

Summary
Cells respond to intrinsic and extrinsic stresses by reducing global protein synthesis and activating gene programs necessary for survival. Here, we show the integrated stress response is driven by the non-canonical cap-binding protein eIF3d which acts as a critical effector to control core stress response orchestrators, the translation factor eIF2a and the transcription factor ATF4. We find that during persistent stress, eIF3d activates translation of the kinase GCN2, inducing eIF2ɑ phosphorylation and inhibiting general protein synthesis. In parallel, eIF3d upregulates the m6A demethylase ALKBH5 to drive 5′ UTR-specific demethylation of stress response genes, including ATF4. Ultimately, this cascade converges on ATF4 expression by increasing mRNA engagement of translation machinery and enhancing ribosome bypass of upstream open reading frames. Our results reveal that eIF3d acts in a life-or-death decision point during chronic stress and uncover a synergistic signaling mechanism in which translational cascades complement transcriptional amplification to control essential cellular processes.
Graphical Abstract
eTOC
Mukhopadhyay, et al. demonstrate that sustained ER stress activates a translation adaptation program mediated by the cap-binding protein eIF3d. eIF3d controls the persistent integrated stress response, including expression of critical transcription, translation, and RNA methylation regulators essential for cellular survival.
Introduction
Diverse cellular stresses activate a conserved adaptive pathway called the integrated stress response (ISR)1. The induction of the ISR converges on phosphorylation of eIF2a by a family of four stress-responsive kinases, GCN2, PERK, PKR, and HRI1,2. eIF2ɑ phosphorylation depletes the eIF2ɑ-GTP-methionyl transfer tRNA ternary complex and shuts off general protein synthesis3. Simultaneously, the ISR is accompanied with selective translation of proteins required for return to homeostasis. One example of translational reprogramming comes from studies of the transcription factor ATF4, which is the primary effector upregulated during the ISR to activate a gene expression response that mediates cellular recovery4. Human ATF4 contains two upstream open reading frames (uORFs), which are preferentially engaged by initiating ribosomes under homeostatic conditions, thus blocking translation elongation from the coding sequence. Upon eIF2ɑ phosphorylation by induction of the ISR, low levels of active ternary complex cause ribosomes to bypass the uORFs, translate the ATF4 coding sequence, and mount a pro-survival response5.
While the ISR activates a protective gene program, this occurs simultaneously with expression of pro-apoptotic genes6, and therefore the balance of appropriate gene expression is critical for the cellular decision to adapt or die. In particular, chronic stresses are linked to human pathologies, but how organisms sustain a protective response is poorly understood. For example, long-term ER stress is associated with numerous diseases including diabetes, autoimmunity, neurodegenerative disorders, and cancer1. In the case of diabetes, obesity is linked to chronic ER stress and downstream insulin resistance7, and treatment of mouse models with compounds that block the ISR result in pancreatic β-cell survival8. Paradoxically, while inappropriate chronic activation of the ISR kinases can lead to memory impairment and neuronal cell death, enhancement of the ISR is beneficial in certain mouse models of neurodegeneration, including amyotrophic lateral sclerosis and multiple sclerosis9-11. In addition, both inhibition and activation of the ISR has been shown to block tumor growth12,13, further highlighting the dual roles of the ISR in cell survival versus death. Therefore, understanding the molecular pathways that drive distinct gene programs during the chronic ISR is essential for understanding cellular fates.
However, beyond the critical role of eIF2ɑ phosphorylation, it remains unclear how other mechanisms of mRNA translation contribute to cellular adaptation during the ISR, and how these modes allow for a response specific to stimulus, temporal length, or severity of stress. Eukaryotic translation initiation factor 3 (eIF3a–m) is a 13-subunit complex that functions in numerous steps of global and gene-specific translation. eIF3 regulates general protein synthesis by binding to the small ribosomal subunit and coordinating pre-initiation complex formation on mRNAs. In addition, for a subset of transcripts including membrane-associated genes, eIF3 is retained on the 80S ribosome during early steps of elongation14-17. eIF3 also regulates transcript-specific translation, predominantly through direct targeting of mRNAs as an RNA-binding complex18-21. Notably, the eIF3d subunit of the eIF3 complex is a 5′ cap-binding protein that supports non-canonical translation when the cap-binding protein eukaryotic initiation factor 4E (eIF4E) is inactivated22. eIF3d is phosphorylated under homeostatic conditions, and loss of modification upon metabolic stress increases its cap-binding activity and drives a translation adaptation response during chronic glucose deprivation23. Beyond its role in supporting protein synthesis during eIF4E inhibition, it is unknown if eIF3d is repurposed for other translation adaptation pathways, such as when eIF2ɑ is turned off. Here we report that during chronic ER stress, activation of eIF3d drives translation reprogramming needed for cellular survival. eIF3d controls the core ISR orchestrators, the translation factor eIF2ɑ and the transcription factor ATF4, along with mRNA modification, through the m6A demethylation enzyme ALKBH5. By targeting transcription, translation, and epitranscriptome-based regulation of gene expression, eIF3d supports a pro-survival response during the persistent ISR.
Results
Regulation of eIF3d phosphorylation is critical for the persistent integrated stress response
eIF3d is critical for translation adaptation during sustained shutoff of the canonical cap-binding protein eIF4E, however its role in other adaptive translational gene programs is not known. To determine if eIF3d is involved in stress-responsive translation reprogramming when eIF2ɑ is inhibited, we examined changes to eIF3d activity upon activation of the integrated stress response. Induction of sustained, but not acute, ER stress with thapsigargin (Tg) or tunicamycin (Tn) led to loss of phosphorylation of eIF3d (Fig. 1A, Fig S1A). Loss of modification was temporally mirrored by increased cap-binding to the known eIF3d target mRNA JUN22 (Fig. 1B). To examine the impact of regulation of eIF3d on the cellular response to chronic ER stress, we genetically engineered human embryonic kidney (HEK) 293T cells to generate phosphomimetic (S528D/S529D) eIF3d mutations (Fig. S1B). In agreement to the requirement of active eIF3d for efficient translation initiation complex formation on JUN mRNA 22,23, the phosphomimetic-edited cells cannot support association of the JUN mRNA with polysomes (Fig. S1C).
Figure 1. Regulation of eIF3d phosphorylation is critical for the persistent integrated stress response.

(A) Phosphorylation of eIF3 subunits upon integrated stress response (ISR) induction with DMSO control (veh.) or thapsigargin (Tg). Phosphorylation was detected by [32P]-orthophosphate labeling of HEK293T cells, immunoprecipitation of the eIF3 complex, and resolution of subunits by SDS-polyacrylamide gel electrophoresis (PAGE) gel. Coomassie staining of the gel is shown as a loading control (Coom.). Quantification (Quant.) of orthophosphate signal normalized to Coomassie gel lane intensity is represented as fold change to cells at 0 h. (B) eIF3d binding to the 5′ cap of JUN mRNA upon ISR induction. eIF3d cap-binding is quantified as levels of JUN bound to eIF3d in HIV-1 PR-treated eIF3d immunoprecipitation samples compared to total input JUN RNA. Cap-binding is normalized to untreated (0 h) cells and represented as the mean ± s.d. of three independent experiments. (C) Representative images of cell morphology in HEK293T cells edited to express wild-type (WT) or phosphomimetic eIF3d (S528D/S529D) upon treatment with Tg or DMSO control (veh). (D) Volcano plot comparing fold change in transcript expression levels with adjusted P values in cells expressing phosphomimetic versus WT eIF3d upon ISR induction by 16 h treatment with Tg. (E) Gene ontology analysis of mRNAs downregulated in cells expressing phosphomimetic versus WT eIF3d upon ISR induction. (F) Immunoblot of ISR signaling cascade activation in eIF3d WT and phosphomimetic cells. Phos-tag gel allows electrophoretic separation of phosphorylated (p) and unmodified (0) eIF2ɑ protein. Tg treatment in (D – F) was performed for 16 h. The results of (A), (C) and (F) are representative of three independent experiments. See also Figures S1, S2, and Table S1.
Cells expressing phosphomimetic eIF3d undergo increased cell death upon sustained treatment with Tg, without changes at shorter timepoints of ER stress (Fig. 1C, S1D). Furthermore, phosphomimetic eIF3d-expressing cells are unable to activate partial protein synthesis recovery that is associated with appropriate switching to a sustained ISR gene program (Fig. S1E)24. This is accompanied by inhibited expression of genes required for the unfolded protein response (Fig. 1D,E, S2A, Table S1). Strikingly, both core mediators of the integrated stress response are dysregulated, with induction of the transcription factor ATF4 and phosphorylation of eIF2ɑ inhibited in phosphomimetic eIF3d-expressing cells (Fig. 1F). In agreement, these cells are unable to upregulate genes transcriptionally regulated by ATF4, including those that act in negative feedback signaling to restore homeostasis, such as TRIB3, PPP1R15A (GADD34), and ATF3 (Fig. S2B,C)25-27. eIF3d provides precise temporal control of the ISR, as ATF4 and eIF2ɑ remain appropriately regulated in phosphomimetic eIF3d-expressing cells at shorter ER stress timepoints (Fig. S2D). Thus, activation of eIF3d cap-binding activity by loss of phosphorylation is essential for induction of the integrated stress response during chronic ER stress.
eIF3d controls ISR-dependent upregulation of the m6A demethylase ALKBH5.
The persistent ISR is characterized by translation reprogramming that is independent of the canonical eIF4E-mediated cap-dependent translation initiation mechanism24. To determine the mechanistic basis of how non-canonical eIF3d-mediated cap-dependent translation contributes to the persistent ISR, we performed eIF3dTEV Subunit-Seq in HEK293T cells after induction of chronic ER stress. eIF3dTEV Subunit-Seq is a crosslinking and immunoprecipitation approach which allows isolation of mRNAs bound at the 5′ cap structure by the eIF3d cap-binding domain (Fig. S3A)23. We identified 564 genes that are bound at the 5′ cap structure by eIF3d during chronic ER stress (Fig 2A, Table S2). The majority of genes bound by eIF3d are distinct during chronic ER stress versus glucose deprivation (Fig. 2B), suggesting eIF3d selects different mRNAs dependent on the nature of the upstream stimulus. Gene ontology analysis revealed eIF3d targets are enriched in RNA processing functions, in agreement to widespread changes to rRNA processing, mRNA stability, and RNA modification induced during the ISR28-30 (Fig. S3B). Additionally, eIF3d targets are involved in cell stress response pathways, including PPP1R15A, which is itself an ISR regulator31; FMN2, which is activated in response to hypoxia and DNA damage32; PPP3R1, which regulates autophagy33; and ABL1, which mediates oxidative stress adaptation34 (Fig. S3B,C). We validated that identified targets display increased eIF3d cap-binding upon sustained Tg treatment in a phosphorylation-regulated manner, as this phenotype is abolished in phosphomimetic eIF3d-expressing cells (Fig. S3D).
Figure 2. eIF3d controls ISR-dependent upregulation of the m6A demethylase ALKBH5.

