

Abstract
By employing a chiral bifunctional phosphine ligand, a gold(I)-catalyzed efficient and highly enantioselective dearomatization of phenols is achieved via versatile metal-ligand cooperation. The reaction is proven to be remarkably general in scope, permitting substitutions at all four remaining benzene positions, accommodating electron-withdrawing groups including strongly deactivating nitro, and allowing carbon-based groups of varying steric bulk including tert-butyl at the alkyne terminus. Moreover, besides N-(o-hydroxyphenyl)alkynylamides, the corresponding ynoates and ynones are all suitable substrates. Spirocyclohexadienone-pyrrol-2-ones, spirocyclohexadienone-butenolides, and spirocyclohexadenone-cyclopentenones are formed in yields up to 99% and with ee up to 99%.
Keywords: Dearomatization, gold catalysis, metal-ligand cooperation, enantioselectivity, cyclization
Graphical Abstract
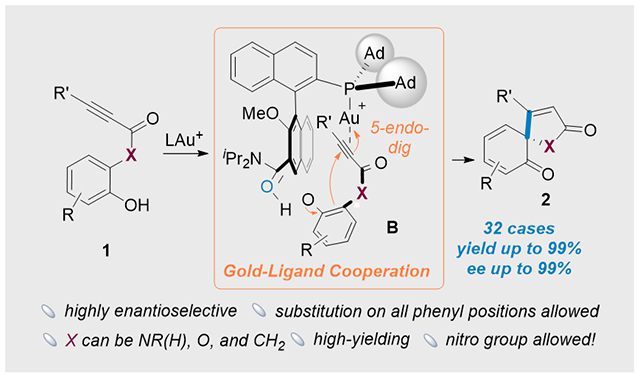
Asymmetric dearomatization of phenols was achieved by employing chiral bifunctional phosphine ligands in cooperative gold catalysis. This transformation demonstrated a remarkable generality, affording the desired product with high yields (up to 99%) and excellent enantioselectivities (ee up to 99%).
Catalytic asymmetric dearomatization (CADA)[1–2] is an efficient method for converting abundant planar aromatic compounds into chiral three-dimension structures featuring challenging quaternary carbon centers, enabling rapid synthesis of natural products[3–5] and offering an expedient venue to escape the ‘flatland’.[6] Over the past two decades, significant developments have been made in the catalytic asymmetric dearomatization of electron-rich aromatic compounds and in particular naphthols, furans, pyrroles, and indoles.[1–2] However, the asymmetric dearomatization of ubiquitous phenols affording chiral quaternary centers remains largely underdeveloped and challenging partly due to their higher aromatic stabilization,[7–9] and most literature reports in this area exhibit moderate enantioselectivity.[10–14] For the ones reporting excellent ee values,[15–24] the substrate scopes are limited (Scheme 1A). For example, in the enantioselective oxidative spiro-lactonization of phenol derivatives reported by Ishihara,[16] an ortho substitution is required to achieve ≥90% ee; in the enantioselective fluorinative dearomatization of phenols reported by Toste, both the R group and the C6 substituents need to be H to achieve ≥87% ee;[15] in the asymmetric hydroxylative phenol dearomatization reported by Pouységu and Quideau,[17] the R group ortho to the phenolic OH group needs to be sterically demanding for high enantioselectivity; several studies focus on specific substrate types including resorcinol-based aryl ketones/aldehydes[18–19] and alkyl-/RO substituted phenols of certain patterns.[20–24] Most of the substituents in these studies are limited to electron-donating alkyl/alkoxy/hydroxy, and no strongly electron-withdrawing groups are tolerated except in the case of resorcinol substrates[18–19] where the additional phenolic OH group counters the deactivating carbonyl group. Only one case possessing a nitro group is reported, albeit exhibiting a low yield (24%).[18] Notwithstanding, we describe in this work a highly enantioselective and high-yielding cyclative dearomatization of phenols that for the first time exhibits an extremely broad substrate scope including the accommodation of substitution at every remaining benzene ring position and the tolerance of ester and even nitro substituents and affords synthetically valuable chiral spirocyclic trienone products or their dimeric products.
Scheme 1.
