

Abstract
Background:
TCF3 is a transcription factor contributing to early lymphocyte differentiation. Germline monoallelic dominant negative (DN) and biallelic loss-of-function (LOF) Null TCF3 mutations cause a fully penetrant severe immunodeficiency. We identified 8 individuals from 7 unrelated families with monoallelic LOF TCF3 variants presenting with immunodeficiency with incomplete clinical penetrance.
Objective:
To define TCF3 haploinsufficiency biology and its association with immunodeficiency.
Methods:
Patient clinical data and blood samples were analyzed. Flow cytometry, western blotting, plasmablast differentiation, immunoglobulin secretion, and transcriptional activity studies were conducted on individuals carrying TCF3 variants. Mice with a heterozygous Tcf3 deletion were analyzed for lymphocyte development and phenotyping.
Results:
Individuals carrying monoallelic LOF TCF3 variants showed B cell defects (e.g., reduced total, class-switched memory and/or plasmablasts), and reduced serum immunoglobulin levels; most but not all presented with recurrent, non-severe infections. These TCF3 LOF variants were either not transcribed or translated, resulting in reduced wildtype TCF3 protein expression, strongly suggesting haploinsufficiency (HI) pathophysiology for the disease. Targeted RNAseq analysis of T cell blasts from TCF3 Null, DN, or HI individuals clustered away from healthy donors, implying that two WT copies of TCF3 are needed to sustain a tightly regulated TCF3 gene-dosage effect. Murine TCF3 HI resulted in a reduction of circulating B cells but overall normal humoral immune responses.
Conclusion:
Monoallelic LOF TCF3 mutations cause a gene-dosage-dependent reduction in wildtype protein expression, B cell defects and a dysregulated transcriptome, resulting in immunodeficiency. Tcf3+−/− mice partially recapitulate the human phenotype underscoring differences between TCF3 in humans and mice.
Keywords: primary immunodeficiency, inborn errors of immunity, E47, E12, E2A, predominantly antibody deficiency, common variable immunodeficiency, gene-dosage, hypogammaglobulinemia, haploinsufficiency
Capsule summary
Germline monoallelic loss-of-function variants manifest as a predominantly antibody deficiency with incomplete clinical penetrance and present a milder form of disease when compared to biallelic LOF and monoallelic DN TCF3 variants. The definition of TCF3 haploinsufficiency adds to the previously known body of literature describing TCF3 variants in PID/IEI and B-ALL cohorts.
Introduction
E proteins are members of the basic helix loop helix (bHLH) family of transcription factors that have essential roles in lymphocyte development.1 They bind to E box CANNTG DNA sequences as heterodimers or homodimers, exerting different transcriptional profiles that establish alternate lymphoid lineages. There are four members of the E protein family: E12, E47, E2–2 (TCF4), and HEB (TCF12). E12 and E47 are generated by alternative splicing of the C’ terminal region of the same gene, E2A (TCF3), and only differ by one exon located within the bHLH region.2 Whilst there is no difference in the DNA binding specificity between E12 and E47, E12 includes a small inhibitory region that interferes with homodimerization and DNA binding efficiency, but does not interfere with heterodimerization with E47 or other bHLH proteins.2 E47 on the other hand, efficiently binds to DNA as a homodimer, in a B lineage-restricted manner.3 Studies in mice show some redundant and some unique roles for E12 and E47 in B cell development.4
In mice, TCF3 is first expressed in developing lymphocytes in the bone marrow, with the earliest expression detected in lymphoid primed progenitors (LMPPs). Here, TCF3 aids in establishing differentiation to common lymphoid progenitors (CLPs) by upregulating lymphoid-specific genes.5 TCF3-deficient mice have no lymphocytes as a consequence of being unable to correctly express the lymphoid gene signature in LMPPs.5–7 Murine studies have shown that during B and T cell development, TCF3 also has essential roles in establishing both B and T cell identity in the bone marrow and thymus respectively. In the bone marrow, TCF3 expression remains high throughout precursor B cell development. During pro-B cell development, TCF3 activates EBF1 to initiate the B cell-specific transcriptional profile, including the up-regulation of PAX5 by EBF1.8–10 Additionally, TCF3 directly modulates RAG expression during immunoglobulin heavy and light chain gene rearrangement11 allowing for progression through both pro- and pre-B cell checkpoints.10 Once BCR check-point signaling has been cleared in the bone marrow, IgM+ cells will migrate to the spleen where they downregulate TCF3 expression10 and complete maturation through the transitional B cell stages (T1-T3). Here, it appears TCF3 expression is not essential in mice, despite it having some effects on mature follicular B cell (FoB) survival.10 In addition, reduced expression of TCF3 appears to promote marginal zone B cell (MZB) development.12 Upon T-dependent activation, TCF3-deficient B cells are able to initiate germinal centers (GC) but at significantly reduced numbers.10, 13, 14 Although E proteins have been shown to be essential for class-switch recombination by initiating Aicda expression,15 this process appears to be redundant in mice as the ability to isotype switch and produce high-affinity antibodies is maintained when TCF3 is conditionally deleted in mature B cells.10
Previously, monoallelic dominant negative (DN) and biallelic loss-of-function (LOF, Null) TCF3 variants have been associated with fully penetrant syndromic and combined immunodeficiency forms of disease, respectively.16–20 A monoallelic DN variant, E555K, that only affects E47, causes an incomplete arrest of B cell development in the bone marrow prior to the pro-B cell stage and agammaglubulinemia.16, 17, 20 The few B cells that are present in the periphery of these patients lack a BCR and have increased expression of CD19.16, 17 Biallelic LOF variants (c.808 C>T, p.Q270* and an out-of-frame exon 5–11 deletion) resulting in Null TCF3 expression also cause immune defects with the clinical phenotype consisting of severely reduced B cells and agammaglobulinemia, recurrent bacterial infections, facial dysmorphia characterized by overfolded ear helixes and prominent antihelix stems, as well as an increased risk for developing pediatric B-ALL.18, 19
Here, we present a cohort of unrelated individuals with heterozygous LOF TCF3 variants defining TCF3 haploinsufficiency at a genetic, transcriptional, protein expression, and clinical level.
Methods
Patients suspected of having a PID/IEI and their relatives were evaluated for this study (Families B, C, F, G and H). Patients known to carry disease-causing TCF3 mutations, and their relatives were also tested (Families A, D and E). All research subjects or their guardians provided written informed consent in accordance with the Declaration of Helsinki under institutional review board−approved protocols at their primary care centers, as the Alfred Health 109/15 and Monash University CF15/771-2015-0344 protocols, or the NIH/NIAID protocol 10-I-0216; ClinicalTrials.gov Identifier: NCT01222741. Peripheral blood samples from patients and their relatives were immunophenotyped and functionally evaluated. Genomic DNA was extracted, tested by whole exome sequencing, and prioritized variants were Sanger confirmed; cDNA was also sequenced, either directly after mRNA/cDNA conversion or after subcloning. PBMCs were collected and used for protein immunoblotting, as well as in-vitro plasmablast differentiation and immunoglobulin secretion. Transcriptomic activity was evaluated by RNASeq on CD4+ T cell blasts; plasmids containing different TCF3 variants were generated, and transcriptional luciferase activity was tested after transfection of HEK293T cells. C57BL6/N Tcf3+/− and Tcf3+/S513* mice were generated by CRISPR/Cas9 methodology, immunophenotyped and functionally evaluated. More detailed materials & methods can be found on the supplementary appendix.
RNAseq data may be found in the GEO repository, accession ID: GSE218787. Tcf3+/− and Tcf3+/S513* mice are available through the Australian Phenome Bank (https://pb.apf.edu.au).
Results
Clinical, immunological, and genetic characterization
Eight different TCF3 variants, including the previously reported DN E555K (Family D), were identified in thirteen individuals from eight unrelated families (Figure 1A,B). While the variants detected in Families A-G affected single nucleotides or small indels at both the N’ and C’ gene termini, the index in Family H had a large, heterozygous chromosomal deletion (Ch37/hg19 19p13.3(chr19: 959646–1649320)x1) that spanned 38 genes including exons ~3–21 of TCF3 (information on the other affected genes can be found in Supplementary Table 1). In-depth clinical summaries for each patient can be found in the supplemental data. Previously, only patients with biallelic LOF/Null and monoallelic DN (E555K) variants of TCF3 have been described.16–20 Here, we observed monoallelic LOF TCF3 mutations (n=8) associated with a milder form of a predominantly antibody deficiency presenting in children or adults with reduced total B cells, switched-memory B cells and/or plasmablasts, hypogammaglobulinemia and increased susceptibility to infections with incomplete penetrance (asymptomatic = 3/8) (Figure 1C,D; Table 1) In contrast, Null (n=4) and DN (n=1) mutations caused a more severe and broader phenotype with facial dysmorphia (Null, Supplemental Figure 1), recurrent severe infections, susceptibility to B-ALL, and reduced circulating B cells, class-switched memory B cells, and plasmablasts (Table 1). Additionally, individuals with HI mutations had normal Igκ usage whilst those with Null mutations had a skewed Igκ:Igλ ratio (Figure 1E); of note, the patient with the DN mutation did not express a BCR.
