

Abstract
Thrombosis is the leading cause of death in many diseased conditions. Oxidative stress is characteristic of these conditions. Yet the mechanisms through which oxidants become prothrombotic are unclear. Recent evidence suggests protein cysteine and methionine oxidation as prothrombotic regulators. These oxidative post-translational modifications occur on proteins that participate in the thrombotic process, including Src family kinases, protein disulfide isomerase, β2 glycoprotein I, von Willebrand factor, and fibrinogen. New chemical tools to identify oxidized cysteine and methionine proteins in thrombosis and hemostasis, including carbon nucleophiles for cysteine sulfenylation and oxaziridines for methionine, are critical to understanding why clots occur during oxidative stress. These mechanisms will identify alternative or novel therapeutic approaches to treat thrombotic disorders in diseased conditions.
Keywords: Benzothiazine, Beta-2 Glycoprotein I, Cysteine, Disulfide, Fibrinogen, Glutathionylation, Hemostasis, Methionine, Methionine Sulfoxide, Methionine Sulfoxide Reductase, Nitrosation, Oxaziridine, Oxidation, Protein Disulfide Isomerase, Src kinase, Sulfenylation, Sulfhydration, Thrombosis, von Willebrand factor
Graphical Abstract
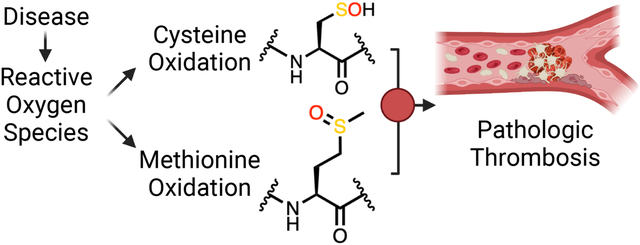
1. Introduction
Oxidative stress is characteristic of many systemic and metabolic disorders. Oxidants generated during oxidative stress promote thrombus formation, the leading cause of organ failure and death in diseased conditions [1]. These clots include arterial thrombotic disorders, which are observed in heart attack and stroke relating to metabolic syndromes [2,3], deep vein thrombosis present in inflammatory conditions and aging [4], and microvascular thrombosis during hemoglobinopathy [4], infection, and other blood disorders. The mechanisms that transduce oxidants into prothrombotic entities are poorly understood. Oxidants target cellular constituents, including lipids, nucleotides, and amino acids [5], and could be the mechanism for their thrombogenicity. Oxidation of the amino acids cysteines and methionines does not initiate clot formation, nor are these oxidative events the final effectors of clotting. Protein oxidation is thus an additional layer of regulation in the thrombotic process. A better understanding of protein oxidation in thrombus formation is critical to identify therapeutic targets whose inhibition will prevent pathogenic clots yet maintain sufficient hemostasis to prevent bleeding. In this review, we discuss the process of thrombosis and hemostasis and focus on recent evidence of redox-sensitive proteins of endothelial, platelet, and plasma origin containing oxidizable cysteines and methionines. Oxidation of cysteines and methionines within these proteins promotes thrombus formation through mechanisms that require further investigation.
2. Thrombosis and Hemostasis
Thrombosis and hemostasis are multifactorial processes regulated by oxidative stress and involve circulating blood platelets, the endothelium, and clotting factors in the plasma (Figure 1). The quiescent endothelium affords an anti-thrombotic surface that prevents thrombus formation [6], involving the potent anti-thrombotic agents nitric oxide [7] and prostacyclins [8]. Vascular damage, such as physical trauma to the vessel wall, exposes a subendothelial extracellular matrix that initiates platelet adhesion, activation, and aggregation [9]. The aggregated platelets form a hemostatic plug to prevent further blood loss. Plasma clotting factors also maintain the integrity of the clot. In pathologic conditions, the regulation of these stages is impaired, and the platelet plug can form a superficial thrombus, potentially occluding the lumen of the vessel. Such occlusion may be defined by multiple signaling modalities that are redox-regulated.
Figure 1. Clot formation in pathologic thrombosis.
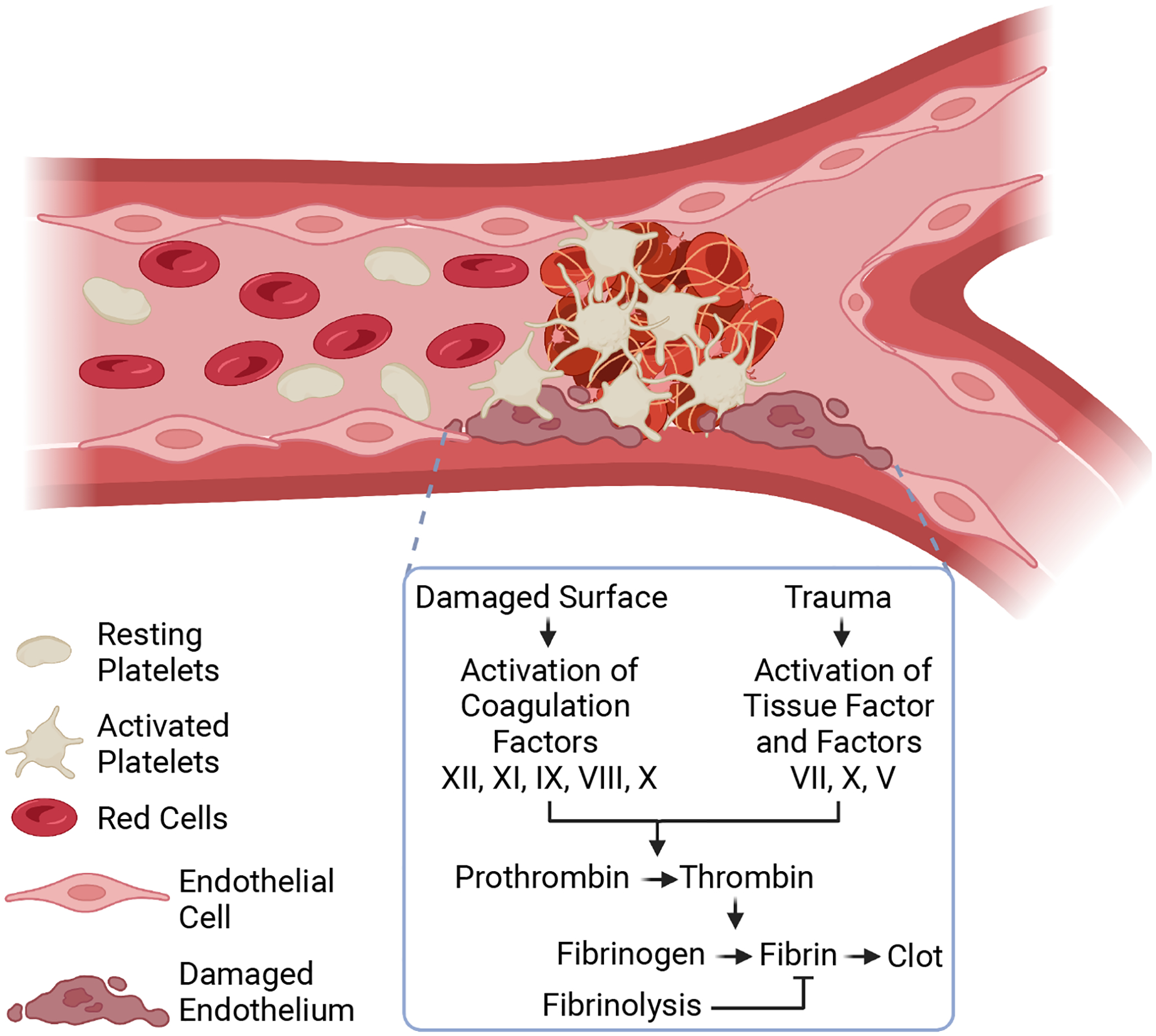
Clot formation is initiated by physical injury to the vessel wall. This injury damages the endothelium and exposes a subendothelial matrix that enhances platelet adhesion, recruitment, and aggregation. Platelet dynamics ultimately form a platelet plug to prevent blood loss (i.e. hemostasis). The damaged surfaces or the injury-induced trauma activates a series of coagulation cascades (via the Factors XII, XI, IX, VIII, X pathway or the Tissue Factor-Factors VII, X, V pathway). The coagulation cascade activates the zymogen prothrombin to thrombin, a protease that cleaves soluble fibrinogen to form fibrin. Fibrin stabilizes the platelet plug by creating a fibrous gel-like structure before degradation by fibrinolysis mechanisms. The formed superficial thrombus could cause clinically significant events, including heart attack, stroke, and organ failure. Created with BioRender.com.
