

Abstract
The BET family protein BRD4, which forms the CDK9-containing BRD4-PTEFb complex, is considered to be a master regulator of RNA Polymerase II (Pol II) pause release. Because its tandem bromodomains interact with acetylated histone lysine residues, it has long been thought that BRD4 requires these bromodomains for its recruitment to chromatin and transcriptional regulatory function. Here, using rapid depletion and genetic complementation with domain deletion mutants, we demonstrate that BRD4 bromodomains are dispensable for Pol II pause release. A minimal, bromodomain-less C-terminal BRD4 fragment containing the PTEFb-interacting C terminal motif (CTM) is instead both necessary and sufficient to mediate Pol II pause release in the absence of full-length BRD4. Although BRD4-PTEFb can associate with chromatin through acetyl recognition, our results indicate that a distinct, active BRD4-PTEFb population functions to regulate transcription independently of bromodomain-mediated chromatin association. These findings may enable more effective pharmaceutical modulation of BRD4-PTEFb activity.
Graphical Abstract:
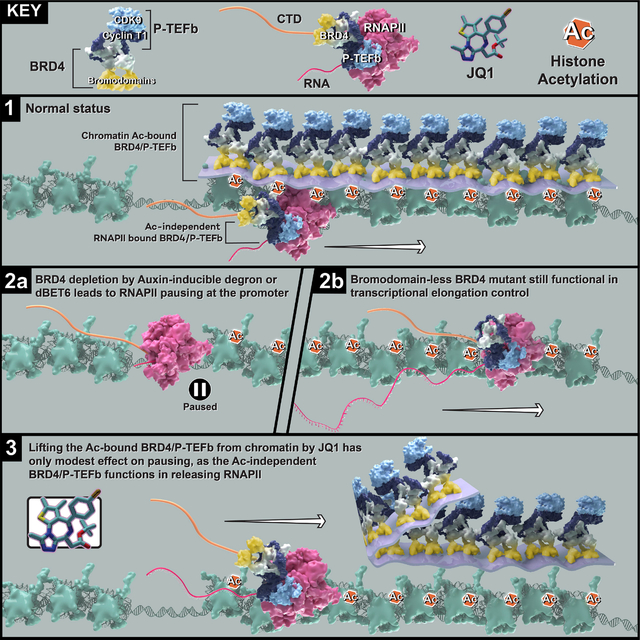
eTOC Blurb
Zheng et al. demonstrated that while the regulation of Pol II pause release by BRD4 does not require bromodomain-mediated BRD4 binding to acetylated histones, a bromodomain-less, C-terminal fragment of BRD4 is both necessary and sufficient to drive the release of paused Pol II into productive elongation.
Introduction
Transcription catalyzed by RNA Polymerase II is tightly regulated at the distinct stages of initiation, pausing, elongation and termination.1–3 Pol II promoter-proximal pausing is a prominent feature at the majority of the genes in human cells, and the positive transcription elongation factor (P-TEFb), which consists of the catalytic subunit CDK9 and the regulatory subunit Cyclin T, is an essential complex that regulates the release of promoter-proximal paused Pol II into elongation in gene bodies.4,5 Mechanistic studies have revealed that PTEFb mainly phosphorylates the serine residues at positions 2 and 5 of the heptapeptide repeats (52, in mammals) within the C-terminal domain (CTD) of RPB1, the main subunit of Pol II.6,7 PTEFb has also been shown to participate in multiple complexes, including the active BRD4-PTEFb complex, the active Super Elongation Complex (SEC) and the inactive 7SK-HEXIM complex, in which a majority of PTEFb is sequestered.8–11 We have previously demonstrated that PTEFb may be recruited to Pol II by either BRD4 under normal conditions or by the SEC under stress conditions such as heat shock.12
BRD4 is a member of the BET protein family, which also includes BRD2, BRD3 and BRDT. Each BET family member has two tandem bromodomains, which can bind with high affinity to acetylated lysines within histone tails, and an ET domain, which can interact with multiple transcription factors. However, only BRD4 and BRDT have C-terminal regions that can interact with PTEFb13,14. Histone acetylation, especially the extensively studied H3K27ac, is a marker of active chromatin15–17. Thus, a dominant model in the field holds that BRD4 bound to H3K27ac at promoters or enhancers recruits PTEFb to release paused Pol II, promoting transcription18,19. In accordance with this model, bromodomain inhibitors have been developed and are now widely used both in research and in clinical trials; JQ1 is the most prominent example of a bromodomain inhibitor compound20. JQ1-based proteolysis targeting chimeras (PROTACs) such as dBET6 target bromodomain-containing proteins for degradation. However, the effects of PROTAC-induced BRD4 depletion differ strikingly from the effects of bromodomain inhibition.21–24 Some attempts to explain these differences focus on incomplete displacement of BRD4 from chromatin after JQ1 treatment. However, accumulating evidence calls in to question the assumed requirement for H3K27ac marks in transcriptional regulation25–27. A recent study suggests that histone acetylation may not always be instructive to transcription, as complementation with catalytic-dead CBP or P300 mutants can restore gene expression just as well as their wild type, catalytic-active counterparts.28 Therefore, in this study we aimed to revise the established model by scrutinizing the functions of individual BRD4 domains using genetic complementation after acute depletion of endogenous BRD4. These rescue experiments revealed that BRD4 participates in Pol II pause release independently of its bromodomains binding to acetylated histones. We tested this phenomenon in distinct human cell lines, gaining confidence in the broad mechanistic implication. A “layer” of BRD4-PTEFB is associated with chromatin through BRD4 bromodomain-mediated interactions with acetylated histones. However, our study reveals that when we remove this layer, either by disrupting the interaction of BRD4 bromodomains with acetylation or through replacement of endogenous BRD4 with bromodomain-less versions, Pol II pause release remains functional genome-wide. Our genetic and molecular studies have also resulted in the identification of a BRD4 region necessary for Pol II pause release that functions independently of any bromodomain-mediated chromatin association. Altogether, this study introduces an important and possibly singular mechanism of histone acetylation-independent transcriptional regulatory function for BRD4, providing a basis for specific pharmaceutical modulation of BRD4-PTEFb activity to control transcription.
Results
Broad genome-wide changes in Pol II occupancy upon BRD4 depletion but not bromodomain inhibition-
It has been demonstrated, both by RNA-seq and by measurement of nascent RNA associated with Pol II on chromatin (NET-seq), that the effect of the BET protein family inhibitor JQ1 differs from that of its PROTAC version, dBET6: while JQ1 bromodomain disruption has only a modest effect on Pol II pausing, dBET6-induced degradation of BRD2, BRD3, BRD4 and BRDT proteins leads to a dramatic, genome-wide increase in paused Pol II at promoters. 21 To better understand the molecular mechanism underlying this disparity, we compared the effects of JQ1 or dBET6 treatment to that of acute and specific depletion of BRD4, the BET family member implicated in the regulation of Pol II pause release12,29,30. To specifically deplete BRD4 without affecting other BET proteins, we generated an updated BRD4 degron line by modifying the published miniIAA7 degron system (hereafter BRD4-IAA7) to reduce basal degradation seen with AID degron-tagged BRD431 (Figures 1A and S1A). BRD4 was rapidly degraded upon auxin treatment in our updated line, and indeed basal degradation appears to be reduced compared to the BRD4-AID line (Figure S1B). Compared to auxin treatment in BRD4-IAA7, dBET6 treatment degrades BRD4 to a similar extent, while JQ1 treatment does not, as expected (Figure 1A). We then performed Pol II ChIP-seq upon auxin, JQ1 or dBET6 treatment in BRD4-IAA7 cells to assess the impacts of BRD4 degradation vs bromodomain inhibition on the Pol II occupancy profile. As expected, dBET6 treatment phenocopied auxin treatment, inducing pausing at the majority of Pol II-transcribed genes, while JQ1 induced modest pausing (Figures 1B, 1C, and 1E). As shown by Estimated Cumulative Density Fraction (ECDF) and boxplot representations, as well as PCA analysis of the pause release ratios (PRR, reported as log2 fold change), JQ1 treatment did not recapitulate the genome-wide Pol II pausing effect seen upon auxin or dBET6 treatment (Figures 1D and 1F–1H). As shown by scatter plot representation, the gene-by-gene log2PRR correlation with auxin treatment was much stronger for dBET6 than for JQ1, for which the change in PRR was poorly correlated with that of auxin treatment on a gene-by-gene basis (Figure S1C). An effect of JQ1 on pausing was pronounced at super enhancer-controlled genes such as Myc22,32 (Figure S1D). Since BRD4 is known to interact with PTEFb to phosphorylate Pol II33,34, we also tested the effect of the potent CDK9 inhibitor NVP231 in DLD1 cells and found that CDK9 inhibition also induced strong Pol II pausing genome-wide (Figure S1E). Taken together, these data demonstrated that bromodomain inhibition was not sufficient to abolish BRD4’s participation in Pol II pause release, suggesting that PTEFb might release Pol II through a BRD4-dependent but bromodomain-independent mechanism.
Figure 1. Broad Pol II pausing upon BRD4 depletion but not bromodomain inhibition.
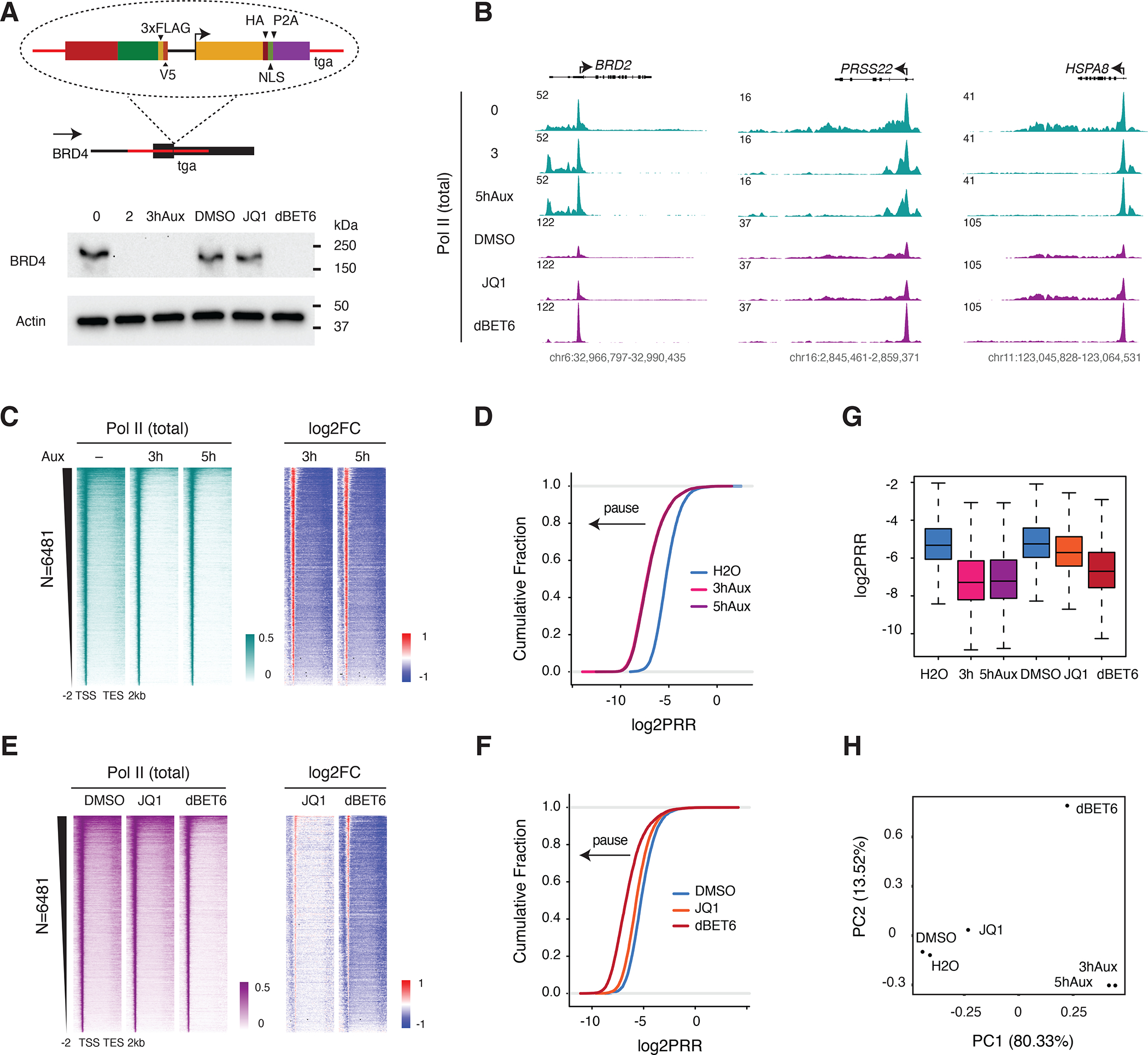
A. Schematic of the updated BRD4-IAA7 degron line. The F-box protein AtAFB2 was integrated into the C terminus of the BRD4 locus under the control of an independent promoter. Western blot showing the acute depletion of BRD4 by auxin (500 uM) or dBET6 (250 nM) treatment but not JQ1 (1 uM) in the BRD4-IAA7 degron line. Cells were treated with auxin for 2 or 3h, or with JQ1 or dBET6 for 3h. These treatment concentrations/durations are relevant to all of Figure 1.
B. Genome browser track examples of total Pol II ChIP-seq signal at the representative genes BRD2, PRSS22 and HSPA8 upon auxin treatment (0, 3 or 5h), JQ1 or dBET6 treatment (3h).
C. Heatmap showing the genome-wide Pol II occupancy profile upon auxin treatment and the profile of fold change in Pol II occupancy compared to control, in which the differential between promoter (high occupancy, red) and gene body (low occupancy, blue) indicates paused Pol II. The gene list (N=6481) in this analysis is used throughout the study (except for Figures 3H–J).
D. Estimated Cumulative Density Function (ECDF) of pause release ratios (PRR, reported as log2 fold change) for auxin treatment vs untreated control. The PRR is the ratio of Pol II occupancy within gene bodies to its occupancy at promoters. A leftward shift of the curve indicates an increase in the frequency and/or duration of promoter-proximal pausing, while a rightward shift indicates reduced pausing and/or more efficient release into gene bodies.