Figure 1:
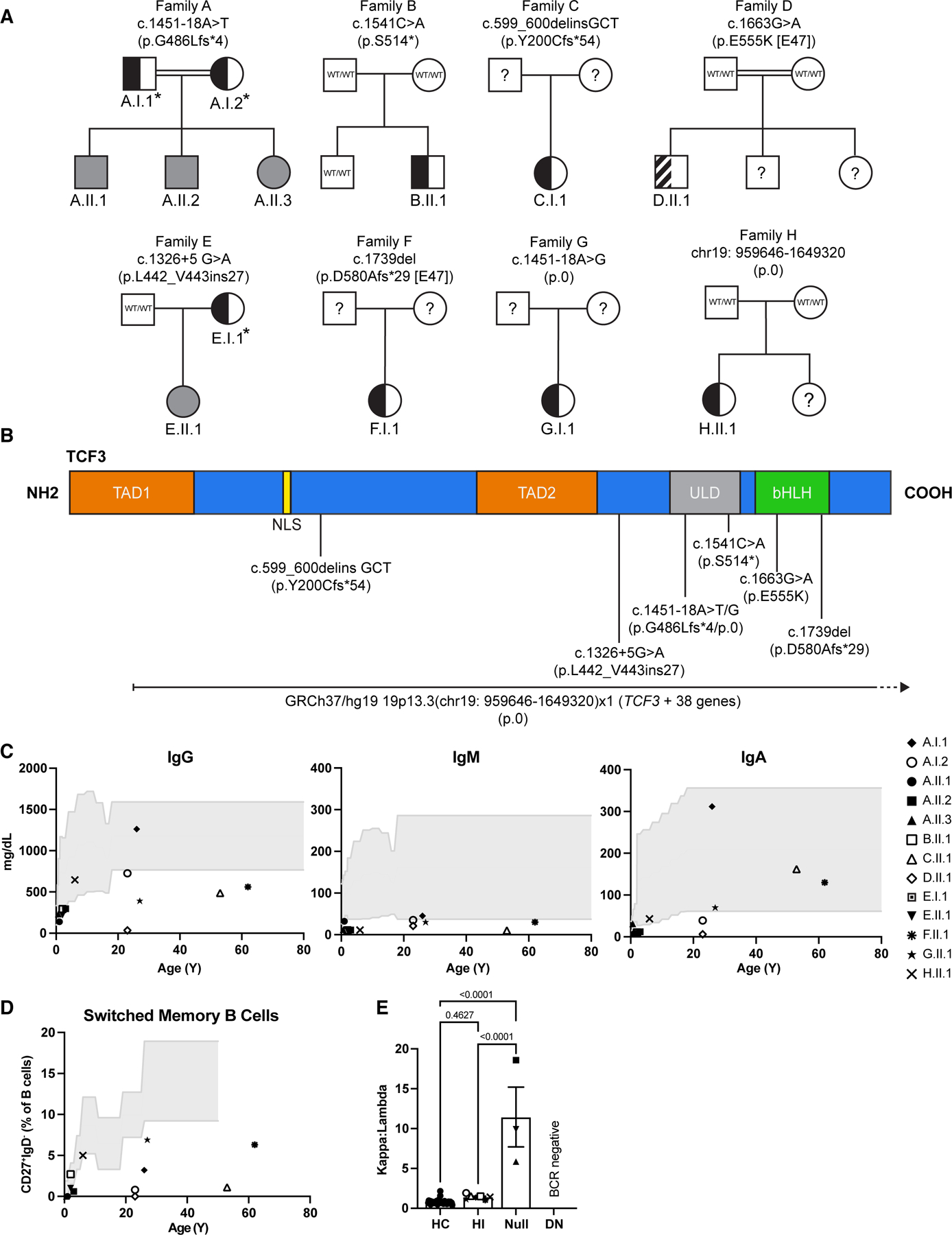
Pedigree analysis and clinical manifestations of families with TCF3 variants. (A) Pedigrees of 8 kindreds with different TCF3 variants (families A–H). Individuals with heterozygous and homozygous variants are shown in semi-shaded (black) and shaded (gray) symbols respectively, question marks indicate unscreened individual, asterisks indicate mutation positive asymptomatic individuals, the striped semi-shaded square refers to the known DN variant. (B) Schematic representation of the TCF3 protein and corresponding domains. Predicted effect of each mutation on the amino acid sequence and its location are shown. Numbers indicate cDNA and amino acid location. TAD1: transactivation domain 1; NLS: nuclear localization sequence; TAD2: transactivation domain 2; ULD: ubiquitin-like domain; bHLH: basic helix-loop-helix. (C) Immunoglobulin levels in the serum for each tested individual as denoted by unique symbols, compared with healthy control ranges over age (gray shaded area). (D) Frequency of switched memory B cells (CD27+IgD−) in the blood, determined by flow cytometry. (E) Igκ to Igλ ratio of B cells in the blood, determined by flow cytometry. Bars show the average for each group ± SEM, one-way ANOVA with Tukey correction, each patient is represented by a unique symbol. HC: healthy control, HI: haploinsufficient, DN: dominant negative.
Table 1:
Clinical and laboratory features of individuals with TCF3 variants.
| Null | HI | DN | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample ID | A.II.1 | A.II.2 | A.II.3 | E.II.1 | A.I.1 | A.I.2 | B.II.1 | C.II.1 | E.I.1 | F.I.1 | G.I.1 | H.II.1 | D.II.1 |
| Age/Sex | 6y/M | 3y/M | 8m/F | 7y/F | 26y/M | 23y/F | 10y/M | 53y/F | 32y/F | 62y/F | 27y/F | 6y/F | 23y/M |
| TCF3 variant | c.1451–18 A>T | c.1451–18 A>T | c.1451–18 A>T | c.1326+5 G>A | c.1451–18 A>T | c.1451–18 A>T | c.1541 C>A | c.599_600delinsGCT | c.1326+5 G>A | c.1739del | c.1451–18 A>G | chr19: 959646–1649320 | c.1663 G>A |
| Predicted protein product | p.G486Lfs*4 | p.G486Lfs*4 | p.G486Lfs*4 | p.L442_V443ins27 | p.G486L fs*4 | p.G486L fs*4 | p.S514* | p.Y200Cfs*54 | p.V433* | p.D580Afs*29 | p.0 | p.0 | p.E555K |
| Zygosity | Hom | Hom | Hom | Hom | Het | Het | Het | Het | Het | Het | Het | Het | Het |
| T cells | |||||||||||||
| CD3 (cells/ul) | 649 (L) | 3723 | 5398 | 4081 (H) | 2953 (H) | 615 (L) | 2744 (H) | 1007 | 2368 (H) | 1229 | 2008 | 2731 | 1138 |
| CD4 (cells/ul) | 306 (L) | 1889 | 3617 | 2154 | 1847 (H) | 369 | 1433 (H) | 721 | 1584 (H) | 904 | 1296 | 2259 | 696 |
| CD8 (cells/ul) | 241 (L) | 1574 (H) | 1430 | 1633 (H) | 937 (H) | 204 | 907 (H) | 247 | 723 | 283 | 595 | 368 (L) | 316 |
| CD3 (%) | 94.1 (H) | 80.4 (H) | 77 | 84.5 (H) | 89.2 (H) | 85.4 (H) | 82.9 (H) | 70.4 | 69.6 | 62.7 | 73.1 | 97.2 (H) | 90.3 (H) |
| CD4 (%) | 44.3 | 40.8 | 51.6 | 44.6 | 55.8 | 51.3 | 43.3 | 50.4 | 46.6 | 45.6 | 42.9 | 80.4 (H) | 55.4 |
| CD8 (%) | 34.9 (H) | 34 (H) | 20.4 | 33.8 | 28.3 | 28.3 | 27.4 | 17.3 | 21.3 | 13.2 | 24.2 | 13.1 (L) | 23.4 |
| CD4/CD8 ratio | 1.27 | 1.2 | 2.53 | 1.32 | 1.97 | 1.81 | 1.58 | 2.91 | 2.19 | 3.45 | 1.77 | 6.14 (H) | 2.2 |
| CD3+CD4−CD8− (DNT) (%) | 15.4 (H) | 6 (L) | 3.2 | 6.1 | 4.4 | 6.4 | 13.4 | 3.4 | ND | 4.6 | 6.2 | 3.2 | 12.1 |
| TCR / CD3+ (%) | 16.7 (H) | 5.6 | 5.7 | 11.7 | 3.1 | 5.4 | 13.4 (H) | ND | ND | 6 | 9.5 | 1.9 (L) | 18.9 (H) |
| CD4+CD31+ CD45RA+ (RTE) (%) | 22.7 (L) | 40.5 (L) | 62.6 | 67.5 | 22.1 | 16.6 | 54.6 | 14.2 | ND | 36.2 | 30.2 | 44.1 | 23.8 |
| CD4+CD62L+CD45RA+ (naive) (%) | 38.5 | 58.3 | 78.9 | 73.7 (H) | 46.6 | 51.1 | 57.9 | 30.3 | ND | 11.8 (L) | 2.2 (L) | 48.3 | 50.7 |
| CD4+CD62L+CD45RA− (central memory) (%) | 45.2 (H) | 33.7 (H) | 18.6 | 15.4 | 38.9 | 39.8 | 33.5 (H) | 54.6 | ND | 8.9 (L) | 2.0 (L) | 20.2 | 36.4 |
| CD4+CD62L−CD45RA− (effector memory) (%) | 15.3 | 7.5 (L) | 2.4 (L) | 9.9 | 13.7 | 8.6 | 8.2 (L) | 15 | ND | 36 (H) | 61.6 (H) | 24.1 (H) | 12.6 |
| CD4+CD62L−CD45RA+ (TEMRA) (%) | 1 (L) | 0.5 (L) | 0.2 (L) | 1 (L) | 0.8 | 0.5 | 0.4 (L) | 0.1 | ND | 43.3 (H) | 34.2 (H) | 7.4 (H) | 0.3 |
| CD8+CD62L+CD45RA+ (naive) (%) | 67.2 | 61.3 | 79.5 (H) | 42 | 46.9 | 66.5 | 52.1 | 34.8 | ND | 4.6 (L) | 3.4 (L) | 53.4 | 35.3 |
| CD8+CD62L+CD45RA− (central memory) (%) | 8.5 | 7.3 | 7.2 | 2.2 | 15.5 | 6.8 | 13.8 | 16.5 (L) | ND | 6.8 (L) | 1.1 (L) | 0.5 (L) | 8.4 |
| CD8+CD62L−CD45RA− (effector memory) (%) | 13.7 (L) | 14.6 | 4 (L) | 25 | 25.7 | 7.6 | 20.7 | 13.8 | ND | 21.7 | 23.5 | 4.6 (L) | 21.6 |
| CD8+CD62L−CD45RA+ (TEMRA) (%) | 10.7 | 16.8 | 9.4 | 30.9 (H) | 11.9 | 19.1 | 13.3 | 34.8 | ND | 66.9 (H) | 72.0 (H) | 41.4 (H) | 34.6 |
| CD4+FoxP3+CD25+ (Treg) (%) | 7.4 (H) | 3.8 (H) | ND | 4.1 (H) | 2.5 | 1.7 | 2.1 | ND | ND | 7.5 | 2.2 (L) | 5.1 (H) | 3.3 |
| CD4+CD45RA−CXCR5+ (Tfh) (%) | 3.2 (L) | 4.8 (L) | 1.8 (L) | 3.8 (L) | 5.7 | 7.4 | 9.2 | 10.2 | ND | 3.8 | 2.8 (L) | 3.9 (L) | 39.5 (H) |
| B cells | |||||||||||||
| CD19+ (cells/ul) | 0 (L) | 116 (L) | 210 (L) | 396 | 175 | 50 (L) | 136 (L) | 124 | 366 | 204 | 166 | 76 (L) | 8 (L) |