3. Oxidative Cysteine Modifications in Thrombosis
Cysteine is a versatile amino acid for redox reactions. The cysteine thiol is in equilibrium with its thiolate anion, and the thiolate form is nucleophilic and reactive to oxidants. The fraction of the thiolate form is controlled by the pKa of the cysteine, which has a wide range dependent on the protein microenvironment surrounding the cysteine. A cysteine favors the deprotonated thiolate form if the pKa is lower than the pH of the local environment. As such, cysteine residues with a pKa less than 7.4 favor the thiolate form at physiologic pH. These thiolates are thus susceptible to oxidation by peroxides and other oxidants. Other factors that regulate thiolate formation are detailed in [10].
Several lines of evidence indicate a functional role for oxidative cysteine modification in thrombotic machinery. The thrombogenicity of cysteine oxidation depends on the type of modification and the protein [11]. In this review, we focus on cysteine sulfenylation (section 3.1; Figure 2A), disulfide formation (section 3.2; Figure 2A), and nitrosation (section 3.3; Figure 2A) as, to date, these modifications are the best-studied cysteine oxoforms implicated in thrombus formation. Other modifications, including glutathionylation and sulfhydration (section 3.4; Figure 2A), are implicated as regulators in thrombosis and hemostasis, but their functional roles require further investigation. Cysteine modifications not studied in detail in thrombosis include sulfinylation, sulfonylation, thiosulfenates, and sulfenamides. As the type of cysteine modification determines whether the oxoform is pro- or anti-thrombotic, further investigation of each oxoform is needed as they relate to clotting.
Figure 2. Oxidative cysteine modification and implications in platelet CD36 and protein disulfide isomerase redox signaling in dyslipidemia.
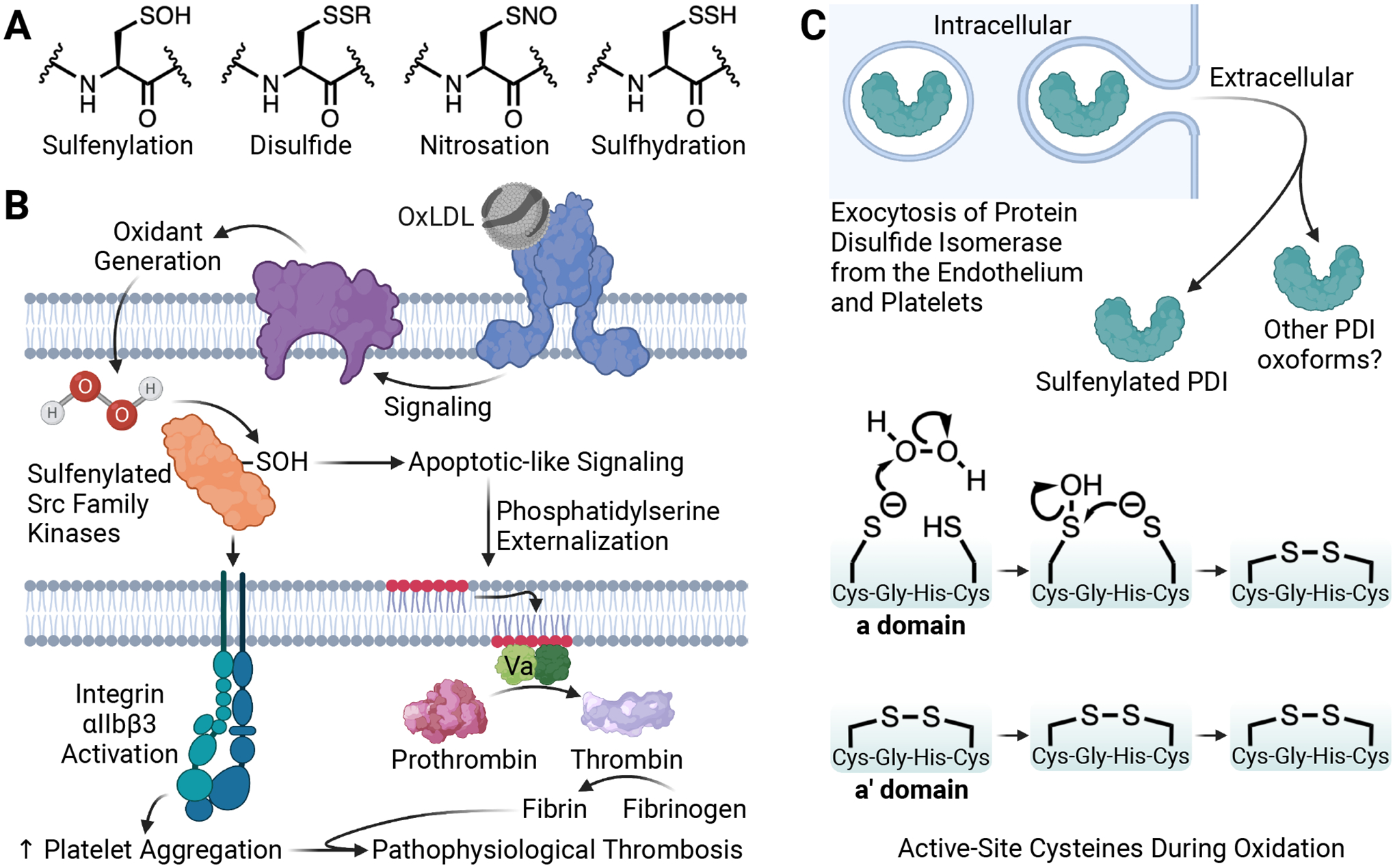
(A) Structure of the oxidative cysteine modifications sulfenylation, disulfides (e.g. glutathionylation), nitrosation, and sulfhydration. (B) Circulating oxidized low-density lipoprotein (oxLDL) particles are present in dyslipidemia. OxLDL is recognized by platelet scavenger receptor CD36 to promote oxidant generation from NADPH oxidase. The two-electron oxidant H2O2 sulfenylates the platelet proteome, including Src family kinases. Src kinase sulfenic acid (SOH) formation promotes Src kinase activity to enhance integrin αIIbβ3 activation for platelet aggregation and externalization of phosphatidylserine for procoagulant potential during pathophysiologic thrombosis. (C) Intracellular PDI from the endothelium and platelets is secreted upon cellular activation. Sulfenylation of extracellular PDI is an oxoform of the secreted pool of PDI; other PDI oxoforms may be present in the extracellular environment. Biochemical analysis of the protein domains revealed that the active-site cysteines in the 397Cys-Gly-His-Cys400 motif of the a′ domain of PDI are already oxidized to disulfides in the basal state and that it is the 57Cys-Gly-His-Cys60 motif in the a domain with the available thiols for H2O2-mediated sulfenic acid generation. The most likely mechanism involves disulfide formation in the a domain with sulfenic acid as a necessary intermediate. Whether Cys57 or Cys60 is sulfenylated is currently unclear. The proposed mechanism is based on Cys57 as the more nucleophilic thiolate. Created with BioRender.com.
