

Abstract
Because of structural and cellular differences (i.e. degrees of matrix abundance and cross linking, mural cell density, and adventitia), large and medium sized vessels, in comparison to capillaries, react in a unique manner to stimuli that induce vascular disease. A stereotypical vascular injury response is extracellular matrix (ECM) remodeling that occurs particularly in larger vessels in response to injurious stimuli such as elevated angiotensin II, hyperlipidemia, hyperglycemia, genetic deficiencies, inflammatory cell infiltration, or exposure to proinflammatory mediators. Even with substantial and prolonged vascular damage, large and medium sized arteries, persist, but become modified by; i) changes in vascular wall cellularity; ii) modifications in the differentiation status of endothelial cells (ECs), vascular smooth muscle cells, or adventitial stem cells (each can become activated); iii) infiltration of the vascular wall by various leukocyte types; iv) increased exposure to critical growth factors and pro-inflammatory mediators; and v) marked changes in the vascular ECM, that remodels from a homeostatic, pro-differentiation ECM environment to matrices that instead promote tissue reparative responses. This latter ECM presents previously hidden matricryptic sites that bind integrins to signal vascular cells and infiltrating leukocytes (in coordination with other mediators) to proliferate, invade, secrete ECM-degrading proteinases and deposit injury-induced matrices (predisposing to vessel wall fibrosis). In contrast, in response to similar stimuli, capillaries can undergo regression responses (rarefaction). In summary, we have described the molecular events controlling ECM remodeling in major vascular diseases as well as the differential responses of arteries versus capillaries to key mediators inducing vascular injury.
Introduction
Vascularization is required for tissue development and maintenance, and blood vessels are critical for the essential exchange of gases, nutrition, and wastes, as well as the rapid circulation of hormones and other signaling molecules within our bodies1–8. A healthy vasculature is suggested to provide key angiocrine signals to adjacent tissues to promote tissue health and differentiation9,10 and, furthermore, a healthy capillary vasculature appears capable of suppressing basic disease mechanisms that prevent pathologic states including edema, inflammation, ischemic damage, thrombosis, hemorrhage, infection, autoimmunity and fibrosis7,11. In contrast, diseased vessels, in particular capillaries, are less able to promote tissue health leading to systemic injuries associated with declining tissue function that is evident in major diseases such as diabetes, hypertension, atherosclerosis, malignant cancers, and aging. A fundamental relationship of vascular structure and function is mediated by vascular wall cells and their surrounding extracellular matrices (ECM), which is necessary for proper communication between blood vessels and tissues, and, also for the propagation of flow forces as well as delivery of blood cells and plasma components throughout the circulatory system8,12. In this review, we highlight how this vascular cell-ECM relationship becomes altered leading to abnormalities (i.e. vascular matrix remodeling and cell phenotypic changes) that alter the function of blood vessels to cause cardiovascular disease.
Organization of ECM in Healthy Vasculature
A single layer of endothelial cells (ECs) lines the entire vascular system, and are important for communication between the circulation and organ-specific cell types4,9,10,13. The major constituent of the vessel wall is extracellular matrix (ECM), collectively known as stroma or matrix12. All EC-lined vessel lumens are anchored to an underlying basement membrane, a thin sheet-like ECM structure containing mainly laminin isoforms, type IV collagen, nidogens, perlecan, fibronectin, and other molecules including growth factors and the matrix metalloproteinase and Adam/Adamts proteinase inhibitor, TIMP-33,7,14–16. EC basement membrane assembly depends on heterotypic interactions with mural cells, in particular, pericytes during capillary network assembly6,17,18. Disruption of EC-mural cell interactions through genetic manipulations or blocking pericyte recruitment with various inhibitors leads to blockade to EC basement membrane assembly in vitro and in vivo18–20. ECs primarily interact with the basement membrane scaffold through adhesive interactions with key integrins on the EC surface, including the fibronectin (FN) receptor, α5β121,22 and the laminin receptors, α6β1 and α3β13,14. Major EC integrins that control vascular morphogenesis in development or postnatal life (i.e. during tissue injury and repair events) appear to be α5β1, α2β1 (a collagen receptor), and αvβ3 (an RGD-binding receptor)23, that can interact with many ECM proteins including vitronectin, fibrinogen, fibronectin, and denatured collagens23–30. In contrast, during vessel maturation and stabilization events, ECs express other β1 integrins including α1β1, α3β1, and α6β1, which are thought to interact with the BM matrix proteins, collagen type IV, laminins, nidogens, and perlecan3,14,17.
In larger arteries and veins, the vessel wall is organized into three layers which are the intima, media and adventitia, each with unique ECM compositions12. The normal arterial intimal ECM is a proteoglycan rich matrix (i.e. versican, hyaluronic acid, decorin and others) with interspersed interstitial collagens such as collagen type I, III, V and VI and adhesive ECM glycoproteins such as FN12,31–33. In contrast, the normal vascular media ECM represents a specialized structure, with circumferential layers involving elastin/ fibrillin/ fibulins/ microfibril glycoprotein-associated matrices (termed elastic lamellae) as well as intermixed and discontinuous basement membrane components (including collagen type IV, laminins, perlecan, nidogens, FN) along with interstitial collagens12,31–36. This specialized medial ECM normally suppresses vascular smooth muscle cell (VSMC) activation and promotes VSMC differentiated functions8,12,33,37. In addition, these medial ECM matrices are also highly cross-linked by lysyl oxidase (LOX) isoforms, and transglutaminases to provide mechanical stability to the highly stressed vascular wall8,12,33–35. The signaling events controlling the differentiation promoting influence of this specialized ECM (and possibly its ECM-associated growth factors) are not well understood. The VSMC receptors involved in maintaining the VSMC differentiated state include integrin and non-integrin ECM receptors, which may include a combination of the elastin receptor complex38, as well as integrins with affinity for components of the fibrillin/fibulin-rich matrix, basement membrane proteins and interstitial collagen components37. The net effect of this ECM signaling process is to promote the VSMC differentiated state maintaining cyclical contractile functions and coordinated ion channel signaling events to control these responses37. The ECM composition of the adventitial layer is an interstitial collagen-rich matrix deposited by adventitial layer fibroblasts that provides structural support for the vascular wall as well as the vasa vasorum capillary vasculature, and maintains Sca1+ progenitor cells in check, but ready to be activated following vessel injury39–42.
ECM Alterations in Vascular Disease States and Vascular Cell Responses
A central manifestation of vascular wall injury is extracellular matrix remodeling, which typically occurs in two phases (like most tissue injuries) which is acute provisional ECM deposition, followed by a chronic phase characterized by replacement of the injured cellular and matrix elements with interstitial collagenous matrices containing collagen type I, III, V, and VI, FN, osteopontin, tenascin C, and various proteoglycans such as versican, aggrecan and decorin (i.e. vessel wall fibrosis response)8,12,32,43–50. Following vascular wall injury, the differentiation and quiescence signals supplied by the healthy vascular ECM are diminished by; i) physical loss of ECM components via ECM proteolysis; ii) exposure of previous hidden integrin-binding sites in the insoluble ECM, termed matricryptic sites51–53; iii) release of ECM fragments with biological activity termed matricryptins52,54–59; iv) deposition of provisional ECM components from plasma leakage14,26,60–63, and; v) synthesis of injury-induced ECM proteins including cellular FN (i.e. EDA splice isoform), osteopontin and tenascin C64–70. In the case of vascular wall media injury, these altered matrices serve to induce VSMC de-dedifferentiation (i.e. synthetic phenotype)71 that stimulates these cells to proliferate, invade, and deposit reparative and pro-fibrotic matrices including proteoglycans, hyaluronic acid, and interstitial collagens (such as type I, III, V, and VI)72. The injured and remodeling matrices also contribute and present key growth factors including TGFβ isoforms that stimulate this pro-fibrotic vascular ECM remodeling response73. Adventitial injury can result in a further expansion of the adventitial ECM with a granulation tissue type of response due to the presence of leaky capillary vessels (from the vasa vasorum) which initiate angiogenesis and provisional ECM deposition39,41,74,75. These altered matrices in conjunction with the availability of growth factors and mediators, leads to activation of fibroblasts, inflammatory cells, and Sca1+ stem cells to stimulate a pro-fibrotic ECM response with substantial interstitial collagen deposition (which further stiffens the adventitial layer)39,40,42,76.
In recent years, there has been an increasing consideration of both outside-in versus inside-out vessel injury responses, and how this is likely to be an important pathologic feature of most vascular wall diseases40,75,77. For example, wall injury can be stimulated by leakage of plasma components into the intimal space secondary to EC layer injury or similar leakage into the adventitial space due to increased vascular permeability from capillaries in the vasa vasorum. Such processes also contribute to the entry of inflammatory cells such as neutrophils, monocytes, and lymphocytes into vessel walls from either side. It is generally assumed that intimal hyperplasia, a common pathologic finding in large and medium sized vessel injuries, is primarily due to VSMC proliferation and invasion from the medial layer into this space5,47,78. However, other work suggests that additional cell types including circulating stem cell fibrocytes, Sca1+ stem cell-derived cells (from the adventitia), ECs that have undergone endothelial mesenchymal transition (endo-MT) from luminal and intimal injury, and pericytes (which are highly invasive and proliferative) from adventitial capillaries may also contribute to the increased intimal cellularity39,79,80. To facilitate vessel wall injury responses, the adventitial layer contains many important vascular reparative cells including fibroblasts, multipotential stem cells carrying the marker Sca1+, various leukocytes including macrophages, dendritic cells, lymphocytes, and mast cells, as well as the vasa vasorum capillary vasculature composed of ECs and pericytes39–41,76,80.
An important general question concerns the nature of ECM remodeling events within vessel walls that occurs following vascular injury responses arising from different disease states (Figure 1). Vascular ECM remodeling events result from; i) leakage and deposition of plasma ECM components into the vessel wall62,72,81; ii) ECM degradation of basement membranes, elastic lamellae or interstitial collagen matrices by proteinases such as MMPs and elastases58,82–85; iii) synthesis and deposition of injury-induced ECM components including cellular FNs, osteopontin, tenascin C and proteoglycans8,34,47,49,65; and iv) exposure of matricryptic integrin binding sites and release of matricryptins52,53,56. These matricryptic sites include an abundance of RGD sites from unfolded collagens51, FNs including plasma FN86,87 and an alternatively spliced FN isoform (EDA) (also “EDGIHEL” site)26,88,89, osteopontin (also “SVVYGLR” and “ELVTDFPTDLPAT” sites)90–93 and tenascin C (also “AEIGDIEL”)94 allowing for cell injury responses mediated by αv integrins like αvβ3, αvβ195 and αvβ5 (which bind RGD sites), and α5β1 (which binds the RGD site within FN), as well as α9β1 and α4β1 (which bind the matricryptic peptide sequences in parentheses from cellular FN, osteopontin, or tenascin C). In chronic vessel injuries, i) increased proteoglycan and interstitial collagen deposition occurs in the expanded intimal space in atherosclerosis96–98; ii) deposition of interstitial collagenous matrices occurs along with hyaluronic acid, versican, aggrecan and decorin, and FN isoforms to replace damaged elastic lamellae in vascular media (due to vasculitis or genetic syndromes with deficient elastin or fibrillin1 matrices)50,99–101; iii) increased collagen deposition occurs in the adventitial ECM in hypertension and syndromes such as aortic coarctation, which constitute vascular wall fibrosis responses102–105; and iv) increased ECM cross-linking occurs due to lysyl oxidases and glycation which are observed in aging and diabetes106–108. Overall, these ECM remodeling changes lead to reduced vessel functional ability and vessel walls that are stiffer and less compliant.
Figure 1. ECM remodeling events occurring following arterial wall injury: Cellular and functional consequences.
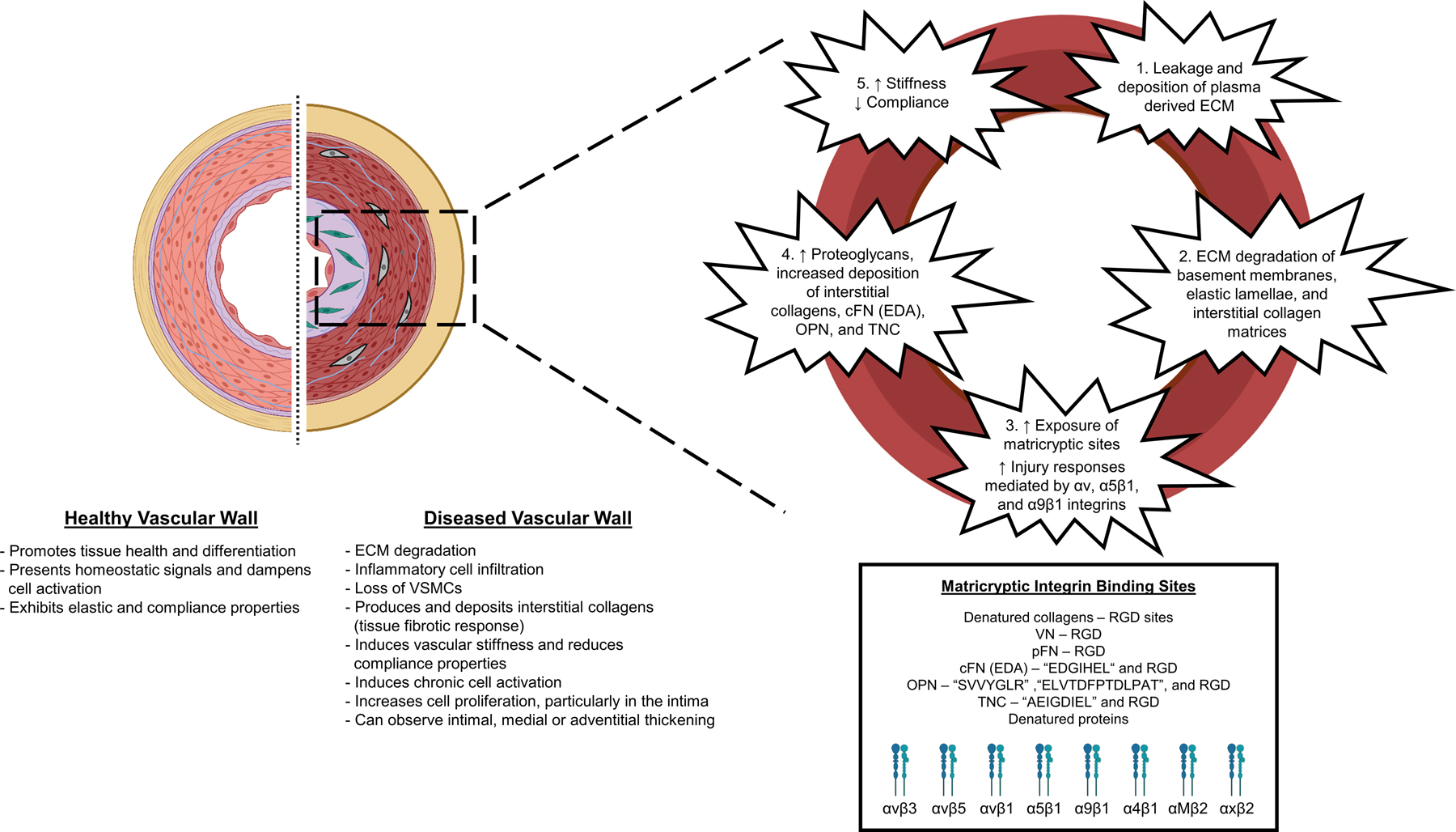
A healthy arterial wall schematic is depicted with its appropriate cellular and ECM components. The concentric elastic lamellae in healthy arterial walls provides elastic and compliant properties that are critical to normal arterial function, along with other key ECM matrices including basement membrane components underlying the ECs and interspersed among VSMCs and interstitial collagen matrices in the media and adventitia. These matrices contribute to vessel wall stabilization, maintenance of cell differentiation status and suppression of cellular activation. In the diseased arterial wall schematic, we depict changes that can occur in various vascular disease states, including intimal expansion with cell accumulation due to disruption of the internal elastic lamina, alterations in the vascular media including disruptions in the concentric elastic lamellae, loss of VSMCs, and ECM remodeling changes due to enhanced proteoglycan and interstitial collagen deposition, and increased adventitial thickening due to further interstitial collagen deposition. Such ECM changes lead to vascular wall injury responses including enhanced vascular cell proliferation and invasion (into the intimal space from the media or from the adventitial space into the media), changes in the differentiation status of ECs, VSMCs or adventitial stem cells, proteolytic destruction of ECM components, accumulation of inflammatory cells such as monocyte/macrophages, neutrophils and lymphocytes, and synthesis and replacement of damaged ECM by new proteoglycan and interstitial collagen matrices. We also describe five key steps which contribute to different stages of vascular ECM remodeling. First, the leakage and deposition of plasma-derived ECM components into the vessel wall (pFN, VN, fibrinogen). Second, ECM degradation of basement membranes, elastic lamellae or interstitial collagen matrices by proteinases such as MMPs and elastases. Third, synthesis and deposition of injury-induced ECM components (i.e. cFNs, OPN, TNC and proteoglycans), which induce exposure of matricryptic integrin binding sites: recognized through αv, α5β1, α9β1, and α4β1 integrins and, also through the macrophage integrins, αMβ2 and αxβ2. Fourth, increased proteoglycan deposition including versican and aggrecan, increased interstitial collagen deposition, and injury-induced matrices cFN (EDA splice variant), OPN, and TNC to replace degraded ECM components. Fifth, the replacement of functional elastic lamellae with rich interstitial collagenous matrices as well as proteoglycans leads to a stiffer and less compliant vessel wall. cFN: cellular fibronectin; EDA: fibronectin extra domain A; pFN: plasma fibronectin; OPN: osteopontin, TNC: tenascin C, RGD: arginine–glycine–aspartic acid, VN: vitronectin.