| CD19+ (%) | 0 (L) | 2.5 (L) | 3 (L) | 8.2 | 5.3 | 7 | 4.1 (L) | 8.7 | 10.8 | 5.7 | 4.8 | 2.7 (L) | 0.6 (L) |
| CD20+IgM+CD27+ (unswitched memory) (%) | 0 (L) | 3.1 (L) | 3.5 | 3.4 | 6.2 | 9.4 | 8.1 | 2.7 (L) | ND | 7 | 10.03 | 0.1 (L) | 0 (L) |
| CD20+IgM− CD27+ (class-switched memory) (%) | 0 (L) | 0.6 (L) | 0 (L) | 1 (L) | 3.2 (L) | 0.8 (L) | 2.7 (L) | 1.1 (L) | ND | 6.3 | 6.9 | 0.1 (L) | 0 (L) |
| CD19+CD24++CD38++ (transitional B cells) (%) | 0 (L) | 25.7 | 29.9 (H) | 30 (H) | 14.3 | 6.4 | 8.8 | 18.3 (H) | ND | 10.1 | 8.3 | 0.1 (L) | 12.2 |
| CD19+CD24−CD38++ (plasmablast) (%) | 0 (L) | 1.7 (L) | 0.4 (L) | 0.4 (L) | 0.4 | 0.2 (L) | 0.2 (L) | 0.3 | ND | 1.8 | 1.2 | 0.1 (L) | 0 (L) |
| CD20+IgM+ (%) | 0 (L) | 95.6 | 97.3 | 98.8 | 95.9 | 96 | 92.3 | 97.3 | ND | 80 | 74.7 | 89.3 | 4.8 |
| NK cells | |||||||||||||
| CD3−CD16+CD56+ (cells/ul) | 44 (L) | 773 (H) | 1388 (H) | 333 | 182 | 53 (L) | 444 | 297 | 645 | 400 | 314 | 14 | 115 (L) |
| CD3−CD16+CD56+ (%) | 6.4 | 16.7 | 19.8 | 6.9 | 5.5 (L) | 7.4 | 13.4 | 20.8 | 19 | 31.1 | 21.8 | 0.5 | 9.1 |
| Serum immunoglobulins (mg/dL) | |||||||||||||
| IgG | 137 (L) | 295 (L) | 230 | 752# | 1262 | 726 | 901# | 486 (L) | ND | 560 (L) | 390 (L) | 646 | 33 (L) |
| IgA | 7 (L) | 12 (L) | 31 | 27 (L) | 312 | 39 (L) | 28 (L) | 162 | ND | 130 | 70 (L) | 43 | 6 (L) |
| IgM | 32 (L) | 11 (L) | 12 (L) | 17 (L) | 45 | 35 (L) | 18.1 (L) | 10 (L) | ND | 30 (L) | 30 (L) | 11 (L) | 21 (L) |
| Lymphocyte proliferation & activation | |||||||||||||
| T cell proliferation (CD3/CD28 beads) | Normal | Normal | ND | ND | Normal | Normal | Normal | Normal | ND | Normal | Normal | ND | Normal |
| T cell activation (CD25) | Normal | Normal | ND | ND | Normal | Normal | Normal | Normal | ND | Normal | Normal | ND | Normal |
| B cell proliferation (CD40L+IL21) | No B cells | Normal | ND | ND | Normal | Normal | ND | Normal | ND | Normal | Normal | ND | No B cells |
| Clinical features | |||||||||||||
| Infections | Recurrent respiratory; pneumonias | No | No | Bacteremic pneumococcal pneumonia; EBVassociated leukocytosis with | No | No | Varicella, impetigo, oral thrush, gastroenteritis (not severe) | Recurrent and chronic sinus infection (Mycobacterium avium complex culture +, once) | No | Recurrent sinusitis, impaired vaccination response to pneumococcus | Pneumonia, bronchitis, vitiligo | Recurrent respiratory infections (RSV) | Gastroenteritis; Haemophilus influenza type B meningitis; otitis media; Rickettsia infection and pneumonia; bronchiectasis |
| Facial dysmorphism | Yes (mild) | Yes (mild) | Yes (mild) | Yes (mild) | No | No | No | No | No | No | No | No | No |
| B-ALL | Yes | No | Yes | No | No | No | No | No | No | No | No | No | No |
| Others | Mild intellectual disability | Asthma; granulomatous lung disease; obesity, type 2 diabetes, diabetes | Cholecystectomy | Vitiligo | Crohn’s, early onset IBS, Peutz-Jeghers syndrome, mild motor and intellectual developmental delay | ||||||||
T cell and B cell subsets are expressed as the percentage among CD4+ or CD8+ T cells, or CD19+ B cells respectively. L and H indicate low and high when compared to age-matched normal ranges. Normal ranges for adults were established through NIH in-house clinical testing; pediatric normal ranges were adopted from Schatorjé et al35 and Morbach et al.36
on IgG replacement therapy. HI, haploinsufficient; DN, dominant negative; Hom, homozygous; Het, heterozygous.
TCF3/E2A protein expression and gene transcription
Protein expression of TCF3 was reduced in all TCF3-mutated individuals, except for the patient with the previously described DN E555K variant (D.II.1), who had normal TCF3 expression (Figure 2A,B). Null mutations in TCF3 completely abolished TCF3 protein expression, whilst monoallelic LOF mutations showed intermediate levels of protein (Figure 2B), strongly suggesting HI of TCF3 as the mode of disease pathophysiology in these individuals. TCF3 mRNA was evaluated either by bulk cDNA or subcloning analysis from T cell blasts derived from all described variants (except for H.II.1). No mutant transcripts were detected in Family G (Supplemental Figure 2), strongly suggesting that the observed reduced protein expression was the result of mutant TCF3 mRNA decay. Although protein accumulation was markedly decreased, mutant cDNA transcripts were detected in members of Families A, B, C, E, and F (Supplemental Figure 2) suggesting that reduced stability of the mutant protein is the likely mechanism underlying this finding. As expected, mutant and WT transcripts were detected in Family D index patient (DN mutation), supporting previously reported results of stably expressed mutant transcript and protein.17 Lastly, although the mutation site identified in Families A and G was the same (c.1451–18A), the different nucleotide substitutions generated a stable mutant transcript in Family A and no mutant transcript in Family G, yet both variants resulted in reduced TCF3 protein expression.
