

Abstract
Astroviruses cause a spectrum of diseases spanning asymptomatic infections to severe diarrhea, but little is understood about their pathogenesis. We previously determined that small intestinal goblet cells were the main cell type infected by murine astrovirus-1. Here, we focused on the host immune response to infection and inadvertently discovered a role for indoleamine 2,3-dioxygenase 1 (Ido1), a host tryptophan catabolizing enzyme, in the cellular tropism of murine and human astroviruses. We identified that Ido1 expression was highly enriched among infected goblet cells, and spatially corresponded to the zonation of infection. Because Ido1 can act as a negative regulator of inflammation, we hypothesized it could dampen host antiviral responses. Despite robust interferon signaling in goblet cells, as well as tuft cell and enterocyte bystanders, we observed delayed cytokine induction and suppressed levels of fecal lipocalin-2. Although we found Ido−/− animals were more resistant to infection, this was not associated with fewer goblet cells nor could it be rescued by knocking out interferon responses, suggesting that IDO1 instead regulates cell permissivity. We characterized IDO1−/− Caco-2 cells and observed significantly reduced human astrovirus-1 infection. Together this study highlights a role for Ido1 in astrovirus infection and epithelial cell maturation.
INTRODUCTION
Globally, astroviruses are a major cause of diarrheal illness in humans. Beyond humans, this family of viruses exhibits a broad host range, and in 2012 it was discovered that immunocompromised mice harbor a plethora of diverse astroviruses1–4. To better understand their pathogenesis, we previously identified two strains of murine astrovirus-1 (MuAstV1) and characterized them by experimentally infecting wild-type C57BL/6 mice5. We and others found that murine astrovirus in mice is restricted to the small intestine5,6, with the highest levels observed in the duodenum5,7, highlighting the distinct biogeography within the gastrointestinal tract. Similar to other astrovirus infections in humans and turkeys8,9, minimal pathology or immune cell infiltration occurs during MuAstV1 infection5,6. The fact that these enteric viruses sustain infection with minimal inflammation and intestinal damage suggests that they have a means of coopting host responses and/or dampening the inflammatory response, potentially including the attenuation of early interferon (IFN) signaling.
Type I, II, and III IFNs have proven key in preventing symptoms and/or lethality in mouse models of other enteric viruses, including noroviruses and reoviruses10–13. However, these viruses also encode IFN antagonists, some of which degrade IFN receptors14, or block signaling molecules along the IFN response pathway, including interferon response factor (IRF)3, IRF5, IRF715,16, and nuclear factor κB (NFκB) activation17. In vitro studies have shown that goblet cells and other epithelial cell types become infected by human astroviruses (i.e. human astrovirus-1 and VA1)18,19 and that IFN can limit virus replication4,18–20. However, it is unclear how these host immune responses are shaped at the mucosal interface in vivo.
We previously identified goblet cells, specialized epithelial cells that produce mucus, as the main population of cells susceptible to MuAstV1 infection21. In addition to maintaining the mucus barrier, goblet cells also function as antigen passages to promote oral tolerance22,23. In this study, we questioned whether infection of the very cells tasked with promoting key gut homeostatic functions would enable MuAstV1 to evade host immune responses. We identified an interferon-induced tryptophan catabolizing enzyme24, indoleamine 2,3-dioxygenase 1 (Ido1), as highly expressed among infected goblet cells. Due to its ability to dampen immune responses during viral infection25, we hypothesized that IDO1 may promote MuAstV1 infection. Instead, this study shows that IDO1 is an important host factor underlying the cellular tropism of astroviruses, and may play a role in the maturation of gut epithelial cells.
RESULTS
MuAstV1 targets a subset of goblet cells expressing Ido1
We previously performed single-cell RNA (scRNA) sequencing on epithelial cells collected from the duodenums of MuAstV1-infected and uninfected C57BL/6 animals (n = 4/group) at 6 days postinfection (dpi)21 (Fig. 1A). To understand which cellular pathways were driven by infection, we performed a Hallmark Gene Set Enrichment Analysis (GSEA) comparing infected to uninfected goblet cells and identified 11 significantly enriched pathways, most notably IFNγ and IFNɑ pathways (Fig. 1B). Among the IFNγ signature in the GSEA, and the top differentially expressed gene within infected cells compared to uninfected goblet cells, was the IFN-inducible gene, Ido1. Ido1 is the main tryptophan catabolizing enzyme in the gut and, when expressed by antigen-presenting cells, can promote T cell anergy and T regulatory cell induction26,27. Among all epithelial cells in the duodenum, we solely observed Ido1 expression among goblet cells (Fig. 1C), which is consistent with two recent studies using scRNA-sequencing and in situ hybridization28,29. Ido1 expression also corresponded with MuAstV1-infected cells when visualized in all epithelial cells (Fig. 1C) or in goblet cell subpopulations (Fig. 1D). Among goblet cells, Ido1 expression positively correlated with MuAstV1 viral gene expression (r = 0.04, Fig. 1E). While Ido1 expression was enriched among infected cells, infection did not drive a significant increase in its expression over baseline (Supplementary Fig. 1). Using Ido1-mCherry reporter animals, expression of Ido1 in goblet cells spatially correlated with MuAstV1 gene expression in the top two-thirds of the villus (Fig. 1F), consistent with the zonation of infection. We demonstrated with dual in situ hybridization (ISH) staining that Ido1 and MuAstV1 expression colocalized to the same individual goblet cells, providing additional support that MuAstV1 targets a subset of goblet cells expressing Ido1 (Fig. 1G).
Fig. 1.