Since integrin-dependent ECM signaling is coordinated with growth factor receptor signaling events (see below), normal vs. injury ECM matrices will present different and unique combinations of integrin-binding sites and growth factors that will dictate whether vascular cell types, such as ECs or VSMCs maintain their differentiated state and function or become activated to participate in vessel reparative events. For example, ECs can be stimulated to undergo angiogenic responses109, undergo endoMT conversion110 or regress111, while VSMC activation leads to a “synthetic phenotype” with increased proliferation, invasion, and loss of SMC differentiation markers112. These cellular responses are attempts to repair or correct the altered vessels, but they also directly contribute to the various vascular pathologies that arise.
Major mediators of vascular wall injury leading to ECM remodeling in disease states
Major scientific efforts over decades have identified key mediators that induce vascular wall injury that stimulate cellular and ECM reparative responses to the injurious stimuli. A major regulator of vascular wall injury responses are growth factors. The vascular ECM is decorated heavily with growth factors113 and growth factor binding proteins (including those that control growth factor activity such as LTBP-1, IGFBP3, CTGF, gremlin, follistatins)73, and when alterations in the vascular ECM occur, this leads to marked changes in growth factor availability and activity3,14,99,114. A critical growth factor that is liberated and exposed following vascular wall injury is TGFβ1. TGFβ1 is normally maintained in an inactive state due to the presence of its latency associated peptide that anchors to latency binding protein (LTBP)-173,115. Furthermore, LTBP-1 binds several ECM and ECM-associated proteins including fibrillin-1, fibulins, fibronectin, and IGFBP-373. TGFβ1 activation can occur through several mechanisms including plasmin-mediated proteolysis, or mechanical activation (causing a conformation change in the TGFβ protein) by integrin binding to the RGD site in the latency associated peptide through the integrins, αvβ1, αvβ6, and αvβ873. Other important growth factors such as VEGF or PDGF-BB, and HB-EGF are also acutely liberated to activate ECs and mural cells, respectively, to stimulate vascular injury responses. EC luminal injury exposing underlying ECM leads to generation of thrombin and platelet activation, leading to release of many important growth factors, such as PDGFs and TGFβ1 and other mediators, including thrombospondin-1, sphingosine-1-phosphate and lysophosphatidic acid that have important signaling and acute provisional ECM assembly effects within the vessel wall. Leakage of fibrinogen, FN and vitronectin (VN) into the injured vessels allows for assembly of fibrin/FN/VN provisional matrices into the injured vessels (from the luminal side or from the adventitial capillaries).
Other key mediators that are important in vascular wall injury are angiotensin II, oxidized LDL, pro-inflammatory mediators including IL-1β, TNFα, and thrombin, circulating proteinase zymogens (plasminogen and plasma prekallikrein) that become activated to plasmin and plasma kallikrein, respectively116–121. Additional mediators are leukocyte proteinases such as neutrophil elastase, cathepsin G, matrix metalloproteinases (MMP)-8 and MMP-9, mast cell proteinases, tryptase and chymase, as well as vascular cell proteinases including various MMPs, MMP-1, MMP-2, MMP-3, MMP-12, MMP-13, Adamts4, and Adamts-582,83,122–125. Further regulators of vascular wall injury include glycated plasma proteins or glycated ECM proteins in diabetes126,127, and genetic mutations in the vascular wall ECM genes including elastin, fibrillin-1, fibulins, microfibril associated glycoproteins, as well as the ECM cross-linking protein, LOX12,82,101,128,129. Other mediators include oxygen radicals130, circulating metabolites like high glucose (in diabetes)127 or homocysteine46 and microbial products (i.e. microbiome), which can contribute toll-like receptor ligands, including lipopolysaccharide (LPS), that affects vascular cell responses in part due to macrophage production of the proinflammatory mediators, IL-1β and TNFα117,118,131–133. In the next sections, we will present specific examples that highlight some of the unique features of these distinct but related vascular disease states where ECM remodeling plays an important pathogenic role.
Vascular ECM Remodeling in Atherosclerosis
Three major cell types play an essential pathogenic role during different stages of the atherosclerotic process, which are ECs, monocyte/macrophages, and VSMCs. As discussed below, ECM interactions with these different cell types during the evolution of atherosclerosis are fundamental regulators of these events. Atherosclerosis begins with damage to the vascular ECs in atheroprone flow-disturbed areas that both facilitates the deposition of lipids in the subendothelial space, and the accumulation of monocyte/macrophages, to co-trigger an arterial proinflammatory process. Atherosclerotic plaque formation involves a series of sequential steps which includes; i) the accumulation of low density lipoprotein (LDL) within the intimal space; ii) conversion of native LDL to oxidized LDL; iii) EC expression of VCAM-1 allowing for monocyte recruitment into the intima and conversion to macrophages; iv) internalization of LDL by the macrophage scavenger receptors, including CD36 and SR-B1134,135, leading to macrophage foam cell transformation; and v) formation of a fibrous cap with associated smooth muscle and remodeled interstitial collagenous matrix136. A pathological diffuse intimal thickening (DIT) response occurs which contains abundant de-differentiated SMCs, and other cell types that were discussed previously108,136. These intimal cells are induced to migrate, proliferate, invade, and deposit ECM during atherogenesis, including interstitial collagens, FNs, osteopontin, tenascin C, and various proteoglycans. Within these active intimal lesions, we speculate that there will be a substantial increase in available matricryptic integrin binding sites (i.e. RGD and others) (Figure 1) from denatured collagens, fibronectin, osteopontin, and tenascin C that are used as matrix substrates for these proliferating and invading SMCs which act in conjunction with available growth factors to promote these cell behaviors. For example, the RGD-binding integrin, αvβ3, heparin-binding epidermal growth factor-like growth factor, and TGFβ are produced by and colocalized with SMCs in DIT97. In addition, cryptic sites from ECM and other denatured proteins in injury sites could serve as adhesion sites for macrophages that are localized in the remodeled intimal ECM while they ingest LDL and accumulate lipids. Macrophages express αMβ2 and αxβ2 integrins as well as α4β1, which bind denatured protein substrates137,138, including osteopontin, an ECM protein that can readily unfold due to a lack of disulfide bonds91,92. Growth factors may also be liberated from ECM binding sites through proteolysis that occurs during cell invasion or liberated from ECM following heparin release from activated mast cells (if they are present in the adventitia of the injured atherosclerotic vessel). Since activated and proliferative SMCs are a major source of proteoglycans, including versican, aggrecan, hyaluronic acid (HA) and decorin, and such proteoglycans are enriched in DIT, these intimal proteoglycans can also facilitate the localization and accumulation of LDL to this expanded ECM space136.
Other pathogenic factors in the development of atherosclerotic lesions are phenotypic and signaling changes that occur in the ECs lining the arterial wall that are exposed to oscillatory flow in combination with a hyperlipidemic metabolic state81,139,140. Under these conditions, the EC integrin, α5β1, a major fibronectin receptor, is central to activating the EC response to these injurious stimuli141,142. The cytoplasmic tail of the α5 integrin subunit is required for this response through its interactions with a phosphodiesterase isoform, PDE4D, which inactivates cyclic AMP141,143. Switching the α5 cytoplasmic tail with the α2 integrin cytoplasmic tail blocks this α5β1 integrin-dependent EC activation response, such that the underlying atherosclerotic process in the ApoE mutant mice model is reduced141,144. These data indicate that EC integrins such as α2β1, through its interactions with basement membrane matrices, suppresses the initiation and propagation of the atherosclerotic process. Perhaps other EC integrins such as the laminin receptors, α6β1 and α3β1, may also possess this ability. Thus, specific integrins, through their signaling ability, contribute to the ability of the basement membrane matrix to support EC monolayer stability and quiescence and suppress EC activation. In contrast, when vessel wall damage occurs, with increased vascular permeability or EC loss, fibronectin leaks into the vessel wall and signals ECs to drive an injury response, which includes the upregulation of NF-κB and Pak kinase signaling81,145–147. The deposition of provisional ECM components can also facilitate the activation of monocyte/macrophages or infiltrating VSMCs in the intimal space through interactions with fibronectin, vitronectin and fibrinogen/fibrin.
Other work supports this concept, where the growth factor, FGF-2, and its major receptor, FGFR1, have been observed to be centrally important for the ability of EC-lined vessels to remain stable and quiescent110,148,149. This FGF-2-dependent pathway also depends on downstream Erk1/2 activation which induces the expression of let-7 miRNAs148,150. These miRNAs regulate endo-MT151, which destabilizes the EC vessel monolayer surface. Endo-MT changes in ECs would be expected to lead to increased vascular permeability, which allows for entry of provisional ECM proteins including FN, VN, fibrinogen/ fibrin, and other mediators, that will facilitate activation of ECs, VSMCs, pericytes, and inflammatory cells. In addition, these interactions will further enhance cellular responses to vessel injurious stimuli such as oxidized LDL, oxygen free radicals, angiotensin II, and proinflammatory mediators such as IL-1β, TNFα, and thrombin. If this FGF-2 pathway is blocked, let-7 miRNAs are reduced which then drive the production of TGFβ2, a growth factor that stimulates endo-MT152. This TGFβ2-dependent endo-MT process has been shown to play an important role in the development of atherosclerotic lesions in mice with the ApoE mutant background152.
This important work raises a critical question over the role of the EC layer in inducing underlying intimal hyperplasia. We recently defined that ECs attract pericytes during capillary assembly by producing a series of five factors that stimulate pericyte invasion, proliferation, elongation on EC tube networks, survival, and pericyte-induced EC basement membrane assembly. These factors include PDGF-BB, PDGF-DD, endothelin-1 (ET-1), TGFβ1, and HB-EGF18,153. Individual blockade of each factor had significant inhibitory effects on pericyte interactions with EC-lined tubes, but only the combined blockade of all five factors caused profound inhibition of pericyte-EC interactions18,153. Under these combined blocking conditions, the pericytes behaved as if the ECs were not present. We speculate that the intimal hyperplasia responses beneath the affected EC monolayer in atherosclerosis, may involve increased production, secretion and availability of these five EC-derived factors that could directly stimulate activated VSMC cell intimal invasion, proliferation, and ECM remodeling responses. In support of this possibility is that the endo-MT conversion of ECs (induced by loss of Erk1/2 signaling and increased TGFβ2 expression) has been reported to increase production of ET-1150. ET-1 is an important stimulator of pericyte invasion and proliferation18, which could contribute to intimal hyperplasia if pericytes can migrate and invade from the adventitia or if activated SMCs similarly respond to ET-1.
Over time, atherosclerotic plaques can rupture leading to life-threatening coronary thrombosis154. Plaque rupture is the most frequent cause of arterial thrombosis155,156 and happens when the fibrous cap is thinnest and most infiltrated by macrophage foam cells154. Mechanisms leading to the thinning of the fibrous cap including loss of VSMCs and reduced interstitial collagenous matrix compared to intact caps. VSMCs are typically absent from the actual site of rupture157,158. In addition, macrophage foam cells infiltrate the cap region and produce proteinases including plasminogen activators and MMPs that contribute to plaque matrix degradation and rupture. Macrophage MMPs, including MMP-1 and MMP-8, can degrade native collagens (i.e. type I and type III collagen)159. In addition, the production of stromelysins such as MMP-3 and MMP-10 from vascular cells can also contribute to MMP-1 activation and plaque rupture since they can also degrade non-collagenous ECM components such as fibronectin and basement membrane components159,160. Another important issue is that these MMPs are secreted as zymogens and can be activated by serine proteinases originating from plasma sources (like plasminogen and plasma pre-kallikrein, which are converted to active plasmin or plasma kallikrein) or from neutrophils, macrophages or mast cells, which secrete a variety of serine proteinases including neutrophil elastase, cathepsin G, plasminogen activators, chymase and tryptase83,159,161,162. Such enzymes will be present when there is vascular injury and could be derived following leakage from the EC luminal surface, activated VSMCs and macrophages within the intimal lesion, activated VSMCs from the media, or from adventitial cell populations.
In addition, there is evidence that plaque rupture events are increased when they are associated with intraplaque angiogenesis109,163. In addition to infiltrating leukocytes like macrophages and neutrophils, ECs are also highly proteolytic cells and can strongly induce ECM proteolysis that could be important in plaque rupture events. In a previous study of human EC tube network regression in collagenous matrices, they were found to secrete proMMP-1 and proMMP-10, which when activated by various serine proteinases including plasmin and plasma kallikrein, induced marked collagen type I matrix destruction and tube collapse161. Interestingly, both MMP-1 and MMP-10 have been strongly implicated in human atherosclerotic plaque rupture160,164,165. MMP-1 is a major interstitial collagenase in humans, while MMP-10 (stromelysin-2), and the related MMP-3, are stromelysins, can strongly target non-collagenase ECM components, such as fibronectin, nidogens, various proteoglycans and many others159,162,166. MMP-3, which is produced by VSMCs, has been implicated in aneurysm formation and is upregulated following IL-1 treatment, a key cytokine involved in atherosclerosis, aneurysms, vasculitis, and capillary regression167 (see below). Thus, targeting of specific MMPs might represent a new therapeutic strategy to reduce plaque rupture, but also other critical vascular pathologies.