Figure 2:
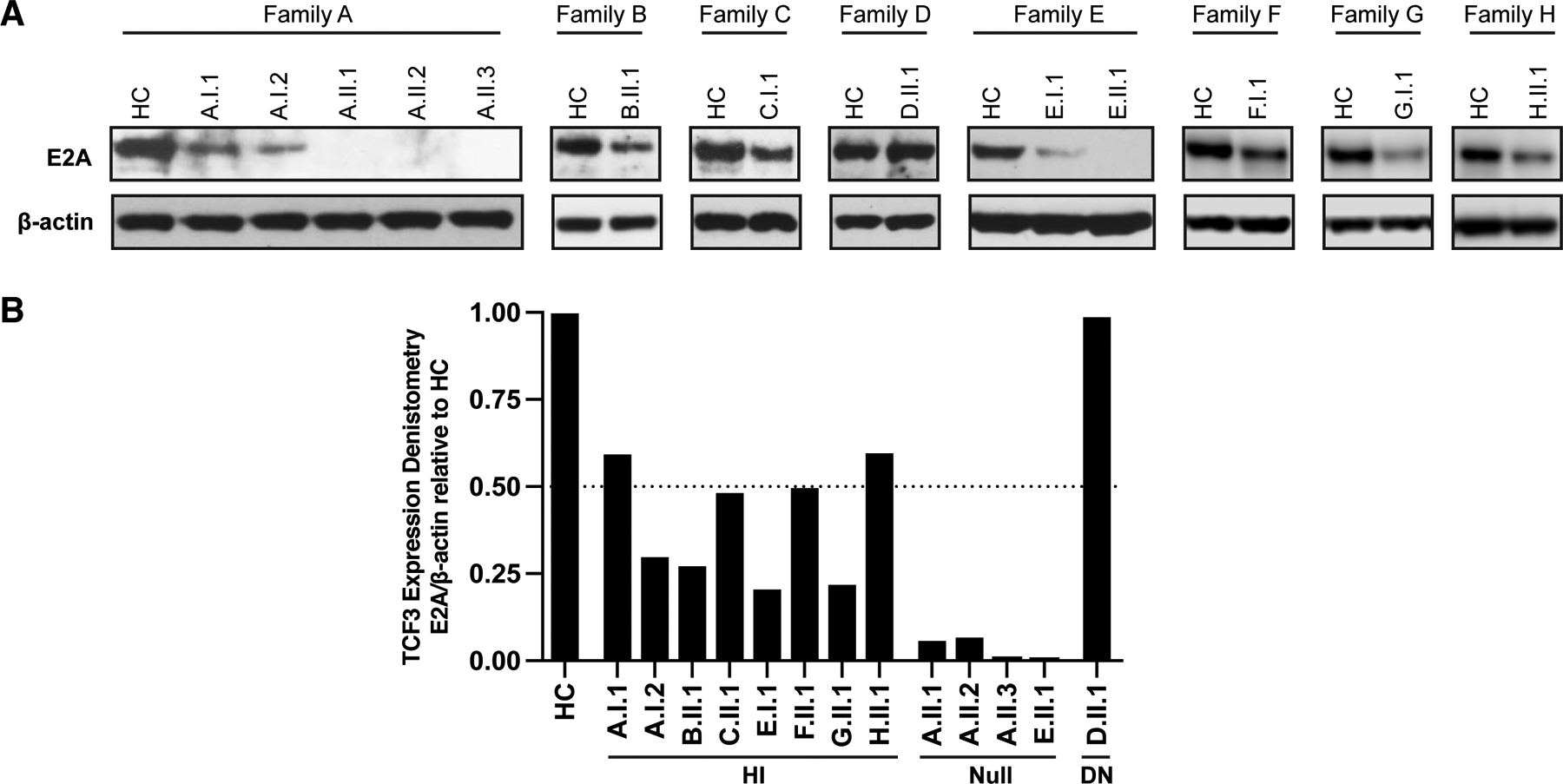
TCF3/E2A protein expression in T cell blasts. (A) TCF3/E2A protein expression from T cells blasts from healthy controls (HC) and TCF3-mutated individuals. PBMCs were stimulated with anti-CD3/anti-CD28 (1μg/ml each) plus IL-2 (20ng/ml) for 7–12 days followed by protein lysate preparation and immunoblotting with a TCF3/E2A antibody that recognizes both E12 and E47 isoforms. Data shown is representative of two independent experiments. (B) Densitometry of E2A protein expression normalized to β-actin and relative to the HC samples run on the same gel, is shown for TCF3 haploinsufficient (HI), null, and dominant negative (DN) individuals.
Functional defects of TCF3 transcript-positive mutations
Although mutant proteins were not detected in any of the 5 Families showing mutant transcripts (Families A, B, C, E and F), we still explored the potential transcriptional role of three of these predicted proteins: the intronic splice mutation leading to p.G486Lfs*4 in Family A, the exonic mutation leading to p.S514* in Family B, and the one nucleotide deletion leading to D580Afs*29 in family F and compared these to the known DN variant, E555K. Mutant constructs coding for predicted proteins p.G486Lfs*4 (Family A), p.S514* (Family B), and p.D580Afs*29 as well as the efficiently-detected DN variant E555K (Family D), were cloned into N-terminal-Flag tagged E47 vectors and tested in a luciferase assay to evaluate the regulation of the TCF3-target gene MCK, in single mutant or WT/mutant co-transfection experiments (Supplemental Figure 3). While all mutants were efficiently overexpressed in the transfection system, they lost their ability to upregulate MCK as compared to the WT vector in the single transfection experiments. When the mutant vector coding for E555K was co-transfected with the WT vector, it exerted a DN effect over the WT protein activity that was not observed when Family A, B, or F variants were co-transfected with the WT allele. Therefore, any hypothetical residual TCF3 protein expression from mutant alleles in Families A, B, or F would result in LOF with no evidence of a DN effect, further supporting HI as a mechanism of disease for these variants (Supplemental Figure 3). Whilst we were unable to clone cDNA constructs from Family E and were unable to detect mutant transcript from Families G and H, the absence of mutant protein detected in these individuals suggest that these variants create unstable transcripts also supporting an HI mechanism of disease.
Plasmablast differentiation and immunoglobulin production in TCF3-mutated individuals
Unlike Null or DN mutations, HI mutations in TCF3 do not appear to cause significant reductions in peripheral B cell numbers in most subjects. Individuals with TCF3 HI do, however, tend to have a reduction in total B cells (CD19+), switched-memory B cells (CD20+CD27+IgM−) and plasmablasts (CD19+CD24−CD38++) as also described in Null and DN individuals (Figure 1D, Table 1). B and T cell activation and proliferation in response to in vitro CpG and IgM, CD40L and IL-21, or αCD3/αCD28 stimulation were normal in all TCF3-mutated individuals (Table 1).
In vitro plasmablast differentiation, however, was impacted in almost all mutated individuals tested. Patients with Null or the monoallelic DN variants, produced virtually no plasmablasts in response to CD40L and IL-21 stimulation (Figure 3A). However, individuals with HI variants were able to produce approximately 50% of the number of plasmablasts compared to healthy controls (Figure 3A). Similarly, supernatant collected from in vitro stimulation with CD40L and IL-21 showed that biallelic TCF3 Null or DN mutations lead to virtually absent immunoglobulin production. In contrast, the heterozygous HI mutations lead to an approximate 50% reduction in immunoglobulin levels when compared to healthy controls (Figure 3B). No statistically significant differences were observed when asymptomatic and symptomatic HI individuals were treated as separate groups (data not shown) suggesting no immunological prediction as to whether two individuals carrying the same heterozygous HI variant will develop clinical symptoms. Taken together, HI variants in TCF3 halve protein expression and are associated with decreased plasmablast differentiation and immunoglobulin production compared to healthy controls, demonstrating a TCF3 gene dosage effect.
Figure 3:
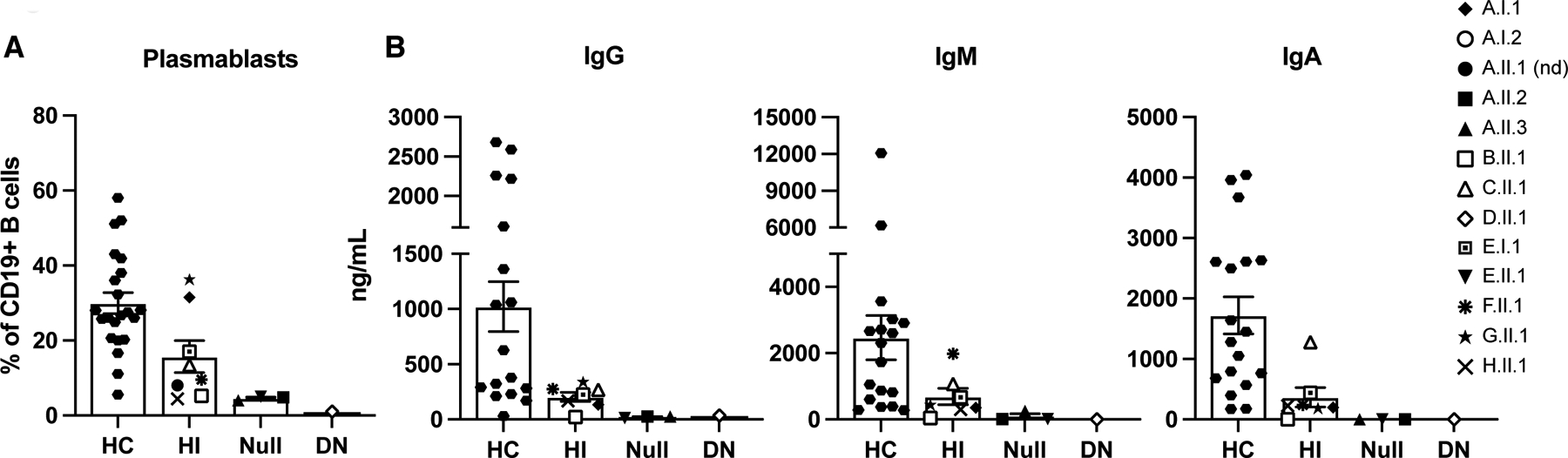
TCF3 mutations affect in vitro plasmablast differentiation and immunoglobulin production. (A and B) PBMCs were stimulated with CD40L (100 ng/ml) and IL-21(100 ng/ml) for 5 days. (A) Plasmablast frequency in healthy controls (HC), haploinsufficient (HI, monoallelic LOF), Null (biallelic LOF), and dominant negative (DN, monoallelic DN) individuals was determined by the percentage of CD27+CD38+ cells from total CD19+ B cells. (B) Concentration (ng/ml) of immunoglobulin production in the cell culture supernatant post CD40L and IL-21 stimulation determined by ELISA. (A and B) Bars show mean ± SEM of the average of two independent experiments with individuals represented by unique symbols as indicated.