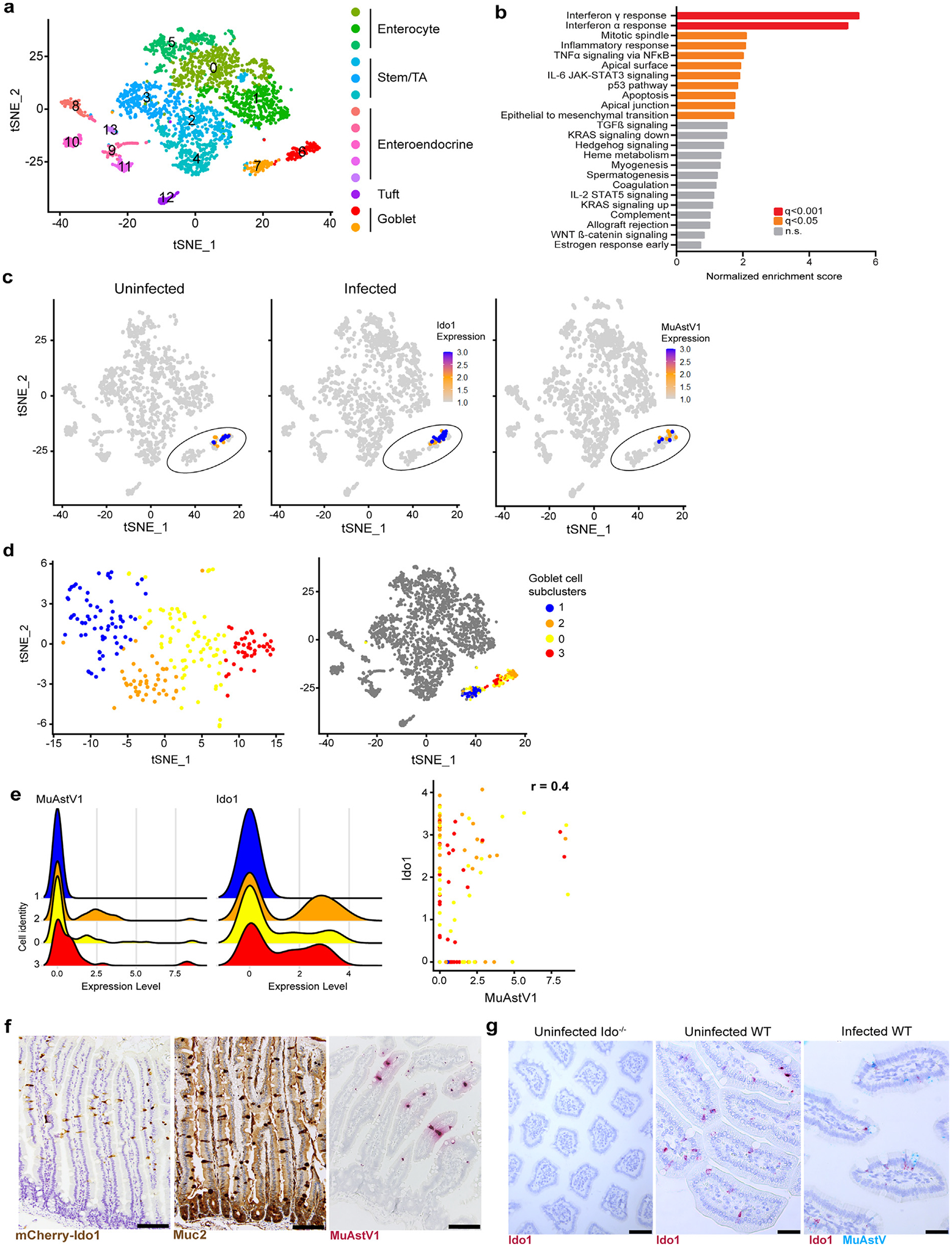
MuAstV1 targets a subset of goblet cells expressing Ido1 and this aligns with the zonation of infection. Analysis of single-cell RNA-sequencing dataset of duodenal epithelial cells collected 6 dpi from infected and mock-infected animals (n = 4/group). (A) Feature plot denoted epithelial cell subclusters previously identified by marker gene sets; (B) Gene Set Enrichment Analysis summarized by normalized enrichment score and colored by q value; (C) Feature plot showing Ido1 selectively expressed in goblet cells (circled) among epithelial cells collected from uninfected and infected animals, followed by MuAstV1 expression within infected animals (6 dpi). Relative Ido1 and MuAstV expression denoted by color scale, with each dot representing a single cell within the dataset; (D) Goblet cell subclusters 0–3 within goblet cells (left) and epithelial cell populations (right); (E) Ridge plot highlighting MuAst1V expression and Ido1 expression levels across the subclusters (left) and correlation plot of MuAstV1 and Ido1 expression in goblet cells (colored according to subcluster) with r value noted at the top (right); (F) Representative images of Ido1 (brown), Muc2 (brown), and MuAstV1 (red) staining in the small intestine < scale bar = 100 μm>; (G) Representative images (n = 2 animals/ group) of Ido1 ISH (red) in the small intestines of uninfected Ido−/− and WT animals and dual ISH staining of Ido1 (red) and MuAstV1 (blue) in infected WT animals (6 dpi) < scale bar = 60 μm >. dpi = days post infection; Ido1 = indoleamine 2,3-dioxygenase 1; ISH = in situ hybridization; MuAstV1 = murine astrovirus-1; WT = wild-type.
MuAstV1 infections are characterized by delayed and suppressed inflammation despite IFN signaling in epithelial cells
Because the immunosuppressive effects of Ido1 can be coopted by viruses25, we next assessed how neighboring epithelial cells responded to the infection. We focused our analysis on the top 30 differentially expressed genes from uninfected and infected animals to identify those involved in IFN signaling. With this compilation of 14 genes, we examined the patterns of expression across the epithelial cell subsets, including enterocyte, stem, enteroendocrine, tuft, and goblet cells, that were previously resolved by marker gene sets21,30 (Fig. 2A). Relative to uninfected goblet cells, infected goblet cells exhibited robust IFN-stimulated gene (ISG) expression, notably Ifi27l2b (Fig. 2A and 2B). In bystander cells, including tuft and a subset of enterocyte cells and to a lesser extent, uninfected goblet cells, a different ISG signature (B2m, Irf7, Oas1a, Bst2, Isg15, Stat1, and Irgm1) was triggered by infection. In the four subpopulations of goblet cells we previously computationally identified21, we observed the same distinct IFN patterns, with higher levels of MuAstV1 replication associated with a stronger induction of Ifi27l2b (Fig. 2A). Together these data indicate that similarly to what was observed in an enteroid model with human astrovirus-1 (HAstV1)19, epithelial cell subsets exhibit distinct IFN programming following astrovirus infection, with neighboring bystander cells coordinating a complementary response.
Fig. 2.
