

Abstract
Background:
ZFHX3, a gene that encodes a large transcription factor, is at the second-most significantly associated locus with AF, but its function in the heart is unknown. This study aims to identify causative genetic variation related to AF at the ZFHX3 locus and examine the impact of Zfhx3 loss on cardiac function in mice.
Methods:
CRISPR-Cas9 genome editing, chromatin immunoprecipitation, and luciferase assays in pluripotent stem cell-derived cardiomyocytes were used to identify causative genetic variation related to AF at the ZFHX3 locus. Cardiac function was assessed by echocardiography, MRI, electrophysiology studies, calcium imaging, and RNA sequencing in mice with heterozygous and homozygous cardiomyocyte-restricted Zfhx3 deletion (Zfhx3 Het and KO, respectively). Human cardiac single-nucleus ATAC-sequencing data was analyzed to determine which genes in atrial cardiomyocytes are directly regulated by ZFHX3.
Results:
We found SNP rs12931021 modulates an enhancer regulating ZFHX3 expression, and the AF risk allele is associated with decreased ZFHX3 transcription. We observed a gene-dose response in AF susceptibility with Zfhx3 KO mice having higher incidence, frequency, and burden of AF than Zfhx3 Het and WT mice, with alterations in conduction velocity, atrial action potential duration, calcium handling and the development of atrial enlargement and thrombus, and dilated cardiomyopathy. Zfhx3 loss results in atrial-specific differential effects on genes and signaling pathways involved in cardiac pathophysiology and AF.
Conclusions:
Our findings implicate ZFHX3 as the causative gene at the 16q22 locus for AF, and cardiac abnormalities caused by loss of cardiac Zfhx3 are due to atrial-specific dysregulation of pathways involved in AF-susceptibility. Together, these data reveal a novel and important role for Zfhx3 in the control of cardiac genes and signaling pathways essential for normal atrial function.
Keywords: Atrial fibrillation, gene expression, electrophysiology, transcription factors
Introduction
Atrial fibrillation (AF) is the most common sustained arrhythmia and is associated with significant morbidity1. Various risk factors for AF have been identified including age, sex, cardiovascular disease, obesity, and family history2. In the last decade, large scale studies in population genetics have identified more than 100 loci associated with AF3. However, the mechanisms by which genetic variation at these loci leads to AF remains largely unknown. For example, the second-most significantly associated locus with AF was identified over a decade ago near the gene ZFHX33–5, yet little is known about the role of this gene in cardiac function or AF development.
Located on human chromosome 16q22, ZFHX3 encodes AT motif binding factor 1 (ATBF1), a large transcription factor with various zinc finger motifs and homeodomains6. Studies have indicated that ZFHX3 functions as both a promoter and suppressor of carcinogenesis and tumor formation in multiple cancers. In breast cancer, ZFHX3 was shown to promote tumor growth through the upregulation of transcription factors such as TBX3, but it has also been shown to bind the AT-rich enhancer regions of alpha-fetoprotein and aminopeptidase-N, as well as the oncoprotein Myb, leading to inhibition of transcriptional activity7–10. Additionally, ZFHX3 has been shown to play a role in regulating myogenic and neuronal differentiation11,12, and is among the genes least tolerant of loss of function variation, implying a critical role in physiological homeostasis13. In vitro experiments performed in murine atrial myocytes have indicated an important role for Zfhx3 in the pathogenesis of AF through altered calcium homeostasis14,15. Moreover, it was recently shown that Zfhx3 plays a role in mitochondrial dysfunction induced with tachypacing in mouse atrial cardiomyocytes16. However, the effects that cardiac ZFHX3 loss may have on the pathophysiology of AF in vivo are not known.
We therefore sought to identify the causative genetic variation related to AF at this locus and then examined the impact of Zfhx3 loss on cardiac structure and function in a mouse knockout (KO) model. Our results reveal an important role for Zfhx3 in cardiac function and through regulation of genes essential for normal atrial function.
Methods
Additional methods are available in Supplemental Material provided. The RNA sequencing data have been uploaded to the Gene Expression Omnibus (GEO) database with free accessibility (GSE229525). The data that support the findings of this study are available from the corresponding author upon reasonable request.
Animal Protocols
All animal studies were approved by the Institutional Animal Care and Use Committee at Massachusetts General Hospital and were in compliance with relevant ethical regulations.
Statistical analyses
Statistical data analysis is included in every figure and described in detail on the respective figure legends. The exact P values and number of cells analyzed per condition (n) are stated in the figures, figure legends, and in Data Set S5. All experimentation and data analyses were conducted blinded to genotype. Animal group sizes were as low as possible and determined based on previous experience. Results are presented as mean values ± s.e.m. All statistical analyses were performed with GraphPad Prism 9 software. A P-value of <0.05 was considered statistically significant. Data were tested for normality using the Shapiro-Wilk test when applicable. For two group comparisons, unpaired two-tailed Student’s t test was used. Multiple group comparisons were performed by one-way or two-way ANOVA with Tukey’s multiple comparisons test (unless otherwise noted) when the data exhibited a normal distribution. For data sets that did not follow a normal distribution or when sample size was smaller than 6, either Mann-Whitney non-parametric analysis (two-group analysis) was performed, or the normal distributions of the variables were externally supported by our previous work17. Multiple testing corrections were performed independently within each hypothesis. Representative images were selected to represent the means of the quantified data.
Results
rs12931021 is a functional genetic variant that modulates expression of ZFHX3
We began by linking the genetic variant(s) associated with AF risk to altered expression of a gene in the region. The sentinel AF SNP at the locus, rs2106261, is located between the first and second exon of ZFHX3. The AF locus, defined as SNPs with linkage disequilibrium r2 > 0.3 to rs2106261 (in population of European ancestry from the 1000 Genomes Project data) spans a ~74 kb region and contains a total of 52 common variants with a minor allele frequency greater than 1% in individuals of European descent. We did not observe any expression quantitative trait loci (eQTL) for this variant in the heart tissues from the Genotype Tissue Expression (GTEx) database18 nor in our prior data eQTL data from human left atrium3.
We narrowed down our search for potential functional variants to those within open chromatin regions by utilizing the digital DNaseI hypersensitivity (DHS) dataset from the Encyclopedia of DNA Elements (ENCODE) project19. Specifically, we intersected the 52 AF-associated SNPs on 16q22 with the DHS signal in human cardiac myocytes. Six candidate SNPs (Table S1) showed colocalization with the DHS signal and overlapped with active chromatin marks as shown by the 18-state model of the Roadmap Epigenomics consortium (Figure 1a)20. We then assessed the effect of candidate SNPs on gene expression by cloning 500–1000 bp regions surrounding each candidate SNP (risk and non-risk alleles) upstream of a minimal promoter driving the firefly luciferase gene. Enhancer activity between the two alleles was compared in human pluripotent stem cell (PSC)-derived cardiomyocytes (PSC-CMs). Out of the six SNPs, only rs12931021 exhibited differential activity dependent on genotype, with the non-risk C allele being 3.9-fold more active than the risk A allele, although the overall luciferase signal was modest (Figure 1b). On the other hand, rs12596810 displayed the highest luciferase signal, suggesting the presence of a strong enhancer which is also evolutionarily conserved. However, no differential allelic activity was observed for rs12596810 (Figure 1b). To confirm the allele-specific regulatory effects of rs12931021, we performed chromatin immunoprecipitation (ChIP) in PSC-CMs heterozygous at rs12931021. Using antibodies against H3K4me1 and H3K27ac, we then quantified the enrichment of each allele by qPCR. While no enrichment for H3K27ac was detected in either allele, a greater enrichment of H3K4me1 at the C allele was observed compared to the A allele (21.7 ± 0.6 versus 15.4 ± 1.2; Figure 1c), verifying the genotype-dependent regulatory activity at rs12931021. We then explored whether the presence of the rs12931021-containing regulatory element effects the expression of ZFHX3. Using CRISPR-Cas9, we deleted a 219 bp region harboring rs12931021 in PSC-CMs (Figure S1a) and measured the ZFHX3 level by qPCR. The PSC-CMs with the deletion of rs12931021 expressed a significantly lower level of ZFHX3, compared to the wild-type PSC-CMs (0.85 ± 0.02 versus 1.01 ± 0.02; Figure 1d).
Figure 1. Identification of a functional variant at the ZFHX3 locus on chromosome 16q22.
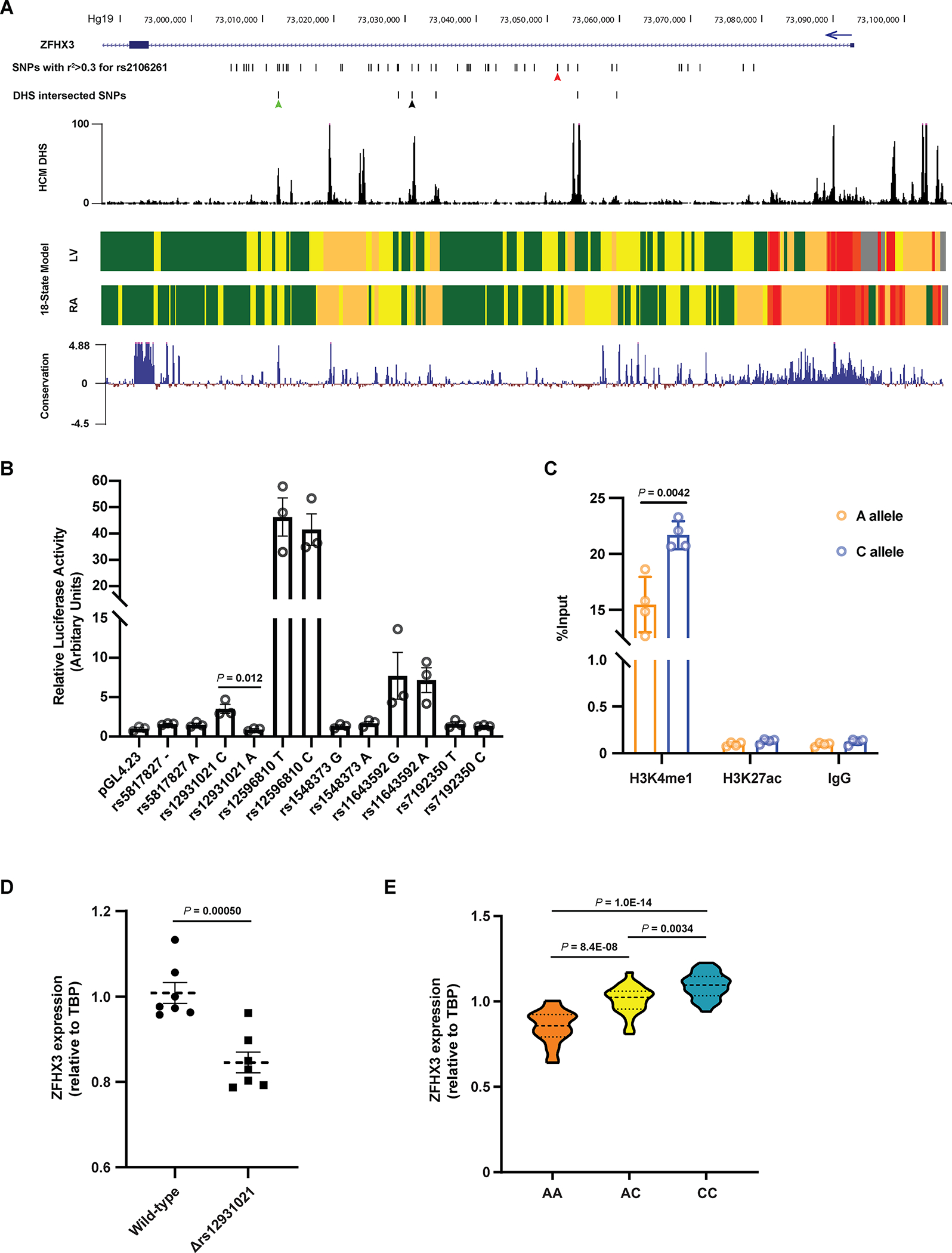
A. The region encompassing all common AF-associated SNPs with r2 > 0.3 with respect to the sentinel SNP rs2106261. The six candidate SNPs intersecting DNaseI hypersensitivity (DHS) signal are shown. The DHS signal in human cardiac myocytes and the mammalian conservation (Phylop) around this region were obtained from ENCODE. The 18-state models from Roadmap Epigenomics marking various regulatory elements in human left ventricles (LV) and right atria (RA) show regions of transcription, weak enhancer, active enhancer, transcription start site and repressed polycomb as green, yellow, orange, red and grey blocks, respectively. The red, green and black arrows indicate the position of rs2106261, rs12596810 and rs12931021, respectively. B. Luciferase data showing allele-specific activities for the six candidate SNPs in PSC-CMs. n = 3. C. Allele-specific ChIP-qPCR results in PSC-CMs heterozygous at rs12931021. The pulldown of H3K4me1 and H3K27ac were conducted to evaluate the enrichment of chromatin fragments containing either allele of rs12931021. n = 3. D. qPCR showing that deletion of rs12931021-containing regulatory element reduces the expression of ZFHX3 in PSC-CMs. n = 7. E. Relative expression of ZFHX3 in isogenic PSC-CMs carrying AA, AC or CC genotype at rs12931021. n = 24. P values are indicated (B-E). Data are mean ± s.e.m. Groups were compared using unpaired t tests (B-D). Groups were compared using ordinary one-way ANOVA with Tukey’s multiple comparisons test (E).
