

Abstract
Cannabis has been used recreationally and medically for centuries, yet research into understanding the mechanisms of its therapeutic effects has only recently garnered more attention. There is evidence to support the use of cannabinoids for the treatment of chronic pain, muscle spasticity, nausea and vomiting due to chemotherapy, improving weight gain in HIV-related cachexia, emesis, sleep disorders, managing symptoms in Tourette syndrome, and patient-reported muscle spasticity from multiple sclerosis. However, tolerance the risk for cannabis use disorder are two significant disadvantages for cannabinoid-based therapies in humans. Recent work has revealed prominent sex differences in the acute response and tolerance to cannabinoids in both humans and animal models. This review will discuss evidence demonstrating cannabinoid tolerance in rodents, non-human primates, and humans and our current understanding of the neuroadaptations occurring at the cannabinoid type 1 receptor (CB1R) that are responsible tolerance. CB1R expression is downregulated in tolerant animals and humans while there is strong evidence of CB1R desensitization in cannabinoid tolerant rodent models. Throughout the review, critical knowledge gaps are indicated and discussed, such as the lack of a neuroimaging probe to assess CB1R desensitization in humans. The review discusses the intracellular signaling pathways that are responsible for mediating CB1R desensitization and downregulation including the action of G protein-coupled receptor kinases, β–arrestin2 recruitment, c-Jun N-terminal kinases, protein kinase A, and the intracellular trafficking of CB1R. Finally, the review discusses approaches to reduce cannabinoid tolerance in humans based on our current understanding of the neuroadaptations and mechanisms responsible for this process.
Keywords: cannabinoid, desensitization, downregulation, THC, tolerance, cannabis use disorder
Graphical Abstract
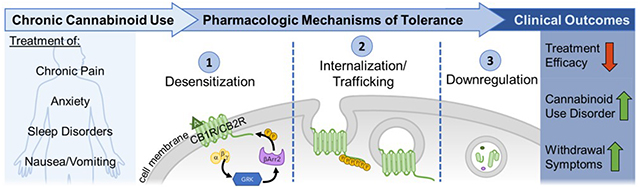
1. Introduction
Cannabinoids have been used for recreational and medical purposes for many centuries with the first reported medical use reported by the Chinese emperor Sheng Nung in 2729 BC. In modern times, cannabis and cannabinoids represent the most widely used illicit drug in the world. Due to the recent legalization of medical and/or recreational use of cannabinoids, the prevalence of cannabis use in the United States (US) has been growing over the last ten years.
The Cannabis plant genus is made up of three species including C. sativa, C. indica, and C. ruderalis. Although the specific constituents vary between specific strains and species, these plants contain more than 560 identified compounds, of which 150 are phytocannabinoids (Hanus et al., 2016; Lewis et al., 2017; Pertwee, 2014). The most abundant of these cannabinoids are (−)-trans-delta-9-tetrahydrocannabinol (Δ9-THC) and (−)-cannabidiol (CBD). The psychoactive effects of cannabis have been attributed to the action of THC at neuronal type 1 cannabinoid receptors (CB1R) while CBD has been shown to exert anti-inflammatory actions via multiple molecular targets. The Δ9-THC content in medical and recreational cannabis strains has increased from 3% weight/volume (w/v) in strains in 1980s compared to more than 12% Δ9-THC that is found in modern cannabis strains. Thus, there is an increasing and urgent need to understand the impact of high Δ9-THC cannabis strains with chronic use and elevated dosage (ElSohly et al., 2016).
1.1. The Endocannabinoid System
Cannabinoids exert their physiological effects primarily through action on the endogenous cannabinoid system (ECS). The ECS is involved in pain, cognition, learning and memory, locomotion, gastrointestinal regulation, sleep, as well as, pulmonary, autonomic, and immunomodulatory control mechanisms (Cinar et al., 2017; Fehr et al., 1976; Gine et al., 2017; Kesner and Lovinger, 2020; Martin et al., 1999; Rossi et al., 2013; Schoch et al., 2018; Sibaev et al., 2009; Storr et al., 2004) (Figure 1). The ECS consists of three main components: the endogenous ligands (endocannabinoids), the cannabinoid receptors, and the enzymes responsible for the synthesis and catabolism of endocannabinoids. CB1R and CB2R are two primary receptors of the endocannabinoid system and both are G protein-coupled receptors (GPCRs) that are coupled to Gi/o proteins (Howlett, 2005). However, CB1R can also couple to Gs and Gq/11 proteins under certain conditions (Glass and Felder, 1997; Lauckner et al., 2005). These Class A, rhodopsin-like GPCRs share less than half of their amino acid sequence identity, resulting in substantial differences in signaling outcomes.
Figure 1. Roles of ECS in physiology and human health.
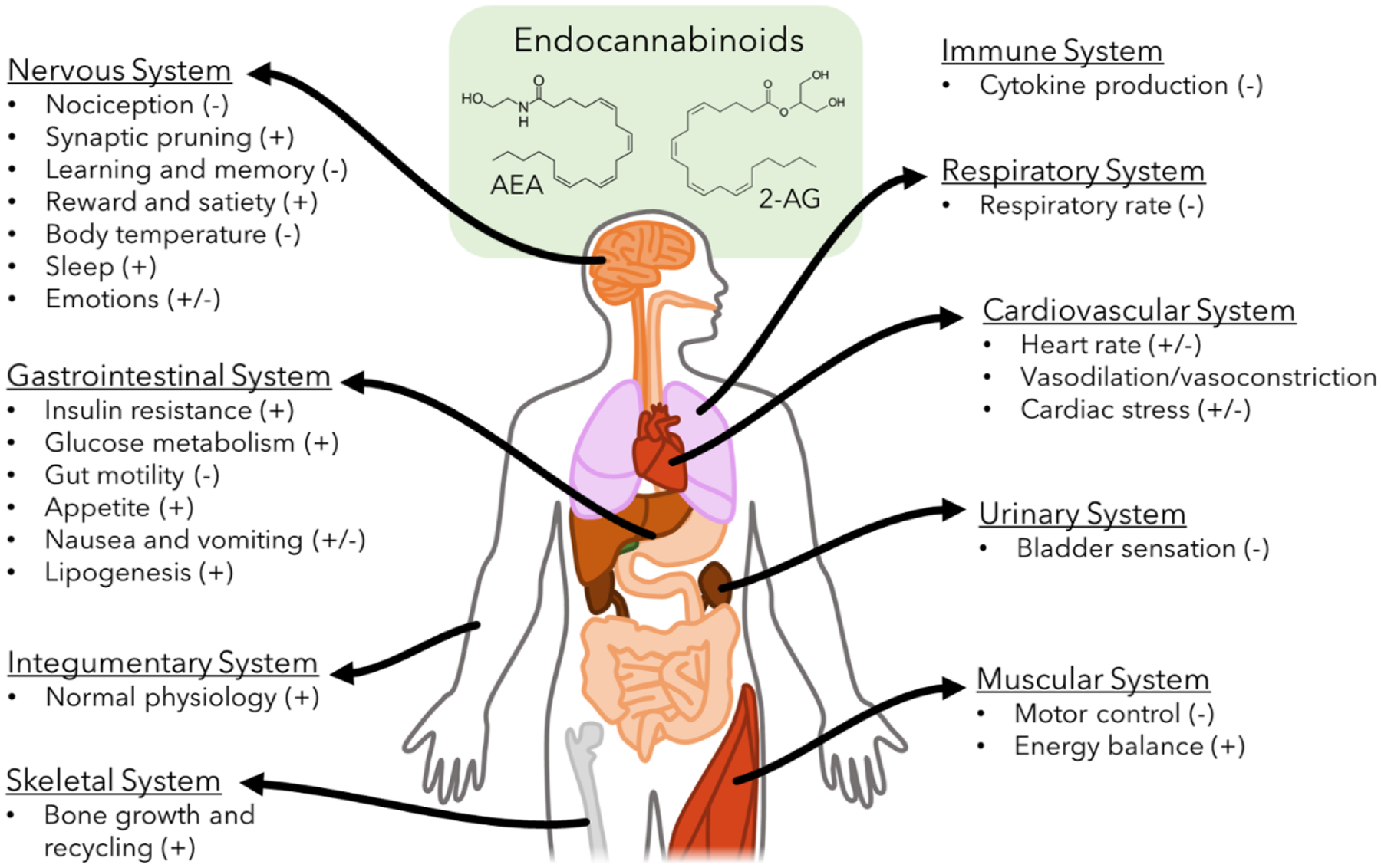
Endocannabinoids, such as the AEA and 2-AG, have been implicated in the modulation and maintenance of many human organ systems. These effects may be induced (+), suppressed (−), or variable (+/−) following endocannabinoid activation.
CB1R is widely expressed throughout the central nervous system where it mediates the psychoactive effects of Δ9-THC (Devane et al., 1988; Herkenham et al., 1991; Herkenham et al., 1990; Mailleux and Vanderhaeghen, 1992; Matsuda et al., 1990; Tsou et al., 1998). However, CB1R is also present in peripheral “non-neuronal” tissues including the liver, adipose tissue, and the pancreas (Cota et al., 2003; Osei-Hyiaman et al., 2005; Ravinet Trillou et al., 2004). Within the brain, CB1R is most densely expressed in the cerebral cortex, amygdala, basal ganglia, hippocampus, and cerebellum (Herkenham et al., 1990; Mackie, 2005). However, CB1R is also found at moderate levels in brain and spinal cord regions associated with pain, such as the dorsal root ganglia, periaqueductal gray (PAG), rostro-ventral medulla, and the limbic system (Ahluwalia et al., 2000; Mitrirattanakul et al., 2006; Wilson-Poe et al., 2021). Interestingly, CB1R is highly expressed in the cortico-limbic brain circuits that are central to the processing of affective components of pain (Burns et al. 2007; Lee et al. 2013). Agonist activation of CB1R has been shown to lead to stimulation of mitogen-activated protein kinases (MAPK) (Bouaboula et al., 1995) and G protein-coupled inward rectifying potassium channels (GIRKs) (Mackie et al., 1995) as well as inhibition of adenylyl cyclase (AC) (Howlett, 1985; Howlett and Fleming, 1984; Howlett et al., 1986) and voltage-gated calcium channels (VGCCs) (Mackie et al., 1993; Mackie and Hille, 1992) (Figure 2). Functional selectivity has been demonstrated for CB1R with specific agonists exerting distinct and differential modulatory effects on these signaling pathways (Ibsen et al., 2017; Khajehali et al., 2015; Laprairie et al., 2016).
Figure 2. CB1R modulation of intracellular signaling pathways.
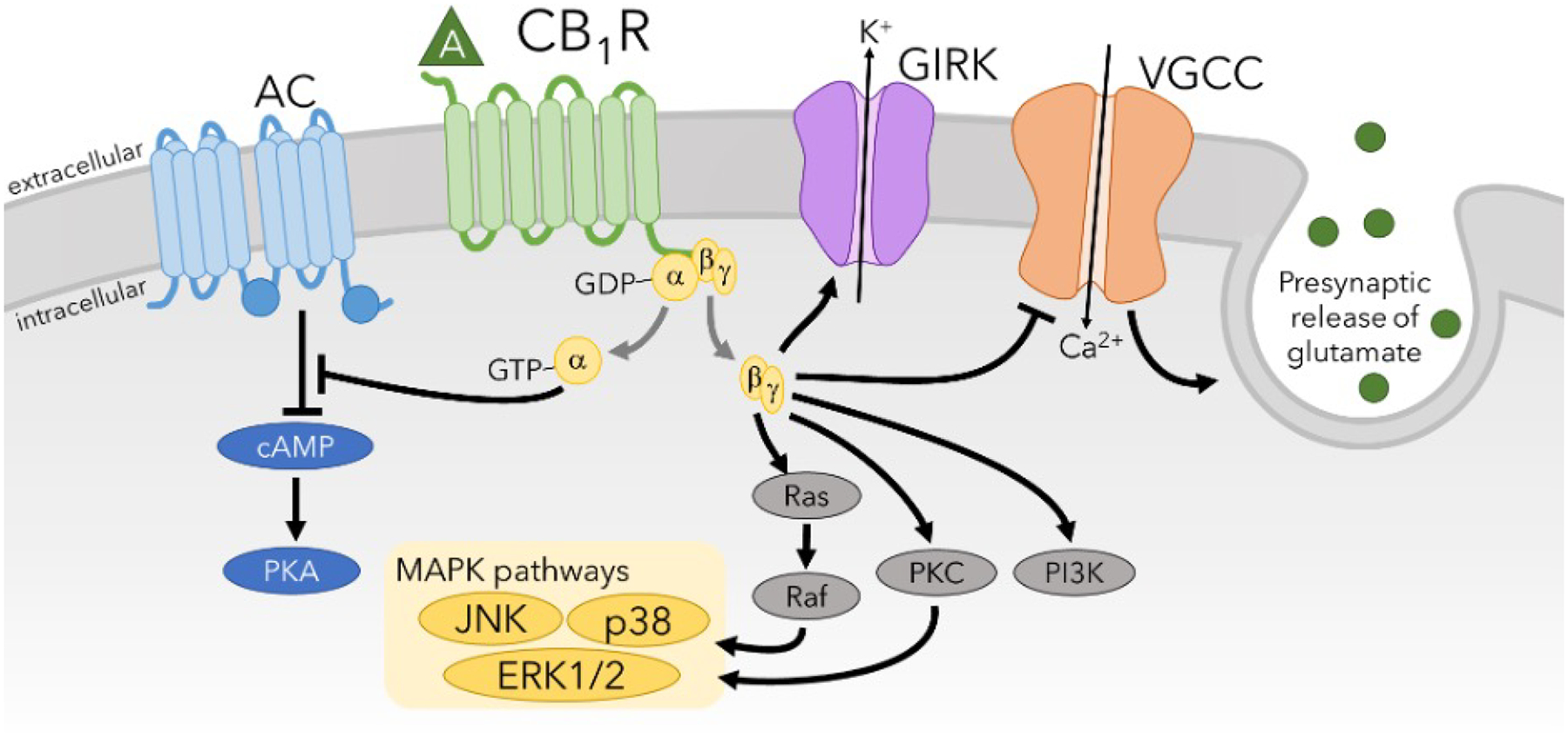
Agonist-induced activation by CB1R causes inhibition of VGCCs and adenylyl cyclase and production of cAMP while stimulating GIRK potassium channels and MAPK signaling pathways such as extracellular-regulated kinase (ERK) 1/2, JNK, and p38. Activation of CB1R also increases PKC and PI3K signaling pathways. A major overall effect of CB1R activation is that it inhibits neuronal signaling by preventing neurotransmitter release (inhibition of VGCCs) and hyperpolarization of the membrane potential (activation of GIRKs).
CB2R is also a Gαi/o-coupled GPCR that is expressed mainly on cells of the immune system although low neuronal expression of CB2R has been reported in the brain (Munro et al., 1993; Van Sickle et al., 2005). However, despite low expression of CB2R in neurons, the analgesic effects of cannabinoids are mediated via action at both CB1R and CB2R. Since CB2R is coupled to Gai/o proteins, it modulates many of the same intracellular signaling pathways as CB1R including inhibition of AC (Felder et al., 1995) and activation of MAPKs (Bouaboula et al., 1996), activation of Phosphoinositide-3 kinase (PI3K)-Akt/Protein Kinase B signaling (Molina-Holgado et al., 2007), and GIRKS (Ho et al., 1999) (Figure 3). It has also been shown that when CB2R is transfected into primary hippocampal neurons, it can also inhibit VGCCs and synaptic transmission (Atwood et al., 2012a). A high degree of functional selectivity for the CB2R has been demonstrated with CP55,940 but not WIN55,212-2 causing robust inhibition of VGCCs and β–arrestin2 recruitment (Atwood et al., 2012b). Several studies have also identified orphan GPCRs such as GPR3, GPR6, GPR12, GPR18, and GPR55 that can mediate the effects of some cannabinoids under certain circumstances (Allende et al., 2020; Morales et al., 2017; Nourbakhsh et al., 2019; Ryberg et al., 2007). Tolerance to cannabinoids is almost entirely due to neuroadaptations occurring at the CB1R with no tolerance occurring for CB2R selective agonists. Therefore, the mechanisms responsible for these neuroadaptations at CB1R that cause tolerance to cannabinoids will be a main focus of this review.
Figure 3. CB2R-mediated signaling pathways.
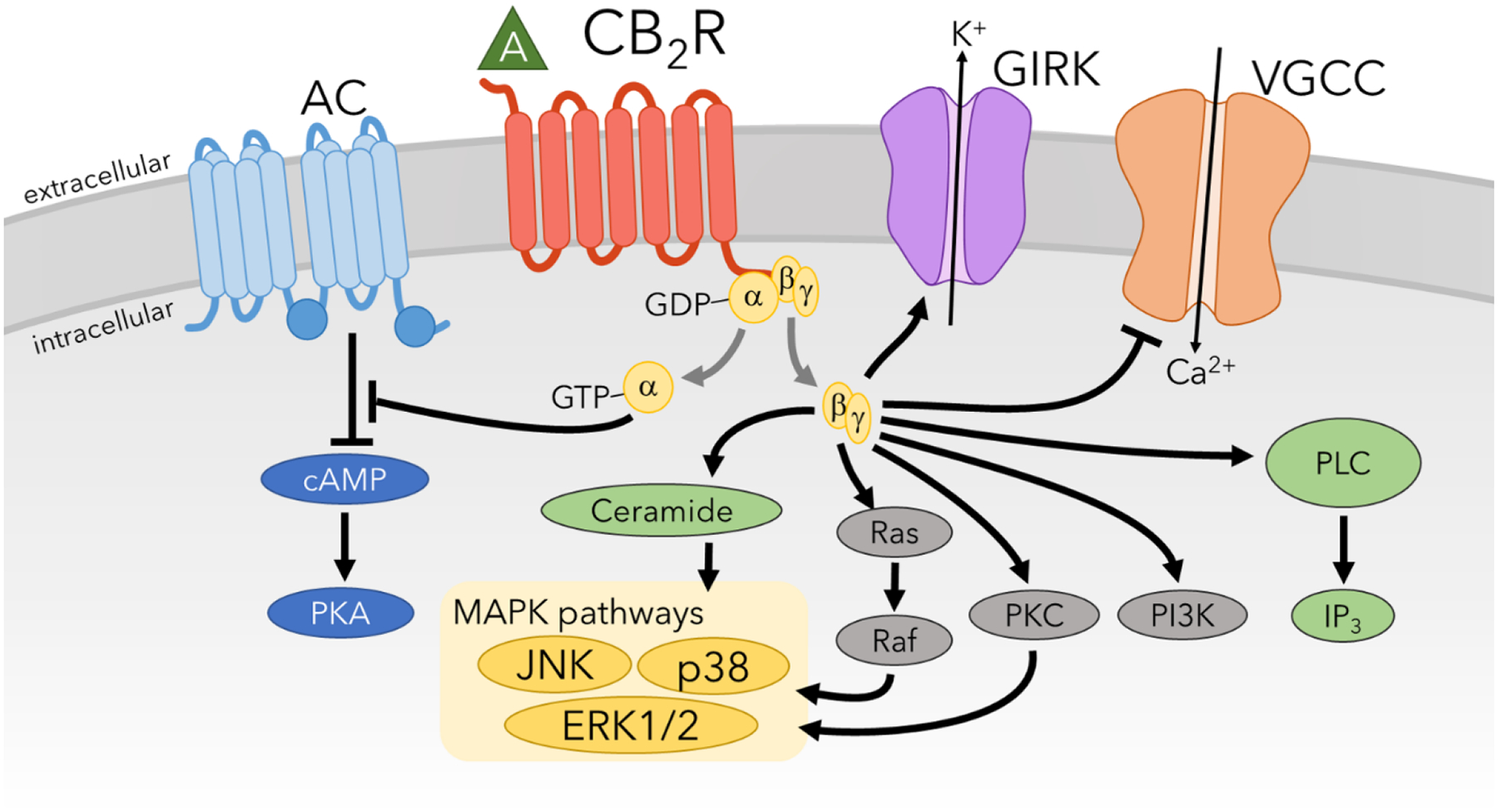
While the targets of CB2R signaling are largely similar to that of CB1R, differences in biases for these pathways account for significant differences in physiology and behaviors when activated. Additionally, CB2R activation has substantial anti-inflammatory components, which are produced through stimulation of ceramide and phospholipase C (PLC) pathways.
Two primary endocannabinoids have been identified, N-arachidonoyl ethanolamine (AEA; anandamide) and 2-arachidonoyl-glycerol (2-AG) (Devane et al., 1992; Stella et al., 1997). In the brain, these endocannabinoids are produced on demand in post-synaptic neurons from plasma membrane phospholipids in response to increased levels of intracellular calcium and/or excitatory post-synaptic potential-induced depolarization of the plasma membrane (Maejima et al., 2001; Maejima et al., 2005). AEA can be produced from phosphatidylethanolamine (PE) can occur multiple synthetic pathways. Most 2-AG is produced through the action of phospholipase C to produce diacylglycerol (DAG) from phosphatidylinositol 4,5-bisphosphate (PIP2), followed by the action of sn-1-diacylglycerol lipase alpha and beta (DAGL α/β) to convert DAG to 2-AG (Bisogno et al., 2003).
The concentration of 2-AG (~10 nM) in the brain has been shown to be almost 1000-fold higher than that of AEA (~50 pM) (Stella et al., 1997). In vitro molecular pharmacological assays have shown that AEA is a partial agonist at cannabinoid receptors while 2-AG acts as a full agonist. Endocannabinoids diffuse in a retrograde manner across the synapse where they act at pre-synaptic CB1R to suppress neurotransmission (Kreitzer and Regehr, 2001; Maejima et al., 2001; Wilson and Nicoll, 2001). Both 2-AG and AEA exert antinociceptive effects on pain through their action at CB1R and CB2R (Guindon et al., 2006; Guindon et al., 2007; Woodhams et al., 2015). AEA can also exert modulatory effects on pain by acting as an endovanillioid signal at the transient receptor potential cation channel subfamily V member 1 (Fischbach et al., 2007; Smart et al., 2000; van der Stelt et al., 2005). The duration of action for endocannabinoid signaling is tightly regulated and transient since these lipid signals are rapidly degraded by hydrolytic enzymes after signaling has occurred (Di Marzo et al., 1994). Hydrolysis of AEA occurs via fatty-acid amide hydrolase (FAAH) (Cravatt et al., 1996). Breakdown of 2-AG is medicated predominately through the action of monoacylglycerol lipase (MAGL; 85% of 2-AG breakdown) (Dinh et al., 2002). The remaining 15% of 2-AG breakdown is mediated predominately by members of the alpha-beta-hydrolase domain family including ABHD6 and ABHD12 (Blankman et al., 2007; Marrs et al., 2010).
1.2. Synthetic Cannabinoids
In addition to cannabis, and oral and inhaled formulations of Δ9-THC, a wide range of synthetic cannabinoids are also commonly used for recreational purposes. These synthetic cannabinoids are highly potent, full agonists at CB1R and the type 2 cannabinoid receptor (CB2R) and bind with a 10–100X higher binding affinity compared to Δ9-THC (Ford et al., 2017; Gatch and Forster, 2016). These synthetic cannabinoids also exhibit increased potency relative to Δ9-THC in preclinical animal models (Gatch and Forster, 2016). Synthetic cannabinoids were originally synthesized as research tools during the 1980s and have been produced by clandestine laboratories since the early 2000s and sold as “legal cannabis”. Recreational synthetic cannabinoid compounds such as JWH-018, are typically sprayed onto organic herbal plant mixtures and have been marketed throughout Europe, Australia and New Zealand, and the US under the trade names “K2”, “Spice”, “Black Mamba”, and “Scooby Snax”. The specific synthetic cannabinoids produced for these products is a constantly evolving subset of compounds that have undergone several generational changes to evade Drug Enforcement Agency scheduling and retain legal status (Ford et al., 2017).
