

Abstract
The incretin system is an essential metabolic axis that regulates postprandial metabolism. The two incretin peptides that enable this effect are the glucose-dependent insulinotropic polypeptide (GIP) and the glucagon-like peptide 1 (GLP-1), which have cognate receptors (GIPR and GLP-1R) on islet β-cells as well as in other tissues. Pharmacologic engagement of the GLP-1R is a proven strategy for treating hyperglycemia in diabetes and for reducing body weight. Tirzepatide is the first monomeric peptide with dual activity at both incretin receptors now available for clinical use, and in clinical trials has shown unprecedented effects to reduce blood glucose and body weight. Here we discuss the foundational science that led to the development of monomeric multi-incretin receptor agonists, culminating in the development of tirzepatide. We also look to the future of this field and comment on how the concept of multi-receptor agonists will continue to progress for the treatment of metabolic disease.
Introduction
For decades now, there has been an urgent need for medicines capable of controlling body fat, managing excess weight and eliminating obesity. The lack of effective, scalable interventions to treat the general increase in human body weight that marks the past 50 years has made obesity one of the major public health concerns worldwide. The discovery of leptin1 revealed the existence of molecular systems controlling mammalian food intake, energy homeostasis and body weight. Yet, despite enormous growth in scientific understanding following this discovery, to date the only therapeutic approach offering weight loss of 20% or more is bariatric surgery. However, in the last twenty years drug development targeting the glucagon-like peptide 1 receptor (GLP-1R) has opened a new window of opportunity for reliable, significant medical weight loss at tolerable doses2 (Figure 1). More recently, the discovery of monomolecular peptides that simultaneously function at the GLP-1R and other family B G-protein coupled receptors (GPCR) as dual and triple receptor co-agonists has demonstrated clinical weight loss at levels not previously approached by medical therapy. As one example, tirzepatide, is a GIPR/GLP-1R co-agonist, and the first drug in this emerging class to achieve registration for treatment of adult-onset diabetes. It also demonstrated more than 20% placebo-corrected, weight loss in participants with obesity, without diabetes3. These results may herald the beginning of pharmaceutical management of excess weight with potential to reduce morbidity and mortality, analogous to treatment of hyperglycemia, dyslipidemia and hypertension.
Figure 1. Timeline of the discovery of seminal incretin-based therapies.
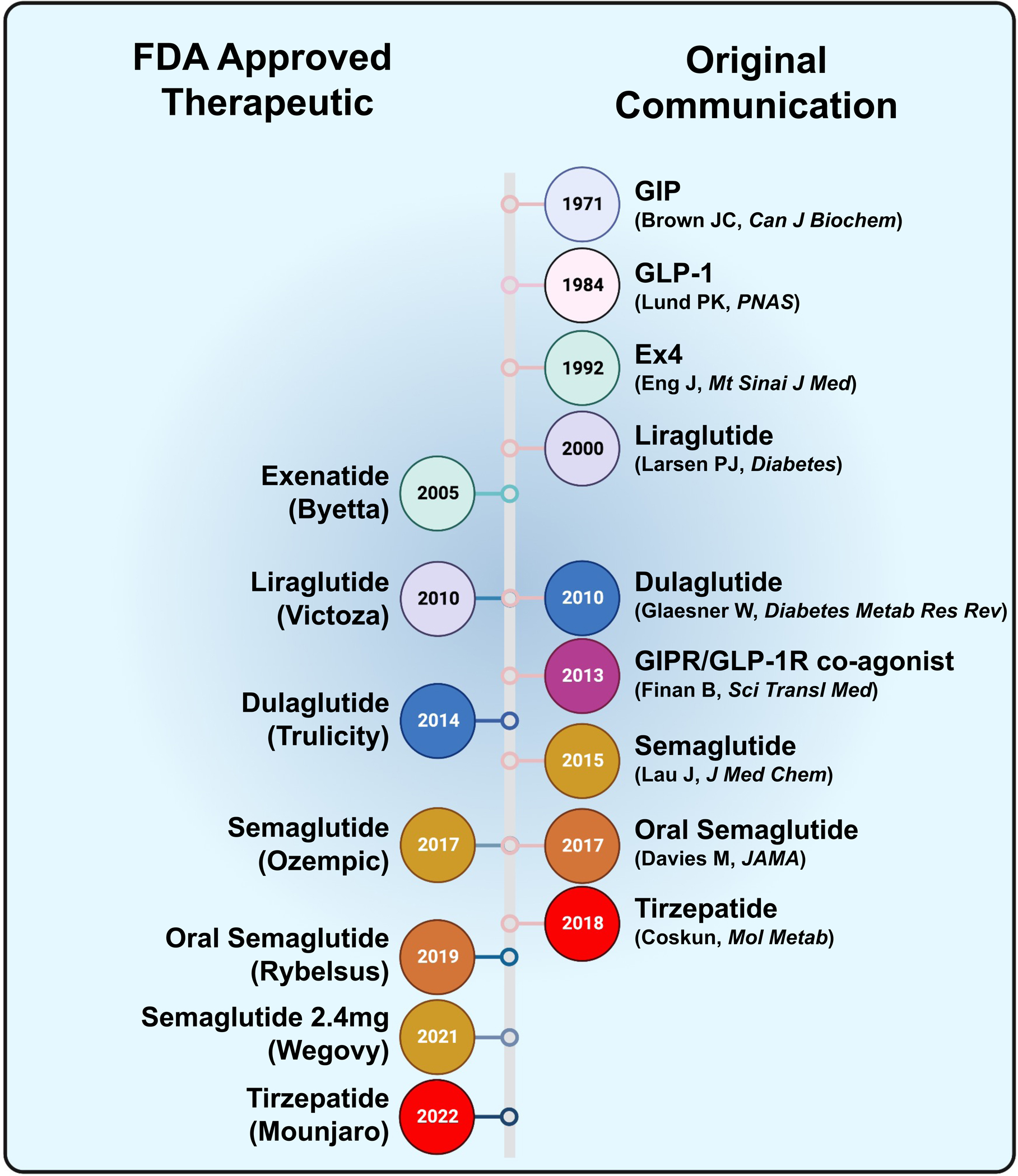
The left side indicates the year each therapy was approved by the United States Food and Drug Administration (FDA). The right side indicates the year the first communication was published.
Endocrine pathways exert broad control over metabolic homeostasis, exemplified by the pleiotropic actions of insulin. The discovery of insulin in 19214 has been fundamental in demonstrating the therapeutic benefit of hormones, but also in unlocking insights into normal physiology and pathophysiology across multiple metabolic networks. Similarly, glucagon was ‘accidentally’ discovered as a contaminant during optimization of insulin purification5, and then largely ignored for decades until the realization of its potential use to reverse insulin-induced hypoglycemia. This renewed interest in the physiology of glucagon and its receptor led to work that was the foundation of three Nobel prizes6–8. The actions of insulin and glucagon have been conceptualized as a dual hormonal balance that is central to the control of glucose metabolism, and provides a model for coordinate signaling in tissues like the liver. Even prior to the discovery of insulin, it had been proposed that hormonal factors produced in the intestine might have glucoregulatory actions9. Following the discovery of insulin, the Belgian physiologist La Barre demonstrated that duodenal extracts stimulate insulin secretion and termed these gut-derived hormones incretins10,11. Glucose-dependent insulinotropic polypeptide (GIP) was identified and validated as an incretin in the 1970s in research led by the Canadian physiologists Brown and Dupre12,13. Subsequent pursuit of additional incretins eventually led to the discovery of glucagon-like peptide 1 (GLP-1) in 198714–16. The relative contribution of GIP versus GLP-1 to mediate postprandial insulin secretion has been extensively studied and appears to differ along the continuum from metabolic health to type 2 diabetes (T2D). However, both preclinical17–20 and clinical 21,22 studies largely agree that GIP and GLP-1 are the two primary physiological incretins that mediate nutrient signaling from the intestine to the pancreatic islets, to properly regulate insulin secretion in control of postprandial glucose metabolism.