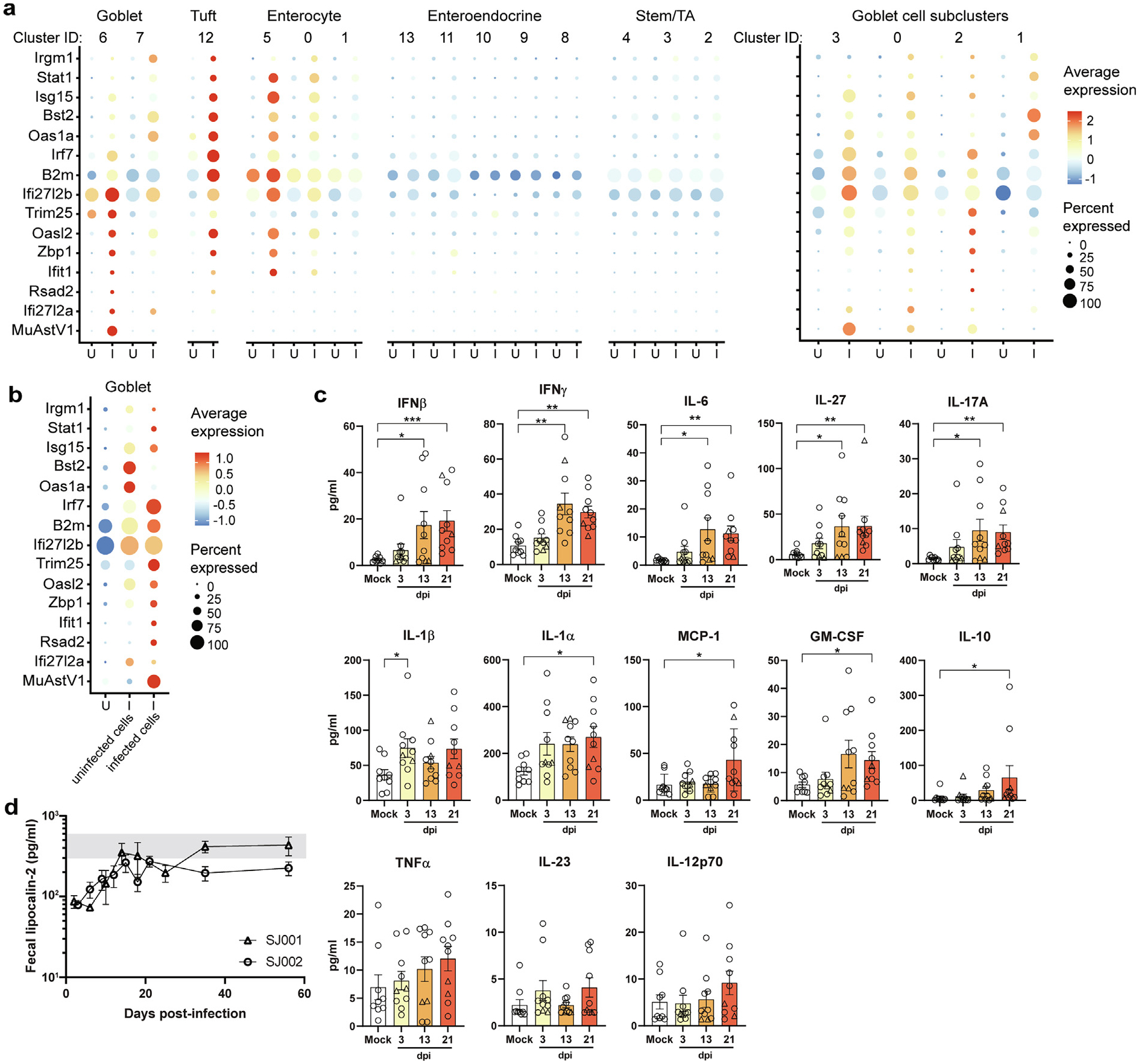
Delayed cytokine induction and evidence of suppressed inflammation despite robust ISG expression within infected cells relative to bystanders. (A) Summary of top differentially expressed ISGs among all epithelial cells and goblet cell subclusters. (B) Differential expression of ISGs in uninfected goblet cells from uninfected animals versus uninfected and infected goblet cells from infected animals. Average expression and percent of cell population expressing each gene are indicated by color gradient and size of dot in plot, respectively (B); Duodenal homogenates from infected and mock-infected animals assayed for 13 proinflammatory cytokines and chemokines by a bead-based flow cytometry analysis. Data shown are from two independent experiments (n = 9–10 mice/group). Samples from mice infected with SJ001 (n = 8) and SJ002 (n = 2) are denoted by circles and triangles, respectively. (C) Fecal lipocalin was measured by enzyme-linked immunosorbent assay at the time points indicated after infection, with the range of values observed in mock-infected mice highlighted in gray. Data shown are combined from two independent experiments (n = 7–8 mice/group). Comparisons between groups were assessed by Kruskal-Wallis Test with Dunn’s multiple comparisons and significant differences noted as indicated: * p < 0.05, ** p < 0.005, or *** p < 0.0001. GM-CSF = granulocyte-macrophage colony-stimulating factor; I = infected; IFN = interferon; IL = interleukin; ISG = IFN-stimulated gene; MCP = monocyte chemoattractant protein-1; TNF = tumor necrosis factor; U = uninfected.
Next, to examine whether these IFN responses at the epithelial barrier translated to tissue-level immune signaling, we assayed 13 proinflammatory cytokines and chemokines in duodenums collected at 3, 13, and 21 days postinfection. We infected mice with two distinct MuAstV1 isolates (SJ001 and SJ002) that differ by their level of persistence5. Consistent with increased IFNβ and IFNγ signaling at 6 dpi in epithelial cells, we observed significant increases of IFNβ and IFNγ at 13 dpi, in addition to interleukin (IL)-6, IL-27, and IL-17A (Fig. 2B). We also observed a slight increase in IL-1β at 3 dpi and elevated levels of IL-1ɑ, MCP-1, GM-CSF, and IL-10 by 21 dpi (Fig. 2C). The early increase in IL-1β is notable since it has been shown to increase intestinal permeability31 and is consistent with the increase in permeability observed by our group previously4. However, the inflammatory effects of this increase are likely limited given the absence of overt pathology observed following infection5,6. As a secondary measure, we quantified the amount of fecal lipocalin-2, a biomarker of inflammation in the gut32. Lipocalin-2 was suppressed within infected animals relative to mock-infected animals at the start of infection (Fig. 2D). However, lipocalin-2 levels in mice infected with SJ001 returned to that of the mock-infected mice after 21 dpi, when viral clearance occurs5. In contrast, levels remained suppressed at 60 dpi for SJ002-infected animals, which does not clear for 70 days5. The limited inflammatory responses at 3 dpi and lower levels of lipocalin-2 suggest delayed immune mobilization and subclinical inflammation, which is consistent with previous studies showing prolonged MuAstV1 infections in mice5,6.
Ido-deficiency reduces virus infection and is not rescued by the loss of interferon
Since it appeared that MuAstV1 was specifically targeting Ido1-expressing goblet cells, we hypothesized that the virus may co-opt the immunomodulatory functions of Ido1 to promote its replication. Indoleamine 2,3-dioxygenase 2 (Ido2) is another tryptophan catabolizing enzyme, and although it is not expressed in the gut33, in order to prevent any compensatory effects that could result in the loss of Ido134, we next used animals deficient for Ido1 and Ido2 (Ido−/−) to test for infection. Loss of IDO led to reduced infection and virus shedding at 3 and 6 dpi (Fig. 3A). Based on MuAstV1 ISH, goblet cells were still the main cell target infected by the virus in Ido−/− infected animals (Supplementary Fig. 2). Because we observed higher baseline levels of lipocalin-2 in the feces of Ido−/− animals relative to wild-type (WT) (Fig. 3B), we questioned whether the tissue environment may be primed for inflammation in the absence of Ido1 thereby preventing the virus from establishing infection. Given the importance of IFN responses in antiviral immunity35 and previous studies showing enhanced IFN signaling in the absence of IDO136,37, we next generated Ido−/− Ifnar−/− and Ido−/− Ifnlr−/− double-knockouts to test whether the loss of either type I or III IFN signaling could rescue the infection. However, we found that both double-knockout animals shed comparable levels of virus as the Ido−/− animals (Fig. 3C). From these data, we conclude that the loss of antiviral responses from either IFNα or IFNλ cannot rescue the virus infection in Ido−/− animals. Because the double-knockout animals did not have reduced infection relative to the single IFN knockouts, we also conclude one of two possibilities: (1) the IDO-deficiency phenotype of reduced virus infection only manifests with a functional IFN response or (2) this phenotype can be mediated by either loss of IDO1 or IFNα/λ responses. In the process of making Ido−/− Ifnar−/− double-knockouts, we crossed Ido−/− animals with persistently infected Ifnar−/− animals and found that while pups from Ifnar−/− × Ifnar−/− and Ifnar−/− × WT crossings remained positive at the time of weaning (3 weeks old), heterozygous pups from Ifnar−/− × Ido−/− breeding pairs had undetectable levels of virus in their feces (Fig. 3D). Together these data suggest that the loss of Ido enables resistance to infection via a mechanism that may require IFN-sufficiency.