E. Heatmap showing the genome-wide Pol II occupancy profile upon JQ1 or dBET6 treatment and the profile of fold change in occupancy vs control.
F. ECDF of the log2PRR upon JQ1 or dBET6 treatment.
G. Boxplot of the log2PRR for auxin, JQ1 and dBET6 treatment.
H. PCA analysis of the log2PRR for auxin, JQ1 and dBET6 treatment.
Bromodomains of BRD4 are dispensable for Pol II pause release in living cells -
The finding that disruption of BRD4 binding to acetylated histone lysines was not sufficient to disrupt BRD4-mediated Pol II pause release (Figure 1) naturally led us to hypothesize that PTEFb could release Pol II in a BRD4-dependent but bromodomain-independent manner. To address this hypothesis, we generated stable lines with Dox-inducible, FLAG-tagged BRD4 constructs, including full-length (FL) BRD4 as well as bromodomains (ΔBD), ET (ΔET) or CTM (ΔCTM) domain deletion mutants, in BRD4-IAA7 cells. We also created a construct for a short, endogenously-expressed BRD4 isoform (BRD4S) that also lacks the CTM (Figure 2A). GFP in the same backbone was used as a construct expression control. We first induced expression of the BRD4 constructs by Dox treatment for two days before rapidly depleting endogenous BRD4 with auxin for three hours and then carried out Pol II ChIP-seq (Figure 2B). Surprisingly, we found that all three of the FL, ΔBD and ΔET constructs could rescue the Pol II pausing caused by depletion of endogenous BRD4, while the ΔCTM and BRD4S constructs could not (Figure 2C). Genome-wide analysis of the Pol II ChIP-seq demonstrated a visibly inverted pattern of Pol II occupancy in cells expressing the ΔBD or ΔET but not ΔCTM constructs, indicating that the ability of BRD4 to release Pol II is affected by CTM deletion but not by bromodomains or ET domain deletion (Figure 2D). As shown by boxplot and ECDF representations of log2PRR, the ΔBD and ΔET mutant BRD4 constructs rescued Pol II release to an extent similar to that of WT (FL) BRD4, but ΔCTM and BRD4S did not (Figures 2E and 2D). Notably, upon addition of Dox, signal from Pol II that is actively transcribing genome-integrated BRD4 constructs is observable in the Pol II ChIP-seq, confirming the expression of our mutant constructs. (Figure S2A). We also tested Pol II serine 2 phosphorylation (Ser2P), a modification at the Pol II CTD that is strongly associated with the elongation stage6,35, by ChIP-seq for the FL, ΔBD and ΔCTM rescue conditions (Figures 2G and 2H). Consistent with the total Pol II ChIP-seq, FL and ΔBD constructs could rescue the loss of Ser2P signal caused by depletion of endogenous BRD4, while ΔCTM could not (Figures 2G and 2H). We did observe several genes (such as MYC) that were not fully rescued by the bromodomain-less BRD4, reflecting the pausing effect of JQ1 treatment at some genes (Figure S2B). We performed genome-wide K-means clustering to identify the “Myc-like” genes, but didn’t observe notable differences between rescue by FL and ΔBD constructs. When calculating the log2FC of the PRR and Pol II occupancy at the genebody, we found that only four genes met a stringent cutoff (log2FC of PRR FLvsΔBD >=1 & log2FC of Pol II occupancy FLvsΔBD >=1). These four genes are MYC, KRT80, CXCL5, and SYT8. However, 49 genes met a less stringent cutoff (>=0.585) (Table S1). Next, we tested the ability of the mutant constructs to rescue cell growth and confirmed that the bromodomain-less constructs can rescue the cell growth defect caused by endogenous BRD4 depletion (Figure S2C). We also carried out RNA-seq for the rescue experiment after 24 hours of BRD4 depletion and found 863 down-regulated genes that were rescued by bromodomain-less mutants but not the ΔCTM mutant, further supporting the transcriptional relevance of our conclusion at the level of Pol II occupancy and paus-release. (Figure S2D).
Figure 2. BRD4 bromodomains are dispensable for Pol II pause release.
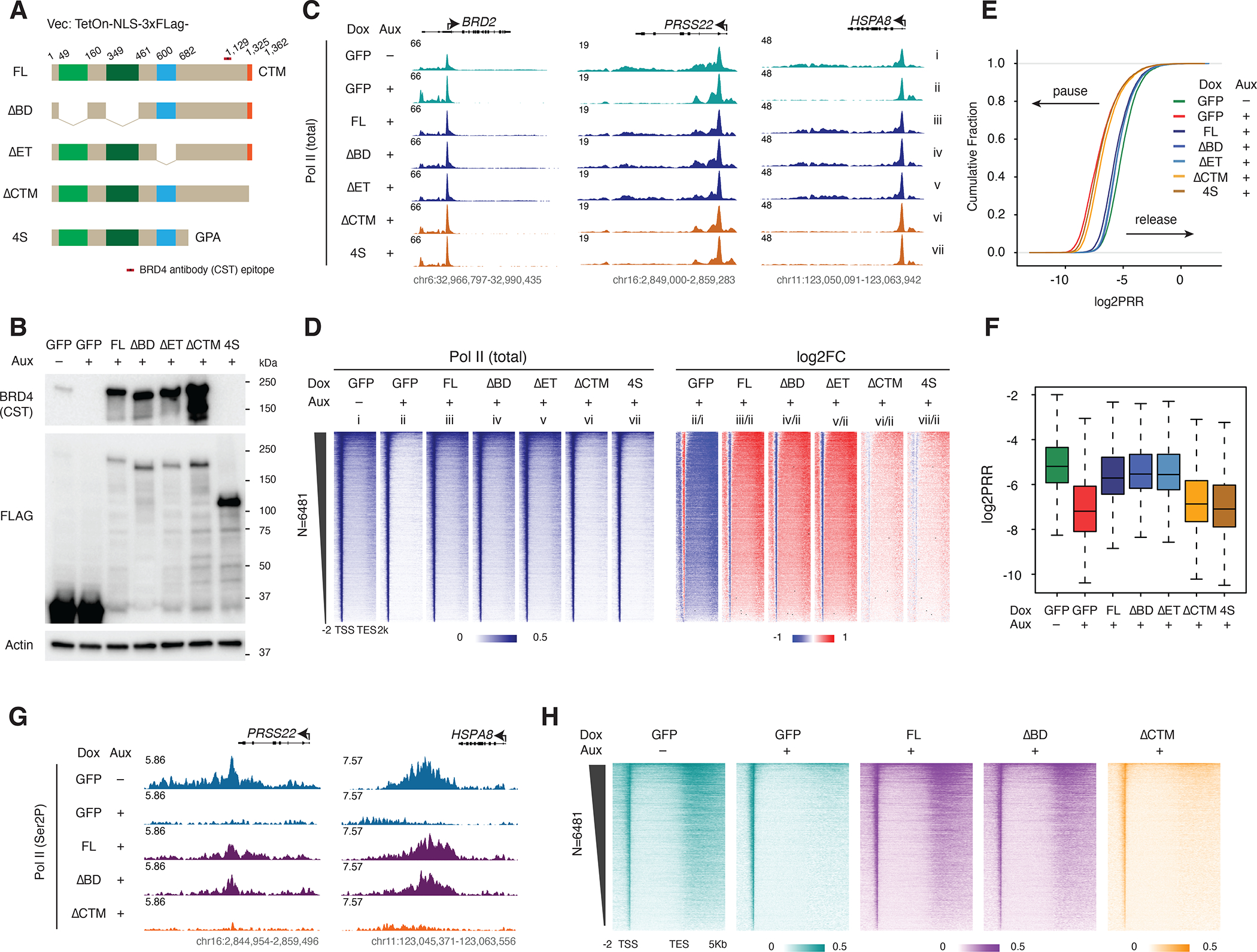
A. Schematic of the FLAG-tagged, Dox-inducible BRD4 mutant constructs, relevant to all of Figure 2 (vector: TetOn-NLS-3xFLAG). The location of the epitope for the commercial BRD4 antibody (CST) is indicated.
B. Western blot showing BRD4 mutant construct expression in the same conditions used for subsequent rescue experiments: auxin-treated (3h) BRD4-IAA7 cells, 2d after Dox treatment (50nM). Blot was probed for BRD4 using antibodies against a C-terminal epitope (see A) and the N-terminal FLAG tags.
C. Track examples showing the total Pol II ChIP-seq signal at the BRD2, PRSS22 and HSPA8 loci for the rescue experiment, in which auxin-induced endogenous BRD4 depletion is complemented by Dox-induced mutant construct expression.
D. Heatmaps showing the genome-wide Pol II occupancy and fold change in occupancy for mutant constructs vs GFP control in the rescue experiment.
E. ECDF comparison of log2PRR in the rescue experiment showing ΔCTM and BRD4S mutant constructs clustered with auxin-treated (BRD4 depleted) GFP control, while FL, ΔBD and ΔET constructs are clustered with the untreated GFP control.
F. Boxplot comparison of log2PRR in the rescue experiment.
G. Track examples of the Pol II Ser2P ChIP-seq signal at the PRSS22 and HSPA8 loci, showing loss of Pol II Ser2P occupancy due to auxin-induced endogenous BRD4 depletion that is rescued by Dox-induced expression of the FL or ΔBD but not ΔCTM mutant constructs.
H. Heatmaps showing the genome-wide Ser2P occupancy under the same rescue experiment conditions.
Bromodomain dispensability is conserved across cell lines-
Since the bromodomain-less mutant BRD4 is resistant to dBET6 treatment (Figure 3A), we treated cells expressing the bromodomain-less mutant with dBET6 to see whether the mutant could reverse the pausing effect normally seen upon dBET6 treatment. As expected, we found that dBET6-induced pausing is abolished by bromodomain-less BRD4 (Figures 3B–3E). We then tested whether this effect holds true beyond DLD1 cells by expressing the bromodomain-less mutant and testing the effect of dBET6 in a very different cell line, NCIH2009. In addition to BRD4, the NCIH2009 cells also endogenously express the BET family member BRDT, but both proteins are degraded by dBET6 (Figures 3F, S3A, and S3B). The genome-wide Pol II profile differs between DLD1 and NCIH20009, but we found that for most genes displaying dBET6-induced pausing in NCIH2009, this effect can be rescued by reconstitution with the bromodomain-less mutant (Figure 3G). Genome-wide analysis in NCIH2009 further supported our conclusion that the bromodomain-less mutant rescues Pol II pause release in NCIH2009 cells (Figure S3C). Out of 6481 genes, a total of 1565 dBET6-paused genes (24.15%) were selected using a dual threshold: 2-fold reduction of Pol II at gene bodies and 2-fold reduction of PRR. We repeated the same analysis with these genes and found that most of the highly paused genes were rescued by the bromodomain-less BRD4 in NCIH2009 cells (Figures 3H–3J). Although the role of BRDT in transcription is largely unknown, it contains a PTEFb-interacting domain and could potentially function similarly to BRD4 in releasing paused Pol II, and bromodomain-less BRD4 may also rescue the effect of BRDT depletion in NCIH2009 cells. Nevertheless, our data indicates that the bromodomain-independent function of BRD4 in Pol II release is not strictly limited to DLD1 cells.
Figure 3. Bromodomain dispensability conserved across cell lines.
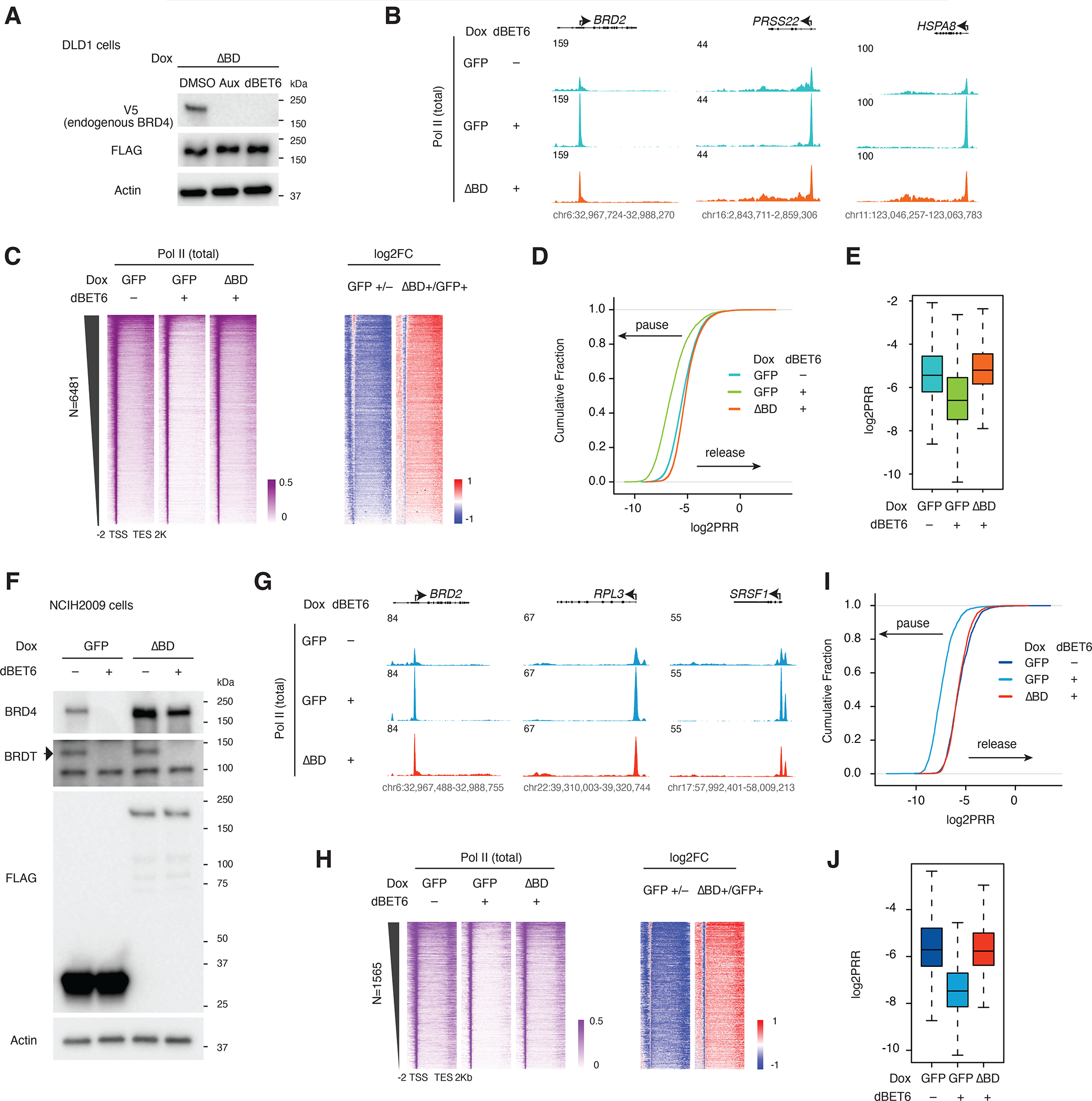
A. Western blot showing degradation of endogenous BRD4 (V5) but not the bromodomain-less BRD4 mutant (FLAG) upon auxin or dBET6 treatment in the BRD4-IAA7 DLD1 degron line.