The incretin effect accounts for as much as 70% of postprandial insulin secretion and defects in the incretin axis significantly contributes to the impaired insulin secretion in T2D23. Consequently, medicinal agents have been designed to activate and enhance incretin receptor activity to reduce hyperglycemia and forestall T2D24,25. Three formative observations in incretin research have shaped this area of drug development, and elevated GLP-1 over GIP as the principal agent. First, infusions of GLP-1 were demonstrated to normalize blood glucose in participants with type 2 diabetes26,27. Second, several studies suggested that patients with T2D were insensitive to the insulinotropic actions of GIP, while GLP-1 was a potent insulin secretagogue in these participants28. Third, transgenic mice with deletion of the GIP receptor (GIPR) were protected from weight gain on a high fat diet, leading to the inference that GIP signaling promotes obesity29. Thus, the last 20 years have seen enormous thought and energy invested in the development of peptidic GLP-1R agonists (GLP-1RA). This work has included pharmacokinetic optimization, with steps to prevent peptide inactivation and to extend the pharmacodynamics to minimize the number of injections required for therapeutic effect. Once daily drugs caused larger reductions in hemoglobin A1c than twice daily agents, and weekly administration had an even bigger impact. One crucial observation made in the early clinical trials with GLP-1RA was that treated patients consistently lost weight relative to controls receiving placebo. This finding coincided with physiologic studies demonstrating that GLP-1R activation reduced food intake30–32. With the creation of more potent GLP-1RA, the weight-reducing effects were enhanced to a point where they were considered for treatment of people with obesity without diabetes2. A defining moment in the progression of GLP-1RA from primarily glucose lowering agents, to drugs used specifically for weight loss were the results from trials with the GLP-1RA semaglutide, that caused weight reduction of ~15% in participants with obesity without diabetes33. It is worth noting that moderate reductions in blood pressure are observed after treatment with GLP-1RAs34. Moreover, the results of cardiovascular outcome trials in persons with T2D given GLP-1RA demonstrating reduced stroke, myocardial infarction, and cardiovascular death35 along with the emerging evidence demonstrating positive effects of GLP-1RAs36,37 on kidney outcomes adds further impetus that these drugs have pleiotropic benefits. However, while the clinical results with semaglutide support indications for treatment of obesity per sé (WegovyR) as well as T2D (OzempicR), weight reduction with these agents remains considerably less than the best surgical results, and is often only a fraction of what most patients require for correction of the co-morbidities of increased body weight. Thus, even with progress in pharmacological weight loss, there remains a significant unmet need for obesity treatment.
Discovery of multi-receptor agonists for the GIPR and GLP-1R
The multifactorial nature of physiological control of metabolism and body weight, coupled with the demonstration that gastric bypass procedures increase secretion of GLP-1 and several other gut-derived hormones, suggested that engineering medicines combining GLP-1R activity with other hormonal stimuli might offer superior clinical outcomes. The first breakthrough confirming this hypothesis pertained to the seemingly antithetical combination of glucagon receptor (GCGR) activity with GLP-1R agonism, as a monomolecular, similarly sized co-agonist38. This dual agonist produced enhanced metabolic and body weight benefits compared to a GLP-1R monoagonist in preclinical models. Moreover, several versions of glucagon/GLP1 co-agonists are now progressing through clinical trials in participants with diabetes and obesity (Table 1), testing the translational potential of this first multi-receptor agonist (MRA) approach.
Table 1 –
Ongoing clinical trials for multi-receptor agonists
| Compound | Stage | Published Results |
|---|---|---|
| GLP1R/GCGR | ||
| LY3305677 | Phase 1b | 84 |
| Cotadutide | Phase 2b | 83 |
| BI-456906 | Phase 1 | |
| Efinopegduite | Phase 2 | 91 |
| ATL-801 | Phase 1 | |
| GLP1R/GIPR | ||
| Semaglutide + GIPR agonist | Phase 1 | |
| GLP1R/GIPR/GCGR | ||
| SAR441255 | Phase 1 | |
| LY3437943 | Phase 2 | 92 |
| HM15211 | Phase 2 | |
| GLPIR/Amylin | ||
| Semaglutide + Cagrilintide | Phase 1b | 90 |
A subsequent iteration on MRAs was the possibility that GIPR activation could bring different but equally valuable metabolic synergy in combination with GLP-1R agonism. Once again, the design strategy was contrary to the mainstream view in that GIPR agonism was thought to add little to glycemic lowering in people with diabetes, and could prove deleterious for weight gain. In fact, GIPR antagonism as a mechanism to improve metabolic outcomes has been proffered by some experts39. A series of studies between 2006 and 2013 led to the creation of active GIPR/GLP-1R co-agonists40. The goal here was to use combinatorial chemistry to create single molecules with multi-receptor potency comparable to the native incretins that might produce large effect sizes in preclinical studies such that effects were likely to be translatable to humans. It was established early on that GLP-1 and GIP are sufficiently homologous to enable a single peptide sequence with high potency agonism at both receptors. A series of rationally designed modifications included the interchange of specific GIP-derived amino acids, the addition of the C-terminally extended residues from the reptilian GLP-1R agonist, exendin-4, and select non-canonical amino acids at position 2 and 20. The result was a single peptide with subnanomolar, balanced activity at both incretin receptors and minimal activity at the glucagon receptor40. Additions of either fatty acyl or polyethylene glycol (PEG) side-chains did not interfere with the relative potencies at each receptor, and provided sustained duration of action in vivo by delaying renal clearance. In obese mice, the GIPR/GLP-1R co-agonist produced greater decreases in body weight and food intake, as well as improved glycemic and lipid outcomes, when compared to equal doses of either exendin-4 or liraglutide. The fatty acylated co-agonist also produced a greater rate of insulin secretion compared to liraglutide during a hyperglycemic clamp in cynomolgus monkeys40.
The original GIPR/GLP-1R agonist was modified with a C-18 acyl group to extend pharmacokinetics, and began Phase 1 clinical trials at Marcadia Biotech with Roche support (RG7697)41,42, leading to subsequent Phase 2 studies that were funded by Novo Nordisk (NNC0090–2746)43. The original drug candidate had pharmacokinetics that were suitable for daily dosing, and also dose limiting GLP-1R mediated GI adverse effects in healthy41 participants and those with T2D42. Phase 2 trials utilized a fixed 1.8 mg, once-daily dosing in participants with T2D for 12-weeks and included a titrated liraglutide (1.8mg) open-label reference arm43. The co-agonist reduced HbA1c by 1.36% and reduced body weight by 3.3%, while in participants receiving the active comparator liraglutide the respective differences were 0.96% and 1.7%. While the effects of the dual incretin receptor agonist were promising, the changes in glycemic control and body weight were not statistically different from liraglutide at the single dose tested. A longer study with an optimized dose in a dose-titrated manner in similar fashion to the unblinded control was planned as a more definitive test of clinical benefits. However, studies conducted concurrently showed unexpected superiority of once-weekly forms of GLP-1 drug candidates to daily forms, and raised questions as to whether the addition of GIP was necessary for a useful incretin agonist. Thus, the dual agonist was shelved, and the two-incretin concept pursued by adding a weekly dose of a novel, high potency GIPR agonist (NNC080–0389) to semaglutide (2.4 mg) in participants with T2D (ClinicalTrials.gov: NCT05144984), while also exploring the action of different molecular forms of longer-action, multimodal GIP-based agonists in preclinical studies.
