

Abstract
Neurons require physiological IFN-γ signaling to maintain central nervous system (CNS) homeostasis, however, pathological IFN-γ signaling can cause CNS pathologies. The downstream signaling mechanisms that cause these drastically different outcomes in neurons has not been well studied. We hypothesized that different levels of IFN-γ signaling in neurons results in differential activation of its downstream transcription factor, signal transducer and activator of transduction 1 (STAT1), causing varying outcomes. Using primary cortical neurons, we showed that physiological IFN-γ elicited brief and transient STAT1 activation, whereas pathological IFN-γ induced prolonged STAT1 activation, which primed the pathway to be more responsive to a subsequent IFN-γ challenge. This is an IFN-γ specific response, as other IFNs and cytokines did not elicit such STAT1 activation nor priming in neurons. Additionally, we did not see the same effect in microglia, suggesting this non-canonical IFN-γ/STAT1 signaling is unique to neurons. Prolonged STAT1 activation was facilitated by continuous janus kinase (JAK) activity, even in the absence of IFN-γ. Finally, although IFN-γ initially induced a canonical IFN-γ transcriptional response in neurons, pathological levels of IFN-γ caused long-term changes in synaptic pathway transcripts. Overall, these findings suggest that IFN-γ signaling occurs via non-canonical mechanisms in neurons, and differential STAT1 activation may explain how neurons have both homeostatic and pathological responses to IFN-γ signaling.
Keywords: Neuroimmunology, interferon-gamma, STAT1, cytokine, neuron
1. INTRODUCTION
IFN-γ is a classical immune cytokine that is critical for CNS health but also associated with many CNS pathologies and diseases (Ellwardt et al., 2016, Filiano et al., 2017, Filiano et al., 2016, Kunis et al., 2013). Specifically, IFN-γ signaling in neurons has become recognized as a major player in both maintaining CNS health and perpetuating CNS disease (Clark et al., 2022). Under homeostatic conditions, neurons require low physiological levels of IFN-γ for proper development, function, and homeostasis (Filiano, Xu, 2016, Flood et al., 2019, Janach et al., 2020, Nagakura et al., 2014). However, higher levels of IFN-γ associated with infection and inflammation can lead to unfavorable outcomes, including altered excitability (Vikman et al., 2003, Vikman et al., 2001, Vikman et al., 2005), morphological differences (Wong et al., 2004), and neurotoxicity (Mizuno et al., 2008). Additionally, human neuronal progenitor cells (NPCs) and neurons, derived from induced pluripotent stem cells (iPSC), treated with pathological levels of IFN-γ exhibited similar gene dysregulation as those differentially expressed in the brains of individuals with autism spectrum disorder (ASD) and schizophrenia (Warre-Cornish et al., 2020). Others have shown that IFN-γ treatment during the differentiation of human pluripotent stem cells (hPSCs) to cortical neurons reduced dendritic spine density (Kathuria et al., 2022). These works suggest differential outcomes downstream of physiological versus pathological IFN-γ signaling in neurons, though it is unclear how these mechanisms differ.
IFN-γ signaling has been well-defined as an anti-viral immune signaling pathway, but the response in neurons has not been well defined. Canonically, IFN-γ induces pro-inflammatory and anti-viral gene expression via activation of the downstream transcription factor STAT1. Upon viral recognition, immune cells produce and release IFN-γ which binds the IFN-γ receptor (IFNGR) on infected cells, leading to activation of JAK1 and JAK2, and phosphorylation of STAT1 (pSTAT1). pSTAT1 translocates to the nucleus and regulates the expression of IFN-γ stimulated genes (ISGs), resulting in a cytolytic response to clear virally infected cells. In peripheral cell types, STAT1 activation is a rapid and transient process, with pSTAT1 levels returning to baseline within 24–48hrs after initial activation (Ramana et al., 2002, Stark et al., 1998). In contrast, neurons exhibit extended JAK activation and delayed STAT1 dephosphorylation compared to peripheral cells (Podolsky et al., 2012), suggesting the pathway may use different mechanisms to activate and inhibit signaling. Infected neurons also utilize the IFN-γ/STAT1 to mount an immune response, however they clear virus in a non-cytolytic manner (Burdeinick-Kerr et al., 2009, Burdeinick-Kerr et al., 2007, O’Donnell et al., 2015), further suggesting that IFN-γ/STAT1 signaling may work through non-canonical mechanisms in neurons. It is worth noting that STAT1 activation in neurons has primarily been investigated in response to pathological levels of IFN-γ, and it is unknown how STAT1 activation occurs downstream of physiological IFN-γ.
Much of the work investigating the role of IFN-γ signaling in neurons has been inconsistent in experimental setups, using varying IFN-γ concentrations for different durations, as well as using various cell sources at different developmental timepoints. In this study, we investigated neuronal STAT1 activation in response to IFN-γ in different contexts, including IFN-γ concentration, duration of treatment, and timing of treatment. We hypothesized that different concentrations of IFN-γ result in differential downstream STAT1 activation, which contributes to the varying neuronal responses previously reported. We aimed to elucidate the underlying mechanisms contributing to non-canonical IFN-γ signaling in neurons in response to physiological versus pathological IFN-γ signaling, and how IFN-γ signaling affects neurons long term, focusing on future IFN-γ signaling and the downstream transcriptional response.
2. METHODS
2.1. Animals
C57BL/6J and CD-1 mice were originally purchased from The Jackson Laboratory and bred in-house. All animals were housed under normal conditions. All experiments were performed in accordance with Duke University Institutional Animal Care and Use Committee’s policies.
2.2. Primary Neuron Cultures
Brains were harvested from P0 pups and kept in cold HBSS. Brains were processed separately so that each plate/well consisted of neurons from one animal. Meninges were removed, then brains were digested for 30 minutes in digestion buffer (500μl Hanks Balanced Salt Solution (Gibco, cat #14025092), 20 U/ml papain (Worthington Biochemical, cat #LK003176), 100 U/ml DNAse (Millipore Sigma, cat #04716728001)) at 37°C. About 15 minutes into digestion, brains were gently triturated 1–2 times using an autoclaved glass pipette, and then allowed to continue digesting. At the end of the 30-minute digestion, brains were gently triturated 10 times using an autoclaved glass pipette. The brain homogenates were spun for 5 minutes at 200 g, then resuspended in 10 ml warm Neurobasal media (Gibco, cat #21103049) and passed through a 70um screen to create a single cell suspension. Cells were spun for 5 minutes at 200 g and resuspended in 1 ml full neuron media (50 ml Neurobasal media, 1 ml B27 Supplement (50x) (Gibco, cat #17504044), 500μl GlutaMAX (100x) (Gibco, cat #35050061), 60 μl penicillin/streptomycin (Sigma-Aldrich, cat #P0781). Cells were counted using a Cellometer (Nexcelom, US) and then resuspended in full neuron media at concentration of 350,000 cells/ml. Cells were plated in 24-well Poly-D-Lysine-coated plates (Corning, cat #354414), in 500 μl (175,000 cells) per well. Cultures were incubated at 37°C, 5% CO2. Media was changed by removing half media (~200 μl) and replacing with 200 μl fresh media every 2–3 days, beginning at day in vitro 7 (DIV7) (DIV5 for experiments beginning treatment on DIV5).
2.3. Primary Microglia Cultures
Brains were harvested from P1-P3 pups and were minced into small pieces with scissors in a petri dish, then digested with digestion buffer (HBSS with 2 mg/ml papain, 50 U/ml DNase-I) and pooled. Microglia were isolated using CD11b microbeads (Miltenyi Biotec, cat #130093634) according to manufacturer’s instructions. Isolated microglia were plated in collagen IV coated or poly-D-lysine coated 24-well plates (Corning cat #354430, #354414) with microglia growth media (DMEM/F12 (Gibco, cat #11330032), 100 U/ml penicillin, 100 μg/ml streptomycin (Sigma-Aldrich, cat #P0781), 2 mM glutamine (Gibco, cat #25030081), 5 μg/ml N-acetyl cysteine (Millipore Sigma, cat #A8199), 5 μg/ml insulin (Gibco, cat #12585014), 100 μg/ml apo-transferrin (Sigma-Aldrich, cat #T1147), 100 ng/ml sodium selenite (Sigma-Aldrich, cat #S5261), 2 ng/ml human TGF-β2 (BioLegend, cat #580702), 100 ng/ml murine IL-34 (R&D Systems, cat #5195), 1.5 μg/ml ovine wool cholesterol (Avanti Polar Lipids), and 1 μg/ml heparin sulfate (Galen Laboratory Supplies). Cultures were incubated at 37°C, 5% CO2. Microglia were treated with IFN-γ beginning on DIV2.
2.4. Primary Astrocyte Cultures
Astrocytes were isolated from P3–4 CD1 mice as previously described (Holt et al., 2019, Holt and Olsen, 2016). Briefly, cortices from 3–6 mice were dissected out, minced, and digested using papain (Worthington, cat #LK003178). After 15 and 30 minutes of digestion, the tissue was triturated 10–15 times. Tissue homogenate was transferred to a 15 mL tube and spun for 10 minutes at 500 g. Cells were resuspended in 5 mL MACS buffer (435.2 mL ddH20, 49.8 mL HBSS, 0.5% glucose, 0.015M HEPES pH 7, 0.2% milk peptone, 0.002M EDTA), filtered with a celltrics filter (Sysmex cat #04–004-2326), then spun for 10 minutes at 300 g. Myelin and microglia were removed (Myelin Removal Beads II, human, mouse, rat (Miltenyi Biotec, cat #130–096-733); CD11b (Microglia) MicroBeads, human and mouse (Miltenyi Biotec cat #130-093-634)) and astrocytes were isolated using Anti-ACSA-2 MicroBead Kit, mouse (Miltenyi Biotec cat #130–097-678) and LS Columns (Miltenyi Biotec, cat #130–042-401) (Holt and Olsen, 2016). Isolated astrocytes were plated in astrocyte media supplemented with Heparin-binding EGF-like growth factor (Foo et al., 2011) (equal parts Neurobasal (Gibco, cat #21103049) and DMEM (Gibco, cat #11960044) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin (Sigma-Aldrich, cat #P0781), 1 mM sodium pyruvate (Gibco, cat #11360070), 2 mM L-glutamine (Gibco, cat #25030081), 5 μg/ml N-acetyl cysteine (Millipore Sigma, cat #A8199), B27 Supplement (50x) (Gibco, cat #17504044), and 5 ng/ml Heparin-binding EGF-like growth factor (Millipore Sigma, cat #SRP6050–10UG)) and incubated at 37°C, 10% CO2.
On DIV1, cells were washed 3 times with room temperature DPBS without calcium or magnesium before adding fresh astrocyte media. On DIV3, a half media change was performed with fresh astrocyte media. On DIV6, cells were trypsinized and frozen in Cryostor CS 10 at 300,000 cells per 500 μl. Cells were frozen in a Mr. Frosty at −80 °C for 4–24 hours before being moved to liquid nitrogen for long term storage.
Cells were thawed quickly in a 37°C water bath and resuspended in 500 μl of warm astrocyte media. Cells were transferred to a 15 mL conical, resuspended in 10 mL astrocyte media, then spun at 300 g for 10 minutes. Cells were resuspended in 2.5 mL astrocyte media and plated in 24-well Poly-D-Lysine-coated plates (Corning, cat #354414), in 500 μl (100,000 cells) per well. Cultures were incubated at 37°C, 5% CO2. Media was changed 2 days after plating, and astrocytes were treated 4 days after plating.
2.5. Cytokine treatment
Half media (~200 μl) was removed from each well to be treated and replaced with 200 μl of fresh media containing the appropriate concentration of cytokine; each cytokine solution was prepared to be double the concentration of the desired final concentration in culture (200 U/ml for 100 U/ml final concentration) for pathological IFN-γ (BioLegend, cat #575302), IFN-β (BioLegend, cat #581302), IFN-α (Biolegend cat #752802), IL-6 (BioLegend, cat #575702), and IL-17a (BioLegend, cat #576002); 0.04 U/ml (0.02 U/ml final concentration) for physiological IFN-γ). Cytokine solutions were added to culture for 30 minutes (acute treatment) or 24 hours (priming treatment). To remove cytokine from cultures, all media was removed, wells were washed with PBS 3 times, and a solution of half fresh full neuron media and half conditioned full neuron media was added back to each well. Conditioned media was only collected from untreated neurons to ensure it was cytokine free.
For IFN-β and IFN-α dose response experiments, half media (~200 μl) was removed from each well to be treated and replaced with 200 μl of fresh media containing the appropriate concentration of cytokine; each cytokine solution was prepared to be double the concentration of the desired final concentration in culture.
2.6. Ruxolitinib treatment
Half media (~200 μl) was removed from each well and replaced with 200 μl of fresh media containing 2 μM Ruxolitinib (Chemscene, cat #941678–49-5) so that the final concentration of Ruxolitinib in the cultures was 1 μM. Ruxolitinib was either left in culture until sample collection, or washed out after 24 hours by removing all media, washing wells 3 times with PBS, then adding 500 μl of half fresh media and half conditioned media. For experiments where Ruxolitinib was left in for 1 week, half media changes were performed without adding additional Ruxolitinib.