B. Track examples of the total Pol II ChIP-seq signal at the BRD2, PRSS22 and HSPA8 loci after dBET6 treatment and rescue by bromodomain-less BRD4 in DLD-1 cells.
C. Heatmap showing the genome-wide Pol II occupancy and fold change in occupancy for the bromodomain-less mutant construct vs GFP control after dBET6 treatment in DLD-1.
D. ECDF of the log2PRR from the total Pol II ChIP-seq for the dBET6 rescue experiment in DLD-1.
E. Boxplot of the log2PRR from the total Pol II ChIP-seq for the dBET6 rescue experiment in DLD-1.
F. Western blot showing the depletion of endogenous BRD4 and BRDT, but not the bromodomain-less mutant, by dBET6 treatment (3h) in NCIH2009 cells.
G. Track examples of the total Pol II ChIP-seq signal upon dBET6 treatment (3h) and rescue by bromodomain-less BRD4 in NCIH2009 cells.
H. Heatmap showing the Pol II occupancy profiles and corresponding fold changes across the 1565 genes for which dBET6 causes strong pausing (2-fold reduction of Pol II at gene bodies and 2-fold reduction of PRR), rescued by bromodomain-less BRD4 in NCIH2009.
I. ECDF of the log2PRR from the Pol II ChIP-seq for the dBET6 rescue experiment in NCIH2009 (N=1565).
J. Boxplot of the log2PRR from the Pol II ChIP-seq for the dBET6 rescue experiment in NCIH2009 (N=1565).
A bromodomain-less BRD4 C-terminal fragment interacts with PTEFb and rescues Pol II pause release –
To investigate which BRD4 regions are required for the function of BRD4 in Pol II pause release, we generated a series of N-terminal truncation mutants retaining the CTM in the same FLAG-tagged vector as above (Figure S3D). After validating the induced expression of these mutants (Figure S3D), we used Pol II ChIP-seq to screen them using the same rescue experiment setup, with the bromodomain deletion mutant (ΔBD) as a negative control. To our surprise, the Pol II profile was similar for all the mutant constructs, with a noticeable decrease in gene body Pol II occupancy for only the shortest mutant construct (Cs), which consists of just 170 residues from the C terminus of BRD4 (Figures S3E and S3F). We calculated the log2PRR and found that all the truncation mutants could rescue Pol II release to an extent equal or greater to that of the ΔBD construct, as indicated in the boxplot representation (Figure S3G). The endogenous BRD4 acquired a 3xFLAG tag upon IAA7 tagging, so to exclude any potential confusion deriving from residual endogenous BRD4, we replaced the 3xFLAG tag with GFP for further characterization of the C-terminal constructs (Figure 4A) (in our previous experience, FLAG ChIP-seq works poorly in DLD-1 cells, while GFP ChIP-seq works well.) We first validated the expression of the GFP-tagged shortest construct (Cs), Cs with CTM deletion (CsΔCTM) and a CTM-only (CTM) construct by western blot (Figure S4A). Then, we carried out immunoprecipitation followed by mass spectrometry (IP-MS) for these mutants. The consistent results of two replicate IP-MS experiments showed that only the Cs mutant retained the ability to interact with PTEFb (Figure 4B). We assessed the Cs interactome, represented by interacting protein Qvalue vs their enrichment over that of the vector and found that two of the most outstanding Cs interacting proteins are components of the PTEFb complex, which we then validated by western blot analysis (Figures 4C and 4D). We further validated these results by carrying out Pol II ChIP-seq for the GFP-tagged mutant constructs in comparison with full-length (FL) BRD4. Consistently, the Pol II occupancy profile shows the rescue of Pol II pause release by the Cs construct (Figures 4E and 4F). Consistent with the FLAG-tagged version, despite noticeable reduction in gene body Pol II occupancy for the Cs mutant there was no corresponding defect in the Cs PRR relative to the FL BRD4 positive control (Figures S4B–S4D). Since the PRR was actually highest for the extended C terminus (C) in our screen of FLAG-tagged mutant constructs (Figure S3G), we also generated a GFP-tagged C construct (Figure 4A) and compared its ability to rescue Pol II release to the FL and ΔBD constructs, finding no obvious defect in rescued Pol II occupancy at the gene body relative to the BRD4-FL positive control (Figures S4D–S4I). We also confirmed the functionality of the C terminus by Ser2P ChIP-seq in the rescue experiment (Figures 4G and 4H). Multiple replicates of IP-MS, Pol II ChIP-seq, and Ser2P ChIP-seq experiments reproducibly consolidated our conclusion that the C terminus of BRD4 is sufficient for the release of paused Pol II.
Figure 4. BRD4 C-terminal fragment interacts with PTEFb and rescues Pol II pause release.
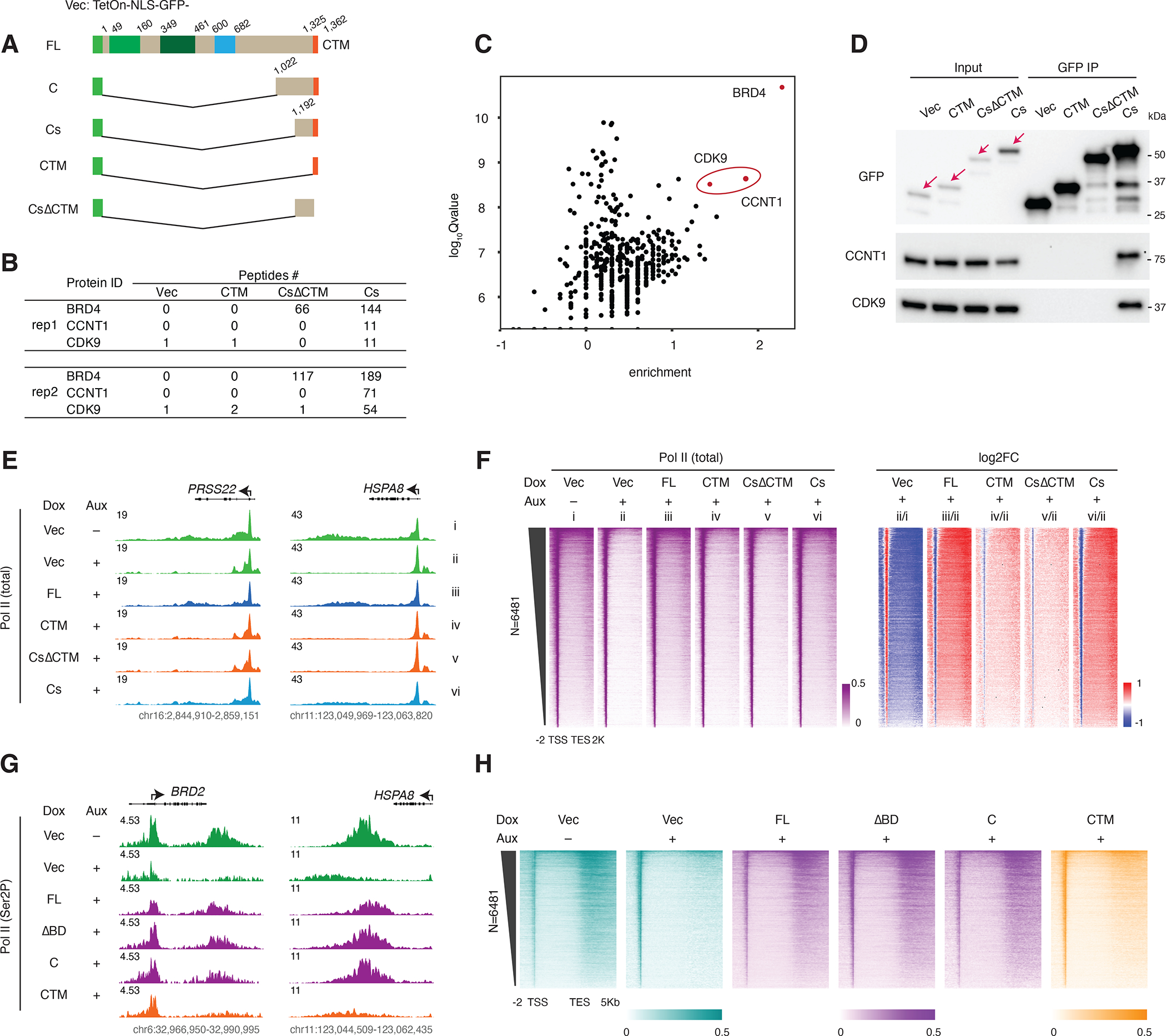
A. Schematic of the GFP-tagged FL BRD4 and BRD4 C-terminus constructs (vector: TetOn-NLS-GFP). Cs: short C terminus.
B. GFP IP-MS results showing peptide counts for the bait (BRD4) and PTEFb complex components (CCNT1 and CDK9) in two replicates after Dox (50nM) induced expression of Vector, CTM, CsΔCTM or Cs mutant constructs for 2 days. Note: the 0 bait peptide count for the CTM construct is probably due to impaired peptide alignment, as the CTM construct is itself a (GFP-tagged) peptide of only 37 residues.
C. Scatter plot showing the log10Qvalue vs the enrichment over Vector (calculated by log10FC of peptide counts +1) for the BRD4-Cs interacting proteins in the second GFP IP-MS replicate.
D. Western blot validating the GFP IP-MS result using the elute from the second replicate.
E. Track examples for total Pol II ChIP-seq signal at the PRSS22 and HSPA8 gene loci upon auxin treatment and rescue by different GFP-tagged mutants.
F. Heatmap showing the genome-wide Pol II occupancy profile and the corresponding fold changes for the GFP-tagged rescue experiment.
G. Track examples for Ser2P ChIP-seq signal at the BRD2 and HSPA8 gene loci upon auxin treatment and rescue by different GFP-tagged mutants.
H. Heatmap showing the genome-wide Pol II occupancy profile and the corresponding fold changes for the GFP-tagged rescue experiment.
Distinct layers of the BRD4-PTEFb complex: identification of a histone acetylation-independent but active BRD4-PTEFb population-
The extent to which BRD4 is required for recruiting PTEFb to chromatin and regulating gene expression remains a subject of some debate21,36,37. Aiming to further investigate this point, we carried out ChIP-seq for Pol II, CCNT1 and CDK9 in BRD4-IAA7 cells with and without prior depletion of endogenous BRD4 by auxin treatment. BRD4 depletion led to a dramatic reduction in the CCNT1 peaks at promoters genome-wide, indicating that CCNT1 recruitment to chromatin relies on BRD4 (Figures 5A and S5A) but not vice versa, as CDK9 depletion leads to increased BRD4 ChIP signal (Figures S5C and S5D). We integrated this data with our previously published ChIP-seq in the BRD4-AID line and compared the peaks of CCNT1 and BRD4 to H3K27ac (Figure 5A). Genome-wide analysis revealed general colocalization of CCNT1 and BRD4 at promoters, which was mostly associated with histone acetylation (Figures 5B and 5C). Signal was generally low in our CDK9 ChIP-seq, but at genes that were highly enriched with Pol II we were able to see CDK9 peaks with the same trend seen for CCNT1 (Figures S5A and S5B). Notably, a few genes show increased Pol II occupancy and thus retain PTEFb upon BRD4 depletion (Figure S5A), which is presumably recruited by SEC (data not shown). Next, we carried out GFP ChIP-seq for the GFP-tagged BRD4-FL and mutants (Figure 4A). Surprisingly, though they could all rescue Pol II occupancy and release, the ΔBD, C and Cs mutants had ChIP-seq signals that were minimal genome-wide compared to that of the FL BRD4 (Figures 5D and S5E). We tried several different ChIP-seq conditions with increasing amounts of chromatin but was not able to obtain clear peaks for all bromodomain-less BRD4 mutants. We then checked CCNT1 occupancy in the rescue experiments and found that CCNT1 signal was restored by BRD4-FL but not the bromodomain-less C mutant (Figure 5E). This result is consistent with the GFP ChIP-seq results and further indicates that the population or “layer” of acetylation-bound BRD4-PTEFb captured by ChIP-seq is mainly associated with BRD4 bromodomains interacting with acetylation sites, while a distinct acetylation-independent layer, which could be too dynamic to be efficiently captured by ChIP-seq, nevertheless functions to release Pol II (Figure 5E). To further confirm that disruption of the acetylation-bound BRD4-PETFb layer alone could not abolish Pol II pause release, we introduced an acetylation-bound but phosphorylation-defective BRD4-PTEFb layer by overexpressing the CTM-less (and therefore PTEFb-blind) BRD4 mutants to displace the population of acetylation-bound BRD4-PTEFb. Indeed, a significant amount of BRD4-PTEFb was knocked off from chromatin by overexpression of either the ΔCTM or 4S CTM-less BRD4 mutants (Figure 5F). However, this displacement did not constitute a dominant negative effect, as the ChIP-seq signals for both total Pol II and Ser2P were not noticeably diminished by CTM-less construct overexpression (Figures S5F–S5H). To further validate this observation with a more severe disruption of the acetylation-bound layer, we then treated cells with increasing concentrations of JQ1. We also included dBET6 treatment as a control for joint disruption of both the acetylation-bound and acetylation-independent layers. Though CCNT1 ChIP-seq signals decreased dramatically with increasing concentrations of JQ1, concomitant change in the Ser2P ChIP-seq signals was minimal, and only with dBET6 disruption of both layers did Ser2P signal fade away (Figures 5H–J). These results suggest that disruption of the acetylation-independent BRD4-PTEFb layer is the underlying mechanism by which dBET6 differs from JQ1 in effectively abolishing Pol II pause release.
