

Abstract
The electrochemical nitrate (NO3ˉ) reduction reaction (NO3RR) to ammonia (NH3) represents a sustainable approach for denitrification to balance global nitrogen cycles and an alternative to traditional thermal Haber-Bosch processes. Here, we present a supramolecular strategy for promoting NH3 production in water from NO3RR by integrating two-dimensional (2D) molecular cobalt porphyrin (CoTPP) units into a three-dimensional (3D) porous organic cage architecture. The porphyrin box CoPB-C8 enhances electrochemical active site exposure, facilitates substrate-catalyst interactions, and improves catalyst stability, leading to turnover numbers and frequencies for NH3 production exceeding 200,000 and 56 s−1, respectively. These values represent a 15-fold increase in NO3RR activity and 200-mV improvement in overpotential for the 3D CoPB-C8 box structure compared to its 2D CoTPP counterpart. Synthetic tuning of peripheral alkyl substituents highlights the importance of supramolecular porosity and cavity size on electrochemical NO3RR activity. These findings establish the incorporation of 2D molecular units into 3D confined space microenvironments as an effective supramolecular design strategy for enhancing electrocatalysis.
Keywords: electrocatalysis, supramolecular chemistry, nitrate reduction, porous organic cage, ammonia electrosynthesis
Graphical Abstract

We present a supramolecular strategy for electrochemical nitrate reduction by augmenting porosity and electrochemically active sites as well as facilitating catalyst-substrate interactions. The resulting 3D archtiecture enables selective ammonia production in water with greater than 90% Faradaic efficiency and turnover numbers exceeding 200,000, representing a 15-fold increase in activity and higher catalytic stability over its 2D counterpart.
Introduction
Denitrification, a microbially-facilitated process that reduces nitrate (NO3ˉ) and nitrite (NO2ˉ) to dinitrogen (N2), plays a pivotal role in balancing the global nitrogen cycle.[1] Rising NO3ˉ levels in soil and water, largely triggered by anthropogenic activities, continue to induce serious environmental and health issues by disrupting endogenous nitrogen stores.[2] Indeed, reactive nitrogen (Nr) generation has increased from ca. 30 Tg N yr−1 in 1968 to 226 Tg N yr−1 in 2020, and continues to climb.[3] Excess Nr in the environment, particularly NO3ˉ, has contributed to hydrosphere deterioration,[4] ozone depletion,[5] and damage to the human endocrine system.[6] As such, developing eco-friendly processes for artificial denitrification and remediation of Nr pollution are highly needed. While physical NO3ˉ removal technologies, including ion exchange,[7] reverse osmosis,[8] and electrodialysis,[9] are effective for wastewater purification, the resulting highly concentrated NO3ˉ solutions require further treatment.[10]
In this context, the electrochemical nitrate reduction reaction (NO3RR) is a potentially attractive approach to nitrogen cycle remediation as it can be powered by sustainable energy sources to form more environmentally benign and/or value-added nitrogen-based chemical products.[11] Moreover, when ammonia is the product of NO3RR,[12] it offers an alternative route to this important industrial material and carbon-free fuel compared to the traditional thermal Haber-Bosch process. However, because the reduction of NO3ˉ to NH3 involves the coupled transfer of multiple electron and proton equivalents, NO3RR selectivity is a significant challenge as various products are accessible (e.g., NO2ˉ, NO, N2O, N2, NH2OH).[11a] Against this background, we sought to contribute to the development of molecular electrocatalysts for NO3RR,[13] which are far less studied relative to heterogeneous materials systems,[14] but offer untapped potential to incorporate molecular design elements to facilitate substrate-catalyst interaction and stabilize downstream reduction intermediates.
Along these lines, integrating molecular catalysts into discrete porous supramolecular architectures, such as organic cages,[15] represents an effective strategy to combine molecularly structural tailorability with material porosity and stability and offers potential to bridge the gap between homogeneous and heterogeneous catalysis; this approach has gained increasing attention in electrocatalysis design.[16] The resulting supramolecular electrocatalyst retains the intrinsic reactivities of the molecular subunits, but augments their properties by embedding them within a confined space microenvironment with size-tunable cavities. As part of a larger program in our laboratory in hybrid bioinorganic catalysis,[16b] we have leveraged individual facets of site isolation or increased surface area for achieving electrochemical oxygen reduction reaction (ORR),[17] carbon dioxide reduction reaction (CO2RR),[18] and hydrogen evolution reaction (HER).[19] Inspired by supramolecular systems for NO3ˉ recognition[20] and transport[21] and noting that porphyrin box organic cages[22] are capable of serving as synthetic ion channels to transport nitrate anions across lipid bilayers,[23] we sought to exploit these dual features of supramolecular porosity and intrinsic NO3ˉ affinity for electrochemical NO3RR. We now report that integrating two-dimensional (2D) molecular cobalt porphyrin (CoTPP) catalyst units into a three-dimensional (3D) porous organic cage structure (CoPB-C8) markedly improves its activity and stability for electrochemical NO3RR (Scheme 1A).[16a] This supramolecular catalyst promotes efficient NO3RR in water with high selectivity for ammonia production (>90% Faradaic efficiency), achieving total turnover number (TON) and turnover frequency (TOF) values that exceed 200,000 (33,858 per [Co]) and 56 s−1 (9.4 s−1 per [Co]), respectively. These values represent a 15-fold improvement in NH3 production relative to parent CoTPP, which can be attributed to an increase in electrochemically active cobalt centers and facilitated interactions between the supramolecular CoPB-C8 catalyst and NO3ˉ substrate. Further synthetic tuning by modifying peripheral alkyl substituents reveals the importance of cage porosity and cavity size on electrochemical NO3RR.
Scheme 1.