(A) Volcano plot comparing the eIF3dTEV Subunit-IP/Input log2(fold enrichment) versus – log10(adjusted P value) in cells with ISR induced by treatment with thapsigargin. Tg, 16 h thapsigargin treatment. Targets of eIF3d cap-binding activity are colored blue. (B) Heat map of eIF3d target mRNAs in cells with ISR induction versus glucose deprivation23. Genes that are not bound by eIF3d in the given condition are colored gray. (C) Read mapping to ALKBH5 from eIF3dTEV Subunit-Seq during ISR induction with thapsigargin. The annotated y-axis maximum is equivalent for all samples. The transcript architecture is represented by the schematics: 5′ and 3′ UTRs (thin line), coding region (thick line). In, input; eIF3dTEV-IP, anti-HA eIF3d immunoprecipitant samples after TEV protease cleavage; Tg, 16 h thapsigargin treatment. (D) eIF3d does not bind to the 5′ cap of FTO mRNA upon ISR induction in HEK293T. Cap-binding is normalized to eIF3d–ALKBH5 cap-binding, as calculated by levels of ALKBH5 enriched by immunoprecipitation of the eIF3d cap-binding domain compared to total input ALKBH5 RNA. PSMB6 is a negative control mRNA that is not bound by eIF3d23. (E) Specificity of eIF3d binding to the ALKBH5 5′ cap structure in cells. Results are normalized to samples that are not treated with an on-bead competitor ligand wash. m7G, competitor cap analog m7GpppG; G, unmethylated GpppG. (F) eIF3d binding to the 5′ cap of ALKBH5 mRNA in HEK293T upon ISR induction with thapsigargin (Tg). Cap-binding is normalized to untreated (0 h) cells. (G) eIF3d binding to the 5′ cap of ALKBH5 mRNA upon ISR induction in HEK293T cells expressing wild-type (WT) and phosphomimetic (DD) eIF3d. Cap-binding is normalized to untreated cells. Results in (D–G) are represented as the mean ± s.d. of three independent experiments. (H) Immunoblot of ALKBH5 levels in eIF3d cell lines upon Tg treatment. The results are representative of three independent experiments. See also Figure S3 and Table S2.
How does eIF3d-mediated translational reprogramming promote cell survival during chronic ISR? Within the eIF3d targets involved in RNA processing, we identified the m6A demethylase enzyme ALKBH5 (Fig. 2C), which was previously found to be induced in hypoxia and amino-acid starvation stresses35,36. N6-methyladenosine (m6A) is the most prevalent post-transcriptional internal RNA modification and can be reversed by the demethylase enzymes ALKBH5 and FTO in response to cellular context37-39. As eIF3d recognizes the 5′ cap of ALKBH5 but not FTO (Fig. 2D), we hypothesized that distinct demethylase regulation could provide a potential mechanism to change m6A modification and alter target-specific RNA function and gene expression during the ISR. Using purified components, we validated that eIF3d crosslinks to the 32P-labeled cap structure of the ALKBH5 5′ UTR in vitro (Fig. S4A). This interaction is specific to the 5′ methylated cap structure, as eIF3d–cap binding can be competed away by methylated (m7GpppG) but not unmethylated (GpppG) cap structure (Fig. S4A), and this selectivity is also conserved in cells (Fig. 2E). In support of regulation by eIF3d during the ISR, eIF3d binding to the ALKBH5 5′ cap structure increased upon chronic treatment with Tg (Fig. 2F), and this is abolished in cells expressing phosphomimetic eIF3d (Fig. 2G). Analysis of ALKBH5 mRNA association with translation complexes showed that ALKBH5 shifts from monosome to highly translating polysome fractions in WT eIF3d-expressing cells upon Tg treatment (Fig. S4B). In contrast, ALKBH5 mRNA is constitutively maintained in non-translating free RNA fractions in phosphomimetic eIF3d-expressing cells (Fig. S4B). In agreement, ALKBH5 protein levels are not induced in phosphomimetic eIF3d-expressing cells upon induction of chronic ER stress (Fig. 2H). Thus, the persistent ISR upregulates expression of ALKBH5 through an eIF3d-directed translation mechanism.
Stress-dependent translational control of ALKBH5 requires a cis-acting 5′ UTR stem loop
Prior studies of eIF3d-specialized translational control of the JUN and RPTOR mRNAs revealed that regulation requires initial recruitment of the eIF3 complex to secondary structure motifs in the 5′ UTR of the target mRNA, which leads to activation of eIF3d cap-binding through allosteric regulation22,23. Using purified recombinant eIF3 complex and in vitro transcribed RNA, we demonstrated that the eIF3 complex binds directly to the 5′ UTR of ALKBH5 by native gel shift analysis (Fig. S4C). By analyzing eIF3 4-thiouridine PAR-CLIP datasets22, we identified a region in the ALKBH5 5′ UTR that crosslinks to the eIF3 complex (Fig. 3A). Deletion of this site blocks translation upregulation of a luciferase reporter mRNA containing the ALKBH5 5′ UTR during sustained Tg treatment (Fig. 3B), confirming that eIF3 binding to the 5′ UTR is critical for its translational control. To examine how RNA secondary structure contributes to specific recognition by the eIF3 complex, we performed selective 2′ hydroxyl acylation analyzed by primer extension (SHAPE)40 analysis of the ALKBH5 5′ UTR. We identified a stem loop (sl3) in the eIF3 interaction site and validated the SHAPE-directed RNA secondary structure prediction using a deletion and map approach (Fig. 3C). Deletion of sl3 or disruption of the stem structure by mutation blocks binding of the eIF3 complex to the ALKBH5 5′ UTR (Fig. 3D,E). In contrast, eIF3 binding can be restored by making compensatory mutations to restore secondary structure, and is unaffected by mutations to the top loop (Fig. 3E). To measure mRNA translation independently of the nuclear and organelle-driven gene regulation changes induced by the ISR, we developed a cell-free protein synthesis system that recapitulates stress-induced translational control in vitro, including shutoff of general protein synthesis (Fig. S4D). ALKBH5 5′ UTR luciferase reporter mRNAs are translationally induced in Tg-treated extracts, while disruption of sl3 abolishes this upregulation (Fig. 3F). Therefore, the mechanism by which ALKBH5 engages ISR/eIF3d-mediated translation requires eIF3–RNA recruitment.
Figure 3. Stress-dependent translational control of ALKBH5 requires a cis-acting 5′ UTR stem loop.

(A) eIF3 PAR-CLIP cluster in the 5′ UTR of the ALKBH5 mRNA. (B) Luciferase activity in cells transfected with ALKBH5 5′ UTR mutant. Results are normalized to untreated cells. Δpar, deletion of eIF3 PAR-CLIP peak. (C) SHAPE-based secondary structure of the ALKBH5 WT 5′ UTR (nt 79-345) or with the eIF3 binding site (Δsl3) deleted. Nucleotides are color coded by SHAPE reactivity, with higher reactivity correlating with single-stranded probability. The results are representative of two independent experiments. Black line, main eIF3 PAR-CLIP peak; gray line, extended peak. (D) Schematic of ALKBH5 5′ UTR luciferase (Luc) reporter constructs. sl3, eIF3-binding stem loop identified by PAR-CLIP analysis. (E) eIF3 RNA-binding to ALKBH55′ UTR mutants during ISR induction. RNA-binding is normalized to eIF3 binding to WT ALKBH 5′ UTR. Δ, deletion of sl3. (F) Luciferase activity in vitro mediated by ALKBH5 5′ UTR mutants. Results are normalized to WT ALKBH5 5′ UTR luciferase activity in in vitro translation extracts made from untreated HEK293T cells. Results in (B), (E) and (F) are represented as the mean ± s.d. of three independent experiments. See also Figure S4.
eIF3d-specialized translational control remodels the m6A epitranscriptome during the persistent ISR
The functional role of ALKBH5 as a m6A demethylase38 suggests its ISR/eIF3d-mediated upregulation could contribute to dynamic reversal of RNA modification during chronic ER stress30,35. To examine if ISR/eIF3d-mediated translational control of ALKBH5 drives stress-specific changes to mRNA methylation, we performed m6A enhanced version of the crosslinking and immunoprecipitation (m6A-eCLIP)41,42 in WT and phosphomimetic eIF3d-expressing HEK293T cells upon sustained treatment with Tg. m6A-eCLIP allows for transcriptome-wide identification of m6A sites by taking advantage of crosslinking-induced transitions or truncations adjacent to the methylated residue. We identified 6590 m6A sites across cellular conditions, with site assignment supported by motif analysis showing enrichment of the DRACH (D=A,U,G;R=A,G;H=A,U,C) m6A consensus sequence43 (Fig. 4A,B, Table S3). Notably, in support of increased ISR/eIF3d-regulated ALKBH5 demethylase activity, on a global level, we identified more m6A sites in Tg-treated phosphomimetic and untreated WT eIF3d-expressing cells (5061 and 3001, respectively) compared to Tg-treated WT cells (1575). This is coupled with a higher fraction of m6A sites conserved between the former cells (Fig. 4A, S5A). Thus, phosphomimetic eIF3d-expressing cells phenocopy the m6A methylome found in untreated WT cells, and ISR/eIF3d-mediated translational control supports dynamic m6A regulation during chronic ER stress.
Figure 4. eIF3d-specialized translational control remodels the m6A epitranscriptome during the persistent ISR.

(A) Venn diagram of overlapping m6A-modified transcripts in wild-type (WT) and phosphomimetic (DD) eIF3d expressing HEK293T. Tg, 16h of thapsigargin treatment. (B) Frequency plot of the canonical m6A DRACH motif in m6A-eCLIP peaks in eIF3d cell lines. (C) Distribution of m6A sites in transcript regions. eIF3d/ISR-regulated m6A sites (gray) are those that exhibit 1.5-fold more methylation in both untreated WT and Tg-treated eIF3d phosphomimetic cell lines when compared to Tg-treated WT eIF3d cells. Blue, sites not regulated by eIF3d/ISR signaling; UTR, untranslated region; CDS, coding sequence. (D) Gene ontology analysis of transcripts with eIF3d/ISR-downregulated m6A sites. (E) Levels of m6A in transcripts identified by m6A-eCLIP, measured by m6A RNA immunoprecipitation (RIP) and quantitative reverse transcription polymerase chain reaction (qRT-PCR). (F) Read mapping to ATF4 from m6A-eCLIP in eIF3d mutant cell lines. The annotated y-axis maximum is equivalent for all samples, and the regulated m6A site as determined by crosslinking-induced truncation sites analysis is annotated by a triangle. (G) Levels of m6A in ATF4 mRNA, measured by m6A-RIP and qRT-PCR. (H) Representative immunoblot showing reduced expression of ALKBH5 and ATF4 upon ISR induction in cells with dysregulated eIF3d-specialized translational control of ALKBH5. Δsl3, cells with the eIF3 binding site in the ALKBH5 5′ UTR deleted (Δsl3). The results are representative of three independent experiments. (I) Levels of m6A in ATF4 mRNA in cells with dysregulated eIF3d-specialized translational control of ALKBH5. Results in (E), (G) and (I) are normalized to untreated WT cells and represent the mean ± s.d. of three independent experiments. See also Figure S5 and Table S3.