2.7. Western Blot
Samples to be analyzed by western blot were lysed in the well. Media was removed and each well was washed 1 time with PBS. 70 μl of lysis buffer (Pierce RIPA buffer (Thermo Scientific, cat #89900), cOmplete Tablets protease inhibitor (Roche, cat #04693159001), PhosSTOP phosphatase inhibitor (Roche, cat #04906845001) was added to each well and incubated at room temperature for 3–5 minutes. Cells were further lysed by scraping the well and lysate was collected into a 1.5 ml Eppendorf tube and incubated on ice for 5–10 minutes. Lysates were spun for 10 minutes at 10,000g at 4°C. Supernatants were collected and stored at −80°C. Protein concentration of each sample was measured using a Bradford Assay (Bio-Rad, cat #5000202EDU) and spectrophotometer (Implen, Denville Scientific Inc) and extrapolated from a BSA standard curve. Samples were then diluted with lysis buffer to load equal total amounts of protein for each sample. Samples were run on SDS-PAGE (4–20% Mini-PROTEAN, Bio-Rad, Cat #4561096) and transferred onto nitrocellulose membranes (Bio-Rad, cat #1620168) overnight at 4°C. Membranes were blocked in TBS-T (TBS (Bio-Rad, Cat #1706435), 0.5% Tween-20 (Aldrich-Sigma, cat #P7949)) with 2% BSA (Gibco, cat #15260037) for 1 hour at room temperature on a shaker. Membranes were incubated with primary antibody (1:2000 pSTAT1 (Santa Cruz Biotechnology, cat #sc-136229), 1:1000 STAT1 (Cell Signaling Technology, cat #14994), 1:1000 GAPDH (Millipore Sigma, cat #MAB374) in blocking buffer overnight at 4°C. Membranes were washed with TBS-T 3 times for 10 minutes on a shaker at room temperature, then incubated with secondary antibody (1:1000 anti-rabbit IgG H&L (HRP) (Abcam, cat #ab6721), 1:1000 anti-mouse IgG (Peroxidase) (Jackson ImmunoResearch, cat #115035003)) in TBS-T for 1 hour on a shaker at room temperature. Membranes were washed with TBS-T 3 times for 10 minutes on a shaker at room temperature, then incubated with ECL solution (Bio-Rad, cat #1705060) for 5 minutes on a shaker at room temperature. Membranes were imaged using a C-Digit Blot Scanner (LI-COR Biosciences) and Image Studio Digits Ver 5.2 software (LI-COR Biosciences). Blots were analyzed using ImageJ (NIH).
2.8. mRNA Isolation
Media was removed from each well and washed once with PBS. 70 μl of TrypLE Select (10x) (Gibco, cat #A1217701) was added to each well, then incubated for 2–3 minutes at 37°C. 1 ml of PBS was added to stop the trypsin reaction, then cells were collected from each well. Cells were pelleted by spinning for 10 minutes at 10,000g at 4°C. Cell pellets were stored at −80°C. mRNA was extracted using the Qiashredder (Qiagen, cat #79654) and RNeasy kit (Qiagen, cat #74106) according to manufacturer protocol and quantified using a nanophotometer (Implen, Denville Scientific Inc).
2.9. RT-qPCR
cDNA was synthesized using Superscript VILO cDNA Synthesis kit (Thermo Scientific, cat #11754250) according to the manufacturer’s protocol, then diluted 1:20 for use in qPCR. qPCR was performed using Taqman Gene expression assays (Stat1 Mm01257286_m1, Irf1 Mm00515192_m1, Cxcl10 Mm00445235_m1, Socs1 Mm00782550_s1, Gapdh Mm99999915_g1, Thermo Fisher Scientific, cat #4331182) and TaqMan Fast Advanced master mix (Thermo Fisher Scientific, cat #4444963), and was run on the CFX96 Real-Time System (Bio-Rad) and Bio-Rad CFX Manager Software. Samples were analyzed in duplicate. Each sample was probed separately for the gene of interest and Gapdh (used as a housekeeping gene).
2.10. RNA sequencing
Libraries were created and sequencing was performed by the Duke Center for Genomic and Computational Biology Shared Resource using the Illumina NovaSeq 6000 platform with 50-bp paired-end reads.
2.9.1. Differential Gene Expression and Motif Analysis
RNA-Seq FASTQ files were aligned to the mm39 mouse reference genome using STAR (version 2.7.3a) (Dobin et al., 2013). Differential gene expression was determined and principal component analysis (PCA) performed using the ‘DESeq2’ Bioconductor package in R (Love et al., 2014). P-values were adjusted for false discovery rate using the Benjamini-Hochberg procedure (Benjamini and Hochberg, 1995). Genes with Padj < 0.05 are reported. Over-represented gene ontology (GO) terms were determined using a series of hypergeometric test with the ‘GOstats’ Bioconductor package in R (Falcon and Gentleman, 2007). UpSet plot was generated using the ‘upsetjs’ package in R (Conway et al., 2017, Lex et al., 2014). Conserved motifs were determined using the Sensitive, Thorough, Rapid, Enriched Motif Elicitation (STREME) tool within the MEME suite (version 5.4.1) (Bailey et al., 2015). STREME output was compared to the HOCOMOCOv11 database using the MEME Tomtom tool. Resulting alignment comparisons are shown.
2.9.2. Venn diagram
Venn diagrams were generated using Venny 2.1.0 (https://bioinfogp.cnb.csic.es/tools/venny/index.html; Oliveros, J.C. (2007–2015) Venny. An interactive tool for comparing lists with Venn’s Diagrams.) online web tool.
2.11. Experimental Design and Statistical Analysis
Male and female mice were used in all experiments. When male cultures were compared to female cultures, we observed no effect of sex in time course experiments on pSTAT1 (Two Way ANOVA, repeated measures: main effect of sex p=0.9462) or STAT1 levels (Two Way ANOVA, repeated measures: main effect of sex p=0.8617), therefore we did not segregate by sex for statistical analyses reported here. N = number of wells; for primary neurons experiments, each well consisted of cultured neurons from one mouse; for primary microglia experiments, each well consisted of pooled microglia from multiple brains within a litter. Each experiment used at least two mice from two different litters. Two-way repeated measures ANOVA were used, unless there were missing values (due to some experiments only looking at some conditions) in which case a Mixed Effects Model was used. If a significant main effect was observed, appropriate post hoc tests were run (Sidak’s or Tukey’s). For time course experiments, repeated measures were used. Raw data, statistics, and N’s are included in extended data. RNA-sequencing data discussed in this publication have been deposited in the NCBI’s Gene Expression Omnibus (GSE232304) (Edgar et al., 2002).
To account for variation across blots, values from each blot were normalized to a consistent treatment condition across blots. Because pSTAT1 levels in untreated neurons were near zero, pSTAT1 levels were reported as “% max pSTAT1 signal” and were calculated within individual blots. Total STAT1 levels were reported as “fold-change – normalized to untreated” and were calculated compared to untreated neurons at time 0 within individual blots. pSTAT1 levels in the Ruxolitinib experiments were calculated as a % of the vehicle treated samples at each corresponding timepoint. For RT-qPCR, the double delta Ct method was used to calculate relative fold change in gene expression. Time course experiments were calculated relative to untreated neurons (at time 0); Ruxolitinib experiments were calculated relative to vehicle treated neurons at each corresponding timepoint.
3. RESULTS
3.1. Neurons have unique STAT1 response to pathological IFN-γ compared to physiological IFN-γ.
Neurons have been reported to have delayed STAT1 dephosphorylation when treated with high levels of IFN-γ at day 5 in culture (DIV5) (Podolsky, Solomos, 2012), a timepoint at which primary neurons do not have fully matured synapses and may be considered “immature”. First, we sought to answer whether this non-canonical STAT1 activation in response to pathological IFN-γ was specific to this early timepoint, or if it is intrinsic to all neurons regardless of developmental age. To answer this, we cultured primary cortical neurons and compared IFN-γ induced STAT1 activation in immature neurons (DIV5) or neurons that are synaptically connected (DIV12) (Rao et al., 1998) (Fig 1A). We used two doses of IFN-γ: a “pathological” dose of IFN-γ (100 U/ml) which is similar to IFN-γ levels found in the CNS during viral infection (Frei et al., 1988), and has been associated with pathological outcomes in vitro, or a lower “physiological” dose (0.02 U/ml) which is similar to IFN-γ levels found in the CNS of healthy patients (Baruch et al., 2014, Deczkowska et al., 2016) and has been shown to have homeostatic functions. Going forward, we will refer to the higher (100 U/ml) dose as pathological IFN-γ and the lower (0.02 U/ml) dose as physiological IFN-γ.
Fig 1. Neurons have prolonged STAT1 response to pathological IFN-γ compared to physiological IFN-γ.
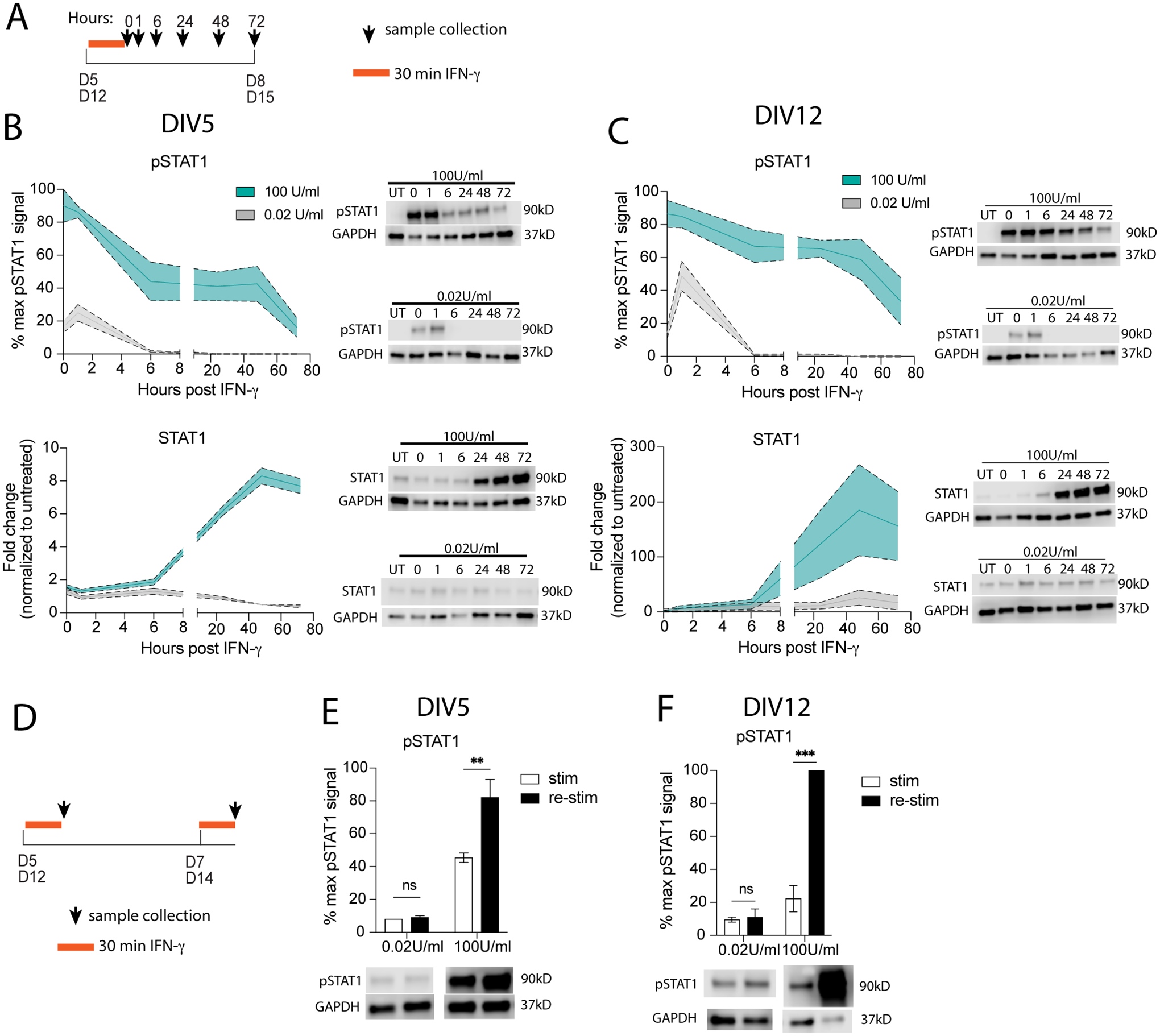
(A) Primary mature neurons were treated on (B) DIV5 or (C) DIV12 with physiological (gray; 0.02 U/mL) or pathological levels (cyan; 100 U/mL) of IFN-γ for 30 minutes then washed out; pSTAT1 and STAT1 were measured by western blot. (D) Primary mature neurons were treated with physiological (gray; 0.02 U/mL) or pathological levels (cyan; 100 U/mL) of IFN-γ for 30 minutes on (E) DIV5 or (F) DIV12, then re-stimulated 48 hours later with physiological or pathological IFN-γ. pSTAT1 levels were measured by western blot. Male and female mice included. Dotted lines represent SEM. (B) Two-Way ANOVA, repeated measures: pSTAT1: main effect of time *p<0.05 and dose **p<0.005, N=2–5; STAT1: main effect of time ***p<0.0005 and dose ***p<0.0005, interaction ***p<0.0005; N=2–5). (C) Mixed Effects Analysis, repeated measures: pSTAT1: main effect of time ***p<0.0005 and dose ***p<0.0005, interaction *p<0.05, N=6; STAT1: main effect of time *p<0.05, dose p=0.06, interaction **p<0.005; N=6). (E) Two Way ANOVA: main effect of dose ***p<0.0005 and restim *p<0.05, interaction *p<0.05; post-hoc Sidak’s multiple comparison: 100U/ml **p<0.005; N=3–5. (F) Two Way ANOVA: main effect of dose ***p<0.0005 and restim ***p<0.0005, interaction ***p<0.0005; post-hoc Sidak’s multiple comparison: 100U/ml ***p<0.0005; N=2–4.