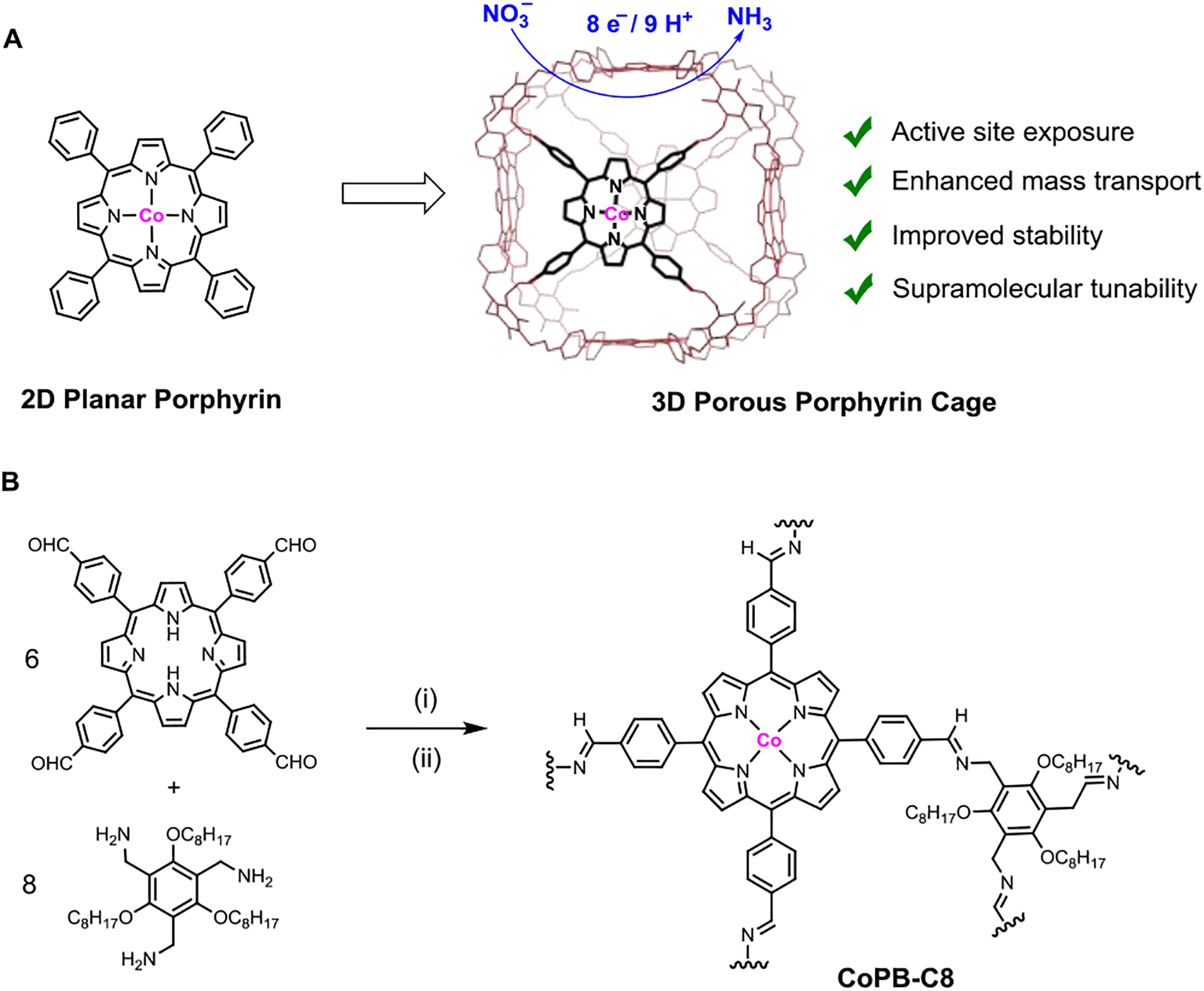
(A) Supramolecular enhancement of electrochemical NO3RR catalyzed by embedding 2D molecular cobalt porphyrin into a 3D porous organic cage architecture. (B) Synthesis of the CoPB-C8. (i) trifluoroacetic acid (TFA), CHCl3, 60 °C, 24 h, 90%; (ii) CoCl2, 2,6-lutidine, THF, 70 °C, 48 h, 95%.
Results and Discussion
Supramolecular Integration of Molecular Cobalt Porphyrin Units Increases Electroactive Metal Centers and Responses to Nitrate
To build a three-dimensional (3D) porous organic cage from two-dimensional (2D) porphyrin building blocks, the free-base porphyrin box (PB-C8) was first self-assembled from six tetraformylphenylporphyrin units and eight tri-amine linkers (Scheme 1B).[24] The selection of PB-C8 was based on its potential as a synthetic ion channel to facilitate the transport of nitrate anions.[23] Metalation of the six porphyrin subunits in PB-C8 with CoCl2 yields the desired cobalt porphyrin box (CoPB-C8)[17a, 19] as confirmed by MALDI mass spectrometry and UV/Vis spectroscopy (Figure S25 and S26). A monomeric cobalt tetraphenylporphyrin (CoTPP) was used as a control compound to evaluate the effects of the supramolecular architecture. To assess electrochemical NO3RR under aqueous conditions, both catalysts were mixed with multi-walled carbon nanotubes (MWNTs) and further deposited onto a glassy carbon electrode.[25] These working electrodes contain a catalyst loading of 5 nmol/cm2 of CoPB-C8 or 30 nmol/cm2 of CoTPP, which normalizes the concentration of cobalt atoms in both electrodes to 30 nmol/cm2. The CV measurements of both electrodes in 0.5 M pH 7.3 sodium sulfate (Na2SO4) electrolyte showed a clear CoIII/CoII redox wave under argon (Ar),[17a, 25–26] which confirms the successful immobilization of catalysts and indicates that the intrinsic electronic properties of molecular porphyrin subunits in the supramolecular structure are not perturbed (Figure 1A and 1B). The slight positive shift of the CoIII/CoII redox couple observed for CoPB-C8 (E1/2 = −0.34 V vs. saturated calomel electrode, SCE) relative to that of CoTPP (E1/2 = −0.39 V vs. SCE) is likely to arise from the electron-withdrawing effect of the imine linkages in the porphyrin box. Plotting the cathodic and anodic peak currents of the CoIII/CoII redox couple versus the scan rate gave linear correlations for both catalysts (Figure S1), enabling us to calculate the electrochemically active Co centers (EACo) for each compound using the corresponding slope (Equation S1).[17a, 27] The calculated EACo concentration normalized per metal center was 2.16 ± 0.17 nmol/cm2 for the CoPB-C8 electrode and 0.44 ± 0.05 nmol/cm2 for CoTPP electrode, corresponding to a 5-fold increase in electrochemically active sites upon embedding the 2D Co porphyrin unit within a 3D porous supramolecular structure (7.2% electroactive Co in CoPB-C8 vs. 1.4% in CoTPP). Upon titration with sodium nitrate (NaNO3) (Figure S2A and S2B), a much larger catalytic current was observed for CoPB-C8 over CoTPP electrode (Figure 1C), indicating superior NO3RR activity for CoPB-C8. As a control experiment, the catalyst-free glassy carbon electrode shows negligible NO3RR activity, confirming that the catalytic currents indeed arise from the cobalt compounds (Figure S2C).
Figure 1.
