

Abstract
Purpose
Previous studies from our lab utilized an ultra-high throughput screening method to identify compound 1 as a small molecule that binds to alpha-synuclein (α-synuclein) fibrils. The goal of the current study was to conduct a similarity search of 1 to identify structural analogs having improved in vitro binding properties for this target that could be labeled with radionuclides for both in vitro and in vivo studies for measuring α-synuclein aggregates.
Methods
Using 1 as a lead compound in a similarity search, isoxazole derivative 15 was identified to bind to α-synuclein fibrils with high affinity in competition binding assays. A photocrosslinkable version was used to confirm binding site preference. Derivative 21, the iodo-analog of 15, was synthesized, and subsequently radiolabeled isotopologs [125I]21 and [11C]21 were successfully synthesized for use in in vitro and in vivo studies, respectively. [125I]21 was used in radioligand binding studies in post-mortem Parkinson’s disease (PD) and Alzheimer’s disease (AD) brain homogenates. In vivo imaging of an α-synuclein mouse model and non-human primates was performed with [11C]21.
Results
In silico molecular docking and molecular dynamic simulation studies for a panel of compounds identified through a similarity search, were shown to correlate excellently with Ki values obtained from in vitro binding studies. Improved affinity of isoxazole derivative 15 for α-synuclein binding site 9 was indicated by photocrosslinking studies with CLX10. Design and successful (radio)synthesis of iodo-analog 21 of isoxazole derivative 15 enabled further in vitro and in vivo evaluation. Kd values obtained in vitro with [125I]21 for α-synuclein and Aβ42 fibrils were 0.48 ± 0.08 nM and 2.47 ± 1.30 nM, respectively. [125I]21 showed higher binding in human postmortem PD brain tissue compared with AD tissue, and low binding in control brain tissue. Lastly, in vivo preclinical PET imaging showed elevated retention of [11C]21 in PFF-injected mouse brain. However, in PBS-injected control mouse brain, slow washout of the tracer indicates high non-specific binding. [11C]21 showed high initial brain uptake in a healthy non-human primate, followed by fast washout that may be caused by rapid metabolic rate (21% intact [11C]21 in blood at 5 min p.i.).
Conclusion
Through a relatively simple structure-based similarity search, we identified a new radioligand that binds with high affinity (<10 nM) to α-synuclein fibrils and PD tissue. Although the radioligand has suboptimal selectivity for α-synuclein towards Aβ and high non-specific binding, we show here that a simple in silico approach is a promising strategy to identify novel ligands for target proteins in the CNS with the potential to be radiolabeled for PET neuroimaging studies.
Introduction
Alpha-synuclein (α-synuclein) is a 140 amino acid protein whose function in cell biology is not entirely understood. Post-translational modification and abnormal aggregation of this protein is a key event in a family of neurodegenerative disorders referred to as the “synucleinopathies” [1-3]. The most common synucleinopathy is Parkinson’s disease (PD), which is characterized by the insoluble aggregates of α-synuclein in the form of Lewy bodies (LBs) and Lewy neurites (LNs) [4, 5]. Another synucleinopathy is multiple system atrophy (MSA), which is characterized by α-synuclein aggregates in the form of glial cell inclusions (GCIs) in oligodendrocytes concentrated in the striatum, cerebellum, pons and medulla [6-8]. Since the abnormal aggregation of α-synuclein is believed to play a causal role in PD and MSA, a major effort has been undertaken to develop novel therapeutic strategies aimed at preventing the aggregation of pathological forms of α-synuclein or facilitating their clearance from the brain [9]. For example, there are currently five ongoing clinical trials using humanized monoclonal antibodies raised towards α-synuclein aggregates as a means of clearing aggregated α-synuclein from the CNS in order to delay disease progression [9]. A clinical trial using UCB0599, a small molecule inhibitor of alpha synuclein misfolding, was recently initiated in the UK [10]. Consequently, the development of a noninvasive imaging technique for measuring α-synuclein aggregates would provide a valuable tool for evaluating the efficacy of novel anti-α-synuclein therapies in PD and MSA patients, as well as in pre-clinical research [2, 11-14].
We previously reported the use of a novel, ultra-high throughput in silico screening method to identify a panel of small molecules with high affinity for α-synuclein aggregates [15]. This method involved the generation of a pseudostructure termed an “exemplar” [16] towards the different putative binding sites for small molecules in the solid state NMR of α-synuclein fibrils [15, 17-19]. The most active “hit” in this series was compound 1, which had a high affinity for site 2 (Ser42 to Thr44) and reduced affinity for site 9 (Gly86 to Lys96) of the α-synuclein structure (Figure 1). Although site 2 is present in recombinant fibrils of α-synuclein [17], we had some concerns regarding its utility as a target for radioligand binding studies in postmortem samples of aggregated α-synuclein, since previous studies have shown a number of post-translational modifications (PTMs) near this region of α-synuclein [3, 20]. These modifications could lead to a reduction in binding affinity in going from recombinant fibrils to α-synuclein aggregates in postmortem samples of PD and MSA brain [20-23]. In particular, Tyr39 phosphorylation has been observed in α-synuclein from PD and MSA patients as well as healthy controls, and previous structural and biophysical studies have shown that this can influence aggregation by altering fibril conformation [24-27]. Since there are no PTMs located near site 9 (Figure 1) [3, 20], we focused on this binding site for radioligand development.
Figure 1.

Structure of compound 1, the “hit” from the ultra-high throughput in silico screen and the binding sites on α-synuclein fibrils. The solid state NMR structure of α-synuclein is shown (PDB ID 2n0a) with site 2 and site 9 indicated.[53] The positions of PTMs near sites 2 and 9 are shown on the sequence diagram at top. No PTMs are known to occur from I88 to V95.
As part of this ongoing research project, we conducted a similarity search of 1 as a means of finding structural congeners with improved binding properties to site 9 over 1, preferably a compound that could be radiolabeled with both a positron emitter for in vivo imaging studies and also iodine-125, a gamma emitter, for in vitro binding studies. In this communication, we report on the progress of in silico ligand discovery methods to identify 15, a novel, high affinity α-synuclein ligand and an iodo-analog 21 that can be radiolabeled with either iodine-125 or carbon-11.
Materials and Methods
In silico compound selection.
In an effort to improve the binding properties of 1 for site 9, a similarity search of compound 1 was executed on the Mcule, Inc. website (https://mcule.com/search/), in which the similarity threshold ranged from >0.8 to >0.6, as previously described by Ferrie et al [15]. The search identified compounds 2-20 (Table 1), which were subsequently ordered from Mcule, Inc. or ChemDiv, Inc. All compounds were subjected to molecular docking and in vitro screening assays as described below.
Table 1.
Docking results and in vitro screening assay testing %Inhibition against [3H]BF-2846 on α-synuclein fibrils
| Compound # | Structure | Site 9 Binding | % Inhibition [3H]BF-2846 |
|
|---|---|---|---|---|
| Probability (%) | Best Binding Energy (kcal/mol) | |||
| 2 |
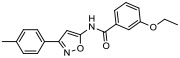
|
23.20% | −7.31 | 13% |
| 3 |
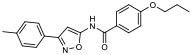
|
29.30% | −7.49 | 39% |
| 4 |
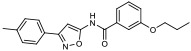
|
19.70% | −7.36 | <0% |
| 5 |
|
29.30% | −7.29 | 4% |
| 6 |
|
30.20% | −7.09 | 20% |
| 7 |
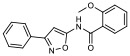
|
29.00% | −6.98 | <0% |
| 8 |
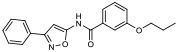
|
19.60% | −7.22 | <0% |
| 9 |
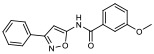
|
28.40% | −6.88 | <0% |
| 10 |
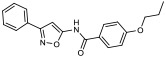
|
33.20% | −7.37 | 22% |
| 11 |
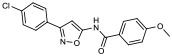
|
29.70% | −7.16 | 56% |
| 12 |
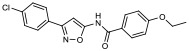
|
29.90% | −7.54 | 69% |
| 13 |
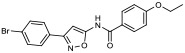
|
28.20% | −7.59 | 14% |
| 14 |

|
22.80% | −7.66 | 70% |
| 15 |
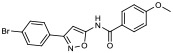
|
29.70% | −7.40 | 80% |
| 16 |
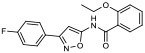
|
20.50% | −6.95 | <0% |
| 17 |
|
29.70% | −6.69 | 46% |
| 18 |
|
16.50% | −6.62 | <0% |
| 19 |
|
28.30% | −6.84 | <0% |
| 20 |
|
21.30% | −7.08 | <0% |
| 21 |
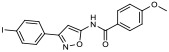
|
27.60% | −7.64 | − |
Molecular Docking.
