). JAK inhibitors impeded OSM-mediated conversion to CD44HI/mesenchymal state, and prevented SNAI1 and ZEB1 induction, CDH1 repression, and migration (Fig. 5B, C, S5A). Moreover, JAK inhibition prevented OSM induction of OSMR feed-forward loop signaling (Fig. 5B). Interestingly, MEK inhibition comparably blocked CD44HI/mesenchymal conversion, induction of ZEB1 and SNAI1, and migration, but did not disrupt OSMR expression (Fig. 5B, C, S5B). Next, we assessed the role of an ERK inhibitor SCH772984 in suppressing OSM/OSMR-mediated stem-like/mesenchymal reprogramming. ERK inhibition reduced ERK phosphorylation, while OSMR-induced MEK and STAT3 phosphorylation and target gene expression (SOCS3 and OSMR) were unaffected (Fig. 5D, E). This result suggests that STAT3 nuclear localization, DNA binding, and transcriptional activity is not inhibited by ERK inhibition. Interestingly however, ERK inhibition significantly repressed OSM-mediated ZEB1, CD44, and SNAI1 induction and prevented CDH1 repression (Fig. 5D, E). Furthermore, ERK inhibition led to a reduced spontaneous CD44HI/mesenchymal population and expanded the CD24HI/epithelial population (Fig. 5F). Next, we assessed whether ERK inhibition could reduce OSM-mediated tumor growth. OSM-expressing HPACs were injected subcutaneously into immunocompromised mice and tumors permitted to grow until 50–100 mm3 before daily ERK inhibitor (or vehicle control) treatment was begun. Over 2 weeks, OSM induced a two-fold increase in tumor size in the vehicle control group. In contrast, two weeks of ERK inhibitor treatment prevented any further OSM-mediated tumor growth (Fig. 5G). Similarly, in HPAC, Panc1, and Panc 04.03, OSM-induced tumorsphere initiation and migration was significantly reduced with ERK inhibitor treatment (S5C, E-G). Finally, a 48-hour pretreatment with ERKi significantly diminished OSM-mediated outgrowth during gemcitabine treatment relative to controls (Fig. 5H).
In breast cancer, OSM induces a mesenchymal/stem-like reprogramming through STAT3 promotion of SMAD3-mediated gene transcription; the identification of a TGFβ response signature in the OSM-expressing cells suggests a similar interaction occurs in response to OSM in PDAC (Table 1). Therefore, to evaluate the role of STAT3 and SMAD3 in CD44HI/mesenchymal reprogramming, CRISPR/Cas9 was used knock-out STAT3 or SMAD3 in HPAC cells. Three small-guide RNAs constructs (sg1–3) were used for each, as well as a scrambled control (sgS). Sg3 targeting STAT3 (sgSTAT3) and Sg2 targeting SMAD2 (SgSMAD2) were most effective at knocking out each protein (Fig. 6A, S6A). Knock-out of STAT3 dramatically repressed the expression of ZEB1, SNAI1, and OSMR after 10 days of recombinant OSM (Fig. 6A, C). Moreover, knock-out of STAT3 inhibited CD44HI induction, migration, and gemcitabine resistance, suggesting an integral role for STAT3 in OSM signaling (Fig. 6B, D, E). Next, the SMAD3 knock-outs were transduced with either vector control or OSM and after two weeks, downstream signaling and CD44HI/mesenchymal conversion were assessed. Interestingly, SMAD3 knock-out reduced STAT3 phosphorylation and STAT3 target genes, SOCS3 and SNAI1, similar to STAT3 knock-out, without preventing CD44HI conversion, ZEB1 or OSMR expression, or OSM-mediated tumor growth and lethality (S6B-E). This finding suggests that while SMAD3 is required for the OSM-mediated transcriptional activity of SNAI1, neither SMAD3 nor SNAI1 are important in conferring tumor initiation.
Figure 6: STAT3 is essential for OSM-induced migration and gemcitabine resistance.
