

Abstract
Melanoma, the cancer of the melanocyte, is the deadliest form of skin cancer with an aggressive nature, propensity to metastasize and tendency to resist therapeutic intervention. Studies have identified that the re-emergence of developmental pathways in melanoma contributes to melanoma onset, plasticity, and therapeutic response. Notably, it is well known that noncoding RNAs play a critical role in the development and stress response of tissues. In this review, we focus on the noncoding RNAs, including microRNAs, long non-coding RNAs, circular RNAs, and other small RNAs, for their functions in developmental mechanisms and plasticity, which drive onset, progression, therapeutic response and resistance in melanoma. Going forward, elucidation of noncoding RNA-mediated mechanisms may provide insights that accelerate development of novel melanoma therapies.
Keywords: non-coding RNAs (ncRNAs), melanoma, plasticity, lineage, developmental state, translational plasticity, microRNAs (miRNAs), long non-coding RNAs (lncRNAs), circular RNAs (circRNAs)
Introduction
As the deadliest form of skin cancer, cutaneous malignant melanoma (CMM) accounts for more than 75% of skin cancer-related deaths, with a 5-year survival rate of 23% in late-stage patients (Rebecca et al., 2020). At an early stage (stages I and II), CMM can be cured by surgical resection with favorable prognosis (Rutkowski et al., 2010). However, once the disease progresses to metastasis (stage III and IV), ultimately ~40% develop resistance to therapy and are incurable (Dimitriou et al., 2018). The American Joint Committee on Cancer (AJCC) staging, recently revised to its eighth edition, has been the principal guide of CMM staging, prognosis, and risk evaluation (Keung and Gershenwald, 2018). Despite these efforts, ~5–10% of fully resected “early stage” melanomas still progress to metastasis (von Schuckmann et al., 2019), suggesting advanced disease was overlooked in this subset of patients. Even among patients of the same stage, survival outcomes and response to treatment can vary greatly, illustrating the heterogeneity of melanoma (Shain and Bastian, 2016).
The classical model of melanoma onset is the mole-to-melanoma model, in which benign melanocyte proliferation forms a melanocytic nevus (Clark et al., 1984). Subsequently, aberrant differentiation results in abnormal cells (dysplasia) and dysplastic nevi. Oncogenic transformation results in progression to the radial growth phase (RGP), in which cancer cells grow horizontally in epidermis and penetrate the epidermal/dermal junction to move into the dermis. Upon progression to the vertical growth phase (VGP), deep invasion and metastasis occur. However, in reality only 20% of melanomas arise from a nevus and many spontaneously occur from any of the steps described above (Damsky and Bosenberg, 2017).
It is known that phenotypic heterogeneity in melanomas, i.e. phenotypic differences between cells of the same tumor - result in poorer patient survival (Wolf et al., 2019). In the past decade, many studies have sought to characterize the heterogeneity of melanoma and its impact on diagnosis and/or prognosis. Based on driver mutations, four subtypes of CMM have been identified: mutant BRAF, mutant RAS, mutant NF1, and BRAF/RAS/NF1-wildtype (triple-WT), which usually exhibits other driver mutations, including KIT and GNAQ/GNA11 (Cancer Genome Atlas, 2015; Kiuru and Busam, 2017). On the other hand, transcriptomic analyses have classified melanoma into four developmental subtypes: undifferentiated (AXL-high, SOX10/NGFR/MITF-low), neural crest-like (SOX10-high, NGFR-high, MITF-low), transitory (SOX10-high, NGFR-medium, MITF-medium), and melanocytic (SOX10-high, NGFR-low, MITF-high) (Comandante-Lou et al., 2022). In addition, melanomas have been stratified according to their intratumoral immune status, including “inflamed”, “T-cell dysfunctional”, “immune depleted”, and “immune exclusion”(Busam et al., 2001; Clemente et al., 1996; Mihm et al., 1996; Tucci et al., 2019). Although these systems help to characterize melanoma, the association of subtypes with therapeutic responses is limited, as illustrated by many efforts to integrate different biomarkers for better predictive power (Havel et al., 2019). Moreover, different regions within a melanoma can exhibit distinct gene expression patterns of developmental states, repertoires of neoantigens, stromal components, and infiltrated immune cells (Grzywa et al., 2017; Jia et al., 2022; Somasundaram et al., 2012).
It has been revealed that the transcriptional states of melanoma cells are closely associated with neural crest developmental programs (Gopalan, 2022; Marie et al., 2020). Interestingly, melanoma cells can change their transcriptional states without genetic mutations in response to environmental cues or therapeutic stress. The ability to switch states to adapt to environmental stress (i.e. plasticity), further complicated our understanding of their behaviors, as well as our treatment options. Studies showed that such phenotype plasticity drives heterogeneity as well as disease progression and therapeutic resistance. For example, sensitivities to BRAF and/or MEK inhibitors were associated with relative expression levels of MITF, NFκB, and AXL, or neural crest stem cell (NCSC) markers within melanoma cell populations (Konieczkowski et al., 2014; Landsberg et al., 2012; Marin-Bejar et al., 2021; Muller et al., 2014; Rambow et al., 2018; Tsoi et al., 2018). Therapeutic resistance was associated with expansion of the NCSC+ population of melanoma cells upon the treatment of glial cell line-derived neurotrophic factor (GDNF) (Marin-Bejar et al., 2021).
The transcriptional state switching in cancer cells is mediated by epigenetic mechanisms, including transcriptional regulation (DNA methylation and histone modification) and post-transcriptional regulation (RNA stability and modification). In a broader perspective, it also includes translational regulation on mRNA level (translation initiation and elongation). Alteration of transcriptional states by these mechanisms can be made rapidly and reversibly, giving the cells plasticity to adapt to environmental change. Among these mechanisms, noncoding RNAs are important components of epigenetic circuits for controlling transcriptional states. Noncoding RNAs (ncRNAs) are comprised of a diverse range of RNA species, including rRNAs and others that can be further categorized into short ncRNAs and long ncRNAs (lncRNAs). These versatile RNA molecules appear to underlie a hidden layer of internal signals that control various levels of gene expression in physiology and development, including chromatin architecture/epigenetic memory, transcription, RNA splicing, editing, translation, and turnover. RNA regulatory networks may determine most of our complex characteristics, play a significant role in diseases, and constitute an unexplored world of genetic variation both within and between species. A regulatory network is composed of a complex web of molecular factors that interact with each other and with genes in order to control gene expression. In fact, noncoding RNAs are mostly known to be involved in the developmental process of many tissues as well as neural plasticity because of their potential roles in regulating individual genes, as well as large gene networks in tissues. They also confer upon cells the capacity to exert very precise control over the spatiotemporal deployment of genes, which is crucial for executing complex biological processes (Liau et al., 2021; Qureshi and Mehler, 2012). Since melanocytes and melanoma cells are derived from the NC, it is not surprising that noncoding RNAs are also involved in the regulation of NC developmental programs in melanoma. Noticeably, their expression responds to environmental stress promptly.
There are several types of noncoding RNAs: (1) microRNAs (miRNAs); (2) long noncoding RNAs (lncRNAs) and circular RNAs (circRNAs); (3) tRNAs and their fragments (tRFs); (4) small nuclear RNAs (snRNAs); (5) small nucleolar RNA (snoRNA); and (6) PIWI-interacting RNAs (piRNA). These noncoding RNAs execute different functions, forming an enormous gene regulatory circuit network. In an oversimplified summary: lncRNAs mediate transcriptional control and regulate miRNA and/or tRFs; miRNA and tRFs mediate post-transcriptional and translational regulation; snRNAs regulate alternative splicing; snoRNAs mediate post-transcriptional regulation; and piRNAs regulate the expression of other major classes of RNAs in germ cells (Panni et al., 2020; Saito and Siomi, 2010; Zhang et al., 2019b). The major types of noncoding RNAs regulating developmental states and plasticity are miRNAs as well as fragments of other small RNAs, lncRNAs, and circRNAs (Box 1). Their computational analysis is explained in Box 2.
Box 1: Type of noncoding RNAs.
Most of the existing RNAs do not code for proteins and are known as non-coding RNAs (ncRNAs). ncRNAs not only affect biological processes such as translation and splicing (ribosomal RNA [rRNA], transfer RNA [tRNA] and small nuclear RNA [snRNA]) as well as the modification process of other RNA molecules providing a more interesting role from an epigenetic point of view. ncRNAs are divided into two main groups: small non-coding (<200 nucleotides) and long non-coding RNAs (>200 nucleotides), and each group is subdivided into many distinct families.
The first described noncoding RNAs were rRNAs and tRNAs in the early days of deciphering the genetic codes, during the 1960’s. After two decades, the discovery of RNA molecules as part of ribonucleoprotein complexes came with the discovery of the small nuclear RNAs (snRNAs) as part of the RNA splicing machinery. Almost at the same time the discovery of introns and the catalytic RNAs draw immediately the attention of researchers in the field and in 1993 the description of small regulatory RNAs that regulate C. elegans development, was followed by the description of the RNA interference pathway and the identification of ribonucleases Drosha and Dicer, as well as the family of Argonaute (AGO) proteins as part of a conserved and highly important mechanism of post-transcriptional regulation of the genetic information. For a more detailed review on the detailed timeline of noncoding RNA discovery we refer to a very thorough and detailed review by (Morris and Mattick, 2014).
Surprisingly, recent advances in next generation sequencing and bioinformatics revealed that even the oldest noncoding RNAs, like tRNAs, can break down into fragments through specific cleavage by important endonucleases, like angiogenin and Dicer, to give rise to hundreds of smaller noncoding RNA species. Today, it has been established by many breakthrough studies that tRNA-derived fragments (tRFs) act like miRNAs through association with AGO proteins and, some of them are stress-induced with either tumor suppressing or promoting function (Schimmel, 2018). Even more interesting is the fact a comprehensive atlas of all noncoding RNAs is far from been complete and recently, thousands of previously uncharacterized RNAs were reported, increasing the number of documented noncoding RNAs by approximately 8% and expanding the universe of the noncoding RNAs that are involved in the regulation of virtually every cellular activity (Lorenzi et al., 2021).
microRNAs (miRNAs) are the most well-studied family of small ncRNAs. They are single stranded RNA molecules and their length is approximately between 18 and 23 nucleotides. miRNAs are produced from primary miRNA transcripts (pri-miRNAs) via several enzymes such as Drosha, XPO5 and Dicer. Mature miRNAs are loaded on and guide the RNA-induced silencing complex (RISC) to control transcriptional and post-transcriptional regulation of coding RNAs.
tRNA fragments (tRFs) are one of the most recently discovered families of sncRNAs, generated by specific cleavage of tRNA transcripts. Based on their length, tRFs are divided into two classes. The first includes the tRNA halves, stressed-induced tRFs that are generated by a specific cleavage from angiogenin at the anticodon loop and their length is approximately between 31 and 40 nucleotides. The second class includes smaller tRNA fragments, starting either from the 5’or the 3’ end of the tRNA and their length is between 14 and 30 nucleotides. Based on their mapping on the mature tRNA, they are divid classified as either in tRF-5 or tRF-3. Of note, if their sequence is mappeding with the 3’-end of the primary tRNAs they are called tRF-1. Many studies provided evidence that tRFs belongning to the second class can interact with AGO proteins and act like miRNAs to regulate gene expression.
P-element-induced wimpy testis (Piwi)-interacting RNAs (piRNAs) form the largest group of sncRNAs. piRNAs are 21–36 nucleotides single stranded RNAs that are mainly expressed in spermatogenic cells. They can bind Piwi proteins and mediate epigenetic silencing, which can affect germline development and functions. It has been demonstrated that piRNAs contribute to genome integrity maintenance via transposons silencing.
Small nucleolar RNAs (snoRNAs) mainly accumulate in the nucleoli. Their length is approximately between 60–300 nucleotides, and they are implicated in post-transcriptional modifications of many RNA molecules. snoRNAs exhibit specific sequence motifs and secondary structures, thus are divided into C/D and H/ACA box snoRNAs as well as small Cajal body-specific RNAs (scaRNAs). C/D box snoRNAs guide the 2’-O-ribose methylation of rRNA residues while the H/ACA box snoRNAs catalyze the pseudouridylation of nucleotides. scaRNAs exhibit both the C/D and H/ACA boxes along with a CAB box motif that acts as a Cajal-body localization signal.
Long non-coding RNAs (lncRNAs) are transcripts longer than 200 nucleotides that exhibiting an emerging role in the pathogenesis of cancer. Circular RNAs (circRNAs) are, single stranded RNA molecules that form a covalently closed continuous loop and are abundant across the evolutionary ladder. They lack a 5’Cap and a poly-A tail and they are resistant to degradation by exonucleases, thus showing high stability and half-life despite of the fact that circRNAs are expressed in much lower levels than their linear counterparts (Chen et al., 2020b; Kristensen et al., 2019; Panda et al., 2017). Their biogenesis is regulated by the combinatorial action of several factors such as Alu or inverted repeated sequences and binding of RBPs near the circularized spice sites, which work to bring them in close proximity and eventually ligation (Ashwal-Fluss et al., 2014; Conn et al., 2015; Ivanov et al., 2015). Canonical splicing generates linear RNAs, whereas back-splicing produces circRNAs and alternatively spliced linear RNAs.
Both LncRNAs and circRNAs can regulate gene expression by participating in processes that alter chromatic conformation, forming triplexes with DNA as well as interfering with transcription enzymes. Of note, several sncRNAs are also implicated in these processes, highlighting the fact that ncRNAs can act synergistically, facilitating our understanding of human diseases.
Since each miRNA has numerous RNA targets, and the vast majority of RNAs harbor several miRNA binding sites and are therefore targeted by different miRNAs, it was hypothesized that that different RNAs compete for limited pools of miRNAs, acting as competitive endogenous RNAs (ceRNA hypothesis). Two consequences could be derived from the ceRNA hypothesis. First, a major function of noncoding RNAs such as lncRNAs and circRNAs may serve as endogenous miRNA decoys. Second, besides serving as a template for protein synthesis, mRNAs may be involved in the posttranscriptional regulation of other transcripts (Karreth and Pandolfi, 2013).
The types of ncRNAs are summarized in the Table of Box 1. Recent advances in computational analyses and genomic engineering (e.g. CRISPR) greatly facilitate the identification of specific ncRNAs and their targets of interest, and vice versa. Such technologies allow elucidation of ncRNAs’ function in the adaptation of melanoma cells to the microenvironment by switching states, i.e. plasticity. For instance, miRNAs and lncRNAs interact with the transcription factors to change the developmental state of melanoma, and tRNAs, tRFs, and snoRNAs may allow some translational plasticity. Many of them may confer melanoma cells with metabolic plasticity. Moreover, given their critical role in melanoma plasticity that drives progression and resistance to therapies, ncRNAs may serve as diagnostic and prognostic markers. These perspectives will be discussed in this review.
Box 1 Table.
Summary of ncRNAs
| Physical Properties | Target | Functions | |
|---|---|---|---|
| miRNA | Single strand, 18–23 nucleotides | messenger RNA, lncRNA | Transcriptional and post transcriptional regulation in conjunction with RISC, regulate transcriptional factors that are important to developmental state |
| tRF | 31–40 or 14–30 nucleotides depending on type | messenger RNA | Transcriptional and post transcriptional regulation in conjunction with AGO |
| piRNAs | 21–36 nucleotides | Transposons | Mediate epigenetic silencing, also silence transposons with PIWI protein |
| snoRNA | 60–300 nucleotides | Ribosomal RNA | Affect RNA splicing, post transcriptional modification of ribosomal RNA to perturb translational profile |
| lncRNA | Longer than 200 nucleotides | Chromatin, transcription enzymes, miRNA, tRFs | Affect chromatin conformation, interfere with transcription enzymes, recruit ribonucleoprotein complex, control access to transcription sites, regulate miRNA and/or tRFs, mediate metabolic changes |
| circRNA | Single stranded RNA loop | Chromatin, transcription enzymes, miRNA | Affect chromatin conformation, interfere with transcription enzymes, sponge or regulate miRNA |
Box 2: Computational methods for analysis of noncoding RNA regulatory networks in melanoma.
