

SUMMARY
Small cell lung cancers (SCLCs) rapidly resist cytotoxic chemotherapy and immune checkpoint inhibitor (ICI) treatments. New, non-cross-resistant therapies are thus needed. SCLC cells are committed into neuroendocrine lineage then maturation arrested. Implicating DNA methyltransferase 1 (DNMT1) in the maturation arrests, we find (1) the repression mark methylated CpG, written by DNMT1, is retained at suppressed neuroendocrine-lineage genes, even as other repression marks are erased; (2) DNMT1 is recurrently amplified, whereas Ten-Eleven-Translocation 2 (TET2), which functionally opposes DNMT1, is deleted; (3) DNMT1 is recruited into neuroendocrine-lineage master transcription factor (ASCL1, NEUROD1) hubs in SCLC cells; and (4) DNMT1 knockdown activated ASCL1-target genes and released SCLC cell-cycling exits by terminal lineage maturation, which are cycling exits that do not require the p53/apoptosis pathway used by cytotoxic chemotherapy. Inhibiting DNMT1/corepressors with clinical compounds accordingly extended survival of mice with chemorefractory and ICI-refractory, p53-null, disseminated SCLC. Lineage commitment of SCLC cells can hence be leveraged into non-cytotoxic therapy able to treat chemo/ICI-refractory SCLC.
In brief
Small cell lung cancers (SCLCs) are typically lethal and require new therapies that are not cross-resistant with present cytotoxic and immune checkpoint inhibitor (ICI) treatments. Biswas et al. show that clinical agents, applied for specific non-cytotoxic molecular-targeted effects, leverage neuroendocrine-lineage commitment into resumed lineage maturation to thereby release p53-null chemo/ICI-refractory SCLCs to cell-cycle exits.
Graphical Abstract
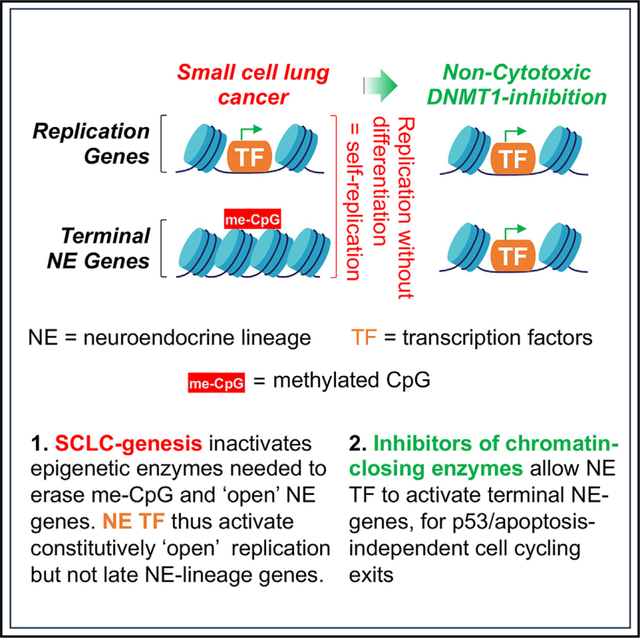
INTRODUCTION
Small cell lung cancers (SCLCs) constitute ~15% of all lung cancers and are increasing in incidence.1 Because clinical presentation is usually with disseminated disease, standard first-line therapy for several decades has been intense systemic combination chemotherapy (platinum drugs and etoposide), supplemented sometimes with local radiation and/or prophylactic cranial irradiation. Addition to chemotherapy of the immune checkpoint inhibitors (ICIs) atezolizumab or durvalumab (antibodies against programmed cell death 1 ligand [anti-PDL1]) generated modest improvements in overall survivals to 12.3 and 13 months, respectively, vs. 10.3 months with chemotherapy alone. That is, 5-year survival rates are ~5% even with ICI, and SCLC has been designated a “recalcitrant cancer” by the United States Congress. New treatments that are not cross-resistant with chemotherapy, radiation, and ICI are thus needed.
Chemotherapy and radiation aim to damage DNA as a stimulus to upregulate the “guardian of the genome” p53, that in turn forces cell-cycle exits followed by orderly cell self-destructions (apoptosis also known as cytotoxicity). The p53 gene TP53 is, however, mutated and deleted (biallelic inactivation) in >95% of SCLCs,2 a loss shown to mediate resistance of SCLC cells to multiple chemotherapeutics and radiation,3–5 even as the same treatments are cytotoxic to normal dividing cells including immune effectors, causing major clinical toxicities and immune suppression. SCLC cells are committed into neuroendocrine lineage, seen by pathology assessment of morphology and lineage markers,6–9 and confirmed by dependency of the cells on specific neuroendocrine-lineage master transcription factors, e.g., achaete-scute family basic-helix-loop-helix (bHLH) transcription factor 1 (ASCL1) and/or neuronal differentiation 1 (NEUROD1).10–14 ASCL1 and NEUROD1 normally activate and cooperate with MYC (or its paralogues MYCL or MYCN), master transcription factor regulators of cell growth and division, to drive cell replications every ~24 H.14,15 Simultaneously, ASCL1/NEUROD1 activate a series of neuroendocrine-lineage gene expression modules that culminate after ~4 replications in the activation of final programs that antagonize MYC/MYCL/MYCN, conclude replications, and focus on specialized neuroendocrine functions instead (terminal lineage differentiation).14,15 This lineage maturation driving function of ASCL1/NEUROD1 is, however, compromised in SCLCs, since terminal neuroendocrine-lineage programs are suppressed persistently through successive cell replications, shown by comprehensive gene-expression profiles.6–9
Encouraging investigation of the mechanisms for the lineage-maturation arrests, cell-cycling exits that occur with maturation into specialized cells do not require the p53/apoptosis pathway used by chemotherapy and radiation (reviewed in Velcheti et al.16). Therefore, toward the overall goal of relieving suppression in SCLC cells of terminal neuroendocrine-lineage programs, as a potential alternative to replication control by p53/apoptosis, we first analyzed the epigenetic landscape at suppressed ASCL1-target genes. “Epigenetic” as we use it here refers to modifications to DNA (methylation of deoxycytidine residues in the DNA sequence that precede deoxyguanosines [CpG]) and post-translational modifications to histones that regulate whether genes are activated vs. repressed, in a manner that can be inherited through cell divisions. We found preservation of the epigenetic repression (transcription “off”) mark methylated-CpG (me-CpG) at neuroendocrine-lineage genes, even as another repression mark histone 3 lysine 27 trimethylation (H3K27me3) was appropriately erased. Further studies implicated DNA methyltransferase 1 (DNMT1), which writes me-CpG, in this aberrant repression, identifying it as a candidate target for therapy. DNMT1 can be depleted from cells using the clinical agents decitabine and 5-azacytidine, including by regimens that are non-cytotoxic.17–19 Neither agent, however, had been described previously to treat SCLC, pre-clinically or clinically (reviewed in Nervi et al.20 and Zavras et al.21). We evaluated the activity of decitabine/5-azacytidine in SCLC cells in vitro and in vivo. We incorporated and extended lessons learned from the clinical application of these DNMT1-targeting pyrimidine nucleoside pro-drugs to treat other cancers, including the value of combination with non-cytotoxic clinical modulators of pyrimidine metabolism,21,22 such as inhibitors of de novo pyrimidine synthesis.23 In this way, we leveraged neuroendocrine lineage commitment of SCLC cells to create p53-independent cell-cycle exits, able to treat p53-null, chemorefractory and ICI-refractory disseminated SCLC. This approach, distinct from chemotherapy/radiation/ICI, is feasible for clinical evaluation, because we used clinically available agents at human-equivalent exposures known to be safe, applied for pre-defined non-cytotoxic pharmacodynamic effects.
RESULTS
Neuroendocrine-lineage gene repression in SCLC cells is mediated by me-CpG
Target genes of the neuroendocrine-lineage master transcription factor ASCL1 have been identified by chromatin immunoprecipitation sequencing (ChIP-seq) (Table S1).14 For comparison, we also analyzed target genes of the master transcription factor regulator of cell growth and division, MYC (Table S1).24 In normal pulmonary neuroendocrine cells, ASCL1-and MYC-target genes were activated to similar levels (Figure 1A). In SCLC cells, however, ASCL1-target genes were suppressed several-fold relative to MYC-target genes (Figure 1A), with lower ASCL1-target gene expression significantly correlating with higher MYC-target gene expression (Figure 1B). We therefore examined distributions of repression (transcription “off”) marks H3K27me3 and me-CpG at ASCL1-and MYC-target genes in SCLC cells. These epigenetic marks are inherited through cell divisions, hence for baseline configurations, we looked in embryonic stem cells (ESCs): in ESCs, H3K27me3 and me-CpG were enriched at ASCL1 targets and depleted at MYC targets (Figures 1C and 1D). In SCLC cells, H3K27me3 was extensively erased from ASCL1 targets and other neuroendocrine genes, yielding a sparse distribution similar to that observed at MYC-target genes (Figures 1C and S1A); me-CpG remained conspicuously enriched, however, at ASCL1 targets and other neuroendocrine genes (Figures 1D and S1B) and inversely correlated with their expression (Figures 1E and S1C).
Figure 1. SCLC cells erase the epigenetic repression mark H3K27me3, but not me-CpG, from ASCL1 targets, recurrently delete me-CpG eraser TET2 (and TET1), and recurrently amplify me-CpG writer DNMT1.
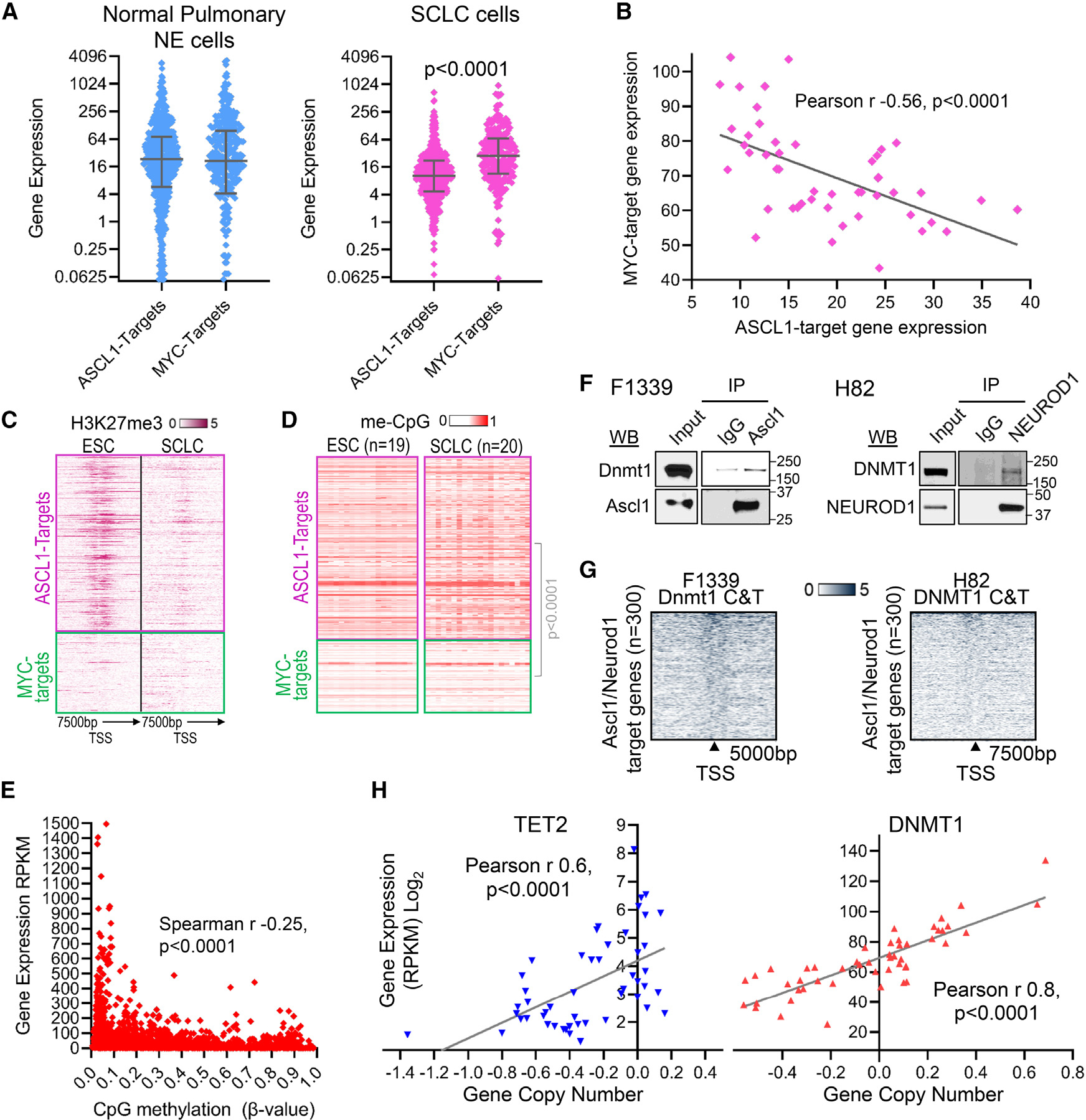
(A) Normal pulmonary neuroendocrine (NE) cells express high levels of both ASCL1-target and MYC-target genes (public single-cell RNA sequencing [RNA-seq] gene expression data are from Human Lung Cell Atlas:25 https://hlca.ds.czbiohub.org/); however, SCLC cells, although also enriched for ASCL1, display several-fold suppression of ASCL1-target vs. MYC-target genes. Average expression of individual ASCL1- and MYC-target genes in 32 SCLC cell lines with ASCL1 expression values > 3 (gene expression by RNA-seq reads per kilobase per million mapped reads (RPKM), Cancer Cell Line Encyclopedia [CCLE]). p value, Mann-Whitney test two-sided. ASCL1-target genes were identified by ChIP-seq by Borromeo et al.14 and with expression value > 0 in pulmonary NE cells. Related to Table S1.
(B) MYC-target gene expression in SCLC cells inversely correlates with ASCL1-target gene expression. Pearson correlation coefficient, two-sided. Average expression of each gene category in individual SCLC cell lines (CCLE). Related to Table S1.
(C) H3K27me3 is enriched at ASCL1 targets, but not MYC targets, at baseline in embryonic stem cells (ESCs) and erased in SCLC cells. ChIP-seq H1 ESCs (GSM733748 Encode) and H69 SCLC cells (GSM513363926).
(D) me-CpG is enriched at ASCL1 targets over MYC targets at baseline in ESCs, a pattern that is preserved into SCLC cells. Mann-Whitney test, two-sided. DNA methylation (β values), CpG in CpG islands linked to these genes, 450K Illumina array (ESC cells, GEO: GSE3184827; SCLC cell lines, GEO: GSE6629528). Related to Table S2.
(E) Negative correlation between me-CpG and gene expression at ASCL1-target genes in SCLC cells. The expression of these genes measured by RNA-seq (RPKM, CCLE) was correlated with average methylation levels at CpG islands linked to the gene (β values, 450K Illumina array, GEO: GSE6629528) in individual SCLC cell lines (n = 17). p value, two-sided.