As others have previously reported (Podolsky, Solomos, 2012), we observed prolonged STAT1 phosphorylation (pSTAT1) levels in neurons treated for 30 minutes with pathological IFN-γ on DIV5 (Fig 1B). Like in most cell types, pSTAT1 levels peaked at the time of IFN-γ washout (t=0). However, whereas in most cell types pSTAT1 levels return to baseline by 24–48 hours after IFN-γ stimulation (Podolsky, Solomos, 2012), we observed that pSTAT1 levels had still not returned to baseline in neurons at 72 hours post-washout (t=72). Since STAT1 regulates its own expression, we also measured total STAT1 protein expression after pathological IFN-γ. Total STAT1 levels began to increase after IFN-γ treatment and continued to increase for up to 72 hours post IFN-γ washout (Fig 1B).
We wanted to know if this prolonged STAT1 response was unique to young developing neurons. To test this, we also treated primary neuron cultures on DIV12, at which they have been reported to have mature synapses (Rao, Kim, 1998). We observed a similar prolonged STAT1 response as we did in immature cultures, with pSTAT1 levels peaking immediately after IFN-γ treatment and remaining elevated for up to 72 hours post IFN-γ washout (Fig 1C). Total STAT1 levels also increased after IFN-γ treatment and continued to increase for up to 72 hours post IFN-γ washout. To note STAT1 levels were approximately 20 times higher in DIV12 compared to DIV5. These data suggest that this unique STAT1 response is present in neurons at all stages and is not due to developmental differences.
While many studies have tested the effects of pathological IFN-γ on neurons (Kathuria, Lopez-Lengowski, 2022, Mizuno, Zhang, 2008, Vikman, Hill, 2003, Vikman, Owe-Larsson, 2001, Vikman, Siddall, 2005, Warre-Cornish, Perfect, 2020, Wong, Goldshmit, 2004), the effects of physiological IFN-γ on neurons is not well understood. Physiological levels (pg/ml) of IFN-γ are always present in the CNS and are important for maintaining proper neuron function and inhibitory tone (Baruch, Deczkowska, 2014, Deczkowska, Baruch, 2016, Filiano, Xu, 2016). So far, it is unclear how IFN-γ can have diverse effects on neurons. We hypothesized that physiological IFN-γ would result in a different STAT1 response than that seen in response to pathological levels of IFN-γ. To test this, we treated primary neurons (immature (DIV5) or mature (DIV12)) for 30 minutes with physiological levels of IFN-γ (0.02 U/ml; 20 pg/ml) (Fig 1A). Interestingly, neurons had a very different response to physiological levels of IFN-γ. After IFN-γ washout in both immature and mature neurons, pSTAT1 levels continued to increase, with peak levels observed at 1hr post-washout. Surprisingly, pSTAT1 levels returned to baseline by 6hr post-washout. Total STAT1 levels never increased above baseline untreated levels (Fig 1B, 1C). These results indicate that the length of the STAT1 response in neurons differs depending on the concentration of IFN-γ eliciting the response.
Since the levels of STAT1 remained elevated after a pathological exposure, we sought out to determine whether the prolonged STAT1 response after pathological IFN-γ would influence subsequent IFN-γ/STAT1 responses. To test this, we treated immature or mature neurons with pathological IFN-γ for 30 minutes, then 48 hours post IFN-γ washout re-challenged the neurons with pathological IFN-γ for another 30 minutes (Fig 1D). We observed a priming effect in both immature and mature neurons after re-challenging with pathological IFN-γ. In immature neurons, pSTAT1 levels were 2-fold higher after re-challenging compared to the initial treatment (Fig 1E). Even more striking, in mature neurons pSTAT1 levels were 5-fold higher after rechallenging compared to after the initial treatment (Fig 1F). These data suggest that pathological levels of IFN-γ lead to prolonged STAT1 responses that are also able to prime future IFN-γ insults. Interestingly, physiological levels of IFN-γ did not induce a priming effect, with pSTAT1 levels reaching similar levels after both initial treatment and re-challenge (Fig 1E, 1F).
3.2. Prolonged pathological IFN-γ induces persistent STAT1 response in neurons.
To better model a prolonged IFN-γ response that may occur during infection or neuroinflammation (El-Ansary and Al-Ayadhi, 2012, Olsson, 1992, Ottum et al., 2015, Soltani Khaboushan et al., 2022), we treated developing neurons with IFN-γ for 24 hours before measuring STAT1 activation (Fig 2A). Like the 30-minute treatment, we observed maximum pSTAT1 levels immediately after IFN-γ washout (t=0). Interestingly, pSTAT1 levels were decreased at 6 hours but then rebounded again at 24 hours and remained stable for up to 72 hours (Fig 2B). Surprisingly, pSTAT1 levels remained well above baseline for 144 hours, a full week after removing IFN-γ from culture, and still had not completely returned to baseline by 3 weeks (480 hours) after IFN-γ washout. Total STAT1 levels followed a very similar pattern, with STAT1 levels peaking after washout and remaining high for at least 72 hours before finally declining, beginning 1 week after IFN-γ washout (Fig 2B). Treatment with physiological IFN-γ for 24 hours did not elicit a prolonged STAT1 response, with pSTAT1 levels nearly undetectable at every timepoint after IFN-γ washout (Fig 2B). There was a brief increase of total STAT1 which returned to baseline by 24 hours (Fig 2B).
Fig 2. Pathological IFN-γ primes immature neurons to have a heightened STAT1 response in mature neurons.
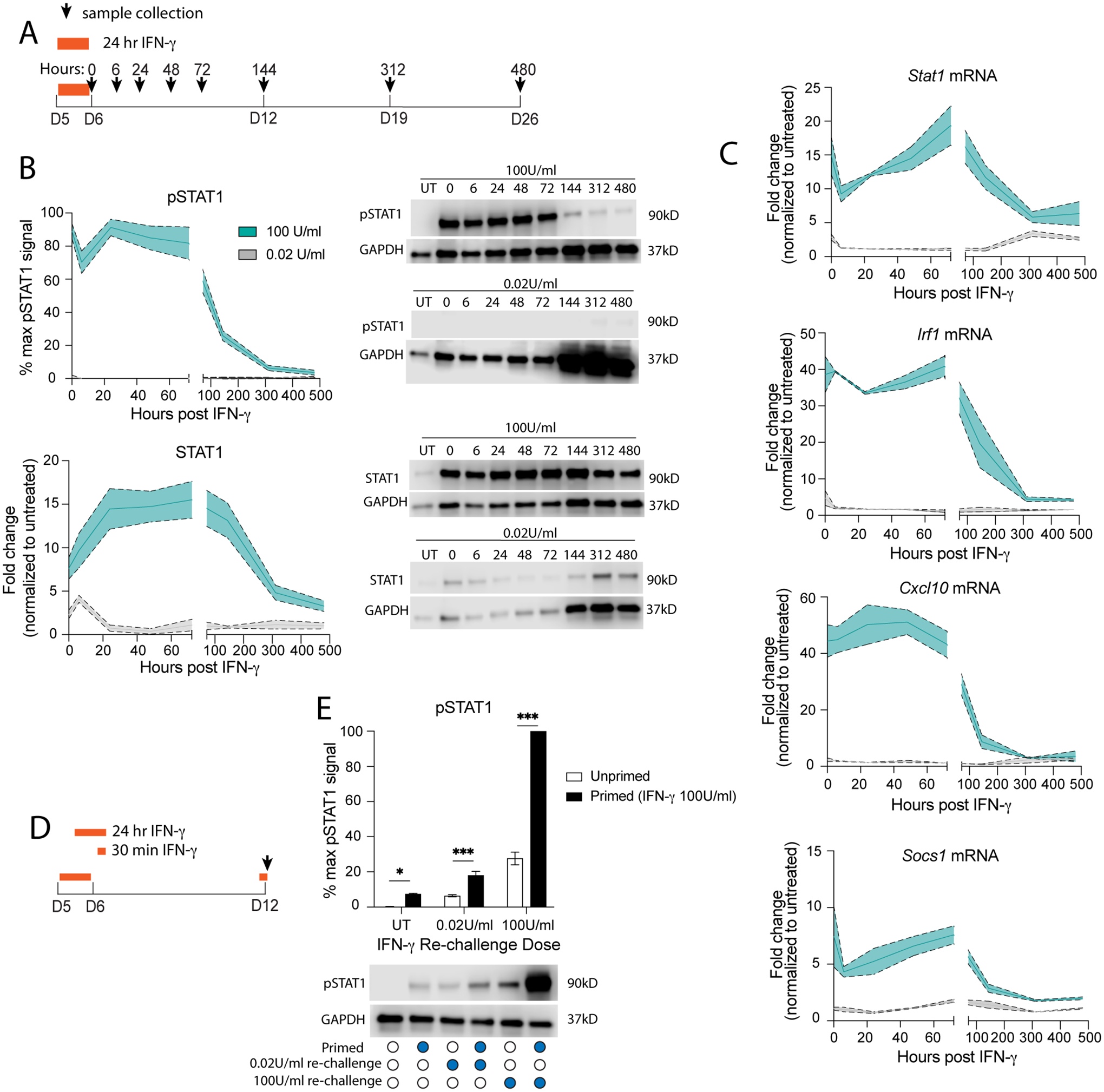
(A) Primary neurons (DIV5) were treated with physiological (gray; 0.02 U/mL) or pathological levels (cyan; 100 U/mL) of IFN-γ for 24 hours then washed out; (B) pSTAT1 and STAT1 were measured by western blot. (C) mRNA was collected and Stat1, Irf1, Cxcl10, and Socs1 mRNA levels were measured by qRT-PCR. (D) Primary neurons were treated with pathological IFN-γ for 24 hours on DIV5, then rechallenged with physiological or pathological IFN-γ for 30 minutes on DIV12. (E) pSTAT1 levels were measured by western blot. Male and female mice included. Dotted lines represent SEM. (B) Mixed Effects Model, repeated measures: pSTAT1: main effect of time ***p<0.0005 and dose ***p<0.0005, interaction ***p<0.0005, N=2–8; STAT1: main effect of time *p<0.05 and dose ***p<0.0005, interaction ***p<0.0005; N=2–12. (C) Mixed Effects Model, repeated measures: Stat1: main effect of time ***p<0.0005 and dose **p<0.005, interaction ***p<0.0005, N=2–5; Irf1: main effect of time ***p<0.0005 and dose ***p<0.0005, interaction ***p<0.0005, N=2–4; Cxcl10: main effect of time ***p<0.0005 and dose ***p<0.0005, interaction ***p<0.0005, N=2–4; Socs1: main effect of time **p<0.005 and dose ***p<0.0005, interaction **p<0.005, N=2–4. (E) Two Way ANOVA: main effect of priming ***p<0.0005 and restim dose ***p<0.0005, interaction ***p<0.0005; post-hoc Sidak’s multiple comparison: *p<0.05, ***p<0.0005; N=5.
To determine if prolonged STAT1 activation after pathological IFN-γ affected STAT1’s function as a transcription factor, we measured gene expression of canonical ISGs by qRT-PCR. We found that gene expression of Stat1, Irf1, Cxcl10, and Socs1 all followed a similar pattern as pSTAT1 and total STAT1 levels after pathological IFN-γ, with a robust increase in expression following IFN-γ treatment, which persisted until 1–2 weeks post IFN-γ washout. Physiological IFN-γ resulted in a transient initial increase in gene expression of Stat1 and Irf1, but not Cxcl10 or Socs1 (Fig 2C). This suggests that prolonged STAT1 activation does affect the function of STAT1 as a transcription factor and has consequences for downstream genes regulated by STAT1.