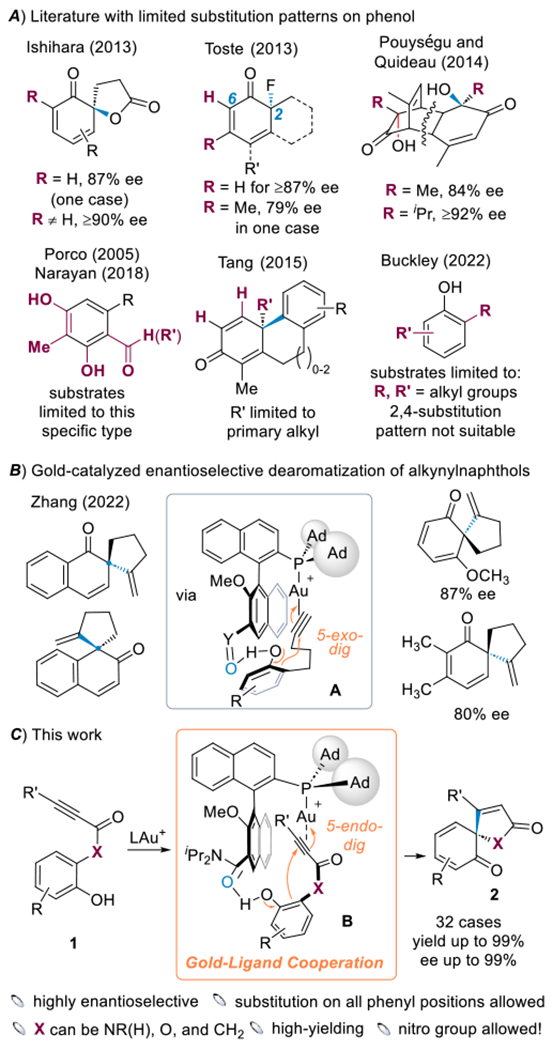
Prior studies of asymmetric dearomatization of phenols and this work.
In the past several years, we introduced the concept of gold-ligand cooperation to homogeneous gold catalysis via the development of biaryl-2-ylphosphine ligands featuring a remote basic group.[25] This metal-ligand cooperation can achieve dramatic rate acceleration[26] and offers a unique and novel approach to realizing asymmetric gold-catalyzed cyclization.[27–28] For example, we achieved an enantioselective dearomatization of alkynylnaphthols via a 5-exo-dig cyclization (Scheme 1B).[28] The H-bonding interaction between the ligand basic substituent and the phenolic hydrogen shown in structure A is key for the observed chiral induction. However, for more challenging phenol derivatives, the few reported substrates are limited to electron-rich ones and the enantioselectivity is moderate to good. We reasoned this type of metal-ligand cooperation, due to the rigid ligand framework and the potentially optimal special positioning of the metal center and the ligand H-bond acceptor group, would be uniquely equipped to deliver dearomatization with a general tolerance of various substitutions. In this context, we considered the phenolic compound of type 1 (Scheme 1C) as suitable substrates for this endeavor as its alkynylcarbonyl moiety would render a) the C-C triple bond more electrophilic and hence reactive towards variously-substituted phenols including electronically deactivated ones and b) the more rigid nature of the connection between the C-C triple bond and the phenol moiety – due mostly to the carbonyl group – more conducive to enantiocontrol by the gold complex, as hypothesized in the structure B. Notably, the cyclization would be 5-endo-dig, which is unique as our prior works on asymmetric cooperative gold catalysis all involve exo cyclization.[27–28]
We initiated our study using N-benzyl-N-(2-hydroxyphenyl)hept-2-ynamide 1a as the substrate (Table 1). It is readily prepared in two steps from 2-benzoxazolinone. The dearomative spirocyclization of this type of 2-alkynamide-functionalized phenols or protected phenols has been realized via gold catalysis[29–31] or the activation of stoichiometric electrophilic halogen or chalcogen species.[32–35] But, no enantioselective variants have been documented. With WangPhosAuCl (5 mol%) as the precatalyst, the reaction went to completion in less than 15 minutes at room temperature in DCM, affording the spirobicycle 2a in 90% yield (Table 1, entry 1). 2a contains one cyclohexa-2,4-dien-1-one ring and one γ-lactam ring, which are fused through a chiral quaternary carbon center. The fast reaction is consistent with the rate acceleration phenomena observed in our previous gold-ligand cooperative catalysis.[26, 28] Encouraged by this racemic result, we explored several chiral bifunctional ligands prepared from (S)-binol[28] for chiral induction. While the phosphonate ligand L1 (entry 2) performed poorly, the amide-functionalized ligands L2-L4 (entries 3-5) led to rapid reactions (<15 min) and excellent enantioselectivities, with (S)-L4 featuring a N,N-diisopropylamide group being the best and affording 2a with 98% ee (entry 5). In contrast, when the ligand (S)-L5 missing the remote amide functional group was employed, the reaction was notably slower and resulted in a moderate ee with the opposite sense (entry 6). 1H NMR monitoring of the reactions using L4 and L5 (0.5 mol %) separately as the ligand establishes >30 times of rate acceleration by the former. (see Supporting Information for more details). These results highlight the essential role of the amide functionality as a rate-accelerating directing group that inverts the inherent stereoinduction by the chiral ligand framework. Among the solvents tested, DCM and DCE gave the best result (entry 5, entries 7-10). Moreover, replacing the halogen scavenger NaBARF (10 mol%) with AgNTf2 (5 mol%) or AgSbF5 (5 mol%) did not affect the enantioselectivity (entries 11 and 12).
