

Abstract
Background:
Staphylococcus aureus is a common gram-positive bacterium that is the causative agent for several human diseases, including sepsis. A key virulence mechanism is pathogen binding to host fibrinogen through the C-terminal region of the γ-chain. Previous work demonstrated that FggΔ5 mice expressing mutant fibrinogen γΔ5 lacking a S. aureus binding motif had significantly improved survival following S. aureus septicemia. Fibrinogen γ′ is a human splice variant that represents about 10% to 15% of the total fibrinogen in plasma and circulates as a fibrinogen γ′-γ heterodimer (phFibγ′-γ). The fibrinogen γ′-chain is also expected to lack S. aureus binding function.
Objective:
Determine if human fibrinogen γ′-γ confers host protection during S. aureus septicemia.
Methods:
Analyses of survival and the host response following S. aureus septicemia challenge in FggΔ5 mice and mice reconstituted with purified phFibγ′-γ or phFibγ-γ.
Results:
Reconstitution of fibrinogen-deficient or wildtype mice with purified phFibγ′-γ prior to infection provided a significant prolongation in host survival relative to mice reconstituted with purified phFibγ-γ, which was superior to that observed with heterozygous FggΔ5 mice. Improved survival could not be accounted for by quantitative differences in fibrinogen-dependent adhesion or clumping, but phFibγ′-γ-containing mixtures generated notably smaller bacterial aggregates. Importantly, administration of phFibγ′-γ after infection also provided a therapeutic benefit by prolonging host survival relative to administration of phFibγ-γ.
Conclusion:
These findings provide the proof-of-concept that changing the ratio of naturally occurring fibrinogen variants in blood could offer significant therapeutic potential against bacterial infection and potentially other diseases.
Keywords: bacteremia, fibrinogen, S. aureus, sepsis
1 |. INTRODUCTION
Staphylococcus aureus is a gram-positive pathogen and causative agent for illnesses ranging from minor skin infections to serious and potentially life-threatening conditions [1–3]. These infections are particularly problematic in hospital settings where individuals are often immunocompromised. Indeed, prominent vehicles for infection by S. aureus are foreign bodies such as catheters, surgical implants, and sutures [4,5]. The transition from a local to a systemic infection occurs once S. aureus enter the blood (ie, bacteremia), which can result in sepsis, a major life-threatening disease [6]. The emergence of antibiotic-resistant strains of S. aureus (eg, methicillin- and vancomycin-resistant S. aureus) are particularly problematic for treatment and has driven the need for novel strategies distinct from classic antibiotic approaches.
Fibrinogen is a major soluble plasma glycoprotein and a dimeric molecule, consisting of 2 pairs of 3 polypeptide chains designated Aα, Bβ, and γ that are connected by disulfide bridges. Beyond playing a key role in clot formation and controlling hemorrhage, fibrinogen can serve as an early line of host defense by limiting pathogen growth and mediating antimicrobial mechanisms against pathogens. However, S. aureus has evolved to counteract fibrinogen-mediated antimicrobial function by producing several virulence factors that engage and activate host clotting proteins [7–11]. Fibrinogen-binding proteins are a particularly prominent class of S. aureus virulence factors [12–14]. S. aureus fibrinogen binding proteins engage multiple domains on fibrinogen, but the C-terminal portion of the fibrinogen γ-chain is a core binding motif for several important S. aureus virulence factors. For example, clumping factor A (ClfA) is a cell wall anchored protein that binds the C-terminal portion of the γ-chains and promotes fibrinogen-mediated bacterial clumping in suspension and bacterial adhesion to fibrinogen-coated surfaces [10,13,15]. ClfA further promotes pathogen virulence in sepsis by inhibiting complement activation and neutrophil and macrophage phagocytosis [10,16,17]. Previous work demonstrated that FggΔ5 mice carrying a mutant form of fibrinogen lacking the final 5 amino acids of the fibrinogen γ-chain, residues essential for ClfA binding, had significantly prolonged survival compared to wildtype (WT) mice following an S. aureus bacteremia challenge and that purified fibrinogen γΔ5 failed to support S. aureus clumping and adhesion [18].
Circulating fibrinogen is a heterogeneous mixture of several variants that occur in the blood of all healthy individuals and are the result of an alteration in either the Aα chains or γ chains. The major form of human fibrinogen in circulation consists of Aα chains that are 610 amino acids in length, Bβ chains of 461 amino acids and γ chains of 411 amino acids. Fibrinogen γ′ is a naturally occurring fibrinogen variant that circulates at concentrations ranging from 8% to 15% of total fibrinogen in plasma [19]. It is the product of alternative splicing of FGG mRNA and results in the substitution of the final 4 amino acids of the fibrinogen γ-chain with a 20 amino acid sequence [20,21]. Fibrinogen γ′ shares some structural and functional similarities with fibrinogen γΔ5, namely the loss of key residues required for ClfA binding [10,15]. Here, we investigated the hypothesis that fibrinogen γ′ would confer host protection in a manner similar to that observed with FggΔ5 mice following a S. aureus bloodstream infection.
2 |. MATERIAL AND METHODS
2.1 |. Bacteria and growth conditions
WT and ClfA- USA300 as well as WT green fluorescent protein (GFP)-expressing Newman S. aureus were kindly provided by Magnus Höök [22]. Stationary phase bacteria were grown in tryptic soy broth (Difco Laboratories) at 37°C overnight, washed, and resuspended in phosphate-buffered solution (PBS) and diluted to an optical density (OD) at 600 nm of 0.4, 1.0, or 6.0, based on the assay. The precise number of bacteria for each assay were determined by a colony forming units (CFUs) assay.
2.2 |. Fibrinogen purification and recombinant fibrinogen production
Mouse fibrinogen was purified from citrate plasma isolated from naïve WT, heterozygous, and homozygous FggΔ5 mice by ammonium sulfate or glycine precipitation, as previously described [23]. In each case, the final fibrinogen pellet was resuspended in HEPES-buffered saline (20 mM HEPES pH 7.4, 150 mM NaCl, and 5 mM ε-amino-n-caproic acid). Notably, fibrinogen purified using each of the methods performed identically in the in vitro bacterial adhesion and clumping assays. Human plasma fibrinogen variants (phFibγ-γ and phFibγ′-γ) were purified from commercially available plasma fibrinogen (FIB3, Enzyme Research Laboratories) by anion exchange chromatography using a trimethylaminoethyl resin. The resulting subfractions were concentrated and diafiltered to formulation buffer (either PBS or 5 mM citrate, 50 mM L-arginine with 100 mM NaCl at pH 7.3) by tangential flow filtration using a 100 kDa cut-off filter and sterile filtered. Purity was estimated at >99% by Sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and western blot analysis using a sheep anti-human fibrinogen γ′ antibody (Santa Cruz).
Recombinant human fibrinogen variants (rec-hFibγ-γ and rechFibγ′-γ′) were produced by Chinese hamster ovary cells in stirred tank reactors, using fed batch cell culture and serum-free cell culture media. Following a 10-day production time, cell culture harvest was clarified by depth filtration, and the recombinant fibrinogen protein was purified using a custom-made affinity resin (Life Technologies Corporation) based on the Gly-Pro-Arg-Pro peptide, additional chromatography and filtration steps (details not disclosed for proprietary reasons). The final formulation was performed identical to the formulation of the plasma-derived fibrinogen variants as described above. Purity was >99% as assessed by SDS-PAGE analysis and host cell protein enzyme-linked immunosorbent assay (ELISA) (CHO HCP ELISA Kit, 3G, F550–1 Cygnus).
2.3 |. Fibrinogen–S. aureus adhesion
Bacterial adhesion was analyzed in 96-well NUNC plates (Thermo Fisher) coated with dilution buffer (15 mM Na2HCO3, 35 mM NaHCO3, 3.2 mM NaN3) and incubated overnight at 4°C. Plates were then washed 3 times with wash buffer (150 mM NaCl and 0.01% Tween-20), blocked with 1% BSA, 0.05% Tween-20 solution in PBS for 1 hour at 37°C, and subsequently washed 3 times with wash buffer. S. aureus was added and incubated for 2 hours at 37°C. Plates were washed 3 times, fixed for 30 minutes with 25% formaldehyde solution, washed, and stained with 0.1% crystal violet. Bound crystal violet was solubilized in 10% acetic acid and quantified in a plate reader at 570 nm.
2.4 |. Fibrinogen–S. aureus clumping
Bacterial clumping in solution was analyzed with purified mouse or human fibrinogen diluted in PBS at concentrations ranging from 0.25 to 25 μg/mL. A suspension of stationary phase cultures of WT or ClfA-S. aureus USA300 with an OD at 600 nm of 6.0 were added to 96-well tissue culture plates containing fibrinogen solutions. The plates were agitated using an orbital shaker for 5 minutes followed by measurement at 570 nm to quantify the level of bacterial clumping. For imaging studies, stationary phase WT USA300 or Newman GFP-expressing S. aureus were similarly prepared and analyzed in 48-well plates. After the 5 minute clumping reaction, images were captured by either standard brightfield or fluorescent microscopy.
2.5 |. Bacteremia infection model and treatment of mice with purified fibrinogen
Animal studies were approved by the Institutional Animal Care and Use Committees of Cincinnati Children’s Hospital Medical Center or the University of North Carolina at Chapel Hill. Age matched (>8 weeks) male and female mice on a C57Bl/6J background were used. Human fibrinogen levels in mice were determined following retroorbital (RO) injection of 2 mg of purified human fibrinogen into C57Bl/6 mice. Blood was collected from the inferior vena cava into citrate at defined time points. Platelet poor plasma was analyzed by ELISA using a monoclonal anti-human fibrinogen (Y-18) described previously [24] as capture antibody and a goat anti-human fibrinogen conjugated to peroxidase as detecting antibody. Fibrinogen-deficient (Fga−/−) and FggΔ5 mice have been previously described [25,26]. Mice were injected with S. aureus USA300 and 6 mg of purified fibrinogen via RO injection in the orbital plexus opposite that for which S. aureus was delivered. In studies of prophylactic fibrinogen treatment, Fga−/− mice were injected 24 hours prior to S. aureus challenge to ensure these animals tolerated the bolus of fibrinogen. WT mice were injected 30 minutes prior to S. aureus challenge. For analyses of bacterial burden and host responses, Fga−/− mice were similarly treated with 6 mg of fibrinogen or vehicle control 24 hours prior to RO infection with S. aureus USA300, and mice were euthanized 8 hours after infection for collection of plasma and organs. Complete blood count analyses on whole blood were performed with an Element HT5 (Heska). Bacterial burden was determined for whole blood or tissue homogenates by serial dilution and CFU analysis. Cardiac troponin I (cTnI) levels in plasma were determined by ELISA using a high-density mouse cTnI kit (Life Diagnostics, Inc). Plasma alanine aminotransferase levels were determined using an enzyme assay kit (Labs Biotechnology). In studies where fibrinogen was given as a therapeutic treatment, mice were injected 30 minutes after S. aureus challenge. In all survival studies, moribundity defined as a state of nonrecovery was used as a humane endpoint.