3.3. Pathological IFN-γ primes immature neurons to have a heightened STAT1 response as mature neurons.
We demonstrated above that pathological IFN-γ has a priming effect on neurons, and re-challenge resulted in increased pSTAT1 levels in both immature and mature neurons. Since treating neurons with pathological IFN-γ resulted in a prolonged STAT1 response persisting for weeks after IFN-γ washout, we hypothesized that these neurons would also exhibit a persistent priming effect. To test this, we primed developing neurons with pathological IFN-γ for 24 hours, washed out IFN-γ, and then re-challenged one week later (DIV12; t=144 hours) with either pathological or physiological IFN-γ for 30 minutes (Fig 2D). Neurons re-challenged with pathological IFN-γ had significantly increased pSTAT1 levels, suggesting that even a week after IFN-γ washout, the priming effect is still intact. Surprisingly, re-challenging with physiological IFN-γ also resulted in increased pSTAT1 levels compared to unprimed neurons receiving only physiological IFN-γ for 30 minutes on DIV12. In fact, pSTAT1 levels after re-challenge with physiological IFN-γ resembled pSTAT1 levels observed in mature neurons treated with pathological IFN-γ for 30 minutes (Fig 2E). These data suggest that pathological IFN-γ exerts a priming effect which persists into maturity, making neurons more sensitive to future IFN-γ re-challenge, even at low physiological doses.
3.4. Other cytokines do not elicit a prolonged STAT1 response in neurons.
Next, we tested whether other cytokines can induce prolonged STAT1 activation in neurons. Like IFN-γ, type I-IFNs also result in pSTAT1 but utilize pSTAT1/pSTAT2 heterodimers (coupled with IRF9) for a downstream transcriptional response (Stark, Kerr, 1998). We treated developing neurons with IFN-α or IFN-β (100U/ml) for 24 hours, washed out and then measured pSTAT1 and STAT1 levels. IFN-α treatment resulted in a transient increase in pSTAT1 levels, which returned to baseline by 24 hours following washout (Sup. Fig 1a). Total STAT1 levels were initially increased approximately 8-fold compared to untreated neurons, a similar increase to that observed in the IFN-γtreated neurons. However, IFN-α did not induce a prolonged STAT1 response, with total STAT1 levels returning to baseline by 72 hours (Fig 3A). We were unable to detect pSTAT1 after 24 hour treatment with IFN-β (extended data 1); we predicted that pSTAT1 was present initially following treatment, but then quickly recovered back to baseline prior to the end of the 24 hour treatment period, similar to pSTAT1 levels we observed after physiological levels of IFN-γ (Fig 2B). To confirm this, we treated neurons with IFN-β for 30 minutes and then probed for pSTAT1. We observed that IFN-β did induce a transient increase in pSTAT1, with levels returning to baseline by 24 hours (Sup. Fig 1B). Total STAT1 levels were 5-fold higher than untreated at the time of IFN-β washout, and quickly returned to baseline levels by 48 hours (Fig 3A). To confirm that type I IFNs do not induce a prolonged STAT1 response, we treated neurons with higher doses of IFN-α or IFN-β (500 U/mL and 5000 U/mL). Neither dose was sufficient to induce a prolonged STAT1 response (Sup. Fig 2).
Fig 3. Other cytokines do not elicit a prolonged STAT1 response in neurons.
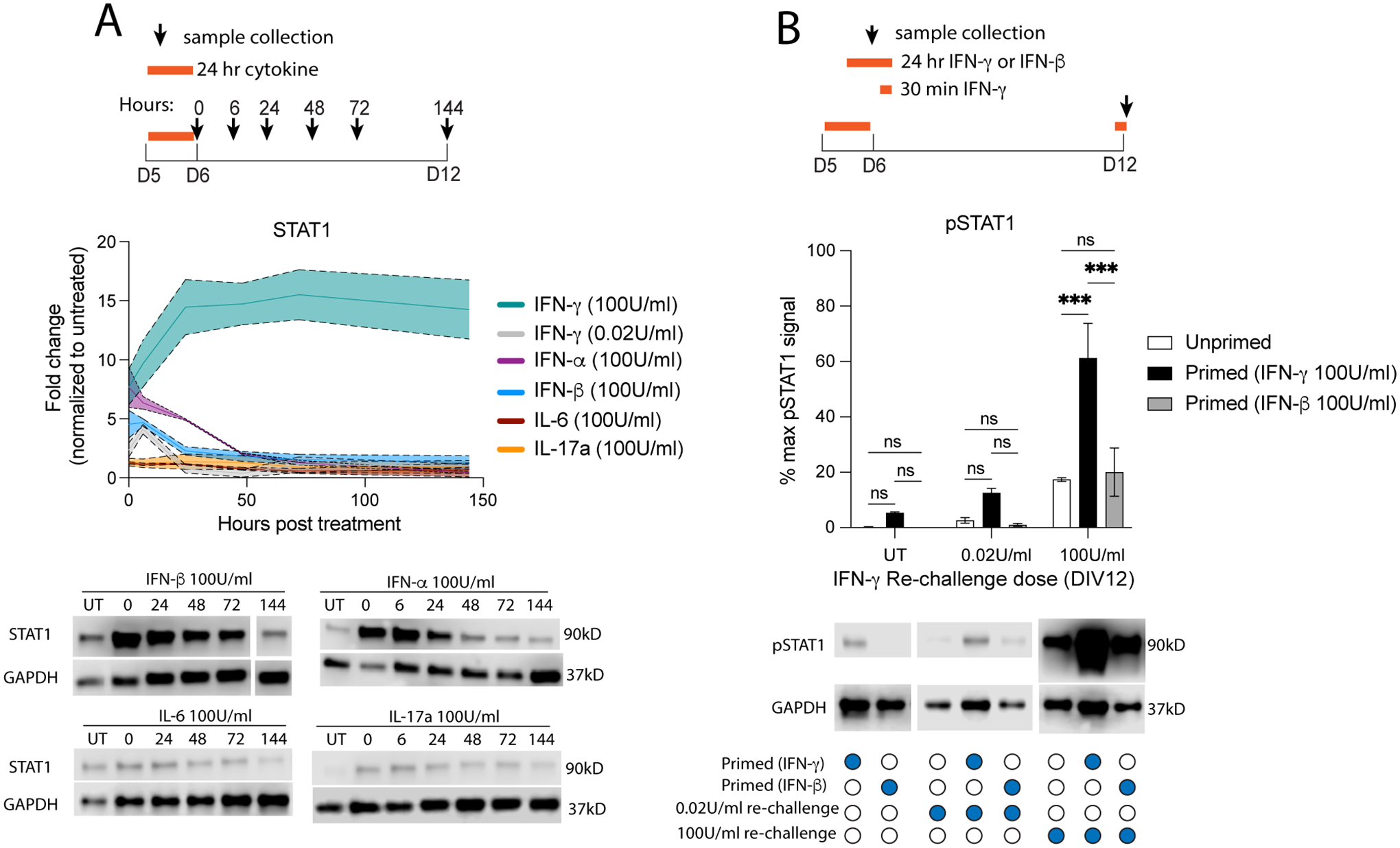
(A) Primary neurons were treated with IFN-α, IFN-β, IL-6, or IL-17a for 24 hours then washed out. Total STAT1 protein expression was quantified by western blot. We then compared these data with data from Fig 2B where neurons were treated with IFN-γ. (B) Primary neurons were treated with IFN-γ or IFN-β for 24 hours (primed) or not (unprimed) on DIV5, then re-challenged with IFN-γ for 30 minutes on DIV12. Male and female mice included. Dotted lines represent SEM. (A) Mixed Effects Model, repeated measures: main effect of cytokine ***p<0.0005, interaction (cytokine × time) ***p<0.0005, N=2–8. (B) Two Way ANOVA: main effect of priming ***p<0.0005 and restim dose ***p<0.0005, interaction ***p<0.0005; post-hoc Tukey’s multiple comparison: ***p<0.0005; N=3–10.
We also tested the STAT1 response to other cytokines implicated in various CNS pathologies (Alves de Lima et al., 2020, Choi et al., 2016, Jones et al., 2017, Kathuria, Lopez-Lengowski, 2022, Lau and Yu, 2001, Li et al., 2009, Orellana et al., 2005, Reed et al., 2020, Smith et al., 2007, Sukoff Rizzo et al., 2012, Wei et al., 2012, Wong and Hoeffer, 2018). Treatment with either IL-6 or IL-17a for 24 hours elicited no STAT1 response, with no detectable pSTAT1 and STAT1 levels remaining the same as in untreated neurons (Fig 3A, extended data 1). These data suggest that other cytokines do not induce prolonged STAT1 activation in neurons.
We also tested whether the priming effect observed after pathological IFN-γ was IFN-γ specific or independent of the type of ligand. Neurons primed with IFN-β for 24 hours on DIV5 and re-challenged with IFN-γ for 30 minutes on DIV12 did not have increased pSTAT1 levels (Fig 3B). However, it is worth noting that at the time of re-challenge, STAT1 levels in neurons primed with IFN-β were lower than those in neurons primed with IFN-γ (Fig 3A).
3.5. Prolonged STAT1 response is unique to neurons.
Next, we tested whether prolonged STAT1 activation is a trait intrinsic to other resident CNS cell types as well. As the resident macrophages of the CNS, microglia can respond to IFN-γ and have also been implicated in CNS health and disease (Ben-Yehuda et al., 2020, Butovsky et al., 2006, Butturini et al., 2019, Cowan et al., 2022, Di Liberto et al., 2018, Ding et al., 2015, Neher and Cunningham, 2019, Ziv et al., 2006). Similarly, IFN-γ signaling in astrocytes plays both protective and detrimental roles in infection and various CNS disorders (Halonen et al., 2001, Hashioka et al., 2009, Hindinger et al., 2012, Smith et al., 2020). To compare the microglial, astrocyte, and neuronal IFN-γ/STAT1 responses, we treated primary microglial cultures and primary astrocyte cultures with pathological IFN-γ (100U/ml) for 24 hours, then measured pSTAT1 and total STAT1 protein levels. In both microglia and astrocytes. pSTAT1 levels increased after 24 hours of treatment and quickly returned to baseline, with microglia pSTAT1 reaching baseline by 24 hours, and astrocyte pSTAT1 reaching baseline by 48 hours, as has been observed in many other peripheral cell types (Podolsky, Solomos, 2012) (Fig 4A). In microglia, total STAT1 protein levels increased 2-fold compared to untreated levels which persisted for 48 hours after IFN-γ washout (Fig 4A). Interestingly, total STAT1 levels in astrocytes reached levels similar to those observed in neurons, with a 13-fold compared to untreated levels. However, unlike neurons, total STAT1 levels began to decrease immediately following IFN-γ washout. These data suggest that prolonged STAT1 activation is specific to neurons, and not an intrinsic feature of resident CNS cell types.
Fig 4. Prolonged STAT1 response is unique to neurons.