In silico molecular docking studies were performed following a previously published protocol [18]. Briefly, all compound structures were drawn in ChemDraw Professional 15.1 (PerkinElmer Informatics, Inc.) and imported in Chem3D Ultra 15.1 (PerkinElmer Informatics, Inc.) to calculate individual minimum energy structures of compounds 2-21 by MMFF94 force field in preparation for molecular docking. Molecular blind docking studies were performed via the AutoDock 4.2 [28] plug-in on PyMOL (pymol.org). The solid-state NMR structure of a full-length α-synuclein fibril (PDB ID: 2N0A) was obtained from the RCSB protein data bank (https://www.rcsb.org/) as a target protein for blind docking. The 2N0A structure was chosen as it was the only α-synuclein fibril structure available when the work was undertaken and is still representative of in vitro fibrils used in initial compound screening. Although many cryo-EM structures are now available and differ in aspects like the number of strands and their packing, we have shown that the local folds of the protein remain similar in the sites of interest [15]. Thus, we continue to use it for docking analysis. Non-polar hydrogens were removed from both protein and compound structures. A grid box with a dimension of 95 × 50 × 95 Å3 was applied to the α-synuclein fibril structure. The Lamarckian Genetic Algorithm with a maximum of 2,500,000 energy evaluations was used to calculate 1,000 protein-ligand binding poses for each compound. The % probability and predicted best binding energy of each binding site that was determined from blind docking are reported in Table 1.
Molecular Dynamics.
In order to investigate the interaction between compounds and site 9 of the α-synuclein fibril, molecular dynamics simulations (MDS) were performed using NAMD 2.12 [29] with CHARMM22 force field, and analyzed using the visualization program VMD [30]. The binding pose of compounds in site 9 with the best binding energy was used to perform MDS. The disorder N-terminus and C-terminus were truncated from α-synuclein fibril structure, and the core from residue Lys32 to Lys102 was used for further MDS. The ligand-protein complex was solvated in a TIP3P water box with a minimized volume of 86 × 77 × 60 Å3, and then 40 sodium ions were randomly added to the solvated system for charge neutralization. Periodic boundary conditions were used. A cut-off of 12 Å was set to non-bonded interactions, and long-range electrostatic interactions were calculated using the Particle Mesh Ewald (PME) method. The SHAKE algorithm was used to constrain bonds involving hydrogen atoms. A 50,000 steps of energy minimization was implemented. Then, the minimized system was heated in NVT (constant temperature and constant volume) ensemble with constant volume at 310 K for 100 picoseconds (ps) with a time step of 1 femtosecond (fs). The system was then equilibrated in NPT (constant temperature and constant pressure) ensemble at 310 K and 1 atm for 200 ps with a 1 fs time step. The Langevin dynamics and Langevin piston method were used to maintain the pressure at 1 atm and temperature at 310 K. A 5 nanosecond production run was performed in a NPT ensemble with a time step of 1 fs. The trajectories were saved every 0.5 ps.
Compound screening.
In order to screen compounds 2-20 for α-synuclein binding, 100 nM of each compound was incubated for 1 hour at 37 °C with 100 nM α-synuclein fibrils and [3H]BF2846 (3 nM) in 50 mM Tris-HCl, pH 7.4. α-synuclein fibrils were prepared as described previously [19]. Briefly, human α-synuclein with a C-terminal intein fusion was transformed into Escherichia coli BL21(DE3). Following induced protein expression, cells were lysed and α-synuclein protein was purified. Purity was confirmed by SDS-PAGE and protein concentration was determined by measuring the absorbance at 280 nm and using the calculated (ExPASy) extinction coefficient of 5960 M−1cm−1. Total binding was measured in the absence of competitor and nonspecific binding was determined in reactions containing unlabeled BF2846 (0.5 μM). After incubation, bound and free radioligand were separated by vacuum filtration through Whatman GF/C filters (Brandel, Gaithersburg, MD, USA) in a 24-sample harvester system (Brandel), followed by washing with buffer containing 10 mM Tris-HCl (pH 7.4) and 150 mM NaCl. Filters containing the bound ligand were mixed with 3 mL of scintillation cocktail (MicroScint-20, PerkinElmer, Shelton, CT, USA) and counted after 12 hours of incubation on a MicroBeta System (PerkinElmer). All data points were performed in triplicates. Percentage of bound radioligand relative to total binding was plotted and data was analyzed by One-Way ANOVA, comparing the mean of each data set to the mean of total binding.
Compounds displaying a percent inhibition >40% were submitted for full competition binding assays. Compound 6 showed a percent inhibition of only 20% and was expected to have a low affinity for site 9, and was therefore included in the binding assay as a “negative” control.
Competition binding assay.
α-synuclein, Aβ42, and Aβ40 fibrils were prepared as described in the prior studies [19, 31, 32]. α-synuclein fibrils (50 nM) or Aβ42 fibrils (100 nM) were mixed with site 9 ligand [3H]BF2846 (3 nM) and varying concentrations of unlabeled compounds 2-20. Compounds were diluted in 50 mM Tris-HCl (pH 7.4) and mixed with fibrils and radioligand in a total volume of 150 μL. Total binding was measured in the absence of competitor and non-specific binding was determined in reactions containing unlabeled BF2846 (0.5 μM). In a duplicate set of binding reactions, fibrils were replaced with equal volume of buffer to measure the amount of radioligand binding to the filter paper. Reactions were incubated at 37 °C for 1 hour. After incubation bound and free radioligand were separated by vacuum filtration through Whatman GF/C filters (Brandel) in a 24-sample harvester system (Brandel), followed by washing with buffer containing 10 mM Tris-HCl (pH 7.4) and 150 mM NaCl. Filters containing the bound ligand were mixed with 3 mL of scintillation cocktail (MicroScint-20, PerkinElmer; Waltham, MA, USA) and counted after 12 hours of incubation on a MicroBeta System (PerkinElmer). Counting of all samples was performed in triplicate and mean values computed for radioligand binding analyses. Ki values were calculated by fitting the data to the equation below by nonlinear regression, using GraphPad Prism software (version 8.3.0; GraphPad Software LLC, San Diego, CA, USA):
Where logIC50 is the log of the concentration of competitor that results in binding half-way between Bottom and Top; logKi is the log of the molar equilibrium dissociation constant of unlabeled ligand; [radioligand] is the concentration of radiolabeled ligand in nM; Kd is the equilibrium dissociation constant of the radioligand in nM; Top and Bottom are plateaus in the units of Y axis (i.e., counts per min).
Intact Protein Crosslinking and Mass Spectrometry Analysis.
Covalent photo-crosslinking of small molecule ligands generates a covalent bind between the ligand and the protein target, enabling the precise identification of binding sites via subsequent bottom-up tandem MS experiments. In order to obtain this information fibrils were incubated with photocrosslinker at 37 °C and irradiated with 365 nm light in a 1.5 mL tube. After irradiation, 50 μL of 250 mM aqueous SDS solution (250 mM) was added, for a final SDS concentration of 125 mM. Samples were then incubated at 100 °C for 30 min. Resulting monomeric crosslinked α-synuclein samples were subjected to a chloroform/methanol/water precipitation (4:3:1), as previously described [15, 33]. The resulting protein pellet was then re-solubilized in 25 μL H2O, 0.1%TFA. The sample was prepared for MALDI-TOF by spotting 1 μL of analyte in 1 μL of sinapic acid matrix (saturated solution in 50:50 CH3CN:H2O, 0.1% TFA) on a ground steel MALDI plate. The sample was then analyzed using linear positive mode with scanning between 5 and 20 kDa at 23% laser power on a Bruker UltraFlex III MALDI-TOF-TOF instrument (Billerica, MA, USA).