To examine the relationship more precisely between rs12931021 genotype and ZFHX3 expression, we generated two isogenic CRISPR PSC lines with the AA and CC genotype at rs12931021, respectively, from the wild-type heterozygous (AC) PSCs (Figure S1b). We differentiated these isogenic PSC lines to PSC-CMs and evaluated the effect of rs12931021 genotype on ZFHX3 expression. As a result, we observed a dose relationship between non-risk C allele number and greater ZFHX3 expression (Figure 1e). Taken together, these data support ZFHX3 as the causal gene at the 16q22 locus for AF association and suggests that rs12931021 is a functional SNP mediating the genetic association with increased AF risk correlated to reduced ZFHX3 expression.
Cardiac-restricted loss of Zfhx3 increases arrhythmia susceptibility
The cardiovascular effects of Zfhx3 loss in vivo were examined using mice with cardiomyocyte-restricted heterozygous or homozygous knockout of Zfhx3 (denoted Zfhx3 Het or Zfhx3 KO, respectively) created by crossbreeding heterozygous or homozygous floxed Zfhx3 (Zfhx3fl/+or Zfhx3fl/fl, respectively) mice with Zfhx3 heterozygous or homozygous KO expressing α-myosin heavy chain-cre (Myh6-cre- Zfhx3fl/+ or Myh6-cre-Zfhx3fl/fl, respectively). Age-matched Zfhx3fl/fl littermates were used as controls (denoted WT). Organ specific genotyping confirmed that deletion of the critical exons was restricted to cardiac tissues in Zfhx3 KO mice (Figure S2) and that expression of Zfhx3 in the adult WT mouse heart was seen primarily in the atria (Figure 2a). No differences in offspring survival between genotypes were seen however, we observed a high incidence of premature death in both Zfhx3 Het and Zfhx3 KO mice: by age 10 months 32 out of 42 Zfhx3 KO mice had died, while 20 out of 36 Zfhx3 Het mice had died by 12 months of age. In contrast, there was minimal mortality in WT mice with only 4 out of 42 dead by 9 months of age (Figure 2b).
Figure 2. Zfhx3 Het and KO mice have diminished survival.
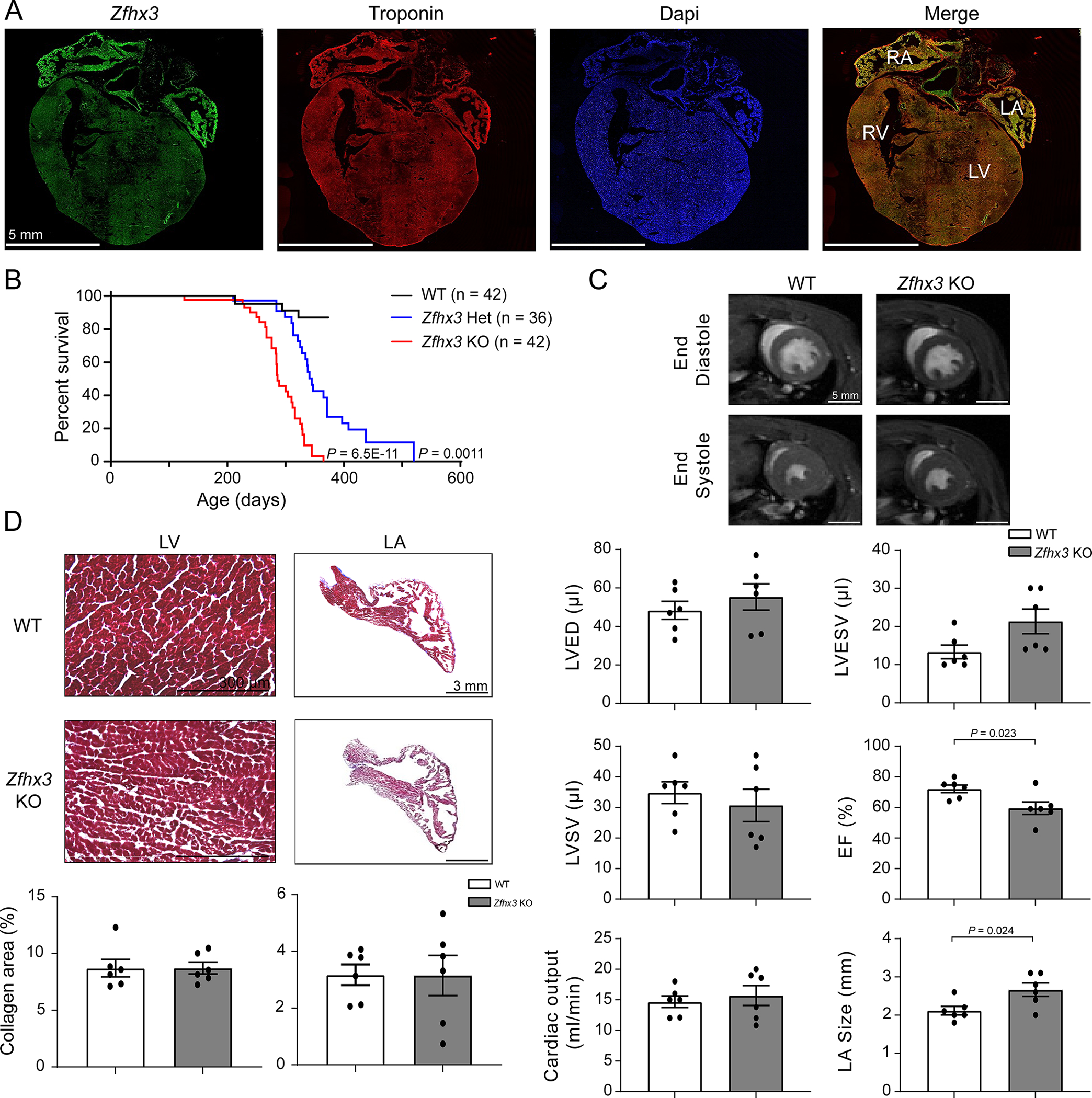
A. Confocal images of Zfhx3 (green), troponin (red), and DAPI (blue) in adult WT mouse heart tissue. B. Percent survival of WT (n = 42), Zfhx3 Het (n = 36), and Zfhx3 KO (n = 42) mice. C. Representative left ventricular end- diastolic (top) and systolic (bottom) frames of short axis myocardium slices for WT (left) and Zfhx3 KO (right) mice at 3 months of age. Quantitative comparison (bottom) of left ventricular end- diastolic volume (LVEDV), left ventricular end systolic volume (LVESV), left ventricular stroke volume (LVSV), ejection fraction (EF), and cardiac output, and left atrial (LA) size between WT and Zfhx3 KO mice. n = 6 per genotype. D. Masson’s trichrome staining in WT (top) and Zfhx3 KO (bottom) heart sections from 3-month-old mice. Bar graphs show quantification of fibrotic areas in histological sections (LV, left; LA, right; n = 6 samples per genotype). LV indicates left ventricle; RV, right ventricle; LA, left atrium; RA, right atrium. P values are indicated. Percent survival was compared using log-rank Mantel-Cox test, WT versus Zfhx3 Het and Zfhx3 KO (B). Data are mean ± s.e.m. Groups were compared using unpaired t tests (C and D).
To assess the effects of Zfhx3 loss on cardiac function, we performed two dimensional and M-mode echocardiography to screen for ventricular and atrial structural and functional abnormalities in 3-month-old WT, Zfhx3 Het, and Zfhx3 KO mice. No differences were observed among genotypes in left ventricular internal dimension in systole (LVIDs), left ventricular end dimension in diastole (LVIDd), interventricular septal thickness in systole (IVSs), interventricular septal thickness in diastole (IVSd), posterior wall thickness in systole (PWs), posterior wall thickness in diastole (PWd), left atrial diameter or left ventricular function (Table S2).
To further evaluate cardiac structure and to exclude subtle anomalies that might have been missed by echocardiography, we performed cardiac magnetic resonance imaging (MRI) in WT and Zfhx3 KO mice at 3 months of age and found significantly reduced ejection fraction (WT vs Zfhx3 KO mice: 72.2 ± 2.47 vs 59.5 ± 4.05 %, P = 0.023) and significantly increased left atrial size (WT vs Zfhx3 KO mice: 2.12 ± 0.11 vs 2.67 ± 0.17 mm, P = 0.024) in Zfhx3 KO mice (no differences in LV end diastolic volume (EDV), end systolic volume (ESV), or stroke volume between the groups; Figure 2c). Finally, we assessed trichrome-stained heart sections for cardiac fibrosis and found no differences in fibrosis between 3-month-old Zfhx3 KO and WT mice (Figure 2d).
To examine the susceptibility of Zfhx3 Het and Zfhx3 KO mice to arrhythmias, we then performed in vivo cardiac electrophysiology testing at the same age. Despite no overt differences in baseline electrophysiological parameters measured (Table S3), we observed a gene-dose response in inducible atrial arrhythmias (AA)/AF by programmed stimulation. Zfhx3 KO mice had a higher incidence of AA/AF than WT mice (13/17 vs 6/15, respectively,) and Zfhx3 Het mice (13/17 vs 4/6 mice, respectively) at shorter atrial cycle lengths lasting both at least 1 sec after burst pacing (Figure 3a, b) and at least 15 sec after burst pacing with spontaneous return to baseline or termination by overdrive pacing (Zfhx3 KO, 13/17; WT, 2/15; and Zfhx3 Het 4/6; Figure S3). Zfhx3 KO mice were also more prone to arrhythmia induction and had increased frequency of AA/AF compared to WT and Zfhx3 Het mice (Figure 3c, d). In some cases, arrhythmias in the Zfhx3 KO mice persisted until termination by either burst pacing or introduction of programmed extra stimuli. These arrhythmias presented as spontaneous termination of atrial tachycardia in Zfhx3 KO mice manifest by an abrupt reduction in the cycle length and inversion of the P wave axis on the surface EKG. This was followed by spontaneous resumption of the atrial tachycardia. (Figure 3e).