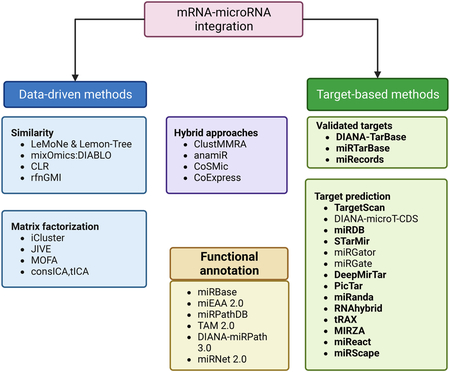
Recent advances in high-throughput sequencing technologies and computational platforms have been pivotal towards the discovery and classification of the noncoding RNAs, including rRNAs, short ncRNAs (miRNAs, tRNAs, tRFs, snoRNA, snRNAs) and long ncRNAs (lncRNAs and circRNAs).
MicroRNAs regulate the expression of the target messenger RNAs (mRNAs), usually by binding to a short complementary sequence often located in the 3′ UTR region of the mRNA. TargetScan predicts microRNA targets from the presence of 6–8 nucleotides in the 3′ UTR of the mRNA, matching nucleotides 2–7 of the microRNA. Other algorithms based on similar considerations are PicTar and miRanda. Similarly, RNAhybrid provides a variation of RNA secondary structure prediction methods and calculates the most favorable hybridization site between the microRNA and the mRNA. Advances in gene sequencing techniques and the development of bioinformatics databases have allowed analytical research using public databases for secondary data integration and analysis. tRAX (tRNA Analysis of eXpression) is a user-friendly analytic package for streamlined processing and graphic presentation of small-RNA sequencing data, including transfer tRNAs (tRNAs) and tRNA-derived small RNAs (tDRs).
In silico analysis of datasets representing only one RNA species is well established and a variety of tools and pipelines are available. However, attaining a more systematic view of how different players come together to regulate the expression of a gene or a group of genes requires a more intricate approach to data analysis. To fully understand complex transcriptional networks, datasets representing different RNA species need to be integrated. Pearson’s correlation test is utilized to calculate the correlation coefficients between microRNA and mRNAs. Deconvolution and deep learning approaches are promising new approaches to improve miRNA targetome predictions.
A critical step in understanding the function of miRNAs is to first identify their regulatory targets. Several databases such as Tarbase, miRTarbase, miRecords, etc. provide a comprehensive resource for the experimentally validated miRNA-target gene interactions (Huang et al., 2020; Karagkouni et al., 2018; Xiao et al., 2009). However, due to challenges in experimental ascertainment of miRNA targets, these resources are limited in the number of miRNA:mRNA interactions and generally lack target information for non-model organisms. To address this limitation, computational tools for identifying miRNA targets have been proposed. These tools commonly rely on: (1) seed matching (complementary matching of the first 2–7 nt miRNA); (2) evolutionary conservation; (3) thermodynamic stability; and (4) target site accessibility. Among these, TargetScan is one of the most widely used tools, providing both precomputed results and source code for custom analysis (Agarwal et al., 2015). Another popular tool, miRanda, considers the whole miRNA sequence instead of just the seed region (Enright et al., 2003).
There is, however, low agreement between the tools and databases, suggesting strong contextual effects. Ultimately, experimental validation of computational prediction has been limited. Notably, D’Arcangelo et al. experimentally validated the TargetScan’s putative target of miR-503, CXCL10/IP-10, showing that downregulation of miR-503 affects endothelial and melanoma cells proliferation in a CXCL10/IP-10 dependent way (D’Arcangelo et al., 2016). More recently, several tools have attempted to integrate high-throughput CLIP-seq data with machine learning to predict miRNA targets, such as MIRZA, STarMir, miRDB, DeepMirTar, etc. (Khorshid et al., 2013; Rennie et al., 2014; Wang, 2016; Wen et al., 2018). For a more detailed review of computational approaches to miRNA target prediction, we refer the readers to these references (Bottini et al., 2018; Fan and Kurgan, 2015; Riffo-Campos et al., 2016; Thomas et al., 2010; Zheng et al., 2013).
Given the importance of cellular heterogeneity and the microenvironment in oncogenesis, to understand the role of miRNAs in cancer initiation and progression one needs to profile miRNA activities at a cellular resolution. However, experimental technologies to profile miRNA expression at single cell resolution are currently lacking, with only a handful of available datasets, thus limiting our understanding of the cellular dynamics of miRNAs (Faridani et al., 2016; Wang et al., 2019). To overcome this technical limitation, a few computational methods for inferring the miRNA activity at cellular resolution have been proposed (Nielsen and Pedersen, 2021; Olgun et al., 2022). miReact predicts the miRNA activity based on the assumption that a lower expression of putative target genes, inferred from seed sequence matching, is indicative of high miRNA activity in a cell (Nielsen and Pedersen, 2021). This, however, ignores the indirect downstream targets of the miRNA. A recent machine learning-based method, miRSCAPE, makes use of the complex direct and downstream indirect regulatory links between miRNAs and the global gene expression profile to infer miRNAs at a single-cell level, resulting in a superior performance based on several benchmarks (Olgun et al., 2022). These methods are summarized in the figure of this Box.
As plasticity is the ability to switch among transcriptional states of cells, several features of noncoding RNAs make them effective regulators. First, these RNA effectors can regulate RNAs directly, changing their levels efficiently. This keeps the flexibility of gene circuits without altering the transcriptional mechanism. Second, each RNA effector can have multiple targets and be regulated by multiple upstream effectors. These effectors and targets include both RNA and proteins such as transcriptional factors, so they can form versatile regulatory circuits for different functions. Third, their expression can be induced and reduced promptly, making them efficient responders to environmental changes (Cursons et al., 2018). In this review, we will discuss the functions of noncoding RNAs in regulating transcriptional and metabolic reprogramming in melanoma, as well as their impact on disease progression and therapeutic resistance. Importantly, these noncoding RNAs could serve as therapeutic targets for preventing melanoma from recurrence or sensitizing them to current therapies. As RNA therapy technology continues to mature, the potential of noncoding RNAs in the development of melanoma therapeutics will become even more significant.
Waddington landscape and melanoma plasticity
In 1957, Conrad Waddington described the development of embryonic stem cells into a mature differentiated state as a ball rolling down from the top of a mountain to the bottom of a valley (Waddington, 1957). It demonstrated visually the natural restriction of cell differentiation potential during normal development. Later, the concept was extended so that the ball at the top of the mountain is a stem cell, and individual valleys are distinct differentiated states of different tissues. How the stem cell differentiates to matured cells in a specific tissue depends on the path it rolls down. Pioneering work by Stuart Kauffman suggested that this process of differentiation may be realized via gene regulatory networks driven by interactions between transcription factors (Kauffman, 1969), such that each cell type represents a stable equilibrium state of genome-wide expression (Huang et al., 2009).
However, a series of landmark experiments showed that cell fate is flexible and reversible. With the monumental discovery that induced pluripotent stem cells (iPSCs) can be generated by reprogramming diverse types of somatic cells and that these iPSCs can be differentiated into other somatic cell types, the pluripotent state was identified as a hub that connects different lineage routes at the top of the Waddington model. Moreover, the notion of whether cell fates are interconvertible had already been posited decades earlier, outside the context of pluripotency. Subsequent studies then provided additional evidence for successful cell fate conversion between related lineages (Li et al., 2010; Park et al., 2008a; Yu et al., 2007). Recent work showing that a Waddington landscape-type model may be mathematically fitted to transcriptional data from embryos and even predict fate biases in stem cells (Saez et al., 2022). A key feature in landscape models, however, is the notion of gene expression fluctuation (Ross et al., 1994). Such noise can lead to remarkable phenotypic variation in the absence of genetic variation (Ozbudak et al., 2004). Evolution has led to regulatory strategies to reduce the influence of such noise during development (Simon et al., 2018). In contrast, for adult organism, high expression fluctuation can allow adaptation to stress without necessarily acquiring additional genetic mutations and traverse Waddington landscapes with fewer constraints than in development (Brock et al., 2009; Pisco and Huang, 2015). Plasticity, therefore, can be thought of as a “shortcut” between the states in the landscape.
The neural crest (NC) is a multipotent cell population that gives rise to several cell lineages including melanocytes, Schwann cells, and peripheral neurons. Its development in embryo has the following stages: (1) induction, when NC cells are induced from neural plate at the early stage of embryonic development; (2) epithelial-to mesenchymal transition (EMT), when NC cells become capable of migrating; (3) delamination, when NC cells migrate away from the neural tube region; (4) migration, when NC cells migrate toward different destinations while differentiating to specific lineages; and (5) colonization, when NC-derived cells invade the destinated tissue and maturate there (Hovland et al., 2020).
During the migration stage, a subset of NC cells differentiates to become the melanocyte precursors, melanoblasts. They long distances in the body to ultimately reach the skin and fully differentiate into pigment-producing melanocytes. A second population of melanoblasts can arise from an embryonic multipotent population called Schwann Cell Precursors (SCPs) and are associated with peripheral nerves Hence, phenotypic plasticity and a high migratory potential are characteristics of the melanocyte lineage that can be traced back to its neural-crest origin (Mort et al., 2015). Recent studies revealed a hierarchical model of melanoma growth that mirrors the cell-fate specification during NC differentiation (Karras et al., 2022; Shakhova, 2014; White and Zon, 2008). Conceptually, these data paint a picture of melanoma evolution like a ball rolling on a NC-style Waddington landscape. On the top of this melanoma landscape are the stem-like cells. As these stem-like cells proliferate, some may “roll” down the hill to pass the plateau of precursors (NC-like and mesenchymal cells), and eventually spread all over the valleys of melanocytic cells (Figure. 1). Such developmentally hierarchical growth would account for the heterogeneity of melanoma, where each differentiation state exhibits a unique function; for example, melanocytic cells are proliferative and mesenchymal-like cells are invasive. These results effectively show that transcriptional states of melanoma cells are defined by the developmental programs in the NC-style Waddington landscape of NC differentiation. Plasticity, therefore, is the “shortcut” between the states in the landscape. For example, the frequently observed EMT in melanoma cells involves upregulation of N-cadherin, ZEB1, and SNAI2 (Garg, 2013; Loh et al., 2019). Such change directly corresponds to the switching from melanocytic to mesenchymal status (Gopalan, 2022).
Figure 1.
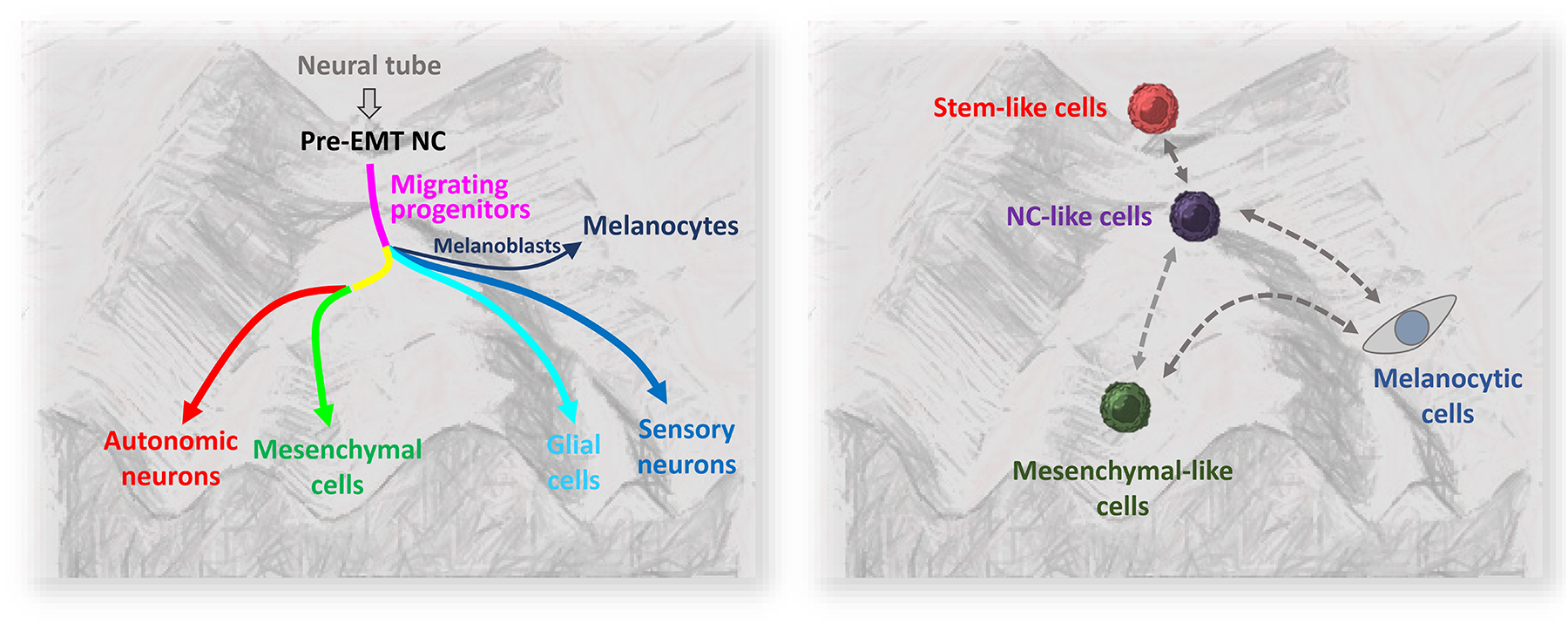
Comparison of NC differentiation and melanoma development on a Waddington-style landscape. Left panel, NC differentiation. NC stem cells (pre-EMT NC) are derived from neural tube. They delaminate, migrate (migrating progenitors), and start specification. Upon reaching the destination, they become committed cell types (autonomic neurons, mesenchymal cells, glial cells, sensory neurons, melanocytes). Each cell type is a stable transcriptional state against perturbation (“valley”) unless receiving specific signals. Right panel, melanoma development. Melanoma is initiated by the stem-like cells that correspond to pre-EMT NC state. As expanding, they differentiate to NC-like cells, mesenchymal-like cells, and melanocytic cells (corresponding to migrating NC progenitors, mesenchymal cells, and melanocytes in the NC differentiation). Like NC development landscape, each differentiated type represents a transcriptional state. However, in contrast to the NC landscape, the plasticity of melanoma cells allows them to switch between transcriptional state (dashed arrows). This can be mediated by genetic circuits that involve noncoding RNAs; see Figure 2 to 5. The left panel is adopted from Marie et al (Marie et al., 2022).
As discussed above, alteration of transcriptional states by these mechanisms can be made rapidly and reversibly, giving the cells plasticity for adapting to environmental change. Studies have shown that, among these mechanisms, noncoding RNAs are important components of epigenetic circuits for controlling developmental programs of melanoma cells, forming the switches between transcriptional states in the NC-differentiating landscape. They will be discussed in the following sections.
MicroRNAs and other small RNAs form regulatory circuits with lineage determining factors in melanoma plasticity
miRNA circuitry in Epilthelial-to-Mesenchymal Transitions
EMT is an epithelial to mesenchymal transition. This distinct feature of early-stage NC cells allows the melanoma cells to invade and interact with the environment. In melanoma, miRNAs are a common regulator of EMT genes, including ZEB1, ZEB2, SNAIL, SNAIL2. miRNAs can regulate gene expression by either mediating degradation of mRNA or blocking translation from mRNA. Moreover, several different miRNAs can target a specific mRNA synergistically, enhancing their flexibility in gene regulation. In recent years, it was found that miRNAs and transcriptional factors can form a negative feedback loop, generating two or more transactional states. One of the most extensively studied cases is the regulation of EMT by ZEB1 and the miR-200 family, which inhibit each other by transcriptional and post-transcriptionally mechanisms, respectively. Pro-mesenchymal signals such as TGF-β increase the expression of ZEB1, tipping the system to shut down miR-200 family members and fully turn on Zeb1 and thus its downstream mesenchymal effectors (Bracken et al., 2008). This circuitry forms a system with two stable equilibrium states (bistability) (Figure 2a).
Figure 2.