Tirzepatide was the second dual incretin receptor agonist advanced to clinical study. While NNC0090–2746 was generated by chemical modification of a glucagon/GLP-1 co-agonist, tirzepatide was developed by selective alterations in the native GIP sequence to provide agonism at the GLP-1R 44. It possesses high affinity for both the GIPR (Ki = 0.135 nM) and the GLP-1R (Ki = 4.23 nM) in human cell lines. These binding affinities compare similarly to native GIP for its receptor but are approximately 5-fold lower when compared to native GLP-1 for its receptor 44. It is important to note that these values are based on the human GIPR (hGIPR), while tirzepatide is much less potent (30–100-fold reduced) at the mouse GIPR (mGIPR)45. The human GIP sequence, which differs from the mouse sequence, is a less potent and a partial agonist at the mGIPR46. This may be the basis of the relatively low affinity of tirzepatide at the mGIPR since it is derived from the human GIP sequence. Still, high-doses of tirzepatide stimulates insulin secretion and lowers glycemia in mice with selective deletion of either the Gipr or Glp1r, supporting in vivo activity at both receptors44,45. Receptor pharmacology studies suggest that tirzepatide engages the hGIPR in a manner similar to GIP but displays biased signaling at the GLP-1R to favor cAMP generation over beta-arrestin recruitment (Figure 2)47–49. Thus, in comparison to NNC0090–2746, which has balanced activity at both incretin receptors, tirzepatide potency is distorted towards hGIPR. Whether this imbalance is optimal and enhances the efficacy of tirzepatide remains to be determined. Support for this idea may come from an ongoing clinical trial where increased relative amounts of GIPR agonism (NNC0090–0389; 1:1 to 1:9) are provided with semaglutide. On the other hand, the pharmacology of NNC0090–0389 and semaglutide at their respective receptors differ from tirzpetide at the incretin receptors, providing an alternative reason if the results of various ratios are inconclusive. Remarkably, the structural differences between tirzepatide and NNC0090–2746 are modest (Figure 3). Both peptides start with a 28 amino acid sequence that is extended by the addition of an 11 or 12 amino acid C-terminal tail based on exendin-4. Twenty of the 28 amino acids are common to both peptides, with the greatest difference residing in the middle sequence where tirzepatide possesses homology with hGIP, while NNC0090–2764 is similar to GLP-1. Interestingly, the mid-section of tirzepatide also possesses a lysine residue to facilitate the 20-carbon fatty diacid, while NNC0090–2764 has a fatty-acyl group with a monoacid at the C-terminus. The location and composition of the fatty acid modification appears to be a crucial determinant in how tirzepatide engages both the GIPR and GLP-1R50. In addition to providing a pharmacokinetic profile that enables once-weekly dosing, the acylation moiety of tirzepatide also contributes to the interaction with both the GIPR and GLP-1R, as well as possibly the cell membrane50. The acylation does not appear to influence the potency of action at the GIPR, enabling tirzepatide to act as a full agonist at the human GIPR. However, this addition drives biased agonism at the GLP-1R, reducing beta-arrestin recruitment and subsequently leading to less internalization47,50. These studies reveal that the pharmacology profile of tirzepatide is not only determined by the peptide sequence, but also the location and composition of the acyl side chain.
Figure 2. Tirzepatide action in islet endocrine cells.
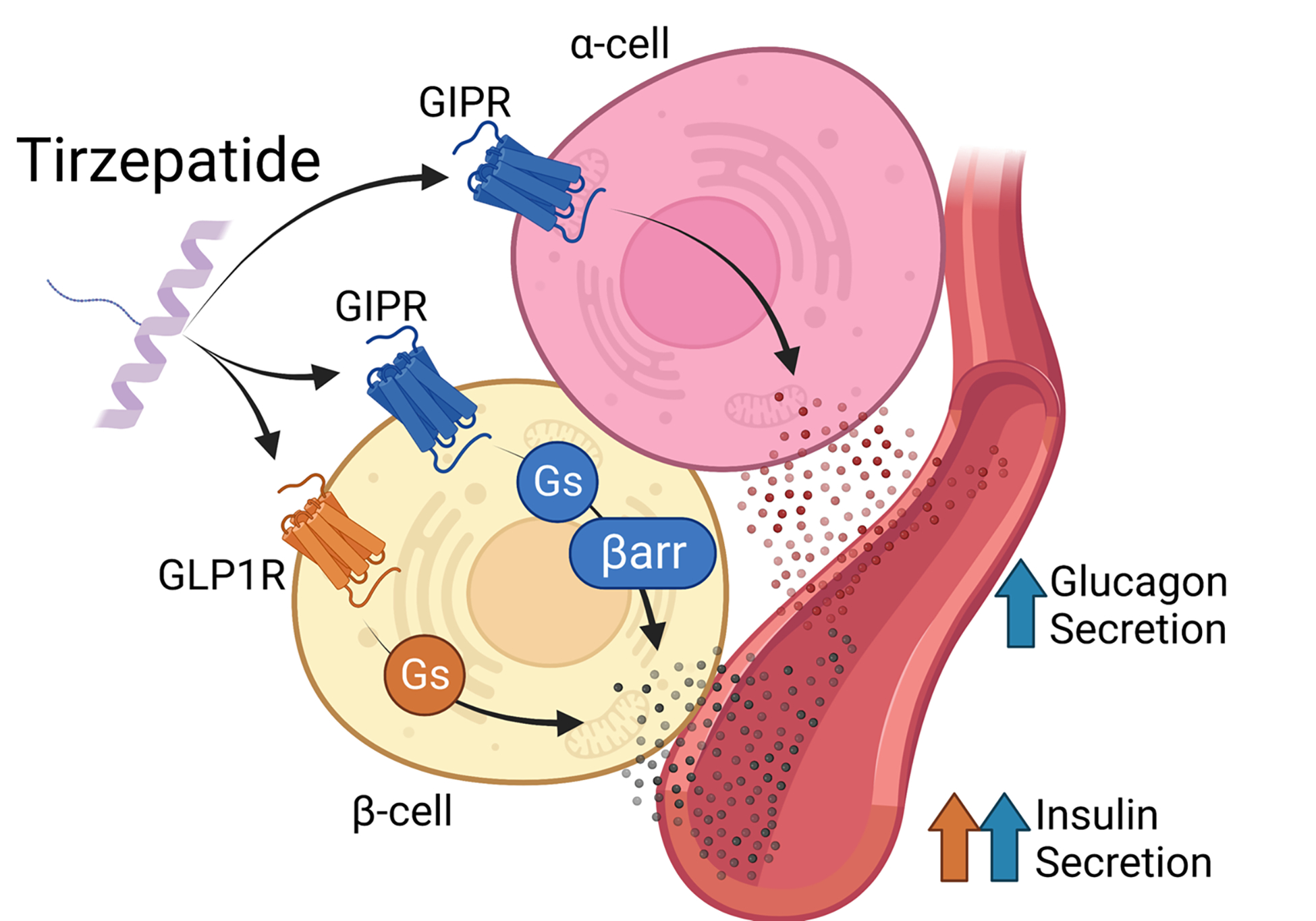
Tirzepatide stimulates insulin secretion through both the GLP-1R and GIPR in beta-cells. It engages the GIPR receptor similar to native GIP, acting as a full agonist to signal through both Gs/cAMP and β-arrestin (βarr) pathways. On the other hand, tirzpetide engages the GLP-1R in a biased manner, favoring Gs/cAMP signaling over β-arrestin. Tirzpeatide also stimulates glucagon secretion from alpha-cells through the GIPR through undefined mechanisms.
Figure 3. Amino acid sequence of human GIP, GLP-1, glucagon (GCG), NNC0090–2746, and tirzepatide (TZP).

Amino acids are colored according to their specificity for GIP (red), GLP-1 (blue), or glucagon (orange). Amino acids common to all three peptides are shown in gray, while amino acids shared between two peptides are shown with split colors. Aib (green) is a non-proteinogenic amino, while the c-terminal extension of exendin-4 (CEX) is shown in yellow.
A phase 2 clinical study with tirzepatide included four different once-weekly doses (1, 5, 10 and 15 mg) and used the GLP-1R monoagonist dulaglutide (1.5 mg), as an active comparator in people with T2D followed for 26 weeks43. The results of this first extended study were impressive, with the 15 mg dose of tirzepatide producing a 2.4% reduction in HbA1c (compared to 1.1% for dulaglutide) and 11.3 kg weight loss (compared to 4.8 kg for dulaglutide). Phase 3 clinical trials compared tirzepatide at 5, 10 and 15 mg doses to semaglutide (1 mg) in a 40-week trial of patients with T2D51. All doses of tirzepatide produced greater reductions in HbA1c, fasting glycemia, lipid levels, and weight loss compared to semaglutide. Remarkably, the 15 mg dose of tirzepatide decreased HbA1c by 2.46% and body weight by 12.4 kg at the end of the study. Tirzepatide was approved by the Food and Drug Administration in May 2022 for the treatment of T2D, and later in October received Fast Track designation for treatment of obesity. The remarkable impact on weight loss initiated the investigation of tirzepatide for the treatment of obesity independent of diabetes. Phase 3 clinical trials tested 5, 10 and 15 mg doses of tirzepatide for 72 weeks (Table 2)3. Placebo subtracted weight loss at the end of the study was 13.6% (5 mg), 19% (10mg), and 20.1% (15mg), and over 50% of the participants treated with 10 or 15 mg doses achieved greater than 20% weight loss. Pooled analysis of the tirzepatide-treated groups demonstrated clinically meaningful improvements in blood pressure, lipid profiles, and fasting insulin values. Whether the improvement in these outcomes is secondary to weight loss, or attributable to direct actions of GIPR/GLP1R agonism in key tissues (Figure 4) is currently the focus of ongoing mechanistic studies. This clinical trial did not include patients with T2D, and the emerging data suggests that the weight-loss effects of tirzepatide are reduced in patients with diabetes, a difference also observed in trials with 2.4 mg of semaglutide2. These results from longer trials with tirzepatide have elevated the performance standard for drug therapy directed at T2D and obesity. Most importantly, clinical assessment of cardiovascular safety of tirzepatide in patients with T2D at low, medium, and high cardiovascular risk are ongoing (Table 2), and it is not yet clear whether effects will be comparable to selective GLP-1R agonists52,53.