Vascular ECM remodeling in aortic aneurysms: Genetic and pro-inflammatory predispositions
Over the past decades, considerable progress has been made investigating and elucidating the cellular and molecular basis for thoracic and abdominal aortic aneurysms. There are also differences in the pathogenic and genetic basis for thoracic vs. abdominal aortic aneurysms82,168–171. In general terms, thoracic aortic aneurysms have a frequent association with genetic mutations (or familial association) including Marfan’s syndrome (fibrillin-1 loss of function)101,172,173, Loeys-Dietz syndrome (with loss of function mutations in TGFβ and TGFβ receptor or downstream signaling genes including TGFBR2, TGFBR1, TGFβ2, or Smad2/3)174–176, and Ehlers-Danlos syndrome (with loss of function of collagen type III)169 and can occur in younger or older patients. In contrast, abdominal aortic aneurysms have strong association with risk-factors such as smoking, with some degree of association with a family history, and a lesser association with risk factors related to atherosclerotic disease, such as hypertension and hypercholesterolemia, and typically are observed in older patients from 65–75 years of age168. However, experimental models of abdominal aortic aneurysms in the mouse can be induced by combining the administration of angiotensin II with hypercholesterolemia (i.e. apoE or LDL receptor knockouts), although angiotensin II alone also is capable of causing these aneurysms, but less frequently 177–179. Common pathogenic features of these two major types of aortic aneurysms include the loss of ECM integrity in the arterial media or adventitia in conjunction with the loss of VSMCs in the arterial media, which lead to vessel ballooning and enlargement over time due to the continuous pulsatile flow and pressure forces8,12,170,171. Normally, the elastic vascular media ECM that is reinforced with interstitial collagenous matrices (that are also covalently cross-linked), the arterial VSMC-rich media, as well as the adventitial collagenous matrix resist these forces to maintain the structural integrity, diameter, and function of the aorta throughout life12. Many experimental animal models have been created to investigate the development and therapeutics of aortic aneurysms. These models include, i) genetic deletion of vascular ECM components (e.g. fibrillin-1, fibulin4) or growth factor receptors (TGFβR2); ii) administration of angiotensin II usually in combination with hypercholesterolemia; and iii) supplying ECM degrading proteinases such as elastases including the elastolytic MMP-12 enzyme; and iv) disruption of ECM cross-links using depletion of copper to inhibit LOX or via genetic deletion of LOX family enzymes33,35,82,99,179–183. Furthermore, there is a clear association between proinflammatory signals and inflammatory cell infiltration with aneurysm development, particularly with abdominal aortic aneurysms117,184. A variety of MMPs have been strongly implicated in the pathogenic development and progression of aortic aneurysms including MMP-1, MMP-3, MMP-13, MMP-9, MMP-2, MMP-12, and MT1-MMP (MMP-14)168,185–190. Thus, inducing inflammation and inflammatory infiltration enhances aneurysm development by stimulation of vascular ECM degradation mediated through inflammatory cells such as macrophages, and neutrophils, but also VSMCs, invading pro-fibrogenic cells, and pro-angiogenic ECs.
The depletion or loss of cross-links in the elastic lamellae or interstitial collagenous matrices within the arterial media represents a major predisposing factor to develop aneurysms, which also leads to marked vascular ECM remodeling responses33,35,82. These critical ECM structures are then replaced by a reparative fibrosis-like matrix response, involving predominantly the deposition of interstitial collagenous matrices (type I, III, V, VI collagens) with associated proteoglycans, including versican and aggrecan (which accumulate in aneurysms), FNs, osteopontin, and tenascin C (as detailed above). This reparative ECM environment will stimulate previously quiescent SMCs to become activated to invade and proliferate in conjunction with the availability of growth factors including PDGFs, TGFβ isoforms, and HB-EGF. An important marker of activated and proliferative SMCs is αvβ3, a major RGD-binding integrin191,192, and the transcription factor, KLF4, an important regulator of cell activation, injury responses, and stem cell proliferation71,193. Interestingly, in mouse models of intimal injury and proliferation, conditional SMC knockout of KLF4 leads to increased intimal proliferative responses, suggesting that KLF4 has an inhibitory role in this response, despite its ability to induce SMC dedifferentiation events193. Other studies have implicated the role of the integrin α9β1 in SMC responses to arterial media damage194, which is a receptor for the injury induced ECM proteins, cellular FN (EDA splice variant)88, osteopontin90 and tenascin C94, which express matricryptic binding sites for both RGD-binding integrins and separately, α9β1 (Figure 1).
Of great interest, are findings in the genetic disease, Williams-Beuren’s syndrome, which demonstrate elastin gene deficiency195. In this disease, highly proliferative VSMCs invade the aortic intimal space and occlude the lumen of the aorta causing supravalvular aortic stenosis195. In addition, this elastin deficiency can also result in stenosis of other large and small arteries through the same intimal hyperplasia mechanism. Mouse knockouts of the elastin gene were developed to mimic this elastin deficiency state, and several studies demonstrated that they were able to recapitulate these findings in the mouse181,191,196. Genetic and phenotypic analysis of this proliferative VSMC population which expands the aortic intima and occludes the luminal space, demonstrates strong inductions of the integrin αvβ3 and blockade of this integrin in vivo using RGD peptide reagents or knockout of the β3 integrin subunit reduced the VSMC proliferative response191. The same group has now identified that VSMC Notch3 and Jagged-1 also control this VSMC proliferative response following elastin knockout in the mouse192. Elastin knockout leads to upregulation of jagged-1 in both VSMCs and ECs to activate Notch3 expressed on VSMCs leading generation of the Notch3 intracellular domain (NICD3), a transcriptional regulator192. They also provided evidence that NICD3 activates the expression of the β3 integrin subunit in VSMCs, and that blockade of the Notch3 pathway inhibited the expression of the β3 integrin as well as the VSMC intimal proliferative response192. It is interesting to speculate whether the ECM remodeling events (with increased availability of RGD containing matrix binding sites) that occur following genetic or pathologic loss of the vascular elastin matrix, might further enhance this Jagged-1 and Notch-3 signaling response to promote VSMC proliferative and invasion responses.
Finally, there is considerable interest and effort in evaluating the importance of cell-mediated mechanical forces in the development of aortic aneurysms8,82,170,197. Cell-matrix interactions, particularly from the vascular media VSMCs and the adventitial fibroblasts, generate forces that affect the structure and functional integrity of the aortic wall and promote mechanosensory homeostasis. The vessel wall ECM bears the great majority of stress that is exerted by blood pressure forces and protects the vascular cells from stress-induced damage170. In contrast, when ECM disruptions or ECM remodeling events occur, decreased mechanosensing may ensue, leading to adaptations resulting from reduced wall stress that induce VSMC death, increased production of ECM-degrading proteinases, and thinning of the vessel wall (all characteristics of aneurysms)170. In a detailed biomechanical analysis and comparison of 10 different mouse models of ascending thoracic aortic aneurysms, two major conclusions were observed which are: 1) increased circumferential material stiffness correlated well with the degree of aortic dilation in the aneurysm, and 2) decreased mechanical functionality of the aorta did not appear to contribute directly to the aneurysmal dilation198. Overall, these results suggested that dysfunctional cellular sensing and/or the regulation of the altered vascular ECM environment underlies the development and progression of thoracic aortic aneurysms in the mouse198.
Vascular ECM remodeling in vasculitis syndromes
There are a wide variety of human vasculitides which affect vessels of all sizes and involve the infiltration of the vessels with inflammatory cells including neutrophils, monocyte/macrophages, and lymphocytes, such as various T cell subtypes199–203. The causative etiologies vary and include autoimmune reactions to vessel antigens from circulating antibodies or cytotoxic T cells, immune complex deposition, infections, drug reactions, and genetic mutations. Destruction of vessel walls occurs due to ECM degradation which can occur in the intimal, medial or adventitial matrices in large and medium sized vessels, and which can also be accompanied by cell loss due to destruction of ECs, VSMCs or adventitial cells. The vessel wall matrix destruction leads to marked ECM remodeling which is similar to that observed in the other vessel wall diseases discussed above with a vessel wall fibrosis-like response. Of great interest is that proinflammatory mediators appear to play a central role in these vascular destruction processes and include varying roles for IL-1β, IL-1α, TNFα, IL-6 and IFNγ133,199,201,202. Blockade of such mediators is demonstrating significant clinical benefits in specific types of vasculitis. Other types of therapeutics such as corticosteroids and intravenous immunoglobulin (IVIG) have been also useful. Briefly, we will highlight several key vasculitis conditions including giant cell arteritis, Takayasu’s arteritis, and Kawasaki’s disease.
Giant cell arteritis is a common form of vasculitis in elderly patients, and typically observed in large and medium sized vessels of the head, and, in particular, is observed in the temporal artery, but also can involve the ophthalmic and vertebral arteries200,203. The histopathology observed shows intimal hyperplasia with luminal narrowing and occasional thrombosis. These cellular and the ECM remodeling events occur downstream of elastin lamellae destruction secondary to infiltrating leukocytes and proteinase-dependent matrix degradation. There is now considerable evidence for an autoimmune cytotoxic T cell response to the vascular media in this disease200. The vascular media is normally an immune privileged site and alterations in the ability of PD-1/ PD-L1 signaling to suppress cytotoxic T cell function leads to enhanced T cell mediated cytotoxicity within these arteries occurs in this type of vasculitis133,200. In addition, the infiltration and activation of lymphocytes and macrophages results in the production of proinflammatory cell mediators and ECM degrading proteinases to damage the arterial media. Takayasu’s arteritis is a large and medium sized vessel vasculitis that is observed in patient’s typically younger than 50 years202. The disease can also occur in children. It typically involves the aortic arch as well as the great vessels including the brachiocephalic, carotid, and subclavian arteries. Marked luminal narrowing can occur in these vessels due to arterial media destruction combined with intimal hyperplasia and, thus, the disease process has been referred to as “pulseless disease”. Infiltration of mononuclear cells including macrophages, lymphocytes, and multinucleated giant cells are observed along with medial wall destruction, extensive fibrosis, intimal hyperplasia, and luminal narrowing202. Some therapeutic successes have occurred with corticosteroids and more recently with blocking antibodies directed to TNFα or IL-6, where disease remissions can occur in some cases202.
Kawasaki’s disease is a vasculitis that involves medium and small sized vessels and has a predilection to involve the coronary arteries, with transmural inflammation involving monocyte/macrophages and activated T cells (both CD8 and CD4), and the development of aneurysms204. As with the other vasculitides, there is significant ECM remodeling that can be observed throughout the vessel wall which represents a fibrosis type of response in conjunction with observed focal disruptions of the elastin/fibrillin matrices in the arterial media. The coronary artery aneurysms can rupture causing thrombosis, and, in addition, result in myocardial infarction in pediatric patients that are typically 4 years old or younger204. A mouse model of Kawasaki’s disease has been developed by intraperitoneal injection of a cell wall extract from the bacterium, Lactobacillus casei (LCWE)204–206. This extract is known to activate the toll-like receptor, TLR2, which can stimulate macrophages to produce and secrete the key proinflammatory mediators, IL-1β, IL-1α, TNFα, and IL-6205. In this model, marked vasculitis of coronary arteries occurs as well as the development of coronary artery aneurysms205,206. In addition, abdominal aortic aneurysms develop in a high frequency of mice injected with this bacterial extract184. These profound vascular changes in either the coronary vessels or abdominal aorta in the response to LCWE administration are markedly blocked by genetic deletion of IL-1 isoforms or the IL-1 receptor gene, IL-1R1184,199,206. In addition, administration of IL-1 blocking antibodies or IL-1 receptor antagonist (IL-1RA), which interferes with the activities of IL-1β or IL-1α, also leads to blockade of the vascular damage199,206,207. In contrast, administration of IL-6 blocking agents fails to inhibit the vascular damage response but does interfere with the induction of plasma acute phase reactants following injection of LCWE208. Studies in humans also supports the concept that IL-1 is an important cytokine mediator of Kawasaki’s disease. Genetic analysis of these patients shows a strong induction of IL-1 and its downstream effector pathways compared to controls implying its potential pathogenic role in the disease209. In support of this conclusion, administration of IL-1 antagonists such as IL-1RA has resulted in significant clinical improvement in children (including regression of coronary artery aneurysms) with Kawasaki’s disease210–212. Interestingly, other studies in children have also revealed clinical improvement by administration of TNFα blocking antibodies213. Perhaps combining blocking reagents for IL-1 and TNFα would further improve the clinical response and outcome (see discussion below on capillary regression).
Vascular ECM remodeling in capillary regression (rarefaction)
There is an increasing appreciation that capillary regression, which is also termed capillary rarefaction, is an important regulator of major human disease states including hypertension, diabetes, heart failure (particularly heart failure with preserved ejection fraction), ischemia and infarction events, sepsis, malignant cancers, neurodegenerative diseases including Alzheimer’s disease, and aging111,214–223. In large, medium and small arteries, the presence of an intima, media and adventitia layer, which is structurally delineated by an EC basement membrane, by the elastic lamellae and other intervening matrices in the media, the circumferentially oriented VSMC layers, and adventitial interstitial collagenous matrices provide a highly stabilized and cross-linked ECM structure that maintains vessel structure and function despite significant vascular pathologic disease processes (Figure 2). Exposure of these larger arteries and veins to injurious stimuli in comparison to capillaries, results in both cellular and ECM remodeling responses that we have described above in atherosclerosis, aneurysms, hypertension, and vasculitis syndromes. The larger vessels survive and persist despite the injurious insults and markedly change their cellular composition, and functional abilities as they acquire inflammatory or reparative cell types, and at the same time change their ECM compositions (Figure 2). By contrast, the smallest and most numerous blood vessels are capillary tube networks, which consist of a single layer of ECs, and a single layer of pericytes present on the EC abluminal tube surface which are both attached to an intervening basement membrane matrix. These capillaries are much less stable and more dynamic, by comparison with larger arteries and veins, due to a lack of an intimal, media or adventitial layer and diminished ECM cross-linking. Exposure of less stabilized capillaries to similar injurious stimuli (compared to larger vessels) results in capillary regression or loss (Figure 2), which can lead to increased vascular permeability, hemorrhage, thrombosis, and tissue perfusion deficits with low oxygenation and nutrient delivery.
Figure 2. Comparative structure and injury responses of the arterial versus capillary vasculatures under healthy and disease states.