3.1. Cysteine sulfenylation during dyslipidemia
The thiolate (-S−) of a cysteine residue can be oxidized to a sulfenic acid (-SOH). Further oxidation can lead to a sulfinic acid (-SO2H) and, subsequently, a sulfonic acid (-SO3H). Sulfenic acids can also react with other cysteine or small molecule thiols (e.g. glutathione) to form disulfides. A detailed review of these mechanisms is found in [12]. Signaling transducers link oxidative stress in diseased conditions to a prothrombotic response. Sulfenylation (Figure 2A) of these effectors controls protein function and thus lowers the threshold for platelet activation. Src family kinases are abundantly expressed in platelets and are examples of signaling transducers that regulate platelet function [13]. Cysteine sulfenylation of Src kinases promotes kinase activity by oxidation of Cys185 [14,15]. During the inflammatory process of dyslipidemia, oxidized low-density lipoprotein (oxLDL) particles are abundantly present in the circulation and are recognized by a platelet membrane protein called cluster of differentiation 36 (CD36) [16,17]. CD36 is a scavenger receptor of the innate immune system that is highly expressed on the platelet surface to initiate downstream oxidant generation from NADPH oxidase 2 (NOX2) [18,19]. Hydrogen peroxide (H2O2) is the two-electron oxidant generated, which sulfenylates platelet protein cysteine residues [20]. Determining the identity of the entire sulfenylated proteome in this context requires further proteomic studies; however, proteomic analysis during platelet pathogen-inactivation indicated many oxidizable proteins [21]. These protein families include integrin signaling proteins and cytoskeletal regulatory proteins. Src family kinases were also sulfenylated in this screen. In dyslipidemia, Src family kinases link CD36 signaling at the membrane level to intracellular signaling cascades that ultimately cause pro-aggregatory integrin αIIbβ3 activation and externalization of procoagulant phosphatidylserine [16,22]. Src family kinases are a target of CD36-mediated H2O2 generation (Figure 2B). Sulfenylation of Src family kinases promotes their kinase activity [14] and is a positive feedback loop to lower the threshold for platelet activation in the CD36 signaling pathway. In addition, using a benzothiazine-based (BTD) carbon nucleophile identified in the Carroll laboratory to target sites of sulfenylation [20] selectively, we found that this benzothiazine probe decreases platelet and fibrin deposition in vivo on an injured arterial vessel wall in a mouse model of dyslipidemia [20]. The mechanism for reduced thrombus formation is unclear, as the alkylation of sulfenic acids with the benzothiazine probe results in a stable thioether that prevents the cysteines from being reduced or further oxidized. Notably, the benzothiazine probe did not prevent thrombus formation in control non-dyslipidemic conditions. Further analytical work is required to understand the bioavailability and reactivity profile of the benzothiazine probe in vivo. These data suggest that in vivo, cysteine sulfenylation or subsequent oxidative modifications control the thrombogenicity of platelets.
In addition to Src kinases, we recently found that thiol isomerases are redox-sensitive enzymes sulfenylated in dyslipidemia [23]. Thiol isomerases are endoplasmic reticulum-resident enzymes classically known for their roles in oxidative protein folding. Vascular thiol isomerases escape the endoplasmic reticulum upon platelet and endothelial cell activation and are found extracellularly to support thrombus formation [24,25]. Protein disulfide isomerase (PDI) is the archetypal thiol isomerase and the most studied in thrombosis and hemostasis compared to the other thiol isomerase family members [26]. PDI has an a-b-b′-a′-c domain configuration with its catalytic redox-sensitive cysteines within a Cys-Gly-His-Cys (CGHC) motif in the a and a′ domain [27]. PDI has multiple functions, including the ability to reduce cysteine disulfides (reductase activity), oxidize cysteines to disulfides (oxidase activity), and rearrange disulfides (isomerase activity) [11,26]. The enzymatic activity of PDI is controlled by redox-regulation of its horseshoe-shaped structure, where the reduced form of the protein adopts a more closed configuration and the oxidized form a more open configuration [28]. Extracellular oxidized PDI is implicated in platelet reactivity and aggregation [29]. We found that the CGHC motif in the a domain is sensitive to sulfenylation by peroxides and that sulfenylation is an intermediate modification to disulfide formation, thus converting extracellular PDI from a reductase into an oxidase during oxidative stress (Figure 2C) [23]. The finding that PDI sulfenylation is an intermediate to disulfide formation supports previous biochemical data of sulfenic acids as oxidized sulfur intermediates toward disulfide formation [30]. Surprisingly, the a′ domain was found to be already oxidized when purified. In dyslipidemia, circulating oxidized lipoprotein particles enhance PDI sulfenylation, likely through lipid and amino acid hydroperoxides, and promote platelet accumulation in vivo after experimentally injuring the arteriolar vessel wall in mice [23]. These studies suggest that PDI is sensitive to oxidative stress in dyslipidemia and promotes thrombosis.
Although dyslipidemia is an oxidative stress model with sulfenylation implicated in thrombus formation, whether sulfenylation of Src family kinases and thiol isomerases is required for clot formation in other oxidative stress-diseased conditions will be critical to determine in future studies.
3.2. Allosteric disulfides in thrombus formation
Oxidants during oxidative stress induce disulfide bond formation, and protein cysteine disulfides are a sensitive marker of oxidative stress in blood disorders [31]. Oxidant-induced disulfides are more promiscuous than enzyme-mediated disulfide formation by the thiol isomerase family members. Identifying proteins that are reduced or oxidized by thiol isomerases is an active area of research with a current list of thrombotic proteins published in [32]. We highlight recent biochemical data on β2-glycoprotein I (β2GPI), a plasma protein whose disulfide redox switches are regulated by thiol isomerases to enhance thrombus formation.
β2GPI clears necrotic cells, neutralizes cell debris, binds to anionic phospholipids, and participates in the clotting cascade of antiphospholipid syndrome [33], an autoimmune blood disorder with clots in various vascular beds [34]. Oxidative stress is associated with antiphospholipid syndrome [35], and oxidized β2GPI is the hypothesized redox form of the protein that is the immunogenic target of pathogenic antibodies in the disorder [36]. Allosteric disulfide bonds of oxidized β2GPI were identified over 10 years ago as a substrate of thiol isomerase in vitro and in vivo [37,38]. Specifically, thioredoxin 1 and PDI regulate the disulfide on β2GPI [38]. Yet, recent structural insights through mutagenesis studies revealed that loss of the allosteric Cys288-Cys326 disulfide in Domain V of β2GPI is sufficient to impair binding to anionic phospholipids [39]. The Cys32-Cys60 disulfide in domain I of the protein is a target of pathogenic anti-β2GPI antibodies; however, this disulfide is more critical structurally than as an allosteric regulator. These studies complemented the findings of Bucholz and coworkers, who found that chemical or enzymatic reduction of Cys288-Cys326 affords flexibility to β2GPI, allowing β2GPI to adopt a more open configuration for pathogenic antibodies to bind [40]. In non-pathogenic blood clotting, β2GPI domain V regulates thrombus formation as β2GPI deficiency in mice shows decreased platelet and fibrin accumulation in vivo after vessel injury; this defect was rescued by the infusion of recombinant domain V [41]. Infusion of recombinant proteins or specific domains, such as described for β2GPI in experimental thrombosis in mice, can determine if these proteins participate in oxidative stress clots. These studies underscore the importance of allosteric disulfides regulated by thiol isomerases to tune the thrombotic response during thrombus formation.
3.3. Anti-thrombotic cysteine nitrosation
Nitric oxide is potently antithrombotic. The anti-thrombogenic potential of nitric oxide is beneficial in preventing thrombosis but could increase the risk of bleeding complications. Nitric oxide produced enzymatically from nitric oxide synthases regulates vascular tone and prevents platelet activation by nitrosylating the ferrous heme of soluble guanylate cyclase [42]. Soluble guanylate cyclase nitrosylation promotes antithrombotic signaling through protein kinase G signaling. Higher-order nitric oxide species can also oxidize cysteine residues resulting in cysteine nitrosation (Figure 2A) [43,44] and subsequent regulation of thrombotic machinery. We focus again on thiol isomerases, as thiol isomerases also support anti-thrombotic signaling through cysteine nitrosation. PDI is sensitive to enzymatic regulation by cysteine nitrosation in the active site motif [45]. PDI can also transnitrosate membrane proteins on platelets, including integrin αIIbβ3. Nitrosated PDI prevents platelet aggregation in vitro, and infusion of nitrosated PDI limits platelet and fibrin accumulation in vivo. These findings suggest that, in addition to the classic role of nitric oxide in regulating platelet activation through soluble guanylate cyclase, PDI coordinates nitric oxide signaling within the vasculature to prevent pathogenic thrombosis.
3.4. Other cysteine modifications in thrombosis and hemostasis
Other oxidative cysteine modifications could be either pro- or anti-thrombotic, depending on the cysteine oxoform and protein site(s) modified. As such, these other cysteine modifications may regulate thrombosis and contribute to thrombotic disorders. Like nitrosation, cysteine glutathionylation and sulfhydration (Figure 2A) regulate platelet activation and aggregation [46,47]. Cysteine glutathionylation of the platelet proteome is expectedly linked to a decrease in the available reduced glutathione levels [48]. Although the mechanism requires further investigation, it involves glutathionylation of actin cytoskeletal proteins [48], which could be linked to the decreased cytoskeletal mobilization needed for platelet shape change and granule secretion. As reduced glutathione is critical for the redox buffering capacity of the cell, determining if a difference in the intracellular redox buffering capacity is related to intracellular protein glutathionylation and either platelet pro- or anti-thrombotic potentials during oxidative stress would be of great interest.
