

Abstract
Background:
Fewer than 50% of patients who develop aortic valve calcification suffer from concomitant atherosclerosis, implying differential pathogenesis. While circulating extracellular vesicles (EVs) act as biomarkers of cardiovascular diseases, tissue-entrapped EVs associate with early mineralization but their cargoes, functions, and contributions to disease remain unknown.
Methods:
Disease stage-specific proteomics was performed on human carotid endarterectomy specimens (n=16) and stenotic aortic valves (n=18). Tissue EVs were isolated from human carotid arteries (normal: n=6, diseased: n=4) and aortic valves (normal: n=6; diseased: n=4) by enzymatic digestion, (ultra)centrifugation, and a 15-fraction density gradient validated by proteomics, CD63-immunogold electron microscopy, and nanoparticle tracking analysis. Vesiculomics, comprising vesicular proteomics and small RNA-sequencing, was conducted on tissue EVs. TargetScan identified miRNA targets. Pathway network analyses prioritized genes for validation in primary human carotid artery smooth muscle cells (hCtASMCs) and aortic valvular interstitial cells (hVICs).
Results:
Disease progression drove significant convergence (p<0.0001) of carotid artery plaque and calcified aortic valve proteomes (2,318 proteins). Each tissue also retained a unique subset of differentially-enriched proteins (381 in plaques, 226 in valves, q<0.05). Vesicular GO terms increased 2.9x (p<0.0001) amongst proteins modulated by disease in both tissues. Proteomics identified 22 EV markers in tissue digest fractions. Networks of proteins and miR targets changed by disease progression in both artery and valve EVs revealed shared involvement in intracellular signaling and cell cycle regulation. Vesiculomics identified 773 proteins and 80 miRs differentially-enriched by disease exclusively in artery or valve EVs (q<0.05); multi-omics integration found tissue-specific EV cargoes associated with pro-calcific Notch and Wnt signaling in carotid arteries and aortic valves, respectively. Knockdown of tissue-specific EV cargoes FGFR2, PPP2CA, and ADAM17 in hCtASMCs and WNT5A, APP, and APC in hVICs significantly modulated calcification.
Conclusions:
The first comparative proteomics study of human carotid artery plaques and calcified aortic valves identifies unique drivers of atherosclerosis vs. aortic valve stenosis and implicates EVs in advanced cardiovascular calcification. We delineate a vesiculomics strategy to isolate, purify, and study protein and RNA cargoes from EVs entrapped in fibro-calcific tissues. Integration of vesicular proteomics and transcriptomics by network approaches revealed novel roles for tissue EVs in modulating cardiovascular disease.
Keywords: Extracellular Vesicles, Proteomics, MicroRNAs, Atherosclerosis, Aortic Valve Stenosis
Introduction
Cardiovascular calcification correlates with incidence of myocardial infarction, stroke, and heart failure, and strongly predicts morbidity and mortality.1 While pathological mineralization of arteries and heart valves share numerous risk factors, only 25–50% of patients with valvular calcification also develop vascular calcification, implying differential disease drivers.2 Histopathological studies described these two conditions as grossly comparable3 and there has been little subsequent study of this apparent paradox. Despite histological similarities, pharmacotherapies such as statins successfully mitigate inflammation and lower cholesterol in patients with atherosclerosis but fail to improve outcomes for aortic valve stenosis – leaving patients without effective therapeutics.4 Calcification-prone extracellular vesicles (EVs) offer a possible explanation; they are important drivers and building blocks of cardiovascular mineralization.5–8 EVs are small membrane-bound structures secreted by all human cell types and found throughout the body’s tissues and fluids.9 They contain actively-selected bioactive cargoes (lipids, proteins, and noncoding microRNAs [miRs]), and mediate cell-cell communication.10 Circulating levels of EVs rise in patients with coronary artery disease and associate with a higher risk of cardiovascular death, revascularization, and occurrence of major adverse cardiovascular events.11 This has spurred a focus on circulating EV cargoes as biomarkers in cardiovascular disease, however EVs entrapped within cardiovascular tissue remain understudied.
In vitro, EVs from cell types found in diseased vessels and/or valves directly drive calcification. Cultured macrophages release EVs with high aggregation potential that nucleate hydroxyapatite, and EVs from vascular smooth muscle cells (SMCs) under calcifying conditions are loaded with pro-mineralizing annexins, phosphatidylserine, and reduced levels of calcification inhibitors.6,12 These EVs act as sites of mineral nucleation and modulate the formation of early plaque microcalcifications.8 Recent work has localized EVs in both human atherosclerotic plaques6,8 and calcified vessels from patients with chronic kidney disease;13,14 however, while tissue-entrapped EVs presumably mediate intercellular signaling within the plaque, they have not been previously characterized. Unlike straightforward isolation of EVs from biofluids, extracting tissue EVs of sufficient purity, quality, and quantity for downstream applications presents substantial challenges. Tissue EV contents have thus rarely been studied, and only in more readily-disrupted brain or tumor samples.15 Tissue EVs are therefore a rich, untapped source of biological insight into putative disease mechanisms and therapeutic targets.
Here, we addressed the persistent uncertainty surrounding differential pathogenesis of calcification in human atherosclerotic arteries and stenotic aortic valves (detailed study roadmap: Figure S1). Due to pathological EV secretion by cell types implicated in vascular and valvular disease, we hypothesized that tissue-entrapped vesicular cargoes contribute to shared/tissue-specific modulation of cardiovascular calcification. Using global proteomics, we examined three distinct stages of disease within human carotid artery plaques and calcified aortic valves, which identified unique drivers of atherosclerosis vs. aortic stenosis and implicated EV-associated pathways in disease progression of both tissues. Guided by proteomics, we developed an EV isolation approach that employed enzymatic digestion, sequential centrifugation, ultracentrifugation, and density gradient separation to enrich EVs from fibro-calcific human tissues (Figure 1). Lastly, we interrogated the normal and diseased cardiovascular tissue vesiculome by applying proteomics, small RNA-sequencing, multi-omics integration, multi-dimensional network analysis, and systems approaches to derive biological insights from tissue EV cargoes. In doing so, we assess EVs as regulators of the distinctive pathogenesis of vascular and valvular calcification.
Figure 1: Experimental Overview – Isolation and Analysis of Cardiovascular Tissue-Entrapped Extracellular Vesicles.
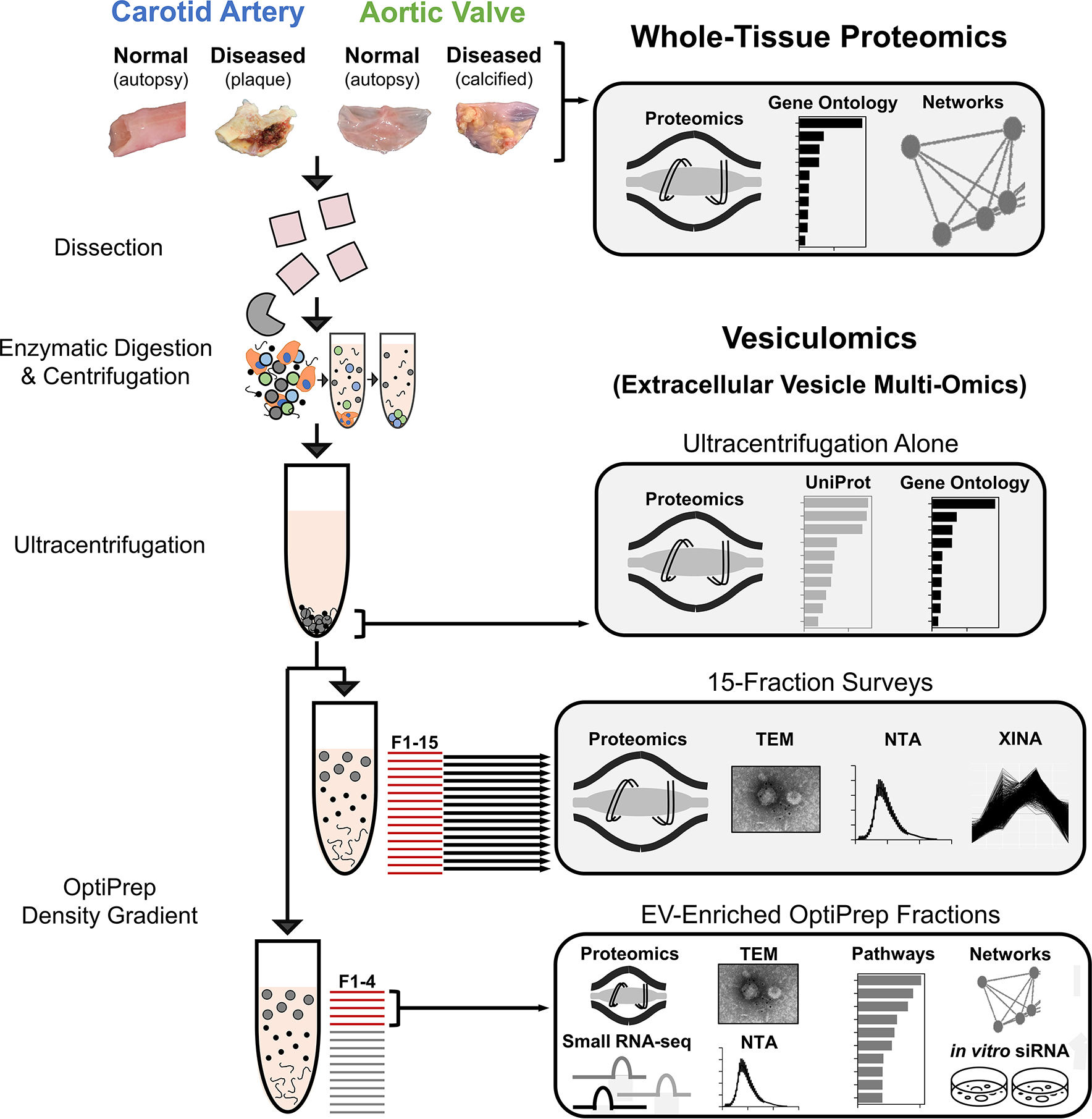
Label-free proteomics was conducted on whole-tissue samples of human normal carotid arteries, diseased carotid artery atherosclerotic plaques (from carotid endarterectomies), normal aortic valves, and diseased calcified aortic valves (from valve replacement surgeries for aortic valve stenosis). These sample types also underwent enzymatic digestion and serial low- and high-speed centrifugation. The high-speed supernatant then underwent ultracentrifugation to wash/pellet EVs. 15-fraction survey experiments utilized OptiPrep density gradient separation in concert with mass spectrometry, transmission electron microscopy (TEM), nanoparticle tracking analysis (NTA), and coabundance profiling (XINA) to identify which fractions were enriched in EVs and which contained non-EV contaminants. Enrichment performance was compared to that of ultracentrifugation alone by performing mass spectrometry on the pellet resulting from ultracentrifuged tissue digests. Tissue EV cargoes were then assessed by pooling EV-enriched OptiPrep fractions (F1–4) together and employing mass spectrometry, transcriptomics, TEM, NTA, and bioinformatics approaches to interrogate and classify these vesicles.
Methods
Detailed methods are provided in the Supplemental Materials. Human carotid arteries were acquired from i) patients undergoing carotid endarterectomies due to carotid artery stenosis (diseased calcified carotid artery atherosclerotic plaques) and ii) autopsy (normal carotid arteries). Human aortic valve (AV) leaflets were obtained from i) valve replacement surgeries due to AV stenosis (diseased calcified AVs), ii) autopsy (normal AVs), and iii) heart transplant recipients (normal AVs). Human samples were handled under Institutional Review Board (IRB) protocols to Dr. Peter Libby (1999P001348), E.A. (2014P001505), and S.C.B/J.D.M. (2011P001703), approved by the Partners Human Research Committee IRB. For 1999P001348, the IRB approved a waiver of informed consent and authorization for the use of excess human material from procedures that would otherwise be discarded. The IRB determined 2014P001505 (autopsy) did not meet the definition of human subjects research. Collection of aortic valves under 2011P001703 included written consent from all donors. Proteomics datasets are deposited to the ProteomeXchange Consortium/PRIDE repository (Table S1). Bioinformatics datasets and raw small RNA-seq counts are included in the Supplemental Materials; small RNA-seq fastq files are not publicly available to protect research participant privacy/consent. All other data that support the findings of this study are available from the corresponding author upon reasonable request. Tissues underwent collagenase digestion, (ultra)centrifugation, and a 15-fraction density gradient to enrich EVs. Label-free liquid-chromatography-tandem mass spectrometry (LC-MS/MS) proteomics were undertaken on intact tissue samples, each fraction, and EV-enriched pooled fractions; the latter also underwent small RNA-sequencing. Knockdown of EV-derived calcification targets was performed in primary human adult carotid artery smooth muscle cells (hCtASMCs) and human adult valvular interstitial cells (hVICs) cultured in normal or pro-calcifying media.