Table 2 –
Summary of clinical trials for tirzepatide
| Participants | Intervention | Primary Outcome | Results | |
|---|---|---|---|---|
| SURPASS | ||||
| 1 | T2D | Tirzepatide 5, 10 and 15 mg | Change in A1C | 93 |
| 2 | T2D | Tirzepatide vs 1.0 mg semaglutide | Change in A1C | 51 |
| 3 | T2D | Tirzepatide vs insulin degludec | Change in A1C | 94 |
| 4 | T2D | Tirzepatide vs insulin glargine | Change in A1C | 52 |
| 5 | T2D | Tirzepatide with insulin glargine | Change in A1C | 95 |
| 6 | T2D | Tirzepatide vs insulin lispro | Change in A1C | Anticipated end of 2023 |
| CVOT | T2D with CV risk | Tirzepatide vs dulaglutide | MACE-3 (Myocardial infarction, Stroke, CV death) | Anticipated 202453 |
| EARLY | T2D < 4 years | Tirzepatide vs. antihyperglycemic care | Change in A1C | Anticipated 2025 |
| SWITCH | T2D | Dulaglutide treatment continued or switched to tirzepatide | Change in A1C | Anticipated 2024 |
| SWITCH-2 | T2D | GLP-1R agonist treatment continued or switched to tirzepatide | Change in A1C | Anticipated end of 2023 |
| SURMOUNT | ||||
| 1 | Overweight/Obese | Tirzepatide 5, 10, and 15 mg | Change in body weight | 3 |
| 2 | Overweight/Obese T2D | Tirzepatide 5, 10, and 15 mg | Change in body weight | Anticipated 2023 |
| 3 | Overweight/Obese | Tirzepatide in combination with lifestyle intervention | Change in body weight | Anticipated 2023 |
| 4 | Overweight/Obese | Tirzepatide continuation versus switch to placebo | Change in body weight | Anticipated 2023 |
| 5 | Overweight/Obese | Tirzepatide vs 2.4 mg semaglutide | Change in body weight | Anticipated 2025 |
| MMO | Overweight/Obese | Tirzepatide vs placebo | Time to first occurrence of allcause death, nonfatal MI, nonfatal stroke, or heart failure | Anticipated 2027 |
| SUMMIT | Heart failure with preserved ejection fraction | Tirzepatide 5, 10 and 15 mg | All-cause mortality, hearth failure events, 6-minute walk test | Anticipated 2024 |
| SYNERGY-NASH | Overweight/Obese with NASH | Tirzepatide 5, 10 and 15 mg | NASH scoring | Anticipated 2024 |
Figure 4. Key metabolic sites of action for GLP-1 (blue) and GIP (red).
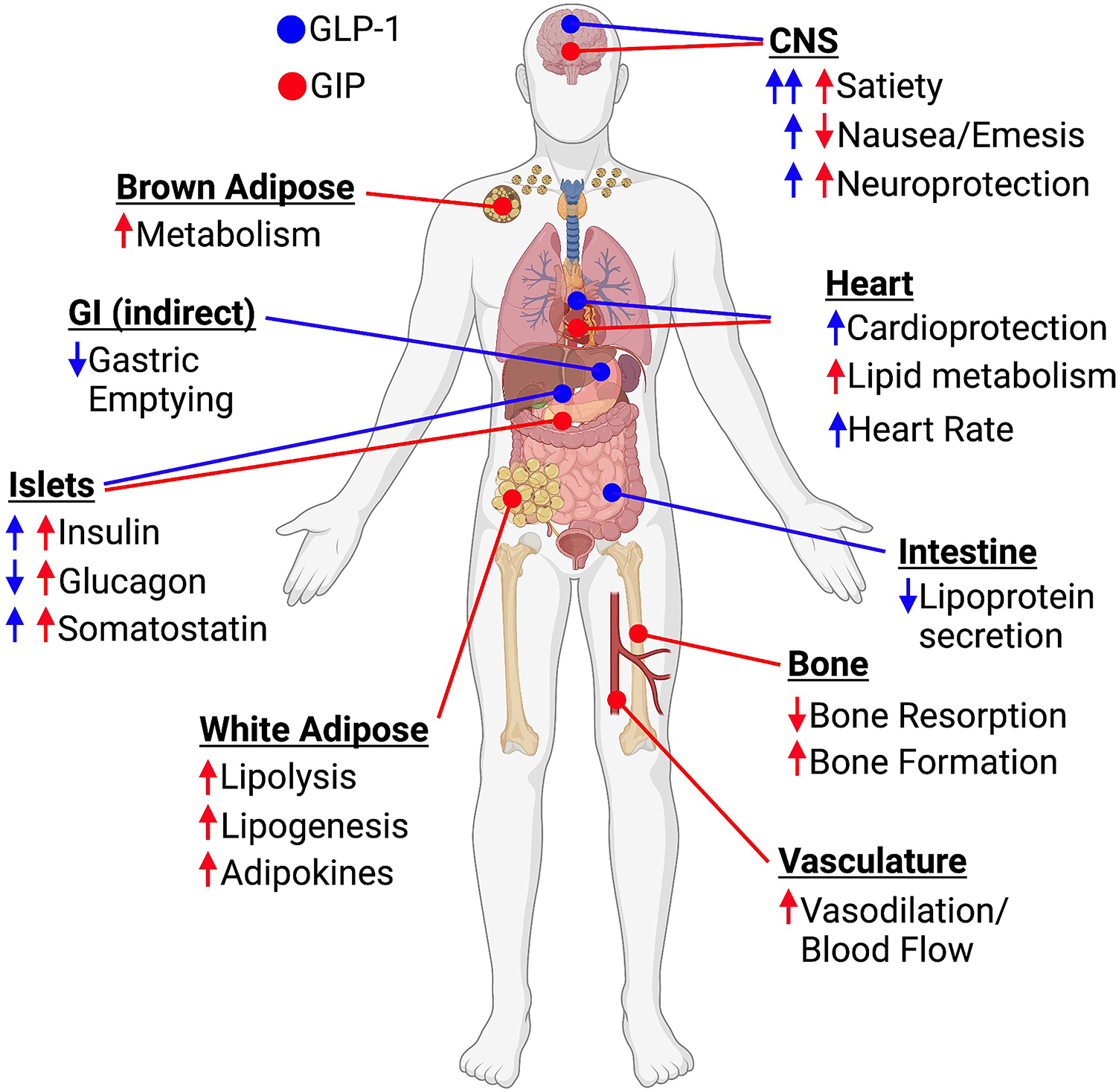
NNC0090–2764 and tirzepatide are potent agonists at both the GIPR and GLP-1R, and yet have reported different degrees of efficacy in glycemic control and weight loss. There are several potential explanations for this which may be useful in guiding development of next generation drug candidates with biological action at similar and different receptors. The longest duration study of NNC0090–2764 was only 12-weeks, while the most impressive results with tirzepatide were obtained at 26-weeks and longer. Twelve-week studies were initially designed to obtain a quality assessment of the impact on HbA1C but have proven to be of insufficient duration to confidently assess weight loss relative to placebo, and especially differential efficacy to other weight lowering drugs. Improvements in HbA1c and weight loss for the different doses of tirzepatide did not separate until past the 12-week period3,51, which is partially a function of the scheduled dose titration vital to achieving optimal clinical outcomes. Such a titration was not employed with NNC0090–2746 in the 12-week study, as commonly employed with once-daily liraglutide (the unblinded comparator). Another difference of considerable importance is the once-weekly form of action, where semaglutide at 10% the weekly dose of once daily liraglutide demonstrates twice the weight lowering efficacy. The difference in pharmacokinetics could account for unique receptor engagement that impacts the biological outcomes or more likely increased access to privileged sites that control appetite. In this latter regard, it is important to note that in obese rodents, daily administration of NNC0090–2746 and tirzepatide have effects that are much more similar than the clinical results would predict. In rodents, both peptides have abbreviated pharmacokinetics compared to humans as albumin clearance is nearly ten-times faster. In addition, the differences between the two co-agonists may be attributable to unique receptor pharmacology with tirzepatide engaging the GLP-1R in a manner different from GLP-1, including reduced beta-arrestin recruitment and internalization47,48,50. Furthermore, the relative balance of GIPR versus GLP-1R agonism is different between NNC0090–2764 and tirzepatide, with the latter favoring GIPR agonism. In mice, NNC0090–2764 demonstrates superior body weight reduction relative to pharmacokinetically-matched GLP-1R monoagonism and this difference was lost with ablation of CNS-GIPR expression54. These collective results support GIPR agonism as a key component for the enhanced efficacy of dual-agonists. However, how much relative GIPR to GLP-1R agonism is optimal, and whether biased agonism is rending a unique virtue to the clinical efficacy of tirzepatide are challenging, but tractable, questions requiring additional study.