For binding site determination, crosslinked monomeric α-synuclein was re-solubilized in 100 μL ammonium bicarbonate buffer (50 mM NH4HCO3, pH 7.4) and digested overnight at 37 °C via the addition of 1 μg of trypsin (Promega; Madison, WI, USA). Digestion was quenched by adding 1.0 μL of formic acid, and the resulting peptide mixture was cleaned up using custom C18 stage tips [15, 33]. The eluted mixture from the stage tip was then concentrated via Speedvac (Savant), and re-solubilized in 20 μL of H2O with 0.1% TFA. 2 μL of this sample was then analyzed directly via Orbitrap mass spectrometry on a Thermo QE-HF instrument. The resulting data was searched using Proteome Discoverer with the mass of the photocrosslinker included as a variable modification on all residues. These data are summarized in Figure 2 (see MS/MS Data Acquisition and Analysis section in Supporting Information for details).
Figure 2.

Structure of photocrosslinker CLX10 and sites of α-synuclein fibril crosslinking. Crosslinked peptides from trypsin digest shown colored in red on the sequence and on the 2n0a fibril structure.[53] Crosslinked residues indicated with * in the sequence and shown as spheres in the structure. Tyr39 is also shown as spheres.
General chemistry
Chemicals were obtained from commercial sources and used without further purification. Solvents were purchased from commercial sources and used as received unless stated otherwise. Reactions were performed at room temperature unless stated otherwise. Reactions were monitored by thin layer chromatography (TLC) on pre-coated silica 60 F254 aluminum plates (MilliporeSigma, Burlington, MA, USA), spots were visualized by UV light. Evaporation of solvents was performed under reduced pressure at 40 °C using a rotary evaporator. Flash column chromatography was performed on a Biotage® (Charlotte, NC, USA) Isolera One system equipped with Biotage® SNAP KP-Sil cartridges. Nuclear magnetic resonance (NMR) spectroscopy was performed on a Bruker (Billerica, MA, USA) DMX 500 (500 MHz for 1H and 125 MHz for 13C) a Bruker Avance Neo 400 (400 MHz for 1H and 101 MHz for 13C) with chemical shifts (δ) reported in parts per million (ppm) relative to the solvent (CDCl3, 1H 7.26 ppm, 13C 77.16 ppm; dimethyl sulfoxide (DMSO)-d6, 1H 2.50 ppm, 13C 39.52 ppm). Liquid Chromatography Mass Spectrometry (LCMS) was carried out using a Waters SQD equipped with an Acquity UPLC instrument in positive ion mode. High resolution LCMS (HRMS) for accurate mass measurement was carried out using a Waters LCT Premier XE instrument in positive ion mode.
N-(3-(4-bromophenyl)isoxazol-5-yl)-4-methoxybenzamide (15).
A solution of 4-methoxybenzoyl chloride (285 mg, 1.7 mmol) in anhydrous CH2Cl2 (2 mL) was added dropwise to a solution of 3-(4-bromophenyl)isoxazol-5-amine (200 mg, 0.8 mmol) and triethylamine (0.12 mL, 1.7 mmol) in anhydrous CH2Cl2 (2 mL) at 0 °C. After warming to rt and stirring for 48 h, the reaction mixture was diluted with CH2Cl2 (30 mL), poured into water (15 mL) and layers were separated. The water layer was extracted once more with CH2Cl2 (20 mL) and combined organic layers were dried over Na2SO4 and concentrated in vacuo. Flash column chromatography (gradient of 25-98% EtOAc in hexanes) followed by recrystallization from EtOAc/hexanes yielded 15 (58 mg, 19%). 1H NMR (400 MHz, DMSO-d6) δ 11.95 (s, 1H), 8.06 (d, J = 8.7 Hz, 2H), 7.86 (d, J = 8.4 Hz, 2H), 7.73 (d, J = 8.4 Hz, 2H), 7.10 (d, J = 8.7 Hz, 2H), 6.93 (s, 1H), 3.86 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 163.5, 162.2, 132.6, 130.7, 129.0, 124.9, 124.1, 115.2, 114.4, 100.0, 87.1, 56.0; LRMS (ESI) m/z calcd. for C17H14BrN2O3+ [M+H]+, 373.0; found: 373.4.
N-(3-(4-iodophenyl)isoxazol-5-yl)-4-methoxybenzamide (21).
A solution of 3-(4-iodophenyl)isoxazol-5-amine (178 mg, 0.6 mmol), 4-dimethylaminopyridine (DMAP; 49 mg, 0.4 mmol) and triethylamine (0.5 mL, 3.6 mmol) in anhydrous CH2Cl2 (6 mL) was stirred for 10 min. 4-methoxybenzoyl chloride (60 mg, 0.35 mmol) was added in one portion and the reaction mixture was stirred for 20 h. The reaction mixture was diluted with CH2Cl2 (30 mL), poured into water (15 mL) and layers were separated. The water layer was extracted with CH2Cl2 (20 mL) and combined organic layers were washed with 0.5 M NaHCO3, dried over Na2SO4 and concentrated in vacuo. Flash column chromatography (20-30% EtOAc in hexanes) yielded 21 (85 mg, 33% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 11.94 (s, 1H), 8.06 (d, J = 8.5 Hz, 2H), 7.89 (d, J = 8.0 Hz, 2H), 7.69 (d, J = 8.0 Hz, 2H), 7.10 (d, J = 8.5 Hz, 2H), 6.91 (s, 1H), 3.85 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 163.0, 162.8, 161.9, 137.9, 130.2, 128.4, 128.3, 124.4, 113.9, 97.1, 86.6, 55.5; LRMS (ESI) m/z calcd. for C17H14IN2O3+[M+H]+, 421.0; found: 421.3 [M+H]+; HRMS (ESI) m/z calcd. for C17H14IN2O3+[M+H]+, 421.0044; found 421.0056.
4-methoxy-N-(3-(4-(tributylstannyl)phenyl)isoxazol-5-yl)benzamide (23).
A solution of 15 (50 mg, 0.135 mmol) was dissolved in toluene (5 mL). Bis(tributyl)tin (0.1 mL) was added, followed by the addition of XPhos Pd G2 (5 mg). The mixture was stirred in a sealed vessel charged with nitrogen at 80 °C for 1 h. Volatiles were removed in vacuo and the mixture was purified by flash chromatography (hexanes/ethanol, 0-30%) to yield 23 as a light-yellow solid (30 mg, 39%). 1H NMR (500 MHz, CDCl3) δ 8.78 (s, 1H), 7.90 (d, J = 8.8 Hz, 2H), 7.77 (d, J = 7.9 Hz, 2H), 7.56 (d, J = 7.9 Hz, 2H), 6.99 (d, J = 8.8 Hz, 2H), 6.86 (s, 1H), 3.88 (s, 3H), 1.66 – 1.53 (m, 6H), 1.38 – 1.25 (m, 8H), 1.10 – 1.09 (m, 4H), 0.94 – 0.89 (m, 9H); 13C NMR (125 MHz, CDCl3) δ 164.1, 163.4, 162.4, 160.8, 145.4, 136.9, 129.4, 128.5, 125.9, 124.4, 114.3, 87.0, 55.5, 29.1, 29.0, 27.9, 27.8, 27.6, 27.3, 27.1, 26.8, 17.5, 13.7, 13.6, 9.6; LRMS (ESI) m/z calcd. for C17H15N2O3+[M – Bu3Sn + H + H]+, 295.1; found: 295.3.
4-hydroxy-N-(3-(4-iodophenyl)isoxazol-5-yl)benzamide (24).
To a suspension of 3-(4-iodophenyl)-1,2-oxazole-5-amine 200 mg, 0.67 mmol) and 4-acetoxy-benzoyl chloride (280 mg, 1.4 mmol) in dichloromethane (3 mL) was added trimethylamine (1.2 eq.) and the mixture was stirred at ambient temperature for 20 h. Volatiles were removed in vacuo and the mixture was purified by flash chromatography (hexanes/ethyl acetate, 0-60%) to yield a white solid (85 mg, 33% yield) as a white solid.1H NMR (400 MHz, DMSO-d6) δ 8.09 (d, J = 8.7 Hz, 2H), 7.89 (d, J = 8.4 Hz, 2H), 7.70 (d, J = 8.4 Hz, 2H), 7.33 (d, J = 8.7 Hz, 2H), 6.95 (s, 1H), 2.31 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 169.4, 163.5, 163.3, 162.5, 138.4, 130.5, 130.3, 128.9, 128.7, 122.6, 97.6, 87.4, 21.4; LRMS (ESI) m/z calcd. for C18H14IN2O4+ [M+H]+, 449.0; found, 449.4.