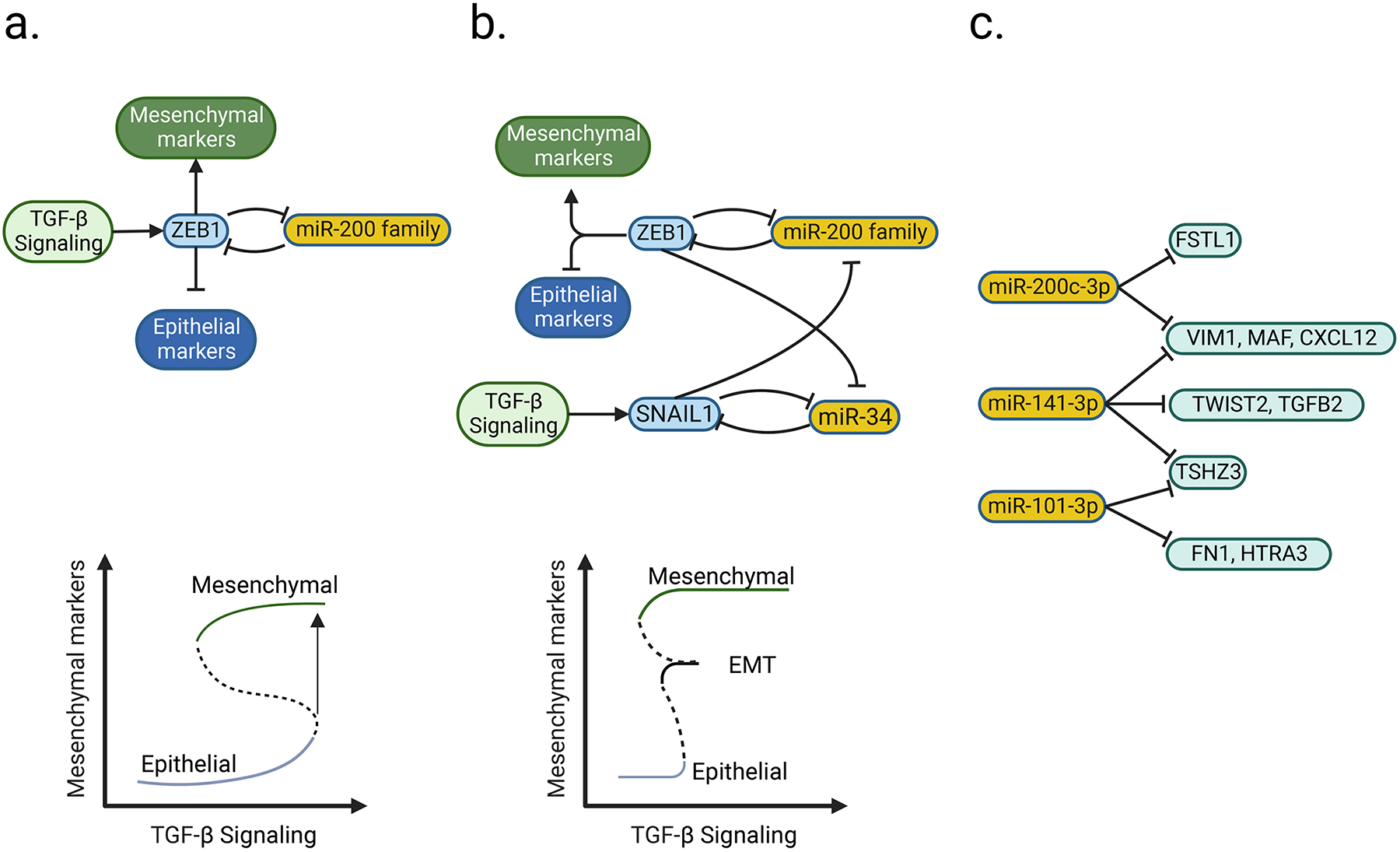
Examples of microRNAs and transcription factors forming a dynamic control circuit for switching between transcriptional states. a, ZEB1 activates and suppresses the expression of mesenchymal and epithelial marker genes, respectively. It and miR-200 family reciprocally inhibit each other, constituting a negative feedback loop (upper panel). When the input signal (TGF-β) reaches over a threshold level, the system will switch from epithelial-high to mesenchymal-high state (lower panel; adopted from Stallaert et al (Stallaert et al., 2019)). Solid and dashed curve, stable and unstable state, respectively. This figure is adopted from b, Incorporation of another negative feedback loop (SNAIL1 and miR-34) to ZEB1/miR-200 family circuit (upper panel) results in an additional state (EMT, lower panel; adopted from Zhang et al (Zhang et al., 2014)). As the input signal (TGF-β) keeps increasing, the system will switch from epithelial-high to EMT, and then to mesenchymal-high state (lower panel). c, An example of the collaborative behavior of miRNA. A gene can be targeted by multiple miRNA (e.g. VIM1, MAF, and CXCL12 are targeted by both miR-200c-3p and miR-141–3p), but each miRNA can target multiple genes in which some of them might not be shared with other miRNAs (e.g. miR-200–3p targeting VIM1 and FSTL1) (Guo et al., 2014). Part of the figure is generated using birender.com (2022).
Other mutually inhibitory factors can be added to the system, such as the mesenchymal transcriptional factor, SNAIL1, and miRNA family, miR-34(Migault et al., 2022). Theoretical models have predicted this creates three stable states (epithelial, partial EMT, and mesenchymal) that respond to different threshold levels of TGF-β (a multifunctional cytokine associated with mesenchymal lineage commitment) (Figure 2b). This model demonstrates how transcriptional factors and miRNAs can serve to fine-tune multi-state signaling systems in response to different environmental stresses (Zhang et al., 2014).
This fine-tunability of miRNAs can be amplified, for instance, TGF-β-induced gene expression can be targeted by eight different miRNAs. However, only when more than four of the miRNAs were expressed simultaneously, the full suppression of the targeted genes could be achieved. (Cursons et al., 2018). This suggests that their diverse while cooperative behaviors enable the miRNAs to form flexible and responsive regulatory networks. Conceptually, the regulation is achieved, on one hand, by a group of miRNAs targeting different sites in a mRNA collectively, so each one of them does not need to be expressed at high level; on the other hand, each miRNA may target various different mRNA (Figure 2c). Therefore, subtle alterations of expression levels of some collaborative miRNAs would be sufficient for prompt but subtle modulation of gene expression in response to environment change.
miRNA opposing feedback loops in phenotypic switching
Transformation of melanocytes can result in a reprogramming of developmental status, including de-differentiation. Therefore, neural crest lineage specification genes are frequent regulatory targets of noncoding RNAs in melanoma. These include: (1) melanocytic specification genes, such as MITF; (2) neural specification genes, such as SOX2, POU3F2 (BRN2), NUAK1, and pathway genes; (3) mesenchymal specification genes, such as ZEB1; (4) genes in neural crest stem cell status, such as TFAP2B (Bell et al., 2014; Boyle et al., 2011; Brombin et al., 2022; Mazar et al., 2010; Zhang et al., 2020).
The two features of miRNAs, feedback loop and collaborative action, make them suitable effectors to mediate the fast switching between cellular states. Bell et al, characterized invasive and proliferative subtypes of human melanomas, two phenotypes typically considered to be mutually exclusive of each other. Interestingly, miRNAs in the former targets specifically expressed genes in the latter, and vice versa. For example, in the proliferative subtype of melanoma, MITF drives the expression of miR-211, which inhibits NUAK1, a gene specifically expressed in the invasive subtype of melanoma (Bell et al., 2014). In support of this, two big-scale multi-omic studies have subsequently demonstrated the existence of such regulatory networks in melanoma. One of them has shown that the functional phenotypes of melanoma are controlled by two opposing feedback loops of miRNAs and expression of neural crest lineage genes. In that study, miR-221-3p inhibits expression of key transcription factors associated with melanocytic differentiation from the neural crest, MITF, SOX10, and TFAP2A. Inhibition of this gene expression causes a melanoma switch to the invasive phenotype. In contrast, miR-211–5p inhibits NLGN1 and FXYD5 that mediate neural differentiation and EMT, respectively, switching melanoma to the proliferative phenotype (Rambow et al., 2015). In another study, two groups of miRNAs were differentially expressed in melanocytic-like (MEL) or mesenchymal-like (MES) melanomas. Interestingly, miRNAs enriched in MES melanoma target genes highly expressed in MEL melanoma, and vice versa. For example, miR-211–5p, which is more highly expressed in MEL melanoma, targets ZEB1 and Cadherin 2 (CDH2), genes required for a mesenchymal-like transition. In contrast, miR-218, which is more highly expressed in MES melanoma, targets MITF and HPS4, which regulate melanosomal genes (Andrews et al., 2022).
The feedback loop between miRNA and Transcription Factors to control developmental states has been experimentally validated. MITF drives the expression of TRPM1, an ion channel gene whose intron hosts the gene for miR-211; hence, miR-211 is a direct transcriptional “co-product” of MITF activity (Margue et al., 2013). It has been shown that miR-211 inhibits POU3F2, which in turn represses MITF activity. As MITF and POU3F2 drive melanocytic and mesenchymal gene modules, respectively, MITF/miR-211/POU3F2 forms a feedback loop to control the switch between “MEL/proliferative” and “MES/invasive” states (Fane et al., 2019). Further investigation is required for identifying other “switch-pulling” factors. Similarly, miR-218 is an intronic miRNA located within introns of the axon guidance genes SLIT2 and SLIT3, which are more highly expressed in undifferentiated or NC-like melanoma. miR-218 inhibits MITF, whose activation induces the melanocytic differentiation from the stem-like state (Martinez et al., 2008). Therefore, developmental factors that enhance the expression of SLIT genes, such as miR-218, can reduce the level of MITF, blocking the melanocytic differentiation. Recently, an alternative promoter activated by the inflammatory regulator, NF-kB, was identified in miR-218 genes (Rheinheimer, 2020). Since NF-kB is activated by inflammatory signals, the latter may override the SLIT gene-controlled expression of miR-218, which in turn suppresses MITF and switches the melanoma cells to NC-like states during immune response/inflammation. In fact, recent studies have shown that resistance of melanoma to immunotherapies is associated with NC-like phenotypes (Diener and Sommer, 2021; Wessely et al., 2021).
As stated in the Introduction, more developmental states in melanoma were identified recently, and they help explain some of the responses to different environmental cues. Previous studies have implied the existence of a miRNA-lineage factor feedback loop for switching between these states. For example, miR-221-3p inhibits NAB1, resulting in the inhibition of Schwann cell maturation and myelination of peripheral nerves in the adult. In the embryo, a population of peripheral nerve-associated Schwann cell progenitors (SCPs) are known to also provide a reservoir or melanocytic progenitors/stem cells during a second wave of melanocyte development (Adameyko and Lallemend, 2010). Since NAB1 is a transcriptional repressor, it is a candidate to form a feedback loop with miR-221-3p for switching between stem-like and mature Schwann cell states (Zhao et al., 2018), although studies are required to test this hypothesis. Recently, adoption of a SCP-like state in melanom was shown to associate with resistance from immunotherapy. The NAB1/miR-221-3p feedback loop could offer a plausible mechanisms as to how melanomas switch to an SCP phenotype to become resistant to immunotherapy (Gopalan, 2022). Interestingly, in other cell types, miR-221-3p has been shown to block interferon β, suggesting another resistant mechanism of innate immunity. These results are summarized in Figure 3; the two transcriptional states, invasive/MES phenotype and proliferative/MEL phenotype, reciprocally inhibit each other through miRNAs and Transcription Factors. Importantly, inflammatory signals (including interferons and others through NF-kB) could tip the homeostasis, driving the melanoma cells toward the invasive/MES phenotype. This model explains how chronic inflammation induces resistance of melanoma to immunotherapies (Du et al., 2018; Zhao et al., 2016).
Figure 3.
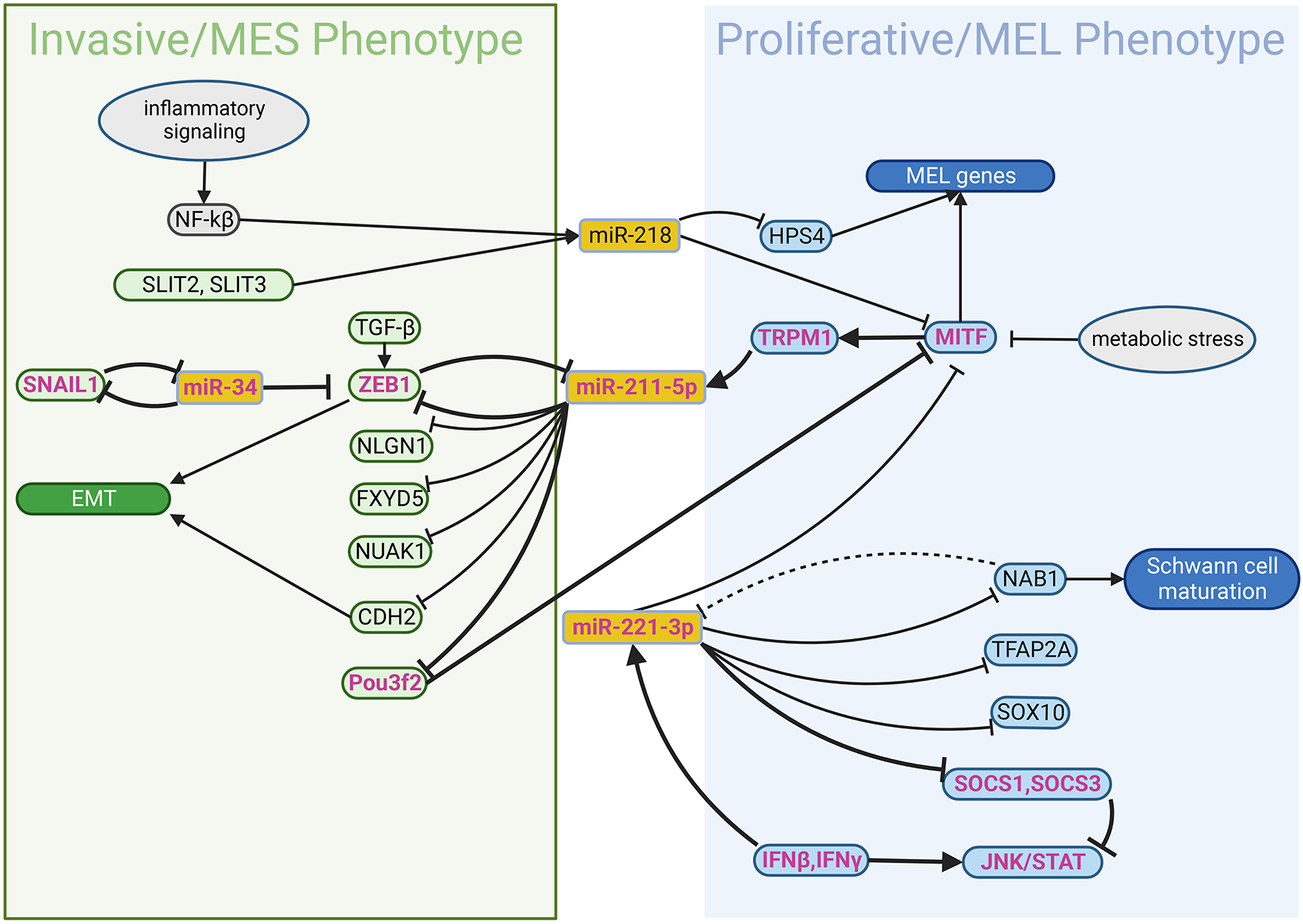
The miRNA-protein factor circuit for control the transcriptional states and mediate plasticity in melanoma. In melanoma cells, similar to Fig. 2A and B, miRNAs and protein factors form a negative-feedback circuit for switching between proliferative/MEL and invasive/MES states, putatively in response to input signal such inflammation. miRNAs can be activated from their own promoters by transcriptional factors via input signals (e.g. NFkB and inflammatory signal, respectively), or expressed from the introns of the coding genes (e.g. TRPM1, SLIT1, SLIT3) during their transcription. The function of miRNAs is to suppress expression of coding genes. MEL and MES, melanocytic and mesenchymal phenotype, respectively. Part of the figure is generated using birender.com (2022).
We expect that more and more feedback loops between miRNA and lineage factors for switching between novel states of melanoma will be identified and their function in adaptation of environmental stress should be further investigated. Moreover, signals that trigger the up- and down-regulation of these miRNAs would be critical for understanding what drives melanoma plasticity. Notably, they could also serve as biomarkers or therapeutic targets.