Table 1.
Reaction Conditions Optimization.
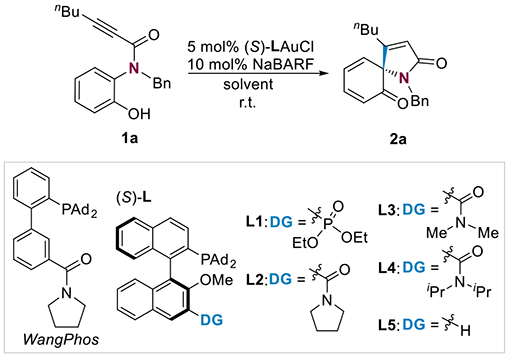
| |||||
|---|---|---|---|---|---|
|
| |||||
| Entry | Ligand | Solvent | Time | Yield[a] | ee[b] |
| 1 | WangPhos | DCM | <15 min | 90% | NA |
| 2 | (S)-L1 | DCM | 2 h | 91% | −26% |
| 3 | (S)-L2 | DCM | <15 min | 95% | 94% |
| 4 | (S)-L3 | DCM | <15 min | 86% | 94% |
| 5 | (S)-L4 | DCM | <15 min | 90% | 98% |
| 6 | (S)-L5 | DCM | 30 min | 87% | −64% |
| 7 | (S)-L4 | THF | 24 h | 14% | NA |
| 8 | (S)-L4 | PhCF3 | <15min | 83% | 97% |
| 9 | (S)-L4 | toluene | 30 min | 95% | 92% |
| 10 | (S)-L4 | DCE | <15 min | 89% | 98% |
| 11 | (S)-L4[c] | DCM | <15 min | 88% | 98% |
| 12 | (S)-L4[d] | DCM | <15 min | 89% | 98% |
Determined by 1H NMR using diethyl phthalate as the internal standard.
Determined by chiral HPLC.
5 mol% AgNTf2 was used instead of 10 mol% NaBARF.
5 mol% AgSbF6 was used instead of 10 mol% NaBARF.