Transcriptomic analysis of TCF3-deficient T cells
To understand how mutations in TCF3 confer immune dysregulation in a gene dosage-dependent manner, we compared the transcriptomic landscape from individuals carrying Null, DN, and heterozygous HI TCF3 mutations (Figure 4). As B cells were markedly reduced or virtually absent in patients with Null and DN TCF3 mutations, we studied activated CD4+ T cells, a cell lineage present and responsive to activation in all TCF3 allelic variants. When filtered down to genes that were differentially expressed across the TCF3-mutated individuals tested, all TCF3 mutation positive individuals clustered together regardless of their underlying genetic variant and separated from the healthy controls, suggesting that two WT TCF3 alleles are required for normal gene transcription (Figure 4A). Pathway analysis of differentially expressed genes showed that Null and DN individuals overexpressed genes involved in the recruitment of leukocytes, activation of cells, hypersensitivity reactions, and proliferation of immune cells, whereas the HI individuals underexpressed genes involved in those functions. In contrast, all three allelic variants similarly overexpressed genes involved in accumulation of leukocytes, invasion of cells, leukocyte migration, chemotaxis, homing of cells, hematologic cancer, lymphocyte homeostasis, and T cell development (Figure 4B). This shows that TCF3 allelic variants can regulate specific pathways in similar or different directions and, as shown in T-cell blasts, two TCF3 wildtype copies are needed to sustain normal transcriptomic activity. Moreover, transcriptomic abnormalities did not cosegregate with clinical penetrance as asymptomatic individuals (A.I.1 and A.I.2) demonstrated no unique transcriptomic patterns when compared with symptomatic HI individuals (B.II.1 and C.I.1) or Null (A.II.2) and DN (D.II.1) individuals (Figure 4A).
Figure 4:
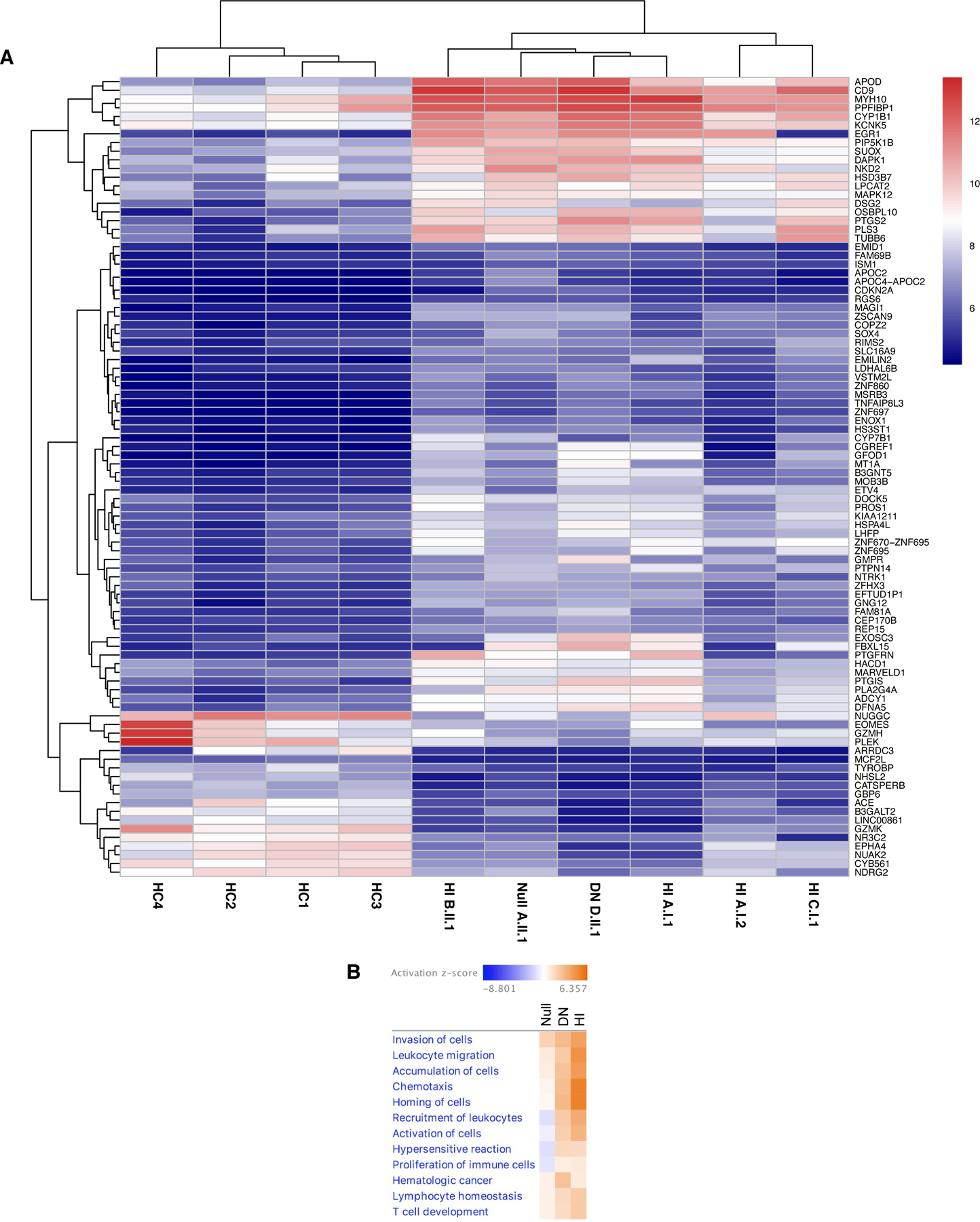
Null, DN, and HI TCF3 variants change the mRNA landscape of T cell blasts compared with healthy controls. (A) Heatmap of commonly differentially expressed genes in healthy controls (HC), haploinsufficient (HI), null, and dominant negative (DN) individuals. (B) Ingenuity Pathway Analysis comparing diseases & biofunctions in null, DN, and HI TCF3 individuals. (A-B) Healthy controls (n=4), TCF3 null (n=1; A.II.1), TCF3 HI (n=4; A.I.1, A.I.2, B.II.1 and C.II.2), TCF3 DN (n=1; D.II.1). Two technical replicates were performed for each individual.