Many of the adverse clinical effects associated with synthetic cannabinoid consumption also occur for cannabis and natural cannabinoids. For example, acute intoxication with synthetic cannabinoids typically causes euphoria, dizziness, somnolence, dry mouth, and nausea. Users of synthetic cannabinoids present to emergency departments for clinical care much more frequently than users of natural cannabis. The typical symptoms associated with acute synthetic cannabinoid intoxication include anxiety, panic, agitation, tremors, tachycardia, and emesis (Cooper, 2016). However, in some cases, serious, potentially life-threatening cardiovascular, renal, neurological, and gastrointestinal conditions arise and require immediate treatment (Tait et al., 2016). Synthetic cannabinoid-induced tachycardia can often be associated with chest pain, dyspnea, and at least 8 cases of myocardial infarction have been reported (Armenian et al., 2018). Hypertension resulting from synthetic cannabinoid use has also been reported (Ford et al., 2017). Acute kidney injury is relatively rare, but overdose injury from synthetic cannabinoids has resulted in several deaths (Tait et al., 2016). Although less common, clinically significant hyperemesis and seizures have also been reported (Tait et al., 2016).
2. Clinical Use of Cannabinoids
2.1. Medical Cannabinoids
Medical cannabinoids refer to the use of cannabis or cannabinoids as medical therapy to treat disease or alleviate symptoms. These compounds can be natural such as herbal cannabis, cannabis extracts, or synthetics such as nabilone. Medically prescribed cannabinoids include dronabinol capsules, nabilone capsules, and the oromucosal spray nabiximols (Hazekamp et al., 2013). Some countries have legalized medicinal-grade herbal cannabis for chronically ill patients. Canada and the Netherlands have government-run programs in which specialized companies supply quality-controlled herbal cannabis (Hazekamp et al., 2013). In the United States, 37 states plus the territories of Guam and Puerto Rico, and the District of Columbia, have introduced laws to permit the medical use of cannabis. There is evidence to support the use of cannabinoids for the treatment of chronic pain, muscle spasticity, nausea and vomiting due to chemotherapy, to improve weight gain in HIV infection, treatment of sleep disorders, and symptom management in Tourette syndrome (Whiting et al., 2015). The National Academies of Science published a report in January 2017 outlining the health effects of cannabinoids and cannabis in medicine (National Academies of Sciences and Medicine, 2017). The committee of experts tasked with this report resolved there was conclusive evidence that cannabis or cannabinoids are effective for the treatment of chronic pain, emesis, and patient-reported muscle spasticity from multiple sclerosis (MS). The report concludes there is moderate evidence that cannabis or cannabinoids are effective for sleep disturbances associated with obstructive sleep apnea, fibromyalgia, chronic pain, or MS. There is limited evidence that cannabis or cannabinoids are effective for the treatment of anxiety disorders, post-traumatic stress disorder, clinician-reported MS muscle spasticity, cachexia and anorexia associated with HIV infection. Limited evidence also suggests that cannabinoids are not effective for improving dementia, intraocular pressure associated with glaucoma, depressive symptoms in chronic pain and MS patients.
2.2. Cannabinoid Use Disorder
However, the long-term use of cannabinoid for recreational purposes or as medicines for chronic diseases is associated with limitations and disadvantages including tolerance, dependence, and/or the risk for cannabis use disorder (CUD). The lifetime risk of developing CUD in individuals that have used cannabis is estimated to be ~9.1%. However, this value has been challenged as an underestimate by other research that projects the prevalence of CUD to be closer to 22% in non-naive individuals that have used cannabis more than 5 times (Leung et al., 2020). For daily or weekly cannabis users the risk for CUD increases to 33%. As defined by the Diagnostic and Statistical Manual (DSM-5) from the American Psychiatric Association, the criteria for diagnosis with CUD include tolerance (the primary topic of this review), escalation of use, unsuccessful attempts to stop use, craving, usage despite adverse consequences, and withdrawal symptoms during early abstinence (American Psychiatric Association, 2013). CUD is responsible for 16% of admissions to drug treatment facilities which use combinations of cognitive-behavior therapies, motivational enhancement, and contingency management approaches to promote abstinence (Elkashef et al., 2008). Other treatment approaches include the management of cannabis withdrawal syndrome using cannabinoid replacement therapies.
2.3. Cannabis Withdrawal Syndrome
Cannabis withdrawal syndrome (CWS) is defined as somatic and behavioral symptoms associated with abstinence from cannabinoid consumption in dependent users. CWS represents the hallmark of cannabis dependence and is one of the DSM-V diagnostic criteria for CUD. Physical dependence resulting from heavy use is a hallmark of CUD and may underlie the poor success rates in its treatment (Budney et al., 2008; Ramesh et al., 2011). Symptoms of CWS include anger, anxiety, decreased appetite, dysphoria, craving irritability, and sleep disturbances (Budney et al., 2007; Curran et al., 2016). The onset of CWS starts ~24 hours after abstinence, peaks at ~2–3 days post abstinence, and continues for ~2–3 weeks (Budney et al., 2004).
Decreased levels of striatal dopamine have been demonstrated in patients with CUD and the degree of striatal dopamine deficits correlated with the frequency of cannabis use and the severity of neurocognitive problems including poor working memory and inattention (van de Giessen et al., 2017). Inhalation of vaporized Δ9-THC reduced binding of [11C]raclopride, a PET ligand for dopamine D2 and D3 receptors, by 4% in the striatum of healthy controls suggesting that cannabinoids such as Δ9-THC causes modest increases in striatal dopamine release, a neurotransmitter change associated with drug reward. Although, this 4% decrease in D2/D3 availability demonstrates that Δ9-THC shares addictive properties with other drugs of abuse, it was relatively modest compared to the 10–30% reductions observed for other drugs of abuse such as alcohol, nicotine, cocaine, and amphetamine (Bossong et al., 2009).
Sustained use and abuse of synthetic cannabinoids can also result in a withdrawal syndrome consisting of headache, tremors, emesis, anxiety, nightmares, and tachycardia that has been treated effectively with the atypical antipsychotic medication, quetiapine (Castaneto et al., 2014; Cooper, 2016). A recent emergency department case report described the use of lorazepam to treat tonic-clonic seizures in a heavy user of the synthetic cannabinoid, K2 (Sampson et al., 2015). A survey of synthetic cannabinoid users found that a subset of chronic users report a desire for clinical treatment to help reduce their consumption (Winstock and Barratt, 2013).
There are limited pharmacotherapies that have been tested for the treatment of CUD and none have been approved by the Food and Drug Administration. The use of oral Δ9-THC to treat CUD has yielded the most promising results and has been shown to attenuate CWS in a dose-dependent manner. However, the highest and most effective doses of oral Δ9-THC also produced intoxication and subjective “liking” effects that are indicative of potential abuse liability (Budney et al., 2007). Smoked cannabis has also been shown to reduce shown to reduce CWS (Haney et al., 1999). Other studies have shown that divalproex (Levin et al., 2004), buproprion (Haney et al., 2001), buspirone (McRae-Clark et al., 2015), fluoxetine (Cornelius et al., 2010), or lithium (Johnston et al., 2014) were not effective for treatment of CUD or reducing CWS.
Animal studies have also demonstrated CWS in rodents (Aceto et al., 1995; Cook et al., 1998; Hutcheson et al., 1998; Lichtman et al., 2001a; Tsou et al., 1995; Wilson et al., 2006) or dogs (Lichtman et al., 1998) exposed to injected Δ9-THC or smoked cannabis. Rats exposed to repeated treatment with WIN55,212-2 exhibited signs of spontaneous withdrawal when drug treatment was stopped (Aceto et al., 2001). However, the existence of spontaneous CWS in animal models is controversial and has been difficult to demonstrate reliably. Therefore, most studies of CWS in animal models have used rimonabant (SR141716A), an inverse agonist at CB1R, to precipitate a CWS in mice chronically exposed to cannabinoids. Somatic symptoms of precipitated withdrawal in rodents include wet dog shakes, forepaw fluttering, head shakes, chewing, tongue rolling, scratching, retropulsion and ptosis (Aceto et al., 1995; Tsou et al., 1995). Precipitated withdrawal from Δ9-THC in mice also causes impairments of spatial memory (Wise et al., 2011).
Precipitated withdrawal from Δ9-THC was absent in CB1R knock-out (KO) mice (Ledent et al., 1999) and reduced in mu opioid receptor (MOR) KO mice (Lichtman et al., 2001b) and pre-proenkephalin KO mice lacking endogenous ligands for the MOR (Valverde et al., 2000). Precipitated cannabinoid withdrawal in mice has been shown to elicit increases in adenylyl cyclase activity and cAMP levels in the cerebellum, implicating neuroadaptations in cAMP/Protein Kinase A (PKA) signaling as important modulators of cannabinoid withdrawal (Hutcheson et al., 1998; Tzavara et al., 2000). Treatment with RP-8Br-cAMP, a cAMP antagonist, reduced some somatic signs of precipitated withdrawal in Δ9-THC dependent mice providing further support for cAMP/PKA signaling in cannabinoid withdrawal (Tzavara et al., 2000). Corticotropin-releasing factor (CRF) signaling and c-fos expression, a biochemical marker of neuronal activation, are increased in the central nucleus of the amygdala of mice undergoing precipitated withdrawal suggesting engagement of stress and negative affect pathways. Similar to human clinical trials, treatment with Δ9-THC reduces withdrawal signs occurring during precipitated withdrawal. In addition, enhancement of endocannabinoid tone, using inhibitors of the FAAH and MAGL, the enzymes that hydrolyze AEA and 2-AG, respectively, also can reduce precipitated withdrawal somatic signs (Schlosburg et al., 2009). This finding raises the intriguing possibility that FAAH or MAGL inhibitors might be useful as a therapeutic in humans to reduce CWS.
3. Cannabinoid Tolerance
3.1. Cannabinoid Tolerance in Humans
Multiple studies have documented tolerance to the effects of Δ9-THC or inhaled cannabis in human subjects (Benowitz and Jones, 1981; D’Souza et al., 2008; Gorelick et al., 2013; Hunt and Jones, 1980; Jones et al., 1976; Jones et al., 1981; Mason et al., 2021). Tolerance to the psychomimetic, amnestic, perceptual effects, anxiogenic, and cortisol increasing effects of intravenous Δ9-THC has been demonstrated in frequent cannabis users (D’Souza et al., 2008; Ranganathan et al., 2009). Interestingly, the euphoric effects of intravenous (IV) Δ9-THC were not blunted in cannabis users providing evidence that tolerance and the neuroadaptations associated with this process might develop in a response- and brain region-specific manner (D’Souza et al., 2008). Other studies have shown that tolerance develops to the intoxicating “high” effects of oral Δ9-THC (Gorelick et al., 2013) as well as the acute effects of Δ9-THC on functional connectivity between regions of the brain involved in reward signaling such as nucleus accumbens, ventral palladium, and cortex (Mason et al., 2021).
Clinical studies done in the 1970s and 80s demonstrated that acute intake of cannabis or Δ9-THC caused increased heart rate, orthostatic hypotension, decreased skin temperature (hypothermia) and increased body weight in healthy controls (Benowitz and Jones, 1981; Hunt and Jones, 1980; Jones et al., 1981). However, the effects of Δ9-THC or cannabis on heart rate (Benowitz and Jones, 1981; Hunt and Jones, 1980; Jones et al., 1981), blood pressure (Benowitz and Jones, 1981; Jones et al., 1981), hypothermic (Hunt and Jones, 1980), intoxicating “high” effects (Hunt and Jones, 1980), and body weight (Hunt and Jones, 1980) were attenuated in frequent cannabis users suggesting the development of tolerance. These studies typically used relatively high daily doses of Δ9-THC (60 mg/kg/day or greater) that caused partial 50% tolerance within 4 days and near complete 80% tolerance by 10–12 days of chronic treatment (Jones et al., 1981). One possible mechanistic explanation was that increases in Δ9-THC metabolism and clearance might account for this observed tolerance. However, the very modest changes in the Δ9-THC metabolism and renal clearance of Δ9-THC metabolites that were detected are not sufficient to account for the magnitude of tolerance observed in clinical studies (Benowitz and Jones, 1981; Hunt and Jones, 1980). These early findings suggested that cannabinoid tolerance was mediated by pharmacodynamic changes in CB1R function rather than differences in cannabinoid pharmacokinetics.
Indeed, as the ability to probe pharmacodynamic changes in CB1R availability in humans using neuroimaging became possible as effective positron emission tomography (PET) radioligands were developed, it became evident that pharmacodynamic neuroadaptations in CB1R played a critical role in cannabinoid tolerance. Multiple studies have demonstrated that CB1R is downregulated in cannabis users meeting the diagnostic criteria for CUD using CB1R selective PET radioligands. Downregulation of CB1R in cannabis users was less profound in subcortical brain regions compared to cortical regions. The finding of brain-region specific CB1R downregulation in cortical brain areas but not subcortical regions in humans is consistent with studies assessing CB1R down-regulation in animal models that will be discussed later in this review.
PET neuroimaging found that CB1R was downregulated by 20% in chronic cannabis users and that these decreases occurred primarily in cortical versus subcortical brain regions (Hirvonen et al., 2012). This study also found that decreased CB1R levels were reversed after 28 days of abstinence suggesting that chronic cannabis use doesn’t cause long-term changes. A subsequent study probed the amount of time required for CB1R downregulation to recover in chronic cannabis users by performing PET neuroimaging after two and 28 days of abstinence. This work found that CB1R availability decreased by ~15% across most brain regions in dependent cannabis users but these differences in CB1R levels were no longer detectable after two days of abstinence (D’Souza et al., 2016). There was also a strong negative correlation between CB1R levels and the severity of withdrawal symptoms, suggesting that CB1R down-regulation is strongly involved in CWS. Other studies using different PET neuroimaging ligands found that CB1R availability was decreased by ~11% in chronic cannabis users compared to healthy controls with little evidence of CB1R down-regulation in sub-cortical brain regions (Ceccarini et al., 2015).
3.2. Cannabinoid Tolerance in Animal Studies
Cannabinoid agonists have been shown to elicit well-characterized “tetrad” effects in animals that include tail-flick antinociception, decreased body temperature, decreased locomotor activity, and catalepsy (Janoyan et al., 2002; Moore and Weerts, 2022; Ryan et al., 1995). Tetrad effects are absent in CB1R KO mice or in mice treated with CB1R antagonists such as rimonabant (Rinaldi-Carmona et al., 1994). As mentioned above, the antinociceptive effects of cannabinoids in the context of pathological and/or chronic pain is also partially mediated by CB2R. However, CB2R agonists have been shown to have minimal effects on tail-flick tests of antinociception that are mediated predominately by spinal mechanisms (Lin et al., 2018; Yuill et al., 2017).
Tolerance occurs with repeated, chronic drug administration resulting in a progressive decrease in response. Tolerance to the “tetrad” effects (antinociception, hypothermia, catalepsy, and hypoactivity) of twice-daily injections of 10 m/kg Δ9-THC has been demonstrated in mice (Abood et al., 1993; Bass and Martin, 2000; Oviedo et al., 1993). This dosing paradigm caused a 27-fold rightward shift in the dose response curve (ED50) for Δ9-THC due to the development of tolerance (Abood et al., 1993). Tolerance to antinociceptive and hypoactive effects of Δ9-THC developed rapidly with mice demonstrating the onset of robust partial tolerance after only 3 injections of 10 mg/kg Δ9-THC with compete tolerance occurring following 7 injections (Abood et al., 1993). This study also found that tolerance to the motility and antinociceptive effects of Δ9-THC were fully reversed by 7.5 days and 11.5 days of abstinence, respectively (Abood et al., 1993). Another study found that Δ9-THC tolerance developed in a manner that was dose-dependent in mice with higher doses producing increased tolerance (McKinney et al., 2008). Tolerance has also been demonstrated for the inhibitory effects of Δ9-THC on electrically-evoked contractions of the vas deferens (Pertwee et al., 1993), for the diuretic effects of Δ9-THC (Chopda et al., 2016), and for the effects of Δ9-THC on brain glucose utilization (Whitlow et al., 2003). Interestingly, tolerance to the effects of cannabinoids on memory and neuroendocrine functions took much longer (weeks to months) to occur suggesting that cannabinoid tolerance develops in a response-specific manner (de Miguel et al., 1998; Gonzalez et al., 1999; Hampson et al., 2003).
While tolerance to many of the effects of Δ9-THC have been demonstrated across many studies, tolerance to the endocannabinoids, AEA and 2-AG, has only been investigated more recently. Chronic administration of exogenous AEA to FAAH KO mice caused modest shifts in the dose-response curve for AEA compared to the dose response curves for Δ9-THC in FAAH KO mice chronically treated with Δ9-THC (Falenski et al., 2010). Neuroadaptations such as CB1R downregulation and desensitization have been demonstrated to occur with chronic cannabinoid drug treatment and will be discussed in detail later in this review. Interestingly chronic AEA treatment produced less CB1R downregulation and desensitization compared to chronic treatment with Δ9-THC. Treatment with the MAGL inhibitor JZL184 has been shown to dramatically increase 2-AG levels and elicit “tetrad” effects (Long et al., 2009). However, repeated treatment with 40 mg/kg JZL184 resulted in tolerance to these effects while also causing CB1R neuroadaptations such as downregulation and downregulation (Schlosburg et al., 2014). Supporting the finding that tolerance develops for the effects of 2-AG, the effects of WIN55,212-2 on cannabinoid “tetrad” behaviors and synaptic plasticity in the hippocampus were blunted in MAGL KO mice (Pan et al., 2011; Schlosburg et al., 2010). Tolerance to the antinociceptive and gastroprotective effects of JZL184 can be prevented using lower drug doses (4 mg/kg) that don’t cause CB1R downregulation and desensitization, suggesting that tolerance to 2-AG is concentration/dose-dependent (Kinsey et al., 2013). Taken together these results suggest that tolerance develops for 2-AG to a much greater extent than it does for AEA. However, tolerance to both endocannabinoids seems to be less pronounced than tolerance to chronically administered exogenous cannabinoids such as Δ9-THC, WIN55,212-2, and CP55,940.
Tolerance to the “tetrad” effects of synthetic cannabinoids such as CP55,940 and WIN55–212,2 have also been demonstrated (Fan et al., 1996; Gomez et al., 2021; Nealon et al., 2019; Sim-Selley and Martin, 2002). Tolerance to the hypothermic effects of commonly abused synthetic cannabinoids found in “K2” or “Spice” including AB-PINACA, 5F-AB-PINACA, 5F-ABD-PINACA, and JWH-018 has also demonstrated (Wilson et al., 2022). Interestingly, tolerance was much less for 5F-AB-PINACA, 5F-ABD-PINACA than for JWH-018 suggesting agonist-specific differences in tolerance. Indeed, we and others have demonstrated tolerance develops at different rates for many cannabinoid agonists with synthetic, high-potency, full cannabinoids such as WIN55,212-2 and CP55,940 often showing slower tolerance compared to Δ9-THC (Figure 4) (Henderson-Redmond et al., 2020; Nealon et al., 2019; Wilson et al., 2022). These observations suggest the possibility of agonist-specific mechanisms of cannabinoid tolerance that will be elaborated on in detail later in this review.
Figure 4. Wildtype mice display faster tolerance to Δ9-THC compared to CP55,940, a strongly internalizing cannabinoid agonist.
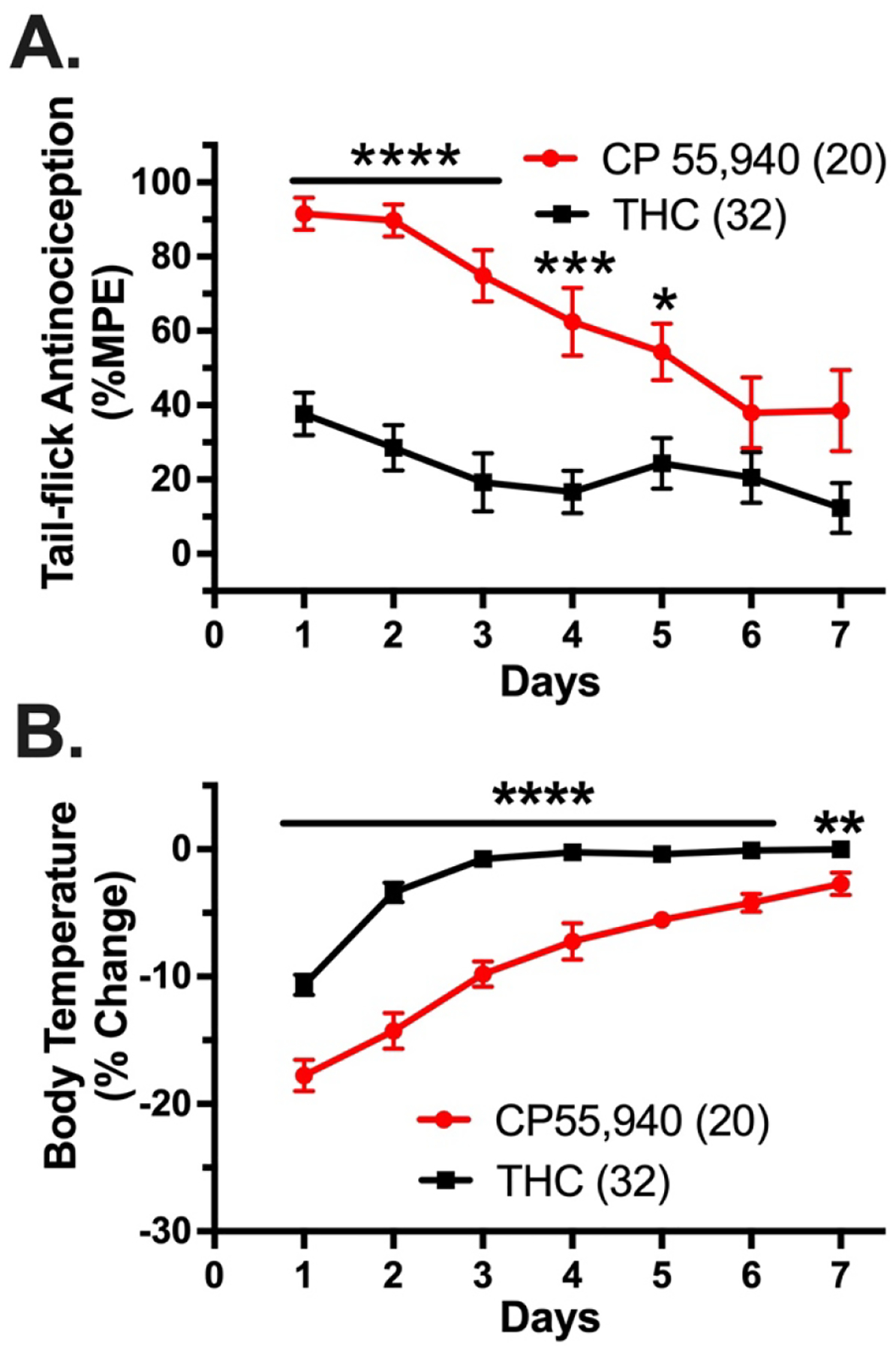
Wild-type mice were treated with once-daily intraperitoneal injections of 0.3 mg/kg CP55,940 (red circles and line) or 30 mg/kg Δ9-THC (THC; black squares and line) once daily and tested for tail-flick antinociception and body temperature 1 hour after drug treatment. To measure antinociception, a Columbus Instruments TF-1 tail-flick analgesia meter (Columbus, OH) was calibrated to an intensity of 5. To avoid potential tissue damage to the tail, the instrument was programmed to a 10 s cut-off time. Tail-flick measurements were recorded between 2–5 times for each time point. The recorded measurements were used to calculate the antinociceptive response as a percent of the maximum possible effect (%MPE) using the following equation: %MPE = [(post-drug latency)-(pre-drug latency)]/[pre-determined cut-off time (10 s)-(pre-drug latency)]x100. Hypothermia was assessed by taking each subject’s body temperature using a mouse rectal thermometer (Physiotemp Instruments, Clifton, NJ) prior to and 60 minutes following injection. Recorded values, in °C, were used to calculate the percent change in body temperature (%Δ) = [(post-body temperature)-(pre-body temperature)/(pre-body temperature)]x100. Data represent mean values ± SEM and were analyzed by mixed two-way ANOVA with Bonferroni post-hoc tests (*p<0.05, **P<0.01, ***p<0.001, ****p<0.0001). For this experiment, tolerance is defined as the decrease in antinociceptive response each day that occurs with repeated dosing. There were significant effects of drug treatment (F1,46=28.56, p<0.0001), time (F4.417,195.8=11.74, p<0.0001), and a time × drug treatment interaction effect (F6,266=4.530, p<0.001) for the antinociceptive effects of CP55,940 versus Δ9-THC. There were significant effects of drug treatment (F1,50=112.3, p<0.0001), time (F3.858,187.1=94.36, p<0.0001), and a time × drug treatment interaction effect (F6,291=9.038, p<0.0001) for the hypothermic effects of CP55,940 versus Δ9-THC. The sample size of tested animals is designated in parentheses.