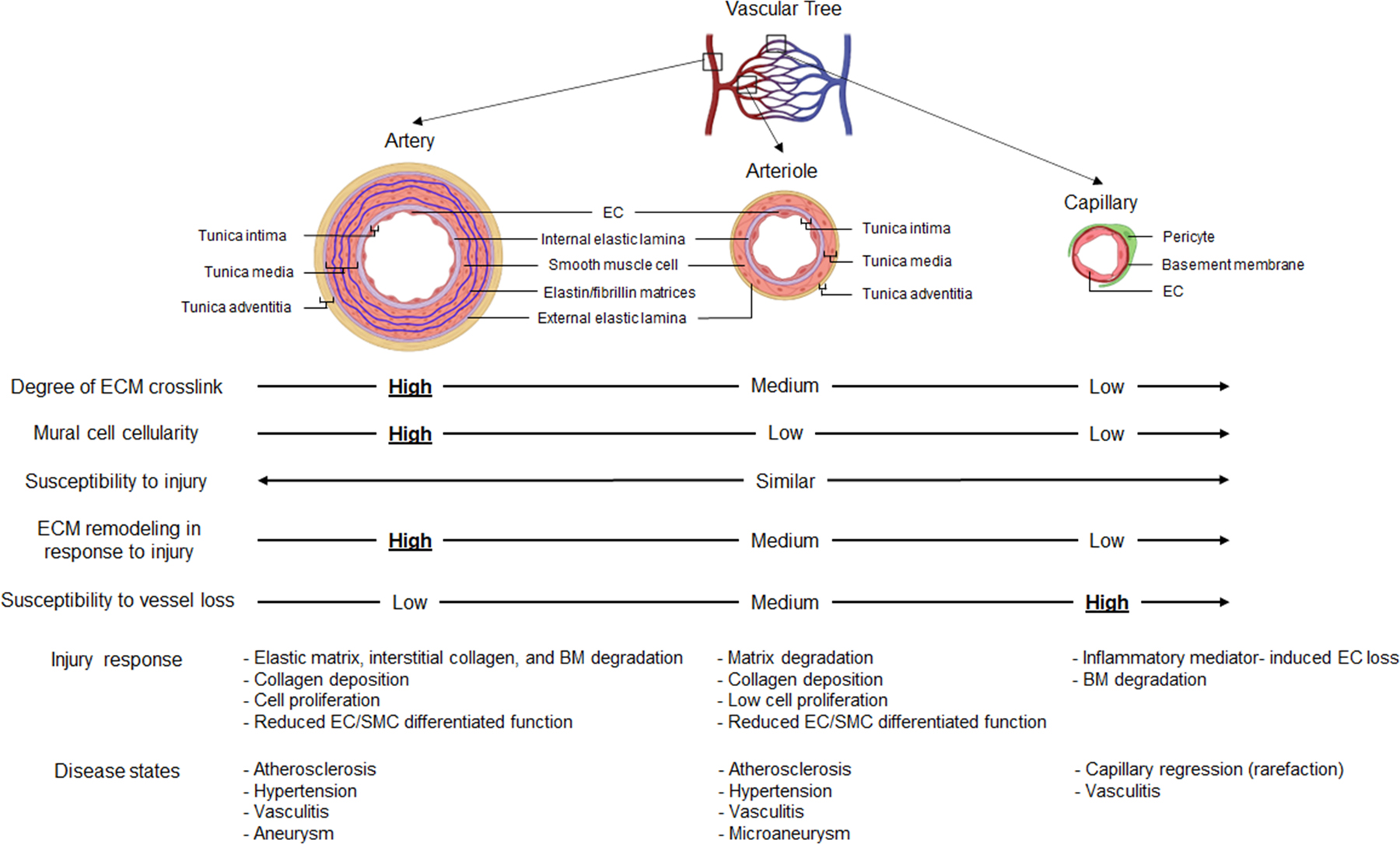
A single layer of endothelial cells (ECs) lines the entire vascular system, while the ECM is the major constituent of the vessel wall, particularly in larger vessels. Arteries and arterioles both have three ECM layers: tunica intima (a single layer of ECs attached to the subendothelial layer and internal elastic lamina), tunica media (circumferential layers of vascular smooth muscle cells and elastic lamellae with interspersed basement membrane and interstitial matrix components), and tunica adventitia (separated from tunica media by external elastic lamina and contains fibroblasts, progenitor cells, immune cells, and a capillary vasculature embedded in interstitial collagenous matrices). On the contrary, capillaries consist only of a single layer of ECs, a thin sheet of basement membrane, and surrounding pericytes. Compared to arteries and arterioles, capillaries have the least amount of matrix crosslinking and mural cell cellularity, which is represented by smooth muscle in arteries/arterioles, and pericytes in capillaries. Because of the structural and ECM differences between arteries/arterioles compared to capillaries, the two vessel groups react in a markedly distinct manner when exposed to injurious stimuli. ECM remodeling in response to injury occurs mostly in arteries and least in capillaries. Under disease states, capillaries are most prone to vascular regression due to inherently less interstitial collagen matrices (and a lack of an elastin-rich matrix), fewer mural cells, and reduced matrix cross-linking compared to arteries and arterioles. During injury responses, arteries and arterioles undergo matrix degradation (including degradation of elastic lamellae, interstitial collagens, and basement membrane), physical and functional cell loss with SMC dedifferentiation events, reactive ECM remodeling that includes provisional matrix deposition, exposure of previously hidden matricryptic integrin binding sites, and synthesis of new injury-induced ECM components as well as interstitial collagens and proteoglycans (i.e. vessel fibrosis response); and intimal hyperplasia of activated SMCs as well as other cell types which accumulate from the circulation or the adventitia. Key growth factors, such as TGFβ isoforms, and proinflammatory mediators such as IL-1β, TNFα and thrombin are liberated during these injuries to regulate these processes. These adverse changes in arteries and arterioles lead to mostly chronic disease states (the larger more stable vessels persist) including atherosclerosis, hypertension, aneurysms (microaneurysms in very small arteries and arterioles), and vasculitis. In contrast, capillaries readily undergo inflammatory mediator-induced tube regression with EC loss and accompanying degradation of the basement membrane. A spectrum of responses occurs downstream of capillary regression including increased vascular permeability, tissue hemorrhage, and parenchymal cell dysfunction or loss due to ischemia, lack of nutrients, or reduced removal of waste products. EC: endothelial cell, ECM: extracellular matrix, BM: basement membrane, SM: smooth muscle cell.
A number of past and recent studies have been performed to address the underlying basis for capillary tube network regression. Three distinct categories appear to underlie this process, and include i) ECM proteolysis that degrades the matrix scaffolds in which capillary tubes are embedded that involves MMP-1 and MMP-10, and activating serine proteinases such as plasmin and plasma kallikrein161,224–227; ii) proinflammatory mediators such as IL-1β, TNFα, thrombin and IFNγ, which appear to directly induce regression of blood capillaries111, but also lymphatic capillaries216; and iii) growth factor deficiency states which have been described in the context of the declining availability of VEGF during aging228.
ECM proteolysis is a key mechanism by which capillary vessels can regress. ECs are remarkably proteolytic and can readily degrade the matrices in which they are embedded. A series of in vitro as well as in vivo findings support this conclusion. Using a serum-free defined system of EC tubulogenesis in 3D collagen matrices, the addition of plasminogen led to conversion to plasmin by EC plasminogen activators, which further activated pro-MMP-1 and pro-MMP-10, which degraded the matrix causing tube regression224. Other serine proteinases including plasma kallikrein, the neutrophil-derived serine proteinases, neutrophil elastase and cathepsin G, and the mast cell serine proteinases, chymase and tryptase, are all capable of activating pro-MMP-1 and pro-MMP-10 to induce EC tube regression161. Of great interest are findings that EC-pericyte interactions decrease this mechanism of MMP-dependent tube regression by the production of TIMP-2 and TIMP-3, the latter is primarily made by pericytes225. Several genetic knockout studies in mice also support these findings. Hdac7 knockouts in mice lead to marked elevations of MMP-10 and decreased levels of TIMP-1, leading to an embryonic lethal phenotype caused by vascular network breakdown with hemorrhage227. In addition, genetic deletion of the epigenetic modifier, CHD4, resulted in plasminogen and plasmin-dependent vascular network regression that also was embryonic lethal226. Finally, mouse knockout of the MMP inhibitor, RECK, caused ECM degradation which again resulted in vessel breakdown and embryonic lethality during vascular development229.
Another mechanism of capillary regression occurs following exposure of capillary tubes to pro-inflammatory mediators111,214. Using a serum-free defined human capillary tube assembly model, a broad screen of defined factors identified that the major proinflammatory mediators, IL-1β, IL-1α, TNFα and thrombin had potent pro-regressive activity which was further enhanced when the mediators were combined111. Macrophage activation by the toll-like receptor agonists, Pam3CSK4 (which activates TLR2), or lipopolysaccharide (which activates TLR4), both lead to the secretion of pro-regressive activity that was completely neutralized by blocking antibodies to IL-1β and TNFα, but not IL-1α or IL-6111. Addition of IL-6 to capillary tubes failed to induce regression. This study also identified other factors with pro-regressive activity including IFNγ, BMP-9, BMP-10, IL-4, TGFβ1 and TGFβ2 while many other members of the IL-1 and TNF protein families did not possess pro-regressive activity111. Additional work demonstrated that the physiological process of hyaloid vascular regression in the postnatal mouse eye could also be abrogated by blocking antibodies to IL-1β and TNFα or by administration of the anti-inflammatory mediator, IL-10111. An important point is that these pro-regressive mediators selectively induced EC tube collapse and apoptosis, while the pericytes remained intact, but appeared to be activated during and following the regression process111. Perhaps these pericytes could then participate in regenerative processes after injury to promote angiogenesis and tissue repair events including matrix remodeling (i.e. tissue fibrosis responses). The observed pericyte responses that we have described111 are very analogous to the VSMC responses observed in the large vessel wall injuries discussed above. It is remarkable, but perhaps not surprising, that separate studies evaluating the molecular basis for capillary regression have identified many of the same mediators and causative etiologies (e.g. hypertension) that are known to regulate the development of key large and medium sized vessel disease processes in humans including atherosclerosis, aneurysms, and vasculitis (Figure 2).
Conclusions
In this review, we have addressed how vascular injuries of varying types (and which manifest as major human vascular disease states) lead to pathological vessel wall changes which include ECM remodeling. The key ECM remodeling events that occur involve; i) entry and deposition of plasma-derived ECM components; ii) synthesis and deposition of injury-induced ECM components from vascular wall cells or inflammatory cells; iii) ECM degradation from MMPs, Adamts, and serine proteinases such as elastase; iv) ECM deposition to remodel and fill in injured vessel areas due to ECM degradation or vascular cell loss (i.e. VSMC loss); v) exposure of matricryptic sites within insoluble ECM proteins (such as RGD) to facilitate vascular cell and inflammatory cell activation within the injured wall; vi) release of soluble ECM fragments (matricryptins) which can further activate these cells; and vii) with sustained vessel injury, ECM remodeling involving interstitial collagen deposition along with the deposition of proteoglycans and FNs, representing a vessel wall fibrosis response (Figure 1). We highlighted specific vascular diseases, including atherosclerosis, aneurysms, vasculitides, and capillary regression, and discussed how the unique events within the vascular wall are affected by cellular infiltration, proliferation, and invasion, cell-mediated ECM proteolysis, and how growth factors, peptides and proinflammatory mediators differentially affect arteries compared to capillaries. It is intriguing to consider just how stable large and medium sized are, even when exposed to significant injurious stimuli over time (Figure 2). These vessels can thicken and become mechanically stiffer due to intimal hyperplasia, media, or adventitial injury with substantial interstitial collagen deposition, or they can undergo aneurysmal dilation or undergo dissections due to focal ECM loss, particularly of the elastic lamellae. In contrast, capillaries, being much less stable, are prone to regress after exposure to the same injurious agents (Figure 2).
It has become increasingly apparent that particular growth factors, peptides, and proinflammatory mediators are fundamentally important in these vascular diseases and for the development of ECM remodeling events within the different vessel types. Such vascular disease-inducing factors include angiotensin II, ET-1, oxidized LDL, hyperglycemia, TGFβ isoforms, IL-1β, IL-1α, TNFα, thrombin IFNγ, MMP-1, MMP-3, MMP-10, MMP-12, Adamts4, Adamts-5, and various serine proteinases including plasmin, plasma kallikrein and neutrophil elastase. In addition, therapeutic blockade of some of these molecules has substantially aided in the clinical treatment of these vascular diseases. For example, the identification of IL-1β as an important mediator in many vascular diseases has led to improved therapeutics for atherosclerosis, and vasculitides, and in the future could be used for preventing capillary regression. In most clinical studies to date, only one of the proinflammatory mediators is targeted at a time. It would be interesting to combine blocking agents directed to multiple mediators to see how much further the clinical benefit might be. In our studies of capillary regression, we have shown that thrombin enhances the pro-regressive impact of molecules such as IL-1β and TNFα111,216; we suggest that adding thrombin inhibition (by inhibiting Factor Xa), in combination with antagonists of IL-1β and TNFα, might also aid in such therapeutic approaches. Finally, the development of more specific MMP or Adamts inhibitors, or greater use of the combined MMP and Adamts inhibitor, TIMP-3, might further enhance the therapeutics of many vascular wall injuries.
Highlights.
Vascular wall extracellular matrix remodeling is an important pathogenic feature of many vascular disease states.
Vascular injuries induce changes in vascular wall cells from a homeostatic, more differentiated state to an activated, more proliferative, invasive, and de-differentiated state that occurs in part due to their responses to an altered ECM environment.
Vascular ECM remodeling events include accumulation of provisional ECM proteins from vascular leakage, exposure of matricryptic sites from ECM proteolysis and deposition of injury-induced ECM proteins, and replacement of degraded elastic lamellae or basement membrane matrices with proteoglycan- and interstitial collagen-rich matrices.
Large and medium sized arteries can withstand considerable cellular and ECM remodeling changes that persist and progress over time due to exposure of injurious stimuli to the vascular wall, while capillaries will more readily undergo regression responses (rarefaction) in response to the same stimuli.
Sources of funding:
This work was supported by NIH grants HL149748 and HL126518 (GED) and an AHA predoctoral fellowship award (PKL).
Abbreviations
- ECM
Extracellular matrix
- EC
Endothelial cell
- Adam
A disintegrin and metalloproteinase
- Adamts
A disintegrin and metalloproteinase with thrombospondin motifs
- BM
Basement membrane
- VSMC
Vascular smooth muscle
- EDA
fibronectin extra domain A
- TGFβ
Transforming growth factor beta
- Sca
Stem cells antigen
- EndoMT
Endothelial to mesenchymal transition
- LTBP-1
Latent transforming growth factor beta binding protein 1
- IGFBP3
Insulin-like growth factor binding protein 3
- CTGF
Connective tissue growth factor
- RGD
Arginine-glycine-aspartic acid
- VEGF
Vascular endothelial growth factor
- PDGF-BB
Platelet-Derived Growth Factor-BB
- HB-EGF
Heparin-binding EGF-like growth factor
- FN
Fibronectin
- VN
Vitronectin
- LDL
Low-density lipoprotein
- IL
Interleukin
- TNFα
Tumour necrosis factor alpha
- MMP
Matrix metalloproteinase
- LOX
Lysyl oxidases
- LPS
Lipopolysaccharide
- DIT
Diffuse intimal thickening
- HA
Hyaluronic acid
- PDE4
Phosphodiesterase type 4
- Cyclic AMP
Cyclic adenosine monophosphate
- ApoE
Apolipoprotein E
- NF-κB
Nuclear factor kappa B
- FGF-2
Fibroblast growth factor 2
- Erk1/2
Extracellular signal‑regulated protein kinase
- miRNA
MicroRNA
- S1P
Sphingosine-1-phosphate
- KLF4
Krüppel-like factor 4
- NICD3
Notch3 intracellular domain
- IFNγ
Interferon γ
- IVIG
Intravenous immunoglobulin
- PD-1
Programmed cell death 1
- PD-L1
Programmed death ligand-1
- CD
Cluster of differentiation
- LCWE
Lactobacillus casei wall extract
- TLR2
Toll-like receptor 2
- IL-1RA
Interleukin -1 receptor antagonist
- IL-1R
Interleukin -1 receptor
- Hdac7
Histone deacetylase 7
- CHD4
Chromodomain-helicase-DNA-binding protein 4
- RECK
Reversion-inducing, cysteine-rich protein with Kazal motifs
- Pam3CSK4
Pam3CysSerLys4
- BMP
Bone morphogenetic protein
Footnotes
Disclosures: The authors declare no competing financial interests related to this review.