2.6 |. Statistical analyses
All analyses were performed using Prism 9. Comparisons of multiple groups were performed using analysis of variance (anova) and Tukey’s multiple comparison test. Analyses of survival were performed using the Kaplan-Meier method. Results were considered significant when p < .05.
3 |. RESULTS
3.1 |. Host protection from S. aureus septicemia in mice expressing fibrinogen γΔ5
In previous studies, WT mice were shown to rapidly succumb to an intravenous S. aureus infection whereas homozygous fibrinogen γΔ5 (ie, FggΔ5/Δ5) mice displayed markedly improved host survival. The molecular basis for this observation was linked to elimination of the C-terminal fibrinogen γ ‘AGDV’ binding motif on fibrinogen that was shown to be required for binding to the S. aureus virulence factor ClfA [18]. Here, we sought to determine whether a protective benefit would be conferred if only a fraction of the circulating fibrinogen was in the form of fibrinogen γΔ5. FggWT/WT, FggWT/Δ5, and FggΔ5/Δ5 mice were administered an intravenous infection with S. aureus USA300 and monitored. At an infection dose of 2×108 CFUs, both FggWT/Δ5 and FggΔ5/Δ5 mice displayed similar survival phenotypes, showing a significant advantage over infected FggWT/WT mice (Figure 1A). At a higher infection dose of 6×108 CFUs, FggWT/Δ5 and FggΔ5/Δ5 mice still displayed a significant survival advantage over FggWT/WT mice, although all the FggWT/Δ5 mice eventually succumbed to the infection (Figure 1B). At the highest infection dose analyzed of 7×108 CFUs, all mice succumbed to the infection with no genotype-dependent difference in survival times (Figure 1C). These findings indicate that there can be a significant benefit to the host based on infection dose with S. aureus sepsis, even if only a portion of the circulating fibrinogen lacks the C-terminal AGDV motif of the γ-chain.
FIGURE 1. Partial elimination of the ClfA-binding motif results in protection from S. aureus septicemia associated with a reduction in fibrinogen-mediated clumping.
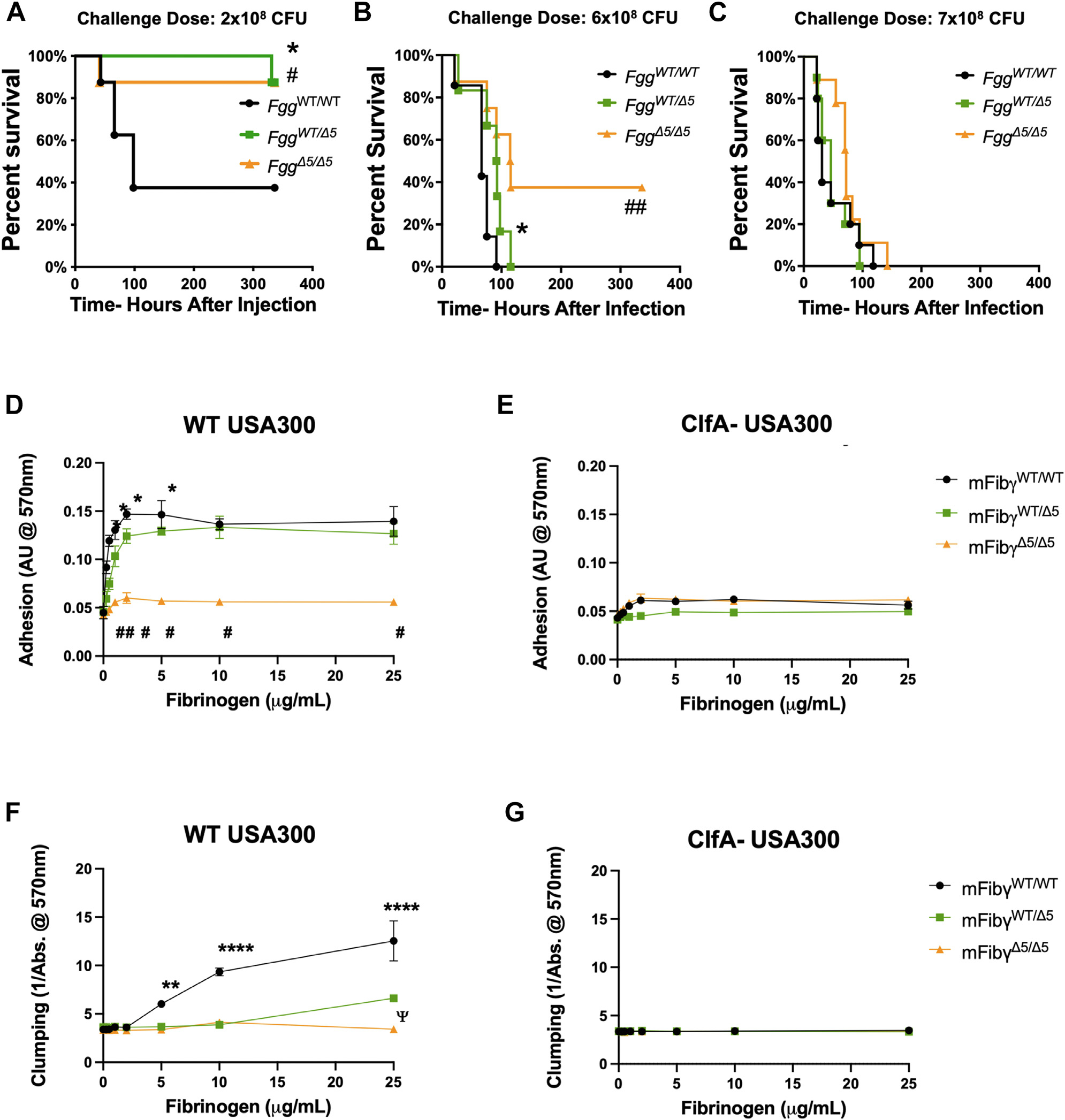
(A) FggWT/WT, FggWT/Δ5 and FggΔ5/Δ5 mice (n=10 per group) were infected via retroorbital injection with 2×108 S. aureus USA300, and host survival was monitored over time. Data were analyzed by Kaplan-Meier log-rank analysis with *p < .05 for FggWT/WT vs FggWT/Δ5 and #p < .05 for FggWT/WT vs FggΔ5/Δ5 mice. (B) FggWT/WT, FggWT/Δ5, and FggΔ5/Δ5 mice (n=10 per group) were infected with 6×108 CFUs of S. aureus USA300, and host survival was monitored over time. Data were analyzed by Kaplan-Meier log-rank analysis with *p < .05 for FggWT/WT vs FggWT/Δ5 and #p < .05 for FggWT/WT vs FggΔ5/Δ5 mice. (B) FggWT/WT, FggWT/Δ5 , and FggΔ5/Δ5 mice (n=10 per group) were infected with 6×108 CFUs of S. aureus USA300, and host survival was monitored over time. Data were analyzed by Kaplan-Meier log-rank analysis with *p < .05 for FggWT/WT vs FggWT/Δ5 and ##p < .01 for FggWT/WT vs FggΔ5/Δ5. (C) FggWT/WT, FggWT/Δ5, and FggΔ5/Δ5 mice (n=10 per group) were infected with 7×108 CFUs of S. aureus USA300, and host survival was monitored over time. Data were analyzed by Kaplan-Meier log-rank analysis. Adhesion of (D) WT USA300 or (E) ClfA- USA300 bacteria to immobilized mFibγWT/WT, mFibγWT/Δ5, and mFibγΔ5/Δ5. Data are presented as the mean ± SEM and analyzed by 2-way analysis of variance with Tukey’s multiple comparisons test. #p < .0001 for mFibγΔ5/Δ5 vs mFibγWT/WT and mFibγWT/Δ5; *p < .05 for mFibγWT/WT vs mFibγWT/Δ5. Clumping of (F) WT USA300 or (G) ClfA- USA300 mediated by mFibγWT/WT, mFibγWT/Δ5, and mFibγΔ5/Δ5. Data are presented as the mean ± SEM and analyzed by 2-way analysis of variance with Tukey’s multiple comparisons test. **p < .01 and ****p < .0001 for mFibγWT/WT vs mFibγWT/Δ5 and mFibγΔ5/Δ5. Ψp < .05 for mFibγWT/Δ5 vs mFibγΔ5/Δ5.
We next evaluated whether the protection in FggWT/Δ5 and Fgg Δ5/Δ5 mice was linked to altered interactions between fibrinogen and the bacteria. Adhesion of WT S. aureus USA300 to immobilized fibrinogen was analyzed. A modest but statistically significant reduction was observed in S. aureus adhesion to fibrinogen from FggWT/Δ5 (mFibγWT/Δ5) relative to that from FggWT/WT (mFibγWT/WT) mice at low plating concentrations of fibrinogen (ie, 0.25 to 5 μg/mL), but no differences were observed between mFibγWT/WT and mFibγWT/Δ5 at higher plating concentrations (ie, 10 to 25 μg/mL) once binding became saturable (Figure 1D). Little to no adhesion was observed for WT USA300 to fibrinogen from FggΔ5/Δ5 (mFibγΔ5/Δ5) (Figure 1D), as shown previously [18,23]. In addition, ClfA- USA300 did not bind to fibrinogen from any of the mouse strains (Figure 1E), similar to previous results [18,23]. Clumping analyses of USA300 in fibrinogen solutions were also analyzed. Here, mFibγWT/WT supported a dose-dependent increase in clump formation of WT USA300 with no clumping observed in solutions of mFibγΔ5/Δ5 (Figure 1F), similar to previous findings [23]. Notably, clumping was significantly reduced in solutions of mFibγWT/Δ5 relative to mFibγWT/WT with only the highest concentration of fibrinogen analyzed (ie, 25 μg/mL) displaying a signal above background (ie, that detected for mFibγΔ5/Δ5), but clumping even at this concentration of fibrinogen was significantly less than that observed for mFibγWT/WT (Figure 1F). ClfA- USA300 did not display clumping with fibrinogen purified from any of the mouse genotypes analyzed (Figure 1G).