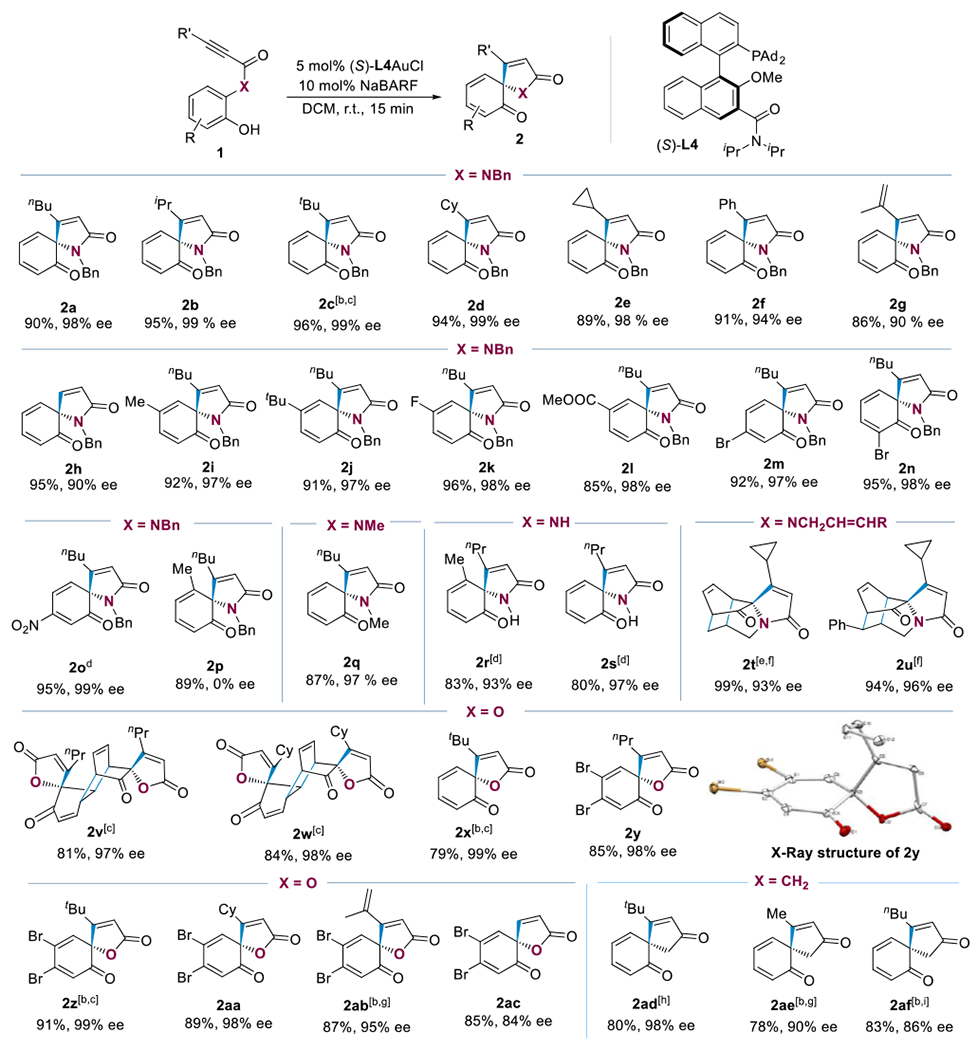
With the optimized reaction condition in hand (Table 1, entry 5), we carried out the scope study (Table 2). The reaction readily tolerates various alkyne substituents including iPr, tBu, different ring structures, and unsaturated groups. The spirocyclization products 2a-2g were isolated in good to excellent yields and exhibited excellent enantiomeric excesses – in most cases ≥98%. The reaction forming 2c required a longer reaction time due to steric hindrance caused by the tBu substituent. Despite this, the product was formed with near enantiopurity (i.e., ee = 99%). The propiolamide substrate (i.e., R1 = H) also reacted smoothly, affording 2h in 95% yield and with 90% ee. The reaction also showed excellent tolerance of different substituents on the phenol ring including Me, tBu, F, Br, CO2Me, and strongly electron-withdrawing NO2. The nitro case is remarkable in asymmetric phenol dearomatization and confirms the versatility of the cooperative catalysis. All of the corresponding dearomatized cyclization products (2i-2o) were obtained in good yields and high enantioselectivities (97%-99% ee). Interestingly, the reaction forming 2p, which has a methyl ortho to the amide group, was not enantioselective at all. This is likely due to the existence of racemic non-interconvertible atropisomers of the substrate 1p at the ambient reaction temperature, which eventually led to the equal ratio of opposite enantiomeric products. The restricted rotation of the phenyl-N bond in this case is avoided by replacing the N-benzyl group with a hydrogen atom. As such, the spirocyclization led to the desired 2r in 93% ee. Moreover, removing the ortho-methyl group led to 2s with an NH group with a 97% ee. The N-benzyl group was also readily replaced by a N-Me group without consequence in the case of 2q. When an N-allyl group was present in the reaction, the reaction proceeded to completion in 15 minutes under the standard reaction conditions, and the expected spirobicyclic product was observed by NMR measurement (see the Supporting Information for details). However, attempts to obtain the pure product were unsuccessful. As it undergoes the intramolecular Diels-Alder reaction (IMDA) [31] during workup and purification to form the adduct 2t featuring a tetracyclic bridged skeleton. To this end, we performed the reaction at 40 °C for 12 hours. The starting material was fully converted to 2t, which was isolated in 99% yield with 93% ee. Additionally, the tetracyclic product 2u was obtained in 96% ee after the substrate containing a N-cinnamyl group was subjected to gold catalysis. In this case, the IMDA was faster as the reaction was performed at ambient temperature. In light of the afore-discussed scope limitations in the literature reports, it is remarkable that this chemistry tolerates various substituents at all four available positions on the benzene ring and of highly electron-withdrawing characteristics.