(F) Endogenous ASCL1 and NEUROD1 recruit DNMT1. Immunoprecipitation-western blot of Ascl1 and NEUROD1 from F1339 and H82 SCLC cells, respectively.
(G) DNMT1 localized at the proximal promoters of ~300 ASCL1- and NEUROD1-target genes by DNMT1 Cut and Tag analyses in F1339 and H82 cells. Experiment was performed in triplicate.
(H) Recurrent deletions of TET2 in SCLC cells with corresponding lower gene expression and recurrent DNMT1 copy number (CN) gains with corresponding higher expression. Gene expression by RNA-seq (RPKM, log2). Normalized CN log2 ratios (log2(CN/2)) segmented by the circular binary segmentation algorithm, with values <−0.15 considered as deletion and >0.15 as amplification (CCLE, SCLC cell lines n = 50).29
DNMT1 writes me-CpG and is a corepressor known to be recruited by other lineage master transcription factors.30–34 We therefore examined whether the neuroendocrine-lineage master transcription factors ASCL1 and/or NEUROD1, highly expressed in F1339 and H82 SCLC cells, respectively, recruit DNMT1. DNMT1 was recruited by endogenous ASCL1 or NEUROD1, shown by immunoprecipitation-western blot (Figure 1F) and by DNMT1 localization at the proximal promoter regions of 300 ASCL1 and NEUROD1-target genes, shown by the DNMT1 Cut and Tag method (Figure 1G). We then examined whether DNMT1 is selected for recurrent genetic alterations in SCLCs. DNMT1 was amplified in 22% of human SCLC cell lines (n = 50), correlating with higher expression (Figure 1H). Conversely, the me-CpG eraser TET2 (Ten-Eleven-Translocation 2) that functionally opposes DNMT1 was recurrently deleted, with copy number loss in 62% of the SCLC cell lines correlating with lower expression (Figure 1H).
DNMT1 knockdown by small interfering RNA (siRNA) or small-molecule drugs activated ASCL1-target genes/terminal lineage fates in SCLC cells
We used siRNA to knock down DNMT1 from human SCLC cells H82 and H146, confirmed by western blot (Figure 2A). This knockdown, but not control, siRNA decreased SCLC cell proliferation (Figure 2B). The cytoreduction was not via apoptosis, measured by Annexin staining and flow cytometry (Figure 2C). Neuroendocrine-lineage gene signatures of normal pulmonary neuroendocrine cells were identified from a human lung single-cell RNA sequencing (RNA-seq) atlas25: consistent with cytoreduction via terminal lineage maturation, siDNMT1, but not control siRNA, significantly upregulated pulmonary neuroendocrine signature genes gastrin-related peptide (GRP), neuronatin (NNAT), chromogranin B (CHGB), and secretogranin II (SCG2) (also known as chromogranin C [CHGC]) (Figure 2D). The DNMT1 knockdown also induced morphologic changes expected with terminal lineage maturation (decreased nuclear/cytoplasmic ratio, increased cell size) (Figure 2E).
Figure 2. DNMT1 knockdown using siRNA or small-molecule drugs activated pulmonary NE signature genes and decreased SCLC growth without activating apoptosis.
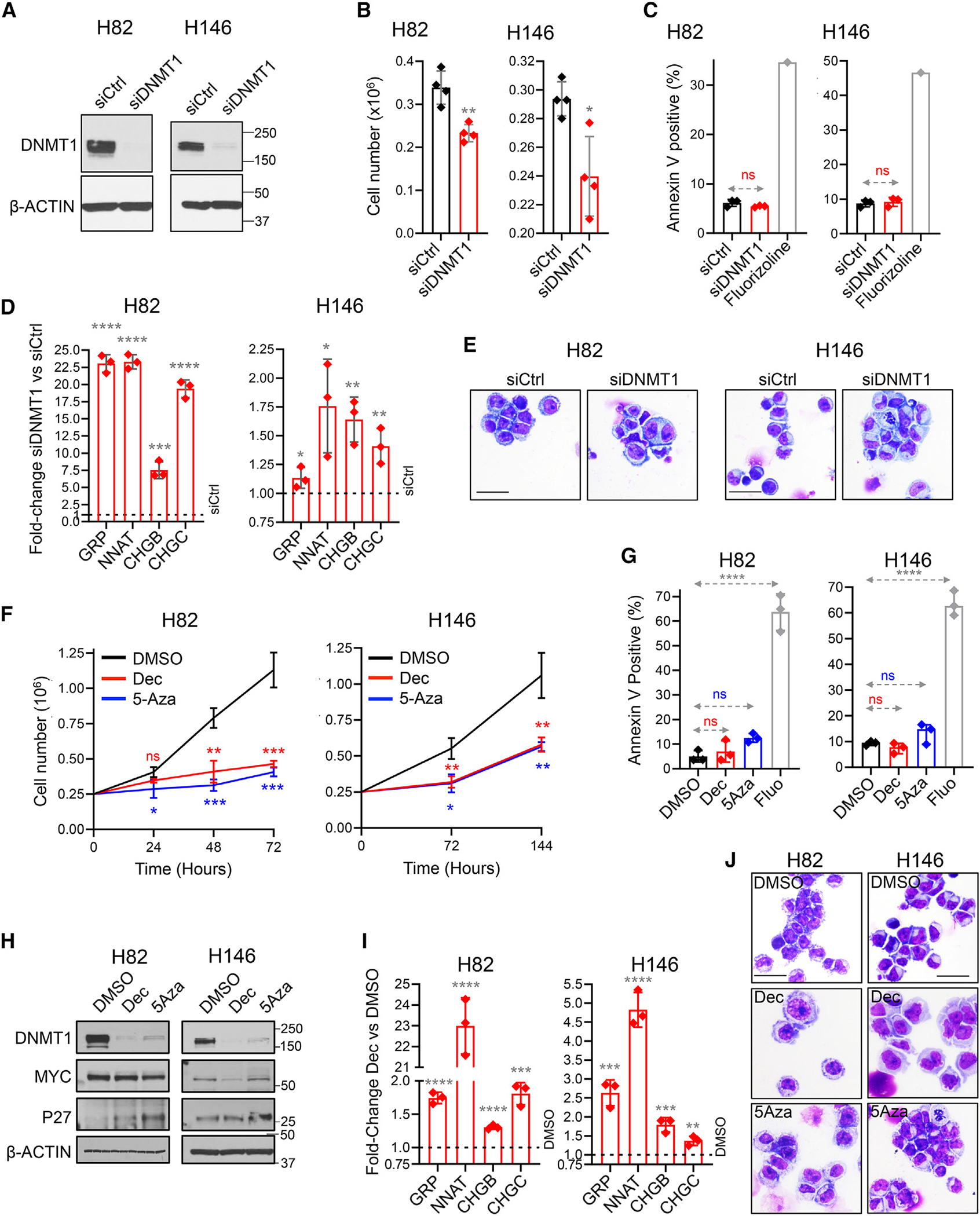
Human SCLC cells H82 and H146 were transfected with DNMT1 siRNA (siDNMT1) or scrambled siRNA (siCtrl).
(A) DNMT1 knockdown. DNMT1 protein was measured by western blot 72 h after transfection.
(B) Cell growth inhibition. Cell counts by automated counter 72 h after transfection. Mean ± SD of four independent experiments.
(C) Apoptosis not activated. Apoptosis was measured by Annexin V staining and flow cytometry analysis 24 h after transfection. Fluorizoline (10 μM) was used as a positive control for apoptosis. Mean ± SD of three independent experiments.
(D) Activation of pulmonary NE-lineage signature genes. Gastrin-related peptide (GRP), neuronatin (NNAT), chromogranin B (CHGB), and secretogranin II (SCG2), also known as chromogranin C (CHGC), were increased by DNMT1 knockdown. Quantitative reverse polymerase chain reaction (qPCR) 72 h after transfection. Internal control was β-ACTIN; relative gene expression was calculated by the Livak-Schmittgen method. Mean ± SD of three independent experiments. ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05, p > 0.05. t test two-sided vs. siCtrl.
(E) Cell morphology. Giemsa-stained cell imaged by Leica Upright Microscope-Orion. Magnification ×400; scale bar, 10 μm.
(F) DNMT1 depletion using decitabine (Dec) or 5-azacytidine (5Aza) decreased SCLC proliferation. SCLC cells H82 and H146 were treated with DMSO vehicle control, Dec 0.5 μM, or 5Aza 1 μM added once at time 0 h. Cell counts by automated counter.
(G) The cytoreduction was not by apoptosis. Apoptosis was measured by Annexin V staining and flow cytometry at 24 h. Positive control fluorizoline (10 μM).
(H) Dec and 5Aza treatment decreased DNMT1 and MYC and increased p27/CKDN1B (mediator of cell-cycle exits by terminal lineage differentiation). Western blot.
(I) Dec and 5Aza treatment increased expression of pulmonary NE-lineage signature genes. Measured at 72 h in H82 cells and 96 h in H146 cells, as per (D). Experiment in triplicate. Statistics are as per (D) treated vs. vehicle.
(J) Cell morphology. Giemsa-stained cytospin preparations of cells harvested after 72–96 h. Leica Upright Microscope-Orion; magnification ×400; scale bar, 10 μm.
DNMT1 can also be depleted from dividing cells using non-cytotoxic concentrations of the pyrimidine nucleoside analog pro-drugs decitabine and 5-azacytidine.17–19 We thus treated SCLC cells H82 and H146 in vitro with clinically relevant concentrations of decitabine (0.5 μM) or 5-azacytidine (1 μM). Both decitabine and 5-azacytidine decreased proliferation of the cells (Figure 2F). Apoptosis, measured by Annexin staining and flow cytometry, was not detected (Figure 2G). DNMT1, measured by western blot, was depleted (Figure 2H). Consistent with neuroendocrine-lineage maturation as the pathway for cell-cycle exits, decitabine or 5-azacytidine treatment (1) downregulated MYC and upregulated p27/CDKN1B protein (Figure 2H); (2) significantly upregulated >100 neuroendocrine signature genes in H146 cells by unbiased differential gene expression analyses by RNA-seq (DESeq2), whereas MYC-target genes were simultaneously suppressed (Figure S2; Table S4) (qRT-PCR was used to corroborate these data for GRP, NNAT, CHGB, and SCG2 in both H146 and H82 cells; Figure 2I); and (3) induced morphologic changes consistent with maturation induction (decreased nuclear cytoplasmic ratio, increased cytoplasmic complexity) shown by Giemsa staining (Figure 2J).
SCLC cells display rapid adaptive metabolic responses to decitabine or 5-azacytidine
To evaluate efficacy of DNMT1 targeting to treat SCLC in vivo, alone or in combination with ICI, we employed a syngeneic, immunocompetent, genetically engineered mouse model of SCLC, established in vivo by intra-tracheal instillation of Adeno-CMV-Cre in Rblox/p53lox mice,35 recapitulating key genetic characteristics of human SCLC and used also by others to answer therapy questions in SCLC.23,36 We confirmed and selected an iteration of this model that captured aggressiveness of metastatic spread of human disease by rapidly invading the liver, the most frequent site of dissemination in extensive stage human SCLC (Figure S3A).37
In clinical experience with decitabine and 5-azacytidine to treat myeloid malignancies, resistance emerges rapidly from adaptive responses of the pyrimidine metabolism network that dampen pro-drug processing into DNMT1-depleting nucleotide (Figure 3A).22 We therefore examined whether metabolism in SCLC cells reacts similarly, because this would guide in vivo regimen design. F1339 cells were treated in vitro with clinically relevant concentrations of decitabine (0.5 μM), 5-azacytidine (5 μM), or equimolar natural deoxycytidine or cytidine. Decitabine and 5-azacytidine, but not deoxycytidine or cytidine, decreased SCLC cell proliferation (Figure 3B). Decitabine or 5-azacytidine did not increase apoptosis, measured by Annexin staining and flow cytometry; fluorizoline, which induces apoptosis independent of p53, was used as a positive control (Figure 3C). Of 69 genes significantly upregulated by decitabine treatment in unbiased differential gene expression analyses by RNA-seq (DESeq2), 13 were Ascl1/Neurod1-target neuroendocrine genes, whereas none were Myc-target genes (Figure 3D; Table S5). Upregulation of neuroendocrine lineage-differentiation signature genes Grp, Nnat, Chgb and Chgc by both decitabine and 5-azacytidine was corroborated by qRT-PCR (Figure 3E). Major histocompatibility complex I (MHC class I) molecules, potentially relevant to combination therapy with ICI, are generally upregulated with lineage maturation, and decitabine and 5-azacytidine also significantly increased MHC class I expression, measured by flow cytometry (Figure 3F). Also consistent with maturation induction as the pathway for cytoreduction, decitabine and 5-azacytidine decreased Dnmt1 protein, decreased levels of Myc and its paralog L-Myc, and simultaneously increased p27/Cdkn1b (Cdkn1b is a canonical cyclin-dependent kinase inhibitor that normally antagonizes Myc to force cell-cycle exits by differentiation) (Figures 3G and S3B). Decitabine and 5-azacytidine also induced morphologic changes consistent with resumed lineage maturation (decreased nuclear cytoplasmic ratio, increased cytoplasmic complexity) (Figure 3H).
Figure 3. Non-cytotoxic, DNMT1-targeting concentrations of Dec (Aza-dC) and 5Aza (5Aza, Aza-C) that suppressed p53-null, chemorefractory SCLC cells also triggered automatic adaptive responses from the pyrimidine metabolism network.