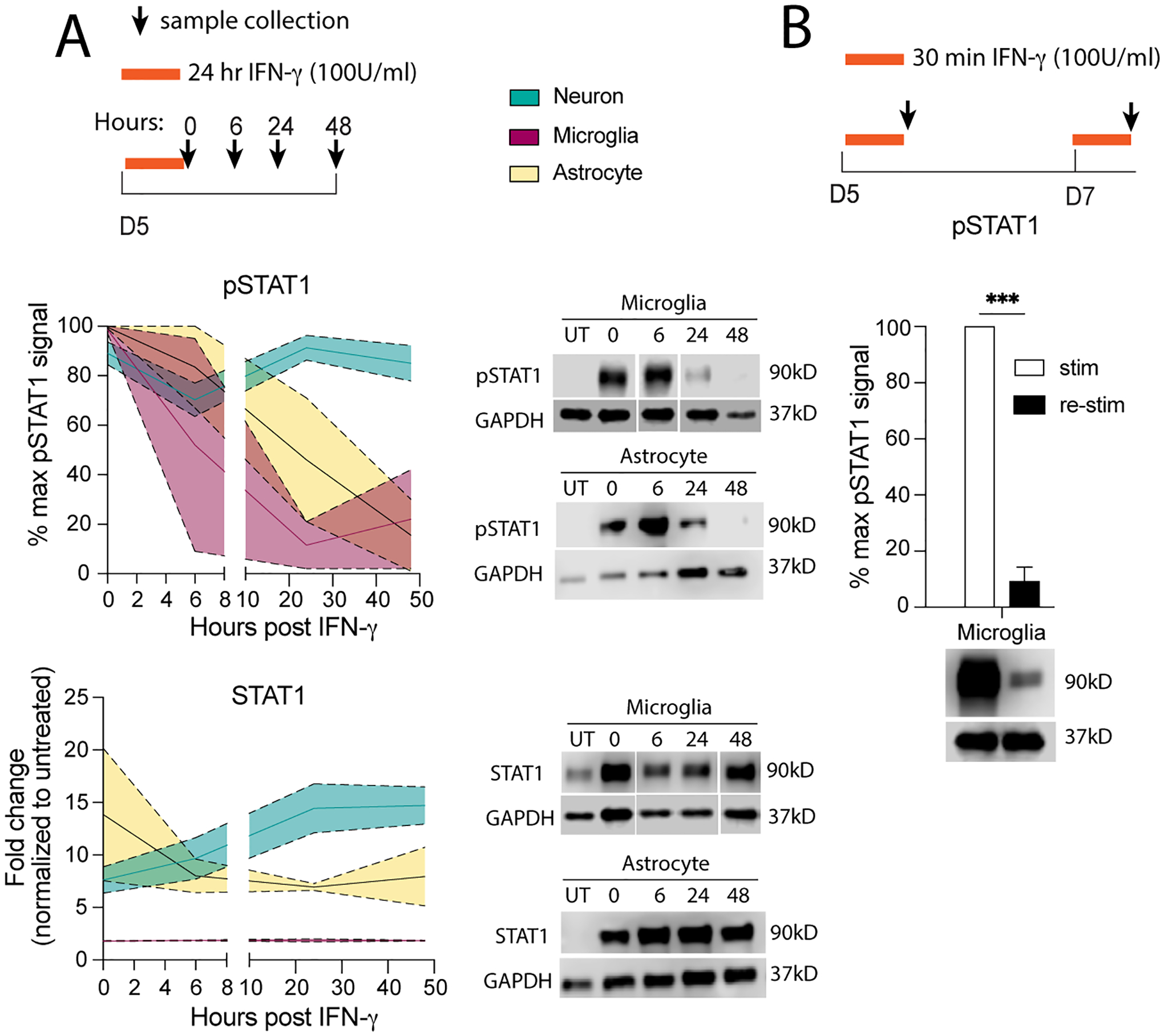
(A) Primary microglia or astrocytes were treated for 24 hours with 100 U/mL IFN-γ then washed out; pSTAT1 and STAT1 protein was measured by western blot. pSTAT1 and STAT1 protein levels from neurons treated in Fig 2B, were used to compare to pSTAT1 and STAT1 levels in microglia and astrocytes. (B) Microglia were treated with 100 U/mL IFN-γ for 30 minutes, then washed and 48 hours later restimulated with 100 U/mL IFN-γ for 30 minutes; pSTAT1 was measured by western blot and compared to pSTAT1 levels from neurons in Fig 1E. Male and female mice included. Dotted lines represent SEM. (A) Mixed Effects Model, repeated measures: pSTAT1: main effect of time **p<0.005, cell type ***p<0.0005, and interaction **p<0.005, N=2–12; STAT1: cell type p=0.06, interaction (time × cell type) *p<0.05, N=2–12. (B) Two Way ANOVA: main effect of restim **p<0.005, interaction (restim × cell type) ***p<0.0005; post-hoc Sidak’s multiple comparison: **p<0.005, ***p<0.0005 N=2–5.
IFN-γ is known to have a priming effect in macrophages, making them more responsive to future encounters with a pathogen (Pace et al., 1983). Similarly, microglia are prone to immune memory, however this memory can cause microglia to become either primed or more tolerant to future inflammatory insults (Neher and Cunningham, 2019). To determine if IFN-γ induces priming memory in microglia as we have observed in neurons (Fig 1E, 1F), we treated primary microglia with pathological IFN-γ for 30 minutes, then re-challenge the cultures with pathological IFN-γ for 30 minutes, 48 hours post IFN-γ priming (as described in neurons). Unlike neurons, the STAT1 response in re-challenge of microglia was blunted, with pSTAT1 levels about 10-fold lower than pSTAT1 levels after the initial IFN-γ priming (Fig 4B). This suggests that different cells within the CNS have different responses to IFN-γ, and that prolonged STAT1 activation and IFN-γ priming is unique to neurons in the CNS.
3.6. Continuous JAK activity perpetuates prolonged STAT1 response in immature neurons.
So far, we have demonstrated that neurons have prolonged STAT1 activation in response to pathological levels of IFN-γ, but the underlying mechanism is unclear. Others have demonstrated that JAK1 and JAK2 are phosphorylated for up to 48 hours after IFN-γ treatment in neurons and that inhibiting JAK activity immediately after IFN-γ treatment results in pSTAT1 levels decreasing at a quicker rate (although not as quickly as in MEFs) (Podolsky, Solomos, 2012). These data suggest that JAK1/2 or some other upstream player may be responsible for prolonged STAT1 activation, but this has not been tested. To determine if prolonged STAT1 activation is caused by extended JAK activity, we utilized Ruxolitinib, a JAK inhibitor. Ruxolitinib acts by competitive inhibition of the JAK active site where STAT1 is phosphorylated (Mascarenhas and Hoffman, 2012); therefore, Ruxolitinib should not affect the activation state of JAK1/2 itself. First, we tested whether inhibiting JAK1/2 could suppress the prolonged neuronal STAT1 response. We treated developing neurons with pathological IFN-γ for 24 hours, then added Ruxolitinib to the cultures immediately after IFN-γ washout and analyzed 3 (D9) and 6 (D12) days after the addition of Ruxolitinib (Fig 5Ai). Ruxolitinib decreased pSTAT1 levels by 74% and 82% at D9 and D12, respectively, compared to vehicle controls analyzed at the same timepoints (Fig 5B). Total STAT1 levels were also decreased by 72% and 51% compared to vehicle treated samples (Fig 5B). These data suggest that JAK activity immediately following IFN-γ treatment contributes to the prolonged STAT1 activation in neurons.
Fig 5. Continuous JAK activity perpetuates prolonged STAT1 response in neurons.
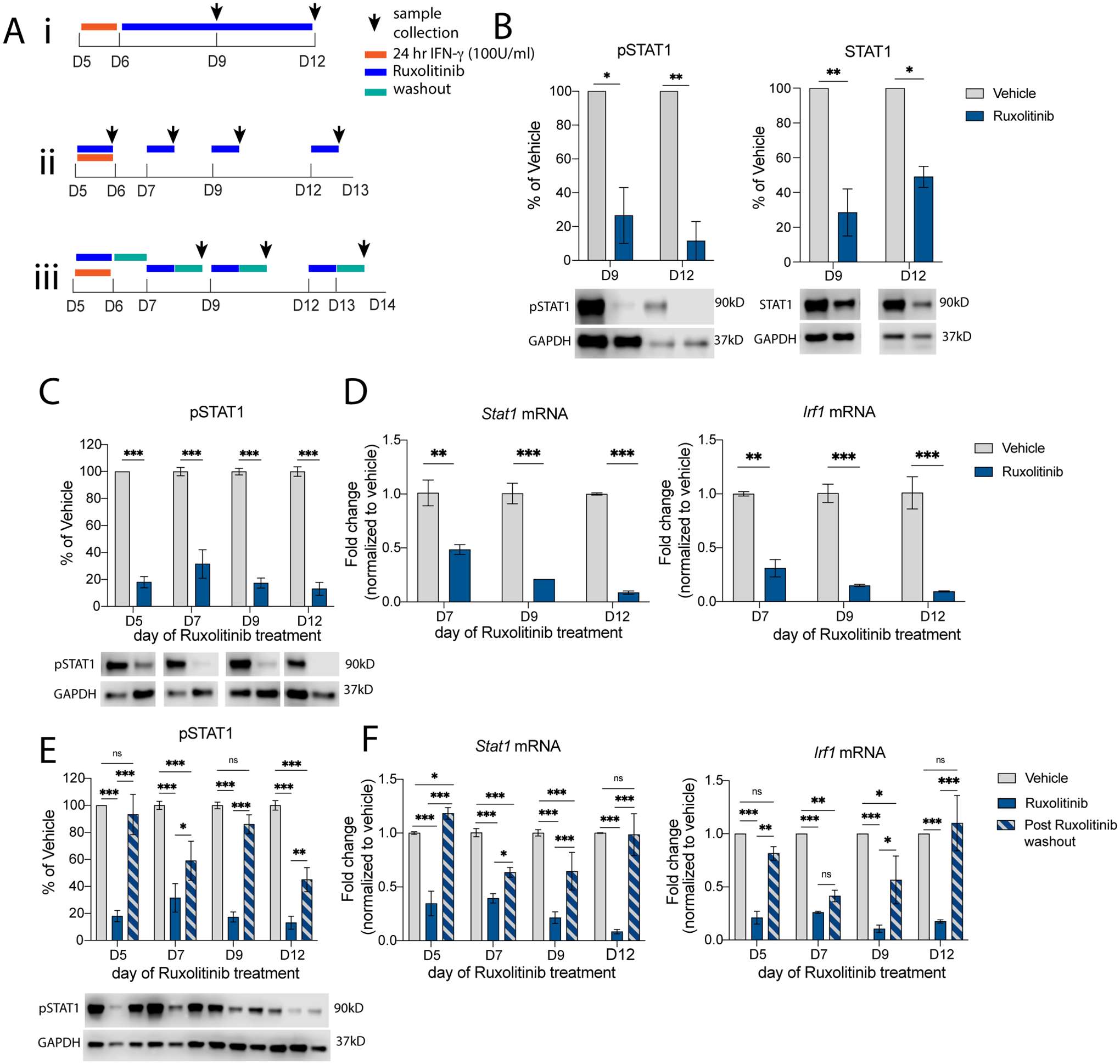
(Ai) Primary neurons (DIV5) were treated for 24 hours with 100 U/mL IFN-γ, Ruxolitinib (1μM) was added on DIV6 and left in cultures until DIV12, with samples collected at DIV9 and DIV12. (B) pSTAT1 and STAT1 were measured by western blot. (Aii) Primary neurons (DIV5) were treated for 24 hours with 100 U/mL IFN-γ, Ruxolitinib (1μM) was added for 24 hours on DIV5, DIV7, DIV9, or DIV12, with samples collected after 24 hours of Ruxolitinib treatment. (C) pSTAT1 was measured by western blot; (D) Stat1 and Irf1 mRNA expression were measured by qRT-PCR. (Aiii) Primary neurons (DIV5) were treated for 24 hours with 100 U/mL IFN-γ, Ruxolitinib (1μM) was added for 24 hours on DIV5, DIV7, DIV9, or DIV12, then washed out and samples were collected 24 hours after Ruxolitinib washout. (E) pSTAT1 was measured by western blot; (F) Stat1 and Irf1 mRNA expression were measured by qRT-PCR. Male and female mice included. (B) Two Way ANOVA: pSTAT1: main effect of ruxo treatment **p<0.005; post-hoc Sidak’s *p<0.05, **p<0.005, N=2; STAT1: main effect of ruxo treatment **p<0.005; post-hoc Sidak’s *p<0.05, **p<0.005, N=2. (C) Two Way ANOVA: pSTAT1: main effect of ruxo treatment ***p<0.0005, post-hoc Sidak’s ***p<0.0005, N=7–8; (D) Two Way ANOVA: Stat1 mRNA: main effect of ruxo treatment ***p<0.0005, interaction p=0.06; post-hoc Sidak’s **p<0.005, ***p<0.0005, N=2; Irf1 mRNA: main effect of ruxo treatment ***p<0.0005; post-hoc Sidak’s **p<0.005, ***p<0.0005; N=2. (E) Two Way ANOVA: pSTAT1: main effect of ruxo treatment ***p<0.0005, day of treatment *p<0.05, interaction **p<0.005; post-hoc Sidak’s *p<0.05, **p<0.005, ***p<0.0005; N=4–8 (F) Two Way ANOVA: Stat1 mRNA: main effect of ruxo treatment ***p<0.0005, day of treatment ***p<0.0005, and interaction ***p=0.0005; post-hoc Tukey’s *p<0.05, ***p<0.0005, N=2–5; Irf1 mRNA: main effect of ruxo treatment ***p<0.0005, interaction *p<0.05; post-hoc Tukey’s *p<0.05, **p<0.005, ***p<0.0005; N=2.
Next, we wanted to know if JAK activity contributed to prolonged STAT1 activation by phosphorylating STAT1 at later timepoints when pSTAT1 levels are still increased, but IFN-γ is not present. To test this, we treated developing neurons with IFN-γ for 24 hours and then treated the cultures with Ruxolitinib for 24 hours at a time, beginning on D7, D9, or D12 (1-, 3-, or 6-days post IFN-γ washout, respectively); samples were analyzed at D8, D10, or D13 (Fig 5Aii). Surprisingly, Ruxolitinib treatment decreased pSTAT1 at every time point (Fig 5C). Of note, addition of Ruxolitinib on D12, one week after IFN-γ washout, pSTAT1 levels were decreased by 90% compared to vehicle treated samples. STAT1–mediated transcription of ISGs was also affected, as mRNA expression of Stat1 and Irf1 were both decreased at every timepoint (Fig 5D). These data suggest that JAK activity extends to at least a week after IFN-γ washout in neurons and contributes to both prolonged STAT1 activation and expression of STAT1–mediated ISGs.