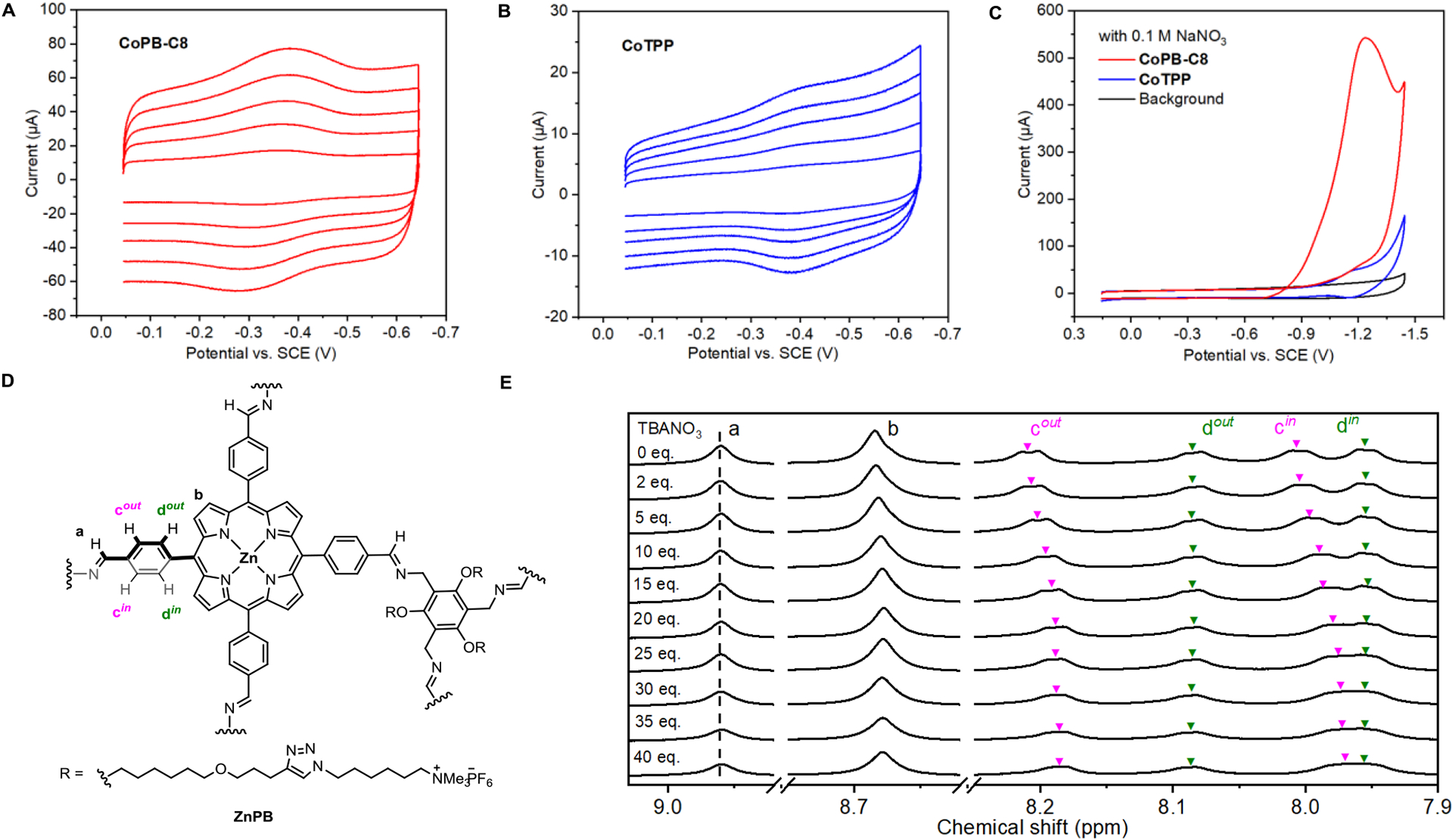
The supramolecular cobalt porphyrin box organic cage CoPB-C8 shows an increase in electroactive metal sites and superior electrochemical nitrite reduction reaction (NO3RR) activity relative to the parent cobalt tetraphenylporphyrin (CoTPP) control analog. Scan rate dependence from 0.1 to 0.5 V/s of the CoIII/CoII redox couples for (A) supramolecular CoPB-C8 and (B) molecular CoTPP catalyst electrodes in 0.5 M pH 7.3 aqueous Na2SO4 solution under an Ar atmosphere. Each catalyst electrode was prepared by depositing 30 nmol/cm2 Co (5.0 nmol/cm2 of CoPB-C8 and 30.0 nmol/cm2 of CoTPP). Plotting the corresponding cathodic and anodic peak currents against the scan rate indicated 1.4% of Co centers are electroactive in the CoTPP film and 7.2% of the Co centers are electroactive in the CoPB-C8 film. (C) CV traces for CoPB-C8 (red), CoTPP (blue), and electrode background (black) obtained in 0.5 M Na2SO4/0.1 M NaNO3 saturated with Ar. (D) Chemical structure of the zinc porphyrin cage (ZnPB) modified for solubility used for 1H-NMR titration experiments. (E) 1H NMR titration of a diamagnetic analog of the supramolecular porphyrin catalyst, ZnPB (1 eq.), with varying concentrations of tetrabutylammonium nitrate (TBANO3) (0–40 eq.), revealing a host-guest interaction between the porphyrin box host and nitrate anion guest.
We hypothesized that compared to gaseous CO2 and O2 substrates, studies of potential substrate-catalyst interactions within supramolecular electrocatalyst systems would be more feasible with solution NO3ˉ via spectroscopic techniques such as nuclear magnetic resonance (NMR) spectroscopy. Indeed, we were able to observe direct interactions between the supramolecular catalyst platform and NO3ˉ substrate by 1H-NMR spectroscopy titration experiments (Figure S3). The addition of tetrabutylammonium nitrate (TBANO3) to a diamagnetic and soluble zinc porphyrin box[28] congener (Figure 1D) in CD3CN solvent led to apparent upfield shifts of protons on the porphyrinic phenyl rings, whereas no shift was observed for the imine protons (a) or the pyrrolic protons (b) (Figure 1E). Interestingly, only the protons at the ortho-position (c) of the imine substituent shifted while the chemical shift of protons at the meta-position (d) remained unchanged, which indicates a possible anion-π interaction of the negative charged NO3ˉ and partially positive charged porphyrinic phenyl rings induced by the adjacent electron-withdrawing imine group.[29] We reasoned that the interaction between the supramolecular cage and NO3ˉ substrate could potentially facilitate mass transport and thus enhance catalytic performance.[23]
Supramolecular Cobalt Porphyrin Box Catalyst Shows Enhanced Reactivity and Stability Over Parent Porphyrin for Electrochemical NO3RR With Ammonia as the Major Product
We next performed controlled potential electrolysis (CPE) experiments with product detection in 0.5 M Na2SO4 with added 0.1 M NaNO3 under an Ar atmosphere. Under these conditions, both CoPB-C8 and CoTPP selectively produced NH3 with Faradaic efficiencies (FE) reaching greater than 90% over a range of applied potentials (−1.04 to −1.54 V vs. SCE) (Figure 2A). Importantly, CoPB-C8 showed superior reactivity for NH3 production, which starts to generate NH3 at an onset potential of −0.94 V vs. SCE with an NH3 yield rate of 16.7 μg h−1 (Figure 2B), while CoTPP required a 200 mV more negative applied potential (-1.14 V vs. SCE) to achieve a similar NH3 production rate (14.2 μg h−1). These results are in line with the CV data (Figure 1C) and establish a supramolecular enhancement for electrochemical NO3RR. Along the applied potentials, CoPB-C8 showed superior NH3 partial current density than CoTPP (Figure S6), of which the largest difference in catalytic activity for the two compounds was observed at −1.14 V vs. SCE with a 15-fold increase in NH3 yield rate for the CoPB-C8 electrode (221.5 μg h−1) over the CoTPP electrode (14.2 μg h−1), thus demonstrating the advantages of integrating a molecular electrocatalyst into a supramolecular matrix. After electrolysis at −1.54 V vs. SCE for 1 h, CoPB-C8 produced 2,729 μg of NH3 with a turnover number (TON) per cobalt center of 5,343 (Figure 2D). Further optimization of catalyst loading showed that the concentration of CoPB-C8 can be lowered to 0.5 nmol/cm2 while still maintaining good electrochemical NO3RR activity, resulting in TON values reaching 208,998 (TON = 34,833 per Co) after 1 h, corresponding to a turnover frequency (TOF) of 56 s−1 (TOF = 9.4 s−1 per Co) (Figure 2E), rendering CoPB-C8 a highly efficient electrochemical NO3RR catalyst under aqueous conditions (Table S2).
Figure 2.