Fig. 3.
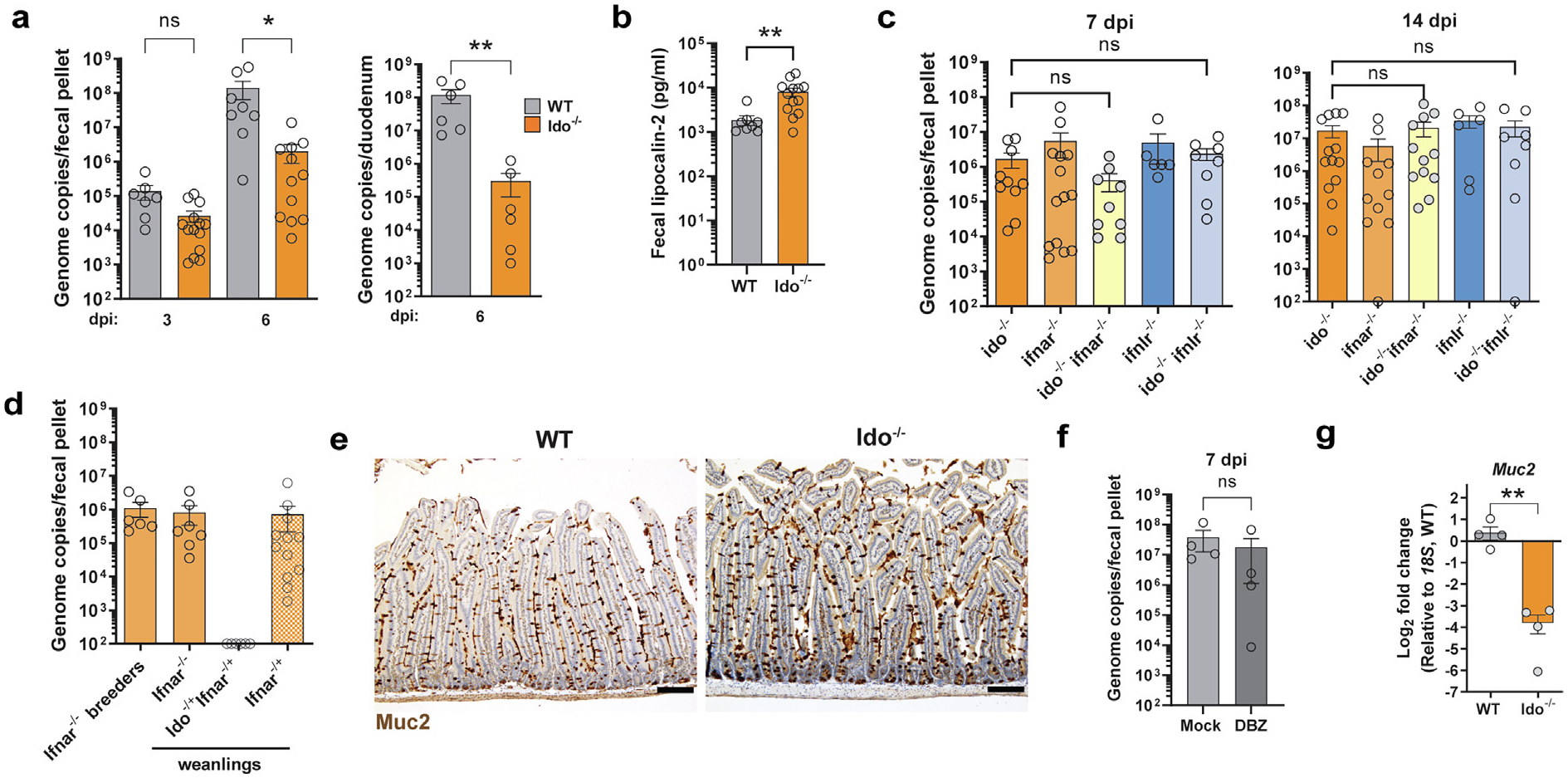
Ido-deficiency results in reduced virus infection and is not rescued by the loss of interferon. (A) MuAstV1 infection quantification in feces and duodenums at time points indicated is lower in Ido−/− animals compared to WT at 3 and 6 dpi; (B) Higher baseline fecal lipocalin-2 levels observed in Ido−/− animals compared to WT; (C) MuAstv1 fecal shedding is comparable in Ido−/−, Ifnar−/−, and Ifnlr−/− single knockouts compared to Ido−/− Ifnar−/− and Ido−/− Ifnlr−/− double-knockouts at 7 and 14 dpi; (D) Ifnar−/− and Ifnar−/+ pups remain positive at weaning whereas Ido−/+Ifnar−/+ are negative despite exposure from a persistently infected Ifnar−/− parent; (E) Muc2 staining of duodenal tissue from WT and Ido−/− animals, scale bar = 100 μm; (F) DBZ treatment prior to MuAstV1 infection does not enhance infection levels; (G) Baseline Muc2 expression is lower in Ido−/− animals compared to WT. Data on graphs are represented by group means ± standard error of mean. Comparisons between groups were assessed by Mann-Whitney U (A, B, F) or Kruskal-Wallis Test with Dunn’s multiple comparisons (C) or unpaired t test with Welch’s correction (G). Significant differences noted as indicated * p < 0.05, ** p < 0.005. Data represent at least two independent experiments (A–D) or a single experiment (E–G). dpi = days post infection; Ido1 = indoleamine 2,3-dioxygenase 1; IFN = interferon; MuAstV1 = murine astrovirus-1; ns = non-significant; WT = wild-type.
We next examined whether there were fewer goblet cells, and therefore fewer target cells for MuAstV1 to infect, in Ido−/− compared to WT animals. We found comparable numbers of goblet cells per villus based on Muc2 staining of the duodenum (Fig. 3E), with an average of 7.16 versus 6.26 goblets/villus in Ido−/− and WT animals, respectively (n = 2–4 animals/group, n = 4 10X-field views/animal). The lack of difference in total goblet cell numbers does not preclude a difference in susceptible goblet cells, as we previously observed that only a subset of actively secreting goblet cells are susceptible to infection21 (Fig. 1D goblet subpopulations 3, 0, and 2). Consistent with this idea, we found that treatment with DBZ, an inhibitor of γ -secretase that blocks Notch signaling and drives goblet cell hyperplasia38 (Supplementary Fig. 2A), did not result in higher levels of Ido1-expressing goblet cells (Supplementary Fig. 2B) or a higher level of infection (Fig. 3F). Instead, we found that Muc2 expression was lower in the Ido−/− animals relative to WT (Fig. 3G), consistent with a previous study examining the role of IDO1 in goblet cell secretion39. Collectively, these data indicate that the loss of IDO alters the permissivity of goblet cells and that lower Muc2 expression may reflect fewer actively secreting goblet cells, which is important for productive virus replication21.