In mRNAs, m6A modifications are enriched in 3′ UTRs and stop codons43,44. However, regional localization of m6A modification within a transcript can direct distinct outcomes, indicating that cellular regulation of specific sites could be a mechanism for gene regulation19,45-47. As expected, we found that m6A sites are preferentially installed around the stop codon and in the 3′ UTR, and this global regional distribution is not regulated by eIF3d mutant or cell treatment (Fig. S5B). In contrast, analysis of sites that are specifically demethylated through an ISR/eIF3d-dependent mechanism display a striking enrichment in the 5′ UTR (Fig. 4C). In support that regulated demethylation supports establishment of the persistent ISR, transcripts carrying ISR/eIF3d-regulated m6A sites are enriched in biological functions involved in cellular stresses (Fig. 4D,E). Notably, we identified three m6A sites in the transcription factor ATF4, including one in the 5′ UTR at the transcription start site48 that undergoes ISR/eIF3d-dependent demethylation (Fig. 4F). We confirmed quantitative changes to m6A levels on the ATF4 mRNA in WT versus phosphomimetic eIF3d-expressing cells by m6A immunoprecipitation and qRT-PCR (Fig. 4G). To examine the functional consequence of regulated ATF4 methylation, we engineered HEK293T cells to decouple ALKBH5 translational control from the eIF3d/ISR response (ALKBH5-Δsl3) (Fig. S5C). In agreement with its critical role in engaging eIF3-mediated translation, genomic deletion of sl3 blocks ALKBH5 protein upregulation during sustained ER stress (Fig. 4H), without changes to mRNA stability (Fig. S5D). ALKBH5-Δsl3 cells exhibit shared dysregulation of m6A to phosphomimetic eIF3d-expressing cells (Fig. S5E), including the inability to demethylate ATF4 upon Tg treatment (Fig. 4I). Furthermore, incorrect removal of m6A is associated with reduced ATF4 protein levels in ALKBH5-Δsl3 cells (Fig. 4H, S5F). In agreement, in vitro translation of cap-proximal methylated ATF4 5′ UTR luciferase reporter mRNA is inhibited in extracts from Tg-treated HEK293T cells (Fig. S5G). Thus, during chronic ER stress eIF3d-mediated translation induction of ALKBH5 results in decreased m6A modification of ATF4 mRNA and subsequent increased ATF4 protein synthesis. Interestingly, in mouse embryonic fibroblasts, ATF4 is also translationally regulated during the ISR through methylation, but instead through a m6A site in uORF2 that alters start-codon recognition35. This is distinct from the cap-proximal site found in human cells48,49, suggesting a convergence of m6A-directed control of ATF4 expression through divergent translation mechanisms.
eIF3d regulates GCN2 kinase during the persistent integrated stress response
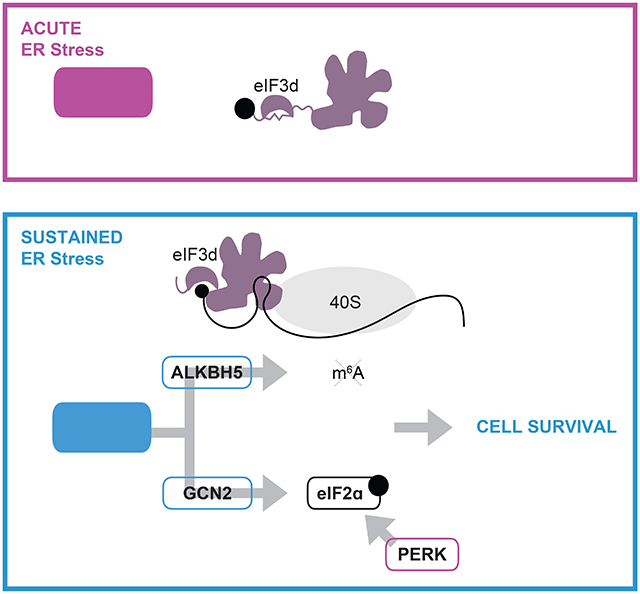
While ALKBH5-Δsl3 cells have downregulated ATF4 expression during chronic ER stress (Fig. 4H), they do not fully phenocopy phosphomimetic eIF3d-expressing cells such as inhibited eIF2ɑ phosphorylation (Fig. 1F, S2D), indicating other mechanisms driven by eIF3d are important for the persistent ISR. We bioinformatically analyzed if other mRNAs bound at the 5′ cap by eIF3d during sustained ER stress could contribute to this phenotype (Fig. 2A,B) and identified GCN2 as an eIF3d target (Fig. 5A). GCN2 is one of the four kinases that directly phosphorylates eIF2ɑ during the ISR50. We validated that eIF3d binds to the 5′ cap of the GCN2 mRNA in vitro and in cells (Fig 5B,C), and that both cap-binding and in vitro translation of a GCN2 5′ UTR luciferase reporter mRNA is regulated by eIF3d phosphorylation status (Fig. 5B, D). By contrast, PSMB6 mRNA translation is inhibited in lysates derived from Tg-treated cells regardless of eIF3d genotype, highlighting the role of eIF3d and specific 5′ UTR recruitment in sustaining GCN2 translation during the persistent ISR (Fig. 5D). Furthermore, GCN2 mRNA can only associate with translation complexes in cells expressing eIF3d that can appropriately undergo a phosphorylation switch during chronic ER stress (Fig. 5E). Correspondingly, during sustained Tg treatment, appropriate control of eIF3d phosphorylation is required for activation and autophosphorylation of GCN2 protein (Fig. 5F, S5H). While PERK is the primary eIF2ɑ kinase activated during the unfolded protein response51, our results suggest that during sustained ER stress, GCN2 additionally contributes to eIF2ɑ phosphorylation. To examine the temporal contribution of eIF2ɑ kinases, we pharmacologically disrupted GCN2 or PERK activity after activation of acute or persistent ISR signaling. PERK inhibition (PERKi, GSK2656157)52 is sufficient to disrupt ISR signaling at 8 h of Tg treatment, while suppression of GCN2 (GCNiB)53 has no effect (Fig. 5G). In contrast, during chronic ER stress, treatment with either kinase inhibitor leads to reduced eIF2ɑ phosphorylation and ATF4 expression (Fig. 5G). Therefore, these findings reveal a role for concurrent kinase activation during chronic ER stress and provide a mechanistic basis for how eIF3d regulates eIF2ɑ phosphorylation during the persistent ISR (Fig. 5F).
Figure 5. eIF3d regulates GCN2 kinase during the persistent integrated stress response.

(A) Read mapping to GCN2 from eIF3dTEV Subunit-Seq during ISR induction with thapsigargin. Tg, 16 h treatment with thapsigargin; In, input; eIF3dTEV-IP, anti-HA eIF3d immunoprecipitant samples after TEV protease cleavage. (B) eIF3d binding to the 5′ cap of GCN2 mRNA during chronic ER stress in HEK293T cells expressing wild-type (WT) and phosphomimetic (DD) eIF3d. Cap-binding is normalized to untreated cells. (C) Specificity of in vitro eIF3d crosslinking to the 5′ cap structure of the GCN2 5′ UTR. Binding is specific to the methylated cap structure of addition of cap analog competes away eIF3d binding. m7G, competitor cap analog m7GpppG; G, unmethylated GpppG. (D) Luciferase activity in vitro mediated by the GCN2 5′ UTR is eIF3d/ISR-responsive. PSMB6 is a control transcript not targeted by eIF3. Results are normalized to luciferase activity from in vitro translation extracts made from WT eIF3d-expressing HEK293T cells. Results in (B) and (D) are represented as the mean ± s.d. of three independent experiments. (E) Association of GCN2 mRNA with translating ribosomes in eIF3d cell lines. GCN2 abundance is expressed as a percentage of total transcripts and plotted as the mean ± s.d. Blue, Tg-treated cells; gray, untreated cells. The results are representative of two biological replicates. (F) Immunoblot of GCN2 kinase activation in WT and phosphomimetic eIF3d-expressing cells. (G) Immunoblot of ISR signaling cascade activation upon pharmacological inhibition of PERK or GCN2 reveals contribution of both kinases during chronic ER stress. HEK293T cells were treated with DMSO or Tg for 8 or 16 h, followed by addition of indicated inhibitor for 45 min. PERKi, PERK inhibitor; GCN2iB, GCN2 inhibitor. The results in (F) and (G) are representative of three independent experiments. (H) Model for gene cascade controlled by eIF3d-mediated translation that drives synergized regulation of the persistent integrated stress response. See also Figure S5.
Discussion
Our results reveal that eIF3d is critical for translational adaptation during the persistent ISR. We present a model where activation of eIF3d cap-binding through a phosphorylation switch triggers a gene regulatory cascade where direct translational control of ALKBH5 and GCN2 results in synergistic downstream regulation of the core ISR effectors ATF4 and eIF2ɑ (Fig. 5H). Notably, unlike the ubiquitous gene regulatory cascades well characterized as driven by transcription factors, this molecular network drives the temporal gene regulation needed for cell survival through integration of translational control. As eIF3d also supports distinct adaptive gene programs in cellular conditions where eIF4E is inhibited23,24, these findings reveal that eIF3d serves as a key regulator to interpret modes of global protein synthesis repression and allow dynamic translational activation of the appropriate response.
How can eIF3d mediate different gene programs dependent on physiological condition? Interestingly, previous mass spectrometry studies have identified putative phosphorylation sites on eIF3 subunits54. Structural data reveals that the eIF3d cap-binding domain contains a loop termed the “RNA gate” that occludes the cap-binding pocket22. Due to this auto-inhibited conformation of eIF3d, cap-binding requires initial recruitment of RNA to the larger eIF3 complex, which leads to allosteric changes to open the eIF3d cap-binding pocket. Therefore, modification of other eIF3 subunits is a potential mechanism for stress-specific delivery of mRNAs to eIF3d. Alternatively, regulation of eIF3 subunit expression could contribute to state-dependent gene regulation by altering levels of fully assembled eIF3 complex that is capable of translation activation. Indeed, during HCMV viral infection, eIF3d expression is upregulated and harnessed for both viral protein synthesis and pro-viral remodeling of the host proteome55.
We find that during chronic ER stress PERK is not sufficient to fully support ISR signaling and GCN2 must be concurrently activated. Therefore, the appropriate temporal cellular response to ISR activation may be further tuned by kinase-specific downstream gene programs. Notably, prior research has shown that multiple eIF2ɑ kinases can be simultaneously activated dependent on stress24,56. For example, all four eIF2ɑ kinases can be regulated during oxidative stress2, while both PERK and PKR are turned on in response to heat shock 57,58. Intriguingly, GCN2 is the most ancient and conserved of the ISR kinases and displays broader activation specificity including by diverse stresses from amino acid deprivation, glucose deprivation, ribosome collision, to UV irradiation50,59-62. It will be of future interest to understand if temporal cooperativity between GCN2 and the other eIF2ɑ kinases is a conserved paradigm during prolonged stress and how this contributes to distinct cellular outcomes.
Our findings demonstrate that ALKBH5 is translationally induced by eIF3d in response to sustained ER stress and this results in gene- and location-specific m6A demethylation. Notably, ALKBH5 is not important for general gene expression, as knockout mice are viable and do not display abnormalities beyond impaired spermatogenesis38. However, ALKBH5 is inhibited by post-translational modification in response to DNA damage63, and upregulated upon viral infection64 and in malignancies including glioblastoma, breast, and pancreatic cancers65,66 67. Therefore, our findings support a role of ALKBH5 in cellular state-specific reprogramming of the epitranscriptome, and reveal that dysregulation of m6A demethylation has a significant impact on viability during prolonged stress. Furthermore, we demonstrated that the ALKBH5 target mRNA, ATF4, is regulated by both methylation and eIF2ɑ phosphorylation. Thus, our results reveal how multifaceted control by RNA modification together with other forms of gene regulation can contribute to reversible or titratable expression needed for dynamic stress signaling.
Limitations of the Study
Here we find that activation of eIF3d cap-binding leads to increased GCN2 levels. It remains unknown why upregulated GCN2 synthesis is needed for its kinase function during chronic ER stress and will be of interest in future studies. In addition, cellular treatment with GCN2iB during chronic ER stress leads to downregulated GCN2 protein levels. Further experiments are needed to determine if this is due to a feedback loop that leads to eIF3d inactivation, an uncharacterized connection between GCN2 kinase activity and protein stability, or alternative modes of regulation. In addition, we observe that ISR signaling is dependent on eIF3d cap-binding activity only upon chronic ER stress. Notably, prior studies find siRNA knockdown of eIF3a, c, d, or g subunits all lead to decreased levels of ATF4 protein at early timepoints of ER stress, with no alteration of eIF2ɑ phosphorylation24. However, given the different experimental approaches of phosphomimetic inhibition of eIF3d cap-binding activity versus eIF3 subunit knockdown, along with the multifunctionality of the eIF3 complex68; further experiments are likely to reveal novel eIF3 complex functions during shorter induction of ER stress.
