

Summary
Assembly of tau into beta-sheet-rich amyloids dictates the pathology of a diversity of diseases. Lysine acetylation has been proposed to promote tau amyloid assembly but no direct mechanism has emerged. Using tau fragments, we identify patterns of acetylation that flank amyloidogenic motifs on the tau fragments that drive rapid fibril assembly. We determined a 3.9 Å cryo-EM amyloid fibril structure assembled from an acetylated tau fragment uncovering how lysine acetylation can mediate gain-of-function interactions. Comparison of the structure to an ex vivo tauopathy fibril reveals regions of structural similarity. Finally, we show that fibrils encoding disease-associated patterns of acetylation are active in cell-based tau aggregation assays. Our data uncover the dual role of lysine residues in limiting tau aggregation while their acetylation leads to stabilizing pro-aggregation interactions. Design of tau sequence with specific acetylation patterns may lead to controllable tau aggregation to direct folding of tau into distinct amyloid folds.
eTOC
Post-translational modifications on the microtubule-associated protein tau are linked to disease pathogenesis. Li Li et al show that lysine acetylation on tau in proximity of amyloidogenic sequences is sufficient to mediate rapid tau fibrillization. Structural analysis of the acetyl-lysine containing fibrils uncovers gain-of-interactions that stabilize the fibril.
Graphical Abstract
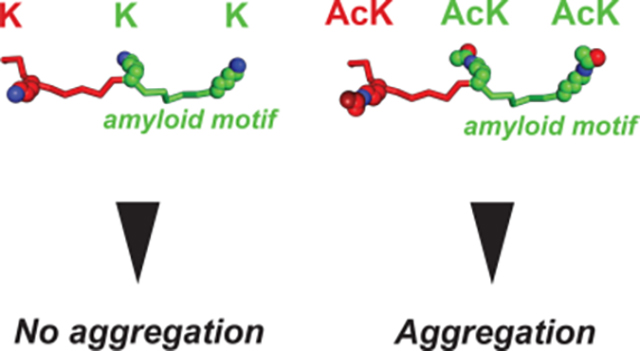
Introduction
The deposition of the microtubule-associated protein tau in beta-sheet-rich amyloid conformations is a hallmark of many neurodegenerative diseases. These diseases are classified as tauopathies and include Alzheimer’s disease (AD), Corticobasal degeneration (CBD), Pick’s disease (PiD), chronic traumatic encephalopathy (CTE) and frontotemporal dementia with tau (FTD-tau)1. Recent ex vivo cryo-EM structures of tau fibrils isolated from tauopathy patient material have resolved a series of distinct structures (i.e. structural polymorphs) each linked to a different disease2–6. Moreover, each structural polymorph of tau has been proposed to encode the capacity to replicate their shape in a prion-like manner, spread their conformation throughout the brain, and to cause progressive neurodegeneration7–10. Despite this strong link to disease, the intrinsically disordered protein tau under normal conditions is aggregation-resistant and its aggregation must be induced using polyanions or pathogenic seeds to form beta-sheet-rich amyloids11–14. It thus remains unknown how tau converts into pathogenic states capable of initiating conformation-specific tauopathies.
Short amyloid-forming motifs (275VQIINK280, 306VQIVYK311 and 337VEVKSE342) control tau assembly into fibrils by engaging in hetero-typic interactions with other parts of the sequence. The amyloid motifs in tau localize to the beginning of each of the four repeats (i.e. R1, R2, R3 and R4) in the repeat domain1. Each of the amyloid forming motifs is preceded by a P-G-G-G sequence motif that stabilizes beta-turn conformations15. These beta-turn motifs have been shown to regulate the aggregation capacity of tau by engaging side-chain interactions with the adjacent amyloid motifs15,16. We and others have shown that perturbation of beta-turn-based regulatory elements in a monomer can lead to the formation of a seed-competent monomer13,14,17. The seeding-competent monomer species can be isolated from AD patients and tauopathy mouse brains but are absent in control tissues13,14,17. These regulatory elements can be recapitulated with wildtype tau peptides that span the amyloid motif and the preceding sequence including the P-G-G-G motif in 4 repeat (4R) tau (i.e. R1R2, R2R3, R3R4 and R4R’) as well as 3 repeat (3R) tau in which the second repeat is spliced out creating a new junction between repeats 1 and 3 (i.e. R1R3). Disease-associated mutations in tau linked to FTD-tau localize to these regulatory elements and increase aggregation capacity into fibrils compared to wildtype18. We hypothesized that dysregulation of these elements may preferentially expose different amyloid motifs enabling the combinatorics of their exposure to stabilize intermediates towards distinct structural polymorphs.
For many years, post-translational modifications (PTMs) such as phosphorylation have been implicated in conversion of tau into pathogenic conformations19,20. However, recent data have suggested that phosphorylation may not be the emergent perturbation that drives the formation of the first pathogenic tau species but rather accumulates once the assemblies are formed17. Detection of phosphorylated tau is the de facto method for disease diagnosis in blood19 but also for detection of tau pathology in post-mortem tissues20,21. Phosphorylation on tau is relatively ubiquitous but excluded from the repeat domain with the exception of AD-specific modifications on serine S262 that lie outside of the AD fibril core17. Consistent with this observation, phosphor-modifications in the repeat domain inhibit tau assembly22. Other common PTMs such as acetylation and ubiquitination have similarly been implicated in driving progression of disease through promoting aggregation23,24 but the mechanism remains unknown17. A recent structural and proteomic analysis of AD and CBD fibrils proposed that acetylation and ubiquitination may be important for control of structural polymorph assembly25. Additionally, others have shown that acetylation-specific antibodies can detect early pathogenic species26,27. It is not known how acetylation may influence tau aggregation. It is possible that acetylation on lysines directly influences aggregation by stabilizing pro-aggregation interactions, disrupting protective interactions or reducing interaction with microtubules28–30. Moreover, it is possible that acetylation may influence the process by blocking ubiquitination sites required for subsequent degradation. Sites of acetylation on tau have been reported throughout the entire sequence including in the repeat domain surrounding the amyloid motifs including lysines K267, K274, K280 and K281 in repeats 1 and 224. Such sites have also been detected in repeats 3 and 4 at lysines K311 and K35324. The acetyl transferase enzymes involved in this process are not known but histone deacetylases (HDACs) and SIRT1 have been implicated in their removal. HDAC6 has been implicated as the deacetylase that may regulate removal of acetylation from lysines on tau28. Specific modifications surrounding the amyloid motifs (i.e. K274, K280 and K311) have been proposed to alter Hsp70 recruitment thus reducing chaperone-mediated autophagy31. Furthermore, tau encoding acetylated K280 generated using semi-synthetic methods increases heparin-induced aggregation propensity 3-fold compared to unmodified tau yielding a mixture of oligomers and short fibrils22. Thus the proximity of disease-associated acetylation sites on tau to our established aggregation regulatory elements15 suggests that we may be able to test the role of lysine acetylation in mediating early conformational changes to drive tau assembly. Finally, lysine acetylation on tau at key sites (including K280, K281 and K311)24 may be markers for neurodegeneration28,32 and their reduction may be neuroprotective28,32. Thus understanding how acetylation may drive tau fibril formation may yield insight into mechanisms of pathogenesis.
Here we show that disease-associated patterns of tau acetylation on minimal tau fragments encoding regulatory elements can drive amyloid assembly. We first use a chemical approach to modify tau fragments that span the R1R2, R2R3, R3R4 and R4R’ interrepeat regions in 4R tau and R1R3 in 3R tau. We show that saturating chemical acetylation of R1R2, R2R3 and R1R3 leads to rapid fibrillar assembly while the unmodified sequences remain inert. We next use synthetic sequences with site specific acetylation to uncover patterns of modification that regulate the assembly of tau fragments. We also test commonly used lysine to glutamine substitutions and show that similar patterns of modifications can lead to their assembly with drastically different fibril morphologies. We use cryo-EM to determine a fibril structure of one of our R1R2 acetylated peptides encoding disease-associated acetylation sites at residues K274 and K280. The structure uncovers interactions mediated by acetylation that balance hydrogen bonding and nonpolar contacts to stabilize the structure not possible by a lysine side chain. The structure also revealed that specific patterns of acetylation stabilize the regulatory elements in an extended conformation similar to what has been observed in ex vivo patient structures. Finally, we show that a fibril with disease-associated modifications has the capacity to promote assembly in a cell model of tau aggregation. Together, our data highlight that disease-associated patterns of tau acetylation can drive tau assembly by creating new interactions not possible by a lysine residue. This study lays the groundwork for understanding how specific patterns of acetylation may mediate control of tau assembly into the various structural polymorphs seen in disease.
Results
Acetylation adjacent to amyloidogenic sequence regulates tau aggregation
The frequency of acetylation on the repeat domain of tau correlates with the formation of pathological species24, particularly when the modifications occur in proximity to amyloidogenic sequences 306VQIVYK311 and 275VQIINK280 (Figure 1A). Using our peptide model system that leverages the regulatory elements to limit aggregation behavior15, we assessed whether disease-associated acetylation patterns on tau can destabilize the protective structures and drive assembly of these fragments into amyloids. We systematically probed elements that span the interrepeat regions including R1R2, R2R3, R3R4 as well as the 3R specific isoform fragment R1R3 (Figure 1B). The R1R2 (i.e. TENLKHQPGGGKVQIINK) and R1R3 (i.e. TENLKHQPGGGKVQIVYK) sequences are highly homologous differing at only two positions in the amyloid motifs. Conversely, the R2R3 (i.e. KDNIKHVPGGGSVQIVYK) and R1R3 (i.e. TENLKHQPGGGKVQIVYK) sequences have the same amyloid motif but differ at five positions N-terminal to the amyloid motif. Our previous work on this model system uncovered that the WT sequences are aggregation resistant due to the stabilizing interactions involving the conserve P-G-G-G beta-turn stabilizing motif, and that disease-associated mutations in the R2R3 sequence drive aggregation15. Interestingly, testing the most dominant proline to serine mutation in the P-G-G-G motif dramatically increased R2R3 aggregation but had no effect on R1R2 and R1R3 assembly suggesting that the regulatory elements in each sequence have different capacities to reduce aggregation15. We hypothesize that post-translational modifications, such as acetylation, could drive these differences in aggregation.
Figure 1. Chemical acetylation of lysines on amyloid motif-containing tau fragments drives fibril assembly.