The product above (80 mg, 0.18 mmol) was dissolved in methanol (2 mL) and aqueous sodium hydroxide (2N; 2.5 mL) was added. The mixture was stirred at 60 °C for 3 h, then cooled to 0 °C (ice bath) and acidified with aqueous HCl (6N). The white solid was filtered out, dried on vacuum. The target compound was obtained as a white solid (31 mg, 43%) and used in the radiolabeling reactions.1H NMR (400 MHz, DMSO-d6) δ 11.82 (s, 1H), 10.30 (s, 1H), 7.94 (d, J = 8.7 Hz, 2H), 7.88 (d, J = 8.4 Hz, 2H), 7.68 (d, J = 8.4 Hz, 2H), 6.90 – 6.88 (m, 3H). 13C NMR (101 MHz, DMSO-d6) δ 163.1, 163.1, 161.9, 161.6, 137.9, 130.4, 128.4, 128.4, 122.9, 115.2, 97.0, 86.4; LRMS (ESI) m/z calcd. for C16H12IN2O3+ [M+H]+, 407.0; found, 407.4.
Radiochemistry
Synthesis of [125I]21.
70 MBq (1.9 mCi) of [125I]NaI solution in 0.1 M NaOH (PerkinElmer), was added to 50 μL of a solution of stannyl precursor 23 (2.0 mg/mL solution in MeOH), directly followed by 50 μL of a pre-mixed solution of H2O2 (30%)/AcOH (1:3). The reaction mixture was vortexed and left to react at room temperature for 30 min, while shortly vortexed every 5 min. The reaction mixture was then diluted with HPLC mobile phase (4 mL, MeCN/50 mM NH4CO2 (pH 4.5) (70:30 v/v)) and subjected to preparative HPLC on an Agilent (Santa Clara, CA, USA) 1200 Series HPLC system equipped with a Luna C18 (5 μm, 100 Å, 250x10 mm) column (Phenomenex, Torrance, CA, USA) and Eckert & Ziegler (Hopkinton, MA, USA) FC-3300 radioactivity detector using the following gradient: 0-10 min 100% 0.1 M NH4CO2 (pH 4.5); 10-35 min MeCN/0.1 M NH4CO2 (pH 4.5) (70:30, v/v); 35-60 min 100% MeCN (to elute unreacted precursor 23). The collected product fraction (tR = 25 min, 10 mL collected) was diluted with H2O (40 mL) and trapped on a solid phase extraction (SPE) cartridge (SepPak tC18 Light, Waters, Milford, MA, USA) pre-conditioned with 1 mL of EtOH and 10 mL of H2O. The SPE cartridge was washed with H2O (10 mL) and [125I]21 (35-48 MBq, 0.96-1.31 mCi, 64 ± 12% radiochemical yield, n=3) was eluted with ethanol (300 μL) for use in in vitro studies. Identity of the radiolabeled product was confirmed by co-injection of [125I]21 and unlabeled 21 on analytical HPLC using an Agilent Eclipse XDB C18 5 μm, 4.6 x 150 mm column, with MeCN/0.1 M NH4CO2 (pH 4.5) (60:40 v/v) as mobile phase (tR = 10 min). The radiochemical purity of [125I]21 was >99% (n=3) with a molar activity of 80 GBq·μmol−1.
Synthesis of [11C]21.
[11C]CO2 was produced by a 14N(p,α)11C nuclear reaction performed in a 0.5% O2/N2 gas mixture using an IBA Cyclone 18/9 cyclotron (IBA, Louvain-la-Neuve, Belgium). Subsequently, [11C]CO2 was transferred to a commercial Synthra MeI Plus module (Synthra GmbH, Hamburg, Germany) and reacted with H2 at 425 °C to form [11C]CH4 that was trapped at −150 °C. [11C]CH4 was then released and recirculated through a high temperature oven in the presence of sublimed I2 (720-730 °C), and formed [11C]CH3I was trapped on a Porapak Q filled column. [11C]CH3I was then released by heating the Porapak column to 200 °C and subsequently trapped in a reaction vial containing a solution of precursor 24 (0.5 mg, 1.3 μmol) and K2CO3 (5 mg, 36 μmol) in 0.25 mL of anhydrous DMSO. The reaction mixture was heated at 70 °C for 5 min, after which it was diluted with 1.3 mL of mobile phase and purified by HPLC on a Zorbax SB-C18 5 μm (4.6 x 100 mm) column (Agilent Technologies, Inc.) using 100 mM NH4COOH (pH 4.5)/acetonitrile (42:58, v/v) as mobile phase at a flow rate of 3 mL·min−1. The fraction containing [11C]21 (tR = 10 min) was collected, diluted with 35 mL of water and trapped on a SepPak tC18 Plus Light cartridge (Water, Milford, MA, USA), preconditioned with 1 mL of EtOH and 10 mL of water. The cartridge was washed with 10 mL of water. For mouse studies, [11C]21 was eluted with 0.5 mL EtOH and 4.5 mL of saline. For non-human primate scans, [11C]21 was eluted with 0.7 mL of EtOH and immediately pushed over a sterile filter (Millex-FG, 0.20 μm, hydrophobic PTFE, 25 mm; MilliporeSigma, Burlington, MA, USA), followed by 9 mL of saline. Occasional filter blockage was overcome by pushing over an extra 0.2 mL of EtOH. [11C]21 was obtained with a decay corrected radiochemical yield (RCY) of 12 ± 2% (n=4), calculated from [11C]CO2 at end of bombardment (EOB), with a radiochemical purity > 90%, a molar activity (Am) of 344 ± 235 GBq·μmol−1 (n=4) at end of synthesis (EOS) and an overall synthesis time of 45-50 min. The identity of the product was confirmed by co-injection of [11C]21 and unlabeled 21 on analytical UPLC (using a Waters BEH C18 x 1.7μm (2.1 x 50 mm) column (Waters, Ireland) with mobile phase of MeCN/H2O (60/40 v/v) at a flow rate of 0.5 mL·min−1 (tR =1.23 min).
Saturation binding assay of [125I]21.
To measure the binding affinity (Kd) of compound 21, saturation binding assays were performed using synthetic α-synuclein (50 nM) or Aβ42 (100 nM) fibrils. Solutions containing fibrils were incubated for 1 hour at 37 °C with increasing concentrations of [125I]21 in 50 mM Tris-HCl, pH 7.4, in a total volume of 150 μL. Non-specific binding was determined in a duplicate set of binding reactions containing 2 μM unlabeled 21. To measure the amount of radioligand binding to the filter paper, fibrils were replaced with an equal volume of buffer in a duplicate set of binding reactions. After incubation, bound and free radioligand were separated by vacuum filtration through Whatman GF/C filters (Brandel) in a 24-sample harvester system (Brandel), followed by washing with buffer containing 10 mM Tris-HCl (pH 7.4) and 150 mM NaCl. Filters containing the bound ligand were counted immediately on a 2470 WIZARD Automatic Gamma Counter (Perkin Elmer). All assays were performed in triplicate and mean values computed for radioligand binding analyses. The equilibrium dissociation constant (Kd) and the maximal number of binding sites (Bmax) were determined by fitting the data to the equation Y = Bmax*X/(Kd+X), using GraphPad Prism software.
Radioligand binding assays of [125I]21 in postmortem brain samples, α-synuclein fibrils, and Aβ40 fibrils.
Fresh-frozen, autopsy-confirmed brain tissues were obtained from the University of Pittsburgh Alzheimer Disease Research Center Brain Bank. Samples of cortical gray matter were dissected from 1-cm-thick coronal slices taken at autopsy and frozen at −80°C or fixed in formalin for extensive histological and immunohistochemistry examinations using established AD and PD neuropathological criteria to ensure the absence of potentially confounding co-pathologies [34, 35]. The postmortem AD brain tissue was taken from the medial frontal cortex (MFC) of an 84 year-old male subject with end stage AD and provided the following neuropathology scores: ABC=3,3,3; Braak neurofibrillary tangle (NFT) stage=6; MFC NFT score=3; MFC neuritic plaque (NP) score=3; Lewy body (LB) score=0; TDP-43=negative. The postmortem PD brain tissue was taken from the MFC of an 55 year-old male subject with extensive Lewy body pathology and provided the following neuropathology scores: NIA-RI diagnosis=low; Braak NFT stage=1; Braak PD stage=6; LB score=10; MFC LB score=3; MFC NFT score=0; MFC NP score=0; MFC TDP-43=negative. The young control brain was taken from the MFC of a 32-year-old male accident victim, and neuropathological examination demonstrated no detectable aggregated amyloid or TDP-43 pathologies. The frozen brain tissue samples were subsequently thawed and homogenized in ice-cold sodium phosphate buffered saline (PBS; pH 7.0) at 300 mg/mL using a glass homogenizer and then diluted 30-fold with PBS to 10 mg/mL and homogenized a second time with a Brinkmann Polytron homogenizer before storage at −80°C until used for binding assays. Aβ40 and α-synuclein fibrils were prepared as described previously [32, 36].