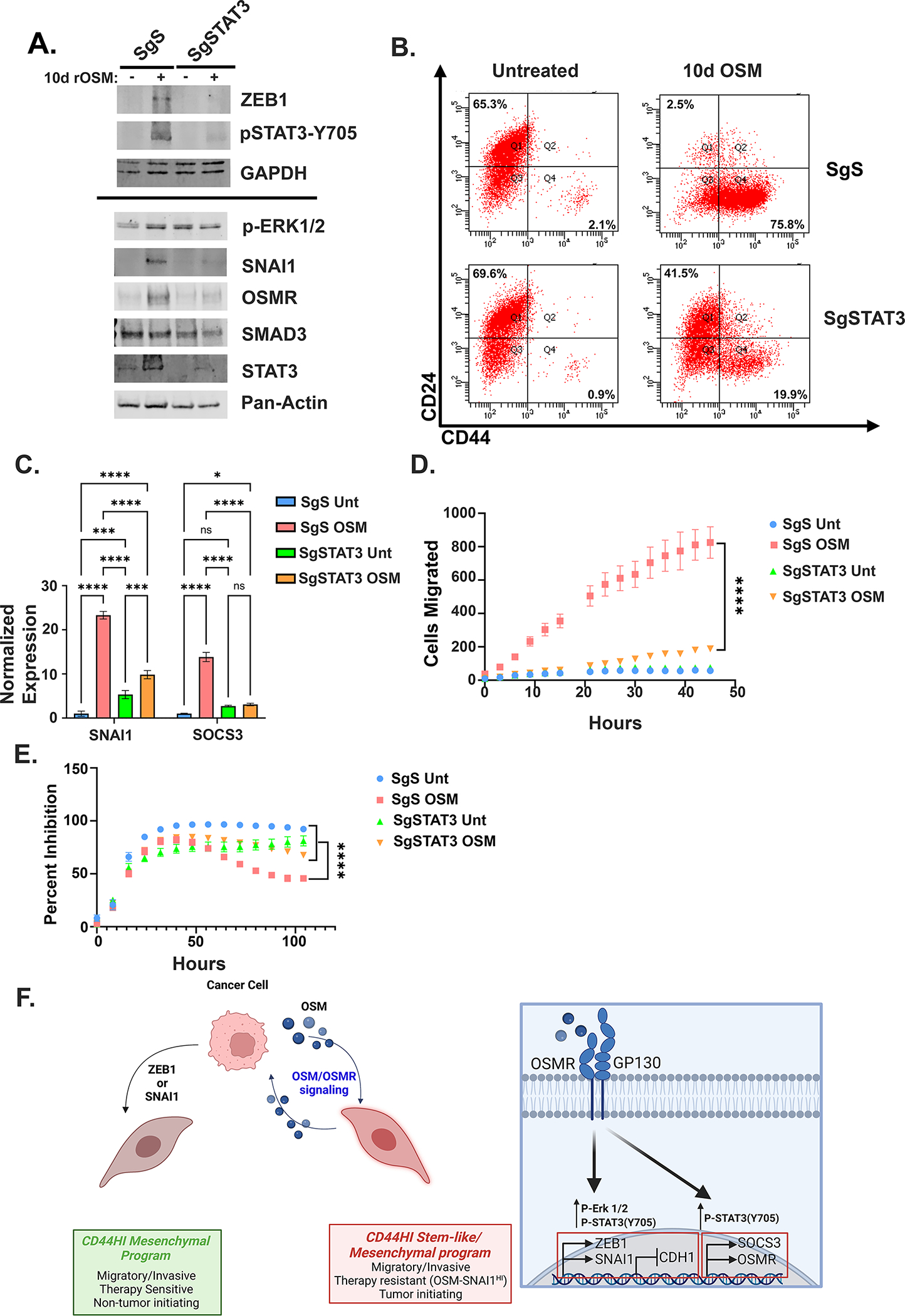
A Western blot, B flow cytometry, and C q-PCR of HPAC STAT3 knock-out cells after 10 days of OSM treatment. Statistical significance was determined by two-way ANOVA where *P<0.05, ***P<0.001, ****P<0.0001. D Transwell migration of HPAC STAT3 knock-out cells after 7 days of OSM treatment with the number of cell migrated was quantified by an Incucyte imager. Data are plotted as cells migrated from initial timepoint. Statistical significance was determined by two-way ANOVA where ****P<0.0001. E HPAC STAT3 knock-out cells pre-treated with OSM for 48 hours before the addition of 16 nM of gemcitabine. Data are plotted as cell number in gemcitabine at a specific timepoint divided by cell number of the corresponding untreated control at the same timepoint. Statistical significance was determined by two-way ANOVA where ****P<0.0001.