Statistical analysis:
Proteomics were quantified in Proteome Discoverer v2.2, normalized by total peptide amount and median abundance (Figures S2–S4), and differential enrichment was assessed in Qlucore Omics Explorer v3.7 via ANOVA (mixed-effects as appropriate) at a Benjamini-Hochberg false discovery rate (FDR, q/adjusted p-value) ≤0.05. Transcriptomics were processed by bcbio.nextgen and miRs were annotated against miRbase. miRs were tested for differential expression using DESeq216 defaults at an FDR≤0.05. miR targets were identified by TargetScan v7.2 (≥95th percentile weighted context++ score). BioCarta/KEGG/Reactome pathway analyses were tested for enrichment by a hypergeometric test and the Benjamini-Hochberg FDR, with p≤0.05 considered to be significantly-enriched in a gene set of interest. Pathway networks were based on Jaccard index gene overlap, while Louvain modularity optimization17 was utilized to cluster pathway nodes. Protein-protein interaction networks were built in STRING v11.0.18 Statistical analyses of non-omics datasets was performed via Student’s t-test (two-tailed, (un)paired as appropriate) for comparisons between two groups; one-way or two-way ANOVA with/without mixed-effects as appropriate with Holm-Sidak post-hoc multiple comparisons tests were used to evaluate statistically significant differences in multiple group comparisons (GraphPad Prism v9). Fisher’s exact test or multiple logistic regression assessed prevalence as appropriate.
Results
Whole-Tissue Proteomics Identifies Unique Drivers of Vascular and Valvular Disease Progression and Conservation of EV-Associated Protein Expression
While proteomics has been previously performed separately on atherosclerotic plaques19,20 or calcified stenotic aortic valves,21 the proteomes of these cardiovascular tissues have never been directly compared. To identify shared or deviating mechanisms underlying pathogenesis, we performed label-free LC-MS/MS on non-diseased, fibrotic, and calcified segments of age, sex, and histological disease burden-matched (Figures S5–S6, Table S2) human carotid artery atherosclerotic plaques (n=16×3 stages) and calcified aortic valves (n=18×3 stages). The proteomes of intact normal tissues and non-diseased regions are distinct from those of fibrotic or calcified regions (Figure S7). Principal component analysis of 2,318 proteins found clear tissue- and disease stage-specific clustering, with a significant convergence of carotid and valve tissue proteomes during disease progression (Figure 2A). Despite this similarity in advanced disease stage, differential-enrichment analysis identified unique drivers of atherosclerosis vs. aortic stenosis, as 381 and 226 proteins were altered exclusively by disease progression in carotid arteries or aortic valves, respectively (FDR≤0.05; Figure 2B, Tables S3/S4). Disease-altered proteins in carotid artery plaques (Figure 2C, Table S5) were dominated by associations with cell adhesion, migration, and cytoskeletal force generation (SYNPO2, PLP2, CALD1, NEXN, SMTN, COTL1); SMC plasticity (LMOD1); inflammation and lipid metabolism (ORM2, CXCL16, CASP1, CLIC1, CD84, GRN, CTSG, DDAH1, NPC2); and calcification (OSCAR, ADAM10). We investigated the global impact of the 381 proteins altered only in carotid artery disease via pathway enrichment analysis: 413 KEGG, BioCarta, and Reactome pathways (Figure 2F, Figure S8, Tables S6/S7) were significantly-enriched. Differentially-enriched pathway networks with unbiased Louvain clustering17 linked functional network communities to mechanotransduction, platelet activation, lipoprotein and energy metabolism, and FcεRI signaling and immune responses. In contrast, proteins significantly-enriched by aortic valve calcification (Figure 2E, Table S8) were associated with calcification/amyloidosis, (GAS6, SMOC1, PRPSAP1, TTR, CA1, CLEC11A, TNFRSF11B); apoptosis (ANP32A, PDCD6); inflammation and complement cascades (CRP, C4A, HBB, HBD, PROC, MASP1, SERPINE1); and histone/nucleosome function (H2BC17, U2AF2, NAP1L1). 169 pathways were significantly-enriched amongst the 226 disease-altered proteins unique to the aortic valve (Figure 2H, Figure S9, Tables S9/S10), including those involved in nonsense-mediated decay, complement cascades, carboxylation/hemostasis, apoptosis, and regulation of cell division. Lastly, 120 proteins were changed by disease progression in both carotid arteries and aortic valves (FDR≤0.05; Figure 2B, Table S11). These were associated with a 2.9-fold increase in vesicle-associated gene ontology (GO) terms vs. the total GO term database (p<0.0001; Figure 2D, Table S12). Furthermore, 70% of the most significantly-enriched GO terms in this shared proteome associated with EVs, exosomes, exocytosis, or secretion. Pathway network communities (Figure 2G, Figure S10, Tables S13/S14) were linked to lipoprotein remodeling, adaptive immunity/antigen presentation, autophagy/phagocytosis, and various receptor-mediated signaling cascades. Together, these observations implied that EV-related functions were highly present in carotid artery plaque and calcified aortic valve pathogenesis, prompting us to isolate and directly interrogate the contents of these tissue EVs.
Figure 2: Whole-Tissue Proteomics Finds Conservation of Extracellular Vesicle-Associated Processes Between Vascular and Valvular Disease Progression.
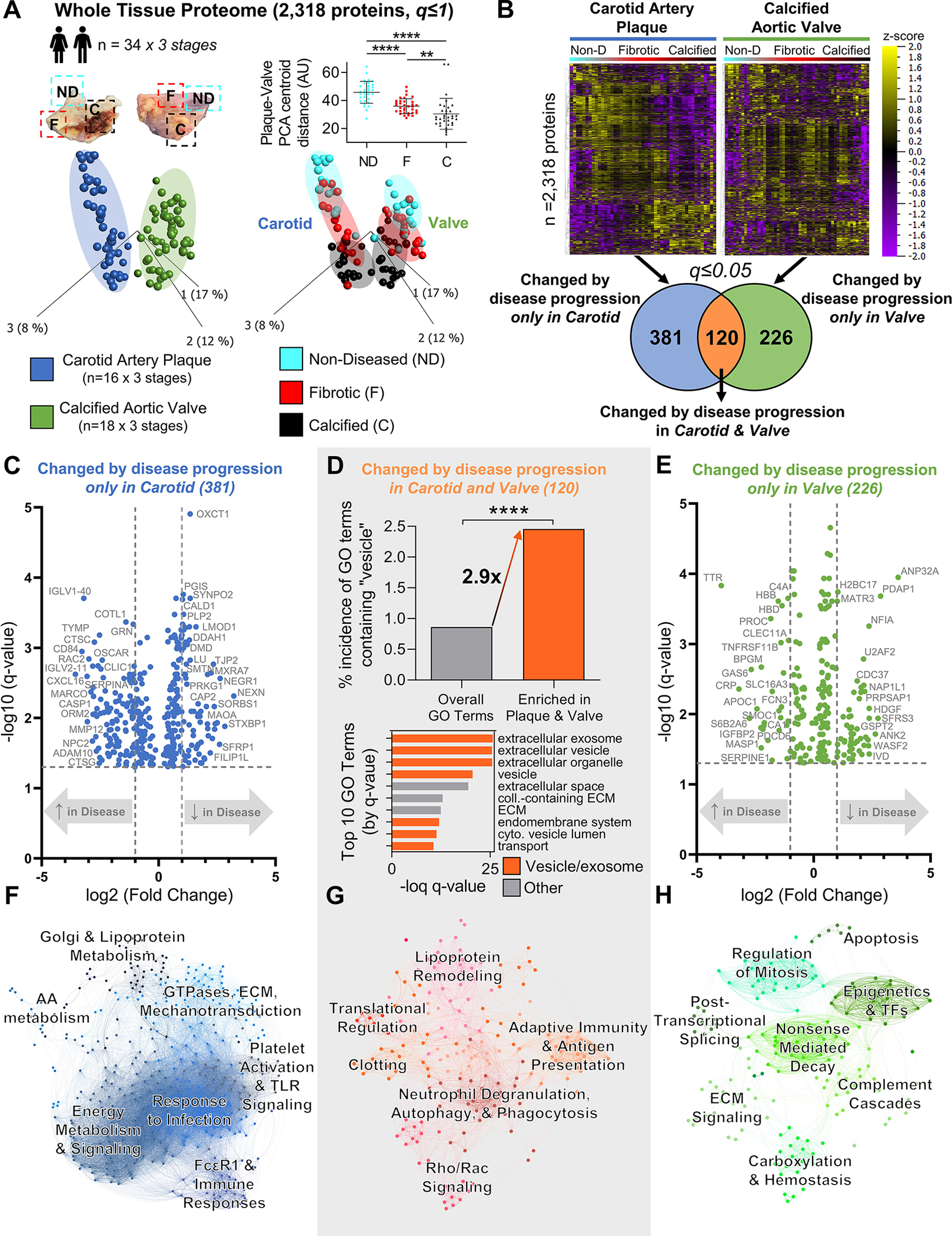
A, Principal component analysis of 2,318 proteins (q≤1) quantified by disease stage-specific whole-tissue proteomics of human carotid artery plaques and calcified aortic valves revealed significant convergence of carotid and valve tissues during disease pathogenesis; n=16 carotid artery plaque and 18 calcified aortic valve donors x 3 stages of disease (non-diseased, fibrotic, calcified) per donor; mean±SD; *p<0.05, ****p<0.0001. B, Unfiltered heat map analyses (q≤1; ordered by hierarchical clustering) demonstrated disease stage-specific alterations of protein abundances in carotid artery plaques (left) and calcified aortic valves (right). Enrichment analysis identified 381 proteins whose abundances were significantly differentially-enriched only by disease progression in carotid artery plaques, 226 proteins in calcified aortic valves, and 120 proteins in both tissue types (significantly-enriched proteins filtered at q≤0.05). C and E, Volcano plots of proteins significantly differentially-enriched by disease progression only in carotid artery plaques (381 proteins) or only in calcified aortic valves (226 proteins); cutoffs at a fold-change of 2 and a q-value of 0.05. D, In proteins significantly differentially-enriched in both carotid artery plaques and calcified aortic valves (120 proteins), we found a found a statistically significant 2.9-fold increase in the incidence of vesicle-associated GO terms vs. the total GO term database (****p<0.0001). In addition, 7 of the top 10 most-significant GO terms were associated with extracellular vesicles, exosomes, exocytosis, or secretion (orange bars). F-H, Networks based on KEGG, Reactome, and BioCarta pathway enrichment among proteins significantly differentially-enriched by disease progression only in carotid artery plaques (381 proteins, F), in both carotid artery plaques and calcified aortic valves (120 proteins, G), or only in calcified aortic valves (226 proteins, H) with pathways as the nodes (node size corresponds to -log(q-value)) and shared detected proteins between pathways as the edges (edge thickness matches the Jaccard index of overlap between detected proteins of the two connected pathway nodes). Unbiased clustering of pathways into real network communities by the Louvain method revealed shared- and tissue-specific drivers of disease pathogenesis.