Haploinsufficiency of TCF3 in mice
To further explore the gene dose-dependent effects of TCF3 mutations on B cell function, we used CRISPR/Cas9 to delete Tcf3 in the germline of C57 BL/6 mice. A complete absence of Tcf3 in the germline is incompatible with life, with homozygous pups dying soon after birth6, 7, 21 and as such, mice were only bred to heterozygosity (Tcf3+/−). Haploinsufficiency of TCF3 caused a significant reduction in the frequency of circulating B cells in the blood (Figure 5A). Total cell numbers in the bone marrow (BM) were unaffected, but there was a significant reduction of B cells, including precursor, immature, and mature B cells due to a partial blockage of cells progressing to the pro-B cell stage (Figure 5B). B cell frequencies in the spleen were reduced in Tcf3+/− mice, including a reduction in follicular B cells (FoB) and in immature B cells migrating from the bone marrow (Figure 5C). B cell maturation through transitional B cell stages was however, unaffected, suggesting normal B cell maturation. Switched (IgM−IgD−) B cells were normal in Tcf3+/− mice but marginal zone (MZ) B cells were slightly increased. The proportion of B cells expressing Igλ was very significantly reduced in Tcf3+/− B cells (Figure 5C) confirming previous reports that TCF3 affects Igλ locus accessibility.4, 12 Total and CD4+ T cells in the spleen were unaffected in Tcf3+/− mice. These results confirm that murine TCF3 haploinsufficiency results in a significant impact on B cell biology stemming from an inability for B cells to correctly progress to the pro-B cell stage within the BM.5, 12, 22
Figure 5:
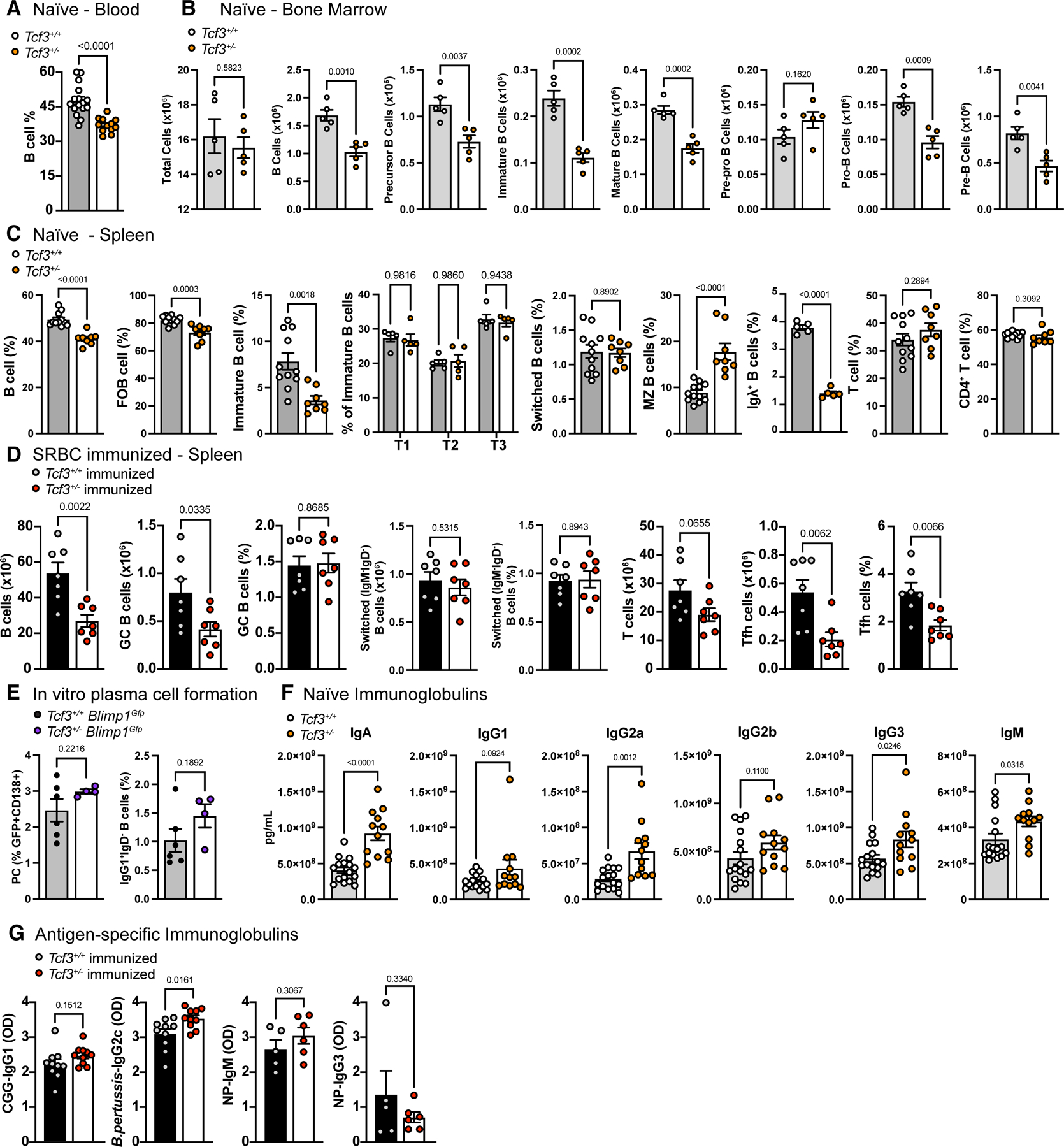
Haploinsufficiency of TCF3 in mice affects B cell development, but not the humoral immune response. (A) Frequency of total B cells (B220+) out of lymphocytes in the peripheral blood of wildtype (Tcf3+/+) and heterozygous (Tcf3+/−) mice, determined by flow cytometry. (B) Total cell number of B cell populations in the bone marrow of naïve mice, determined by flow cytometry. B cells (B220+), precursor B (B220+IgM−IgD−), immature B (B220+IgM+IgD−), mature B (B220+IgM+IgD+), pre-pro B (precursor, CD43+CD24−), pro-B (precursor, CD43+CD24low), pre-B (precursor, CD43−CD24high). (C) Frequencies of B and T cell populations in the spleen of naïve mice, determined by flow cytometry. B cells (B220+), follicular B cells (FoB; B220+CD93CD23+CD21/35low, immature B (B220+CD93+), transitional 1 (T1; immature B, IgM+CD23−), transitional 2 (T2; (immature B, IgM+CD23+), transitional 3 (T3; immature B, IgMlowCD23+), switched B cells (B220+IgM−IgD−,), marginal zone B cells (MZ; B220+CD93−CD23−CD21/35high), Igλ+ B cells (B220+Igλ+, shown as % of B cells), T cells (CD3+B220−), CD4+ T cells (T cells, CD4+). (D) Total cell number and frequencies of B and T cell populations of the spleen of sheep red blood cell (SRBC) immunized mice 8 days post immunization, determined by flow cytometry. B cells (B220+), germinal center B cells (GC; B220+GL7+CD95+, shown as total cell number and % of B cells), switched B cells (B220+IgM−IgD−, shown as total cell number and % of B cells), T cells (CD3+B220−), T follicular helper cells (Tfh; CD3+CD4+CXCR5+PD1+, shown as total cell number and % of CD4+ T cells). (E) Frequency of plasma cells (PC; CD19+CD138+GFP+, shown as % of B cells) and class-switched B cells (IgG1+IgD−, shown as % of B cells) in Tcf3 Blimp1Gfp reporter mice post in vitro stimulation with LPS, or LPS, IL-4, and IL-5 for 4 days, determined by flow cytometry. (F) Concentration of immunoglobulin isotypes in the plasma of naïve mice, determined by Mesoscale analysis. (G) Optical density (OD) of antigen-specific immunoglobulins in the plasma post immunization, determined by ELISA. Chicken gamma globulin (CGG) and B. pertussis antibodies determined 2 weeks post immunization, NP-specific antibodies determined 6 days post immunization. (A-G) Bars represent the mean of the group ± SEM with individual mice represented by colored circles. Statistics show unpaired student’s T test assuming for normal distribution and an α of 0.05. (C) Data combined from two independent experiments. (A-E) All populations pre-gated on live, single-cell, lymphocytes.
Additionally, we used CRISPR/Cas9 to mimic the S514* mutation found in Family B and created mice heterozygous for the homologous S513* mutation (Tcf3+/S513*). This showed a similar reduction in circulating B cells and a reduced frequency of Igλ+ B cells (Supplemental Figure 4) confirming the deleterious nature of a monoallelic Tcf3 nonsense mutation.
To determine the impact of TCF3 HI on the humoral immune response, the ability to form germinal centers after immunization with sheep red blood cells (SRBCs) was analyzed (Figure 5D). Surprisingly, the frequency of GC B cells was not affected in Tcf3+/− mice, yet total numbers were significantly reduced due to lower numbers of total B cells. Both the frequency and number of switched (IgM−IgD−) B cells were normal in Tcf3+/− mice. Moreover, T follicular helper cells (Tfh) were significantly reduced in both total number and frequency, despite comparable total T cell numbers.
Next, we tested the ability for Tcf3+/− B cells to differentiate into plasma cells in vitro by crossing Tcf3+/− with Blimp1Gfp reporter mice.23 This showed normal plasma cell frequencies in Tcf3+/− Blimp1Gfp mice as well as normal isotype switching, as frequencies of plasma cells (CD138+GFP+) and IgG1+IgD− B cells were comparable to wildtype following 4 days of in vitro stimulation with LPS ± IL-4 and IL-5 (Figure 5E).
Finally, to test antibody production in the context of murine TCF3 HI, we measured immunoglobulin concentrations in the plasma of naïve mice (Figure 5F). Surprisingly, IgA, IgG2a, IgG3, and IgM were significantly increased in Tcf3+/− mice compared with Tcf3+/+ mice, potentially implying a slightly increased activation state in naïve conditions. When challenged with foreign immunogens, immunized Tcf3+/− mice produced antigen-specific antibodies comparable to Tcf3+/+ mice (Figure 5G). Immunization with T-dependent chicken gamma globulin (CGG) and B. pertussis showed normal CGG-specific IgG1 and slightly increased B. pertussis-specific IgG2c in Tcf3+/− mice two weeks post immunization. Immunization with T-independent NP-Ficoll showed normal NP-specific IgM and switched NP-specific IgG3 6 days post immunization in Tcf3+/− mice. Together, this data demonstrates that murine TCF3 HI has significant effects on B cell development stemming from the bone marrow and alters B cell subsets in the periphery, but this loss of mature B cells in the periphery does not impede the humoral immune response.