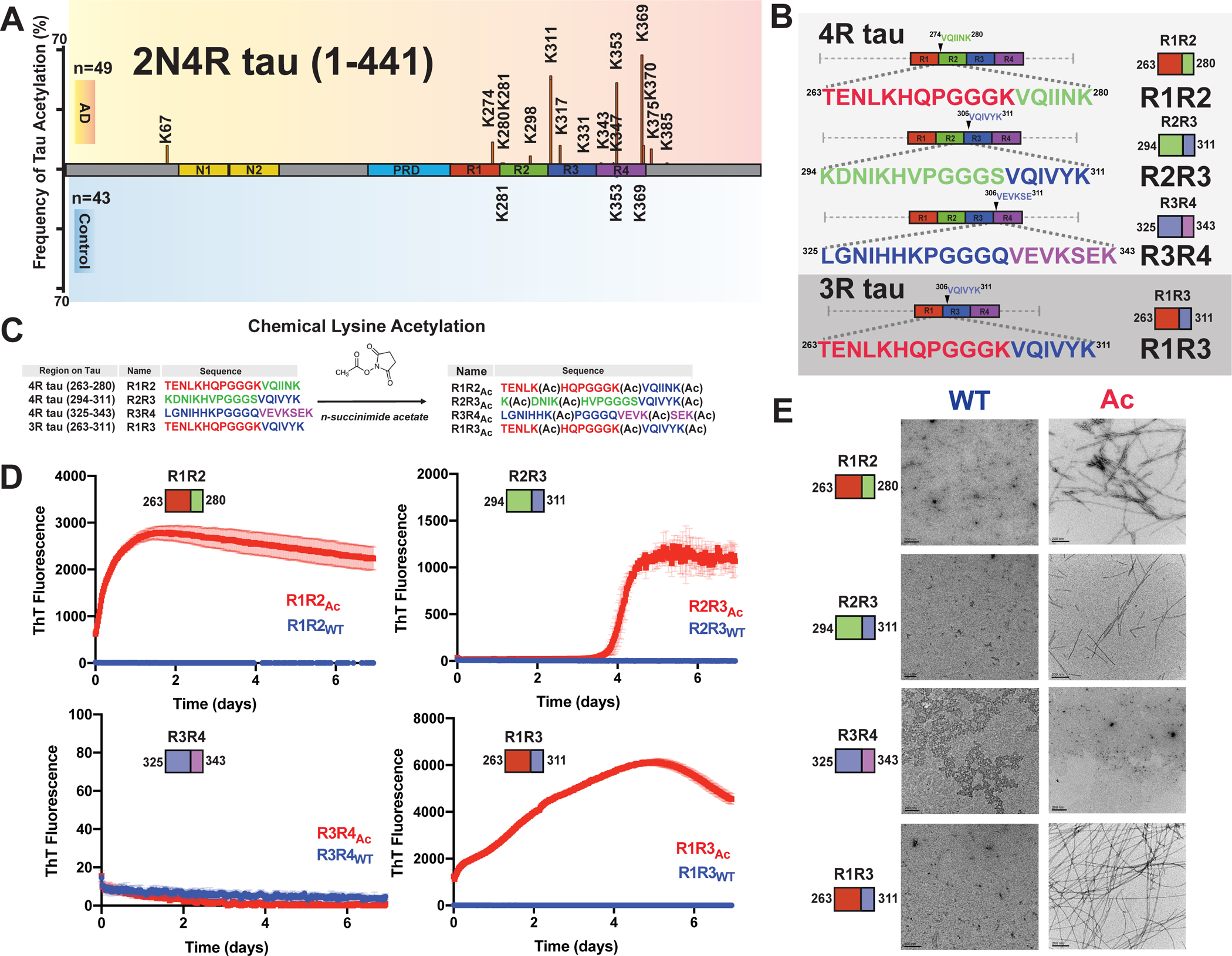
A. Literature reported acetylation sites on 2N4R tau in AD and control patients; the modification frequency is noted on the y axis. N-terminal extension domains, N1 and N2, are colored in orange. Proline-rich domain (PRD) is colored in blue. Repeat domains are colored in red, blue, green and magenta for repeats 1, 2, 3 and 4, respectively. B. Schematic illustrating the four tau model peptides (R1R2, R2R3, R3R4 and R1R3). Their sequence and relative location in the repeat domain are shown. Repeat domain elements are colored as in Figure 1A. C. Schematic illustrating acetylation modification reaction with n-succinimide acetate of the tau peptides. D. ThT fluorescence aggregation assay of acetylated peptides (red) and their corresponding non-acetylated controls (blue). Aggregation experiments were performed in triplicate and the averages are shown with standard deviation. The data were fit to a non-linear regression model fitting in GraphPad Prism to estimate an average t1/2max with a standard deviation. E. TEM images of acetylation modified peptides (red, right column) and their corresponding non-modified controls (blue, left column). Scale bars indicate 0.2 μm.
To test the effect of acetylation on the assembly of these peptides, we utilized N-succinimidyl acetate to chemically modify lysine residues to acetyl-lysine (AcK) (Figure 1C). Each peptide contains three lysine residues that were distributed throughout the sequence and includes lysine residues that have been observed to be acetylated in disease including K267, K274, K280 in R1R2, K311 in R2R3 and K331, K340 and K343 in R3R4 and K369 and K370 in R4R’24. All peptides contained a lysine residue at the C-terminus of the amyloid motif (i.e. 306VQIVYK(Ac)311 or 275VQIINK(Ac)280) while the other two lysines were distributed either in the N-terminus (R2R3) or one at the N-terminus and the other flanking the N-terminus of the amyloid motif (R1R2, R3R4 and R1R3). We first used mass spectrometry to confirm that the acetylation reactions proceeded to completion (Figure 1C). We found the reaction products to be heterogenous with the most abundant species containing all three lysines acetylated but also included double and single modifications (Figure S1A–D). The modified peptides were purified, and their aggregation was monitored in a Thioflavin T (ThT) fluorescence aggregation assay for 7 days. Three acetylated peptides R1R2Ac, R1R3Ac, R2R3Ac showed significant increase in ThT fluorescence with t1/2max values of 0.20 ± 0.04, 1.66 ± 0.08 and 4.12 ± 0.01 days, respectively, while their unmodified counterparts did not aggregate (Figure 1D). The acetylated R3R4Ac and the unmodified control also did not aggregate (Figure 1D). The presence of fibrils in the samples was confirmed with negative stain Transmission Electron Microscopy (TEM) (Figure 1E). The R1R2Ac fibrils appeared to have the most consistent twist. Our data support that chemical modification of our model tau peptides can disrupt the regulatory elements and drive amyloid motif-dependent assembly into beta-sheet-rich amyloids. We next set out to identify specific patterns of acetylation on our peptides that promote the formation of fibrils.
Defined patterns of acetylation drive tau into amyloid fibrils
With the observation that chemical acetylation of our peptides could dramatically change the aggregation propensity, we used a synthetic modification approach to generate all possible combinations of lysine acetylation modifications. R1R2, R2R3 and R1R3 peptides were synthesized with discrete modifications at each site individually (i.e. 1Ac, 2Ac, and 3Ac), as pairs (i.e. 12Ac, 23Ac, and 13Ac) and at all three sites (i.e. 123Ac) yielding 7 different combinations of modifications for each peptide (Figure 2A). Additionally, we tested a subset of variants for the R3R4 and R4R’ fragments (Figure 2B). Within this series we include acetylation modifications that have been observed in disease including K267, K274, K280 in R1R2; K311 in R2R3; K331, K340, K343 in R3R4; and K369, K370 in R4R’24. Aggregation of the above peptides was evaluated in a ThT aggregation assay over 8 days. For both the R1R2 and R1R3 series that only vary by two positions in the amyloid motifs (Figure 1B), we observed that modification on the second and third lysines (R1R2_23Ac and R1R3_23Ac) or all three lysines (R1R2_123Ac and R1R3_123Ac) aggregated rapidly with t1/2max values of 0.23 ± 0.02, 1.98 ± 0.13, 0.19 ± 0.04 and 0.56 ± 0.01 days, respectively (Figures 2C,D and S2A). All other R1R2 and R1R3 modified peptides and their unmodified controls did not aggregate (Figures 2C,D and 2A). The data on the R1R2 and R1R3 peptides suggest that modification on the second and third lysines on the peptide have the largest impact on the kinetics of its fibrillization. By contrast, the R2R3 series yielded much different results. While the control R2R3 peptide did not aggregate, all except for one modified peptide aggregated within the time frame of the experiment (Figure 2E). Fitting the data to quantify the t1/2max uncovers general rules in which the propensity to aggregate appear to be similarly site dependent but that the R2R3 sequence has a lower capacity to limit assembly compared to the R1R2 and R1R3 peptides. For the R2R3 peptides, we find that the 23Ac, 123Ac and 2Ac modifications yield the fastest aggregation with t1/2max values of 1.8 ± 0.18, 2.56 ± 0.03 and 3.18 ± 0.31 days, respectively, suggesting that modifications on the second lysine may be the most important for mediating aggregation (Figures 2E and S2A). The remaining R2R3 peptides formed aggregates within the time scale of the experiment with t1/2max values that ranged from 3.80 to 5.35 days. The unmodified R2R3 and R2R3_3Ac did not aggregate (Figures 2E and S2A). Finally, none of the R3R4 and R4R’ peptides aggregated (Figure 2F). For all samples, the formation of fibrils was confirmed by negative stain TEM (Figures 2F and S2B). Our experiments have uncovered general rules for how the regulatory tau peptides may be disrupted by acetylation sites observed in disease to drive aggregation. We find that modifications closest to the amyloid motifs have the largest effect on disruption of the protective structures.
Figure 2. Site-specific acetylation on tau peptides reveals patterns of modification that regulate aggregation propensity.