Supporting this hypothesis is recent work that assessed tolerance for PNR-4-20, a G protein biased CB1R agonist. While PNR-4-20 produced similar tolerance to Δ9-THC for its antinociceptive effects, tolerance was lower than Δ9-THC for the hypothermic and cataleptic effects of this biased agonist. In addition, cross-tolerance between Δ9-THC and PNR-4-20 was unidirectional with chronic Δ9-THC treatment causing tolerance to the effects of PNR-4-20, but chronic PNR-4-20 did not cause tolerance for Δ9-THC (Ford et al., 2019). In addition, tolerance was rapidly reserved by abstinence for PNR-4-20 but tolerance for the effects of Δ9-THC and JWH-018 persisted following 7–12 days of abstinence. Importantly, chronic PNR-4-20 caused much less downregulation of CB1R compared to chronic treatment with either Δ9-THC or JWH-018.
Treatment of rhesus monkeys with Δ9-THC caused tolerance to the discriminative and locomotor effects of Δ9-THC (Hruba et al., 2012). Treatment with Δ9-THC also produced cross-tolerance to subjective discriminative effects of the synthetic cannabinoids, CP55,940, JWH-018, and JWH-073 in rhesus monkeys (Hruba et al., 2012). Other studies demonstrated that tolerance developed to the sedative and cognitive effects of 1 mg/kg Δ9-THC in squirrel monkeys (Withey et al., 2021). However, tolerance to these effects of Δ9-THC was not altered by co-administration of 3 mg/kg CBD. Interestingly, this study found that tolerance didn’t develop for the ability of Δ9-THC to cause fragmented sleep patterns. Taken together, these data demonstrate robust tolerance to cannabinoids across a wide range of species including rodents, non-human primates, and humans.
Many drugs of abuse including cannabinoids and opioids cause dopaminergic neurons of the ventral tegmental area to release dopamine within the ventral striatum (Gomez et al., 2021). This dopaminergic signaling between the VTA and striatum is an essential neurochemical signal for drug-induced reward and reinforcement. Tolerance to the effects of cannabinoids on striatal dopamine release has also been recently demonstrated. Chronic exposure to WIN55,212-2 produced tolerance to the effects of WIN55,212-2 on striatal dopamine release as well as cross-tolerance for the ability of heroin to elicit dopamine signaling (Gomez et al., 2021). Other studies have demonstrated that acute Δ9-THC causes decreased food intake as well as sleep disturbance including increased non-rapid eye movement (NREM) sleep and decreased REM sleep. This work found that chronic exposure to Δ9-THC caused tolerance to the effects on sleep and feeding behavior (Kesner et al., 2022). Recent work has assessed tolerance to the antinociceptive effects of orally consumed Δ9-THC in mice with chronic neuropathic pain caused by spared nerve ligation surgery (Abraham et al., 2020). This study demonstrated that tolerance developed for the ability of morphine but not Δ9-THC to alleviate mechanical sensitivity and hyperalgesia associated in mice with neuropathic pain. Interestingly, non-neuropathic mice given oral Δ9-THC also did not display tolerance to the “tetrad” effects of challenge injection of Δ9-THC suggesting that the route of administration (oral vs. systemic injection) likely plays an important role in the development of tolerance.
Historically, tolerance to cannabinoid compounds has been associated with downregulation and desensitization of CB1R (Martin et al., 2004). The role of CB2R in tolerance, conversely, is thought to be minimal. Numerous in vivo models of neuropathic pain have found that CB2R agonists provide significant antinociception without the development of tolerance (Blanton et al., 2019; Deng et al., 2015; Li et al., 2019; Lin et al., 2018). Similar findings of CB2R-mediated antinociception without tolerance are observed using inflammatory, postoperative, osteoarthritic, and hyperalgesia pain models (Yao et al., 2009; Yao et al., 2008; Yuill et al., 2017). However, tolerance to the immunosuppressive effects of the mixed cannabinoid agonist CP55,940 was found to be associated with CB2R, rather than CB1R, activity (Yao et al., 2008). In vitro studies have also found that CB2R agonists can induce desensitization and downregulation of the CB2R receptor in Chinese hamster ovary and HL-60 cells (Shoemaker et al., 2005a; Shoemaker et al., 2005b).
Recent investigation into the mechanisms of cannabinoid tolerance has revealed possible mechanisms for CB2R downregulation. However, this critical question remains largely unanswered, and this knowledge gap requires additional investigation. The c-Jun N-terminal kinase (JNK) signaling pathway can mediate cannabinoid tolerance to mixed cannabinoid agonists in both inflammatory and neuropathic pain models (Henderson-Redmond et al., 2020) and this mechanism will be discussed in detail later in this review. Inhibition of this JNK pathway produced antinociception in the formalin model of pain that was mediated by CB2R alone in female mice and was associated with upregulation in the mRNA for CB2R and decreased levels of β-arrestin1 transcript (Blanton et al., 2021). Research on the role of β-arrestin2 in cannabinoid tolerance revealed a role in CB1R and CB2R internalization that was only modestly increased by overexpression of GRK (Ibsen et al., 2019). Furthermore, this internalization was caused by 2-AG and CP55,940 but not by Δ9-THC. The role of β-arrestin in CB2R downregulation is further supported by the finding that the β-arrestin-biased CB2R agonist GW833972A produced greater tolerance than the cAMP-biased CB2R agonist, JWH-133 (Mlost et al., 2021). Recent in vitro studies suggest that the mechanism of CB2R-mediated β-arrestin2 recruitment is not through the classical GRK-mediated pathway, but rather through phosphomimetic C-terminal aspartate residues (Patel et al., 2022).
CB2R agonists are being explored as clinical analgesics due to the wealth of preclinical data demonstrating that they don’t cause tolerance and might also reduce opioid tolerance (Bie et al., 2018). CB2R agonists can delay or reverse morphine tolerance, possibly by reversing opioid-mediated inflammation, through a mechanism that involves activation of MAPK phosphatases 1 and 3 and reversal of opioid-mediated MAPK phosphorylation (Carey et al., 2023; Kong et al., 2022; Reichenbach et al., 2022). Whether CB2R agonists might also reduce tolerance for other GPCR agonists, including cannabinoid agonists acting through CB1R, has not been investigated to date and should be addressed in future studies. Two independent Phase 2 clinical trials on the analgesic potential of CB2R agonist Olorinab for abdominal pain due to Crohn’s disease and irritable bowel syndrome revealed significant relief of abdominal pain without the development of tolerance (Chang et al., 2023; Yacyshyn et al., 2021). The anti-inflammatory potential of the CB2R agonist, Lenabasum, was evaluated in patients with either cystic fibrosis or dermatomyositis, and was found to produce significant reductions in inflammatory markers without evidence of tolerance (Chmiel et al., 2021; Werth et al., 2022). While some preclinical data suggests the possibility of tolerance for CB2R agonists, current clinical data has yet to show such an effect and the numerous ongoing and planned clinical trials for CB2R -mediated analgesics will help bring further clarity to this critical question.
4. Mechanisms of Cannabinoid Tolerance
4.1. CB1R Neuroadaptations Involved in Cannabinoid Tolerance
As mentioned earlier, chronic cannabinoid treatment causes multiple CB1R neuroadaptations such as desensitization and downregulation that lead to tolerance. In this review, we define the process of desensitization as the functional uncoupling of the receptor from its G protein signaling moieties, a process that typically involves phosphorylation of the receptor by GRKs and the recruitment of β–arrestin protein (Figure 5). In contrast, downregulation corresponds to the permanent removal of receptor from the plasma member due to internalization and trafficking to lysosomal pathways for degradation. Both processes cause reductions in agonist-stimulated receptor signaling that are, at least partially, responsible for tolerance.
Figure 5. CB1R desensitization.
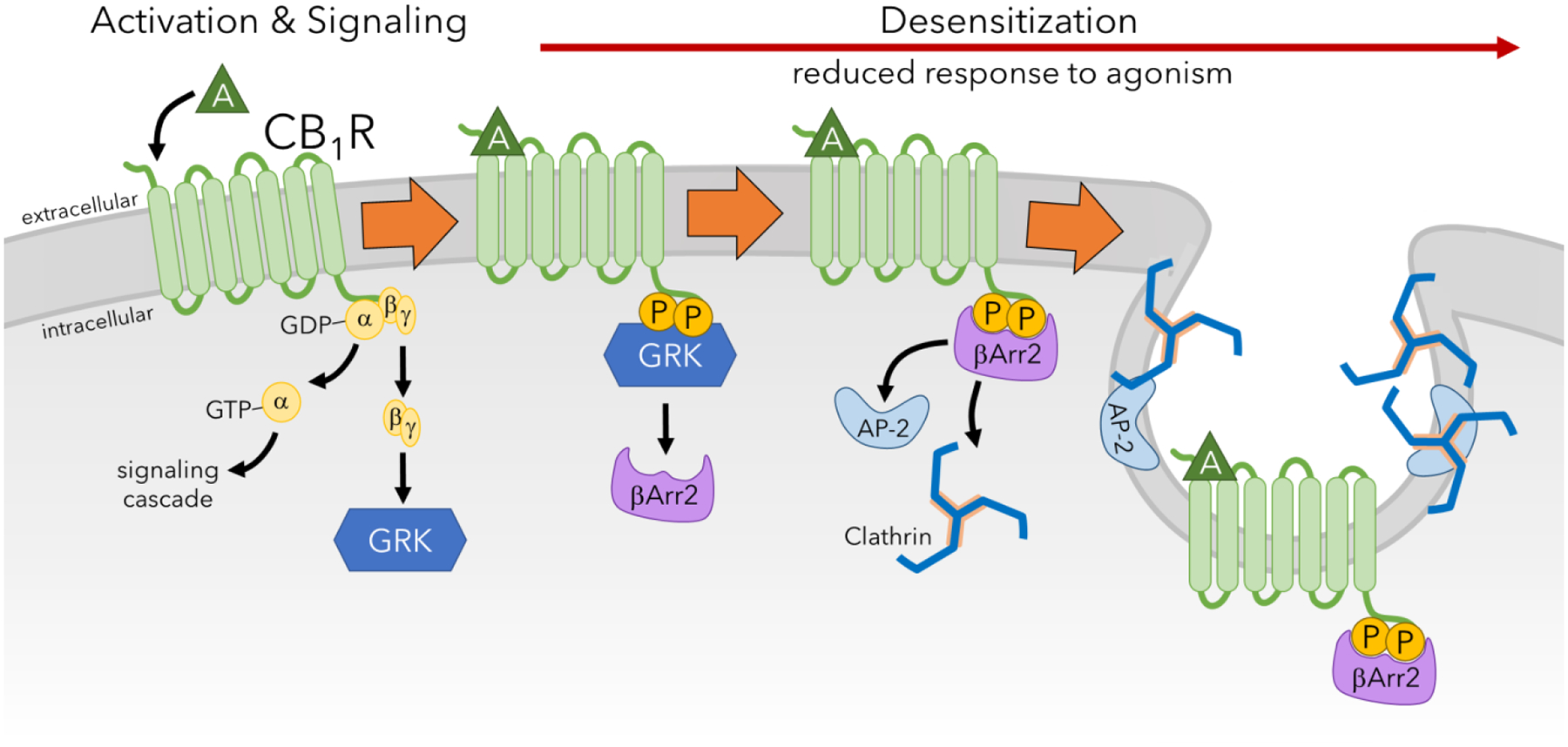
Upon activation of CB1R by an agonist (A), the coupled G protein disassociates. While the alpha subunit participates in signaling mechanisms, the beta and gamma complex recruits GRK. Phosphorylation of the C-terminus of CB1R by GRK results in the recruitment of β–arrestin2 (βArr2) which causes steric inhibition and prevents the G protein from interacting with CB1R. The association of β–arrestin2 with CB1R also causes recruitment of adaptor protein-2 (AP-2) and clathrin which facilitate endocytosis of the receptor. This mechanism of desensitization reduces response to CB1R activation and signaling, and repetitions of this process leads to downregulation and tolerance.
Early work demonstrated that tolerance due to chronic cannabinoid treatment was associated with robust down-regulation of CB1R. For example, chronic treatment with WIN55,212-2, CP55,940, or Δ9-THC resulted in a loss of cannabinoid receptor binding sites throughout the brains of tolerant mice and rats (Breivogel et al., 1999; Di Marzo et al., 2000; Romero et al., 1997). Chronic treatment with Δ9-THC caused time- and region-dependent downregulation of [3H]-WIN55,212-2 binding sites corresponding to ~20% loss of CB1R in the cerebellum, hippocampus, and striatum, with the hippocampus displaying the highest level of downregulation. Downregulation of CB1R was detected in the hippocampus and striatum after only three days of treatment with 10 mg/kg Δ9-THC, reaching maximal levels after 14–21 days of chronic Δ9-THC treatment (Breivogel et al., 1999). Importantly, chronic Δ9-THC treatment did not cause CB1R down-regulation in subcortical areas of the brain such as the globus pallidus, consistent with reports of CB1R availability in human cannabis users. In general, this study and others did not observe any change in radioligand binding affinity in Δ9-THC-tolerant rodents.
Other studies examining CB1R downregulation in Δ9-THC-tolerant rats also found that CB1R was decreased in the cerebellum, striatum, cerebral cortex, hippocampus, and brainstem, but not in the limbic forebrain (Di Marzo et al., 2000). Tolerance and CB1R downregulation in Δ9-THC and CP55,940-treated rodents was found to be dose-dependent with higher cannabinoid doses producing stronger tolerance and greater magnitudes of CB1R downregulation (McKinney et al., 2008; Oviedo et al., 1993). While some studies have found that Cnr1 transcript is decreased in the whole brains of Δ9-THC-tolerant rats, other studies have found no difference or increases in CB1R mRNA levels (Abood et al., 1993; Sim-Selley et al., 2006). Recent work has used Stochastic Optical Reconstruction Microscopy super-resolution imaging to assess CB1R distribution at a nanoscale level in the brains of Δ9-THC-tolerant mice (Dudok et al., 2015). This study found that chronic Δ9-THC caused 74% downregulation of CB1R and an increase in the ratio of internalized CB1R in the perisomatic boutons of hippocampal GABAergic interneurons. CB1R downregulation was partially rescued after 11 days of abstinence, although complete recovery of CB1R downregulation took 6 weeks. These results demonstrate that downregulation of CB1R might be persistent and long-lasting in Δ9-THC-tolerant rodents.
CB1R downregulation in the hippocampus and striatum of Δ9-THC-tolerant mice is generally longer acting than desensitization with recovery of CB1R levels taking 7 or 14 days of sustained abstinence in striatum and hippocampus, respectively, to achieve. This finding demonstrates that recovery of CB1R in tolerant animals occurs faster in some brain regions (striatum) than others (hippocampus) and these differences might explain response-specific differences in the development of and recovery from tolerance. In contrast, the recovery of CB1R desensitization generally takes between 3–7 days to occur depending on brain region (Sim-Selley et al., 2006). Interestingly, the recovery of CB1R downregulation during Δ9-THC abstinence did not correspond to and could not be explained by concurrent compensatory changes in CB1R transcript expression.
Chronic treatment with either Δ9-THC or WIN55,212-2 produced robust tolerance to the “tetrad” effects of these agonists. While chronic Δ9-THC and WIN55,212-2 treatments produced equivalent levels of CB1R downregulation, these agonists produced differing amounts of desensitization. Treatment with Δ9-THC produced greater (~2-fold) CB1R desensitization compared to WIN55,212-2, providing early evidence that cannabinoid tolerance might be mediated by agonist-specific neuroadaptations. As mentioned earlier in this review, the G protein-biased cannabinoid agonist, PNR-4-20, produced less tolerance than unbiased cannabinoid agonists such as Δ9-THC and JWH-018 (Ford et al., 2019). This study found that PNR-4-20 produced less CB1R downregulation in the hypothalamus and thalamus compared to Δ9-THC. Although chronic treatment with AEA did not induce CB1R downregulation (Falenski et al., 2010), chronic enhancement of 2-AG in MAGL KO mice did cause robust downregulation of CB1R, similar to the amount of downregulation that is observed in the brains of Δ9-THC-tolerant mice (Schlosburg et al., 2010). These findings raise the possible that G protein-biased agonists and therapeutics that enhance AEA levels might be able to yield sustained therapeutic efficacy due to reduced CB1R downregulation and tolerance for these agonists.
Many of the studies described above examining CB1R downregulation also assessed CB1R desensitization. These studies typically use agonist-stimulated [35S]-GTPγS binding assays to measure G protein activation and coupling in ex vivo tissue preparations to assess CB1R desensitization. For GPCRs, including CB1R, desensitization is linked to the phosphorylation of the GPCR by a G protein receptor kinase (GRK) and interaction of the phosphorylated receptor with an arrestin protein, such as β–arrestin2 (DeWire et al., 2007; Moore et al., 2007). GRKs phosphorylate serine and threonine residues, typically at residues in the 3rd intracellular loop or the C-terminus of the GPCR. For the CB1R, transfection of Xenopus oocytes with GRK 3 and β-arrestin2 was sufficient for CB1R desensitization, suggesting that these GRK and β-arrestin isoforms are sufficient for this process (Jin et al., 1999). However, it is likely that other GRKs and arrestin proteins can mediate CB1R desensitization in certain cell types where these other forms are expressed.
Chronic treatment with 10 mg/kg Δ9-THC for 21 days resulted in region-specific CB1R desensitization in mouse brain sections and membrane homogenates subjected to autoradiographic analysis of WIN55,212-2-stimulated [35S]-GTPγS binding (Sim et al., 1996). This study found widespread CB1R desensitization in most brain regions of Δ9-THC-tolerant mice that were examined including the hippocampus, cortex, striatum, and cerebellum. Notably, CB1R desensitization was not detected in the PAG, an area of the brain involved in antinociception and pain processing. Additional studies using a shorter 5 day duration of chronic Δ9-THC treatment detected CB1R desensitization in the substantia nigra but not in the globus pallidus or striatum (Romero et al., 1998). Furthermore, chronic Δ9-THC treatment can also produce neuroadaptations in the opioid system including increased agonist-stimulated mu opioid receptor signaling (Corchero et al., 1999). Multiple studies have found CB1R desensitization is rapid and can be detected after only 1–3 days of chronic Δ9-THC, while the magnitude of desensitization increases progressively with longer durations of Δ9-THC treatments (Breivogel et al., 1999; Corchero et al., 1999). Similar to CB1R downregulation in humans and animal models, desensitization was less pronounced in subcortical regions of the brain including the globus pallidus and striatum compared to cortical structures such as the hippocampus (Breivogel et al., 1999). Also similar to CB1R downregulation in animal models, desensitization was found to be both dose- and agonist-dependent (McKinney et al., 2008; Sim-Selley and Martin, 2002). While chronic treatment with Δ9-THC or WIN55,212-2 produced equivalent amounts of CB1R downregulation, Δ9-THC produced greater desensitization in some brain regions providing early evidence of agonist differences in CB1R neuroadaptations associated with cannabinoid tolerance (Sim-Selley and Martin, 2002). Abstinence from chronic Δ9-THC resulted in more rapid recovery of desensitized receptors in striatum compared to hippocampus (Sim-Selley et al., 2006). The restoration of WIN55,212-2-stimulated [35S]-GTPγS binding to control levels in Δ9-THC-tolerant mice took 14 days in the hippocampus compared to 3 days in the striatum. This finding demonstrates that the development of CB1R desensitization, as well as the recovery of this process can occur in a region-specific way that might underly response-specific differences in cannabinoid tolerance.
Other studies have assessed CB1R desensitization by measuring the desensitization of cannabinoid-mediated synaptic plasticity in hippocampal neurons. Depolarization of post-synaptic hippocampal neurons has been shown to suppress pre-synaptic glutamatergic signaling through a mechanism mediated by endocannabinoid release that causes a process known as depolarization-induced suppression of excitation (DSE). However, pretreatment with cannabinoid agonists such as WIN55,212-2 caused partial desensitization of DSE within 2 hours, while 24 hours of treatment resulted in complete desensitization of DSE (Kouznetsova et al., 2002). Desensitization of DSE was reduced by dominant negative forms of GRK2 and β–arrestin2, demonstrating that GRK signaling and β–arrestin signaling are critical for the process of CB1R desensitization (Kouznetsova et al., 2002).
Subsequent work demonstrated that the acute response to Δ9-THC was enhanced while tolerance for Δ9-THC was decreased in β–arrestin2 KO mice (Breivogel et al., 2008; Nguyen et al., 2012). This finding was similar to work demonstrating that the acute response to agonists such as morphine at MOR, another GPCR that is dependent on β–arrestin2, was enhanced in β–arrestin2 KO mice (Bohn et al., 1999). Deletion of β–arrestin2 enhanced WIN55,212-2 and CP55,940 [35S]-GTPγS binding but not binding of the CB1R selective antagonist, [3H]-SR141716A, demonstrating that CB1R -stimulated G protein signaling was enhanced in β–arrestin2 KO tissues without increases in the number of CB1R binding sites (Nguyen et al., 2012). Furthermore, deletion of β–arrestin2 reduced tolerance to the effects of Δ9-THC and reduced downregulation and desensitization of CB1R in the cerebellum, spinal cord, and PAG. Consistent with previous findings in humans and animal models, CB1R downregulation and desensitization in wild-type mice was less in subcortical brain regions compared to cortical areas. Cannabinoid tolerance was not changed in mice lacking βa-rrestin1 suggesting that β–arrestin2 is the primary arrestin isoform responsible for this process (Breivogel and Vaghela, 2015).
Our work and others have demonstrated the critical role of several putative protein kinase phosphorylation sites in CB1R that are critical for desensitization. Protein kinase C (PKC) has been shown to attenuate the effects of cannabinoid agonists on ion channels including the activation of GIRK channels and inhibition of VGCCs (Garcia et al., 1998). The effect of PKC on CB1R ion channel signaling is mediated by phosphorylation at S317 on the 3rd intracellular loop of the receptor. Phosphorylation of S426 and S430 by GRKs and β–arrestin2 recruitment to these phosphorylated residues is essential for desensitizing the effects of CB1R on GIRK channels and MAPK signaling (Daigle et al., 2008a; Jin et al., 1999). CB1R desensitization occurred rapidly (within ~5 minutes) and the expression of alanine point mutations at residues S426 and S430 had no effect on internalization despite reducing desensitization by ~75% (Daigle et al., 2008a).
4.2. Mechanism of GRK/β–arrestin2-Mediated Cannabinoid Tolerance
The expression of the S426A/S430A point mutations in mice caused an enhancement in the acute duration of “tetrad” effects for a single injection of Δ9-THC while causing a decrease in tolerance for the tetrad effects of chronically administered Δ9-THC (Morgan et al., 2014). Desensitization and downregulation of CB1R was reduced in S426A/S430A versus wild-type mice chronically treated with Δ9-THC, demonstrating that the neuroadaptations responsible for cannabinoid tolerance were partially prevented in mice lacking the putative GRK phosphorylation sites responsible for CB1R desensitization. Tolerance was also decreased for the antinociceptive effects of cannabinoids on formalin pain (LaFleur et al., 2018) and chemotherapy-evoked neuropathic pain (Nealon et al., 2019). Tolerance to the effects of WIN55,212-2 was much more profoundly affected than tolerance to Δ9-THC, suggesting that the role of GRKs and β–arrestin in cannabinoid tolerance is agonist-dependent (Nealon et al., 2019). These findings in mice with point mutations in GRK phosphorylation sites that mediate desensitization closely resemble the effects on acute cannabinoid response and tolerance that were observed in β–arrestin2 KO mice (Nguyen et al., 2012).