Discussion
TCF3 deficiency was originally described as an autosomal dominant PID/IEI disease caused by a monoallelic DN mutation (E555K) leading to a virtual absence of B cells and agammaglobulinemia.16, 17, 20 Later, an autosomal recessive variant of the disease was described, where biallelic LOF mutations leading to Null protein expression were linked to a similar immune phenotype, but also included mild facial dysmorphisms and increased B-ALL susceptibility.18, 19 Here, we establish TCF3 HI as a new trait for immune deficiency and compare this with previously described TCF3 allelic variants. By studying TCF3-mutated patients and their relatives we determined that TCF3 HI appears to have complete immunologic but incomplete clinical penetrance, as observed in several other PID/IEI based on HI.24–26 While the reasons underlying the incomplete clinical penetrance for such diseases are not yet fully understood, factors intrinsic to the affected gene (e.g., related to particular cell lineages and thresholds for function), as well extrinsic to the gene, may likely be involved. Whilst all HI individuals described in this report had reduced WT TCF3 and no mutant protein expression, the mechanisms underlying this finding could result from either partial gene deletion, mutant mRNA decay and/or mutant protein instability. Symptomatic TCF3 HI causes a milder immunologic and clinical phenotype than the other allelic variants, with symptomatic individuals presenting as children or adults with hypogammaglobulinemia, reduced total B cells, switched-memory B cells and/or plasmablasts, as well as recurrent, although not opportunistic or life threatening, infections. Of note, while no malignancies were reported in our TCF3 HI cohort, germline heterozygous LOF variants were detected in 12/4,183 (0.29%) pediatric B-ALL patients.27 This finding not only suggests that TCF3 HI can include hematologic malignancies as part of its phenotype, but also reinforces TCF3 HI as a symptomatic allelic variant and should be considered when determining risk of B-ALL during follow-up evaluation.
Studying individuals with TCF3 HI together with those carrying DN or Null genotypes and phenotypes allowed us to delineate the role of TCF3 in human B cell biology including a gene-dosage perspective. Besides the reduction of total B cells, switched-memory B cells and plasmablasts associated with all three allelic variants (although not in all TCF3 LOF monoallelic mutation carriers), TCF3 HI causes an intermediate reduction in in vitro plasmablast generation and serum immunoglobulin secretion without affecting B cell activation or proliferation when compared to healthy controls (Figure 1C–E, Table 1). In addition to its known role in B cell development in humans, TCF3 is also upregulated during plasma cell differentiation.28 It is possible that reduced TCF3 affects the expression of the transcription factor network required for induction of the plasma cell program. Moreover, other TCF3-extrinsic factors such as epigenetic methylation during terminal differentiation of human B cells into plasma cells, also play a role in this function and may even affect penetrance of the disease.29
Whilst the immune phenotype of the individual with the chr19 microdeletion that involves 38 genes including TCF3 exons ~3–21 fits the immune parameters described in the other TCF3 HI individuals, it also includes additional clinical features (e.g., developmental delay, Crohn’s-like disease, and Peutz-Jeghers Syndrome) that may depend on other genes compromised in this contiguous gene disorder. In this setting, we can neither absolutely nor formally exclude the potential contributions of some other genes in this micro-deleted region to the immune phenotype of this patient (Supplementary Table 2). Altogether, TCF3 variants resulting in no mutant protein expression/reduced WT protein expression either by partial gene deletion, mRNA decay, or protein instability strongly support TCF3 HI as the mechanism of disease in the reported individuals.
Heterozygous LOF mutations resulting in TCF3 HI appear to affect immunoglobulin production in humans more than in mice, as Tcf3+/− mice had normal frequencies of GCBs, in vitro plasmablast differentiation and immunoglobulin, and specific response to antigen stimulation (Figure 5D–G). Whilst B cell development does not appear to be consistently affected by HI mutations in humans, a mild but significant reduction in B cells was observed both in our Tcf3+/− mice and those previously described by other groups.10, 12, 22, 30, 31 Additionally, the ability to mount a normal humoral response was observed in the murine model (Figure 5D–G) despite a lack of normal immunoglobulin production in the human HI cohort.
Secondary Igλ rearrangements are sensitive to TCF3 dosage in mice,12 but this does not appear to be the case in humans as those with TCF3 HI had normal usage of the Igλ light chain. Individuals with DN or null variants however, had skewed κ:λ ratios corroborating murine studies that TCF3 is essential for chromatin accessibility at the Igλ locus.4, 12
The lethality observed in Tcf3−/− neonatal mice6, 7, 21 is also not reflected in human cohorts as evidenced by TCF3 Null patients in ours and others’ previous studies.18, 19 The loss of TCF3 therefore has markedly dissimilar impacts on neonatal survival in mice compared with humans, reinforcing TCF3 human/murine differences with human B cells being more sensitive to TCF3 dosage than murine B cells.
Interestingly, two recent papers explored heterozygous TCF3 mutations that caused B cell defects and reduced antibody production.32, 33 In one publication, two family members carrying a heterozygous variant in TCF3 were diagnosed with CVID.32 Whilst one of the family members also carried a heterozygous C104R variant in TACI, the other only carried the TCF3 T168fs*191 variant. This mutation resulted in nonsense mediated decay with reduced WT protein expression, suggesting HI of TCF3. TCF3 T168fs*191 was also associated with lower IgG production in vitro, normal B cell proliferation and a normal ability for cells to undergo isotype switching. In another report, a patient with a history of recurrent infections, low IgG, slightly increased B cells, and normal numbers of T cells, as well as progressive multifocal leukoencephalopathy, was diagnosed with combined immune deficiency and was reported to carry a heterozygous TCF3 variant (c.807 C>G, p.H269Q). In this case, TCF3 HI was suspected to be the underlying cause of the disease, although no functional tests were conducted.33
In summary, TCF3 HI is an autosomal dominant genetic trait presenting with complete immunologic but incomplete clinical penetrance that underlies a predominantly antibody deficiency.34 Exploring TCF3 HI in a murine model allowed us to corroborate its impact on B cell development and function, although with substantial differences compared to the human disease. TCF3 HI will likely contribute to elucidate underlying genetic diagnoses and segregation in PID/IEI patients.
Supplementary Material
Clinical Implications.
Germline monoallelic loss-of-function variants in TCF3 should be considered as a genetic diagnosis underlying predominantly antibody deficiency in PID/IEI patients.
Acknowledgements
We thank the patients and their families for their contributions to the study. MAW and SMH want to acknowledge the National Institute of Allergy and Infectious Diseases (NIAID) Centralized Sequencing Program (CSP) including Morgan Similuk, Jia Yan, Rajarshi Ghosh, Bryce Seifert, Michael Setzer, Michael Kamen, Colleen Jodarski, Kathleen Jevtich, Yunting Yu, and Rylee Duncan, the Department of Laboratory Medicine (DLM) staff members including Adrienne Borges, and Josephine Geh, NIAID Collaborative Bioinformatics Resource (NCBR) and Frederick National Laboratory for Cancer Research (FNLCR) including Justin B. Lack and Vasudev Kuram and the Genomic Research Integration System (GRIS) team including Sandhya Xirasagar, Jason Barnett, Xi Cheng, Yongjie Fan, Ke Huang, Krishnaveni Kaladi, Eric Karlins, Zhiwen Li, Joseph Mackey, Andrew Oler, Daniel Veltri, Lingwen Zhang, Satishkumar Ranganathan, Nikita Vlassenko, Smilee Samuel, and Robert Gilmore, Office of Cyber Infrastructure and Computational Biology, NIAID, NIH, Rockville, Maryland. The content of this article does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government.
AE would like to acknowledge the Phenogenomics Translational Initiative and Australian Phenomics for support of the Gene Targeting Facility (Ms. Rachael Milne, Dr. Harish Padmanabhan and Mr. Neel Thakkar) at the Australian Phenomics Facility. This project was supported by technical assistance from staff (Koula Diamand, Ainsely Davies, Fei-Ju, and Kristy Kwong) of the Phenomics Translation Initiative. We thank Zhijia Yu and Fiona Ballard for their technical assistance with the mouse experiments. MCvZ and EE would like to acknowledge the ARAflowcore at Monash University.
Declaration of Funding:
These studies were supported by the National Institutes of Health (NIH) Intramural Research Program, NIH Clinical Center and NIAID (SDR and HSK). Also by: the Jeffrey Modell Foundation (MCvZ) and the Australian National Medical and Health Research Council (NHMRC; Senior Research Fellowship 1117687 to MCvZ); the Genomic Research Integration System (GRIS) system was developed in part with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services under BCBB Support Services Contract HHSN316201300006W/HHSN27200002 to MSC, Inc.; the Phenomics Translation Initiative, an MRFF funded program (EPCD000035); the ISCIII through the project (FIS PI21/01642) to LIG and co-funded by the European Union.