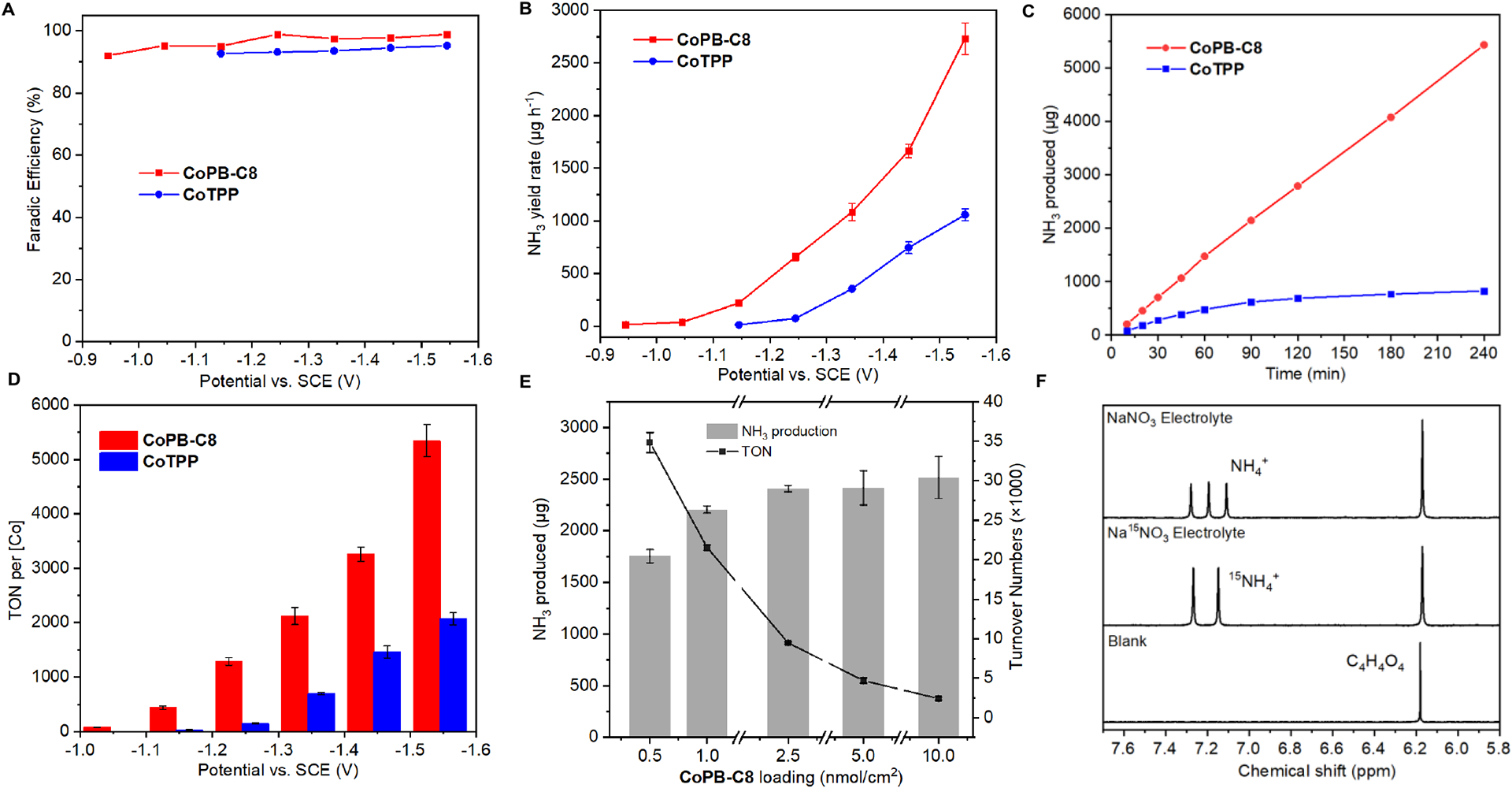
Supramolecular cobalt porphyrin box organic cage shows superior electrochemical nitrate reduction (NO3RR) activity relative to a parent cobalt tetraphenylporphyrin control analog. (A) Faradaic efficiency (FE) for electrochemical nitrate reduction to ammonia, plotted by NH3 production, catalyzed by CoPB-C8 (red) and CoTPP (blue) over a range of applied potentials after 1 h electrolysis. (B) NH3 yield rate for CoPB-C8 (red), and CoTPP (blue). (C) NH3 production during 4 h electrolysis at −1.34 V vs. SCE for CoPB-C8 (red), and CoTPP (blue). (D) Comparison of turnover number (TON) values per Co over a range of applied potentials after 1 h electrolysis. (E) NH3 production and TON for various amounts of CoPB-C8 loading at −1.54 V vs. SCE. (F) 1H NMR spectra showing direct detection of NH3 product with 15N labeling of substrate for electrolysis runs, after three independent nitrate reduction tests at −1.44 V vs SCE, 0.5 M Na2SO4/0.1 M NaNO3, 0.5 M Na2SO4/0.1 M Na15NO3, and 0.5 M Na2SO4. Maleic acid (C4H4O4) was added as an internal standard.
Moreover, embedding the molecular cobalt porphyrin unit into a supramolecular porous cage increases catalyst stability. Stable NH3 production was observed for CoPB-C8 over longer-term CPEs at −1.34 V vs. SCE (Figure 2C), where this catalyst kept producing NH3 at a relatively constant rate of 1,358 μg/h to produce a total of 5,432 μg of NH3 during the 4-h electrolysis, while maintaining a high FE of 95%. In contrast, CoTPP loses significant activity after 1.5 h with the NH3 yield nearing a plateau, resulting in only 825 μg of NH3 being produced after 4 h with a lower FE of 78% (Figures S7 and S8). We speculate that this improved stability likely arises from the isolated cobalt site in the PB architecture, which can prevent catalyst deactivation pathways.[17a, 30] Next, we used 1H-NMR to validate the generation of NH3 by electrochemical NO3RR catalysis, including with 15N-labeled substrate. Indeed, a set of three symmetric peaks with a spacing of 51 Hz corresponding to 14NH4+ were observed after electrolysis at −1.44 V vs. SCE in NaNO3 electrolyte (Figure 2F). Moreover, a doublet peak (JN-H = 72 Hz) attributed to 15NH4+ was detected when using isotopic 15N labeled Na15NO3 electrolyte (Figure 2F), thus confirming that NO3ˉ is indeed the nitrogen source for the observed NH3 product.