Binding of [125I]21 to PD, AD, control brain, and Aβ40 synthetic fibrils to determine the displaceable amount at a single concentration of [125I]21 (0.0679 nM; Figure 4) was performed as described previously [37]. Briefly, frozen brain aliquots were thawed and diluted in PBS to 1 mg/mL. Unlabeled 21 was dissolved in DMSO at 400 μM (to yield <1% DMSO in the final assay). A small aliquot of no carrier-added [125I]21 (80 GBq·μmol−1) in ethanol containing ~330,000 cpm was diluted to a volume of 900 μL of tris buffer (pH 7.2) containing 5% ethanol and 0.1% bovine serum albumin (BSA) with or without the addition of 1 μM unlabeled 21. Then, 100 μL of the 1 mg/mL brain homogenate was added to achieve a final concentration of 100 μg tissue per mL or a concentration of 200 nM synthetic fibrils. After incubation for 60 min at room temperature, the binding mixture was filtered through a Whatman GF/B glass filter via a Brandel M-24R cell harvester (Gaithersburg, MD) and rapidly washed four times with 3 mL of the tris buffer/5% ethanol/0.1% BSA solution. The filters were counted in Cytoscint-ES after thorough vortexing and sitting overnight. Results were corrected for non-specific, non-displaceable binding in the presence of 1 μM unlabeled 21 and expressed as picomoles of [125I]21 bound per gram of wet tissue weight, or picomoles of [125I]21 bound per 100 nmol of synthetic fibrils in the homogenate. All assays were performed in triplicate.
Figure 4.

Comparison of [125I]21 binding to homogenates from Parkinson's disease (PD) brain, No tissue (filter only), Alzheimer's disease (AD) brain, young control brain, synthetic α-synuclein fibrils, and synthetic Aβ40 fibrils. All assays were performed with [125I]21 at a concentration of 0.07 nM. Bars represent the mean of three independent assays, and error bars represent the standard deviation. The specific binding of [125I]21 was significantly higher in PD tissue than in young control tissue (p<0.01), while specific binding in AD tissue and No Tissue were not significantly different than young control tissue (p=0.15 and p=0.88, respectively), using Welch’s T-test (two-tailed).
The Kd and Bmax values of [125I]21 binding to PD brain homogenates were similarly determined using a Scatchard plot as described previously [18], with concentrations of unlabeled 21 competitor ranging from 1 nM to 1000 nM and a constant radioligand concentration of [125I]21 (~0.07 nM). Results were corrected for non-specific, non-displaceable binding in the presence of 1 μM 21, and all assays were performed in triplicate.
Animals.
Synthetic α-synuclein preformed fibrils (PFFs, 5 μg) injected (n=1) and sham control PBS injected (n=2) male A53T mice were obtained from Dr. Luk’s laboratory at the Department of Pathology and Laboratory Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA. The procedures of PFF and PBS injection followed the prior published protocols [38]. All animal studies were performed under protocols approved by the University of Pennsylvania Institutional Animal Care and Use Committee.
Immunohistochemistry.
Paraffin-embedded sections (6 um) containing mouse brain tissue were rehydrated by immersion in xylene, ethanol (100%, 95%, 70%, 50%) followed by deionized water. After blocking (3% normal goat serum, 3% bovine serum albumin, 0.3% Triton X-100 in PBS), sections were incubated overnight with 81A (1:8,000), washed and incubated with biotinylated anti-mouse IgG (Vector Laboratories). Signal was visualized using the ABC kit and 3, 3 -diaminobenzidine (Vector Laboratories) as per manufacturer’s instructions and counterstained with hematoxylin. Digitization was performed using a Perkin-Elmer Lamina P250 scanner.
Preclinical PET imaging
PET imaging in rodents.
Rodent PET imaging was performed in PFF-injected (n=1), and PBS-injected sham (n=2) using the β-Cube preclinical PET scanner (Molecubes, Ghent, Belgium). Mice were anesthetized with 1-2% isoflurane, a tail vein catheter was placed for PET tracer administration, and the animal was placed on the scanner bed. Dynamic scans of 0-60 min post-injection were acquired after injection of 7.8-9.9 MBq of [11C]21. After completion of the PET scan, the animal was transferred to the X-Cube CT scanner (Molecubes, Ghent, Belgium) and a low resolution CT scan was acquired for anatomical reference and attenuation correction. PET images were reconstructed with a matrix size of 192 x 192 x 384, and a voxel size of 0.4 x 0.4 x 0.4 mm with frame lengths of 6 x 10 sec, 9 x 60 sec, and 10 x 300 sec for 60 minutes dynamic scans. All corrections were applied using a manufacturer supplied reconstruction program. CT images were reconstructed with a matrix size of 200 x 200 x 550, and a voxel size of 0.2 x 0.2 x 0.2 mm with a manufacturer supplied reconstruction program.
All [11C]21 PET and CT imaging data in rodents were processed and analyzed by using PMOD software (version 3.7, PMOD Technologies Ltd., Zurich, Switzerland). Rigid body matching was manually performed on individual micro-CT images to co-register to the Mirrione mouse MR-T2 weighted brain template [39]. Then, the resulting transformation parameters were applied to the corresponding micro-PET image. Two volumes of interest (VOIs) including cortex and brain stem were selected from the Mirrione atlas [39]. Time activity curves were extracted from each VOI in kBq/cm3 and standard uptake value (SUV) were calculated by normalizing kBq/cm3 to body weight (g).
PET imaging in non-human primates.
Male rhesus macaques (21 years old) were sedated with ketamine/dexdomidor and anesthesia was maintained for imaging with 1% isoflurane. Body temperature was maintained with a recirculating water warming pad and vital signs such as blood pressure, pulse oximetry, and EKG were monitored continuously. Two rhesus macaques were scanned on the G-PET (Physics and Instrumentation group, the University of Pennsylvania), a high sensitivity, high resolution PET scanner using gadolinium orthosilicate crystals incorporated into an Anger-logic detector developed for brain imaging [40]. Data was acquired for 90 min (12×10 sec, 2x30 sec, 4×60 sec, 2×120 sec, 3×180 sec, 2×300 sec, 2x600 sec and 2×1200 sec) in list mode after an intravenous injection of 121-135 MBq of [11C]21. For metabolite analysis, a venous blood sample was drawn into a heparinized syringe at 5- and 30-min post tracer injection. The acquired data were sorted into sinograms and reconstructed using the fully 3D LOR-RAMLA [41-43] iterative reconstruction algorithm. Seven VOIs including caudate, putamen, thalamus, frontal cortex, occipital cortex, cerebellum, and whole brain were manually delineated on a T1-weighted MR image with PMOD image analysis software (version 3.7, PMOD Technologies LLC). Time activity curves were extracted from all the VOIs and expressed as percent injected dose per cm3 (%ID/ cm3) and SUV.
Metabolite analysis in non-human primates.
Venous blood samples were drawn into a heparinized syringe at 5- and 30-min post tracer injection during PET acquisition. Samples were centrifuged at 3,000 g for 10 min to separate plasma and red blood cells. Acetonitrile (2 mL) was added to the plasma sample (1 mL) for protein precipitation and radiotracer extraction. The plasma solution was vortexed, followed by centrifugation at 3,000 g for 10 min. Supernatant and pellet were separated and subsequently counted with a gamma counter (PerkinElmer Wizard 2480) to determine the extraction efficiency. The plasma supernatant was diluted with water (1 mL) and passed through a 0.45 μm nylon filter. The filtered solution (200 μL) was subjected to HPLC analysis using an Agilent 1200 series instrument equipped with an Agilent XDB-C18 column (150×10 mm) for analysis. The mobile phase used was 16% methanol in 0.1 M ammonium formate buffer (volume) and the HPLC system was operated at a flow rate was 1 mL·min−1. Radioactivity was measured using a Posi-RAM™ radioHPLC detector (LabLogic, UK). Unlabelled reference standard 21 was co-injected with the processed plasma sample and monitored with a UV detector set at 254 nm to confirm the identity of the parent tracer (tR = 7 min).