STAR Methods
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Amy S.Y. Lee (amysy_lee@dfci.harvard.edu).
Materials Availability
Reagents and materials produced in this study are available from the lead contact pending a completed Materials Transfer Agreement.
Data and Code Availability
All sequencing data have been deposited in the Gene Expression Omnibus under accession number GSE236188 and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-N6-methyladenosine | AbCam | Cat#151230; RRID:AB_2753144 |
| Mouse monoclonal anti-hsp90 | BD Biosciences | Cat#610418; RRID:AB_397798 |
| Rabbit polyclonal anti-eIF3b | Bethyl | Cat#A301-761A; RRID:AB_1210995 |
| Mouse monoclonal anti-ATF4 | Cell Signaling Technology | Cat#11815; RRID:AB_2616025 |
| Rabbit monoclonal anti-phospho-GCN2 | Cell Signaling Technology | Cat#94668; RRID:n/a |
| Rabbit polyclonal anti-GCN2 | Invitrogen | Cat#PA5-17523; RRID:AB_10983800 |
| Rabbit monoclonal anti-PERK | Cell Signaling Technology | Cat#C33E10; RRID:n/a |
| Rabbit polyclonal anti-rpS19 | Proteintech | Cat#15085-1-AP; RRID:AB_2180202 |
| Rabbit polyclonal anti-eIF2α | Proteintech | Cat#11170-1-AP; RRID:AB_2096489 |
| Rabbit monoclonal anti-ALKBH5 | Invitrogen | Cat#10H24L9; RRID:n/a |
| Goat anti-Rabbit HRP | Invitrogen | Cat#31462; RRID:AB_228338 |
| Goat anti-Mouse HRP | Invitrogen | Cat#31432; RRID:AB_228302 |
| Chemicals, peptides, and recombinant proteins | ||
| Fetal Bovine Serum | Biowest | Cat#S1620 |
| Thapsigargin | Cayman Chemical | Cat#10522 |
| Tunicamycin | Cayman Chemical | Cat#11445 |
| PERKi (GSK2656157) | MedChemExpress | Cat#HY-13820 |
| GCN2iB | MedChemExpress | Cat#HY-112654 |
| Cap-Clip Acid Pyrophosphatase | Cellscript | Cat#C-CC15011H |
| Bovine-Serum Albumin | Chemimpex | Cat#00577 |
| DTT | Chemimpex | Cat#00127 |
| Hybond-N+ membrane | Cytiva | Cat#RPN303B |
| Trypsin | Genesee | Cat#25-510 |
| Penicillin-streptomycin | Genesee | Cat#25-512 |
| Random Hexamer | Integrated DNA Technologies | Cat#51-01-18-01 |
| Murine RNase inhibitor | New England Biolabs | Cat#M0314L |
| Luna Universal qPCR master mix | New England Biolabs | Cat#M3003E |
| m7GpppG | New England Biolabs | Cat#S1411L |
| GpppG | New England Biolabs | Cat#S1407L |
| Q5 polymerase | New England Biolabs | Cat#M0491L |
| Vaccinia capping enzyme | New England Biolabs | Cat#M2080S |
| 2′ O-Methyltransferase enzyme | New England Biolabs | Cat#M0366S |
| Antarctic phosphatase | New England Biolabs | Cat#MO289L |
| T4 PNK | New England Biolabs | Cat#M0201L |
| XRN-1 | New England Biolabs | Cat#M0338L |
| 32P Radionuclide | Perkin-Elmer | Cat#NEX053H001MC |
| [ɑ-32P]-GTP | Perkin-Elmer | Cat#BLU006H250UC |
| Polyethylenimine, Linear, MW 25,000 | Polysciences, Inc | Cat#23966-1 |
| PhosSTOP | Sigma Aldrich | Cat#4906837001 |
| DMEM | Gibco | Cat#11995073 |
| OptiMEM | Gibco | Cat#11058021 |
| DMSO | ThermoFisher Scientific | Cat#BP231100 |
| Puromycin Dihydrochloride | ThermoFisher Scientific | Cat#AAJ61278MC |
| Sera-Mag Protein A/G beads | Cytiva | Cat#09-981-921 |
| Anti-HA magnetic beads | ThermoFisher Scientific, Pierce | Cat#PI88837 |
| SeraMag Oligo dT magnetic beads | Cytiva | Cat#09-981-145 |
| G-25 column | Cytiva | Cat#45 001 397 |
| Heparin | ThermoFisher Scientific | Cat#BP2425 |
| Cycloheximide | MP Biomedicals, Inc. | Cat#ICN10018301 |
| Phos-Tag acrylamide | Apexbio Technology | Cat#NC1646902 |
| SUPERase-In RNase Inhibitor | Invitrogen | Cat#AM2696 |
| Proteinase K | ThermoFisher Scientific | Cat#BP1700-100 |
| Phenol Chloroform | VWR | Cat#97064-712 |
| Glycogen | VWR | Cat#97063-256 |
| HIV-1 Protease | C. Schiffer, University of Massachusetts Medical School | |
| NEBuilder HiFi DNA Assembly | NEB | Cat#E2621L |
| EXPRE35S35S Protein Labeling Mix | Perkin Elmer | Cat#NEG072002MC |
| Critical commercial assays | ||
| NEBNext Ultra II Directional RNA Library Prep Kit for Illumina | New England Biolabs | Cat#E7760L |
| m6A eCLIP Kit | Eclipse Bioinnovations | Cat#143651 |
| Deposited data | ||
| RNA-Seq (WT versus phosphomimetic eIF3d HEK293T, veh. versus 16 h Tg) | This paper | GSE236188 |
| eIF3dTEV Subunit-Seq (HEK293T 16h Tg) | This paper | GSE236188 |
| m6A eCLIP (WT versus phosphomimetic eIF3d HEK293T, veh. versus 16 h Tg) | This paper | GSE236188 |
| Experimental models: Cell lines | ||
| Human: HEK293T | ATCC | CRL-11268 |
| Human: HEK293T eIF3dTEV | Lamper et al23 | |
| Human: HEK293T eIF3d S528D/S529D | This paper | |
| Human: HEK293T ALKBH5-ΔSL3 | This paper | |
| Oligonucleotides | ||
| DNA primers for RT-qPCR, see Table S4 | This paper | Table S4 |
| DNA primers for ALKBH5 cloning, see Table S4 | This paper | Table S4 |
| DNA primers for ATF4 cloning, see Table S4 | This paper | Table S4 |
| DNA primers for GCN2 cloning, see Table S4 | This paper | Table S4 |
| sgRNA sequences (eIF3d and ALKBH5), see Table S4 | This paper | Table S4 |
| Donor oligos (eIF3d and ALKBH5), see Table S4 | This paper | Table S4 |
| Recombinant DNA | ||
| Plasmid: pLBH269 SpCas9 sgRNA | L. Harrington and J. Doudna (UC Berkeley) | |
| Plasmid: pcDNA4-ALKBH5 5′ UTR-RLuc | This paper | |
| Plasmid: pcDNA4-ALKBH5 5′ UTR-RLuc – stem | This paper | |
| Plasmid: pcDNA4-ALKBH5 5′ UTR-RLuc – comp | This paper | |
| Plasmid: pcDNA4-ALKBH5 5′ UTR-RLuc – loop | This paper | |
| Plasmid: pcDNA4-GCN2 5′ UTR-RLuc | This paper | |
| Plasmid: pcDNA4-ATF4 5′ UTR-RLuc | This paper | |
| Plasmid: pGEX-4T1-PCIF1 | Boulias et al.48 | E. Greer, Washington University, St. Louis |
| Software and algorithms | ||
| CFX Maestro | Biorad | |
| Windaq | DataQ instruments | |
| RNAStructure | Reuter et al.75 | https://rna.urmc.rochester.edu/RNAstructure.html |
| VARNA | Darty et al.74 | https://varna.lri.fr |
| DAVID | Dennis et al.90 | https://david.ncifcrf.gov |
| ShinyGO | Ge et al.89 | http://bioinformatics.sdstate.edu/go/ |
| DESeq2 | Love et al.82 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Bowtie2 | Langmead et al.78 | https://bowtie-bio.sourceforge.net/bowtie2 |
| bwa | Li et al.86 | https://bio-bwa.sourceforge.net |
| Cutadapt | Martin77 | https://pypi.org/project/cutadapt/ |
| SAMtools | Li et al.80 | http://www.htslib.org |
| STAR | Dobin et al.79 | https://github.com/alexdobin/STAR |
| UMItools | Smith et al.84 | https://github.com/CGATOxford/UMI-tools |
| pyCRAC | Webb et al.85 | https://sandergranneman.bio.ed.ac.uk/pycrac-software |
| CTK | Shah et al.87 | https://zhanglab.c2b2.columbia.edu/index.php/CTK_Documentation |
Experimental model and study participant details
Cell culture
All cell lines were grown in complete media (DMEM, high glucose and L-glutamine (Gibco) supplemented with 10 % FBS (Biowest) and 500 U mL−1 penicillin-streptomycin (GenClone). For ISR induction, cells were seeded to be at ~80 % confluency at time of treatment and then treated with 600 nM thapsigargin (Cayman Chemical), 3 μM tunicamycin (Cayman Chemical), or equal volume of DMSO vehicle (Alfa Aesar) for 16 h or annotated timepoints. For kinase inhibitor experiments, cells were first treated with thapsigargin for 8 or 16 h, followed by addition of 500 nM PERKi (GSK2656157, MedChemExpress), 2 μM GCN2iB (MedChemExpress), or equal volume of DMSO vehicle for 45 min. To measure viability, cells were trypsinized and resuspended in complete media at 4 × 106 cells mL−1. 20 μL of cells were mixed with 20 μL of 0.4 % (w/v) trypan blue solution (Gibco), loaded onto chamber slides, and the percentage of dead cells were determined using a Countess 3 Automated Cell Counter (Invitrogen). For HA-eIF3d immunoprecipitation assays, HEK293T cells were transfected with 10 μg of pcDNA5/FRT eIF3d HA plasmid23 and incubated for 48 h to incorporate HA-tagged eIF3d into the endogenous eIF3 complex. RNA transfections were performed using TransIT mRNA reagent (Mirius) according to the manufacturer’s protocol. Briefly, cells in a 96-well plate were transfected with 500 ng of RNA and harvested at 6 h after transfection. HEK293T cell lines expressing eIF3d or ALKBH5 mutants were generated by CRISPR/Cas9-induced homology directed repair as previously described69,70. Briefly, cells in a 6-well plate were co-transfected with 3.2 μg pLBH269 SpCas9 sgRNA plasmid (L. Harrington and J. Doudna (UC Berkeley)), which expresses Cas9 and the targeting sgRNA, and 2.5 nM HDR donor double-stranded oligo (IDT) in 200 uL volume using PEI (1 mg ml−1 linear polyethylenimine (Polysciences)). After 24 h post-transfection, media was supplemented with 1 μg ml−1 puromycin to select for transfected cells. At 48 h post-selection, cells were trypsinized and seeded in 96-well plates for single cell cloning by serial dilution. Single colony clones were genotyped by Sanger sequencing. sgRNA and donor oligo sequences are listed in Table S4.