Abbreviations
- B-ALL
B cell acute lymphoblastic leukemia
- BCR
B cell receptor
- bHLH
basic helix loop helix
- BM
bone marrow
- CGG
chicken gamma globulin
- CLP
common lymphoid progenitor
- CVID
common variable immunodeficiency
- DN
dominant negative
- ELISA
enzyme linked immunosorbent assay
- FACS
fluorescence activated cell sorting
- FBS
fetal bovine serum
- FCS
fetal calf serum
- FoB
follicular B cell
- GC
germinal center
- HI
haploinsufficiency
- IEI
inborn errors of immunity
- LMPP
lymphoid primed progenitor
- LN
lymph nodes
- LOF
loss-offunction
- LPS
lipopolysaccharide
- MZB
marginal zone B cell
- NK
natural killer cell
- PBMC
peripheral blood mononuclear cells
- PID
primary immune deficiency
- PSG
penicillinstreptomycin-glutamine
- SRBC
sheep red blood cells
- T1
transitional 1
- T2
transitional 2
- T3
transitional 3
- Tfh
T follicular helper cells
- WES
whole exome sequencing
- WT
wildtype
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
References
- 1.Massari ME, Murre C. Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol. 2000;20(2):429–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sun XH, Baltimore D. An inhibitory domain of E12 transcription factor prevents DNA binding in E12 homodimers but not in E12 heterodimers. Cell. 1991;64(2):459–70. [DOI] [PubMed] [Google Scholar]
- 3.Shen CP, Kadesch T. B-cell-specific DNA binding by an E47 homodimer. Mol Cell Biol. 1995;15(8):4518–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beck K, Peak MM, Ota T, Nemazee D, Murre C. Distinct roles for E12 and E47 in B cell specification and the sequential rearrangement of immunoglobulin light chain loci. J Exp Med. 2009;206(10):2271–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dias S, Månsson R, Gurbuxani S, Sigvardsson M, Kee BL. E2A proteins promote development of lymphoid-primed multipotent progenitors. Immunity. 2008;29(2):217–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhuang Y, Soriano P, Weintraub H. The helix-loop-helix gene E2A is required for B cell formation. Cell. 1994;79(5):875–84. [DOI] [PubMed] [Google Scholar]
- 7.Bain G, Maandag EC, Izon DJ, Amsen D, Kruisbeek AM, Weintraub BC, et al. E2A proteins are required for proper B cell development and initiation of immunoglobulin gene rearrangements. Cell. 1994;79(5):885–92. [DOI] [PubMed] [Google Scholar]
- 8.Seet CS, Brumbaugh RL, Kee BL. Early B cell factor promotes B lymphopoiesis with reduced interleukin 7 responsiveness in the absence of E2A. J Exp Med. 2004;199(12):1689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lazorchak AS, Wojciechowski J, Dai M, Zhuang Y. E2A promotes the survival of precursor and mature B lymphocytes. J Immunol. 2006;177(4):2495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kwon K, Hutter C, Sun Q, Bilic I, Cobaleda C, Malin S, et al. Instructive Role of the Transcription Factor E2A in Early B Lymphopoiesis and Germinal Center B Cell Development. Immunity. 2008;28(6):751–62. [DOI] [PubMed] [Google Scholar]
- 11.Miyazaki K, Watanabe H, Yoshikawa G, Chen K, Hidaka R, Aitani Y, et al. The transcription factor E2A activates multiple enhancers that drive Rag expression in developing T and B cells. Sci Immunol. 2020;5(51). [DOI] [PubMed] [Google Scholar]
- 12.Quong MW, Martensson A, Langerak AW, Rivera RR, Nemazee D, Murre C. Receptor editing and marginal zone B cell development are regulated by the helix-loop-helix protein, E2A. J Exp Med. 2004;199(8):1101–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gloury R, Zotos D, Zuidscherwoude M, Masson F, Liao Y, Hasbold J, et al. Dynamic changes in Id3 and E-protein activity orchestrate germinal center and plasma cell development. J Exp Med. 2016;213(6):1095–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wöhner M, Tagoh H, Bilic I, Jaritz M, Poliakova DK, Fischer M, et al. Molecular functions of the transcription factors E2A and E2–2 in controlling germinal center B cell and plasma cell development. J Exp Med. 2016;213(7):1201–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sayegh CE, Quong MW, Agata Y, Murre C. E-proteins directly regulate expression of activation-induced deaminase in mature B cells. Nat Immunol. 2003;4(6):586–93. [DOI] [PubMed] [Google Scholar]
- 16.Dobbs AK, Bosompem A, Coustan-Smith E, Tyerman G, Saulsbury FT, Conley ME. Agammaglobulinemia associated with BCR− B cells and enhanced expression of CD19. Blood. 2011;118(7):1828–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boisson B, Wang Y-D, Bosompem A, Ma CS, Lim A, Kochetkov T, et al. A recurrent dominant negative E47 mutation causes agammaglobulinemia and BCR–B cells. The Journal of clinical investigation. 2013;123(11):4781–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ben-Ali M, Yang J, Chan KW, Ben-Mustapha I, Mekki N, Benabdesselem C, et al. Homozygous transcription factor 3 gene (TCF3) mutation is associated with severe hypogammaglobulinemia and B-cell acute lymphoblastic leukemia. Journal of Allergy and Clinical Immunology. 2017;140(4):1191–4. e4. [DOI] [PubMed] [Google Scholar]
- 19.Qureshi S, Sheikh MDA, Qamar FN. Autosomal Recessive Agammaglobulinemia-first case with a novel TCF3 mutation from Pakistan. Clinical Immunology. 2019;198:100–1. [DOI] [PubMed] [Google Scholar]
- 20.Al Sheikh E, Arkwright PD, Herwadkar A, Hussell T, Briggs TA. TCF3 Dominant Negative Variant Causes an Early Block in B-Lymphopoiesis and Agammaglobulinemia. J Clin Immunol. 2021;41(6):1391–4. [DOI] [PubMed] [Google Scholar]
- 21.Bain G, Robanus Maandag EC, te Riele HP, Feeney AJ, Sheehy A, Schlissel M, et al. Both E12 and E47 allow commitment to the B cell lineage. Immunity. 1997;6(2):145–54. [DOI] [PubMed] [Google Scholar]
- 22.Herblot S, Aplan PD, Hoang T. Gradient of E2A activity in B-cell development. Mol Cell Biol. 2002;22(3):886–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kallies A, Hasbold J, Tarlinton DM, Dietrich W, Corcoran LM, Hodgkin PD, et al. Plasma cell ontogeny defined by quantitative changes in blimp-1 expression. The Journal of experimental medicine. 2004;200(8):967–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuehn HS, Boisson B, Cunningham-Rundles C, Reichenbach J, Stray-Pedersen A, Gelfand EW, et al. Loss of B Cells in Patients with Heterozygous Mutations in IKAROS. N Engl J Med. 2016;374(11):1032–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tuijnenburg P, Lango Allen H, Burns SO, Greene D, Jansen MH, Staples E, et al. Loss-of-function nuclear factor κB subunit 1 (NFKB1) variants are the most common monogenic cause of common variable immunodeficiency in Europeans. J Allergy Clin Immunol. 2018;142(4):1285–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klemann C, Camacho-Ordonez N, Yang L, Eskandarian Z, Rojas-Restrepo JL, Frede N, et al. Clinical and Immunological Phenotype of Patients With Primary Immunodeficiency Due to Damaging Mutations in NFKB2. Front Immunol. 2019;10:297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Escherich CS, Chen W, Miyamoto S, Namikawa Y, Yang W, Teachey DT, et al. Identification of TCF3 germline variants in pediatric B-cell acute lymphoblastic leukemia. Blood Advances. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kassambara A, Herviou L, Ovejero S, Jourdan M, Thibaut C, Vikova V, et al. RNAsequencing data-driven dissection of human plasma cell differentiation reveals new potential transcription regulators. Leukemia. 2021;35(5):1451–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caron G, Hussein M, Kulis M, Delaloy C, Chatonnet F, Pignarre A, et al. Cell-Cycle-Dependent Reconfiguration of the DNA Methylome during Terminal Differentiation of Human B Cells into Plasma Cells. Cell Reports. 2015;13(5):1059–71. [DOI] [PubMed] [Google Scholar]
- 30.O’Riordan M, Grosschedl R. Coordinate regulation of B cell differentiation by the transcription factors EBF and E2A. Immunity. 1999;11(1):21–31. [DOI] [PubMed] [Google Scholar]
- 31.Bradney C, Hjelmeland M, Komatsu Y, Yoshida M, Yao TP, Zhuang Y. Regulation of E2A activities by histone acetyltransferases in B lymphocyte development. J Biol Chem. 2003;278(4):2370–6. [DOI] [PubMed] [Google Scholar]
- 32.Ameratunga R, Koopmans W, Woon ST, Leung E, Lehnert K, Slade CA, et al. Epistatic interactions between mutations of TACI (TNFRSF13B) and TCF3 result in a severe primary immunodeficiency disorder and systemic lupus erythematosus. Clinical & translational immunology. 2017;6(10):e159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Q, Tang C, Zhu J, Zhang L. A case of progressive multifocal leukoencephalopathy with hypogammaglobulinemia and a TCF3 mutation. J Neurovirol. 2022. [DOI] [PubMed] [Google Scholar]
- 34.Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J Clin Immunol. 2020;40(1):66–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schatorjé EJ, Gemen EF, Driessen GJ, Leuvenink J, van Hout RW, de Vries E. Paediatric reference values for the peripheral T cell compartment. Scand J Immunol. 2012;75(4):436–44. [DOI] [PubMed] [Google Scholar]
- 36.Morbach H, Eichhorn EM, Liese JG, Girschick HJ. Reference values for B cell subpopulations from infancy to adulthood. Clin Exp Immunol. 2010;162(2):271–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.