Figure 3. Gene-dose response in susceptibility to AA/AF at 3-months-old.
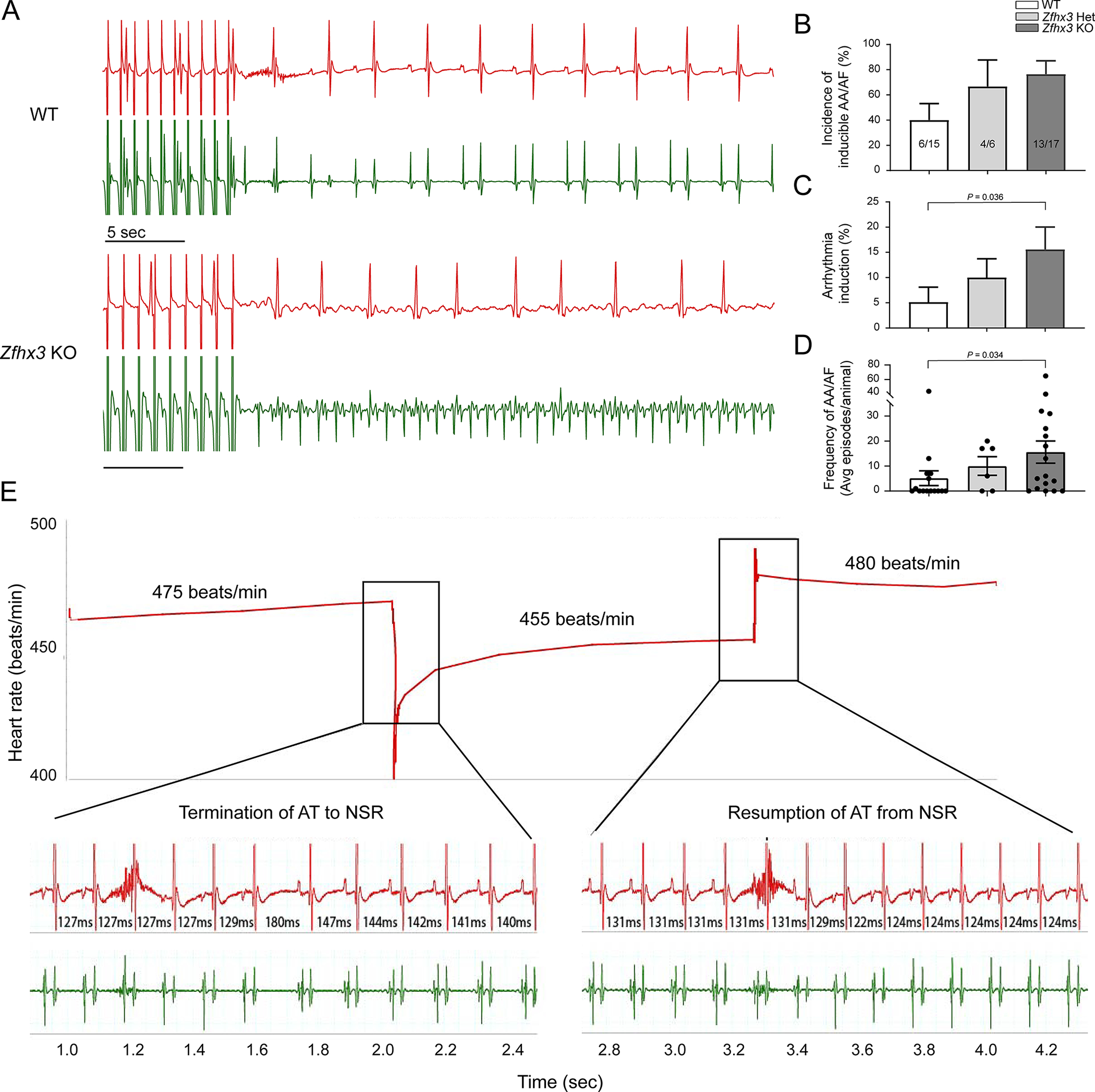
A. Representative surface (red) and intracardiac atrial (green) ECGs (mV) in WT (top) and Zfhx3 KO mice (bottom) after rapid atrial pacing (cycle length of 25 ms). B. Percentage of mice with inducible AA/AF (6/15 WT, 4/6 Zfhx3 Het, and 13/17 Zfhx3 KO mice). C. Comparison of arrhythmia burden by genotype (n = 15 WT, 6 Zfhx3 Het, and 17 Zfhx3 KO mice). D. Frequency of AA/AF in each group at 3-months-old (n = 15 WT, 6 Zfhx3 Het, and 17 Zfhx3 KO mice). E. Representative surface (red) and intracardiac atrial (green) ECGs (mV) in Zfhx3 KO mice showing atrial tachycardia termination and resumption. Time between beats indicated in ms. AA indicates atrial arrhythmia; AF, atrial fibrillation; ECGs, electrocardiogram; AT, atrial tachycardia, NSR, normal sinus rhythm. Data are mean ± s.e.m. P values indicated. Significance of arrhythmia induction and frequency of AA/AF were determined with Mann-Whitney test.
Zfhx3 loss increases arrhythmia susceptibility through alterations in cardiac conduction velocity, action potential duration, and calcium handling
To identify potential mechanisms by which Zfhx3 loss increases susceptibility to AA/AF, we performed ex vivo optical mapping in isolated 3-month-old Langendorff-perfused hearts. Zfhx3 KO mouse hearts demonstrated significantly slower right atrial (RA) conduction velocity (CV: 0.81 ± 0.044 vs. 0.59 ± 0.63 m/s, P = 0.016; Figure S4), and no differences in APD90 in the RA or LA were observed (Figure S4).
Next, we performed calcium imaging as calcium dysregulation plays a critical role in the pathophysiology of AF21. Removal of Zfhx3 caused abnormal calcium handling in murine left atrial cardiomyocytes. Notably, we observed afterdepolarization-associated calcium transients in the left atrial cardiomyocytes of Zfhx3 KO mice but not in WT mice (Figure 4a), which potentially provide a trigger that might contribute to arrhythmogenesis in Zfhx3 KO mice. Additionally, we saw significant increases in both decay time and spontaneous calcium release events compared to control (Decay time: 82.7 ± 4.4 vs 60.6 ± 2.8 ms, P = .0063; Spontaneous calcium release events per second: 0.914 ± 0.15 vs 0.437 ± 0.08, P = .0063, respectively; Figure 4a-f). Together, these data indicate loss of Zfhx3 results in conditions that predispose to increased automaticity resulting in the observed increased susceptibility to AA/AF.
Figure 4. Zfhx3 loss results in abnormal calcium handling at 3-months-old.
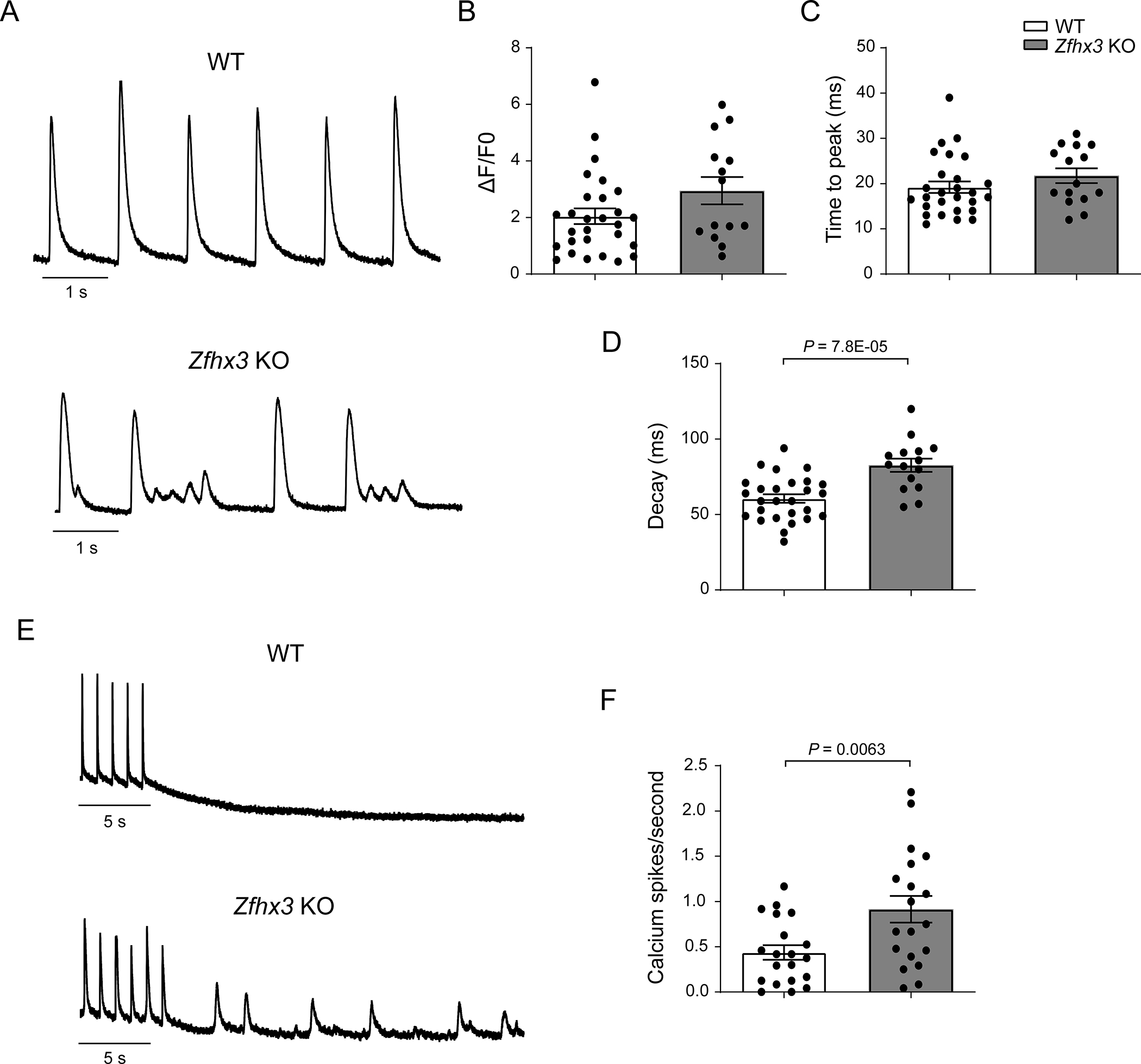
A. Representative traces of calcium transients recorded from WT (top) and Zfhx3 KO (bottom) mice left atrial cardiomyocytes with fluo-3AM and paced at 1 Hz. Mean amplitude (B, n = 28 WT and 14 Zfhx3 KO cells), time to peak (C, n = 27 WT and 15 Zfhx3 KO cells), and decay time constants (D, n = 27 WT and 15 Zfhx3 KO cells) of calcium transients. E. Representative traces of spontaneous calcium events after 1 min of pacing at 1 Hz. F. Mean spontaneous calcium events per second without pacing (n = 20 WT and 19 Zfhx3 KO cells). Data are mean ± s.e.m. P values are indicated. Groups were compared using unpaired t tests
Loss of Zfhx3 disrupts expression of genes and signaling pathways involved in cardiac pathophysiology
To explore any molecular mechanisms underlying AA/AF, we identified transcripts altered by Zfhx3 by RNA sequencing of the LA and RA of 6-month-old Zfhx3 KO, Zfhx3 Het, and WT mice (Figure 5a). Pearson’s correlation analysis and clustering revealed that both Zfhx3 Het and KO atria were distinguishable from WT samples, thus we grouped them together for all further analysis (Figure 5b, see methods). Next, we performed differential expression analysis and found that there were 613 total significantly differentially expressed genes in the RA (Figure 5c, Data Set S1) and 1246 significantly differentially expressed genes in the LA (Figure 5d, Data Set S2). To determine the overlap of differentially expressed genes between each atrial chamber we combined all significantly affected genes (n=1541, adjusted p-value < 0.05) together (Figure 5e). Overall, we observed that the major gene expression patterns were concordant. In mutant animals we found a consistent upregulation of genes associated with dendritic cells, inflammation, and cardiac injury, including Cd209a, Sfrp5, and Nppb22–24. Among the commonly downregulated genes we found Myl2, Cacna1s, Hcn1, and Scn4b. There were more discordantly expressed genes found upregulated in the LA (n=45) than in the RA (n=29). Genes elevated in expression in the LA but not the RA included inflammatory markers such as Ifit1, Mx1, and Il2ra.