3.2 |. Fibrinogen γ′-γ improves survival following septicemia challenge in mice
Based on the important finding that a 50% reduction in S. aureus fibrinogen γ chain binding motifs provided a significant survival benefit at some infection dosages with S. aureus, we hypothesized that human fibrinogen γ′-γ would offer the same host protective effects as those observed in FggΔ5 mice. To test this hypothesis, we separated total human plasma fibrinogen into purified fibrinogen γ-γ (phFibγ-γ) and fibrinogen γ′-γ (phFibγ′-γ) fractions (Figure 2A). Next, we determined how long phFibγ-γ and phFibγ′-γ would persist in mouse circulation. Each human fibrinogen variant was readily detected in isolated mouse plasma following injection, and both variants displayed a similar half-life of ~15 hours (Figure 2B). Next, a prophylactic approach in which all fibrinogen would be human was tested. Fga−/− mice were administered 6 mg of fibrinogen prior to infection and survival was monitored (Figure 2C). Fga−/− mice receiving only vehicle (ie, untreated) or phFibγ-γ prior to challenge with an aggressive 1×109 CFUs of S. aureus USA300 displayed rapid mortality with ~75% of the mice eliminated in less than 24 hours (Figure 2D), and all these animals succumbed to the infection by ~50 hours. In contrast, Fga−/− mice administered phFibγ′-γ displayed a significant survival advantage compared to each of the other groups (Figure 2D). In a second study, WT mice were prophylactically treated with fibrinogen and challenged with dose of 5×108 CFUs of USA300. Even in the presence of normal levels of WT fibrinogen, a similar result was observed in that phFibγ′-γ-treated mice displayed significantly prolonged survival compared with untreated or phFibγ-γ-treated mice (Figure 2E). Finally, an analysis to directly compare FggWT/Δ5 and the human fibrinogen variants was performed. Here, Fga−/− mice administered phFibγ′-γ displayed a significant survival advantage over the 48-hour observation window relative to both FggWT/Δ5 mice and Fga−/− mice carrying phFibγ-γ (Figure 2F). Collectively, these findings suggest that exogenous human fibrinogen γ′ heterodimer can provide significant host protection from S. aureus septicemia, even in the context of circulating WT fibrinogen.
FIGURE 2. Prophylactic treatment of WT or fibrinogen-deficient mice with fibrinogen γ′ improves survival following septicemia challenge.
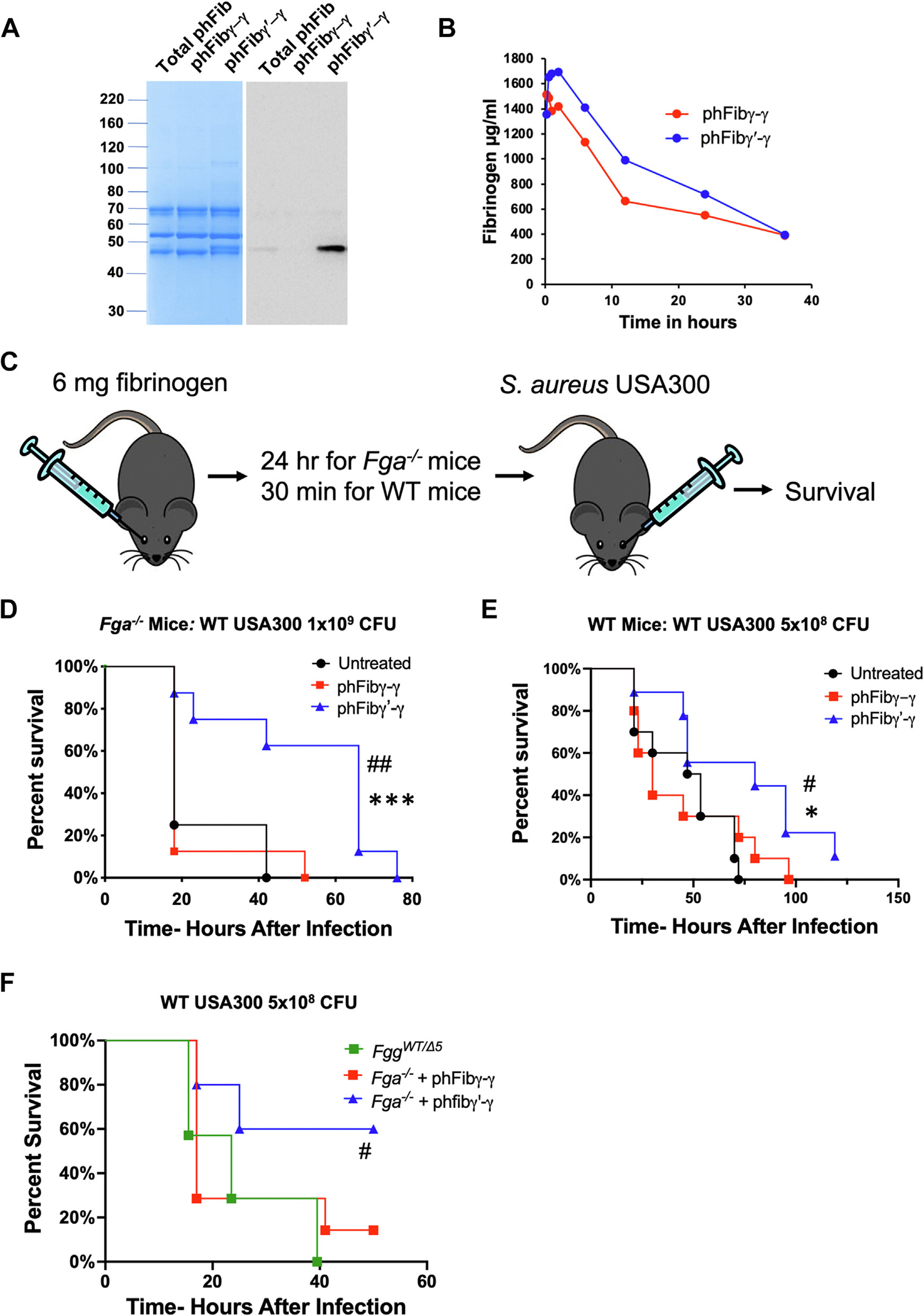
(A) Purified total phFib, phFibγ-γ, and phFibγ′-γ were analyzed by SDS-PAGE and Coomassie staining (left) and western blot analysis for the fibrinogen γ′ chain (right). (B) ELISA analysis of citrate plasma collected at various time points from C57Bl/6 mice that were injected with 2 mg of phFibγ-γ or phFibγ′-γ (n=3 mice per time point for each fibrinogen). (C) Model of prophylactic human fibrinogen treatment followed by intravenous infection with S. aureus USA300 in mice. (D) Fga−/− mice (n=7 per group) were either untreated or injected with 6 mg of phFibγ-γ or phFibγ′-γ followed by retroorbital infection with 1×109 CFUs of S. aureus USA300, and survival was monitored over time. Data were analysed by Kaplan-Meier log-rank analysis, with ##p < .01 for untreated vs phFibγ′-γ and ***p < .001 phFibγ-γ vs phFibγ′-γ. (E) WT mice (n=10 per group) were untreated or injected with 6 mg of phFibγ-γ or phFibγ′-γ followed by retroorbital infection with 5×108 CFUs of S. aureus, and survival was monitored over time. Data were analyzed by Kaplan-Meier log-rank analysis with #p < .05 for untreated vs phFibγ′-γ and *p < .05 for phFibγ-γ vs phFibγ′-γ. (F) FggWT/Δ5 and Fga−/− mice treated with phFibγ-γ or phFibγ′-γ (n=6–7 mice per group) were injected with 5×108 CFUs of S. aureus USA300, and survival was monitored over time. Data were analyzed by Kaplan-Meier log-rank analysis with #p < .05 for FggWT/Δ5 vs phFibγ′-γ.
3.3 |. Fibrinogen γ′-γ reduces S. aureus organ colonization, protects against reactive changes in circulating blood cells, and suppresses cardiac damage
To determine the underlying mechanism by which fibrinogen γ′-γ prolongs host survival, Fga−/− were again treated with either vehicle, phFibγ-γ, or phFibγ′-γ prior to infection with 5×108 CFUs of S. aureus USA300. Mice were euthanized 8 hours after infection, a time point immediately preceding the first overt symptoms of the septicemia challenge in control mice. Analysis of bacterial burden in blood revealed no differences regardless of treatment (Figure 3A). Similarly, no fibrinogen treatment-dependent differences were observed in the bacterial burden of liver tissue homogenates (Figure 3B). However, mice treated with phFibγ-γ had significantly higher bacterial burdens than mice administered phFibγ′-γ in tissue homogenates from kidney (Figure 3C), heart (Figure 3D), and lung (Figure 3E). Intriguingly, Fga−/−mice administered only vehicle also had significantly reduced numbers of CFUs relative to phFibγ-γ-treated mice, levels nearly identical to phFibγ′-γ-treated mice in these same tissues (Figure 3C–E). These data suggest that WT fibrin(ogen) supports the accumulation and/or proliferation of S. aureus in host tissues, whereas eliminating a key virulence factor binding domain on fibrin(ogen) suppresses this activity.
FIGURE 3. Fga−/− mice reconstituted with fibrinogen γ′-γ and challenged with an intravenous S. aureus infection show less organ colonization, protection from reactive changes in complete blood counts, and reduced evidence of end organ damage.