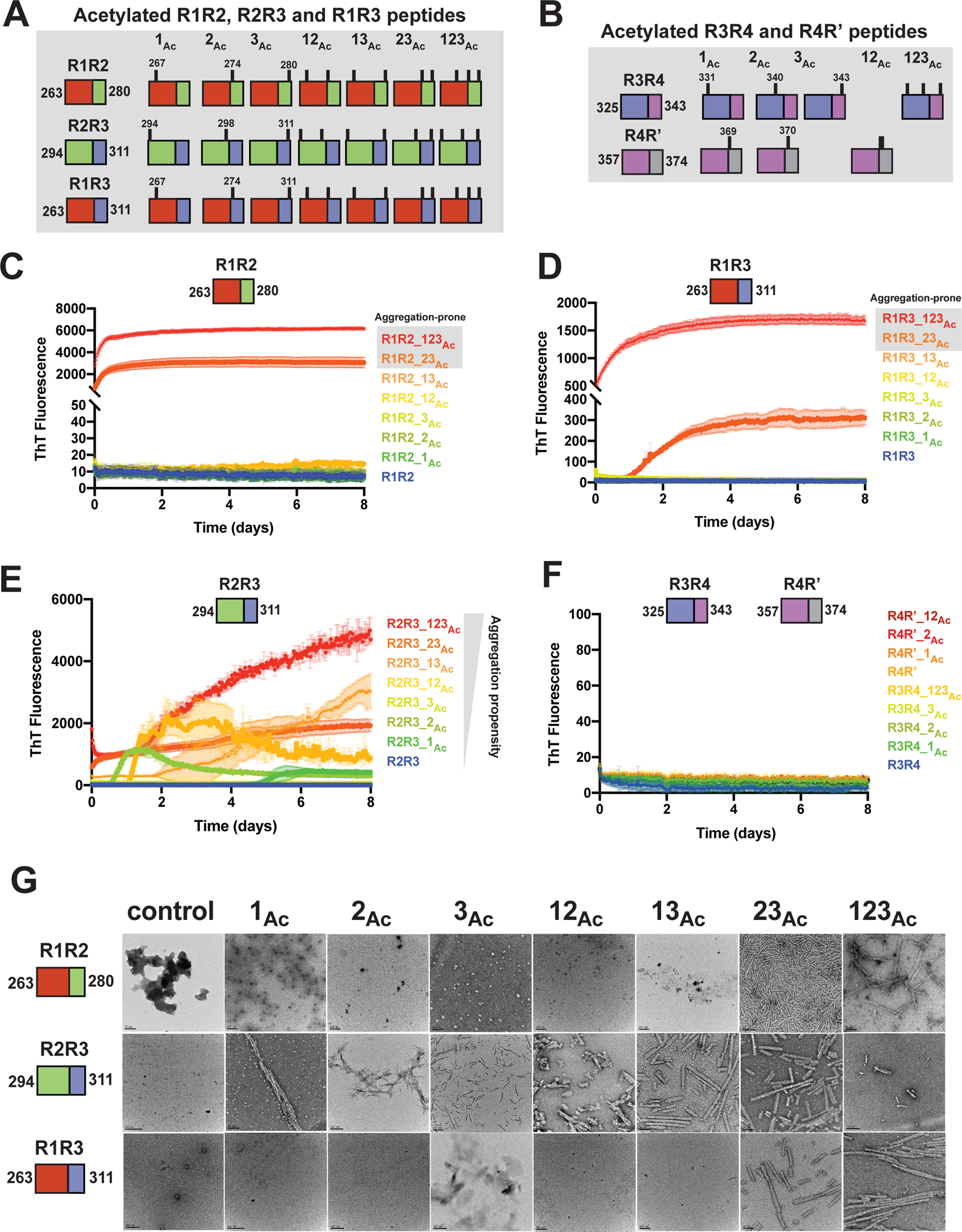
Illustration of the tau peptide series with all combinatorial acetylation patterns for the R1R2, R2R3 and R1R3 (A) and the R3R4 and R4R’ (B) tau peptides. Sequences are colored by repeat domain as in Figure 1A. Acetylation sites are indicated by ticks above the cartoon for each peptide. ThT fluorescence aggregation experiments of the R1R2 (C), R2R3 (D), R1R3 (E) and R3R4/R4R’4 (F) unmodified and acetylation modified series. Curves are colored by the number of modifications from blue (control) to red (fully modified). Aggregation experiments were performed in triplicate and the averages are shown with standard deviation. The data were fit to a non-linear regression model fitting in GraphPad Prism to estimate an average t1/2max with a standard deviation. G. TEM images of ThT fluorescence aggregation assay end products from control (WT) and each acetylated peptide. Scale bars indicate 0.05–0.2 μm.
Lysine to glutamine mutations mimic acetylation in aggregation experiments
The two possible routes of lysine acetylation on proteins involve either chemical or enzymatic modification33. Methods to genetically encode acetylation using orthogonal incorporation at amber codons is possible but yields and efficiency remain an issue34. Genetically, the closest amino acid that mimics an acetylated lysine is glutamine but the length of the side chain and geometry of the polar groups is different33. Nonetheless, this is the closest approximation that has been employed in both recombinant, cellular and animal systems. Previous work on tau has shown that a lysine to glutamine substitution at position 280 promoted aggregation of full-length tau in vivo, in cells and in vitro28,32, however, this modification alone in our peptide system (Figure 2C, R1R2_3Ac) did not aggregate. To understand the relationship between acetylation and glutamine substitutions, we generated a series of R1R2, R2R3 and R1R3 peptides with lysine to glutamine mutations at singly (i.e. 1Q, 2Q, and 3Q), doubly (i.e. 12Q, 23Q, and 13Q) and triply (i.e. 123Q) substituted sites (Figure S2C). Similar to the above experiments with acetylated peptides, the peptides were aggregated in a ThT fluorescence aggregation assay over 8 days. We found that the R1R2, R2R3 and R1R3 peptides with single modifications of lysine to glutamine on the first and second lysine positions (i.e. 1Q and 2Q) did not aggregate with the exception of R2R3_2Q which had a t1/2max of 3 ± 0.3 days (Figure S2D–G). Glutamine substitution on the third lysine for all peptides yielded rapid aggregation that ranged from 0.16 to 1.5 days with the R2R3_3Q peptide aggregating the fastest (Figure S2D–G). For double substitutions in the R1R3 sequence, the R1R3_23Q mutant aggregated the fastest (t1/2max < 0.25 day), followed by R1R3_12Q (t1/2max < 1.5 days) while the R1R3_13Q mutant was the slowest (Figure S2D,F). The R2R3 double mutants behaved similarly except for R2R3_13Q which started with fluorescence signal at time zero (Figure S2E). Finally, the triple substituted peptides all aggregated rapidly with t1/2max values that ranged from 0.12 to 1.2 days (Figure S2D–F). For all incubated peptides, we confirmed the presence of fibrils using negative stain TEM (Figure S2H). Interestingly, the fibril morphology of the lysine to glutamine fibrils was wider than the acetylated peptides. Overall, we find that our “Q” peptide series largely follows the behavior of the acetylated peptides with perturbations on the second position being the most important for driving fibril formation. Our data also suggest that glutamine mutations appear to strongly increase aggregation propensity consistent with asparagine and glutamine residues increasing amyloidogenicity of sequences35. More importantly, these data point to the role of positively charged lysines in slowing down assembly while polar groups of acetyl-lysine (AcK) and glutamines can promote fibril assembly. Despite being implicated in driving aggregation in disease, it has thus far remained unknown how acetylation can promote fibril assembly.
Cryo-EM structure of an acetylated tau peptide
To gain insight into how acetylation may regulate formation of fibrils in tau, we set out to determine a cryo-EM structure of an acetylated tau fragment fibril assembly. The R1R2_23Ac peptide aggregated rapidly and TEM imaging revealed fibrils with a defined and reproducible twist (Figure 2D,G). We then optimized cryo-EM grids with R1R2_23Ac fibrils so that the fibrils were dispersed to facilitate automatic picking of fibril filaments (Figure S3A). We collected 6,209 images on a Titan Krios, used crYOLO to auto-pick filaments, and used Relion 3.136 to reconstruct 2D and 3D maps of the R1R2_23Ac fibril (Figure S3B,C, Table 1 and methods). In parallel, we also acquired cryo-EM data on triple acetylated R1R2 fibrils (i.e. R1R2_123Ac) but 2D reconstruction revealed little twist prohibiting structure determination (Figure S3D). The core of the double acetylated R1R2_23Ac fibril is well resolved with ~3.5 Å resolution with the periphery being less ordered including the N-terminal portions of each peptide in the left and right edges of the layer more poorly resolved >4.5 Å (Figure S3E). The fibril exhibited a crossover distance of 855 Å, equivalent to a twisting angle of −1o and a helical raise of 4.75 Å. The fibril was assumed to have a left-handed twist since the resolution of the map, 3.9 Å, was not sufficiently high to determine the handedness unambiguously (Figure S3F). Each layer is comprised of 12 different fragments (Figure S3G,H) that fit the density well (Figure 3C). Each fibril layer exhibits several features that are related by symmetry. The central core of the fibril is comprised of two full-length R1R2_23Ac fragments (i.e. TENLKHQPGGGK(Ac)VQIINK(Ac)) that are related by a 2-fold symmetry (Figure S3H; i.e. A interface). This core is stabilized by a network of hydrogen bonds across two fully extended 18 residue R1R3_23Ac fragments (Figure S3H; i.e. A interface). Focusing on the AcK274 and Q269, we find that they form a hydrogen bond network with the 2-fold related Q269 and AcK274 of the opposite peptide fragment (Figure S3I). This tetravalent hydrogen bond network is the first description of a gain-of-function mediated by acetylated lysine in a cryo-EM fibril structure. Moving from this central core, the remainder of the structure is comprised of 274K(Ac)VQIINK(Ac)280 trimers that are centered on the acetylated K280 and each side contains two trimers that are related by a two-fold symmetry (Figure S3H,I; i.e. AcK cluster). These interactions are stabilized by contacts between a single acetyl K280 (i.e. AcK280) that forms a hydrogen bond with N279 but also nonpolar contacts to I278 and V276 (Figure S3I). We also find four trimeric “AcK clusters”, in which pairs of these trimers are related by a 2-fold symmetry in which two 274K(Ac)VQIINK(Ac)280 form stabilizing interactions centered on I278 (Figures 3D and S3I; Interface B). Similar interactions were described in x-ray crystal structures of the VQIINK amyloid motif (Figure S4A,B)37. The periphery of these contacts is stabilized by contacts between acetyl K274 and Q276 (Figure S3I). In all, a single layer in our R1R2_23Ac fibril structure is composed of 12 fragments that are stabilized by a combination of polar and nonpolar contacts and highlighted by the gain-of-function mediated by acetylated lysine.
Table 1.
Data collection and refinement statistics.
| Data collection | |
|---|---|
| Microscope | Titan Krios |
| Acceleration Voltage (kV) | 300 |
| Detector | K3 |
| Software | SerialEM 3.8 |
| Magnification | 105,000x |
| Pixel size at detector (Å/px) | 0.83 |
| Defocus range (μm) | −1.20 to −2.40 |
| Total dose (e−/ Å2) | 52 |
| Exposure time (sec) | 4.50 |
| Number of movie frames | 50 |
| Reconstruction | |
| Usable micrograph | 5901 |
| Box size (pixel) | 180 |
| Total extracted segments | 1,696,844 |
| Number of segments after 2D | 125,446 |
| Number of segments after 3D | 45,674 |
| Symmetry imposed | C1 |
| Helical rise (Å) | 4.75 |
| Helical twist (°) | −1.00 |
| Crossover length (Å) | t 855 |
| Map sharpening B-factor (Å2) | −143.25 |
| Map resolution (Å; FSC=0.143) | 3.88 |
| Model composition | |
| Non-hydrogen atoms | 4,096 |
| Protein residues | 504 |
| Number of chains | 4.00 |
| Water | 0.00 |
| Ligands (Acetylated) | 8.00 |
| Model validation | |
| Map CC (mask) | 0.74 |
| MolProbity score | 2.14 |
| Clash score | 25.23 |
| R.M.S deviations bonds (Å) | 0.007 |
| R.M.S deviations angle (°) | 1.291 |
| Rotamer outliers (%) | 0.00 |
| Ramachandran plot (favored/allowed/outliers) | (96.30/3.70/0.00) |
| Cβ outliers | 0.00 |
| CaBLAM outliers (%) | 2.56% |
Figure 3. Cryo-EM structure of the R1R2_23Ac fibril.