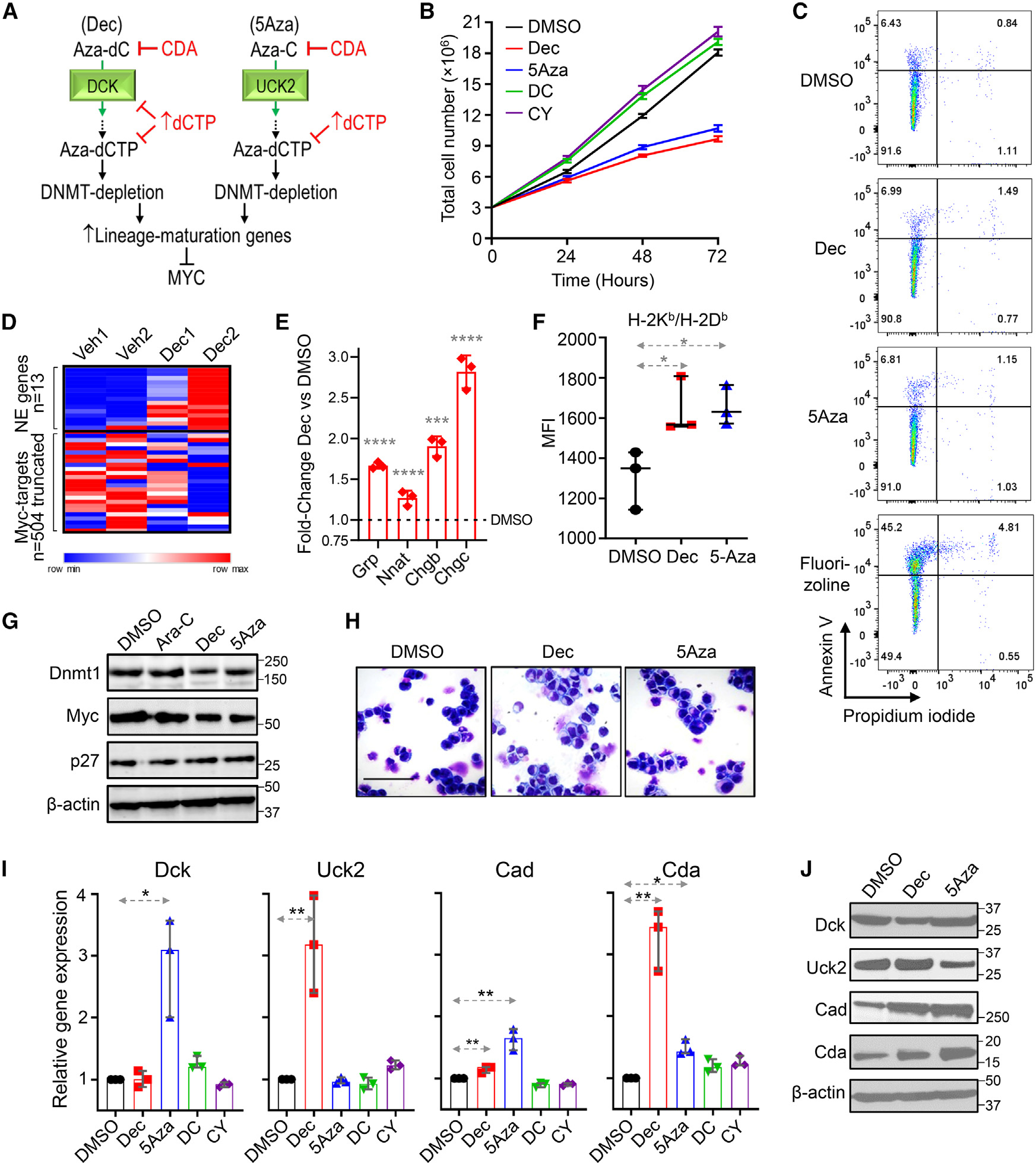
(A) Dec and 5Aza are deoxycytidine (DC) and cytidine (CY) analogs, respectively, and are processed by pyrimidine metabolism into the DC nucleotide analog Aza-dCTP, which after incorporation into the newly synthesized DNA strand during S-phase can deplete DNMT1 without terminating DNA chain synthesis. Shown are key pyrimidine metabolism enzymes that dictate Dec and 5Aza processing into DNMT1-depleting Aza-dCTP. CDA is a key enzyme that decreases DNMT1 depletion by catabolizing Dec and 5Aza. De novo pyrimidine synthesis competes with Aza-dCTP by synthesizing dCTP de novo from amino acid building blocks.
(B) Dec and 5Aza, but not equimolar DC or CY, were anti-proliferative to p53-null SCLC cells. F1339 cells were seeded at day (D) 0 and treated with DMSO, DC (0.5 μM), CY (5 μM), Dec (0.5 μM), and 5Aza (5 μM). Cell counts (mean ± SEM) by automated counter.
(C) These anti-proliferative actions of Dec and 5Aza were not via apoptosis. Cell membrane Annexin V staining measured by flow cytometry after 24 h of treatment. Fluorizoline 10 μM positive control.
(D) Of 69 genes significantly upregulated by Dec treatment in unbiased differential gene-expression analyses by RNA-seq (DESeq2), 13 were NE genes, whereas none were of the 504 known Myc-target genes. F1339 cells were treated with DMSO vehicle or Dec 0.5 μM for 72 h (two biological replicates) (Table S5).
(E) Pulmonary NE signature genes Grp, Nnat, Chgb, and Scg2 (also known as Chgc) were increased after Dec treatment. After 72 h of Dec (0.5 μM) treatment, gene expression in F1339 cells was measured by qPCR. Internal control was β-actin; relative gene expression was calculated by the Livak-Schmittgen method. Mean ± SD of three independent experiments. ****p < 0.0001, ***p < 0.001, t test two-sided vs. DMSO.
(F) Dec and 5Aza increased MHC class 1 (H-2Kb/H-2Db) expression by F1339 cells. Measured by flow cytometry. Bar and whiskers, median ± interquartile range [IQR]; *p < 0.05 vs. DMSO, two-sided Mann-Whitney test.
(G) Dec and 5-Aza, but not the dC analog cytarabine, decreased Dnmt1 and Myc, and increased p27/Cdkn1B (mediator of cell-cycle exits by terminal lineage differentiation). Western blot D5 (drug added D0). Western blot.
(H) Dec and 5-Aza treatment induced morphologic changes of differentiation (decreased nuclear cytoplasmic ratio, increased cytoplasmic complexity). Drug was added D0, and Giemsa-stained cytospins D5. Magnification ×400; scale bar, 10 μm.
(I) Dec and 5Aza trigger adaptive pyrimidine metabolism shifts that auto-dampen their activity (auto-resistance) (rapid suppression of Dck and Uck2, respectively, upregulating Cda [catabolism] and Cad [de novo pyrimidine synthesis]); also, Dec upregulated Uck2, and 5Aza upregulated Dck (each agent cross-primed for activity of the other). qPCR 72 h after Dec or 5Aza addition. Median ± IQR. *p < 0.05, **p < 0.01; Mann-Whitney test two-sided.
(J) Protein expression tracked the mRNA expression changes. Western blot 96 h after Dec or 5Aza addition.
Expression of key pyrimidine metabolism enzymes was measured by qRT-PCR and western blot; deoxycytidine kinase (Dck) executes the initial phosphorylation of decitabine: decitabine decreased Dck protein, whereas 5-azacytidine upregulated Dck mRNA and protein up to 4-fold (Figures 3I and 3J). Uridine cytidine kinase 2 (Uck2) executes the initial phosphorylation of 5-azacytidine: 5-azacytidine decreased Uck2 protein, whereas decitabine upregulated Uck2 mRNA and protein up to 4-fold (Figures 3I and 3J). Carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase (Cad) executes the first step in de novo pyrimidine synthesis, which manufactures cytidine or deoxycytidine nucleobases endogenously that can then compete with exogenous decitabine or 5-azacytidine: 5-azacytidine and decitabine increased Cad mRNA and protein (Figures 3I and 3J). Cytidine deaminase (Cda) rapidly catabolizes decitabine and 5-azacytidine into uridine counterparts that cannot deplete DNMT1: both decitabine and 5-azacytidine upregulated Cda mRNA and protein (some discrepancies between the extent of change in the same direction are because of different timings of the mRNA and protein analyses, at 72 and 96 h, respectively; Figures 3I and 3J). Thus, SCLC cells, like malignant myeloid cells, demonstrate adaptive metabolic responses (“auto-resistance”) upon decitabine or 5-azacytidine exposures, warranting in vivo regimen design as described below.22 Further encouraging evaluation and optimization of decitabine and 5-azacytidine to treat SCLCs in particular, SCLCs express DCK and UCK2 at levels several-fold higher than other solid cancer histologies, although lower than in myeloid and lymphoid malignancies (Figure S4).
Dnmt1 targeting to treat chemorefractory SCLC in vivo
To counter the adaptive metabolic barriers to decitabine and 5-azacytidine activity and achieve Dnmt1 targeting in vivo, we alternated decitabine with 5-azacytidine every 3–4 days (timed to exploit the metabolic cross-priming of each agent for the other), and both agents were combined with tetrahydrouridine (THU) to inhibit the catabolic enzyme Cda that is especially enriched in the liver site of metastases and that is auto-upregulated upon decitabine or 5-azacytidine exposures (Figure 4A).22,38 We also evaluated ICI alone (anti-Pd1) and combination anti-Pd1/Dnmt1 targeting, because standard SCLC therapy now incorporates ICI. As an additional control, we evaluated intense etoposide and cisplatin, the mainstay treatment for human SCLC.
Figure 4. A non-cytotoxic Dnmt1-targeting regimen (Dnmt1i), but not anti-Pd1 ICI or intense chemotherapy, was active against p53-null, chemorefractory SCLC in vivo.
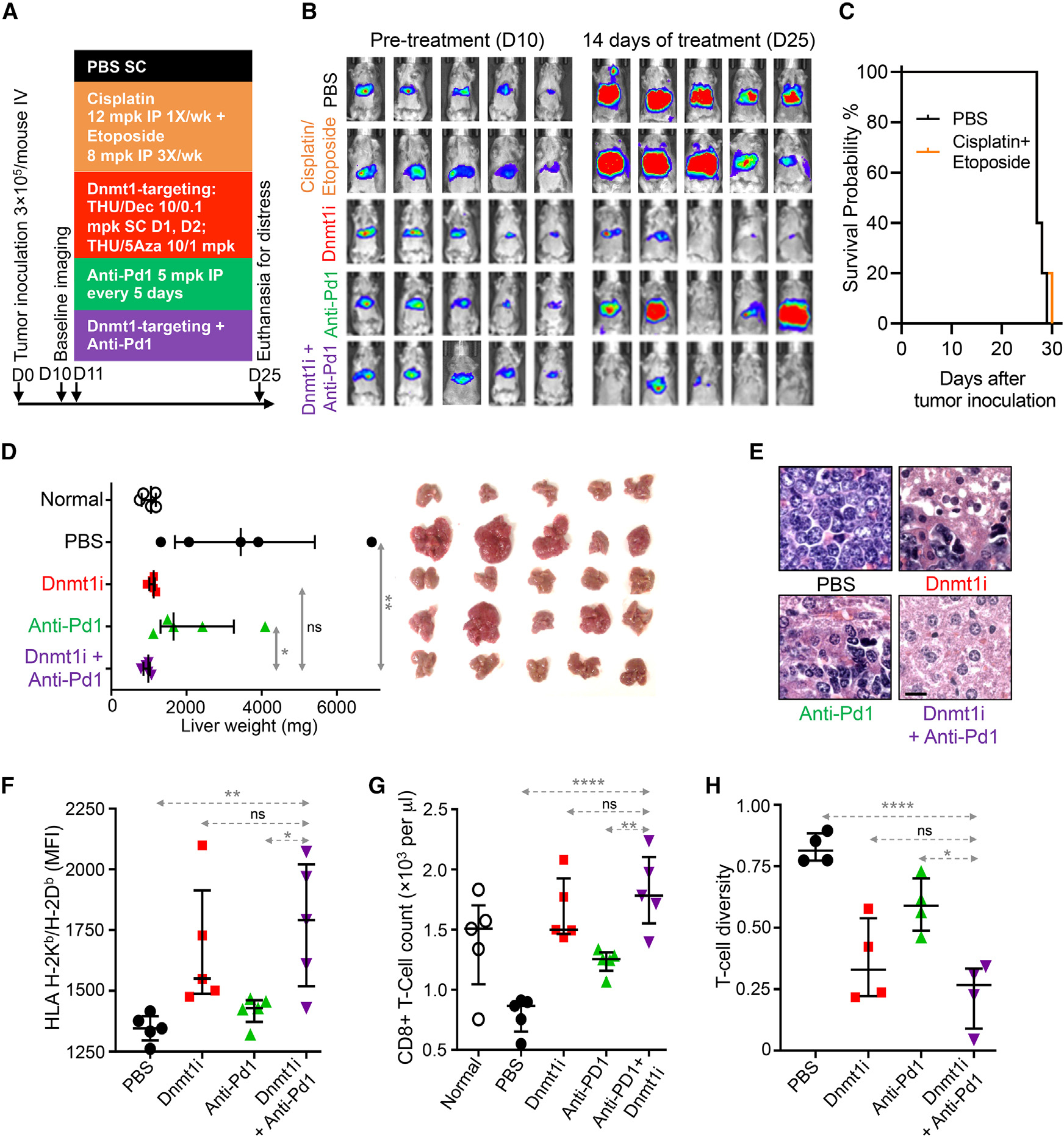
(A) High doses of etoposide and cisplatin were ineffective in vivo. B6/129 SF1 mice were inoculated via tail vein with p53-null F1339-luc SCLC cells (0.3 × 106 cells/mouse). After documentation of lung invasion by live imaging, mice were distributed to PBS or combination cisplatin 12 mg/kg intraperitoneal (IP) once per week + etoposide 8 mg/kg IP three times per week, Dnmt1 targeting with THU-Dec/5Aza, anti-PD1, or combination Dnmt1i + anti-PD1 (n = 5/group).
(B) No benefit of cisplatin + etoposide by tumor burden measured by chemo-luminescence (the SCLC cells are liver tropic).
(C) No benefit of cisplatin + etoposide measured by time to distress (“survival”). Mice were euthanized for signs of distress.
(B and D) Non-cytotoxic Dnmt1-targeting therapy (Dnmt1i), but not anti-Pd1 ICI, benefitted mice with chemorefractory SCLC (analysis after 14 days of treatment). Bar and whiskers = median ± IQR. *p < 0.05; **p < 0.01; ****p < 0.0001; nsp > 0.05. Mann-Whitney test, two-sided. (B) Tumor burden by bioluminescence-imaging.
(D) Tumor burden by liver weights. The SCLC cells are hepatotropic.
(E) Tumor histological sections after 14 days of Dnmt1i showed decreased nuclear/cytoplasmic ratio and increased necrosis. H&E stain; magnification ×400; scale bar, 10 μm.
(F) MHC class I (H-2Kb/H-2Db) expression on tumor tissue. Flow cytometry.
(G) Dnmt1i and combination Dnmt1i + ICI similarly increased peripheral blood CD8+ T cells. Median ± IQR. **p < 0.01; ****p < 0.0001; nsp > 0.05; Mann-Whitney test two-sided.
(H) Diversity of tumor infiltrating T cells. T cell diversity: 1 – percentage of the CD8+ T cell population represented by the 1,000 most abundant T cell clones, measured by T cell receptor (TCR) RNA-seq.