Stress-induced translational plasticity: a case study of tRNAs, tRFs and snoRNAs
Translational control allows cells in skin to promptly and dynamically adapt to a variety of stimuli (Grafanaki et al., 2019). In response to environmental signals or stress, cancer can switch cell state or metabolic activity by several mechanisms of controlling translational activities (Fabbri et al., 2021). tRNAs and snoRNAs, as well as their fragments, are involved in the translational plasticity of cancer cells at multiple levels. In melanoma, tRNA’s can be modified at the monophosphate level to alter translation efficiency of different codons. 5-methoxycarbonyl methyl-2-thiouridine (mcm5s2U34) modification of tRNA is necessary for the efficient translation of the AAA (lysine), GAA (glutamate), and CAA (glutamine) codons. As these three amino acids are highly used in protein modification and energy metabolism, a high level of mcm5s2U34 is required for melanoma cells to survive. The HIF1α protein, which is enriched with these codons, requires the mcm5s2U34 modification enzymes ELP3, cytoplasmic tRNA 2-thiolation proteins 1, and 2 (CTU1 and CTU2) to be efficiently translated and to exert HIF1α-dependent metabolic reprogramming in melanoma. The acquired resistance to anti-BRAF therapy is associated with PI3K signaling that induces high levels of U34 enzymes and HIF1α (Rapino et al., 2018).
Stress-induced cleavage of tRNA within the anticodon loops to produce tRFs impairs the formation of translation initiation complex, inhibiting translation of mRNAs (Ivanov et al., 2011). Alternatively, CCA tail-degraded tRNAs can be incorporated into ribosomes, causing ribosome stalling and translation arrest (Chen and Tanaka, 2018). Recently, the tRFs have been shown to act like miRNAs that bind to mRNA to block translation (Xiao et al., 2021).
snoRNAs are metabolically stable, 60–300-nucleotide-long RNAs abundant in the nucleolus and involved in the post-transcriptional modification (2′-O-ribose methylation or pseudouridylation) of ribosomal RNAs, which regulates the function and fidelity of the ribosome and change the balance between different ribosome conformational states, modifying translational profile in a cancer cells (Khoshnevis et al., 2022). It has been shown that snoRNAs are also involved in the regulation of RNA splicing. Stress alters the expression of snoRNAs, changing the repertoire of translated genes. For example, knockdown of snoRNA genes promotes the binding of ribosomal protein L5 (RPL5) and ribosomal protein L11 (RPL11) with MDM2, resulting in the stabilization and thus accumulation of p53 (Liang et al., 2019). Similar to the cleavage of tRNAs, it has been found that stress can induce snoRNAs processing to generate smaller miRNA-like molecules that are capable of binding argonaute (AGO) and exerting miRNA-like effects (Liang et al., 2019).
In recent years, more new tools of data analysis have been developed for identifying tRNA, snoRNA, and their derived fragments in cancers, including melanoma. The studies of the function of these small RNAs in melanoma, as summarized in Figure 4, have just begun, and we anticipate that they will grow rapidly (Fabbri et al., 2021).
Figure 4.
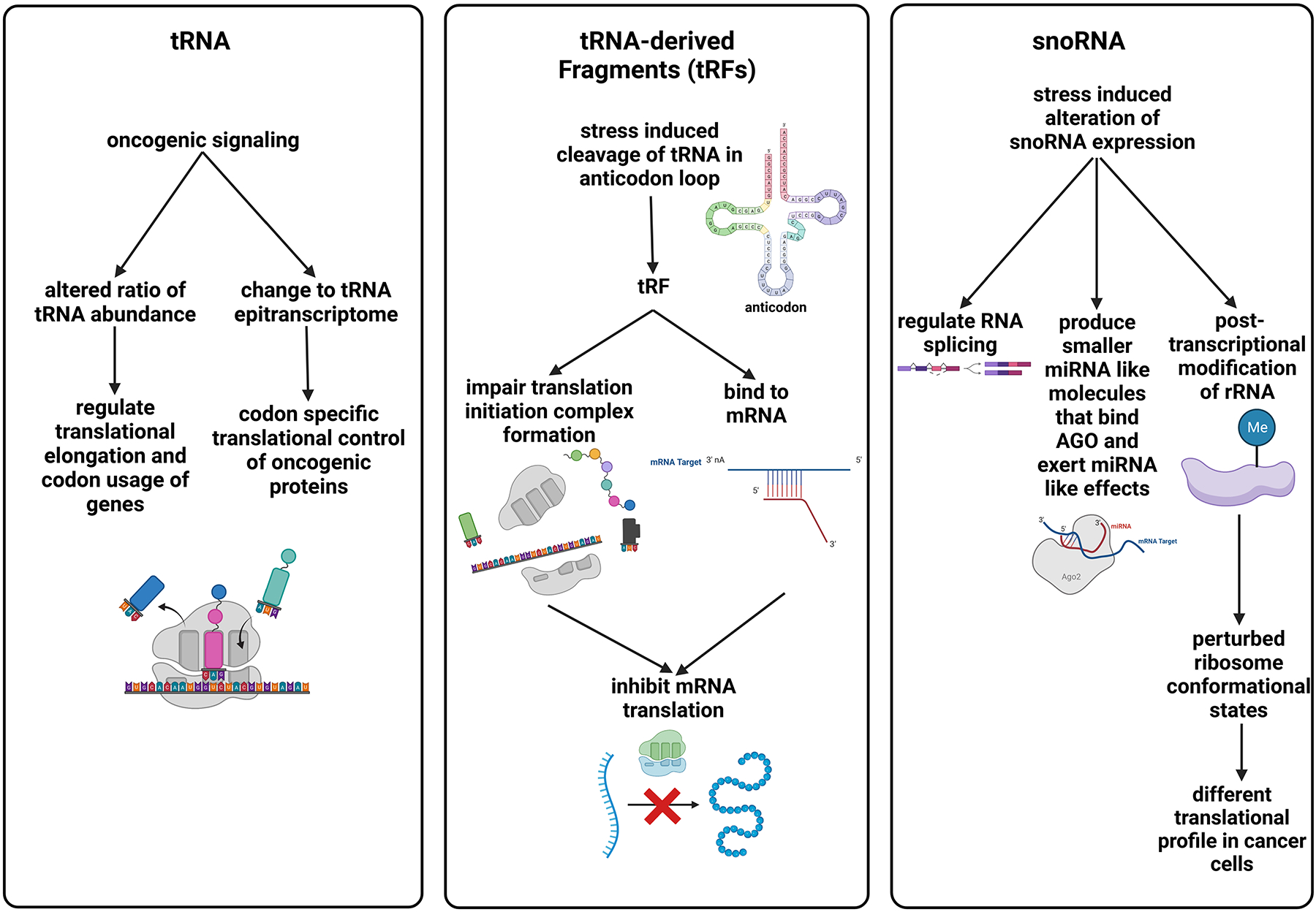
Summary of actions of other small RNAs involved in the control of plasticity in melanoma. Part of the figure is generated using birender.com (2022).
lncRNAs for lineage determination and metabolic plasticity in melanoma cells
Metabolic reprogramming is critical for melanoma cells to adapt to their environment during disease progression. These include the MYC pathway and energy metabolism pathways. Like the cases of EMT genes above, the progression of melanoma is frequently associated with the downregulation of miRNAs that target these genes, and this can be achieved by activating lncRNAs through some of the mechanisms and examples described. In recent years, more and more lncRNA loci have been identified. They have been shown to regulate expression of nearby genes by various mechanisms, including by: (1) guiding chromatin modifying enzymes to promoters; (2) forming scaffolds to recruit ribonucleoprotein complex; (3) serving as a decoy to remove proteins from the transcriptional sites; (4) sponging miRNAs; (5) contributing as the precursor of miRNAs; (6) locking enhancers by chromatin looping; and (7) blocking translation from mRNA. Multiple lncRNAs gene regulation mechanisms are implemented in metabolic reprogramming in response to environmental stress, following expression of oncogenic MYC and/or following hypoxic stimulation.
Intriguingly, many highly expressed lncRNAs, such as PVT1, H19 and MIAT are transcriptionally activated by MYC in melanoma. Some of them regulate the expression of the oncogene, MYC, at various levels. For example, PVT1 binds to MYC to block its phosphorylation, preventing it from degradation, and H19 acts as a sponge of miRNAs that would otherwise suppress MYC (e.g., let-7); both form positive feedback loops to increase MYC level (Figure 5) (Tseng et al., 2014). While MYC has multiple functions, considering most lncRNAs involved in these genetic regulatory circuits are induced by environmental stress (e.g., hypoxia activates H19), it is likely lncRNAs center around regulation of metabolic plasticity of melanoma (Avagliano et al., 2020; Wu et al., 2017). In fact, MYC and AKT signaling pathways stabilize Hypoxia Inducible Factor 1 (HIF1), increasing glucose uptake and lactate production by tumors. In such a response, MYC activates several genes involved in glucose metabolism, including LDHA (converting pyruvate to lactate), GLUT1 (glucose transporter 1), and HK2 (the first rate-limiting enzyme in the glycolytic pathway), resulting in increased glucose uptake and glycolytic activity. Meanwhile, MYC will suppress mitochondrial respiration (Abildgaard and Guldberg, 2015). With respect to the clinical relevance of metabolic regulation in this pathway, H19 is overexpressed in melanoma tissues relative to other skin tissue from the same patient and overexpression also correlates with advanced tumor invasion, metastasis and poor patient survival.
Figure 5.
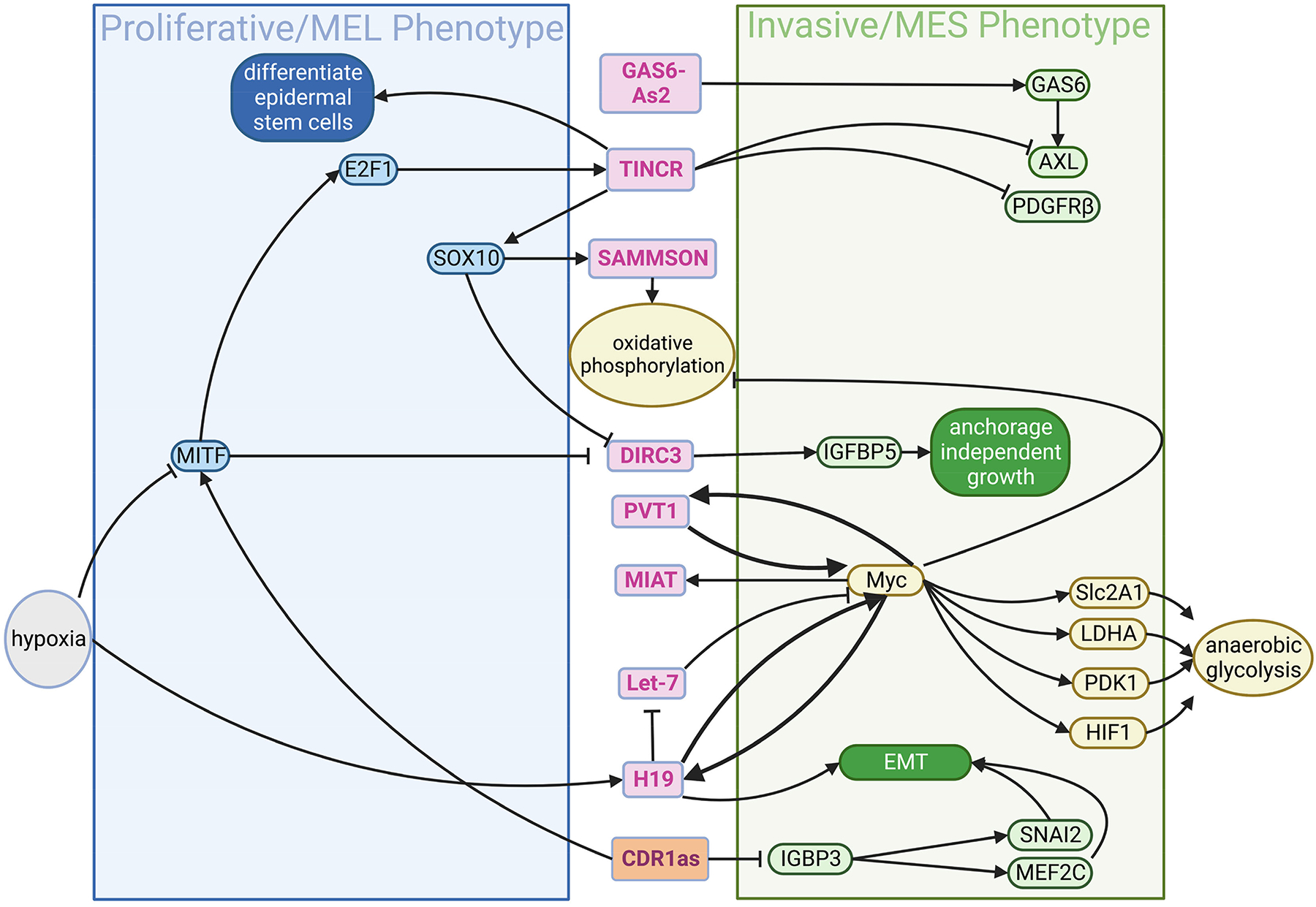
The lncRNA-protein circuit for control the transcriptional states and metabolic plasticity in melanoma. lncRNAs (marked by the magenta font in the pink circles) can be activated from their own promoters by some transcriptional factors (e.g. MYC) in response to environmental signals (e.g. hypoxia). In contrast, their expression could be suppressed by other transcriptional factors (e.g. MITF). Once expressed, lncRNAs can bind proteins or target genes for their activation or suppression. In melanoma cells, lncRNAs and protein factors form a circuit for the reciprocal inhibition between proliferative/MEL and invasive/MES states, putatively in response to input signal such hypoxia. MEL and MES, melanocytic and mesenchymal phenotype, respectively. Part of the figure is generated using birender.com (2022).
Metabolic reprogramming has been shown to impact cell state switching between lineage-derived cell states in melanoma. lncRNAs can directly impact lineage factors such as MITF and SOX10 by “locking in” developmental gene programs. The lncRNA, SAMMSON, whose gene is located 30 kb downstream from MITF, is induced by SOX10. It interacts with p32 protein to activate mitochondrial biogenesis and oxidative phosphorylation. Melanoma cells develop resistance to BRAF inhibition by increasing oxidative phosphorylation, and suppression of SAMMSON reverses such resistance (Leucci et al., 2016). In many cases, lncRNAs regulate expression of the nearby genes. Therefore, they can be regarded as the “guard” of the transcription for committing cells to a specific fate. In neural stem cells, lncRNA, TUNA, facilitates chromatin binding of polypyrimidine tract binding protein 1 (PTBP1), heterogeneous nuclear ribonucleoprotein K (hnRNP-K), and nucleolin (NCL) to the promoters of NANOG, SOX2 and FGF4, promoting the expression of these pluripotency genes in ESCs and thus their self-renewal (Lin et al., 2014).
circRNAs for lineage determination and metabolic plasticity in melanoma cells
circRNAs are a class of covalently closed non-coding RNAs which are generated with the process of backsplicing where a splice donor is ligated with an upstream acceptor to form a circularized RNA molecule. Although circRNAs were discovered several decades ago, their function had remained largely unexplored due to the fact that they were considered by-products of splicing. They gained major attention by the scientific community when a new role in regulating gene expression was attributed to them through sponging and sequestering miRNAs (Hansen et al., 2013; Memczak et al., 2013). Since then, other functions of circRNAs have been identified including binding and sequestering RNA-Binding Proteins (RBPs) (Abdelmohsen et al., 2017), regulating splicing and transcription (Ashwal-Fluss et al., 2014; Li et al., 2015; Liang et al., 2017) and generating short peptides from Open Reading Frames (ORFs) formed after circularization (Begum et al., 2018; Pamudurti et al., 2017). The role of circRNAs in cancer has also started to emerge as increasing numbers of studies show that they are differentially expressed in various cancer types, and they are involved in processes such as tumorigenesis, metastasis, tumor suppression, and lineage determination (Kristensen et al., 2018).
Recently, new discoveries have shown that circRNAs are involved in melanoma as well (Mecozzi et al., 2021), with the most characteristic example being the circRNA, CDR1as. CDR1as is derived from a transcript from the lncRNA LINC00632 which contains Alu repeats to mediate circularization (Hanniford et al., 2020). This was the first circRNA to draw major attention and to be implicated in the ceRNA theory (BOX1) where it was originally identified to sponge miR-7 (it contains 63 sites for miR-7) and modulates its activity (Hansen et al., 2013; Memczak et al., 2013). A recent study showed that loss of CDR1as was able to drive invasion and metastasis of melanoma cells, through binding and sequestering the protein IGF2BP3 (Hanniford et al., 2020). Depletion of CDR1as was able to upregulate the IGF2BP3-mediated expression of SNAI2 and MEF2C, which induced invasion and neural crest gene expression, respectively. This promising finding initiated the idea of CDR1as being an indicator of transcriptional cell state in melanoma and, indeed, melanoma cell lines with low expression of CDR1as showed higher sensitivity to GPX4 inhibitors and correlation with high AXL and low MITF expression (Hanniford et al., 2020) (Figure 5), a result consistent with the study of Tsoi et al (Tsoi et al., 2018).