Hydrogen sulfide is a reactive sulfur species that is formed enzymatically by three human enzymes — cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE), and 3-mercaptopyruvate sulfurtransferase (3MST) — and is present in nano- to low micro-molar ranges in human plasma (reviewed in [49]). Hydrogen sulfide is nucleophilic and can react with oxidized thiols [50]. For example, hydrogen sulfide reaction with disulfides or sulfenic acids can result in cysteine persulfides (i.e. sulfhydration). As such, the redox mechanisms of hydrogen sulfide is multifaceted [50]. Evidence suggests that hydrogen sulfide is an anti-thrombotic agent [47]. GYY4137, a hydrogen sulfide donor, was used to investigate the mechanism of protein cysteine sulfhydration in the thrombotic machinery [47]. Although GYY4137 promoted protein sulfhydration in platelets, the sulfhydrated proteins were not identified. Functionally, GYY4137 concentration-dependently prevented platelet activation and prolonged the time to thrombus formation [47]. Implementing proteomic methods to identify proteins sulfhydrated by hydrogen sulfide [51,52] would help explain why GYY4137 decreases platelet activation and prolongs thrombus formation. In addition, assessing if the physiologic generation of hydrogen sulfide impacts thrombus formation in the setting of inflammation would be of great interest. In this condition, endogenous hydrogen sulfide levels are expected to increase [49].
4. Methionine Oxidation in Thrombosis and Hemostasis
Like cysteines, the sulfur atom of methionine is prone to oxidation and is important in regulating pathogenic thrombosis. Methionine oxidation is akin to cysteine oxidation. The first oxidation results in oxygen added to the sulfur of the methionine thioether sidechain in two different stereoisomers, methionine-R-sulfoxide and methionine-S-sulfoxide, which can induce structural and functional changes to a protein. This is a reversible mechanism back to a thioether similar to the reversibility of a cysteine sulfenic acid to a free reduced thiol. Methionine sulfoxides can be enzymatically reduced to methionine by methionine sulfoxide reductase. Like sulfenic acids, further oxidation of methionine sulfoxides generates irreversible sulfones. Vascular thrombotic proteins are susceptible to methionine oxidation and have been reviewed in detail [53], including a disintegrin and metalloproteinase with a thrombospondin type 1 motif member 13 (ADAMTS13) [54], thrombomodulin, protein C, and others [55]. Based on recent publications, we focus on two major clotting-related proteins, von Willebrand factor (vWF), a large multimeric glycoprotein released from vascular endothelial cells, and fibrinogen, a plasma protein that mediates platelet aggregation and fibrin clot formation. We also focus on methionine sulfoxidation of actin as it relates to platelet cytoskeletal dynamics during a growing thrombus.
vWF is a high molecular weight plasma protein assembled in multiple domains with the configuration D1-D2-D′-D3-A1-A2-A3-D4-B1-B2-B3-C1-C2-CK [56]. vWF supports platelet adhesion to the subendothelial connective tissue and helps solubilize clotting Factor VIII. vWF is a redox-sensitive cysteine-rich protein—the cysteines are required for disulfide bond formation to support multimerization for clotting [56]. In the blood, multimerization of vWF into high molecular weight species is necessary for hemostasis, and vWF is regulated by proteolytic cleavage by ADAMTS13 (Figure 3A). This circulating zinc metalloprotease cleaves vWF between Tyr1605 and Met1606 into less-reactive species. Several methionine residues within the vWF A1-A2-A3 domains are susceptible to oxidation. Specifically, Met1606 in the A2 domain at the ADAMTS13 cleavage site can be oxidized to a methionine sulfoxide [57]. Met1606 sulfoxidation in the large multimer prevents ADAMTS13-catalyzed cleavage (Figure 3A). As proteolysis by ADAMTS13 limits vWF assembly to large multimers, the Met1606 oxidation event promotes thrombosis by enhancing platelet binding by preventing ADAMTS13-catalyzed cleavage. Met1606 sulfoxidation within vWF is thus a thrombogenic mechanism during inflammation and in patients with sickle cell disease. In addition, efficient ADAMTS13 cleavage of vWF at Met1606 is controlled by auto-inhibition with the adjacent A1 and A3 domains. Recent molecular dynamic simulations suggest that Met1606 sulfoxidation inhibits auto-regulation by the A1 and A3 domains [58]. These in silico studies were validated experimentally using dynamic flow chambers to simulate blood rheology where Met1606 sulfoxidation in the A2 domain prevents A1 and A3 binding. vWF methionine sulfoxidation is thus a prothrombotic mechanism to control thrombosis.
Figure 3. Oxidative methionine modification on the plasma proteins von Willebrand factor (vWF) and fibrinogen.
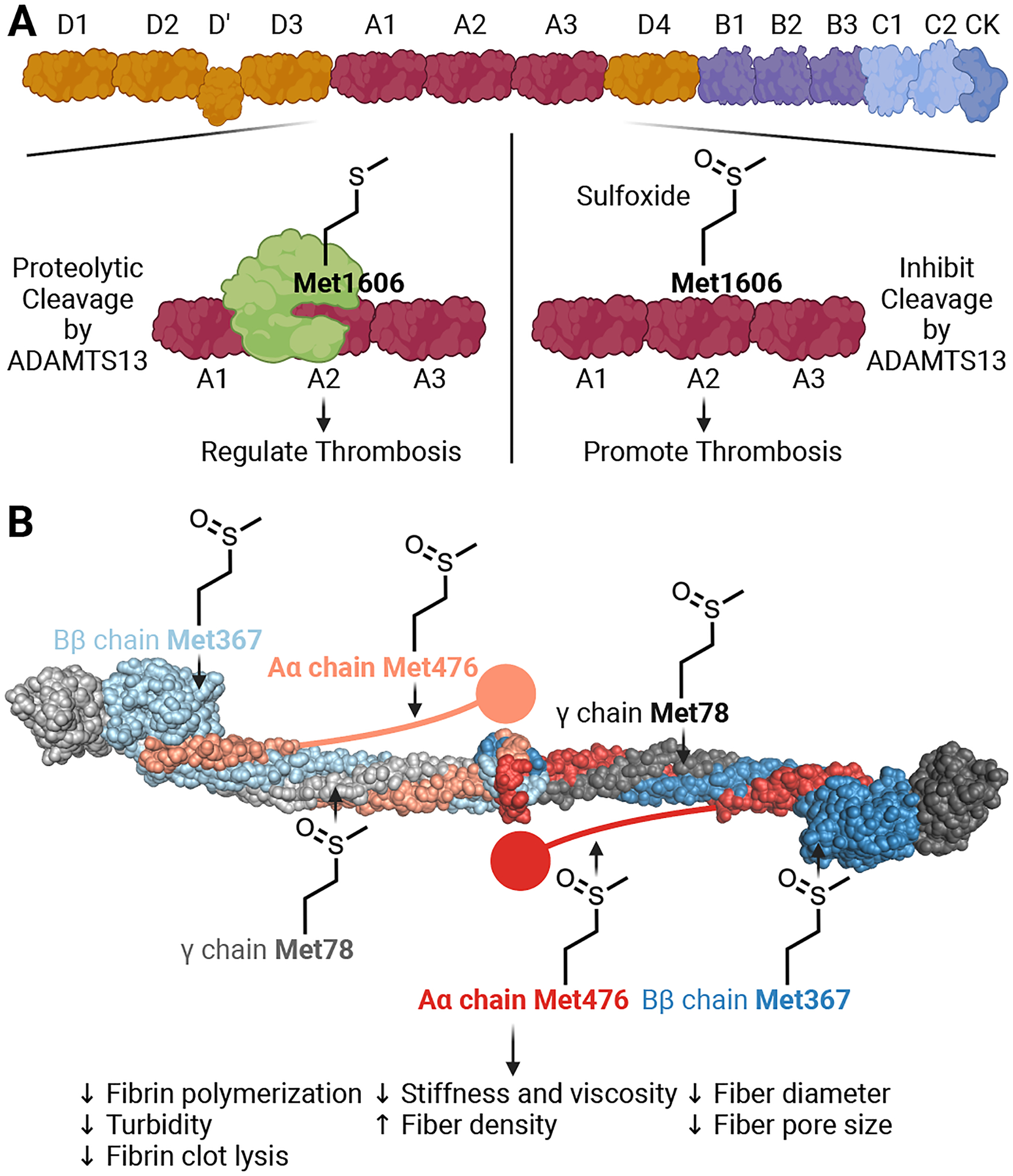
(A) The domain structure of vWF. Oxidative methionine modification occurs on the A1, A2, and A3 domains of the protein. In the A2 domain of vWF, the metalloprotease a disintegrin and metalloproteinase with a thrombospondin type 1 motif member 13 (ADAMTS13) cleaves the protein between Tyr1605 and Met1606. This cleavage regulates thrombosis by preventing vWF from multimerizing. During oxidative stress, sulfoxidation occurs on Met1606, preventing ADAMTS13 cleavage of vWF. This mechanism potentiates thrombosis by maintaining vWF in a large multimeric state. (B) The domain structure of fibrinogen. Fibrinogen consists of homodimeric Aα, Bβ, and γ chains. Fibrinogen polymerizes to form a fibrin mesh to support clot stability. Methionine sulfoxidation occurs on all chains of the protein. Specifically, sulfoxidation of Met78 in the γ chains, Met367 in the Bβ chains, and Met476 in the Aα chains decreases fibrin polymerization, diameter, pore size, total turbidity, clot lysis, stiffness, and viscosity. The fibrin densities are larger in size compared to the unoxidized form. Created with BioRender.com.