Nitrite and Hydroxylamine Are Competent Intermediates in Ammonia Electrosynthesis Catalyzed by the Supramolecular Cobalt Porphyrin Box
Nitrite (NO2ˉ), the two-electron reduction product of NO3ˉ, is another major water pollutant that is even more potentially harmful than NO3ˉ.[11d] Thus, efficient catalysts for the electrochemical NO2ˉ reduction reaction (NO2RR) are desired for developing effective denitrification and chemical production. While several types of molecular catalysts have shown activity towards electrochemical NO2RR, achieving NH3 selectivity with a sufficiently high rate of product generation remains challenging.[31] Recently, a graphite conjugated diimine macrocyclic cobalt catalyst has been reported to reduce NO2ˉ selectively to NH3 with TOF up to 14.4 s−1 when using a glassy carbon electrode.[32] We performed electrochemical NO2RR with the CoPB-C8 catalyst to compare its reactivity with reported systems and determine whether NO2ˉ could be a competent intermediate during our electrochemical NO3RR system. Electrochemical NO2RR was assayed in a similar manner to electrochemical NO3RR except for the use of NaNO2 as a substrate (0.1 M). As shown in Figure 3A, a greater catalytic current was observed for the CoPB-C8 electrode compared to the CoTPP electrode by CV (Figure S9), indicating higher electrochemical NO2RR for the former catalyst. Indeed, CPE experiments revealed that CoPB-C8 showed superior activity to CoTPP for NO2RR (Figures 3B and S10), with NH3 detected as the major product (Figure S11). Under optimized conditions, the highest NO2RR TON of 238932 (39822 per Co center) was obtained with 0.5 nmol/cm2 CoPB-C8 loading at −1.44 vs. SCE after 1 h, corresponding to a TOF of 66.6 s−1 (11.1 s−1 per [Co]) (Figure S12). These data establish that CoPB-C8 is an efficient catalyst for electrochemical NO2ˉ reduction, comparable to the high-performing NO2RR systems reported in the literature.[33] The magnitude of the observed activity enhancement for CoPB-C8 over CoTPP is smaller for NO2RR than for NO3RR. For example, at the applied potential of −1.14 V vs. SCE, CoPB-C8 exhibited a ca. 2.6-fold increase in NH3 yield rate over CoTPP for NO2RR, relative to the 15-fold enhancement for NO3RR, thus demonstrating that the porous supramolecular structure of CoPB-C8 more strongly facilitates the rate-determining step of nitrate to nitrite reduction in the electrochemical NO3RR.
Figure 3.
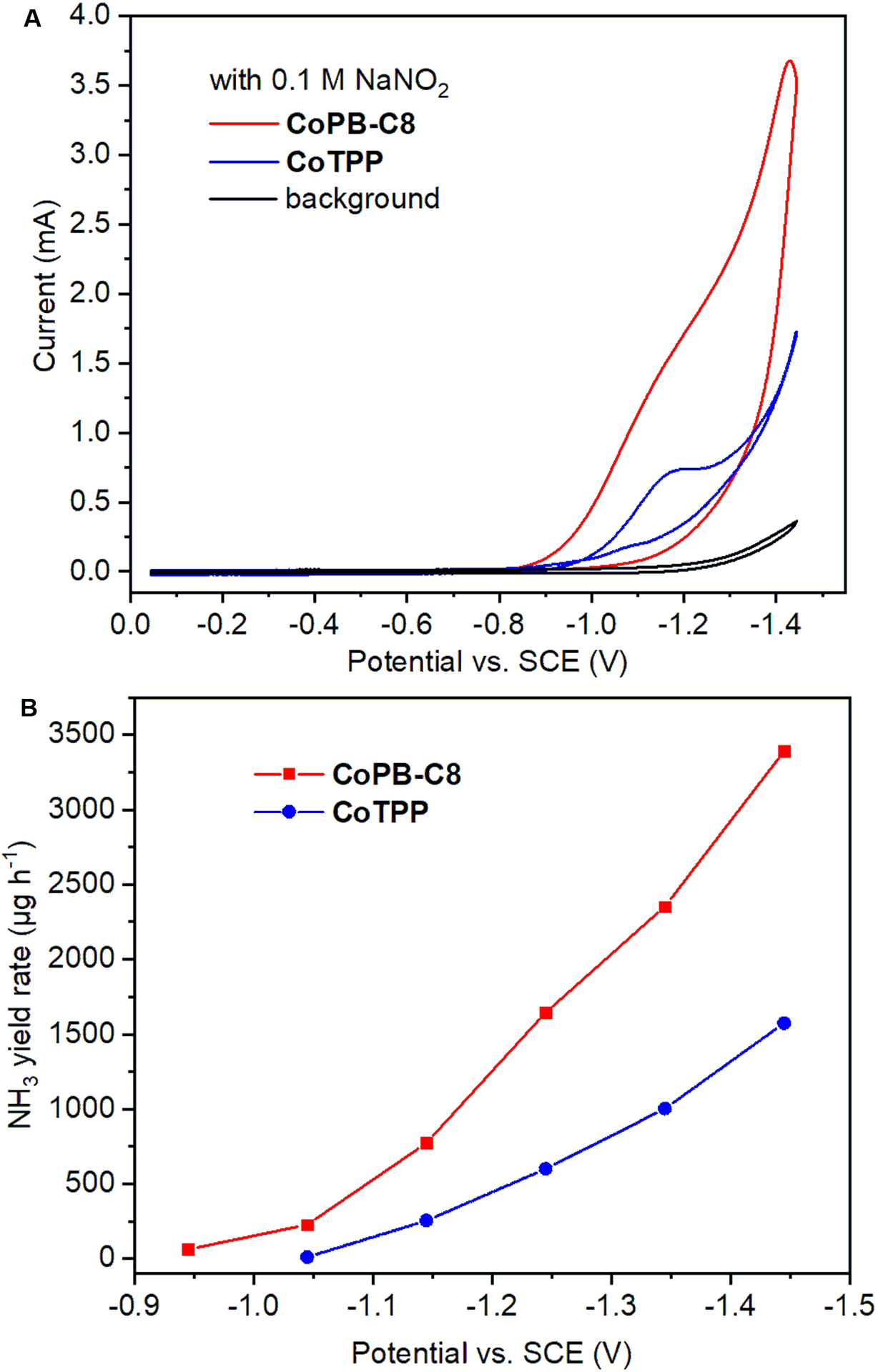
Supramolecular cobalt porphyin box organic cage shows superior electrochemical nitrite reduction activity relative to a cobalt tetraphenylporphyrin control analog. (A) CV traces for CoPB-C8 (red), CoTPP (blue), and electrode background (black) obtained in 0.5 M Na2SO4/0.1 M NaNO2 saturated with Ar. (B) NH3 yield rate for CoPB-C8 (red), and CoTPP (blue).
Finally, hydroxylamine (NH2OH) has been proposed as another key intermediate in both electrochemical NO3RR and NO2RR but often requires the use of a special metal electrode, such as Hg or Cu, to ultimately form NH3.[11c] For our CoPB-C8 electrode, a CPE experiment in 0.5 M Na2SO4/0.1 M NH2OH electrolyte with potential holding at −1.34 V vs. SCE exhibited >99% FE for NH3 production, confirming that NH2OH can also behave as a substrate for electrocatalytic NH3 generation in our platform (Figure S13). The collective results show that the supramolecular porphyrin box catalyst platform is capable of sequential electrochemical reductions to convert NO3− to NH3, but under catalytic conditions does not release partially reduced NO2− or NH2OH intermediates. These findings highlight the ability of this supramolecular architecture to funnel intermediates to desirable product pathways.