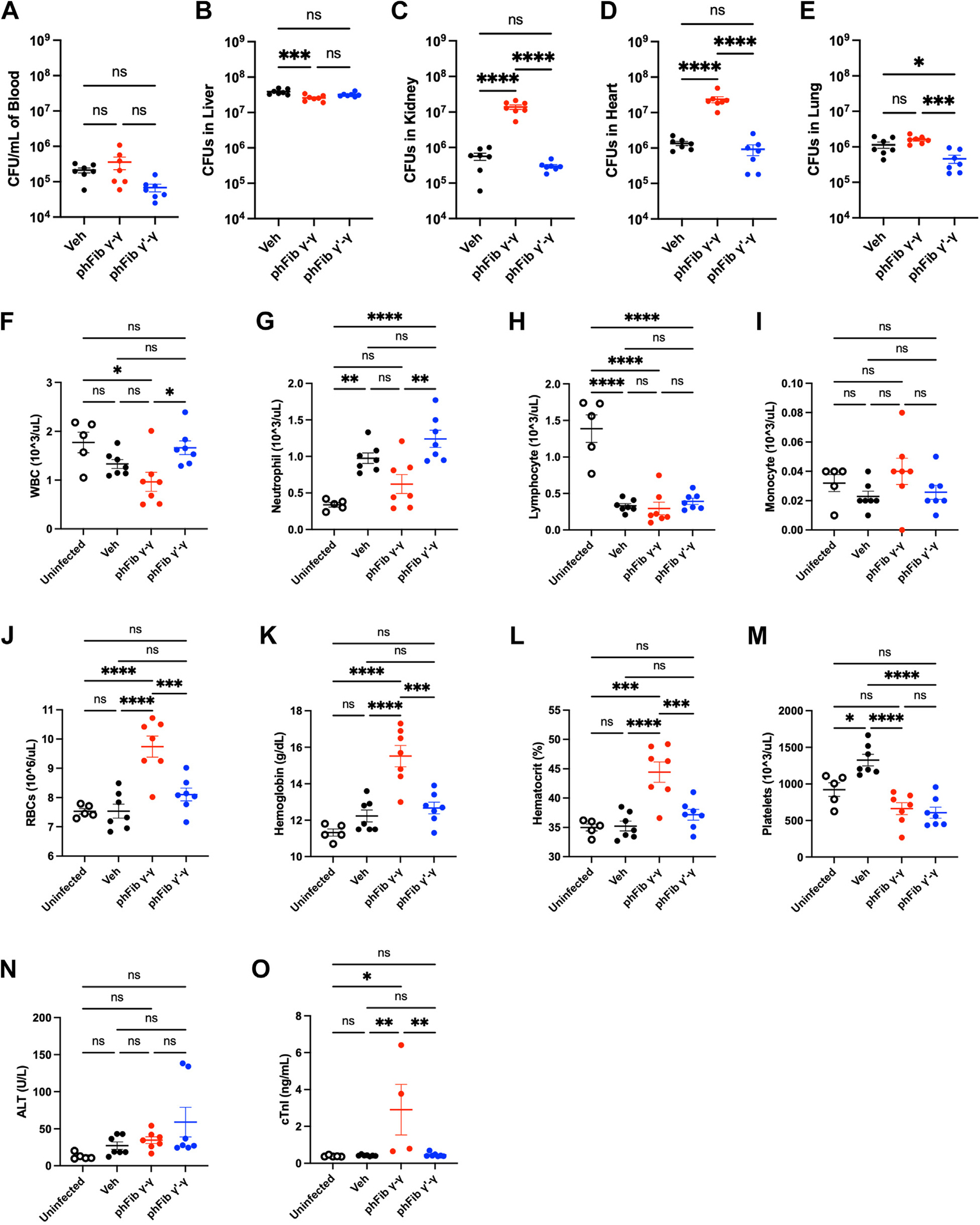
Fga−/− mice were administered a retroorbital injection of either a vehicle (Veh) control, 6 mg phFibγ-γ, or 6 mg phFibγ′-γ followed by an intravenous infection with 5×108 CFUs of S. aureus USA300 24 h later and collection of blood and organ tissues 8 h after infection. Bacterial CFU analyses were performed on (A) blood and tissue homogenates of (B) liver, (C) kidney, (D) heart, and (E) lung. Complete blood counts were performed on uninfected as well as infected mice for analysis of (F) white blood cells (WBCs), (G) neutrophils, (H) lymphocytes, (I) monocytes, (J) red blood cells (RBCs), (K) hemoglobin, (L) hematocrit, and (M) platelets. Plasma was used to analyze circulating tissue damage markers, including (N) alanine aminotransferase (ALT) and (M) cardiac troponin I (cTnI). Data presented as the mean ± SEM and were analyzed by one-way analysis of variance with Tukey’s multiple comparisons test. *p < .05, **p < .01, ***p < .001, and ****p < .0001.
Potential reactive changes in complete blood count were also evaluated. Mice administered phFibγ-γ and challenged with S. aureus USA300 showed a significant reduction in white blood cells (WBCs) relative to uninfected mice or mice administered phFibγ′-γ (Figure 3F). The basis for the differences in WBCs was linked to neutrophils and lymphocytes. Whereas all infected animals had higher neutrophil counts relative to uninfected animals, infected mice administered phFibγ-γ had significantly lower neutrophil counts than infected vehicle- or phFibγ′-γ-treated mice (Figure 3G). In addition, lymphocyte counts were significantly higher for uninfected animals relative to all infected groups (Figure 3H). Monocyte counts were not different among all groups (Figure 3I). Intriguingly, S. aureus-infected mice administered phFibγ-γ had significantly elevated red blood cells, hemoglobin, and hematocrit (Figure 3J–L) relative to all other groups, including uninfected control mice. Platelet counts were significantly elevated in S. aureus-infected mice receiving vehicle control relative to uninfected mice and mice that received either phFibγ-γ or phFibγ′-γ (Figure 3M). The platelet count in mice receiving either fibrinogen was not significantly different from each other or from those in uninfected mice (Figure 3M). There were no differences in the circulating alanine aminotransferase activity among all groups (Figure 3N), while cTnI levels were elevated in phFibγ-γ-treated mice compared with the other groups (Figure 3O), suggesting phFibγ-γ promoted cardiac myocyte damage while phFibγ′-γ was protective against heart injury.
3.4 |. Fibrinogen γ′-γ supports ClfA-mediated S. aureus USA300 adhesion to immobilized fibrinogen and fibrinogen-dependent clumping
We postulated the protection conferred by fibrinogen γ′-γ was linked to an alteration in S. aureus–fibrinogen interactions. A previous report indicated that fibrinogen γ′ failed to bind S. aureus through ClfA [18]. To confirm and expand on that observation, in vitro analyses of bacterial adhesion to immobilized fibrinogen were performed. As shown in Figure 4A, WT S. aureus bacteria showed a dose-dependent adhesion at low coating concentrations (ie, <10 μg/mL) that was saturable at higher concentrations (ie, >10 μg/mL) of phFibγ-γ, phFibγ′-γ, and mFibγWT/WT. As a negative control, mFibγΔ5/Δ5 was used, and no adhesion to this fibrinogen mutant was observed with WT S. aureus USA300 (Figure 4A). Notably, bacterial adhesion to immobilized fibrinogen was dependent on the S. aureus fibrinogen receptor ClfA, as ClfA- S. aureus showed little, if any, appreciable adhesion to any of the fibrinogen species at any coating concentration (Figure 4B). To determine if an increase in the relative percentage of phFibγ′-γ in the presence of total phFib altered S. aureus adhesion, experiments were performed with total purified human fibrinogen (total phFib) and phFibγ′-γ mixes at different ratios ranging from 100% total phFib to 100% phFibγ′-γ. Adhesion of WT S. aureus to fibrinogen was detected in all samples (Figure 4C). Although modest differences were detected, no clear distinctions related to the percentage of phFibγ′-γ in the reaction mixture were observed (Figure 4C). The adhesion of S. aureus that did occur was dependent on ClfA expression (Figure 4D).
FIGURE 4. Fibrinogen γ′-γ supports ClfA-mediated adhesion and clumping with S. aureus USA300 that is not altered by the presence of total human purified plasma fibrinogen.
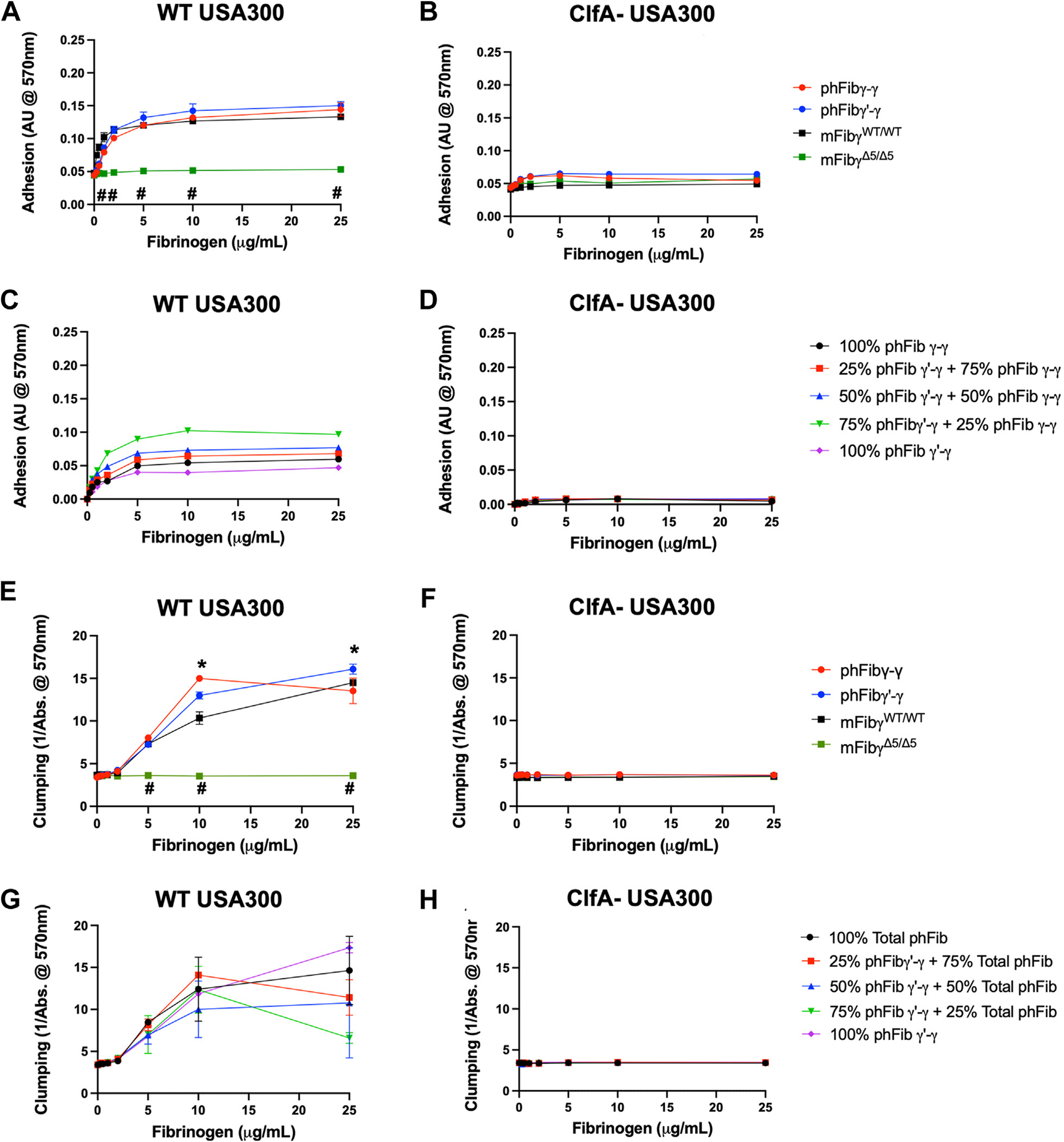
Adhesion of bacteria to immobilized phFibγ-γ, phFibγ′-γ, mFibγWT/WT, and mFibγΔ5/Δ5 was determined for stationary phase (A) wildtype (WT) S. aureus USA300 and (B) ClfA- S. aureus USA300. Adhesion of bacteria to immobilized mixes of purified total phFib and phFibγ′-γ was determined (C) WT USA300 S. aureus and (D) ClfA- S. aureus USA300. Clumping of bacteria in suspension mediated by solutions of phFibγ-γ, phFibγ′-γ, mFibγWT/WT, and mFibγΔ5/Δ5 was determined for (E) WT S. aureus USA300 and (F) ClfA- S. aureus USA300. Clumping of bacteria in suspension mediated by solutions containing mixtures total phFib and phFibγ′-γ was determined for (E) WT S. aureus USA300 and (F) ClfA- S. aureus USA300. For each fibrinogen concentration, n=3 replicates were performed. Data are expressed as the mean ± SEM and analyzed by 2-way analysis of variance with Tukey’s multiple comparison test. #p < .001 for mFibγWT/WT vs mFibγΔ5/Δ5.