To further examine the role of IDO1 during MuAstV1 infection, we turned to a neonatal model after previous research showed Ido1 expression is low during early development40. Using Ido1-mCherry reporter animals, we confirmed that Ido1 is minimally expressed at postnatal day (PND) 3 compared to PND 7 (Fig. 4A) and that was not due to a difference in goblet cell abundance (3.34 vs. 3.99 goblets/villus in PND 3 and PND 7 animals, respectively; n = 2 animals/group, n = 4 10X-field views/animal). We hypothesized that neonatal animals infected at these two developmental stages would show differential levels of infection. Like adult animals5, neither group of neonates exhibited signs of pathology based on clinical review by a veterinary pathologist of hematoxylin & eosin staining of the small intestines collected at 3 and 6 dpi. With respect to infection, we noted a much higher virus load in the tissue from animals infected at PND 7 compared to PND 3 based on ISH staining at 7 dpi (Fig. 4A). Quantifying the virus in the tissue by quantitative reverse transcription-polymerase chain reaction (RT-PCR) showed that peak infection occurred on different days, with levels peaking at 14 dpi in PND 3 animals and 7 dpi in PND 7 animals (Fig. 4B). In contrast, no significant difference in virus levels was detected in the small intestines of PND 3 and PND 7 Ido−/− animals at 7 dpi (Supplementary Fig. 4). However, it is notable that neonatal Ido−/− animals exhibit a high level of infection, which may indicate that other biological factors enhance the infection during this early developmental stage, such as the establishment the microbiota and mucosal immunity. Together, these data indicate that the level of infection in neonates may in part be modulated by the expression of Ido1 in goblet cells during development; however, it is likely that other factors related to this development period (e.g. microbial establishment) are at play.
Fig. 4.
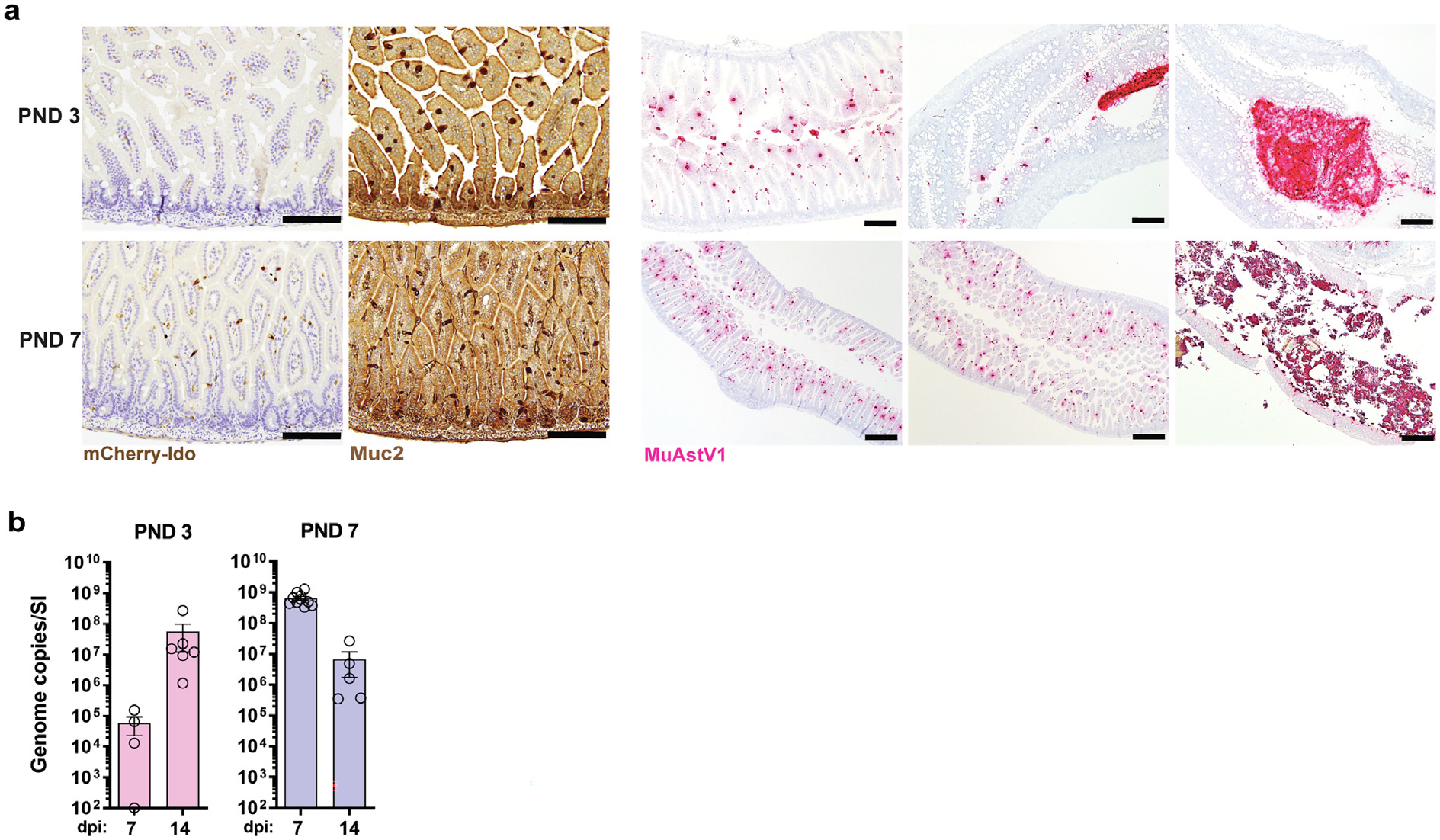
Ido1 expression increases during neonatal development and this corresponds with the level of MuAstV1 infection. (A) Duodenal tissue sections collected from animals at PND 3 and 7 with Ido-expressing cells (brown) indicated in panel 1 and goblet cells stained by Muc2 (brown) in panel 2 < scale bar = 200 μm >. Panels 3–5 show MuAstV1 in situ hybridization staining (red) across the duodenum, jejunum, and colon in animals from PND 3 < scale bar = 200 μm > and the duodenum, jejunum, and colon at PND 7 < scale bar = 500 μm>; (B) The level of infection throughout the small intestine at indicated time points in animals infected at PND 3 as compared to animals infected at PND 7 (n = 4–10 animals/group/time point). Data collectively from two independent experiments. dpi = days post infection; Ido1 = indoleamine 2,3-dioxygenase 1; MuAstV1 = murine astrovirus-1; PND = postnatal day.