Figure 5. Identification of a distinct BRD4-PTEFb population: active but not histone acetylation-bound.
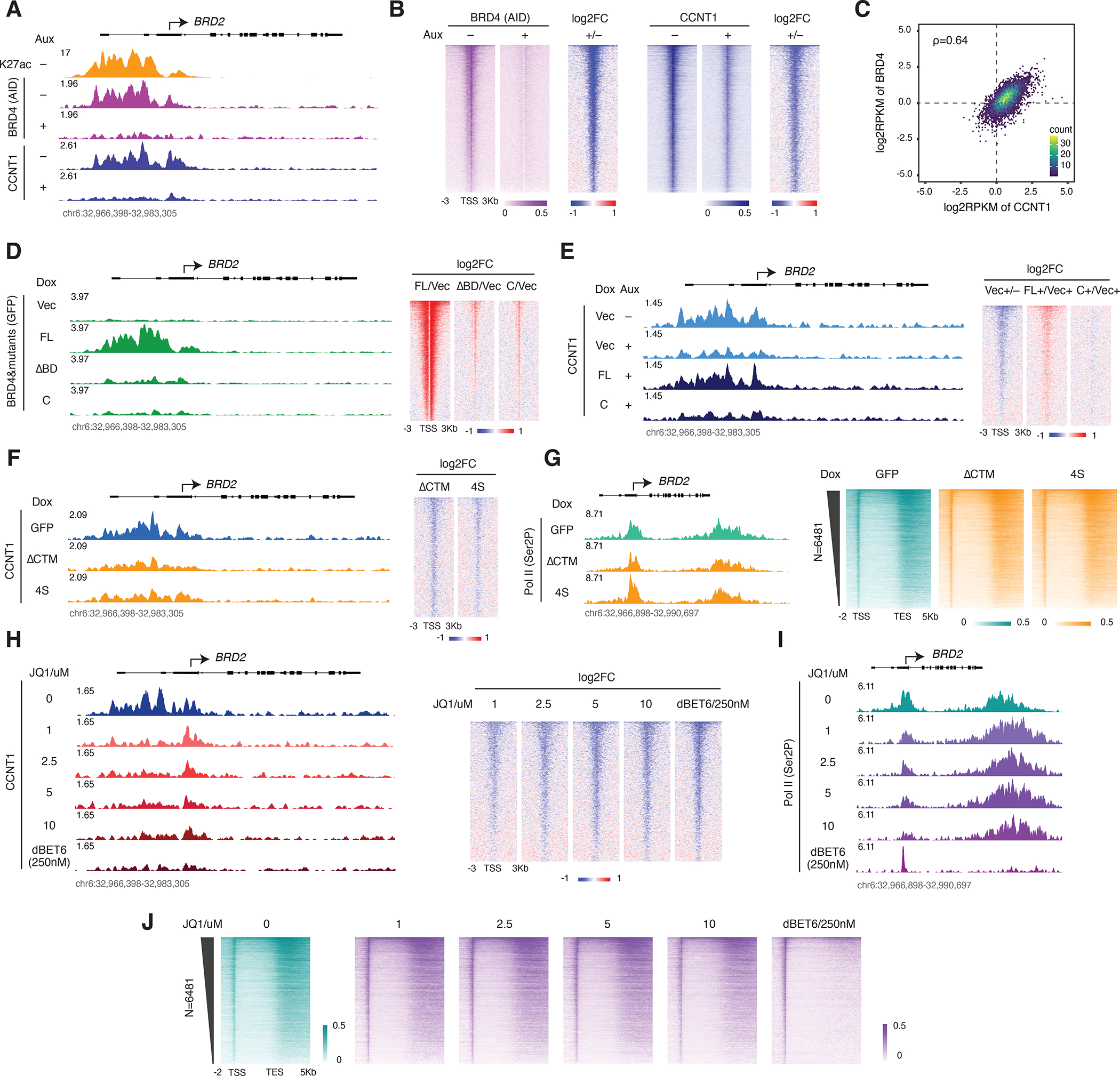
A. Track examples for AID ChIP-seq in the BRD4-AID line and CCNT1 ChIP-seq in the BRD4-IAA7 line with/without auxin treatment (3h). H3K27ac ChIP-seq signal in the BRD4-AID line is shown for comparison.
B. Heatmap showing the genome-wide BRD4 and CCNT1 occupancy profile and the corresponding fold change upon auxin treatment for the conditions in A. Genes ranked by BRD4 signal in control.
C. Scatter plot showing the relative promoter density of CCNT1 vs BRD4, calculated by the log2RPKM of the ChIP-seq signals at the regions flanking (±1Kb) the Pol II pausing sites.
D. Track examples for GFP ChIP-seq signal upon induction of GFP-tagged BRD4-FL or the indicated mutants (left). Heatmap showing the genome-wide fold changes of the GFP-tagged BRD4 mutants relative to the vector (right). The endogenous BRD4 was depleted by auxin treatment (3h) in all samples. Genes are ranked by GFP signal in the GFP-BRD4-FL condition.
E. Track examples for CCNT1 ChIP-seq signal upon endogenous BRD4 depletion and rescue with indicated GFP-tagged mutants (left). Heatmap showing the genome-wide CCNT1 fold changes (right). Genes are ranked by CCNT1 signal in the untreated vector control.
F. Track examples for the CCNT1 ChIP-seq signal upon induction of the FLAG-tagged, CTM-less BRD4 mutants by Dox treatment for 2 days (left). Heatmap showing the genome-wide profile of fold changes in CCNT1 occupancy (right). Genes are ranked by CCNT1 signal in the GFP control.
G. Track examples for the Ser2P ChIP-seq from the same samples in F (left). Heatmap showing the corresponding genome-wide Ser2P occupancy profile (right).
H. Track examples for the CCNT1 ChIP-seq in a series concentration of JQ1 and 250nM dBET6 treatment for 3h in the BRD4-IAA7 cells (left). Heatmap showing the corresponding genome-wide CCNT1 fold changes relative to DMSO treatment (right). Genes ranked by CCNT1 signal in the DMSO control.
I. Track examples for the Ser2P ChIP-seq from the same JQ1 or dBET6-treated samples in H.
J. Heatmap showing the corresponding genome-wide Ser2P occupancy profile.
The BRD4 C-terminal disordered region is required for CyclinT1 binding in the BRD4-PTEFb complex-
The fact that the GFP-tagged CTM cannot pull down PTEFb, while the extended Cs can, prompted us to investigate how the C terminus of BRD4 mediates its interaction with PTEFb. We first carried out GFP IP for the GFP-tagged, full-length BRD4 upon dCDK9 treatment. CDK9 degradation also impairs BRD4-CCNT1 interaction, indicating the requirement of CDK9 for stable BRD4-PTEFb complex formation (Figure 6A). We then generated GFP-tagged partial (ΔCp) and entire (ΔCd) C-terminus deletion mutants (Figure 6B and S6A). Ser2P ChIP-seq in the rescue experiment revealed that though Pol II-releasing activity was largely impaired in the partial deletion mutant, the entire deletion almost completely abolished activity (Figure S6B and S6C). We carried out GFP IP for the ΔCd mutant and found a severe loss of binding to CCNT1 but not CDK9 compared to the FL and C mutants (Figure 6C), which could explain why the ΔCd mutant cannot rescue Pol II release, as was further confirmed by the total Pol II ChIP-seq (Figures 6D–6G). Finally, we computed the predicted structure of the CTM of BRD4 C (37 residues) in association with the PTEFb complex (initial N-terminal 344 residues of CDK9 and the initial 293 N-terminal residues of CCNT1, as the corresponding crystal structure has been published38) using AlphaFold integrated in ChimeraX 39. In line with the results of this study, the predicted structure highlights a critical role for the CTM in mediating the interaction between BRD4 and PTEFb, with CTM protruding into the pocket created by CDK9 and CCNT1 (Figure 6H).
Figure 6. The BRD4 C-terminus stabilizes CyclinT1 in the BRD4-PTEFb complex.
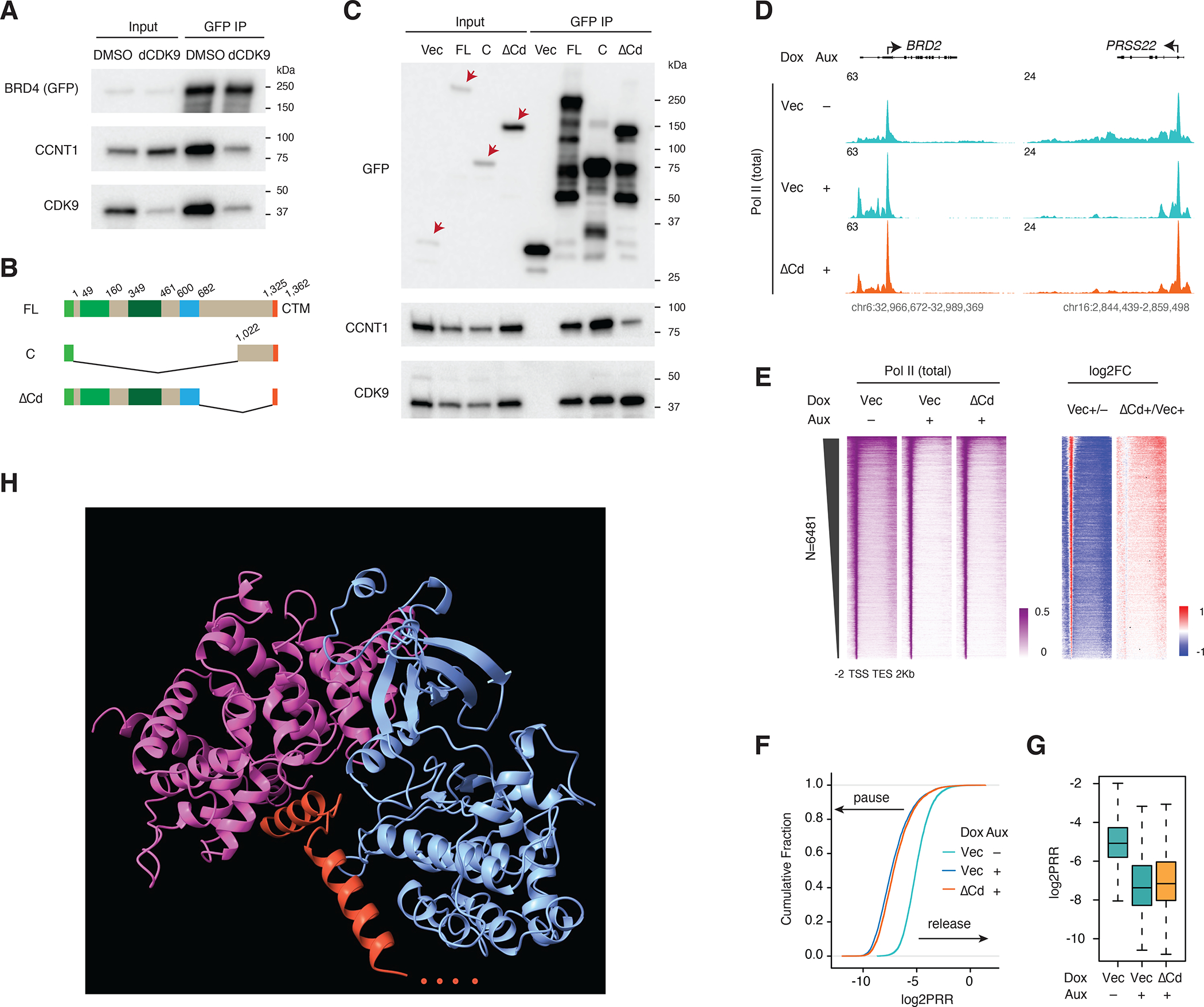
A. Western blot of GFP IP showing the PTEFb complex components associated with BRD4 upon dCDK9 (2.5uM) treatment for 3h. GFP-tagged BRD4 -FL was induced by Dox treatment for 2 days.
B. Schematic of the GFP-tagged constructs: BRD4-FL, BRD4-C and BRD4-ΔCd (deletion of the C-terminal disordered region).
C. Western blot of GFP IP showing PTEFb complex components associated with BRD4 and its C terminal mutants. GFP-tagged BRD4-FL and mutant constructs were induced by Dox treatment for 2 days. Endogenous BRD4 was depleted in all samples by auxin treatment (3h) prior to IP.
D. Track examples for the total Pol II ChIP-seq signal upon BRD4 depletion and rescue by BRD4-ΔCd.
E. Heatmap showing the genome-wide Pol II occupancy and the corresponding fold changes for the rescue experiment in D.
F. ECDF showing the log2PRR for the Pol II ChIP-seq for the rescue experiment in D.
G. Boxplot showing the log2PRR for the Pol II ChIP-seq for the rescue experiment in D.
H. AlphaFold-predicted structure for CTM of BRD4 and the PTEFb complex (344 CDK9 N-terminal residues, 293 N-terminal CCNT1 residues and 37 C-terminal BRD4 residues were used for the computational prediction.
Discussion
In this study, we sought to determine why broad genome-wide changes in Pol II occupancy are seen upon BRD4 depletion but not bromodomain inhibition. Our depletion and rescue strategy allowed us to directly compare exogenous wildtype BRD4 with mutant BRD4 constructs, including bromodomain-less mutant BRD4 constructs that are unable to bind acetylated histone lysine residues. By characterizing the effects of these constructs on the chromatin-associated profiles of Pol II and its elongating Ser2P form, we determined that while the C terminus of BRD4 is required for its participation in Pol II pause release, BRD4 bromodomains are dispensable for this function. We further validated the essential role for the BRD4 C terminus, isolating a minimal C-terminal fragment which lacks bromodomains but physically interacts with PTEFb, and showing that cellular expression of this C-terminal fragment is sufficient to replicate the role of full-length BRD4 in Pol II pause release. Displacing endogenous BRD4 from chromatin by overexpressing a PTEFb-blind mutant reduced PTEFb chromatin occupancy without affecting Pol II pause release, phenocopying the effect of bromodomain inhibitor treatment. Importantly, this result indicates that endogenous bromodomain-containing BRD4 can release paused Pol II irrespective of its association with acetylated histones. The endogenous expression of the BRD4 short isoform (4S), which lacks the CTM required for PTEFb interaction and thus cannot participate in release of paused Pol II, suggests that this displacement is physiologically relevant.