Synthetic Tuning of Peripheral Alkyl Substituents on the Supramolecular Box Structure Highlight the Importance of Box Porosity and Cavity Size on Ammonia Electrosynthesis by NO3RR
With these results in hand, we sought to further optimize the catalytic performance of this supramolecular electrocatalyst platform for NO3RR by tuning peripheral structure. We have achieved gains in CO2RR by appending remote positive charges into supramolecular scaffolds.[28] In this instance, we hypothesized that tuning the lengths of peripheral alkyl chains on these CoPB catalysts with a conserved supramolecular core would show measurable effects on electrochemical NO3RR activity and provide direct structure-activity relationships to assess contributions of supramolecular porosity. To this end, we synthesized CoPB-C4 bearing shorter butyl (-C4H9) substituents and CoPB-C13’ bearing longer 6-(hexyloxy)hexyl (-(CH2)6OC6H13) substituents relative to the octyl (-C8H17) groups in CoPB-C8. The electrochemical NO3RR activities of these CoPBs are summarized in Figure 4. While CoPB-C4 showed a similar supramolecular enhancement for NH3 production, the porous cage effect on electrochemical NO3RR of CoPB-13’ was totally suppressed, resulting in a similar reactivity to the nonporous CoTPP. We suggest that the loss of the supramolecular enhancement for NO3RR in CoPB-13’ is likely due to substantial blocking of the cage windows and access to the porous cavity by the longer alkyl chains, with subsequent suppression of nitrate binding and transport. To further assess the effects of alkyl substituent lengths on box porosity and cavity size, we performed molecular dynamics (MD) simulations to obtain the lowest energy conformers of these CoPBs (Figure S14).[17a, 34] We then used a python package, named pywindow,[35] to calculate the average window diameter (Dwindow) and intrinsic void diameter (Dvoid) of this family of CoPBs (Table S1). As expected, the inclusion of longer alkyl chains in the CoPB structures results in decreased Dwindow values as listed in Table S1. While CoPB-C8 (4.2 Å) still retains comparable Dwindow to CoPB-C4 (6.0 Å), the Dwindow of CoPB-C13’ decreased to 2.1 Å, which is much smaller than the hydrodynamic radius of nitrate (3.16 Å),[36] thus suppressing NO3RR for this catalyst. It is noteworthy that the calculated Dwindow for CoPB-C4 (6.0 Å) is close to the window size obtained from the crystal structure of the metal free PB-C4 (6.6 × 8.5 Å),[24] thus demonstrating the reliability of current calculation and indicating negligible structure change of PBs after metalation. The observed correlation between NO3RR reactivity and the cage window size indicates that, despite the presence of multiple potential catalytic sites and significant differences in molecular weight, these CoPBs can be considered as single molecular catalyst entities. Importantly, this correlation suggests that it is feasible to manipulate and adjust the reactivity of these catalysts through synthetic chemistry. Interestingly, the calculated Dvoid values for CoPB-C4 (18.92 Å) and CoPB-C8 (18.71 Å) remained constant, indicating that even the octyl substituent is not long enough to occupy the space inside the cage. However, the calculated Dvoid value decreased significantly for CoPB-C13’ (11.85 Å), presumably due to its much longer alkyl substituents. Taken together, these findings reveal the importance of box porosity and cavity size on electrochemical NO3RR, which will be helpful in establishing design rules for broader exploration of supramolecular electrocatalysis.
Figure 4.
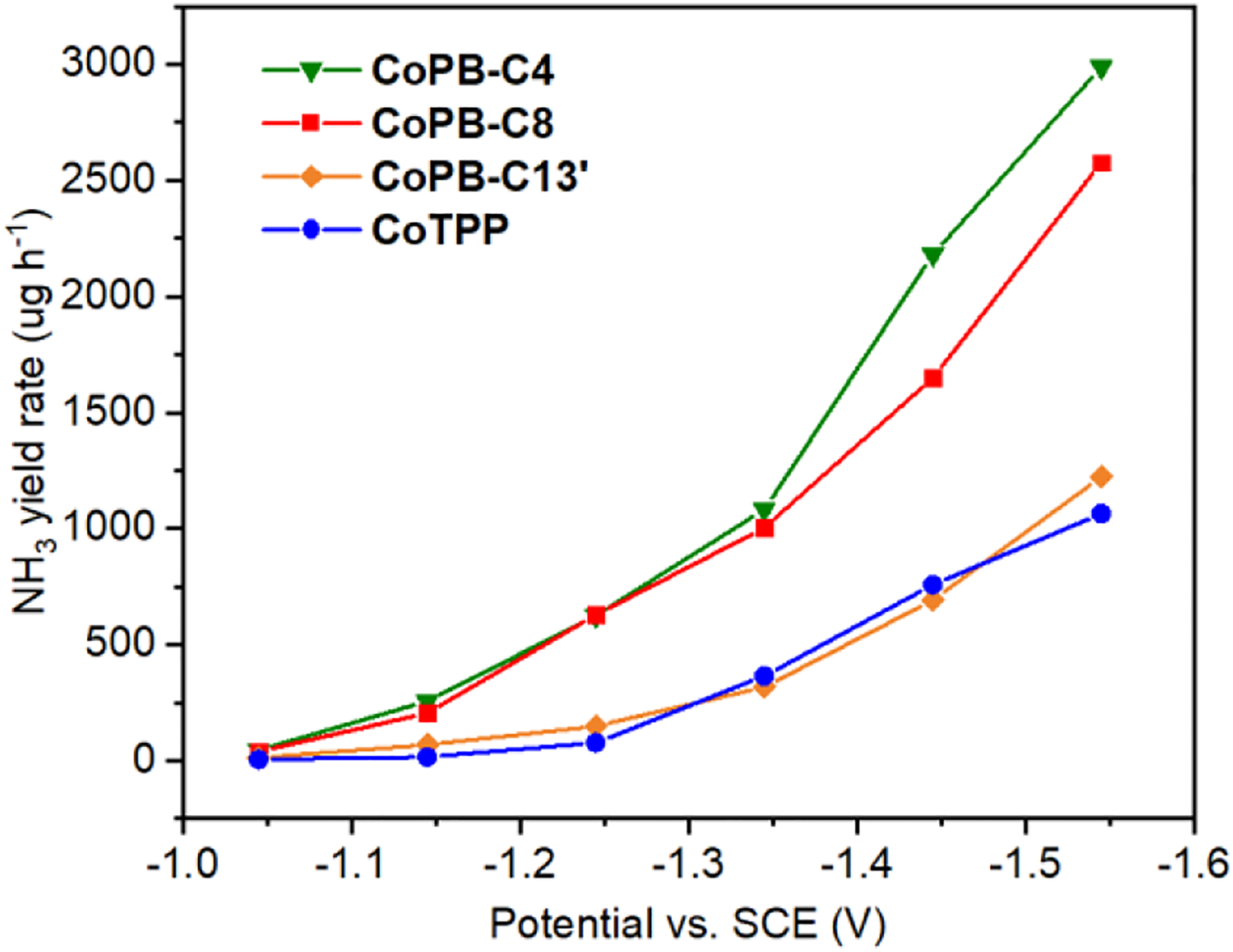
The effect of peripheral alkyl substituents on the eNO3RR activity of CoPBs. NH3 yield rate comparison for CoPB-C4 (green), CoPB-C6 (purple), CoPB-C8 (red), CoPB-C13’ (orange), and CoTPP (blue).