Discussion
In cancer, epithelial-mesenchymal (EM) plasticity and differentiation status are key factors that allow cancer cells to enter a stem-like/mesenchymal state that helps them survive cancer therapies and metastasize to secondary organs (38–40). Numerous studies, including our own, link adaptive EMT and dedifferentiation to a stem-like state (41–44). Concomitant EMT and dedifferentiation result in cancer cells that are more migratory and invasive, have enhanced tumor-initiating capacity important for outgrowth at metastatic sites, and have a decreased sensitivity to DNA-damaging chemotherapies. OSM is a known TME cytokine capable of imparting a stem-like/mesenchymal program in multiple cancers (PDAC, TNBC, glioblastomas, non-small-cell lung carcinomas, and ovarian cancer); thus, identifying proteins and pathways that influence this stem-like/mesenchymal reprogramming is critically important for fostering novel therapies that deplete these pools of problematic cells and improve patient outcomes (25, 32, 45–48). Here, we demonstrate that OSMR signaling induces a tumor-initiating capacity, that is dependent on MAPK activation, and outgrowth in gemcitabine, that is dependent on cooperative activation of ERK, STAT3.
Our studies utilized parental or sorted populations of cells harboring distinct CD24HI/epithelial and CD44HI/mesenchymal phenotypes (including spontaneously-developed CD44HI/mesenchymal cells or those induced to a CD44HI/mesenchymal state by EMT transcription factors ZEB1 or SNAI1). We observed that OSM can enhance a tumor-initiating capacity and drug resistance that are not inherent to CD44HI/mesenchymal cells. While exposure of spontaneously developed CD44HI/mesenchymal HPAC cells to OSM doesn’t alter their CD44HI/mesenchymal state or migratory capacity, OSM does enhance tumor-initiating capacity and survival in gemcitabine (Fig. 6F). Yet, in other cell models where CD44HI expression was similarly un-altered by OSM (Panc1 and Panc 04.03), there was no increase in gemcitabine resistance (S1C and S1H). Rather, among the 4 cell lines tested, we noted a correlation between the OSM-induced SNAI1 transcription (which requires MAPK-ERK activity) and gemcitabine resistance, as OSM did not induce SNAI1 in either Panc 04.03 or Panc1 cells, and neither line showed an increase in gemcitabine resistance, unlike in HPAC and Panc 05.04 (S1C, D, G, H, J, K). Thus, we suggest that stem-like reprogramming can be separable from the CD44HI/mesenchymal program; in addition, our data suggest that tumor initiation can be separable from therapy resistance. Others have similarly reported a loss of stem-like behaviors in fully converted mesenchymal cells and postulate that the epigenetic modifications controlling the accessibility of stem-cell related genes can become entrenched and un-responsive over time, resulting in a loss of cellular plasticity (49–51). If true, we hypothesize that OSM may increase plasticity by loosening the epigenetic constraints that limit the accessibility of stem-like genes in tumor cells, allowing a greater adaptive response when cancer cells encounter stress.
The OSM-mediated reprogramming of either sorted CD24HI/epithelial or CD44HI/mesenchymal cells to a stem-like, tumor-initiating state (CD44HI/mesenchymal-TIHI cells) result in a similar gene expression profile, including genes involved in interleukin-1 beta and vascular endothelial growth factor production (Table 1). However, some differences in stem-cell associated genes were noted depending on whether OSM was expressed in CD24HI/epithelial or CD44HI/mesenchymal cells. For example, OSM induced the expression of aldehyde dehydrogenase 1 and aldehyde dehydrogenase 2 (ADLHA1 and ALDH2, respectively) only when expressed in CD24HI/epithelial cells, but not spontaneous CD44HI/mesenchymal cells, despite both populations ultimately converting to a comparable CD44HI/mesenchymal phenotype and having comparable tumor-initiating capability. In contrast, OSM induced the expression of fibroblast growth factor 1 (FGF1) and amphiregulin (AREG) in spontaneous CD44HI/mesenchymal only (Table 2). ALDH1/2, AREG, and FGF1 are reported to enhance stem-like behaviors in a wide range of cancers, suggesting that the accessibility of genes in distinct cell states can determine how they respond to common stimuli, such as TME cytokines like OSM (52–56).
Table 2.