Methods details
Cloning
pLBH269 SpCas9 sgRNA vectors for targeting eIF3d and ALKBH5 were prepared by digestion with BbsI and ligation of annealed oligos to insert the sgRNA sequences. The wild-type (WT) ALKBH5, ATF4, and GCN2 5′ UTR luciferase reporter plasmids were prepared by amplification of the UTR sequence from HEK293T cDNA and Gibson cloning into pcDNA4 Rluc18. The ALKBH5 5′ UTR Δpar (eIF3 PAR-CLIP site deletion reporter) plasmid was generated by around-the-horn PCR and ligation. The ALKBH5 5′ UTR stem mutant reporter plasmids were generated from the WT plasmid by digestion with SacI and NheI and two-insert Gibson cloning to introduce the mutated sequence. Primers used for cloning are listed in Table S4.
Reverse transcription and quantitative PCR
cDNA was generated from RNA by reverse transcription using random hexamers and MMLV M5 RT under standard conditions as previously described23. Quantitative PCR was performed using Luna Universal qPCR master mix (NEB) and primers used are listed in Table S4.
eIF3–RNA interaction assays
eIF3 complex immunoprecipitation and eIF3d cap-binding were performed as previously described18,22,23. Briefly, for eIF3 complex immunoprecipitation, HEK239T cells were lysed in NP-40 lysis buffer (50 mM HEPES-KOH pH 7.5, 150 mM KCl, 5 mM MgCl2, 0.5 % v/v NP-40, 0.25 mM DTT), nuclei were pelleted by centrifugation for 10 min at 16,000 × g at 4 °C, and 10 % of the supernatant was retained as an input sample. eIF3 was immunoprecipitated using rabbit anti-eIF3b antibody (Bethyl A301-761A) with magnetic Sera-Mag Protein A/G beads (Cytiva), beads were washed 3× with high salt NP-40 wash buffer (50 mM HEPES-KOH pH 7.5, 500 mM KCl, 5 mM MgCl2, 0.5 % v/v NP-40, 0.25 mM DTT), and RNA was isolated from beads and input samples by phenol-chloroform extraction and ethanol precipitation.
For eIF3d cap-binding, supernatant and input samples from HA-eIF3d-transfected cells were made as described above. eIF3d was immunoprecipitated using anti-HA magnetic beads (Pierce) and beads were washed 3× with NP-40 lysis buffer. To isolate the eIF3d cap-binding domain separately of the eIF3 complex, beads were mixed with 10 μM HIV-1 PR in Cech-Ca buffer (25 mM Tris-HCl pH 7.5, 5 mM Mg(OAc)2, 70 mM KCl, 0.1 mg ml−1 BSA, 2 mM DTT) for 30 min at room temperature, beads were washed 3× with high salt NP-40 wash, and RNA was isolated from beads and input samples by phenol-chloroform extraction and ethanol precipitation. 50 μM m7GpppG or GpppG (NEB) was added to the wash buffer for the final 2 washes for the cap competition assays. cDNA synthesis and quantitative PCR was performed as described above.
Native eIF3–RNA gel shift analysis was performed as previously described18. Recombinant eIF3 was expressed and purified from Escherichia coli as previously described71. Briefly, eIF3 and [32P] cap-labeled RNA were incubated in Binding Buffer (25 mM Tris-HCl pH 7.5, 5 mM Mg(OAc)2, 70 mM KCl, 0.1 mg ml−1 BSA, 2 mM DTT, 0.1 μg μl−1 yeast tRNA, 20 μg ml−1 heparin) in a final volume of 5 μl at 25 °C for 30 min. One microliter of 6× non-denaturing loading dye (40 % w/v sucrose, with bromophenol blue) was added, reactions were loaded on a 0.7 % agarose gel made with a buffer consisting of 1× TBE supplemented with 75 mM KCl, and gel electrophoresis was performed in the same buffer composition. The gel was incubated in fixing buffer (10 % acetic acid, 40 % ethanol) for 5 min, placed on a positively charged nylon Hybond-N+ membrane (Cytiva), dried and imaged using a phosphoimager.
Metabolic Labeling
Prior to [32P]-orthophosphate labeling, HA-eIF3d-transfected cells were treated for denoted timepoints with 600 nM thapsigargin, 3 μM tunicamycin, or an equivalent volume of DMSO. For labeling, cells were incubated with 12.5 μCi ml−1 of 32P Radionuclide (Perkin Elmer) for 5 h. Cells were harvested and HA-eIF3d immunoprecipitation was performed as described above. Isolated proteins were separated by SDS-PAGE and the gel was dried and imaged using a phosphorimager. Gel lane intensity was calculated using ImageJ72.
Prior to [35S]-labeling, cells were treated for indicated timepoints with 600 nM thapsigargin or an equivalent volume of DMSO in a 6-well plate at ~70 % confluency. On the day of the experiment, the media was switched to DMEM without methionine or cysteine (Invitrogen) supplemented with 10 μg mL−1 actinomycin D and incubated for 30 min after which 5 μL of EXPRE35S35S Protein Labeling Mix (Perkin Elmer) was added to each well. After 1 hr, the cells were harvested and lysed by incubation for 5 min on ice with 3× the pellet volume of NP-40 lysis buffer. Nuclei were pelleted by centrifugation for 5 min at 16,000 × g at 4 °C, and equal amounts of cell lysates were separated by SDS-PAGE. The gel was stained with Coomassie Blue for examining equal loading and then dried and imaged using a phosphoimager. Gel lane intensity was calculated using ImageJ72.
In vitro transcription
RNAs for luciferase reporter assays were transcribed using T7 RNA polymerase and a purified PCR product as template, and capped using vaccinia virus capping enzymes (NEB). RNAs for in vitro binding assays were radiolabeled at the 5′ cap using [ɑ-32P]-GTP and vaccinia virus capping enzymes, and free nucleotide was removed by one round of phenol-chloroform extraction and ethanol precipitation followed by two purifications through a G-25 column (Cytiva). m6A modification of the ATF4 luciferase reporter mRNA was performed as described previously48. pGEX-4T1-PCIF1 was a kind gift from E. Greer (Washington University in St. Louis), and PCIF1 recombinant protein expression and purification was performed as described previously48. Briefly, in vitro transcribed ATF4 RNA was methylated using mRNA Cap 2′-O-Methyltransferase (NEB), incubated with 50 nM PCIF1 protein in a final reaction containing 50 mM Tris-HCl pH 8.0, 1 mM DTT, 1 mM EDTA, 5 % (v/v) glycerol, and 160 mM SAM (NEB) for 10 min at 37 °C, and purified by ethanol precipitation. For RNA structure analysis of ALKBH5, the 5′ UTR was in vitro transcribed with T7 RNA polymerase as described above and SHAPE was performed as previously described using the SHAPE primer in Table S418. SHAPE reactivity was calculated using RiboCAT73 and structure analysis and visualization was performed with RNAStructure and VARNA74,75.
Luciferase assays
Cells were treated for 16 h with DMSO or Tg, as described above, and in vitro translation extracts were made from cells as previously described22. Each reaction contained 50 % (v/v) in vitro translation extracts and 500 ng capped and polyadenylated in vitro transcribed mRNA in a final reaction containing 0.84 mM ATP, 0.21 mM GTP, 21 mM creatine phosphate (Roche), 45 U ml−1 creatine phosphokinase (Roche), 10 mM HEPES-KOH pH 7.6, 2 mM DTT, 2 mM Mg(OAc)2, 150 mM KOAc, 8 mM amino acids (Promega), 255 mM spermidine and 1 U ml−1 murine RNase inhibitor (NEB). Translation reactions were incubated for 1 h at 30 °C, luciferase assay was performed (Genecopoeia), and relative luminescence units was measured with a Glomax Multi+ plate reader (Promega). For in cell assays, cells were treated for 16 h with DMSO or Tg and mRNAs were transfected as described above. Cells were harvested 6 h after transfection and luciferase activity was measured as described above.
Polysome profiling
Polysome profiling was performed as previously described76. Briefly, cells were treated for 5 min with 100 μg ml−1 cycloheximide (MP Biomedicals) and then harvested into PBS with 100 μg ml−1 cycloheximide. Cells were pelleted by centrifugation for 5 min at 100 × g for 5 min at 4°C, lysed in polysome lysis buffer (10 mM HEPES-KOH pH 7.4, 150 mM KOAc, 5 mM Mg(OAc)2, 1 % v/v Triton-X 100, 100 μg ml−1 cycloheximide, 1 mM DTT), and triturated through a 21-g needle five times. Nuclei were removed by centrifugation at 10,000 × g for 5 min at 4°C and equal A260 units of lysates were loaded on 10–50 % (w/v) sucrose gradient made in polysome lysis buffer without cycloheximide and detergent. Gradients were centrifuged for 2 h at 4°C at 35,000 rpm in a SW41 Ti Rotor and separated using a Brandel gradient fractionator with A254 profile monitoring by a Dataq data acquisition system. RNA was purified from fractions by phenol-chloroform extraction and ethanol precipitation and cDNA synthesis and quantitative PCR was performed as described above.
Immunoblot
For analysis of phosphorylated proteins, samples were resolved on a 12 % SDS-PAGE with 50 μM phos-tag (Apexbio) and 50 μM MnCl. Immunoblot analysis was performed with antibodies listed in the Key Resources Table.
m6A RNA immunoprecipitation
Total RNA was isolated from cells and polyadenylated RNA was purified using SeraMag Oligo dT magnetic beads (Cytiva). For each sample, 25 μl of magnetic Sera-Mag Protein A/G beads (Cytiva) was prepared by washing twice with 1 ml of IPP buffer (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.1 % v/v NP-40, 1 mM DTT). For the m6A RNA immunoprecipitation, 1 μg of polyadenylated RNA, 4 μl of anti-N6-methyladenosine rabbit monoclonal antibody (AbCam 151230), 2 μl of murine RNase inhibitor (NEB), and 25 μl washed beads in a final volume of 1 ml were incubated at 4 °C overnight with rotating head-over-head. Beads were washed three times with 1 ml of IPP buffer, RNA bound to beads was extracted using phenol chloroform and ethanol precipitation, and cDNA synthesis and quantitative PCR was performed as described above.
eIF3dTEV Subunit-Seq
eIF3dTEV Subunit-Seq was performed as previously described23. Briefly, for each experiment, 10× 15-cm plates of eIF3dTEV-293T cells were treated with 600 nM of Tg or DMSO for 16 h prior to harvest. Cells were crosslinked with UV 254 nm light, harvested, and lysed in NP-40 lysis buffer (50 mM HEPES-KOH pH 7.4, 150 mM KCl, 5 mM Mg(OAc)2, 1 % v/v NP-40, 0.25 mM DTT). The eIF3 complex was immunoprecipitated using anti-HA magnetic beads (Pierce), and the eIF3d cap-binding domain was isolated by TEV protease digestion. Contaminating RNA where the 5′ cap is not protected by eIF3d was removed by sequential treatment with Antarctic Phosphatase (NEB), Cap-Clip Acid Pyrophosphatase (Cellscript), T4 PNK (NEB), and XRN-1 (NEB). Proteins were removed by proteinase K (Fisher) digestion and RNA from IP and input samples were extracted by phenol-chloroform extraction and ethanol precipitation. cDNA libraries were constructed from polyadenylated RNA using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (NEB) according to the manufacturers protocol and sequenced using an Illumina NextSeq 500.
m6A-eCLIP
m6A-eCLIP libraries were constructed using the m6A-eCLIP kit from Eclipse Bio. 6× 15-cm plates of HEK293T WT or S528D/S529D eIF3d-expressing cells were treated with 600 nM of Tg or DMSO for 16 h prior to harvest. Cells were lysed with NP-40 lysis buffer, total RNA was isolated by phenol chloroform extraction and ethanol precipitation, and polyadenylated RNA was isolated from total RNA using SeraMag Oligo dT magnetic beads (Cytiva). Briefly, 500 ng of polyadenylated RNA from each sample was heat-fragmented and UV cross-linked to an anti-m6A antibody. Methylated RNAs were immunoprecipitated using Protein G-coupled magnetic beads and on-bead RNA ligation of an indexed 3′ RNA adapter was performed. The RNP complex was isolated by SDS-PAGE and membrane transfer, the bound antibody was digested by Proteinase K, and the RNA was converted to cDNA by reverse transcription. A 3′ single-stranded DNA adapter was ligated onto the cDNA and libraries were amplified by PCR. Libraries from two biological replicates were sequenced using an Illumina NextSeq 500.