Conclusion
To close, we have presented a supramolecular strategy to enhance electrochemical NO3RR catalysis through integrating a monomeric two-dimensional (2D) cobalt porphyrin electrocatalyst into a three-dimensional (3D) porphyrin-based porous organic cage architecture. The resulting supramolecular electrocatalyst, CoPB-C8, is capable of increasing active site exposure and electrochemically active sites, as well as facilitating catalyst-substrate interactions. These combined features enable selective NO3RR to NH3 product in water with greater than 90% Faradaic efficiency and turnover numbers exceeding 200,000, representing a 15-fold increase in activity, a 200-mV improvement in overpotential, and higher catalytic stability for ammonia electrosynthesis catalyzed by the 3D structure over its 2D counterpart. Moreover, this 3D box platform is competent for selective nitrate-to-ammonia conversion via sequential multi-electron reduction processes, without releasing nitrite or hydroxylamine intermediates. Finally, synthetic tuning of peripheral alkyl substituents reveal the importance of box porosity and cavity size on electrochemical NO3RR activity. This work provides a starting point for using bioinspired, supramolecular design principles for developing catalysts for electrochemical reduction processes related to the nitrogen cycle and a broader range of small-molecule substrates, particularly oxyanions that are important in water remediation and energy storage.
Supplementary Material
Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Science, Office of Advanced Scientific Computing, Office of Basic Energy Sciences, via the Division of Chemical Sciences, Geosciences, and Bioscience of the U.S. Department of Energy at Lawrence Berkeley National Laboratory (Grant No. DE-AC02-05CH11231 to C.J.C.). C.J.C. is a CIFAR Fellow. L.A. thanks SIOC/Pharmaron for a postdoctoral fellowship. M.R.N. acknowledges NSERC (Canada) for a postdoctoral fellowship. P.T.S. and P.D.T. acknowledge the NSF for Graduate Research Fellowships. We thank Dr. Hasan Celik in the UC Berkeley College of Chemistry NMR facility (CoC-NMR) for technical assistance and Prof. Kimoon Kim (POSTECH) for helpful discussions. Instruments in the CoC-NMR are supported in part by NIH (S10OD024998). We also thank the College of Chemistry’s Molecular Graphics and Computation Facility, which is supported by the NIH (S10OD023532), for computational resources.
References
- [1].Kuypers MMM, Marchant HK, Kartal B, Nat. Rev. Microbiol 2018, 16, 263–276. [DOI] [PubMed] [Google Scholar]
- [2].Zhang X, Ward BB, Sigman DM, Chem. Rev 2020, 120, 5308–5351. [DOI] [PubMed] [Google Scholar]
- [3].a) Galloway JN, Bleeker A, Erisman JW, Annu. Rev. Environ. Resour 2021, 46, 255–288; [Google Scholar]; b) Galloway JN, Townsend AR, Erisman JW, Bekunda M, Cai Z, Freney JR, Martinelli LA, Seitzinger SP, Sutton MA, Science 2008, 320, 889–892. [DOI] [PubMed] [Google Scholar]
- [4].Bijay S, Craswell E, SN Appl. Sci 2021, 3, 518. [Google Scholar]
- [5].Lehnert N, Dong HT, Harland JB, Hunt AP, White CJ, Nat. Rev. Chem 2018, 2, 278–289. [Google Scholar]
- [6].Ward MH, Jones RR, Brender JD, De Kok TM, Weyer PJ, Nolan BT, Villanueva CM, Van Breda SG, Int. J. Env. Res. Public Health 2018, 15, 1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Werth CJ, Yan CX, Troutman JP, Acs Es&T Engineering 2021, 1, 6–20. [Google Scholar]
- [8].Scholes RC, Vega MA, Sharp JO, Sedlak DL, Environ. Sci.: Water Res. Technol 2021, 7, 650–661. [Google Scholar]
- [9].Rivera FF, Rivero EP, Castaneda-Zaldivar F, Ind. Eng. Chem. Res 2021, 60, 5014–5023. [Google Scholar]
- [10].Barrabés N, Sá J, Appl. Catal., B 2011, 104, 1–5. [Google Scholar]
- [11].a) Rosca V, Duca M, de Groot MT, Koper MTM, Chem. Rev 2009, 109, 2209–2244; [DOI] [PubMed] [Google Scholar]; b) Duca M, Koper MTM, Energy Environ. Sci 2012, 5, 9726–9742; [Google Scholar]; c) Zeng Y, Priest C, Wang G, Wu G, Small Methods 2020, 4, 2000672; [Google Scholar]; d) Min B, Gao Q, Yan Z, Han X, Hosmer K, Campbell A, Zhu H, Ind. Eng. Chem. Res 2021, 60, 14635–14650. [Google Scholar]
- [12].a) van Langevelde PH, Katsounaros I, Koper MTM, Joule 2021, 5, 290–294; [Google Scholar]; b) Qing G, Ghazfar R, Jackowski ST, Habibzadeh F, Ashtiani MM, Chen C-P, Smith MR, Hamann TW, Chem. Rev 2020, 120, 5437–5516; [DOI] [PubMed] [Google Scholar]; c) Valera-Medina A, Amer-Hatem F, Azad AK, Dedoussi IC, de Joannon M, Fernandes RX, Glarborg P, Hashemi H, He X, Mashruk S, McGowan J, Mounaim-Rouselle C, Ortiz-Prado A, Ortiz-Valera A, Rossetti I, Shu B, Yehia M, Xiao H, Costa M, Energy Fuels 2021, 35, 6964–7029; [Google Scholar]; d) Hao D, Liu Y, Gao S, Arandiyan H, Bai X, Kong Q, Wei W, Shen PK, Ni B-J, Mater. Today 2021, 46, 212–233. [Google Scholar]
- [13].a) Taniguchi I, Nakashima N, Matsushita K, Yasukouchi K, J. Electroanal. Chem 1987, 224, 199–209; [Google Scholar]; b) Chebotareva N, Nyokong T, J. Appl. Electrochem 1997, 27, 975–981; [Google Scholar]; c) Shen J, Birdja YY, Koper MTM, Langmuir 2015, 31, 8495–8501; [DOI] [PubMed] [Google Scholar]; d) Xu S, Ashley DC, Kwon HY, Ware GR, Chen CH, Losovyj Y, Gao XF, Jakubikova E, Smith JM, Chem. Sci 2018, 9, 4950–4958; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Braley SE, Ashley DC, Jakubikova E, Smith JM, Chem. Commun 2020, 56, 603–606; [DOI] [PubMed] [Google Scholar]; f) Partovi S, Xiong ZQ, Kulesa KM, Smith JM, Inorg. Chem 2022, 61, 9034–9039. [DOI] [PubMed] [Google Scholar]