Table 2.
Reaction Scope[a]
|
Standard reaction conditions: 5 mol% (S)-L4AuCl, 10 mol% NaBARF, DCM (0.05 M), room temperature, 15 min.
5 mol% AgNTf2 was used instead of 10 mol % NaBARF.
24 h.
2 h.
40 °C.
12 h.
−40 °C.
10 mol % (S)-L4AuCl and 10 mol % AgNTf2 was used, 24 h.
−78 °C, 48 h.
We were delighted to discover that the substrate scope can be expanded to 2-hydroxyphenyl ynoates (X = O). For example, the reaction of 2-hydroxyphenyl hex-2-ynoate (1v) resulted in the formation of the dimeric product 2v in 81% yield and with 97% ee. The facile dimerization of the initially-formed spirocyclohexadienone-butenolide via the D-A reaction is consistent with the literature report.[36–38] The structure of 2v was confirmed by X-ray diffraction studies.[39] Changing the alkyne terminal nPr group to sterically bulkier cyclohexyl group also resulted in the formation of the dimer product 2w. However, a tBu group does suppress the D-A reaction, and the spirodienone-butenolide 2x was isolated in a 79% yield. In all these cases, the reactions were highly enantioselective, with ee values ranging from 97% to 99%. The facile D-A dimerization can be prevented by the presence of 4,5-dibromo substitutions on the substrate phenol ring, and several spirocyclohexadienone-butenolides, i.e., 2y-2ab, were obtained in good to excellent yields (85%-94%) and with excellent enantioselectivities (95%-98%). In the case of the propiolate substrate containing a terminal alkyne, the enantioselectivity is moderate (ee = 84%). The absolute configuration of the chiral quaternary carbon center of the dearomatization product was determined to be R by analyzing the X-ray structure of 2y.[39]
Moreover, o-hydroxybenzyl ynones (i.e., X = CH2) are also suitable substrates for this asymmetric dearomatization chemistry.[40–43] In the case of 2ad possessing a bulky t-butyl group at the alkyne terminus, the gold catalysis is efficient and highly enantioselective, affording the spirodienone-cyclopentenone structure in 80% yield and with 98% ee. For the cases with a methyl (2ae) and an n-butyl (2af) group instead, the reactions were run at cryogenic conditions, and good to excellent enantioselectivities (90% and 86%, respectively) were achieved.
This asymmetric gold catalysis can be readily scaled up (Scheme 2). For example, 2m was prepared in 1.40 g (91% isolated yield and 99% ee) by using only 1 mol % catalyst loading. We investigated the preliminary synthetic utilities of these chiral spiro structures. For example, 2m underwent the Suzuki coupling to afford the phenylated product 3 in 93% yield and 98% ee. The dimer 2v can undergo retro-D-A reaction to generate the spirobicycle cyclohexadienone-butenolide in situ upon heating at 180 °C in mesitylene in a sealed tube, which was trapped in situ by norbornene to afford the adduct 4 in 60% yield. The ee of 4 dropped to 75%, which might be due to a racemization process at a high temperature. In a similar vein, 2y reacts smoothly with norbornene at room temperature, generating 5 in 87% yield and 99% ee, the structures of which were confirmed by X-ray crystallographic.[39] Likewise, its D-A reaction with styrene was uneventful, affording 6 in 91% yield and 98% ee and with an endo/exo ratio of 13:1.
Scheme 2.

Gram-scale synthesis and preliminary synthetic applications.