Contribution of GIPR agonism
The GIPR is expressed in numerous locations throughout the body that potentially impact the metabolic processes that govern glycemic control and body weight (Figure 4)55. There is evidence for expression of the GIPR by alpha-, beta-, and delta-cells in pancreatic islets56–58, white and brown adipose tissue59,60, and various regions of the CNS54,61,62. Understanding how pharmacological GIPR activation in these cell types regulates physiological and clinical outcomes independently and in concert with GLP-1R activation is necessary to delineate the mechanism of actions for MRA that incorporate GIPR activity.
Islets:
The GIPR is expressed to a similar degree in all three major endocrine cell types of the pancreatic islets. In beta-cells, GIP stimulates insulin secretion in a glucose-dependent manner56, and in alpha-cells, GIP stimulates glucagon in an amino-acid and glucose-dependent manner57. Activity of GIP in both alpha- and beta-cells is needed for insulin secretion and glycemic control following a meal57. GIP stimulates somatostatin secretion, however, the factors that govern this, and the implications for metabolism have not been worked out. GIP and GLP-1 are known to have additive effects on insulin secretion when infused at physiological levels into healthy humans63, but it is unclear if GIP can provide additive effects to pharmacological levels of GLP-1R agonism. Moreover, the additive effects between GIP and GLP-1 are lost when given to participants with T2D64, potentially due to the observation that the insulin response to physiological levels of infused GIP are decreased in participants with T2D28. Interestingly, maintaining a period of euglycemia restores some of the insulinotropic activity of GIP in patients with T2D65. Thus, combining GIPR and GLP-1R agonism may enhance insulin secretion in patients with T2D by first rescuing dysfunctional beta-cells with GLP-1R agonism followed by additive effects on insulin secretion provided by both receptors. Moreover, GIP has been shown to stimulate insulin secretion through both direct actions on beta-cells and indirect actions mediated by alpha- to beta-cell communication57 and GIP is considered the predominant physiological incretin with respect to insulin secretion21. However, direct evidence that the GIPR activity of tirzepatide contributes to the improved glycemic control in patients with T2D has not been established, although antagonizing the GIPR reduced tirzepatide-stimulated insulin secretion in isolated human islets to a greater degree than antagonizing the GLP-1R45. With respect to the alpha-cell, GLP-1R agonists reduce glucagon secretion, GIPR agonists enhance glucagon secretion and the combined actions of GIP and GLP-1 infusion on glucagon secretion is offset to produce a neutral outcome64. To this end, tirzepatide robustly stimulated glucagon secretion in isolated human islets, further emphasizing a meaningful contribution of tirzepatide activity at the GIPR (Figure 2)45.
Central nervous system (CNS):
Chronic peripheral54,66 and central54 GIPR agonism decreases body weight by reduction in food intake, suggesting a CNS-mediated mechanism. In line with this notion, GIPR is expressed in the hypothalamus and hindbrain54,61,62,67,68, and a single 3rd ventricle bolus injection of GIP is sufficient to decrease body weight and food intake in diet-induced obese mice54. When injected into the brain or the periphery, GIP activates neurons in the hypothalamus54, and targeted (DREADD-mediated) activation of hypothalamic GIPR neurons decreases food intake in mice61. Confirming the metabolic relevance of central GIPR signaling, neuronal loss of GIPR renders mice resistant to GIP-induced body weight loss and GIPR/GLP-1R co-agonism is more potent than selective GLP-1R agonism for weight loss54. Interestingly, intracerebroventricular administration of an antibody directed against the GIPR decreases body weight, along with an increase in hypothalamic leptin signaling62. Future studies will have to address how that finding can be reconciled with the abundance of clinical studies showing superior metabolic benefits of molecules activating the human GIP receptor, including dissection of weight loss components into fluid as well as bone-, fat-, and muscle mass. Other groups have demonstrated that chronic systemic GIPR antagonism produces a moderate69,70 to null66 effect on body weight in obese mice, suggesting this is not an efficacious mechanism to pursue alone. However, GIPR antagonism seems to synergize with GLP-1R agonism to produce robust effects on body weight66,69, although the mechanism of action has not been described, raising the question as to how both pharmacological agonism and antagonism of the GIPR can produce similar decreases in weight loss. Interestingly, neither peripheral antagonism of the GIPR or knockout of the GIPR gene produce strong reductions in food intake, suggesting that the reduction in body weight stems from a separate, and yet to be fully resolved mechanism. On the other hand, GIPR agonism does reduce food intake in a manner that requires GIPR expression in the CNS54. Proposed mechanisms for this include activation of hypothalamic neurons that suppress food intake61 and activation of hindbrain neurons that engage an anti-emetic effect67,71. The latter could have important implications for the mechanism of tirzepatide, as GIPR neurons in the hindbrain could temper the nausea induced by activation of neighboring GLP-1R neurons67, enabling greater systemic GLP-1R activity with manageable gastrointestinal effects. However, there is a clear need for a better understanding of the effects of GIPR in the CNS, including the translation of these preclinical results.
Insulin sensitivity:
In addition to increasing insulin secretion, there is emerging evidence that GIPR agonism could also increase peripheral insulin sensitivity. Chronic treatment with tirzepatide enhanced insulin sensitivity in obese mice through a mechanism that was independent of weight loss and the GLP-1R72. Moreover, these insulin-sensitizing effects of tirzepatide could be replicated with chronic GIPR agonism alone. Interestingly, the enhanced insulin-mediated glucose disposal was attributed to a greater degree of glucose uptake into adipose tissue but not skeletal muscle; nor were there changes in hepatic glucose production. Adipose tissue expresses the GIPR, whereas muscle and liver do not, arguing for a direct action of GIPR agonism in adipose tissue to improved insulin sensitivity. GIPR agonism in adipose tissue has been argued to enhance the lipid-buffering capacity of white adipose tissue, promoting advantageous storage of excess fat and limiting ectopic storage in less efficient storage sites like muscle or liver73. Healthy storage of adipose tissue delays the progressive insulin resistance associated with obesity74, providing a plausible hypothesis that if GIPR agonism in adipose tissue can promote lipid storage in the correct location, this could result in positive effects on insulin sensitivity. This hypothesis is supported by previous reports that argue for beneficial effects of GIP activity in adipose tissue75,76, while others also argue that GIPR has negative effects in adipose tissue77,78. Further complicating these interpretations are recent reports that the GIPR is expressed predominantly in the non-adipocyte fractions of both mouse and human adipose tissue60, illustrating the need to clarify how GIPR agonists regulate adipose tissue function and how this might impact insulin sensitivity.