IDO1−/− Caco-2 cells have impaired differentiation and reduced HAstV1 infection
To determine whether IDO1 also plays a role during HAstV1 infection, we next focused on an in vitro model using Caco-2 cells. We first examined whether IDO1 is expressed as the cells differentiate in culture. For HAstV1 infections, Caco-2 cells are typically allowed to differentiate for 5 to 7 days before infection41. Relative to cells collected after 1 day post-plating, IDO1 expression significantly increased by 6 days post-plating (Fig. 5A). We also observed widespread IDO1 staining in Caco2 cells and co-localization of HAstV1 capsid in IDO1+ and IDO1-cells (Fig. 5B). Interestingly, we stained Caco2 with MUC2 and found a subset to be expressing this mucin and detection of infected MUC2+ cells (Supplementary Fig. 5A and 5B), highlighting a common feature between the in vivo and in vitro models. Next, using CRISPR-Cas9, we generated a Caco-2 cell line deficient in IDO1 and characterized three key markers of differentiation. First, we examined the expression of sucrase-isomaltase, a brush-border enzyme, and found significantly lower levels relative to WT cells (Fig. 5C). Second, we measured the ability of the cells to form a tight monolayer by measuring transepithelial electrical resistance and found that IDO1−/− cells had significantly lower levels relative to WT cells at 6 days post-plating (Fig. 5D). Finally, we examined the polarity of the IDO1−/− cell monolayer relative to WT by visualizing the localization of ezrin, an apically expressed protein, and sodium–potassium ATPase, which is expressed on the basolateral surface. While the WT monolayer exhibited a uniform expression of these two markers, IDO1−/− cells showed a disrupted monolayer, decreased presence of ezrin at the apical surface, as well as mislocalized sodium–potassium ATPase (Fig. 5E). Western blot quantification showed significantly less ezrin in IDO1−/− cells relative to WT as well as lower levels of the junctional proteins ZO-1 and occludin (Fig. 5F). These data collectively indicate that the typical differentiation and maturation process of Caco-2 cells was disrupted by the loss of IDO1.
Fig. 5.
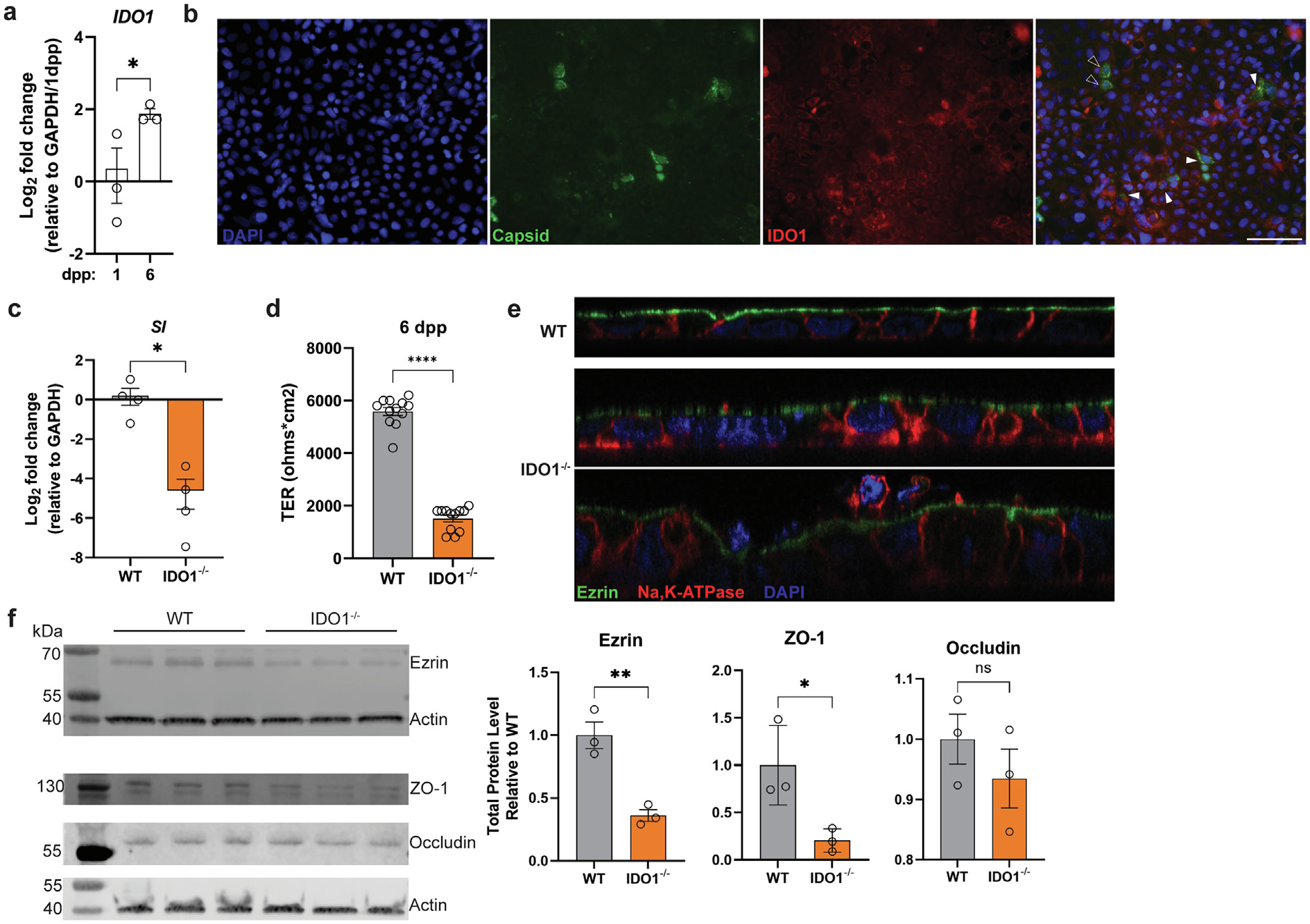
IDO1 is expressed in Caco-2 cells that are infected by HAstV1 and IDO1-deficient cells exhibit impaired differentiation. (A) Relative IDO1 expression in Caco-2 cells harvested at 1 and 6 dpp; (B) HAstV1 infection of Caco-2 cells harvested at 1 dpi and stained for IDO1 (red) and capsid (green). Infected cells expressing IDO1 (closed arrowheads) and infected cells not expressing IDO1 (open arrowheads) are indicated. WT and IDO1−/− cells were compared by (C) relative SI expression at 6 dpp; (D) transepithelial electrical resistance at 6 dpp in transwell inserts; (E) confocal microscopy of ezrin (green), Na, K-ATPase (red) and DAPI (blue) staining, and (F) comparative infection of HAstV1 and Reovirus T1L infection with quantification at 1 dpi, and (F) comparative binding of spike-green fluorescent protein (GFP) binding. Comparisons between groups were assessed by an unpaired t-test with Welch’s correction and one-tailed p values indicated, * p < 0.05, ** p < 0.001, **** p < 0.0001. Data represent individual wells run in at least two independent experiments (B, D) or a single experiment (A, C, E, F). dpi = days post infection; dpp = days post plating; Ido1 = indoleamine 2,3-dioxygenase 1; HAstV1 = human astrovirus-1; ns = non-significant; SI = sucrase-isomaltase; WT = wild-type.