Proteomics, Enzymatic Digestion, Serial (ultra)Centrifugation, and Density Gradient Separation Isolate and Enrich Tissue EVs
We dissociated intact human carotid artery plaques (n=3) and calcified aortic valves (n=4) by bacterial collagenase digestion. To enrich EVs from crude tissue digests, we coupled sequential centrifugation and ultracentrifugation to 15-fraction density gradient separation, an EV enrichment approach used with success in biofluids (Figure S11). We employed label-free proteomics to survey every density gradient fraction to assess where EVs had collected, quantified 1,491 proteins, and found that 22 EV marker proteins (e.g., annexins, CD63/81, flotillins, HSP70s, TSG101, tetraspanins)9 were enriched in the four least-dense fractions from both carotid artery plaques and calcified aortic valves, peaking in fraction 3 (Figure 3A,C, Figure S12A). Using clustering-to-networks (XINA22), protein co-abundance profiling across the 15 fractions demonstrated further indications of EV enrichment in fractions 1–4: the five clusters whose profiles mimicked those of the 22 EV markers were highly significant for GO terms associated with vesicular processes (Figure S12B, Tables S15/S16). Fractionation successfully separated EVs from non-EV contaminants (Figure 3A,C): globular collagens (e.g., Collagen VIA1, VIA2, VIA3) and other extracellular matrix (ECM) components (e.g. Fibrillin 1, Versican) were enriched in fractions 5–10, and fibrillar collagens (e.g., Collagen IA1, IA2, IIIA1) were found primarily in fractions 11–15. This phenotypic separation was also visualized by CD-63 immunogold transmission electron microscopy (TEM), which confirmed CD63+ membrane-bound EVs in low-density fractions from vascular and valvular tissues, globular collagens in fractions 5–10, and fibrillar collagens in the densest fractions (Figure 3E; Figures S13/S14). We then employed nanoparticle tracking analysis (NTA) to quantify the size and presence of tissue EVs (Figure 3B,D). In fractions 1–4, NTA identified particles ~200 nm in diameter with a size distribution characteristic of EVs and which agree with previous reports of EVs released in vitro from cultured macrophages, vascular SMCs, and VICs under calcifying conditions.6,14,23
Figure 3: Tissue EVs are Highly Enriched in the Least-Dense Fractions from Density Gradient Separation.
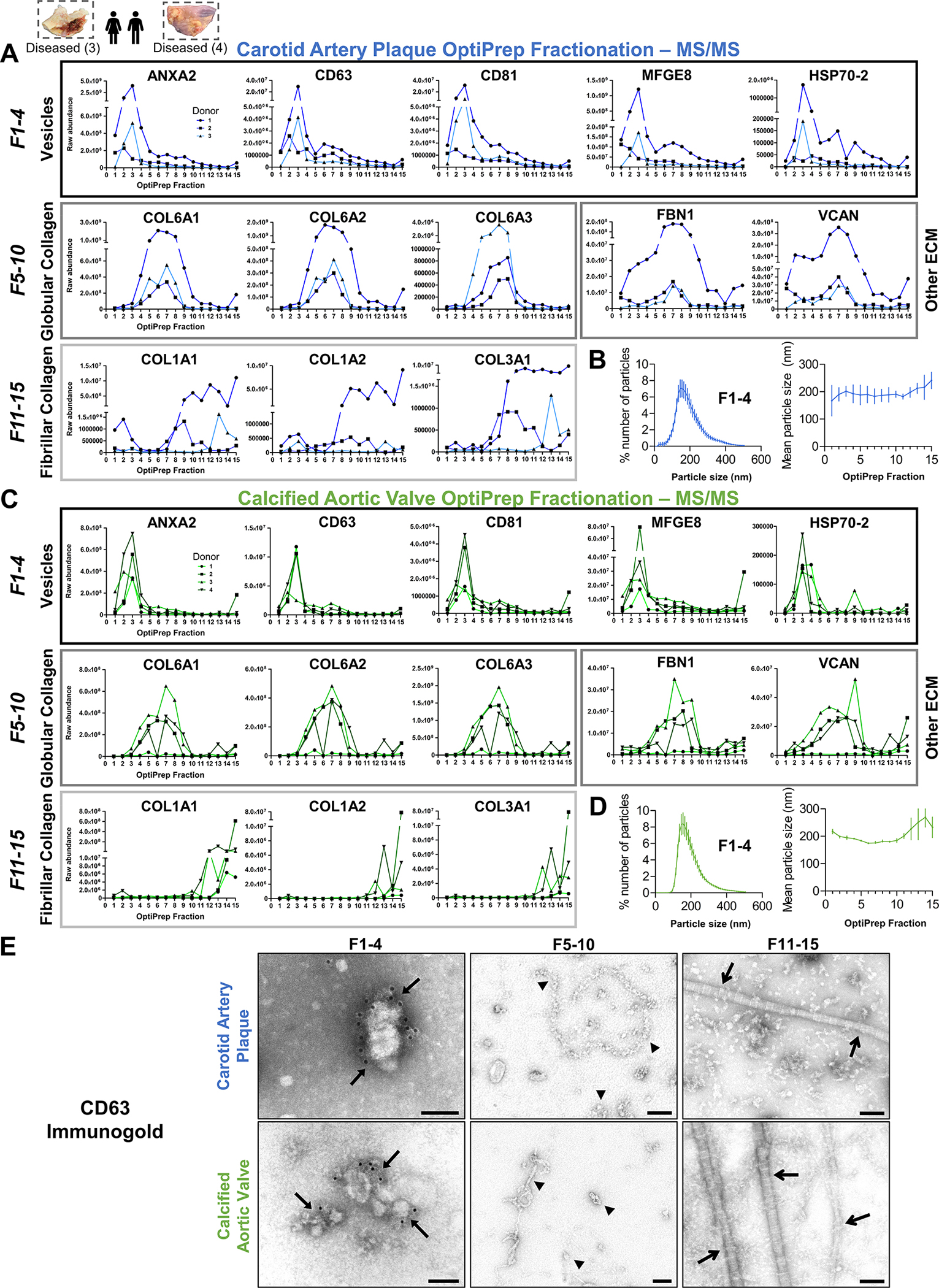
A and C, From 15-fraction survey experiments, raw abundance by mass spectrometry of selected proteins from intact human carotid artery plaques and calcified aortic valves demonstrated that extracellular vesicle marker proteins (Annexin A2, CD63, CD81, MFGE8 [lactadherin], HSP70–2) were highly enriched in the four least-dense fractions of both tissue types; n=3 carotid artery plaque and 4 calcified aortic valve donors. Enrichment of globular collagens (Collagen VIA1, VIA2, VIA3), fibrillar collagens (Collagen IA1, IA2, IIIA1), and other components of the extracellular matrix (ECM; FBN1, VCAN) was identified in denser fractions. B and D, Nanoparticle tracking analysis confirmed the characteristic presence of particles ~150–200 nm in diameter in fractions 1–4 of both intact carotid artery plaques and calcified aortic valves (n=3 per tissue type), while mean particle size remained consistent between fractions (mean±SEM). E, Representative CD63-labelled immunogold transmission electron microscopy (TEM) identified CD63+ membrane-bound EVs in fractions 1–4 (arrows) from intact carotid artery plaques (top) and calcified aortic valves (bottom); bar=100 nm. Consistent with mass spectrometry-derived protein abundance in A/C, TEM showed abundant globular collagens in fractions 5–10 (arrowheads) and fibrillar collagens in the most-dense fractions (open arrows).
Ultracentrifugation has been used in isolation to enrich EVs from biofluids and culture media.24 As tissues are inherently rich in ECM vs. cell culture and biofluids, we performed proteomics to compare calcified carotid artery plaque and aortic valve tissue EVs enriched by our fractionation pipeline vs. ultracentrifugation alone (Figure S15, Tables S17–S20). The fractionation-derived EV proteome was enlarged by nearly 40% vs. ultracentrifugation-alone in both tissues, and loaded with cellular components that may contribute to cell-cell communication and known to be sorted and loaded into EVs during vesicle biogenesis. In contrast, the proteome produced uniquely by ultracentrifugation was rich in ECM and endoplasmic reticulum components.9 Lastly, we developed a targeted mass exclusion strategy that mitigated the impact of iodixanol, improved the size of the fractionation-derived tissue EV proteome by 9.5%, and contributed to detection of pathologically-relevant EV cargoes (Figures S16–S17, Table S21).
Tissue EV Protein Cargoes in the Normal and Diseased Vascular and Valvular Niches
EVs carry bioactive protein cargoes that mediate intercellular signaling, act as early nucleation sites of microcalcification, and reflect pathobiological processes occurring within EV-producing cells themselves.8 We applied our toolkit for enrichment of tissue EVs to study whether tissue-specific alterations of entrapped EV cargoes exist in cardiovascular disease, and examined contributions of these cargoes to the pathogenesis of vascular and valvular calcification. To this end, we extracted tissue EVs from intact normal carotid arteries (n=6 donors), diseased carotid artery atherosclerotic plaques (n=4), normal aortic valves (n=6), and diseased calcified aortic valves (n=4) by pooling density gradient fractions 1–4 and confirmed EV enrichment by TEM and NTA (Figure 4A, Figure S18). Proteomics on these EVs quantified 1,942 proteins, and principal component analyses displayed strong tissue- and disease state-specific clustering (Figure 4B). We identified 525 tissue EV proteins that were differentially-enriched only between normal and diseased carotid arteries, 248 whose abundances differed only between normal and diseased aortic valves, and 404 EV proteins altered by disease in both tissues (FDR≤0.05; Figure 4B, Tables S22–S24). To examine how EV cargoes reflect tissue-level processes, we assessed overlap of the whole-tissue and tissue EV proteomes (Figure 4C): 45.9% of proteins detected overall were found in both datasets and abundances of these shared proteins were significantly correlated between the whole-tissue and EV proteomes. EVs are thus reflective of some, but not all, phenotypes found at the whole-tissue level and may have important biological functions missed by whole-tissue omics (and vice-versa). To obtain further biological insights, we examined the tissue EV proteome through pathway enrichment analyses (Figure 4D, Tables S25–S28); 749 significantly-enriched KEGG pathways were detected in total. Pathways linked to EV proteins changed by disease in both tissues described roles previously implicated in cardiovascular calcification, including leukocyte transendothelial migration, lipolysis/fatty acid degradation, gluconeogenesis, and pyruvate metabolism. Of note, disease-altered carotid EV cargoes were specifically tied to pathways involved in complement and coagulation cascades, platelet activation, and mechanotransduction (including focal adhesions, cytoskeletal regulation, ECM-receptor interactions, and cell adhesion). In contrast, EV proteins enriched by aortic valve pathogenesis associated with amino acid metabolism, calcium reabsorption, PI3K-Akt signaling, and cGMP signaling. Differentially-enriched tissue EV proteins were confirmed by immunogold-TEM (Figure S19, Table S29).
Figure 4: Proteomics of EV-Enriched Pooled Fractions to Quantify EV Protein Cargoes in Normal and Diseased Vascular and Valvular Tissue.