Fibrinogen is an additional plasma protein whose methionine oxidation regulates clot formation. Fibrinogen is a hexameric homodimer of Aα, Bβ, and γ chains made in the liver and supports clotting by its proteolytic conversion to fibrin by thrombin [59]. Fibrin increases clot stability by forming a fibrous gel before fibrinolysis for its degradation. Sulfoxidation on all three chains of fibrinogen occurs preferentially on γ-Met78, Bβ-Met367, and Aα-Met476 and reduces the fiber size making the fibrous gel weaker (Figure 3B). Some studies suggest that fibrinogen oxidation prevents lateral fibrin polymerization needed for the fibrin mesh to form [60–62]. Oxidation also prolongs fibrinolysis time, making it difficult to degrade a clot during oxidative stress [62]. The net effect of fibrinogen oxidation is complex. Fibrinogen oxidation may weaken the clot and increase the potential for embolism. Prolonged fibrinolysis increases the risk of bleeding during trauma, as evidenced by plasminogen activator inhibitor 1-deficiency, a serine protease that regulates fibrinolysis [63]. Hyperfibrinolysis correlates with unstable clots and a rebleeding phenotype [64]. The net effect of methionine sulfoxidation on fibrinogen requires further investigation, especially in clot resolution.
The platelet cytoskeleton is highly dynamic. During cellular activation, cytoskeletal rearrangement is required for platelet formation, shape change, activation, aggregation, and the formation of the hemostatic plug [65]. Actin is essential to these dynamics and is an abundant protein within the cell. Actin can undergo reversible methionine sulfoxidation on Met44 and Met47 through MICAL (molecule interacting with CasL) family enzymes, a conserved family of flavin oxidoreductases that oxidize specific methionines of actin [66]. Met44 and Met47 are located in the D-loop of the subdomain 2 of the protein, which is an actin-actin contact site [67]. These oxidation events promote filamentous actin disassembly and regulate actin repolymeraization. Met44 and Met47 could also be oxidized chemically by reactive oxygen species [66]. Although actin oxidation has not been studied in the platelets, the importance of cytoskeletal dynamics during a hemostatic plug formation in oxidative stress warrants further investigation.
5. Conclusions and Knowledge Gaps
Cysteine and methionine oxidation during oxidative stress is a potential mechanism connecting oxidants to a prothrombotic phenotype. The studies highlighted above underscore the importance of cysteine and methionine oxidative signaling as potential therapeutic targets to regulate thrombosis in diseased conditions. However, these studies are primarily within the bounds of in vitro and ex vivo experimentation. Critical knowledge gaps remain, including whether oxidative cysteine and methionine modifications are present in vivo during a growing thrombus and whether specific proteins or a network of proteins are required. The availability of chemical probes that target particular sulfur oxoforms of cysteine [68] and methionine residues [69] allows investigation of oxidation during thrombosis. Intravital microscopy could also connect oxidative cysteine and methionine modification to phenotypic function. Infusing reductases to specific cysteine and methionine sulfur oxoforms and thrombotic proteins harboring specific cysteine and methionine oxidative modifications will connect these oxidative events to thrombus formation in vivo if the enzymes or proteins impair or enhance the clot. Lastly, probes with appropriate permeability and pharmacokinetics for in vivo use will afford chemoproteomic strategies to identify new protein targets of cysteine- and methionine-oxidization. Rapid-reacting carbon nucleophiles could detect and probe the function of cysteine sulfenic acids by alkylation (Figure 4A). Oxaziridine-based probes [69] that selectively label methionines may be used to identify both reactive methionines and oxidative methionine modification as oxaziridine labeling is competitively blocked by oxidation (Figure 4B). In the latter case, a loss of labeling would be used as a readout of methionine sulfur oxoforms, similar to probes that selectively react with reduced cysteines (e.g. maleimides or haloacetamides) to detect the loss of free thiols as a readout of cysteine oxidation [70]. Identifying the oxidized proteins and the consequence of oxidation on protein function will provide mechanistic information on why pathogenic clots occur in diseased conditions, allowing for alternative or new therapeutic approaches.
Figure 4. Chemical probes to study oxidative cysteine and methionine modification in thrombotic disorders.
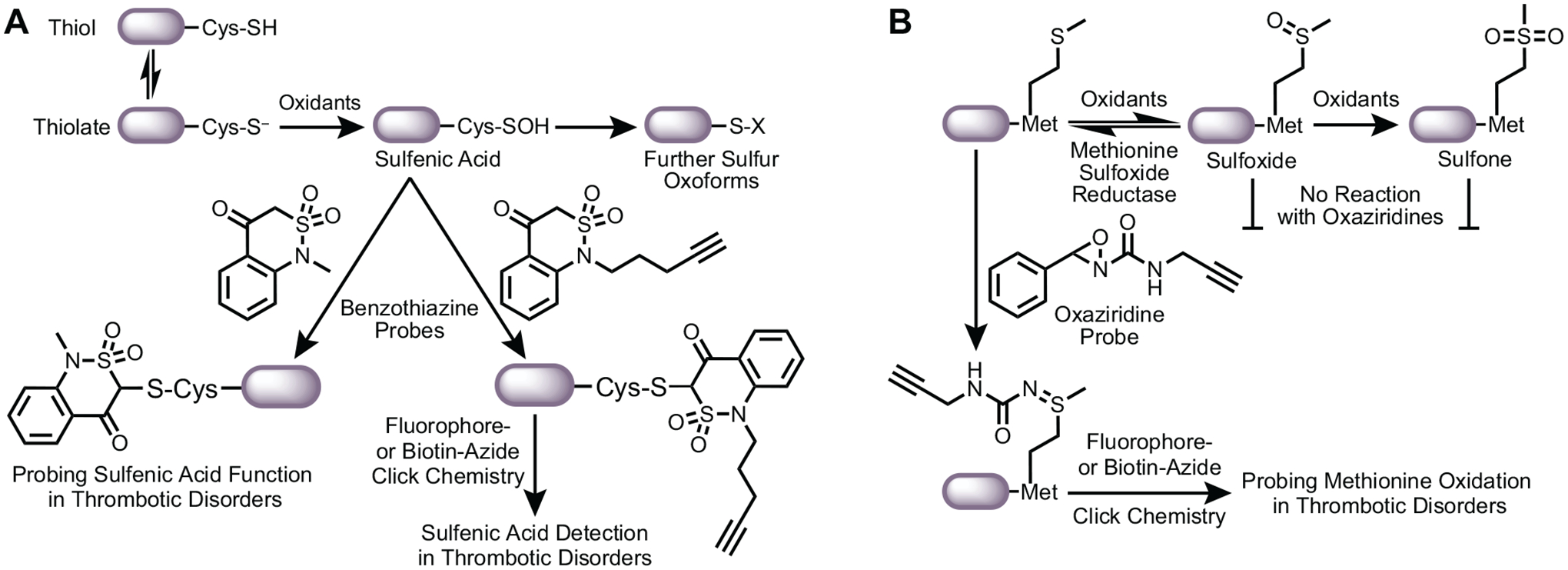
(A) Using carbon nucleophiles to probe for sulfenic acid detection and function in thrombosis. The cysteine thiol is in equilibrium with its deprotonated thiolate anion. The nucleophilic thiolate anion is then oxidized to a sulfenic acid by oxidants generated during oxidative stress. Sulfenic acid is both a nucleophile and an electrophile and is a precursor to further sulfur oxoforms (e.g. sulfinic and sulfonic acids). Sulfenic acids can also be converted to disulfides in the presence of proximal thiols. Carbon nucleophiles, such as benzothiazine-based probes developed by the Carroll laboratory, covalently and selectively target sulfenic acids. Benzothiazine (BTD) with an alkyne arm detects cysteine sulfenylation on proteins using click chemistry with azides in the context of thrombotic disorders. In addition, an alkyneless BTD can probe the function of the sulfenic acid as alkylation prevents further oxidation and reduction of the cysteine. Additional studies are essential to determine what proteins are sulfenylated and the impacts on protein function and subsequent thrombotic activity. (B) Methionine residues can be oxidized to methionine sulfoxides. This oxidative event is reversible with the enzyme methionine sulfoxide reductase. Further oxidation of the sulfoxide yields methionine sulfone, thought to be irreversible. Recent oxaziridine-based probes can label unoxidized methionines. Labeling of the methionine via a sulfimide linkage can be coupled with click chemistry to identify proteins that undergo oxidative methionine modification, as shown by a loss of probe labeling.