Early studies only assessed cannabinoid tolerance in male S426A/S430A mutant mice. Subsequent work assessing tolerance in female S426A/S430A mutant mice suggest that the effect of these point mutations might be more pronounced in male versus female mice, raising the possibility of sex differences in cannabinoid tolerance that will be discussed in detail later in this review (Henderson-Redmond et al., 2021; Henderson-Redmond et al., 2022). Recently published work has examined cannabinoid response tolerance in S426A/S430A × β–arrestin2 KO double mutant mice and found that deletion of β-arrestin2 did not significantly change acute cannabinoid response or tolerance relative to S426A/S430A mutants (Piscura et al., 2023). This finding suggests that phosphorylation at S426 and S430 accounts for most of the effects of β–arrestin2 on cannabinoid response and tolerance. However, this study did not assess CB1R desensitization or downregulation in double mutant mice, so it’s not possible to know whether these two mutations might have additive effects preventing CB1R neuroadaptations associated with tolerance.
4.3. Role of CB1R Internalization in Cannabinoid Tolerance
Agonist-induced internalization leads to two predominant outcomes for the CB1R: either internalization followed by rapid recycling back to the cell surface or sorting to lysosomal pathways and degradation (Figure 6). These two distinct fates have disparate molecular and functional consequences. For example, lysosomal degradation depletes surface receptors from the membrane, a process that drives tolerance. On the contrary, trafficking and restoration of receptors to the plasma membrane is essential for CB1R resensitization. CB1R internalization is a tightly controlled mechanism that is initiated with ligand binding followed by activation of heterotrimeric G proteins, recruitment of GRKs that phosphorylate serine and threonine residues on the C-terminus of the, and β-arrestin recruitment to those phosphorylated residues.
Figure 6. CB1R downregulation.
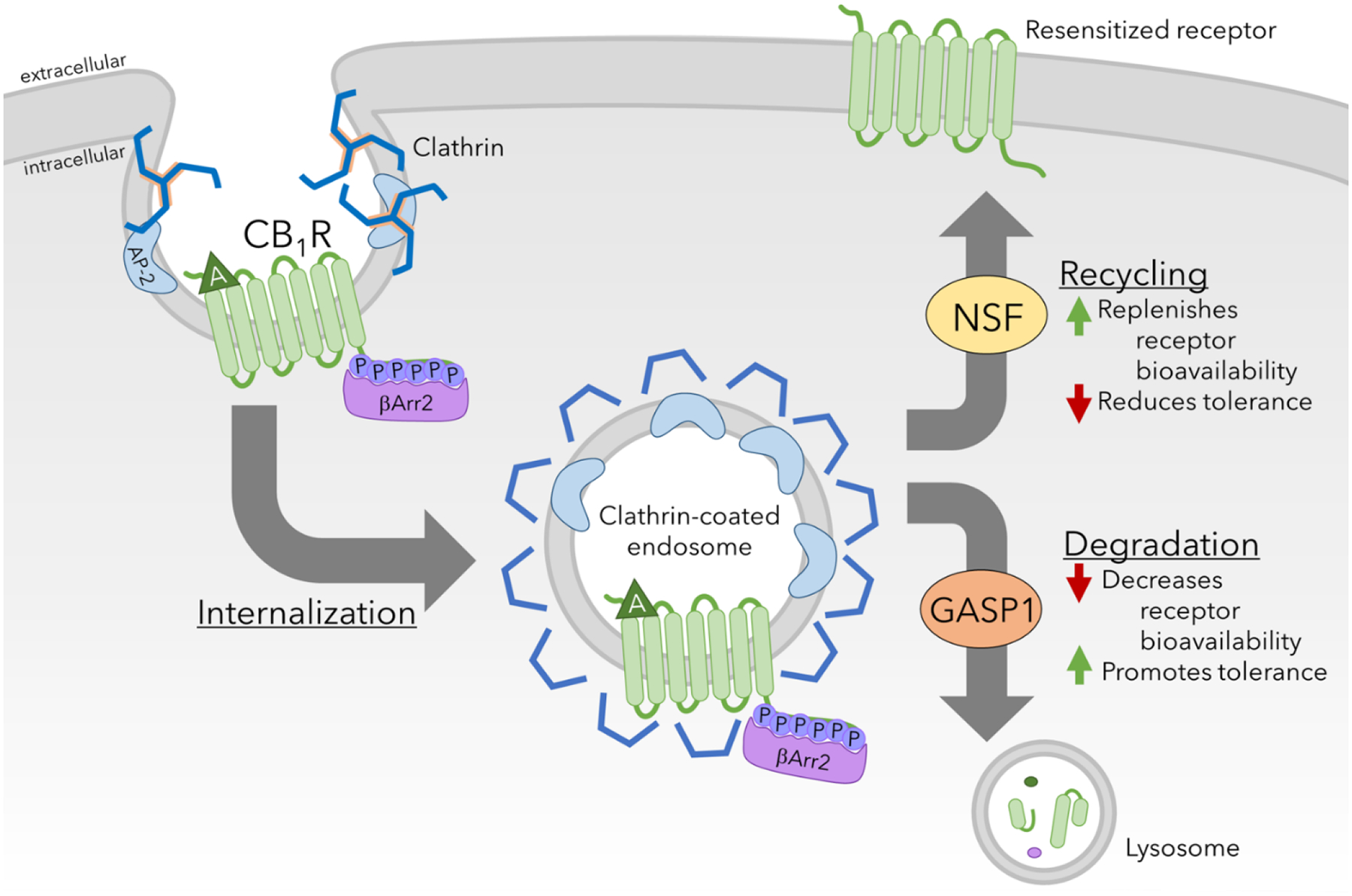
Clathrin-mediated endocytosis results in internalization of CB1Rs, which is sorted to be degraded or recycled. Increased bioavailability of resensitized receptors may reduce tolerance, while degradation of the receptor and reduction of receptor bioavailability promotes tolerance.
Work in heterologous cells found that truncation of CB1R at residue 460 prevented agonist-stimulated internalization in AtT20 cells (Hsieh et al., 1999). WIN55,212-2 treatment causes robust internalization and intracellular trafficking of CB1R in hippocampal neurons (Coutts et al., 2001). Subsequent work found that agonist-stimulated internalization of CB1R is mediated by phosphorylation and recruitment of β–arrestin2 to a second set of more distal C-terminal GRK phosphorylation sites (Daigle et al., 2008b). These studies indicate that specific residues differentially mediate CB1R desensitization (S426 and S430) and internalization (T461, S463, S465, T466, T468, and T469) (Daigle et al., 2008a). CB1R internalization also likely plays important roles in cannabinoid tolerance since this process is required for the trafficking and re-sensitization in early endosomes where CB1R is de-phosphorylated, allowing it to be recycled to the membrane. In contrast, trafficking of CB1R to the lysosome mediates the opposing process of degradation. The mechanism responsible for directing internalized GPCRs including CB1R to the degradative lysosomal pathways versus endosomal re-sensitization pathways are not fully understood. However, G protein associated sorting protein 1 is critical for sorting internalized CB1R for degradation and downregulation, a process critical for the development of tolerance to WIN55,212-2 (Martini et al., 2007). It is not known whether G protein associated sorting protein 1 deletion also modulates tolerance for other cannabinoid agonists such as Δ9-THC and CP55,940 and this knowledge gap should be addressed in future work. A critical determinant of receptor internalization is “endocytic dwell time”, which is defined as the time taken by the receptors to cluster with β–arrestins in clathrin pits. Agonists with smaller dwell times such as WIN55, 212–2 elicit little to no β-arrestin signaling whereas agonists with longer dwell times such as 2-AG produce high β-arrestin activation (Flores-Otero et al., 2014). Such agonist-driven differences in endocytic dwell time and CB1R regulation of could have implications for understanding how tolerance is produced by CB1R ligands.
Despite convincing cell culture studies demonstrating distinctive actions of these residues for desensitization versus internalization, the role that these six putative “internalization” residues play in tolerance and dependence for CB1R agonists in animals is not known. We propose the novel hypothesis that blocking CB1R internalization in mice will cause more rapid and pronounced cannabinoid tolerance and dependence since internalization is essential for the re-sensitization and recycling of inactive, desensitized CB1R by allowing the receptor to be trafficked and de-phosphorylated inside the cell and recycled to the cell surface. Although the effects of CB1R internalization on cannabinoid tolerance have not been studied in vivo, there is good evidence from the opioid field to support our prediction. For example, expression of rapidly internalizing and recycling forms of the mu-opioid receptor delays tolerance to mu agonists in vivo and in cell culture systems (Finn and Whistler, 2001; Kim et al., 2008).
Two alternatively spliced CB1R interacting proteins, 1A (CRIP1a) and 1B, were identified and found to bind to the C-terminal tail of CB1R. Early work found that CRIP1a could co-immunoprecipitate with CB1R, suggesting that these two proteins interact as part of the same protein-protein complex (Niehaus et al., 2007). Co-injection of CRIP1a and CB1R into cervical ganglia was found to suppress tonic inhibition of VGCCs by CB1R, providing early evidence that CRIP1a modulates CB1R signaling. Molecular modeling suggests that CRIP1a interacts with the last 9 residues of CB1R including several putative GRK phosphorylation sites that mediate CB1R internalization (S464, T465, and T467) and are mutated in the six point mutant mice described above (Ahmed et al., 2014). Subsequent work found that CRIP1a competes with β–arrestin for binding at this site on CB1R that includes the six residues responsible for CB1R internalization (Blume et al., 2017). Overexpression of CRIP1a in HEK293 cells attenuated the agonist-stimulated internalization and downregulation of CB1R with no effect on desensitization (Blume et al., 2016; Smith et al., 2015). Based on the ability of CRIP1a to modulate agonist-stimulated CB1R down-regulation in cell lines by competing with β–arrestin2 for binding at C-terminal phosphorylated residues on CB1R, it seems likely that this protein might also be involved in cannabinoid tolerance. In order to address this knowledge gap, cannabinoid tolerance should be assessed in recently produced CRIP1a KO mice in future studies.
4.4. Opioid Tolerance
Opioid receptors belong to the rhodopsin subfamily of Gi/o-coupled GPCRs. Similar to CB1R, activation of opioid receptors results in reduced cellular levels of cAMP, increased MAPK signaling and modulation of GIRK channels (Fukuda et al., 1996; Hampson et al., 2000). Moreover, CB1R and opioid receptors are generally localized to presynaptic terminals where their activation inhibits neurotransmitter release. Additionally, there is evidence of asymmetric cross-modulation of pharmacological responses induced by opioids and cannabinoids such as antinociception, hypolocomotion, catalepsy, and hypothermia (Cichewicz et al., 1999). Cross-tolerance has been reported for the acute antinociceptive effects of Δ9-THC in mice or non-human primates treated with morphine (Gerak et al., 2015; Maguma and Taylor, 2011; Thorat and Bhargava, 1994b). Reciprocal experiments have shown that chronic exposure to Δ9-THC or CP55,940 produces cross-tolerance to the effects of morphine exposure (Garzon et al., 2009; Hine, 1985; Thorat and Bhargava, 1994b). Mice lacking pre-proenkephalin exhibit a decrease in the development of tolerance to the antinociceptive properties of Δ9-THC (Valverde et al., 2000). However, tolerance to opioid-induced antinociception was not altered in CB1R KO mice (Ledent et al., 1999).
Multiple studies have shown that MOR desensitization, phosphorylation, endocytosis, and β-arrestin recruitment are involved in the development of opioid tolerance (Zhou et al., 2021). Prolonged agonist stimulation causes phosphorylation of MOR at specific residues in the intracellular domains of the receptor by GRKs. This phosphorylation causes β-arrestin recruitment to these phosphorylated residues that results in steric inhibition of the ability of G proteins to associate with and couple to MOR (Williams et al., 2013). Both GRK2 and GRK3 have been shown to desensitize MOR in vitro (Kovoor et al., 1998). GRK3 KO mice exhibit reduced tolerance to the antinociceptive effects of fentanyl, but not morphine, suggesting that opioid tolerance occurs via agonist-specific mechanisms (Terman et al., 2004). Studies have also demonstrated an essential role of β-arrestin2 in opioid tolerance. Morphine-induced antinociception is prolonged and enhanced in mice lacking β-arrestin2 (Bohn et al., 1999). Interestingly, tolerance to the antinociceptive effects and desensitization of MOR does not occur in β-arrestin2 knockout mice following chronic morphine treatment (Bohn et al., 2000; Bohn et al., 2002).
Additionally, non-GRK kinases such as JNK, PKC, PKA, CAMKII and MAPK have been shown to phosphorylate the receptor (Liu and Anand, 2001). Phosphorylation by these kinases might contribute to homologous or heterologous desensitization and tolerance (Williams et al., 2013). Studies have demonstrated that inhibition of JNK prevented tolerance to the antinociceptive and antiallodynic effects of the morphine but not fentanyl (Marcus et al., 2015; Melief et al., 2010; Yuill et al., 2016). The effect of JNK on morphine tolerance was found to be mediated by JNK2-mediated desensitization of MOR (Melief et al., 2010). Additional work demonstrated that PRDX6 modulates acute tolerance for the antinociceptive effects of morphine via a pathway that involves JNK signaling and recruitment of PRDX6 to the plasma membrane, where it regulates Gαi palmitoylation through the generation of reactive oxygen species (ROS) (Schattauer et al., 2017).
5. Signaling Pathways Involved In Cannabinoid Tolerance
5.1. c-Jun N-Terminal Kinase (JNK)-mediated Cannabinoid Tolerance
Treatment with Δ9-THC has been shown to stimulate JNK signaling in heterologous cells (Bosier et al., 2008). However, experiments assessing the activation of JNK by Δ9-THC treatment in animals has not been examined. Additional work measuring JNK activation in mice treated with cannabinoids including Δ9-THC, CP55,940, and WIN55,212-2 is necessary to address this knowledge gap and determine whether JNK signaling might be activated by cannabinoids in an agonist-dependent manner. Tolerance to the effects of morphine but not fentanyl is reduced by pre-treatment with the selective JNK inhibitor, SP600125, suggesting that opioid tolerance is mediated by JNK signaling in an agonist-specific manner (Marcus et al., 2015; Melief et al., 2010). Similar to morphine tolerance, pretreatment with the same JNK inhibitor prevented tolerance to the effects of Δ9-THC but not WIN55,212-2, providing additional evidence that cannabinoid tolerance is mediated through agonist-specific mechanisms (Henderson-Redmond et al., 2020).
However, several important questions and knowledge gaps exist regarding the role of JNK signaling on cannabinoid tolerance. First, the isoform of JNK responsible for cannabinoid tolerance is not known. Three isoforms of JNK that have been detected in neurons including JNK1, JNK2, and JNK3 that could potentially be involved in cannabinoid tolerance. Tolerance to morphine was disrupted in KO mice for JNK1, JNK2, or JNK3 (Yuill et al., 2016), although JNK2 has been implicated as the primary contributor to this process suggesting that this might also be the case for cannabinoids (Melief et al., 2010). Second, the impact of JNK inhibition on CB1R desensitization and downregulation has not been assessed making it unclear which of these processes, or both, are responsible for JNK-mediated cannabinoid tolerance. Desensitization of MOR was reduced in JNK2 KO mice treated with morphine demonstrating that JNK signaling modulates opioid tolerance by impacting receptor desensitization, raising the likelihood that JNK-mediated cannabinoid tolerance might also occur via CB1R desensitization (Melief et al., 2010). Third, the other components of the signaling pathway through which JNK impacts cannabinoid tolerance are not known. JNK-mediated morphine tolerance has been shown to be mediated by the recruitment of PRDX6 to the plasma membrane where it regulates Gαi palmitoylation through generation of ROS (Schattauer et al., 2017). Therefore, additional studies are needed to determine whether PRDX6, G protein palmitoylation, and/or ROS might also be involved in cannabinoid tolerance. Fourth, it is not known whether JNK mediates cannabinoid tolerance though direct phosphorylation of CB1R or by indirect phosphorylation of other signaling pathway components involved in this process. Fifth, although we observe evidence of sex differences in the role of GRK/β–arrestin2 signaling on cannabinoid tolerance in S426A/S430A mutant mice, it is not known whether sex differences exist for the role of JNK in cannabinoid tolerance. Taken together, it is evident that many unanswered questions exist regarding the mechanism/s of JNK-mediated cannabinoid tolerance that need to be addressed by additional research.
5.2. Role of PKA Signaling in Cannabinoid Tolerance
Other signaling pathways may also contribute to the process of cannabinoid tolerance. Activation of CB1R and Gi/o proteins coupled to this receptor causes a reduction in cAMP formation and PKA signaling. The use of PKA inhibitors has been shown to reverse tolerance to the “tetrad” effects of Δ9-THC in tolerant mice (Bass et al., 2004; Lee et al., 2003). In contrast, a subsequent study found that tolerance to Δ9-THC is not associated with any changes in PKA activity (Dalton et al., 2005). However, dynorphin levels in cerebrospinal fluid was increased in Δ9-THC-tolerant rats and this increase was also reversed using PKA inhibitors. Although, cAMP-PKA signaling does not appear to play a role in the development of cannabinoid tolerance, these studies demonstrate this signaling pathway plays a critical role in maintaining tolerance once it has been established.
5.3. Role of PKC Signaling in Cannabinoid Tolerance
As mentioned earlier in this review, PKC-mediated phosphorylation of the third intracellular loop of CB1R at S317 modulates cannabinoid-induced calcium and potassium currents (Garcia et al., 1998). Furthermore, mutant mice lacking PKC epsilon (PKCε) isoform are more sensitive to the antinociceptive and hypothermic effects of WIN55,212-2 and display increased tolerance to this cannabinoid with no effect on response or tolerance to CP55,940 (Wallace et al., 2009). This finding provides additional evidence that cannabinoid tolerance occurs via agonist-specific mechanisms. It would be interesting to test whether Δ9-THC tolerance is disrupted in PKCε mutant mice, and this question should be answered. Other studies demonstrated that, unlike PKA, inhibition of PKC had no effect on the reversal of cannabinoid tolerance. These results suggest a complicated role for PKC signaling in cannabinoid tolerance that requires further investigation. The production and assessment of cannabinoid response and tolerance in point mutant mice where the putative PKC phosphorylation site at S317 is mutated to alanine would be a valuable tool and approach for examining this question. It is likely that S317A point mutant mice would exhibit disrupted tolerance and an increased response for WIN55,212-2 but not CP55,940, consistent with previous studies (Garcia et al., 1998; Wallace et al., 2009). However, it is important to point out that point mutation of S317 would disrupt all PKC signaling not just that done by the PKCε isoform.
5.4. Role of NMDA Receptor Signaling in Cannabinoid Tolerance
Glutamate serves as a primary excitatory neurotransmitter in the central nervous system and it’s release from presynaptic neuronal terminals can be suppressed by CB1R activation (Kano et al., 2009; Szabo and Schlicker, 2005). Work suggests that glutamatergic signaling can also modulate cannabinoid tolerance. For example, activation of the glutamate transporter-1 (GLT-1) by ceftriaxone, a beta-lactam antibiotic, dose-dependently mitigates tolerance to WIN 55,212-2 in mice (Gunduz et al., 2011). In addition, inhibition of N-methyl-D-aspartate receptors (NMDARs), a calcium-permeable ion channel activated by glutamate, reduced sensitivity to WIN55,212-2 and WIN55,212-2-stimulated CB1R internalization. Other studies found that MK-801 (dizocilpine), a NMDAR antagonist, suppressed Δ9-THC-induced antinociception but had no effect on the development of Δ9-THC tolerance (Thorat and Bhargava, 1994a). However, several studies have found that NMDAR antagonism attenuates the development opioid tolerance (Deng et al., 2019; Manning et al., 1996; Trujillo and Akil, 1994). Nitric oxide is a retrograde messenger that is produced in response to NMDAR activation (Garthwaite et al., 1988) and has been shown to be involved in opioid tolerance. Inhibition of nitric oxide synthesis (NOS) reduced morphine tolerance while administration of L-Arginine, the precursor for nitric oxide enhanced tolerance (Babey et al., 1994; Bhargava, 1995). Studies have produced conflicting results regarding whether cannabinoid tolerance is modulated by nitric oxide signaling (Banafshe et al., 2005; Spina et al., 1998; Thorat and Bhargava, 1994a). Early work found that pre-treatment with NG-monomethyl-L-arginine (L-NMMA), an inhibitor of NOS, had no effect on tolerance to Δ9-THC (Thorat and Bhargava, 1994a). However, other studies have shown that pre-treatment with either N(omega)-nitro-L-arginine methyl ester (L-NAME), another NOS inhibitor, or cyclosporin, a calcineurin inhibitor, that also inhibits NOS, both prevent tolerance to some WIN55,212-2 tetrad effects (Banafshe et al., 2005; Spina et al., 1998). These findings suggest that nitric oxide signaling might modulate tolerance to WIN55,212-2, but not other cannabinoid agonists, providing additional evidence to support the premise that cannabinoid tolerance develops via agonist-specific mechanisms.
5.5. Role of Other Protein Kinases in Cannabinoid Tolerance
Some studies have shown that inhibition of the receptor tyrosine kinase Src by PP1 could reverse tolerance for “tetrad” effects of Δ9-THC (Bass et al., 2004) while other work found that Src inhibition only reversed tolerance for the motor effects of cannabinoids (Lee et al., 2003). Inhibition of PKG, PI3K, or GRK using selective kinase inhibitors also had no effect on the reversal of cannabinoid tolerance (Lee et al., 2003).
6. Sex Differences in Cannabinoid Response, Tolerance and Use Disorder
6.1. Sex Differences in Cannabinoid Use and Response
Evidence suggests that biological sex influences multiple cannabinoid-related outcomes including the prevalence of CUD (Hernandez-Avila et al., 2004; Khan et al., 2013) abuse liability (Cooper and Haney, 2014), withdrawal severity (Copersino et al., 2006; Herrmann et al., 2015; Levin et al., 2010; Sherman et al., 2017), treatment outcomes (Cuttler et al., 2020), and neuronal activity in those with CUDs (Wetherill et al., 2015). Presently, more men than women report using marijuana recreationally in their lifetime (Cuttler et al., 2016; Johnson-Davis et al., 2016). However, more women than men use medical cannabis for pain relief (Cuttler et al., 2016). Women also report a greater incidence and severity of chronic pain compared to men (Dahlhamer et al., 2018; Nahin, 2015). Therefore, it is imperative that we understand how gender and biological sex (denoted as men and women in clinical studies and males and females in preclinical studies) may influence various cannabinoid-mediated effects that in turn can influence prolonged use and subsequent tolerance.
The results of clinical studies assessing sex differences in the acute effects of are mixed. Some studies show that women may be more sensitive to cannabinoids (Cooper and Haney, 2014; Roser et al., 2009; Wardle et al., 2015) while others found that women are less sensitive (Haney, 2007; Penetar et al., 2005) than men to the subjective (“high”) effects of cannabinoids. Some other studies found no sex differences in cannabinoid response (Anderson et al., 2010; Cooper and Haney, 2016). The effects of cannabis may also be impacted by the route of administration in a sex-dependent manner. Men and women reported that inhaled cannabis decreased their subjective pain ratings for headache and migraine; however, pain relief was greater in men than women (Cuttler et al., 2020). Similarly, in regular cannabis smokers, while cannabis was shown to increase pain tolerance for both men and women in the cold pressor test, antinociception was greater in men (Cooper and Haney, 2016). In contrast, only women were found to display antinociception following oral administration of 1 mg/kg nabilone, a synthetic Δ9-THC analog, in the heat pressor test (Redmond et al., 2008). Additional studies in experienced cannabis users found that women are less sensitive to the tachycardic (Cooper and Haney, 2016; Penetar et al., 2005) effects of cannabinoids. A study assessing sex-differences in acute cannabis effects found that there was no difference in subjective drug effects, mood, heart rate, blood pressure, or cognitive effects in men and women given a cannabis cigarette containing 12.5% Δ9-THC content (Matheson et al., 2020). Interestingly, women smoked less of the cannabis cigarette than men and had lower peak concentrations of Δ9-THC and its inactive metabolite, 11-Nor-carboxy-THC leading to the conclusion that women experienced the same acute effects of smoked cannabis as men at lower doses of Δ9-THC. Taken together, these results suggest that women and men may be able to titrate their cannabis intake adding an additional confounder to determineing how sex may mediate the acute effects of cannabis.