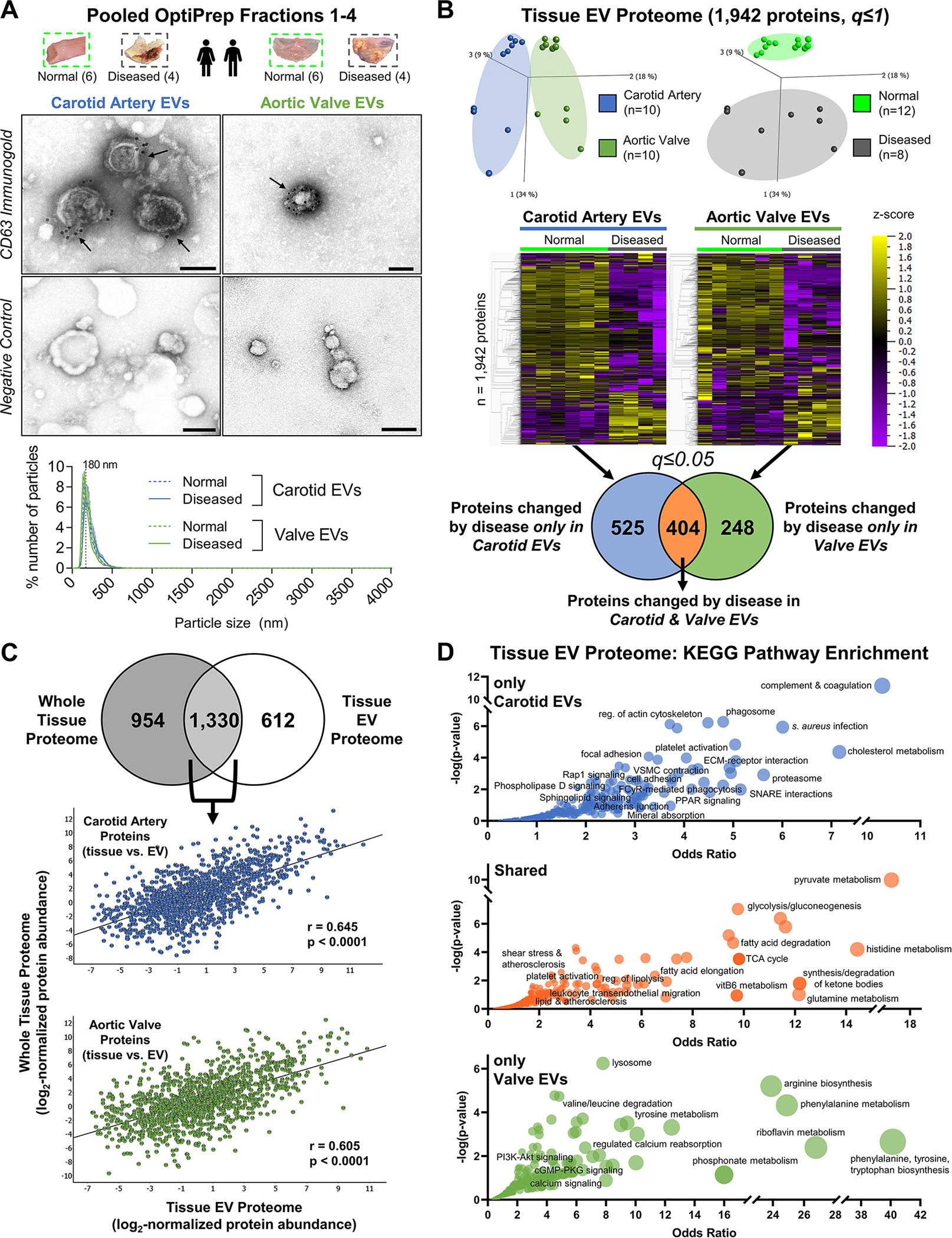
A, Tissue EVs were isolated by density gradient separation from intact normal carotid arteries (n=6 donors), intact diseased carotid artery atherosclerotic plaques (n=4), intact normal aortic valves (n=6), and intact diseased calcified aortic valves (n=4). Representative CD63-labelled immunogold transmission electron microscopy (TEM) and negative control images from pooled fractions 1–4 of intact human carotid artery (left) and aortic valve (right) demonstrated EV enrichment and purification; bar=100 nm. Nanoparticle tracking analysis (bottom, all donors, mean±SEM) found that EVs from intact normal and diseased carotid arteries and aortic valves all had similar mean diameters with a single peak around 200nm, and were isolated without co-enrichment of larger microparticles or apoptotic bodies. B, Isolated tissue EVs from every donor underwent both proteomics and small RNA-seq. The tissue EV proteome was composed of 1,942 proteins. Unfiltered principal component analyses (q≤1) identified tissue- (left) and disease state-specific (right) clustering. Unfiltered heat map analyses (q≤1; ordered by hierarchical clustering) illustrated alterations in individual tissue EV protein abundances between normal and diseased carotid arteries and aortic valves. Abundances of 525 tissue EV proteins were significantly differentially-enriched only between normal and diseased carotid arteries, 248 tissue EV proteins differed only between normal and diseased aortic valves, and 404 tissue EV proteins were significantly altered by disease pathogenesis in both tissue types (significantly-enriched proteins filtered at q≤0.05). C, 68.5% of the tissue EV proteome (1,330 proteins) was also found in the whole-tissue proteome; linear regression identified moderate and statistically significant correlations of per-protein abundances between the whole-tissue and diseased tissue EV proteomes in both carotid artery (Pearson’s r = 0.645, p<0.0001) and aortic valve (Pearson’s r = 0.605, p<0.0001). D, Bubble plots of KEGG pathways that were significantly-enriched amongst tissue EV proteins changed by disease only in carotid arteries (top, 525 proteins), in both carotid arteries and aortic valves (middle, 404 proteins), or only in aortic valves (bottom, 248 proteins) identified shared- and tissue-specific roles of disease-altered cardiovascular tissue EV cargoes. Bubble size corresponds to the percentage of differentially-enriched proteins amongst all pathway constituents.
Sequencing Reveals the Tissue EV Non-Coding miRNAome
Along with protein cargoes, noncoding miRs are also incorporated into EVs and can impact post-transcriptional regulation in recipient cells.10 We extracted miRNA from the same tissue EV samples that underwent proteomics (n=20, reported in Figure 4) and performed small RNA-sequencing to assess the cardiovascular tissue EV miRNAome (Figure 5A, Table S30). 1,083 EV-derived miRs were mapped to miRbase; their expression clustered by tissue-type and disease-state. 44 EV miRs were differentially-enriched by disease only in carotid arteries, 36 miRs changed only in aortic valves, and 26 miRs were altered in EVs from both tissue types (Figure 5B, Tables S31–S33). To determine the biological roles of the EV miRNAome, we used TargetScan 7.2 to predict high-confidence mRNA targets.
Figure 5: Sequencing Reveals the Human Tissue EV non-coding miRNAome from Vessels and Valves.
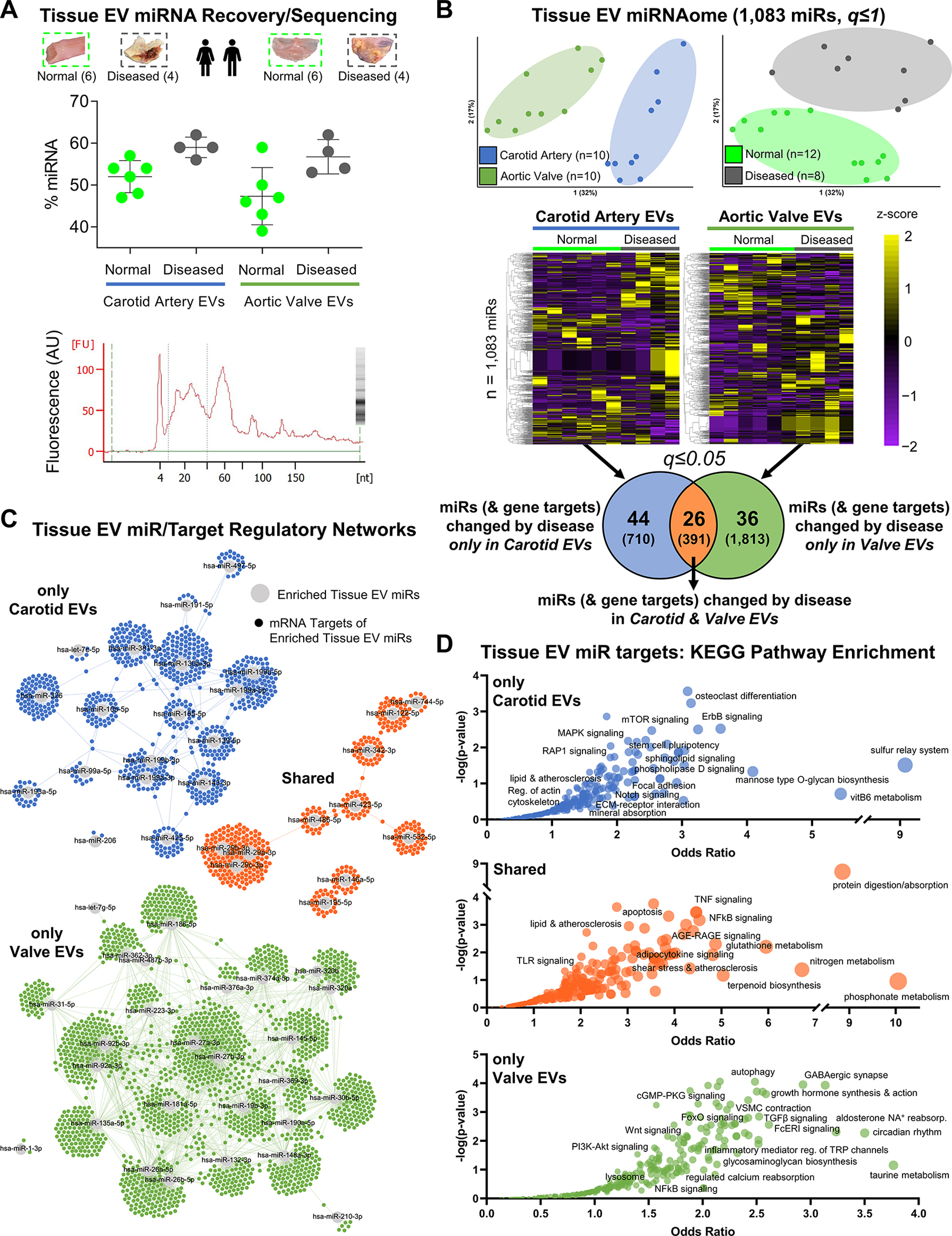
A, Along with proteomics, small RNA-seq was also performed on those tissue EVs collected from intact normal carotid arteries (n=6 donors), intact diseased carotid artery atherosclerotic plaques (n=4), intact normal aortic valves (n=6), and intact diseased calcified aortic valves (n=4). Top: RNA fragment analysis found that density gradient-enriched EVs contained enhanced levels of miRNA (mean miRNA content=53.0% of all small RNA; mean±SD). Bottom: representative fragment analysis tracing showing large miRNA-associated fragment peak at 24 nucleotides (nt). B, Unfiltered principal component analyses (q≤1) identified tissue- (left) and disease state-specific (right) clustering of 1,083 miRs sequenced in the tissue EV miRNAome. Unfiltered heat map analyses (q≤1; ordered by hierarchical clustering) characterized tissue EV miR cargoes altered between normal and diseased carotid arteries and aortic valves: 44 tissue EV miRs (with 710 unique high-confidence gene targets) were significantly differentially-enriched by disease pathogenesis only in carotid arteries, 36 tissue EV miRs (1,813 unique targets) changed only in aortic valves, and 26 tissue EV miRs (391 unique targets) were altered in both tissues (significantly-enriched miRs filtered at q≤0.05). C, miR/target regulatory networks of TargetScan-predicted gene targets (≥95th percentile weighted context++ score) of tissue EV miRs altered by disease only in carotid arteries (top; blue), in both carotid arteries and aortic valves (middle; orange), or only in aortic valves (bottom; green). D, Bubble plots of KEGG pathways significantly-enriched in the unique gene targets of tissue EV miRs changed by disease only in carotid arteries (top), in both carotid arteries and aortic valves (middle) or only in aortic valves (bottom) reveals the regulatory landscape associated with disease-altered tissue EV miR cargoes. Bubble size corresponds to the percentage of unique targets of differentially-enriched miRs amongst all pathway constituents.
miRs impacted by disease exclusively in vascular and valvular EVs targeted 710 and 1,813 genes, respectively (Tables S34–S36). We then built EV miRNA/mRNA target regulatory networks (Figure 5C, Figures S20–S22, Tables S37–S39), which exhibited clustering and segregation of target genes by tissue type, and leveraged pathway enrichment analysis of EV miR gene targets to examine roles of tissue EV contents (Figure 5D, Tables S40–S43). Target genes of the disease-altered EV miRNAome were significantly-enriched among 821 KEGG pathways, with a shared alteration of miRs targeting protein digestion and TNF, NFκB, and TLR signaling. EV miRs changed by carotid atherogenesis targeted the constituents of pathways associated with osteoclasts and mineral reabsorption, mechanotransduction, and Rap1/Notch/sphingolipid signaling pathways. Meanwhile, targets of miRs altered with disease in aortic valve EVs targeted regulatory programs associated with autophagy, proteoglycan biosynthesis, and calcium metabolism in addition to cGMP, Wnt, TGFβ, and FcεRI signaling that play complementary roles in inflammation and calcification.
Vesiculomics Integration Identifies Conserved Cardiovascular Tissue EV Cargoes
While the protein and noncoding miR contents of EVs are both bioactive and modulate recipient cell phenotype and function, their impact has previously only been studied separately. To assess the unified effect of these two types of cargo, we completed pathway-level integration on EV multi-omics data to delineate the predicted impact of the vesiculome as a whole. We first integrated EV cargoes altered by disease in both intact carotid arteries and aortic valves (Figure 4B/5B; orange Venn segments), which represent putatively conserved cardiovascular tissue EV-borne drivers of disease. Pathogenesis modified 50 pathways (Figure 6A, Tables S28, S43) in both the EV proteome and gene targets of the EV miRNAome. To dissect their conserved biological impact, we assembled them into a pathways network based on the Jaccard index of overlap between pathway members that were detected in our multi-omics. Unbiased Louvain clustering17 identified functional communities within the pathway network, revealing that disease-altered EV cargoes shared 4 distinct annotations including modulation of intracellular signaling cascades, cell cycle regulation/apoptosis, protein processing and vesicular transport, and ECM synthesis and organization (Figure 6B, Figure S23, Table S44). These data indicated that cardiovascular tissue EV cargoes are predicted to modulate intracellular signaling of recipient cells in diseased tissues, while pathway network betweenness-centrality scores implicated epidermal growth factor receptor (EGFR) and estrogen signaling as a key shared disease-driven molecular constituent of tissue EVs.