The p53-null F1339 SCLC cells were transduced to express luciferase (F1339-luc) and then inoculated into tail veins of syngeneic B6/129 SF1 mice. Treatments began on day 11, after confirmation of tumor engraftment by bioluminescent live imaging on day 10 (Figure 4A). High-dose etoposide and cisplatin did not reduce tumor burden (Figure 4B) or improve time to distress (“survival”) vs. vehicle (Figure 4C). Dnmt1-and anti-Pd1 treatments were continued until day 25 (14 days of treatment), the time point at which vehicle-treated mice became distressed. At this point, the experiment was terminated to enable analyses of tumor tissue after similar durations of the different treatments: Dnmt1 targeting alone and combination Dnmt1 targeting/anti-Pd1 decreased tumor burden by similar extents (Figures 4B and 4D), with a prominent benefit over anti-Pd1 alone or vehicle (Figures 4B and 4D). Tumor tissue histological sections showed decreased nuclear/cytoplasmic ratio and increased necrosis with Dnmt1 targeting alone or combination Dnmt1 targeting/anti-Pd1 vs. anti-Pd1 or vehicle (Figure 4E). Dnmt1 targeting alone and combination Dnmt1 targeting/anti-Pd1, but not anti-Pd1 or vehicle, significantly upregulated antigen-presenting MHC class I (H-2Kb/H-2Db) levels in tumor tissue (Figure 4F). All treatments increased peripheral blood CD8+ and CD4+ T cells compared with vehicle, with the greatest elevations produced by combination Dnmt1 targeting/anti-Pd1 (Figures 4G and S5). Peripheral blood granulocyte-like myeloid-derived suppressor cells (MDSCs; G-MDSCs) were decreased by Dnmt1 targeting and combination Dnmt1 targeting/anti-Pd1, but not by anti-Pd1 or vehicle alone (Figure S5). T cell diversity of tumor-infiltrating lymphocytes (TILs), measured by RNA-seq of the T cell receptor, was also most restricted by Dnmt1 targeting or combination Dnmt1 targeting/anti-Pd1 vs. anti-Pd1 or vehicle (Figure 4H).
The Dnmt1-targeting and anti-Pd1 treatments were evaluated again but with euthanasia for distress as the endpoint. Treatment was initiated on day 13, after growth of tail-vein-inoculated F1339-luc SCLC cells in liver of B6/129 SF1 mice was confirmed by live imaging (Figures 5A and 5B). Dnmt1 targeting alone, and combination Dnmt1 targeting/anti-Pd1, similarly decreased tumor burden, measured by chemiluminescence imaging on day 28 (Figure 5B), and similarly increased time to distress (Figure 5C), with large advantages over anti-Pd1 alone or vehicle. Consistent with a non-cytotoxic mechanism of action of the Dnmt1-targeting regimen, peripheral blood CD4+ and CD8+ T cells were not decreased by this therapy, measured at the time of euthanasia (Figures 5D and S6A), nor were peripheral blood G-MDSCs or monocyte-like MDSCs (M-MDSCs) significantly decreased (Figures 5E and S6B).
Figure 5. Dnmt1 targeting and ICI with anti-Pd1 to treat the chemorefractory SCLC.
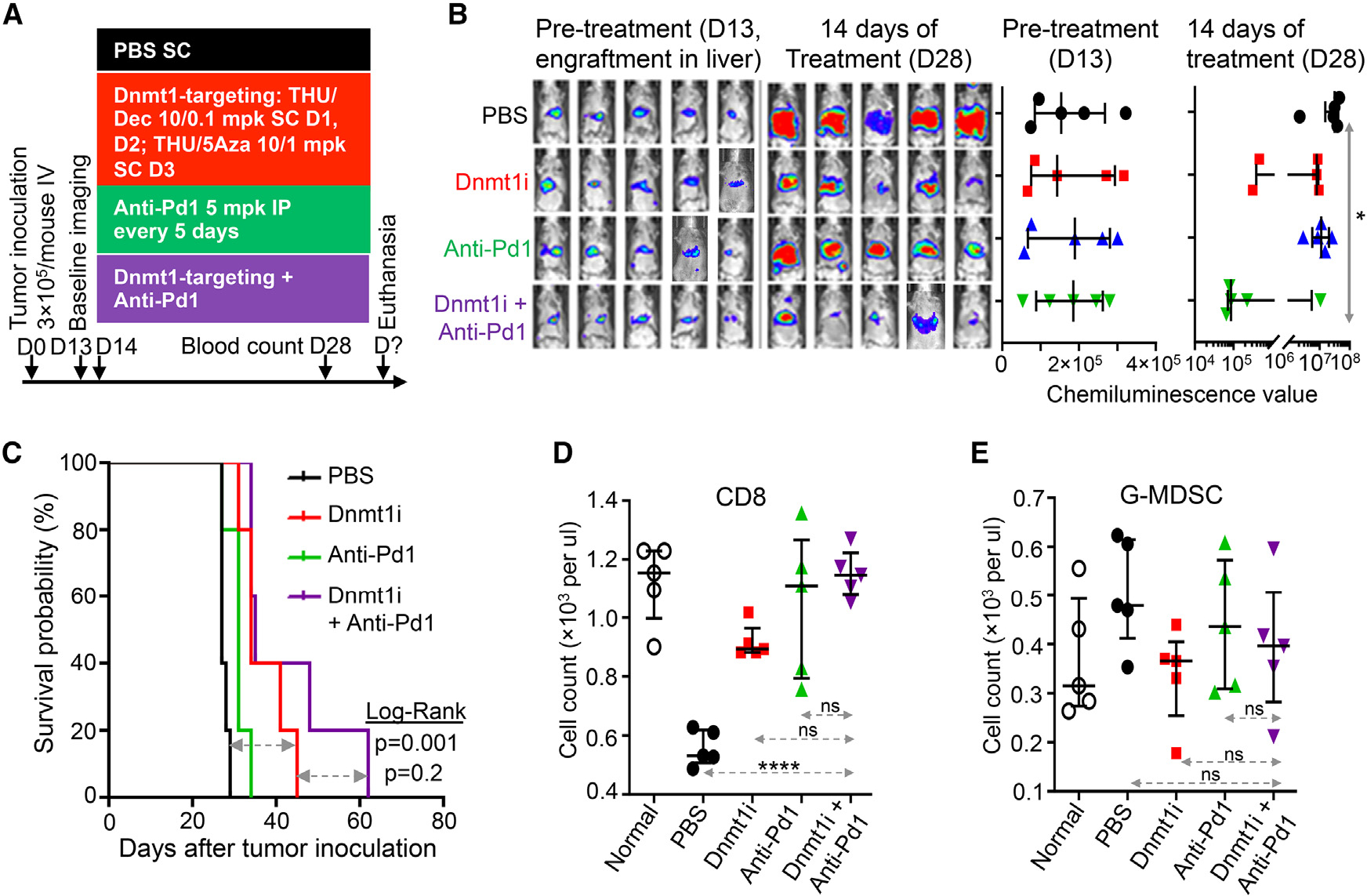
(A) Experiment schema. To target Dnmt1 in the chemorefractory SCLC in vivo, we alternated Dec with 5Aza every 3–4 days, timed to exploit the metabolic cross-priming of each agent for the other, and tetrahydrouridine (THU) was incorporated into the regimen to inhibit the catabolic enzyme Cda that otherwise severely limits Dec/5Aza plasma half-life and tissue distribution. B6/129 SF1 mice were tail-vein inoculated with F1339-luc SCLC cells (0.3 × 106 cells/mouse). ICI with anti-Pd1 was also evaluated alone or with Dnmt1i. Mice were randomized to treatments after documentation of tumor engraftment by live imaging on D13 (n= 5/group). mpk, mg/kg; SC, subcutaneous.
(B) Tumor burden by live imaging.
(C) Time to distress (“survival”). Mice were euthanized for signs of distress.
(D) Peripheral blood CD8+ T cells. Measured by Hemavet and flow cytometry at time of euthanasia. Bar and whiskers = median ± IQR. *p < 0.05; ****p < 0.0001; nsp > 0.05. Two-sided Mann-Whitney test.
(E) Peripheral blood granulocyte myeloid-derived suppressor cells (G-MDSCs). Measured by Hemavet and flow cytometry at time of euthanasia.
We measured the expression of immune checkpoint genes in TILs isolated magnetically from SCLC tumor tissue (Pd1, Lag3, Ctla4, Tim3) and in dissociated tumor tissue itself (Pdl1, Cd80, Cd86, Cd276, Galectin-9, Cd74, Cd155). Anti-Pd1 induced several-fold increases in expression of all these immune checkpoints in both TILs and tumor tissue (Figures S7A and S7B). However, Dnmt1 targeting alone did not upregulate checkpoints in TILs and, when combined with anti-Pd1, markedly downmodulated the checkpoint upregulation that was observed with anti-Pd1 alone (Figure S7A). Dnmt1 targeting upregulated Pd1, Cd80, Galectin-9, and Cd276 in tumor tissue but did not upregulate Cd86 or Cd155 that were upregulated by anti-Pd1 alone (Figure S7B). When combined with anti-Pd1, Dnmt1 targeting did not down-modulate the global upregulation of immune checkpoints in tumor tissue that was produced by anti-Pd1 (Figure S7B). Human SCLC tissue (n = 50) expresses MHC class I at several-fold lower levels than non-SCLC tissue (n = 121) (public database The Cancer Genome Atlas [TCGA]) (Figure S8). In summary, both anti-Pd1 alone and Dnmt1 targeting alone produced measurable effects on a range of immune biomarkers, but anti-Pd1 was nevertheless not efficacious, and the combination of anti-Pd1 with Dnmt1 targeting did not enhance efficacy over Dnmt1 targeting alone.
Incorporation of an inhibitor of de novo pyrimidine synthesis into therapy
We then incorporated an inhibitor of de novo pyrimidine synthesis into the therapy for three reasons. First, de novo pyrimidine synthesis is upregulated in SCLC cells and competes with exogeneous 5-azacytidine/decitabine (Figure 3) (shown also for other cancers22). Second, dihydroorotate dehydrogenase (DHODH) inhibitors trigger p53-independent cell-cycling exits by terminal lineage differentiation in other cancers, an effect that depends on reducing cytidine triphosphate (CTP) levels (reviewed in Saunthararajah39). CTP may be a cofactor for condensin corepressors, e.g., SMC4 that is amplified in 58% of SCLC cell lines; meanwhile, cohesin coactivators that oppose condensins, e.g., SMC3, are recurrently deleted (62%) from SCLC cells (Figure S9). Third, DHODH inhibitors have shown activity against p53-null SCLC in pre-clinical in vivo models even as single agents, although lineage maturation was not examined as a potential mediator of the cell-cycling exits.23 To inhibit DHODH, we used teriflunomide, a generic drug US Food and Drug Administration (FDA)-approved to treat multiple sclerosis, at clinically relevant, non-cytotoxic concentrations (clinical plasma Cmax typically exceeds 50 μM). Teriflunomide 10 μM decreased proliferation of SCLC cells F1339, H82, and H146 (Figure 6A). The teriflunomide treatment did not activate apoptosis, measured by Annexin staining and flow cytometry (Figure 6B), but did activate ASCL1 targets and signature neuroendocrine-lineage genes GRP, NNAT, CHGB, and CHGC several-fold in all three cell lines (Figure 6C). Teriflunomide treatment also induced morphology changes of terminal differentiation, e.g., decrease in nuclear/cytoplasmic ratio (Figure 6D).
Figure 6. Teriflunomide activated pulmonary NE signature genes and decreased SCLC growth without activating apoptosis.
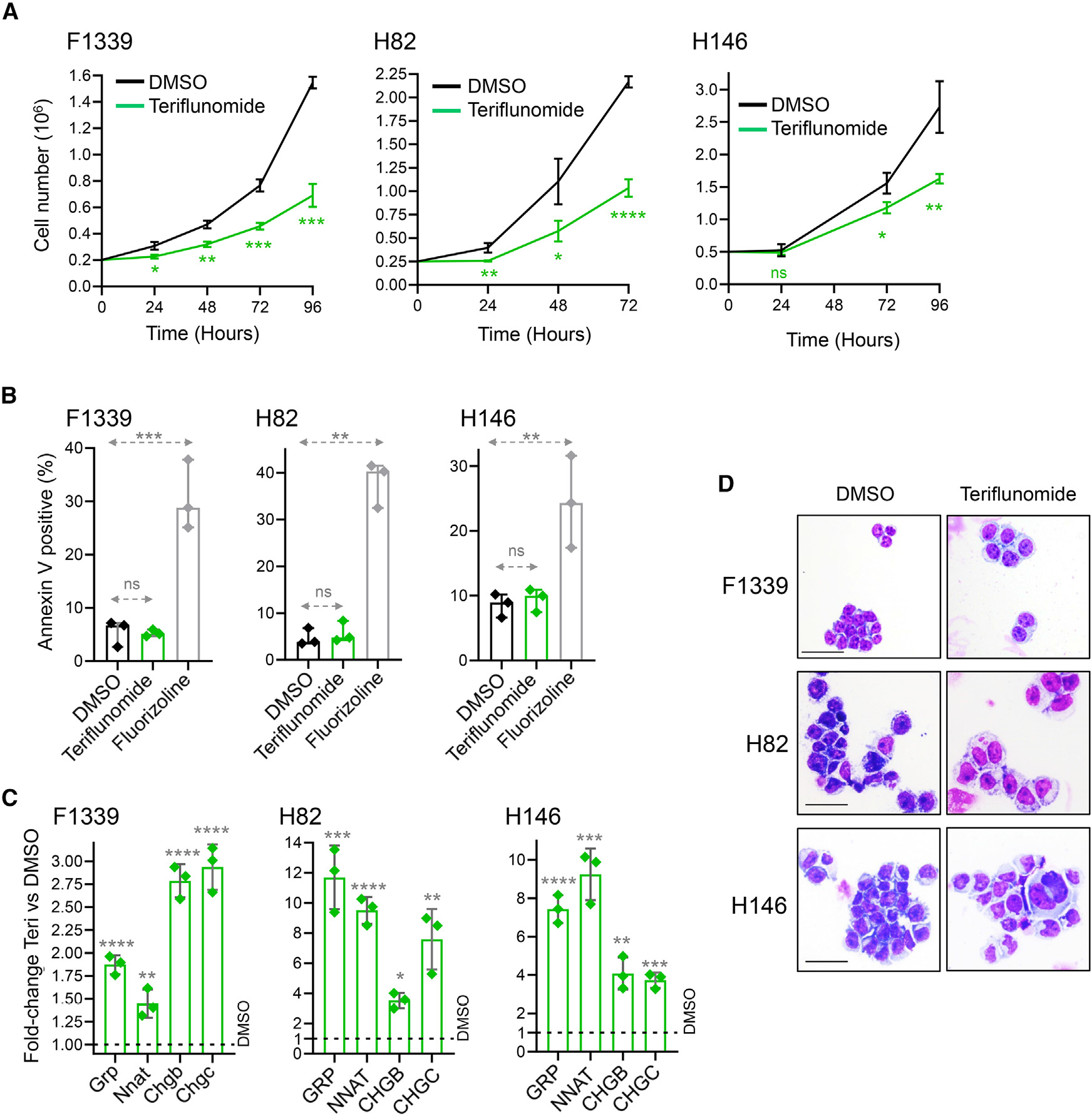
F1339 (p53-null murine SCLC cells) and human SCLC cells NCI-H82 and NCI-H146 were treated with vehicle (DMSO) or teriflunomide 10 μM (single exposure at time 0). ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05, nsp > 0.05; t test two-sided vs. DMSO.
(A) Cell counts. Automated counter. Mean ± SD of three independent experiments.
(B) Apoptosis. Fluorizoline 10 mM was used as a positive control (p53-independent inducer of apoptosis). Annexin V staining measured by flow cytometry at 24 h. Mean ± SD, three independent experiments.
(C) Activation of pulmonary NE-lineage signature genes GRP, NNAT, CHGB, and SCG2, also known as CHGC. qPCR at 48–72 h. Internal control was β-Actin; relative gene expression was calculated by the Livak-Schmittgen method. Mean ± SD of three independent experiments.