Interestingly, the majority of circRNAs found in melanoma have been shown to have oncogenic activity. The circRNA derived from the FOXM1 gene, circ_0025039, inhibits miR-198 to increase expression of Cyclin dependent Kinase-4 (CDK4), and regulate proliferation, invasion and glucose metabolism (Bian et al., 2018). circ_0020710, promotes melanoma progression and contributes to melanoma immune evasion by sponging miR-370–3p and subsequently upregulating levels of the chemokine, CXCL12 that is known to stimulate tumor growth(Wei et al., 2020). miR331–3p is inhibited by the oncogenic circ_0002770 (derived from the MDM2 gene, which encodes a nuclear E3 ubiquitin ligase and negatively regulates p53) to increase expression of the MAPK pathway regulators Dual-specific phosphatase 5 (DUSP5) and Transforming Growth Factor beta Receptor 1 (TGFBR1) (Qian et al., 2020), while circ_0001591 activates the PI3K/AKT pathway by sequestering miR-431–5p and promoting Rho-associated coiled -coil containing protein kinase 1 (ROCK1) expression (Yin et al., 2021).
In addition, there are examples of circRNAs in melanoma which do not involve inhibition of miRNA activity. circGLI1 has the ability to phosphorylate and inactivate the glycogen synthase kinase 3-β (GSK3β) through physical interaction with p70S6K2, subsequently leading to elevated expression of Cyr61, a proangiogenic factor whose upregulation promotes melanoma metastasis (Chen et al., 2020a).
In a different study in glioma, CDR1as was found to stabilize p53 by disrupting its interaction with MDM2 and its subsequent ubiquitination. Although it is not clear whether this is a universal mechanism for other tissues, it may be a mechanism in melanoma cells as well in order to protect p53 from degradation (Lou et al., 2020).
However, skepticism exists regarding the importance and the impact of the ceRNA theory and the ability of circRNAs to sponge other molecules. Some circRNAs have only predicted binding sites for miRNAs. Most of them contain only a limited number of these sequences raising doubts of whether the sponging occurs or not, especially considering the low abundance of these molecules. An explanation can be given with the notion that circRNAs function to fine-tune the miRNA activity and play a small role in larger gene expression regulation networks depending on the states or needs of the cell.
Interaction among noncoding RNAs in the plasticity of melanoma
The interaction between lncRNAs and miRNAs has been extensively studied. To stably maintain the transcriptional state, the lncRNA can secure the expression of lineage-specific genes by serving as a sponge to sequester miRNAs that target those genes. In contrast, miRNAs can target specific lncRNAs to promote its degradation, reversing the lineage-specific gene expression that they secured. Together these form reciprocally inhibitory loops that generate two or more states, as described in the previous sections (Riefolo et al., 2019).
In melanoma, several lncRNAs have been shown to secure specific gene expression by sequestering miRNAs. For example, the STAT3-induced lncRNA, LHFPL3-AS1, serves as a sponge to sequester miR-580-3p that would otherwise inhibit the translation of STAT3 in human melanoma cells. Therefore, LHFPL3-AS1 secured the IL6-induced STAT3 activation and downstream signaling via the feedback loop (Peng et al., 2020). It has been shown that miR-580 can be induced in T cells by IL-2 treatment, and its function is to inhibit TWIST1 and mesenchymal phenotype (Ranji et al., 2015). Taken together, the STAT3/LHFPL3-AS1/miR-580 could form the circuit for responding to different types of inflammatory signals and thus altering cell state. In a related pathway, the hypoxia-inducible lncRNA, NEAT1, is significantly upregulated in melanoma cells as compared to melanocytes (Zhang et al., 2019a). NEAT1 promotes EMT by sequestering miR-200b-3p that targets SMAD2, resulting in the upregulation of SNAIL, MMP2, and MMP9 (Zhou et al., 2020). These studies suggest that the combination of lncRNAs and miRNAs in the genetic circuit can increase the range of modulation in response to external stimuli. For example, in Figure 2b, two reciprocal inhibitory loops, ZEB1/miR-200 and SNAIL1/miR-34, interact with each other to form a modulator of three states (epithelial, EMT, and mesenchymal). TGF-β signal can trigger the switching among states by acting on SNAIL1 in this model. Since TGF-β-responsive lncRNAs, such as MALAT1, ATB, LINC00115, can suppress miR-200 family by sponging them, they allow TGF-β signal to modulate the cell states from different levels. Moreover, lncRNAs can serve as “conductors” between environment stimuli (e.g., hypoxia) and such genetic circuits (Figure 6). In kidney cells, lncRNAs can host miRNA genes that function in the same direction to increase the expression of TGF-β. lncRNA PVT1 induces the expression of miR-1207–5p, derived from the intron of PVT1. miR-1207–5p suppresses PMEPA1, PDPK1, and SMAD7, three genes that block the activation of TGF-β/Smad signaling. Therefore, PVT1/TGF-β/miR-1207–5p form a self-enforcing feedback loop, resulting in the accumulation of extracellular matrix and ultimately fibrosis (Alvarez et al., 2013). These mechanisms could also be relevant to melanoma, indeed PVT1 has been shown to be elevated in melanoma patients (Chen et al., 2017a).
Figure 6.
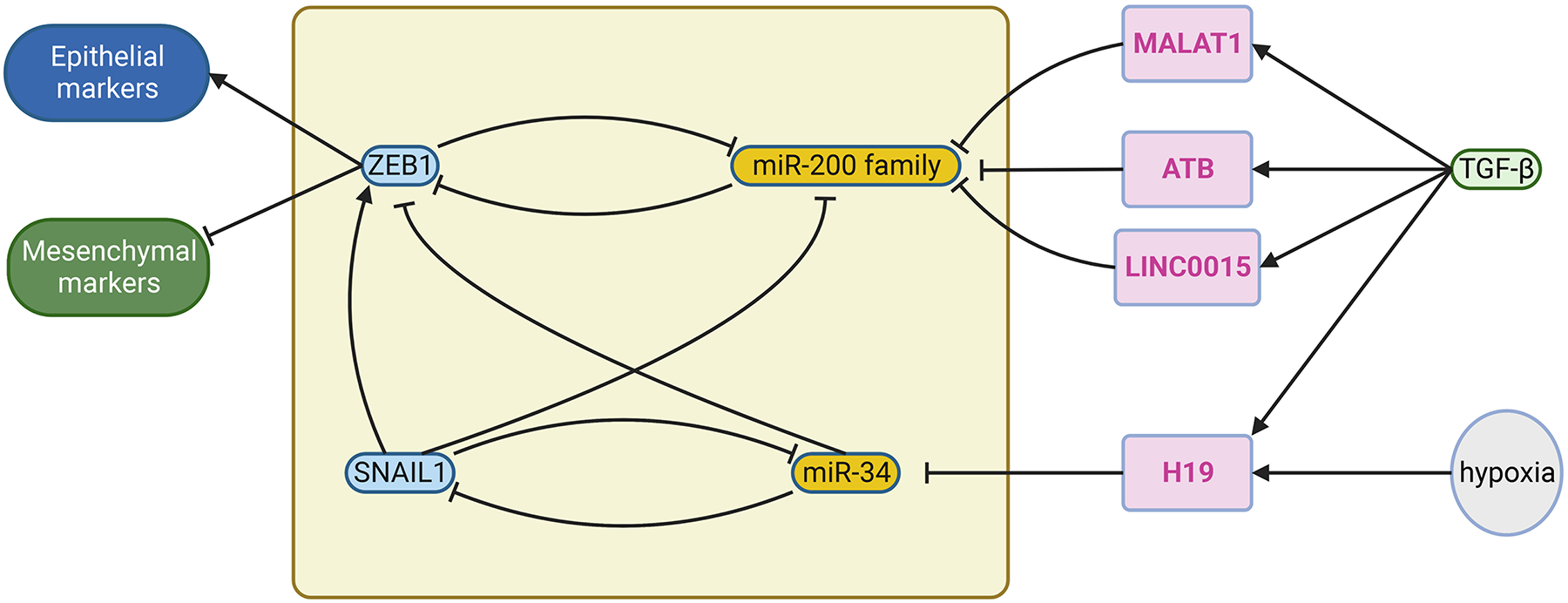
Examples of interaction among noncoding RNAs to control the transcriptional state switch. In Fig. 2b, two interacting negative-feedback loops composed of transcriptional factors and miRNA form a genetic circuits for three cell states (epithelial, EMT, and mesenchymal), and SNAIL1 receives the input signal TGFβ. Alternatively, input signal, such as TGFβ and hypoxia, can control this circuit by inducing lncRNAs that suppress miR-200 family (Li et al., 2016; Liu et al., 2018; Raveh et al., 2015; Yuan et al., 2014) or miR-34 by sponging them. This adds another level of regulation of melanoma plasticity. Part of the figure is generated using birender.com (2022).
Opportunities for identifying biomarkers and therapeutic targets
microRNAs.
Since resistance of melanoma to therapies, including both targeted therapies and immunotherapies, is driven by specific NC developmental states, miRNAs that mediate their switching have great potential to serve as diagnostic or prognostic biomarkers, as well as therapeutic targets. The miR-200 family has previously demonstrated a role in regulating EMT, underscoring the potential significance of miRNA-mediated plasticity in clinical outcomes (Gregory et al., 2008; Korpal et al., 2008; Park et al., 2008b). Therefore, the miR-200 family can serve as a good example of prognostic value of microRNAs in melanoma. Indeed, the expression of miR-211 was significantly decreasing during the course of melanoma progression from nevi to primary tumors, and then metastases. Moreover, delivery of miR-211 mimetics into melanoma cells resulted in the delayed growth in culture and significant reduction in size and number of cell clusters in colony-forming assay (Bell et al., 2014; Xu et al., 2012). In contrast, the expression of miR-221 was significantly increasing from normal melanocytes to primary tumor, reaching a maximum level in metastatic diseases (Mueller et al., 2009). The circulating level of microRNA-221 in patients with malignant melanoma correlated with disease progression or recurrence, suggesting its function as a prognostic marker (Kanemaru et al., 2011). miR-200a may also be useful for tracking melanoma progression and predicting response to therapy. Relative to primary melanomas, regional lymph nodes and distant organ metastases had significantly lower miR-200a. Moreover, primary melanoma patients with low miR-200a had inferior survival rates relative to those with high miR-200a (Bustos et al., 2017).
Overexpression of miR-200a in metastatic melanoma cells attenuated CDK6 expression and phosphorylated Rb levels, thereby slowing cell proliferation at the G1/S checkpoint. The inverse correlation between miR-200a and CDK6 expression may explain differential responses to CDK4/6 inhibitors: metastatic melanoma cells with diminished miR-200a permitted higher CDK6 and subsequently responded better to CDK4/6 inhibitors, such as palbociclib. On the other hand, those with higher miR-200a express less CDK6, leading to a less inhibitory response to the same drug (Bustos et al., 2017). Therefore, miR-200a may also serve as a predictive biomarker for therapeutic responses.
Although many studies have explored using these miRNAs biomarker or therapeutic targets, there is little or none shown to achieve clinical application. Such outcomes may be attributed to two features of miRNAs. First, miRNAs work in a combinatorial manner. Targeting or delivering a specific miRNA may not be sufficient to alter the overall effects of the group of miRNAs. Recently, the studies in long noncoding RNAs (lncRNAs) and miRNA sponge have shown promise to overcome this issue, which will be discussed in the next section. Second, miRNAs work as “dimmers” in gene regulatory circuits. Significant changes of the miRNA levels can switch the cell states but not kill the cells. In this sense, miRNA therapy could sensitize melanoma cells to the immunotherapies or targeted therapies. This potential requires further studies to validate.
lncRNAs.
Although the abundance of lncRNAs is generally low among all RNA species, their size and special expression pattern still allow their detection from standard RNA sequencing data by special alignment pipelines (Zheng et al., 2019). Many lncRNAs have been mined from the data in the public domain (e.g., TCGA mRNA data), increasing the chance of identifying them as diagnostic or prognostic biomarkers. Traditionally, diagnosis of malignancies is based on tissue samples containing tumor cells. Studies have shown the diagnostic and prognostic value of lncRNAs involved in the regulation of melanoma plasticity. For example, twelve lncRNAs are upregulated in melanoma compared with normal tissue, and three of them could be melanoma specific: RMEL3, LLME23, and SAMMSON. Interestingly, both SAMMSON and RMEL3 have been shown to be a metabolic switch, and LLMEL23 promotes the expression of RAB23, which is involved in the differentiation of neural patterning (Seabra et al., 2002).
Such lncRNAs may also function as indicators of disease progression to metastasis. When comparing matched pairs of primary melanomas with lymph node metastases, HOTAIR was significantly overexpressed in the latter. Furthermore, siRNA knockdown of HOTAIR in vitro reduced motility and invasive potential of a human melanoma line (Tang et al., 2013). A separate study examined benign/borderline melanocytic lesions, nonulcerated primary melanomas (pT1a), primary melanomas (pT3/pT4) with metastases, and visceral metastases. Despite probing HOTAIR expression of dysplastic nevi, intradermal nevi, and atypical Spitz nevi, none of these benign melanocytic lesions had measurable HOTAIR expression. Conversely, primary and metastatic melanomas had detectable expression in both the tumor cells and the adjacent tumor microenvironment, where closer lymphocytes were more likely to be HOTAIR positive. While pT1 lesions had relatively low HOTAIR expression, pT3/pT4 lesions and their matched metastases had significant expression. Serum samples from the pT1a revealed baseline HOTAIR expression whereas pT3/pT4 groups had elevated HOTAIR expression (Cantile et al., 2017).
IGF2AS and ZEB2NAT were specifically identified as independent prognostic variables in patients on vemurafenib (Kolenda et al., 2019). Previous studies indicate that these genes have a role in controlling melanoma plasticity and invasive potential, respectively, which suggests that they may help confer resistance to therapy in mutant melanoma (Kolenda et al., 2019; Zhang et al., 2017; Zhuang et al., 2015). Other studies have found similar prognostic value between lncRNA expression signatures and overall survival, tumor size, and other clinical endpoints (Chen et al., 2017b; Guo et al., 2016; Xiao and Yin, 2019). Expression of MIAT in melanoma has been correlated with T cell, CD8+ T cell, and NK infiltration, which could suggest a role of lncRNAs in immune responses to melanoma or immunotherapy (Liu et al., 2019).
Alternatively, melanoma-specific lncRNAs could be released into circulation, so their serum levels may serve as a more accessible diagnostic. For example, serum PVT1 levels are significantly increased in melanoma patients compared with age and gender-matched healthy controls with melanocytic nevi. Therefore, it may be used to evaluate the melanoma risk of individuals carrying nevi (Chen et al., 2017a). As another example, plasma levels of SPRY4IT were significantly higher in the melanoma patient group, as compared to the healthy control group. Moreover, patients with high SPRY4IT had worse survival rates than low SPRY4IT patients. SPRY4IT also increased with advanced tumor stage and proximity to the tumor site (Liu et al., 2016). Monitoring expression of multiple lncRNAs with composite risk scores in circulation may be especially powerful (Tian et al., 2020).
When the lncRNA risk score was combined with clinically evident risk factors, including tumor stage and Breslow thickness, prognostic value was further improved. LncRNA based risk scores may even offer value in triaging patients with advanced or resistant melanomas (Tian et al., 2020). Since lncRNAs can regulate gene expression more directly (e.g. by acting on transcriptional machinery), or more efficiently (e.g. for example, by serving as miRNA sponge), they are supposed to be more effective therapeutic targets. Indeed, knockdown of ANRIL by siRNA in a human melanoma cell line resulted in significant increase of CDKN2A and CDKN2B, the tumor suppressor loci that is frequently suppressed during melanoma progression, change in cell morphology, and decrease in growth and invasiveness in vitro and in vivo (Xu et al., 2016). This study demonstrates the proof-of-concept of targeting lncRNAs for melanoma treatment.