Figure 6: Pathway Networks Shared Across Omics Layers Identify Conserved Roles of Cardiovascular Tissue EV Cargoes Altered by Disease.
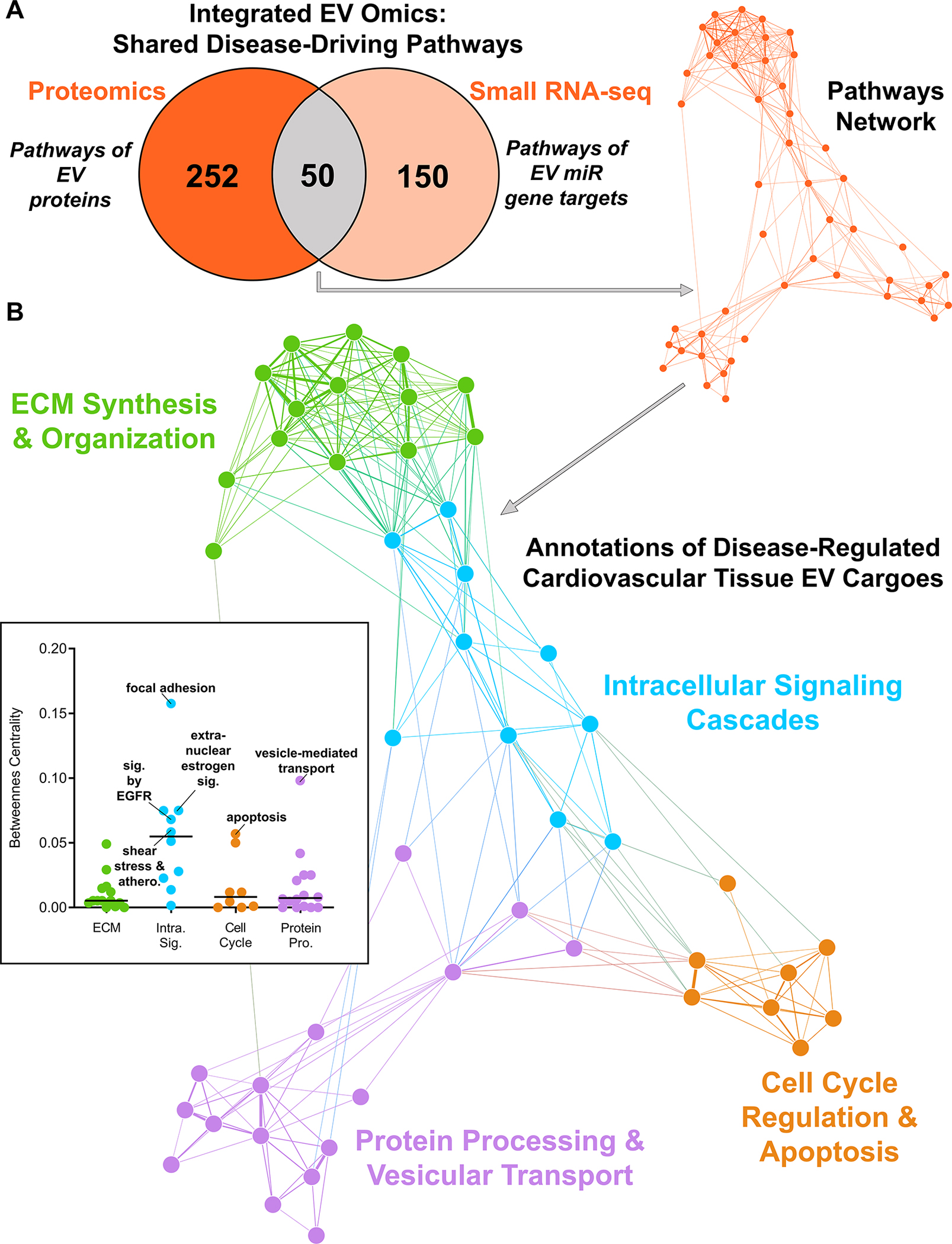
A, The network of 50 overlapping KEGG, Reactome, and BioCarta pathways that were significantly-enriched in the proteome and gene targets of miRs altered by disease in both intact carotid artery and aortic valve tissue EVs (n=6 normal carotid arteries, n=4 diseased carotid artery atherosclerotic plaques, n=6 normal aortic valves, n=4 diseased calcified aortic valves). Pathways are nodes (node size corresponds to -log(q-value)) and shared detected genes between pathways are edges (edge thickness matches the Jaccard index of overlap between detected genes of the two connected pathway nodes). B, Louvain clustering revealed 4 distinct annotations shared by disease-altered cardiovascular tissue-derived EV cargoes, including modulation of intracellular signaling cascades and cell cycle regulation and apoptosis. Pathway network betweenness-centrality scores (inset) implicate estrogen/epidermal growth factor signaling as a key common molecular constituent of tissue EVs.
Network Analyses Across Omics Layers Identify EV-Borne Modulators of Calcification
Vesiculomics clustering and differential enrichment analyses indicated that large subsets of EV protein and miR cargoes were uniquely altered by disease pathogenesis solely in one tissue type (Figures 4B/5B; blue and green segments of Venn diagrams). We therefore also performed tissue-specific multi-omics integration of EV cargoes to assess overall tissue-specific bioactivity of EV contents. 27 and 29 overlapping KEGG/Reactome/BioCarta pathways were enriched amongst both disease-altered EV proteins and gene targets of EV miRs in intact carotid arteries or aortic valves, respectively (Figure 7A/B). Protein-protein interaction networks were generated to assess the integrated impact of those overlapping pathways for which there was robust literature evidence of their involvement in calcification (Figure 7C/D, Tables S45, S46). Among other pathways, EVs carried tissue-specific cargoes associated with pro-calcific Notch and Wnt signaling in carotid arteries and aortic valves, respectively. Carotid artery EV cargoes that i) we identified as disease-regulated and ii) had high betweenness-centrality scores in their respective networks included FGFR2 (Fibroblast Growth Factor Receptor 2; a critical mediator of osteogenic fate in mesenchymal stem cells (MSCs) via ERK/PKC signaling25), PPP2CA (Protein Phosphatase 2 Catalytic Subunit Alpha; an ERK/p38-dependent regulator of MSC osteogenesis/bone formation26), and ADAM17 (ADAM metallopeptidase domain 17; a NOTCH signaling activator regulated by RUNX2 in osteoblasts27). In aortic valve EVs, our network approach identified WNT5A (Wnt Family Member 5A; controller of non-canonical Wnt-mediated osteogenesis28), APP (Amyloid Beta Precursor Protein; basis for amyloid plaques found in AV stenosis and a promoter of osteoblast survival and bone formation29–31), and APC (Adenomatous Polyposis Coli Regulator of WNT Signaling; antagonizes Wnt signaling via degradation of β-catenin32). siRNA-mediated knockdown (Figure 7E/F/I) of FGFR2, PPP2CA, or ADAM17 mRNA significantly reduced Alizarin Red calcification of primary human carotid artery smooth muscle cells (hCtASMCs, n=3 donors) when cultured for 14–21 days in pro-calcifying media. In contrast, knockdown of WNT5A, APP, and APC (Figure 7G/H/J) significantly altered calcification in primary human aortic valvular interstitial cells (hVICs, n=3 donors). Reciprocal knockdown experiments (Figures S24–S25) found that PPP2CA, ADAM17, APP, and APC significantly inhibited calcification in hCtASMCs and VICs, while the effects of FGFR2 and WNT5A were cell-type-specific. These data suggest that cardiovascular EV cargoes regulated in a tissue-specific manner by disease include potent modulators of SMC or VIC calcification, respectively.
Figure 7: Integration of EV Multi-Omics Identifies Modulators of Calcification.
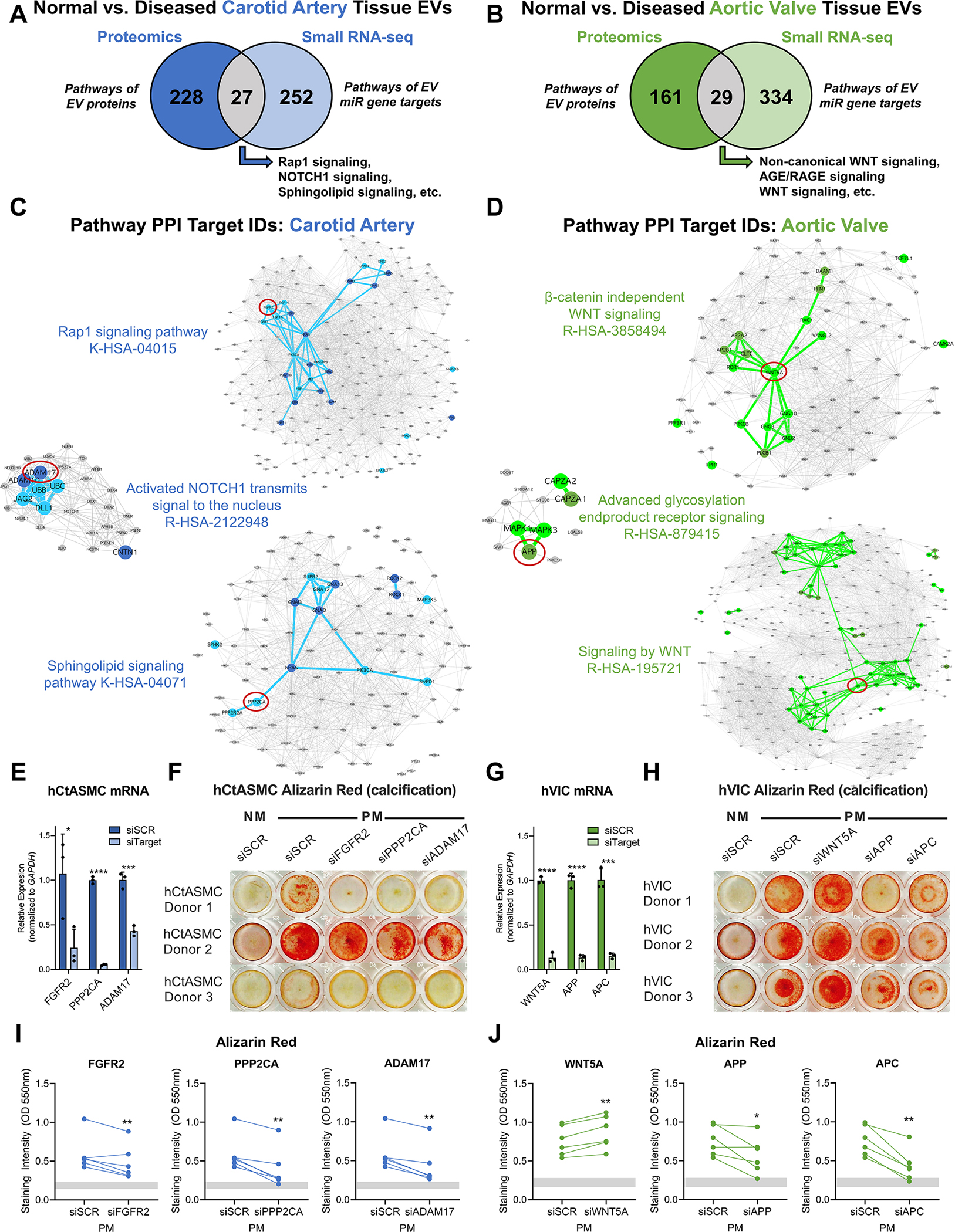
A and B, Overlap of KEGG, Reactome, and BioCarta pathways enriched amongst tissue EV proteins and unique gene targets of tissue EV miRs that were differentially-enriched between intact normal and diseased carotid arteries (A) and aortic valves (B). N=6 normal carotid arteries, n=4 diseased carotid artery atherosclerotic plaques, n=6 normal aortic valves, n=4 diseased calcified aortic valves. C and D,Protein-protein interaction networks further prioritized candidate EV-derived calcification modulators: constituents of selected overlapping carotid artery (C, blue nodes; FGFR2, PPP2CA, ADAM17) and aortic valve pathways (D, green nodes; WNT5A, APP, APC) that were significantly altered by disease progression in EV multi-omics had high betweenness-centrality scores in their respective networks. Node diameter corresponds to node degree. E and G, Relative mRNA expression levels of FGFR2, PPP2CA, ADAM17, WNT5A, APP, and APC vs. GAPDH in primary human carotid artery smooth muscle cells (hCtASMCs, E) and human aortic valvular interstitial cells (hVICs, G) after 6 days in normal medium (NM) incubated with scrambled siRNA (siSCR) or siRNA targeting FGFR2, PPP2CA, ADAM17, WNT5A, APP, and APC (siFGFR2, siPPP2CA, siADAM17, siWNT5A, siAPP, siAPC) demonstrated robust knockdown of target gene expression; n=1 donor per cell type, triplicate wells per donor, duplicate qPCR reactions averaged per well; mean±SD; *p<0.05, ***p<0.001, ****p<0.0001. F and H, Representative Alizarin red staining of target knockdown in hCtASMCs (F) and hVICs (H) at days 14–21 in NM or pro-calcifying medium (PM) culture. I and J, Quantification of solubilized Alizarin red stain in hCtASMCs (I) and hVICs (J) treated as in F and H for 14 and 21 days confirmed that inhibition of molecules identified by integration of EV multi-omics in A-D significantly modulated calcification of human vascular and valvular cells; n=3 hCtASMC donors and 3 hVIC donors, duplicate wells averaged per timepoint per donor; *p<0.05, **p<0.01. Grey bars indicate the minimum to maximum intensity range of the NM siSCR condition.