GIPR agonism versus antagonism
The utility of GIPR antagonism paired with GLP-1R agonism is also being explored79, with preclinical compounds utilizing this strategy producing similar effects on weight loss to what has been reported for GIPR/GLP-1R agonism69. Rationale for this approach is founded on the phenotype of loss-of-function models for the GIPR that produce a protective effect against diet-induced obesity including germline deletion of Gipr29 and deletion of Gip80. Moreover, loss of function mutations in the human GIPR are associated with reductions in BMI81. Interestingly, germline deletion of Glp1r also produced protection against diet-induced obesity17,82 which can be mimicked with pharmacological antagonism of the GLP-1R70, providing a similar paradox of gain- and loss-of-function studies for the GLP-1 system on body weight. The mechanism by which antagonism of the incretin system can reduce body weight remains unclear, but seems like an area where understanding would be valuable. In addition, it is essential to understand the full implications of chronic GIPR antagonism on parameters beyond body weight, in areas where GIPR has been shown to have positive effects including islet function, glucose tolerance, bone health, and immune function. As the clinical investigation of GIPR antagonism continues, it will be essential to monitor these outcomes.
What’s next for multi-receptor agonists
The recent success and clinical implementation of tirzepatide has accelerated interest in the pursuit of MRA. Every current candidate continues to utilize GLP-1R agonism as a cornerstone component (Table 1). There is considerable interest in pairing GLP-1R agonism with glucagon receptor agonism to enhance both energy expenditure to maximize weight loss and potentially leverage the lipid oxidative properties of glucagon in hepatocytes to target nonalcoholic fatty liver disease. Early clinical phase 1 data has provided mixed results on body weight, with some compounds failing to outperform GLP-1R agonists83, while others have shown greater efficacy84. Some GLP-1R/GCGR agonists have produced greater than 10% decreases in weight loss in a 12-week period, which matches the rate of tirzepatide and illustrates the potential of this strategy. Separate from body weight, the outcomes on markers of hepatic steatosis, fibrosis and inflammation will test the utilization of these compounds for nonalcoholic steatohepatitis (NASH); tirzepatide is also being tested for use in NASH (Table 2). In contrast single molecule MRA, the combination of semaglutide and a long-acting GIPR agonists is an alternative application of dual incretin agonist signaling under investigation. There is also motivation for combining activity at both incretin receptors with glucagon receptor activity through the development of triagonists. Recent phase 1 trial results of the GIP/GLP-1/glucagon triple co-agonist LY3437943 showed promising reductions in body weight and blood glucose as well as a safety profile consistent with those of other incretin-based therapeutic agents in early phases of development85,86. Phase 2 trials showed 24.2% weight loss with the highest dose (12 mg) of LY3437943 in 48 weeks, with 26% of the participants in this group losing greater than 30% BW87. There is also tremendous activity in the development of small molecule agonists targeting the incretin system, to produce drugs that are orally available and more economical to produce. The most advanced compounds are small molecule GLP-1R agonists which have progressed through to clinical trials88,89. There is considerable interest in the development of small molecule GIPR and GCGR agonists, most likely to be paired with GLP-1R agonists. Whether this area could expand to have single small molecules that target multiple receptors remains to be seen. Finally, the combination of GLP-1R agonism with a long-acting amylin analogue has demonstrated promising effects on body weight. The highest dose (2.4 mg of each agonist) decreased body weight in participants with overweight by 17% in 20 weeks90. Whether these results demonstrate the ceiling for pharmacologically targeting body weight has yet to be met.
Conclusion
Historically, pharmacological targeting of obesity has been largely unproductive, with interventions that produced safe weight loss reducing body weight by 5% or less. Almost three decades ago, the discovery of leptin triggered a new era of molecular obesity research. Since then, a multitude of signals have been uncovered that are involved in the regulation of human body weight, energy homeostasis and fat mass. However, until recently targeting any single pathway has not conferred benefits comparable to those of bariatric surgery. The discovery of human gut hormone dual and triple agonists has opened new possibilities for medical weight loss. Combining GIP and GLP-1R agonism was first discovered and reported a decade ago40 and has now fully evolved as the GIP/GLP-1R co-agonist tirzepatide that can achieve more than 20% weight loss in obesity as well as offering superior benefits in diabetes. Understanding the mechanisms that drive the efficacy of this combination will no doubt be aggressively pursued in the coming years, along with the continued emergence of additional combinatorial approaches, further expanding pharmacological options for a more personalized metabolic medicine of the future.
Acknowledgements
This work was co-funded by the NIH (DK123075, DK125353, DK046492) and the European Research Council ERC-CoG Trusted no.101044445. TDM further received funding from German Research Foundation (DFG TRR296, TRR152, SFB1123 and GRK 2816/1) and the German Center for Diabetes Research (DZD e.V.). MHT received funding from the European Research Council ERC AdG HypoFlam no. 695054.
Footnotes
Declaration of Interests
JEC and DAD receive funding to carry out basic research from Eli Lilly, Novo Nordisk, and Merck MSD. JEC has served in an advisory role for Boehringer Ingelheim and Structure Therapeutics. DAD has served in an advisory role for Structure Therapeutics and Eli Lilly.
In this review, Campbell et al. discuss the development of multireceptor therapeutics that target incretin receptors to improve glycemic control and induce weight loss. This is a twenty-year perspective of how dual incretin agonists have evolved, culminating in the clinical use of tirzepatide, along with a discussion of potential mechanisms by which GIPR agonism can enhance the clinical efficacy of GLP-1R agonists.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhang Y, et al. Positional cloning of the mouse obese gene and its human homologue. Nature 372, 425–432 (1994). [DOI] [PubMed] [Google Scholar]
- 2.Kumar N & D’Alessio DA Slow and Steady Wins the Race: 25 Years Developing the GLP-1 Receptor as an Effective Target for Weight Loss. J Clin Endocrinol Metab 107, 2148–2153 (2022). [DOI] [PubMed] [Google Scholar]
- 3.Jastreboff AM, et al. Tirzepatide Once Weekly for the Treatment of Obesity. N Engl J Med 387, 205–216 (2022). [DOI] [PubMed] [Google Scholar]
- 4.Banting FG Early Work on Insulin. Science 85, 594–596 (1937). [DOI] [PubMed] [Google Scholar]
- 5.Kimball CP & Murlin JR AQUEOUS EXTRACTS OF PANCREAS: III. SOME PRECIPITATION REACTIONS OF INSULIN. Journal of Biological Chemistry 58, 337–346 (1923). [Google Scholar]
- 6.Capozzi ME, D’Alessio DA & Campbell JE The past, present, and future physiology and pharmacology of glucagon. Cell Metab 34, 1654–1674 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scheen AJ & Lefebvre PJ Glucagon, from past to present: a century of intensive research and controversies. Lancet Diabetes Endocrinol (2022). [DOI] [PubMed] [Google Scholar]
- 8.Sutherland EW & De Duve C Origin and distribution of the hyperglycemic-glycogenolytic factor of the pancreas. J Biol Chem 175, 663–674 (1948). [PubMed] [Google Scholar]
- 9.Bayliss WM & Starling EH The mechanism of pancreatic secretion. J Physiol 28, 325–353 (1902). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.LaBarre J Sur les possibilités d’un traitement du diabète par I’incrétine. Bull Acad R Med Belg 12, 620–634 (1932). [Google Scholar]
- 11.Zunz E & Barre JL Contributions à l’étude des variations physiologiques de la sécrétion interne du pancréas. Archives Internationales de Physiologie 31, 162–179 (1929). [Google Scholar]
- 12.Brown JC & Dryburgh JR A gastric inhibitory polypeptide. II. The complete amino acid sequence. Can J Biochem 49, 867–872 (1971). [DOI] [PubMed] [Google Scholar]