References
- 1.Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol 2007;8:464–478. [DOI] [PubMed] [Google Scholar]
- 2.Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell 2011;146:873–887. doi: 10.1016/j.cell.2011.08.039 [DOI] [PubMed] [Google Scholar]
- 3.Senger DR, Davis GE. Angiogenesis. Cold Spring Harb Perspect Biol 2011;3:a005090. doi: 10.1101/cshperspect.a005090 a005090 [pii] cshperspect.a005090 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Augustin HG, Koh GY. Organotypic vasculature: From descriptive heterogeneity to functional pathophysiology. Science 2017;357. doi: 10.1126/science.aal2379 [DOI] [PubMed] [Google Scholar]
- 5.Mazurek R, Dave JM, Chandran RR, Misra A, Sheikh AQ, Greif DM. Vascular Cells in Blood Vessel Wall Development and Disease. Adv Pharmacol 2017;78:323–350. doi: 10.1016/bs.apha.2016.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kemp SS, Lin PK, Sun Z, Castano MA, Yrigoin K, Penn MR, Davis GE. Molecular basis for pericyte-induced capillary tube network assembly and maturation. Front Cell Dev Biol 2022;10:943533. doi: 10.3389/fcell.2022.943533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davis GE, Kemp SS. Extracellular Matrix Regulation of Vascular Morphogenesis, Maturation, and Stabilization. Cold Spring Harb Perspect Med 2022. doi: 10.1101/cshperspect.a041156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Humphrey JD, Schwartz MA. Vascular Mechanobiology: Homeostasis, Adaptation, and Disease. Annu Rev Biomed Eng 2021;23:1–27. doi: 10.1146/annurev-bioeng-092419-060810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rafii S, Butler JM, Ding BS. Angiocrine functions of organ-specific endothelial cells. Nature 2016;529:316–325. doi: 10.1038/nature17040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramasamy SK, Kusumbe AP, Adams RH. Regulation of tissue morphogenesis by endothelial cell-derived signals. Trends Cell Biol 2015;25:148–157. doi: 10.1016/j.tcb.2014.11.007 S0962-8924(14)00210-4 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis GE, Norden PR, Bowers SL. Molecular control of capillary morphogenesis and maturation by recognition and remodeling of the extracellular matrix: functional roles of endothelial cells and pericytes in health and disease. Connect Tissue Res 2015;56:392–402. doi: 10.3109/03008207.2015.1066781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wagenseil JE, Mecham RP. Vascular extracellular matrix and arterial mechanics. Physiol Rev 2009;89:957–989. doi: 10.1152/physrev.00041.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rocha SF, Adams RH. Molecular differentiation and specialization of vascular beds. Angiogenesis 2009;12:139–147. doi: 10.1007/s10456-009-9132-x [DOI] [PubMed] [Google Scholar]
- 14.Davis GE, Senger DR. Endothelial extracellular matrix: biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ Res 2005;97:1093–1107. doi: 10.1161/01.RES.0000191547.64391.e3 [DOI] [PubMed] [Google Scholar]
- 15.Pozzi A, Yurchenco PD, Iozzo RV. The nature and biology of basement membranes. Matrix Biol 2017;57–58:1–11. doi: 10.1016/j.matbio.2016.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yurchenco PD. Basement membranes: cell scaffoldings and signaling platforms. Cold Spring Harb Perspect Biol 2011;3. doi: 10.1101/cshperspect.a004911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stratman AN, Malotte KM, Mahan RD, Davis MJ, Davis GE. Pericyte recruitment during vasculogenic tube assembly stimulates endothelial basement membrane matrix formation. Blood 2009;114:5091–5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kemp SS, Aguera KN, Cha B, Davis GE. Defining Endothelial Cell-Derived Factors That Promote Pericyte Recruitment and Capillary Network Assembly. Arterioscler Thromb Vasc Biol 2020;40:2632–2648. doi: 10.1161/ATVBAHA.120.314948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stratman AN, Schwindt AE, Malotte KM, Davis GE. Endothelial-derived PDGF-BB and HB-EGF coordinately regulate pericyte recruitment during vasculogenic tube assembly and stabilization. Blood 2010;116:4720–4730. doi: 10.1182/blood-2010-05-286872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stratman AN, Pezoa SA, Farrelly OM, Castranova D, Dye LE, 3rd, Butler MG, Sidik H, Talbot WS, Weinstein BM. Interactions between mural cells and endothelial cells stabilize the developing zebrafish dorsal aorta. Development 2017;144:115–127. doi: 10.1242/dev.143131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruoslahti E Fibronectin and its receptors. Annu Rev Biochem 1988;57:375–413. doi: 10.1146/annurev.bi.57.070188.002111 [DOI] [PubMed] [Google Scholar]
- 22.Pytela R, Pierschbacher MD, Ruoslahti E. Identification and isolation of a 140 kd cell surface glycoprotein with properties expected of a fibronectin receptor. Cell 1985;40:191–198. doi: 10.1016/0092-8674(85)90322-8 [DOI] [PubMed] [Google Scholar]
- 23.Pytela R, Pierschbacher MD, Ruoslahti E. A 125/115-kDa cell surface receptor specific for vitronectin interacts with the arginine-glycine-aspartic acid adhesion sequence derived from fibronectin. Proc Natl Acad Sci U S A 1985;82:5766–5770. doi: 10.1073/pnas.82.17.5766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Francis SE, Goh KL, Hodivala-Dilke K, Bader BL, Stark M, Davidson D, Hynes RO. Central roles of alpha5beta1 integrin and fibronectin in vascular development in mouse embryos and embryoid bodies. Arterioscler Thromb Vasc Biol 2002;22:927–933. doi: 10.1161/01.atv.0000016045.93313.f2 [DOI] [PubMed] [Google Scholar]
- 25.Bayless KJ, Salazar R, Davis GE. RGD-dependent vacuolation and lumen formation observed during endothelial cell morphogenesis in three-dimensional fibrin matrices involves the alpha(v)beta(3) and alpha(5)beta(1) integrins. Am J Pathol 2000;156:1673–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Astrof S, Hynes RO. Fibronectins in vascular morphogenesis. Angiogenesis 2009;12:165–175. doi: 10.1007/s10456-009-9136-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.San Antonio JD, Zoeller JJ, Habursky K, Turner K, Pimtong W, Burrows M, Choi S, Basra S, Bennett JS, DeGrado WF, et al. A key role for the integrin alpha2beta1 in experimental and developmental angiogenesis. Am J Pathol 2009;175:1338–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davis GE, Stratman AN, Sacharidou A, Koh W. Molecular basis for endothelial lumen formation and tubulogenesis during vasculogenesis and angiogenic sprouting. Int Rev Cell Mol Biol 2011;288:101–165. doi: 10.1016/B978-0-12-386041-5.00003-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science 1994;264:569–571. doi: 10.1126/science.7512751 [DOI] [PubMed] [Google Scholar]
- 30.Davis GE, Camarillo CW. An alpha 2 beta 1 integrin-dependent pinocytic mechanism involving intracellular vacuole formation and coalescence regulates capillary lumen and tube formation in three-dimensional collagen matrix. Exp Cell Res 1996;224:39–51. [DOI] [PubMed] [Google Scholar]
- 31.Dingemans KP, Teeling P, Lagendijk JH, Becker AE. Extracellular matrix of the human aortic media: an ultrastructural histochemical and immunohistochemical study of the adult aortic media. Anat Rec 2000;258:1–14. doi: [DOI] [PubMed] [Google Scholar]
- 32.Xu J, Shi GP. Vascular wall extracellular matrix proteins and vascular diseases. Biochim Biophys Acta 2014;1842:2106–2119. doi: 10.1016/j.bbadis.2014.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mecham RP, Ramirez F. Extracellular Determinants of Arterial Morphogenesis, Growth, and Homeostasis. Curr Top Dev Biol 2018;130:193–216. doi: 10.1016/bs.ctdb.2018.02.001 [DOI] [PubMed] [Google Scholar]
- 34.Yanagisawa H, Yokoyama U. Extracellular matrix-mediated remodeling and mechanotransduction in large vessels during development and disease. Cell Signal 2021;86:110104. doi: 10.1016/j.cellsig.2021.110104 [DOI] [PubMed] [Google Scholar]
- 35.Ramirez F, Dietz HC. Fibrillin-rich microfibrils: Structural determinants of morphogenetic and homeostatic events. J Cell Physiol 2007;213:326–330. [DOI] [PubMed] [Google Scholar]
- 36.Papke CL, Yanagisawa H. Fibulin-4 and fibulin-5 in elastogenesis and beyond: Insights from mouse and human studies. Matrix Biol 2014;37:142–149. doi: 10.1016/j.matbio.2014.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hill MA, Meininger GA. Arteriolar vascular smooth muscle cells: mechanotransducers in a complex environment. Int J Biochem Cell Biol 2012;44:1505–1510. doi: 10.1016/j.biocel.2012.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tembely D, Henry A, Vanalderwiert L, Toussaint K, Bennasroune A, Blaise S, Sartelet H, Jaisson S, Gales C, Martiny L, et al. The Elastin Receptor Complex: An Emerging Therapeutic Target Against Age-Related Vascular Diseases. Front Endocrinol (Lausanne) 2022;13:815356. doi: 10.3389/fendo.2022.815356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Majesky MW, Weiser-Evans MCM. The adventitia in arterial development, remodeling, and hypertension. Biochem Pharmacol 2022;205:115259. doi: 10.1016/j.bcp.2022.115259 [DOI] [PubMed] [Google Scholar]
- 40.Tinajero MG, Gotlieb AI. Recent Developments in Vascular Adventitial Pathobiology: The Dynamic Adventitia as a Complex Regulator of Vascular Disease. Am J Pathol 2020;190:520–534. doi: 10.1016/j.ajpath.2019.10.021 [DOI] [PubMed] [Google Scholar]
- 41.Stenmark KR, Yeager ME, El Kasmi KC, Nozik-Grayck E, Gerasimovskaya EV, Li M, Riddle SR, Frid MG. The adventitia: essential regulator of vascular wall structure and function. Annu Rev Physiol 2013;75:23–47. doi: 10.1146/annurev-physiol-030212-183802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Majesky MW, Dong XR, Hoglund V, Mahoney WM, Jr., Daum G. The adventitia: a dynamic interface containing resident progenitor cells. Arterioscler Thromb Vasc Biol 2011;31:1530–1539. doi: 10.1161/ATVBAHA.110.221549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herrera J, Henke CA, Bitterman PB. Extracellular matrix as a driver of progressive fibrosis. J Clin Invest 2018;128:45–53. doi: 10.1172/JCI93557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol 2014;15:786–801. doi: 10.1038/nrm3904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Didangelos A, Yin X, Mandal K, Saje A, Smith A, Xu Q, Jahangiri M, Mayr M. Extracellular matrix composition and remodeling in human abdominal aortic aneurysms: a proteomics approach. Mol Cell Proteomics 2011;10:M111 008128. doi: 10.1074/mcp.M111.008128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stepien KL, Bajdak-Rusinek K, Fus-Kujawa A, Kuczmik W, Gawron K. Role of Extracellular Matrix and Inflammation in Abdominal Aortic Aneurysm. Int J Mol Sci 2022;23. doi: 10.3390/ijms231911078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jain M, Chauhan AK . Role of Integrins in Modulating Smooth Muscle Cell Plasticity and Vascular Remodeling: From Expression to Therapeutic Implications. Cells 2022;11. doi: 10.3390/cells11040646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma Z, Mao C, Jia Y, Fu Y, Kong W. Extracellular matrix dynamics in vascular remodeling. Am J Physiol Cell Physiol 2020;319:C481–C499. doi: 10.1152/ajpcell.00147.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koch CD, Lee CM, Apte SS. Aggrecan in Cardiovascular Development and Disease. J Histochem Cytochem 2020;68:777–795. doi: 10.1369/0022155420952902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cikach FS, Koch CD, Mead TJ, Galatioto J, Willard BB, Emerton KB, Eagleton MJ, Blackstone EH, Ramirez F, Roselli EE, et al. Massive aggrecan and versican accumulation in thoracic aortic aneurysm and dissection. JCI Insight 2018;3. doi: 10.1172/jci.insight.97167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Davis GE. Affinity of integrins for damaged extracellular matrix: alpha v beta 3 binds to denatured collagen type I through RGD sites. Biochem Biophys Res Commun 1992;182:1025–1031. doi: 10.1016/0006-291x(92)91834-d [DOI] [PubMed] [Google Scholar]
- 52.Davis GE, Bayless KJ, Davis MJ, Meininger GA. Regulation of tissue injury responses by the exposure of matricryptic sites within extracellular matrix molecules. Am J Pathol 2000;156:1489–1498. doi: 10.1016/S0002-9440(10)65020-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Davis GE. Matricryptic sites control tissue injury responses in the cardiovascular system: relationships to pattern recognition receptor regulated events. J Mol Cell Cardiol 2010;48:454–460. doi: 10.1016/j.yjmcc.2009.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lindsey ML, Iyer RP, Zamilpa R, Yabluchanskiy A, DeLeon-Pennell KY, Hall ME, Kaplan A, Zouein FA, Bratton D, Flynn ER, et al. A Novel Collagen Matricryptin Reduces Left Ventricular Dilation Post-Myocardial Infarction by Promoting Scar Formation and Angiogenesis. J Am Coll Cardiol 2015;66:1364–1374. doi: 10.1016/j.jacc.2015.07.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Okada M, Imoto K, Sugiyama A, Yasuda J, Yamawaki H. New Insights into the Role of Basement Membrane-Derived Matricryptins in the Heart. Biol Pharm Bull 2017;40:2050–2060. doi: 10.1248/bpb.b17-00308 [DOI] [PubMed] [Google Scholar]
- 56.de Castro Bras LE, Frangogiannis NG. Extracellular matrix-derived peptides in tissue remodeling and fibrosis. Matrix Biol 2020;91–92:176–187. doi: 10.1016/j.matbio.2020.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Senior RM, Griffin GL, Mecham RP. Chemotactic responses of fibroblasts to tropoelastin and elastin-derived peptides. J Clin Invest 1982;70:614–618. doi: 10.1172/jci110654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Elastases Heinz A. and elastokines: elastin degradation and its significance in health and disease. Crit Rev Biochem Mol Biol 2020;55:252–273. doi: 10.1080/10409238.2020.1768208 [DOI] [PubMed] [Google Scholar]
- 59.Mogford JE, Davis GE, Platts SH, Meininger GA. Vascular smooth muscle alpha v beta 3 integrin mediates arteriolar vasodilation in response to RGD peptides. Circ Res 1996;79:821–826. doi: 10.1161/01.res.79.4.821 [DOI] [PubMed] [Google Scholar]
- 60.Preissner KT, Seiffert D. Role of vitronectin and its receptors in haemostasis and vascular remodeling. Thromb Res 1998;89:1–21. doi: 10.1016/s0049-3848(97)00298-3 [DOI] [PubMed] [Google Scholar]