Results
In silico studies and in vitro binding assays.
In silico molecular docking studies and MDS studies were conducted using the solid-state NMR structure of α-synuclein as described previously [17] to determine the probability of binding and relative binding energy of the ordered compounds for site 9. All compounds had a probability of binding to site 9 of approximately 20 – 30% with best binding energies ranging from −6.7 to −7.66 kcal/mol (Table 1).
The results of the competition binding studies are shown in Table 2. An MDS study was also conducted to compare with the results of the in vitro binding assay. The results of this analysis indicated that there was an excellent correlation (r=0.9567, p=0.0007) between the root mean square fluctuation (RMSF) measures of ligand from the MDS studies and Ki values from the in vitro binding studies. These data suggest that MDS simulation studies may be useful in predicting the relative affinity of a compound for binding to site 9 of α-synuclein. Increased affinity for site 9 was also supported by the observation of photo-crosslinking of CLX10, an analog of 15, to residues in site 9 (Figure 2). Although CLX10 still also crosslinks to site 2, only crosslinking to site 2 was observed in previous studies of photoaffinity analogs of 1,[15, 33] indicating that compounds such as 15 indeed have improved affinity for site 9.
Table 2.
Binding affinities of selected compounds against [3H]BF-2846 on α-synuclein and Aβ42 fibrils and the Site 9 RMSF values from MDS
| Compound # | α-synuclein fibrils (Ki; nM) a | Aβ42 fibrils (Ki; nM) a | Site 9 RMSF |
|---|---|---|---|
| 3 | 4.25 (2.55 - 7.17) | 15.2 (9.36 - 24.9) | 0.54 |
| 6 | 252 (86.7 - 731) | 107 (24.4 - 58.3) | 1.82 |
| 11 | 9.73 (3.20 - 30.3) | 52.8 (20.2 -142) | 0.40 |
| 12 | 8.28 (3.25 - 21.3) | 162 (79.6 - 321) | 0.48 |
| 14 | 6.15 (3.34 - 11.5) | 31.6 (9.7 - 106) | 0.27 |
| 15 | 11.0 (6.43 - 18.9) | 36.1 (23.4 - 56.0) | 0.69 |
| 17 | 36.1 (24.5 - 53.0) | 261 (149 - 466) | 1.25 |
Value present as mean (95% confidence intervals)
Design and synthesis of 21.
The high in vitro binding affinity of bromo-aminoisoxazole 15 prompted us to synthesize the corresponding iodo-analog 21, since this analog could be radiolabeled with iodine-125 and used for in vitro binding and autoradiography studies. The synthesis of the target compound and the precursor for radiolabeling are shown in Scheme 1. Condensation of 5-amino-isoxazole compounds 22a, b with 4-methoxybenzoyl chloride gave the corresponding benzamide analogs 21 and 15 in a yield of 33% and 19%, respectively. Conversion of the bromo substituent of 15 to the stannylated precursor for radioiodination (23) was achieved using standard palladium chemistry. Radioiodination was achieved by oxidative iodo-destannylation chemistry to give [125I]21 in a radiochemical yield of 64 ± 12% (n=3).
Scheme 1: Synthesis of [125I]21.

Reagents and conditions: a) 4-methoxybenzoyl chloride, DMAP, TEA, CH2Cl2, 0 °C to rt., 20 h, 21 33%; 4-methoxybenzoyl chloride, TEA, CH2Cl2, 0 °C to rt., 48 h 15 19%; b) 15, (SnBu3)2, XPhos Pd G2, toluene, 80 °C, 1 h, 39%; c) [125I]NaI, H2O2, AcOH, MeOH, 30 min, radiochemical yield = 64 ± 12%, radiochemical purity >99%, molar activity = 80 GBq·μmol−1(n=3).
In vitro binding studies of [125I]21.
Full competition binding curves for displacing [3H]BF2846 from α-synuclein fibrils were conducted with 15 and 21. Compound 21 demonstrated a higher affinity for α-synuclein than did 15 (Ki = 3.6 vs. 11 nM) (Figure 3A and B). Scatchard analyses of [125I]21 in both recombinant α-synuclein fibrils (Figure 3C) and recombinant Aβ42 fibrils (Figure 3D) indicated that [125I]21 has a subnanomolar affinity for α-synuclein, which is ~10-fold higher than what was predicted from the competition studies with [3H]BF2846. However, [125I]21 was also found to have an affinity of ~2.5 nM for Aβ42 (Figure 4B), giving a selectivity ratio for α-synuclein versus Aβ42 of only ~5.2. In vitro binding studies of brain tissue homogenates obtained from a PD patient, an Alzheimer’s disease (AD) patient and a young control subject indicate that [125I]21 demonstrates high binding in PD brain and recombinant α-synuclein fibrils. Binding was also observed in AD brain and Aβ40 fibrils, but little binding was observed in young control brain (Figure 4). Homogenate binding assays of [125I]21 with PD brain tissues resulted in a Kd value of 3.5 nM and a Bmax of 80 nM. Thus, [125I]21 binds with high affinity to α-synuclein, but binding to Aβ indicates that this radioligand may lack the selectivity needed for in vivo imaging studies of synucleinopathies. However, this radioligand may be useful for in vitro studies to screen candidate ligands for binding to site 9 in α-synuclein fibrils.
Figure 3. In vitro binding studies.

Competition binding curves in alpha-synuclein fibrils (100 nM) for (A) 15 and (B) 21 with site 9 ligand [3H]BF2846. The Ki values were 11 nM for 15 and 3.6 nM for 21. Saturation binding curves for [125I]21. Kd values obtained for (C) α-synuclein fibrils (0.48 ± 0.08 nM) and (D) Aβ42 fibrils (2.47 ± 1.30 nM). Black: total binding, red: specific binding, and blue: non-specific binding.
In vivo imaging studies of [11C]21.
The above in vitro binding studies encouraged us to evaluate [11C]21 in preclinical PET imaging studies of A53T mice. The synthesis of the precursor for radiolabeling is shown in Scheme 2. The 2-aminoisoxazole compound was condensed with 4-acetoxybenzoyl chloride to give the corresponding amide intermediate, which was immediately hydrolyzed under basic conditions to give the phenol derivative (24). Radiolabeling with [11C]methyl iodide was achieved in the presence of potassium carbonate/DMSO to give [11C]21 in a decay corrected radiochemical yield of ~12% and suitable molar activity for in vivo studies.
Scheme 2: Synthesis of [11C]21.

Reagents and conditions: a) 4-acetoxybenzoyl chloride, TEA, CH2Cl2, 0 °C to rt, 20 h, 33%; b) NaOH3, MeOH/H2O, 60 °C, 3 h, 43%; c) [11C]CH3I, K2CO3, DMSO, 70 °C, 5 min, radiochemical yield (decay corrected) = 12 ± 2% (n=4), radiochemical purity = 94 ± 4% (n=4), molar activity (at end of synthesis) = 344 ± 235 GBq·μmol−1 (n=4).
PET imaging studies were conducted in PFF injected and PBS injected (sham control). We observed elevated retention of [11C]21 in brain regions showing high levels of AsynPS129 staining in the PFF injected mouse (Figure 5). In contrast, relatively slow rate of washout in the sham control mouse brain is consistent with a high level of non-specific binding of [11C]21.
Figure 5.

Micro-PET images using [11C]21 in PFF injected, sham control, and the normal aging A53T mouse. (A) 35-60 min post [11C]21 injection images of PBS injected and PFF injected A53T mice in sagittal section. (B) Coronal brain section at the level of brainstem and cerebellum for PFF or PBS injected A53T mouse micro-PET image and the representative α-synuclein staining image of the littermates. (C) Time activity curves in SUV of brainstem and cortex. The peak (~7min) to 30 min ratio of whole brain is 1.5-1.7 for PBS injected and 1.6 for PFF injected mice.