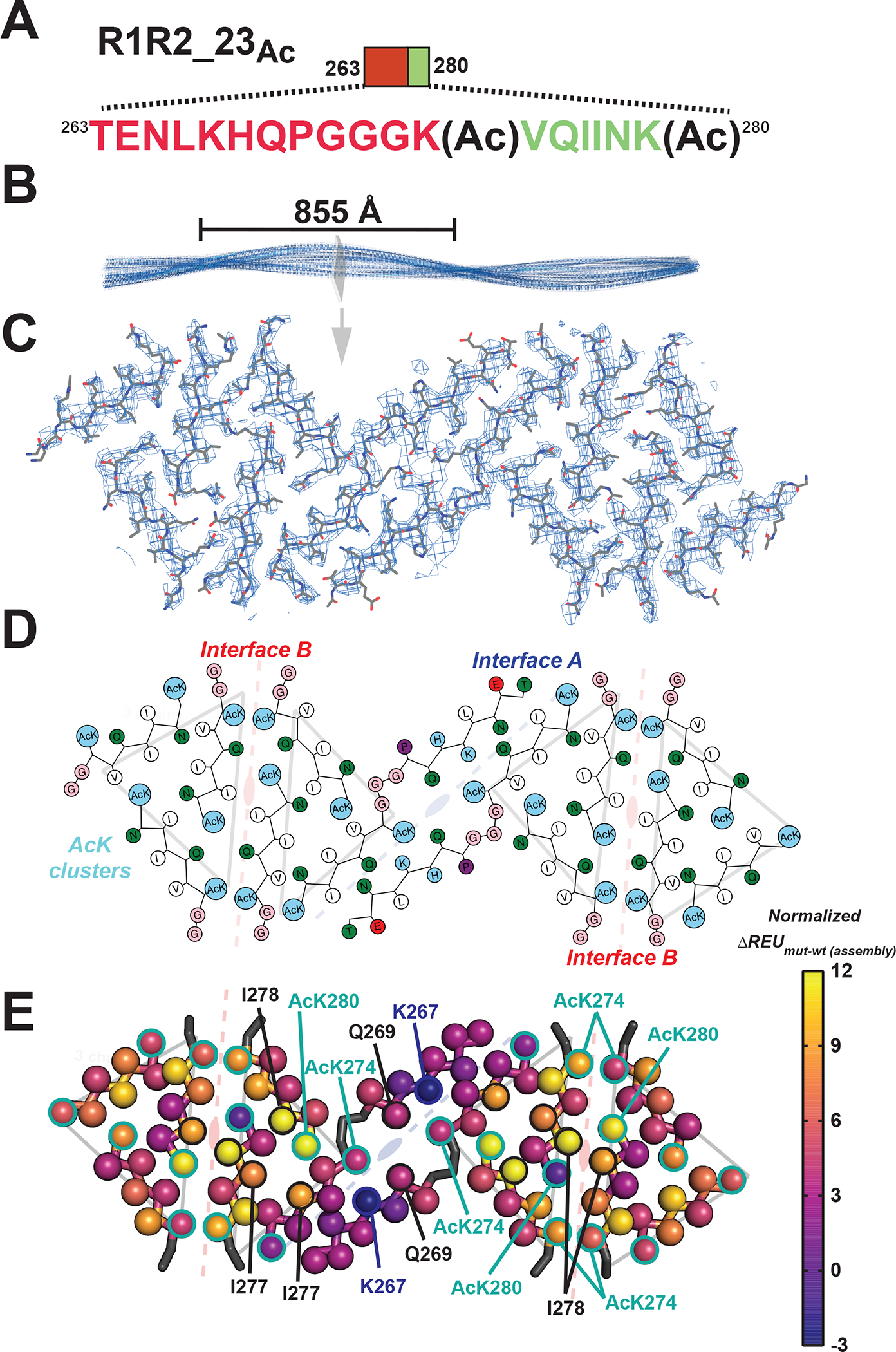
A. Schematic of the R1R2_23Ac sequence and the acetylation sites. The sequence is colored as in Figure 1A. Lysine acetylation sites are indicated by “Ac” following the modified lysine residue. B. 3D reconstruction of the fibril illustrating the cross-over distance. The fibril is shown in cartoon and is colored in blue. A single layer of the fibril is illustrated as a slice. C. Fit between the model and map of a single layer is contoured at 7 σ. Structure is shown as sticks with backbone and side chains. D. 2D illustration of the amino acid composition of the 12 molecules in the layer. AcK and lysine residues are colored light blue, nonpolar residues are white, polar residues are green, acidic residues are red and glycines are colored in pink. The description of the symmetry of the different molecules described in Figure 3E are shown in the background. E. Mapping per residue profiles onto our cryo-EM structure uncovers residues that contribute more to stability of the monolayer assembly. Structures are shown as a single layer shown in ribbons with C-beta atoms shown in spheres for each amino acid. The C-beta is colored by changes in energy due to individual amino acid mutations to alanine using the plasma color scheme, yellow (12 ΔREUs, important) to blue (−3 ΔREUs, not important). Key residues in each interface are indicated by arrows. The description of the symmetry of the different molecules described in Figure 3E are shown in the background.
Energetics of amino acid contributions in the R1R2_23Ac fibril structure
We next interpreted the putative stability of these interactions. We leveraged a recently described computational method that captures the energetic contribution of interactions in a fibril based on changes in calculated energy of the assembly following alanine substitution38. Application of this method allows us to begin interpreting which interactions are important in our fibril assembly. Starting at the central “interface A”, the interactions between AcK274 and Q269 are only defined by hydrogen bonds which do not contribute significantly to the calculated energy scores (Figures 3E and S3G; Interface A). In this core interface, mutation of K267 to alanine is stabilizing, indicating that burial of the lysine charge may be destabilizing. Interestingly, for the R1R2 fragment, the only other acetylation modification pattern that drove assembly included acetylation on all three lysine residues, including K267 but did not yield fibrils with consistent morphologies suggesting that placement of an acetyl K267 in the context of our structure may be sterically incompatible. The other two interfaces contribute more to the stabilization. In the “interface B” site, the symmetric I278 contacts are both important for stability while the adjacent acetylated K280 contribute unevenly with one being important and the other not (Figures 3E and S3G; Interface B). Finally, pairs of acetylated K274’s contact each other across the interface at both ends and all four contribute modestly to the energetics (Figures 3E and S3G; Interface B). The ”AcK cluster” interface centered on acetylated K280 which forms a hydrogen bond to N279 and nonpolar van der Waals interactions with I277 and I278 (Figures 3E and S3G; Ack cluster). In parallel, we used the solvation energy calculations39,40 to assess congruence with the alanine mutational data. The calculated energetics are similar between the two methods, despite the fact that the previous method is unable to capture the contribution of acetylated lysine (Figure S4C,D). Thus, our data support that specific patterns of acetylation proximal to amyloid motifs may dysregulate aggregation inhibition to drive rapid fibril assembly by stabilizing new interactions not possible with lysine.
Finally, we relate our structure to previously deposited structures of tau fragments and ex vivo tauopathy assemblies. As described above, the interactions observed in the “interface B” of our fibril structure, resemble previously observed interactions in the x-ray crystallographic structure of KVQIINKKLD peptides (Figure S4A)37. In the crystal structure of KVQIINKKLD peptides, V275 and I277 form 2-fold symmetric interactions (Figure S4A) while in a crystal structure of the minimal amyloid motif, VQIINK, I277 and I278 form stacking interactions (Figure S4B, i.e. translational)37. In our structure and within the “AcK cluster”, two I277s interact with the aliphatic chain of AcK280 while N279 makes a hydrogen bond to AcK280 (Figure S3I). In interface B, I278-I278 make symmetric interactions similar to VQIINK alone (Figures S3G and S4B)37. We next compared the conformation of our structure to ex vivo tauopathy structures. From our structure the only region that is compatible with longer fragments of tau is the extended 18 residue fragment that forms “Interface A” (Figure 4A). We find that the most similar element observed previously is derived from the PiD structure in which the region spanning repeats 1 and 3 similarly adopts a bulge around the P-G-G-G motif (Figure 4B). Indeed, the alignment of the fibril core of R1R2_23Ac and the PiD tau structure results in an root mean square deviation (rmsd) of 1.9 Å and the VQIINK segment from our structure superimposes to the VQIVYK element in the PiDs structure (Figure 4C). Interestingly, V275 and I277 face inward to make heterotypic interactions to the 337VEVKSE342 motif in PiD while in our structure they face outward and stabilize nonpolar contacts with an acetylated lysine and I277 from a different fragment (Figure 4D). Thus, the exposure of nonpolar residues in these elements may nucleate fibril formation as proposed previously38.
Figure 4. Similarity of the R1R2_23Ac structure to the disease-derived Pick’s disease fibril.
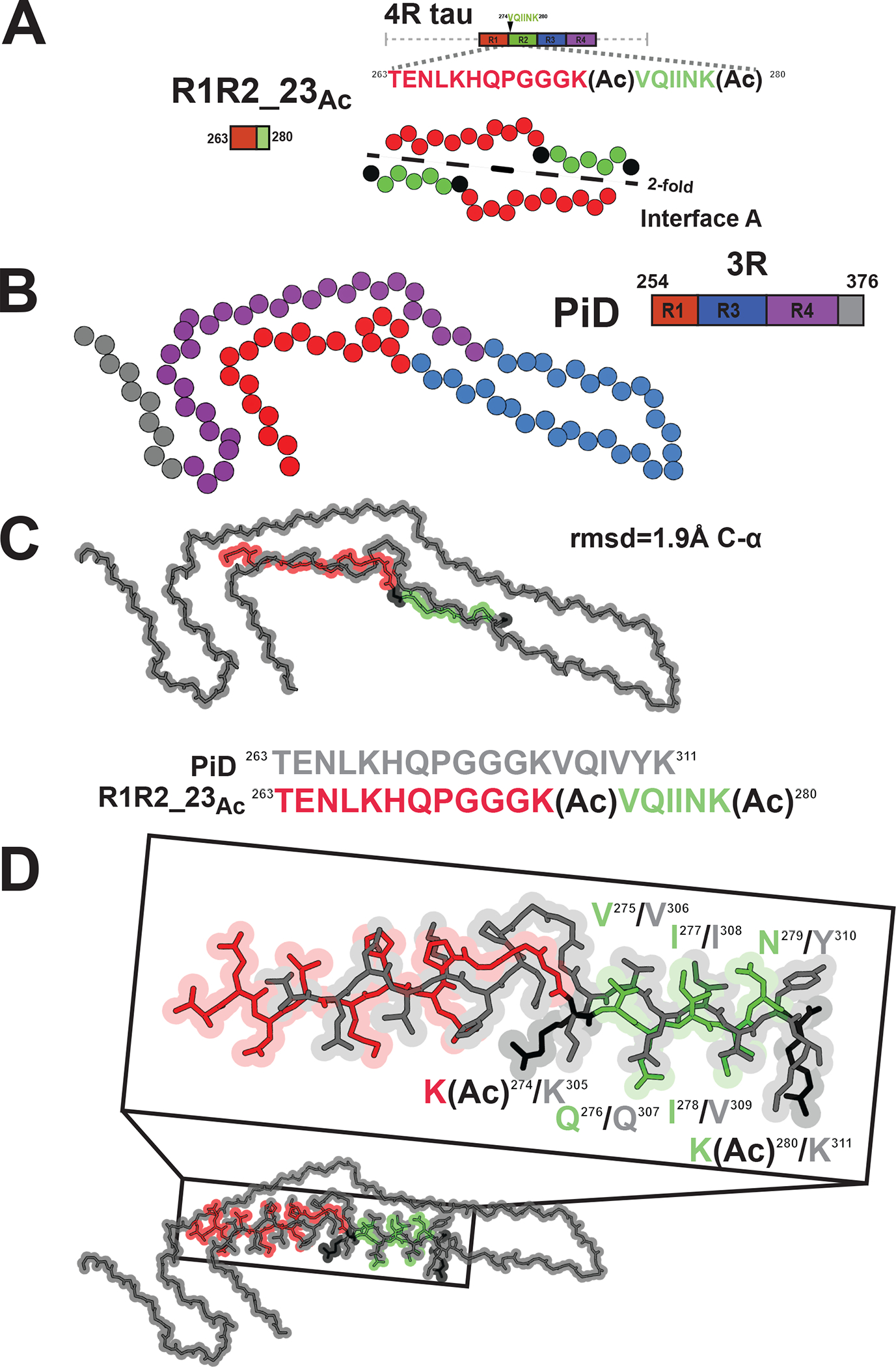
A. Schematic illustrating the R1R2_23Ac core sequence (top). View of the core of the R1R2_23Ac fragment highlighting the 2-fold “interface A” interaction (bottom). Acetylation sites are shown in black. Fragments are colored as in Figure 1A. Structure is shown as spheres with Ca and is colored as in Figure 1A. B. Illustration of a monomer layer from a protofilament of a PiD fibril (PDB id 6gx5) encoding residues 254–376. Structure and schematic is colored as in Figure 1A. Structure is shown as spheres with C-a. C. Overlay of the R1R2 23Ac core fragment (one chain) with the equivalent sequence in the PiD structure reveals 1.8 Å C-a atom rmsd (top). Comparison of the equivalent sequences in PiD and R1R2_23 Ac that were aligned (bottom). PiD and R1R2_23Ac structures/sequence are colored grey and red/green, respectively. Acetylated lysines are colored in black. The structures are shown as spheres with backbone-only. D. Full-atom alignment of the R1R2_23Ac to the homologous region in the PiD structure. Key residues from the 275VQIINK280 and 306VQIVYK311 motifs are highlighted. Residues are numbered according to the 4R nomenclature and colored as in Figure 4C. Structures are shown in space fill with all-atoms.