Figure 5. Zfhx3 loss leads to differentially expressed genes in left and right atria at 6 months of age.
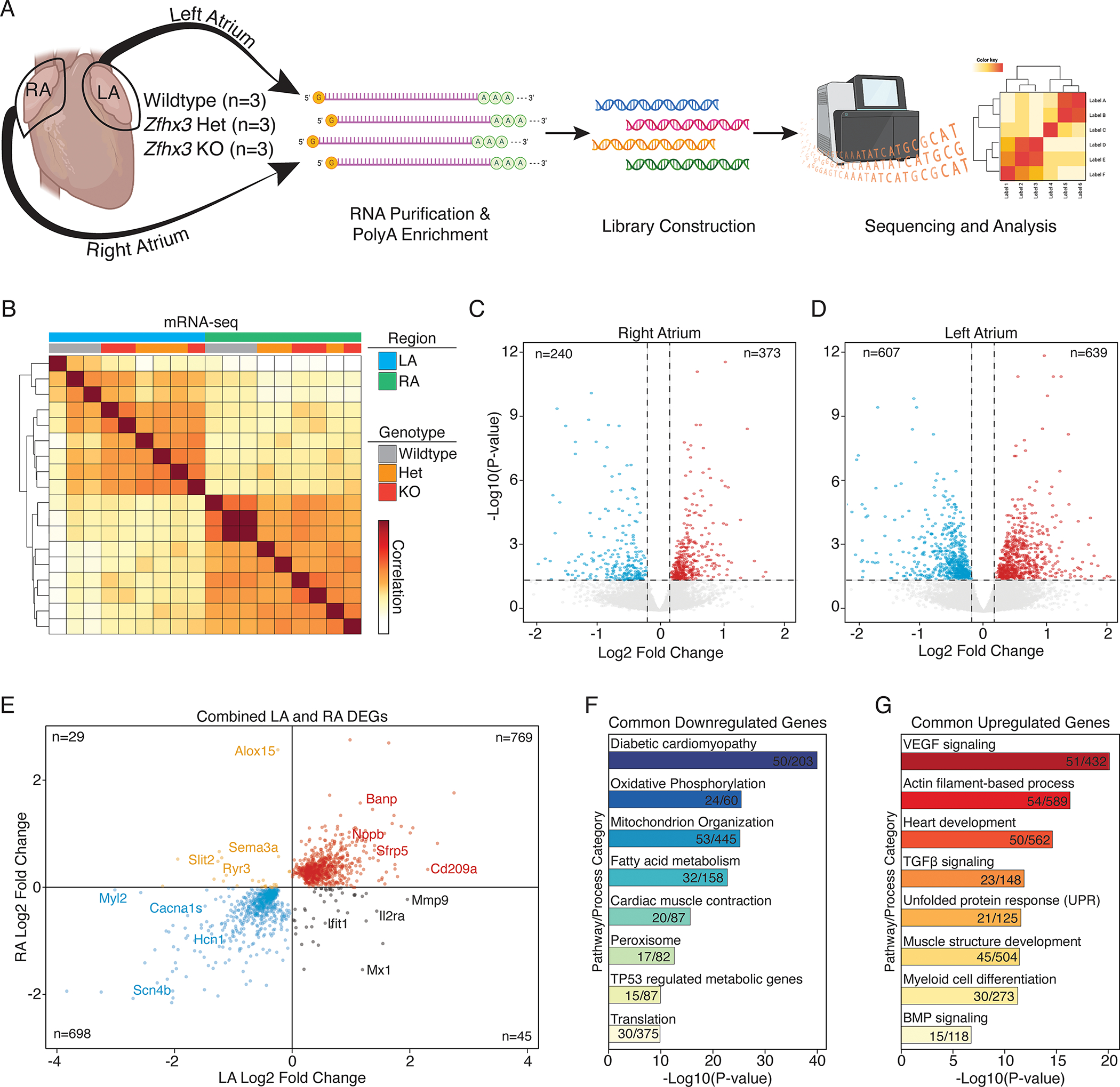
A. Experimental overview for RNA-seq. Both the LA and RA were profiled from 3 different animals of each genotype (total animals profiled = 9 and, mRNA-seq libraries = 18). B. Correlation heatmap of RNA-seq sample distances. Individual libraries annotated by region, and genotype. C, D. Volcano plot of total RNA sequencing of LA (C) and RA (D) in WT vs Zfhx3 KO and Het mouse hearts. E. Scatterplot of all combined significantly differentially expressed genes (DEGs) compared across the left atria (LA) and right atria (RA). F, G. GO and pathway for common downregulated (F) and upregulated (G) genes. LA indicates left atrium; RA, right atrium; GO; gene ontology.
Next, we performed gene ontology (GO) and pathway enrichment analysis of the common differentially expressed genes to determine the global transcriptional signatures associated with Zfhx3 loss of function (Figure 5f, and 5g). We found that in Zfhx3 deficient atria many genes associated with diabetic cardiomyopathy, oxidative phosphorylation, and fatty acid metabolism were downregulated (Figure 5f). Further, genes associated with cardiac muscle contraction and protein translation were also significantly downregulated. Upregulated signaling pathways included, VEGF, TGFB, and BMP (Figure 5g), possible indications of interstitial cardiac fibrosis and endothelial dysfunction. Moreover, we found evidence for cell stress and the unfolded protein response in mutant atrial tissue. We also found numerous genes involved in the regulation of Wnt signaling were significantly differentially expressed in our dataset. Of the Wnt signaling-related dysregulated transcripts in the atria of Zfhx3 KO mice, secreted frizzled-related protein 5 (Sfrp5; Figure 5e). Sfrps act to inhibit Wnt pathway signaling, which has been shown to have positive effects on cardiac function, left ventricular remodeling, and fibrosis25. Taken together, these findings establish ZFXH3 in the regulation of genes signaling pathways involved in cardiac pathophysiology.
Zfhx3 directly regulates genes related to cardiac conduction and AF
The DNA binding motif of ZFHX3 has been characterized in vitro and is known to bind AT rich sequences26–28. However, no ChIP-seq datasets exist to precisely characterize the consensus binding motif and cistrome for ZFHX3, as there are no antibodies available of sufficient quality. To overcome this issue and to determine which genes are directly regulated by ZFXH3 in vivo, we generated a consensus ZFHX3 motif derived from published AT sequences of genes regulated by Zfhx3, as has been done previously29 (Figure 6a). This approach takes advantage of in vitro luciferase validated ZFHX3 binding sites that share sequence conservation across mammals in order to develop a ZFHX3 motif that can be used to assess identify putative target sites in silico. To determine which genes in atrial cardiomyocytes (CMs) are putatively regulated directly by ZFHX3, we analyzed human cardiac single nucleus ATAC-seq data (snATAC) obtained from each of the 4 cardiac chambers (LV, RV, LA, RA)30. Firstly, we evaluated the enrichment of ZFHX3 binding sites across all identified cardiac regulatory elements (n= 287,415 peaks from 9 cell clusters) and identified 6,779 high confidence binding sites (log-odds score > 10). We then determined the cell type-specific enrichment across these peaks and found that atrial CMs had the greatest signal when compared to ventricular CMs and cardiac fibroblasts (Figure 6b). Secondly, we looked at all the putative ZFHX3 binding sites in atrial CMs alone and performed Genomic Regions Enrichment Annotation (GREAT) analysis (Figure 6c)31. Among the top enriched terms for the putative ZFHX3 cistrome were cardiac conduction, heart contraction, and Ca2+ transport. Next, we extracted all the atrial-enriched snATAC peaks and performed motif enrichment analysis for ZFHX3, identifying 4,174 ZFHX3 atrial CM-specific binding sites (Figure 6d, Data Set S3). Of these, 306 genes were identified as differentially expressed from our RNA-seq analysis (adjusted p-value < 0.05). GO analysis of these 306 direct ZFHX3 target genes uncovered enrichment for genes with roles in cardiac development, VEGF signaling, Wnt signaling, calcium binding, and heart contraction (Figure 6e). Further, we intersected the direct ZFHX3 target genes with all known AF GWAS loci (Figure 6d, Data Set S4)32. We found 6 genes located at AF GWAS loci that are directly regulated by ZFHX3, including PFDN1, PLN, CAV1, TBX5, SYNE2, and CYS1 (Figure 6d, Data Set S4). The directionality of expression of these and other genes was consistent with both positive and negative regulation by ZFHX3 in cardiomyocytes (Figure 6f). For example, Tbx5 expression increases in Zfhx3-deficient CMs, while Pln and Pfdn1 decreases. These results are consistent with previous studies evaluating the repressive role of Zfhx3 on other cardiac-specific transcription factors, including Tbx5 (Figure 6g)33.
Figure 6. ZFHX3 directly regulates genes related to atrial conduction and AF.