Since Ruxolitinib inhibits pSTAT1 by negatively regulating JAK activity by competitive inhibition at the active sites, and not by dephosphorylation or by deactivating JAK1/2, the JAK/STAT1 signaling pathway may still be engaged despite being actively inhibited. We hypothesized that removing Ruxolitinib from the cultures would allow JAK1/2 to resume phosphorylation activity, resulting in increased pSTAT1 levels. To test this, we treated developing neurons with IFN-γ for 24 hours then subsequently treated neurons with Ruxolitinib for 24 hours at a time as previously shown; after 24 hours, Ruxolitinib was washed out and we waited another 24 hours before analysis (Fig 5Aiii). After removing Ruxolitinib from the cultures, pSTAT1, Stat1, and IRF1 levels increased, with most timepoints returning to pre-Ruxolitinib levels (Fig 5E, 5F). These data further suggest that JAK1/2 are active in the absence of IFN-γ (up to a week after IFN-γ removal) and capable of continuously activating STAT1 and driving the prolonged STAT1 response seen in neurons. Thus, once developing neurons are primed with pathological levels of IFN-γ, pSTAT1 is prolonged due to continue activation of the JAK/STAT1 pathway. This could have profound effects on downstream neuronal function.
3.7. Pathological priming with IFN-γ results in a distinct and lasting transcriptional response.
We have shown that pathological levels of IFN-γ caused a prolonged STAT1 response in developing neurons that primes a rechallenge. To determine how priming affects the function of STAT1 as a transcription factor, we treated developing neurons with pathological IFN-γ for 24 hours, then re-challenged on DIV12 with physiological or pathological levels of IFN-γ for 30 minutes (acute treatment). After the acute re-challenge, we removed IFN-γ and collected mRNA 24 hours later for bulk sequencing. We also treated unprimed neurons acutely on DIV12 with physiological or pathological levels of IFN-γ for 30 minutes (Fig 6A).
Fig 6. Pathological priming with IFN-γ results in a distinct and lasting transcriptional response.
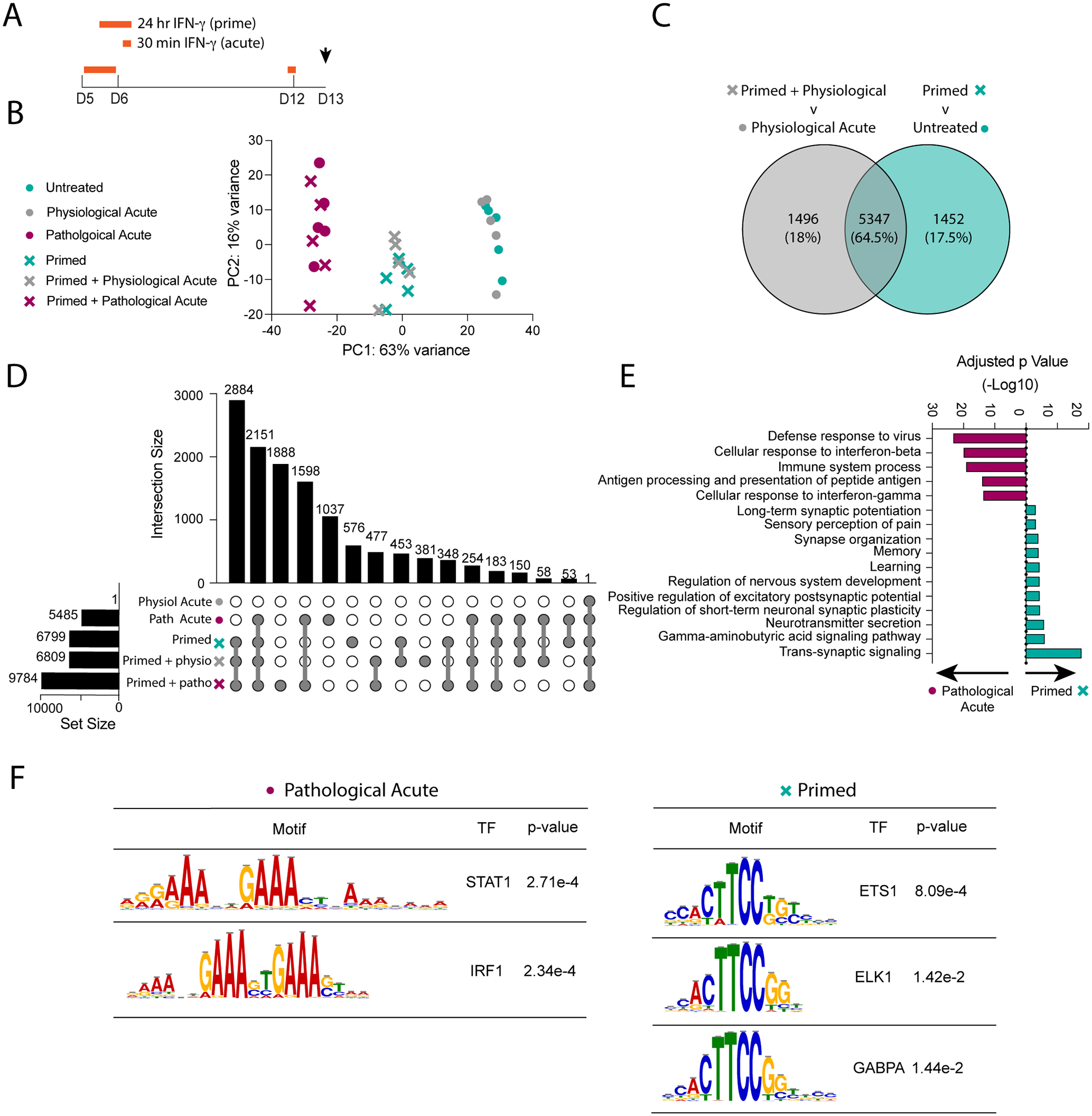
(A) Primary neurons were primed with pathological IFN-γ for 24 hours on DIV5 and then re-stimulated acutely with physiological or pathological IFN-γ for 30 minutes on DIV12. mRNA was collected on DIV13 for RNA-sequencing. (B) Principal Component Analysis of top two principal components comprising 79% of total variance. (C) Venn Diagram comparing the significant differentially expressed gene (DEG) sets (p<0.05) from the “primed + physio” versus “physiological acute” comparison and the “primed” v untreated comparison. (D) UpSet plot comparing the significant DEGs (p<0.05) identified from comparing each condition to untreated neurons. (E) Gene Ontology Analysis comparing significant differentially expressed genes (p<0.05) from the pathological acute versus primed conditions. (F) Motif analysis of the regions upstream of significant DEGs (p<0.05) from the pathological acute versus primed conditions.
We identified 3 clusters along the first principal component accounting for 63% of variability (Fig 6B). The first cluster included unprimed neurons that received physiological IFN-γ “physiological acute” and untreated neurons. When comparing physiological acute to untreated neurons, we observed only one upregulated gene (Cxcl9) and no downregulated genes, suggesting that physiological levels of IFN-γ have little to no impact on transcription.
The second cluster consisted of primed neurons, with or without re-challenge. “Primed” neurons had 6799 DEGs compared to untreated neurons, whereas primed neurons rechallenged with physiological IFN-γ “primed + physiological acute” had 6809 DEGs compared to untreated neurons. Interestingly, we observed no differentially expressed genes when directly comparing the two conditions, suggesting that acute physiological treatment after priming does not have an additive effect to priming at the transcriptional level.
To directly assess whether priming influenced a rechallenge with physiological IFN-γ, we compared the “primed + physiological acute” to “physiological acute” conditions. We found 6843 DEGs between the two conditions. This is perhaps not surprising since physiological acute had a very minimal transcriptional effect, unlike priming. To rule out the possibility that these differences are purely a result of the initial priming effect and not the additional physiological IFN-γ rechallenge, we compared this set of DEGs to the set of DEGs specific to the priming effect “primed v untreated”. Although we found a large group of DEGs that overlap between the two sets (Fig 6C), rechallenging primed neurons with physiological IFN-γ did induce different DEGs, including Tlr2, Ccl2, and Mbd5 all of which have been associated with CNS pathologies (Di Liberto, Pantelyushin, 2018, Dzamko et al., 2017, Tang et al., 2023). This suggests that pathological IFN-γ primes neurons to have a dysregulated response to later physiological IFN-γ signaling.
The third cluster consisted of neurons treated with pathological IFN-γ with or without priming (Fig 6B). We hypothesized that these samples were clustered based on the strength of a pathological acute response independent on when the neurons were treated (i.e., primed at DIV5 or acute at DIV12). Therefore, to better understand the impact specific to priming, we compared looked at the DEGs between the two groups and identified 4261 DEGs unique to the primed response and 2947 DEGs unique to the acute response. There was only a 26% overlap in DEGs (2538 genes), emphasizing how distinct these two responses are to each other. To further investigate this, we identified the DEGs between each condition compared to untreated neurons, then used an UpSet Plot to find intersections between each set of DEGs. We found only 53 DEGs shared between the primed response and acute response, that were not found in any other intersections, further emphasizing how distinct these two responses are. Of note, the largest intersection (2884 DEGs) included all conditions in which neurons were primed, regardless of additional rechallenge (“primed”, “primed + physiological acute”, “primed + pathological acute”). This supports the idea that priming has a profound effect on neuronal transcription (Fig 6D).
Next, we identified the differences in biological processes enriched in the primed and acute response. Using GO enrichment analysis, we identified canonical immune signaling and IFN signaling pathways, including “cellular response to IFN-γ” and “defense response to virus”, increased in both responses, however they were more highly enriched in the acute response. DEGs in primed neurons were enriched for neuro-centric pathways, including “synapse organization”, “learning”, and “trans-synaptic signaling” (Fig 6E).
Along with canonical ISGs, STAT1 is also known to regulate many other transcription factors (Ramana et al., 2000). We hypothesized there may be other transcription factors, downstream of STAT1, that drive the primed transcriptional response in neurons. We used motif enrichment analysis to compare the transcription factor binding sites enriched in the “pathological acute” versus “primed” responses (Fig 6F). We found 52 motifs enriched in the “pathological acute” response and 75 motifs enriched in the “primed” response. Of these 31 were shared between the two. Of the 21 “pathological acute” response motifs, 9 of the corresponding transcription factors were also differentially expressed compared to untreated neurons. As expected, these comprised of many canonical IFN-γ signaling transcription factors including STAT1, STAT2, IRF1, IRF2, IRF7, and IRF9. Of the 44 “primed” response motifs, 11 of the corresponding transcription factors were also differentially expressed compared to untreated neurons. Many of these genes are part of the ETS transcription factor family, including ETS-1, ELK1, and GAPBα. Overall, these data suggest that neurons can have drastically varying and persistent responses to pathological IFN-γ, which can affect many neuronal pathways aside from canonical immune signaling pathways.
4. DISCUSSION
Neurons have both physiological and pathological responses to IFN-γ/STAT1 signaling, however it is unclear how this immune signaling pathway leads to each unique outcome. Here, we demonstrated that physiological IFN-γ signaling induces brief and transient STAT1 activation, while pathological IFN-γ drives a prolonged STAT1 activation, primes subsequent IFN-γ signaling, and induces long-term transcriptional effects.
Here, we demonstrated that pathological levels of IFN-γ induce a prolonged STAT1 response in neurons. It is worth emphasizing how surprising these results are considering the absence of IFN-γ in the cultures at the time of analysis. It was previously demonstrated that neurons have extended JAK1 and JAK2 phosphorylation after IFN-γ treatment, compared to MEFs treated with pathological IFN-γ (Podolsky, Solomos, 2012). This suggests that JAK1 and JAK2 may also have extended functional capacity after IFN-γ washout in neurons and that prolonged STAT1 signaling may indeed be due to upstream factors. We demonstrated that inhibiting JAK1 and JAK2 up to a week after IFN-γ washout resulted in decreased pSTAT1 levels. pSTAT1 levels returned to their previously high levels after removing the JAK inhibitor from cultures, suggesting JAK1 and JAK2 can continuously phosphorylate STAT1 and do indeed contribute to prolonged STAT1 activation in neurons. It is unclear how and why the IFN-γ/JAK/STAT1 pathway behaves this way in neurons, though others have demonstrated various non-canonical signaling events and functions of this pathway in other cell-types which may also contribute to this unique signaling in neurons. For example, components of the entire IFNGR signaling complex (IFN-γ, IFNGR1, JAK1, and JAK2) have all been found in the nucleus following IFN-γ stimulation in other cell-types (Dawson et al., 2009, Johnson and Ahmed, 2016, Noon-Song et al., 2011, Subramaniam et al., 1999). Additionally, kinases in other signaling pathways activated by IFN-γ in neurons, such as extracellular signal-regulated kinase (ERK), are known to translocate from distal processes to the nucleus via clathrin coated vesicles in association with AMPA receptor endocytosis (O’Donnell, Henkins, 2015, Trifilieff et al., 2009). IFNGR also associates with AMPA receptors (Mizuno, Zhang, 2008), making it possible that the IFNGR signaling complex may also utilize an endocytic-trafficking mechanism, which could contribute to the prolonged STAT1 activation, however this is outside the scope of our work presented here.