To further characterize the interactions between S. aureus and fibrinogen γ′-γ, in vitro experiments were performed to determine the ability of fibrinogen in solution to support the formation of S. aureus aggregates or ‘clumps.’ Dose-dependent clumping of WT S. aureus with phFibγ-γ and mFibγWT/WT was observed (Figure 4E). No clumping was observed with mFibγΔ5/Δ5 (Figure 4E). Surprisingly, clumping was observed with phFibγ′-γ, indicating that loss of only one of the AGDV motifs on the human fibrinogen molecule is not sufficient to eliminate clumping (Figure 4E). Consistent with previous observations [23], ClfA-S. aureus did not support clumping with any of the fibrinogen variants analyzed (Figure 4F). In clumping reactions, altering the relative percentage of phFibγ′-γ did not quantitatively change the overall amount of bacterial clumping with S. aureus in the OD assay, but results were variable at the highest concentration of fibrinogen (ie, 25 μg/mL) analyzed (Figure 4G). ClfA- S. aureus did not form clumps regardless of the phFibγ′-γ ratio (Figure 4H). Collectively, these data highlight the requirement for ClfA to support clumping and that a human fibrinogen molecule lacking only one AGDV motif still supports clumping.
The observations that phFibγ′-γ conferred a S. aureus-infected host a survival benefit and yet supported quantitatively similar levels of fibrinogen-mediated adhesion and clumping to the microbe suggested there might be qualitative differences in the interactions of S. aureus with phFibγ′-γ compared with phFibγ-γ. Given the focus on a blood-stream infection, we postulated that phFibγ′-γ supports the formation of clumps with properties distinct from that observed with phFibγ-γ. Accordingly, fibrinogen-mediated clumps were generated with S. aureus USA300 and imaged with standard brightfield microscopy. Here, S. aureus clumps generated with phFibγ′-γ appeared smaller and more diffuse overall than clumps generated with total phFib or phFibγ-γ (Figure 5A). Similar studies were performed with GFP-labeled S. aureus Newman strain. Here, S. aureus clumps formed with phFibγ′-γ again appeared smaller and more diffuse than those formed with total phFib or phFibγ-γ (Figure 5B).
FIGURE 5. Incubation of S. aureus with fibrinogen γ′-γ results in the formation of smaller clumps compared to fibrinogen γ-γ.
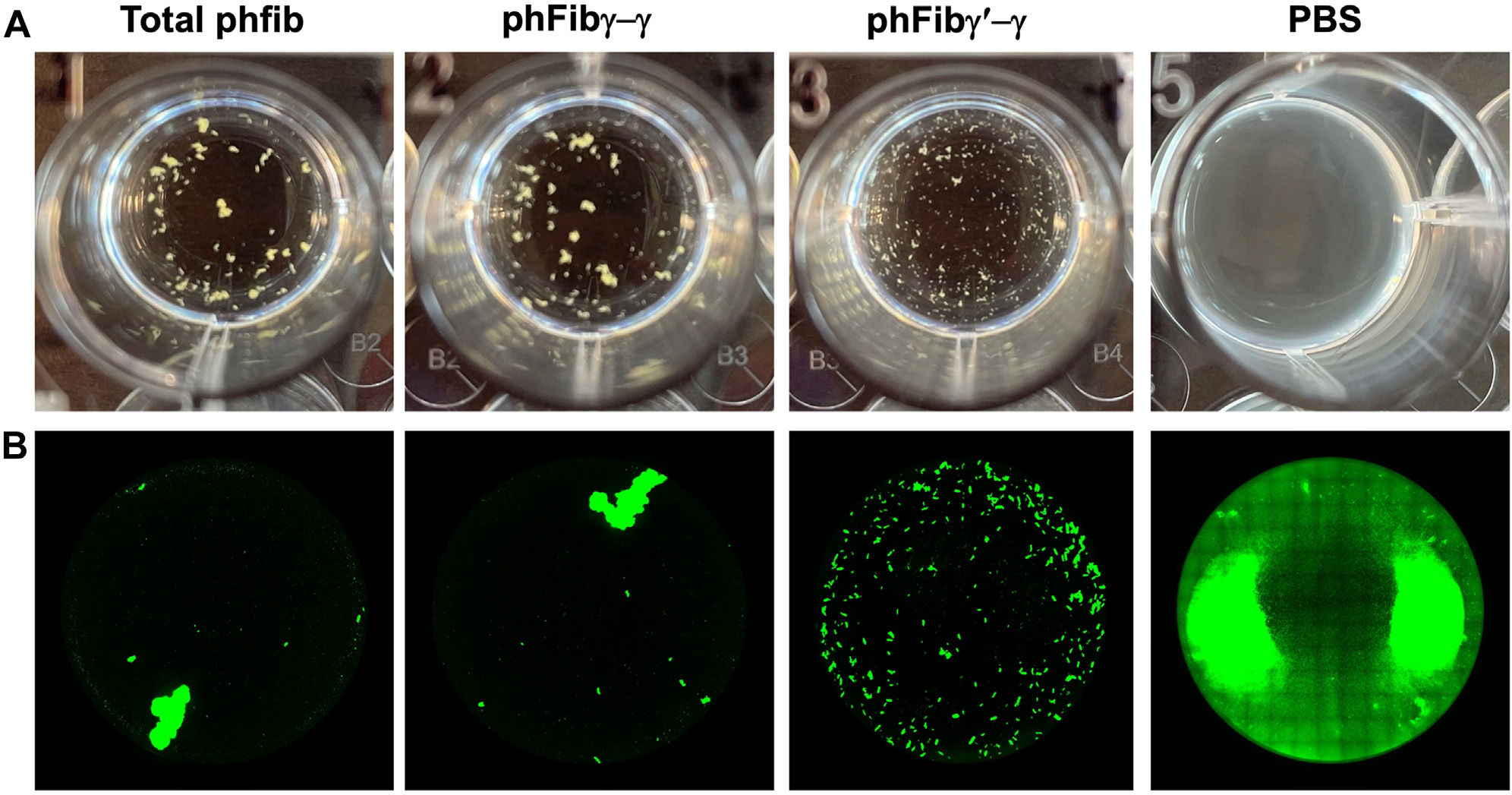
(A) Clumping reactions of S. aureus USA300 in solutions of total phFib, phFibγ-γ, phFibγ′-γ, or phosphate-buffered saline (PBS) vehicle were imaged by brightfield microscopy. (B) Clumping reactions of GFP-expressing S. aureus Newman in solutions of total phFib, phFibγ-γ, phFibγ′-γ, or PBS vehicle were imaged by fluorescent microscopy. Note in each case the large clumps generated by total phFib and phFibγ-γ compared to smaller clumps generated by phFibγ′-γ.
Analyses of plasma-derived phFibγ′-γ′ homodimer are limited by the fact this fibrinogen variant represents <1% of total plasma fibrinogen [20], and thus purification is impractical. To overcome this limitation, we generated sufficient quantities of recombinant fibrinogen γ-γ (rec-hFibγ-γ) and fibrinogen γ′-γ′ (rec-hFibγ′-γ′) for S. aureus adhesion and clumping assays (Figure S1A). Here, immobilized rec-hFibγ-γ supported ClfA-mediated adhesion of WT USA300 similar to plasma-purified total hFib, but rec-hFibγ′ -γ′ did not support S. aureus adhesion at any coating concentration (Figure S1B–C). Similarly, solutions of rec-hFibγ-γ promoted ClfA-dependent clumping of WT USA300 in suspension similar to total hFib, but no clumping was observed at any fibrinogen concentration evaluated for rec-hFibγ′ -γ′ (Supplementary Figure S1D–E).