Discussion
A common set of risk factors predispose patients to cardiovascular calcification, though only a subset suffer from calcification of both the vessels and valves.2 Our comparison of whole-tissue proteomes across a spectrum of disease stages revealed novel insights into tissue-specific modes of calcification and identified a set of differentially-enriched proteins that have potential usage as therapeutic targets for tailored treatments of vascular vs. valvular calcification, or as putative biomarkers to discriminate between these two pathologies. We revealed enrichment of cell adhesion, migration, and cytoskeletal force generation, inflammation, and lipid metabolism in carotid artery atherogenesis. In calcific aortic valves, our whole-tissue results demonstrate over-representation of amyloid formation, apoptosis and complement cascade-associated components vs. atherosclerotic plaques – hallmarks of valvular stenosis in humans.21,30,31,33 We also identified a previously-unreported enhancement of nonsense-mediated mRNA decay (NMD) pathways in valvular pathogenesis. NMD is a critical cellular surveillance mechanism whose enrichment underscores potential genetic contributions to AV stenosis and represents a novel avenue for repurposing therapeutic NMD inhibition.34 Electron microscopy has shown calcified EVs to be present in mineralized regions of both tissues,8,14 consistent with the substantial enrichment of vesicle ontologies shared by the disease-altered proteomes of carotid arteries and aortic valves.
EVs regulate a spectrum of biological processes, act as mediators of cell-cell communication,10 predict cardiovascular events,11 and form the building blocks of cardiovascular microcalcification.8 This work represents, to the best of our knowledge, the first comprehensive study of EV cargoes from tissues other than easy-to-dissociate brain or tumors.15 Extraction of EVs from diseased cardiovascular tissues is particularly challenging due to the extensive ECM-rich fibrosis and severe calcific burden present. Loss of cellular integrity due to mechanical tissue homogenization leads to co-isolation of intracellular vesicles or artifactual membranous particles,35 and the apoptotic/necrotic nature of these diseases can also leave the extracellular microenvironment replete with large apoptotic bodies. Previous attempts to extract large microparticles from atherosclerotic human plaques by mincing and centrifugation alone36 or from calcified aorta by tissue grinding, enzymatic digestion, and ultracentrifugation13 were prone to these potential sources of contamination. Our use of bacterial collagenase circumvented mechanical homogenization and enabled digestion of collagen-rich cardiovascular tissues, maintenance of cell integrity/viability, and effective in silico elimination of the enzymatic protein. Our approach removes non-EV contaminants that are present should tissue digests solely undergo high-speed and/or ultracentrifugation prior to downstream analyses. Prior studies in brain have relied on sucrose-based gradients which can cause leaching of vesicle cargoes; here we employ iodixanol gradients while accounting for the impact of this compound on proteomics. By combining ultracentrifugation with density gradients, we enrich EVs from cardiovascular tissues and thoroughly classify them based on their size, density, morphology, and protein/nucleic acid cargoes as per MISEV guidelines.35
Overall, we found that the cardiovascular EV vesiculome was associated with pathways that included intercellular vesicle-mediated communication and processes that are incriminated in cardiovascular calcification: fatty acid degradation, macrophage activation, TNF signaling, NFκB signaling, and TLR signaling. In vascular vs. valvular calcification, ~13–27% of the EV proteome was differentially-enriched in a tissue-specific manner, indicating that this set of proteins may delineate pathophysiology of these two diseases. The disease-modulated carotid artery EV proteome linked to complement and coagulation cascades, mechanotransduction, SNARE interactions, and PPAR signaling, while EV cargoes implicated in valvular pathogenesis were associated with alternatively activated M2 macrophage polarization, cholesterol efferocytosis, calcium reabsorption, and cGMP/PKG signaling. Notably, cGMP/PKG signaling has been shown to control valvular myofibrogenesis, osteogenesis, calcification, and stenosis.37 We also identified disease-driven tissue-specific enrichment of 80 EV miRs, some of which have been previously implicated in cardiovascular disease, though not in the context of EV-mediated delivery. EV levels of miR-199b-5p and miR-381–3p were lowered in plaque-derived EVs vs. normal controls; reduction of miR-199b-5p is associated with production of reactive oxygen species, mitochondrial fission, and endothelial apoptosis38 while miR-381–3p protects against ox-LDL-induced endothelial inflammatory damage and inhibits hyperglycemia-driven SMC oxidative stress, and proliferation.39 In contrast, miR-27a-3p was depleted in diseased valvular EVs; this miR reduces calcium deposition via the osteogenic transcription factor ATF3 and protects against VIC inflammation.40 Disease also drove valvular EV enrichment of miR-181a-5p, which is shear-regulated, highly-enriched in the disease-prone fibrosa layer of the aortic valve, and promotes osteogenesis through CREB1.41 Pathway analysis of these 80 miRs strongly implicated osteoclast differentiation, neuregulin growth factor signaling, mTOR signaling, and o-linked glycosylation as key downstream pathological effectors of vascular EVs, while valvular EV miRs were linked to autophagy, TGF-β signaling, and Wnt signaling pathways involved in osteogenesis/calcification.42
Integration of these multiple omics layers allows for more coherent biological insight to be drawn than from the perspective provided by any one omics modality,21 and offered the first means to predict the combined impact of protein and RNA EV cargoes. Disease altered components of the tissue EV vesiculome connected to a host of signaling cascades, including a potential bi-directional role for EGFR signaling in EV-associated cardiovascular disease: vascular EGFR activation promotes inflammation and oxidative stress, while pharmaceutical inhibition prevents atherogenesis. In contrast, disrupted EGFR signaling induces aortic valve hyperplasia and stenosis – likely via induction of pro-osteogenic BMP signaling.43 Disease-regulated carotid artery plaque EV protein and miR contents also revealed a strong tissue-specific signal for Rap1, sphingolipid, and Notch signaling, all of which play fundamental roles in atheroprogression. EV cargoes FGFR2, PPP2CA, and ADAM17 were selected by network-based prioritization and promoted SMC calcification. FGFR2 drives osteogenic transitions via ERK/PKC signaling, while inhibition of FGFR2 in vivo attenuates atherosclerosis in ApoE-deficient mice by reducing SMC proliferation and macrophage infiltration.25 PPP2CA, a Ser/Thr phosphatase that inhibits MSC osteogenesis and long bone formation, prevents macrophage accumulation, lipid uptake, and foam cell formation.44 Meanwhile, RUNX2 regulates osteoblastic expression of the Notch signaling activator ADAM17; this metalloprotease controls endochondral ossification and regulates atherogenesis via TNFα.27 We also identified a link between aortic valve-derived EV cargoes, Wnt signaling, and advanced glycosylation endproduct receptor signaling (AGE-RAGE), and validated the impact of WNT5A, APP, and APC on hVIC mineralization. Aberrant Wnt signaling is implicated in osteogenic cardiovascular calcification, and WNT5A controls osteogenesis via non-canonical PKC/JNK upregulation and β-catenin antagonization.28 It is found focally around areas of calcification in human aortic valves,42 and hVIC treatment with the calcification inhibitor Fetuin A reduces WNT5A expression.45 APC acts as Wnt signaling antagonist via degradation of β-catenin.32 Notably, peripheral blood APC is associated with aortic stiffness and cardiovascular disease, raising EV-bound APC as an intriguing potential biomarker of AV stenosis.46 The AGE-RAGE axis promotes progression of valvular calcification and RAGE serves as a receptor for amyloid-β.47 Amyloid plaque co-localizes with apoptosis and calcific nodule formation in AVs, increases mineralization of hVICs,31 and multi-omics meta-analyses of AV stenosis suggest a role for the amyloid-β precursor APP in the formation of amyloid structures.30 Our whole-tissue proteomics provide some of the first evidence that amyloid contributes predominantly to valvular, not vascular, calcification.48 Knockdown of PPP2CA, ADAM17, APP, and APC significantly inhibited calcification in both cell types, suggesting that the pro-calcifying potential of these pathways is not specific to carotid arteries or aortic valves. Rather, differential EV loading may induce tissue-specific levels of these molecules and thus a calcification-prone environment. In contrast, the cell type-specific calcification effect of FGFR2 and WNT5A marks these cargoes as high-priority candidates for future translational efforts aimed at targeted treatment of vascular vs. valvular calcification. While further studies will be needed to delineate the mechanistic contributions of EVs to these processes, our data indicate that tissue-specific EV cargoes include powerful regulators of SMC or VIC calcification and may act as differential disease drivers in carotid arteries and aortic valves.
Though our findings are robust and represent important progress in the understanding of vascular/valvular pathobiology, there are limitations to this study. Despite participants being similar in age, sex distribution, and comparable for histological measures of inflammation, fibrosis, and calcification, carotid artery and aortic valve specimens would ideally be obtained from the same patient. While our knockdown experiments confirm the pathobiological relevance of key tissue EV cargoes, future studies will need to examine how these cargoes reflect biology of the cells from which EVs originate or their extracellular impact (e.g., on recipient cell function or matrix pathobiology).
Conclusions
The present study delves into the long-standing question of analogous pathobiology in vascular and valvular fibro-calcification processes. We identify unique drivers of these diseases, implicate tissue EVs in both conditions, and motivate development of distinct therapeutic strategies. By tightly integrating advancements in sample processing, proteomics, transcriptomics, and network medicine, we establish vesiculomics: interrogation and quantification of the biological impact of tissue EVs. We tie the vesiculome to tissue-specific regulation of key pathogenic mechanisms including Notch and Wnt signaling. This work opens an avenue towards examination of the critical roles that vesicles play as mediators of cell-cell communication, cellular differentiation, and cell-matrix interactions in the circulatory system. Both circulating and tissue EV pools likely contain sub-populations of vesicles that are secreted by different cell types (Figure S26), have exosomal or microvesicular origins, and function in protective or pathological ways.49 Here, we potentiate the examination of tissue EV populations and their complex interplay, which can be applied to future investigations of fibro-calcific processes in human, animal, and engineered tissues.8,50
Supplementary Material
Clinical Perspective.
What is New?
The first comparative proteomics study of pathogenesis in calcified human carotid arteries and aortic valves identifies unique drivers of atherosclerosis vs. aortic valve stenosis and implicates extracellular vesicles (EVs) in both diseases.
Vesiculomics: an efficient, cost-effective, accessible, and highly-validated strategy for isolating, quantifying, and assessing EV protein and small RNA cargoes directly from fibro-calcific human tissues which is applicable to other disease contexts or evaluation of animal/engineered tissues.