As developmental states are associated with responses to therapies, including both targeted and immuno-therapies, an alternative approach is to target the lncRNAs that controls cell states, sensitizing melanoma cells to those therapies. This concept has been demonstrated by a study of TINCR, which promotes the differentiation of melanoma cells. Delivery of TINCR (3.7kb) into melanoma cells reduces the expression of AXL but increases the expression of SOX10 and MITF, indicating a more differentiated state. Moreover, the TINCR-transfectant melanoma cells became more sensitive to MEK inhibitor trametinib and reduced metastatic capacity (Melixetian et al., 2021). Further studies are needed to address if TINCR expression can overcome the resistance of melanoma to immunotherapy, especially immune checkpoint blockade (ICB) therapies.
circRNA.
Since the discovery of circRNAs as contributors to gene expression regulation, several attempts have been made to identify their role in melanoma. Many circRNAs have been shown to be differentially expressed in melanoma cells compared to melanocytes (Wang et al., 2018). However, only a few have been extensively studied. Most of these circRNAs have been associated with sponging and regulating miRNA activity. Several examples have recently come to surface such as circRNA_0084043, which regulates proliferation and migration of melanoma cells by sequestering miR-429 and preventing it from inhibiting expression of TRIB2 (Chen et al., 2020a). Another miRNA regulated by circRNA_0084043 is miR-153–3p, a tumor suppressor capable of regulating EMT by targeting SNAIL (Luan et al., 2018). circ_0079593 is overexpressed in human melanoma tissues and cell lines and it also has an effect on two miRNAs, miR-516b, and miR-573 (Lu and Li, 2020; Zhao et al., 2021). Moreover, circ_0016418, interferes with the activity of more than one miRNA, miR-605–5p and miR-625, to modulate the expression of GLS and YY1 respectively, and promotes proliferation and migration/invasion of melanoma cells (Lu et al., 2020; Zou et al., 2019). These circRNAs may serve as diagnostic markers for premalignant lesion of melanocytes.
Some studies have shown that circRNAs are secreted via exosomes and can be found in patient serum (O’Brien et al., 2020). The purpose of the secretion of the circRNAs is not known but it provides a field of investigation to establish circRNAs as biomarkers in melanoma and other cancer types despite their low abundance. So far, several of the circRNAs discussed above have been studied with respect to their ability to serve as melanoma prognosis markers and if they are connected to melanoma patients survival and metastasis. For example, high expression of circ_0084043 predicted poor overall survival for melanoma patients as its expression correlated with clinical stage of melanoma and decreased overall survival (Luan et al., 2018). Similarly, patients with high expression of circ_0025039, circ_0001591, or circ_0079593 had a shorter survival time shown in three independent studies (Bian et al., 2018; Lu and Li, 2020; Yin et al., 2021), while circ_0002770 levels were increased in metastatic melanoma tissue, also implying poor prognosis (Qian et al., 2020).
Thus far, most of the circRNAs that are good candidates for biomarkers are mostly associated with poor prognosis and decreased overall survival. As CDR1as was identified to inhibit metastasis in melanoma, it would be interesting to investigate whether it can be established as a predictive biomarker for early-stage patients. Indeed, lower CDR1as expression was identified in metastatic melanoma cell lines and this loss correlated with shorter overall survival of patients. Nevertheless, the study published by Hanniford et al. indicates that it can aid to select appropriate treatment for melanoma patients since it can indicate the cell state of melanoma cells and subsequently, which inhibitors (MAPK or GPX4) may be more suitable for use (Hanniford et al., 2020).
Perspectives in therapeutic development.
Due to their dynamic nature and complex nature, as well as their central role in directly affecting the robustness of the expression of the genetic information, several attempts have been made to target several noncoding RNAs, as part of effective and tailor-made therapeutic effects. The major approaches include either targeting with specific antisense oligonucleotides (ASOs) or the design of specific small molecule inhibitors that could possibly interfere with RNA-protein interactions. ASOs are single-stranded DNA molecules with full complementarity to one select target mRNA and may act by blocking protein translation (via steric hindrance), causing mRNA degradation (via RNase H cleavage) or changing pre-mRNA splicing (via interference with cis-splicing elements causing exon inclusion or exclusion). Using antisense oligonucleotides (ASOs) that can block complementary RNA molecules has already been used successfully to target and repair defective mRNAs in diseases such as spinal muscular atrophy and Duchenne muscular dystrophy. In the same line, ASOs can also be used to target miRNAs and lncRNAs and several trials are currently evaluating the efficacy and safety of their use in various diseases (Winkle et al., 2021). Another approach to targeting noncoding RNAs is the use of small molecule inhibitors that can either bind to specific structural motifs or proteins involved in noncoding RNA regulation, such as RNase H, an RNA-binding protein involved in the degradation of certain noncoding RNAs, or directly target specific miRNAs(Warner et al., 2018).
Despite the potential of these approaches, targeting noncoding RNAs is challenging. One of the major challenges is the delivery of therapeutic agents to the specific tissues where noncoding RNAs are expressed and also their ability to induce adverse immune responses (reviewed in Winkle et al). Achieving specificity and avoiding off-target effects is also difficult because noncoding RNAs have multiple targets and functions. Moreover, the dynamic nature of noncoding RNAs, which is influenced by various factors such as environmental cues and cellular stress, further complicates therapeutic targeting. It is therefore imperative that further in-depth research will shed light in a more specific, exclusive and effective delivery and targeting of noncoding RNAs with pivotal role that would open new avenues in RNA-based therapeutics.
Conclusion
It has been recognized that melanoma cells exhibit distinct transcriptional states that are adopted from NC differentiation programs. In response to environmental signaling or stress, melanoma cells are able to switch among these cell states for adaptation; such capacity is referred to as plasticity. Why discrete states are used by cells, instead of proportional changes in response to signals, can be understood by the analogy of the transmission gears in cars; that is, shifting gears is for operating the engine at optimal output to get the speed desired to run on the road. Similarly, changing the cell state allows cells to perform the required function at the best energy efficiency.
In theory, in response to stress promptly changing melanoma cell states would be more efficient to adapt than increasing or decreasing individual functions would. As many cell states have been identified, how they are induced and maintained is not clear. Moreover, the switching between cell states is putatively in a discrete (binary) manner. These observations suggest that the transcriptional states of melanoma cells are determined by genetic regulatory circuits that include feedback loops. In this review we have delineated the regulatory circuits operated by the noncoding RNAs and collaborating factors, determining melanoma cell states in response to the environmental signals. For example, feedback loops between miRNAs and lineage-determining transcription factors can form discrete cell states of the “valleys” in a Waddington landscape. lncRNAs can secure the transcriptional status by recruiting or sequestering the factors required for transcription. Other small RNAs like tRFs or snoRNAs could amplify the inhibitory signals. Which types of regulatory circuits that a specific kind of ncRNAs is involved may depends on the nature of the ncRNAs. As mentioned in the sections above, miRNAs can target multiple RNAs simultaneously, and lncRNA can regulate the function of transcription factors by direct binding.
These features make them effective modulators of transcriptional states that define cell lineages. In comparison, snoRNAs and tRNAs regulate protein synthesis directly, and many of them are induced by MYC, the “master regulator” of cell growth. Therefore, it is expected that snoRNAs and tRFs are highly involved in the translational plasticity. However, it could not be excluded that our understanding of the connection between each type of noncoding RNAs and an aspect of plasticity is partially determined by the current focuses of research. This illustrates the importance of further development of computational and genomic analyses for ncRNAs.
It is well recognized that melanoma becomes resistant to therapies by switching cell states, including both targeted therapies and immunotherapies. Interestingly, many noncoding RNAs can be induced by inflammatory signals, such as interferons and other cytokines, and metabolic stress, such as hypoxia or reactive oxygen species. Such noncoding RNAs could be the essential components of the gene regulatory circuits for melanoma cell states and plasticity, and therefore having important therapeutic implication. For example, suppression of such noncoding RNAs may prevent melanoma cells from switching states, sensitizing them to therapies. Thanks to the development of RNA vaccines, the technology of RNA therapies has advanced rapidly in recent years. Therefore, targeting noncoding RNAs in combination with current therapies should be an important direction for the therapeutic development of melanoma in the coming years.
Acknowledgment
We would like to thank Dr. Ashish Lal (Cancer Genetics Branch, National Cancer Institute, NIH, Bethesda, MD, USA) for his advice and support. This research was supported in part by funds from the NIH intramural research program and a FLEX Synergy Award from the NCI Center for Cancer Research. Kerrie Marie was supported by The Wellcome Trust [204796/Z/16/Z] (KLM) and The University of Manchester FBMH Dean’s Prize award. Katerina Grafanaki is a recipient of a Research Fellowship from Fulbright Foundation Greece, which is gratefully acknowledged.
Abbreviations
- ceRNA
Competitive endogenous RNA
- circRNA
Circular RNA
- CMM
Cutaneous malignant melanoma
- EMT
Epithelial to mesenchymal transition
- iPSC
Induced pluripotent stem cells
- lncRNA
Long non-coding RNA
- MEL
Melanocytic-like
- MES
Mesenchymal-like
- miRNA
microRNA
- NC
Neural crest
- ncRNA
Non-coding RNA
- piRNA
PIWI-interacting RNAs
- rRNA
Ribosomal RNAs
- scaRNA
Small Cajal-body specific RNAs
- SCP
Schwann Cell Progenitor
- snoRNA
Small nucleolar RNA
- snRNA
Small nuclear RNA
- tDR
tRNA-derived small RNAs
- tRAX
tRNA Analysis of eXpression
- tRFs
tRNA’s and their fragments
- tRNA
Transfer RNA
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Reference
- Abdelmohsen K, Panda AC, Munk R, Grammatikakis I, Dudekula DB, De S, Kim J, Noh JH, Kim KM, Martindale JL, et al. (2017). Identification of HuR target circular RNAs uncovers suppression of PABPN1 translation by CircPABPN1. RNA Biol 14, 361–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abildgaard C, and Guldberg P (2015). Molecular drivers of cellular metabolic reprogramming in melanoma. Trends Mol Med 21, 164–171. [DOI] [PubMed] [Google Scholar]
- Adameyko I, and Lallemend F (2010). Glial versus melanocyte cell fate choice: Schwann cell precursors as a cellular origin of melanocytes. Cell Mol Life Sci 67, 3037–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal V, Bell GW, Nam JW, and Bartel DP (2015). Predicting effective microRNA target sites in mammalian mRNAs. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez ML, Khosroheidari M, Eddy E, and Kiefer J (2013). Role of microRNA 1207-5P and its host gene, the long non-coding RNA Pvt1, as mediators of extracellular matrix accumulation in the kidney: implications for diabetic nephropathy. PLoS One 8, e77468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews MC, Oba J, Wu CJ, Zhu H, Karpinets T, Creasy CA, Forget MA, Yu X, Song X, Mao X, et al. (2022). Multi-modal molecular programs regulate melanoma cell state. Nat Commun 13, 4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwal-Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M, Evantal N, Memczak S, Rajewsky N, and Kadener S (2014). circRNA biogenesis competes with pre-mRNA splicing. Mol Cell 56, 55–66. [DOI] [PubMed] [Google Scholar]
- Avagliano A, Fiume G, Pelagalli A, Sanita G, Ruocco MR, Montagnani S, and Arcucci A (2020). Metabolic Plasticity of Melanoma Cells and Their Crosstalk With Tumor Microenvironment. Front Oncol 10, 722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begum S, Yiu A, Stebbing J, and Castellano L (2018). Novel tumour suppressive protein encoded by circular RNA, circ-SHPRH, in glioblastomas. Oncogene 37, 4055–4057. [DOI] [PubMed] [Google Scholar]
- Bell RE, Khaled M, Netanely D, Schubert S, Golan T, Buxbaum A, Janas MM, Postolsky B, Goldberg MS, Shamir R, et al. (2014). Transcription factor/microRNA axis blocks melanoma invasion program by miR-211 targeting NUAK1. J Invest Dermatol 134, 441–451. [DOI] [PubMed] [Google Scholar]
- Bian D, Wu Y, and Song G (2018). Novel circular RNA, hsa_circ_0025039 promotes cell growth, invasion and glucose metabolism in malignant melanoma via the miR-198/CDK4 axis. Biomed Pharmacother 108, 165–176. [DOI] [PubMed] [Google Scholar]
- Bottini S, Pratella D, Grandjean V, Repetto E, and Trabucchi M (2018). Recent computational developments on CLIP-seq data analysis and microRNA targeting implications. Brief Bioinform 19, 1290–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle GM, Woods SL, Bonazzi VF, Stark MS, Hacker E, Aoude LG, Dutton-Regester K, Cook AL, Sturm RA, and Hayward NK (2011). Melanoma cell invasiveness is regulated by miR-211 suppression of the BRN2 transcription factor. Pigment Cell Melanoma Res 24, 525–537. [DOI] [PubMed] [Google Scholar]
- Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, and Goodall GJ (2008). A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res 68, 7846–7854. [DOI] [PubMed] [Google Scholar]
- Brock A, Chang H, and Huang S (2009). Non-genetic heterogeneity--a mutation-independent driving force for the somatic evolution of tumours. Nat Rev Genet 10, 336–342. [DOI] [PubMed] [Google Scholar]
- Brombin A, Simpson DJ, Travnickova J, Brunsdon H, Zeng Z, Lu Y, Young AIJ, Chandra T, and Patton EE (2022). Tfap2b specifies an embryonic melanocyte stem cell that retains adult multifate potential. Cell Rep 38, 110234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busam KJ, Antonescu CR, Marghoob AA, Nehal KS, Sachs DL, Shia J, and Berwick M (2001). Histologic classification of tumor-infiltrating lymphocytes in primary cutaneous malignant melanoma. A study of interobserver agreement. Am J Clin Pathol 115, 856–860. [DOI] [PubMed] [Google Scholar]
- Bustos MA, Ono S, Marzese DM, Oyama T, Iida Y, Cheung G, Nelson N, Hsu SC, Yu Q, and Hoon DSB (2017). MiR-200a Regulates CDK4/6 Inhibitor Effect by Targeting CDK6 in Metastatic Melanoma. J Invest Dermatol 137, 1955–1964. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas N (2015). Genomic Classification of Cutaneous Melanoma. Cell 161, 1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantile M, Scognamiglio G, Marra L, Aquino G, Botti C, Falcone MR, Malzone MG, Liguori G, Di Bonito M, Franco R, et al. (2017). HOTAIR role in melanoma progression and its identification in the blood of patients with advanced disease. J Cell Physiol 232, 3422–3432. [DOI] [PubMed] [Google Scholar]
- Chen CW, and Tanaka M (2018). Genome-wide Translation Profiling by Ribosome-Bound tRNA Capture. Cell Rep 23, 608–621. [DOI] [PubMed] [Google Scholar]
- Chen J, Zhou X, Yang J, Sun Q, Liu Y, Li N, Zhang Z, and Xu H (2020a). Circ-GLI1 promotes metastasis in melanoma through interacting with p70S6K2 to activate Hedgehog/GLI1 and Wnt/beta-catenin pathways and upregulate Cyr61. Cell Death Dis 11, 596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Gao G, Liu S, Yu L, Yan D, Yao X, Sun W, Han D, and Dong H (2017a). Long Noncoding RNA PVT1 as a Novel Diagnostic Biomarker and Therapeutic Target for Melanoma. Biomed Res Int 2017, 7038579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Guo W, Xu XJ, Su F, Wang Y, Zhang Y, Wang Q, and Zhu L (2017b). Melanoma long non-coding RNA signature predicts prognostic survival and directs clinical risk-specific treatments. J Dermatol Sci 85, 226–234. [DOI] [PubMed] [Google Scholar]