RNA-Sequencing
Total RNA was isolated from cells and polyadenylated RNA was purified using SeraMag Oligo dT magnetic beads (Cytiva) as described previously. 100 ng of polyadenylated RNA was processed for library generation with NEBNext Ultra II RNA Library Prep Kit for Illumina (NEB) and sequenced using an Illumina NextSeq 500. Libraries were made from two biological replicates for each cellular condition.
Bioinformatics
For RNA-Sequencing and eIF3dTEV Subunit-Seq analysis, reads were quality filtered and trimmed using Cutadapt77. Reads derived from rRNA and repetitive elements were removed with bowtie278, and remaining reads were mapped to the human genome (GRCh38) using STAR79. Mapping was converted to indexed bam files using SAMtools80 and gene-level counts were obtained in R Studio using summarizeOverlaps from GenomicAlignments81. For the RNA-Sequencing, differential enrichment of gene expression in HEK293T expressing WT eIF3d versus phosphomimetic eIF3d during Tg treatment in two biological replicates was calculated using DESeq282 and P-value correction was applied using fdrtool83. Genes transcriptionally dysregulated in phosphomimetic eIF3d cells were identified as those with a p-adj value less than 0.1 and fold change greater than 2. For the eIF3dTEV Subunit-Seq, genes with less than 60 reads were discarded, and differential enrichment of IP versus input reads in two biological replicates was calculated using DESeq2. eIF3d targets were identified as those with a p-adj value less than 0.1 and fold change greater than 2.
For analysis of m6A-eCLIP, the unique molecular identifier sequences were extracted and appended to the read names using UMItools84. Reads were quality filtered and trimmed twice using Cutadapt, sorted by read identifier using fastq-sort, and mapped to repeat elements using bowtie2. PCR duplicates were collapsed using pyCRAC85, read names were reformatted to be used with CTK using a custom python script, and reads were mapped to the human genome (GRCh38) using bwa86. Mapped reads were parsed to collapse PCR duplicates and used to create a mutation file using CTK87 parseAlignment, tag2collapse, and joinWrapper.py, keeping only unique mapping reads and a minimal mapping size of 18 nt. Taking advantage that RNA–antibody cross-linking causes adducts that result in sequence transitions or truncations, CIMS and CITS analysis was performed to identify the m6A sites. For CIMS analysis, the m6A site was extracted by adjusting the position −1 from the sequence transition using bedtools88. m6A sites were defined as those shared between two biological replicates and methylation levels at each site were quantified by using bedtools multicov with the bwa bam output and normalizing mapped reads at each site to counts per million reads. Motif enrichment analysis was performed using homer and metagene analysis of the m6A sites along the transcript architecture was performed using metaPlotR. Read mapping was plotted using wiggleplotR. Gene ontology analyses were performed using ShinyGO89 or DAVID90 with a FDR cutoff of 0.05 and enrichment score cutoff of 1.5.
Quantification and statistical analysis
Results were presented as the mean ± s.d. from the number of replicates indicated in the corresponding figure legends. Details of exact statistical analyses and procedures can be found in the main text, figure legends, and STAR Methods.
Supplementary Material
Supplementary Table 1 ∣ Table of genes dysregulated in phosphomimetic eIF3d-expresssing cells during chronic ER stress, related to Figure 1.
Supplementary Table 2 ∣ Table of eIF3d cap-binding target mRNAs during chronic ER stress, related to Figure 2.
Supplementary Table 3 ∣ Table of m6A sites in eIF3d cell lines, related to Figure 2.
Highlights.
eIF3d regulates integrated stress response effectors, eIF2ɑ and ATF4
eIF3d supports temporal activation of GCN2 kinase and m6A RNA demethylation
eIF3d-specialized translation mediates cellular survival during sustained ER stress
Acknowledgements:
The authors thank K. Chat, E. Greer, N. Bellono, and members of the Lee lab for discussions. We thank N. Phan and R. Tomaino for experimental advice.
Funding:
This work was funded by the Searle Scholars Program, the Pew Biomedical Scholars Program, the V Foundation, and the National Institutes of Health (1R35GM142527) to A.S.Y.L. S.M. is supported as a Helen Gurley Brown Presidential Initiative Fellow.
Inclusion and diversity:
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Declaration of interests: The authors declare no competing interests.
Data and materials availability: All sequencing data have been deposited in the Gene Expression Omnibus under accession number GSE236188.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, and Gorman AM (2016). The integrated stress response. EMBO reports 17, 1374–1395. 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Molecular cell 11, 619–633. 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 3.Krishnamoorthy T, Pavitt GD, Zhang F, Dever TE, and Hinnebusch AG (2001). Tight binding of the phosphorylated alpha subunit of initiation factor 2 (eIF2ɑlpha) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Molecular and cellular biology 21, 5018–5030. 10.1128/MCB.21.15.5018-5030.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, and Ron D (2000). Regulated translation initiation controls stress-induced gene expression in mammalian cells. Molecular cell 6, 1099–1108. 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 5.Starck SR, Tsai JC, Chen K, Shodiya M, Wang L, Yahiro K, Martins-Green M, Shastri N, and Walter P (2016). Translation from the 5' untranslated region shapes the integrated stress response. Science 351, aad3867. 10.1126/science.aad3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Costa-Mattioli M, and Walter P (2020). The integrated stress response: From mechanism to disease. Science 368. 10.1126/science.aat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, and Hotamisligil GS (2004). Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461. 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 8.Engin F, Yermalovich A, Nguyen T, Hummasti S, Fu W, Eizirik DL, Mathis D, and Hotamisligil GS (2013). Restoration of the unfolded protein response in pancreatic beta cells protects mice against type 1 diabetes. Science translational medicine 5, 211ra156. 10.1126/scitranslmed.3006534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Way SW, Podojil JR, Clayton BL, Zaremba A, Collins TL, Kunjamma RB, Robinson AP, Brugarolas P, Miller RH, Miller SD, and Popko B (2015). Pharmaceutical integrated stress response enhancement protects oligodendrocytes and provides a potential multiple sclerosis therapeutic. Nature communications 6, 6532. 10.1038/ncomms7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dash PK, Hylin MJ, Hood KN, Orsi SA, Zhao J, Redell JB, Tsvetkov AS, and Moore AN (2015). Inhibition of Eukaryotic Initiation Factor 2 Alpha Phosphatase Reduces Tissue Damage and Improves Learning and Memory after Experimental Traumatic Brain Injury. Journal of neurotrauma 32, 1608–1620. 10.1089/neu.2014.3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bond S, Lopez-Lloreda C, Gannon PJ, Akay-Espinoza C, and Jordan-Sciutto KL (2020). The Integrated Stress Response and Phosphorylated Eukaryotic Initiation Factor 2alpha in Neurodegeneration. Journal of neuropathology and experimental neurology 79, 123–143. 10.1093/jnen/nlz129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ishizawa J, Kojima K, Chachad D, Ruvolo P, Ruvolo V, Jacamo RO, Borthakur G, Mu H, Zeng Z, Tabe Y, et al. (2016). ATF4 induction through an atypical integrated stress response to ONC201 triggers p53-independent apoptosis in hematological malignancies. Science signaling 9, ra17. 10.1126/scisignal.aac4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suresh S, Chen B, Zhu J, Golden RJ, Lu C, Evers BM, Novaresi N, Smith B, Zhan X, Schmid V, et al. (2020). eIF5B drives integrated stress response-dependent translation of PD-L1 in lung cancer. Nature cancer 1, 533–545. 10.1038/s43018-020-0056-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin Y, Li F, Huang L, Polte C, Duan H, Fang J, Sun L, Xing X, Tian G, Cheng Y, et al. (2020). eIF3 Associates with 80S Ribosomes to Promote Translation Elongation, Mitochondrial Homeostasis, and Muscle Health. Molecular cell 79, 575–587 e577. 10.1016/j.molcel.2020.06.003. [DOI] [PubMed] [Google Scholar]
- 15.Bohlen J, Fenzl K, Kramer G, Bukau B, and Teleman AA (2020). Selective 40S Footprinting Reveals Cap-Tethered Ribosome Scanning in Human Cells. Molecular cell 79, 561–574 e565. 10.1016/j.molcel.2020.06.005. [DOI] [PubMed] [Google Scholar]
- 16.Wagner S, Herrmannova A, Hronova V, Gunisova S, Sen ND, Hannan RD, Hinnebusch AG, Shirokikh NE, Preiss T, and Valasek LS (2020). Selective Translation Complex Profiling Reveals Staged Initiation and Co-translational Assembly of Initiation Factor Complexes. Molecular cell 79, 546–560 e547. 10.1016/j.molcel.2020.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hronová V, Mohammad MP, Wagner S, Pánek J, Gunišová S, Zeman J, Poncová K, and Valášek LS (2017). Does eIF3 promote reinitiation after translation of short upstream ORFs also in mammalian cells? RNA biology 14, 1660–1667. 10.1080/15476286.2017.1353863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee AS, Kranzusch PJ, and Cate JH (2015). eIF3 targets cell-proliferation messenger RNAs for translational activation or repression. Nature 522, 111–114. 10.1038/nature14267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian SB, and Jaffrey SR (2015). 5' UTR m(6)A Promotes Cap-Independent Translation. Cell 163, 999–1010. 10.1016/j.cell.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kieft JS, Zhou K, Jubin R, and Doudna JA (2001). Mechanism of ribosome recruitment by hepatitis C IRES RNA. Rna 7, 194–206. 10.1017/s1355838201001790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma S, Liu JY, and Zhang JT (2023). eIF3d: A driver of noncanonical cap-dependent translation of specific mRNAs and a trigger of biological/pathological processes. The Journal of biological chemistry 299, 104658. 10.1016/j.jbc.2023.104658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee AS, Kranzusch PJ, Doudna JA, and Cate JH (2016). eIF3d is an mRNA cap-binding protein that is required for specialized translation initiation. Nature 536, 96–99. 10.1038/nature18954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lamper AM, Fleming RH, Ladd KM, and Lee ASY (2020). A phosphorylation-regulated eIF3d translation switch mediates cellular adaptation to metabolic stress. Science 370, 853–856. 10.1126/science.abb0993. [DOI] [PubMed] [Google Scholar]
- 24.Guan BJ, van Hoef V, Jobava R, Elroy-Stein O, Valasek LS, Cargnello M, Gao XH, Krokowski D, Merrick WC, Kimball SR, et al. (2017). A Unique ISR Program Determines Cellular Responses to Chronic Stress. Molecular cell 68, 885–900 e886. 10.1016/j.molcel.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma Y, and Hendershot LM (2003). Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. The Journal of biological chemistry 278, 34864–34873. 10.1074/jbc.M301107200. [DOI] [PubMed] [Google Scholar]
- 26.Ohoka N, Yoshii S, Hattori T, Onozaki K, and Hayashi H (2005). TRB3, a novel ER stress-inducible gene, is induced via ATF4–CHOP pathway and is involved in cell death. The EMBO journal 24, 1243–1255. 10.1038/si.emboj.7600596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang HY, Wek SA, McGrath BC, Lu D, Hai T, Harding HP, Wang X, Ron D, Cavener DR, and Wek RC (2004). Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Molecular and cellular biology 24, 1365–1377. 10.1128/MCB.24.3.1365-1377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.English AM, Green KM, and Moon SL (2022). A (dis)integrated stress response: Genetic diseases of eIF2ɑlpha regulators. Wiley interdisciplinary reviews. RNA 13, e1689. 10.1002/wrna.1689. [DOI] [PubMed] [Google Scholar]