3.5 |. Treatment of mice with fibrinogen γ′-γ extends host survival following infection with S. aureus USA300
To determine if fibrinogen γ′-γ could provide therapeutic host protection following a systemic S. aureus infection, mice were given 6 mg of fibrinogen 30 minutes after S. aureus infection (Figure 6A). Fga−/− mice were challenged with S. aureus USA300 intravenously and left untreated or treated with phFibγ-γ or phFibγ′-γ. Animals treated with phFibγ-γ displayed a significant survival advantage over infected untreated animals, consistent with a recent study showing that elevated fibrinogen levels improve host survival in sepsis [27]. Importantly, mice treated with phFibγ′-γ had a significant survival advantage over the first ~90 hours compared with both untreated and phFibγ-γ-treated mice (Figure 6B). At 96 hours after the infection, surviving mice were redosed with an additional 6 mg of fibrinogen. Following retreatment, mice administered phFibγ′-γ showed prolonged survival relative to the remaining mice treated with phFibγ-γ (Figure 6C). Together, these data suggest that therapeutic fibrinogen γ′ treatment can extend host survival following S. aureus septicemia and that continued administration offers extended support to the host.
FIGURE 6. Therapeutic treatment of fibrinogen-deficient mice with fibrinogen g’ improves mouse survival following septicemia.
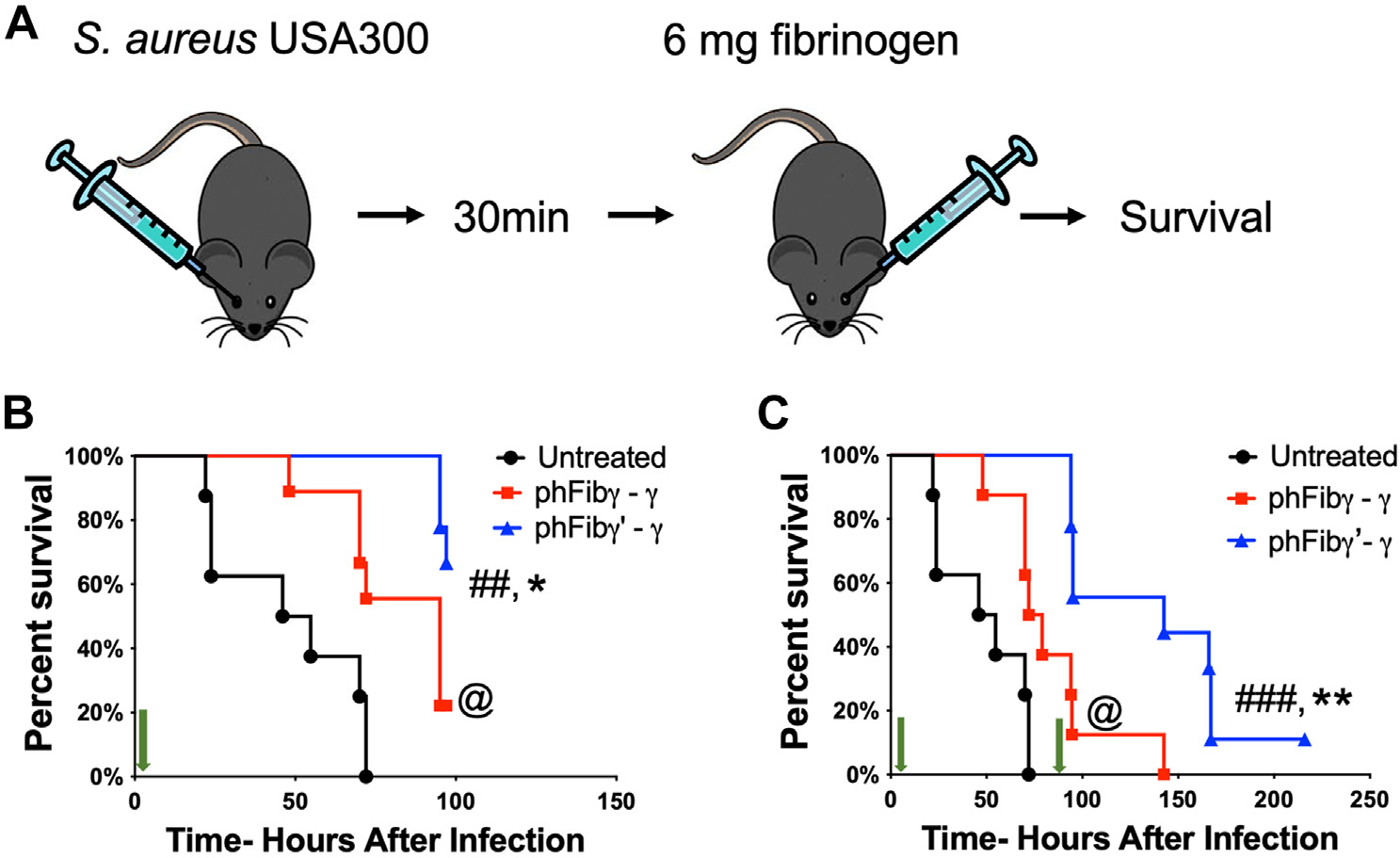
(A) Mouse model of therapeutic fibrinogen treatment after S. aureus USA300 infection. Fga−/− mice were infected with 5×108 CFUs of S. aureus USA300 and were untreated (n=8) or treatment with 6 mg of phFibγ-γ (n=8) or phFibγ′-γ (n=9) 30 min after the infection. (B) Survival analysis at 90 h after infection and fibrinogen treatment. Data were analyzed by Kaplan-Meier log-rank analysis with ##p < .01 for untreated vs phFibγ′-γ, @p < .01 for untreated vs phFibγ-γ, and *p < .05 phFibγ-γ vs. phFibγ′-γ. (C) After 96 h infection, the remaining mice in the study were treated with a second dose of 6 mg of phFibγ-γ or phFibγ′-γ. Data were analyzed by Kaplan-Meier log-rank analysis with ###p < .001 for untreated vs phFibγ′-γ, @p < .05 for untreated vs phFibγ-γ, and **p < .01 for phFibγ’-γ vs. phFibγ′-γ. For each graph, fibrinogen dosing time is marked with green arrows.
4 |. DISCUSSION
In this study, we postulated that a significant increase in host survival following S. aureus infection could be appreciated by introducing fibrinogen variants lacking a key S. aureus virulence factor binding motif. Building on previous studies, improved survival was observed in mice heterozygous for the γΔ5 mutation in which the binding capacity of fibrinogen to S. aureus was reduced. Based on the fibrinogen (AαBβγ)2 molecular structure, FggWT/Δ5 mice are expected to have a heterogeneous population of fibrinogen molecules with approximately 25% (AαBβγ)2, 50% [(AαBβ)2γγΔ5], and 25% (AαBβγΔ5)2. Thus, we speculated that a fibrinogen heterodimer with half the molecule lacking the final 5 amino acids of the normal γ-chain would be sufficient to confer a benefit to the host. To both test this concept and translate our findings to human fibrinogen variants, we revealed that mice reconstituted with the naturally occurring human fibrinogen γ′-γ displayed significantly improved survival over control animals that was associated with reduced bacterial burden in organ systems, preservation of circulating WBCs/neutrophils, and suppression of tissue damage.
Previous studies documented that fibrinogen γ′-γ can form a fibrin matrix similar to fibrinogen γ-γ [28]. It was also shown that residues critical for binding to several S. aureus virulence receptors (eg, ClfA, FnbpA, FnbpB) are absent in fibrinogen γ′ [29–33]. ClfA is of particular interest as this receptor has been shown to directly promote agglutination in blood and thromboembolic lesions in the heart following a bloodstream S. aureus infection [9]. In mouse studies, ClfA-deficient S. aureus are less virulent following bloodstream infection than WT S. aureus [9,18]. A direct link between ClfA, the C-terminal portion of the fibrinogen γ-chain, and the development of S. aureus sepsis was also previously documented [18]. Whereas FggΔ5 mice show a significant survival advantage over WT mice following infection with WT S. aureus, survival of WT and FggΔ5 mice is equivalent following infection with ClfA-deficient S. aureus [18]. Collectively these previous studies and our current findings suggest that following S. aureus bloodstream infection, fibrinogen γ′-γ is sufficient to support hemostasis and maintenance of vascular integrity but has a reduced capacity to bind S. aureus bacteria and support virulence.
Our in vitro data show that fibrinogen γ′-γ can support S. aureus adhesion to immobilized fibrinogen. This observation is not unexpected as fibrinogen γ′-γ encodes one WT γ-chain with a preserved ClfA-binding motif. It was notable that even at fibrinogen coating concentrations as low as 0.25 μg/mL, no significant quantitative difference in binding was observed. This observation may suggest that a fibrinogen molecule coated on a surface is only able to engage bacteria on one half of the fibrinogen (AαBβγ)2 molecule and that once binding occurs, bacteria engagement with the other half of the molecule is precluded. It is notable that the binding mechanism between ClfA and fibrinogen is complex and involves residues in the γ-chain beyond the terminal AGDV motif. Recent studies suggest that adhesive function of ClfA for fibrinogen is regulated by mechanical tension as would be experienced under blood flow [34]. This mechanism has been proposed as part of a bridging mechanism between S. aureus, fibrinogen, and integrin αVβ3 on endothelial cells that contributes to sepsis [35]. In this way, ClfA acts as a type of mechanosensor, but this function could be interrupted with fibrinogen γ′-γ. The bacterial adhesion studies performed here were under static conditions. Conducting similar S. aureus binding studies to immobilized fibrinogen under flow would help to resolve whether binding differences to fibrinogen γ′-γ may be appreciated under shear stress conditions.
A net result of the S. aureus fibrinogen clumping is to form a ‘shield’ around bacteria protecting the pathogen from host antimicrobial mechanisms and promoting virulence [36–40]. Supporting this concept, S. aureus with ClfA genetically eliminated have significantly reduced agglutination in plasma, are less pathogenic, and support the formation of smaller and fewer abscesses in a mouse bacteremia/sepsis model [9,41]. Accordingly, the loss of a ClfA-binding motif and smaller clumps mediated by fibrinogen γ′-γ would be expected to diminish the shielding function of fibrinogen. The reduction in overall bacterial burden in mice with phFibγ′-γ (Figure 3C–E) is consistent with this concept. The mechanism by which fibrinogen γ′-γ potentially perturbs the fibrinogen shield is unknown as it could be a function of an overall reduction in the amount of fibrin(ogen) surrounding S. aureus in circulation, an altered fibrin(ogen) shield structure, or both. Moreover, we found that fibrinogen γ′-γ′ is fully deficient in supporting adhesion and clumping of S. aureus. With future large-scale production of recombinant fibrinogen, including fibrinogen γ-γ, fibrinogen γ′-γ, and fibrinogen γ′-γ′, it will be possible to determine whether the heterodimer or homodimer confers a greater benefit to the host and the possible mechanisms through which the protective benefit is conferred for each variant.