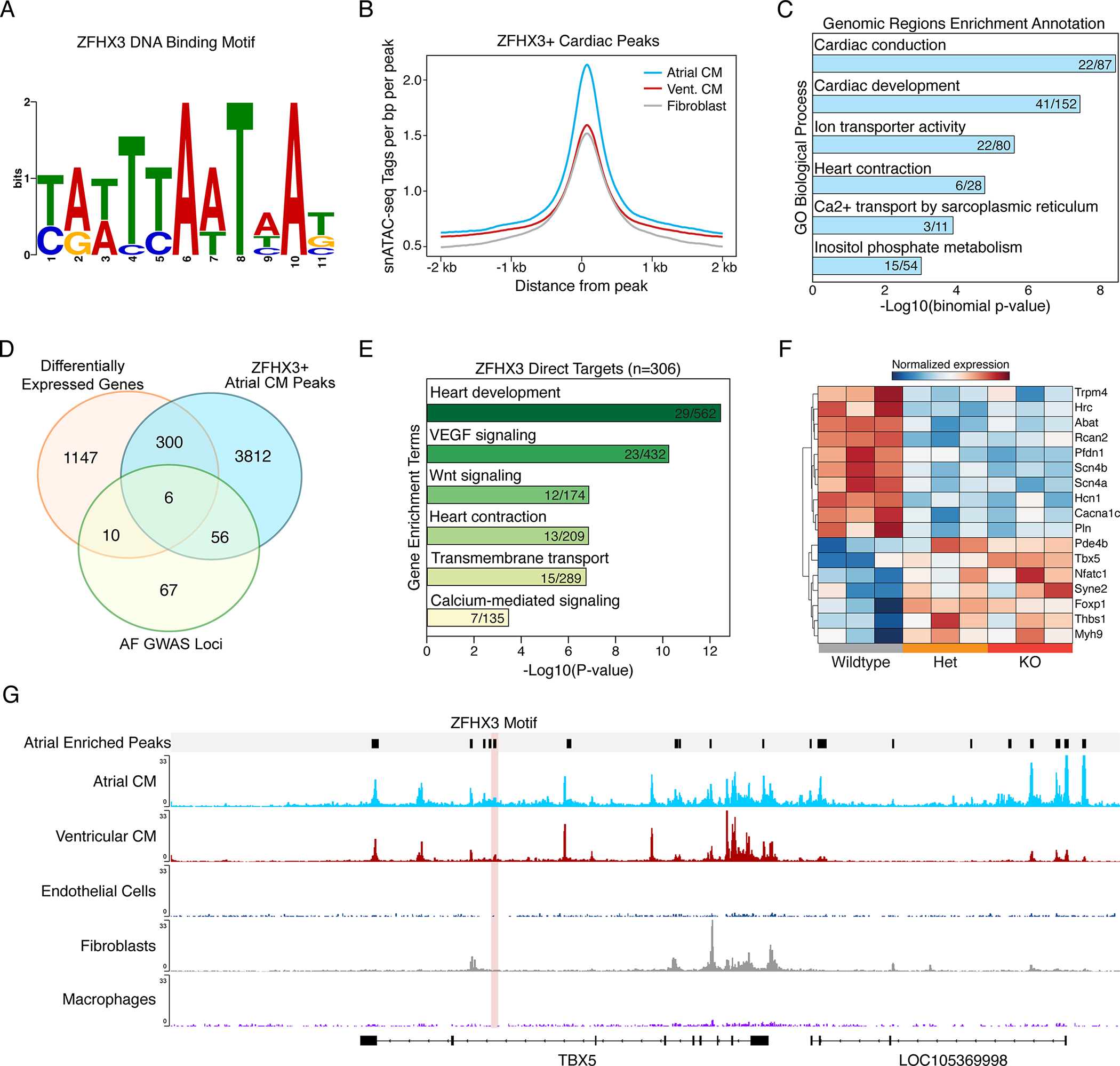
A. The consensus ZFHX3 motif derived from published AT sequences of genes regulated by Zfhx3. B. snATAC-seq fragment depth per cell cluster across high confidence ZFHX3 motif containing peaks (logs ratio >10). C. GREAT analysis for top atrial CM ZFHX3+ peaks. D. Venn diagram displaying the overlap of differentially expressed genes from Zfhx3 KO mice (adjusted P-value < 0.05) combined from both left and right atria, human ZFHX3 target genes identified from atrial CMs via snATAC and known AF GWAS loci. E. Gene ontology analysis for ZFHX3 target genes differentially expressed in Zfhx3-deficient hearts. F. Heatmap displaying genes expression of cardiac genes, identified in panel E. G. Genome browser track showing snATAC from the human heart separated by cardiac cell type. Atrial-enriched peak with a ZFHX3 motif is highlighted in light red. CM indicates cardiomyocyte; GREAT indicates genomic regions enrichment annotations.
Zfhx3 loss ultimately leads to the development of a dilated cardiomyopathy, atrial enlargement, and intra-atrial thrombus formation
Finally, we investigated whether cardiac abnormalities were responsible for the observed diminished survival in Zfhx3 KO mice. Indeed, both gross and histologic examination of the Zfhx3 KO hearts between 9 and 11 months of age confirmed cardiac enlargement and revealed the formation of a large left atrial thrombus in ~67% of Zfhx3 KO mice (8/12 Zfhx3 KO mice) and large thrombi in both the left and right atria in ~17% of Zfhx3 KO mice (2/12 Zfhx3 KO mice; Figure 7a) with significantly increased fibrosis of the atrial wall and left ventricle (Figure 7b). Supporting the observation of thrombus formation in mice lacking Zfhx3, we found significantly increased levels of Vcam1 in both the LA and RA, a cell surface protein upregulated in patients with left atrial appendage thrombi (Data Set S1)34,35. Analysis of the serial in vivo MRI-acquired images revealed significant atrial enlargement in Zfhx3 KO mice beginning at 3-months-old (WT vs Zfhx3 KO mice: 2.12 ± 0.11 vs 2.67 ± 0.17 mm in Zfhx3 KO mice, P = 0.024; Figure S5). Further, MRI analysis in Zfhx3 KO mice at 9 months of age was notable for severely impaired left ventricular function (significantly increased left ventricular end systolic volume, WT vs Zfhx3 KO mice: 55.0 ± 4.33 vs 108.8 ± 5.94 μl, P = 2.50E-05 and left ventricular end systolic volume: 17.8 ± 1.97 vs 86.33 ± 3.40 μl, P = 1.10E-08; Figure 7c), and significantly reduced stroke volume (WT vs Zfhx3 KO mice: 36.0 ± 3.68 vs 21.2 ± 3.74 μl, P = 0.018; Figure 7c), ejection fraction (WT vs Zfhx3 KO mice: 67.7 ± 2.40 vs 24.5 ± 0.56 %, P = 7.9E-09; Figure 7c), and cardiac output (WT vs Zfhx3 KO mice: 16.3 ± 1.06 vs 11.4 ± 0.94 ml/min, P = 0.0065; Figure 7c), with significant LA enlargement (WT vs Zfhx3 KO mice: 2.42 ± 0.080 vs 3.22 ± 0.17 mm, P = 0.0017; Figure 7c). In addition to massively dilated hearts, Zfhx3 KO mice displayed premorbid phenotype indicating advanced heart failure including diffuse edema/anasarca, abdominal distention presumably from fluid retention, tachypnea, and muscle wasting (data not shown).
Figure 7. 9-month-old Zfhx3 KO mice have dilated cardiomyopathy and atrial thrombus.
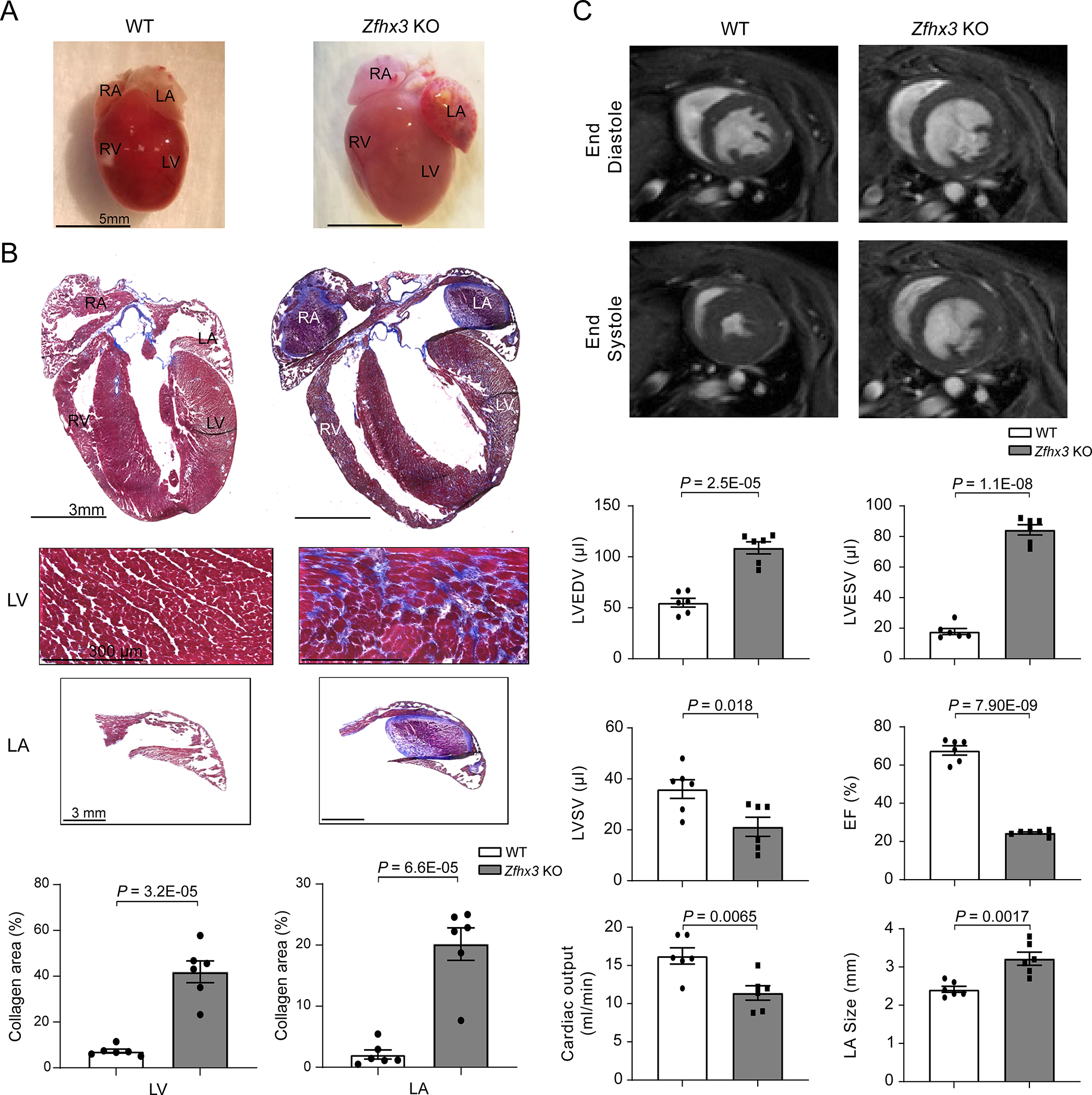
A. Representative gross heart size in WT (left) and Zfhx3 KO (right) mice. B. Representative Masson’s trichrome staining of whole hearts, WT (top left) and Zfhx3 KO mice (top right), and heart sections from LV (top) and LA (bottom). Bar graph shows quantification of left ventricular and left atrial fibrotic areas in histological sections. n = 6 samples per genotype. C. Representative left ventricular end- diastolic (top) and systolic (bottom) frames of short axis myocardium slices for WT (left) and Zfhx3 KO (right). Quantitative comparison (bottom) of left ventricular end- diastolic volume (LVEDV), left ventricular end systolic volume (LVESV), left ventricular stroke volume (LVSV), ejection fraction (EF), and cardiac output, and left atrial (LA) size between WT and Zfhx3 KO mice. n = 6 per genotype. LV indicates left ventricle; RV, right ventricle; LA, left atrium; RA, right atrium. Data are mean ± s.e.m. P values are indicated. Groups were compared using unpaired t tests
Discussion
To date, more than 100 genetic loci have been associated with the risk of AF3; however, the causative genes and the mechanisms by which they lead to AF remains unknown for most of these loci. The disconnect between the rapid pace of genetic discovery and our mechanistic understanding of AF-related genes is exemplified by the ZFHX3 locus. An association between AF genetic variants located intergenic to ZFHX3 was first described over a decade ago36, yet our understanding of the role of this gene in the heart remains limited. In the present study, we provide evidence strongly implicating ZFHX3 as the causative gene at the 16q22 locus for AF and that the rs12931021 SNP at this locus modulates the expression of ZFHX3. Further, we show that complete loss of Zfhx3 in cardiomyocytes results in an increased incidence of AA/AF through atrial specific transcriptional differences. Ultimately, cardiac restricted Zfhx3 deletion in mice results in atrial enlargement, atrial thrombus formation, and dilated cardiomyopathy.