OSM induces stem-like genes
| CD24-VEC vs CD24-OSM | CD44-VEC vs. CD44-OSM | ||||
|---|---|---|---|---|---|
| Gene | Fold Change | P-value | Gene | Fold Change | P-value |
| ALDH1A1 | 2.97268 | 7.37E-04 | FGF1 | 10 | 7.37E-04 |
| ALDH2 | 4 | 7.37E-04 | AREG | 1.7 | 7.37E-04 |
| EGFR | −0.802 | 7.37E-04 | EGFR | −1.7 | 7.37E-04 |
OSM signals through two different heterodimer complexes, OSMR-GP130 or LIFR-GP130. While both receptor complexes are reported to promote mesenchymal and stem-like properties, the unique ability of OSMR to enhance MAPK signaling through a SHC-binding domain sets it apart from other IL-6 family receptor complexes and may explain why OSMR associates with a worse patient outcome (37, 57, 58). In addition, the OSM-OSMR feed forward loop, whereby OSMR activation results in increased OSMR protein production, is strongly implicated in the persistent signaling noted in other metastatic and drug tolerant cancers, such as glioblastoma, TNBC, small-cell lung cancer, ovarian cancer, and squamous cell carcinomas (20, 21, 26, 30, 47). In glioblastoma, OSMR also interacts with wild-type and mutant EGFR to propagate feed-forward activation of STAT3 (25). Importantly, EGFR is expressed on >90% of PDAC cells and propagates MAPK and STAT3 signaling, with EGFR inhibitors remaining an important component of current treatment strategies (59). Whether OSMR and EGFR interact or cooperate in PDAC cells remains unclear, but we would predict that EGFR/OSMR signaling complexes would negatively impact the efficacy of EGFR inhibitors. In addition, our RNA-seq data identified a significant reduction in EGFR expression in response to OSM treatment and stem-like/mesenchymal reprogramming (Table 2). In tumors with elevated OSM-OSMR signaling, the reduction in EGFR expression when cells convert to a more stem-like/mesenchymal state would potentially de-sensitize cancer cells to EGFR targeted therapies. Additional studies examining OSMR and EGFR integrations and expression changes are ongoing.
We propose that disrupting the OSM-OSMR feedforward loop is a novel way to therapeutically target EMT and stem-like reprogramming, particularly in PDAC where OSM was recently shown to orchestrate tumor microenvironment interactions between PDAC cells, TAMs, and CAFs. M2 polarized macrophages secrete OSM to re-shape the stromal compartment through CAFs, which express abundant OSMR, causing a phenotypic switch in the CAF population, from myofibroblast to inflammatory CAFs (iCAFs) (23). The iCAFs alter tumor cell secretion of GM-CSF, which recruits and polarizes additional macrophages to a pro-tumorigenic, OSM-secreting M2 state, reinforcing the entire cycle. Taken together, the increases in OSM expression within the TME, increases in OSMR expression in cancer and stromal cells, and the consequences of OSMR signaling (stem-like/mesenchymal cancer cell reprogramming, iCAF reprogramming, and immune suppression) make targeting the OSM-OSMR axis particularly attractive (Fig. 6). Recently, OSMR-targeting antibodies were created that have a robust efficacy in pre-clinical ovarian cancer models, demonstrating a reduction of STAT3 phosphorylation, growth, and metastasis in vivo by inducing the internalization and degradation of OSMR (30). These finding are consistent with a reported role of OSM-OSMR signaling in promoting persistent inflammation in inflammatory bowel disease (IBD). The set of chemokines and cytokines responsible for inducing and maintaining IBD are reduced following neutralization of OSM; in addition, the severity of the disease is significantly reduced suggesting that OSM is a keystone of the persistent and damaging inflammation (60). Many of these OSM-regulated cytokines have also been implicated in increasing the aggressiveness of cancer cells (migration, invasion, therapy failure, and stem-like markers) and acting to modulate the immune system.