The use of preclinical models allows more precise control of experimental variables to study sex differences in cannabinoid response. Studies in our lab using wild-type mice on a C57BL/6 background have found that male mice are more sensitive to the antinociceptive effects of Δ9-THC and CP55,940 compared to female littermates (Henderson-Redmond et al., 2021; Henderson-Redmond et al., 2022; LaFleur et al., 2018; Piscura et al., 2023). This finding was consistent across models of acute tail-flick (Henderson-Redmond et al., 2022; Piscura et al., 2023) and inflammatory (LaFleur et al., 2018) antinociception and chronic chemotherapy-induced neuropathic pain (Henderson-Redmond et al., 2021). In contrast, a different study found no sex differences in WIN55,212-2-mediated antinociception between male and female wild-type mice (Blednov et al., 2003).
In contrast to the paucity of studies examining cannabinoid-mediated analgesia in mice, there is a much greater depth of literature examining this phenomenon in rats. The observation of decreased sensitivity to cannabinoids in female mice seems to vary by species. For example, work in both adult Sprague-Dawley and Wistar rats finds that females are more sensitive than males to the antinociceptive and antiallodynic effects of cannabinoids including Δ9-THC and CP55,940 (Craft et al., 2013a; Craft et al., 2013b; Craft et al., 2012; Romero et al., 2002; Wakley and Craft, 2011; Wakley et al., 2014b). This effect of increased sensitivity to cannabinoids in female rats has been consistent across multiple pain modalities, including the acute tail immersion and paw withdrawal tests (Craft et al., 2012; Romero et al., 2002; Tseng and Craft, 2001; Wakley et al., 2014b) and complete Freund’s adjuvant (CFA) induced model of persistent inflammatory pain (Craft et al., 2013a). However, not all studies in rats find females to be more sensitive to the antinociceptive effects of cannabinoids than males. For example, male Sprague-Dawley rats displayed increased sensitivity to the anti-inflammatory effects of arachidonylcyclopropylamide, a selective CB1R agonist, in a CFA-induced orofacial pain model (Niu et al., 2012). The methylated cannabinoid, cannabidiolic acid, elicited anti-hyperalgesic effects in male rats yet had little to no effect in female rats (Zhu et al., 2020).
In contrast to cannabinoid-induced antinociception, female mice are more sensitive to the locomotor effects of Δ9-THC than male littermates (Wiley, 2003). Female Sprague-Dawley rats also display increased locomotor effects of Δ9-THC than male rats (Tseng and Craft, 2001; Tseng et al., 2004). Other studies have found that male rats are more sensitive to the hyperphagic effect of cannabinoids than female rats (Diaz et al, 2009). Female Sprague-Dawley were able to discriminate lower doses of Δ9-THC more easily from vehicle compared to male rats, indicating that female rats are more sensitive to lower doses of Δ9-THC than their male counterparts (Wiley et al., 2021). In contrast, this study did not detect sex differences in drug discrimination for Δ9-THC in C57BL/6 mice.
6.2. Sex Differences in Cannabinoid Tolerance
Chronic cannabinoid use, which leads to tolerance, varies based on sex. Despite an increased lifetime use and a greater incidence of developing CUDs in men, women display an accelerated advancement (also called telescoping) from casual use to the development of CUD (Farmer et al., 2015; Kerridge et al., 2018). One possible interpretation of this enhanced telescoping in women is that they might show more rapid tolerance to cannabis compared to men (Towers et al., 2023). Women progress faster to seeking treatment for CUD than men (Hernandez-Avila et al., 2004) and show increased escalation of usage compared to men (Ehlers et al., 2010; Kerridge et al., 2018; Khan et al., 2013). Women experience greater severity of CWS compared to men (Copersino et al., 2006; Herrmann et al., 2015; Sherman et al., 2017) and some studies report that women exhibit worse treatment outcomes compared to men (McRae-Clark et al., 2015). These data suggest that women may be more predisposed to develop tolerance to cannabis and CUD than men.
Studies examining sex differences in cannabinoid tolerance in human subjects are limited and the ones that exist have typically assessed this question using a retrospective analysis. In a study assessing the analgesic effects of cannabis in regular cannabis users, researchers found that men showed greater cannabis-induced antinociception in the cold pressor test compared to women (Cooper and Haney, 2016). One possible interpretation of the result that women displayed reduced antinociceptive effects of cannabis is that they were more tolerant compared to men. Another study assessing the cardiovascular effects of cannabis cigarettes containing 1% Δ9-THC found that naive users, both men and women, showed robust tachycardia following the first cigarette. However, the decrease in tachycardia following the second Δ9-THC cigarette due to tolerance was more pronounced in women than men, suggesting tolerance to the cardiovascular effects of Δ9-THC develops faster in women (Cocchetto et al., 1981). Since the neuroadaptations that lead to tolerance (CB1R desensitization and downregulation) also cause withdrawal, the observation of greater CWS in women supports the possibility that cannabinoid tolerance is greater in women compared to men. However, this question requires much more thorough and controlled investigation. In particular, additional studies where the dose of cannabinoid is adjusted for body weight are desperately needed since the body weight of women is generally less than men. Therefore, past studies examining cannabinoid response and tolerance in subjects given standard doses (not adjusted for body weight) may not fully capture critical and significant sex differences.
By administering equal doses (based on gram per kilogram of body weight), preclinical studies are better able to assess the effectiveness of a single dose of cannabinoid agonist over time allowing investigators to gain a better understanding of sex differences in cannabinoid. Studies using both rats and mice have consistently shown that female rodents develop tolerance faster than their male counterparts for the antinociceptive (Henderson-Redmond et al., 2021; Henderson-Redmond et al., 2022; Nguyen et al., 2020; Nguyen et al., 2018; Parks et al., 2020; Wakley et al., 2014b), locomotor (Wiley, 2003), and hypothermic (Nguyen et al., 2020; Nguyen et al., 2018; Piscura et al., 2023; Wakley et al., 2014b) effects of cannabinoid administration.
Some rodent studies have attempted to assess tolerance using cannabinoid doses that were equally efficacious in males and females. For example, since female rats have been shown to be more sensitive than male rats to the equivalent doses of Δ9-THC, Wakley and colleagues, opted to use doses that were approximately 30% less potent (5.4 mg/kg) in female Sprague-Dawley rats compared to males (7.6 mg/kg). This work found that following 9.5 days of twice-daily Δ9-THC injections, that female rats showed significantly greater ED50 values for tail withdrawal and paw pressure than male littermates, indicating greater tolerance to antinociceptive effects of Δ9-THC in female rats (Wakley et al., 2014b). Similar experiments using equally efficacious doses of Δ9-THC were used to assess sex differences in the antiallodynic effects of Δ9-THC using doses that produced the same acute effects in male (6 mg/kg) and female (10 mg/kg) mice. This work found that tolerance to the antiallodynic effects of Δ9-THC developed faster in female C57BL/6 mice with chemotherapy-evoked neuropathic pain compared to male littermates (Henderson-Redmond et al., 2021). In Wistar rates repeatedly exposed to Δ9-THC vapor inhalation, only females but not males, showed any evidence of any tolerance to the antinociceptive and hypothermic effects of Δ9-THC (Nguyen et al., 2020; Nguyen et al., 2018). These data suggest that congruent with clinical observations of “telescoping” in the advancement of first use to CUD development, female rodents show faster development of tolerance to the effects of cannabinoids and may cause them to be predisposed for cannabinoid dependence.
Differences in CB1R density, downregulation, and/or desensitization are mechanisms that have been proposed as potential reasons for sex differences in cannabinoid response and/or tolerance. Previous work found differences in CB1R density and desensitization between male and female rodents (Castelli et al., 2014; Farquhar et al., 2019; Gonzalez et al., 2005; Wiley et al., 2021). Although, multiple studies have demonstrated sex differences in Δ9-THC-induced antinociception, these same Δ9-THC doses did not cause sex differences in the acute hypothermic response raising the possibility that cannabinoid sex differences might be response-specific. This finding could be due to underlying differences in cannabinoid signaling within the specific brain regions such as the periaqueductal gray, spinal cord, and hypothalamus that mediate these different responses.
Although there are no sex differences in CB1R levels across some regions of the mouse brain including the cerebellum (Wiley et al., 2021), less is known about possible sex differences in CB1R levels in areas that control antinociception. That said, a recent study determined that females have higher levels of CB1R on GABAergic neurons in the ventrolateral PAG compared to males and that this could be responsible for sex differences in cannabinoid-mediated antinociception (Jiang et al., 2022). It is also possible that response-specific sex differences in cannabinoid response could be due to differences in other important endocannabinoid signaling components such endocannabinoid levels, G protein coupling, CB2R levels, and β–arrestin expression. For example, previous work has demonstrated that endocannabinoid levels vary as a function of sex (males had lower levels of 2-AG and AEA than females) and estrous cycle (females had greater levels of 2-AG and AEA in diestrus compared to all cycle stages) in the hypothalamus of Sprague–Dawley rats (Bradshaw et al., 2006).
Our own work found that tolerance to the antinociceptive effects of Δ9-THC and CP55,940 was reduced in male but not female mice expressing a desensitization-resistant form of CB1R (Henderson-Redmond et al., 2020; LaFleur et al., 2018), suggesting that the S426A/S430A mutation plays a sex-specific role in cannabinoid tolerance. As previously described, the S426A/S430A mutation prevents β-arrestin2 recruitment and CB1R desensitization. Mice lacking β–arrestin2 display enhanced Δ9-THC-induced antinociception, delayed tolerance to Δ9-THC-mediated antinociception, and decreased CB1R downregulation and desensitization in the PAG and spinal cord (Breivogel et al., 2008; Nguyen et al., 2012). However, it is not known whether β–arrestin2 KO deletion impacts these cannabinoid effects in a sex-specific manner and this question should be addressed by future work.
Previous work has also shown inhibition of JNK signaling in male mice disrupts cannabinoid tolerance in an agonist-specific manner (Henderson-Redmond et al., 2020). It is not known whether sex differences might exist for JNK-mediated cannabinoid tolerance. However, recent studies assessing the antinociceptive effects of the JNK inhibitor, SU-3327, provide some important clues. Our work assessing the antinociceptive effects of SU-3327 in formalin-induced inflammatory pain found that lower doses that produced antinociception in male mice had no effect in female mice (Blanton et al., 2021). Of note, the effects of SU-3327 were mediated by both CB1R and CB2R in male mice while they were mediated almost exclusively through CB2R in female mice.
Estrogen has been shown to interfere with the ability of Δ9-THC to bind to CB1R (Wakley et al., 2014b), and hormone fluctuations across phases of the estrous cycle impact G protein coupling to CB1R (Riebe et al., 2010). Additional work in rats found that the levels of endocannabinoids also fluctuate across the estrous cycle in several brain regions (Bradshaw et al., 2006; Gonzalez et al., 2000). Acute treatment of ovariectomized (OVX) female rats with a bolus injection of estradiol has been shown to decrease CP55,940-stimulated [35S]-GTPγS binding in the cortex and hippocampus, suggesting that estradiol can suppress CB1R signaling (Mize and Alper, 2000). However, another study in rats found that CB1R protein and WIN55,212-2-stimulated [35S]-GTPγS binding in the striatum is decreased in OVX females, suggesting an opposite finding that estrogen and/or progesterone enhance CB1R expression and signaling (Winsauer et al., 2011).
OVX enhanced the antinociceptive (but not cataleptic) effects of WIN55,212-2, and this effect was blocked by pretreatment with estradiol, but not progesterone, suggesting that estradiol could modulate sex differences in cannabinoid-induced antinociception (Kalbasi Anaraki et al., 2008). The finding that changes in catalepsy were not modulated by hormonal manipulations suggests the effects of sex hormones might be response-specific for antinociception. A second study showed that mifepristone and metyrapone, two anti-progesterone drugs, enhanced the acute hypothermic effects of 25 mg/kg Δ9-THC in female mice (Pryce et al., 2003). In contrast to studies suggesting that estrous cycle may modulate Δ9-THC-induced antinociception, administration of estradiol, progesterone, or the combination of both in gonadally intact Sprague-Dawley rats did not alter Δ9-THC-induced antinociception (Wakley et al., 2014a). Inhalation of vapor containing 100 mg/mL Δ9-THC caused antinociception in Wistar rats that did not vary across estrous cycle stage (Falenski et al., 2010)
7. Conclusions and Future Directions
The results summarized in this review demonstrate that tolerance does develop to the effects of cannabinoids in both humans and animal models. We describe current knowledge about the mechanisms and signaling pathways that are responsible for cannabinoid tolerance. Those mechanisms include neuroadaptations at CB1R such as downregulation and desensitization that are mediated by a range of signaling pathways and components including GRKs, β–arrestin2, JNKs, nitric oxide, and PKA. Some of the neuroadaptations such as CB1R downregulation have been demonstrated in human cannabis users while the process of desensitization has only been demonstrated in animal models where appropriate tools for assessing this process exist. Development of a neuroimaging tool to visualize CB1R desensitization in humans would represent a major advance that would allow investigators to assess whether this process contributes to cannabinoid tolerance in humans. Such a breakthrough would also likely be relevant to the assessment of desensitization for other GPCR including opioid receptors.
Many studies have demonstrated that phosphorylation of CB1R by GRKs and subsequent recruitment of β–arrestin2 play a critical role in the CB1R neuroadaptations responsible for cannabinoid tolerance. In addition to putative phosphorylation sites at S317, S426, S430A, T461, S463, S465, T466, T468, and S469 that have been studied so far, there are possible phosphorylation sites at S401, S411, S415, S442, S449, S453, and T454 that have not been studied. Therefore, given the complexity of possible phosphorylation events in the C-terminal region of CB1R that regulate desensitization, downregulation, and trafficking, it is important to determine whether the “bar code” phosphorylation state of CB1R might be agonist specific. Another significant knowledge gap surrounding cannabinoid tolerance in preclinical studies is that most animal studies of tolerance has focused on purified cannabinoid compounds such Δ9-THC, CP55,940, WIN55,212-2, and CBD. Whether the mechanisms of tolerance discovered for these isolated cannabinoids also extends and is relevant to tolerance for cannabis, which contains many cannabinoid and terpene components, has been largely unstudied. This question is important because many studies and reports have documented entourage effects for cannabis or cannabinoid mixtures where the different constituents exert modulatory effects on each other. Therefore, the mechanisms of tolerance might be divergent depending on the specific mixture of compounds and signaling pathways that are activated by those components.
The topic of this review also raises the important clinical question of which approaches might be useful for the reduction of cannabinoid tolerance in humans. There are several possibilities arising from our current understanding of cannabinoid tolerance. First, early work on cannabinoid tolerance demonstrated that this process is dose dependent. Therefore, it is possible that the use of sufficiently low cannabinoid doses may be able to produce therapeutic effects without tolerance. Another possibility is that combination polypharmacy with therapeutics such as nonsteroidal anti-inflammatory drugs, opioids, anticonvulsants (gabapentin), and others might be useful for keeping cannabinoid doses low enough to prevent tolerance. Second, the use of cannabinoid agonists that promote rapid-recycling and resensitization of desensitized CB1R might reduce the development of tolerance. Third, animal studies indicate tolerance is undetectable for AEA and can be kept at a modest level for 2-AG, if sufficiently low doses of MAGL inhibitors (JZL184) are used. Therefore, it is possible that the use of endocannabinoid hydrolysis enzyme inhibitors might be able to elicit positive therapeutic effects without the development of tolerance. However, it is important to point out that clinical trials testing beneficial effects of FAAH inhibitors have been unsuccessful in yielding positive results. Fourth, preclinical studies have shown less tolerance for G protein-biased cannabinoid agonists. This finding is consistent with the idea that cannabinoid agonists producing minimal β–arrestin2 signaling and recruitment, a protein involved in CB1R desensitization and downregulation, might produce less tolerance. Fifth, although not discussed so far in this review, the use of cannabinoid receptor allosteric modulators, rather than direct agonists represents another approach that might prevent tolerance. Allosteric modulators bind to allosteric sites on the receptor where they enhance the activity of endogenous or exogenous agonists bound at the orthosteric site. Thus, cannabinoid receptor allosteric modulators exert their effect by enhancing the effects of endocannabinoids, which have been shown to cause minimal (AEA) or modest (2-AG) tolerance.
Acknowledgements:
This work was supported by National Institute of Health grant DA044999.