A major finding of our study was a gene-dose response in susceptibility to AA/AF with Zfhx3 KO mice having higher incidence, frequency, and burden of AA/AF than Zfhx3 Het and WT mice. We can attribute the increased vulnerability to AA/AF in ZFHX3 Het and KO mice to basic mechanisms of AF substrate formation including the observed reductions in conduction velocity in Zfhx3 KO mice, as well as dysregulated calcium handling with frequently observed afterdepolarization-associated calcium transients in left atrial cardiomyocytes from Zfhx3 KO mice30.
The cardiac gene regulatory network driven by Zfhx3 discovered here offers additional clues to the molecular mechanisms that lead to the development of arrhythmia susceptibility in Zfhx3 Het and KO mice. In mutant animals, we observed consistent upregulation of genes associated with inflammation and cardiac injury, both of which are suggested to contribute to the development of AF37. Upregulated signaling pathways included VEGF, TGFB, and BMP, possible indications of interstitial cardiac fibrosis and endothelial dysfunction. Conversely, the downregulation of genes encoding membrane currents associated with the generation of the cardiac action potential, as well as genes involved in calcium handling, such as Cacna1c, in Zfhx3 KO mice are also substrates for arrythmias. Reduced left atrial expression of the calcium subunit modulating ICa,L has been associated with promoting atrial APD shortening in animal models of AF and in patients with chronic AF and thus, AF maintenance38. Additional downregulated genes in mutant mice included Myl2, Cacna1s, Hcn1, and Scn4b. Defects in the sodium channel subunit gene Scn4b are associated with long QT syndrome as well as familial AF39,40. Moreover, pathways such as diabetic cardiomyopathy, oxidative phosphorylation, and fatty acid metabolism were associated with downregulated genes in Zfhx3 deficient atria. This dramatic shift in metabolic gene expression is consistent with what has been previously observed in Pitx2 knockout cardiomyocytes41, and with the protective role played by ZFHX3 in neurons42. Further, genes associated with cardiac muscle contraction and protein translation were also significantly downregulated38,43,44. Human snATAC-seq data, revealed atrial target genes regulated directly by ZFHX3 in cardiomyocytes, and these genes were involved in molecular pathways related to cardiac development, VEGF signaling, Wnt signaling, calcium binding, and heart contraction. These findings are consistent with the impaired calcium handling observed in Zfhx3 KO mice, and further implicate ZFHX3 as a regulator of cardiac conduction. Overall, these results indicate loss of cardiac Zfhx3 results in disrupted expression of genes and transcription networks essential for normal atrial function likely forming a substrate for AF, and ultimately increasing arrhythmia susceptibility.
Our study also revealed AF-associated genes were directly regulated by ZFHX3 such as Tbx5 and CAV1. In prior work, Nadadur and colleagues showed Tbx5 and Pitx2 co-regulate atrial rhythm in a gene regulatory network45. We found Zfhx3 directly regulates Tbx5, suggesting Zfhx3 may play a role in this regulatory network important for maintaining atrial rhythm homeostasis. Moreover, studies revealing that ZFHX3 functions in a transcription network in breast cancer to promote tumor growth through upregulation of transcription factors including TBX3 and MYC7, as well as those studies indicating that the gene-gene interactions of ZFHX3 with PITX2 may be involved in the generation of AF26, support the hypothesis that ZFHX3 functions as part of an AF gene regulatory network.
Our findings show Sfrp5 was significantly upregulated in the atria of Zfhx3 KO mice. SFRP5 is a member of the secreted frizzled-related protein family that act primarily by inhibiting the Wnt signaling pathway46. Wnt signaling is critically important during development, and while it is quiescent in the healthy adult cardiovascular system, the pathway is reactivated in disease states46. Dysregulation of the Wnt signaling pathway has been seen in various cardiovascular disease states including hypertrophy, fibrosis, myocardial infarction, heart failure47–51, and inhibition of non-canonical Wnt signaling has been shown to prevent the onset of familial DCM52. Although studies have investigated the role of Wnt signaling in arrhythmia generation, the exact mechanisms involved are also largely unknown46. In addition to the role of this pathway in regulating known substrates of arrhythmogenesis such as fibrosis and inflammation53, Wnt signaling has also been shown to regulate expression of Cx43 to facilitate the formation of functional gap junctions, and disruption of this signaling can lead to arrhythmia generation and maintenance54. Moreover, recent work identified differentially expressed genes in paired human LA and RA appendages of healthy and AF patients and found gene expression in LA tissue had greater involvement in Wnt signaling than RA tissue in patients with AF55. Through inhibition of Wnt pathway signaling, Sfrps have been shown to have beneficial effects on infarct size and cardiac function after myocardial infarction56, antifibrotic effects57, and improved wound healing58. Specifically, Sfrp5 has been shown in mice to inhibit inflammation after ischemia/reperfusion injury, and treatment with recombinant Sfrp5 in rat ventricular myocytes lessened ischemic myocardial injuries59. In humans, abnormal serum levels of SFRP5 have been associated with coronary artery disease60 and increased incidence of major adverse cardiovascular events61. Together, these data support the hypothesis that altered Sfrp5 expression resulting from loss of Zfhx3 disrupts Wnt signaling and contributes to the development of arrhythmogenesis, DCM, and atrial thrombus, suggesting that Sfrp5 may serve as future biomarker and/or therapeutic target for cardiovascular diseases.
Finally, we observed an age dependent progression in LV and atrial dilation, fibrosis, and thrombus formation, and a premorbid phenotype indicating advanced heart failure including diffuse edema/anasarca, abdominal distention presumably from fluid retention, tachypnea, and muscle wasting with loss of Zfhx3. Similar findings have been observed in elderly patients with AF. For example, atrial size is well-known to increase with age and AF status62–64. Increasing fibrosis is a hallmark of AF progression65, and stroke risk due to atrial thrombus formation increases with age64. We also observed a significant upregulation of Vcam-1 in the atria of Zfhx3 KO mice. Evidence suggests VCAM-1 is involved in atrial remodeling, clot formation, and AF34. Increased expression of VCAM-1 has been observed in patients with paroxysmal and persistent AF, with the highest expression levels observed in patients with left atrial appendage thrombosis. Additionally, during atrial pacing in pigs, Vcam1 has been shown to be upregulated, contributing to inflammation and generating a prothrombotic environment within atrial tissue34,35,66–68. Together, these findings correlate with the thrombus formation observed in mice lacking Zfhx3.
Despite the relatively low expression of Zfhx3 in the ventricles, the striking LV phenotype in older Zfhx3 KO mice may be related to total ablation of Zfhx3 expression in cardiomyocytes compared to the human phenotype where much ZFHX3 expression is retained. AF and heart failure/cardiomyopathy often co-exist and the direction of cause and effect remains elusive69. Indeed, a polymorphism in the ZFHX3 gene increased the AF risk in patients with HF70, however, the possibility of undetected AF makes the temporal relationship between AF and HF somewhat uncertain. And while AF is often the first manifestation of HF, many patients hospitalized with HF go on to develop AF71. As such, a common genetic basis cannot be excluded. There is often overlap between ventricular dysfunction, increased left ventricular mass, and atrial dilation for AF risk, as we observed in mice with complete and partial loss of Zfhx3. While it is intriguing to speculate that loss of Zfhx3 may provide a unifying mechanism for these features of AF in patients, these observations will require further study in the future.
It is important to address limitations of our work. First, the maturity of the PSC-CMs used in our study. It has been established that most differentiation protocols produce cardiomyocytes resembling early fetal hearts. Compared to mature cardiomyocytes, PSC-CMs are lacking the well-established myofibrils and T-tubules, are composed of a rather heterogeneous population, and display a distinct electrophysiological profile72. Despite these limitations, differentiation of PSC-CMs is an efficient, well-established process, generating a physiologically relevant in vitro system for studying cardiovascular conditions72,73. Second, extrapolating the results obtained in our mouse model to what is seen in large animal models of and in clinical AF. The higher basal heart rate, smaller body and heart size, and shorter APD of mice may not only limit reentrant circuits but additionally, make data interpretation and accuracy within the electrophysiological study more difficult. Compared to large animal models of AF, we do not often observe in mice the structural and electrical remodeling associated with sustained AF. Additionally, mice do not generally develop AF without programmed electrical stimulation, and compared to humans, have differences in ionic currents and channel distribution responsible for the time of repolarization, which may also make it more difficult to characterize abnormalities in cardiac conduction and arrhythmic phenotypes. Nevertheless, genetically engineered mice have improved our understanding of the genetic basis and signaling pathways contributing to cardiac arrhythmias74. Finally, the results of our study reflect constitutive deletion of Zfhx33. While reduction of ZFHX3 expression in humans with the culprit SNP is present from earliest development, conditional deletion of Zfhx3 in the adult mouse remains an interesting area for future study and would help discern a developmental effect of Zfhx3.
In conclusion, our data demonstrates that an AF associated SNP, rs12931021, modulates expression of ZFHX3. Furthermore, loss of cardiac Zfhx3 is sufficient to provoke AA/AF in adult mice through alterations in conduction velocity, atrial action potential duration, calcium handling, and through dysregulation of genes involved in both AF-susceptibility and cardiac contractile function. Taken together, these data reveal a novel and important role for Zfhx3 in the control of cardiac genes and signaling pathways essential for atrial homeostasis.
Supplementary Material
Novelty and Significance.
What is known?
Genome wide association studies have identified a susceptibility locus for atrial fibrillation (AF) at ZFHX3, a gene that encodes a large transcription factor.
Despite being the second-most significantly associated locus with AF, the function of ZFHX3 in the heart is unknown.
What New Information Does This Article Contribute?
We provide evidence strongly implicating ZFHX3 as the causative gene at the 16q22 locus for AF and that SNP rs12931021 SNP modulates the expression of ZFHX3.
Loss of Zfhx3 in cardiomyocytes results in a gene-dose response in AF susceptibility with Zfhx3 KO mice having higher incidence, frequency, and burden of AF than Zfhx3 Het and WT mice.
Cardiac abnormalities caused by loss of cardiac Zfhx3 are due to atrial-specific differential effects on genes essential for normal atrial function.
The disconnect between the rapid pace of genetic discovery and the mechanistic understanding of AF-related genes identified by genome-wide association studies is exemplified by the ZFHX3 locus. An association between AF genetic variants located intergenic to ZFHX3 was first described over a decade ago, yet our understanding of the role of this gene in the heart remains limited. Here, we provide evidence strongly implicating ZFHX3 as the causative gene at the 16q22 locus for AF and that the rs12931021 SNP at this locus modulates the expression of ZFHX3. Further, we show that complete loss of Zfhx3 in cardiomyocytes results in an increased incidence of arrythmias through atrial specific transcriptional differences, atrial enlargement and thrombus formation, and dilated cardiomyopathy. In aggregate, these findings provide a causal link between a genetic locus associated with AF and ZFHX3.