Highlights.
Oxidative stress promotes thrombosis in diseased conditions.
The mechanisms linking oxidants to thrombosis are poorly understood.
Cysteine and methionine residues are prothrombotic targets of oxidants.
Cysteine and methionine modification are potential antithrombotic drug targets.
Acknowledgments
This work was supported by the National Institutes of Health [grant numbers R35 GM128840 to BCS and K99 HL164888 to MY] and the American Society of Hematology Scholar Award [to MY]. These funding sources had no involvement in writing this review or submitting this review for publication.
Abbreviations:
- ADAMTS13
a disintegrin and metalloproteinase with a thrombospondin type 1 motif member 13
- BTD
benzothiazine-based carbon nucleophile
- CD36
cluster of differentiation 36
- CDK4
cyclin-dependent kinase 4
- CGHC
cysteine-glycine-histidine-cysteine
- MICAL
molecule interacting with CasL
- NOX2
NADPH oxidase 2
- PDI
protein disulfide isomerase
- NADPH
reduced nicotinamide adenine dinucleotide phosphate
- vWF
von Willebrand factor
- β2GPI
β2 glycoprotein I
- oxLDL
oxidized low-density lipoprotein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
Papers of particular interest, published within the period of review, have been highlighted as:
* of special interest
** of outstanding interest
- [1].Wendelboe AM, Raskob GE: Global Burden of Thrombosis: Epidemiologic Aspects. Circ Res 2016, 118:1340–1347. 10.1161/CIRCRESAHA.115.306841. [DOI] [PubMed] [Google Scholar]
- [2].Vaidya AR, Wolska N, Vara D, Mailer RK, Schroder K, Pula G: Diabetes and Thrombosis: A Central Role for Vascular Oxidative Stress. Antioxidants (Basel) 2021, 10. 10.3390/antiox10050706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Yang M, Silverstein RL: CD36 signaling in vascular redox stress. Free Radic Biol Med 2019. 10.1016/j.freeradbiomed.2019.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wang Q, Zennadi R: Oxidative Stress and Thrombosis during Aging: The Roles of Oxidative Stress in RBCs in Venous Thrombosis. Int J Mol Sci 2020, 21. 10.3390/ijms21124259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sies H, Belousov VV, Chandel NS, Davies MJ, Jones DP, Mann GE, Murphy MP, Yamamoto M, Winterbourn C: Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat Rev Mol Cell Biol 2022, 23:499–515. 10.1038/s41580022-00456-z. [DOI] [PubMed] [Google Scholar]
- [6].Neubauer K, Zieger B: Endothelial cells and coagulation. Cell Tissue Res 2022, 387:391–398. 10.1007/s00441-021-03471-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gambaryan S: The Role of NO/sGC/cGMP/PKG Signaling Pathway in Regulation of Platelet Function. Cells 2022, 11. 10.3390/cells11223704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Braune S, Kupper JH, Jung F: Effect of Prostanoids on Human Platelet Function: An Overview. Int J Mol Sci 2020, 21. 10.3390/ijms21239020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Scridon A: Platelets and Their Role in Hemostasis and Thrombosis-From Physiology to Pathophysiology and Therapeutic Implications. Int J Mol Sci 2022, 23. 10.3390/ijms232112772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wible RS, Sutter TR: Soft Cysteine Signaling Network: The Functional Significance of Cysteine in Protein Function and the Soft Acids/Bases Thiol Chemistry That Facilitates Cysteine Modification. Chem Res Toxicol 2017, 30:729–762. 10.1021/acs.chemrestox.6b00428. [DOI] [PubMed] [Google Scholar]
- [11].Yang M, Flaumenhaft R: Oxidative Cysteine Modification of Thiol Isomerases in Thrombotic Disease: A Hypothesis. Antioxid Redox Signal 2021, 35:1134–1155. 10.1089/ars.2021.0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Paulsen CE, Carroll KS: Cysteine-mediated redox signaling: chemistry, biology, and tools for discovery. Chem Rev 2013, 113:4633–4679. 10.1021/cr300163e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Senis YA, Nagy Z, Mori J, Lane S, Lane P: Platelet Src family kinases: A tale of reversible phosphorylation. Res Pract Thromb Haemost 2021, 5:376–389. 10.1002/rth2.12495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Heppner DE, Dustin CM, Liao C, Hristova M, Veith C, Little AC, Ahlers BA, White SL, Deng B, Lam YW, et al. : Direct cysteine sulfenylation drives activation of the Src kinase. Nat Commun 2018, 9:4522. 10.1038/s41467-018-06790-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Heppner DE: Structural insights into redox-active cysteine residues of the Src family kinases. Redox Biol 2021, 41:101934. 10.1016/j.redox.2021.101934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zheng TJ, Kohs TCL, Mueller PA, Pang J, Reitsma SE, Parra-Izquierdo I, Melrose A, Yang L, Choi J, Zientek KD, et al. : Effect of antiplatelet agents and tyrosine kinase inhibitors on oxLDL-mediated procoagulant platelet activity. Blood Adv 2022. 10.1182/bloodadvances.2022007169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Podrez EA, Byzova TV, Febbraio M, Salomon RG, Ma Y, Valiyaveettil M, Poliakov E, Sun M, Finton PJ, Curtis BR, et al. : Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nature Medicine 2007, 13:1086–1095. 10.1038/nm1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Magwenzi S, Woodward C, Wraith KS, Aburima A, Raslan Z, Jones H, McNeil C, Wheatcroft S, Yuldasheva N, Febbriao M, et al. : Oxidised LDL activates blood platelets through CD36-NADPH oxidase-mediated inhibition of the cGMP/Protein kinase G signalling cascade. Blood 2015, 125:2693–2703. 10.1182/blood-2014-05574491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yang M, Cooley BC, Li W, Chen Y, Vasquez-Vivar J, Scoggins NO, Cameron SJ, Morrell CN, Silverstein RL: Platelet CD36 promotes thrombosis by activating redox sensor ERK5 in hyperlipidemic conditions. Blood 2017, 129:2917–2927. 10.1182/blood-2016-11-750133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *[20].Yang M, Li W, Harberg C, Chen W, Yue H, Ferreira RB, Wynia-Smith SL, Carroll KS, Zielonka J, Flaumenhaft R, et al. : Cysteine sulfenylation by CD36 signaling promotes arterial thrombosis in dyslipidemia. Blood Adv 2020, 4:4494–4507. 10.1182/bloodadvances.2020001609. [DOI] [PMC free article] [PubMed] [Google Scholar]; We showed that platelet CD36 signaling promotes generation of reactive-oxygen species resulting in sulfenylation of Src family kinases as a positive feed-forward signaling to enhance platelet activation and coagulation potential.
- **[21].Sonego G, Le TM, Crettaz D, Abonnenc M, Tissot JD, Prudent M: Sulfenylome analysis of pathogen-inactivated platelets reveals the presence of cysteine oxidation in integrin signaling pathway and cytoskeleton regulation. J Thromb Haemost 2021, 19:233–247. 10.1111/jth.15121. [DOI] [PubMed] [Google Scholar]; The authors used dimedones to identify all the proteins sulfenylated in platelets. The most notable proteins identified include integrin signaling pathway proteins and cytoskeletal proteins.