Another important aspect of our findings implicate both OSMR-activated MEK/ERK and STAT3 signaling as important regulators of select OSM-activated genes, including ZEB1, SNAI1, and OSM-repressed genes, including CDH1 (Fig. 6F). Other OSM-induced genes do not require MAPK signaling, relying solely on JAK/STAT3 activation (including SOCS3 and OSMR). Since JAK and MEK/ERK inhibitors and STAT3 knock-out prevented conversion to a stem-like/mesenchymal state (22), we conclude that both signaling arms are important contributors in OSMR induction of CD44HI/mesenchymal state and hypothesize that STAT3 induces feed-forward increase in OSMR expression, resulting in increased and sustained hyperactivation of ERK, which is essential for tumor initiating capacity. Hyperactivation of MAPK (MEK and ERK) in PDAC is common, given that KRAS is mutated in 95% of cases and EGFR is often highly expressed. Targeting MAPK signaling has had limited impact, only moderately improving patient survival (61, 62). MEK and ERK inhibitors have failed in pre-clinical trials as single agents due to the activation of compensatory signaling pathways, including STAT3 (63). Likewise, we observed a hyperactivation of STAT3 following treatment with an ERK inhibitor in Panc1 and HPACs (Fig. 5D and S5D). Single agent use of MEK inhibitors in breast, ovarian, and lung cancers also result in the compensatory activation of PI3K/mTOR signaling cascades; combining PI3K and MEK inhibitors more effectively inhibits cancer growth (64). In melanoma, treatment with a MEK inhibitor induces the differentiation of cancer cells to an epithelial state, similar to our observations with both MEK and ERK inhibitors (65). Furthermore, MEK/ERK inhibition induced the expansion of the CD24HI/epithelial population and resenstitized cells to gemcitabine (Fig. 5F, H). Combining ERK inhibitors with cytotoxic agents may shift cancer cells towards an epithelial, therapy-sensitive state and/or prevent the cells from undergoing EMT or de-differentiation. Interestingly, MEK and ERK inhibitors may also undermine the elevated OSM-OSMR signaling in stromal cells within a tumor, such as CAFs and immune cells, potentially preventing the reprogramming of iCAFs as well as M2 polarization (66, 67). Taken together, our data supports the future assessment of strategies that target OSM-OSMR signaling, including MAPK signaling, in PDAC as a means of preventing the reprogramming of cancer cells, fibroblasts, and immune cells to variants that support tumor survival and their potential for metastatic dissemination.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Implications Statement:
Small molecule MAPK inhibitors may effectively target the OSM/OSMR-axis that leads to EMT and tumor initiating properties that promote aggressive PDAC.
Acknowledgments
This research was funded by R01CA252224 (Jackson); T32CA059366 (Polak); and DoD BCRP awards W81XWH-20-1-0464 (Jackson) and W81XWH-18-1-0552 (Jackson). The Cytometry and Microscopy Core, Genetics and Genomics Core, and small animal research core were supported by the Case Comprehensive Cancer Center (P30 CA043703).
Footnotes
Competing Interests
The authors declare no competing financial interests
References
- 1.Adamska A, Domenichini A, Falasca M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int J Mol Sci. 2017;18(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Society AC. Cancer Facts & Figures 2021. 2021.
- 3.Takikawa T, Kikuta K, Hamada S, Kume K, Miura S, Yoshida N, et al. Clinical features and prognostic impact of asymptomatic pancreatic cancer. Sci Rep. 2022;12(1):4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hosein AN, Brekken RA, Maitra A. Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat Rev Gastroenterol Hepatol. 2020;17(8):487–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369(18):1691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hurwitz H, Van Cutsem E, Bendell J, Hidalgo M, Li CP, Salvo MG, et al. Ruxolitinib + capecitabine in advanced/metastatic pancreatic cancer after disease progression/intolerance to first-line therapy: JANUS 1 and 2 randomized phase III studies. Invest New Drugs. 2018;36(4):683–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Babaei G, Aziz SG, Jaghi NZZ. EMT, cancer stem cells and autophagy; The three main axes of metastasis. Biomed Pharmacother. 2021;133:110909. [DOI] [PubMed] [Google Scholar]