In summary, we report an efficient and highly enantioselective gold(I)-catalyzed dearomatization of phenols. The chiral binaphthylphosphine ligand featuring a 3’-N,N-diisopropylcarbamoyl group enables asymmetric gold-ligand cooperation in a 5-endo-dig cyclization via the formation of H-bonding interaction with the phenolic proton. The ligand rigidity and the optimal positioning of ligand basic group and Au enable this asymmetric phenol dearomatization to exhibit unprecedented generality in reaction scope. In contrast to prior arts, this chemistry tolerates substitution at all four remaining benzene positions and can accommodate the strongly electron-withdrawing nitro group, which is rare. It allows carbon-based groups of varying steric bulk including tert-butyl at the alkyne terminus. Moreover, the connecting group between the phenol moiety and the alkynylacyl group can be variously substituted amino group, oxygen, and methylene.
Supplementary Material
Acknowledgements
L.Z. thanks NIH (R35GM139640) for financial support and NSF MRI-1920299 for the acquisition of two Bruker NMR instruments. We thank Dr. Rachel Behrens from UCSB and Dr. Felix Grun from UCI for their help with HRMS analysis. We thank Dr. Guang Wu for his help with the X-Ray crystal measurement. The research reported here made use of shared facilities of the UC Santa Barbara MRSEC (NSF DMR-1720256), a member of the Materials Research Facilities Network (www.mrfn.org).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Zheng C, You SL, ACS Cent. Sci 2021, 7, 432–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cheng YZ, Feng Z, Zhang X, You SL, Chem. Soc. Rev 2022, 51, 2145–2170. [DOI] [PubMed] [Google Scholar]
- [3].Pouységu L, Deffieux D, Quideau S, Tetrahedron 2010, 66, 2235–2261. [Google Scholar]
- [4].Roche SP, Porco JA Jr., Angew. Chem., Int. Ed 2011, 50, 4068–4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zheng C, You SL, Nat. Prod. Rep 2019, 36, 1589–1605. [DOI] [PubMed] [Google Scholar]
- [6].Lovering F, Bikker J, Humblet C, J. Med. Chem 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]
- [7].Sun W, Li G, Hong L, Wang R, Org. Biomol. Chem 2016, 14, 2164–2176. [DOI] [PubMed] [Google Scholar]
- [8].Wu WT, Zhang L, You SL, Chem. Soc. Rev 2016, 45, 1570–1580. [DOI] [PubMed] [Google Scholar]
- [9].Quideau S, Pouységu L, Deffieux D, Synlett 2008, 2008, 467–495. [Google Scholar]
- [10].Nemoto T, Ishige Y, Yoshida M, Kohno Y, Kanematsu M, Hamada Y, Org. Lett 2010, 12, 5020–5023. [DOI] [PubMed] [Google Scholar]
- [11].Yang L, Zheng H, Luo L, Nan J, Liu J, Wang Y, Luan X, J. Am. Chem. Soc 2015, 137, 4876–4879. [DOI] [PubMed] [Google Scholar]
- [12].Lee E, Hwang Y, Kim YB, Kim D, Chang S, J. Am. Chem. Soc 2021, 143, 6363–6369. [DOI] [PubMed] [Google Scholar]
- [13].Xu RQ, Gu Q, Wu WT, Zhao ZA, You SL, J. Am. Chem. Soc 2014, 136, 15469–15472. [DOI] [PubMed] [Google Scholar]
- [14].Rousseaux S, García-Fortanet J, Del Aguila Sanchez MA, Buchwald SL, J. Am. Chem. Soc 2011, 133, 9282–9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Phipps RJ, Toste FD, J. Am. Chem. Soc 2013, 135, 1268–1271. [DOI] [PubMed] [Google Scholar]
- [16].Uyanik M, Yasui T, Ishihara K, 2013, 52, 9215–9218. [DOI] [PubMed] [Google Scholar]