Multi-omics reveals that cardiovascular EV cargoes are regulated in a tissue-specific manner during progression of disease; these EVs carry potent modulators of calcification in vascular smooth muscle and valvular interstitial cells.
What Are the Clinical Implications?
Despite shared risk factors and similarities in advanced disease phenotypes, anti-atherosclerosis pharmacotherapies fail to improve outcomes in aortic valve stenosis.
Probing tissue-entrapped EV cargoes opens a new avenue for precision medicine treatments in cardiovascular disease.
Discovery of EV-derived therapeutic targets using multi-omics-based network medicine approaches is a promising avenue for the future tailored treatment of vascular and valvular calcification.
Acknowledgments
We thank Dr. Peter Libby (P.L.) for carotid endarterectomies; Maria Ericsson at the Harvard Medical School Electron Microscopy Facility; Johana Barrientos, Katelyn A. Perez, and Daniel Deppe for assistance with histology; Svetlana Gorbatov, James Gosnell, Amanda Pang, Thy Nguyen, and Marie McGourty for collecting clinical samples; Dr. Gary Sommerville and Dr. Zachary Herbert from the Molecular Biology Core Facilities at Dana-Farber Cancer Institute; Dr. Sergey Naumenko of the Harvard Chan Bioinformatics Core for assistance with sequencing alignment.
Sources of Funding
This study was supported by a research grant from Kowa Company, Ltd. (M.A.), NIH grants R01 HL136431, R01 HL141917, R01 HL147095 and a Harvard Catalyst Advanced Microscopy Pilot grant (E.A.), NIH grants R01 HL150401 and HL149998 (J.D.M.), and NIH K25 HL150336 (A.H.). L.P. was supported by NIH UL 1TR002541 and financial contributions from Harvard University/affiliated academic healthcare centers. P.L. is supported by the NIH (R01 HL080472, R01 HL134892), American Heart Association (18CSA34080399), and RRM Charitable Fund.
Non-standard Abbreviations and Acronyms
- ADAM17
ADAM metallopeptidase domain 17
- APC
adenomatous polyposis coli regulator of wnt signaling
- APP
amyloid beta precursor protein
- AV
aortic valve
- ECM
extracellular matrix
- EV
extracellular vesicle
- FDR
false discovery rate
- FGFR2
fibroblast growth factor receptor 2
- GO
gene ontology
- hCtASMC
human carotid artery smooth muscle cell
- hVICs
human valvular interstitial cel
- LC-MS/MS
label-free liquid-chromatography-tandem mass spectrometry
- miR
microRNA
- NMD
nonsense-mediated mRNA decay
- NTA
nanoparticle tracking analysis
- PPP2CA
protein phosphatase catalytic subunit alpha
- SMC
smooth muscle cell
- TEM
transmission electron microscopy
- WNT5A
wnt family member 5A
Footnotes
Disclosures
H.H. is an employee of Kowa Company, Ltd and was a visiting scientist at Brigham and Women’s Hospital during this study. Kowa had no role in study design/data collection/analysis/manuscript preparation. E.A. is a member of the scientific board of Elastrin Therapeutics Inc. G.C. is a member of the scientific board of Unicyte AG. P.L. is an unpaid consultant to/involved in clinical trials for Amgen, AstraZeneca, Esperion Therapeutics, Ionis Pharmaceuticals, Kowa Pharmaceuticals, Novartis, Pfizer, Sanofi-Regeneron, and XBiotech, Inc. and a member of scientific advisory boards for Amgen, Corvidia Therapeutics, DalCor Pharmaceuticals, IFM Therapeutics, Kowa Pharmaceuticals, Olatec Therapeutics, Medimmune, Novartis, and XBiotech, Inc. P.L. serves on the Board of XBiotech, Inc. P.L.’s laboratory has received research funding in the last 2 years from Novartis. The other authors declare no competing interests.
References
- 1.Chen J, Budoff MJ, Reilly MP, Yang W, Rosas SE, Rahman M, Zhang X, Roy JA, Lustigova E, Nessel L, et al. Coronary Artery Calcification and Risk of Cardiovascular Disease and Death Among Patients With Chronic Kidney Disease. JAMA cardiology. 2017;2:635–643. doi: 10.1001/jamacardio.2017.0363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rapp AH, Hillis LD, Lange RA, Cigarroa JE. Prevalence of coronary artery disease in patients with aortic stenosis with and without angina pectoris. The American journal of cardiology. 2001;87:1216–1217; A1217. doi: 10.1016/s0002-9149(01)01501-6 [DOI] [PubMed] [Google Scholar]
- 3.Otto CM, Kuusisto J, Reichenbach DD, Gown AM, O’Brien KD. Characterization of the early lesion of ‘degenerative’ valvular aortic stenosis. Histological and immunohistochemical studies. Circulation. 1994;90:844–853. doi: 10.1161/01.cir.90.2.844 [DOI] [PubMed] [Google Scholar]
- 4.Rossebo AB, Pedersen TR, Boman K, Brudi P, Chambers JB, Egstrup K, Gerdts E, Gohlke-Barwolf C, Holme I, Kesaniemi YA, et al. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med. 2008;359:1343–1356. doi: 10.1056/NEJMoa0804602 [DOI] [PubMed] [Google Scholar]
- 5.Itoh S, Mizuno K, Aikawa M, Aikawa E. Dimerization of sortilin regulates its trafficking to extracellular vesicles. The Journal of biological chemistry. 2018;293:4532–4544. doi: 10.1074/jbc.RA117.000732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.New SE, Goettsch C, Aikawa M, Marchini JF, Shibasaki M, Yabusaki K, Libby P, Shanahan CM, Croce K, Aikawa E. Macrophage-derived matrix vesicles: an alternative novel mechanism for microcalcification in atherosclerotic plaques. Circulation research. 2013;113:72–77. doi: 10.1161/CIRCRESAHA.113.301036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goettsch C, Hutcheson JD, Aikawa M, Iwata H, Pham T, Nykjaer A, Kjolby M, Rogers M, Michel T, Shibasaki M, et al. Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. The Journal of clinical investigation. 2016;126:1323–1336. doi: 10.1172/JCI80851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hutcheson JD, Goettsch C, Bertazzo S, Maldonado N, Ruiz JL, Goh W, Yabusaki K, Faits T, Bouten C, Franck G, et al. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nature materials. 2016;15:335–343. doi: 10.1038/nmat4519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nature reviews Molecular cell biology. 2018;19:213–228. doi: 10.1038/nrm.2017.125 [DOI] [PubMed] [Google Scholar]
- 10.Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nature cell biology. 2007;9:654–659. doi: 10.1038/ncb1596 [DOI] [PubMed] [Google Scholar]
- 11.Sinning JM, Losch J, Walenta K, Bohm M, Nickenig G, Werner N. Circulating CD31+/Annexin V+ microparticles correlate with cardiovascular outcomes. European heart journal. 2011;32:2034–2041. doi: 10.1093/eurheartj/ehq478 [DOI] [PubMed] [Google Scholar]
- 12.Kapustin AN, Davies JD, Reynolds JL, McNair R, Jones GT, Sidibe A, Schurgers LJ, Skepper JN, Proudfoot D, Mayr M, et al. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circulation research. 2011;109:e1–12. doi: 10.1161/CIRCRESAHA.110.238808 [DOI] [PubMed] [Google Scholar]
- 13.Kapustin AN, Chatrou ML, Drozdov I, Zheng Y, Davidson SM, Soong D, Furmanik M, Sanchis P, De Rosales RT, Alvarez-Hernandez D, et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circulation research. 2015;116:1312–1323. doi: 10.1161/CIRCRESAHA.116.305012 [DOI] [PubMed] [Google Scholar]
- 14.Cui L, Rashdan NA, Zhu D, Milne EM, Ajuh P, Milne G, Helfrich MH, Lim K, Prasad S, Lerman DA, et al. End stage renal disease-induced hypercalcemia may promote aortic valve calcification via Annexin VI enrichment of valve interstitial cell derived-matrix vesicles. Journal of cellular physiology. 2017;232:2985–2995. doi: 10.1002/jcp.25935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crescitelli R, Lasser C, Lotvall J. Isolation and characterization of extracellular vesicle subpopulations from tissues. Nat Protoc. 2021;16:1548–1580. doi: 10.1038/s41596-020-00466-1 [DOI] [PubMed] [Google Scholar]
- 16.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology. 2014;15:550. doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blondel VD, Guillaume J-L, Lambiotte R, Lefebvre E. Fast unfolding of communities in large networks. Journal of Statistical Mechanics: Theory and Experiment. 2008;2008:P10008. doi: 10.1088/1742-5468/2008/10/p10008 [DOI] [Google Scholar]
- 18.Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic acids research. 2019;47:D607–D613. doi: 10.1093/nar/gky1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Langley SR, Willeit K, Didangelos A, Matic LP, Skroblin P, Barallobre-Barreiro J, Lengquist M, Rungger G, Kapustin A, Kedenko L, et al. Extracellular matrix proteomics identifies molecular signature of symptomatic carotid plaques. The Journal of clinical investigation. 2017;127:1546–1560. doi: 10.1172/JCI86924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herrington DM, Mao C, Parker SJ, Fu Z, Yu G, Chen L, Venkatraman V, Fu Y, Wang Y, Howard TD, et al. Proteomic Architecture of Human Coronary and Aortic Atherosclerosis. Circulation. 2018;137:2741–2756. doi: 10.1161/CIRCULATIONAHA.118.034365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schlotter F, Halu A, Goto S, Blaser MC, Body SC, Lee LH, Higashi H, DeLaughter DM, Hutcheson JD, Vyas P, et al. Spatiotemporal Multi-Omics Mapping Generates a Molecular Atlas of the Aortic Valve and Reveals Networks Driving Disease. Circulation. 2018;138:377–393. doi: 10.1161/CIRCULATIONAHA.117.032291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee LH, Halu A, Morgan S, Iwata H, Aikawa M, Singh SA. XINA: A Workflow for the Integration of Multiplexed Proteomics Kinetics Data with Network Analysis. Journal of proteome research. 2019;18:775–781. doi: 10.1021/acs.jproteome.8b00615 [DOI] [PubMed] [Google Scholar]
- 23.Hutcheson JD, Goettsch C, Pham T, Iwashita M, Aikawa M, Singh SA, Aikawa E. Enrichment of calcifying extracellular vesicles using density-based ultracentrifugation protocol. Journal of extracellular vesicles. 2014;3:25129. doi: 10.3402/jev.v3.25129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Linares R, Tan S, Gounou C, Arraud N, Brisson AR. High-speed centrifugation induces aggregation of extracellular vesicles. Journal of extracellular vesicles. 2015;4:29509. doi: 10.3402/jev.v4.29509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miraoui H, Oudina K, Petite H, Tanimoto Y, Moriyama K, Marie PJ. Fibroblast growth factor receptor 2 promotes osteogenic differentiation in mesenchymal cells via ERK1/2 and protein kinase C signaling. The Journal of biological chemistry. 2009;284:4897–4904. doi: 10.1074/jbc.M805432200 [DOI] [PubMed] [Google Scholar]
- 26.Xie F, Li F, Li R, Liu Z, Shi J, Zhang C, Dong N. Inhibition of PP2A enhances the osteogenic differentiation of human aortic valvular interstitial cells via ERK and p38 MAPK pathways. Life Sci. 2020;257:118086. doi: 10.1016/j.lfs.2020.118086 [DOI] [PubMed] [Google Scholar]
- 27.Araya HF, Sepulveda H, Lizama CO, Vega OA, Jerez S, Briceno PF, Thaler R, Riester SM, Antonelli M, Salazar-Onfray F, et al. Expression of the ectodomain-releasing protease ADAM17 is directly regulated by the osteosarcoma and bone-related transcription factor RUNX2. J Cell Biochem. 2018;119:8204–8219. doi: 10.1002/jcb.26832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keller KC, Ding H, Tieu R, Sparks NR, Ehnes DD, Zur Nieden NI. Wnt5a Supports Osteogenic Lineage Decisions in Embryonic Stem Cells. Stem Cells Dev. 2016;25:1020–1032. doi: 10.1089/scd.2015.0367 [DOI] [PubMed] [Google Scholar]