(D) Cell morphology. Giemsa-stained cytospin preparations imaged by Leica Upright Microscope-Orion at 72 h (F1339, H82) or 120 h (H146); magnification ×400; scale bar, 10 μm.
We therefore evaluated teriflunomide in vivo, alone and together with Dnmtl1 targeting, to treat chemorefractory SCLC (Figure 7A). F1339-luc SCLC cells were tail-vein inoculated into syngeneic B6/129 SF1 mice. After baseline blood counts on day 7, mice were randomly distributed to treatment with (1) vehicle, (2) Dnmt1 targeting with THU-decitabine/THU-5-azacytidine, (3) Dhodh targeting with teriflunomide, or (4) combination Dnmt1/Dhodh targeting (five mice per group) (Figure 7A). Dnmt1 or Dhodh targeting alone similarly decreased tumor burden vs. vehicle treatment as measured by chemiluminescence imaging on day 28 (Figure 7B), but the greatest tumor-burden reduction was with combination Dnmt1/Dhodh targeting (Figure 7B). Dnmt1 or Dhodh targeting alone similarly increased time to distress vs. vehicle treatment by ~20 days, whereas the combination treatment extended time to distress vs. vehicle by ~40 days (replicated also in a separate experiment) (Figures 7C and 7D). Even at time of euthanasia ~40 days later than in vehicle-treated mice, tumor burden remained significantly lower with Dnmt1/Dhodh-targeting treatment (Figure 7E). Consistent with a non-cytotoxic mechanism of action, none of the treatments decreased peripheral blood white cell, red cell, or platelet counts vs. vehicle treatment (Figure 7F), and murine body weights were similar in control and combination treatment groups (Figure 7G).
Figure 7. Combination non-cytotoxic Dnmt1- and Dhodh-targeting therapy in vivo.
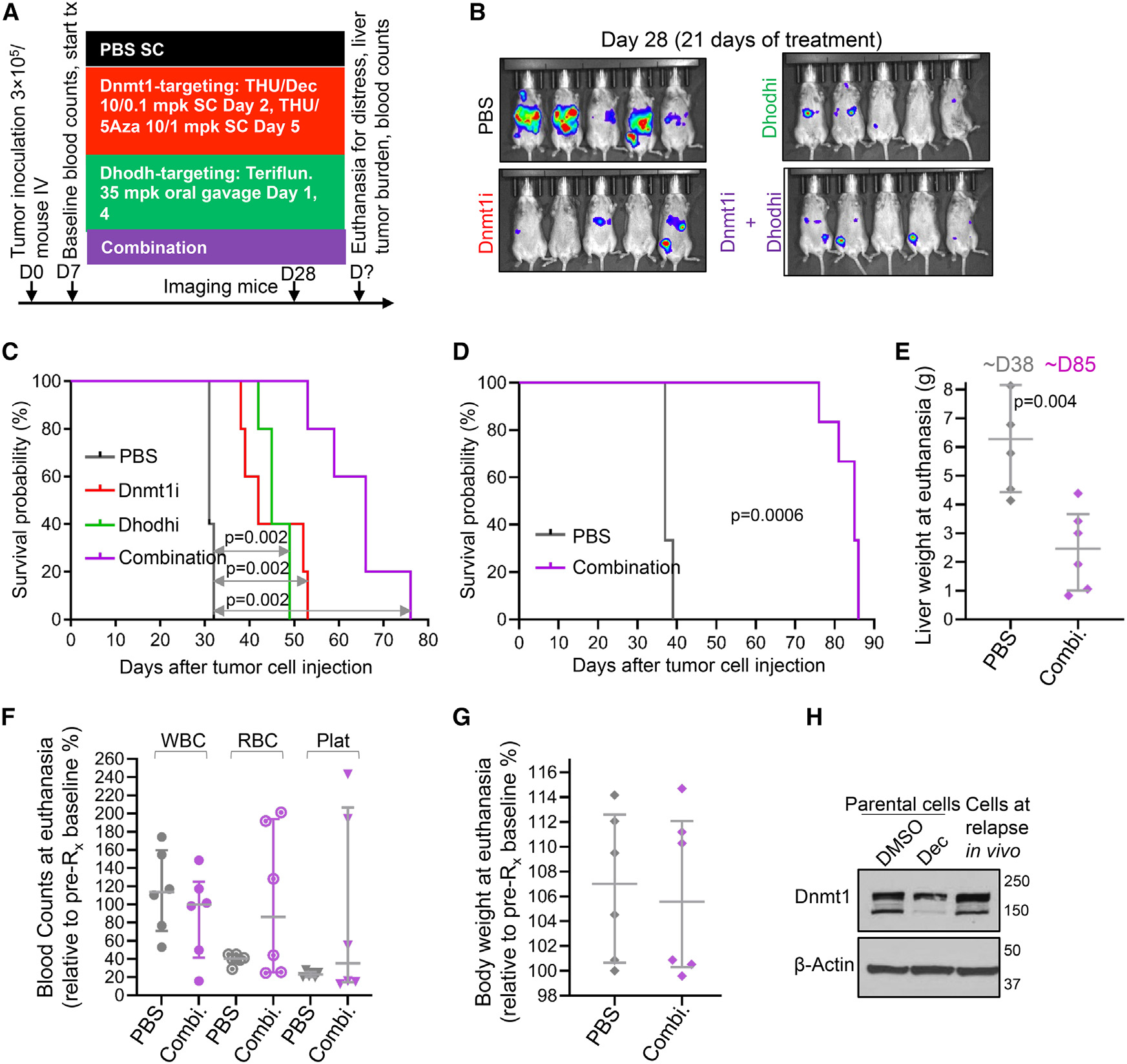
(A) Experiment schema. B6/129 SF1 mice were tail-vein inoculated with F1339-luc SCLC cells (0.3 × 106 cells/mouse). At D7, mice (n = 5/group) were distributed to PBS, THU-Dec/5Aza, teriflunomide, or combination therapy. Median ± IQR.
(B) Tumor burden. Bioluminescence imaging at D28. Treatment was started at D7 when tumor was not yet visible by bioluminescence imaging.
(C) Survival (time to distress). Log rank tests.
(D) Survival (time to distress) replicate experiment using F1339 cells (no luciferase), n = 6 per treatment group, vehicle vs. combination Dnmt1i + Dhodhi treatment as per (A). Log rank test.
(E) Liver weights at time of euthanasia. Mann-Whitney test two-sided. Median ± IQR.
(F) Blood counts at time of euthanasia, relative to pre-treatment baseline blood counts in individual animals.
(G) Body weight at time of euthanasia, relative to pre-treatment baseline body weight.
(H) Tumor cells harvested at relapse demonstrated preserved Dnmt1 despite continuous therapy. Dnmt1 measured by western blot. Controls were F1339 parental SCLC cells treated with vehicle or 0.5 μM Dec in vitro for 72 h. Tumor cells were collected at time of euthanasia for distress, after ~80 days of therapy with combination Dnmt1/Dhodh-targeted therapy.
SCLC tumor that progressed through the every-week therapy to eventually cause murine distress was found to have evaded Dnmt1 depletion, contrasting with Dnmt1 depletion in the positive control (Figure 7H). Failure to deplete DNMT1 is observed also in malignant myeloid cells that resist and proliferate through clinical decitabine or 5-azacytidine therapy.22
DISCUSSION
SCLCs express and depend on neuroendocrine-lineage master transcription factors, e.g., ASCL1 and/or NEUROD1,10–14,40 but these fail to completely activate neuroendocrine lineage-differentiation programs. Neuroendocrine-lineage genes, identified from single-cell gene expression databases, have a “closed” chromatin configuration in ESCs, enriched for the epigenetic repression marks me-CpG and H3K27me3. This contrasts with “open” configurations at replication (MYC-target) and house-keeping genes in ESCs and beyond. In SCLC cells, H3K27me3 is erased from neuroendocrine-lineage genes as is expected from neuroendocrine-lineage commitment; however, me-CpG is retained and correlates negatively with expression of the genes. The key epigenetic regulator DNMT1 writes me-CpG, is the maintenance methyltransferase, and is a corepressor recruited by ASCL1 and NEUROD1, as it is also by other lineage master transcription factors in other lineage contexts.30–34 DNMT1 is recurrently amplified in SCLCs, with corresponding higher gene expression, whereas TET2 that erases me-CpG and functionally opposes DNMT1 is recurrently deleted. Demonstrating a cause-effect contribution of DNMT1 to stalled neuroendocrine-lineage maturation in SCLC cells, its knockdown by siRNA activated ASCL1 targets, resumed lineage maturation, and thus terminated SCLC cell replications, without requiring or activating apoptosis. Other corepressors recruited by transcription factors, e.g., condensin SMC4, are also recurrently amplified in SCLCs, and other coactivators, e.g., cohesin SMC3 and histone acetyltransferases EP300 or CREBBP (~30% of SCLC cases), are also recurrently inactivated by deletions and/or mutations2: continuous selection for unbalanced corepressor/coactivator amounts in lineage master transcription factor hubs has been shown to occur in neoplastic evolution of other tissue lineages, and it seems likely that these dynamics operate in SCLCs also.30–32,41–44
To translate DNMT1 target-validation data into in vivo therapy, we used the pyrimidine nucleoside analog pro-drugs decitabine and 5-azacytidine that are standard clinical treatments for myeloid malignancies, including by non-cytotoxic regimens.17–19 No pre-clinical or clinical results to treat SCLC had been described (no results reported from an SCLC clinical trial of oral 5-azacytidine alone, ClinicalTrials.gov: NCT02223052, or guadecitabine [decitabine analog] combined with carboplatin, ClinicalTrials.gov: NCT03913455) (reviewed in Nervi et al.20 and Zavras et al.21). However, results are available from clinical trials to treat several other solid cancers and have disappointed despite scientific DNMT1 target validation in these indications also. Reasons for this have been investigated. In patients with treatment-refractory thoracic malignancies, decitabine infusions sustained decitabine plasma concentrations of >40 nM for ~72 h and caused grade 3/4 myelosuppression, but even so, solid cancer tissue DNA hypomethylation was observed in <25% of ontherapy tumor samples, suggesting target-engagement failure in the tumor tissue.45 One reason for decitabine/5-azacytidine activity in some tissue compartments but not others is the catabolic enzyme CDA. CDA is highly expressed in liver and other solid tissues and deaminates decitabine and 5-azacytidine within minutes into uridine counterparts that do not deplete DNMT1. The murine SCLC cells we used in our experiments recapitulated the most frequent site of spread of human SCLCs by metastasizing extensively to liver.37 CDA in the liver can protect cancer cells from decitabine treatment effects: decitabine alone was ineffective against myeloid malignancy located in the liver, but co-administration of lower doses of decitabine with the CDA-inhibitor THU enabled decitabine activity in this sanctuary.38 Therefore, to treat SCLC in vivo, we combined THU with decitabine and 5-azacytidine, adjusting decitabine and 5-azacytidine doses to ensure a non-cytotoxic profile of activity in sensitive normal myeloid tissue22,38 (oral combination formulations of decitabine or 5-azacytidine with CDA inhibitors are available for clinical evaluation). Another reason that the myeloid compartment is the most sensitive to decitabine or 5-azacytidine treatment effects46 is ~4-fold higher expression here than in other tissues of the pyrimidine metabolism salvage enzymes DCK and UCK2 that rate-limit decitabine and 5-azacytidine processing into DNMT1-depleting nucleotide. To surmount these barriers to treatment of SCLC, we alternated decitabine with 5-azacytidine, to exploit adaptive upregulation of UCK2 by decitabine and of DCK by 5-azacytidine22: normal myelopoiesis proliferates and then terminally differentiates in waves and hence was not expected to be cross-primed by the regimen. Accordingly, tumor cytoreduction, but not myelosuppression, was observed in this and previous pre-clinical in vivo studies of alternating THU/decitabine with THU/5-azacytidine (a decitabine/5-azacytidine alternating regimen is being evaluated in a myeloid malignancy clinical trial, ClinicalTrials.gov: NCT04187703).22,31,47 Notably, among solid tumor malignancies, SCLCs have the highest baseline expression of DCK and UCK2, further encouraging evaluation of decitabine and/or 5-azacytidine to treat SCLC.
Yet another pyrimidine metabolism barrier to decitabine or 5-azacytidine activity to treat SCLC is automatic adaptive responses by this metabolic network that dampen pro-drug processing into DNMT1-depleting nucleotide: these adaptive responses include shifts in CDA, DCK, and UCK2 expression that can be countered as described above.22 In addition, however, there are automatic upregulations of de novo pyrimidine synthesis that manufactures CTP and deoxycytidine triphosphate (dCTP) that compete with exogenous 5-azacytidine and decitabine (reviewed in Saunthararajah39). This mechanism of resistance is one reason to incorporate an inhibitor of de novo pyrimidine synthesis into decitabine/5-azacytidine therapy. Another reason is that inhibitors of DHODH or CTP synthase 2 (CTPS2) release terminal differentiations in liquid and solid tumor cancers; this effect occurs via CTP reduction, because CTP restoration with exogenous cytidine prevented this effect of the DHODH inhibitor leflunomide (the pro-drug for teriflunomide).39 It is possible that a corepressor that mediates repression of lineage-differentiation genes uses CTP as a cofactor: prokaryotic DNA packaging proteins that are phylogenetically related to eukaryote condensins are CTP dependent.48 Moreover, condensins are recurrently amplified in SCLCs, whereas opposing cohesion coactivators are recurrently deleted. Motivated by these observations, we used the DHODH inhibitor teriflunomide to inhibit de novo pyrimidine synthesis: we chose teriflunomide because it is a known safe, non-cytotoxic drug approved and taken daily to treat multiple sclerosis in multi-year treatment durations. Teriflunomide, at clinically relevant concentrations and dosages, induced terminal neuroendocrine lineage differentiation in SCLC cells without activating apoptosis or impacting normal blood counts. Combination Dhodh and Dnmt1 targeting extended time to distress by ~40 days in mice with chemorefractory disseminated p53-null SCLC. The resistance and progression that eventually occurred was characterized by Dnmt1 recovery despite ongoing therapy. There thus remains a need to further optimize countermeasures to adaptive responses by pyrimidine metabolism. DHODH inhibitors as single agents have been shown also by others to cytoreduce p53-null SCLC in vivo, although these authors did not specifically evaluate for neuroendocrine-lineage maturation as the pathway for cell-cycling exits.23
ICIs have modest activity against SCLCs. One mechanism by which cancers can resist ICI is suppression of MHC class I molecules that present antigen to cytotoxic T cells.49 Consistent with such suppression being epigenetically mediated, non-cytotoxic Dnmt1 depletion by decitabine upregulated MHC class I expression by SCLC cells in vitro and in vivo. Moreover, the Dnmt1-targeting regimen preserved immune effectors. However, there was no tumor cytoreduction or time-to-distress benefit from anti-Pd1 incorporation into treatment in the syngeneic model of SCLC, even though the anti-Pd1 treatment was immune modulating, as indicated by an increase in circulating T cells, restricted T cell diversity of TILs, immune checkpoint (Pd1, Lag3, Ctla4, Tim3) upregulation in TILs, and immune checkpoint (Pdl1, Cd80, Cd86, Cd276, Galectin-9, Cd74, Cd155) upregulation in tumor tissue. One possible cause is very low baseline MHC class I expression in SCLCs,49 such that even upregulation by Dnmt1 targeting is insufficient to achieve thresholds needed for functionally impactful antigen presentation. Another possible cause is the observed upregulations of multiple immune checkpoints upon anti-Pd1 exposure, which may preserve the check on immune attack, as observed by others in other cancers (immune checkpoint redundancy).50,51 Human SCLCs are smoking related and have extensive somatic mutation burden; thus, mutation burden per se seems unlikely to explain their limited ICI responsiveness vs. non-SCLCs.2
In conclusion, with 5-year survival probabilities of <5%, there is a need for new treatment modalities for SCLC that are not cross-resistant with chemotherapy, radiation, and ICI. We show that clinically available pro-drugs and drugs, applied for safe and specific molecular-targeted effects without cytotoxicity, can leverage neuroendocrine-lineage commitment of SCLC cells into resumed lineage maturation, capable of forcing cell-cycling exits in chemorefractory and ICI-refractory SCLC.