Defined acetylation patterns on tau fragments promote seeding in cells
We also tested the biological activity of the in vitro aggregated peptides, R1R2, R2R3 and R1R3 peptides series (acetylated, glutamine mutants and WT), in a cell-based tau aggregation experiment (Figure 5A). In this system, the tau repeat domain is expressed in HEK293T cells as fusion to FRET compatible cyan and yellow fluorescent proteins41. Transduction of exogenous, recombinant or patient derived, tau seeds into these tau biosensor cells converts the endogenous tau into inclusions that can be quantified by FRET41. For R1R2, R2R3 and R1R3 peptide series, we evaluated whether in vitro formed aggregates containing acetyl-lysine modifications are able to seed the intracellular tau. Incubated peptides were transfected into tau biosensors and the FRET was quantified using flow cytometry to assess the extent of tau seeding (Figure S5A). We find that the R1R2_123Ac (i.e. AcK267, AcK274 and AcK280) was the only peptide in the R1R2 series that seeded significantly with 10.5±0.7% of the cells containing aggregates (Figure 5B). In the R2R3 peptide series, the R2R3_2Ac yielded signal above baseline with ~1.1% of cells containing aggregates (Figure 5C). Unsurprisingly, the R1R3 peptides did not induce seeding, consistent with a 4R vs 3R isoform seeding barrier (Figure 5D)42. In parallel, we also tested activity of our “Q” peptide series and found that the R1R2Q and R1R3Q series showed seeding below the 1% baseline, while in the R2R3 series, R2R3_2Q seeded 2.2±3.5 cells and for R2R3_123Q is 1.5±0.5%. From these data, the R1R2_123Ac peptide had the most seeding activity while nearly all other fragments were below 1% or slightly above this threshold (Figure S5B–D). Our data shows that acetylation patterns on our model peptides are important for fibril formation, however not all fibrils lead to tau aggregation in cells. Interestingly, in vitro fibril formation is not exactly linked to seeding activity in cells suggesting that other factors may be involved in propagation of seed conformation when modifications are present on the seed.
Figure 5. Defined patterns of acetylation on tau fragments promote cellular tau seeding.
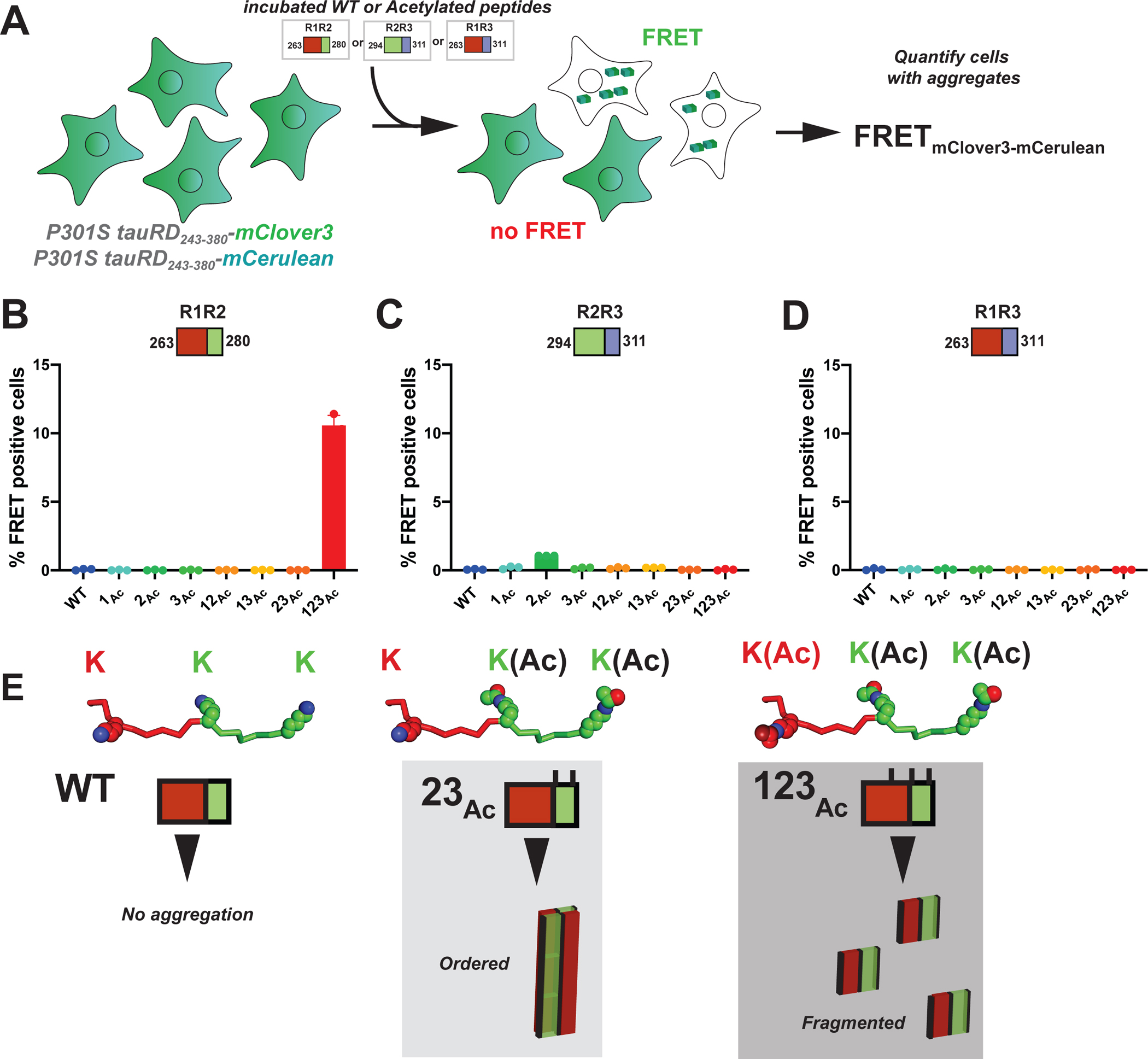
A. Schematic of the cell-based tau aggregation assay. Tau repeat domain is expressed as a fusion to mClover3 and mCerulean. Incubated peptides (WT or Acetylated) are transduced into the tau expressing cells and evaluated for conversion of the intracellular tau into aggregates as measured by FRET on a flow cytometer. Quantification of FRET signal across all peptides transduced into the tau biosensor cells: R1R2 (B), R2R3 (C) and R1R3 (D). Bar plots are colored as in Figure 2. Data is shown as averages across three experiments with error bars representing a 95% CI of each condition. E. Model for how specific acetylation patterns on the R1R2 drive formation of fibrils but not all lead to seeding cells. The WT R1R2 sequence does not aggregate (left). The R1R2_23Ac pattern (middle) leads to large ordered aggregates while the R1R2_123Ac modified peptide (right) leads to more fragmented seeds which promote rapid aggregation in cells.
Discussion
Here we present a new tau aggregation system to interpret the role of pathological acetylation patterns driving their aggregation. We identify specific acetyl-lysine sites that can promote tau assembly into fibrils and show that lysine to glutamine substitutions have largely similar effects by position but lead to different fibril morphologies. We next determined a structure of a tau fibril encoding two pathological acetylation sites uncovering exciting new interactions. Consistent with our previously described tau model system15, the fragments regulate tau aggregation via formation of local structures through a P-G-G-G motif and our model peptide encoding acetylation patterns uncovered an “open” conformation in the fibril of the protective hairpin. We additionally, find that acetylation on lysines promotes the formation of new interactions that balance polar and nonpolar interactions to stabilize the fibrils suggesting that the positive charge of lysine residues is inhibitory to aggregation. We also find new interactions involving the VQIINK amyloid-forming motif that interact with acetyl-lysine that further stabilize the fibrils. A comparison of our new structure to pathological conformations observed in disease reveals similarities, highlighting our ability to engineer sequences that begin to control tau assembly into disease-like conformations.
The dual roles of lysine-based regulation of tau aggregation
Under normal healthy conditions, tau is aggregation resistant and our prior work suggested that local structural rearrangements around amyloid motifs15 plays a role in converting tau to adopt pathological conformations even as a monomer13,14. Using these conformational changes as a guide, we developed a tau fragment model system that highlights the role of these regulatory conformations and how pathogenic mutations disrupt them to drive aggregation. In this study, we further leverage this system to show that specific acetyl-lysines can also perturb protective structures to promote amyloid motif-dependent fibrillization. These data showcase specifically that lysine residues proximal to amyloid motifs slow down aggregation and their acetylation rapidly switches them into pathogenic conformations. Furthermore, our structure reveals that while the loss of charge on the lysine is important for fibrillization, the gain of interactions also plays a role in fibril stabilization. It is interesting to note that in all existing structures of tau, recombinant and ex vivo, lysine residues often face outward toward solvent and do not stabilize the aggregates indicating that lysine residues may play protective roles. This is perhaps why tau aggregation inducers are often polyanionic, including RNA or heparin, in which the negative charges on the inducer help align the basic residues on tau to stabilize aggregation-prone conformations11,12,14. In the structures, we also observe that lysine residues are inhibitory to tau aggregation – evidence that acetylation can promote peptide assembly into fibrils faster. In disease-derived fibrils, lysine residues are often solvent exposed and do not contribute to the stability of fibrils38 again indicating that under normal conditions they are protective but reorienting them through polyanion binding may preferentially expose the amyloid motifs to promote assembly.