Abbreviations:
- AC
adenylyl cyclase
- AEA
anandamide
- 2-AG
2-arachidonylglycerol
- CBD
cannabidiol
- CB1R
cannabinoid type 1 receptor
- CB2R
cannabinoid type 2 receptor
- CRIP
CB1R interacting proteins
- CUD
cannabis use disorder
- CWS
cannabinoid withdrawal syndrome
- DAG
diaglycerol
- DAGL
diaglycerol lipase
- DSE
depolarization-induced suppression of excitation
- ECS
endocannabinoid signaling
- FAAH
fatty acid amide hydrolase
- GIRK
inward-rectifying potassium channel
- GPCR
G protein-coupled receptor
- KO
knock-out
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- MS
multiple sclerosis
- MAGL
monoacylglycerol lipase
- MOR
mu opioid receptor
- NOS
nitric oxide synthesis
- OVX
ovariectomized
- PAG
periaqueductal gray
- PET
positron emission tography
- PI3K
phosphatidyl-inositol-3-kianse
- PKA
protein kinase A
- PKC
protein kinase C
- Δ9-THC
delta-9-tetrahydrocannabinol
- VGCC
voltage-gated calcium channel
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: none
References
- Abood ME, Sauss C, Fan F, Tilton CL and Martin BR (1993) Development of behavioral tolerance to delta 9-THC without alteration of cannabinoid receptor binding or mRNA levels in whole brain. Pharmacol Biochem Behav 46:575–579. [DOI] [PubMed] [Google Scholar]
- Abraham AD, Leung EJY, Wong BA, Rivera ZMG, Kruse LC, Clark JJ and Land BB (2020) Orally consumed cannabinoids provide long-lasting relief of allodynia in a mouse model of chronic neuropathic pain. Neuropsychopharmacology 45:1105–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aceto MD, Scates SM, Lowe JA and Martin BR (1995) Cannabinoid precipitated withdrawal by the selective cannabinoid receptor antagonist, SR 141716A. Eur J Pharmacol 282:R1–2. [DOI] [PubMed] [Google Scholar]
- Aceto MD, Scates SM and Martin BB (2001) Spontaneous and precipitated withdrawal with a synthetic cannabinoid, WIN 55212-2. Eur J Pharmacol 416:75–81. [DOI] [PubMed] [Google Scholar]
- Ahluwalia J, Urban L, Capogna M, Bevan S and Nagy I (2000) Cannabinoid 1 receptors are expressed in nociceptive primary sensory neurons. Neuroscience 100:685–688. [DOI] [PubMed] [Google Scholar]
- Ahmed MH, Kellogg GE, Selley DE, Safo MK and Zhang Y (2014) Predicting the molecular interactions of CRIP1a-cannabinoid 1 receptor with integrated molecular modeling approaches. Bioorg Med Chem Lett 24:1158–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allende G, Chavez-Reyes J, Guerrero-Alba R, Vazquez-Leon P and Marichal-Cancino BA (2020) Advances in Neurobiology and Pharmacology of GPR12. Front Pharmacol 11:628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association D-TF (2013) Diagnostic and statistical manual of mental disorders: DSM-5, American Psychiatric Publishing, Inc. [Google Scholar]
- Anderson BM, Rizzo M, Block RI, Pearlson GD and O’Leary DS (2010) Sex differences in the effects of marijuana on simulated driving performance. J Psychoactive Drugs 42:19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armenian P, Darracq M, Gevorkyan J, Clark S, Kaye B and Brandehoff NP (2018) Intoxication from the novel synthetic cannabinoids AB-PINACA and ADB-PINACA: A case series and review of the literature. Neuropharmacology 134:82–91. [DOI] [PubMed] [Google Scholar]
- Atwood BK, Straiker A and Mackie K (2012a) CB(2) cannabinoid receptors inhibit synaptic transmission when expressed in cultured autaptic neurons. Neuropharmacology 63:514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, Wager-Miller J, Haskins C, Straiker A and Mackie K (2012b) Functional selectivity in CB(2) cannabinoid receptor signaling and regulation: implications for the therapeutic potential of CB(2) ligands. Mol Pharmacol 81:250–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babey AM, Kolesnikov Y, Cheng J, Inturrisi CE, Trifilletti RR and Pasternak GW (1994) Nitric oxide and opioid tolerance. Neuropharmacology 33:1463–1470. [DOI] [PubMed] [Google Scholar]
- Banafshe HR, Ghazi-Khansari M and Dehpour AR (2005) The effect of cyclosporine on the development and expression of cannabinoid tolerance in mice. Pharmacol Biochem Behav 82:658–663. [DOI] [PubMed] [Google Scholar]
- Bass CE and Martin BR (2000) Time course for the induction and maintenance of tolerance to Delta(9)-tetrahydrocannabinol in mice. Drug Alcohol Depend 60:113–119. [DOI] [PubMed] [Google Scholar]
- Bass CE, Welch SP and Martin BR (2004) Reversal of delta 9-tetrahydrocannabinol-induced tolerance by specific kinase inhibitors. Eur J Pharmacol 496:99–108. [DOI] [PubMed] [Google Scholar]
- Benowitz NL and Jones RT (1981) Cardiovascular and metabolic considerations in prolonged cannabinoid administration in man. J Clin Pharmacol 21:214S–223S. [DOI] [PubMed] [Google Scholar]
- Bhargava HN (1995) Attenuation of tolerance to, and physical dependence on, morphine in the rat by inhibition of nitric oxide synthase. Gen Pharmacol 26:1049–1053. [DOI] [PubMed] [Google Scholar]
- Bie B, Wu J, Foss JF and Naguib M (2018) An overview of the cannabinoid type 2 receptor system and its therapeutic potential. Curr Opin Anaesthesiol 31:407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno T, Howell F, Williams G, Minassi A, Cascio MG, Ligresti A, Matias I, Schiano-Moriello A, Paul P, Williams EJ, Gangadharan U, Hobbs C, Di Marzo V and Doherty P (2003) Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol 163:463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankman JL, Simon GM and Cravatt BF (2007) A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol 14:1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanton HL, Brelsfoard J, DeTurk N, Pruitt K, Narasimhan M, Morgan DJ and Guindon J (2019) Cannabinoids: Current and Future Options to Treat Chronic and Chemotherapy-Induced Neuropathic Pain. Drugs 79:969–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanton HL, Pietrzak A, McHann MC and Guindon J (2021) Sex and dose-dependent antinociceptive effects of the JNK (c-Jun N-terminal kinase) inhibitor SU 3327 are mediated by CB2 receptors in female, and CB1/CB2 receptors in male mice in an inflammatory pain model. Brain Res Bull 177:39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Stoffel M, Alva H and Harris RA (2003) A pervasive mechanism for analgesia: activation of GIRK2 channels. Proc Natl Acad Sci U S A 100:277–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume LC, Leone-Kabler S, Luessen DJ, Marrs GS, Lyons E, Bass CE, Chen R, Selley DE and Howlett AC (2016) Cannabinoid receptor interacting protein suppresses agonist-driven CB1 receptor internalization and regulates receptor replenishment in an agonist-biased manner. J Neurochem 139:396–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume LC, Patten T, Eldeeb K, Leone-Kabler S, Ilyasov AA, Keegan BM, O’Neal JE, Bass CE, Hantgan RR, Lowther WT, Selley DE and Howlett AL (2017) Cannabinoid Receptor Interacting Protein 1a Competition with beta-Arrestin for CB1 Receptor Binding Sites. Mol Pharmacol 91:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ and Caron MG (2000) Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature 408:720–723. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ and Caron MG (2002) Differential mechanisms of morphine antinociceptive tolerance revealed in (beta)arrestin-2 knock-out mice. J Neurosci 22:10494–10500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG and Lin FT (1999) Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 286:2495–2498. [DOI] [PubMed] [Google Scholar]
- Bosier B, Lambert DM and Hermans E (2008) Reciprocal influences of CB1 cannabinoid receptor agonists on ERK and JNK signalling in N1E-115 cells. FEBS Lett 582:3861–3867. [DOI] [PubMed] [Google Scholar]
- Bossong MG, van Berckel BN, Boellaard R, Zuurman L, Schuit RC, Windhorst AD, van Gerven JM, Ramsey NF, Lammertsma AA and Kahn RS (2009) Delta 9-tetrahydrocannabinol induces dopamine release in the human striatum. Neuropsychopharmacology 34:759–766. [DOI] [PubMed] [Google Scholar]
- Bouaboula M, Poinot-Chazel C, Bourrie B, Canat X, Calandra B, Rinaldi-Carmona M, Le Fur G and Casellas P (1995) Activation of mitogen-activated protein kinases by stimulation of the central cannabinoid receptor CB1. Biochem J 312 (Pt 2):637–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouaboula M, Poinot-Chazel C, Marchand J, Canat X, Bourrie B, Rinaldi-Carmona M, Calandra B, Le Fur G and Casellas P (1996) Signaling pathway associated with stimulation of CB2 peripheral cannabinoid receptor. Involvement of both mitogen-activated protein kinase and induction of Krox-24 expression. Eur J Biochem 237:704–711. [DOI] [PubMed] [Google Scholar]
- Bradshaw HB, Rimmerman N, Krey JF and Walker JM (2006) Sex and hormonal cycle differences in rat brain levels of pain-related cannabimimetic lipid mediators. Am J Physiol Regul Integr Comp Physiol 291:R349–358. [DOI] [PubMed] [Google Scholar]
- Breivogel CS, Childers SR, Deadwyler SA, Hampson RE, Vogt LJ and Sim-Selley LJ (1999) Chronic delta9-tetrahydrocannabinol treatment produces a time-dependent loss of cannabinoid receptors and cannabinoid receptor-activated G proteins in rat brain. J Neurochem 73:2447–2459. [DOI] [PubMed] [Google Scholar]
- Breivogel CS, Lambert JM, Gerfin S, Huffman JW and Razdan RK (2008) Sensitivity to delta9-tetrahydrocannabinol is selectively enhanced in beta-arrestin2 −/− mice. Behav Pharmacol 19:298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breivogel CS and Vaghela MS (2015) The effects of beta-arrestin1 deletion on acute cannabinoid activity, brain cannabinoid receptors and tolerance to cannabinoids in mice. J Recept Signal Transduct Res 35:98–106. [DOI] [PubMed] [Google Scholar]
- Budney AJ, Hughes JR, Moore BA and Vandrey R (2004) Review of the validity and significance of cannabis withdrawal syndrome. Am J Psychiatry 161:1967–1977. [DOI] [PubMed] [Google Scholar]
- Budney AJ, Vandrey RG, Hughes JR, Moore BA and Bahrenburg B (2007) Oral delta-9-tetrahydrocannabinol suppresses cannabis withdrawal symptoms. Drug Alcohol Depend 86:22–29. [DOI] [PubMed] [Google Scholar]
- Budney AJ, Vandrey RG, Hughes JR, Thostenson JD and Bursac Z (2008) Comparison of cannabis and tobacco withdrawal: severity and contribution to relapse. J Subst Abuse Treat 35:362–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey LM, Xu Z, Rajic G, Makriyannis A, Romero J, Hillard C, Mackie K and Hohmann AG (2023) Peripheral sensory neuron CB2 cannabinoid receptors are necessary for both CB2-mediated antinociceptive efficacy and sparing of morphine tolerance in a mouse model of anti-retroviral toxic neuropathy. Pharmacol Res 187:106560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaneto MS, Gorelick DA, Desrosiers NA, Hartman RL, Pirard S and Huestis MA (2014) Synthetic cannabinoids: epidemiology, pharmacodynamics, and clinical implications. Drug Alcohol Depend 144:12–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelli MP, Fadda P, Casu A, Spano MS, Casti A, Fratta W and Fattore L (2014) Male and female rats differ in brain cannabinoid CB1 receptor density and function and in behavioural traits predisposing to drug addiction: effect of ovarian hormones. Curr Pharm Des 20:2100–2113. [DOI] [PubMed] [Google Scholar]
- Ceccarini J, Kuepper R, Kemels D, van Os J, Henquet C and Van Laere K (2015) [18F]MK-9470 PET measurement of cannabinoid CB1 receptor availability in chronic cannabis users. Addict Biol 20:357–367. [DOI] [PubMed] [Google Scholar]
- Chang L, Cash BD, Lembo A, Kunkel DC, English BA, Lindstrom B, Gu G, Skare S, Gilder K, Turner S, Cataldi F, Lipkis D and Jan T (2023) Efficacy and safety of olorinab, a full agonist of the cannabinoid receptor 2, for the treatment of abdominal pain in patients with irritable bowel syndrome: Results from a phase 2b randomized placebo-controlled trial (CAPTIVATE). Neurogastroenterol Motil:e14539. [DOI] [PubMed] [Google Scholar]
- Chmiel JF, Flume P, Downey DG, Dozor AJ, Colombo C, Mazurek H, Sapiejka E, Rachel M, Constantine S, Conley B, Dgetluck N, Dinh Q, White B, Elborn JS and Lenabasum JBTCFSG (2021) Safety and efficacy of lenabasum in a phase 2 randomized, placebo-controlled trial in adults with cystic fibrosis. J Cyst Fibros 20:78–85. [DOI] [PubMed] [Google Scholar]
- Chopda GR, Parge V, Thakur GA, Gatley SJ, Makriyannis A and Paronis CA (2016) Tolerance to the Diuretic Effects of Cannabinoids and Cross-Tolerance to a kappa-Opioid Agonist in THC-Treated Mice. J Pharmacol Exp Ther 358:334–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichewicz DL, Martin ZL, Smith FL and Welch SP (1999) Enhancement mu opioid antinociception by oral delta9-tetrahydrocannabinol: dose-response analysis and receptor identification. J Pharmacol Exp Ther 289:859–867. [PubMed] [Google Scholar]
- Cinar R, Gochuico BR, Iyer MR, Jourdan T, Yokoyama T, Park JK, Coffey NJ, Pri-Chen H, Szanda G, Liu Z, Mackie K, Gahl WA and Kunos G (2017) Cannabinoid CB1 receptor overactivity contributes to the pathogenesis of idiopathic pulmonary fibrosis. JCI Insight 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocchetto DM, Cook LF and Cato AE (1981) A critical review of the safety and antiemetic efficacy of delta-9-tetrahydrocannabinol. Drug Intell Clin Pharm 15:867–875. [DOI] [PubMed] [Google Scholar]
- Cook SA, Lowe JA and Martin BR (1998) CB1 receptor antagonist precipitates withdrawal in mice exposed to Delta9-tetrahydrocannabinol. J Pharmacol Exp Ther 285:1150–1156. [PubMed] [Google Scholar]
- Cooper ZD (2016) Adverse Effects of Synthetic Cannabinoids: Management of Acute Toxicity and Withdrawal. Curr Psychiatry Rep 18:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper ZD and Haney M (2014) Investigation of sex-dependent effects of cannabis in daily cannabis smokers. Drug Alcohol Depend 136:85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper ZD and Haney M (2016) Sex-dependent effects of cannabis-induced analgesia. Drug Alcohol Depend 167:112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copersino ML, Boyd SJ, Tashkin DP, Huestis MA, Heishman SJ, Dermand JC, Simmons MS and Gorelick DA (2006) Cannabis withdrawal among non-treatment-seeking adult cannabis users. Am J Addict 15:8–14. [DOI] [PubMed] [Google Scholar]
- Corchero J, Romero J, Berrendero F, Fernandez-Ruiz J, Ramos JA, Fuentes JA and Manzanares J (1999) Time-dependent differences of repeated administration with Delta9-tetrahydrocannabinol in proenkephalin and cannabinoid receptor gene expression and G-protein activation by mu-opioid and CB1-cannabinoid receptors in the caudate-putamen. Brain Res Mol Brain Res 67:148–157. [DOI] [PubMed] [Google Scholar]
- Cornelius JR, Bukstein OG, Douaihy AB, Clark DB, Chung TA, Daley DC, Wood DS and Brown SJ (2010) Double-blind fluoxetine trial in comorbid MDD-CUD youth and young adults. Drug Alcohol Depend 112:39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cota D, Marsicano G, Tschop M, Grubler Y, Flachskamm C, Schubert M, Auer D, Yassouridis A, Thone-Reineke C, Ortmann S, Tomassoni F, Cervino C, Nisoli E, Linthorst AC, Pasquali R, Lutz B, Stalla GK and Pagotto U (2003) The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. J Clin Invest 112:423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutts AA, Anavi-Goffer S, Ross RA, MacEwan DJ, Mackie K, Pertwee RG and Irving AJ (2001) Agonist-induced internalization and trafficking of cannabinoid CB1 receptors in hippocampal neurons. J Neurosci 21:2425–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft RM, Kandasamy R and Davis SM (2013a) Sex differences in anti-allodynic, anti-hyperalgesic and anti-edema effects of Delta(9)-tetrahydrocannabinol in the rat. Pain 154:1709–1717. [DOI] [PubMed] [Google Scholar]
- Craft RM, Marusich JA and Wiley JL (2013b) Sex differences in cannabinoid pharmacology: a reflection of differences in the endocannabinoid system? Life Sci 92:476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft RM, Wakley AA, Tsutsui KT and Laggart JD (2012) Sex differences in cannabinoid 1 vs. cannabinoid 2 receptor-selective antagonism of antinociception produced by delta9-tetrahydrocannabinol and CP55,940 in the rat. J Pharmacol Exp Ther 340:787–800. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA and Gilula NB (1996) Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 384:83–87. [DOI] [PubMed] [Google Scholar]
- Curran HV, Freeman TP, Mokrysz C, Lewis DA, Morgan CJ and Parsons LH (2016) Keep off the grass? Cannabis, cognition and addiction. Nat Rev Neurosci 17:293–306. [DOI] [PubMed] [Google Scholar]
- Cuttler C, Mischley LK and Sexton M (2016) Sex Differences in Cannabis Use and Effects: A Cross-Sectional Survey of Cannabis Users. Cannabis Cannabinoid Res 1:166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuttler C, Spradlin A, Cleveland MJ and Craft RM (2020) Short- and Long-Term Effects of Cannabis on Headache and Migraine. J Pain 21:722–730. [DOI] [PubMed] [Google Scholar]
- D’Souza DC, Cortes-Briones JA, Ranganathan M, Thurnauer H, Creatura G, Surti T, Planeta B, Neumeister A, Pittman B, Normandin M, Kapinos M, Ropchan J, Huang Y, Carson RE and Skosnik PD (2016) Rapid Changes in CB1 Receptor Availability in Cannabis Dependent Males after Abstinence from Cannabis. Biol Psychiatry Cogn Neurosci Neuroimaging 1:60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza DC, Ranganathan M, Braley G, Gueorguieva R, Zimolo Z, Cooper T, Perry E and Krystal J (2008) Blunted psychotomimetic and amnestic effects of delta-9-tetrahydrocannabinol in frequent users of cannabis. Neuropsychopharmacology 33:2505–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlhamer J, Lucas J, Zelaya C, Nahin R, Mackey S, DeBar L, Kerns R, Von Korff M, Porter L and Helmick C (2018) Prevalence of Chronic Pain and High-Impact Chronic Pain Among Adults - United States, 2016. MMWR Morb Mortal Wkly Rep 67:1001–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle TL, Kearn CS and Mackie K (2008a) Rapid CB1 cannabinoid receptor desensitization defines the time course of ERK1/2 MAP kinase signaling. Neuropharmacology 54:36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle TL, Kwok ML and Mackie K (2008b) Regulation of CB1 cannabinoid receptor internalization by a promiscuous phosphorylation-dependent mechanism. J Neurochem 106:70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton GD, Smith FL, Smith PA and Dewey WL (2005) Protein Kinase A activity is increased in mouse lumbar spinal cord but not brain following morphine antinociceptive tolerance for 15 days. Pharmacol Res 52:204–210. [DOI] [PubMed] [Google Scholar]
- de Miguel R, Romero J, Munoz RM, Garcia-Gil L, Gonzalez S, Villanua MA, Makriyannis A, Ramos JA and Fernandez-Ruiz JJ (1998) Effects of cannabinoids on prolactin and gonadotrophin secretion: involvement of changes in hypothalamic gamma-aminobutyric acid (GABA) inputs. Biochem Pharmacol 56:1331–1338. [DOI] [PubMed] [Google Scholar]
- Deng L, Guindon J, Cornett BL, Makriyannis A, Mackie K and Hohmann AG (2015) Chronic cannabinoid receptor 2 activation reverses paclitaxel neuropathy without tolerance or cannabinoid receptor 1-dependent withdrawal. Biol Psychiatry 77:475–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng M, Chen SR, Chen H, Luo Y, Dong Y and Pan HL (2019) Mitogen-activated protein kinase signaling mediates opioid-induced presynaptic NMDA receptor activation and analgesic tolerance. J Neurochem 148:275–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devane WA, Dysarz FA 3rd, Johnson MR, Melvin LS and Howlett AC (1988) Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol 34:605–613. [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A and Mechoulam R (1992) Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258:1946–1949. [DOI] [PubMed] [Google Scholar]
- DeWire SM, Ahn S, Lefkowitz RJ and Shenoy SK (2007) Beta-arrestins and cell signaling. Annu Rev Physiol 69:483–510. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Berrendero F, Bisogno T, Gonzalez S, Cavaliere P, Romero J, Cebeira M, Ramos JA and Fernandez-Ruiz JJ (2000) Enhancement of anandamide formation in the limbic forebrain and reduction of endocannabinoid contents in the striatum of delta9-tetrahydrocannabinol-tolerant rats. J Neurochem 74:1627–1635. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC and Piomelli D (1994) Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 372:686–691. [DOI] [PubMed] [Google Scholar]
- Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, Kathuria S and Piomelli D (2002) Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci U S A 99:10819–10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudok B, Barna L, Ledri M, Szabo SI, Szabadits E, Pinter B, Woodhams SG, Henstridge CM, Balla GY, Nyilas R, Varga C, Lee SH, Matolcsi M, Cervenak J, Kacskovics I, Watanabe M, Sagheddu C, Melis M, Pistis M, Soltesz I and Katona I (2015) Cell-specific STORM super-resolution imaging reveals nanoscale organization of cannabinoid signaling. Nat Neurosci 18:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers CL, Gizer IR, Vieten C, Gilder DA, Stouffer GM, Lau P and Wilhelmsen KC (2010) Cannabis dependence in the San Francisco Family Study: age of onset of use, DSM-IV symptoms, withdrawal, and heritability. Addict Behav 35:102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkashef A, Vocci F, Huestis M, Haney M, Budney A, Gruber A and el-Guebaly N (2008) Marijuana neurobiology and treatment. Subst Abus 29:17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ElSohly MA, Mehmedic Z, Foster S, Gon C, Chandra S and Church JC (2016) Changes in Cannabis Potency Over the Last 2 Decades (1995–2014): Analysis of Current Data in the United States. Biol Psychiatry 79:613–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falenski KW, Thorpe AJ, Schlosburg JE, Cravatt BF, Abdullah RA, Smith TH, Selley DE, Lichtman AH and Sim-Selley LJ (2010) FAAH−/− mice display differential tolerance, dependence, and cannabinoid receptor adaptation after delta 9-tetrahydrocannabinol and anandamide administration. Neuropsychopharmacology 35:1775–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan F, Tao Q, Abood M and Martin BR (1996) Cannabinoid receptor down-regulation without alteration of the inhibitory effect of CP 55,940 on adenylyl cyclase in the cerebellum of CP 55,940-tolerant mice. Brain Res 706:13–20. [DOI] [PubMed] [Google Scholar]
- Farmer RF, Kosty DB, Seeley JR, Duncan SC, Lynskey MT, Rohde P, Klein DN and Lewinsohn PM (2015) Natural course of cannabis use disorders. Psychol Med 45:63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farquhar CE, Breivogel CS, Gamage TF, Gay EA, Thomas BF, Craft RM and Wiley JL (2019) Sex, THC, and hormones: Effects on density and sensitivity of CB(1) cannabinoid receptors in rats. Drug Alcohol Depend 194:20–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehr KA, Kalant H and LeBlanc AE (1976) Residual learning deficit after heavy exposure to cannabis or alcohol in rats. Science 192:1249–1251. [DOI] [PubMed] [Google Scholar]
- Felder CC, Joyce KE, Briley EM, Mansouri J, Mackie K, Blond O, Lai Y, Ma AL and Mitchell RL (1995) Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol Pharmacol 48:443–450. [PubMed] [Google Scholar]
- Finn AK and Whistler JL (2001) Endocytosis of the mu opioid receptor reduces tolerance and a cellular hallmark of opiate withdrawal. Neuron 32:829–839. [DOI] [PubMed] [Google Scholar]
- Fischbach T, Greffrath W, Nawrath H and Treede RD (2007) Effects of anandamide and noxious heat on intracellular calcium concentration in nociceptive drg neurons of rats. J Neurophysiol 98:929–938. [DOI] [PubMed] [Google Scholar]
- Flores-Otero J, Ahn KH, Delgado-Peraza F, Mackie K, Kendall DA and Yudowski GA (2014) Ligand-specific endocytic dwell times control functional selectivity of the cannabinoid receptor 1. Nat Commun 5:4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford BM, Cabanlong CV, Tai S, Franks LN, Penthala NR, Crooks PA, Prather PL and Fantegrossi WE (2019) Reduced Tolerance and Asymmetrical Crosstolerance to Effects of the Indole Quinuclidinone Analog PNR-4-20, a G Protein-Biased Cannabinoid 1 Receptor Agonist in Mice: Comparisons with Delta(9)-Tetrahydrocannabinol and JWH-018. J Pharmacol Exp Ther 369:259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford BM, Tai S, Fantegrossi WE and Prather PL (2017) Synthetic Pot: Not Your Grandfather’s Marijuana. Trends Pharmacol Sci 38:257–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda K, Kato S, Morikawa H, Shoda T and Mori K (1996) Functional coupling of the delta-, mu-, and kappa-opioid receptors to mitogen-activated protein kinase and arachidonate release in Chinese hamster ovary cells. J Neurochem 67:1309–1316. [DOI] [PubMed] [Google Scholar]