- 8.Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527(7579):525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kroger C, Afeyan A, Mraz J, Eaton EN, Reinhardt F, Khodor YL, et al. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc Natl Acad Sci U S A. 2019;116(15):7353–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Theodoropoulos PA, Polioudaki H, Agelaki S, Kallergi G, Saridaki Z, Mavroudis D, et al. Circulating tumor cells with a putative stem cell phenotype in peripheral blood of patients with breast cancer. Cancer Lett. 2010;288(1):99–106. [DOI] [PubMed] [Google Scholar]
- 11.Jolly MK, Somarelli JA, Sheth M, Biddle A, Tripathi SC, Armstrong AJ, et al. Hybrid epithelial/mesenchymal phenotypes promote metastasis and therapy resistance across carcinomas. Pharmacol Ther. 2019;194:161–84. [DOI] [PubMed] [Google Scholar]
- 12.Chen YC, Sahoo S, Brien R, Jung S, Humphries B, Lee W, et al. Single-cell RNA-sequencing of migratory breast cancer cells: discovering genes associated with cancer metastasis. Analyst. 2019;144(24):7296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Semaan A, Bernard V, Kim DU, Lee JJ, Huang J, Kamyabi N, et al. Characterisation of circulating tumour cell phenotypes identifies a partial-EMT sub-population for clinical stratification of pancreatic cancer. Br J Cancer. 2021;124(12):1970–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu X, Taftaf R, Kawaguchi M, Chang YF, Chen W, Entenberg D, et al. Homophilic CD44 Interactions Mediate Tumor Cell Aggregation and Polyclonal Metastasis in Patient-Derived Breast Cancer Models. Cancer Discov. 2019;9(1):96–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature. 2015;526(7571):131–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khaled N, Bidet Y. New Insights into the Implication of Epigenetic Alterations in the EMT of Triple Negative Breast Cancer. Cancers (Basel). 2019;11(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Unver N, McAllister F. IL-6 family cytokines: Key inflammatory mediators as biomarkers and potential therapeutic targets. Cytokine Growth Factor Rev. 2018;41:10–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Torres C, Perales S, Alejandre MJ, Iglesias J, Palomino RJ, Martin M, et al. Serum cytokine profile in patients with pancreatic cancer. Pancreas. 2014;43(7):1042–9. [DOI] [PubMed] [Google Scholar]
- 19.Chen M, Ren R, Lin W, Xiang L, Zhao Z, Shao B. Exploring the oncostatin M (OSM) feed-forward signaling of glioblastoma via STAT3 in pan-cancer analysis. Cancer Cell Int. 2021;21(1):565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shien K, Papadimitrakopoulou VA, Ruder D, Behrens C, Shen L, Kalhor N, et al. JAK1/STAT3 Activation through a Proinflammatory Cytokine Pathway Leads to Resistance to Molecularly Targeted Therapy in Non-Small Cell Lung Cancer. Mol Cancer Ther. 2017;16(10):2234–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Araujo AM, Abaurrea A, Azcoaga P, Lopez-Velazco JI, Manzano S, Rodriguez J, et al. Stromal oncostatin M cytokine promotes breast cancer progression by reprogramming the tumor microenvironment. J Clin Invest. 2022;132(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smigiel JM, Parameswaran N, Jackson MW. Potent EMT and CSC Phenotypes Are Induced By Oncostatin-M in Pancreatic Cancer. Mol Cancer Res. 2017;15(4):478–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee BY, Hogg EKJ, Below CR, Kononov A, Blanco-Gomez A, Heider F, et al. Heterocellular OSM-OSMR signalling reprograms fibroblasts to promote pancreatic cancer growth and metastasis. Nat Commun. 2021;12(1):7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rose-John S. Interleukin-6 Family Cytokines. Cold Spring Harb Perspect Biol. 2018;10(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jahani-Asl A, Yin H, Soleimani VD, Haque T, Luchman HA, Chang NC, et al. Control of glioblastoma tumorigenesis by feed-forward cytokine signaling. Nat Neurosci. 2016;19(6):798–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kucia-Tran JA, Tulkki V, Scarpini CG, Smith S, Wallberg M, Paez-Ribes M, et al. Anti-oncostatin M antibody inhibits the pro-malignant effects of oncostatin M receptor overexpression in squamous cell carcinoma. J Pathol. 2018;244(3):283–95. [DOI] [PubMed] [Google Scholar]