- 29.Szaflarski W, Leśniczak-Staszak M, Sowiński M, Ojha S, Aulas A, Dave D, Malla S, Anderson P, Ivanov P, and Lyons SM (2021). Early rRNA processing is a stress-dependent regulatory event whose inhibition maintains nucleolar integrity. Nucleic acids research 50, 1033–1051. 10.1093/nar/gkab1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engel M, Eggert C, Kaplick PM, Eder M, Roh S, Tietze L, Namendorf C, Arloth J, Weber P, Rex-Haffner M, et al. (2018). The Role of m(6)A/m-RNA Methylation in Stress Response Regulation. Neuron 99, 389–403 e389. 10.1016/j.neuron.2018.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Novoa I, Zeng H, Harding HP, and Ron D (2001). Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2ɑlpha. The Journal of cell biology 153, 1011–1022. 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Belin BJ, Lee T, and Mullins RD (2015). DNA damage induces nuclear actin filament assembly by Formin −2 and Spire-(1/2) that promotes efficient DNA repair. [corrected]. eLife 4, e07735. 10.7554/eLife.07735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, et al. (2015). Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nature cell biology 17, 288–299. 10.1038/ncb3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sourbier C, Ricketts CJ, Matsumoto S, Crooks DR, Liao PJ, Mannes PZ, Yang Y, Wei MH, Srivastava G, Ghosh S, et al. (2014). Targeting ABL1-mediated oxidative stress adaptation in fumarate hydratase-deficient cancer. Cancer cell 26, 840–850. 10.1016/j.ccell.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou J, Wan J, Shu XE, Mao Y, Liu XM, Yuan X, Zhang X, Hess ME, Bruning JC, and Qian SB (2018). N(6)-Methyladenosine Guides mRNA Alternative Translation during Integrated Stress Response. Molecular cell 69, 636–647 e637. 10.1016/j.molcel.2018.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, He X, and Semenza GL (2016). Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m(6)A-demethylation of NANOG mRNA. Proceedings of the National Academy of Sciences of the United States of America 113, E2047–2056. 10.1073/pnas.1602883113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG, and He C (2011). N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nature chemical biology 7, 885–887. 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, Vagbo CB, Shi Y, Wang WL, Song SH, et al. (2013). ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Molecular cell 49, 18–29. 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mauer J, and Jaffrey SR (2018). FTO, m(6) A(m) , and the hypothesis of reversible epitranscriptomic mRNA modifications. FEBS letters 592, 2012–2022. 10.1002/1873-3468.13092. [DOI] [PubMed] [Google Scholar]
- 40.Merino EJ, Wilkinson KA, Coughlan JL, and Weeks KM (2005). RNA structure analysis at single nucleotide resolution by selective 2'-hydroxyl acylation and primer extension (SHAPE). Journal of the American Chemical Society 127, 4223–4231. 10.1021/ja043822v. [DOI] [PubMed] [Google Scholar]
- 41.Van Nostrand EL, Nguyen TB, Gelboin-Burkhart C, Wang R, Blue SM, Pratt GA, Louie AL, and Yeo GW (2017). Robust, Cost-Effective Profiling of RNA Binding Protein Targets with Single-end Enhanced Crosslinking and Immunoprecipitation (seCLIP). Methods in molecular biology 1648, 177–200. 10.1007/978-1-4939-7204-3_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, and Jaffrey SR (2015). Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nature methods 12, 767–772. 10.1038/nmeth.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, and Jaffrey SR (2012). Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell 149, 1635–1646. 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206. 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 45.Choi J, Ieong KW, Demirci H, Chen J, Petrov A, Prabhakar A, O'Leary SE, Dominissini D, Rechavi G, Soltis SM, et al. (2016). N(6)-methyladenosine in mRNA disrupts tRNA selection and translation-elongation dynamics. Nature structural & molecular biology 23, 110–115. 10.1038/nsmb.3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, and He C (2015). N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 161, 1388–1399. 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mao Y, Dong L, Liu XM, Guo J, Ma H, Shen B, and Qian SB (2019). m(6)A in mRNA coding regions promotes translation via the RNA helicase-containing YTHDC2. Nature communications 10, 5332. 10.1038/s41467-019-13317-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boulias K, Toczydlowska-Socha D, Hawley BR, Liberman N, Takashima K, Zaccara S, Guez T, Vasseur JJ, Debart F, Aravind L, et al. (2019). Identification of the m(6)Am Methyltransferase PCIF1 Reveals the Location and Functions of m(6)Am in the Transcriptome. Molecular cell 75, 631–643 e638. 10.1016/j.molcel.2019.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sendinc E, Valle-Garcia D, Dhall A, Chen H, Henriques T, Navarrete-Perea J, Sheng W, Gygi SP, Adelman K, and Shi Y (2019). PCIF1 Catalyzes m6Am mRNA Methylation to Regulate Gene Expression. Molecular cell 75, 620–630 e629. 10.1016/j.molcel.2019.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hinnebusch AG, and Fink GR (1983). Positive regulation in the general amino acid control of Saccharomyces cerevisiae. Proceedings of the National Academy of Sciences of the United States of America 80, 5374–5378. 10.1073/pnas.80.17.5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ma K, Vattem KM, and Wek RC (2002). Dimerization and release of molecular chaperone inhibition facilitate activation of eukaryotic initiation factor-2 kinase in response to endoplasmic reticulum stress. The Journal of biological chemistry 277, 18728–18735. 10.1074/jbc.M200903200. [DOI] [PubMed] [Google Scholar]
- 52.Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, Li WHH, Heerding DA, Minthorn E, Mencken T, et al. (2012). Discovery of 7-Methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a Potent and Selective First-in-Class Inhibitor of Protein Kinase R (PKR)-like Endoplasmic Reticulum Kinase (PERK). Journal of medicinal chemistry 55, 7193–7207. 10.1021/jm300713s. [DOI] [PubMed] [Google Scholar]
- 53.Nakamura A, Nambu T, Ebara S, Hasegawa Y, Toyoshima K, Tsuchiya Y, Tomita D, Fujimoto J, Kurasawa O, Takahara C, et al. (2018). Inhibition of GCN2 sensitizes ASNS-low cancer cells to asparaginase by disrupting the amino acid response. Proceedings of the National Academy of Sciences of the United States of America 115, E7776–E7785. 10.1073/pnas.1805523115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Andaya A, Villa N, Jia W, Fraser CS, and Leary JA (2014). Phosphorylation stoichiometries of human eukaryotic initiation factors. International journal of molecular sciences 15, 11523–11538. 10.3390/ijms150711523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thompson L, Depledge DP, Burgess HM, and Mohr I (2022). An eIF3d-dependent switch regulates HCMV replication by remodeling the infected cell translation landscape to mimic chronic ER stress. Cell reports 39, 110767. 10.1016/j.celrep.2022.110767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hamanaka RB, Bennett BS, Cullinan SB, and Diehl JA (2005). PERK and GCN2 contribute to eIF2ɑlpha phosphorylation and cell cycle arrest after activation of the unfolded protein response pathway. Molecular biology of the cell 16, 5493–5501. 10.1091/mbc.e05-03-0268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Elvira R, Cha SJ, Noh GM, Kim K, and Han J (2020). PERK-Mediated eIF2ɑlpha Phosphorylation Contributes to The Protection of Dopaminergic Neurons from Chronic Heat Stress in Drosophila. International journal of molecular sciences 21. 10.3390/ijms21030845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao M, Tang D, Lechpammer S, Hoffman A, Asea A, Stevenson MA, and Calderwood SK (2002). Double-stranded RNA-dependent protein kinase (pkr) is essential for thermotolerance, accumulation of HSP70, and stabilization of ARE-containing HSP70 mRNA during stress. The Journal of biological chemistry 277, 44539–44547. 10.1074/jbc.M208408200. [DOI] [PubMed] [Google Scholar]
- 59.Deng J, Harding HP, Raught B, Gingras AC, Berlanga JJ, Scheuner D, Kaufman RJ, Ron D, and Sonenberg N (2002). Activation of GCN2 in UV-irradiated cells inhibits translation. Current biology : CB 12, 1279–1286. 10.1016/s0960-9822(02)01037-0. [DOI] [PubMed] [Google Scholar]
- 60.Yang R, Wek SA, and Wek RC (2000). Glucose limitation induces GCN4 translation by activation of Gcn2 protein kinase. Molecular and cellular biology 20, 2706–2717. 10.1128/MCB.20.8.2706-2717.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu CC, Peterson A, Zinshteyn B, Regot S, and Green R (2020). Ribosome Collisions Trigger General Stress Responses to Regulate Cell Fate. Cell 182, 404–416 e414. 10.1016/j.cell.2020.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harding HP, Ordonez A, Allen F, Parts L, Inglis AJ, Williams RL, and Ron D (2019). The ribosomal P-stalk couples amino acid starvation to GCN2 activation in mammalian cells. eLife 8. 10.7554/eLife.50149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu F, Wei J, Cui X, Yu C, Ni W, Bungert J, Wu L, He C, and Qian Z (2021). Post-translational modification of RNA m6A demethylase ALKBH5 regulates ROS-induced DNA damage response. Nucleic acids research 49, 5779–5797. 10.1093/nar/gkab415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rubio RM, Depledge DP, Bianco C, Thompson L, and Mohr I (2018). RNA m(6) A modification enzymes shape innate responses to DNA by regulating interferon beta. Genes & development 32, 1472–1484. 10.1101/gad.319475.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fry NJ, Law BA, Ilkayeva OR, Carraway KR, Holley CL, and Mansfield KD (2018). N(6)-methyladenosine contributes to cellular phenotype in a genetically-defined model of breast cancer progression. Oncotarget 9, 31231–31243. 10.18632/oncotarget.25782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cho SH, Ha M, Cho YH, Ryu JH, Yang K, Lee KH, Han ME, Oh SO, and Kim YH (2018). ALKBH5 gene is a novel biomarker that predicts the prognosis of pancreatic cancer: A retrospective multicohort study. Annals of hepato-biliary-pancreatic surgery 22, 305–309. 10.14701/ahbps.2018.22.4.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dixit D, Xie Q, Rich JN, and Zhao JC (2017). Messenger RNA Methylation Regulates Glioblastoma Tumorigenesis. Cancer cell 31, 474–475. 10.1016/j.ccell.2017.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Farache D, Antine SP, and Lee ASY (2022). Moonlighting translation factors: multifunctionality drives diverse gene regulation. Trends in cell biology 32, 762–772. 10.1016/j.tcb.2022.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, and Church GM (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Farache D, Liu L, and Lee ASY (2022). Eukaryotic Initiation Factor 5A2 Regulates Expression of Antiviral Genes. Journal of molecular biology 434, 167564. 10.1016/j.jmb.2022.167564. [DOI] [PubMed] [Google Scholar]
- 71.Sun C, Todorovic A, Querol-Audi J, Bai Y, Villa N, Snyder M, Ashchyan J, Lewis CS, Hartland A, Gradia S, et al. (2011). Functional reconstitution of human eukaryotic translation initiation factor 3 (eIF3). Proceedings of the National Academy of Sciences of the United States of America 108, 20473–20478. 10.1073/pnas.1116821108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nature methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cantara WA, Hatterschide J, Wu W, and Musier-Forsyth K (2017). RiboCAT: a new capillary electrophoresis data analysis tool for nucleic acid probing. Rna 23, 240–249. 10.1261/rna.058404.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Darty K, Denise A, and Ponty Y (2009). VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics 25, 1974–1975. 10.1093/bioinformatics/btp250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Reuter JS, and Mathews DH (2010). RNAstructure: software for RNA secondary structure prediction and analysis. BMC bioinformatics 11, 129. 10.1186/1471-2105-11-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee AS, Burdeinick-Kerr R, and Whelan SP (2013). A ribosome-specialized translation initiation pathway is required for cap-dependent translation of vesicular stomatitis virus mRNAs. Proceedings of the National Academy of Sciences of the United States of America 110, 324–329. 10.1073/pnas.1216454109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011 17, 3. 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 78.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lawrence M, Huber W, Pages H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, and Carey VJ (2013). Software for computing and annotating genomic ranges. PLoS computational biology 9, e1003118. 10.1371/journal.pcbi.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Strimmer K (2008). fdrtool: a versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics 24, 1461–1462. 10.1093/bioinformatics/btn209. [DOI] [PubMed] [Google Scholar]
- 84.Smith T, Heger A, and Sudbery I (2017). UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome research 27, 491–499. 10.1101/gr.209601.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Webb S, Hector RD, Kudla G, and Granneman S (2014). PAR-CLIP data indicate that Nrd1-Nab3-dependent transcription termination regulates expression of hundreds of protein coding genes in yeast. Genome biology 15, R8. 10.1186/gb-2014-15-1-r8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shah A, Qian Y, Weyn-Vanhentenryck SM, and Zhang C (2017). CLIP Tool Kit (CTK): a flexible and robust pipeline to analyze CLIP sequencing data. Bioinformatics 33, 566–567. 10.1093/bioinformatics/btw653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ge SX, Jung D, and Yao R (2020). ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36, 2628–2629. 10.1093/bioinformatics/btz931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dennis G Jr., Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, and Lempicki RA (2003). DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome biology 4, P3. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1 ∣ Table of genes dysregulated in phosphomimetic eIF3d-expresssing cells during chronic ER stress, related to Figure 1.