- Chen Z, Chen J, Wa Q, He M, Wang X, Zhou J, and Cen Y (2020b). Knockdown of circ_0084043 suppresses the development of human melanoma cells through miR-429/tribbles homolog 2 axis and Wnt/beta-catenin pathway. Life Sci 243, 117323. [DOI] [PubMed] [Google Scholar]
- Clark WH Jr., Elder DE, Guerry D.t., Epstein MN, Greene MH, and Van Horn M (1984). A study of tumor progression: the precursor lesions of superficial spreading and nodular melanoma. Hum Pathol 15, 1147–1165. [DOI] [PubMed] [Google Scholar]
- Clemente CG, Mihm MC Jr., Bufalino R, Zurrida S, Collini P, and Cascinelli N (1996). Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer 77, 1303–1310. [DOI] [PubMed] [Google Scholar]
- Comandante-Lou N, Baumann DG, and Fallahi-Sichani M (2022). AP-1 transcription factor network explains diverse patterns of cellular plasticity in melanoma cells. Cell Rep 40, 111147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA, Roslan S, Schreiber AW, Gregory PA, and Goodall GJ (2015). The RNA binding protein quaking regulates formation of circRNAs. Cell 160, 1125–1134. [DOI] [PubMed] [Google Scholar]
- Cursons J, Pillman KA, Scheer KG, Gregory PA, Foroutan M, Hediyeh-Zadeh S, Toubia J, Crampin EJ, Goodall GJ, Bracken CP, et al. (2018). Combinatorial Targeting by MicroRNAs Co-ordinates Post-transcriptional Control of EMT. Cell Syst 7, 77–91 e77. [DOI] [PubMed] [Google Scholar]
- D’Arcangelo D, Facchiano F, Nassa G, Stancato A, Antonini A, Rossi S, Senatore C, Cordella M, Tabolacci C, Salvati A, et al. (2016). PDGFR-alpha inhibits melanoma growth via CXCL10/IP-10: a multi-omics approach. Oncotarget 7, 77257–77275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damsky WE, and Bosenberg M (2017). Melanocytic nevi and melanoma: unraveling a complex relationship. Oncogene 36, 5771–5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diener J, and Sommer L (2021). Reemergence of neural crest stem cell-like states in melanoma during disease progression and treatment. Stem Cells Transl Med 10, 522–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitriou F, Krattinger R, Ramelyte E, Barysch MJ, Micaletto S, Dummer R, and Goldinger SM (2018). The World of Melanoma: Epidemiologic, Genetic, and Anatomic Differences of Melanoma Across the Globe. Curr Oncol Rep 20, 87. [DOI] [PubMed] [Google Scholar]
- Du H, Cui S, Li Y, Yang G, Wang P, Fikrig E, and You F (2018). MiR-221 negatively regulates innate anti-viral response. PLoS One 13, e0200385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright AJ, John B, Gaul U, Tuschl T, Sander C, and Marks DS (2003). MicroRNA targets in Drosophila. Genome Biol 5, R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbri L, Chakraborty A, Robert C, and Vagner S (2021). The plasticity of mRNA translation during cancer progression and therapy resistance. Nat Rev Cancer 21, 558–577. [DOI] [PubMed] [Google Scholar]
- Fan X, and Kurgan L (2015). Comprehensive overview and assessment of computational prediction of microRNA targets in animals. Brief Bioinform 16, 780–794. [DOI] [PubMed] [Google Scholar]
- Fane ME, Chhabra Y, Smith AG, and Sturm RA (2019). BRN2, a POUerful driver of melanoma phenotype switching and metastasis. Pigment Cell Melanoma Res 32, 9–24. [DOI] [PubMed] [Google Scholar]
- Faridani OR, Abdullayev I, Hagemann-Jensen M, Schell JP, Lanner F, and Sandberg R (2016). Single-cell sequencing of the small-RNA transcriptome. Nat Biotechnol 34, 1264–1266. [DOI] [PubMed] [Google Scholar]
- Garg M (2013). Epithelial-mesenchymal transition - activating transcription factors - multifunctional regulators in cancer. World J Stem Cells 5, 188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalan V, Day C-P, Pérez-Guijarro E, Chin S, Ebersole J, Smith C, Simpson M, Sassano A, Alves Constantino M, Wu E, Yang HH, Lee MP, Hannenhalli S, Merlino G, Marie KL (2022). Comprehensive single-cell transcriptomic analysis of embryonic melanoblasts uncovers lineage-specific mechanisms of melanoma metastasis and therapy resistance (bioRxiv). [Google Scholar]
- Grafanaki K, Anastasakis D, Kyriakopoulos G, Skeparnias I, Georgiou S, and Stathopoulos C (2019). Translation regulation in skin cancer from a tRNA point of view. Epigenomics 11, 215–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, and Goodall GJ (2008). The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 10, 593–601. [DOI] [PubMed] [Google Scholar]
- Grzywa TM, Paskal W, and Wlodarski PK (2017). Intratumor and Intertumor Heterogeneity in Melanoma. Transl Oncol 10, 956–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Parker Kerrigan BC, Yang D, Hu L, Shmulevich I, Sood AK, Xue F, and Zhang W (2014). Post-transcriptional regulatory network of epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions. J Hematol Oncol 7, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Yao L, and Jiang Y (2016). A novel integrative approach to identify lncRNAs associated with the survival of melanoma patients. Gene 585, 216–220. [DOI] [PubMed] [Google Scholar]
- Hanniford D, Ulloa-Morales A, Karz A, Berzoti-Coelho MG, Moubarak RS, Sanchez-Sendra B, Kloetgen A, Davalos V, Imig J, Wu P, et al. (2020). Epigenetic Silencing of CDR1as Drives IGF2BP3-Mediated Melanoma Invasion and Metastasis. Cancer Cell 37, 55–70 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, and Kjems J (2013). Natural RNA circles function as efficient microRNA sponges. Nature 495, 384–388. [DOI] [PubMed] [Google Scholar]
- Havel JJ, Chowell D, and Chan TA (2019). The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer 19, 133–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovland AS, Rothstein M, and Simoes-Costa M (2020). Network architecture and regulatory logic in neural crest development. Wiley Interdiscip Rev Syst Biol Med 12, e1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HY, Lin YC, Li J, Huang KY, Shrestha S, Hong HC, Tang Y, Chen YG, Jin CN, Yu Y, et al. (2020). miRTarBase 2020: updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res 48, D148–D154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Ernberg I, and Kauffman S (2009). Cancer attractors: a systems view of tumors from a gene network dynamics and developmental perspective. Semin Cell Dev Biol 20, 869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A, Memczak S, Wyler E, Torti F, Porath HT, Orejuela MR, Piechotta M, Levanon EY, Landthaler M, Dieterich C, et al. (2015). Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep 10, 170–177. [DOI] [PubMed] [Google Scholar]
- Ivanov P, Emara MM, Villen J, Gygi SP, and Anderson P (2011). Angiogenin-induced tRNA fragments inhibit translation initiation. Mol Cell 43, 613–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Q, Wang A, Yuan Y, Zhu B, and Long H (2022). Heterogeneity of the tumor immune microenvironment and its clinical relevance. Exp Hematol Oncol 11, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanemaru H, Fukushima S, Yamashita J, Honda N, Oyama R, Kakimoto A, Masuguchi S, Ishihara T, Inoue Y, Jinnin M, et al. (2011). The circulating microRNA-221 level in patients with malignant melanoma as a new tumor marker. J Dermatol Sci 61, 187–193. [DOI] [PubMed] [Google Scholar]
- Karagkouni D, Paraskevopoulou MD, Chatzopoulos S, Vlachos IS, Tastsoglou S, Kanellos I, Papadimitriou D, Kavakiotis I, Maniou S, Skoufos G, et al. (2018). DIANA-TarBase v8: a decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res 46, D239–D245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karras P, Bordeu I, Pozniak J, Nowosad A, Pazzi C, Van Raemdonck N, Landeloos E, Van Herck Y, Pedri D, Bervoets G, et al. (2022). A cellular hierarchy in melanoma uncouples growth and metastasis. Nature 610, 190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karreth FA, and Pandolfi PP (2013). ceRNA cross-talk in cancer: when ce-bling rivalries go awry. Cancer Discov 3, 1113–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman SA (1969). Metabolic stability and epigenesis in randomly constructed genetic nets. J Theor Biol 22, 437–467. [DOI] [PubMed] [Google Scholar]
- Keung EZ, and Gershenwald JE (2018). The eighth edition American Joint Committee on Cancer (AJCC) melanoma staging system: implications for melanoma treatment and care. Expert Rev Anticancer Ther 18, 775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khorshid M, Hausser J, Zavolan M, and van Nimwegen E (2013). A biophysical miRNA-mRNA interaction model infers canonical and noncanonical targets. Nat Methods 10, 253–255. [DOI] [PubMed] [Google Scholar]
- Khoshnevis S, Dreggors-Walker RE, Marchand V, Motorin Y, and Ghalei H (2022). Ribosomal RNA 2’-O-methylations regulate translation by impacting ribosome dynamics. Proc Natl Acad Sci U S A 119, e2117334119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiuru M, and Busam KJ (2017). The NF1 gene in tumor syndromes and melanoma. Lab Invest 97, 146–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolenda T, Rutkowski P, Michalak M, Kozak K, Guglas K, Rys M, Galus L, Wozniak S, Lugowska I, Gos A, et al. (2019). Plasma lncRNA expression profile as a prognostic tool in BRAF-mutant metastatic melanoma patients treated with BRAF inhibitor. Oncotarget 10, 3879–3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konieczkowski DJ, Johannessen CM, Abudayyeh O, Kim JW, Cooper ZA, Piris A, Frederick DT, Barzily-Rokni M, Straussman R, Haq R, et al. (2014). A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov 4, 816–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpal M, Lee ES, Hu G, and Kang Y (2008). The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem 283, 14910–14914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, and Kjems J (2019). The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet 20, 675–691. [DOI] [PubMed] [Google Scholar]
- Kristensen LS, Hansen TB, Veno MT, and Kjems J (2018). Circular RNAs in cancer: opportunities and challenges in the field. Oncogene 37, 555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landsberg J, Kohlmeyer J, Renn M, Bald T, Rogava M, Cron M, Fatho M, Lennerz V, Wolfel T, Holzel M, et al. (2012). Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 490, 412–416. [DOI] [PubMed] [Google Scholar]
- Leucci E, Vendramin R, Spinazzi M, Laurette P, Fiers M, Wouters J, Radaelli E, Eyckerman S, Leonelli C, Vanderheyden K, et al. (2016). Melanoma addiction to the long non-coding RNA SAMMSON. Nature 531, 518–522. [DOI] [PubMed] [Google Scholar]
- Li Q, Zhang C, Chen R, Xiong H, Qiu F, Liu S, Zhang M, Wang F, Wang Y, Zhou X, et al. (2016). Disrupting MALAT1/miR-200c sponge decreases invasion and migration in endometrioid endometrial carcinoma. Cancer Lett 383, 28–40. [DOI] [PubMed] [Google Scholar]
- Li Y, Zhao H, Lan F, Lee A, Chen L, Lin C, Yao Y, and Li L (2010). Generation of human-induced pluripotent stem cells from gut mesentery-derived cells by ectopic expression of OCT4/SOX2/NANOG. Cell Reprogram 12, 237–247. [DOI] [PubMed] [Google Scholar]
- Li Z, Huang C, Bao C, Chen L, Lin M, Wang X, Zhong G, Yu B, Hu W, Dai L, et al. (2015). Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol 22, 256–264. [DOI] [PubMed] [Google Scholar]
- Liang D, Tatomer DC, Luo Z, Wu H, Yang L, Chen LL, Cherry S, and Wilusz JE (2017). The Output of Protein-Coding Genes Shifts to Circular RNAs When the Pre-mRNA Processing Machinery Is Limiting. Mol Cell 68, 940–954 e943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Wen J, Huang Z, Chen XP, Zhang BX, and Chu L (2019). Small Nucleolar RNAs: Insight Into Their Function in Cancer. Front Oncol 9, 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liau WS, Samaddar S, Banerjee S, and Bredy TW (2021). On the functional relevance of spatiotemporally-specific patterns of experience-dependent long noncoding RNA expression in the brain. RNA Biol 18, 1025–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin N, Chang KY, Li Z, Gates K, Rana ZA, Dang J, Zhang D, Han T, Yang CS, Cunningham TJ, et al. (2014). An evolutionarily conserved long noncoding RNA TUNA controls pluripotency and neural lineage commitment. Mol Cell 53, 1005–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Liu Z, Liu X, and Chen H (2019). Comprehensive Analysis of a Competing Endogenous RNA Network Identifies Seven-lncRNA Signature as a Prognostic Biomarker for Melanoma. Front Oncol 9, 935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Shen SK, Xiong JG, Xu Y, Zhang HQ, Liu HJ, and Lu ZG (2016). Clinical significance of long noncoding RNA SPRY4-IT1 in melanoma patients. FEBS Open Bio 6, 147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Li Y, Xu Q, Yao W, Wu Q, Yuan J, Yan W, Xu T, Ji X, and Ni C (2018). Long non-coding RNA-ATB promotes EMT during silica-induced pulmonary fibrosis by competitively binding miR-200c. Biochim Biophys Acta Mol Basis Dis 1864, 420–431. [DOI] [PubMed] [Google Scholar]
- Loh CY, Chai JY, Tang TF, Wong WF, Sethi G, Shanmugam MK, Chong PP, and Looi CY (2019). The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzi L, Chiu HS, Avila Cobos F, Gross S, Volders PJ, Cannoodt R, Nuytens J, Vanderheyden K, Anckaert J, Lefever S, et al. (2021). The RNA Atlas expands the catalog of human non-coding RNAs. Nat Biotechnol 39, 1453–1465. [DOI] [PubMed] [Google Scholar]
- Lou J, Hao Y, Lin K, Lyu Y, Chen M, Wang H, Zou D, Jiang X, Wang R, Jin D, et al. (2020). Circular RNA CDR1as disrupts the p53/MDM2 complex to inhibit Gliomagenesis. Mol Cancer 19, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, and Li Y (2020). Circ_0079593 facilitates proliferation, metastasis, glucose metabolism and inhibits apoptosis in melanoma by regulating the miR-516b/GRM3 axis. Mol Cell Biochem 475, 227–237. [DOI] [PubMed] [Google Scholar]
- Lu R, Zhang X, Li X, and Wan X (2020). Circ_0016418 promotes melanoma development and glutamine catabolism by regulating the miR-605–5p/GLS axis. Int J Clin Exp Pathol 13, 1791–1801. [PMC free article] [PubMed] [Google Scholar]
- Luan W, Shi Y, Zhou Z, Xia Y, and Wang J (2018). circRNA_0084043 promote malignant melanoma progression via miR-153–3p/Snail axis. Biochem Biophys Res Commun 502, 22–29. [DOI] [PubMed] [Google Scholar]
- Margue C, Philippidou D, Reinsbach SE, Schmitt M, Behrmann I, and Kreis S (2013). New target genes of MITF-induced microRNA-211 contribute to melanoma cell invasion. PLoS One 8, e73473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie KL, Merlino G, and Day CP (2022). The Hitchhiker’s Guide across a Waddington’s landscape of melanoma. Dev Cell 57, 2447–2449. [DOI] [PubMed] [Google Scholar]
- Marie KL, Sassano A, Yang HH, Michalowski AM, Michael HT, Guo T, Tsai YC, Weissman AM, Lee MP, Jenkins LM, et al. (2020). Melanoblast transcriptome analysis reveals pathways promoting melanoma metastasis. Nat Commun 11, 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Bejar O, Rogiers A, Dewaele M, Femel J, Karras P, Pozniak J, Bervoets G, Van Raemdonck N, Pedri D, Swings T, et al. (2021). Evolutionary predictability of genetic versus nongenetic resistance to anticancer drugs in melanoma. Cancer Cell 39, 1135–1149 e1138. [DOI] [PubMed] [Google Scholar]
- Martinez I, Gardiner AS, Board KF, Monzon FA, Edwards RP, and Khan SA (2008). Human papillomavirus type 16 reduces the expression of microRNA-218 in cervical carcinoma cells. Oncogene 27, 2575–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazar J, DeYoung K, Khaitan D, Meister E, Almodovar A, Goydos J, Ray A, and Perera RJ (2010). The regulation of miRNA-211 expression and its role in melanoma cell invasiveness. PLoS One 5, e13779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecozzi N, Vera O, and Karreth FA (2021). Squaring the circle: circRNAs in melanoma. Oncogene 40, 5559–5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melixetian M, Bossi D, Mihailovich M, Punzi S, Barozzi I, Marocchi F, Cuomo A, Bonaldi T, Testa G, Marine JC, et al. (2021). Long non-coding RNA TINCR suppresses metastatic melanoma dissemination by preventing ATF4 translation. EMBO Rep 22, e50852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, et al. (2013). Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495, 333–338. [DOI] [PubMed] [Google Scholar]