Our data also demonstrated that pathological IFN-γ had a priming effect in neurons and enhanced STAT1 activation in response to subsequent IFN-γ signaling. We hypothesized that this priming response was at least in part due to increased pools of total STAT1 at the time of re-challenge in these primed neurons. Another possibility is that the priming effect is specific to the IFN-γ signaling pathway. Others have demonstrated that while IFN-β activates STAT1 and confers antiviral activity in neurons, it does not result in pSTAT1 translocation to the nucleus (Song et al., 2016). We did not observe prolonged pSTAT1 or total STAT1 expression after IFN-β treatment, suggesting the signaling cascades downstream of IFN-γ and type I IFNs may activate STAT1 in different ways. JAK1, which is utilized in both pathways, has modestly extended phosphorylation in neurons compared to MEFs, while JAK2, which is only utilized in IFN-γ signaling, appears to have more dramatically enhanced phosphorylation (Podolsky, Solomos, 2012). These data suggest that the IFN-γ specific priming effect may be due to an upstream signaling component specific to the IFN-γ pathway, such as JAK2, though more work is needed to make any conclusions.
To the best of our knowledge, STAT1 activation in neurons in response to physiological IFN-γ has not been studied. Here we demonstrated that unlike pathological IFN-γ, physiological IFN-γ induced brief and transient STAT1 activation. Additionally, physiological IFN-γ did not induce a priming effect when neurons were primed and re-challenged with physiological IFN-γ. Because pSTAT1 levels decreased so quickly, we did not investigate whether physiological IFN-γ also results in continuous JAK activation, though we hypothesize that it is unlikely. Our data suggests that the continuous JAK activity and prolonged STAT1 activation in neurons described above is specifically induced by pathological levels of IFN-γ. More studies are needed to discern how physiological and pathological IFN-γ induce differential STAT1 activation at the molecular level.
It is well documented that neurons mount an immunological response when stimulated with IFN-γ (Binder and Griffin, 2001, Burdeinick-Kerr, Govindarajan, 2009, Chesler and Reiss, 2002, Komatsu et al., 1996, O’Donnell, Henkins, 2015, Song, Koyuncu, 2016). We investigated how a pathological acute IFN-γ response compares to the primed IFN-γ response in neurons. As expected, neurons treated acutely were enriched for pathways relating to an interferon-mediated immune response. Primed neurons were also enriched for immune-related pathways compared to untreated neurons, though to a lower extent than the acute response, as shown when directly comparing the primed to the acute response. This is likely because primed neurons were analyzed one week after treatment, a timepoint that we’ve demonstrated the STAT1 response is still high but beginning to decline (Fig 2B), whereas the acutely treated neurons were analyzed one day after treatment. Others have also reported a persistent immune-related transcriptional response in neurons primed with high levels of IFN-γ (Kathuria, Lopez-Lengowski, 2022, Warre-Cornish, Perfect, 2020). Surprisingly, we observed primed neurons were also enriched for neuro-centric pathways, which could contribute to many pathological responses reported. These results demonstrate that prolonged exposure to IFN-γ can change neurons transcriptomic profiles long-term, even after the initial IFN-γ stimulus has been resolved.
We also investigated the effect of pathological IFN-γ priming on downstream transcriptional responses. Others have demonstrated IFN-γ induced transcriptional dysregulation in neurons primed and re-challenged with high levels of IFN-γ (Warre-Cornish, Perfect, 2020). Because low physiological levels of IFN-γ are always present in the CNS, we investigated how priming neurons with pathological IFN-γ would affect their response to physiological IFN-γ. Interestingly, we saw that in unprimed neurons, physiological IFN-γ had very little effect on transcription, with only one gene being differentially expressed compared to untreated neurons. This was somewhat surprising considering physiological IFN-γ elicits a functional response in neurons (Filiano, Xu, 2016). It is possible that physiological IFN-γ induced gene expression is transient and had returned to baseline in the 24 hours between treatment and sample collection. Additionally, physiological IFN-γ may have a transcription-independent role in neurons, though this will need to be studied further. When comparing the “physiological acute” response to the “primed + physiological acute” response, we found a large set of DEGs between the two conditions. However, considering there was a minimal transcriptional response in the physiological response, we thought these differences may simply be due to priming alone response. To investigate this further, we directly compared the “primed” response to the “primed + physiological acute” response and found that while there was a large overlap in DEGs, there were also DEGs specific to either response. A few genes of note specific to the “primed + physiological acute” response include Tlr2, Ccl2, and Mbd5, all of which have been associated with CNS pathologies (Di Liberto, Pantelyushin, 2018, Dzamko, Gysbers, 2017, Tang, Zhang, 2023). This suggests that priming influences the transcriptional response to physiological IFN-γ signaling and may skew the physiological response to behave more pathologically.
We also investigated whether the primed response might be regulated by transcriptions factors other than STAT1. As expected, the pathological acute response was enriched for STAT and IRF family binding motifs. The primed response, however, was enriched for many binding motifs associated with the ETS transcription factor family. Therefore, it is possible that the IFN-γ induced priming response may not solely be due to transcripts regulated by STAT1, but additionally regulated by secondary transcription factors that are expressed downstream of STAT1. Alternatively, the primed response may be regulated by an IFN-γ-dependent/STAT1-independent mechanism. For example, IFN-γ can activate NFκB (Lin et al., 2012); however, we did not identify NFκB binding sites as significantly enriched in our motif analysis. Others have demonstrated that IFN-γ can also activate the ERK signaling pathway in neurons which results in a protective phenotype against viral infection. (O’Donnell, Henkins, 2015). Interestingly, ETS members such as ELK1, are also known to be activated by ERK (Besnard et al., 2011) and have been shown to have pro-apoptotic functions in neurons (Besnard, Galan, 2011, Demir et al., 2011, Oikawa and Yamada, 2003). In our dataset, we see that Ets1, Elk1, and Gabpa are significantly downregulated in the priming response, therefore, it is possible that the primed response is facilitated by IFN-γ/ERK signaling and contributes to the protective anti-viral response in neurons, however this is out of the scope of our work presented here.
Overall, our work takes an important step in understanding the underlying mechanisms in neuronal IFN-γ/STAT1 signaling and how STAT1 activation under different conditions can lead to varying outcomes.
Supplementary Material
Supplemental Fig 1. IFN-α and IFN-β induce transient increase in pSTAT1. (A) Neurons were treated with IFN-α (100 U/mL) for 24 hours then washed out. pSTAT1 protein was measured by western blot. (n = 2) (B) Neurons were treated with IFN-β (100 U/mL) for 30 minutes then washed out. pSTAT1 protein was measured by western blot. (n = 2)
Supplemental Fig 2. Type 1 IFNs do not induce prolonged STAT1 response. (A) Neurons were treated with 100, 500, or 5000 U/mL of IFN-α then washed out. STAT1 protein was measured by western blot. IFN-γ data is from Fig. 2B. (B) Neurons were treated with 100, 500, or 5000 U/mL of IFN-β then washed out. STAT1 protein was measured by western blot. IFN-γ data is from Fig. 2B. Male and female mice included. Dotted lines represent SEM. (A) Mixed Effects Model, repeated measures: main effect of time: ns, main effect of cytokine: ns; N=2–8. (B) Mixed Effects Model, repeated measures: main effect of time: ns, main effect of cytokine: ns; N=4–8.
Highlights.
Pathological levels of IFN-γ induce a prolonged STAT1 response in neurons.
A prolonged STAT1 response in neurons is due to a persistent activation of the IFN-γ pathway.
Neurons initially induce canonical IFN-γ genes; however, the persistent response drives transcriptional changes in neuron-specific pathways.
Pathological IFN-γ primes neurons to enhance STAT1 activation in response to subsequent IFN-γ signaling.
Acknowledgements
The authors would like to thank all members of the Filiano lab and the Marcus Center for Cellular Cures for their feedback on data presented in the manuscript. We would also like to thank Dr. Cagla Eroglu and her lab for providing cryopreserved primary astrocytes and assisting with the setup of the astrocyte culture experiments. This work was supported by grants from the NIH R01NS123084, 2T32-AI052077-19, and funding from the Marcus Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
AJF has intellectual property that has been licensed to Cryocell.
REFERENCES
- Alves de Lima K, Rustenhoven J, Da Mesquita S, Wall M, Salvador AF, Smirnov I, et al. Meningeal γδ T cells regulate anxiety-like behavior via IL-17a signaling in neurons. Nature Immunology. 2020;21:1421–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Johnson J, Grant CE, Noble WS. The MEME Suite. Nucleic Acids Research. 2015;43:W39–W49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, et al. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science. 2014;346:89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Yehuda H, Matcovitch-Natan O, Kertser A, Spinrad A, Prinz M, Amit I, et al. Maternal Type-I interferon signaling adversely affects the microglia and the behavior of the offspring accompanied by increased sensitivity to stress. Molecular Psychiatry. 2020;25:1050–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society: Series B (Methodological). 1995;57:289–300. [Google Scholar]
- Besnard a, Galan B, Vanhoutte p, Caboche J. Elk-1 a Transcription Factor with Multiple Facets in the Brain. Frontiers in Neuroscience. 2011;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder GK, Griffin DE. Interferon-gamma-Mediated Site-Specific Clearance of Alphavirus from CNS Neurons. Science. 2001;293:303–6. [DOI] [PubMed] [Google Scholar]
- Burdeinick-Kerr R, Govindarajan D, Griffin DE. Noncytolytic Clearance of Sindbis Virus Infection from Neurons by Gamma Interferon Is Dependent on Jak/Stat Signaling. J Virol. 2009;83:3429–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdeinick-Kerr R, Wind J, Griffin DE. Synergistic Roles of Antibody and Interferon in Noncytolytic Clearance of Sindbis Virus from Different Regions of the Central Nervous System. J Virol. 2007;81:5628–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Ziv Y, Schwartz A, Landa G, Talpalar AE, Pluchino S, et al. Microglia activated by IL-4 or IFN-γ differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Molecular and Cellular Neuroscience. 2006;31:149–60. [DOI] [PubMed] [Google Scholar]
- Butturini E, Boriero D, Carcereri de Prati A, Mariotto S. STAT1 drives M1 microglia activation and neuroinflammation under hypoxia. Archives of Biochemistry and Biophysics. 2019;669:22–30. [DOI] [PubMed] [Google Scholar]
- Chesler DA, Reiss CS. The role of IFN-γ in immune responses to viral infections of the central nervous system. Cytokine & Growth Factor Reviews. 2002;13:441–54. [DOI] [PubMed] [Google Scholar]
- Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science. 2016;351:933–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark DN, Begg LR, Filiano AJ. Unique aspects of IFN-γ/STAT1 signaling in neurons. Immunological Reviews. 2022;311:187–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway JR, Lex A, Gehlenborg N. UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics. 2017;33:2938–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan MN, Sethi I, Harris TH. Microglia in CNS infections: insights from Toxoplasma gondii and other pathogens. Trends in Parasitology. 2022;38:217–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MA, Bannister AJ, Göttgens B, Foster SD, Bartke T, Green AR, et al. JAK2 phosphorylates histone H3Y41 and excludes HP1α from chromatin. Nature. 2009;461:819–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deczkowska A, Baruch K, Schwartz M. Type I/II Interferon Balance in the Regulation of Brain Physiology and Pathology. Trends in Immunology. 2016;37:181–92. [DOI] [PubMed] [Google Scholar]
- Demir O, Aysit N, Onder Z, Turkel N, Ozturk G, Sharrocks AD, et al. ETS-domain transcription factor Elk-1 mediates neuronal survival: SMN as a potential target. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2011;1812:652–62. [DOI] [PubMed] [Google Scholar]
- Di Liberto G, Pantelyushin S, Kreutzfeldt M, Page N, Musardo S, Coras R, et al. Neurons under T Cell Attack Coordinate Phagocyte-Mediated Synaptic Stripping. Cell. 2018;175:458–71.e19. [DOI] [PubMed] [Google Scholar]