Our results all support a previously unappreciated role for BRD4 in acetyl recognition-independent release of paused Pol II. In the model emerging from our results, there are two distinct populations or “layers” of BRD4-PTEFb: a first layer of acetylation-bound BRD4-PTEFb complemented by a second layer of acetylation-independent BRD4-PTEFb that effectively releases paused Pol II. One question emerging from this model is what role the prominent acetylation-bound BRD4-PTEFb layer plays in transcription. Further studies are needed to dissect the endogenous functions of full-length BRD4 and its short isoform, which could potentially balance the two layers of BRD4-PTEFb. Nevertheless, this angle on BRD4 function brings clarity to the long-running debate concerning the causes of insensitivity or resistance to BET inhibitors, which has previously been observed in multiple studies and trials40–42. Notably, other studies have also shown that BRD4 is required for survival even in BET inhibitor-resistant cells, similarly implicating a function for BRD4 beyond that mediated by its bromodomains43–45. Our findings represent a major step towards mechanistic understanding of this function, as they reveal the crucial importance of BRD4’s C terminus for CDK9/CCNT1 interaction and regulation of Pol II pause release. Future studies will be focused on determining the structural basis of the interaction of BRD4 with CDK9 and CCNT1 and developing BRD4 C-terminal specific inhibitors, which (unlike pan-BET inhibitors 46) would leave the acetylation-bound layer of BRD4 and other BET family proteins unaffected. We predict that this class of inhibitors will be highly efficient in abolishing the function of BRD4 in Pol II pause release, resulting in potent therapeutic benefit.
In line with the bromodomain-independent function of BRD4, Sankar et al. mutated all 28 H3 alleles from K27 to R27 to show that H3K27 modification is not required either for proper loading of the Pol II machinery onto chromatin or for transcriptional activation during cell fate transition in mESCs26. Similarly, Zhang et al. found that abolishing H3K27ac was not sufficient to disrupt enhancer activity27. Narita et al. used nascent RNA-seq to characterize the immediate transcriptional outcome of inhibiting p300/CBP enzymatic activity via the small molecule A-485. For the majority of nascent transcripts, abundance remained unaltered by enzymatic inhibition even at high A-485 doses that induce an observable decrease in H3K27ac genome wide.47 Most recently, Ciabrelli et al. reported that catalytic dead CBP/Gcn5 recapitulates the function of the WT protein in driving zygotic genome activation.28 Our finding, that BRD4-dependent transcription has no requirement for bromodomain-mediated interaction with histone acetylation, may offer an explanation. H3K27ac may be better considered as an active transcription marker that reflects gene expression status rather than as a determinant for transcription or enhancer activity. The advantage for the cell to dispense with any requirement for histone acetylation would be that during conditions such as cell division and differentiation, rapid and specific gene expression changes could proceed undelayed by the process of histone modification establishment. Indeed, the transcriptional kinetics of up- and down-regulation in response to heat shock are largely independent of the histone modification context at heat shock-responsive genes.48
We did find a handful of genes that are upregulated upon depletion of endogenous BRD4, and for which BRD4 or mutant construct rescue had very different (or opposite) effects from those seen at the vast majority of genes. Because these genes were so few, they were not excluded from and did not affect our genome-wide analyses, but they are certainly worth considering separately. Future studies will be needed to address the mechanism of transcriptional activation for these genes. We also observed several genes that were not fully rescued by the bromodomain-less BRD4 mutants, including the gene encoding the important and well-characterized transcription factor Myc. Since the MYC loci is known to be regulated by super enhancers, and because super enhancers are bound by Mediator and BRD4 through bromodomains32, we suspect that the defect in MYC rescue could reflect a requirement for the histone-bound layer of BRD4-PTEFb in the maintenance of super enhancer activity. BRD2 has also been shown by our lab and others to be essential for transcriptional regulation49–51, so it will be interesting to investigate whether this function is conserved for bromodomains among different BET family members or if the bromodomains of different BET family members have distinct functional roles.
Limitations of the study
BRD4 mutants were induced prior to auxin depletion of IAA7-tagged, endogenous BRD4, and the result that bromodomain-less BRD4 mutants could rescue the genome-wide Pol II pausing caused by three hours of endogenous BRD4 depletion, while the same mutants could only rescue transcript levels for a subset of the genes downregulated by 24 hours of depletion, is consistent with potential bromodomain-mediated gene expression regulatory roles beyond pause release such as the regulation of transcription initiation47,52 and RNA processing.30,53 However, endogenous BRD4 activity prior to auxin treatment could potentially establish or poise Pol II machinery in such a way that persistence of such a state after endogenous BRD4 depletion might theoretically enable the full functionality observed upon complementation with the bromodomain-less BRD4. This is a design limitation inherent in the acute depletion setting, which was chosen to avoid the lethality of long-term or total BRD4 depletion in our rescue experiments. Due to the use of pooled lentivirus-infected cells to avoid clonal expression effects, another limitation of the rescue experiments is that the potential contribution of expression level to the differential effects of mutant constructs on the Pol II profile (for example by ΔCTM and BRD4S at the Myc locus) is not addressed.
From a conceptual standpoint, our data indicates that either BRD4 or its functional C-terminal fragments can form stable complexes with PTEFb that are capable of CTD phosphorylation to release paused Pol II, regardless of the acetylation status of nearby histones. However, it remains unclear whether a complete loss of all histone acetylation is a survivable condition, because even in the study by Ciabrelli et al., the H3K27ac, H3K9ac, and H3K18ac deposited by CBP/GCN5 were not simultaneously absent.28 It therefore remains to be determined whether BRD4 could function normally in the total absence of any histone acetylation in living cells. It is entirely possible that acetyl-bound and unbound BRD4-PTEFb populations could act to co-regulate transcription through interdependent mechanisms, but this study does not indicate whether or how this co-regulation might occur.
STAR ★ Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for reagents should be directed to and will be fulfilled by the lead contact, Ali Shilatifard (ash@northwestern.edu).
Materials Availability
Unique reagents generated in this study are available from the lead contact without restriction.
Data and Code Availability:
All genomic data generated in this study have been deposited to GEO under the accession number GSE232869 and are publicly available as of the date of publication. Accession number is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| BRD4 (for WB) | Cell Signaling Technology | Cat#13440 |
| β-Actin (for WB) | Cell Signaling Technology | Cat#3700 |
| V5-Tag (for WB) | Cell Signaling Technology | Cat#13202 |
| HA-Tag (for WB) | Cell Signaling Technology | Cat#3724S |
| CDK9 (for WB) | Cell Signaling Technology | Cat#2316 |
| Cyclin T1 (for WB and ChIP) | Cell Signaling Technology | Cat#81464 |
| Tubulin (for WB) | Developmental Studies Hybridoma Bank | Cat#E7 |
| FLAG (for WB) | Millipore Sigma | Cat#F1804 |
| GFP (for WB) | Santa Cruz Biotechnology | Cat#sc-9996 |
| H3 (for WB) | This study | NA |
| BRDT (for WB) | This study | NA |
| Rpb1 (for ChIP) | Cell Signaling Technology | Cat#D8L4Y |
| Ser2p (for ChIP) | Active Motif | Cat#61984 |
| CDK9 (for ChIP) | Santa Cruz Biotechnology | Cat#sc-13130 X |
| GFP (for ChIP) | Developmental Studies Hybridoma Bank | Cat#GFP-G1 |
| GFP (for ChIP) | Developmental Studies Hybridoma Bank | Cat#GFP-12A6 |
| GFP (for ChIP) | Developmental Studies Hybridoma Bank | Cat#GFP-12E6 |
| GFP (for ChIP) | Developmental Studies Hybridoma Bank | Cat#GFP-8H11 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Auxin (3-Indole-acetic acid sodium salt) | Abcam | Cat#ab146403 |
| JQ1 | Tocris | Cat#4499 |
| dBET6 | Selleckchem | Cat#S876202 |
| NVP-2 | MedChemExpress | Cat#HY-12214A |
| THAL-SNS-032 (dCDK9) | MedChemExpress | Cat#HY-123937 |
| Doxycycline | Stem Cell Technologies | Cat#72742 |
| Benzonase | Millipore Sigma | Cat#E1014 |
| Dynabeads Protein G | Invitrogen | Cat#10004D |
| ChromoTek GFP-Trap Magnetic Agarose | Proteintech | Cat#gtma |
| Deposited Data | ||
| Genomics data | This study | GSE232869 |
| Experimental Models: Cell Lines | ||
| DLD-1 | ATCC | Cat#CCL-221 |
| NCI-H2009 | ATCC | Cat#CRL-5911 |
| Mouse embryonic fibroblasts | STEMCELL Technologies | Cat#00325 |
| BRD4-IAA7 DLD-1 #G3/D4 | This study | NA |
| Recombinant DNA | ||
| Cas9 plasmid for BRD4-IAA7 (sgRNA: CACCGAATCTTTTCTGAGCGCACCT) | Zheng et al., 2021 | NA |
| Cas9 plasmid for BRD4-IAA7 (sgRNA: CACCGATCAAAGTCAGAAGCCACCT) | Zheng et al., 2021 | NA |
| BRD4-IAA7-Neo donor plasmid | This study | pBZ14 |
| BRD4-IAA7-Hygro donor plasmid | This study | pBZ15 |
| Lenti-TetON-Sox2 | Addgene | Cat#110280 |
| p6344 pcDNA4-TO-HA-Brd4FL | Addgene | Cat#31351 |
| pFlag-CMV2-Brd4 del ET | Addgene | Cat#21938 |
| pTet-On-sNLS-3xFlag-EGFP-UBC-rtTeR-IRES-BSD | This study | pBZ113 |
| pTet-On-sNLS-3xFlag-BRD4-FL-UBC-rtTeR-IRES-BSD | This study | pBZ111 |
| pTet-On-sNLS-3xFlag-BRD4-ΔBD-UBC-rtTeR-IRES-BSD | This study | pBZ160 |
| pTet-On-sNLS-3xFlag-BRD4-ΔET-UBC-rtTeR-IRES-BSD | This study | pBZ136 |
| pTet-On-sNLS-3xFlag-BRD4-ΔCTM-UBC-rtTeR-IRES-BSD | This study | pBZ158 |
| pTet-On-sNLS-3xFlag-BRD4S-UBC-rtTeR-IRES-BSD | This study | pBZ137 |
| pTet-On-sNLS-3xFlag-BRD4-ΔN-UBC-rtTeR-IRES-BSD | This study | pBZ176 |
| pTet-On-sNLS-3xFlag-BRD4-Cm-UBC-rtTeR-IRES-BSD | This study | C1 |
| pTet-On-sNLS-3xFlag-BRD4-C-UBC-rtTeR-IRES-BSD | This study | C2 |
| pTet-On-sNLS-3xFlag-BRD4-Cs-UBC-rtTeR-IRES-BSD | This study | C3 |
| pTet-On-sNLS-GFP-UBC-rtTeR-IRES-BSD (Vector) | This study | MS1 |
| pTet-On-sNLS-GFP-BRD4-FL-UBC-rtTeR-IRES-BSD | This study | MS0 |
| pTet-On-sNLS-GFP-BRD4-CTM-UBC-rtTeR-IRES-BSD | This study | MS2 |
| pTet-On-sNLS-GFP-BRD4-CsΔCTM-UBC-rtTeR-IRES-BSD | This study | MS3 |
| pTet-On-sNLS-GFP-BRD4-Cs-UBC-rtTeR-IRES-BSD | This study | MS4 |
| pTet-On-sNLS-GFP-BRD4-C-UBC-rtTeR-IRES-BSD | This study | MS6 |
| pTet-On-sNLS-GFP-BRD4-FL-ΔBD-UBC-rtTeR-IRES-BSD | This study | MS7 |
| pTet-On-sNLS-GFP-BRD4-FL-ΔCd-UBC-rtTeR-IRES-BSD | This study | pBZ196 |
| pTet-On-sNLS-GFP-BRD4-FL-ΔCp-UBC-rtTeR-IRES-BSD | This study | pBZ197 |
| Software and Algorithms | ||
| bowtie 2.2.6.0 | Langmead et al., 2009 | NA |
| TopHat 2.1.0 | Trapnell et al., 2009 | NA |
| DESeq2 | Love et al., 2014 | NA |
| deepTools 3.1.1 | Ramírez et al., 2016 | NA |
| featureCounts 2.0.1 | Liao et al., 2014 | NA |
| AlphaFold integrated in ChimeraX | Pettersen et al., 2021 | NA |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
DLD1 (CCL-221) and NCIH2009 (CRL-5911) human cells were purchased from ATCC. BRD4-IAA7 DLD1 degron cells were generated as described in the following section. All cell lines were cultured in DMEM (Corning, #10013CV) and PRMI 1640 (ThermoFisher, #A1049101) respectively, supplemented with 10% FBS (Sigma-Aldrich, #F2442), 1% Glutmax (Gibco, #35050061), and 1% PS (Gibco, #15140122), in 37°C incubator with 5% CO2.
METHOD DETAILS
Drug treatments:
Auxin (#ab146403) was purchased from abcam. Doxycycline (#72742) was obtained from Stem Cell Technologies. JQ1 (#4499) was purchased from Tocris. dBET6 (#S876202) was purchased from Selleckchem. NVP-2 (#HY-12214A) and dCDK9 (#HY-123937) were purchased from MedChemExpress.
Generation of degron cell lines:
BRD4-IAA7 DLD1 degron cells were generated similarly to previously described.12 Specifically, PX330 (Addgene, #42230) altered with the insertion of sgRNA (#1 AATCTTTTCTGAGCGCACCT, or #2 ATCAAAGTCAGAAGCCACCT) targeting the stop codon area of the BRD4 genomic locus was co-transfected with a donor plasmid using the Lipofectamine 3000 Transfection Reagent (Invitrogen, #L3000001) to trigger donor integration at the target site via homologous recombination repair. The donor plasmid, which we designed to integrate the IAA7 tag after the final BRD4 exon and introduce the F-box protein AtAFB2 under the EFIa promoter, was made using the pBlueScript II SK (+) backbone and synthesized gBlocks for the AA7-AtAFB2 pair and antibiotic selection marker. Upon transfection, DLD1 cells were selected for colony formation in the presence of either Geneticin (Gibco, #10131027) or hygromycin B (Invitrogen, #10687010) for 2 weeks. Single clone colonies were picked and verified by PCR and western blot analysis.