- Migault M, Sapkota S, and Bracken CP (2022). Transcriptional and post-transcriptional control of epithelial-mesenchymal plasticity: why so many regulators? Cell Mol Life Sci 79, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihm MC Jr., Clemente CG, and Cascinelli N (1996). Tumor infiltrating lymphocytes in lymph node melanoma metastases: a histopathologic prognostic indicator and an expression of local immune response. Lab Invest 74, 43–47. [PubMed] [Google Scholar]
- Morris KV, and Mattick JS (2014). The rise of regulatory RNA. Nat Rev Genet 15, 423–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mort RL, Jackson IJ, and Patton EE (2015). The melanocyte lineage in development and disease. Development 142, 620–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller DW, Rehli M, and Bosserhoff AK (2009). miRNA expression profiling in melanocytes and melanoma cell lines reveals miRNAs associated with formation and progression of malignant melanoma. J Invest Dermatol 129, 1740–1751. [DOI] [PubMed] [Google Scholar]
- Muller J, Krijgsman O, Tsoi J, Robert L, Hugo W, Song C, Kong X, Possik PA, Cornelissen-Steijger PD, Geukes Foppen MH, et al. (2014). Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat Commun 5, 5712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen MM, and Pedersen JS (2021). miRNA activity inferred from single cell mRNA expression. Sci Rep 11, 9170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien K, Breyne K, Ughetto S, Laurent LC, and Breakefield XO (2020). RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat Rev Mol Cell Biol 21, 585–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olgun G, Gopalan V, and Hannenhalli S (2022). miRSCAPE - inferring miRNA expression from scRNA-seq data. iScience 25, 104962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozbudak EM, Thattai M, Lim HN, Shraiman BI, and Van Oudenaarden A (2004). Multistability in the lactose utilization network of Escherichia coli. Nature 427, 737–740. [DOI] [PubMed] [Google Scholar]
- Pamudurti NR, Bartok O, Jens M, Ashwal-Fluss R, Stottmeister C, Ruhe L, Hanan M, Wyler E, Perez-Hernandez D, Ramberger E, et al. (2017). Translation of CircRNAs. Mol Cell 66, 9–21 e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda AC, Grammatikakis I, Munk R, Gorospe M, and Abdelmohsen K (2017). Emerging roles and context of circular RNAs. Wiley Interdiscip Rev RNA 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panni S, Lovering RC, Porras P, and Orchard S (2020). Non-coding RNA regulatory networks. Biochim Biophys Acta Gene Regul Mech 1863, 194417. [DOI] [PubMed] [Google Scholar]
- Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, Lerou PH, Lensch MW, and Daley GQ (2008a). Reprogramming of human somatic cells to pluripotency with defined factors. Nature 451, 141–146. [DOI] [PubMed] [Google Scholar]
- Park SM, Gaur AB, Lengyel E, and Peter ME (2008b). The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev 22, 894–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Q, Liu L, Pei H, Zhang J, Chen M, and Zhai X (2020). A LHFPL3-AS1/miR-580-3p/STAT3 Feedback Loop Promotes the Malignancy in Melanoma via Activation of JAK2/STAT3 Signaling. Mol Cancer Res 18, 1724–1734. [DOI] [PubMed] [Google Scholar]
- Pisco AO, and Huang S (2015). Non-genetic cancer cell plasticity and therapy-induced stemness in tumour relapse: ‘What does not kill me strengthens me’. Br J Cancer 112, 1725–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian P, Linbo L, Xiaomei Z, and Hui P (2020). Circ_0002770, acting as a competitive endogenous RNA, promotes proliferation and invasion by targeting miR-331-3p in melanoma. Cell Death Dis 11, 264. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Qureshi IA, and Mehler MF (2012). Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nat Rev Neurosci 13, 528–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambow F, Job B, Petit V, Gesbert F, Delmas V, Seberg H, Meurice G, Van Otterloo E, Dessen P, Robert C, et al. (2015). New Functional Signatures for Understanding Melanoma Biology from Tumor Cell Lineage-Specific Analysis. Cell Rep 13, 840–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambow F, Rogiers A, Marin-Bejar O, Aibar S, Femel J, Dewaele M, Karras P, Brown D, Chang YH, Debiec-Rychter M, et al. (2018). Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 174, 843–855 e819. [DOI] [PubMed] [Google Scholar]
- Ranji N, Sadeghizadeh M, Karimipoor M, Shokrgozar MA, Nakhaei Sistani R, and Paylakhi SH (2015). MicroRNAs Signature in IL-2-Induced CD4+ T Cells and Their Potential Targets. Biochem Genet 53, 169–183. [DOI] [PubMed] [Google Scholar]
- Rapino F, Delaunay S, Rambow F, Zhou Z, Tharun L, De Tullio P, Sin O, Shostak K, Schmitz S, Piepers J, et al. (2018). Codon-specific translation reprogramming promotes resistance to targeted therapy. Nature 558, 605–609. [DOI] [PubMed] [Google Scholar]
- Raveh E, Matouk IJ, Gilon M, and Hochberg A (2015). The H19 Long non-coding RNA in cancer initiation, progression and metastasis - a proposed unifying theory. Mol Cancer 14, 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebecca VW, Somasundaram R, and Herlyn M (2020). Pre-clinical modeling of cutaneous melanoma. Nat Commun 11, 2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennie W, Liu C, Carmack CS, Wolenc A, Kanoria S, Lu J, Long D, and Ding Y (2014). STarMir: a web server for prediction of microRNA binding sites. Nucleic Acids Res 42, W114–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rheinheimer BA V. L; Futscher BW; Heimark RL (2020). Identification and transcriptional regulation of the mir-218-1 alternative promoter. bioRxiv. [Google Scholar]
- Riefolo M, Porcellini E, Dika E, Broseghini E, and Ferracin M (2019). Interplay between small and long non-coding RNAs in cutaneous melanoma: a complex jigsaw puzzle with missing pieces. Mol Oncol 13, 74–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riffo-Campos AL, Riquelme I, and Brebi-Mieville P (2016). Tools for Sequence-Based miRNA Target Prediction: What to Choose? Int J Mol Sci 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross IL, Browne CM, and Hume DA (1994). Transcription of individual genes in eukaryotic cells occurs randomly and infrequently. Immunol Cell Biol 72, 177–185. [DOI] [PubMed] [Google Scholar]
- Rutkowski P, Zdzienicki M, Nowecki ZI, and Van Akkooi AC (2010). Surgery of primary melanomas. Cancers (Basel) 2, 824–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez M, Blassberg R, Camacho-Aguilar E, Siggia ED, Rand DA, and Briscoe J (2022). Statistically derived geometrical landscapes capture principles of decision-making dynamics during cell fate transitions. Cell Syst 13, 12–28 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, and Siomi MC (2010). Small RNA-mediated quiescence of transposable elements in animals. Dev Cell 19, 687–697. [DOI] [PubMed] [Google Scholar]
- Schimmel P (2018). The emerging complexity of the tRNA world: mammalian tRNAs beyond protein synthesis. Nat Rev Mol Cell Biol 19, 45–58. [DOI] [PubMed] [Google Scholar]
- Seabra MC, Mules EH, and Hume AN (2002). Rab GTPases, intracellular traffic and disease. Trends Mol Med 8, 23–30. [DOI] [PubMed] [Google Scholar]
- Shain AH, and Bastian BC (2016). From melanocytes to melanomas. Nat Rev Cancer 16, 345–358. [DOI] [PubMed] [Google Scholar]
- Shakhova O (2014). Neural crest stem cells in melanoma development. Curr Opin Oncol 26, 215–221. [DOI] [PubMed] [Google Scholar]
- Simon CS, Hadjantonakis AK, and Schroter C (2018). Making lineage decisions with biological noise: Lessons from the early mouse embryo. Wiley Interdiscip Rev Dev Biol 7, e319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somasundaram R, Villanueva J, and Herlyn M (2012). Intratumoral heterogeneity as a therapy resistance mechanism: role of melanoma subpopulations. Adv Pharmacol 65, 335–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallaert W, Kedziora KM, Chao HX, and Purvis JE (2019). Bistable switches as integrators and actuators during cell cycle progression. FEBS Lett 593, 2805–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L, Zhang W, Su B, and Yu B (2013). Long noncoding RNA HOTAIR is associated with motility, invasion, and metastatic potential of metastatic melanoma. Biomed Res Int 2013, 251098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M, Lieberman J, and Lal A (2010). Desperately seeking microRNA targets. Nat Struct Mol Biol 17, 1169–1174. [DOI] [PubMed] [Google Scholar]
- Tian J, Yang Y, Li MY, and Zhang Y (2020). A novel RNA sequencing-based prognostic nomogram to predict survival for patients with cutaneous melanoma: Clinical trial/experimental study. Medicine (Baltimore) 99, e18868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng YY, Moriarity BS, Gong W, Akiyama R, Tiwari A, Kawakami H, Ronning P, Reuland B, Guenther K, Beadnell TC, et al. (2014). PVT1 dependence in cancer with MYC copy-number increase. Nature 512, 82–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, Wong DJL, Atefi M, Shirazi R, Wang X, et al. (2018). Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 33, 890–904 e895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucci M, Passarelli A, Mannavola F, Felici C, Stucci LS, Cives M, and Silvestris F (2019). Immune System Evasion as Hallmark of Melanoma Progression: The Role of Dendritic Cells. Front Oncol 9, 1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Schuckmann LA, Hughes MCB, Ghiasvand R, Malt M, van der Pols JC, Beesley VL, Khosrotehrani K, Smithers BM, and Green AC (2019). Risk of Melanoma Recurrence After Diagnosis of a High-Risk Primary Tumor. JAMA Dermatol 155, 688–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington CH (1957). The Strategy of the Genes (Routledge; ). [Google Scholar]
- Wang N, Zheng J, Chen Z, Liu Y, Dura B, Kwak M, Xavier-Ferrucio J, Lu YC, Zhang M, Roden C, et al. (2019). Single-cell microRNA-mRNA co-sequencing reveals non-genetic heterogeneity and mechanisms of microRNA regulation. Nat Commun 10, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Chen J, Wang A, Sun L, Qian L, Zhou X, Liu Y, Tang S, Chen X, Cheng Y, et al. (2018). Differentially expressed circRNAs in melanocytes and melanoma cells and their effect on cell proliferation and invasion. Oncol Rep 39, 1813–1824. [DOI] [PubMed] [Google Scholar]
- Wang X (2016). Improving microRNA target prediction by modeling with unambiguously identified microRNA-target pairs from CLIP-ligation studies. Bioinformatics 32, 1316–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner KD, Hajdin CE, and Weeks KM (2018). Principles for targeting RNA with drug-like small molecules. Nat Rev Drug Discov 17, 547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CY, Zhu MX, Lu NH, Liu JQ, Yang YW, Zhang Y, Shi YD, Feng ZH, Li JX, Qi FZ, et al. (2020). Circular RNA circ_0020710 drives tumor progression and immune evasion by regulating the miR-370–3p/CXCL12 axis in melanoma. Mol Cancer 19, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen M, Cong P, Zhang Z, Lu H, and Li T (2018). DeepMirTar: a deep-learning approach for predicting human miRNA targets. Bioinformatics 34, 3781–3787. [DOI] [PubMed] [Google Scholar]
- Wessely A, Steeb T, Berking C, and Heppt MV (2021). How Neural Crest Transcription Factors Contribute to Melanoma Heterogeneity, Cellular Plasticity, and Treatment Resistance. Int J Mol Sci 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RM, and Zon LI (2008). Melanocytes in development, regeneration, and cancer. Cell Stem Cell 3, 242–252. [DOI] [PubMed] [Google Scholar]
- Winkle M, El-Daly SM, Fabbri M, and Calin GA (2021). Noncoding RNA therapeutics - challenges and potential solutions. Nat Rev Drug Discov 20, 629–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf Y, Bartok O, Patkar S, Eli GB, Cohen S, Litchfield K, Levy R, Jimenez-Sanchez A, Trabish S, Lee JS, et al. (2019). UVB-Induced Tumor Heterogeneity Diminishes Immune Response in Melanoma. Cell 179, 219–235 e221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Hu Q, Nie E, Yu T, Wu Y, Zhi T, Jiang K, Shen F, Wang Y, Zhang J, et al. (2017). Hypoxia induces H19 expression through direct and indirect Hif-1alpha activity, promoting oncogenic effects in glioblastoma. Sci Rep 7, 45029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao F, Zuo Z, Cai G, Kang S, Gao X, and Li T (2009). miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res 37, D105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Q, Gao P, Huang X, Chen X, Chen Q, Lv X, Fu Y, Song Y, and Wang Z (2021). tRFTars: predicting the targets of tRNA-derived fragments. J Transl Med 19, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, and Yin A (2019). LINC0638 lncRNA is involved in the local recurrence of melanoma following surgical resection. Oncol Lett 18, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Wang H, Pan H, Shi Y, Li T, Ge S, Jia R, Zhang H, and Fan X (2016). ANRIL lncRNA triggers efficient therapeutic efficacy by reprogramming the aberrant INK4-hub in melanoma. Cancer Lett 381, 41–48. [DOI] [PubMed] [Google Scholar]
- Xu Y, Brenn T, Brown ER, Doherty V, and Melton DW (2012). Differential expression of microRNAs during melanoma progression: miR-200c, miR-205 and miR-211 are downregulated in melanoma and act as tumour suppressors. Br J Cancer 106, 553–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin D, Wei G, Yang F, and Sun X (2021). Circular RNA has circ 0001591 promoted cell proliferation and metastasis of human melanoma via ROCK1/PI3K/AKT by targeting miR-431–5p. Hum Exp Toxicol 40, 310–324. [DOI] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, et al. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920. [DOI] [PubMed] [Google Scholar]
- Yuan JH, Yang F, Wang F, Ma JZ, Guo YJ, Tao QF, Liu F, Pan W, Wang TT, Zhou CC, et al. (2014). A long noncoding RNA activated by TGF-beta promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell 25, 666–681. [DOI] [PubMed] [Google Scholar]
- Zhang J, Tian XJ, Zhang H, Teng Y, Li R, Bai F, Elankumaran S, and Xing J (2014). TGF-beta-induced epithelial-to-mesenchymal transition proceeds through stepwise activation of multiple feedback loops. Sci Signal 7, ra91. [DOI] [PubMed] [Google Scholar]
- Zhang P, Cao L, Zhou R, Yang X, and Wu M (2019a). The lncRNA Neat1 promotes activation of inflammasomes in macrophages. Nat Commun 10, 1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Wu W, Chen Q, and Chen M (2019b). Non-Coding RNAs and their Integrated Networks. J Integr Bioinform 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Xiong X, and Sun Y (2020). Functional characterization of SOX2 as an anticancer target. Signal Transduct Target Ther 5, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhang X, Hu R, and Hao L (2017). Prognostic implication and functional role of long noncoding RNA IGF2AS in human non-small cell lung cancer. J Cell Biochem. [DOI] [PubMed] [Google Scholar]
- Zhao D, Zhuang N, Ding Y, Kang Y, and Shi L (2016). MiR-221 activates the NF-kappaB pathway by targeting A20. Biochem Biophys Res Commun 472, 11–18. [DOI] [PubMed] [Google Scholar]
- Zhao F, Jia Z, Feng Y, Li Z, and Feng J (2021). Circular RNA circ_0079593 enhances malignant melanoma progression by the regulation of the miR-573/ABHD2 axis. J Dermatol Sci 102, 7–15. [DOI] [PubMed] [Google Scholar]
- Zhao L, Yuan Y, Li P, Pan J, Qin J, Liu Y, Zhang Y, Tian F, Yu B, and Zhou S (2018). miR-221-3p Inhibits Schwann Cell Myelination. Neuroscience 379, 239–245. [DOI] [PubMed] [Google Scholar]
- Zheng H, Brennan K, Hernaez M, and Gevaert O (2019). Benchmark of long non-coding RNA quantification for RNA sequencing of cancer samples. Gigascience 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Fu R, Wang JT, Liu Q, Chen H, and Jiang SW (2013). Advances in the techniques for the prediction of microRNA targets. Int J Mol Sci 14, 8179–8187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou WJ, Wang HY, Zhang J, Dai HY, Yao ZX, Zheng Z, Meng-Yan S, and Wu K (2020). NEAT1/miR-200b-3p/SMAD2 axis promotes progression of melanoma. Aging (Albany NY) 12, 22759–22775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang J, Lu Q, Shen B, Huang X, Shen L, Zheng X, Huang R, Yan J, and Guo H (2015). TGFbeta1 secreted by cancer-associated fibroblasts induces epithelial-mesenchymal transition of bladder cancer cells through lncRNA-ZEB2NAT. Sci Rep 5, 11924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Wang SS, Wang J, Su HL, and Xu JH (2019). CircRNA_0016418 expedites the progression of human skin melanoma via miR-625/YY1 axis. Eur Rev Med Pharmacol Sci 23, 10918–10930. [DOI] [PubMed] [Google Scholar]