- 27.Junk DJ, Bryson BL, Smigiel JM, Parameswaran N, Bartel CA, Jackson MW. Oncostatin M promotes cancer cell plasticity through cooperative STAT3-SMAD3 signaling. Oncogene. 2017;36(28):4001–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu X, Li J, Fu M, Zhao X, Wang W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther. 2021;6(1):402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Irey EA, Lassiter CM, Brady NJ, Chuntova P, Wang Y, Knutson TP, et al. JAK/STAT inhibition in macrophages promotes therapeutic resistance by inducing expression of protumorigenic factors. Proc Natl Acad Sci U S A. 2019;116(25):12442–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geethadevi A, Nair A, Parashar D, Ku Z, Xiong W, Deng H, et al. Oncostatin M Receptor-Targeted Antibodies Suppress STAT3 Signaling and Inhibit Ovarian Cancer Growth. Cancer Res. 2021;81(20):5336–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Junk DJ, Cipriano R, Bryson BL, Gilmore HL, Jackson MW. Tumor microenvironmental signaling elicits epithelial-mesenchymal plasticity through cooperation with transforming genetic events. Neoplasia. 2013;15(9):1100–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doherty MR, Parvani JG, Tamagno I, Junk DJ, Bryson BL, Cheon HJ, et al. The opposing effects of interferon-beta and oncostatin-M as regulators of cancer stem cell plasticity in triple-negative breast cancer. Breast Cancer Res. 2019;21(1):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lanczky A, Gyorffy B. Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J Med Internet Res. 2021;23(7):e27633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bryson BL, Tamagno I, Taylor SE, Parameswaran N, Chernosky NM, Balasubramaniam N, et al. Aberrant Induction of a Mesenchymal/Stem Cell Program Engages Senescence in Normal Mammary Epithelial Cells. Mol Cancer Res. 2021;19(4):651–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hojo N, Huisken AL, Wang H, Chirshev E, Kim NS, Nguyen SM, et al. Snail knockdown reverses stemness and inhibits tumour growth in ovarian cancer. Sci Rep. 2018;8(1):8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han Y, Villarreal-Ponce A, Gutierrez G Jr., Nguyen Q, Sun P, Wu T, et al. Coordinate control of basal epithelial cell fate and stem cell maintenance by core EMT transcription factor Zeb1. Cell Rep. 2022;38(2):110240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hermanns HM, Radtke S, Schaper F, Heinrich PC, Behrmann I. Non-redundant signal transduction of interleukin-6-type cytokines. The adapter protein Shc is specifically recruited to rhe oncostatin M receptor. J Biol Chem. 2000;275(52):40742–8. [DOI] [PubMed] [Google Scholar]
- 38.Gener P, Seras-Franzoso J, Callejo PG, Andrade F, Rafael D, Martinez F, et al. Dynamism, Sensitivity, and Consequences of Mesenchymal and Stem-Like Phenotype of Cancer Cells. Stem Cells Int. 2018;2018:4516454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Enane FO, Saunthararajah Y, Korc M. Differentiation therapy and the mechanisms that terminate cancer cell proliferation without harming normal cells. Cell Death Dis. 2018;9(9):912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Celia-Terrassa T, Jolly MK. Cancer Stem Cells and Epithelial-to-Mesenchymal Transition in Cancer Metastasis. Cold Spring Harb Perspect Med. 2020;10(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marine JC, Dawson SJ, Dawson MA. Non-genetic mechanisms of therapeutic resistance in cancer. Nat Rev Cancer. 2020;20(12):743–56. [DOI] [PubMed] [Google Scholar]
- 42.Doherty MR, Jackson MW. Using interferon-beta to combat cancer stem cell properties in triple negative breast cancer. Oncoscience. 2018;5(5–6):169–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Molejon MI, Tellechea JI, Loncle C, Gayet O, Gilabert M, Duconseil P, et al. Deciphering the cellular source of tumor relapse identifies CD44 as a major therapeutic target in pancreatic adenocarcinoma. Oncotarget. 2015;6(10):7408–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanaka M, Mihaljevic AL, Probst P, Heckler M, Klaiber U, Heger U, et al. Meta-analysis of recurrence pattern after resection for pancreatic cancer. Br J Surg. 2019;106(12):1590–601. [DOI] [PubMed] [Google Scholar]
- 45.Dinca SC, Greiner D, Weidenfeld K, Bond L, Barkan D, Jorcyk CL. Novel mechanism for OSM-promoted extracellular matrix remodeling in breast cancer: LOXL2 upregulation and subsequent ECM alignment. Breast Cancer Res. 2021;23(1):56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tawara K, Bolin C, Koncinsky J, Kadaba S, Covert H, Sutherland C, et al. OSM potentiates preintravasation events, increases CTC counts, and promotes breast cancer metastasis to the lung. Breast Cancer Res. 2018;20(1):53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sharanek A, Burban A, Laaper M, Heckel E, Joyal JS, Soleimani VD, et al. OSMR controls glioma stem cell respiration and confers resistance of glioblastoma to ionizing radiation. Nat Commun. 2020;11(1):4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang ML, Pan CM, Chiou SH, Chen WH, Chang HY, Lee OK, et al. Oncostatin m modulates the mesenchymal-epithelial transition of lung adenocarcinoma cells by a mesenchymal stem cell-mediated paracrine effect. Cancer Res. 2012;72(22):6051–64. [DOI] [PubMed] [Google Scholar]