Generation and expression of BRD4 mutant constructs:
TetOn lentiviral vector (#110280), BRD4-FL cDNA (#31351), and BRD4-ΔET cDNA (#21938) were obtained from Addgene. The BRD4S mutant was generated from TetOn-FLAG-BRD4-FL using the Q5 Site-Directed Mutagenesis Kit (NEB, #E0554S). All other BRD4 mutant constructs were generated via NEBuilder HiFi DNA assembly reaction (NEB, #E2621S) with synthesized gBlocks (IDT and Twist Biosciences). TetOn-BRD4 lentiviral constructs were amplified by transformation of stable competent E. coli (NEB, #C3040H). All BRD4 plasmid insertions were verified by Sanger sequencing (ACGT). Lentivirus for transducing mammalian expression of the BRD4 mutants was generated by co-transfecting BRD4 mutant expressing plasmids with pspax2/pmd2.g lentiviral packaging plasmids in 293T cells in 6-well format. Lentivirus was collected and filtered for infection of BRD4-IAA7 DLD1 cells in 6-well plates. Infected cells were selected with Blasticidin (Gibco, #A1113903) for two weeks. BRD4 mutant expression was achieved by adding 50nM Dox into the medium and incubating for 48h.
Cell growth assay:
0.1 million FLAG-tagged mutants transfected and Blasticidin selected BRD4-IAA7 cells were seeded in 12-well plates on Day 0. Dox was added to induce the mutants’ expression. On Day 2, changed to fresh medium and added Dox to maintain the mutants’ expression while adding auxin to deplete the endogenous BRD4. On day 4, changed to fresh medium and maintained with Dox and auxin. On Day 2, 4 and 6, when collected, each duplicated plate was fixed with 4% paraformaldehyde solution (Fisher Scientific, #50–980-495) in PBS for 20 min at room temperature under shaking. Then plates were washed with tap water for 3 times and dried overnight. Violet solution (Millipore Sigma, #HT90132) was used to stain the fixed cell.
Western blot:
Whole cell lysates for western blot were prepared by directly lysing the cells with Laemmli sample buffer (Bio-Rad, #1610747) and boiling for 10 minutes. BRD4 (#13440), β-Actin (#3700), V5-Tag (#13202), HA-Tag (#3724S), CDK9 (#2316), and Cyclin T1 (#81464) antibodies were purchased from Cell Signaling Technology. Tubulin (#E7) antibody was obtained from Developmental Studies Hybridoma Bank (DSHB). FLAG antibody (#F1804) was obtained from Millipore Sigma. GFP Antibody (#sc-9996) was purchased from Santa Cruz Biotechnology. H3 and BRDT antibodies were generated in-house. Western blot quantification was carried out with the ImageLab from Bio-Rad.
GFP IP-MS:
To prepare the GFP IP-MS samples, cells were released with trypsin and washed with PBS before incubation in cold Buffer A (10mM HEPES pH 7.9, 10mM KCl, 1.5mM MgCl2, DTT) (Thermo Scientific, #A39255), Protease inhibitor (Thermo Scientific, #PIA32963), and Phosphatase inhibitor (Thermo Scientific, #PIA32957)) for nuclear isolation. Nuclear pellets were lysed by the addition of cold Triton X-100 lysis buffer (50mM Tris HCl, pH 8.0, 150mM NaCl, 1.5mM MgCl2, 10% Glycerol, 0.5% Triton X-100, DTT, Benzonase, Protease inhibitor, and Phosphatase inhibitor) followed by rotation for 45min and centrifugation at 20,000g for 15 minutes (all at 4°C.) The supernatant was incubated with ChromoTek GFP-Trap magnetic beads (Proteintech, #gtma) for > 4h at 4°C for immunoprecipitation, then beads were washed 5x with cold Triton X-100 lysis buffer and 1x with cold Triton X-100 wash buffer (50mM Tris HCl, pH 8.0, 150mM NaCl, 1.5mM MgCl2, 1.5mM EDTA, DTT). Glycine 2.5M pH 2.0 was used to elute the immunoprecipitated proteins and Tris buffer pH>10 was used to neutralize the eluate at RT. Neutralized eluate was snap frozen for MS or mixed with Laemmli sample buffer and boiled for 5 minutes for western blot. MS was carried out by the Proteomics Core Facility at the University of Arkansas for Medical Sciences.
RNA-seq:
Total RNAs from 6-well plate were extracted using the RNeasy mini kit from Qiagen (Qiagen, #74106). mRNAs were enriched using a NEBNext poly(A) mRNA magnetic isolation module (NEB, #E7490L). RNA libraries were prepared using NEBNext Ultra II Directional RNA library preparation kit (NEB, #E7760L) and sequenced on the Illumina NovaSeq 6000.
ChIP-seq:
DLD1 cells were crosslinked with 15ml 1% PFA (ThermoFisher, #28908) in PBS on 15cm plates for 10 minutes at RT and quenched by the addition of 2ml 2.5M glycine followed by shaking for 5 minutes. Crosslinked cells were collected by scraping. Chromatin sonication was performed for 10 minutes using a Covaris E200 set to 10% duty factor, 200 cycles per burst, and 140 peak intensity power. For endogenous protein ChIP, 10–20% of mouse embryonic fibroblasts (MEF) chromatin (prepared the same way as in DLD1 cells) was added to each sample as a spike-in control. For overexpressed protein ChIP, such as GFP-ChIP, 20ng spike-in chromatin (Active Motif, #53083) and 2ug spike-in antibody (Active Motif, #61686) were added to each sample. Immunoprecipitation was carried out at 4°C overnight using 5ul of Rpb1 (Cell Signaling Technology, #D8L4Y), 5ul of Ser2p (Active Motif, #61984), 10ul of CCNT1 (Cell Signaling Technology, #81464), 10ul of CDK9 (Santa Cruz Biotechnology, #sc-13130 X) antibodies, or 200ul of a cocktail consisting of 50ul of each GFP antibody (DSHB, #GFP-G1, #GFP-12A6, #GFP-12E6, #GFP-8H11). Immune complexes were enriched with Protein G-coupled Dynabeads (Invitrogen, #10004D) at 4°C for >= 4h, and incubated with proteinase K (Roche, #3115828001) to reverse crosslink at 65°C overnight. Eluted DNA was purified with the QIAquick PCR Purification Kit (Qiagen, #28106), and libraries were prepared using the KAPA HTP library preparation kit (Roche, #07961901001) for sequencing on the NovaSeq 6000.
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-seq data analysis:
Reads were aligned to the human genome (hg38) using Bowtie version 2.2.6.054 and a TopHat run (v2.1.0)55. RNA-Seq read counts were corrected for technical differences and normalized between samples with R package RUVseq56 using upper-quartile (UQ) approach. Differential gene expression analysis was performed with DESeq257 with significance thresholds of adj.p<0.05 and |log2FC|>0.585.
ChIP-seq analysis:
Reads were aligned to hg38 with bowtie 1/258. Genes (N=6,481) with pausing site and TES annotation were obtained from a previously published study59. Promoter regions were designated as spanning from 100bp upstream to 300bp downstream of the pausing site. Gene body regions were designated as spanning from 300bp downstream of the pausing site to the TES. FeatureCounts 2.0.160 was used to calculate the total mapped reads from Pol II ChIP-seq at promoters and within gene bodies. PRR is calculated as the ratio of Pol II signal density within the gene body to Pol II signal density at the promoter. BamCoverage in deeptools 3.1.161 was used to extend ChIP-seq reads to 150bp. Log2FC of ChIP-seq was calculated using bigwigCompare in deeptools with the following nondefault options: binSize 10, pseudocount 0.1. Heatmaps and metagene plots were also generated in deeptools. ECDFs and boxplots/scatterplots for the log2PRR and MS were generated using R. Tracks were visualized in igv 2.13.2 (Broad Institute).
Supplementary Material
Figure 7. The model for transcriptional regulation by a distinct layer of histone acetylation-unbound BRD4-PTEFb complex.
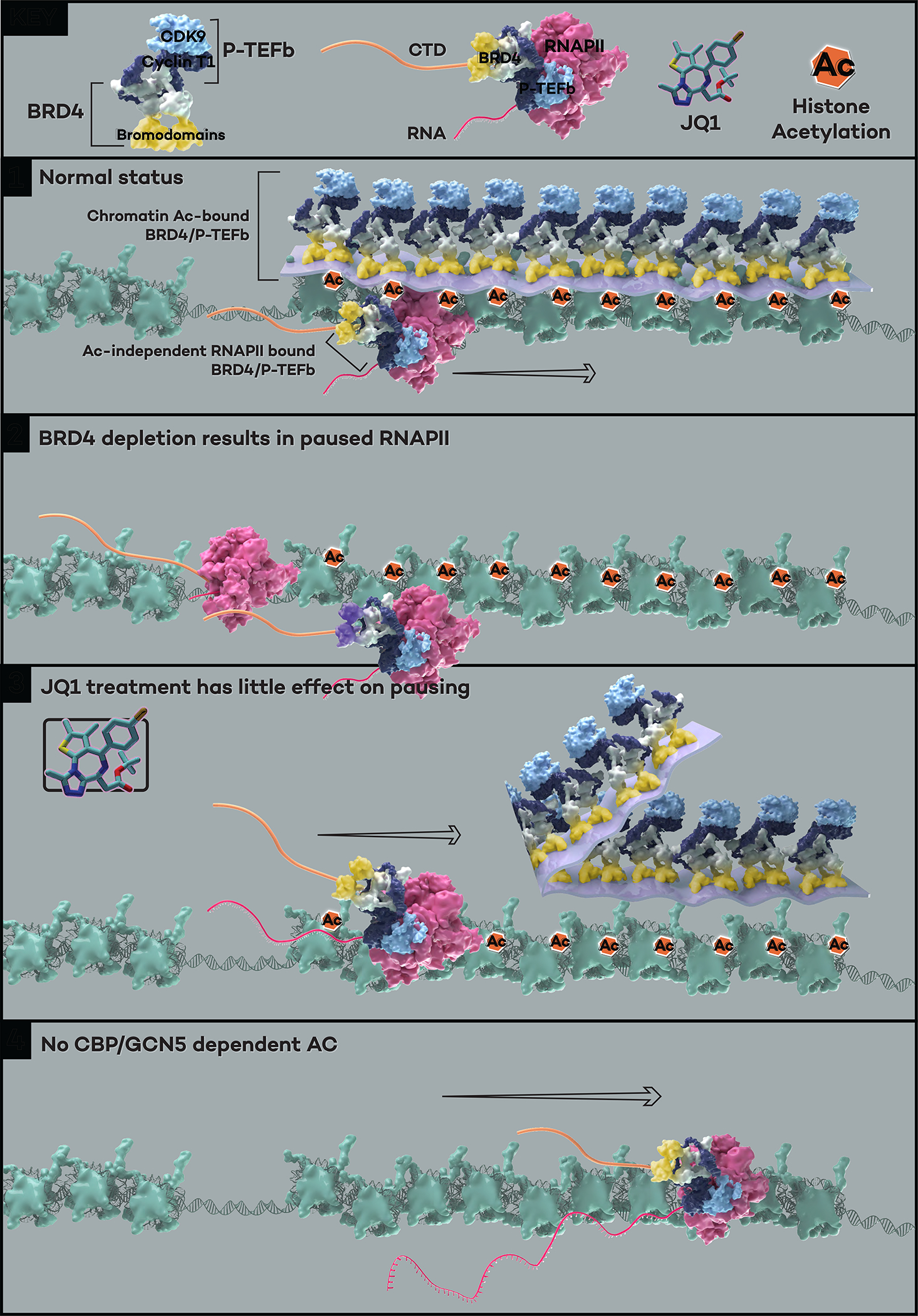
Under normal cellular conditions, the majority of BRD4 molecules are associated with chromatin through binding to acetylated histone tails, and the PTEFb complex is recruited by its interaction with the C terminal of BRD4 to form the acetylation-bound “layer” of BRD4-PTEFb. A small portion of the remaining BRD4-PTEFb complex interacts with the Pol II CTD regardless of histone acetylation, forming a critical layer of BRD4-PTEFb that phosphorylates the Ser2 and Ser5 positions of the CTD heptapeptide repeats (Panel 1). BRD4 depletion achieved either by auxin in the BRD4 degron cells or dBET6 treatment results in genome-wide Pol II pausing (Panel 2). The acetylation-bound BRD4-PTEFb layer is displaced from the chromatin when cells are treated with the bromodomain inhibitor JQ1, but the acetylation-independent layer of BRD4-PTEFb can still function to release Pol II without histone recognition-mediated chromatin association (Panel 3). In the absence of acetylation (e.g., H3K27ac, H3K9ac, and H3K18ac deposited by CBP/GCN528), the acetylation-independent, Pol II-bound BRD4-PTEFb complex retains the ability in releasing Pol II (Panel 4).
Highlights.
Unlike BETi (JQ1), BRD4 degradation causes profound Pol II pausing genome wide
The bromodomains of BRD4 are dispensable for Pol II pause release in living cells
A BET-less C-terminal BRD4 fragment interacts with PTEFb and releases paused Pol II
The C-terminal disordered region of BRD4 secures Cyclin T1 in the BRD4-PTEFb complex
Acknowledgement
We thank Brianna Monroe for illustrating the findings of this study in the Model. We want to thank Emily Rendleman, Sheetal Ganesan, Cassy Philips, and Jacob Zeidner for initial NGS sequencing and Yue He for her participation in the cloning for GFP-tagged ΔBD and C. We also want to thank Drs. Edwin Smith, Marc Morgan, Yuki Aoi and Karen Adelman for in-depth discussions and suggestions for the study. We are grateful for all the current and past Shilatifard lab members for their support. Studies in Shilatifard’s laboratory are supported by generous funding through the Outstanding Investigator Award mechanism by the National Cancer Institute grant R35-CA197569. LW was supported by NIH grant R35-GM146979.