- [14].a) Li J, Zhan GM, Yang JH, Quan FJ, Mao CL, Liu Y, Wang B, Lei FC, Li LJ, Chan AWM, Xu LP, Shi YB, Du Y, Hao WC, Wong PK, Wang JF, Dou SX, Zhang LZ, Yu JC, J. Am. Chem. Soc 2020, 142, 7036–7046; [DOI] [PubMed] [Google Scholar]; b) Chen GF, Yuan YF, Jiang HF, Ren SY, Ding LX, Ma L, Wu TP, Lu J, Wang HH, Nat. Energy 2020, 5, 605–613; [Google Scholar]; c) Wu ZY, Karamad M, Yong X, Huang Q, Cullen DA, Zhu P, Xia C, Xiao Q, Shakouri M, Chen FY, Kim JYT, Xia Y, Heck K, Hu Y, Wong MS, Li Q, Gates I, Siahrostami S, Wang H, Nat Commun 2021, 12, 2870; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Gao Z, Lai YL, Tao Y, Xiao LH, Zhang LX, Luo F, ACS Cent. Sci 2021, 7, 1066–1072; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Gao Q, Yao B, Pillai HS, Zang W, Han X, Liu Y, Yu S-W, Yan Z, Min B, Zhang S, Zhou H, Ma L, Xin H, He Q, Zhu H, Nat. Synth 2023, 10.1038/s44160-023-00258-x; [DOI] [Google Scholar]; f) Lv Y, Ke SW, Gu YM, Tian BL, Tang LY, Ran P, Zhao Y, Ma J, Zuo JL, Ding MN, Angew. Chem. Int. Ed 2023, 10.1002/anie.202305246. [DOI] [PubMed] [Google Scholar]
- [15].a) Tozawa T, Jones JTA, Swamy SI, Jiang S, Adams DJ, Shakespeare S, Clowes R, Bradshaw D, Hasell T, Chong SY, Tang C, Thompson S, Parker J, Trewin A, Bacsa J, Slawin AMZ, Steiner A, Cooper AI, Nature Mater 2009, 8, 973–978; [DOI] [PubMed] [Google Scholar]; b) Diercks CS, Yaghi OM, Science 2017, 355, eaal1585. [DOI] [PubMed] [Google Scholar]
- [16].a) Banerjee S, Anayah RI, Gerke CS, Thoi VS, ACS Cent. Sci 2020, 6, 1671–1684; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Smith PT, Nichols EM, Cao Z, Chang CJ, Acc. Chem. Res 2020, 53, 575–587; [DOI] [PubMed] [Google Scholar]; c) Proppe AH, Li YGC, Aspuru-Guzik A, Berlinguette CP, Chang CJ, Cogdell R, Doyle AG, Flick J, Gabor NM, van Grondelle R, Hammes-Schiffer S, Jaffer SA, Kelley SO, Leclerc M, Leo K, Mallouk TE, Narang P, Schlau-Cohen GS, Scholes GD, Vojvodic A, Yam VWW, Yang JY, Sargent EH, Nat. Rev. Mater 2020, 5, 828–846. [Google Scholar]
- [17].a) Smith PT, Kim Y, Benke BP, Kim K, Chang CJ, Angew. Chem. Int. Ed 2020, 59, 4902–4907; [DOI] [PubMed] [Google Scholar]; b) Oldacre AN, Friedman AE, Cook TR, J. Am. Chem. Soc 2017, 139, 1424–1427. [DOI] [PubMed] [Google Scholar]
- [18].a) Smith PT, Benke BP, Cao Z, Kim Y, Nichols EM, Kim K, Chang CJ, Angew. Chem. Int. Ed 2018, 57, 9684–9688; [DOI] [PubMed] [Google Scholar]; b) Hu Y, Huang S, Wayment LJ, Wu J, Xu Q, Chang T, Chen Y-P, Li X, Andi B, Chen H, Jin Y, Zhu H, Du M, Lu S, Zhang W, Cell Rep. Phys. Sci 2023, 4, 101285. [Google Scholar]
- [19].Smith PT, Benke BP, An L, Kim Y, Kim K, Chang CJ, ChemElectroChem 2021, 8, 1653–1657. [Google Scholar]
- [20].a) Barendt TA, Docker A, Marques I, Felix V, Beer PD, Angew. Chem. Int. Ed 2016, 55, 11069–11076; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lauer JC, Bhat AS, Barwig C, Fritz N, Kirschbaum T, Rominger F, Mastalerz M, Chem.Eur.J 2022, 28, e202201527; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Langton MJ, Beer PD, Chem. Commun 2014, 50, 8124–8127. [DOI] [PubMed] [Google Scholar]
- [21].Wu X, Howe ENW, Gale PA, Acc. Chem. Res 2018, 51, 1870–1879. [DOI] [PubMed] [Google Scholar]
- [22].Mukhopadhyay RD, Kim Y, Koo J, Kim K, Acc. Chem. Res 2018, 51, 2730–2738. [DOI] [PubMed] [Google Scholar]
- [23].Benke BP, Aich P, Kim Y, Kim KL, Rohman MR, Hong S, Hwang IC, Lee EH, Roh JH, Kim K, J. Am. Chem. Soc 2017, 139, 7432–7435. [DOI] [PubMed] [Google Scholar]
- [24].Hong S, Rohman MR, Jia J, Kim Y, Moon D, Kim Y, Ko YH, Lee E, Kim K, Angew. Chem. Int. Ed 2015, 54, 13241–13244. [DOI] [PubMed] [Google Scholar]
- [25].Hu XM, Ronne MH, Pedersen SU, Skrydstrup T, Daasbjerg K, Angew. Chem. Int. Ed 2017, 56, 6468–6472. [DOI] [PubMed] [Google Scholar]
- [26].Cheng SH, Su YO, Inorg. Chem 1994, 33, 5847–5854. [Google Scholar]
- [27].Wang M, Torbensen K, Salvatore D, Ren S, Joulié D, Dumoulin F, Mendoza D, Lassalle-Kaiser B, Işci U, Berlinguette CP, Robert M, Nat. Commun 2019, 10, 3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].An L, De La Torre P, Smith PT, Narouz MR, Chang CJ, Angew. Chem. Int. Ed 2023, 62, e202209396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].a) Adriaenssens L, Estarellas C, Jentzsch AV, Belmonte MM, Matile S, Ballester P, J. Am. Chem. Soc 2013, 135, 8324–8330; [DOI] [PubMed] [Google Scholar]; b) Wang DX, Wang MX, Acc. Chem. Res 2020, 53, 1364–1380; [DOI] [PubMed] [Google Scholar]; c) Watt MM, Zakharov LN, Haley MM, Johnson DW, Angew. Chem. Int. Ed 2013, 52, 10275–10280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ali A, Akram W, Liu H-Y, Molecules 2019, 24, 78. [Google Scholar]
- [31].a) Xu S, Kwon HY, Ashley DC, Chen CH, Jakubikova E, Smith JM, Inorg. Chem 2019, 58, 9443–9451; [DOI] [PubMed] [Google Scholar]; b) Guo YX, Stroka JR, Kandemir B, Dickerson CE, Bren KL, J. Am. Chem. Soc 2018, 140, 16888–16892; [DOI] [PubMed] [Google Scholar]; c) Stroka JR, Kandemir B, Matson EM, Bren KL, ACS Catal. 2020, 10, 13968–13972. [Google Scholar]
- [32].Braley SE, Xie JZ, Losovyj Y, Smith JM, J. Am. Chem. Soc 2021, 143, 7203–7208. [DOI] [PubMed] [Google Scholar]
- [33].Zhang X, Wang YT, Wang YB, Guo YM, Xie XY, Yu YF, Zhang B, Chem. Commun 2022, 58, 2777–2787. [DOI] [PubMed] [Google Scholar]
- [34].Berardo E, Greenaway RL, Turcani L, Alston BM, Bennison MJ, Miklitz M, Clowes R, Briggs ME, Cooper AI, Jelfs KE, Nanoscale 2018, 10, 22381–22388. [DOI] [PubMed] [Google Scholar]
- [35].Miklitz M, Jelfs KE, J. Chem. Inf. Model 2018, 58, 2387–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Marcus Y, Chem. Rev 1988, 88, 1475–1498. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.