- 49.Celia-Terrassa T, Meca-Cortes O, Mateo F, Martinez de Paz A, Rubio N, Arnal-Estape A, et al. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J Clin Invest. 2012;122(5):1849–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ocana OH, Corcoles R, Fabra A, Moreno-Bueno G, Acloque H, Vega S, et al. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell. 2012;22(6):709–24. [DOI] [PubMed] [Google Scholar]
- 51.Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. 2012;22(6):725–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ruscito I, Darb-Esfahani S, Kulbe H, Bellati F, Zizzari IG, Rahimi Koshkaki H, et al. The prognostic impact of cancer stem-like cell biomarker aldehyde dehydrogenase-1 (ALDH1) in ovarian cancer: A meta-analysis. Gynecol Oncol. 2018;150(1):151–7. [DOI] [PubMed] [Google Scholar]
- 53.Ferreira-Teixeira M, Parada B, Rodrigues-Santos P, Alves V, Ramalho JS, Caramelo F, et al. Functional and molecular characterization of cancer stem-like cells in bladder cancer: a potential signature for muscle-invasive tumors. Oncotarget. 2015;6(34):36185–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu Q, Long Q, Zhu D, Fu D, Zhang B, Han L, et al. Targeting amphiregulin (AREG) derived from senescent stromal cells diminishes cancer resistance and averts programmed cell death 1 ligand (PD-L1)-mediated immunosuppression. Aging Cell. 2019;18(6):e13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carter EP, Coetzee AS, Tomas Bort E, Wang Q, Kocher HM, Grose RP. Dissecting FGF Signalling to Target Cellular Crosstalk in Pancreatic Cancer. Cells. 2021;10(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yao SB, Yin XX, Chen TT, Chen WJ, Zuo H, Bi ZR, et al. ALDH2 is a prognostic biomarker and related with immune infiltrates in HCC. Am J Cancer Res. 2021;11(11):5319-+. [PMC free article] [PubMed] [Google Scholar]
- 57.Persson E, Souza PPC, Floriano-Marcelino T, Conaway HH, Henning P, Lerner UH. Activation of Shc1 Allows Oncostatin M to Induce RANKL and Osteoclast Formation More Effectively Than Leukemia Inhibitory Factor. Front Immunol. 2019;10:1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hara T, Chanoch-Myers R, Mathewson ND, Myskiw C, Atta L, Bussema L, et al. Interactions between cancer cells and immune cells drive transitions to mesenchymal-like states in glioblastoma. Cancer Cell. 2021;39(6):779–92 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Navas C, Hernandez-Porras I, Schuhmacher AJ, Sibilia M, Guerra C, Barbacid M. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22(3):318–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.West NR, Hegazy AN, Owens BMJ, Bullers SJ, Linggi B, Buonocore S, et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat Med. 2017;23(5):579–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kun E, Tsang YTM, Ng CW, Gershenson DM, Wong KK. MEK inhibitor resistance mechanisms and recent developments in combination trials. Cancer Treat Rev. 2021;92:102137. [DOI] [PubMed] [Google Scholar]
- 62.Infante JR, Somer BG, Park JO, Li CP, Scheulen ME, Kasubhai SM, et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur J Cancer. 2014;50(12):2072–81. [DOI] [PubMed] [Google Scholar]
- 63.Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ, Settleman J. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell. 2014;26(2):207–21. [DOI] [PubMed] [Google Scholar]
- 64.Brown WS, McDonald PC, Nemirovsky O, Awrey S, Chafe SC, Schaeffer DF, et al. Overcoming Adaptive Resistance to KRAS and MEK Inhibitors by Co-targeting mTORC1/2 Complexes in Pancreatic Cancer. Cell Rep Med. 2020;1(8):100131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sale MJ, Balmanno K, Cook SJ. Resistance to ERK1/2 pathway inhibitors; sweet spots, fitness deficits and drug addiction. Cancer Drug Resist. 2019;2(2):365–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu F, Yang J, Liu J, Wang Y, Mu J, Zeng Q, et al. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct Target Ther. 2021;6(1):218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.He L, Jhong JH, Chen Q, Huang KY, Strittmatter K, Kreuzer J, et al. Global characterization of macrophage polarization mechanisms and identification of M2-type polarization inhibitors. Cell Rep. 2021;37(5):109955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6(1):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All RNA-sequencing data was generated by Case Western Reserve University Genetics and Genomics Core facility and is available upon request from corresponding author.