Gain of interactions mediated by acetyl-lysine groups
Classically, lysine acetylation is thought to control histone modifications that regulate DNA binding and thus exposure of sequences for subsequent transcription and gene activation43. It has also been proposed that enzymatic acetylation and subsequent deacetylation may modify lysines to regulate activity by destabilizing or conversely stabilizing interactions. For example, this mechanism has been proposed to regulate activity of a J-domain protein chaperone where lysine residues on the C-terminal domain may be modified to regulate its substrate activity or possibly recruitment of other chaperones such as Hsp7044. This type of regulation has similarly been observed with phosphorylation that controls activity of kinases. The role of acetylation on tau remains unknown but similarly to phosphorylation, these PTMs have been proposed to increase tau’s aggregation propensity. Despite this, there are no clear patterns of PTMs for tau that drive its aggregation directly and it may simply be a modification as the protein ages or conversely a mark on the pathological assemblies following their conversion into amyloid structures. Our work has shown that tau phosphorylation may occur on tau prior to disease but there is no clear signature that marks its conversion into pathological conformations that seed in a tauopathy mouse model17. Interestingly, phosphorylation on tau often occurs on the periphery outside of the repeat domain required for fibril assembly while acetylation occurs on the repeat domain but its role remains unknown. Basic residues within the repeat domain of tau, including lysines, dictate tau binding to negatively charged microtubules, it is possible that acetylation at lysine sites may reduce tau’s microtubule stabilizing physiological function. Modification of lysine 280 and 281 with acetyl groups has also been linked to the appearance of pathological tau species. Work from Lashuel et al used semi-synthetic methods to produce acetylated tau at position K280 yielding modest increases in heparin-induced tau aggregation with mixed oligomer and fibril morphologies linking acetylation to increased aggregation propensity22. Acetylation of tau may reduce its clearance through both the ubiquitin proteosome and autophagy pathways thus leading to the accumulation of aggregation-prone conformations31,45. Interestingly, reduction of tau acetylation may also be neuroprotective46. Guided by these data and the possibility that tau acetylation is a hallmark of tau-dependent neurodegeneration has also spurred efforts to develop biologics to detect acetylated forms of tau seeds and as a possible therapeutic strategy26. To date, lysine acetylation on tau has suggested loss-of-function activity with respect to microtubule binding or clearance and perhaps gain-of-function through amyloid-based proteotoxicity, but the details of these mechanisms remain unclear. Our cryo-EM structure highlights how acetylation of lysines surrounding tau amyloid motifs yields new interactions not possible with lysine residues hinting towards the formation of alternate structures with perhaps higher toxicity.
Design of tau elements to regulate tau aggregation
Our prior work has proposed that control of amyloid motif exposure may explain tau’s capacity to assemble into distinct structural polymorphs observed in tauopathies. In a more recent analysis of tauopathy fibrils, we showed that amyloid motif interactions with other nonpolar elements in the tau sequence are essential for tau folding into different shapes38. We developed ways to probe the importance of these interactions with peptides in vitro but also in cells leveraging tau’s capacity to replicate conformation in a prion-like manner38. While our understanding of how these different fibrillar shapes are formed remains unknown, more general rules have emerged. We have found that the core amyloid motif interactions are modular in the ex vivo structures followed by more peripheral secondary interactions38. In the present work, we demonstrate a new way dysregulate local structures and promote amyloid motif exposure using lysine acetylation, which promotes gain-of-function interactions. Interestingly, as we have shown previously the R2R3 element at the interface between repeats 2 and 3 was extremely sensitive to aggregation in response to pathogenic mutations while the R1R2 and R1R3 were aggregation resistant even in the context of the proline to serine mutations in the P-G-G-G motif. We do not yet understand how to perturb these with mutations, but our chemical modification strategy suggests that it is possible through acetylation of lysines or alternatively with mutations such as glutamine. Thus, we may be able to regulate exposure of amyloid motifs in a controlled manner to build more complex folds. Indeed, this concept was recently highlighted by Scheres et al where they used fragments of tau in combination with different salts to assemble structures reminiscent of ex vivo structures46. Thus, regulation of assembly may be further controlled by explicit exposure of amyloid motifs. Many more experiments are required to demonstrate this capacity, but the present works sets the stage to fully regulate tau assembly into conformations observed in disease.
Our work supports an exciting new mechanism for how specific disease-associated lysine acetylation signatures may trigger assembly of tau towards pathogenic conformations. Structural analysis of the fibrils reveals gain of interactions mediated through lysine acetylation proximal to amyloid motifs that stabilize fibrillar conformations that partly mimic states observed in disease. Lysine residues show inhibitory effect in fibril aggregation, however modifying these lysines with acetyl groups can alter such effect to allow a mix of polar and nonpolar contacts. We envision that even though acetylation may not control final folding, it promotes interactions with amyloid motifs and perhaps plays a role in initiating tau assembly into pathological conformations. Future efforts must be focused on the design of tau elements that regulate exposure of amyloid motifs to drive formation of distinct structural polymorphs.
STAR Methods
RESOURCE AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Lukasz A. Joachimiak (Lukasz.Joachimiak@utsouthwestern.edu)
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The structural datasets generated during the current study are available in the Electron Microscopy Data Bank repository (https://www.ebi.ac.uk/emdb/) under accession number, EMD-28721 [https://www.ebi.ac.uk/emdb/EMD-28721]. The structural model that was fit into the density is available in the Protein Data Bank under PDB id 8FNZ (https:/doi.10.2210/pdb8FNZ/pdb). Raw ThT and cell-based aggregation data are available as Data S1 (Related to Figure 1 and 2) and Data S2 (Related to Figure 5), respectively. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell lines used in the study are listed in the key resources table.
ADDITIONAL RESOURCES.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| PBS | Sigma | Cat#P3813 |
| Succinimidyl acetate | TCI | Cat#S0878 |
| DMSO | Sigma | Cat#276855 |
| NH4HCO3 | Sigma | Cat#09830 |
| Peptide desalting spin column | Pierce | Cat#89851 |
| TFA | Sigma | Cat#302031 |
| Thioflavin T | EMD Millipore | Cat#596200 |
| Lipofectamine 2000 | Invitrogen | Cat#11668500 |
| Opti-MEM | Gibco | Cat#31985088 |
| Trypsin-EDTA | Fischer Scientific | Cat#25300120 |
| 2% paraformaldehyde | EMS | Cat#15710 |
| See Table S1 for sequences of tau peptides | This study | N/A |
| Deposited data | ||
| EM images and deposited map of the R1R2_23Ac fibril structure | This study | EMD-28721 |
| Coordinates of the R1R2_23Ac fibril structure | This study | 8FNZ |
| Experimental models: Cell lines | ||
| Tau RD P301S HEK293T biosensors | ATCC | Cat#CRL-3275 |
| Software and algorithms | ||
| SerialEM | Mastronarde, D. N. 200547 | https://bio3d.colorado.edu/SerialEM |
| CTFFIND v4.1 | Rohou, A. & Grigorieff, N. 201548 | https://grigoriefflab.umassmed.edu/ctffind4 |
| RELION v3.1 | Scheres, S. H. W. 202036 | https://www3.mrc-lmb.cam.ac.uk/relion/index.php/Main_Page |
| EMAN v2.31 | Tang, G. et al. 200749 | https://blake.bcm.edu/emanwiki/EMAN2/Install/BinaryInstallAnaconda/2.31 |
| crYOLO v1.8.0 | Wagner, T. et al. 202050 | https://github.com/MPI-Dortmund/cryolo/blob/7b138d25f573be097662b0e0fc696b56c5486113/source/installation.rst] |
| COOT v9.4 | Emsley, P. et al. 201053 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| ddG fibril analysis | Mullapudi, V. et al. 202338 | https://git.biohpc.swmed.edu/s184069/flex_ddg_ala_scn_runner |
| Rosetta v3.12 | Leman, J. K. et al. 202058 | https://www.rosettacommons.org/ |
| FlowJo v10 | BD | https://www.flowjo.com/solutions/flowjo |
| GraphPad Prism 9.4.1 | Treestar | https://www.graphpad.com/ |
| Other | ||
| Formvar-coated 300-mesh copper grids | EMS | Cat# FCF200CU-50 |
| R1.2/1.3, 300 mesh copper grids | EMS | Cat# Q350CR1.3 |
METHOD DETAILS
Chemical acetylation of peptides with N-Succinimidyl Acetate
A 1 mM stock in a volume of 0.5 mL of wild type synthetic peptides R1R2, R1R3, R2R3 and R3R4 was prepared in PBS (Sigma). The pH was adjusted to pH 7.4. 20 μL of 250 mM of N-succinimidyl acetate (TCI) in anhydrous DMSO (Sigma) was added to each tube and the reactions were then incubated at room temperature RT for 30 mins. The reactions were quenched by the addition 50 μL 1 M NH4HCO3 (Sigma) followed by an additional 30 min incubation. The product mixtures were purified with peptide desalting spin column (Pierce) and the elutions were subsequently frozen and lyophilized (LabConco).
Both the control and acetylated peptides were reconstituted in 1mL of PBS pH 7.4 to obtain a concentration at 500μM for subsequent ThT fluorescence aggregation assays. The concentration was determined by the starting mass of peptide. In facilitate comparisons between modified and unmodified peptides, the unmodified peptides were subject to the same desalting and lyophilizing procedure to minimize the concentration differences among reconstituted unmodified and modified samples.
Peptides with customized acetylation
Peptides with site specific acetylation were obtained from Genscript (>95% purity) with N-terminal acetylation and C-terminal amidation. Overall, 30 peptides were synthesized with single, double and triple modifications (R1R2, R2R3, R1R3, R3R4 and R4R’, see Table S1) including unmodified controls.
ThT fluorescence aggregation assays
Peptides were treated with 100 μL of concentrated Trifluoroacetic acid TFA (Pierce) for 1 hr at RT, in order to disaggregate any preformed aggregates. TFA was removed under nitrogen, and then lyophilized to remove any additional residue. After TFA treatment, peptides were resuspended in 1 × PBS (Sigma) to make 2 mM of stock, the pH was adjusted to 7.4 with NaOH. A final concentration of 500 μM of each peptide was used in 96 well plate assay with addition of 25 μM ThT (pH adjusted to 7) in a total 60 μL volume for each well. All conditions were done in triplicates. Kinetic scans were run every 30 min on a TecanSPARK plate reader at 446 nm Ex (5 nm bandwidth), 482 nm Em (5 nm bandwidth). ThT fluorescent readings of site-specific acetylated peptides and their controls were plotted in Figure 2. The t1/2 values were derived from a non-linear regression model fitting in GraphPad Prism. Similar method was used for K to Q mutated peptides series.
Transmission Electron Microscopy
5 μL sample was loaded onto a glow-discharged Formvar-coated 300-mesh copper grids (Sigma) for 1 minute and was blotted by filter paper followed by washing the grid with 5 μL ddH2O. After another 30 seconds, 2% uranyl acetate was loaded on the grids for 1 minute and blotted again. The grid was dried and loaded into a FEI Tecnai G2 Spirit Biotwin TEM. All images were captured using a Gatan 2K×2K multiport readout post column CCD at the UT Southwestern EM Core Facility.