We next measured the levels of HAstV1 infection in WT and IDO1−/− cells to determine whether our findings would mirror what we observed in vivo with MuAstV1. Using a multiplicity of infection of 0.1, we observed reduced infection of HAstV1 based on capsid staining at 24 hours post-inoculation (Fig. 6A). To determine whether the loss of IDO1 alters the permissivity to another enteric virus, we tested Reovirus T1L at the same multiplicity of infection but did not observe a significant difference between WT and IDO1−/− cells (Fig. 6A). We next asked whether the lower HAstV1 infection in IDO1−/− cells was due to reduced virus binding to the surface of the cell. Using HAstV1 capsid spike protein conjugated to green fluorescent protein (GFP), we found that IDO1−/− cells showed fewer particles bound as compared to WT cells (Fig. 6B). Overall, these data show that the loss of IDO1 in Caco-2 cells results in reduced markers of differentiation, leading to altered expression and/or localization of binding partners for HAstV1 that results in reduced infectivity.
Fig. 6.
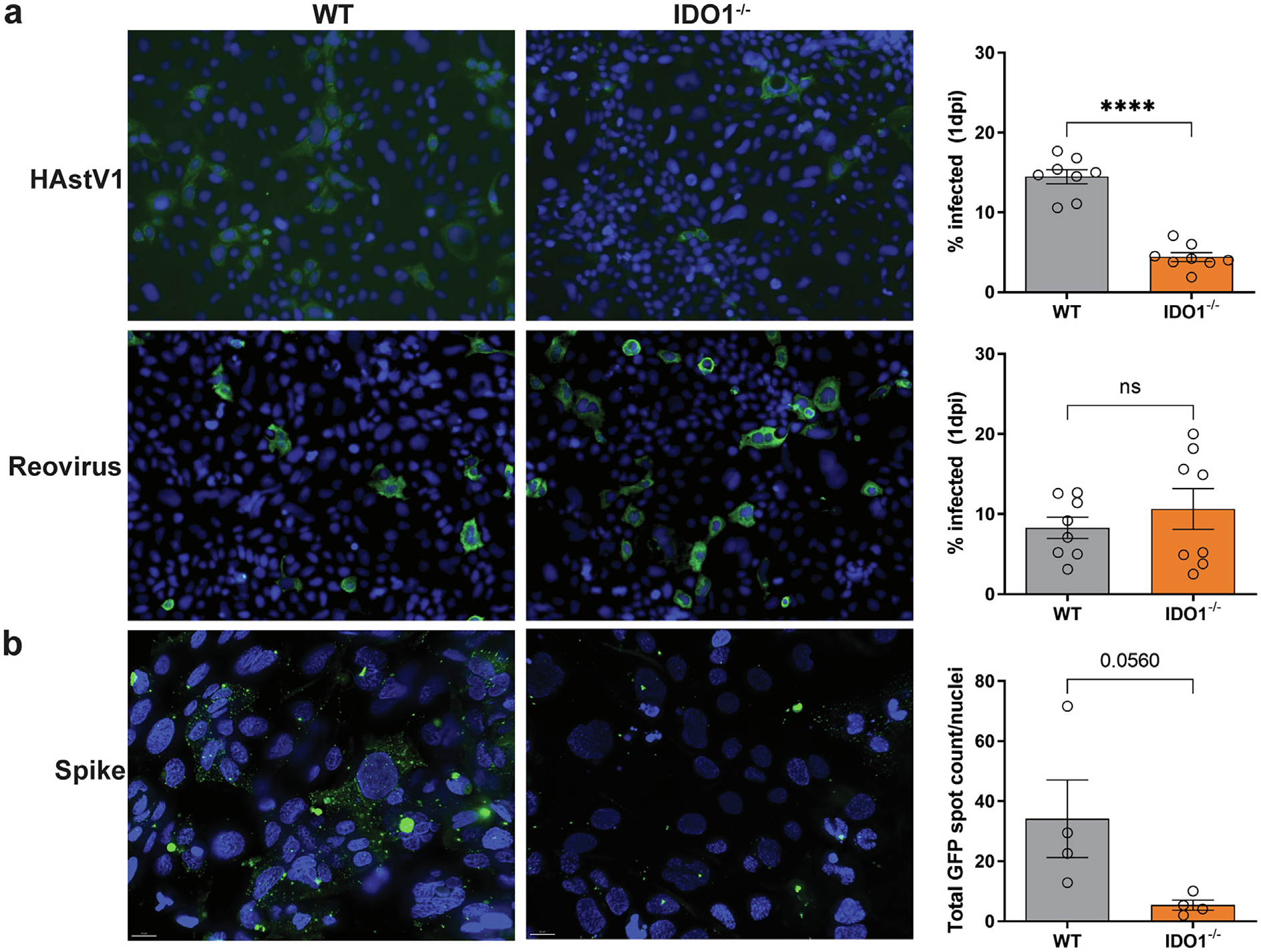
IDO1-deficient Caco-2 cells have reduced HAstV1 infection and capsid spike protein binding. (A) Comparative infection of HAstV1 and Reovirus T1L infection with quantification at 1 dpi and (B) comparative binding of spike-GFP in WT and IDO1−/− cells. Comparisons between groups were assessed by an unpaired t-test with Welch’s correction and one-tailed p values indicated: ns, **** p < 0.0001. Data represent individual wells run in at least two independent experiments. dpi = days post infection; Ido1 = indoleamine 2,3-dioxygenase 1; GFP =; HAstV1 = human astrovirus-1; ns = non-significant; WT = wild-type.
DISCUSSION
Astroviruses remain one of the least characterized enteric RNA viruses despite their broad host range and high prevalence in children. To better understand their pathogenesis, we evaluated the gut epithelial response and subsequent immune signaling following infection. Most notable among the IFN signaling in goblet cells was the expression of Ido1, which can act as a negative regulator of inflammation. We detected delayed inflammatory signaling and evidence of early immunosuppression at the tissue level but robust IFN signatures at the cellular level, particularly in bystander cells. However, in the absence of Ido1, we observed reduced levels of MuAstV1 infection, but notably in a manner that was not mediated by type I or III IFN signaling. Instead, our data support a role for Ido1 in the tropism of murine and human astroviruses, highlighting a new clue to understanding their pathogenesis.