- 13.Dupre J, Ross SA, Watson D & Brown JC Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J Clin Endocrinol Metab 37, 826–828 (1973). [DOI] [PubMed] [Google Scholar]
- 14.Holst JJ, Orskov C, Nielsen OV & Schwartz TW Truncated glucagon-like peptide I, an insulin-releasing hormone from the distal gut. FEBS Lett 211, 169–174 (1987). [DOI] [PubMed] [Google Scholar]
- 15.Kreymann B, Williams G, Ghatei MA & Bloom SR Glucagon-like peptide-1 7–36: a physiological incretin in man. Lancet 2, 1300–1304 (1987). [DOI] [PubMed] [Google Scholar]
- 16.Mojsov S, Weir GC & Habener JF Insulinotropin: glucagon-like peptide I (7–37) co-encoded in the glucagon gene is a potent stimulator of insulin release in the perfused rat pancreas. J Clin Invest 79, 616–619 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hansotia T, et al. Extrapancreatic incretin receptors modulate glucose homeostasis, body weight, and energy expenditure. J Clin Invest 117, 143–152 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miyawaki K, et al. Glucose intolerance caused by a defect in the entero-insular axis: a study in gastric inhibitory polypeptide receptor knockout mice. Proc Natl Acad Sci U S A 96, 14843–14847 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Preitner F, et al. Gluco-incretins control insulin secretion at multiple levels as revealed in mice lacking GLP-1 and GIP receptors. J Clin Invest 113, 635–645 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scrocchi LA, et al. Glucose intolerance but normal satiety in mice with a null mutation in the glucagon-like peptide 1 receptor gene. Nat Med 2, 1254–1258 (1996). [DOI] [PubMed] [Google Scholar]
- 21.Gasbjerg LS, et al. Separate and Combined Glucometabolic Effects of Endogenous Glucose-Dependent Insulinotropic Polypeptide and Glucagon-like Peptide 1 in Healthy Individuals. Diabetes 68, 906–917 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Salehi M, Aulinger B, Prigeon RL & D’Alessio DA Effect of endogenous GLP-1 on insulin secretion in type 2 diabetes. Diabetes 59, 1330–1337 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nauck M, Stockmann F, Ebert R & Creutzfeldt W Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 29, 46–52 (1986). [DOI] [PubMed] [Google Scholar]
- 24.Baggio LL & Drucker DJ Glucagon-like peptide-1 receptor co-agonists for treating metabolic disease. Mol Metab 46, 101090 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Drucker DJ GLP-1 physiology informs the pharmacotherapy of obesity. Mol Metab 57, 101351 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nauck MA, et al. Normalization of fasting hyperglycaemia by exogenous glucagon-like peptide 1 (7–36 amide) in type 2 (non-insulin-dependent) diabetic patients. Diabetologia 36, 741–744 (1993). [DOI] [PubMed] [Google Scholar]
- 27.Rachman J, et al. Normalization of insulin responses to glucose by overnight infusion of glucagon-like peptide 1 (7–36) amide in patients with NIDDM. Diabetes 45, 1524–1530 (1996). [DOI] [PubMed] [Google Scholar]
- 28.Nauck MA, et al. Preserved incretin activity of glucagon-like peptide 1 [7–36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest 91, 301–307 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miyawaki K, et al. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med 8, 738–742 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Tang-Christensen M, et al. Central administration of GLP-1-(7–36) amide inhibits food and water intake in rats. Am J Physiol 271, R848–856 (1996). [DOI] [PubMed] [Google Scholar]
- 31.Turton MD, et al. A role for glucagon-like peptide-1 in the central regulation of feeding. Nature 379, 69–72 (1996). [DOI] [PubMed] [Google Scholar]
- 32.Van Dijk G, et al. Central infusions of leptin and GLP-1-(7–36) amide differentially stimulate c-FLI in the rat brain. Am J Physiol 271, R1096–1100 (1996). [DOI] [PubMed] [Google Scholar]
- 33.Wilding JPH, et al. Once-Weekly Semaglutide in Adults with Overweight or Obesity. N Engl J Med 384, 989–1002 (2021). [DOI] [PubMed] [Google Scholar]
- 34.Hernandez AF, et al. Albiglutide and cardiovascular outcomes in patients with type 2 diabetes and cardiovascular disease (Harmony Outcomes): a double-blind, randomised placebo-controlled trial. Lancet 392, 1519–1529 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Drucker DJ The Ascending GLP-1 Road From Clinical Safety to Reduction of Cardiovascular Complications. Diabetes 67, 1710–1719 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Drucker DJ & Holst JJ The expanding incretin universe: from basic biology to clinical translation. Diabetologia (2023). [DOI] [PubMed] [Google Scholar]
- 37.Sato T, et al. Possible Advantage of Glucagon-Like Peptide 1 Receptor Agonists for Kidney Transplant Recipients With Type 2 Diabetes. J Clin Endocrinol Metab (2023). [DOI] [PubMed] [Google Scholar]
- 38.Day JW, et al. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat Chem Biol 5, 749–757 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Killion EA, et al. Glucose-Dependent Insulinotropic Polypeptide Receptor Therapies for the Treatment of Obesity, Do Agonists = Antagonists? Endocr Rev 41(2020). [DOI] [PubMed] [Google Scholar]
- 40.Finan B, et al. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci Transl Med 5, 209ra151 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Portron A, Jadidi S, Sarkar N, DiMarchi R & Schmitt C Pharmacodynamics, pharmacokinetics, safety and tolerability of the novel dual glucose-dependent insulinotropic polypeptide/glucagon-like peptide-1 agonist RG7697 after single subcutaneous administration in healthy subjects. Diabetes Obes Metab 19, 1446–1453 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Schmitt C, Portron A, Jadidi S, Sarkar N & DiMarchi R Pharmacodynamics, pharmacokinetics and safety of multiple ascending doses of the novel dual glucose-dependent insulinotropic polypeptide/glucagon-like peptide-1 agonist RG7697 in people with type 2 diabetes mellitus. Diabetes Obes Metab 19, 1436–1445 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Frias JP, et al. The Sustained Effects of a Dual GIP/GLP-1 Receptor Agonist, NNC0090–2746, in Patients with Type 2 Diabetes. Cell Metab 26, 343–352 e342 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Coskun T, et al. LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: From discovery to clinical proof of concept. Mol Metab 18, 3–14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.El K, et al. The incretin co-agonist tirzepatide requires GIPR for hormone secretion from human islets. Nat Metab (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sparre-Ulrich AH, et al. Species-specific action of (Pro3)GIP - a full agonist at human GIP receptors, but a partial agonist and competitive antagonist at rat and mouse GIP receptors. Br J Pharmacol 173, 27–38 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Novikoff A, et al. Spatiotemporal GLP-1 and GIP receptor signaling and trafficking/recycling dynamics induced by selected receptor mono- and dual-agonists. Mol Metab 49, 101181 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Willard FS, et al. Tirzepatide is an imbalanced and biased dual GIP and GLP-1 receptor agonist. JCI Insight 5(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou H, Zhang T, Harmon JS, Bryan J & Robertson RP Zinc, not insulin, regulates the rat alpha-cell response to hypoglycemia in vivo. Diabetes 56, 1107–1112 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Sun B, et al. Structural determinants of dual incretin receptor agonism by tirzepatide. Proc Natl Acad Sci U S A 119, e2116506119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frias JP, et al. Tirzepatide versus Semaglutide Once Weekly in Patients with Type 2 Diabetes. N Engl J Med 385, 503–515 (2021). [DOI] [PubMed] [Google Scholar]
- 52.Del Prato S, et al. Tirzepatide versus insulin glargine in type 2 diabetes and increased cardiovascular risk (SURPASS-4): a randomised, open-label, parallel-group, multicentre, phase 3 trial. Lancet 398, 1811–1824 (2021). [DOI] [PubMed] [Google Scholar]
- 53.Sattar N, et al. Tirzepatide cardiovascular event risk assessment: a pre-specified meta-analysis. Nat Med 28, 591–598 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Q, et al. The glucose-dependent insulinotropic polypeptide (GIP) regulates body weight and food intake via CNS-GIPR signaling. Cell Metab 33, 833–844 e835 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Campbell JE & Drucker DJ Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab 17, 819–837 (2013). [DOI] [PubMed] [Google Scholar]