- Ding X, Yan Y, Li X, Li K, Ciric B, Yang J, et al. Silencing IFN-γ Binding/Signaling in Astrocytes versus Microglia Leads to Opposite Effects on Central Nervous System Autoimmunity. The Journal of Immunology. 2015;194:4251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzamko N, Gysbers A, Perera G, Bahar A, Shankar A, Gao J, et al. Toll-like receptor 2 is increased in neurons in Parkinson’s disease brain and may contribute to alpha-synuclein pathology. Acta Neuropathol. 2017;133:303–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Research. 2002;30:207–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Ansary A, Al-Ayadhi L. Neuroinflammation in autism spectrum disorders. Journal of Neuroinflammation. 2012;9:265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellwardt E, Walsh JT, Kipnis J, Zipp F. Understanding the Role of T Cells in CNS Homeostasis. Trends in Immunology. 2016;37:154–65. [DOI] [PubMed] [Google Scholar]
- Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics. 2007;23:257–8. [DOI] [PubMed] [Google Scholar]
- Filiano AJ, Gadani SP, Kipnis J. How and why do T cells and their derived cytokines affect the injured and healthy brain? Nature Reviews Neuroscience. 2017;18:375–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filiano AJ, Xu Y, Tustison NJ, Marsh RL, Baker W, Smirnov I, et al. Unexpected role of interferon-γ in regulating neuronal connectivity and social behaviour. Nature. 2016;535:425–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood L, Korol SV, Ekselius L, Birnir B, Jin Z. Interferon-γ potentiates GABAA receptor-mediated inhibitory currents in rat hippocampal CA1 pyramidal neurons. Journal of Neuroimmunology. 2019;337:577050. [DOI] [PubMed] [Google Scholar]
- Foo LC, Allen NJ, Bushong EA, Ventura PB, Chung WS, Zhou L, et al. Development of a method for the purification and culture of rodent astrocytes. Neuron. 2011;71:799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frei K, Leist TP, Meager A, Gallo P, Leppert D, Zinkernagel RM, et al. Production of B cell stimulatory factor-2 and interferon gamma in the central nervous system during viral meningitis and encephalitis. Evaluation in a murine model infection and in patients. J Exp Med. 1988;168:449–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halonen SK, Taylor GA, Weiss LM. Gamma Interferon-Induced Inhibition ofToxoplasma gondii in Astrocytes Is Mediated by IGTP. Infection and Immunity. 2001;69:5573–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashioka S, Klegeris A, Schwab C, McGeer PL. Interferon-γ-dependent cytotoxic activation of human astrocytes and astrocytoma cells. Neurobiology of Aging. 2009;30:1924–35. [DOI] [PubMed] [Google Scholar]
- Hindinger C, Bergmann CC, Hinton DR, Phares TW, Parra GI, Hussain S, et al. IFN-γ Signaling to Astrocytes Protects from Autoimmune Mediated Neurological Disability. PLoS One. 2012;7:e42088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt LM, Hernandez RD, Pacheco NL, Torres Ceja B, Hossain M, Olsen ML. Astrocyte morphogenesis is dependent on BDNF signaling via astrocytic TrkB.T1. eLife. 2019;8:e44667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt LM, Olsen ML. Novel Applications of Magnetic Cell Sorting to Analyze Cell-Type Specific Gene and Protein Expression in the Central Nervous System. PLoS One. 2016;11:e0150290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janach GMS, Reetz O, Döhne N, Stadler K, Grosser S, Byvaltcev E, et al. Interferon-γ acutely augments inhibition of neocortical layer 5 pyramidal neurons. Journal of Neuroinflammation. 2020;17:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson HM, Ahmed CM. Noncanonical IFN Signaling: Mechanistic Linkage of Genetic and Epigenetic Events. Mediators Inflamm. 2016;2016:9564814-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KL, Croen LA, Yoshida CK, Heuer L, Hansen R, Zerbo O, et al. Autism with intellectual disability is associated with increased levels of maternal cytokines and chemokines during gestation. Molecular Psychiatry. 2017;22:273–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathuria A, Lopez-Lengowski K, Roffman JL, Karmacharya R. Distinct effects of interleukin-6 and interferon-γ on differentiating human cortical neurons. Brain, Behavior, and Immunity. 2022;103:97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu T, Bi Z, Reiss CS. Interferon-γ induced type I nitric oxide synthase activity inhibits viral replication in neurons. Journal of Neuroimmunology. 1996;68:101–8. [DOI] [PubMed] [Google Scholar]
- Kunis G, Baruch K, Rosenzweig N, Kertser A, Miller O, Berkutzki T, et al. IFN-γ-dependent activation of the brain’s choroid plexus for CNS immune surveillance and repair. Brain. 2013;136:3427–40. [DOI] [PubMed] [Google Scholar]
- Lau LT, Yu AC-H. Astrocytes Produce and Release Interleukin-1, Interleukin-6, Tumor Necrosis Factor Alpha and Interferon-Gamma Following Traumatic and Metabolic Injury. Journal of Neurotrauma. 2001;18:351–9. [DOI] [PubMed] [Google Scholar]
- Lex A, Gehlenborg N, Strobelt H, Vuillemot R, Pfister H. UpSet: Visualization of Intersecting Sets. IEEE Transactions on Visualization and Computer Graphics. 2014;20:1983–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Chauhan A, Sheikh AM, Patil S, Chauhan V, Li X-M, et al. Elevated immune response in the brain of autistic patients. Journal of Neuroimmunology. 2009;207:111–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Jamison S, Lin W. Interferon-γ activates nuclear factor-κ B in oligodendrocytes through a process mediated by the unfolded protein response. PLoS One. 2012;7:e36408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascarenhas J, Hoffman R. Ruxolitinib: The First FDA Approved Therapy for the Treatment of Myelofibrosis. Clinical Cancer Research. 2012;18:3008–14. [DOI] [PubMed] [Google Scholar]
- Mizuno T, Zhang G, Takeuchi H, Kawanokuchi J, Wang J, Sonobe Y, et al. Interferon-γ directly induces neurotoxicity through a neuron specific, calcium-permeable complex of IFN-γ receptor and AMPA GluR1 receptor. The FASEB Journal. 2008;22:1797–806. [DOI] [PubMed] [Google Scholar]
- Nagakura I, Van Wart A, Petravicz J, Tropea D, Sur M. STAT1 Regulates the Homeostatic Component of Visual Cortical Plasticity via an AMPA Receptor-Mediated Mechanism. The Journal of Neuroscience. 2014;34:10256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher JJ, Cunningham C. Priming Microglia for Innate Immune Memory in the Brain. Trends in Immunology. 2019;40:358–74. [DOI] [PubMed] [Google Scholar]
- Noon-Song EN, Ahmed CM, Dabelic R, Canton J, Johnson HM. Controlling nuclear JAKs and STATs for specific gene activation by IFNγ. Biochem Biophys Res Commun. 2011;410:648–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell LA, Henkins KM, Kulkarni A, Matullo CM, Balachandran S, Pattisapu AK, et al. Interferon gamma induces protective non-canonical signaling pathways in primary neurons. Journal of Neurochemistry. 2015;135:309–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa T, Yamada T. Molecular biology of the Ets family of transcription factors. Gene. 2003;303:11–34. [DOI] [PubMed] [Google Scholar]
- Olsson T Cytokines in neuroinflammatory disease: role of myelin autoreactive T cell production of interferon-gamma. Journal of Neuroimmunology. 1992;40:211–8. [DOI] [PubMed] [Google Scholar]
- Orellana DI, Quintanilla RA, Gonzalez-Billault C, Maccioni RB. Role of the JAKs/STATs pathway in the intracellular calcium changes induced by interleukin-6 in hippocampal neurons. Neurotoxicity Research. 2005;8:295–304. [DOI] [PubMed] [Google Scholar]
- Ottum PA, Arellano G, Reyes LI, Iruretagoyena M, Naves R. Opposing Roles of Interferon-Gamma on Cells of the Central Nervous System in Autoimmune Neuroinflammation. Frontiers in Immunology. 2015;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace JL, Russell SW, Torres BA, Johnson HM, Gray PW. Recombinant mouse gamma interferon induces the priming step in macrophage activation for tumor cell killing. The Journal of Immunology. 1983;130:2011–3. [PubMed] [Google Scholar]
- Podolsky MA, Solomos AC, Durso LC, Evans SM, Rall GF, Rose RW. Extended JAK activation and delayed STAT1 dephosphorylation contribute to the distinct signaling profile of CNS neurons exposed to interferon-gamma. Journal of Neuroimmunology. 2012;251:33–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramana CV, Chatterjee-Kishore M, Nguyen H, Stark GR. Complex roles of Stat1 in regulating gene expression. Oncogene. 2000;19:2619–27. [DOI] [PubMed] [Google Scholar]
- Ramana CV, Gil MP, Schreiber RD, Stark GR. Stat1-dependent and -independent pathways in IFN-γ-dependent signaling. Trends in Immunology. 2002;23:96–101. [DOI] [PubMed] [Google Scholar]
- Rao A, Kim E, Sheng M, Craig AM. Heterogeneity in the molecular composition of excitatory postsynaptic sites during development of hippocampal neurons in culture. J Neurosci. 1998;18:1217–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed MD, Yim YS, Wimmer RD, Kim H, Ryu C, Welch GM, et al. IL-17a promotes sociability in mouse models of neurodevelopmental disorders. Nature. 2020;577:249–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BC, Sinyuk M, Jenkins JE, Psenicka MW, Williams JL. The impact of regional astrocyte interferon-γ signaling during chronic autoimmunity: a novel role for the immunoproteasome. Journal of Neuroinflammation. 2020;17:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SEP, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27:10695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltani Khaboushan A, Yazdanpanah N, Rezaei N. Neuroinflammation and Proinflammatory Cytokines in Epileptogenesis. Molecular Neurobiology. 2022;59:1724–43. [DOI] [PubMed] [Google Scholar]
- Song R, Koyuncu OO, Greco TM, Diner BA, Cristea IM, Enquist LW, et al. Two Modes of the Axonal Interferon Response Limit Alphaherpesvirus Neuroinvasion. mBio. 2016;7:e02145–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark GR, Kerr IM, Williams BRG, Silverman RH, Schreiber RD. How Cells Respond to Interferons. Annual Review of Biochemistry. 1998;67:227–64. [DOI] [PubMed] [Google Scholar]
- Subramaniam PS, Mujtaba MG, Paddy MR, Johnson HM. The Carboxyl Terminus of Interferon-γ Contains a Functional Polybasic Nuclear Localization Sequence*. Journal of Biological Chemistry. 1999;274:403–7. [DOI] [PubMed] [Google Scholar]
- Sukoff Rizzo SJ, Neal SJ, Hughes ZA, Beyna M, Rosenzweig-Lipson S, Moss SJ, et al. Evidence for sustained elevation of IL-6 in the CNS as a key contributor of depressive-like phenotypes. Translational Psychiatry. 2012;2:e199–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang FL, Zhang XG, Ke PY, Liu J, Zhang ZJ, Hu DM, et al. MBD5 regulates NMDA receptor expression and seizures by inhibiting Stat1 transcription. Neurobiol Dis. 2023;181:106103. [DOI] [PubMed] [Google Scholar]
- Trifilieff P, Lavaur J, Pascoli V, Kappès V, Brami-Cherrier K, Pagès C, et al. Endocytosis controls glutamate-induced nuclear accumulation of ERK. Molecular and Cellular Neuroscience. 2009;41:325–36. [DOI] [PubMed] [Google Scholar]
- Vikman KS, Hill RH, Backström E, Robertson B, Kristensson K. Interferon-γ induces characteristics of central sensitization in spinal dorsal horn neurons in vitro. Pain. 2003;106:241–51. [DOI] [PubMed] [Google Scholar]
- Vikman KS, Owe-Larsson B, Brask J, Kristensson KS, Hill RH. Interferon-γ-induced changes in synaptic activity and AMPA receptor clustering in hippocampal cultures. Brain Research. 2001;896:18–29. [DOI] [PubMed] [Google Scholar]
- Vikman KS, Siddall PJ, Duggan AW. Increased responsiveness of rat dorsal horn neurons in vivo following prolonged intrathecal exposure to interferon-γ. Neuroscience. 2005;135:969–77. [DOI] [PubMed] [Google Scholar]
- Warre-Cornish K, Perfect L, Nagy R, Duarte RRR, Reid MJ, Raval P, et al. Interferon-γ signaling in human iPSC–derived neurons recapitulates neurodevelopmental disorder phenotypes. Science Advances. 2020;6:9506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Chadman KK, McCloskey DP, Sheikh AM, Malik M, Brown WT, et al. Brain IL-6 elevation causes neuronal circuitry imbalances and mediates autism-like behaviors. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2012;1822:831–42. [DOI] [PubMed] [Google Scholar]
- Wong G, Goldshmit Y, Turnley AM. Interferon-γ but not TNFα promotes neuronal differentiation and neurite outgrowth of murine adult neural stem cells. Experimental Neurology. 2004;187:171–7. [DOI] [PubMed] [Google Scholar]
- Wong H, Hoeffer C. Maternal IL-17A in autism. Exp Neurol. 2018;299:228–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N, et al. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nature Neuroscience. 2006;9:268–75. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig 1. IFN-α and IFN-β induce transient increase in pSTAT1. (A) Neurons were treated with IFN-α (100 U/mL) for 24 hours then washed out. pSTAT1 protein was measured by western blot. (n = 2) (B) Neurons were treated with IFN-β (100 U/mL) for 30 minutes then washed out. pSTAT1 protein was measured by western blot. (n = 2)
Supplemental Fig 2. Type 1 IFNs do not induce prolonged STAT1 response. (A) Neurons were treated with 100, 500, or 5000 U/mL of IFN-α then washed out. STAT1 protein was measured by western blot. IFN-γ data is from Fig. 2B. (B) Neurons were treated with 100, 500, or 5000 U/mL of IFN-β then washed out. STAT1 protein was measured by western blot. IFN-γ data is from Fig. 2B. Male and female mice included. Dotted lines represent SEM. (A) Mixed Effects Model, repeated measures: main effect of time: ns, main effect of cytokine: ns; N=2–8. (B) Mixed Effects Model, repeated measures: main effect of time: ns, main effect of cytokine: ns; N=4–8.