Limitations of the study
The in vitro drug treatment studies used two human and one murine cell line model of SCLC, and the in vivo treatment studies used one genetically engineered, syngeneic mouse model of SCLC. Although we used clinically available pro-drugs and drugs for the treatments, at dosages and schedules expected to be human-equivalent safe, inter-species differences in drug pharmacology, tumor metabolism, and/or growth-kinetics can singly or collectively defeat achievement of intended molecular pharmacodynamic effects in human tumor tissue. Efficacy and safety to treat human disease can be definitively determined only via conduct of clinical trials.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Yogen Saunthararajah (saunthy@ccf.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
RNA-Seq data and Cut&Tag data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available nfrom the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-NeuroD1 | Cell Signaling | Cat# 4373S (D35G2); RRID: AB_1054907 |
| Anti-DNMTI | Cell Signaling | Cat# 5032S (D63A6); RRID: AB_1054819 |
| Anti-ASCLI | Cell Signaling | Cat# 43666S (E7N9C) |
| anti-p27 | Cell Signaling | Cat# 3686S (D69C12); RRID: AB_2077850 |
| anti-c-Myc | Cell Signaling | Cat# 5605S (D84C12); RRID: AB_1903938 |
| anti-CAD | Cell Signaling | Cat# 11933S; RRID: AB_2797772 |
| Anti-L-Myc | Cell Signaling | Cat# 76266S (E3M5P) |
| HRP-tagged anti-Rabbit secondary | Cell Signaling | Cat# 7074P2; RRID: AB_2099233 |
| HRP-tagged anti-Mouse secondary | Cell signaling | Cat# 7076P2; RRID: AB_330924 |
| Anti-DCK | Santa Cruz Biotechnology | Cat# sc-393098, Clone: H5 |
| anti-ASCLI | Santa Cruz Biotechnology | Cat# sc-374104; RRID: AB_1091856 |
| anti-NeuroD1 | Abcam | Cat# ab213725; RRID: AB_2801303 |
| anti-UCK2 | Abcam | Cat# ab104731; RRID: AB_1071246 |
| anti-CDA | Abcam | Cat# ab82347; RRID: AB_1658630 |
| FITC-anti-mouse CD4 antibody | BD Biosciences | Cat# 553046, Clone: RM4-5; RRID: AB_394582 |
| PerCP anti-mouse CD8 antibody | BD Biosciences | Cat# 561092, Clone 53–6.7; RRID: AB_1056168 |
| PE anti-mouse FoxP3 antibody | BD Biosciences | Cat# 560408, Clone: MF-23; RRID: AB_1645251 |
| PE-Cy7 anti-mouse CD11b antibody | BD Biosciences | Cat# 561098, Clone: M1/70; RRID: AB_2033994 |
| APC anti-mouse Ly6C antibody | BD Biosciences | Cat# 560595, Clone: AL-21; RRID: AB_1727554 |
| PE-CF594 anti-mouse Ly6G antibody | BD Biosciences | Cat# 562700, Clone: 1A8; RRID: AB_2737730 |
| anti-β-Actin antibody | Sigma-Aldrich | Cat# A5441; RRID: AB_476744 |
| Mouse IgG1 isotype control | Abcam | Cat# ab18443; RRID: AB_2736846 |
| Rabbit IgG isotype control | Abcam | Cat# ab172730; RRID: AB_2687931 |
| Anti-DNMT1 | Active Motif | Cat# 39204; RRID: AB_2614950 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Decitabine | BOC Sciences | Cat# 2353-33-5 |
| 5-Azacytidine | BOC Sciences | Cat# 320-67-2 |
| Teriflunomide | MedChem Express | Cat# HY-15405 |
| Fluorizoline | Sigma-Aldrich | Cat# SML2379 |
| Lipofectamine RNAiMax | Invitrogen | Cat# 1000144720 |
| Fast SYBR Green | Applied Biosciences | Cat# 4385612 |
| Pierce Rapid Gold BCA Protein Assay Kit | Thermo-Scientific | Cat# A53226 |
| Giemsa stain | Sigma-Aldrich | Cat # 48900 |
| 4–12% precast Tris-bis gradient gels | Invitrogen | Cat# NP0335 |
| Protein A/G plus agarose beads | Santa Cruz Biotechnology | Cat# sc-2003 |
| gentleMACS dissociator | Miltenyi Biotec | Cat# 130-093-235 |
| gentleMACS tubes | Miltenyi Biotec | Cat# 130-093-237 |
| 70-μm cell strainer | Corning | Cat# 352350 |
| eBioscience™ IC Fixation Buffer | ThermoFisher Scientific | Cat# 00-8222-49 |
| Monensin | ThermoFisher Scientific | Cat# 00-4505-51 |
| NuPAGE LDS Sample Buffer (4X) | Invitrogen | Cat# NP0007 |
| NuPAGE MOPS SDS Running Buffer | Invitrogen | Cat# NP0001 |
| NuPAGE Transfer Buffer | Invitrogen | Cat# NP0006 |
| Precision Plus Protein Standards | Bio Rad | Cat# 161-0394 |
|
| ||
| Critical commercial assays | ||
|
| ||
| RNeasy Mini Kit | Qiagen | Cat# 74104 |
| Cut & Tag Assay kit | Active Motif | Cat# 53165 |
| iScript cDNA Synthesis kit | Bio-Rad | Cat# 1708891 |
| FITC Annexin V Detection Kit Annexin | BD Biosciences | Cat# 556547 |
| MycoAlert™ Mycoplasma Detection Kit | Lonza | Cat# LT07-118 |
|
| ||
| Deposited data | ||
|
| ||
| RNA-Seq | This paper | GEO Accession ID: GSE238007 |
| Cut &Tag | This paper | GEO Accession ID: GSE238007 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| F1339 | Dr David MacPherson | N/A |
| NCI-H82 | ATCC | HTB-175™ |
| NCI-H146 | ATCC | HTB-173 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse B6/129SF1/J | Jackson Laboratory | RRID:IMSR_JAX:101043 |
|
| ||
| Oligonucleotides | ||
|
| ||
| DNMT1 siRNA | Invitrogen | Cat# 4390825 |
| Control siRNA | Invitrogen | Cat# 4390844 |
| Primers, see Table S6 | Table S6 | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ | NIH | https://ImageJ.nih.gov/ij/ |
| FlowJo V10. | BD Biosciences | https://www.flowjo.com |
| GraphPad Prism 7.0 | GraphPad Software, Inc | https://www.graphpad.com |
| EASEQ | Website | https://easeq.net |
| UseGalaxy | Website | https://usegalaxy.org |
| Adobe Photoshop 2023 | Adobe creative cloud | https://www.adobe.com/creativecloud.html |
EXPERIMENTAL MODEL AND SUBJECT PARTICIPATION DETAILS
Study design
Objectives were to understand and therapeutically remedy lineage-maturation arrests of SCLC cells. The central hypothesis was that DNMT1-depletion from SCLC cells would release cell cycle exits by terminal-maturation, a pathway non-cross-resistant with the p53/apoptosis pathway utilized by chemo-radiation. Secondary hypotheses were that mechanisms-of-resistance to DNMT1-targeting by the clinical small molecules decitabine or 5-azacytidine, documented in other cancers, would also operate in SCLC and require solutions, and that inhibiting de novo pyrimidine synthesis, in addition to cooperating with decitabine or 5-azacytidine therapy, could induce p53-independent cancer cell cycling exits in its own right. For pre-clinical in vivo proof-of-principle, we used a model of syngeneic, p53-null, disseminated murine SCLC. Bioluminescence imaging was used to verify tumor engraftment prior to initiation of therapy, with mice distributed to treatment groups to balance baseline tumor burden. Each in vivo experiment was powered to show statistically significant results between treatment groups, possible with five mice per treatment group because of effect-sizes. Several independent in vivo treatment experiments were conducted. All mice were female, to avoid confounding of interpretation by sex-differences in pro-drug metabolism. All experiments used known safe clinical compounds, to ensure translational relevance.
Cell lines and culture
Murine F1339 small cell lung cancer cells were provided by Dr David MacPherson, and were produced by tracheal infection of Rblox/p53lox mice with Adeno-CMV-Cre. The F1339 cells are not in the database of commonly misidentified cell lines (ICLAC and NCBI Biosample). All cell lines regularly tested negative for Mycoplasma contamination. NCI-H82 and NCI-H146 cell lines were purchased from ATCC and were authenticated by LabCorp by STR profiling at the time-of-experimentation. Cells were maintained in RPMI1640 supplemented with 10% fetal bovine serum (FBS, Gibco), 100 units/ml penicillin, 100 μg/mL streptomycin (Gibco), and cultured at 37°C in 5% CO2.
Syngeneic tumor model
Female and male B6/129SF1/J mice were purchased from Jackson Lab (Bar Harbor, ME, USA) and maintained under specific pathogen-free conditions, with free access to food and water. All procedures were performed in compliance with the legislation on the use and care of laboratory animals, and according to protocols (2013–1137 and 2017–1863) approved by the Institutional Animal Care and Use Committee (IACUC) of Cleveland Clinic. Mice were tail-vein inoculated with 0.3 × 106 F1339 cells at 4–6 weeks of age. Assignment to different treatments was done after documentation of tumor engraftment by bioluminescence imaging or by randomization if treatment was initiated before tumor was visible by imaging. Peripheral blood monitoring on-treatment was by tail-vein phlebotomy. Mice were euthanized for signs of distress as defined in the Animal Protocols, determined by animal facility staff blind to the experimental groups, and blood and tumor were collected for further analyses.
METHOD DETAILS
Nuclear extract preparation and co-immunoprecipitation
Cells were washed with ice-cold PBS and then with a buffer solution containing 10 mM Tris–HCl, pH 7.8, 1.5 mM MgCl2, and 10 mM KCl, supplemented with a protease inhibitor mixture containing 0.5 mM dithiothreitol, 0.4 mM phenylmethyl-sulfonyl fluoride and 1X protease-inhibitor cocktail. After incubation on ice for 10 min, cells were lysed by 10 strokes with a Dounce homogenizer and nuclei pelleted. The pellet was resuspended in a buffer solution containing 420 mM KCl, 20 mM Tris–HCl, pH 7.8, 1.5 mM MgCl2 and 20% glycerol, supplemented with protease inhibitor mixture described above and incubated on ice with gentle agitation. The nuclear extract was centrifuged at 10,000 ×g for 10 min, and the supernatant was dialyzed twice against a solution of 20 mM Tris–HCl, pH 7.8, 100 mM KCl, 0.2 mM EDTA, and 20% glycerol. Protein concentration was determined using BCA reagent with bovine serum albumin as standard. 1–1.5mg total protein was used for immunoprecipitation. Agarose A/G Plus beads were bound with antibody or IgG isotype control with gentle rotation for 1h at RT. Then the beads were incubated with 1% BSA for 1h at RT. Nuclear extract was precleared with isotype control IgG bound beads for 1h at 4°C with gentle rotation. After 3 washes with pre-chilled 1XPBS antibody or IgG isotype control bound beads were incubated with nuclear extract overnight at 4°C with gentle rotation. After washing 4 times with 1XPBS containing 1% NP-40 beads were boiled with sample buffer and subjected to SDS-PAGE and Western blot analysis.
Giemsa staining of cells
Cytospins of cells were fixed for 2 min in methanol, air-dried, and stained for 20 min with filtered modified solution of Giemsa stain (Sigma Aldrich, Cat # 48900, St Louis, MO). Images were captured at 400X using Leica Upright Microscope-Orion.
DNMT1 knockdown
Cells were transfected with 25 nM of either DNMT1 siRNA (Invitrogen, Cat # 4390825) or control siRNA (Invitrogen, Cat # 4390844) using lipofectamine RNAiMAX per manufacturer’s protocol. At 72 h knock-down was evaluated by Western blot.
Histological analysis
Tumors were fixed in 10% buffered formalin phosphate (Thermo Fisher Scientific) for 12 h and embedded in paraffin. Sections were stained using H&E.
T cell receptor (TCR) sequencing (T cell oligoclonality analyses)
RNA was isolated from PBMC or CD8+ TILs using the AllPrep RNA Mini Kit (Qiagen) – 200 ng of total RNA was used to construct TCR alpha and beta chain (a/b) libraries using the SMARTer Mouse TCR a/b Profiling Kit (Takara) per manufacturer’s instruction. Samples were pooled to a final concentration of 4 nM and then the pooled libraries were further diluted to a final concentration of 13.5 p.m. including a 7% PhiX Control v3 (Illumina) spike-in. Sequencing was performed on an Illumina MiSeq sequencer (Illumina) using the 600-cycle MiSeq Reagent Kit V3 (Illumina) with paired-end, 2 × 300 base pair reads. The data was analyzed with MiXCR 1.1.0 (Illumina).
Flow cytometry analysis
Tumor tissue was first digested by cutting into small fragments then incubated with mouse or human tumor enzyme cocktail per the manufacturer’s protocol with GentleMACS tubes and the GentleMACS dissociator (Miltenyi Biotec). After filtering through a 70-μm cell strainer the single-cell tumor suspension was enriched for mononuclear cells by centrifugation using percoll gradient. Cells were stimulated with PMA/ionomyocin in the presence of Golgi stop and Monensin for 4 h, then washed with PBS and stained with Live/Dead stain and antibodies specific to cell surface markers CD3, CD4 and CD8 (BD Biosciences). Flow cytometry analysis was performed on BD LSRFortessa and data was analyzed by FlowJo V10.
Apoptosis assay
Cells were treated with different drugs or vehicle or positive control fluorizoline (p53-independent apoptosis inducer) for 24 h. Cells were washed with 1x phosphate-buffered saline (PBS) and stained with Annexin V or propidium iodide (PI) using FITC Annexin V Apoptosis Detection Kit (BD Pharmingen, cat # 556547) per manufacturer’s instruction. Annexin positive cells were acquired by flow cytometry using BD FACS Versa or BD LSRFortessa (BD Biosciences) and data was analyzed by FlowJo V10.