Supplementary Table 2 ∣ Table of eIF3d cap-binding target mRNAs during chronic ER stress, related to Figure 2.
Supplementary Table 3 ∣ Table of m6A sites in eIF3d cell lines, related to Figure 2.
Data Availability Statement
All sequencing data have been deposited in the Gene Expression Omnibus under accession number GSE236188 and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-N6-methyladenosine | AbCam | Cat#151230; RRID:AB_2753144 |
| Mouse monoclonal anti-hsp90 | BD Biosciences | Cat#610418; RRID:AB_397798 |
| Rabbit polyclonal anti-eIF3b | Bethyl | Cat#A301-761A; RRID:AB_1210995 |
| Mouse monoclonal anti-ATF4 | Cell Signaling Technology | Cat#11815; RRID:AB_2616025 |
| Rabbit monoclonal anti-phospho-GCN2 | Cell Signaling Technology | Cat#94668; RRID:n/a |
| Rabbit polyclonal anti-GCN2 | Invitrogen | Cat#PA5-17523; RRID:AB_10983800 |
| Rabbit monoclonal anti-PERK | Cell Signaling Technology | Cat#C33E10; RRID:n/a |
| Rabbit polyclonal anti-rpS19 | Proteintech | Cat#15085-1-AP; RRID:AB_2180202 |
| Rabbit polyclonal anti-eIF2α | Proteintech | Cat#11170-1-AP; RRID:AB_2096489 |
| Rabbit monoclonal anti-ALKBH5 | Invitrogen | Cat#10H24L9; RRID:n/a |
| Goat anti-Rabbit HRP | Invitrogen | Cat#31462; RRID:AB_228338 |
| Goat anti-Mouse HRP | Invitrogen | Cat#31432; RRID:AB_228302 |
| Chemicals, peptides, and recombinant proteins | ||
| Fetal Bovine Serum | Biowest | Cat#S1620 |
| Thapsigargin | Cayman Chemical | Cat#10522 |
| Tunicamycin | Cayman Chemical | Cat#11445 |
| PERKi (GSK2656157) | MedChemExpress | Cat#HY-13820 |
| GCN2iB | MedChemExpress | Cat#HY-112654 |
| Cap-Clip Acid Pyrophosphatase | Cellscript | Cat#C-CC15011H |
| Bovine-Serum Albumin | Chemimpex | Cat#00577 |
| DTT | Chemimpex | Cat#00127 |
| Hybond-N+ membrane | Cytiva | Cat#RPN303B |
| Trypsin | Genesee | Cat#25-510 |
| Penicillin-streptomycin | Genesee | Cat#25-512 |
| Random Hexamer | Integrated DNA Technologies | Cat#51-01-18-01 |
| Murine RNase inhibitor | New England Biolabs | Cat#M0314L |
| Luna Universal qPCR master mix | New England Biolabs | Cat#M3003E |
| m7GpppG | New England Biolabs | Cat#S1411L |
| GpppG | New England Biolabs | Cat#S1407L |
| Q5 polymerase | New England Biolabs | Cat#M0491L |
| Vaccinia capping enzyme | New England Biolabs | Cat#M2080S |
| 2′ O-Methyltransferase enzyme | New England Biolabs | Cat#M0366S |
| Antarctic phosphatase | New England Biolabs | Cat#MO289L |
| T4 PNK | New England Biolabs | Cat#M0201L |
| XRN-1 | New England Biolabs | Cat#M0338L |
| 32P Radionuclide | Perkin-Elmer | Cat#NEX053H001MC |
| [ɑ-32P]-GTP | Perkin-Elmer | Cat#BLU006H250UC |
| Polyethylenimine, Linear, MW 25,000 | Polysciences, Inc | Cat#23966-1 |
| PhosSTOP | Sigma Aldrich | Cat#4906837001 |
| DMEM | Gibco | Cat#11995073 |
| OptiMEM | Gibco | Cat#11058021 |
| DMSO | ThermoFisher Scientific | Cat#BP231100 |
| Puromycin Dihydrochloride | ThermoFisher Scientific | Cat#AAJ61278MC |
| Sera-Mag Protein A/G beads | Cytiva | Cat#09-981-921 |
| Anti-HA magnetic beads | ThermoFisher Scientific, Pierce | Cat#PI88837 |
| SeraMag Oligo dT magnetic beads | Cytiva | Cat#09-981-145 |
| G-25 column | Cytiva | Cat#45 001 397 |
| Heparin | ThermoFisher Scientific | Cat#BP2425 |
| Cycloheximide | MP Biomedicals, Inc. | Cat#ICN10018301 |
| Phos-Tag acrylamide | Apexbio Technology | Cat#NC1646902 |
| SUPERase-In RNase Inhibitor | Invitrogen | Cat#AM2696 |
| Proteinase K | ThermoFisher Scientific | Cat#BP1700-100 |
| Phenol Chloroform | VWR | Cat#97064-712 |
| Glycogen | VWR | Cat#97063-256 |
| HIV-1 Protease | C. Schiffer, University of Massachusetts Medical School | |
| NEBuilder HiFi DNA Assembly | NEB | Cat#E2621L |
| EXPRE35S35S Protein Labeling Mix | Perkin Elmer | Cat#NEG072002MC |
| Critical commercial assays | ||
| NEBNext Ultra II Directional RNA Library Prep Kit for Illumina | New England Biolabs | Cat#E7760L |
| m6A eCLIP Kit | Eclipse Bioinnovations | Cat#143651 |
| Deposited data | ||
| RNA-Seq (WT versus phosphomimetic eIF3d HEK293T, veh. versus 16 h Tg) | This paper | GSE236188 |
| eIF3dTEV Subunit-Seq (HEK293T 16h Tg) | This paper | GSE236188 |
| m6A eCLIP (WT versus phosphomimetic eIF3d HEK293T, veh. versus 16 h Tg) | This paper | GSE236188 |
| Experimental models: Cell lines | ||
| Human: HEK293T | ATCC | CRL-11268 |
| Human: HEK293T eIF3dTEV | Lamper et al23 | |
| Human: HEK293T eIF3d S528D/S529D | This paper | |
| Human: HEK293T ALKBH5-ΔSL3 | This paper | |
| Oligonucleotides | ||
| DNA primers for RT-qPCR, see Table S4 | This paper | Table S4 |
| DNA primers for ALKBH5 cloning, see Table S4 | This paper | Table S4 |
| DNA primers for ATF4 cloning, see Table S4 | This paper | Table S4 |
| DNA primers for GCN2 cloning, see Table S4 | This paper | Table S4 |
| sgRNA sequences (eIF3d and ALKBH5), see Table S4 | This paper | Table S4 |
| Donor oligos (eIF3d and ALKBH5), see Table S4 | This paper | Table S4 |
| Recombinant DNA | ||
| Plasmid: pLBH269 SpCas9 sgRNA | L. Harrington and J. Doudna (UC Berkeley) | |
| Plasmid: pcDNA4-ALKBH5 5′ UTR-RLuc | This paper | |
| Plasmid: pcDNA4-ALKBH5 5′ UTR-RLuc – stem | This paper | |
| Plasmid: pcDNA4-ALKBH5 5′ UTR-RLuc – comp | This paper | |
| Plasmid: pcDNA4-ALKBH5 5′ UTR-RLuc – loop | This paper | |
| Plasmid: pcDNA4-GCN2 5′ UTR-RLuc | This paper | |
| Plasmid: pcDNA4-ATF4 5′ UTR-RLuc | This paper | |
| Plasmid: pGEX-4T1-PCIF1 | Boulias et al.48 | E. Greer, Washington University, St. Louis |
| Software and algorithms | ||
| CFX Maestro | Biorad | |
| Windaq | DataQ instruments | |
| RNAStructure | Reuter et al.75 | https://rna.urmc.rochester.edu/RNAstructure.html |
| VARNA | Darty et al.74 | https://varna.lri.fr |
| DAVID | Dennis et al.90 | https://david.ncifcrf.gov |
| ShinyGO | Ge et al.89 | http://bioinformatics.sdstate.edu/go/ |
| DESeq2 | Love et al.82 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Bowtie2 | Langmead et al.78 | https://bowtie-bio.sourceforge.net/bowtie2 |
| bwa | Li et al.86 | https://bio-bwa.sourceforge.net |
| Cutadapt | Martin77 | https://pypi.org/project/cutadapt/ |
| SAMtools | Li et al.80 | http://www.htslib.org |
| STAR | Dobin et al.79 | https://github.com/alexdobin/STAR |
| UMItools | Smith et al.84 | https://github.com/CGATOxford/UMI-tools |
| pyCRAC | Webb et al.85 | https://sandergranneman.bio.ed.ac.uk/pycrac-software |
| CTK | Shah et al.87 | https://zhanglab.c2b2.columbia.edu/index.php/CTK_Documentation |