- 61.Senger DR. Molecular framework for angiogenesis: a complex web of interactions between extravasated plasma proteins and endothelial cell proteins induced by angiogenic cytokines. Am J Pathol 1996;149:1–7. [PMC free article] [PubMed] [Google Scholar]
- 62.Barker TH, Engler AJ. The provisional matrix: setting the stage for tissue repair outcomes. Matrix Biol 2017;60–61:1–4. doi: 10.1016/j.matbio.2017.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van Hinsbergh VW, Collen A, Koolwijk P. Role of fibrin matrix in angiogenesis. Ann N Y Acad Sci 2001;936:426–437. doi: 10.1111/j.1749-6632.2001.tb03526.x [DOI] [PubMed] [Google Scholar]
- 64.Glukhova MA, Frid MG, Shekhonin BV, Vasilevskaya TD, Grunwald J, Saginati M, Koteliansky VE. Expression of extra domain A fibronectin sequence in vascular smooth muscle cells is phenotype dependent. J Cell Biol 1989;109:357–366. doi: 10.1083/jcb.109.1.357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jain M, Dhanesha N, Doddapattar P, Chorawala MR, Nayak MK, Cornelissen A, Guo L, Finn AV, Lentz SR, Chauhan AK. Smooth muscle cell-specific fibronectin-EDA mediates phenotypic switching and neointimal hyperplasia. J Clin Invest 2020;130:295–314. doi: 10.1172/JCI124708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.deBlois D, Lombardi DM, Su EJ, Clowes AW, Schwartz SM, Giachelli CM. Angiotensin II induction of osteopontin expression and DNA replication in rat arteries. Hypertension 1996;28:1055–1063. doi: 10.1161/01.hyp.28.6.1055 [DOI] [PubMed] [Google Scholar]
- 67.Wang X, Louden C, Ohlstein EH, Stadel JM, Gu JL, Yue TL. Osteopontin expression in platelet-derived growth factor-stimulated vascular smooth muscle cells and carotid artery after balloon angioplasty. Arterioscler Thromb Vasc Biol 1996;16:1365–1372. doi: 10.1161/01.atv.16.11.1365 [DOI] [PubMed] [Google Scholar]
- 68.Srivatsa SS, Fitzpatrick LA, Tsao PW, Reilly TM, Holmes DR, Jr., Schwartz RS, Mousa SA. Selective alpha v beta 3 integrin blockade potently limits neointimal hyperplasia and lumen stenosis following deep coronary arterial stent injury: evidence for the functional importance of integrin alpha v beta 3 and osteopontin expression during neointima formation. Cardiovasc Res 1997;36:408–428. doi: 10.1016/s0008-6363(97)00184-3 [DOI] [PubMed] [Google Scholar]
- 69.Jones PL, Crack J, Rabinovitch M. Regulation of tenascin-C, a vascular smooth muscle cell survival factor that interacts with the alpha v beta 3 integrin to promote epidermal growth factor receptor phosphorylation and growth. J Cell Biol 1997;139:279–293. doi: 10.1083/jcb.139.1.279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Imanaka-Yoshida K, Yoshida T, Miyagawa-Tomita S. Tenascin-C in development and disease of blood vessels. Anat Rec (Hoboken) 2014;297:1747–1757. doi: 10.1002/ar.22985 [DOI] [PubMed] [Google Scholar]
- 71.Liu Y, Sinha S, McDonald OG, Shang Y, Hoofnagle MH, Owens GK. Kruppel-like factor 4 abrogates myocardin-induced activation of smooth muscle gene expression. J Biol Chem 2005;280:9719–9727. doi: 10.1074/jbc.M412862200 [DOI] [PubMed] [Google Scholar]
- 72.Wight TN. Provisional matrix: A role for versican and hyaluronan. Matrix Biol 2017;60–61:38–56. doi: 10.1016/j.matbio.2016.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rifkin D, Sachan N, Singh K, Sauber E, Tellides G, Ramirez F. The role of LTBPs in TGF beta signaling. Dev Dyn 2022;251:95–104. doi: 10.1002/dvdy.331 [DOI] [PubMed] [Google Scholar]
- 74.Wu J, Montaniel KR, Saleh MA, Xiao L, Chen W, Owens GK, Humphrey JD, Majesky MW, Paik DT, Hatzopoulos AK, et al. Origin of Matrix-Producing Cells That Contribute to Aortic Fibrosis in Hypertension. Hypertension 2016;67:461–468. doi: 10.1161/HYPERTENSIONAHA.115.06123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mulligan-Kehoe MJ, Simons M. Vasa vasorum in normal and diseased arteries. Circulation 2014;129:2557–2566. doi: 10.1161/CIRCULATIONAHA.113.007189 [DOI] [PubMed] [Google Scholar]
- 76.Passman JN, Dong XR, Wu SP, Maguire CT, Hogan KA, Bautch VL, Majesky MW. A sonic hedgehog signaling domain in the arterial adventitia supports resident Sca1+ smooth muscle progenitor cells. Proc Natl Acad Sci U S A 2008;105:9349–9354. doi: 10.1073/pnas.0711382105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Phillippi JA. On vasa vasorum: A history of advances in understanding the vessels of vessels. Sci Adv 2022;8:eabl6364. doi: 10.1126/sciadv.abl6364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Deglise S, Bechelli C, Allagnat F. Vascular smooth muscle cells in intimal hyperplasia, an update. Front Physiol 2022;13:1081881. doi: 10.3389/fphys.2022.1081881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen PY, Schwartz MA, Simons M. Endothelial-to-Mesenchymal Transition, Vascular Inflammation, and Atherosclerosis. Front Cardiovasc Med 2020;7:53. doi: 10.3389/fcvm.2020.00053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Crisan M, Corselli M, Chen WC, Peault B. Perivascular cells for regenerative medicine. J Cell Mol Med 2012;16:2851–2860. doi: 10.1111/j.1582-4934.2012.01617.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Orr AW, Sanders JM, Bevard M, Coleman E, Sarembock IJ, Schwartz MA. The subendothelial extracellular matrix modulates NF-kappaB activation by flow: a potential role in atherosclerosis. J Cell Biol 2005;169:191–202. doi: 10.1083/jcb.200410073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Michel JB, Jondeau G, Milewicz DM. From genetics to response to injury: vascular smooth muscle cells in aneurysms and dissections of the ascending aorta. Cardiovasc Res 2018;114:578–589. doi: 10.1093/cvr/cvy006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang X, Khalil RA. Matrix Metalloproteinases, Vascular Remodeling, and Vascular Disease. Adv Pharmacol 2018;81:241–330. doi: 10.1016/bs.apha.2017.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Back M, Ketelhuth DF, Agewall S. Matrix metalloproteinases in atherothrombosis. Prog Cardiovasc Dis 2010;52:410–428. doi: 10.1016/j.pcad.2009.12.002 [DOI] [PubMed] [Google Scholar]
- 85.Duca L, Blaise S, Romier B, Laffargue M, Gayral S, El Btaouri H, Kawecki C, Guillot A, Martiny L, Debelle L, et al. Matrix ageing and vascular impacts: focus on elastin fragmentation. Cardiovasc Res 2016;110:298–308. doi: 10.1093/cvr/cvw061 [DOI] [PubMed] [Google Scholar]
- 86.Pierschbacher M, Hayman EG, Ruoslahti E. Synthetic peptide with cell attachment activity of fibronectin. Proc Natl Acad Sci U S A 1983;80:1224–1227. doi: 10.1073/pnas.80.5.1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pierschbacher MD, Hayman EG, Ruoslahti E. The cell attachment determinant in fibronectin. J Cell Biochem 1985;28:115–126. doi: 10.1002/jcb.240280205 [DOI] [PubMed] [Google Scholar]
- 88.Liao YF, Gotwals PJ, Koteliansky VE, Sheppard D, Van De Water L. The EIIIA segment of fibronectin is a ligand for integrins alpha 9beta 1 and alpha 4beta 1 providing a novel mechanism for regulating cell adhesion by alternative splicing. J Biol Chem 2002;277:14467–14474. doi: 10.1074/jbc.M201100200 [DOI] [PubMed] [Google Scholar]
- 89.Astrof S, Crowley D, Hynes RO. Multiple cardiovascular defects caused by the absence of alternatively spliced segments of fibronectin. Dev Biol 2007;311:11–24. doi: 10.1016/j.ydbio.2007.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Smith LL, Cheung HK, Ling LE, Chen J, Sheppard D, Pytela R, Giachelli CM. Osteopontin N-terminal domain contains a cryptic adhesive sequence recognized by alpha9beta1 integrin. J Biol Chem 1996;271:28485–28491. [PubMed] [Google Scholar]
- 91.Bayless KJ, Davis GE. Identification of dual alpha 4beta1 integrin binding sites within a 38 amino acid domain in the N-terminal thrombin fragment of human osteopontin. J Biol Chem 2001;276:13483–13489. doi: 10.1074/jbc.M011392200 [DOI] [PubMed] [Google Scholar]
- 92.Bayless KJ, Meininger GA, Scholtz JM, Davis GE. Osteopontin is a ligand for the alpha4beta1 integrin. J Cell Sci 1998;111 ( Pt 9):1165–1174. doi: 10.1242/jcs.111.9.1165 [DOI] [PubMed] [Google Scholar]
- 93.Senger DR, Perruzzi CA. Cell migration promoted by a potent GRGDS-containing thrombin-cleavage fragment of osteopontin. Biochim Biophys Acta 1996;1314:13–24. doi: 10.1016/s0167-4889(96)00067-5 [DOI] [PubMed] [Google Scholar]
- 94.Yokosaki Y, Matsuura N, Higashiyama S, Murakami I, Obara M, Yamakido M, Shigeto N, Chen J, Sheppard D. Identification of the ligand binding site for the integrin alpha9 beta1 in the third fibronectin type III repeat of tenascin-C. J Biol Chem 1998;273:11423–11428. doi: 10.1074/jbc.273.19.11423 [DOI] [PubMed] [Google Scholar]
- 95.Vogel BE, Tarone G, Giancotti FG, Gailit J, Ruoslahti E. A novel fibronectin receptor with an unexpected subunit composition (alpha v beta 1). J Biol Chem 1990;265:5934–5937. [PubMed] [Google Scholar]
- 96.Katsuda S, Kaji T. Atherosclerosis and extracellular matrix. J Atheroscler Thromb 2003;10:267–274. doi: 10.5551/jat.10.267 [DOI] [PubMed] [Google Scholar]
- 97.Nakashima Y, Wight TN, Sueishi K. Early atherosclerosis in humans: role of diffuse intimal thickening and extracellular matrix proteoglycans. Cardiovasc Res 2008;79:14–23. doi: 10.1093/cvr/cvn099 [DOI] [PubMed] [Google Scholar]
- 98.Sluimer JC, Kolodgie FD, Bijnens AP, Maxfield K, Pacheco E, Kutys B, Duimel H, Frederik PM, van Hinsbergh VW, Virmani R, et al. Thin-walled microvessels in human coronary atherosclerotic plaques show incomplete endothelial junctions relevance of compromised structural integrity for intraplaque microvascular leakage. J Am Coll Cardiol 2009;53:1517–1527. doi: 10.1016/j.jacc.2008.12.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ramirez F, Sakai LY, Rifkin DB, Dietz HC. Extracellular microfibrils in development and disease. Cell Mol Life Sci 2007;64:2437–2446. doi: 10.1007/s00018-007-7166-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mecham RP, Gibson MA. The microfibril-associated glycoproteins (MAGPs) and the microfibrillar niche. Matrix Biol 2015;47:13–33. doi: 10.1016/j.matbio.2015.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cavinato C, Chen M, Weiss D, Ruiz-Rodriguez MJ, Schwartz MA, Humphrey JD. Progressive Microstructural Deterioration Dictates Evolving Biomechanical Dysfunction in the Marfan Aorta. Front Cardiovasc Med 2021;8:800730. doi: 10.3389/fcvm.2021.800730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Humphrey JD. Mechanisms of Vascular Remodeling in Hypertension. Am J Hypertens 2021;34:432–441. doi: 10.1093/ajh/hpaa195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Spronck B, Latorre M, Wang M, Mehta S, Caulk AW, Ren P, Ramachandra AB, Murtada SI, Rojas A, He CS, et al. Excessive adventitial stress drives inflammation-mediated fibrosis in hypertensive aortic remodelling in mice. J R Soc Interface 2021;18:20210336. doi: 10.1098/rsif.2021.0336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Funata N, Tanaka M, Hirokawa K. Coarctation of the abdominal aorta--case report with autopsy. Acta Pathol Jpn 1981;31:117–127. doi: 10.1111/j.1440-1827.1981.tb00990.x [DOI] [PubMed] [Google Scholar]
- 105.Fujinami M, Morimoto S, Nishikawa T, Sato A, Yamada Y, Kajita A. Primary pulmonary hypertension. Study of eight autopsy cases. Acta Pathol Jpn 1987;37:401–412. [PubMed] [Google Scholar]
- 106.Lampi MC, Reinhart-King CA. Targeting extracellular matrix stiffness to attenuate disease: From molecular mechanisms to clinical trials. Sci Transl Med 2018;10. doi: 10.1126/scitranslmed.aao0475 [DOI] [PubMed] [Google Scholar]
- 107.Reiser KM. Nonenzymatic glycation of collagen in aging and diabetes. Proc Soc Exp Biol Med 1991;196:17–29. doi: 10.3181/00379727-196-43158c [DOI] [PubMed] [Google Scholar]
- 108.VanderBurgh JA, Reinhart-King CA. The Role of Age-Related Intimal Remodeling and Stiffening in Atherosclerosis. Adv Pharmacol 2018;81:365–391. doi: 10.1016/bs.apha.2017.08.008 [DOI] [PubMed] [Google Scholar]
- 109.Parma L, Baganha F, Quax PHA, de Vries MR. Plaque angiogenesis and intraplaque hemorrhage in atherosclerosis. Eur J Pharmacol 2017;816:107–115. doi: 10.1016/j.ejphar.2017.04.028 [DOI] [PubMed] [Google Scholar]
- 110.Chen PY, Qin L, Baeyens N, Li G, Afolabi T, Budatha M, Tellides G, Schwartz MA, Simons M. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Invest 2015;125:4514–4528. doi: 10.1172/JCI82719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Koller GM, Schafer C, Kemp SS, Aguera KN, Lin PK, Forgy JC, Griffin CT, Davis GE. Proinflammatory Mediators, IL (Interleukin)-1beta, TNF (Tumor Necrosis Factor) alpha, and Thrombin Directly Induce Capillary Tube Regression. Arterioscler Thromb Vasc Biol 2020;40:365–377. doi: 10.1161/ATVBAHA.119.313536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Miano JM, Fisher EA, Majesky MW. Fate and State of Vascular Smooth Muscle Cells in Atherosclerosis. Circulation 2021;143:2110–2116. doi: 10.1161/CIRCULATIONAHA.120.049922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hynes RO. The extracellular matrix: not just pretty fibrils. Science 2009;326:1216–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ramirez F, Rifkin DB. Extracellular microfibrils: contextual platforms for TGFbeta and BMP signaling. Curr Opin Cell Biol 2009;21:616–622. doi: 10.1016/j.ceb.2009.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Robertson IB, Horiguchi M, Zilberberg L, Dabovic B, Hadjiolova K, Rifkin DB. Latent TGF-beta-binding proteins. Matrix Biol 2015;47:44–53. doi: 10.1016/j.matbio.2015.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wagner DD, Heger LA. Thromboinflammation: From Atherosclerosis to COVID-19. Arterioscler Thromb Vasc Biol 2022;42:1103–1112. doi: 10.1161/ATVBAHA.122.317162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Takahashi M NLRP3 Inflammasome as a Common Denominator of Atherosclerosis and Abdominal Aortic Aneurysm. Circ J 2021;85:2129–2136. doi: 10.1253/circj.CJ-21-0258 [DOI] [PubMed] [Google Scholar]
- 118.Melton E, Qiu H. Interleukin-1beta in Multifactorial Hypertension: Inflammation, Vascular Smooth Muscle Cell and Extracellular Matrix Remodeling, and Non-Coding RNA Regulation. Int J Mol Sci 2021;22. doi: 10.3390/ijms22168639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jackson SP, Darbousset R, Schoenwaelder SM. Thromboinflammation: challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood 2019;133:906–918. doi: 10.1182/blood-2018-11-882993 [DOI] [PubMed] [Google Scholar]