Quantitative polymerase chain reaction
RNA was extracted using Trizol reagent (Invitrogen) or using RNA isolation kit (Qiagen, cat # 74104) per manufacturer’s protocol. cDNA was synthesized using a cDNA synthesis kit (BioRad, cat # 1708891) per manufacturer’s instructions. In analyses of multiple genes, equal amounts of DNA were used for each reaction using gene-specific primers and SYBR Green from Applied Biosystems. The relative gene expression was analyzed following the Livak-Schmittgen method. Primer sequences are in Table S6.
Western blot analysis
Tumor cells were lysed with RIPA lysis buffer and protein concentrations determined by BCA Gold protein assay kit (Thermo Fisher Scientific). Samples with equal quantity (40 μg) of total protein were mixed with 4× loading buffer and 10× sample reducing reagent (Thermo Fisher Scientific), subjected to SDS-PAGE (SDS-polyacrylamide gel electrophoresis) using 4–12% gradient gel (Invitrogen, cat # NP0335), then transferred onto polyvinylidene fluoride (PVDF) membranes. After blocking with 5% dried skimmed milk, the membranes were washed three times and incubated with primary antibodies at 4°C overnight. After washing, the membranes were further incubated with corresponding horseradish peroxidase-conjugated secondary antibodies. Immunodetection was performed by enhanced chemiluminescence (ECL, GE Healthcare/Amersham, Buckinghamshire, UK) according to the manufacturer’s protocol and using X-ray film.
DNA methylation analyses
DNA methylation array data (Illumina, 450K array) for ESCs and SCLC cell lines, GSE3184827 and GSE6629528 respectively, were analyzed. Analyses were restricted to CpG methylation β-values less likely to be confounded by noise from single-nucleotide polymorphisms, insertion-deletion mutations, and repeats, as described in,52 and to CpG islands (features characterized by high local concentration of CpG sites, typically low levels of methylation, and methylation impacts on gene expression53) as per genome build GRCh37/hg19. A list of the analyzed CpGs, incorporating positions and annotations per GRCh37/hg19, is provided as Table S2.
DNMT1/Dnmt1 Cut and Tag and data processing
The Cleavage Under Targets and Tagmentation (CUT&Tag) procedure in triplicate, following the manufacturer’s protocol for the CUT&Tag-IT Assay Kit from Active Motif (cat no # 53165), and using DNMT1 antibody (Active Motif cat no # 39204), was applied to murine F1339 and human H82 SCLC cells. Libraries were amplified and normalized with the Illumina Nextera DNA Library prep kit (FC-121–1031) according to the manufacturer’s protocols. Sequencing on NextSeq 550 followed the manufacturer’s protocols through the Genomics Core Facility at Case Western Reserve University. Data processing was on the Galaxy platform (usegalaxy. org).54 To trim the adapters, we used Cutadapt. Sequencing reads were mapped to mm10 or GRCh37/hg19 genomes using Bowtie2.55 Heatmaps of coverage around gene transcription start sites were created using EASEQ.56
RNA-seq, data processing and identification of differentially expressed genes
Human SCLC (H146) and murine SCLC (F1339) were treated with decitabine 0.5 μM for 96 and 72 h respectively in duplicate for gene expression analyses by RNA-sequencing. RNA-extraction and library preparation from the cell pellet, sequencing and data processing was performed by an external vendor, Azenta Life Sciences. Sequence reads were trimmed to remove possible adapter sequences and nucleotides with poor quality using Trimmomatic v.0.36. The trimmed reads were mapped to the Homo sapiens GRCh38 reference genome available on ENSEMBL using the STAR aligner v.2.5.2b. The STAR aligner is a splice aligner that detects splice junctions and incorporates them to help align the entire read sequences. BAM files were generated as a result of this step.
Unique gene hit counts were calculated by using featureCounts from the Subread package v.1.5.2. The hit counts were summarized and reported using the gene_id feature in the annotation file. Only unique reads that fell within exon regions were counted. Since strand-specific library preparation was performed, the reads were strand-specifically counted.
After extraction of gene hit counts, the gene hit counts table, provided as Table S5, was used for downstream differential expression analysis. Using DESeq2,57 a comparison of gene expression between vehicle and decitabine-treated H146 and F1339 SCLC cells was performed. The Wald test was used to generate p values and log2 fold changes. Genes with an adjusted p value <0.05 and absolute log2 fold change >1 were called as differentially expressed genes.
The raw and processed RNA-seq data are available at GEO.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
Statistical analysis was performed with Prism 7.0 software (GraphPad, San Diego, CA). Survival differences among the treatment groups were analyzed by the Kaplan-Meier method and p values were calculated with log rank test. Two-sided Mann–Whitney U or one-way Annova test was used to compare medians and unpaired t test to compare means. Bonferroni’s correction to p < 0.05 was used to determine statistical significance.
Supplementary Material
Highlights.
SCLC cells are committed into neuroendocrine lineage then maturation arrested
The key corepressor DNMT1 mediates the maturation arrests
Non-cytotoxic corepressor-inhibiting drugs relieve maturation-gene repression
Resulting p53-independent cell-cycling exits cytoreduce chemorefractory SCLC
ACKNOWLEDGMENTS
We thank Shannon Hanmer for technical assistance. V.V. was supported by a 2015 ASCO Career Development Award and CARES Award; V.V. and K.K. were supported by a 2018 VeloSano Award. Y.S. was supported by National Heart, Lung, and Blood Institute PO1 HL146372; National Cancer Institute P30 CA043703, RO1 CA204373, and R21 CA263430; philanthropic funds from Robert and Jennifer McNeil, Leszek and Jolanta Czarnecki, and Dane and Louise Miller, and the James Oberle family; and NIH Shared Instrument award S10OD018205.
Footnotes
DECLARATION OF INTERESTS
Ownership: Y.S., EpiDestiny. Consultancy: K.S., Merck, Takeda Pharmaceuticals, Shattuck Labs, Pierre-Fabre, and Celgene; Y.S., EpiDestiny; V.V., Bristol Myers, AstraZeneca, Genentech, Merck, Taekeda, Reddy Labs, Celgene, Foundation Medicine, Nektar Therapeutics, Alkermes, and Amgen. Research support: V.V., Genentech, Alkermes, BMS, Merck, Esai, Navigate Biopharma, Leap Therapeutics, and AstraZeneca; K.S., Vasculox/Tioma, Navigate Biopharma, Tesaro Inc., Onkaido Therapeutics/Moderna, Takeda Pharmaceuticals, Surface Oncology, Pierre-Fabre, Merck, and Bristol-Myers Squibb. Intellectual property: Y.S., patents around tetrahydrouridine, decitabine, and 5-azacytidine (US 9,259,469 B2, US 9,265,785 B2, and US 9,895,391 B2).
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113016.
REFERENCES
- 1.Rindi G, Klimstra DS, Abedi-Ardekani B, Asa SL, Bosman FT, Brambilla E, Busam KJ, de Krijger RR, Dietel M, El-Naggar AK, et al. (2018). A common classification framework for neuroendocrine neoplasms: an International Agency for Research on Cancer (IARC) and World Health Organization (WHO) expert consensus proposal. Mod. Pathol. 31, 1770–1786. 10.1038/s41379-018-0110-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.George J, Lim JS, Jang SJ, Cun Y, Ozretić L, Kong G, Leenders F, Lu X, Fernández-Cuesta L, Bosco G, et al. (2015). Comprehensive genomic profiles of small cell lung cancer. Nature 524, 47–53. 10.1038/nature14664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fujiwara T, Grimm EA, Mukhopadhyay T, Zhang WW, Owen-Schaub LB, and Roth JA (1994). Induction of chemosensitivity in human lung cancer cells in vivo by adenovirus-mediated transfer of the wild-type p53 gene. Cancer Res. 54, 2287–2291. [PubMed] [Google Scholar]
- 4.Zandi R, Selivanova G, Christensen CL, Gerds TA, Willumsen BM, and Poulsen HS (2011). PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin. Cancer Res. 17, 2830–2841. 10.1158/1078-0432.CCR-10-3168. [DOI] [PubMed] [Google Scholar]
- 5.Do PM, Varanasi L, Fan S, Li C, Kubacka I, Newman V, Chauhan K, Daniels SR, Boccetta M, Garrett MR, et al. (2012). Mutant p53 co-operates with ETS2 to promote etoposide resistance. Genes Dev. 26, 830–845. 10.1101/gad.181685.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park KS, Liang MC, Raiser DM, Zamponi R, Roach RR, Curtis SJ, Walton Z, Schaffer BE, Roake CM, Zmoos AF, et al. (2011). Characterization of the cell of origin for small cell lung cancer. Cell Cycle 10, 2806–2815. 10.4161/cc.10.16.17012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Z, Zhou Y, Qian H, Shao G, Lu X, Chen Q, Sun X, Chen D, Yin R, Zhu H, et al. (2013). Stemness and inducing differentiation of small cell lung cancer NCI-H446 cells. Cell Death Dis. 4, e633. 10.1038/cddis.2013.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sarvi S, Mackinnon AC, Avlonitis N, Bradley M, Rintoul RC, Rassl DM, Wang W, Forbes SJ, Gregory CD, and Sethi T (2014). CD133+ cancer stem-like cells in small cell lung cancer are highly tumorigenic and chemoresistant but sensitive to a novel neuropeptide antagonist. Cancer Res. 74, 1554–1565. 10.1158/0008-5472.CAN-13-1541. [DOI] [PubMed] [Google Scholar]
- 9.Poirier JT, Gardner EE, Connis N, Moreira AL, de Stanchina E, Hann CL, and Rudin CM (2015). DNA methylation in small cell lung cancer defines distinct disease subtypes and correlates with high expression of EZH2. Oncogene 34, 5869–5878. 10.1038/onc.2015.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Augustyn A, Borromeo M, Wang T, Fujimoto J, Shao C, Dospoy PD, Lee V, Tan C, Sullivan JP, Larsen JE, et al. (2014). ASCL1 is a lineage oncogene providing therapeutic targets for high-grade neuroendocrine lung cancers. Proc. Natl. Acad. Sci. USA 111, 14788–14793. 10.1073/pnas.1410419111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borges M, Linnoila RI, van de Velde HJ, Chen H, Nelkin BD, Mabry M, Baylin SB, and Ball DW (1997). An achaete-scute homologue essential for neuroendocrine differentiation in the lung. Nature 386, 852–855. 10.1038/386852a0. [DOI] [PubMed] [Google Scholar]
- 12.Jiang T, Collins BJ, Jin N, Watkins DN, Brock MV, Matsui W, Nelkin BD, and Ball DW (2009). Achaete-scute complex homologue 1 regulates tumor-initiating capacity in human small cell lung cancer. Cancer Res. 69, 845–854. 10.1158/0008-5472.CAN-08-2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Osada H, Tatematsu Y, Yatabe Y, Horio Y, and Takahashi T (2005). ASH1 gene is a specific therapeutic target for lung cancers with neuroendocrine features. Cancer Res. 65, 10680–10685. 10.1158/0008-5472.CAN-05-1404. [DOI] [PubMed] [Google Scholar]
- 14.Borromeo MD, Savage TK, Kollipara RK, He M, Augustyn A, Osborne JK, Girard L, Minna JD, Gazdar AF, Cobb MH, and Johnson JE (2016). ASCL1 and NEUROD1 Reveal Heterogeneity in Pulmonary Neuroendocrine Tumors and Regulate Distinct Genetic Programs. Cell Rep. 16, 1259–1272. 10.1016/j.celrep.2016.06.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gehart H, van Es JH, Hamer K, Beumer J, Kretzschmar K, Dekkers JF, Rios A, and Clevers H (2019). Identification of Enteroendocrine Regulators by Real-Time Single-Cell Differentiation Mapping. Cell 176, 1158–1173.e16. 10.1016/j.cell.2018.12.029. [DOI] [PubMed] [Google Scholar]
- 16.Velcheti V, Schrump D, and Saunthararajah Y (2018). Ultimate Precision: Targeting Cancer but Not Normal Self-Replication (American Society of Clinical Oncology educational book. American Society of Clinical Oncology. Annual Meeting), pp. 950–963. 10.1200/EDBK_199753. [DOI] [PubMed] [Google Scholar]
- 17.Ng KP, Ebrahem Q, Negrotto S, Mahfouz RZ, Link KA, Hu Z, Gu X, Advani A, Kalaycio M, Sobecks R, et al. (2011). p53 independent epigenetic-differentiation treatment in xenotransplant models of acute myeloid leukemia. Leukemia 25, 1739–1750. 10.1038/leu.2011.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saunthararajah Y, Sekeres M, Advani A, Mahfouz R, Durkin L, Radivoyevitch T, Englehaupt R, Juersivich J, Cooper K, Husseinzadeh H, et al. (2015). Evaluation of noncytotoxic DNMT1-depleting therapy in patients with myelodysplastic syndromes. J. Clin. Invest. 125, 1043–1055. 10.1172/JCI78789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Awada H, Mahfouz RZ, Kishtagari A, Kuzmanovic T, Durrani J, Kerr CM, Patel BJ, Visconte V, Radivoyevitch T, Lichtin A, et al. (2020). Extended experience with a non-cytotoxic DNMT1-targeting regimen of decitabine to treat myeloid malignancies. Br. J. Haematol. 188, 924–929. 10.1111/bjh.16281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nervi C, De Marinis E, and Codacci-Pisanelli G (2015). Epigenetic treatment of solid tumours: a review of clinical trials. Clin. Epigenet. 7, 127. 10.1186/s13148-015-0157-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zavras PD, Shastri A, Goldfinger M, Verma AK, and Saunthararajah Y (2021). Clinical Trials Assessing Hypomethylating Agents Combined with Other Therapies: Causes for Failure and Potential Solutions. Clin. Cancer Res. 27, 6653–6661. 10.1158/1078-0432.CCR-21-2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gu X, Tohme R, Tomlinson B, Sakre N, Hasipek M, Durkin L, Schuerger C, Grabowski D, Zidan AM, Radivoyevitch T, et al. (2021). Decitabine- and 5-azacytidine resistance emerges from adaptive responses of the pyrimidine metabolism network. Leukemia 35, 1023–1036. 10.1038/s41375-020-1003-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li L, Ng SR, Coló n CI, Drapkin BJ, Hsu PP, Li Z, Nabel CS, Lewis CA, Romero R, Mercer KL, et al. (2019). Identification of DHODH as a therapeutic target in small cell lung cancer. Sci. Transl. Med. 11, eaaw7852. 