- [22].Yang M, Kholmukhamedov A, Schulte ML, Cooley BC, Scoggins NO, Wood JP, Cameron SJ, Morrell CN, Jobe SM, Silverstein RL: Platelet CD36 signaling through ERK5 promotes caspase-dependent procoagulant activity and fibrin deposition in vivo. Blood Adv 2018, 2:2848–2861. 10.1182/bloodadvances.2018025411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *[23].Yang M, Chiu J, Scartelli C, Ponzar N, Patel S, Patel A, Ferreira RB, Keyes RF, Carroll K, Pozzi N, et al. : Sulfenylation links oxidative stress to protein disulfide isomerase oxidase activity and thrombus formation. J Thromb Haemost 2023. 10.1016/j.jtha.2023.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]; We found that the thiol oxidoreductase member PDI is sensitive to various oxidants. PDI cysteine sulfenylation is an intermediate to its disulfide state. Disulfides on oxidized PDI could be enzymatically transferred to substrates. Sulfenylation is part of the pool of PDI secreted from activated platelets and endothelial cells and is important for the prothrombotic effects of oxidized lipids in dyslipidemia.
- [24].Cho J, Furie BC, Coughlin SR, Furie B: A critical role for extracellular protein disulfide isomerase during thrombus formation in mice. J Clin Invest 2008, 118:1123–1131. 10.1172/JCI34134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Reinhardt C, von Bruhl ML, Manukyan D, Grahl L, Lorenz M, Altmann B, Dlugai S, Hess S, Konrad I, Orschiedt L, et al. : Protein disulfide isomerase acts as an injury response signal that enhances fibrin generation via tissue factor activation. J Clin Invest 2008, 118:1110–1122. 10.1172/JCI32376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gaspar RS, Gibbins JM: Thiol Isomerases Orchestrate Thrombosis and Hemostasis. Antioxid Redox Signal 2021, 35:1116–1133. 10.1089/ars.2021.0086. [DOI] [PubMed] [Google Scholar]
- [27].Sousa HR, Gaspar RS, Sena EM, da Silva SA, Fontelles JL, AraUjo TL, Mastrogiovanni M, Fries DM, Azevedo-Santos AP, Laurindo FR, et al. : Novel antiplatelet role for a protein disulfide isomerase-targeted peptide: evidence of covalent binding to the C-terminal CGHC redox motif. J Thromb Haemost 2017, 15:774–784. 10.1111/jth.13633. [DOI] [PubMed] [Google Scholar]
- *[28].Chinnaraj M, Flaumenhaft R, Pozzi N: Reduction of protein disulfide isomerase results in open conformations and stimulates dynamic exchange between structural ensembles. J Biol Chem 2022, 298:102217. 10.1016/j.jbc.2022.102217. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors used a novel fluorescence resonance energy transfer method to show that the archetypal thiol isomerase family member PDI adopts multiple distinct conformations in solution dictated by their redox environment. The reduced form of PDI is in an open conformation contrary to the reduced oxoform crystal structure.
- **[29].Wang L, Wang X, Lv X, Jin Q, Shang H, Wang CC, Wang L: The extracellular Ero1alpha/PDI electron transport system regulates platelet function by increasing glutathione reduction potential. Redox Biol 2022, 50:102244. 10.1016/j.redox.2022.102244. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors showed that oxidized PDI resulting from Ero1a-mediated oxidation enhances platelet aggregation potential. This is one of the first lines of evidence that oxidized thiol isomerases potentiate thrombosis.
- [30].Rehder DS, Borges CR: Cysteine sulfenic acid as an intermediate in disulfide bond formation and nonenzymatic protein folding. Biochemistry 2010, 49:7748–7755. 10.1021/bi1008694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Fu X, Cate SA, Dominguez M, Osborn W, Ozpolat T, Konkle BA, Chen J, Lopez JA: Cysteine Disulfides (Cys-ss-X) as Sensitive Plasma Biomarkers of Oxidative Stress. Sci Rep 2019, 9:115. 10.1038/s41598-018-35566-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chiu J, Hogg PJ: Allosteric disulfides: Sophisticated molecular structures enabling flexible protein regulation. J Biol Chem 2019, 294:2949–2960. 10.1074/jbc.REV118.005604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].McDonnell T, Wincup C, Buchholz I, Pericleous C, Giles I, Ripoll V, Cohen H, Delcea M, Rahman A: The role of beta-2-glycoprotein I in health and disease associating structure with function: More than just APS. Blood Rev 2020, 39:100610. 10.1016/j.blre.2019.100610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Tektonidou MG: Cardiovascular disease risk in antiphospholipid syndrome: Thromboinflammation and atherothrombosis. J Autoimmun 2022, 128:102813. 10.1016/j.jaut.2022.102813. [DOI] [PubMed] [Google Scholar]
- [35].Nocella C, Bartimoccia S, Cammisotto V, D’Amico A, Pastori D, Frati G, Sciarretta S, Rosa P, Felici C, Riggio O, et al. : Oxidative Stress in the Pathogenesis of Antiphospholipid Syndrome: Implications for the Atherothrombotic Process. Antioxidants (Basel) 2021, 10. 10.3390/antiox10111790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Buttari B, Profumo E, Capozzi A, Saso L, Sorice M, Rigano R: Post-translational modifications of proteins in antiphospholipid antibody syndrome. Crit Rev Clin Lab Sci 2019, 56:511–525. 10.1080/10408363.2019.1650714. [DOI] [PubMed] [Google Scholar]
- [37].Ioannou Y, Zhang JY, Passam FH, Rahgozar S, Qi JC, Giannakopoulos B, Qi M, Yu P, Yu DM, Hogg PJ, et al. : Naturally occurring free thiols within beta 2-glycoprotein I in vivo: nitrosylation, redox modification by endothelial cells, and regulation of oxidative stress-induced cell injury. Blood 2010, 116:1961–1970. 10.1182/blood2009-04-215335. [DOI] [PubMed] [Google Scholar]
- [38].Passam FH, Rahgozar S, Qi M, Raftery MJ, Wong JW, Tanaka K, Ioannou Y, Zhang JY, Gemmell R, Qi JC, et al. : Beta 2 glycoprotein I is a substrate of thiol oxidoreductases. Blood 2010, 116:1995–1997. 10.1182/blood-2010-02-271494. [DOI] [PubMed] [Google Scholar]
- *[39].Kumar S, Chinnaraj M, Planer W, Zuo X, Macor P, Tedesco F, Pozzi N: An allosteric redox switch in domain V of beta(2)-glycoprotein I controls membrane binding and anti-domain I autoantibody recognition. J Biol Chem 2021, 297:100890. 10.1016/j.jbc.2021.100890. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors show through site-directed mutagenesis studies that the allosteric disulfide cysteines in domain V of β2GPI is a redox swich to regulate pathogenic antibody recognition in antiphospholipid syndrome. This regulation may control the thrombogenicitiy during the oxidative stress of antiphospholipid syndrome.
- *[40].Buchholz I, McDonnell T, Nestler P, Tharad S, Kulke M, Radziszewska A, Ripoll VM, Schmidt F, Hammer E, Toca-Herrera JL, et al. : Specific domain V reduction of beta-2-glycoprotein I induces protein flexibility and alters pathogenic antibody binding. Sci Rep 2021, 11:4542. 10.1038/s41598-021-84021-2. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors performed biochemical studies using atomic force microscopy to show that chemical reduction of the cysteine redox states of β2GPI leads to an open and flexible structural conformation. The reduced protein potentiates pathogenic autoantibody binding.
- **[41].Passam FH, Chen G, Chen VM, Qi M, Krilis SA, Giannakopoulos B: Beta-2-glycoprotein I exerts antithrombotic function through its domain V in mice. J Autoimmun 2022, 126:102747. 10.1016/j.jaut.2021.102747. [DOI] [PubMed] [Google Scholar]; The authors investigated the enhanced thrombus formation in β2GPI-deficient mice. They found that domain V of the protein is anti-thrombotic in non-pathogenic blood clotting. Future studies to determine if the allosteric disulfides within domain V mediate anti-thrombotic effects are warranted.