- 56.Campbell JE, et al. TCF1 links GIPR signaling to the control of beta cell function and survival. Nat Med 22, 84–90 (2016). [DOI] [PubMed] [Google Scholar]
- 57.El K, et al. GIP mediates the incretin effect and glucose tolerance by dual actions on alpha cells and beta cells. Sci Adv 7(2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gray SM, et al. Discordance between GLP-1R gene and protein expression in mouse pancreatic islet cells. J Biol Chem 295, 11529–11541 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Beaudry JL, et al. Physiological roles of the GIP receptor in murine brown adipose tissue. Mol Metab 28, 14–25 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Campbell JE, et al. GIPR Is Predominantly Localized to Nonadipocyte Cell Types Within White Adipose Tissue. Diabetes 71, 1115–1127 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Adriaenssens AE, et al. Glucose-Dependent Insulinotropic Polypeptide Receptor-Expressing Cells in the Hypothalamus Regulate Food Intake. Cell Metab 30, 987–996 e986 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kaneko K, et al. Gut-derived GIP activates central Rap1 to impair neural leptin sensitivity during overnutrition. J Clin Invest 129, 3786–3791 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nauck MA, Bartels E, Orskov C, Ebert R & Creutzfeldt W Additive insulinotropic effects of exogenous synthetic human gastric inhibitory polypeptide and glucagon-like peptide-1-(7–36) amide infused at near-physiological insulinotropic hormone and glucose concentrations. J Clin Endocrinol Metab 76, 912–917 (1993). [DOI] [PubMed] [Google Scholar]
- 64.Mentis N, et al. GIP does not potentiate the antidiabetic effects of GLP-1 in hyperglycemic patients with type 2 diabetes. Diabetes 60, 1270–1276 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hojberg PV, et al. Four weeks of near-normalisation of blood glucose improves the insulin response to glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes. Diabetologia 52, 199–207 (2009). [DOI] [PubMed] [Google Scholar]
- 66.Mroz PA, et al. Optimized GIP analogs promote body weight lowering in mice through GIPR agonism not antagonism. Mol Metab 20, 51–62 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Borner T, et al. GIP Receptor Agonism Attenuates GLP-1 Receptor Agonist-Induced Nausea and Emesis in Preclinical Models. Diabetes 70, 2545–2553 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ludwig MQ, Todorov PV, Egerod KL, Olson DP & Pers TH Single-Cell Mapping of GLP-1 and GIP Receptor Expression in the Dorsal Vagal Complex. Diabetes 70, 1945–1955 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Killion EA, et al. Anti-obesity effects of GIPR antagonists alone and in combination with GLP-1R agonists in preclinical models. Sci Transl Med 10(2018). [DOI] [PubMed] [Google Scholar]
- 70.Svendsen B, et al. Pharmacological antagonism of the incretin system protects against diet-induced obesity. Mol Metab 32, 44–55 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Samms RJ, et al. GIPR Agonism Inhibits PYY-Induced Nausea-Like Behavior. Diabetes 71, 1410–1423 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Samms RJ, et al. GIPR agonism mediates weight-independent insulin sensitization by tirzepatide in obese mice. J Clin Invest 131(2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Samms RJ, Coghlan MP & Sloop KW How May GIP Enhance the Therapeutic Efficacy of GLP-1? Trends Endocrinol Metab 31, 410–421 (2020). [DOI] [PubMed] [Google Scholar]
- 74.Klein S, Gastaldelli A, Yki-Jarvinen H & Scherer PE Why does obesity cause diabetes? Cell Metab 34, 11–20 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ceperuelo-Mallafre V, et al. Disruption of GIP/GIPR axis in human adipose tissue is linked to obesity and insulin resistance. J Clin Endocrinol Metab 99, E908–919 (2014). [DOI] [PubMed] [Google Scholar]
- 76.Mantelmacher FD, et al. GIP regulates inflammation and body weight by restraining myeloid-cell-derived S100A8/A9. Nat Metab 1, 58–69 (2019). [DOI] [PubMed] [Google Scholar]
- 77.Ahlqvist E, et al. Link between GIP and osteopontin in adipose tissue and insulin resistance. Diabetes 62, 2088–2094 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen S, Okahara F, Osaki N & Shimotoyodome A Increased GIP signaling induces adipose inflammation via a HIF-1alpha-dependent pathway and impairs insulin sensitivity in mice. Am J Physiol Endocrinol Metab 308, E414–425 (2015). [DOI] [PubMed] [Google Scholar]
- 79.Lu SC, et al. GIPR antagonist antibodies conjugated to GLP-1 peptide are bispecific molecules that decrease weight in obese mice and monkeys. Cell Rep Med 2, 100263 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nasteska D, et al. Chronic reduction of GIP secretion alleviates obesity and insulin resistance under high-fat diet conditions. Diabetes 63, 2332–2343 (2014). [DOI] [PubMed] [Google Scholar]
- 81.Kizilkaya HS, et al. Loss of Function Glucose-Dependent Insulinotropic Polypeptide Receptor Variants Are Associated With Alterations in BMI, Bone Strength and Cardiovascular Outcomes. Front Cell Dev Biol 9, 749607 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ayala JE, et al. Glucagon-like peptide-1 receptor knockout mice are protected from high-fat diet-induced insulin resistance. Endocrinology 151, 4678–4687 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nahra R, et al. Effects of Cotadutide on Metabolic and Hepatic Parameters in Adults With Overweight or Obesity and Type 2 Diabetes: A 54-Week Randomized Phase 2b Study. Diabetes Care 44, 1433–1442 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jiang H, et al. A phase 1b randomised controlled trial of a glucagon-like peptide-1 and glucagon receptor dual agonist IBI362 (LY3305677) in Chinese patients with type 2 diabetes. Nat Commun 13, 3613 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Coskun T, et al. LY3437943, a novel triple glucagon, GIP, and GLP-1 receptor agonist for glycemic control and weight loss: From discovery to clinical proof of concept. Cell Metab 34, 1234–1247 e1239 (2022). [DOI] [PubMed] [Google Scholar]
- 86.Urva S, et al. LY3437943, a novel triple GIP, GLP-1, and glucagon receptor agonist in people with type 2 diabetes: a phase 1b, multicentre, double-blind, placebo-controlled, randomised, multiple-ascending dose trial. Lancet 400, 1869–1881 (2022). [DOI] [PubMed] [Google Scholar]
- 87.Jastreboff AM, et al. Triple-Hormone-Receptor Agonist Retatrutide for Obesity - A Phase 2 Trial. N Engl J Med (2023). [DOI] [PubMed] [Google Scholar]
- 88.Kawai T, et al. Structural basis for GLP-1 receptor activation by LY3502970, an orally active nonpeptide agonist. Proc Natl Acad Sci U S A 117, 29959–29967 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Saxena AR, et al. Danuglipron (PF-06882961) in type 2 diabetes: a randomized, placebo-controlled, multiple ascending-dose phase 1 trial. Nat Med 27, 1079–1087 (2021). [DOI] [PubMed] [Google Scholar]
- 90.Enebo LB, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of concomitant administration of multiple doses of cagrilintide with semaglutide 2.4 mg for weight management: a randomised, controlled, phase 1b trial. Lancet 397, 1736–1748 (2021). [DOI] [PubMed] [Google Scholar]
- 91.Alba M, Yee J, Frustaci ME, Samtani MN & Fleck P Efficacy and safety of glucagon-like peptide-1/glucagon receptor co-agonist JNJ-64565111 in individuals with obesity without type 2 diabetes mellitus: A randomized dose-ranging study. Clin Obes 11, e12432 (2021). [DOI] [PubMed] [Google Scholar]
- 92.Coskun T, et al. LY3437943, a novel triple glucagon, GIP, and GLP-1 receptor agonist for glycemic control and weight loss: From discovery to clinical proof of concept. Cell Metab (2022). [DOI] [PubMed] [Google Scholar]
- 93.Rosenstock J, et al. Efficacy and safety of a novel dual GIP and GLP-1 receptor agonist tirzepatide in patients with type 2 diabetes (SURPASS-1): a double-blind, randomised, phase 3 trial. Lancet 398, 143–155 (2021). [DOI] [PubMed] [Google Scholar]
- 94.Ludvik B, et al. Once-weekly tirzepatide versus once-daily insulin degludec as add-on to metformin with or without SGLT2 inhibitors in patients with type 2 diabetes (SURPASS-3): a randomised, open-label, parallel-group, phase 3 trial. Lancet 398, 583–598 (2021). [DOI] [PubMed] [Google Scholar]
- 95.Dahl D, et al. Effect of Subcutaneous Tirzepatide vs Placebo Added to Titrated Insulin Glargine on Glycemic Control in Patients With Type 2 Diabetes: The SURPASS-5 Randomized Clinical Trial. JAMA 327, 534–545 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]