Cryo-Electron Microscopy
The screening of cryoEM condition was performed with FEI Talos™ Arctica transmission electron microscope with K2 direct electron detector. 3 μL sample was applied to glow-discharged R1.2/1.3, 300 mesh copper grids (EMS) that were plunge-frozen in liquid ethane using Vitrobot Mark IV, a Thermo Fisher Scientific Vitrobot. The optimized Vitrobot setting was: blotting force −5, blotting time 4s, humidity 98–100% at RT. Cryo-EM data were acquired at the FEI Titan Krios 300kV transmission electron microscope with Bioquantum energy filter, K3 direct electron detector with fringe free illustration at UT Southwestern Cryo-Electron Microscopy Facility. The prepared grids were loaded for a 24 h SerialEM run47. Each grid was loaded to the stage for session set-up, grid square selection. During the fully automated SerialEM runs, each grid was loaded onto the stage, grid squares were brought to eucentric height, and holes were selected with the stored ice filter settings. All images were recorded at a dose rate of ~8 e-/pixel/s, 105,000x magnification, spot size 7, CDS mode using SerialEM software47, and converted to tiff format using relion_convert_to_tiff prior to processing. 6209 movies were collected with a 10eV energy filter slit width. Images were collected in EER mode using the aberration free image shift method in SerialEM v3.847 (See Table 1 for more details).
Helical Reconstruction of Fibrils
Movie frames were gain-corrected, aligned and dose weighted using RELION’s motion correction program36. Contrast transfer function (CTF) parameters were estimated using CTFFIND-4.148. Helical reconstructions were performed in RELION-3.136. A total of 210 filaments from 24 motion-corrected movies were manually picked using EMAN2.3149 and were used to train the crYOLO 1.8.0 neural network for auto-picking50. The entire dataset of 5901 movies was auto-picked and the coordinates were imported into Relion. Images of filament segments were extracted at different boxsizes of 686 and 180 pixels to perform reference free 2D classification. The best looking 2D classes of 686 pixel segments were used to estimate the cross-over distance of the filaments. The best looking 2D classes of 180 pixel segments were used to generate a de novo initial 3D reference with relion_helix_inimodel2d script36 and for further 3D processing. Fibril helix is assumed to be left-handed. After multiple rounds of 3D classification, helical segments potentially leading to the best reconstructed map were chosen for 3D auto-refinements. The final 3D map was post-processed in Relion with a 6-pixel extended initial binary mask. The final overall resolution estimate was evaluated based on the FSC at 0.143 threshold between two independently refined half-maps51. Due to the low resolution of the map for the 3D reconstruction, we cannot unambiguously assign the handedness of the twist to be left- or right-handed but importantly independent of the handedness of the twist, the fold and interactions are identical.
Model building and refinement
The refined map was further sharpened using phenix.auto_sharpen at the cutoff resolution of 3.88 Å52. Previously published model of tau’s segments VQIINK37 (PDB id: 5V5C) [http://doi.org/10.2210/pdb5V5C/pdb] was used as the initial template to build the filament model in COOT53. Acetyl moieties were added to Lys residues by using PyTMs – a PyMOL plugin54. Multiple rounds of model refinement were carried out using the default settings in phenix.real_space_refine with non-crystallographic symmetry constraints, minimization_global, rigid_body, and local_grid_search55. Model geometry was evaluated using MolProbity, a built-in tool in Phenix56. After each round of refinement, problematic or poorly fitting regions in the model were manually modified using COOT. This procedure was repeated until we achieved an acceptable stereochemistry model and a high overall correlation coefficients of model:map.
In Silico Rosetta and Calculation with Backrub Sampling
A four layer assembly of our cryo-EM structure was prepared using Pymol (version 2.4). Briefly, we used structural alignment to superimpose the top two chains of the deposited fibril with the bottom two chains from a duplicated fibril assembly, preserving the geometry of the assembly while extending the fibril length. Overlapping chains were removed and chains were renamed to produce assemblies of the desired number of layers with chain lettering increasing from the top to the bottom layer. To interpret energetics of acetylated lysine modified parameter files were generated for acetylated lysine. These assemblies were then used as input for the subsequent mutagenesis and minimization using the RosettaScripts57 interface to Rosetta58 in framework similar to previously described38.
Changes in assembly energy were calculated using a method adapted from the Flex ddG59 that were recently implemented for amyloid fibrils38. For both the calculations, all chains were mutated. From the input assembly, a set of pairwise atom constraints with a maximum distance of 9 Å were generated with a weight of 1, using the fa_talaris2014 score function. Using this constrained score function, the structure then underwent minimization. After minimization, the residues within 8 Å of the mutation site underwent backrub sampling to better capture backbone conformational variation. These sampled structures were either only repacked and minimized, or the alanine mutation was introduced, followed by repacking and minimization. For calculations, the bound wild-type and bound mutant structures reported by the interface ddG mover were used for estimating the change in assembly energy due to an alanine substitution. This is repeated for thirty-five independent replicates. The lowest energy bound mutant and bound wild-type structure energies from each replicate were extracted, and the change in energy as calculated by subtracting the wild-type, non-mutagenized assemblies’ energy from the mutant assemblies’ energy. The mean change in energy over the 35 replicates was reported as that residue’s .
The . for a given residue were then averaged over 35 replicates to yield the final values for the residue. This procedure is repeated for every residue in the structure to generate a set of values for all residues in each fibril structure.
Flow cytometry
The HEK293T tauRD FRET biosensor cell line developed by Diamond and colleagues can sensitively and specifically detect pathogenic tau seeds from recombinant or tissues sources41. For all experiments, HEK293T tauRD biosensor cells co-expressing tauRD-mCerulean and tauRD-mClover (see key resources table) were plated in a 96-well plate at 20,000 cells per well in 130 μL of media, and used after 24 h of plating. Peptide incubation end product (from ThT assay) were sonicated at amplitude 75 for 20 min on Q700 Sonicator 600 (QSonica). 2 μL Lipofectamine 2000 (Invitrogen) and 77uL Opti-MEM™ (Gibco) was incubated for 5 min, then 1 μL of sonicated peptide was added. The mixture was set still in cell culture hood for 20 min. 20 μL of the peptide mixture (seed mixture) was added to each well with cells. The final concentration of the peptide in each well was 0.5 μM. The seeding activity (FRET fluorescent) were visualized under fluorescent microscope after 48 h of peptide treatment, images were taken. Then the cells were harvested by 0.05% trypsin digestion and then fixed in PBS with 2% paraformaldehyde (EMS).
A BD LSRFortessa was used to quantify FRET in flow cytometry. To measure mCerulean and FRET, cells were excited with the 405 nm laser, and fluorescence was captured with a 405/50 nm and 525/50 nm filter, respectively. To measure mClover3, cells were excited with a 488 laser and fluorescence was captured with a 525/50 nm filter. To quantify FRET, we used a gating strategy where mCerulean bleed-through into the mClover3 and FRET channels was compensated using FlowJo analysis software (see Figure S5A for gating strategy details). As described previously, FRET signal is defined as the percentage of FRET-positive cells in all analyses. For each experiment, 10,000 cells per replicate were analyzed and each condition was analyzed in triplicate. Data analysis was performed using FlowJo v10 software (Treestar).
QUANTIFICATION AND STATISTICAL ANALYSIS
Fourier shell correlations were calculated using RELION. PDB models were validated using PDB software; available at the PDB. The ThT aggregation curves were fit to a non-linear regression model in GraphPad Prism 9.4.1 to estimate a t1/2max. The t1/2max values are reported as an average with standard deviation. See methods for details.
Supplementary Material
Data S1. Raw ThT fluorescence aggregation curves for R1R2, R2R3, R3R4, R1R3 and R4R’ acetylated peptides, related to Figures 1 and 2. Data is organized by figure panel.
Data S2. Raw tau biosensor seeding data using R1R2, R2R3 and R1R3 chemically modified peptides, related to Figure 5. Data is organized by figure panel.
Highlights.