PET imaging studies were also conducted in a nonhuman primate. We observed high uptake of [11C]21 in the monkey brain, with a peak SUV of ~2.3 at 2 min post-i.v. injection. The rate of clearance from nonhuman primate brain was much faster than what was observed in the mouse imaging studies (Figure 6). This may be the result of the rapid rate of metabolism of the radiotracer in nonhuman primates, with ~21% of the parent compound being present in venous blood samples at 5 min post-i.v. injection (Figure S3), whereas 31 ± 11% of parent compound is present in mouse blood and 67 ± 6% in mouse brain at 20 min post-i.v. injection of the radiotracer (Figure S4).
Figure 6.

Representative PET image of [11C]21 in a nonhuman primate. (A) 0-9 min, 9-20 min, and 50-90 min post [11C]21 injected PET and MR fusion images in transverse section. (B) Time activity curves in %ID/cm3 and SUV of thalamus, caudate, putamen, frontal cortex, occipital cortex, cerebellum, and whole brain.
Discussion
The development of a PET radiotracer that could serve as an imaging biomarker of α-synuclein aggregate formation has been a very active area of research in recent years. The need for an α-synuclein imaging agent is obvious since it could serve as a means for measuring target engagement and the efficacy of a number of therapeutic strategies in clinical trials that are aimed at preventing or delaying the progression of the formation of Lewy bodies, Lewy neurites, and glial cell inclusions in synucleinopathies. Although several radiotracers have been reported in the literature [44-51], no radiotracers developed to date possess the required properties for use in PET imaging studies of synucleinopathies.
We previously reported the use of an ultra-high throughput in silico screen to identify lead compounds that bind with high affinity to α-synuclein fibrils [15]. The goal of the current study was to expand our previous results that identified compound 1 as a lead compound for radiotracer development for α-synuclein aggregates. Although compound 1 had a high affinity for site 2, the proximity of site 2 to sites of frequent PTMs of α-synuclein was a concern since there may be a discrepancy in binding results between recombinant fibrils and α-synuclein obtained from postmortem human brain tissue. The affinity of compound 1 for site 9 was thought to be an advantage since this binding site is not near sites of PTMs, but the 100 nM affinity for site 9 proved to be too low for it to be useful as a PET radiotracer. In order to identify analogs of compound 1 having a higher affinity for this binding site, a panel of 19 compounds was purchased and docking studies were conducted to confirm that they had a high probability for interacting with site 9 of the solid-state NMR structure of α-synuclein. A rapid, 1-point screen was conducted to identify ligands capable of displacing [3H]BF2846 from site 9, and a number of compounds were identified having affinities <10 nM. Photocrosslinking studies further supported the increased affinity for site 9. Since [3H]BF2846 also binds potently to Aβ, we also screened the panel of compounds for their affinity for displacing this radioligand from Aβ fibrils. It is interesting to note that many of the compounds showed a low affinity for displacing [3H]BF2846 from Aβ fibrils, indicating that they have a modest selectivity for α-synuclein over Aβ. Based on the promising results for bromo-aminoisoxazole 15, the corresponding iodo analog 21 was synthesized, with the goal of making a probe that could be labeled with either iodine-125 or carbon-11. The in vitro Scatchard plot shows [125I]21 to be a potentially useful radioligand for in vitro binding assays in α-synuclein fibrils. The in vivo imaging studies in transgenic mice indicate that [11C]21 crosses the blood-brain barrier, although uptake in BPS-injected sham control mice is rather low. [11C]21 exhibits higher uptake and slower washout in brain regions having high levels of AsynPS129, the principal form of α-synuclein found in insoluble aggregates, in the PFF-injected mouse as compared with PBS-injected sham controls. Notably, brain uptake of [11C]21 in a non-human primate was significantly higher than in mice. However, the relatively high binding of this ligand to Aβ fibrils and AD brain tissues suggests that it may not be useful for PET imaging studies in PD subjects, since ~50% of PD subjects have significant Aβ plaque pathology at autopsy [52]. The postmortem AD tissue used in this study contained high levels of both Aβ plaques and tau-containing neurofibrillary tangles. Assays to determine the binding of [125I]21 to Aβ plaques or NFT's separately were not conducted. Although the data are encouraging, it is very likely that a PET radiotracer having improved selectivity for α-synuclein versus Aβ and/or tau will be needed for translational imaging studies. However, we believe the results described herein are noteworthy as they demonstrate that a simple structural similarity search in a commercially available compound library can be used in structure-activity relationships aimed at improving the in vitro binding affinity of a lead compound for a target protein.
The main limitations of the current study are that [11C]21 and [125I]21 were characterized in transgenic models of the synucleinopathies and validated in homogenate binding assays using tissue obtained from PD, AD and control subjects. Although these assays suggest that [11C]21 should be capable of visualizing α-synuclein in PET imaging studies of PD subjects, this must be confirmed in vivo. For example, there may be off target binding in human brain that may not be observed in the homogenate binding assays in tissue homogenates, nor observed in nonhuman primate imaging studies.
Conclusion
By means of a relatively simple, structure-based similarity search, we were able to identify an analog of a lead compound for binding to site 9 of α-synuclein fibrils with an order of magnitude higher affinity (~100 nM to <10 nM). Photocrosslinking to site 9 was observed for a probe based on this analog. In vitro binding demonstrated that a 125I-radiolabeled analog of one of the secondary hits, [125I]21, was capable of binding to α-synuclein fibrils and PD tissue homogenates in radioligand binding assays. MicroPET imaging studies with [11C]21 show high uptake and a slower rate of washout from brain regions having AsynPS129, the principal form of α-synuclein found in insoluble protein aggregates. The suboptimal selectivity of this compound versus Aβ combined with high non-specific binding indicates that this is not a promising candidate radioligand for translational PET imaging studies. However, the strategy outlined in this report demonstrates the value of using the described in silico methods in conducting structure-activity relationship studies in radiotracer development.
Supplementary Material
Acknowledgments.
This research was funded by the Michael J. Fox Foundation and NIH grant U19-NS110456. M.G.L. was supported by an Age Related Neurodegenerative Disease Training Grant fellowship (NIH T32-AG000255). H.J.K. was supported by an NIH Predoctoral Fellowship (NIH F31-AG069390).