- [17].Bosset C, Coffinier R, Peixoto PA, El Assal M, Miqueu K, Sotiropoulos JM, Pouysegu L, Quideau S, Angew. Chem., Int. Ed 2014, 53, 9860–9864. [DOI] [PubMed] [Google Scholar]
- [18].Baker Dockrey SA, Lukowski AL, Becker MR, Narayan ARH, Nat. Chem 2018, 10, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhu J, Grigoriadis NP, Lee JP, Porco JA, J. Am. Chem. Soc 2005, 127, 9342–9343. [DOI] [PubMed] [Google Scholar]
- [20].D’Arcy TD, Elsegood MRJ, Buckley BR, Angew. Chem., Int. Ed 2022, 61, e202205278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dong S, Zhu J, Porco JA Jr., J. Am. Chem. Soc 2008, 130, 2738–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Du K, Guo P, Chen Y, Cao Z, Wang Z, Tang W, Angew. Chem., Int. Ed 2015, 54, 3033–3037. [DOI] [PubMed] [Google Scholar]
- [23].Chu W-D, Liang T-T, Ni H, Dong Z-H, Shao Z, Liu Y, He C-Y, Bai R, Liu Q-Z, Org. Lett 2022, 24, 4865–4870. [DOI] [PubMed] [Google Scholar]
- [24].Shao L, Hu X-P, Chem. Commun 2017, 53, 8192–8195. [DOI] [PubMed] [Google Scholar]
- [25].Cheng X, Zhang L, CCS Chem. 2021, 3, 1989–2002. [Google Scholar]
- [26].Wang Y, Wang Z, Li Y, Wu G, Cao Z, Zhang L, Nat. Commun 2014, 5, 3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wang Z, Nicolini C, Hervieu C, Wong YF, Zanoni G, Zhang L, J. Am. Chem. Soc 2017, 139, 16064–16067. [DOI] [PubMed] [Google Scholar]
- [28].Zhao K, Kohnke P, Yang Z, Cheng X, You SL, Zhang L, Angew. Chem., Int. Ed 2022, 61, e202207518. [DOI] [PubMed] [Google Scholar]
- [29].Nechaev AA, Van Hecke K, Zaman M, Kashtanov S, Ungur L, Pereshivko OP, Peshkov VA, Van der Eycken EV, J. Org. Chem 2018, 83, 8170–8182. [DOI] [PubMed] [Google Scholar]
- [30].He Y, Wu D, Li Z, Robeyns K, Van Meervelt L, Van der Eycken EV, Org. Biomol. Chem 2019, 17, 6284–6292. [DOI] [PubMed] [Google Scholar]
- [31].He Y, Liu Z, Wu D, Li Z, Robeyns K, Van Meervelt L, Van der Eycken EV, Org. Lett 2019, 21, 4469–4474. [DOI] [PubMed] [Google Scholar]
- [32].Okitsu T, Nakazawa D, Kobayashi A, Mizohata M, In Y, Ishida T, Wada A, Synlett 2010, 203–206. [Google Scholar]
- [33].Sahoo H, Grandhi GS, Ramakrishna I, Baidya M, Org. Biomol. Chem 2019, 17, 10163–10166. [DOI] [PubMed] [Google Scholar]
- [34].Nair AM, Halder I, Khan S, Volla CMR, Adv. Synth. Catal 2020, 362, 224–229. [Google Scholar]
- [35].Recchi AMS, Rosa PHP, Back DF, Zeni G, Org. Biomol. Chem 2020, 18, 3544–3551. [DOI] [PubMed] [Google Scholar]
- [36].Liu XY, Qin Y, Nat. Prod. Rep 2017, 34, 1044–1050. [DOI] [PubMed] [Google Scholar]
- [37].Liu L, Han Y, Xiao J, Li L, Guo L, Jiang X, Kong L, Che Y, J. Nat. Prod 2016, 79, 2616–2623. [DOI] [PubMed] [Google Scholar]
- [38].Liao CC, Peddinti RK, Acc. Chem. Res 2002, 35, 856–866. [DOI] [PubMed] [Google Scholar]
- [39].The CCDC depository numbers: 2258971 (2v), 2258972 (2y), and 2258973 (5).
- [40].Sahoo SR, Das B, Sarkar D, Henkel F, Reuter H, Eur. J. Org. Chem 2020, 2020, 891–896. [Google Scholar]
- [41].Tcyrulnikov S, Curto JM, Gilmartin PH, Kozlowski MC, J. Org. Chem 2018, 83, 12207–12212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Clarke AK, Liddon JTR, Cuthbertson JD, Taylor RJK, Unsworth WP, Org. Biomol. Chem 2017, 15, 233–245. [DOI] [PubMed] [Google Scholar]
- [43].Clarke AK, James MJ, O’Brien P, Taylor RJK, Unsworth WP, Angew. Chem., Int. Ed 2016, 55, 13798–13802. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.