- 29.Pan JX, Tang F, Xiong F, Xiong L, Zeng P, Wang B, Zhao K, Guo H, Shun C, Xia WF, et al. APP promotes osteoblast survival and bone formation by regulating mitochondrial function and preventing oxidative stress. Cell Death Dis. 2018;9:1077. doi: 10.1038/s41419-018-1123-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heuschkel MA, Skenteris NT, Hutcheson JD, van der Valk DD, Bremer J, Goody P, Hjortnaes J, Jansen F, Bouten CVC, van den Bogaerdt A, et al. Integrative Multi-Omics Analysis in Calcific Aortic Valve Disease Reveals a Link to the Formation of Amyloid-Like Deposits. Cells. 2020;9. doi: 10.3390/cells9102164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Audet A, Cote N, Couture C, Bosse Y, Despres JP, Pibarot P, Mathieu P. Amyloid substance within stenotic aortic valves promotes mineralization. Histopathology. 2012;61:610–619. doi: 10.1111/j.1365-2559.2012.04265.x [DOI] [PubMed] [Google Scholar]
- 32.Ha NC, Tonozuka T, Stamos JL, Choi HJ, Weis WI. Mechanism of phosphorylation-dependent binding of APC to beta-catenin and its role in beta-catenin degradation. Mol Cell. 2004;15:511–521. doi: 10.1016/j.molcel.2004.08.010 [DOI] [PubMed] [Google Scholar]
- 33.Galeone A, Brunetti G, Oranger A, Greco G, Di Benedetto A, Mori G, Colucci S, Zallone A, Paparella D, Grano M. Aortic valvular interstitial cells apoptosis and calcification are mediated by TNF-related apoptosis-inducing ligand. Int J Cardiol. 2013;169:296–304. doi: 10.1016/j.ijcard.2013.09.012 [DOI] [PubMed] [Google Scholar]
- 34.Zhao J, Li Z, Puri R, Liu K, Nunez I, Chen L, Zheng S. Molecular profiling of individual FDA-approved clinical drugs identifies modulators of nonsense-mediated mRNA decay. Mol Ther Nucleic Acids. 2022;27:304–318. doi: 10.1016/j.omtn.2021.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thery C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, Antoniou A, Arab T, Archer F, Atkin-Smith GK, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of extracellular vesicles. 2018;7:1535750. doi: 10.1080/20013078.2018.1535750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mayr M, Grainger D, Mayr U, Leroyer AS, Leseche G, Sidibe A, Herbin O, Yin X, Gomes A, Madhu B, et al. Proteomics, metabolomics, and immunomics on microparticles derived from human atherosclerotic plaques. Circ Cardiovasc Genet. 2009;2:379–388. doi: 10.1161/CIRCGENETICS.108.842849 [DOI] [PubMed] [Google Scholar]
- 37.Blaser MC, Wei K, Adams RLE, Zhou YQ, Caruso LL, Mirzaei Z, Lam AY, Tam RKK, Zhang H, Heximer SP, et al. Deficiency of Natriuretic Peptide Receptor 2 Promotes Bicuspid Aortic Valves, Aortic Valve Disease, Left Ventricular Dysfunction, and Ascending Aortic Dilatations in Mice. Circulation research. 2018;122:405–416. doi: 10.1161/CIRCRESAHA.117.311194 [DOI] [PubMed] [Google Scholar]
- 38.Cui X, Tian Y, Zhao Y, Gao H, Yao D, Liu L, Li Y. miR-199b-5p-AKAP1-DRP1 Pathway Plays a Key Role in ox-LDL-induced Mitochondrial Fission and Endothelial Apoptosis. Curr Pharm Biotechnol. 2022;23:1612–1622. doi: 10.2174/1389201023666220324123224 [DOI] [PubMed] [Google Scholar]
- 39.Zhu XS, Zhou HY, Yang F, Zhang HS, Ma KZ. miR-381–3p inhibits high glucose-induced vascular smooth muscle cell proliferation and migration by targeting HMGB1. J Gene Med. 2021;23:e3274. doi: 10.1002/jgm.3274 [DOI] [PubMed] [Google Scholar]
- 40.Chen H, Zhang Z, Zhang L, Wang J, Zhang M, Zhu B. miR-27a protects human mitral valve interstitial cell from TNF-alpha-induced inflammatory injury via up-regulation of NELL-1. Braz J Med Biol Res. 2018;51:e6997. doi: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rathan S, Ankeny CJ, Arjunon S, Ferdous Z, Kumar S, Fernandez Esmerats J, Heath JM, Nerem RM, Yoganathan AP, Jo H. Identification of side- and shear-dependent microRNAs regulating porcine aortic valve pathogenesis. Sci Rep. 2016;6:25397. doi: 10.1038/srep25397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Albanese I, Yu B, Al-Kindi H, Barratt B, Ott L, Al-Refai M, de Varennes B, Shum-Tim D, Cerruti M, Gourgas O, et al. Role of Noncanonical Wnt Signaling Pathway in Human Aortic Valve Calcification. Arteriosclerosis, thrombosis, and vascular biology. 2017;37:543–552. doi: 10.1161/ATVBAHA.116.308394 [DOI] [PubMed] [Google Scholar]
- 43.Barrick CJ, Roberts RB, Rojas M, Rajamannan NM, Suitt CB, O’Brien KD, Smyth SS, Threadgill DW. Reduced EGFR causes abnormal valvular differentiation leading to calcific aortic stenosis and left ventricular hypertrophy in C57BL/6J but not 129S1/SvImJ mice. Am J Physiol Heart Circ Physiol. 2009;297:H65–75. doi: 10.1152/ajpheart.00866.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nicolaou A, Zhao Z, Northoff BH, Sass K, Herbst A, Kohlmaier A, Chalaris A, Wolfrum C, Weber C, Steffens S, et al. Adam17 Deficiency Promotes Atherosclerosis by Enhanced TNFR2 Signaling in Mice. Arteriosclerosis, thrombosis, and vascular biology. 2017;37:247–257. doi: 10.1161/ATVBAHA.116.308682 [DOI] [PubMed] [Google Scholar]
- 45.Khan K, Yu B, Kiwan C, Shalal Y, Filimon S, Cipro M, Shum-Tim D, Cecere R, Schwertani A. The Role of Wnt/beta-Catenin Pathway Mediators in Aortic Valve Stenosis. Front Cell Dev Biol. 2020;8:862. doi: 10.3389/fcell.2020.00862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuipers AL, Miljkovic I, Barinas-Mitchell E, Nestlerode CS, Cvejkus RK, Wheeler VW, Zhang Y, Zmuda JM. Wnt Pathway Gene Expression Is Associated With Arterial Stiffness. Journal of the American Heart Association. 2020;9:e014170. doi: 10.1161/JAHA.119.014170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saku K, Tahara N, Takaseya T, Otsuka H, Takagi K, Shojima T, Shintani Y, Zaima Y, Kikusaki S, Fukuda T, et al. Pathological Role of Receptor for Advanced Glycation End Products in Calcified Aortic Valve Stenosis. Journal of the American Heart Association. 2020;9:e015261. doi: 10.1161/JAHA.119.015261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sud K, Narula N, Aikawa E, Arbustini E, Pibarot P, Merlini G, Rosenson RS, Seshan SV, Argulian E, Ahmadi A, et al. The contribution of amyloid deposition in the aortic valve to calcification and aortic stenosis. Nature reviews Cardiology. 2023. doi: 10.1038/s41569-022-00818-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blaser MC, Aikawa E. Differential miRNA Loading Underpins Dual Harmful and Protective Roles for Extracellular Vesicles in Atherogenesis. Circulation research. 2019;124:467–469. doi: 10.1161/CIRCRESAHA.119.314596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van der Valk DC, van der Ven CFT, Blaser MC, Grolman JM, Wu PJ, Fenton OS, Lee LH, Tibbitt MW, Andresen JL, Wen JR, et al. Engineering a 3D-Bioprinted Model of Human Heart Valve Disease Using Nanoindentation-Based Biomechanics. Nanomaterials (Basel). 2018;8:296. doi: 10.3390/nano8050296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schlotter F, de Freitas RCC, Rogers MA, Blaser MC, Wu PJ, Higashi H, Halu A, Iqbal F, Andraski AB, Rodia CN, et al. ApoC-III is a novel inducer of calcification in human aortic valves. The Journal of biological chemistry. 2021;296:100193. doi: 10.1074/jbc.RA120.015700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goto S, Rogers MA, Blaser MC, Higashi H, Lee LH, Schlotter F, Body SC, Aikawa M, Singh SA, Aikawa E. Standardization of Human Calcific Aortic Valve Disease in vitro Modeling Reveals Passage-Dependent Calcification. Frontiers in cardiovascular medicine. 2019;6:49. doi: 10.3389/fcvm.2019.00049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Itou T, Maldonado N, Yamada I, Goettsch C, Matsumoto J, Aikawa M, Singh S, Aikawa E. Cystathionine gamma-lyase accelerates osteoclast differentiation: identification of a novel regulator of osteoclastogenesis by proteomic analysis. Arteriosclerosis, thrombosis, and vascular biology. 2014;34:626–634. doi: 10.1161/ATVBAHA.113.302576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Atkins SK, Sonawane AR, Brouwhuis R, Barrientos J, Ha A, Rogers M, Tanaka T, Okui T, Kuraoka S, Singh SA, et al. Induced pluripotent stem cell-derived smooth muscle cells to study cardiovascular calcification. Frontiers in cardiovascular medicine. 2022;9:925777. doi: 10.3389/fcvm.2022.925777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Decano JL, Iwamoto Y, Goto S, Lee JY, Matamalas JT, Halu A, Blaser M, Lee LH, Pieper B, Chelvanambi S, et al. A disease-driver population within interstitial cells of human calcific aortic valves identified via single-cell and proteomic profiling. Cell Rep. 2022;39:110685. doi: 10.1016/j.celrep.2022.110685 [DOI] [PubMed] [Google Scholar]
- 56.Passos LSA, Jha PK, Becker-Greene D, Blaser MC, Romero D, Lupieri A, Sukhova GK, Libby P, Singh SA, Dutra WO, et al. Prothymosin Alpha: A Novel Contributor to Estradiol Receptor Alpha-Mediated CD8(+) T-Cell Pathogenic Responses and Recognition of Type 1 Collagen in Rheumatic Heart Valve Disease. Circulation. 2022;145:531–548. doi: 10.1161/CIRCULATIONAHA.121.057301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Iqbal F, Schlotter F, Becker-Greene D, Lupieri A, Goettsch C, Hutcheson JD, Rogers MA, Itoh S, Halu A, Lee LH, et al. Sortilin enhances fibrosis and calcification in aortic valve disease by inducing interstitial cell heterogeneity. European heart journal. 2023;44:885–898. doi: 10.1093/eurheartj/ehac818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B (Methodological). 1995;57:289–300. [Google Scholar]
- 59.Tsuji J, Weng Z. DNApi: A De Novo Adapter Prediction Algorithm for Small RNA Sequencing Data. PloS one. 2016;11:e0164228. doi: 10.1371/journal.pone.0164228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. 2011;17:3. doi: 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- 61.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li H A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pantano L, Estivill X, Marti E. SeqBuster, a bioinformatic tool for the processing and analysis of small RNAs datasets, reveals ubiquitous miRNA modifications in human embryonic cells. Nucleic acids research. 2010;38:e34. doi: 10.1093/nar/gkp1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pantano L, Estivill X, Marti E. A non-biased framework for the annotation and classification of the non-miRNA small RNA transcriptome. Bioinformatics. 2011;27:3202–3203. doi: 10.1093/bioinformatics/btr527 [DOI] [PubMed] [Google Scholar]
- 65.Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic acids research. 2019;47:D155–D162. doi: 10.1093/nar/gky1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Andrews S FastQC: A quality control tool for high throughput sequence data. Bioinformatics. 2010. doi: citeulike-article-id:11583827 [Google Scholar]
- 67.Kamburov A, Stelzl U, Lehrach H, Herwig R. The ConsensusPathDB interaction database: 2013 update. Nucleic acids research. 2013;41:D793–800. doi: 10.1093/nar/gks1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chowdhury S, Sarkar RR. Comparison of human cell signaling pathway databases--evolution, drawbacks and challenges. Database (Oxford). 2015;2015. doi: 10.1093/database/bau126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic acids research. 2000;28:27–30. doi: 10.1093/nar/28.1.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B, et al. The Reactome Pathway Knowledgebase. Nucleic acids research. 2018;46:D649–D655. doi: 10.1093/nar/gkx1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.