Acknowledgements
Dr. Ellinor is supported by grants from the National Institutes of Health (1RO1HL092577, 1R01HL157635, 5R01HL139731), from the American Heart Association Strategically Focused Research Networks (18SFRN34110082), and from the European Union (MAESTRIA 965286). This research was also supported by the Carol and Roch Hillenbrand and the George L. Nardi, MD, funds at Massachusetts General Hospital. Dr. Hanley was supported by a postdoctoral fellowship from the Irish Cardiac Society; Drs. Jameson and Hucker were supported by NIH grant 5T32HL00720840. Dr. Xiao was supported by an American Heart Association Career Development Award (20CDA35260081). Dr. Tucker is supported by the National Institutes of Health Mentored Research Scientist Career Development Award (7K01HL140187). Dr. Clauss was supported by a Marie Curie International Outgoing Fellowship within the 7th European Community Framework Programme (PIOF-GA-2012-328352 to SC) and by the German Centre for Cardiovascular Research (DZHK; 81X2600210, 81X2600204).
Non-standard Abbreviations and Acronyms
- AF
Atrial fibrillation
- AA
Atrial arrhythmias
- ATBF1
AT motif binding factor 1
- eQTL
Expression quantitative trait loci
- ChIP
Chromatin immunoprecipitation
- PSC-CMs
Pluripotent stem cell-derived cardiomyocytes
- LV
Left ventricle
- RV
Right ventricle
- LA
Left atrium
- RA
Right atrium
- LVIDs
Left ventricular internal dimension in systole
- LVIDs
Left ventricular end dimension in diastole
- IVSs
Interventricular septal thickness in systole
- IVSd
Interventricular septal thickness in diastole
- PWs
Posterior wall thickness in systole
- PWd
Posterior wall thickness in diastole
- EDV
End diastolic volume
- ESV
End systolic volume
- CV
Conduction velocity
- APD90
Action potential duration at 90% repolarization
- snATAC
Single nucleus ATAC-seq data
Footnotes
Disclosures
Dr. Ellinor receives sponsored research support from Bayer AG, IBM Research, Bristol Myers Squibb and Pfizer; he has also served on advisory boards or consulted for Bayer AG, MyoKardia and Novartis.
References
- 1.Zulkifly H, Lip GYH, Lane DA. Epidemiology of atrial fibrillation. Int J Clin Pract. 2018. [DOI] [PubMed] [Google Scholar]
- 2.Lau DH, Nattel S, Kalman JM, Sanders P. Modifiable Risk Factors and Atrial Fibrillation. Circulation. 2017. [DOI] [PubMed] [Google Scholar]
- 3.Roselli C, Chaffin MD, Weng L-C, Aeschbacher S, Ahlberg G, Albert CM, Almgren P, Alonso A, Anderson CD, Aragam KG, et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat Genet. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benjamin EJ, Rice KM, Arking DE, Pfeufer A, Van Noord C, Smith AV., Schnabel RB, Bis JC, Boerwinkle E, Sinner MF, et al. Variants in ZFHX3 are associated with a trial fibrillation in individuals of European ancestry. Nat Genet. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gudbjartsson DF, Holm H, Gretarsdottir S, Thorleifsson G, Walters GB, Thorgeirsson G, Gulcher J, Mathiesen EB, Njølstad I, Nyrnes A, et al. A sequence variant in ZFHX3 on 16q22 associates with a trial fibrillation and ischemic stroke. Nat Genet. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mao L, Huang W, Zou P, Dang X, Zeng X. The unrecognized role of tumor suppressor genes in atrial fibrillation. Gene. 2017. [DOI] [PubMed] [Google Scholar]
- 7.Dong G, Ma G, Wu R, Liu J, Liu M, Gao A, Li X, Jun A, Liu X, Zhang Z, et al. ZFHX3 promotes the proliferation and tumor growth of er-positive breast cancer cells likely by enhancing stem-like features and MYC and TBX3 transcription. Cancers. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim CJ, Song JH, Cho YG, Cao Z, Lee YS, Nam SW, Lee JY, Park WS. Down-regulation of ATBF1 is a major inactivating mechanism in hepatocellular carcinoma. Histopathology. 2008. [DOI] [PubMed] [Google Scholar]
- 9.Sun X, Frierson HF, Chen C, Li C, Ran Q, Otto KB, Cantarel BM, Vessella RL, Gao AC, Petros J, et al. Frequent somatic mutations of the transcription factor ATBF1 in human prostate cancer. Nat Genet. 2005. [DOI] [PubMed] [Google Scholar]
- 10.Kaspar P, Dvoráková M, Králová J, Pajer P, Kozmik Z, Dvorák M. Myb-interacting protein, ATBF1, represses transcriptional activity of Myb oncoprotein. J Biol Chem. 1999. [DOI] [PubMed] [Google Scholar]
- 11.Berry FB, Miura Y, Mihara K, Kaspar P, Sakata N, Hashimoto-Tamaoki T, Tamaoki T. Positive and Negative Regulation of Myogenic Differentiation of C2C12 Cells by Isoforms of the Multiple Homeodomain Zinc Finger Transcription Factor ATBF1. Journal of Biological Chemistry. 2001. [DOI] [PubMed] [Google Scholar]
- 12.Jung CG, Kim HJ, Kawaguchi M, Khanna KK, Hida H, Asai K, Nishino H, Miura Y. Homeotic factor ATBF1 induces the cell cycle arrest associated with neuronal differentiation. Development. 2005. [DOI] [PubMed] [Google Scholar]
- 13.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang Q, Ni B, Shi J, Han Z, Qi R, Xu W, Wang D, Wang DW, Chen M. Down-regulation of ATBF1 activates STAT3 signaling via PIAS3 in pacing-induced HL-1 atrial myocytes. Biochem Biophys Res Commun. 2014. [DOI] [PubMed] [Google Scholar]
- 15.Kao YH, Hsu JC, Chen YC, Lin YK, Lkhagva B, Chen SA, Chen YJ. ZFHX3 knockdown increases arrhythmogenesis and dysregulates calcium homeostasis in HL-1 atrial myocytes. Int J Cardiol. 2016. [DOI] [PubMed] [Google Scholar]
- 16.Lkhagva B, Lin YK, Chen YC, Cheng WL, Higa S, Kao YH, Chen YJ. ZFHX3 knockdown dysregulates mitochondrial adaptations to tachypacing in atrial myocytes through enhanced oxidative stress and calcium overload. Acta Physiol. 2021. [DOI] [PubMed] [Google Scholar]
- 17.Mahida S, Mills RW, Tucker NR, Simonson B, MacRi V, Lemoine MD, Das S, Milan DJ, Ellinor PT. Overexpression of KCNN3 results in sudden cardiac death. Cardiovasc Res. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ardlie KG, DeLuca DS, Segrè AV., Sullivan TJ, Young TR, Gelfand ET, Trowbridge CA, Maller JB, Tukiainen T, Lek M, et al. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomas DJ, Rosenbloom KR, Clawson H, Hinrichs AS, Trumbower H, Raney BJ, Karolchik D, Barber GP, Harte RA, Hillman-Jackson J, et al. The ENCODE project at UC Santa Cruz. Nucleic Acids Res. 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heijman J, Voigt N, Wehrens XHT, Dobrev D. Calcium dysregulation in atrial fibrillation: The role of CaMKII. Front Pharmacol. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dieterlen MT, John K, Reichenspurner H, Mohr FW, Barten MJ. Dendritic Cells and Their Role in Cardiovascular Diseases: A View on Human Studies. J Immunol Res. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu J, Zheng H, Liu X, Chen P, Zhang Y, Luo J, Kuang J, Li J, Yang Y, Ma T, et al. Prognostic Value of Secreted Frizzled-Related Protein 5 in Heart Failure Patients With and Without Type 2 Diabetes Mellitus. Circ Heart Fail. 2020. [DOI] [PubMed] [Google Scholar]
- 24.Van Duijvenboden K, De Bakker DEM, Man JCK, Janssen R, Günthel M, Hill MC, Hooijkaas IB, Van Der Made I, Van Der Kraak PH, Vink A, et al. Conserved NPPB+ Border Zone Switches From MEF2- to AP-1-Driven Gene Program. Circulation. 2019. [DOI] [PubMed] [Google Scholar]
- 25.Foulquier S, Daskalopoulos EP, Lluri G, Hermans KCM, Deb A, Blankesteijn WM. WNT signaling in cardiac and vascular disease. Pharmacol Rev. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang Y, Wang C, Yao Y, Zuo X, Chen S, Xu C, Zhang H, Lu Q, Chang L, Wang F, et al. Molecular Basis of Gene-Gene Interaction: Cyclic Cross-Regulation of Gene Expression and Post-GWAS Gene-Gene Interaction Involved in Atrial Fibrillation. PLoS Genet. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roselli C, Rienstra M, Ellinor PT. Genetics of Atrial Fibrillation in 2020. Circ Res. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.H Y, A M, T T, T M. ATBF1, a multiple-homeodomain zinc finger protein, selectively down-regulates AT-rich elements of the human alpha-fetoprotein gene. Mol Cell Biol. 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parsons MJ, Brancaccio M, Sethi S, Maywood ES, Satija R, Edwards JK, Jagannath A, Couch Y, Finelli MJ, Smyllie NJ, et al. The Regulatory Factor ZFHX3 Modifies Circadian Function in SCN via an AT Motif-Driven Axis. Cell. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hocker JD, Poirion OB, Zhu F, Buchanan J, Zhang K, Chiou J, Wang TM, Zhang Q, Hou X, Li YE, et al. Cardiac cell type-specific gene regulatory programs and disease risk association. Sci Adv. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roselli C, Roselli C, Rienstra M, Ellinor PT, Ellinor PT. Genetics of Atrial Fibrillation in 2020: GWAS, Genome Sequencing, Polygenic Risk, and Beyond. Circ Res. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rubio-Alarcón M, Cámara-Checa A, Dago M, Crespo-García T, Nieto-Marín P, Marín M, Merino JL, Toquero J, Salguero-Bodes R, Tamargo J, et al. Zfhx3 transcription factor represses the expression of SCN5A gene and decreases sodium current density (Ina). Int J Mol Sci. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Willeit K, Pechlaner R, Willeit P, Skroblin P, Paulweber B, Schernthaner C, Toell T, Egger G, Weger S, Oberhollenzer M, et al. Association Between Vascular Cell Adhesion Molecule 1 and Atrial Fibrillation. JAMA Cardiol. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ding WY, Gupta D, Lip GYH. Atrial fibrillation and the prothrombotic state: revisiting Virchow’s triad in 2020. Heart. 2020. [DOI] [PubMed] [Google Scholar]
- 36.Benjamin EJ, Rice KM, Arking DE, Pfeufer A, Van Noord C, Smith AV., Schnabel RB, Bis JC, Boerwinkle E, Sinner MF, et al. Variants in ZFHX3 are associated with a trial fibrillation in individuals of European ancestry. Nat Genet. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou X, Dudley SC. Evidence for Inflammation as a Driver of Atrial Fibrillation. Front Cardiovasc Med. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heijman J, Voigt N, Nattel S, Dobrev D. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ Res. 2014. [DOI] [PubMed] [Google Scholar]
- 39.Medeiros-Domingo A, Kaku T, Tester DJ, Iturralde-Torres P, Itty A, Ye B, Valdivia C, Ueda K, Canizales-Quinteros S, Tusié-Luna MT, et al. SCN4B-encoded sodium channel beta4 subunit in congenital long-QT syndrome. Circulation. 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li RG, Wang Q, Xu YJ, Zhang M, Qu XK, Liu X, Fang WY, Yang YQ. Mutations of the SCN4B-encoded sodium channel β4 subunit in familial atrial fibrillation. Int J Mol Med. 2013. [DOI] [PubMed] [Google Scholar]