The C-terminal portion of the fibrinogen γ-chain also mediates interaction with the platelet integrin αIIbβ3 receptor that drives fibrinogen-dependent platelet aggregation. Activated platelets express a variety of pattern recognition receptors, phagocytose exogenous antigens, interact with other immune cells (eg, neutrophils), and release numerous soluble mediators (eg, chemokines and cytokines) that can influence the host antimicrobial immune response [42–45]. Previous studies documented that fibrinogen γ′-γ shows an approximate 50% reduction in platelet binding and aggregation [46]. One would speculate that the reduction in fibrinogen–platelet activity conferred by fibrinogen γ′-γ would, if anything, impair the ability of platelets to function as immune mediators to promote antimicrobial activity [47,48]. Additional studies are required to further elucidate the crosstalk between fibrin(ogen), platelets, and S. aureus to decipher the potential influence of fibrinogen γ′-γ on infection outcome.
The improved survival observed in fibrinogen γ′-γ treated mice could also be linked to an anticoagulant effect and suppression of thrombin generation or activity. Fibrinogen γ′ can sequester thrombin and thus inhibit its activity, by high-affinity binding of thrombin of exosite 2 to the unique C-terminal γ′ sequence [49–51]. Reconstitution of fibrinogen-deficient plasma with fibrinogen γ′-γ was shown to provide substantially higher thrombin inhibition than reconstitution with normal fibrinogen [52]. Fibrinogen γ′ also exerts anticoagulant effects by diminishing coagulation factors V and VIII activation and increasing the sensitivity to activated protein C [53–55]. Notably, this property is not present in mouse fibrinogen as the similar alternative splicing event of the mouse Fgg gene does not produce a γ′ protein with equivalent properties. Our previous work showed that mice with a mutation resulting in 10% or normal prothrombin levels had a significantly improved survival profile following intravenous infection with S. aureus relative to WT mice with normal prothrombin levels [18]. S. aureus produces 2 coagulases (ie, Coa and Vwbp) that can nonproteolytically activate prothrombin and promote fibrin formation [7]. Mice infected with S. aureus in which these coagulase proteins were genetically eliminated had a better survival profile relative to WT S. aureus-infected mice [9,41]. Notably, mouse fibrinogen γΔ5 does not have the same thrombin modifying activity as human fibrinogen γ′-γ (ie, fibrinogen γΔ5 does not bind and sequester thrombin). Our studies suggest that fibrinogen γWT/Δ5 conferred less host protection than that conferred by fibrinogen γ′-γ. Prophylactic phFibγ′-γ was protective against higher S. aureus challenge doses than those observed with FggWT/Δ5 (compare, Figure 2D to Figure 1C). Moreover, in a head-to-head comparison, a significant number of fibrinogen-deficient mice supplemented with phFibγ′-γ survived the intravenous S. aureus infection, whereas all FggWT/Δ5 mice succumbed to infection with the same suspension of bacteria. The findings suggest that for fibrinogen γΔ5 the dominant mechanism of action is loss of binding between the bacteria and fibrinogen, but that phFibγ′-γ confers protection against S. aureus infection through multiple pathways. However, formal studies of the various possible mechanisms by which fibrinogen γ′-γ is protective against S. aureus infection remain to be performed.
The results presented here highlight the concept that the naturally occurring fibrinogen variant, fibrinogen γ′-γ, confers host protection following a S. aureus blood-borne infection. Most notably, we showed that fibrinogen γ′-γ could enhance host survival even when administered to mice already challenged with a blood-borne S. aureus infection. Although fibrinogen γ′-γ extended the time of host survival and reduced the overall bacteria burden, it did not promote the complete elimination of the microbes. We postulate that fibrinogen γ′-γ administration may be employed as part of a novel therapeutic strategy for patients with S. aureus bacteremia, to extend the therapeutic window of conventional antibiotics and prevent the onset of sepsis in infected patients.
Supplementary Material
Essentials.
Staphylococcus aureus interactions with fibrinogen are a determinant of host defense and pathogen virulence.
Injection of purified human fibrinogen γ′-γ variant increased survival in a mouse model of S. aureus septicemia even in the presence of normal host fibrinogen.
S. aureus adhesion and clumping in vitro are not quantitatively reduced with fibrinogen γ′-γ heterodimer, but clumping is qualitatively altered.
Fibrinogen γ′-γ host protection is associated with reduced S. aureus colonization in tissues, lower reactive changes in blood cell counts, and suppression of end organ damage.
ACKNOWLEDGMENTS
The authors thank Alyssa Dandridge for her technical assistance. This work was supported in part by Canadian Institutes of Health Research (MFE181897 to W.S.H.), a National Institutes of Health grant (R01DK112778 to M.J.F), funding from the European Union’s Horizon Europe research and innovation program under grant agreement No 190183175, and research funding provided by Fibriant BV. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
DECLARATION OF COMPETING INTERESTS
J.K. is the chief executive officer and founder of Fibriant BV. M.W. and J.G. are co-founders of Fibriant BV. The remaining authors have no competing interests to disclose.
Matthew J. Flick @fib390_396A
SUPPLEMENTARY MATERIAL
The online version contains supplementary material available at https://doi.org/10.1016/j.jtha.2023.03.019
REFERENCES
- [1].Lowy FD. Staphylococcus aureus infections. N Engl J Med. 1998;339:520–32. [DOI] [PubMed] [Google Scholar]
- [2].Cheung GYC, Bae JS, Otto M. Pathogenicity and virulence of Staphylococcus aureus. Virulence. 2021;12:547–69. 10.1080/21505594.2021.1878688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kwiecinski JM, Horswill AR. Staphylococcus aureus bloodstream infections: pathogenesis and regulatory mechanisms. Curr Opin Microbiol. 2020;53:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kristinsson KG. Adherence of staphylococci to intravascular catheters. J Med Microbiol. 1989;28:249–57. [DOI] [PubMed] [Google Scholar]
- [5].Aslam S, Vaida F, Ritter M, Mehta RL. Systematic review and meta-analysis on management of hemodialysis catheter-related bacteremia. J Am Soc Nephrol. 2014;25:2927–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].El Atrouni WI, Knoll BM, Lahr BD, Eckel-Passow JE, Sia IG, Baddour LM. Temporal trends in the incidence of Staphylococcus aureus bacteremia in Olmsted County, Minnesota, 1998 to 2005: a population-based study. Clin Infect Dis. 2009;49:e130–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cheng AG, McAdow M, Kim HK, Bae T, Missiakas DM, Schneewind O. Contribution of coagulases towards Staphylococcus aureus disease and protective immunity. PLOS Pathog. 2010;6:e1001036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Loughman A, Fitzgerald JR, Brennan MP, Higgins J, Downer R, Cox D, Foster TJ. Roles for fibrinogen, immunoglobulin and complement in platelet activation promoted by Staphylococcus aureus clumping factor A. Mol Microbiol. 2005;57:804–18. [DOI] [PubMed] [Google Scholar]
- [9].McAdow M, Kim HK, Dedent AC, Hendrickx AP, Schneewind O, Missiakas DM. Preventing Staphylococcus aureus sepsis through the inhibition of its agglutination in blood. PLOS Pathog. 2011;7:e1002307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].McDevitt D, Nanavaty T, House-Pompeo K, Bell E, Turner N, McIntire L, Foster T, Höök M. Characterization of the interaction between the Staphylococcus aureus clumping factor (ClfA) and fibrinogen. Eur J Biochem. 1997;247:416–24. [DOI] [PubMed] [Google Scholar]
- [11].Panizzi P, Nahrendorf M, Figueiredo JL, Panizzi J, Marinelli B, Iwamoto Y, Keliher E, Maddur AA, Waterman P, Kroh HK, Leuschner F, Aikawa E, Swirski FK, Pittet MJ, Hackeng TM, Fuentes-Prior P, Schneewind O, Bock PE, Weissleder R. In vivo detection of Staphylococcus aureus endocarditis by targeting pathogen-specific prothrombin activation. Nat Med. 2011;17:1142–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Foster TJ, Höök M. Surface protein adhesins of Staphylococcus aureus. Trends Microbiol. 1998;6:484–8. [DOI] [PubMed] [Google Scholar]
- [13].Geoghegan JA, Ganesh VK, Smeds E, Liang X, Höök M, Foster TJ. Molecular characterization of the interaction of staphylococcal microbial surface components recognizing adhesive matrix molecules (MSCRAMM) ClfA and Fbl with fibrinogen. J Biol Chem. 2010;285:6208–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Heilmann C Adhesion mechanisms of staphylococci. Adv Exp Med Biol. 2011;715:105–23. [DOI] [PubMed] [Google Scholar]
- [15].Hawiger J, Timmons S, Strong DD, Cottrell BA, Riley M, Doolittle RF. Identification of a region of human fibrinogen interacting with staphylococcal clumping factor. Biochemistry. 1982;21:1407–13. [DOI] [PubMed] [Google Scholar]
- [16].Hair PS, Echague CG, Sholl AM, Watkins JA, Geoghegan JA, Foster TJ, Cunnion KM. Clumping factor A interaction with complement factor I increases C3b cleavage on the bacterial surface of Staphylococcus aureus and decreases complement-mediated phagocytosis. Infect Immun. 2010;78:1717–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Higgins J, Loughman A, van Kessel KP, van Strijp JA, Foster TJ. Clumping factor A of Staphylococcus aureus inhibits phagocytosis by human polymorphonuclear leucocytes. FEMS Microbiol Lett. 2006;258:290–6. [DOI] [PubMed] [Google Scholar]