Footnotes
Declaration of Interests
The authors declare no conflict of interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- 1.Core L, and Adelman K (2019). Promoter-proximal pausing of RNA polymerase II: A nexus of gene regulation. Genes Dev 33, 960–982. 10.1101/gad.325142.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen FX, Smith ER, and Shilatifard A (2018). Born to run: Control of transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol 19, 464–478. 10.1038/s41580-018-0010-5. [DOI] [PubMed] [Google Scholar]
- 3.Adelman K, and Lis JT (2012). Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet 13, 720–731. 10.1038/nrg3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Price DH (2000). P-TEFb, a Cyclin-Dependent Kinase Controlling Elongation by RNA Polymerase II. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peterlin BM, and Price DH (2006). Controlling the Elongation Phase of Transcription with P-TEFb. Mol Cell 23, 297–305. 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 6.Buratowski S (2009). Progression through the RNA Polymerase II CTD Cycle. Mol Cell 36, 541–546. 10.1016/j.molcel.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsin JP, and Manley JL (2012). The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev 26, 2119–2137. 10.1101/gad.200303.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu X, Zhu X, Li Y, Liu M, Yu B, Wang Y, Rao M, Yang H, Zhou K, Wang Y, et al. (2016). Multiple P-TEFbs cooperatively regulate the release of promoter-proximally paused RNA polymerase II. Nucleic Acids Res 44, 6853–6867. 10.1093/nar/gkw571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, Washburn MP, Conaway JW, Conaway RC, and Shilatifard A (2010). AFF4, a Component of the ELL/P-TEFb Elongation Complex and a Shared Subunit of MLL Chimeras, Can Link Transcription Elongation to Leukemia. Mol Cell 37, 429–437. 10.1016/j.molcel.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Q, Price JP, Byers SA, Cheng D, Peng J, and Price DH (2005). Analysis of the large inactive P-TEFb complex indicates that it contains one 7SK molecule, a dimer of HEXIM1 or HEXIM2, and two P-TEFb molecules containing Cdk9 phosphorylated at threonine 186. Journal of Biological Chemistry 280, 28819–28826. 10.1074/jbc.M502712200. [DOI] [PubMed] [Google Scholar]
- 11.Smith E, Lin C, and Shilatifard A (2011). The super elongation complex (SEC) and MLL in development and disease. Genes Dev 25, 661–672. 10.1101/gad.2015411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng B, Aoi Y, Shah AP, Iwanaszko M, Das S, Rendleman EJ, Zha D, Khan N, Smith ER, and Shilatifard A (2021). Acute perturbation strategies in interrogating RNA polymerase II elongation factor function in gene expression. Genes Dev 35, 273–285. 10.1101/GAD.346106.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu SY, and Chiang CM (2007). The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. Journal of Biological Chemistry 282, 13141–13145. 10.1074/jbc.R700001200. [DOI] [PubMed] [Google Scholar]
- 14.Gaucher J, Boussouar F, Montellier E, Curtet S, Buchou T, Bertrand S, Hery P, Jounier S, Depaux A, Vitte AL, et al. (2012). Bromodomain-dependent stage-specific male genome programming by Brdt. EMBO Journal 31, 3809–3820. 10.1038/emboj.2012.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang T, Cooper S, and Brockdorff N (2015). The interplay of histone modifications – writers that read. EMBO Rep 16, 1467–1481. 10.15252/embr.201540945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Atlasi Y, and Stunnenberg HG (2017). The interplay of epigenetic marks during stem cell differentiation and development. Nat Rev Genet 18, 643–658. 10.1038/nrg.2017.57. [DOI] [PubMed] [Google Scholar]
- 17.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proceedings of the National Academy of Sciences 107, 21931–21936. 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Donati B, Lorenzini E, and Ciarrocchi A (2018). BRD4 and Cancer: Going beyond transcriptional regulation. Mol Cancer 17. 10.1186/s12943-018-0915-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanno T, Kanno Y, Leroy G, Campos E, Sun HW, Brooks SR, Vahedi G, Heightman TD, Garcia BA, Reinberg D, et al. (2014). BRD4 assists elongation of both coding and enhancer RNAs by interacting with acetylated histones. Nat Struct Mol Biol 21, 1047–1057. 10.1038/nsmb.2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shorstova T, Foulkes WD, and Witcher M (2021). Achieving clinical success with BET inhibitors as anti-cancer agents. Br J Cancer 124, 1478–1490. 10.1038/s41416-021-01321-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Winter GE, Mayer A, Buckley DL, Erb MA, Roderick JE, Vittori S, Reyes JM, di Iulio J, Souza A, Ott CJ, et al. (2017). BET Bromodomain Proteins Function as Master Transcription Elongation Factors Independent of CDK9 Recruitment. Mol Cell 67, 5–18.e19. 10.1016/j.molcel.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L, and Sims RJ (2011). Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A 108, 16669–16674. 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, Hines J, Winkler JD, Crew AP, Coleman K, et al. (2015). Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem Biol 22, 755–763. 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, and Bradner JE (2015). Phthalimide conjugation as a strategy for in vivo target protein degradation. Science (1979) 348, 1376–1381. 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Z, Chivu AG, Choate LA, Rice EJ, Miller DC, Chu T, Chou SP, Kingsley NB, Petersen JL, Finno CJ, et al. (2022). Prediction of histone post-translational modification patterns based on nascent transcription data. Nat Genet 54, 295–305. 10.1038/s41588-022-01026-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sankar A, Mohammad F, Sundaramurthy AK, Wang H, Lerdrup M, Tatar T, and Helin K (2022). Histone editing elucidates the functional roles of H3K27 methylation and acetylation in mammals. Nat Genet 54, 754–760. 10.1038/s41588-022-01091-2. [DOI] [PubMed] [Google Scholar]
- 27.Zhang T, Zhang Z, Dong Q, Xiong J, and Zhu B (2020). Histone H3K27 acetylation is dispensable for enhancer activity in mouse embryonic stem cells. Genome Biol 21. 10.1186/s13059-020-01957-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ciabrelli F, Rabbani L, Cardamone F, Zenk F, Löser E, Schächtle MA, Mazina M, Loubiere V, and Iovino N (2023). CBP and Gcn5 drive zygotic genome activation independently of their catalytic activity. Sci Adv 9. 10.1126/sciadv.adf2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muhar M, Ebert A, Neumann T, Umkehrer C, Jude J, Wieshofer C, Rescheneder P, Lipp JJ, Herzog VA, Reichholf B, et al. SLAM-seq defines direct gene-regulatory functions of the BRD4-MYC axis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arnold M, Bressin A, Jasnovidova O, Meierhofer D, and Mayer A (2021). A BRD4-mediated elongation control point primes transcribing RNA polymerase II for 3′-processing and termination. Mol Cell 81, 3589–3603.e13. 10.1016/j.molcel.2021.06.026. [DOI] [PubMed] [Google Scholar]
- 31.Li S, Prasanna X, Salo VT, Vattulainen I, and Ikonen E (2019). An efficient auxin-inducible degron system with low basal degradation in human cells. Nat Methods 16, 866–869. 10.1038/s41592-019-0512-x. [DOI] [PubMed] [Google Scholar]
- 32.Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, and Young RA (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Itzen F, Greifenberg AK, Bösken CA, and Geyer M (2014). Brd4 activates P-TEFb for RNA polymerase II CTD phosphorylation. Nucleic Acids Res 42, 7577–7590. 10.1093/nar/gku449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moon KJ, Mochizuki K, Zhou M, Jeong HS, Brady JN, and Ozato K (2005). The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell 19, 523–534. 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 35.Phatnani HP, and Greenleaf AL (2006). Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev 20, 2922–2936. 10.1101/gad.1477006. [DOI] [PubMed] [Google Scholar]
- 36.Moon KJ, Mochizuki K, Zhou M, Jeong HS, Brady JN, and Ozato K (2005). The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell 19, 523–534. 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 37.Yang Z, He N, and Zhou Q (2008). Brd4 Recruits P-TEFb to Chromosomes at Late Mitosis To Promote G 1 Gene Expression and Cell Cycle Progression. Mol Cell Biol 28, 967–976. 10.1128/mcb.01020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tahirov TH, Babayeva ND, Varzavand K, Cooper JJ, Sedore SC, and Price DH (2010). Crystal structure of HIV-1 Tat complexed with human P-TEFb. Nature 465, 747–751. 10.1038/nature09131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, and Ferrin TE (2021). UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Science 30, 70–82. 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM, Janiszewska M, Huh SJ, Liang Y, Ryan J, et al. (2016). Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 529, 413–417. 10.1038/nature16508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fong CY, Gilan O, Lam EYN, Rubin AF, Ftouni S, Tyler D, Stanley K, Sinha D, Yeh P, Morison J, et al. (2015). BET inhibitor resistance emerges from leukaemia stem cells. Nature 525, 538–542. 10.1038/nature14888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rathert P, Roth M, Neumann T, Muerdter F, Roe JS, Muhar M, Deswal S, Cerny-Reiterer S, Peter B, Jude J, et al. (2015). Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 525, 543–547. 10.1038/nature14898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Calder J, Nagelberg A, Luu J, Lu D, and Lockwood WW (2021). Resistance to BET inhibitors in lung adenocarcinoma is mediated by casein kinase phosphorylation of BRD4. Oncogenesis 10. 10.1038/s41389-021-00316-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun Y, Zhang Z, Zhang K, Liu Y, Shen P, Cai M, Jia C, Wang W, Gu Z, Ma P, et al. (2021). Epigenetic heterogeneity promotes acquired resistance to BET bromodomain inhibition in ovarian cancer. [PMC free article] [PubMed] [Google Scholar]
- 45.Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM, Janiszewska M, Huh SJ, Liang Y, Ryan J, et al. (2016). Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 529, 413–417. 10.1038/nature16508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Andrieu G, Belkina AC, and Denis GV (2016). Clinical trials for BET inhibitors run ahead of the science. Drug Discov Today Technol 19, 45–50. 10.1016/j.ddtec.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Narita T, Ito S, Higashijima Y, Chu WK, Neumann K, Walter J, Satpathy S, Liebner T, Hamilton WB, Maskey E, et al. (2021). Enhancers are activated by p300/CBP activity-dependent PIC assembly, RNAPII recruitment, and pause release. Mol Cell 81, 2166–2182.e6. 10.1016/j.molcel.2021.03.008. [DOI] [PubMed] [Google Scholar]
- 48.Mahat DB, Salamanca HH, Duarte FM, Danko CG, and Lis JT (2016). Mammalian Heat Shock Response and Mechanisms Underlying Its Genome-wide Transcriptional Regulation. Mol Cell 62, 63–78. 10.1016/j.molcel.2016.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheung KL, Zhang F, Jaganathan A, Sharma R, Zhang Q, Konuma T, Shen T, Lee JY, Ren C, Chen CH, et al. (2017). Distinct Roles of Brd2 and Brd4 in Potentiating the Transcriptional Program for Th17 Cell Differentiation. Mol Cell 65, 1068–1080.e5. 10.1016/j.molcel.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang C, Xu Q, Zhang X, Day DS, Abraham BJ, Lun K, Chen L, Huang J, and Ji X (2022). BRD2 interconnects with BRD3 to facilitate Pol II transcription initiation and elongation to prime promoters for cell differentiation. Cellular and Molecular Life Sciences 79. 10.1007/s00018-022-04349-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hsu SC, Gilgenast TG, Bartman CR, Edwards CR, Stonestrom AJ, Huang P, Emerson DJ, Evans P, Werner MT, Keller CA, et al. (2017). The BET Protein BRD2 Cooperates with CTCF to Enforce Transcriptional and Architectural Boundaries. Mol Cell 66, 102–116.e7. 10.1016/j.molcel.2017.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Donczew R, and Hahn S (2021). BET family members Bdf1/2 modulate global transcription initiation and elongation in Saccharomyces cerevisiae. Elife 10. 10.7554/eLife.69619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hussong M, Kaehler C, Kerick M, Grimm C, Franz A, Timmermann B, Welzel F, Isensee J, Hucho T, Krobitsch S, et al. (2017). The bromodomain protein BRD4 regulates splicing during heat shock. Nucleic Acids Res 45, 382–394. 10.1093/nar/gkw729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Risso D, Ngai J, Speed TP, and Dudoit S (2014). Normalization of RNA-seq data using factor analysis of control genes or samples. Nat Biotechnol 32, 896–902. 10.1038/nbt.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aoi Y, Shah AP, Ganesan S, Soliman SHA, Cho BK, Goo YA, Kelleher NL, and Shilatifard A (2022). SPT6 functions in transcriptional pause/release via PAF1C recruitment. Mol Cell 82, 3412–3423.e5. 10.1016/j.molcel.2022.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liao Y, Smyth GK, and Shi W (2014). FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 61.Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–W165. 10.1093/NAR/GKW257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All genomic data generated in this study have been deposited to GEO under the accession number GSE232869 and are publicly available as of the date of publication. Accession number is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| BRD4 (for WB) | Cell Signaling Technology | Cat#13440 |