- 120.Wang M, Jiang L, Monticone RE, Lakatta EG. Proinflammation: the key to arterial aging. Trends Endocrinol Metab 2014;25:72–79. doi: 10.1016/j.tem.2013.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang M, Monticone RE, McGraw KR. Proinflammation, profibrosis, and arterial aging. Aging Med (Milton) 2020;3:159–168. doi: 10.1002/agm2.12099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li Y, Wang W, Li L, Khalil RA. MMPs and ADAMs/ADAMTS inhibition therapy of abdominal aortic aneurysm. Life Sci 2020;253:117659. doi: 10.1016/j.lfs.2020.117659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cui N, Hu M, Khalil RA. Biochemical and Biological Attributes of Matrix Metalloproteinases. Prog Mol Biol Transl Sci 2017;147:1–73. doi: 10.1016/bs.pmbts.2017.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Knox JB, Sukhova GK, Whittemore AD, Libby P. Evidence for altered balance between matrix metalloproteinases and their inhibitors in human aortic diseases. Circulation 1997;95:205–212. doi: 10.1161/01.cir.95.1.205 [DOI] [PubMed] [Google Scholar]
- 125.Rienks M, Barallobre-Barreiro J, Mayr M. The Emerging Role of the ADAMTS Family in Vascular Diseases. Circ Res 2018;123:1279–1281. doi: 10.1161/CIRCRESAHA.118.313737 [DOI] [PubMed] [Google Scholar]
- 126.Fukami K, Yamagishi S, Okuda S. Role of AGEs-RAGE system in cardiovascular disease. Current pharmaceutical design 2014;20:2395–2402. doi: 10.2174/13816128113199990475 [DOI] [PubMed] [Google Scholar]
- 127.Tuleta I, Frangogiannis NG. Diabetic fibrosis. Biochim Biophys Acta Mol Basis Dis 2021;1867:166044. doi: 10.1016/j.bbadis.2020.166044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Asano K, Cantalupo A, Sedes L, Ramirez F. The Multiple Functions of Fibrillin-1 Microfibrils in Organismal Physiology. Int J Mol Sci 2022;23. doi: 10.3390/ijms23031892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Karimi A, Milewicz DM. Structure of the Elastin-Contractile Units in the Thoracic Aorta and How Genes That Cause Thoracic Aortic Aneurysms and Dissections Disrupt This Structure. Can J Cardiol 2016;32:26–34. doi: 10.1016/j.cjca.2015.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Madamanchi NR, Vendrov A, Runge MS. Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol 2005;25:29–38. doi: 10.1161/01.ATV.0000150649.39934.13 [DOI] [PubMed] [Google Scholar]
- 131.Rey FE, Pagano PJ. The reactive adventitia: fibroblast oxidase in vascular function. Arterioscler Thromb Vasc Biol 2002;22:1962–1971. doi: 10.1161/01.atv.0000043452.30772.18 [DOI] [PubMed] [Google Scholar]
- 132.Hettwer J, Hinterdobler J, Miritsch B, Deutsch MA, Li X, Mauersberger C, Moggio A, Braster Q, Gram H, Robertson AAB, et al. Interleukin-1beta suppression dampens inflammatory leucocyte production and uptake in atherosclerosis. Cardiovasc Res 2022;118:2778–2791. doi: 10.1093/cvr/cvab337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tellides G, Pober JS. Inflammatory and immune responses in the arterial media. Circ Res 2015;116:312–322. doi: 10.1161/CIRCRESAHA.116.301312 [DOI] [PubMed] [Google Scholar]
- 134.Endemann G, Stanton LW, Madden KS, Bryant CM, White RT, Protter AA. CD36 is a receptor for oxidized low density lipoprotein. J Biol Chem 1993;268:11811–11816. [PubMed] [Google Scholar]
- 135.Acton SL, Scherer PE, Lodish HF, Krieger M. Expression cloning of SR-BI, a CD36-related class B scavenger receptor. J Biol Chem 1994;269:21003–21009. [PubMed] [Google Scholar]
- 136.Tedgui A, Mallat Z . [Atherosclerotic plaque formation]. Rev Prat 1999;49:2081–2086. [PubMed] [Google Scholar]
- 137.Davis GE. The Mac-1 and p150,95 beta 2 integrins bind denatured proteins to mediate leukocyte cell-substrate adhesion. Exp Cell Res 1992;200:242–252. doi: 10.1016/0014-4827(92)90170-d [DOI] [PubMed] [Google Scholar]
- 138.Davis GE, Thomas JS, Madden S. The alpha4beta1 integrin can mediate leukocyte adhesion to casein and denatured protein substrates. J Leukoc Biol 1997;62:318–328. doi: 10.1002/jlb.62.3.318 [DOI] [PubMed] [Google Scholar]
- 139.Yurdagul A, Jr., Green J, Albert P, McInnis MC, Mazar AP, Orr AW. alpha5beta1 integrin signaling mediates oxidized low-density lipoprotein-induced inflammation and early atherosclerosis. Arterioscler Thromb Vasc Biol 2014;34:1362–1373. doi: 10.1161/ATVBAHA.114.303863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Conway DE, Schwartz MA. Flow-dependent cellular mechanotransduction in atherosclerosis. J Cell Sci 2013;126:5101–5109. doi: 10.1242/jcs.138313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Budatha M, Zhang J, Schwartz MA. Fibronectin-Mediated Inflammatory Signaling Through Integrin alpha5 in Vascular Remodeling. J Am Heart Assoc 2021;10:e021160. doi: 10.1161/JAHA.121.021160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Orr AW, Ginsberg MH, Shattil SJ, Deckmyn H, Schwartz MA. Matrix-specific suppression of integrin activation in shear stress signaling. Mol Biol Cell 2006;17:4686–4697. doi: 10.1091/mbc.e06-04-0289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yun S, Hu R, Schwaemmle ME, Scherer AN, Zhuang Z, Koleske AJ, Pallas DC, Schwartz MA. Integrin alpha5beta1 regulates PP2A complex assembly through PDE4D in atherosclerosis. J Clin Invest 2019;129:4863–4874. doi: 10.1172/JCI127692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Budatha M, Zhang J, Zhuang ZW, Yun S, Dahlman JE, Anderson DG, Schwartz MA. Inhibiting Integrin alpha5 Cytoplasmic Domain Signaling Reduces Atherosclerosis and Promotes Arteriogenesis. J Am Heart Assoc 2018;7. doi: 10.1161/JAHA.117.007501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Petzold T, Orr AW, Hahn C, Jhaveri KA, Parsons JT, Schwartz MA. Focal adhesion kinase modulates activation of NF-kappaB by flow in endothelial cells. Am J Physiol Cell Physiol 2009;297:C814–822. doi: 10.1152/ajpcell.00226.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chen J, Green J, Yurdagul A, Jr., Albert P, McInnis MC, Orr AW. alphavbeta3 Integrins Mediate Flow-Induced NF-kappaB Activation, Proinflammatory Gene Expression, and Early Atherogenic Inflammation. Am J Pathol 2015;185:2575–2589. doi: 10.1016/j.ajpath.2015.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Funk SD, Yurdagul A, Jr., Green JM, Jhaveri KA, Schwartz MA, Orr AW. Matrix-specific protein kinase A signaling regulates p21-activated kinase activation by flow in endothelial cells. Circ Res 2010;106:1394–1403. doi: 10.1161/CIRCRESAHA.109.210286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chen PY, Qin L, Barnes C, Charisse K, Yi T, Zhang X, Ali R, Medina PP, Yu J, Slack FJ, et al. FGF regulates TGF-beta signaling and endothelial-to-mesenchymal transition via control of let-7 miRNA expression. Cell Rep 2012;2:1684–1696. doi: 10.1016/j.celrep.2012.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Simons M Fibroblast growth factors: the keepers of endothelial normalcy. J Clin Invest 2021;131. doi: 10.1172/JCI152716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ricard N, Scott RP, Booth CJ, Velazquez H, Cilfone NA, Baylon JL, Gulcher JR, Quaggin SE, Chittenden TW, Simons M. Endothelial ERK1/2 signaling maintains integrity of the quiescent endothelium. J Exp Med 2019;216:1874–1890. doi: 10.1084/jem.20182151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dejana E, Hirschi KK, Simons M. The molecular basis of endothelial cell plasticity. Nat Commun 2017;8:14361. doi: 10.1038/ncomms14361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Chen PY, Qin L, Li G, Wang Z, Dahlman JE, Malagon-Lopez J, Gujja S, Cilfone NA, Kauffman KJ, Sun L, et al. Endothelial TGF-beta signalling drives vascular inflammation and atherosclerosis. Nat Metab 2019;1:912–926. doi: 10.1038/s42255-019-0102-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bowers SLK, Kemp SS, Aguera KN, Koller GM, Forgy JC, Davis GE. Defining an Upstream VEGF (Vascular Endothelial Growth Factor) Priming Signature for Downstream Factor-Induced Endothelial Cell-Pericyte Tube Network Coassembly. Arterioscler Thromb Vasc Biol 2020;40:2891–2909. doi: 10.1161/ATVBAHA.120.314517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circ Res 2014;114:1852–1866. doi: 10.1161/CIRCRESAHA.114.302721 [DOI] [PubMed] [Google Scholar]
- 155.Davies MJ. The pathophysiology of acute coronary syndromes. Heart 2000;83:361–366. doi: 10.1136/heart.83.3.361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Falk E, Shah PK, Fuster V. Coronary plaque disruption. Circulation 1995;92:657–671. doi: 10.1161/01.cir.92.3.657 [DOI] [PubMed] [Google Scholar]
- 157.Jia H, Abtahian F, Aguirre AD, Lee S, Chia S, Lowe H, Kato K, Yonetsu T, Vergallo R, Hu S, et al. In vivo diagnosis of plaque erosion and calcified nodule in patients with acute coronary syndrome by intravascular optical coherence tomography. J Am Coll Cardiol 2013;62:1748–1758. doi: 10.1016/j.jacc.2013.05.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kolodgie FD, Burke AP, Farb A, Gold HK, Yuan J, Narula J, Finn AV, Virmani R. The thin-cap fibroatheroma: a type of vulnerable plaque: the major precursor lesion to acute coronary syndromes. Curr Opin Cardiol 2001;16:285–292. doi: 10.1097/00001573-200109000-00006 [DOI] [PubMed] [Google Scholar]
- 159.Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res 2003;92:827–839. doi: 10.1161/01.RES.0000070112.80711.3D [DOI] [PubMed] [Google Scholar]
- 160.Montero I, Orbe J, Varo N, Beloqui O, Monreal JI, Rodriguez JA, Diez J, Libby P, Paramo JA. C-reactive protein induces matrix metalloproteinase-1 and -10 in human endothelial cells: implications for clinical and subclinical atherosclerosis. J Am Coll Cardiol 2006;47:1369–1378. doi: 10.1016/j.jacc.2005.10.070 [DOI] [PubMed] [Google Scholar]
- 161.Saunders WB, Bayless KJ, Davis GE. MMP-1 activation by serine proteases and MMP-10 induces human capillary tubular network collapse and regression in 3D collagen matrices. J Cell Sci 2005;118:2325–2340. doi: 10.1242/jcs.02360 [DOI] [PubMed] [Google Scholar]
- 162.Clark IM, Swingler TE, Sampieri CL, Edwards DR. The regulation of matrix metalloproteinases and their inhibitors. Int J Biochem Cell Biol 2008;40:1362–1378. doi: 10.1016/j.biocel.2007.12.006 [DOI] [PubMed] [Google Scholar]
- 163.Sedding DG, Boyle EC, Demandt JAF, Sluimer JC, Dutzmann J, Haverich A, Bauersachs J. Vasa Vasorum Angiogenesis: Key Player in the Initiation and Progression of Atherosclerosis and Potential Target for the Treatment of Cardiovascular Disease. Front Immunol 2018;9:706. doi: 10.3389/fimmu.2018.00706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sukhova GK, Schonbeck U, Rabkin E, Schoen FJ, Poole AR, Billinghurst RC, Libby P. Evidence for increased collagenolysis by interstitial collagenases-1 and -3 in vulnerable human atheromatous plaques. Circulation 1999;99:2503–2509. doi: 10.1161/01.cir.99.19.2503 [DOI] [PubMed] [Google Scholar]
- 165.Higashikata T, Yamagishi M, Higashi T, Nagata I, Iihara K, Miyamoto S, Ishibashi-Ueda H, Nagaya N, Iwase T, Tomoike H, et al. Altered expression balance of matrix metalloproteinases and their inhibitors in human carotid plaque disruption: results of quantitative tissue analysis using real-time RT-PCR method. Atherosclerosis 2006;185:165–172. doi: 10.1016/j.atherosclerosis.2005.05.039 [DOI] [PubMed] [Google Scholar]
- 166.Lemaitre V, D'Armiento J. Matrix metalloproteinases in development and disease. Birth Defects Res C Embryo Today 2006;78:1–10. doi: 10.1002/bdrc.20065 [DOI] [PubMed] [Google Scholar]
- 167.Beltrami-Moreira M, Vromman A, Sukhova GK, Folco EJ, Libby P. Redundancy of IL-1 Isoform Signaling and Its Implications for Arterial Remodeling. PLoS One 2016;11:e0152474. doi: 10.1371/journal.pone.0152474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Quintana RA, Taylor WR. Cellular Mechanisms of Aortic Aneurysm Formation. Circ Res 2019;124:607–618. doi: 10.1161/CIRCRESAHA.118.313187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Meester JAN, Verstraeten A, Schepers D, Alaerts M, Van Laer L, Loeys BL. Differences in manifestations of Marfan syndrome, Ehlers-Danlos syndrome, and Loeys-Dietz syndrome. Ann Cardiothorac Surg 2017;6:582–594. doi: 10.21037/acs.2017.11.03 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Humphrey JD, Schwartz MA, Tellides G, Milewicz DM. Role of mechanotransduction in vascular biology: focus on thoracic aortic aneurysms and dissections. Circ Res 2015;116:1448–1461. doi: 10.1161/CIRCRESAHA.114.304936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Humphrey JD, Milewicz DM, Tellides G, Schwartz MA. Cell biology. Dysfunctional mechanosensing in aneurysms. Science 2014;344:477–479. doi: 10.1126/science.1253026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Dietz HC, Saraiva JM, Pyeritz RE, Cutting GR, Francomano CA. Clustering of fibrillin (FBN1) missense mutations in Marfan syndrome patients at cysteine residues in EGF-like domains. Hum Mutat 1992;1:366–374. doi: 10.1002/humu.1380010504 [DOI] [PubMed] [Google Scholar]
- 173.Holm TM, Habashi JP, Doyle JJ, Bedja D, Chen Y, van Erp C, Lindsay ME, Kim D, Schoenhoff F, Cohn RD, et al. Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science 2011;332:358–361. doi: 10.1126/science.1192149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet 2005;37:275–281. doi: 10.1038/ng1511 [DOI] [PubMed] [Google Scholar]
- 175.Lindsay ME, Schepers D, Bolar NA, Doyle JJ, Gallo E, Fert-Bober J, Kempers MJ, Fishman EK, Chen Y, Myers L, et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet 2012;44:922–927. doi: 10.1038/ng.2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Schepers D, Tortora G, Morisaki H, MacCarrick G, Lindsay M, Liang D, Mehta SG, Hague J, Verhagen J, van de Laar I, et al. A mutation update on the LDS-associated genes TGFB2/3 and SMAD2/3. Hum Mutat 2018;39:621–634. doi: 10.1002/humu.23407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest 2000;105:1605–1612. doi: 10.1172/JCI7818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Daugherty A, Cassis LA. Mouse models of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol 2004;24:429–434. doi: 10.1161/01.ATV.0000118013.72016.ea [DOI] [PubMed] [Google Scholar]