References
- 1.Henderson MX, Trojanowski JQ, and Lee VM, α-Synuclein pathology in Parkinson's disease and related α-synucleinopathies. Neurosci Lett, 2019. 709: p. 134316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seibyl JP, Alpha-synuclein PET and Parkinson's Disease Therapeutic Trials: Ever the Twain Shall Meet? J Nucl Med, 2022. [DOI] [PubMed] [Google Scholar]
- 3.Pancoe SX, et al. , Effects of Mutations and Post-Translational Modifications on α-Synuclein In Vitro Aggregation. Journal of Molecular Biology, 2022. 434(23): p. 167859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gibb W and Lees A, The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson's disease. Journal of Neurology, Neurosurgery & Psychiatry, 1988. 51(6): p. 745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gibb W and Lees A, The significance of the Lewy body in the diagnosis of idiopathic Parkinson's disease. Neuropathology and applied neurobiology, 1989. 15(1): p. 27–44. [DOI] [PubMed] [Google Scholar]
- 6.Jellinger KA, Seppi K, and Wenning GK, Grading of neuropathology in multiple system atrophy: proposal for a novel scale. Movement disorders: official journal of the Movement Disorder Society, 2005. 20(S12): p. S29–S36. [DOI] [PubMed] [Google Scholar]
- 7.Dickson D., et al. , Widespread alterations of α-synuclein in multiple system atrophy. The American journal of pathology, 1999. 155(4): p. 1241–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cykowski MD, et al. , Expanding the spectrum of neuronal pathology in multiple system atrophy. Brain, 2015. 138(8): p. 2293–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prasad EM and Hung SY, Current Therapies in Clinical Trials of Parkinson's Disease: A 2021 Update. 2021. 14(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smit JW, et al. , Phase 1/1b Studies of UCB0599, an Oral Inhibitor of α-Synuclein Misfolding, Including a Randomized Study in Parkinson's Disease. Movement Disorders, 2022. 37(10): p. 2045–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buddhala C., et al. , Dopaminergic, serotonergic, and noradrenergic deficits in Parkinson disease. Annals of Clinical and Translational Neurology, 2015: p. n/a-n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Covell DJ, et al. , Novel conformation-selective alpha-synuclein antibodies raised against different in vitro fibril forms show distinct patterns of Lewy pathology in Parkinson's disease. Neuropathol Appl Neurobiol, 2017. 43(7): p. 604–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Y., et al. , Amyloid fibril structure of alpha-synuclein determined by cryo-electron microscopy. Cell Res, 2018. 28(9): p. 897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kotzbauer PT, Tu Z, and Mach RH, Current status of the development of PET radiotracers for imaging alpha synuclein aggregates in Lewy bodies and Lewy neurites. Clinical and Translational Imaging, 2017. 5(1): p. 3–14. [Google Scholar]
- 15.Ferrie JJ, et al. , Identification of a nanomolar affinity α-synuclein fibril imaging probe by ultra-high throughput in silico screening. Chemical Science, 2020. 11(47): p. 12746–12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson DK and Karanicolas J, Ultra-High-Throughput Structure-Based Virtual Screening for Small-Molecule Inhibitors of Protein-Protein Interactions. J Chem Inf Model, 2016. 56(2): p. 399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brundin P, Dave KD, and Kordower JH, Therapeutic approaches to target alpha-synuclein pathology. Exp Neurol, 2017. 298(Pt B): p. 225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsieh CJ, et al. , Alpha Synuclein Fibrils Contain Multiple Binding Sites for Small Molecules. ACS Chem Neurosci, 2018. 9(11): p. 2521–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lengyel-Zhand Z., et al. , Synthesis and characterization of high affinity fluorogenic alpha-synuclein probes. Chem Commun (Camb), 2020. 56(24): p. 3567–3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sonustun B., et al. , Pathological Relevance of Post-Translationally Modified Alpha-Synuclein (pSer87, pSer129, nTyr39) in Idiopathic Parkinson's Disease and Multiple System Atrophy. 2022. 11(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmid AW, et al. , Alpha-synuclein post-translational modifications as potential biomarkers for Parkinson disease and other synucleinopathies. Mol Cell Proteomics, 2013. 12(12): p. 3543–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fauvet B and Lashuel HA, Semisynthesis and Enzymatic Preparation of Post-translationally Modified α-Synuclein. Methods Mol Biol, 2016. 1345: p. 3–20. [DOI] [PubMed] [Google Scholar]
- 23.Kaide S and Watanabe H, Identification and Evaluation of Bisquinoline Scaffold as a New Candidate for α-Synuclein-PET Imaging. 2020. 11(24): p. 4254–4261. [DOI] [PubMed] [Google Scholar]
- 24.Pan B., et al. , Chemoenzymatic Semi-synthesis Enables Efficient Production of Isotopically Labeled α-Synuclein with Site-Specific Tyrosine Phosphorylation. ChemBioChem, 2021. 22(8): p. 1440–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pan B, Rhoades E, and Petersson EJ, Chemoenzymatic Semisynthesis of Phosphorylated α-Synuclein Enables Identification of a Bidirectional Effect on Fibril Formation. ACS Chemical Biology, 2020. 15(3): p. 640–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao K., et al. , Parkinson’s disease-related phosphorylation at Tyr39 rearranges α-synuclein amyloid fibril structure revealed by cryo-EM. Proceedings of the National Academy of Sciences, 2020. 117(33): p. 20305–20315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang S., et al. , Post-translational Modifications of Soluble α-Synuclein Regulate the Amplification of Pathological α-Synuclein. Nature Neuroscience, In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morris GM, et al. , AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem, 2009. 30(16): p. 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Phillips JC, et al. , Scalable molecular dynamics with NAMD. Journal of computational chemistry, 2005. 26(16): p. 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Humphrey W, Dalke A, and Schulten K, VMD: Visual molecular dynamics (1996) Journal of Molecular Graphics, 14 (1). p. 33–38. [DOI] [PubMed] [Google Scholar]
- 31.Riad A., et al. , The sigma-2 receptor/TMEM97, PGRMC1, and LDL receptor complex are responsible for the cellular uptake of Aβ42 and its protein aggregates. Molecular neurobiology, 2020. 57: p. 3803–3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klunk WE, et al. , Uncharged thioflavin-T derivatives bind to amyloid-beta protein with high affinity and readily enter the brain. Life sciences, 2001. 69(13): p. 1471–1484. [DOI] [PubMed] [Google Scholar]
- 33.Lougee MG, et al. , Harnessing the intrinsic photochemistry of isoxazoles for the development of chemoproteomic crosslinking methods. Chemical Communications, 2022. 58(65): p. 9116–9119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Montine TJ, et al. , National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta neuropathologica, 2012. 123: p. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKeith IG, et al. , Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology, 2017. 89(1): p. 88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bagchi DP, et al. , Binding of the radioligand SIL23 to α-synuclein fibrils in Parkinson disease brain tissue establishes feasibility and screening approaches for developing a Parkinson disease imaging agent. PloS one, 2013. 8(2): p. e55031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klunk WE, et al. , The binding of 2-(4′-methylaminophenyl) benzothiazole to postmortem brain homogenates is dominated by the amyloid component. Journal of Neuroscience, 2003. 23(6): p. 2086–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luk KC, et al. , Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science, 2012. 338(6109): p. 949–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mirrione MM, et al. , A novel approach for imaging brain–behavior relationships in mice reveals unexpected metabolic patterns during seizures in the absence of tissue plasminogen activator. Neuroimage, 2007. 38(1): p. 34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karp JS, et al. , Performance of a brain PET camera based on anger-logic gadolinium oxyorthosilicate detectors. Journal of nuclear medicine, 2003. 44(8): p. 1340–1349. [PubMed] [Google Scholar]
- 41.DePierro A., On some nonlinear iterative relaxation methods in remote sensing. Matematica Aplicada e Computacional, 1989. 8: p. 153–166. [Google Scholar]
- 42.Browne J and De Pierro A, A row-action alternative to the EM algorithm for maximizing likelihood in emission tomography. IEEE transactions on medical imaging, 1996. 15(5): p. 687–699. [DOI] [PubMed] [Google Scholar]
- 43.Daube-Witherspoon ME, Matej S, and Karp JS. Assessment of image quality with a fast fully 3D reconstruction algorithm. in 2001 IEEE Nuclear Science Symposium Conference Record (Cat. No. 01CH37310). 2001. IEEE. [Google Scholar]
- 44.Maurer A., et al. , (11) C Radiolabeling of anle253b: a Putative PET Tracer for Parkinson's Disease That Binds to α-Synuclein Fibrils in vitro and Crosses the Blood-Brain Barrier. 2020. 15(5): p. 411–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheng Y and Prusoff WH, Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem.Pharmacol, 1973. 22(23): p. 3099–3108. [DOI] [PubMed] [Google Scholar]
- 46.Pang A., et al. , Interdomain dynamics and ligand binding: molecular dynamics simulations of glutamine binding protein. FEBS Lett, 2003. 550(1-3): p. 168–74. [DOI] [PubMed] [Google Scholar]
- 47.Brettschneider J., et al. , Converging Patterns of alpha-Synuclein Pathology in Multiple System Atrophy. J Neuropathol Exp Neurol, 2018. 77(11): p. 1005–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zarranz JJ, et al. , The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann.Neurol, 2004. 55(2): p. 164–173. [DOI] [PubMed] [Google Scholar]
- 49.Chu W., et al. , Design, Synthesis, and Characterization of 3-(Benzylidene)indolin-2-one Derivatives as Ligands for alpha-Synuclein Fibrils. J Med Chem, 2015. 58(15): p. 6002–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McKeith IG, et al. , Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology, 1996. 47(5): p. 1113–1124. [DOI] [PubMed] [Google Scholar]
- 51.Kikuchi A., et al. , In vivo visualization of alpha-synuclein deposition by carbon-11-labelled 2-[2-(2-dimethylaminothiazol-5-yl)ethenyl]-6-[2-(fluoro)ethoxy]benzoxazole positron emission tomography in multiple system atrophy. Brain, 2010. 133(Pt 6): p. 1772–8. [DOI] [PubMed] [Google Scholar]
- 52.Irwin DJ, Lee VM-Y, and Trojanowski JQ, Parkinson's disease dementia: convergence of α-synuclein, tau and amyloid-β pathologies. Nature Reviews Neuroscience, 2013. 14(9): p. 626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tuttle MD, et al. , Solid-state NMR structure of a pathogenic fibril of full-length human alpha-synuclein. Nature Structural & Molecular Biology, 2016. 23(5): p. 409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.