- 41.Tao G, Kahr PC, Morikawa Y, Zhang M, Rahmani M, Heallen TR, Li L, Sun Z, Olson EN, Amendt BA, et al. Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim TS, Kawaguchi M, Suzuki M, Jung CG, Asai K, Shibamoto Y, Lavin MF, Khanna KK, Miura Y. The ZFHX3 (ATBF1) transcription factor induces PDGFRB, which activates ATM in the cytoplasm to protect cerebellar neurons from oxidative stress. Dis Model Mech. 2010. [DOI] [PubMed] [Google Scholar]
- 43.Rubio-Alarcón M, Cámara-Checa A, Dago M, Crespo-García T, Nieto-Marín P, Marín M, Merino JL, Toquero J, Salguero-Bodes R, Tamargo J, et al. Zfhx3 Transcription Factor Represses the Expression of SCN5A Gene and Decreases Sodium Current Density (I Na). Int J Mol Sci. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Derangeon M, Montnach J, Bard I, Charpentier F. Mouse Models of SCN5A-Related Cardiac Arrhythmias. Front Physiol. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nadadur RD, Broman MT, Boukens B, Mazurek SR, Yang X, Van Den Boogaard M, Bekeny J, Gadek M, Ward T, Zhang M, et al. Pitx2 modulates a Tbx5-dependent gene regulatory network to maintain atrial rhythm. Sci Transl Med. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dawson K, Aflaki M, Nattel S. Role of the Wnt-Frizzled system in cardiac pathophysiology: A rapidly developing, poorly understood area with enormous potential. Journal of Physiology. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blankesteijn WM, van de Schans VAM, ter Horst P, Smits JFM. The Wnt/frizzled/GSK-3β pathway: a novel therapeutic target for cardiac hypertrophy. Trends Pharmacol Sci. 2008. [DOI] [PubMed] [Google Scholar]
- 48.Laeremans H, Hackeng TM, Van Zandvoort MAMJ, Thijssen VLJL, Janssen BJA, Ottenheijm HCJ, Smits JFM, Blankesteijn WM. Blocking of frizzled signaling with a homologous peptide fragment of Wnt3a/Wnt5a reduces infarct expansion and prevents the development of heart failure after myocardial infarction. Circulation. 2011. [DOI] [PubMed] [Google Scholar]
- 49.Oerlemans MIFJ, Goumans MJ, Van Middelaar B, Clevers H, Doevendans PA, Sluijter JPG. Active Wnt signaling in response to cardiac injury. Basic Res Cardiol. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Malekar P, Hagenmueller M, Anyanwu A, Buss S, Streit MR, Weiss CS, Wolf D, Riffel J, Bauer A, Katus HA, et al. Wnt signaling is critical for maladaptive cardiac hypertrophy and accelerates myocardial remodeling. Hypertension. 2010. [DOI] [PubMed] [Google Scholar]
- 51.Ellis KL, Zhou Y, Beshansky JR, Ainehsazan E, Selker HP, Cupples LA, Huggins GS, Peter I. Genetic modifiers of response to glucose-insulin-potassium (GIK) infusion in acute coronary syndromes and associations with clinical outcomes in the IMMEDIATE trial. Pharmacogenomics J. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hou N, Ye B, Li X, Margulies KB, Xu H, Wang X, Li F. Transcription Factor 7-like 2 Mediates Canonical Wnt/β-Catenin Signaling and c-Myc Upregulation in Heart Failure. Circ Heart Fail. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang D, Zhang Y, Shen C. Research update on the association between SFRP5, an anti-inflammatory adipokine, with obesity, type 2 diabetes mellitus and coronary heart disease. J Cell Mol Med. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ai Z, Fischer A, Spray DC, Brown AMC, Fishman GI. Wnt-1 regulation of connexin43 in cardiac myocytes. Journal of Clinical Investigation. 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thomas AM, Cabrera CP, Finlay M, Lall K, Nobles M, Schilling RJ, Wood K, Mein CA, Barnes MR, Munroe PB, et al. Differentially expressed genes for atrial fibrillation identified by RNA sequencing from paired human left and right atrial appendages. Physiol Genomics. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barandon L, Couffinhal T, Ezan J, Dufourcq P, Costet P, Alzieu P, Leroux L, Moreau C, D,Duplàa C. Reduction of Infarct Size and Prevention of Cardiac Rupture in Transgenic Mice Overexpressing FrzA. Circulation. 2003. [DOI] [PubMed] [Google Scholar]
- 57.He W, Zhang L, Ni A, Zhang Z, Mirotsou M, Mao L, Pratt RE, Dzau VJ. Exogenously administered secreted frizzled related protein 2 (Sfrp2) reduces fibrosis and improves cardiac function in a rat model of myocardial infarction. Proc Natl Acad Sci. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alfaro MP, Pagnia M, Vincent A, Atkinson J, Hill MF, Cates J, Davidson JM, Rottman J, Leed E, Young PP. The Wnt modulator sFRP2 enhances mesenchymal stem cell engraftment, granulation tissue formation and myocardial repair. Proc Natl Acad Sci. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nakamura K, Sano S, Fuster JJ, Kikuchi R, Shimizu I, Ohshima K, Katanasaka Y, Ouchi N, Walsh K. Secreted frizzled-related protein 5 diminishes cardiac inflammation and protects the heart from ischemia/reperfusion injury. Journal of Biological Chemistry. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miyoshi T, Doi M, Usui S, Iwamoto M, Kajiya M, Takeda K, Nosaka K, Nakayama R, Okawa K, Takagi W, et al. Low serum level of secreted frizzled-related protein 5, an anti-inflammatory adipokine, is associated with coronary artery disease. Atherosclerosis. 2014. [DOI] [PubMed] [Google Scholar]
- 61.Ji H, Li H, Zhuang J, Su Y, Wen J, Zhang J, Zhao D, Zhang Y, Xu Y. High Serum Level of Secreted Frizzled-Related Protein 5 (sfrp5) is Associated with Future Cardiovascular Events. Cardiovascular Therapy: Open Access. 2017. [Google Scholar]
- 62.Önundarson PT, Thorgeirsson G, Jonmundsson E, Sigfusson N, Hardarson T. Chronic atrial fibrillation- epidemiologic features and 14 year follow-up: A case control study. Eur Heart J. 1987. [DOI] [PubMed] [Google Scholar]
- 63.Vaziri SM, Larson MG, Benjamin EJ, Levy D. Echocardiographic predictors of nonrheumatic atrial fibrillation: The Framingham Heart Study. Circulation. 1994. [DOI] [PubMed] [Google Scholar]
- 64.Benjamin EJ, D’Agostino RB, Belanger AJ, Wolf PA, Levy D. Left atrial size and the risk of stroke and death: The Framingham Heart Study. Circulation. 1995. [DOI] [PubMed] [Google Scholar]
- 65.Nattel S. Molecular and Cellular Mechanisms of Atrial Fibrosis in Atrial Fibrillation. JACC Clin Electrophysiol. 2017. [DOI] [PubMed] [Google Scholar]
- 66.Goette A, Bukowska A, Lendeckel U, Erxleben M, Hammwöhner M, Strugala D, Pfeiffenberger J, Röhl FW, Huth C, Ebert MPA, et al. Angiotensin II receptor blockade reduces tachycardia-induced atrial adhesion molecule expression. Circulation. 2008. [DOI] [PubMed] [Google Scholar]
- 67.Black N, Mohammad F, Saraf K, Morris G. Endothelial function and atrial fibrillation: A missing piece of the puzzle? J Cardiovasc Electrophysiol. 2022. [DOI] [PubMed] [Google Scholar]
- 68.Khan AA, Lip GYH. The prothrombotic state in atrial fibrillation: pathophysiological and management implications. Cardiovasc Res. 2019. [DOI] [PubMed] [Google Scholar]
- 69.Prabhu S, Voskoboinik A, Kaye DM, Kistler PM. Atrial Fibrillation and Heart Failure — Cause or Effect?. Heart Lung Circ. 2017. [DOI] [PubMed] [Google Scholar]
- 70.Smith JG, Melander O, Sjögren M, Hedblad B, Engström G, Newton-Cheh C, Platonov PG. Genetic polymorphisms confer risk of atrial fibrillation in patients with heart failure: a population-based study. Eur J Heart Fail. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Smit MD, Moes ML, Maass AH, Achekar ID, Van Geel PP, Hillege HL, van Veldhuisen DJ, Van Gelder IC. The importance of whether atrial fibrillation or heart failure develops first. Eur J Heart Fail. 2012. [DOI] [PubMed] [Google Scholar]
- 72.Karbassi E, Fenix A, Marchiano S, Muraoka N, Nakamura K, Yang X, Murry CE. Cardiomyocyte maturation: advances in knowledge and implications for regenerative medicine. Nat Rev Cardiol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mills RJ, Titmarsh DM, Koenig X, Parker BL, Ryall JG, Quaife-Ryan GA, Voges HK, Hodson MP, Ferguson C, Drowley L, et al. Functional screening in human cardiac organoids reveals a metabolic mechanism for cardiomyocyte cell cycle arrest. Proc Natl Acad Sci. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schüttler D, Bapat A, Kääb S, Lee K, Tomsits P, Clauss S, Hucker WJ. Animal Models of Atrial Fibrillation. Circ Res. 2020. [DOI] [PubMed] [Google Scholar]
- 75.Lian X, Zhang J, Azarin SM, Zhu K, Hazeltine LB, Bao X, Hsiao C, Kamp TJ, Palecek SP. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat Protoc. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ye J, Tucker NR, Weng LC, Clauss S, Lubitz SA, Ellinor PT. A Functional Variant Associated with Atrial Fibrillation Regulates PITX2c Expression through TFAP2a. Am J Hum Genet. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sun X, Fu X, Li J, Xing C, Martin DW, Zhang HH, Chen Z, Dong J-T. Heterozygous deletion of Atbf1 by the Cre-loxP system in mice causes preweaning mortality. Genesis. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mahida S, Mills RW, Tucker NR, Simonson B, Macri V, Lemoine MD, Das S, Milan DJ, Ellinor PT. Overexpression of KCNN3 results in sudden cardiac death. Cardiovasc Res. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ruifrok AC, Katz RL, Johnston DA. Comparison of quantification of histochemical staining by hue-saturation-intensity (HSI) transformation and color-deconvolution. Applied Immunohistochemistry and Molecular Morphology. 2003. [DOI] [PubMed] [Google Scholar]
- 81.Mahida S, Mills RW, Tucker NR, Simonson B, Macri V, Lemoine MD, Das S, Milan DJ, Ellinor PT. Overexpression of KCNN3 results in sudden cardiac death. Cardiovasc Res. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bayly P V, Kenknight BH, Rogers JM, Hillsley RE, Ideker RE, Smith WM. Estimation of conduction velocity vector fields from epicardial mapping data. IEEE Trans Biomed Eng. 1998. [DOI] [PubMed] [Google Scholar]
- 83.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016. [DOI] [PubMed] [Google Scholar]
- 87.Soneson C, Love MI, Robinson MD. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Durinck S, Spellman PT, Birney E, Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the panther classification system. Nat Protoc. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Parsons MJ, Brancaccio M, Sethi S, Maywood ES, Satija R, Edwards JK, Jagannath A, Couch Y, Finelli MJ, Smyllie NJ, et al. The Regulatory Factor ZFHX3 Modifies Circadian Function in SCN via an AT Motif-Driven Axis. Cell. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bailey TL, Johnson J, Grant CE, Noble WS. The MEME Suite. Nucleic Acids Res. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hocker JD, Poirion OB, Zhu F, Buchanan J, Zhang K, Chiou J, Wang TM, Zhang Q, Hou X, Li YE, et al. Cardiac cell type-specific gene regulatory programs and disease risk association. Sci Adv. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.