- 179.Sawada H, Lu HS, Cassis LA, Daugherty A. Twenty Years of Studying AngII (Angiotensin II)-Induced Abdominal Aortic Pathologies in Mice: Continuing Questions and Challenges to Provide Insight Into the Human Disease. Arterioscler Thromb Vasc Biol 2022;42:277–288. doi: 10.1161/ATVBAHA.121.317058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Vallet SD, Ricard-Blum S. Lysyl oxidases: from enzyme activity to extracellular matrix cross-links. Essays Biochem 2019;63:349–364. doi: 10.1042/EBC20180050 [DOI] [PubMed] [Google Scholar]
- 181.Li DY, Faury G, Taylor DG, Davis EC, Boyle WA, Mecham RP, Stenzel P, Boak B, Keating MT. Novel arterial pathology in mice and humans hemizygous for elastin. J Clin Invest 1998;102:1783–1787. doi: 10.1172/JCI4487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wagenseil JE, Mecham RP. New insights into elastic fiber assembly. Birth Defects Res C Embryo Today 2007;81:229–240. [DOI] [PubMed] [Google Scholar]
- 183.Wagenseil JE, Ciliberto CH, Knutsen RH, Levy MA, Kovacs A, Mecham RP. The importance of elastin to aortic development in mice. Am J Physiol Heart Circ Physiol 2010;299:H257–264. doi: 10.1152/ajpheart.00194.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Wakita D, Kurashima Y, Crother TR, Noval Rivas M, Lee Y, Chen S, Fury W, Bai Y, Wagner S, Li D, et al. Role of Interleukin-1 Signaling in a Mouse Model of Kawasaki Disease-Associated Abdominal Aortic Aneurysm. Arterioscler Thromb Vasc Biol 2016;36:886–897. doi: 10.1161/ATVBAHA.115.307072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Irizarry E, Newman KM, Gandhi RH, Nackman GB, Halpern V, Wishner S, Scholes JV, Tilson MD. Demonstration of interstitial collagenase in abdominal aortic aneurysm disease. J Surg Res 1993;54:571–574. doi: 10.1006/jsre.1993.1087 [DOI] [PubMed] [Google Scholar]
- 186.Newman KM, Malon AM, Shin RD, Scholes JV, Ramey WG, Tilson MD. Matrix metalloproteinases in abdominal aortic aneurysm: characterization, purification, and their possible sources. Connect Tissue Res 1994;30:265–276. doi: 10.3109/03008209409015042 [DOI] [PubMed] [Google Scholar]
- 187.Newman KM, Ogata Y, Malon AM, Irizarry E, Gandhi RH, Nagase H, Tilson MD. Identification of matrix metalloproteinases 3 (stromelysin-1) and 9 (gelatinase B) in abdominal aortic aneurysm. Arterioscler Thromb 1994;14:1315–1320. doi: 10.1161/01.atv.14.8.1315 [DOI] [PubMed] [Google Scholar]
- 188.Newman KM, Jean-Claude J, Li H, Scholes JV, Ogata Y, Nagase H, Tilson MD. Cellular localization of matrix metalloproteinases in the abdominal aortic aneurysm wall. J Vasc Surg 1994;20:814–820. doi: 10.1016/s0741-5214(94)70169-5 [DOI] [PubMed] [Google Scholar]
- 189.Tromp G, Gatalica Z, Skunca M, Berguer R, Siegel T, Kline RA, Kuivaniemi H. Elevated expression of matrix metalloproteinase-13 in abdominal aortic aneurysms. Ann Vasc Surg 2004;18:414–420. doi: 10.1007/s10016-004-0050-5 [DOI] [PubMed] [Google Scholar]
- 190.Shen M, Lee J, Basu R, Sakamuri SS, Wang X, Fan D, Kassiri Z. Divergent roles of matrix metalloproteinase 2 in pathogenesis of thoracic aortic aneurysm. Arterioscler Thromb Vasc Biol 2015;35:888–898. doi: 10.1161/ATVBAHA.114.305115 [DOI] [PubMed] [Google Scholar]
- 191.Misra A, Sheikh AQ, Kumar A, Luo J, Zhang J, Hinton RB, Smoot L, Kaplan P, Urban Z, Qyang Y, et al. Integrin beta3 inhibition is a therapeutic strategy for supravalvular aortic stenosis. J Exp Med 2016;213:451–463. doi: 10.1084/jem.20150688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Dave JM, Chakraborty R, Ntokou A, Saito J, Saddouk FZ, Feng Z, Misra A, Tellides G, Riemer RK, Urban Z, et al. JAGGED1/NOTCH3 activation promotes aortic hypermuscularization and stenosis in elastin deficiency. J Clin Invest 2022;132. doi: 10.1172/JCI142338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Yoshida T, Kaestner KH, Owens GK. Conditional deletion of Kruppel-like factor 4 delays downregulation of smooth muscle cell differentiation markers but accelerates neointimal formation following vascular injury. Circ Res 2008;102:1548–1557. doi: 10.1161/CIRCRESAHA.108.176974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Jain M, Dev R, Doddapattar P, Kon S, Dhanesha N, Chauhan AK. Integrin alpha9 regulates smooth muscle cell phenotype switching and vascular remodeling. JCI Insight 2021;6. doi: 10.1172/jci.insight.147134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Pober BR. Williams-Beuren syndrome. N Engl J Med 2010;362:239–252. doi: 10.1056/NEJMra0903074 [DOI] [PubMed] [Google Scholar]
- 196.Wagenseil JE, Nerurkar NL, Knutsen RH, Okamoto RJ, Li DY, Mecham RP. Effects of elastin haploinsufficiency on the mechanical behavior of mouse arteries. Am J Physiol Heart Circ Physiol 2005;289:H1209–1217. doi: 10.1152/ajpheart.00046.2005 [DOI] [PubMed] [Google Scholar]
- 197.Yamashiro Y, Yanagisawa H. The molecular mechanism of mechanotransduction in vascular homeostasis and disease. Clin Sci (Lond) 2020;134:2399–2418. doi: 10.1042/CS20190488 [DOI] [PubMed] [Google Scholar]
- 198.Bellini C, Bersi MR, Caulk AW, Ferruzzi J, Milewicz DM, Ramirez F, Rifkin DB, Tellides G, Yanagisawa H, Humphrey JD. Comparison of 10 murine models reveals a distinct biomechanical phenotype in thoracic aortic aneurysms. J R Soc Interface 2017;14. doi: 10.1098/rsif.2016.1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Porritt RA, Zemmour D, Abe M, Lee Y, Narayanan M, Carvalho TT, Gomez AC, Martinon D, Santiskulvong C, Fishbein MC, et al. NLRP3 Inflammasome Mediates Immune-Stromal Interactions in Vasculitis. Circ Res 2021;129:e183–e200. doi: 10.1161/CIRCRESAHA.121.319153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Akiyama M, Ohtsuki S, Berry GJ, Liang DH, Goronzy JJ, Weyand CM. Innate and Adaptive Immunity in Giant Cell Arteritis. Front Immunol 2020;11:621098. doi: 10.3389/fimmu.2020.621098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Chung SW. Vasculitis: From Target Molecules to Novel Therapeutic Approaches. Biomedicines 2021;9. doi: 10.3390/biomedicines9070757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Regola F, Uzzo M, Toniati P, Trezzi B, Sinico RA, Franceschini F. Novel Therapies in Takayasu Arteritis. Front Med (Lausanne) 2021;8:814075. doi: 10.3389/fmed.2021.814075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Watanabe R, Hashimoto M. Aging-Related Vascular Inflammation: Giant Cell Arteritis and Neurological Disorders. Front Aging Neurosci 2022;14:843305. doi: 10.3389/fnagi.2022.843305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Noval Rivas M, Arditi M. Kawasaki disease: pathophysiology and insights from mouse models. Nat Rev Rheumatol 2020;16:391–405. doi: 10.1038/s41584-020-0426-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Rosenkranz ME, Schulte DJ, Agle LM, Wong MH, Zhang W, Ivashkiv L, Doherty TM, Fishbein MC, Lehman TJ, Michelsen KS, et al. TLR2 and MyD88 contribute to Lactobacillus casei extract-induced focal coronary arteritis in a mouse model of Kawasaki disease. Circulation 2005;112:2966–2973. doi: 10.1161/CIRCULATIONAHA.105.537530 [DOI] [PubMed] [Google Scholar]
- 206.Lee Y, Wakita D, Dagvadorj J, Shimada K, Chen S, Huang G, Lehman TJ, Fishbein MC, Hoffman HM, Crother TR, et al. IL-1 Signaling Is Critically Required in Stromal Cells in Kawasaki Disease Vasculitis Mouse Model: Role of Both IL-1alpha and IL-1beta. Arterioscler Thromb Vasc Biol 2015;35:2605–2616. doi: 10.1161/ATVBAHA.115.306475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Gorelik M, Lee Y, Abe M, Andrews T, Davis L, Patterson J, Chen S, Crother TR, Aune GJ, Noval Rivas M, et al. IL-1 receptor antagonist, anakinra, prevents myocardial dysfunction in a mouse model of Kawasaki disease vasculitis and myocarditis. Clin Exp Immunol 2019;198:101–110. doi: 10.1111/cei.13314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Porritt RA, Chase Huizar C, Dick EJ, Kumar S, Escalona R, Gomez AC, Marek-Iannucci S, Noval Rivas M, Patterson J, Forsthuber TG, et al. Inhibition of IL-6 in the LCWE Mouse Model of Kawasaki Disease Inhibits Acute Phase Reactant Serum Amyloid A but Fails to Attenuate Vasculitis. Front Immunol 2021;12:630196. doi: 10.3389/fimmu.2021.630196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Hoang LT, Shimizu C, Ling L, Naim AN, Khor CC, Tremoulet AH, Wright V, Levin M, Hibberd ML, Burns JC. Global gene expression profiling identifies new therapeutic targets in acute Kawasaki disease. Genome Med 2014;6:541. doi: 10.1186/s13073-014-0102-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kone-Paut I, Tellier S, Belot A, Brochard K, Guitton C, Marie I, Meinzer U, Cherqaoui B, Galeotti C, Boukhedouni N, et al. Phase II Open Label Study of Anakinra in Intravenous Immunoglobulin-Resistant Kawasaki Disease. Arthritis Rheumatol 2021;73:151–161. doi: 10.1002/art.41481 [DOI] [PubMed] [Google Scholar]
- 211.Wang WT, He M, Shimizu C, Croker BA, Hoffman HM, Tremoulet AH, Burns JC, Shyy JY. Inflammasome Activation in Children With Kawasaki Disease and Multisystem Inflammatory Syndrome. Arterioscler Thromb Vasc Biol 2021;41:2509–2511. doi: 10.1161/ATVBAHA.121.316210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Kessel C, Kone-Paut I, Tellier S, Belot A, Masjosthusmann K, Wittkowski H, Fuehner S, Rossi-Semerano L, Dusser P, Marie I, et al. An Immunological Axis Involving Interleukin 1beta and Leucine-Rich-alpha2-Glycoprotein Reflects Therapeutic Response of Children with Kawasaki Disease: Implications from the KAWAKINRA Trial. J Clin Immunol 2022;42:1330–1341. doi: 10.1007/s10875-022-01301-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Burns JC, Roberts SC, Tremoulet AH, He F, Printz BF, Ashouri N, Jain SS, Michalik DE, Sharma K, Truong DT, et al. Infliximab versus second intravenous immunoglobulin for treatment of resistant Kawasaki disease in the USA (KIDCARE): a randomised, multicentre comparative effectiveness trial. Lancet Child Adolesc Health 2021;5:852–861. doi: 10.1016/S2352-4642(21)00270-4 [DOI] [PubMed] [Google Scholar]
- 214.Schafer CM, Gurley JM, Kurylowicz K, Lin PK, Chen W, Elliott MH, Davis GE, Bhatti F, Griffin CT. An inhibitor of endothelial ETS transcription factors promotes physiologic and therapeutic vessel regression. Proc Natl Acad Sci U S A 2020;117:26494–26502. doi: 10.1073/pnas.2015980117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Davis GE, Senger DR. Extracellular matrix mediates a molecular balance between vascular morphogenesis and regression. Curr Opin Hematol 2008;15:197–203. doi: 10.1097/MOH.0b013e3282fcc321 [DOI] [PubMed] [Google Scholar]
- 216.Kemp SS, Penn MR, Koller GM, Griffin CT, Davis GE. Proinflammatory mediators, TNFalpha, IFNgamma, and thrombin, directly induce lymphatic capillary tube regression. Front Cell Dev Biol 2022;10:937982. doi: 10.3389/fcell.2022.937982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Yoshida Y, Shimizu I, Minamino T. Capillaries as a Therapeutic Target for Heart Failure. J Atheroscler Thromb 2022;29:971–988. doi: 10.5551/jat.RV17064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Paavonsalo S, Hariharan S, Lackman MH, Karaman S. Capillary Rarefaction in Obesity and Metabolic Diseases-Organ-Specificity and Possible Mechanisms. Cells 2020;9. doi: 10.3390/cells9122683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Kida Y Peritubular Capillary Rarefaction: An Underappreciated Regulator of CKD Progression. Int J Mol Sci 2020;21. doi: 10.3390/ijms21218255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Zeng H, Chen JX. Microvascular Rarefaction and Heart Failure With Preserved Ejection Fraction. Front Cardiovasc Med 2019;6:15. doi: 10.3389/fcvm.2019.00015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Afsar B, Afsar RE, Dagel T, Kaya E, Erus S, Ortiz A, Covic A, Kanbay M. Capillary rarefaction from the kidney point of view. Clin Kidney J 2018;11:295–301. doi: 10.1093/ckj/sfx133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Triantafyllou A, Anyfanti P, Pyrpasopoulou A, Triantafyllou G, Aslanidis S, Douma S. Capillary rarefaction as an index for the microvascular assessment of hypertensive patients. Curr Hypertens Rep 2015;17:33. doi: 10.1007/s11906-015-0543-3 [DOI] [PubMed] [Google Scholar]
- 223.Basile DP. Rarefaction of peritubular capillaries following ischemic acute renal failure: a potential factor predisposing to progressive nephropathy. Curr Opin Nephrol Hypertens 2004;13:1–7. doi: 10.1097/00041552-200401000-00001 [DOI] [PubMed] [Google Scholar]
- 224.Davis GE, Pintar Allen KA, Salazar R, Maxwell SA. Matrix metalloproteinase-1 and -9 activation by plasmin regulates a novel endothelial cell-mediated mechanism of collagen gel contraction and capillary tube regression in three-dimensional collagen matrices. J Cell Sci 2001;114:917–930. doi: 10.1242/jcs.114.5.917 [DOI] [PubMed] [Google Scholar]
- 225.Saunders WB, Bohnsack BL, Faske JB, Anthis NJ, Bayless KJ, Hirschi KK, Davis GE. Coregulation of vascular tube stabilization by endothelial cell TIMP-2 and pericyte TIMP-3. J Cell Biol 2006;175:179–191. doi: 10.1083/jcb.200603176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Ingram KG, Curtis CD, Silasi-Mansat R, Lupu F, Griffin CT. The NuRD chromatin-remodeling enzyme CHD4 promotes embryonic vascular integrity by transcriptionally regulating extracellular matrix proteolysis. PLoS Genet 2013;9:e1004031. doi: 10.1371/journal.pgen.1004031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Chang S, Young BD, Li S, Qi X, Richardson JA, Olson EN. Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell 2006;126:321–334. doi: 10.1016/j.cell.2006.05.040 [DOI] [PubMed] [Google Scholar]
- 228.Grunewald M, Kumar S, Sharife H, Volinsky E, Gileles-Hillel A, Licht T, Permyakova A, Hinden L, Azar S, Friedmann Y, et al. Counteracting age-related VEGF signaling insufficiency promotes healthy aging and extends life span. Science 2021;373. doi: 10.1126/science.abc8479 [DOI] [PubMed] [Google Scholar]
- 229.Oh J, Takahashi R, Kondo S, Mizoguchi A, Adachi E, Sasahara RM, Nishimura S, Imamura Y, Kitayama H, Alexander DB, et al. The membrane-anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis. Cell 2001;107:789–800. doi: 10.1016/s0092-8674(01)00597-9 [DOI] [PubMed] [Google Scholar]