- [18].Flick MJ, Du X, Prasad JM, Raghu H, Palumbo JS, Smeds E, Höök M, Degen JL. Genetic elimination of the binding motif on fibrinogen for the S. aureus virulence factor ClfA improves host survival in septicemia. Blood. 2013;121:1783–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Uitte de Willige S, Standeven KF, Philippou H, Ariëns RA. The pleiotropic role of the fibrinogen gamma’ chain in hemostasis. Blood. 2009;114:3994–4001. [DOI] [PubMed] [Google Scholar]
- [20].Wolfenstein-Todel C, Mosesson MW. Human plasma fibrinogen heterogeneity: evidence for an extended carboxyl-terminal sequence in a normal gamma chain variant (gamma′). Proc Natl Acad Sci U S A. 1980;77:5069–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Crabtree GR, Kant JA. Organization of the rat gamma-fibrinogen gene: alternative mRNA splice patterns produce the gamma A and gamma B (gamma′) chains of fibrinogen. Cell. 1982;31:159–66. [DOI] [PubMed] [Google Scholar]
- [22].Monk IR, Shah IM, Xu M, Tan MW, Foster TJ. Transforming the untransformable: application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. mBio. 2012;3:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Negrón O, Hur WS, Prasad J, Paul DS, Rowe SE, Degen JL, Abrahams SR, Antoniak S, Conlon BP, Bergmeier W, Hӧӧk M, Flick MJ. Fibrin(ogen) engagement of S. aureus promotes the host antimicrobial response and suppression of microbe dissemination following peritoneal infection. PLOS Pathog. 2022;18:e1010227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Koppert PW, Huijsmans CM, Nieuwenhuizen W. A monoclonal antibody, specific for human fibrinogen, fibrinopeptide A-containing fragments and not reacting with free fibrinopeptide A. Blood. 1985;66:503–7. [PubMed] [Google Scholar]
- [25].Holmbäck K, Danton MJ, Suh TT, Daugherty CC, Degen JL. Impaired platelet aggregation and sustained bleeding in mice lacking the fibrinogen motif bound by integrin alpha IIb beta 3. EMBO J. 1996;15:5760–71. [PMC free article] [PubMed] [Google Scholar]
- [26].Suh TT, Holmbäck K, Jensen NJ, Daugherty CC, Small K, Simon DI, Potter S, Degen JL. Resolution of spontaneous bleeding events but failure of pregnancy in fibrinogen-deficient mice. Genes Dev. 1995;9:2020–33. [DOI] [PubMed] [Google Scholar]
- [27].Wada H, Kawasugi K, Honda G, Kawano N, Uchiyama T, Madoiwa S, Takezako N, Suzuki K, Seki Y, Ikezoe T, Iba T, Okamoto K. Sepsis-associated DIC with decreased levels of antithrombin and fibrinogen is the target for combination therapy with thrombomodulin alfa and antithrombin. TH Open. 2023;7:e65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Walton BL, Getz TM, Bergmeier W, Lin FC, Uitte de Willige S, Wolberg AS. The fibrinogen γA/γ′ isoform does not promote acute arterial thrombosis in mice. J Thromb Haemost. 2014;12:680–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Cheng AG, Kim HK, Burts ML, Krausz T, Schneewind O, Missiakas DM. Genetic requirements for Staphylococcus aureus abscess formation and persistence in host tissues. FASEB J. 2009;23:3393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Josefsson E, Hartford O, O’Brien L, Patti JM, Foster T. Protection against experimental Staphylococcus aureus arthritis by vaccination with clumping factor A, a novel virulence determinant. J Infect Dis. 2001;184:1572–80. [DOI] [PubMed] [Google Scholar]
- [31].Rothfork JM, Dessus-Babus S, Van Wamel WJ, Cheung AL, Gresham HD. Fibrinogen depletion attenuates Staphyloccocus aureus infection by preventing density-dependent virulence gene up-regulation. J Immunol. 2003;171:5389–95. [DOI] [PubMed] [Google Scholar]
- [32].Siboo IR, Cheung AL, Bayer AS, Sullam PM. Clumping factor A mediates binding of Staphylococcus aureus to human platelets. Infect Immun. 2001;69:3120–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sullam PM, Bayer AS, Foss WM, Cheung AL. Diminished platelet binding in vitro by Staphylococcus aureus is associated with reduced virulence in a rabbit model of infective endocarditis. Infect Immun. 1996;64:4915–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Viela F, Speziale P, Pietrocola G, Dufrene YF. Mechanostability of the fibrinogen bridge between Staphylococcal surface protein ClfA and endothelial cell integrin αVβ3. Nano Lett. 2019;19:7400–10. [DOI] [PubMed] [Google Scholar]
- [35].McDonnell CJ, Garciarena CD, Watkin RL, McHale TM, McLoughlin A, Claes J, Verhamme P, Cummins PM, Kerrigan SW. Inhibition of major integrin αVβ3 reduces Staphylococcus aureus attachment to sheared human endothelial cells. J Thromb Haemost. 2016;14:2536–47. [DOI] [PubMed] [Google Scholar]
- [36].Ko YP, Kang M, Ganesh VK, Ravirajan D, Li B, Höök M. Coagulase and Efb of Staphylococcus aureus have a common fibrinogen binding motif. mBio. 2016;7(1);e01885–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ko YP, Kuipers A, Freitag CM, Jongerius I, Medina E, van Rooijen WJ, Spaan AN, van Kessel KP, Höök M, Rooijakkers SH. Phagocytosis escape by a Staphylococcus aureus protein that connects complement and coagulation proteins at the bacterial surface. PLOS Pathog. 2013;9:e1003816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kuipers A, Stapels DAC, Weerwind LT, Ko YP, Ruyken M, Lee JC, van Kessel KPM, Rooijakkers SHM. The Staphylococcus aureus polysaccharide capsule and Efb-dependent fibrinogen shield act in concert to protect against phagocytosis. Microbiology (Reading). 2016;162:1185–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Crosby HA, Kwiecinski J, Horswill AR. Staphylococcus aureus aggregation and coagulation mechanisms, and their function in host-pathogen interactions. Adv Appl Microbiol. 2016;96:1–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Thomas S, Arora S, Liu W, Churion K, Wu Y, Höök M. vhp is a fibrinogen-binding protein related to vWbp in Staphylococcus aureus. mBio. 2021;12:e0116721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].McAdow M, Missiakas DM, Schneewind O. Staphylococcus aureus secretes coagulase and von Willebrand factor binding protein to modify the coagulation cascade and establish host infections. J Innate Immun. 2012;4:141–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Cognasse F, Aloui C, Anh Nguyen K, Hamzeh-Cognasse H, Fagan J, Arthaud CA, Eyraud MA, Sebban M, Fromont E, Pozzetto B, Laradi S, Garraud O. Platelet components associated with adverse reactions: predictive value of mitochondrial DNA relative to biological response modifiers. Transfusion. 2016;56:497–504. [DOI] [PubMed] [Google Scholar]
- [43].Vardon Bounes F, Memier V, Marcaud M, Jacquemin A, Hamzeh-Cognasse H, Garcia C, Series J, Sie P, Minville V, Gratacap MP, Payrastre B. Platelet activation and prothrombotic properties in a mouse model of peritoneal sepsis. Sci Rep. 2018;8:13536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hamzeh-Cognasse H, Berthelot P, Tardy B, Pozzetto B, Bourlet T, Laradi S, Garraud O, Cognasse F. Platelet toll-like receptors are crucial sensors of infectious danger moieties. Platelets. 2018;29:533–40. [DOI] [PubMed] [Google Scholar]
- [45].Campbell RA, Manne BK, Banerjee M, Middleton EA, Ajanel A, Schwertz H, Denorme F, Stubben C, Montenont E, Saperstein S, Page L, Tolley ND, Lim DL, Brown SM, Grissom CK, Sborov DW, Krishnan A, Rondina MT. IFITM3 regulates fibrinogen endocytosis and platelet reactivity in nonviral sepsis. J Clin Invest. 2022;132:e153014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Peerschke EI, Francis CW, Marder VJ. Fibrinogen binding to human blood platelets: effect of gamma chain carboxyterminal structure and length. Blood. 1986;67:385–90. [PubMed] [Google Scholar]
- [47].Liu E, Chen Y, Xu J, Gu S, An N, Xin J, Wang W, Liu Z, An Q, Yi J, Yin W. Platelets inhibit methicillin-resistant Staphylococcus aureus by inducing hydroxyl radical-mediated apoptosis-like cell death. Microbiol Spectr. 2022;10:e0244121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ali RA, Wuescher LM, Dona KR, Worth RG. Platelets mediate host defense against Staphylococcus aureus through direct bactericidal activity and by enhancing macrophage activities. J Immunol. 2017;198:344–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Mosesson MW. Fibrinogen and fibrin structure and functions. J Thromb Haemost. 2005;3:1894–904. [DOI] [PubMed] [Google Scholar]
- [50].Fredenburgh JC, Stafford AR, Leslie BA, Weitz JI. Bivalent binding to gammaA/gamma’-fibrin engages both exosites of thrombin and protects it from inhibition by the antithrombin-heparin complex. J Biol Chem. 2008;283:2470–7. [DOI] [PubMed] [Google Scholar]
- [51].Fredenburgh JC, Stafford AR, Pospisil CH, Weitz JI. Modes and consequences of thrombin’s interaction with fibrin. Biophys Chem. 2004;112:277–84. [DOI] [PubMed] [Google Scholar]
- [52].de Bosch NB, Mosesson MW, Ruiz-Sáez A, Echenagucia M, Rodriguez-Lemoin A. Inhibition of thrombin generation in plasma by fibrin formation (Antithrombin I). Thromb Haemost. 2002;88:253–8. [PubMed] [Google Scholar]
- [53].Lovely RS, Boshkov LK, Marzec UM, Hanson SR, Farrell DH. Fibrinogen gamma’ chain carboxy terminal peptide selectively inhibits the intrinsic coagulation pathway. Br J Haematol. 2007;139:494–503. [DOI] [PubMed] [Google Scholar]
- [54].Omarova F, Uitte De Willige S, Ariëns RA, Rosing J, Bertina RM, Castoldi E. Inhibition of thrombin-mediated factor V activation contributes to the anticoagulant activity of fibrinogen γ. J Thromb Haemost. 2013;11:1669–78. [DOI] [PubMed] [Google Scholar]
- [55].Omarova F, Uitte de Willige S, Simioni P, Ariëns RA, Bertina RM, Rosing J, Castoldi E. Fibrinogen γ′ increases the sensitivity to activated protein C in normal and factor V Leiden plasma. Blood. 2014;124:1531–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.