- [42].Underbakke ES, Surmeli NB, Smith BC, Wynia-Smith SL, Marletta MA: Nitric Oxide Signaling. In Comprehensive Inorganic Chemistry II, edn 2. Edited by Reedijk J, Poeppelmeier K: Elsevier Ltd; 2013:241–262. [Google Scholar]
- [43].Wynia-Smith SL, Smith BC: Nitrosothiol formation and S-nitrosation signaling through nitric oxide synthases. Nitric Oxide 2017, 63:52–60. 10.1016/j.niox.2016.10.001. [DOI] [PubMed] [Google Scholar]
- [44].Smith BC, Marletta MA: Mechanisms of S-nitrosothiol formation and selectivity in nitric oxide signaling. Curr Opin Chem Biol 2012, 16:498–506. 10.1016/j.cbpa.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bekendam RH, Iyu D, Passam F, Stopa JD, De Ceunynck K, Muse O, Bendapudi PK, Garnier CL, Gopal S, Crescence L, et al. : Protein disulfide isomerase regulation by nitric oxide maintains vascular quiescence and controls thrombus formation. J Thromb Haemost 2018, 16:2322–2335. 10.1111/jth.14291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Rossi R, Giustarini D, Dalle-Donne I, Milzani A: Protein S-glutathionylation and platelet anti-aggregating activity of disulfiram. Biochem Pharmacol 2006, 72:608–615. 10.1016/j.bcp.2006.05.021. [DOI] [PubMed] [Google Scholar]
- [47].Grambow E, Mueller-Graf F, Delyagina E, Frank M, Kuhla A, Vollmar B: Effect of the hydrogen sulfide donor GYY4137 on platelet activation and microvascular thrombus formation in mice. Platelets 2014, 25:166–174. 10.3109/09537104.2013.786823. [DOI] [PubMed] [Google Scholar]
- [48].Dalle-Donne I, Giustarini D, Colombo R, Milzani A, Rossi R: S-glutathionylation in human platelets by a thiol-disulfide exchange-independent mechanism. Free Radic Biol Med 2005, 38:1501–1510. 10.1016/j.freeradbiomed.2005.02.019. [DOI] [PubMed] [Google Scholar]
- [49].Cao X, Ding L, Xie ZZ, Yang Y, Whiteman M, Moore PK, Bian JS: A Review of Hydrogen Sulfide Synthesis, Metabolism, and Measurement: Is Modulation of Hydrogen Sulfide a Novel Therapeutic for Cancer? Antioxid Redox Signal 2019, 31:1–38. 10.1089/ars.2017.7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Cuevasanta E, Moller MN, Alvarez B: Biological chemistry of hydrogen sulfide and persulfides. Arch Biochem Biophys 2017, 617:9–25. 10.1016/j.abb.2016.09.018. [DOI] [PubMed] [Google Scholar]
- [51].Zhang D, Macinkovic I, Devarie-Baez NO, Pan J, Park CM, Carroll KS, Filipovic MR, Xian M: Detection of protein S-sulfhydration by a tag-switch technique. Angew Chem Int Ed Engl 2014, 53:575–581. 10.1002/anie.201305876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Park CM, Macinkovic I, Filipovic MR, Xian M: Use of the “tag-switch” method for the detection of protein S-sulfhydration. Methods Enzymol 2015, 555:39–56. 10.1016/bs.mie.2014.11.033. [DOI] [PubMed] [Google Scholar]
- [53].Gu SX, Stevens JW, Lentz SR: Regulation of thrombosis and vascular function by protein methionine oxidation. Blood 2015, 125:3851–3859. 10.1182/blood-2015-01-544676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wang Y, Chen J, Ling M, Lopez JA, Chung DW, Fu X: Hypochlorous acid generated by neutrophils inactivates ADAMTS13: an oxidative mechanism for regulating ADAMTS13 proteolytic activity during inflammation. J Biol Chem 2015, 290:1422–1431. 10.1074/jbc.M114.599084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Dayal S, Gu SX, Hutchins RD, Wilson KM, Wang Y, Fu X, Lentz SR: Deficiency of superoxide dismutase impairs protein C activation and enhances susceptibility to experimental thrombosis. Arterioscler Thromb Vasc Biol 2015, 35:1798–1804. 10.1161/ATVBAHA.115.305963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sadler JE: von Willebrand factor assembly and secretion. J Thromb Haemost 2009, 7 Suppl 1:24–27. 10.1111/j.1538-7836.2009.03375.x. [DOI] [PubMed] [Google Scholar]
- [57].Fu X, Chen J, Gallagher R, Zheng Y, Chung DW, Lopez JA: Shear stress-induced unfolding of VWF accelerates oxidation of key methionine residues in the A1A2A3 region. Blood 2011, 118:5283–5291. 10.1182/blood-2011-01-331074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *[58].Tsai R, Interlandi G: Oxidation shuts down an auto-inhibitory mechanism of von Willebrand factor. Proteins 2021, 89:731–741. 10.1002/prot.26055. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors showed that vWF methionine sulfoxidation of Met1606 in the A2 domain prevents auto-regulation by the adjacent A1 and A2 domain, thus promoting vWF multimerization. This is a novel mechanism for vWF thrombogenicity as Met1606 sulfoxidation will prevent cleavage of the multimer by ADAMTS13.
- [59].Pieters M, Wolberg AS: Fibrinogen and fibrin: An illustrated review. Res Pract Thromb Haemost 2019, 3:161–172. 10.1002/rth2.12191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lau WH, White NJ, Yeo TW, Gruen RL, Pervushin K: Tracking oxidation-induced alterations in fibrin clot formation by NMR-based methods. Sci Rep 2021, 11:15691. 10.1038/s41598-021-94401-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Weigandt KM, White N, Chung D, Ellingson E, Wang Y, Fu X, Pozzo DC: Fibrin clot structure and mechanics associated with specific oxidation of methionine residues in fibrinogen. Biophys J 2012, 103:2399–2407. 10.1016/j.bpj.2012.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Martinez M, Weisel JW, Ischiropoulos H: Functional impact of oxidative posttranslational modifications on fibrinogen and fibrin clots. Free Radic Biol Med 2013, 65:411–418. 10.1016/j.freeradbiomed.2013.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Fay WP, Parker AC, Condrey LR, Shapiro AD: Human plasminogen activator inhibitor-1 (PAI-1) deficiency: characterization of a large kindred with a null mutation in the PAI-1 gene. Blood 1997, 90:204–208. [PubMed] [Google Scholar]
- [64].Zheng Z, Nayak L, Wang W, Yurdagul A Jr., Wang X, Cai B, Lapping S, Ozcan L, Ramakrishnan R, Pestell RG, et al. : An ATF6-tPA pathway in hepatocytes contributes to systemic fibrinolysis and is repressed by DACH1. Blood 2019, 133:743–753. 10.1182/blood-2018-07-864843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Shin EK, Park H, Noh JY, Lim KM, Chung JH: Platelet Shape Changes and Cytoskeleton Dynamics as Novel Therapeutic Targets for Anti-Thrombotic Drugs. Biomol Ther (Seoul) 2017, 25:223–230. 10.4062/biomolther.2016.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Rouyere C, Serrano T, Fremont S, Echard A: Oxidation and reduction of actin: Origin, impact in vitro and functional consequences in vivo. Eur J Cell Biol 2022, 101:151249. 10.1016/j.ejcb.2022.151249. [DOI] [PubMed] [Google Scholar]
- [67].Dominguez R, Holmes KC: Actin structure and function. Annu Rev Biophys 2011, 40:169–186. 10.1146/annurev-biophys-042910-155359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Shi Y, Carroll KS: Activity-Based Sensing for Site-Specific Proteomic Analysis of Cysteine Oxidation. Acc Chem Res 2020, 53:20–31. 10.1021/acs.accounts.9b00562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Lin S, Yang X, Jia S, Weeks AM, Hornsby M, Lee PS, Nichiporuk RV, Iavarone AT, Wells JA, Toste FD, et al. : Redox-based reagents for chemoselective methionine bioconjugation. Science 2017, 355:597–602. 10.1126/science.aal3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **[70].Gonzalez-Valero A, Reeves AG, Page ACS, Moon PJ, Miller E, Coulonval K, Crossley SWM, Xie X, He D, Musacchio PZ, et al. : An Activity-Based Oxaziridine Platform for Identifying and Developing Covalent Ligands for Functional Allosteric Methionine Sites: Redox-Dependent Inhibition of Cyclin-Dependent Kinase 4. J Am Chem Soc 2022, 144:22890–22901. 10.1021/jacs.2c04039. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors used oxaziridine-based probes to identify novel reactive methionines on cyclin-dependent kinase 4. Met169 was found to be an allosteric redox-regulated mechanism for cyclin-dependent kinase 4. Labeling with the probe on Met169 decreased kinase activity.