| β-Actin (for WB) | Cell Signaling Technology | Cat#3700 |
| V5-Tag (for WB) | Cell Signaling Technology | Cat#13202 |
| HA-Tag (for WB) | Cell Signaling Technology | Cat#3724S |
| CDK9 (for WB) | Cell Signaling Technology | Cat#2316 |
| Cyclin T1 (for WB and ChIP) | Cell Signaling Technology | Cat#81464 |
| Tubulin (for WB) | Developmental Studies Hybridoma Bank | Cat#E7 |
| FLAG (for WB) | Millipore Sigma | Cat#F1804 |
| GFP (for WB) | Santa Cruz Biotechnology | Cat#sc-9996 |
| H3 (for WB) | This study | NA |
| BRDT (for WB) | This study | NA |
| Rpb1 (for ChIP) | Cell Signaling Technology | Cat#D8L4Y |
| Ser2p (for ChIP) | Active Motif | Cat#61984 |
| CDK9 (for ChIP) | Santa Cruz Biotechnology | Cat#sc-13130 X |
| GFP (for ChIP) | Developmental Studies Hybridoma Bank | Cat#GFP-G1 |
| GFP (for ChIP) | Developmental Studies Hybridoma Bank | Cat#GFP-12A6 |
| GFP (for ChIP) | Developmental Studies Hybridoma Bank | Cat#GFP-12E6 |
| GFP (for ChIP) | Developmental Studies Hybridoma Bank | Cat#GFP-8H11 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Auxin (3-Indole-acetic acid sodium salt) | Abcam | Cat#ab146403 |
| JQ1 | Tocris | Cat#4499 |
| dBET6 | Selleckchem | Cat#S876202 |
| NVP-2 | MedChemExpress | Cat#HY-12214A |
| THAL-SNS-032 (dCDK9) | MedChemExpress | Cat#HY-123937 |
| Doxycycline | Stem Cell Technologies | Cat#72742 |
| Benzonase | Millipore Sigma | Cat#E1014 |
| Dynabeads Protein G | Invitrogen | Cat#10004D |
| ChromoTek GFP-Trap Magnetic Agarose | Proteintech | Cat#gtma |
| Deposited Data | ||
| Genomics data | This study | GSE232869 |
| Experimental Models: Cell Lines | ||
| DLD-1 | ATCC | Cat#CCL-221 |
| NCI-H2009 | ATCC | Cat#CRL-5911 |
| Mouse embryonic fibroblasts | STEMCELL Technologies | Cat#00325 |
| BRD4-IAA7 DLD-1 #G3/D4 | This study | NA |
| Recombinant DNA | ||
| Cas9 plasmid for BRD4-IAA7 (sgRNA: CACCGAATCTTTTCTGAGCGCACCT) | Zheng et al., 2021 | NA |
| Cas9 plasmid for BRD4-IAA7 (sgRNA: CACCGATCAAAGTCAGAAGCCACCT) | Zheng et al., 2021 | NA |
| BRD4-IAA7-Neo donor plasmid | This study | pBZ14 |
| BRD4-IAA7-Hygro donor plasmid | This study | pBZ15 |
| Lenti-TetON-Sox2 | Addgene | Cat#110280 |
| p6344 pcDNA4-TO-HA-Brd4FL | Addgene | Cat#31351 |
| pFlag-CMV2-Brd4 del ET | Addgene | Cat#21938 |
| pTet-On-sNLS-3xFlag-EGFP-UBC-rtTeR-IRES-BSD | This study | pBZ113 |
| pTet-On-sNLS-3xFlag-BRD4-FL-UBC-rtTeR-IRES-BSD | This study | pBZ111 |
| pTet-On-sNLS-3xFlag-BRD4-ΔBD-UBC-rtTeR-IRES-BSD | This study | pBZ160 |
| pTet-On-sNLS-3xFlag-BRD4-ΔET-UBC-rtTeR-IRES-BSD | This study | pBZ136 |
| pTet-On-sNLS-3xFlag-BRD4-ΔCTM-UBC-rtTeR-IRES-BSD | This study | pBZ158 |
| pTet-On-sNLS-3xFlag-BRD4S-UBC-rtTeR-IRES-BSD | This study | pBZ137 |
| pTet-On-sNLS-3xFlag-BRD4-ΔN-UBC-rtTeR-IRES-BSD | This study | pBZ176 |
| pTet-On-sNLS-3xFlag-BRD4-Cm-UBC-rtTeR-IRES-BSD | This study | C1 |
| pTet-On-sNLS-3xFlag-BRD4-C-UBC-rtTeR-IRES-BSD | This study | C2 |
| pTet-On-sNLS-3xFlag-BRD4-Cs-UBC-rtTeR-IRES-BSD | This study | C3 |
| pTet-On-sNLS-GFP-UBC-rtTeR-IRES-BSD (Vector) | This study | MS1 |
| pTet-On-sNLS-GFP-BRD4-FL-UBC-rtTeR-IRES-BSD | This study | MS0 |
| pTet-On-sNLS-GFP-BRD4-CTM-UBC-rtTeR-IRES-BSD | This study | MS2 |
| pTet-On-sNLS-GFP-BRD4-CsΔCTM-UBC-rtTeR-IRES-BSD | This study | MS3 |
| pTet-On-sNLS-GFP-BRD4-Cs-UBC-rtTeR-IRES-BSD | This study | MS4 |
| pTet-On-sNLS-GFP-BRD4-C-UBC-rtTeR-IRES-BSD | This study | MS6 |
| pTet-On-sNLS-GFP-BRD4-FL-ΔBD-UBC-rtTeR-IRES-BSD | This study | MS7 |
| pTet-On-sNLS-GFP-BRD4-FL-ΔCd-UBC-rtTeR-IRES-BSD | This study | pBZ196 |
| pTet-On-sNLS-GFP-BRD4-FL-ΔCp-UBC-rtTeR-IRES-BSD | This study | pBZ197 |
| Software and Algorithms | ||
| bowtie 2.2.6.0 | Langmead et al., 2009 | NA |
| TopHat 2.1.0 | Trapnell et al., 2009 | NA |
| DESeq2 | Love et al., 2014 | NA |
| deepTools 3.1.1 | Ramírez et al., 2016 | NA |
| featureCounts 2.0.1 | Liao et al., 2014 | NA |
| AlphaFold integrated in ChimeraX | Pettersen et al., 2021 | NA |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
DLD1 (CCL-221) and NCIH2009 (CRL-5911) human cells were purchased from ATCC. BRD4-IAA7 DLD1 degron cells were generated as described in the following section. All cell lines were cultured in DMEM (Corning, #10013CV) and PRMI 1640 (ThermoFisher, #A1049101) respectively, supplemented with 10% FBS (Sigma-Aldrich, #F2442), 1% Glutmax (Gibco, #35050061), and 1% PS (Gibco, #15140122), in 37°C incubator with 5% CO2.
METHOD DETAILS
Drug treatments:
Auxin (#ab146403) was purchased from abcam. Doxycycline (#72742) was obtained from Stem Cell Technologies. JQ1 (#4499) was purchased from Tocris. dBET6 (#S876202) was purchased from Selleckchem. NVP-2 (#HY-12214A) and dCDK9 (#HY-123937) were purchased from MedChemExpress.
Generation of degron cell lines:
BRD4-IAA7 DLD1 degron cells were generated similarly to previously described.12 Specifically, PX330 (Addgene, #42230) altered with the insertion of sgRNA (#1 AATCTTTTCTGAGCGCACCT, or #2 ATCAAAGTCAGAAGCCACCT) targeting the stop codon area of the BRD4 genomic locus was co-transfected with a donor plasmid using the Lipofectamine 3000 Transfection Reagent (Invitrogen, #L3000001) to trigger donor integration at the target site via homologous recombination repair. The donor plasmid, which we designed to integrate the IAA7 tag after the final BRD4 exon and introduce the F-box protein AtAFB2 under the EFIa promoter, was made using the pBlueScript II SK (+) backbone and synthesized gBlocks for the AA7-AtAFB2 pair and antibiotic selection marker. Upon transfection, DLD1 cells were selected for colony formation in the presence of either Geneticin (Gibco, #10131027) or hygromycin B (Invitrogen, #10687010) for 2 weeks. Single clone colonies were picked and verified by PCR and western blot analysis.
Generation and expression of BRD4 mutant constructs:
TetOn lentiviral vector (#110280), BRD4-FL cDNA (#31351), and BRD4-ΔET cDNA (#21938) were obtained from Addgene. The BRD4S mutant was generated from TetOn-FLAG-BRD4-FL using the Q5 Site-Directed Mutagenesis Kit (NEB, #E0554S). All other BRD4 mutant constructs were generated via NEBuilder HiFi DNA assembly reaction (NEB, #E2621S) with synthesized gBlocks (IDT and Twist Biosciences). TetOn-BRD4 lentiviral constructs were amplified by transformation of stable competent E. coli (NEB, #C3040H). All BRD4 plasmid insertions were verified by Sanger sequencing (ACGT). Lentivirus for transducing mammalian expression of the BRD4 mutants was generated by co-transfecting BRD4 mutant expressing plasmids with pspax2/pmd2.g lentiviral packaging plasmids in 293T cells in 6-well format. Lentivirus was collected and filtered for infection of BRD4-IAA7 DLD1 cells in 6-well plates. Infected cells were selected with Blasticidin (Gibco, #A1113903) for two weeks. BRD4 mutant expression was achieved by adding 50nM Dox into the medium and incubating for 48h.
Cell growth assay:
0.1 million FLAG-tagged mutants transfected and Blasticidin selected BRD4-IAA7 cells were seeded in 12-well plates on Day 0. Dox was added to induce the mutants’ expression. On Day 2, changed to fresh medium and added Dox to maintain the mutants’ expression while adding auxin to deplete the endogenous BRD4. On day 4, changed to fresh medium and maintained with Dox and auxin. On Day 2, 4 and 6, when collected, each duplicated plate was fixed with 4% paraformaldehyde solution (Fisher Scientific, #50–980-495) in PBS for 20 min at room temperature under shaking. Then plates were washed with tap water for 3 times and dried overnight. Violet solution (Millipore Sigma, #HT90132) was used to stain the fixed cell.
Western blot:
Whole cell lysates for western blot were prepared by directly lysing the cells with Laemmli sample buffer (Bio-Rad, #1610747) and boiling for 10 minutes. BRD4 (#13440), β-Actin (#3700), V5-Tag (#13202), HA-Tag (#3724S), CDK9 (#2316), and Cyclin T1 (#81464) antibodies were purchased from Cell Signaling Technology. Tubulin (#E7) antibody was obtained from Developmental Studies Hybridoma Bank (DSHB). FLAG antibody (#F1804) was obtained from Millipore Sigma. GFP Antibody (#sc-9996) was purchased from Santa Cruz Biotechnology. H3 and BRDT antibodies were generated in-house. Western blot quantification was carried out with the ImageLab from Bio-Rad.
GFP IP-MS:
To prepare the GFP IP-MS samples, cells were released with trypsin and washed with PBS before incubation in cold Buffer A (10mM HEPES pH 7.9, 10mM KCl, 1.5mM MgCl2, DTT) (Thermo Scientific, #A39255), Protease inhibitor (Thermo Scientific, #PIA32963), and Phosphatase inhibitor (Thermo Scientific, #PIA32957)) for nuclear isolation. Nuclear pellets were lysed by the addition of cold Triton X-100 lysis buffer (50mM Tris HCl, pH 8.0, 150mM NaCl, 1.5mM MgCl2, 10% Glycerol, 0.5% Triton X-100, DTT, Benzonase, Protease inhibitor, and Phosphatase inhibitor) followed by rotation for 45min and centrifugation at 20,000g for 15 minutes (all at 4°C.) The supernatant was incubated with ChromoTek GFP-Trap magnetic beads (Proteintech, #gtma) for > 4h at 4°C for immunoprecipitation, then beads were washed 5x with cold Triton X-100 lysis buffer and 1x with cold Triton X-100 wash buffer (50mM Tris HCl, pH 8.0, 150mM NaCl, 1.5mM MgCl2, 1.5mM EDTA, DTT). Glycine 2.5M pH 2.0 was used to elute the immunoprecipitated proteins and Tris buffer pH>10 was used to neutralize the eluate at RT. Neutralized eluate was snap frozen for MS or mixed with Laemmli sample buffer and boiled for 5 minutes for western blot. MS was carried out by the Proteomics Core Facility at the University of Arkansas for Medical Sciences.
RNA-seq:
Total RNAs from 6-well plate were extracted using the RNeasy mini kit from Qiagen (Qiagen, #74106). mRNAs were enriched using a NEBNext poly(A) mRNA magnetic isolation module (NEB, #E7490L). RNA libraries were prepared using NEBNext Ultra II Directional RNA library preparation kit (NEB, #E7760L) and sequenced on the Illumina NovaSeq 6000.
ChIP-seq:
DLD1 cells were crosslinked with 15ml 1% PFA (ThermoFisher, #28908) in PBS on 15cm plates for 10 minutes at RT and quenched by the addition of 2ml 2.5M glycine followed by shaking for 5 minutes. Crosslinked cells were collected by scraping. Chromatin sonication was performed for 10 minutes using a Covaris E200 set to 10% duty factor, 200 cycles per burst, and 140 peak intensity power. For endogenous protein ChIP, 10–20% of mouse embryonic fibroblasts (MEF) chromatin (prepared the same way as in DLD1 cells) was added to each sample as a spike-in control. For overexpressed protein ChIP, such as GFP-ChIP, 20ng spike-in chromatin (Active Motif, #53083) and 2ug spike-in antibody (Active Motif, #61686) were added to each sample. Immunoprecipitation was carried out at 4°C overnight using 5ul of Rpb1 (Cell Signaling Technology, #D8L4Y), 5ul of Ser2p (Active Motif, #61984), 10ul of CCNT1 (Cell Signaling Technology, #81464), 10ul of CDK9 (Santa Cruz Biotechnology, #sc-13130 X) antibodies, or 200ul of a cocktail consisting of 50ul of each GFP antibody (DSHB, #GFP-G1, #GFP-12A6, #GFP-12E6, #GFP-8H11). Immune complexes were enriched with Protein G-coupled Dynabeads (Invitrogen, #10004D) at 4°C for >= 4h, and incubated with proteinase K (Roche, #3115828001) to reverse crosslink at 65°C overnight. Eluted DNA was purified with the QIAquick PCR Purification Kit (Qiagen, #28106), and libraries were prepared using the KAPA HTP library preparation kit (Roche, #07961901001) for sequencing on the NovaSeq 6000.
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-seq data analysis:
Reads were aligned to the human genome (hg38) using Bowtie version 2.2.6.054 and a TopHat run (v2.1.0)55. RNA-Seq read counts were corrected for technical differences and normalized between samples with R package RUVseq56 using upper-quartile (UQ) approach. Differential gene expression analysis was performed with DESeq257 with significance thresholds of adj.p<0.05 and |log2FC|>0.585.
ChIP-seq analysis:
Reads were aligned to hg38 with bowtie 1/258. Genes (N=6,481) with pausing site and TES annotation were obtained from a previously published study59. Promoter regions were designated as spanning from 100bp upstream to 300bp downstream of the pausing site. Gene body regions were designated as spanning from 300bp downstream of the pausing site to the TES. FeatureCounts 2.0.160 was used to calculate the total mapped reads from Pol II ChIP-seq at promoters and within gene bodies. PRR is calculated as the ratio of Pol II signal density within the gene body to Pol II signal density at the promoter. BamCoverage in deeptools 3.1.161 was used to extend ChIP-seq reads to 150bp. Log2FC of ChIP-seq was calculated using bigwigCompare in deeptools with the following nondefault options: binSize 10, pseudocount 0.1. Heatmaps and metagene plots were also generated in deeptools. ECDFs and boxplots/scatterplots for the log2PRR and MS were generated using R. Tracks were visualized in igv 2.13.2 (Broad Institute).