10.1126/scitranslmed.aaw7852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim J, Woo AJ, Chu J, Snow JW, Fujiwara Y, Kim CG, Cantor AB, and Orkin SH (2010). A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell 143, 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, Chang S, Conley SD, Mori Y, Seita J, et al. (2020). A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 587, 619–625. 10.1038/s41586-020-2922-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mahadevan NR, Knelson EH, Wolff JO, Vajdi A, Saigí M, Campisi M, Hong D, Thai TC, Piel B, Han S, et al. (2021). Intrinsic Immunogenicity of Small Cell Lung Carcinoma Revealed by Its Cellular Plasticity. Cancer Discov. 11, 1952–1969. 10.1158/2159-8290.CD-20-0913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nazor KL, Altun G, Lynch C, Tran H, Harness JV, Slavin I, Garitaonandia I, Müller FJ, Wang YC, Boscolo FS, et al. (2012). Recurrent variations in DNA methylation in human pluripotent stem cells and their differentiated derivatives. Cell Stem Cell 10, 620–634. 10.1016/j.stem.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mohammad HP, Smitheman KN, Kamat CD, Soong D, Federowicz KE, Van Aller GS, Schneck JL, Carson JD, Liu Y, Butticello M, et al. (2015). A DNA Hypomethylation Signature Predicts Antitumor Activity of LSD1 Inhibitors in SCLC. Cancer Cell 28, 57–69. 10.1016/j.ccell.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 29.Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, Barretina J, Gelfand ET, Bielski CM, Li H, et al. (2019). Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508. 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gu X, Hu Z, Ebrahem Q, Crabb JS, Mahfouz RZ, Radivoyevitch T, Crabb JW, and Saunthararajah Y (2014). Runx1 regulation of Pu.1 corepressor/coactivator exchange identifies specific molecular targets for leukemia differentiation therapy. J. Biol. Chem. 289, 14881–14895. 10.1074/jbc.M114.562447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gu X, Ebrahem Q, Mahfouz RZ, Hasipek M, Enane F, Radivoyevitch T, Rapin N, Przychodzen B, Hu Z, Balusu R, et al. (2018). Leukemogenic nucleophosmin mutation disrupts the transcription factor hub that regulates granulomonocytic fates. J. Clin. Invest. 128, 4260–4279. 10.1172/JCI97117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gu X, Enane F, Tohme R, Schuerger C, Radivoyevitch T, Parker Y, Zuberi E, Przychodzen B, Jha BK, Lindner D, et al. (2021). PBRM1 loss in kidney cancer unbalances the proximal tubule master transcription factor hub to repress proximal tubule differentiation. Cell Rep. 36, 109747. 10.1016/j.celrep.2021.109747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Papageorgiou DN, Karkoulia E, Amaral-Psarris A, Burda P, Kolodziej K, Demmers J, Bungert J, Stopka T, and Strouboulis J (2016). Distinct and overlapping DNMT1 interactions with multiple transcription factors in erythroid cells: Evidence for co-repressor functions. Biochim. Biophys. Acta 1859, 1515–1526. 10.1016/j.bbagrm.2016.09.007. [DOI] [PubMed] [Google Scholar]
- 34.Di Ruscio A, Ebralidze AK, Benoukraf T, Amabile G, Goff LA, Terragni J, Figueroa ME, De Figueiredo Pontes LL, Alberich-Jorda M, Zhang P, et al. (2013). DNMT1-interacting RNAs block gene-specific DNA methylation. Nature 503, 371–376. 10.1038/nature12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meuwissen R, Linn SC, Linnoila RI, Zevenhoven J, Mooi WJ, and Berns A (2003). Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 4, 181–189. [DOI] [PubMed] [Google Scholar]
- 36.Hiatt JB, Sandborg H, Garrison SM, Arnold HU, Liao SY, Norton JP, Friesen TJ, Wu F, Sutherland KD, Rienhoff HY, et al. (2022). Inhibition of LSD1 with Bomedemstat Sensitizes Small Cell Lung Cancer to Immune Checkpoint Blockade and T-Cell Killing. Clin. Cancer Res. 28, 4551–4564. 10.1158/1078-0432.CCR-22-1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elliott JA, Osterlind K, Hirsch FR, and Hansen HH (1987). Metastatic patterns in small-cell lung cancer: correlation of autopsy findings with clinical parameters in 537 patients. J. Clin. Oncol. 5, 246–254. 10.1200/JCO.1987.5.2.246. [DOI] [PubMed] [Google Scholar]
- 38.Ebrahem Q, Mahfouz RZ, Ng KP, and Saunthararajah Y (2012). High cytidine deaminase expression in the liver provides sanctuary for cancer cells from decitabine treatment effects. Oncotarget 3, 1137–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saunthararajah Y (2020). Mysteries of partial dihydroorotate dehydrogenase inhibition and leukemia terminal differentiation. Haematologica 105, 2191–2193. 10.3324/haematol.2020.254482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang YH, Klingbeil O, He XY, Wu XS, Arun G, Lu B, Somerville TDD, Milazzo JP, Wilkinson JE, Demerdash OE, et al. (2018). POU2F3 is a master regulator of a tuft cell-like variant of small cell lung cancer. Genes Dev. 32, 915–928. 10.1101/gad.314815.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Enane FO, Shuen WH, Gu X, Quteba E, Przychodzen B, Makishima H, Bodo J, Ng J, Chee CL, Ba R, et al. (2017). GATA4 loss of function in liver cancer impedes precursor to hepatocyte transition. J. Clin. Invest. 127, 3527–3542. 10.1172/JCI93488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu Z, Gu X, Baraoidan K, Ibanez V, Sharma A, Kadkol S, Munker R, Ackerman S, Nucifora G, and Saunthararajah Y (2011). RUNX1 regulates corepressor interactions of PU.1. Blood 117, 6498–6508. 10.1182/blood-2010-10-312512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.von Knebel Doeberitz N, Paech D, Sturm D, Pusch S, Turcan S, and Saunthararajah Y (2022). Changing paradigms in oncology: toward non-cytotoxic treatments for advanced gliomas. Int. J. Cancer 151, 1431–1446. 10.1002/ijc.34131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sen P, Luo J, Hada A, Hailu SG, Dechassa ML, Persinger J, Brahma S, Paul S, Ranish J, and Bartholomew B (2017). Loss of Snf5 Induces Formation of an Aberrant SWI/SNF Complex. Cell Rep. 18, 2135–2147. 10.1016/j.celrep.2017.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schrump DS, Fischette MR, Nguyen DM, Zhao M, Li X, Kunst TF, Hancox A, Hong JA, Chen GA, Pishchik V, et al. (2006). Phase I study of decitabine-mediated gene expression in patients with cancers involving the lungs, esophagus, or pleura. Clin. Cancer Res. 12, 5777–5785. [DOI] [PubMed] [Google Scholar]
- 46.Terse P, Engelke K, Chan K, Ling Y, Sharpnack D, Saunthararajah Y, and Covey JM (2014). Subchronic oral toxicity study of decitabine in combination with tetrahydrouridine in CD-1 mice. Int. J. Toxicol. 33, 75–85. 10.1177/1091581814524994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hill B, Jagadeesh D, Pohlman B, Dean R, Parameswaran N, Chen J, Radivoyevitch T, Morrison A, Fada S, Dever M, et al. (2021). A pilot clinical trial of oral tetrahydrouridine/decitabine for noncytotoxic epigenetic therapy of chemoresistant lymphoid malignancies. Semin. Hematol. 58, 35–44. 10.1053/j.seminhematol.2020.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Soh YM, Davidson IF, Zamuner S, Basquin J, Bock FP, Taschner M, Veening JW, De Los Rios P, Peters JM, and Gruber S (2019). Self-organization of parS centromeres by the ParB CTP hydrolase. Science 366, 1129–1133. 10.1126/science.aay3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burr ML, Sparbier CE, Chan KL, Chan YC, Kersbergen A, Lam EYN, Azidis-Yates E, Vassiliadis D, Bell CC, Gilan O, et al. (2019). An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer. Cancer Cell 36, 385–401.e8. 10.1016/j.ccell.2019.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H, et al. (2016). Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 7, 10501. 10.1038/ncomms10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shayan G, Srivastava R, Li J, Schmitt N, Kane LP, and Ferris RL (2017). Adaptive resistance to anti-PD1 therapy by Tim-3 upregulation is mediated by the PI3K-Akt pathway in head and neck cancer. OncoImmunology 6, e1261779. 10.1080/2162402X.2016.1261779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Naeem H, Wong NC, Chatterton Z, Hong MKH, Pedersen JS, Corcoran NM, Hovens CM, and Macintyre G (2014). Reducing the risk of false discovery enabling identification of biologically significant genome-wide methylation status using the Human Methylation450 array. BMC Genom. 15, 51. 10.1186/1471-2164-15-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xie W, Schultz MD, Lister R, Hou Z, Rajagopal N, Ray P, Whitaker JW, Tian S, Hawkins RD, Leung D, et al. (2013). Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 153, 1134–1148. 10.1016/j.cell.2013.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Community Galaxy (2022). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Res. 50, W345–W351. 10.1093/nar/gkac247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lerdrup M, Johansen JV, Agrawal-Singh S, and Hansen K (2016). An interactive environment for agile analysis and visualization of ChIP-sequencing data. Nat. Struct. Mol. Biol. 23, 349–357. 10.1038/nsmb.3180. [DOI] [PubMed] [Google Scholar]
- 57.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 12, 1–222, 3rd edition. 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-Seq data and Cut&Tag data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available nfrom the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-NeuroD1 | Cell Signaling | Cat# 4373S (D35G2); RRID: AB_1054907 |
| Anti-DNMTI | Cell Signaling | Cat# 5032S (D63A6); RRID: AB_1054819 |
| Anti-ASCLI | Cell Signaling | Cat# 43666S (E7N9C) |
| anti-p27 | Cell Signaling | Cat# 3686S (D69C12); RRID: AB_2077850 |
| anti-c-Myc | Cell Signaling | Cat# 5605S (D84C12); RRID: AB_1903938 |
| anti-CAD | Cell Signaling | Cat# 11933S; RRID: AB_2797772 |
| Anti-L-Myc | Cell Signaling | Cat# 76266S (E3M5P) |
| HRP-tagged anti-Rabbit secondary | Cell Signaling | Cat# 7074P2; RRID: AB_2099233 |
| HRP-tagged anti-Mouse secondary | Cell signaling | Cat# 7076P2; RRID: AB_330924 |
| Anti-DCK | Santa Cruz Biotechnology | Cat# sc-393098, Clone: H5 |
| anti-ASCLI | Santa Cruz Biotechnology | Cat# sc-374104; RRID: AB_1091856 |
| anti-NeuroD1 | Abcam | Cat# ab213725; RRID: AB_2801303 |
| anti-UCK2 | Abcam | Cat# ab104731; RRID: AB_1071246 |
| anti-CDA | Abcam | Cat# ab82347; RRID: AB_1658630 |
| FITC-anti-mouse CD4 antibody | BD Biosciences | Cat# 553046, Clone: RM4-5; RRID: AB_394582 |
| PerCP anti-mouse CD8 antibody | BD Biosciences | Cat# 561092, Clone 53–6.7; RRID: AB_1056168 |
| PE anti-mouse FoxP3 antibody | BD Biosciences | Cat# 560408, Clone: MF-23; RRID: AB_1645251 |
| PE-Cy7 anti-mouse CD11b antibody | BD Biosciences | Cat# 561098, Clone: M1/70; RRID: AB_2033994 |
| APC anti-mouse Ly6C antibody | BD Biosciences | Cat# 560595, Clone: AL-21; RRID: AB_1727554 |
| PE-CF594 anti-mouse Ly6G antibody | BD Biosciences | Cat# 562700, Clone: 1A8; RRID: AB_2737730 |
| anti-β-Actin antibody | Sigma-Aldrich | Cat# A5441; RRID: AB_476744 |
| Mouse IgG1 isotype control | Abcam | Cat# ab18443; RRID: AB_2736846 |
| Rabbit IgG isotype control | Abcam | Cat# ab172730; RRID: AB_2687931 |
| Anti-DNMT1 | Active Motif | Cat# 39204; RRID: AB_2614950 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Decitabine | BOC Sciences | Cat# 2353-33-5 |
| 5-Azacytidine | BOC Sciences | Cat# 320-67-2 |
| Teriflunomide | MedChem Express | Cat# HY-15405 |
| Fluorizoline | Sigma-Aldrich | Cat# SML2379 |
| Lipofectamine RNAiMax | Invitrogen | Cat# 1000144720 |
| Fast SYBR Green | Applied Biosciences | Cat# 4385612 |
| Pierce Rapid Gold BCA Protein Assay Kit | Thermo-Scientific | Cat# A53226 |
| Giemsa stain | Sigma-Aldrich | Cat # 48900 |
| 4–12% precast Tris-bis gradient gels | Invitrogen | Cat# NP0335 |
| Protein A/G plus agarose beads | Santa Cruz Biotechnology | Cat# sc-2003 |
| gentleMACS dissociator | Miltenyi Biotec | Cat# 130-093-235 |
| gentleMACS tubes | Miltenyi Biotec | Cat# 130-093-237 |
| 70-μm cell strainer | Corning | Cat# 352350 |
| eBioscience™ IC Fixation Buffer | ThermoFisher Scientific | Cat# 00-8222-49 |
| Monensin | ThermoFisher Scientific | Cat# 00-4505-51 |
| NuPAGE LDS Sample Buffer (4X) | Invitrogen | Cat# NP0007 |
| NuPAGE MOPS SDS Running Buffer | Invitrogen | Cat# NP0001 |
| NuPAGE Transfer Buffer | Invitrogen | Cat# NP0006 |
| Precision Plus Protein Standards | Bio Rad | Cat# 161-0394 |
|
| ||
| Critical commercial assays | ||
|
| ||
| RNeasy Mini Kit | Qiagen | Cat# 74104 |
| Cut & Tag Assay kit | Active Motif | Cat# 53165 |
| iScript cDNA Synthesis kit | Bio-Rad | Cat# 1708891 |
| FITC Annexin V Detection Kit Annexin | BD Biosciences | Cat# 556547 |
| MycoAlert™ Mycoplasma Detection Kit | Lonza | Cat# LT07-118 |
|
| ||
| Deposited data | ||
|
| ||
| RNA-Seq | This paper | GEO Accession ID: GSE238007 |
| Cut &Tag | This paper | GEO Accession ID: GSE238007 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| F1339 | Dr David MacPherson | N/A |
| NCI-H82 | ATCC | HTB-175™ |
| NCI-H146 | ATCC | HTB-173 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse B6/129SF1/J | Jackson Laboratory | RRID:IMSR_JAX:101043 |
|
| ||
| Oligonucleotides | ||
|
| ||
| DNMT1 siRNA | Invitrogen | Cat# 4390825 |
| Control siRNA | Invitrogen | Cat# 4390844 |
| Primers, see Table S6 | Table S6 | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ | NIH | https://ImageJ.nih.gov/ij/ |
| FlowJo V10. | BD Biosciences | https://www.flowjo.com |
| GraphPad Prism 7.0 | GraphPad Software, Inc | https://www.graphpad.com |
| EASEQ | Website | https://easeq.net |
| UseGalaxy | Website | https://usegalaxy.org |
| Adobe Photoshop 2023 | Adobe creative cloud | https://www.adobe.com/creativecloud.html |