- Garcia DE, Brown S, Hille B and Mackie K (1998) Protein kinase C disrupts cannabinoid actions by phosphorylation of the CB1 cannabinoid receptor. J Neurosci 18:2834–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite J, Charles SL and Chess-Williams R (1988) Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature 336:385–388. [DOI] [PubMed] [Google Scholar]
- Garzon J, de la Torre-Madrid E, Rodriguez-Munoz M, Vicente-Sanchez A and Sanchez-Blazquez P (2009) Gz mediates the long-lasting desensitization of brain CB1 receptors and is essential for cross-tolerance with morphine. Mol Pain 5:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatch MB and Forster MJ (2016) Delta(9)-Tetrahydrocannabinol-like effects of novel synthetic cannabinoids in mice and rats. Psychopharmacology (Berl) 233:1901–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerak LR, Zanettini C, Koek W and France CP (2015) Cross-tolerance to cannabinoids in morphine-tolerant rhesus monkeys. Psychopharmacology (Berl) 232:3637–3647. [DOI] [PubMed] [Google Scholar]
- Gine E, Echeverry-Alzate V, Lopez-Moreno JA, Rodriguez de Fonseca F, Perez-Castillo A and Santos A (2017) The CB1 receptor is required for the establishment of the hyperlocomotor phenotype in developmentally-induced hypothyroidism in mice. Neuropharmacology 116:132–141. [DOI] [PubMed] [Google Scholar]
- Glass M and Felder CC (1997) Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor. J Neurosci 17:5327–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez DM, Everett TJ, Hamilton LR, Ranganath A, Cheer JF and Oleson EB (2021) Chronic cannabinoid exposure produces tolerance to the dopamine releasing effects of WIN 55,212–2 and heroin in adult male rats. Neuropharmacology 182:108374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez S, Bisogno T, Wenger T, Manzanares J, Milone A, Berrendero F, Di Marzo V, Ramos JA and Fernandez-Ruiz JJ (2000) Sex steroid influence on cannabinoid CB(1) receptor mRNA and endocannabinoid levels in the anterior pituitary gland. Biochem Biophys Res Commun 270:260–266. [DOI] [PubMed] [Google Scholar]
- Gonzalez S, Cebeira M and Fernandez-Ruiz J (2005) Cannabinoid tolerance and dependence: a review of studies in laboratory animals. Pharmacol Biochem Behav 81:300–318. [DOI] [PubMed] [Google Scholar]
- Gonzalez S, Manzanares J, Berrendero F, Wenger T, Corchero J, Bisogno T, Romero J, Fuentes JA, Di Marzo V, Ramos JA and Fernandez-Ruiz J (1999) Identification of endocannabinoids and cannabinoid CB(1) receptor mRNA in the pituitary gland. Neuroendocrinology 70:137–145. [DOI] [PubMed] [Google Scholar]
- Gorelick DA, Goodwin RS, Schwilke E, Schwope DM, Darwin WD, Kelly DL, McMahon RP, Liu F, Ortemann-Renon C, Bonnet D and Huestis MA (2013) Tolerance to effects of high-dose oral delta9-tetrahydrocannabinol and plasma cannabinoid concentrations in male daily cannabis smokers. J Anal Toxicol 37:11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J, De Lean A and Beaulieu P (2006) Local interactions between anandamide, an endocannabinoid, and ibuprofen, a nonsteroidal anti-inflammatory drug, in acute and inflammatory pain. Pain 121:85–93. [DOI] [PubMed] [Google Scholar]
- Guindon J, Desroches J and Beaulieu P (2007) The antinociceptive effects of intraplantar injections of 2-arachidonoyl glycerol are mediated by cannabinoid CB2 receptors. Br J Pharmacol 150:693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunduz O, Oltulu C and Ulugol A (2011) Role of GLT-1 transporter activation in prevention of cannabinoid tolerance by the beta-lactam antibiotic, ceftriaxone, in mice. Pharmacol Biochem Behav 99:100–103. [DOI] [PubMed] [Google Scholar]
- Hampson RE, Mu J and Deadwyler SA (2000) Cannabinoid and kappa opioid receptors reduce potassium K current via activation of G(s) proteins in cultured hippocampal neurons. J Neurophysiol 84:2356–2364. [DOI] [PubMed] [Google Scholar]
- Hampson RE, Simeral JD, Kelly EJ and Deadwyler SA (2003) Tolerance to the memory disruptive effects of cannabinoids involves adaptation by hippocampal neurons. Hippocampus 13:543–556. [DOI] [PubMed] [Google Scholar]
- Haney M (2007) Opioid antagonism of cannabinoid effects: differences between marijuana smokers and nonmarijuana smokers. Neuropsychopharmacology 32:1391–1403. [DOI] [PubMed] [Google Scholar]
- Haney M, Ward AS, Comer SD, Foltin RW and Fischman MW (1999) Abstinence symptoms following smoked marijuana in humans. Psychopharmacology (Berl) 141:395–404. [DOI] [PubMed] [Google Scholar]
- Haney M, Ward AS, Comer SD, Hart CL, Foltin RW and Fischman MW (2001) Bupropion SR worsens mood during marijuana withdrawal in humans. Psychopharmacology (Berl) 155:171–179. [DOI] [PubMed] [Google Scholar]
- Hanus LO, Meyer SM, Munoz E, Taglialatela-Scafati O and Appendino G (2016) Phytocannabinoids: a unified critical inventory. Nat Prod Rep 33:1357–1392. [DOI] [PubMed] [Google Scholar]
- Hazekamp A, Ware MA, Muller-Vahl KR, Abrams D and Grotenhermen F (2013) The medicinal use of cannabis and cannabinoids--an international cross-sectional survey on administration forms. J Psychoactive Drugs 45:199–210. [DOI] [PubMed] [Google Scholar]
- Henderson-Redmond AN, Crawford LC, Sepulveda DE, Hale DE, Lesperance JJ and Morgan DJ (2021) Sex Differences in Tolerance to Delta-9-Tetrahydrocannabinol in Mice With Cisplatin-Evoked Chronic Neuropathic Pain. Front Mol Biosci 8:684115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson-Redmond AN, Nealon CM, Davis BJ, Yuill MB, Sepulveda DE, Blanton HL, Piscura MK, Zee ML, Haskins CP, Marcus DJ, Mackie K, Guindon J and Morgan DJ (2020) c-Jun N terminal kinase signaling pathways mediate cannabinoid tolerance in an agonist-specific manner. Neuropharmacology 164:107847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson-Redmond AN, Sepulveda DE, Ferguson EL, Kline AM, Piscura MK and Morgan DJ (2022) Sex-specific mechanisms of tolerance for the cannabinoid agonists CP55,940 and delta-9-tetrahydrocannabinol (Delta(9)-THC). Psychopharmacology (Berl) 239:1289–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR and Rice KC (1991) Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci 11:563–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, de Costa BR and Rice KC (1990) Cannabinoid receptor localization in brain. Proc Natl Acad Sci U S A 87:1932–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Avila CA, Rounsaville BJ and Kranzler HR (2004) Opioid-, cannabis- and alcohol-dependent women show more rapid progression to substance abuse treatment. Drug Alcohol Depend 74:265–272. [DOI] [PubMed] [Google Scholar]
- Herrmann ES, Weerts EM and Vandrey R (2015) Sex differences in cannabis withdrawal symptoms among treatment-seeking cannabis users. Exp Clin Psychopharmacol 23:415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hine B (1985) Morphine and delta 9-tetrahydrocannabinol: two-way cross tolerance for antinociceptive and heart-rate responses in the rat. Psychopharmacology (Berl) 87:34–38. [DOI] [PubMed] [Google Scholar]
- Hirvonen J, Goodwin RS, Li CT, Terry GE, Zoghbi SS, Morse C, Pike VW, Volkow ND, Huestis MA and Innis RB (2012) Reversible and regionally selective downregulation of brain cannabinoid CB1 receptors in chronic daily cannabis smokers. Mol Psychiatry 17:642–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho BY, Uezono Y, Takada S, Takase I and Izumi F (1999) Coupling of the expressed cannabinoid CB1 and CB2 receptors to phospholipase C and G protein-coupled inwardly rectifying K+ channels. Recept Channels 6:363–374. [PubMed] [Google Scholar]
- Howlett AC (1985) Cannabinoid inhibition of adenylate cyclase. Biochemistry of the response in neuroblastoma cell membranes. Mol Pharmacol 27:429–436. [PubMed] [Google Scholar]
- Howlett AC (2005) Cannabinoid receptor signaling. Handb Exp Pharmacol:53–79. [DOI] [PubMed] [Google Scholar]
- Howlett AC and Fleming RM (1984) Cannabinoid inhibition of adenylate cyclase. Pharmacology of the response in neuroblastoma cell membranes. Mol Pharmacol 26:532–538. [PubMed] [Google Scholar]
- Howlett AC, Qualy JM and Khachatrian LL (1986) Involvement of Gi in the inhibition of adenylate cyclase by cannabimimetic drugs. Mol Pharmacol 29:307–313. [PubMed] [Google Scholar]
- Hruba L, Ginsburg BC and McMahon LR (2012) Apparent inverse relationship between cannabinoid agonist efficacy and tolerance/cross-tolerance produced by Delta(9)-tetrahydrocannabinol treatment in rhesus monkeys. J Pharmacol Exp Ther 342:843–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh C, Brown S, Derleth C and Mackie K (1999) Internalization and recycling of the CB1 cannabinoid receptor. J Neurochem 73:493–501. [DOI] [PubMed] [Google Scholar]
- Hunt CA and Jones RT (1980) Tolerance and disposition of tetrahydrocannabinol in man. J Pharmacol Exp Ther 215:35–44. [PubMed] [Google Scholar]
- Hutcheson DM, Tzavara ET, Smadja C, Valjent E, Roques BP, Hanoune J and Maldonado R (1998) Behavioural and biochemical evidence for signs of abstinence in mice chronically treated with delta-9-tetrahydrocannabinol. Br J Pharmacol 125:1567–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibsen MS, Connor M and Glass M (2017) Cannabinoid CB(1) and CB(2) Receptor Signaling and Bias. Cannabis Cannabinoid Res 2:48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibsen MS, Finlay DB, Patel M, Javitch JA, Glass M and Grimsey NL (2019) Cannabinoid CB1 and CB2 Receptor-Mediated Arrestin Translocation: Species, Subtype, and Agonist-Dependence. Front Pharmacol 10:350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janoyan JJ, Crim JL and Darmani NA (2002) Reversal of SR 141716A-induced head-twitch and ear-scratch responses in mice by delta 9-THC and other cannabinoids. Pharmacol Biochem Behav 71:155–162. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Wang Q, Zhao J, Wang J, Li Y, Dai W, Zhang X, Fang Z, Hou W and Xiong L (2022) Sex-specific cannabinoid 1 receptors on GABAergic neurons in the ventrolateral periaqueductal gray mediate analgesia in mice. J Comp Neurol 530:2315–2334. [DOI] [PubMed] [Google Scholar]
- Jin W, Brown S, Roche JP, Hsieh C, Celver JP, Kovoor A, Chavkin C and Mackie K (1999) Distinct domains of the CB1 cannabinoid receptor mediate desensitization and internalization. J Neurosci 19:3773–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson-Davis KL, Sadler AJ and Genzen JR (2016) A Retrospective Analysis of Urine Drugs of Abuse Immunoassay True Positive Rates at a National Reference Laboratory. J Anal Toxicol 40:97–107. [DOI] [PubMed] [Google Scholar]
- Johnston J, Lintzeris N, Allsop DJ, Suraev A, Booth J, Carson DS, Helliwell D, Winstock A and McGregor IS (2014) Lithium carbonate in the management of cannabis withdrawal: a randomized placebo-controlled trial in an inpatient setting. Psychopharmacology (Berl) 231:4623–4636. [DOI] [PubMed] [Google Scholar]
- Jones RT, Benowitz N and Bachman J (1976) Clinical studies of cannabis tolerance and dependence. Ann N Y Acad Sci 282:221–239. [DOI] [PubMed] [Google Scholar]
- Jones RT, Benowitz NL and Herning RI (1981) Clinical relevance of cannabis tolerance and dependence. J Clin Pharmacol 21:143S–152S. [DOI] [PubMed] [Google Scholar]
- Kalbasi Anaraki D, Sianati S, Sadeghi M, Ghasemi M, Paydar MJ, Ejtemaei Mehr S and Dehpour AR (2008) Modulation by female sex hormones of the cannabinoid-induced catalepsy and analgesia in ovariectomized mice. Eur J Pharmacol 586:189–196. [DOI] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M and Watanabe M (2009) Endocannabinoid-mediated control of synaptic transmission. Physiol Rev 89:309–380. [DOI] [PubMed] [Google Scholar]
- Kerridge BT, Pickering R, Chou P, Saha TD and Hasin DS (2018) DSM-5 cannabis use disorder in the National Epidemiologic Survey on Alcohol and Related Conditions-III: Gender-specific profiles. Addict Behav 76:52–60. [DOI] [PubMed] [Google Scholar]
- Kesner AJ and Lovinger DM (2020) Cannabinoids, Endocannabinoids and Sleep. Front Mol Neurosci 13:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesner AJ, Mateo Y, Abrahao KP, Ramos-Maciel S, Pava MJ, Gracias AL, Paulsen RT, Carlson HB and Lovinger DM (2022) Changes in striatal dopamine release, sleep, and behavior during spontaneous Delta-9-tetrahydrocannabinol abstinence in male and female mice. Neuropsychopharmacology 47:1537–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khajehali E, Malone DT, Glass M, Sexton PM, Christopoulos A and Leach K (2015) Biased Agonism and Biased Allosteric Modulation at the CB1 Cannabinoid Receptor. Mol Pharmacol 88:368–379. [DOI] [PubMed] [Google Scholar]
- Khan SS, Secades-Villa R, Okuda M, Wang S, Perez-Fuentes G, Kerridge BT and Blanco C (2013) Gender differences in cannabis use disorders: results from the National Epidemiologic Survey of Alcohol and Related Conditions. Drug Alcohol Depend 130:101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JA, Bartlett S, He L, Nielsen CK, Chang AM, Kharazia V, Waldhoer M, Ou CJ, Taylor S, Ferwerda M, Cado D and Whistler JL (2008) Morphine-induced receptor endocytosis in a novel knockin mouse reduces tolerance and dependence. Curr Biol 18:129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Wise LE, Ramesh D, Abdullah R, Selley DE, Cravatt BF and Lichtman AH (2013) Repeated low-dose administration of the monoacylglycerol lipase inhibitor JZL184 retains cannabinoid receptor type 1-mediated antinociceptive and gastroprotective effects. J Pharmacol Exp Ther 345:492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Q, Tian S, Ma C, Wang G and Zhang M (2022) Cannabinoid Receptor Type 2 Agonist Reduces Morphine Tolerance via Mitogen Activated Protein Kinase Phosphatase Induction and Mitogen Activated Protein Kinase Dephosphorylation. Neuroscience 480:56–64. [DOI] [PubMed] [Google Scholar]
- Kouznetsova M, Kelley B, Shen M and Thayer SA (2002) Desensitization of cannabinoid-mediated presynaptic inhibition of neurotransmission between rat hippocampal neurons in culture. Mol Pharmacol 61:477–485. [DOI] [PubMed] [Google Scholar]
- Kovoor A, Celver JP, Wu A and Chavkin C (1998) Agonist induced homologous desensitization of mu-opioid receptors mediated by G protein-coupled receptor kinases is dependent on agonist efficacy. Mol Pharmacol 54:704–711. [PubMed] [Google Scholar]
- Kreitzer AC and Regehr WG (2001) Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 29:717–727. [DOI] [PubMed] [Google Scholar]
- LaFleur RA, Wilson RP, Morgan DJ and Henderson-Redmond AN (2018) Sex differences in antinociceptive response to Delta-9-tetrahydrocannabinol and CP 55,940 in the mouse formalin test. Neuroreport 29:447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprairie RB, Bagher AM, Kelly ME and Denovan-Wright EM (2016) Biased Type 1 Cannabinoid Receptor Signaling Influences Neuronal Viability in a Cell Culture Model of Huntington Disease. Mol Pharmacol 89:364–375. [DOI] [PubMed] [Google Scholar]
- Lauckner JE, Hille B and Mackie K (2005) The cannabinoid agonist WIN55,212-2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins. Proc Natl Acad Sci U S A 102:19144–19149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledent C, Valverde O, Cossu G, Petitet F, Aubert JF, Beslot F, Bohme GA, Imperato A, Pedrazzini T, Roques BP, Vassart G, Fratta W and Parmentier M (1999) Unresponsiveness to cannabinoids and reduced addictive effects of opiates in CB1 receptor knockout mice. Science 283:401–404. [DOI] [PubMed] [Google Scholar]
- Lee MC, Smith FL, Stevens DL and Welch SP (2003) The role of several kinases in mice tolerant to delta 9-tetrahydrocannabinol. J Pharmacol Exp Ther 305:593–599. [DOI] [PubMed] [Google Scholar]
- Leung J, Chan GCK, Hides L and Hall WD (2020) What is the prevalence and risk of cannabis use disorders among people who use cannabis? a systematic review and meta-analysis. Addict Behav 109:106479. [DOI] [PubMed] [Google Scholar]
- Levin FR, McDowell D, Evans SM, Nunes E, Akerele E, Donovan S and Vosburg SK (2004) Pharmacotherapy for marijuana dependence: a double-blind, placebo-controlled pilot study of divalproex sodium. Am J Addict 13:21–32. [DOI] [PubMed] [Google Scholar]
- Levin KH, Copersino ML, Heishman SJ, Liu F, Kelly DL, Boggs DL and Gorelick DA (2010) Cannabis withdrawal symptoms in non-treatment-seeking adult cannabis smokers. Drug Alcohol Depend 111:120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis MM, Yang Y, Wasilewski E, Clarke HA and Kotra LP (2017) Chemical Profiling of Medical Cannabis Extracts. ACS Omega 2:6091–6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li AL, Lin X, Dhopeshwarkar AS, Thomaz AC, Carey LM, Liu Y, Nikas SP, Makriyannis A, Mackie K and Hohmann AG (2019) Cannabinoid CB2 Agonist AM1710 Differentially Suppresses Distinct Pathological Pain States and Attenuates Morphine Tolerance and Withdrawal. Mol Pharmacol 95:155–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman AH, Fisher J and Martin BR (2001a) Precipitated cannabinoid withdrawal is reversed by Delta(9)-tetrahydrocannabinol or clonidine. Pharmacol Biochem Behav 69:181–188. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Sheikh SM, Loh HH and Martin BR (2001b) Opioid and cannabinoid modulation of precipitated withdrawal in delta(9)-tetrahydrocannabinol and morphine-dependent mice. J Pharmacol Exp Ther 298:1007–1014. [PubMed] [Google Scholar]
- Lichtman AH, Wiley JL, LaVecchia KL, Neviaser ST, Arthur DB, Wilson DM and Martin BR (1998) Effects of SR 141716A after acute or chronic cannabinoid administration in dogs. Eur J Pharmacol 357:139–148. [DOI] [PubMed] [Google Scholar]
- Lin X, Dhopeshwarkar AS, Huibregtse M, Mackie K and Hohmann AG (2018) Slowly Signaling G Protein-Biased CB(2) Cannabinoid Receptor Agonist LY2828360 Suppresses Neuropathic Pain with Sustained Efficacy and Attenuates Morphine Tolerance and Dependence. Mol Pharmacol 93:49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JG and Anand KJ (2001) Protein kinases modulate the cellular adaptations associated with opioid tolerance and dependence. Brain Res Brain Res Rev 38:1–19. [DOI] [PubMed] [Google Scholar]
- Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavon FJ, Serrano AM, Selley DE, Parsons LH, Lichtman AH and Cravatt BF (2009) Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol 5:37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie K (2005) Distribution of cannabinoid receptors in the central and peripheral nervous system. Handb Exp Pharmacol:299–325. [DOI] [PubMed] [Google Scholar]
- Mackie K, Devane WA and Hille B (1993) Anandamide, an endogenous cannabinoid, inhibits calcium currents as a partial agonist in N18 neuroblastoma cells. Mol Pharmacol 44:498–503. [PubMed] [Google Scholar]
- Mackie K and Hille B (1992) Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci U S A 89:3825–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie K, Lai Y, Westenbroek R and Mitchell R (1995) Cannabinoids activate an inwardly rectifying potassium conductance and inhibit Q-type calcium currents in AtT20 cells transfected with rat brain cannabinoid receptor. J Neurosci 15:6552–6561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maejima T, Hashimoto K, Yoshida T, Aiba A and Kano M (2001) Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron 31:463–475. [DOI] [PubMed] [Google Scholar]
- Maejima T, Oka S, Hashimotodani Y, Ohno-Shosaku T, Aiba A, Wu D, Waku K, Sugiura T and Kano M (2005) Synaptically driven endocannabinoid release requires Ca2+-assisted metabotropic glutamate receptor subtype 1 to phospholipase Cbeta4 signaling cascade in the cerebellum. J Neurosci 25:6826–6835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguma H and Taylor DA (2011) The effect of chronic opioid vs. cannabinoid exposure on the expression of tolerance to morphine- or WIN-55,212–2-induced analgesia and hypothermia in the guinea pig. Eur J Pharmacol 660:334–340. [DOI] [PubMed] [Google Scholar]
- Mailleux P and Vanderhaeghen JJ (1992) Distribution of neuronal cannabinoid receptor in the adult rat brain: a comparative receptor binding radioautography and in situ hybridization histochemistry. Neuroscience 48:655–668. [DOI] [PubMed] [Google Scholar]
- Manning BH, Mao J, Frenk H, Price DD and Mayer DJ (1996) Continuous co-administration of dextromethorphan or MK-801 with morphine: attenuation of morphine dependence and naloxone-reversible attenuation of morphine tolerance. Pain 67:79–88. [DOI] [PubMed] [Google Scholar]
- Marcus DJ, Zee M, Hughes A, Yuill MB, Hohmann AG, Mackie K, Guindon J and Morgan DJ (2015) Tolerance to the antinociceptive effects of chronic morphine requires c-Jun N-terminal kinase. Mol Pain 11:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrs WR, Blankman JL, Horne EA, Thomazeau A, Lin YH, Coy J, Bodor AL, Muccioli GG, Hu SS, Woodruff G, Fung S, Lafourcade M, Alexander JP, Long JZ, Li W, Xu C, Moller T, Mackie K, Manzoni OJ, Cravatt BF and Stella N (2010) The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat Neurosci 13:951–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BR, Mechoulam R and Razdan RK (1999) Discovery and characterization of endogenous cannabinoids. Life Sci 65:573–595. [DOI] [PubMed] [Google Scholar]
- Martin BR, Sim-Selley LJ and Selley DE (2004) Signaling pathways involved in the development of cannabinoid tolerance. Trends Pharmacol Sci 25:325–330. [DOI] [PubMed] [Google Scholar]
- Martini L, Waldhoer M, Pusch M, Kharazia V, Fong J, Lee JH, Freissmuth C and Whistler JL (2007) Ligand-induced down-regulation of the cannabinoid 1 receptor is mediated by the G-protein-coupled receptor-associated sorting protein GASP1. FASEB J 21:802–811. [DOI] [PubMed] [Google Scholar]
- Mason NL, Theunissen EL, Hutten N, Tse DHY, Toennes SW, Jansen JFA, Stiers P and Ramaekers JG (2021) Reduced responsiveness of the reward system is associated with tolerance to cannabis impairment in chronic users. Addict Biol 26:e12870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheson J, Sproule B, Di Ciano P, Fares A, Le Foll B, Mann RE and Brands B (2020) Sex differences in the acute effects of smoked cannabis: evidence from a human laboratory study of young adults. Psychopharmacology (Berl) 237:305–316. [DOI] [PubMed] [Google Scholar]
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC and Bonner TI (1990) Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346:561–564. [DOI] [PubMed] [Google Scholar]
- McKinney DL, Cassidy MP, Collier LM, Martin BR, Wiley JL, Selley DE and Sim-Selley LJ (2008) Dose-related differences in the regional pattern of cannabinoid receptor adaptation and in vivo tolerance development to delta9-tetrahydrocannabinol. J Pharmacol Exp Ther 324:664–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRae-Clark AL, Baker NL, Gray KM, Killeen TK, Wagner AM, Brady KT, DeVane CL and Norton J (2015) Buspirone treatment of cannabis dependence: A randomized, placebo-controlled trial. Drug Alcohol Depend 156:29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melief EJ, Miyatake M, Bruchas MR and Chavkin C (2010) Ligand-directed c-Jun N-terminal kinase activation disrupts opioid receptor signaling. Proc Natl Acad Sci U S A 107:11608–11613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrirattanakul S, Ramakul N, Guerrero AV, Matsuka Y, Ono T, Iwase H, Mackie K, Faull KF and Spigelman I (2006) Site-specific increases in peripheral cannabinoid receptors and their endogenous ligands in a model of neuropathic pain. Pain 126:102–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mize AL and Alper RH (2000) Acute and long-term effects of 17beta-estradiol on G(i/o) coupled neurotransmitter receptor function in the female rat brain as assessed by agonist-stimulated [35S]GTPgammaS binding. Brain Res 859:326–333. [DOI] [PubMed] [Google Scholar]
- Mlost J, Kostrzewa M, Borczyk M, Bryk M, Chwastek J, Korostynski M and Starowicz K (2021) CB2 agonism controls pain and subchondral bone degeneration induced by mono-iodoacetate: Implications GPCR functional bias and tolerance development. Biomed Pharmacother 136:111283. [DOI] [PubMed] [Google Scholar]
- Molina-Holgado F, Rubio-Araiz A, Garcia-Ovejero D, Williams RJ, Moore JD, Arevalo-Martin A, Gomez-Torres O and Molina-Holgado E (2007) CB2 cannabinoid receptors promote mouse neural stem cell proliferation. Eur J Neurosci 25:629–634. [DOI] [PubMed] [Google Scholar]
- Moore CA, Milano SK and Benovic JL (2007) Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol 69:451–482. [DOI] [PubMed] [Google Scholar]