Disease-associated patterns of lysine acetylation promote tau fragment fibrillization
Lysine acetylation proximal to amyloid motifs is sufficient for fibril formation
Structure of lysine acetylated tau fragment fibril reveals acetyl-specific interactions
ACKNOWLEDGEMENTS
L.A.J was supported by grants from the NIH-NCI (1U01CA242115), the Welch Foundation (I-1928-20200401) and the Chan Zuckerberg Initiative (CZI) Collaborative Science Award (2018-191983). Cryo-EM data were acquired at the Cryo-Electron Microscopy Facility (CEMF) and the Structural Biology Laboratory (SBL) at UTSW, which are supported by a grant from the Cancer Prevention & Research Institute of Texas (RP170644). Transmission electron microscopy was carried out at the Electron Microscopy Core Facility at UTSW, which is supported by the National Institutes of Health (NIH) (1S10OD021685-01A1 and 1S10OD020103 01). Computational resources were provided by the BioHPC cluster supported by the Lyda Hill Department of Bioinformatics at UTSW. We would like to thank Dr. Zhe Chen from the UTSW SBL core for their valuable help with cryo-EM data acquisition and analysis. We thank all members of the Joachimiak lab for discussions and input on the manuscript.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vaquer-Alicea J, Diamond MI, and Joachimiak LA (2021). Tau strains shape disease. Acta Neuropathol. (Berl.) 142, 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Crowther RA, Ghetti B, Goedert M, and Scheres SHW (2017). Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Falcon B, Zhang W, Murzin AG, Murshudov G, Garringer HJ, Vidal R, Crowther RA, Ghetti B, Scheres SHW, and Goedert M (2018). Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 561, 137–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang W, Tarutani A, Newell KL, Murzin AG, Matsubara T, Falcon B, Vidal R, Garringer HJ, Shi Y, Ikeuchi T, et al. (2020). Novel tau filament fold in corticobasal degeneration. Nature 580, 283–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Falcon B, Zivanov J, Zhang W, Murzin AG, Garringer HJ, Vidal R, Crowther RA, Newell KL, Ghetti B, Goedert M, et al. (2019). Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 568, 420–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi Y, Zhang W, Yang Y, Murzin AG, Falcon B, Kotecha A, van Beers M, Tarutani A, Kametani F, Garringer HJ, et al. (2021). Structure-based classification of tauopathies. Nature 598, 359–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, Barker SJ, Foley AC, Thorpe JR, Serpell LC, et al. (2014). Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82, 1271–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaufman SK, Sanders DW, Thomas TL, Ruchinskas AJ, Vaquer-Alicea J, Sharma AM, Miller TM, and Diamond MI (2016). Tau Prion Strains Dictate Patterns of Cell Pathology, Progression Rate, and Regional Vulnerability In Vivo. Neuron 92, 796–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clavaguera F, Lavenir I, Falcon B, Frank S, Goedert M, and Tolnay M (2013). “Prion-Like” Templated Misfolding in Tauopathies. Brain Pathol. 23, 342–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo JL, and Lee VM-Y (2014). Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med. 20, 130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, and Crowther RA (1996). Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 383, 550–553. [DOI] [PubMed] [Google Scholar]
- 12.Zwierzchowski-Zarate AN, Mendoza-Oliva A, Kashmer OM, Collazo-Lopez JE, White CL, and Diamond MI (2022). RNA induces unique tau strains and stabilizes Alzheimer’s disease seeds. J. Biol. Chem. 298, 102132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hou Z, Chen D, Ryder BD, and Joachimiak LA (2021). Biophysical properties of a tau seed. Nat. Publ. Group 11, 13602–13609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mirbaha H, Chen D, Morazova OA, Ruff KM, Sharma AM, Liu X, Goodarzi M, Pappu RV, Colby DW, Mirzaei H, et al. (2018). Inert and seed-competent tau monomers suggest structural origins of aggregation. eLife 7, 338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen D, Drombosky KW, Hou Z, Sari L, Kashmer OM, Ryder BD, Perez VA, Woodard DR, Lin MM, Diamond MI, et al. (2019). Tau local structure shields an amyloid-forming motif and controls aggregation propensity. Nat. Commun. 10, 2493–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mukrasch MD, Bergen M. von, Biernat J, Fischer D, Griesinger C, Mandelkow E, and Zweckstetter M. (2007). The “Jaws” of the Tau-Microtubule Interaction. J. Biol. Chem. 282, 12230–12239. [DOI] [PubMed] [Google Scholar]
- 17.Mirbaha H, Chen D, Mullapudi V, Terpack SJ, White CL, Joachimiak LA, and Diamond MI (2022). Seed-competent tau monomer initiates pathology in a tauopathy mouse model. J. Biol. Chem. 298, 102163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kinoshita J, and Clark T (2007). Alzforum. Methods Mol. Biol. Clifton NJ 401, 365–381. [DOI] [PubMed] [Google Scholar]
- 19.Barthélemy NR, Li Y, Joseph-Mathurin N, Gordon BA, Hassenstab J, Benzinger TLS, Buckles V, Fagan AM, Perrin RJ, Goate AM, et al. (2020). A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer’s disease. Nat. Med. 26, 398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, et al. (2012). National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. J. Alzheimers Assoc. 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, and Del Tredici K (2006). Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. (Berl.) 112, 389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haj-Yahya M, and Lashuel HA (2018). Protein Semisynthesis Provides Access to Tau Disease-Associated Post-translational Modifications (PTMs) and Paves the Way to Deciphering the Tau PTM Code in Health and Diseased States. J. Am. Chem. Soc. 140, 6611–6621. [DOI] [PubMed] [Google Scholar]
- 23.Mair W, Muntel J, Tepper K, Tang S, Biernat J, Seeley WW, Kosik KS, Mandelkow E, Steen H, and Steen JA (2016). FLEXITau: Quantifying Post-translational Modifications of Tau Protein in Vitro and in Human Disease. Anal. Chem. 88, 3704–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wesseling H, Mair W, Kumar M, Schlaffner CN, Tang S, Beerepoot P, Fatou B, Guise AJ, Cheng L, Takeda S, et al. (2020). Tau PTM Profiles Identify Patient Heterogeneity and Stages of Alzheimer’s Disease. Cell 183, 1699–1713.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arakhamia T, Lee CE, Carlomagno Y, Duong DM, Kundinger SR, Wang K, Williams D, DeTure M, Dickson DW, Cook CN, et al. (2020). Posttranslational Modifications Mediate the Structural Diversity of Tauopathy Strains. Cell 180, 633–644.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song H-L, Kim N-Y, Park J, Kim MI, Jeon Y-N, Lee S-J, Cho K, Shim Y-L, Lee K-H, Mun Y-S, et al. (2023). Monoclonal antibody Y01 prevents tauopathy progression induced by lysine 280-acetylated tau in cell and mouse models. J. Clin. Invest. 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Irwin DJ, Cohen TJ, Grossman M, Arnold SE, Xie SX, Lee VM-Y, and Trojanowski JQ (2012). Acetylated tau, a novel pathological signature in Alzheimer’s disease and other tauopathies. Brain J. Neurol. 135, 807–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trzeciakiewicz H, Tseng J-H, Wander CM, Madden V, Tripathy A, Yuan C-X, and Cohen TJ (2017). A Dual Pathogenic Mechanism Links Tau Acetylation to Sporadic Tauopathy. Nat. Publ. Group 7, 44102–44113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cohen TJ, Guo JL, Hurtado DE, Kwong LK, Mills IP, Trojanowski JQ, and Lee VM-Y (2011). The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2, 252–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brotzakis ZF, Lindstedt PR, Taylor RJ, Rinauro DJ, Gallagher NCT, Bernardes GJL, and Vendruscolo M (2021). A Structural Ensemble of a Tau-Microtubule Complex Reveals Regulatory Tau Phosphorylation and Acetylation Mechanisms. ACS Cent. Sci. 7, 1986–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Caballero B, Bourdenx M, Luengo E, Diaz A, Sohn PD, Chen X, Wang C, Juste YR, Wegmann S, Patel B, et al. (2021). Acetylated tau inhibits chaperone-mediated autophagy and promotes tau pathology propagation in mice. Nat. Commun. 12, 2238–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gorsky MK, Burnouf S, Dols J, Mandelkow E, and Partridge L (2016). Acetylation mimic of lysine 280 exacerbates human Tau neurotoxicity in vivo. Nat. Publ. Group 6, 22685–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neumann-Staubitz P, Lammers M, and Neumann H (2021). Genetic Code Expansion Tools to Study Lysine Acylation. Adv. Biol. 5, e2100926. [DOI] [PubMed] [Google Scholar]
- 34.Neumann H, Hancock SM, Buning R, Routh A, Chapman L, Somers J, Owen-Hughes T, van Noort J, Rhodes D, and Chin JW (2009). A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol. Cell 36, 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Halfmann R, Alberti S, Krishnan R, Lyle N, O’Donnell CW, King OD, Berger B, Pappu RV, and Lindquist S (2011). Opposing Effects of Glutamine and Asparagine Govern Prion Formation by Intrinsically Disordered Proteins. Mol. Cell 43, 72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scheres SHW (2020). Amyloid structure determination in RELION-3.1. Acta Crystallogr. Sect. Struct. Biol. 76, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seidler PM, Boyer DR, Rodriguez JA, Sawaya MR, Cascio D, Murray K, Gonen T, and Eisenberg DS (2018). Structure-based inhibitors of tau aggregation. Nat. Chem. 10, 170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mullapudi V, Vaquer-Alicea J, Bommareddy V, Vega AR, Ryder BD, White CL, Diamond MI, and Joachimiak LA (2023). Network of hotspot interactions cluster tau amyloid folds. Nat. Commun. 14, 895–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sawaya MR, Hughes MP, Rodriguez JA, Riek R, and Eisenberg DS (2021). The expanding amyloid family: Structure, stability, function, and pathogenesis. Cell 184, 4857–4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eisenberg D, and McLachlan AD (1986). Solvation energy in protein folding and binding. Nature 319, 199–203. [DOI] [PubMed] [Google Scholar]
- 41.Holmes BB, Furman JL, Mahan TE, Yamasaki TR, Mirbaha H, Eades WC, Belaygorod L, Cairns NJ, Holtzman DM, and Diamond MI (2014). Proteopathic tau seeding predicts tauopathy in vivo. Proc. Natl. Acad. Sci. 111, E4376–E4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schoch KMM, DeVos SLL, Miller RLL, Chun SJJ, Norrbom M, Wozniak DFF, Dawson HNN, Bennett CF, Rigo F, and Miller TMM (2016). Increased 4R-Tau Induces Pathological Changes in a Human-Tau Mouse Model. Neuron 90, 941–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gräff J, and Tsai L-H (2013). Histone acetylation: molecular mnemonics on the chromatin. Nat. Rev. Neurosci. 14, 97–111. [DOI] [PubMed] [Google Scholar]
- 44.Hageman J, Rujano MA, van Waarde MAWH, Kakkar V, Dirks RP, Govorukhina N, Oosterveld-Hut HMJ, Lubsen NH, and Kampinga HH (2010). A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol. Cell 37, 355–369. [DOI] [PubMed] [Google Scholar]
- 45.Min S-W, Cho S-H, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, Huang EJ, Shen Y, Masliah E, Mukherjee C, et al. (2010). Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67, 953–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shin M-K, Vázquez-Rosa E, Koh Y, Dhar M, Chaubey K, Cintrón-Pérez CJ, Barker S, Miller E, Franke K, Noterman MF, et al. (2021). Reducing acetylated tau is neuroprotective in brain injury. Cell 184, 2715–2732.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51. [DOI] [PubMed] [Google Scholar]
- 48.Rohou A, and Grigorieff N (2015). CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, and Ludtke SJ (2007). EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46. [DOI] [PubMed] [Google Scholar]
- 50.Wagner T, Lusnig L, Pospich S, Stabrin M, Schönfeld F, and Raunser S (2020). Two particle-picking procedures for filamentous proteins: SPHIRE-crYOLO filament mode and SPHIRE-STRIPER. Acta Crystallogr. Sect. Struct. Biol. 76, 613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen S, McMullan G, Faruqi AR, Murshudov GN, Short JM, Scheres SHW, and Henderson R (2013). High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy 135, 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Terwilliger TC, Sobolev OV, Afonine PV, and Adams PD (2018). Automated map sharpening by maximization of detail and connectivity. Acta Crystallogr. Sect. Struct. Biol. 74, 545–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 54.Warnecke A, Sandalova T, Achour A, and Harris RA (2014). PyTMs: a useful PyMOL plugin for modeling common post-translational modifications. BMC Bioinformatics 15, 370–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Afonine PV, Poon BK, Read RJ, Sobolev OV, Terwilliger TC, Urzhumtsev A, and Adams PD (2018). Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. Sect. Struct. Biol. 74, 531–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fleishman SJ, Leaver-Fay A, Corn JE, Strauch E-M, Khare SD, Koga N, Ashworth J, Murphy P, Richter F, Lemmon G, et al. (2011). RosettaScripts: a scripting language interface to the Rosetta macromolecular modeling suite. PLoS ONE 6, e20161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leman JK, Weitzner BD, Lewis SM, Adolf-Bryfogle J, Alam N, Alford RF, Aprahamian M, Baker D, Barlow KA, Barth P, et al. (2020). Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nat. Methods 17, 665–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barlow KA, Ó Conchúir S, Thompson S, Suresh P, Lucas JE, Heinonen M, and Kortemme T (2018). Flex ddG: Rosetta Ensemble-Based Estimation of Changes in Protein-Protein Binding Affinity upon Mutation. J. Phys. Chem. B 122, 5389–5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Raw ThT fluorescence aggregation curves for R1R2, R2R3, R3R4, R1R3 and R4R’ acetylated peptides, related to Figures 1 and 2. Data is organized by figure panel.
Data S2. Raw tau biosensor seeding data using R1R2, R2R3 and R1R3 chemically modified peptides, related to Figure 5. Data is organized by figure panel.
Data Availability Statement
The structural datasets generated during the current study are available in the Electron Microscopy Data Bank repository (https://www.ebi.ac.uk/emdb/) under accession number, EMD-28721 [https://www.ebi.ac.uk/emdb/EMD-28721]. The structural model that was fit into the density is available in the Protein Data Bank under PDB id 8FNZ (https:/doi.10.2210/pdb8FNZ/pdb). Raw ThT and cell-based aggregation data are available as Data S1 (Related to Figure 1 and 2) and Data S2 (Related to Figure 5), respectively. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.