- Moore CF and Weerts EM (2022) Cannabinoid tetrad effects of oral Delta9-tetrahydrocannabinol (THC) and cannabidiol (CBD) in male and female rats: sex, dose-effects and time course evaluations. Psychopharmacology (Berl) 239:1397–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales P, Hurst DP and Reggio PH (2017) Molecular Targets of the Phytocannabinoids: A Complex Picture. Prog Chem Org Nat Prod 103:103–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan DJ, Davis BJ, Kearn CS, Marcus D, Cook AJ, Wager-Miller J, Straiker A, Myoga MH, Karduck J, Leishman E, Sim-Selley LJ, Czyzyk TA, Bradshaw HB, Selley DE and Mackie K (2014) Mutation of putative GRK phosphorylation sites in the cannabinoid receptor 1 (CB1R) confers resistance to cannabinoid tolerance and hypersensitivity to cannabinoids in mice. J Neurosci 34:5152–5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro S, Thomas KL and Abu-Shaar M (1993) Molecular characterization of a peripheral receptor for cannabinoids. Nature 365:61–65. [DOI] [PubMed] [Google Scholar]
- Nahin RL (2015) Estimates of pain prevalence and severity in adults: United States, 2012. J Pain 16:769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Academies of Sciences E and Medicine (2017) The Health Effects of Cannabis and Cannabinoids: The Current State of Evidence and Recommendations for Research, The National Academies Press, Washington, DC. [PubMed] [Google Scholar]
- Nealon CM, Henderson-Redmond AN, Hale DE and Morgan DJ (2019) Tolerance to WIN55,212-2 is delayed in desensitization-resistant S426A/S430A mice. Neuropharmacology 148:151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen JD, Creehan KM, Kerr TM and Taffe MA (2020) Lasting effects of repeated Δ(9) -tetrahydrocannabinol vapour inhalation during adolescence in male and female rats. Br J Pharmacol 177:188–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen JD, Grant Y, Kerr TM, Gutierrez A, Cole M and Taffe MA (2018) Tolerance to hypothermic and antinoceptive effects of Δ9-tetrahydrocannabinol (THC) vapor inhalation in rats. Pharmacol Biochem Behav 172:33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen PT, Schmid CL, Raehal KM, Selley DE, Bohn LM and Sim-Selley LJ (2012) beta-arrestin2 regulates cannabinoid CB1 receptor signaling and adaptation in a central nervous system region-dependent manner. Biol Psychiatry 71:714–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehaus JL, Liu Y, Wallis KT, Egertova M, Bhartur SG, Mukhopadhyay S, Shi S, He H, Selley DE, Howlett AC, Elphick MR and Lewis DL (2007) CB1 cannabinoid receptor activity is modulated by the cannabinoid receptor interacting protein CRIP 1a. Mol Pharmacol 72:1557–1566. [DOI] [PubMed] [Google Scholar]
- Nourbakhsh M, Miller A, Gofton J, Jones G and Adeagbo B (2019) Cannabinoid Hyperemesis Syndrome: Reports of Fatal Cases. J Forensic Sci 64:270–274. [DOI] [PubMed] [Google Scholar]
- Osei-Hyiaman D, DePetrillo M, Pacher P, Liu J, Radaeva S, Batkai S, Harvey-White J, Mackie K, Offertaler L, Wang L and Kunos G (2005) Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J Clin Invest 115:1298–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oviedo A, Glowa J and Herkenham M (1993) Chronic cannabinoid administration alters cannabinoid receptor binding in rat brain: a quantitative autoradiographic study. Brain Res 616:293–302. [DOI] [PubMed] [Google Scholar]
- Pan B, Wang W, Zhong P, Blankman JL, Cravatt BF and Liu QS (2011) Alterations of endocannabinoid signaling, synaptic plasticity, learning, and memory in monoacylglycerol lipase knock-out mice. J Neurosci 31:13420–13430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks C, Jones BC, Moore BM and Mulligan MK (2020) Sex and Strain Variation in Initial Sensitivity and Rapid Tolerance to Delta9-Tetrahydrocannabinol. Cannabis Cannabinoid Res 5:231–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M, Matti C, Grimsey NL, Legler DF, Javitch JA, Finlay DB and Glass M (2022) Delineating the interactions between the cannabinoid CB(2) receptor and its regulatory effectors; beta-arrestins and GPCR kinases. Br J Pharmacol 179:2223–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penetar DM, Kouri EM, Gross MM, McCarthy EM, Rhee CK, Peters EN and Lukas SE (2005) Transdermal nicotine alters some of marihuana’s effects in male and female volunteers. Drug Alcohol Depend 79:211–223. [DOI] [PubMed] [Google Scholar]
- Pertwee RG (2014) Elevating endocannabinoid levels: pharmacological strategies and potential therapeutic applications. Proc Nutr Soc 73:96–105. [DOI] [PubMed] [Google Scholar]
- Pertwee RG, Stevenson LA and Griffin G (1993) Cross-tolerance between delta-9-tetrahydrocannabinol and the cannabimimetic agents, CP 55,940, WIN 55,212–2 and anandamide. Br J Pharmacol 110:1483–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piscura MK, Sepulveda DE, Maulik M, Guindon J, Henderson-Redmond AN and Morgan DJ (2023) Cannabinoid Tolerance in S426A/S430A × beta-Arrestin 2 Knockout Double-Mutant Mice. J Pharmacol Exp Ther 385:17–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryce G, Giovannoni G and Baker D (2003) Mifepristone or inhibition of 11beta-hydroxylase activity potentiates the sedating effects of the cannabinoid receptor-1 agonist Delta(9)-tetrahydrocannabinol in mice. Neurosci Lett 341:164–166. [DOI] [PubMed] [Google Scholar]
- Ramesh D, Schlosburg JE, Wiebelhaus JM and Lichtman AH (2011) Marijuana dependence: not just smoke and mirrors. ILAR J 52:295–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan M, Braley G, Pittman B, Cooper T, Perry E, Krystal J and D’Souza DC (2009) The effects of cannabinoids on serum cortisol and prolactin in humans. Psychopharmacology (Berl) 203:737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravinet Trillou C, Delgorge C, Menet C, Arnone M and Soubrie P (2004) CB1 cannabinoid receptor knockout in mice leads to leanness, resistance to diet-induced obesity and enhanced leptin sensitivity. Int J Obes Relat Metab Disord 28:640–648. [DOI] [PubMed] [Google Scholar]
- Redmond WJ, Goffaux P, Potvin S and Marchand S (2008) Analgesic and antihyperalgesic effects of nabilone on experimental heat pain. Curr Med Res Opin 24:1017–1024. [DOI] [PubMed] [Google Scholar]
- Reichenbach ZW, DiMattio K, Rajakaruna S, Ambrose D, Cornwell WD, Tallarida RJ, Rogers T, Liu-Chen LY, Tuma RF and Ward SJ (2022) Modulation of Morphine Analgesia, Antinociceptive Tolerance, and Mu-Opioid Receptor Binding by the Cannabinoid CB2 Receptor Agonist O-1966. Front Pharmacol 13:803331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riebe CJ, Hill MN, Lee TT, Hillard CJ and Gorzalka BB (2010) Estrogenic regulation of limbic cannabinoid receptor binding. Psychoneuroendocrinology 35:1265–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi-Carmona M, Barth F, Heaulme M, Shire D, Calandra B, Congy C, Martinez S, Maruani J, Neliat G, Caput D and et al. (1994) SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett 350:240–244. [DOI] [PubMed] [Google Scholar]
- Romero EM, Fernandez B, Sagredo O, Gomez N, Uriguen L, Guaza C, De Miguel R, Ramos JA and Viveros MP (2002) Antinociceptive, behavioural and neuroendocrine effects of CP 55,940 in young rats. Brain Res Dev Brain Res 136:85–92. [DOI] [PubMed] [Google Scholar]
- Romero J, Berrendero F, Garcia-Gil L, Ramos JA and Fernandez-Ruiz JJ (1998) Cannabinoid receptor and WIN-55,212–2-stimulated [35S]GTP gamma S binding and cannabinoid receptor mRNA levels in the basal ganglia and the cerebellum of adult male rats chronically exposed to delta 9-tetrahydrocannabinol. J Mol Neurosci 11:109–119. [DOI] [PubMed] [Google Scholar]
- Romero J, Garcia-Palomero E, Castro JG, Garcia-Gil L, Ramos JA and Fernandez-Ruiz JJ (1997) Effects of chronic exposure to delta9-tetrahydrocannabinol on cannabinoid receptor binding and mRNA levels in several rat brain regions. Brain Res Mol Brain Res 46:100–108. [DOI] [PubMed] [Google Scholar]
- Roser P, Della B, Norra C, Juckel G and Uhl I (2009) No association between chronic cannabis use and loudness dependence of auditory evoked potentials as indicator of central serotonergic neurotransmission. Neurosci Lett 465:113–117. [DOI] [PubMed] [Google Scholar]
- Rossi F, Bernardo ME, Bellini G, Luongo L, Conforti A, Manzo I, Guida F, Cristino L, Imperatore R, Petrosino S, Nobili B, Di Marzo V, Locatelli F and Maione S (2013) The cannabinoid receptor type 2 as mediator of mesenchymal stromal cell immunosuppressive properties. PLoS One 8:e80022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan W, Singer M, Razdan RK, Compton DR and Martin BR (1995) A novel class of potent tetrahydrocannabinols (THCS): 2’-yne-delta 8- and delta 9-THCS. Life Sci 56:2013–2020. [DOI] [PubMed] [Google Scholar]
- Ryberg E, Larsson N, Sjogren S, Hjorth S, Hermansson NO, Leonova J, Elebring T, Nilsson K, Drmota T and Greasley PJ (2007) The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol 152:1092–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson CS, Bedy SM and Carlisle T (2015) Withdrawal seizures seen in the setting of synthetic cannabinoid abuse. Am J Emerg Med 33:1712 e1713. [DOI] [PubMed] [Google Scholar]
- Schattauer SS, Land BB, Reichard KL, Abraham AD, Burgeno LM, Kuhar JR, Phillips PEM, Ong SE and Chavkin C (2017) Peroxiredoxin 6 mediates Galphai protein-coupled receptor inactivation by cJun kinase. Nat Commun 8:743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG, Nguyen PT, Ramesh D, Booker L, Burston JJ, Thomas EA, Selley DE, Sim-Selley LJ, Liu QS, Lichtman AH and Cravatt BF (2010) Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat Neurosci 13:1113–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosburg JE, Carlson BL, Ramesh D, Abdullah RA, Long JZ, Cravatt BF and Lichtman AH (2009) Inhibitors of endocannabinoid-metabolizing enzymes reduce precipitated withdrawal responses in THC-dependent mice. AAPS J 11:342–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosburg JE, Kinsey SG, Ignatowska-Jankowska B, Ramesh D, Abdullah RA, Tao Q, Booker L, Long JZ, Selley DE, Cravatt BF and Lichtman AH (2014) Prolonged monoacylglycerol lipase blockade causes equivalent cannabinoid receptor type 1 receptor-mediated adaptations in fatty acid amide hydrolase wild-type and knockout mice. J Pharmacol Exp Ther 350:196–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch H, Huerta MY, Ruiz CM, Farrell MR, Jung KM, Huang JJ, Campbell RR, Piomelli D and Mahler SV (2018) Adolescent cannabinoid exposure effects on natural reward seeking and learning in rats. Psychopharmacology (Berl) 235:121–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman BJ, McRae-Clark AL, Baker NL, Sonne SC, Killeen TK, Cloud K and Gray KM (2017) Gender differences among treatment-seeking adults with cannabis use disorder: Clinical profiles of women and men enrolled in the achieving cannabis cessation-evaluating N-acetylcysteine treatment (ACCENT) study. Am J Addict 26:136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker JL, Joseph BK, Ruckle MB, Mayeux PR and Prather PL (2005a) The endocannabinoid noladin ether acts as a full agonist at human CB2 cannabinoid receptors. J Pharmacol Exp Ther 314:868–875. [DOI] [PubMed] [Google Scholar]
- Shoemaker JL, Ruckle MB, Mayeux PR and Prather PL (2005b) Agonist-directed trafficking of response by endocannabinoids acting at CB2 receptors. J Pharmacol Exp Ther 315:828–838. [DOI] [PubMed] [Google Scholar]
- Sibaev A, Yuce B, Kemmer M, Van Nassauw L, Broedl U, Allescher HD, Goke B, Timmermans JP and Storr M (2009) Cannabinoid-1 (CB1) receptors regulate colonic propulsion by acting at motor neurons within the ascending motor pathways in mouse colon. Am J Physiol Gastrointest Liver Physiol 296:G119–128. [DOI] [PubMed] [Google Scholar]
- Sim LJ, Hampson RE, Deadwyler SA and Childers SR (1996) Effects of chronic treatment with delta9-tetrahydrocannabinol on cannabinoid-stimulated [35S]GTPgammaS autoradiography in rat brain. J Neurosci 16:8057–8066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim-Selley LJ and Martin BR (2002) Effect of chronic administration of R-(+)-[2,3-Dihydro-5-methyl-3-[(morpholinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxazinyl]-(1-naphthalenyl)methanone mesylate (WIN55,212-2) or delta(9)-tetrahydrocannabinol on cannabinoid receptor adaptation in mice. J Pharmacol Exp Ther 303:36–44. [DOI] [PubMed] [Google Scholar]
- Sim-Selley LJ, Schechter NS, Rorrer WK, Dalton GD, Hernandez J, Martin BR and Selley DE (2006) Prolonged recovery rate of CB1 receptor adaptation after cessation of long-term cannabinoid administration. Mol Pharmacol 70:986–996. [DOI] [PubMed] [Google Scholar]
- Smart D, Gunthorpe MJ, Jerman JC, Nasir S, Gray J, Muir AI, Chambers JK, Randall AD and Davis JB (2000) The endogenous lipid anandamide is a full agonist at the human vanilloid receptor (hVR1). Br J Pharmacol 129:227–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TH, Blume LC, Straiker A, Cox JO, David BG, McVoy JR, Sayers KW, Poklis JL, Abdullah RA, Egertova M, Chen CK, Mackie K, Elphick MR, Howlett AC and Selley DE (2015) Cannabinoid receptor-interacting protein 1a modulates CB1 receptor signaling and regulation. Mol Pharmacol 87:747–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spina E, Trovati A, Parolaro D and Giagnoni G (1998) A role of nitric oxide in WIN 55,212–2 tolerance in mice. Eur J Pharmacol 343:157–163. [DOI] [PubMed] [Google Scholar]
- Stella N, Schweitzer P and Piomelli D (1997) A second endogenous cannabinoid that modulates long-term potentiation. Nature 388:773–778. [DOI] [PubMed] [Google Scholar]
- Storr M, Sibaev A, Marsicano G, Lutz B, Schusdziarra V, Timmermans JP and Allescher HD (2004) Cannabinoid receptor type 1 modulates excitatory and inhibitory neurotransmission in mouse colon. Am J Physiol Gastrointest Liver Physiol 286:G110–117. [DOI] [PubMed] [Google Scholar]
- Szabo B and Schlicker E (2005) Effects of cannabinoids on neurotransmission. Handb Exp Pharmacol:327–365. [DOI] [PubMed] [Google Scholar]
- Tait RJ, Caldicott D, Mountain D, Hill SL and Lenton S (2016) A systematic review of adverse events arising from the use of synthetic cannabinoids and their associated treatment. Clin Toxicol (Phila) 54:1–13. [DOI] [PubMed] [Google Scholar]
- Terman GW, Jin W, Cheong YP, Lowe J, Caron MG, Lefkowitz RJ and Chavkin C (2004) G-protein receptor kinase 3 (GRK3) influences opioid analgesic tolerance but not opioid withdrawal. Br J Pharmacol 141:55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorat SN and Bhargava HN (1994a) Effects of NMDA receptor blockade and nitric oxide synthase inhibition on the acute and chronic actions of delta 9-tetrahydrocannabinol in mice. Brain Res 667:77–82. [DOI] [PubMed] [Google Scholar]
- Thorat SN and Bhargava HN (1994b) Evidence for a bidirectional cross-tolerance between morphine and delta 9-tetrahydrocannabinol in mice. Eur J Pharmacol 260:5–13. [DOI] [PubMed] [Google Scholar]
- Towers EB, Williams IL, Qillawala EI, Rissman EF and Lynch WJ (2023) Sex/Gender Differences in the Time-Course for the Development of Substance Use Disorder: A Focus on the Telescoping Effect. Pharmacol Rev 75:217–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo KA and Akil H (1994) Inhibition of opiate tolerance by non-competitive N-methyl-D-aspartate receptor antagonists. Brain Res 633:178–188. [DOI] [PubMed] [Google Scholar]
- Tseng AH and Craft RM (2001) Sex differences in antinociceptive and motoric effects of cannabinoids. Eur J Pharmacol 430:41–47. [DOI] [PubMed] [Google Scholar]
- Tseng AH, Harding JW and Craft RM (2004) Pharmacokinetic factors in sex differences in Delta 9-tetrahydrocannabinol-induced behavioral effects in rats. Behav Brain Res 154:77–83. [DOI] [PubMed] [Google Scholar]
- Tsou K, Brown S, Sanudo-Pena MC, Mackie K and Walker JM (1998) Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience 83:393–411. [DOI] [PubMed] [Google Scholar]
- Tsou K, Patrick SL and Walker JM (1995) Physical withdrawal in rats tolerant to delta 9-tetrahydrocannabinol precipitated by a cannabinoid receptor antagonist. Eur J Pharmacol 280:R13–15. [DOI] [PubMed] [Google Scholar]
- Tzavara ET, Valjent E, Firmo C, Mas M, Beslot F, Defer N, Roques BP, Hanoune J and Maldonado R (2000) Cannabinoid withdrawal is dependent upon PKA activation in the cerebellum. Eur J Neurosci 12:1038–1046. [DOI] [PubMed] [Google Scholar]
- Valverde O, Maldonado R, Valjent E, Zimmer AM and Zimmer A (2000) Cannabinoid withdrawal syndrome is reduced in pre-proenkephalin knock-out mice. J Neurosci 20:9284–9289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Giessen E, Weinstein JJ, Cassidy CM, Haney M, Dong Z, Ghazzaoui R, Ojeil N, Kegeles LS, Xu X, Vadhan NP, Volkow ND, Slifstein M and Abi-Dargham A (2017) Deficits in striatal dopamine release in cannabis dependence. Mol Psychiatry 22:68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Stelt M, Trevisani M, Vellani V, De Petrocellis L, Schiano Moriello A, Campi B, McNaughton P, Geppetti P and Di Marzo V (2005) Anandamide acts as an intracellular messenger amplifying Ca2+ influx via TRPV1 channels. EMBO J 24:3026–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Sickle MD, Duncan M, Kingsley PJ, Mouihate A, Urbani P, Mackie K, Stella N, Makriyannis A, Piomelli D, Davison JS, Marnett LJ, Di Marzo V, Pittman QJ, Patel KD and Sharkey KA (2005) Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 310:329–332. [DOI] [PubMed] [Google Scholar]
- Wakley AA and Craft RM (2011) Antinociception and sedation following intracerebroventricular administration of Delta(9)-tetrahydrocannabinol in female vs. male rats. Behav Brain Res 216:200–206. [DOI] [PubMed] [Google Scholar]
- Wakley AA, McBride AA, Vaughn LK and Craft RM (2014a) Cyclic ovarian hormone modulation of supraspinal Delta9-tetrahydrocannabinol-induced antinociception and cannabinoid receptor binding in the female rat. Pharmacol Biochem Behav 124:269–277. [DOI] [PubMed] [Google Scholar]
- Wakley AA, Wiley JL and Craft RM (2014b) Sex differences in antinociceptive tolerance to delta-9-tetrahydrocannabinol in the rat. Drug Alcohol Depend 143:22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace MJ, Newton PM, McMahon T, Connolly J, Huibers A, Whistler J and Messing RO (2009) PKCepsilon regulates behavioral sensitivity, binding and tolerance to the CB1 receptor agonist WIN55,212-2. Neuropsychopharmacology 34:1733–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardle MC, Marcus BA and de Wit H (2015) A Preliminary Investigation of Individual Differences in Subjective Responses to D-Amphetamine, Alcohol, and Delta-9-Tetrahydrocannabinol Using a Within-Subjects Randomized Trial. PLoS One 10:e0140501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werth VP, Hejazi E, Pena SM, Haber J, Zeidi M, Reddy N, Okawa J, Feng R, Bashir MM, Gebre K, Jadoo AS, Concha JSS, Dgetluck N, Constantine S and White B (2022) Safety and Efficacy of Lenabasum, a Cannabinoid Receptor Type 2 Agonist, in Patients with Dermatomyositis with Refractory Skin Disease: A Randomized Clinical Trial. J Invest Dermatol 142:2651–2659 e2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetherill RR, Jagannathan K, Hager N, Childress AR and Franklin TR (2015) Sex differences in associations between cannabis craving and neural responses to cannabis cues: Implications for treatment. Exp Clin Psychopharmacol 23:238–246. [DOI] [PubMed] [Google Scholar]
- Whiting PF, Wolff RF, Deshpande S, Di Nisio M, Duffy S, Hernandez AV, Keurentjes JC, Lang S, Misso K, Ryder S, Schmidlkofer S, Westwood M and Kleijnen J (2015) Cannabinoids for Medical Use: A Systematic Review and Meta-analysis. JAMA 313:2456–2473. [DOI] [PubMed] [Google Scholar]
- Whitlow CT, Freedland CS and Porrino LJ (2003) Functional consequences of the repeated administration of Delta9-tetrahydrocannabinol in the rat. Drug Alcohol Depend 71:169–177. [DOI] [PubMed] [Google Scholar]
- Wiley JL (2003) Sex-dependent effects of delta 9-tetrahydrocannabinol on locomotor activity in mice. Neurosci Lett 352:77–80. [DOI] [PubMed] [Google Scholar]
- Wiley JL, Barrus DG, Farquhar CE, Lefever TW and Gamage TF (2021) Sex, species and age: Effects of rodent demographics on the pharmacology of Δ(9)-tetrahydrocanabinol. Prog Neuropsychopharmacol Biol Psychiatry 106:110064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT, Ingram SL, Henderson G, Chavkin C, von Zastrow M, Schulz S, Koch T, Evans CJ and Christie MJ (2013) Regulation of mu-opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol Rev 65:223–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CD, Hiranita T and Fantegrossi WE (2022) Cannabimimetic effects of abused indazole-carboxamide synthetic cannabinoid receptor agonists AB-PINACA, 5F-AB-PINACA and 5F-ADB-PINACA in mice: Tolerance, dependence and withdrawal. Drug Alcohol Depend 236:109468. [DOI] [PubMed] [Google Scholar]
- Wilson DM, Varvel SA, Harloe JP, Martin BR and Lichtman AH (2006) SR 141716 (Rimonabant) precipitates withdrawal in marijuana-dependent mice. Pharmacol Biochem Behav 85:105–113. [DOI] [PubMed] [Google Scholar]
- Wilson RI and Nicoll RA (2001) Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 410:588–592. [DOI] [PubMed] [Google Scholar]
- Wilson-Poe AR, Wiese B, Kibaly C, Lueptow L, Garcia J, Anand P, Cahill C and Moron JA (2021) Effects of inflammatory pain on CB1 receptor in the midbrain periaqueductal gray. Pain Rep 6:e897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winsauer PJ, Daniel JM, Filipeanu CM, Leonard ST, Hulst JL, Rodgers SP, Lassen-Greene CL and Sutton JL (2011) Long-term behavioral and pharmacodynamic effects of delta-9-tetrahydrocannabinol in female rats depend on ovarian hormone status. Addict Biol 16:64–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winstock AR and Barratt MJ (2013) Synthetic cannabis: a comparison of patterns of use and effect profile with natural cannabis in a large global sample. Drug Alcohol Depend 131:106–111. [DOI] [PubMed] [Google Scholar]
- Wise LE, Varvel SA, Selley DE, Wiebelhaus JM, Long KA, Middleton LS, Sim-Selley LJ and Lichtman AH (2011) delta(9)-Tetrahydrocannabinol-dependent mice undergoing withdrawal display impaired spatial memory. Psychopharmacology (Berl) 217:485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withey SL, Kangas BD, Charles S, Gumbert AB, Eisold JE, George SR, Bergman J and Madras BK (2021) Effects of daily Delta(9)-Tetrahydrocannabinol (THC) alone or combined with cannabidiol (CBD) on cognition-based behavior and activity in adolescent nonhuman primates. Drug Alcohol Depend 221:108629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhams SG, Sagar DR, Burston JJ and Chapman V (2015) The role of the endocannabinoid system in pain. Handb Exp Pharmacol 227:119–143. [DOI] [PubMed] [Google Scholar]
- Yacyshyn BR, Hanauer S, Klassen P, English BA, Stauber K, Barish CF, Gilder K, Turner S and Higgins PDR (2021) Safety, Pharmacokinetics, and Efficacy of Olorinab, a Peripherally Acting, Highly Selective, Full Agonist of the Cannabinoid Receptor 2, in a Phase 2a Study of Patients With Chronic Abdominal Pain Associated With Crohn’s Disease. Crohns Colitis 360 3:otaa089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao BB, Hsieh G, Daza AV, Fan Y, Grayson GK, Garrison TR, El Kouhen O, Hooker BA, Pai M, Wensink EJ, Salyers AK, Chandran P, Zhu CZ, Zhong C, Ryther K, Gallagher ME, Chin CL, Tovcimak AE, Hradil VP, Fox GB, Dart MJ, Honore P and Meyer MD (2009) Characterization of a cannabinoid CB2 receptor-selective agonist, A-836339 [2,2,3,3-tetramethyl-cyclopropanecarboxylic acid [3-(2-methoxy-ethyl)-4,5-dimethyl-3H-thiazol-(2Z)-ylidene]-amide], using in vitro pharmacological assays, in vivo pain models, and pharmacological magnetic resonance imaging. J Pharmacol Exp Ther 328:141–151. [DOI] [PubMed] [Google Scholar]
- Yao BB, Hsieh GC, Frost JM, Fan Y, Garrison TR, Daza AV, Grayson GK, Zhu CZ, Pai M, Chandran P, Salyers AK, Wensink EJ, Honore P, Sullivan JP, Dart MJ and Meyer MD (2008) In vitro and in vivo characterization of A-796260: a selective cannabinoid CB2 receptor agonist exhibiting analgesic activity in rodent pain models. Br J Pharmacol 153:390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuill MB, Hale DE, Guindon J and Morgan DJ (2017) Anti-nociceptive interactions between opioids and a cannabinoid receptor 2 agonist in inflammatory pain. Mol Pain 13:1744806917728227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuill MB, Zee ML, Marcus D and Morgan DJ (2016) Tolerance to the antinociceptive and hypothermic effects of morphine is mediated by multiple isoforms of c-Jun N-terminal kinase. Neuroreport 27:392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Ma R, Jin Y, Fang J, Du J, Shao X, Liang Y and Fang J (2021) Molecular mechanisms of opioid tolerance: From opioid receptors to inflammatory mediators (Review). Exp Ther Med 22:1004. [DOI] [PMC free article] [PubMed] [Google Scholar]