Astroviridae encompasses a large and diverse virus family, with over 20 members classified into two genera: Avastrovirus and Mamastrovirus42. Despite their expansive genetic distance, there are both distinct and common features across virus members. While the majority of astroviruses cause gastrointestinal tract infections, they have the potential for expanded tissue tropism43. Here, while focusing on the immune responses to MuAstV1 in laboratory mice, we identified a new common feature between human and murine astroviruses and their cellular tropism. IDO1, the main tryptophan catabolizing enzyme in the gut, is a known biomarker for inflammatory bowel disease44 and is associated with the zonation of goblet cells28,29. The reduced infectivity of mouse tissues as well as Caco-2 cells with MuAstV1 and HAstV1, respectively, indicates that the loss of IDO1 may be important for the permissivity of the cells. Further, the heterogeneity of the infection in Caco-2 cells (Fig. 5B, Supplementary Fig. 5) indicates that there may be different routes of entry in enterocytes and goblet cells. The reduction in binding of the spike protein on Caco-2 cells may also indicate that there is a decreased density of a binding partner and/or receptor required by HAstV1, which are currently uncharacterized for any astrovirus. Further evaluation of these cell lines may aid in their discovery.
Similar to turkeys and consistent with the limited information we have from human infections8,9, we and others have previously shown that there is minimal evidence of pathophysiology, cell death, or immune infiltration in astrovirus-infected mice5,6. However, we show here clear evidence of IFN induction, which has previously been observed with a different strain of MuAstV17, and subclinical inflammation, albeit delayed, that coincides with the initiation of viral clearance. As was found after HAstV1 infection in enteroids19, we observed distinct IFN signatures across the different epithelial cell subsets. In particular, ISG Ifi27l2a stood out as solely being expressed within a small percentage of goblet cells whereas Ifi27l2b was robustly induced in most goblet cells as well as (to a lesser degree) a subset of enterocytes and tuft cells. Ifi27l2a and Ifi27l2b are paralogs and are thought to act via cell death pathways in a cell type specific manner, as it was previously shown that the absence of Ifi27l2a during West Nile Virus infection leads to a higher virus burden in neurons due to their prolonged survival45. Since cell death is not observed following MuAstV1 infection, it is possible that these two ISGs act via different mechanisms in these epithelial cell types or that MuAstV1 is able to modulate their activity to avoid cell death. Future investigations are needed to elucidate their precise effects, especially considering that the observed IFN signatures in enterocytes and tuft cells were distinctive from the goblet cell response to infection. Intestinal tuft cells are not classically known for orchestrating antiviral responses, but recent studies have shown that they can upregulate immune response pathways in response to rotavirus infection that are distinct from those in helminth or bacterial infections46,47 and intestinal tuft cells even respond to influenza infection48. Additional work focused on the crosstalk between these epithelial cell subsets in the context of viral infection is needed to better understand how these signals are coordinated and induce a local response to promote clearance in the absence of major immune cell infiltration.
Overall, while our findings indicate a clear role for Ido1 in viral tropism, this study must be framed with a few limitations, and here we posit additional open questions. First, our focus was on the epithelial response and was not inclusive of other innate and adaptive immune responses that are likely triggered during infection. To what extent these other arms of immunity are mobilized remains to be defined. Second, we focused on IFNs because they are known to shape the overall response to viral infection34. Because we did not observe an increase in viral replication in the Ido-IFN double-knockout animals relative to Ido−/− animals, it remains possible that the global loss of Ido1 may result in other immune enhancements that reduce viral infection as was evidenced by increased baseline levels of lipocalin-2 in Ido−/− animals. However, the lower level of infection in WT animals observed early on at 3 dpi indicates that increased mobilization of adaptive immune cells is unlikely, but to what extent innate cells, including resident T cells such as gamma delta T cells or innate lymphoid cells, could be responding to infection remains to be seen. It is notable that we also did not observe a phenocopy of the decreased virus levels resulting from Ido-deficiency in the double-knockouts relative to the single IFN knockouts (Fig. 3C). This perhaps indicates a cell-intrinsic role for these IFNs in mediating the Ido resistance phenotype. While Ifnar−/− animals still have a functional IFNλ response (and vice versa) to help control viral infection, it is also known that these innate immune responses regulate gut epithelial proliferation and host-microbiota interactions49–51, which could be altering goblet cell development and the efficiency of astrovirus infection. Future studies that tease apart these host-virus-microbe interactions are needed to fully understand these complexities. Lastly, our work indicates that the loss of IDO1 results in altered differentiation or maturation of goblet cells and possibly reduced mucus secretion. However, our work was limited by the use of Muc2 as a goblet cell marker to quantify differences between Ido−/− and WT, as well as PND 3 and 7 animals. Additional work is needed to define additional non-secreted protein markers to quantify goblet cells as well as determine the role(s) of epithelial-derived IDO1, how these findings may translate in vivo, and whether there are shared mechanisms with immune cell-derived IDO1.
METHODS
Ethics
All animal experiments were approved by the St. Jude Children’s Research Hospital (St. Jude) and/or University of California, Santa Cruz (UCSC) Institutional Animal Care and Use Committee (protocol 570). St. Jude and UCSC are fully accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International (AAALAC-I) and have an approved Animal Welfare Assurance Statement on file with the Office of Laboratory Animal Welfare (St. Jude: A3077–01, UCSC: D16–00493). These guidelines were established by the Institute of Laboratory Animal Resources and were approved by the Governing Board of the U.S. National Research Council.
Animals
Adult (7 weeks old) male and female WT C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and used 1 week after arrival for all experiments in adult animals. C57BL/6 breeders for neonate experiments were also purchased from The Jackson Laboratory. Ido−/− and Ido1-mCherry mice were generated by Dr. Peter Murray (Max Planck Institute) and were a kind gift from his laboratory. The Ifnar−/− and Ifnlr−/− colonies were originally obtained from Dr. Laura Knoll (University of Wisconsin) and Dr. Megan Baldridge (Washington University at St. Louis) and rederived at St. Jude. Two St. Jude Ifnar−/− colonies were maintained in separate cubicles for breeding in order to maintain persistently infected and uninfected groups. Ifnar−/− and Ifnlr−/− were bred with Ido−/− animals to generate double-knockout mice. Mouse genotypes were confirmed in-house, except for Ifnlr−/−, which was genotyped via Transnetyx (Cordova, TN, USA).
For all experiments, mice were co-housed for at least 1 week before inoculation on day 0, and co-housed mice of the same genotype (2–5 mice/cage) were confirmed to be negative for MuAstV by quantitative RT-PCR screening of fresh feces. In all experiments with neonates, breeding pairs were screened for MuAstV prior to experimentation as well as on day 0 of the neonatal infection. For all experiments in adult animals, mice aged 8 weeks were orally inoculated with 100 μl of fecal filtrate (100 mg/ml). For experiments in neonatal animals, 7-day-old mice were given 1 μl of filtrate. Mock infections used phosphate buffer saline (PBS) alone. Samples of fresh feces and tissues were obtained at the time points indicated and stored at −80 °C until processed. For dibenzazepine (DBZ) (Axon Medchem, Groningen, Netherlands) treatments, animals were given 30 μmol/kg dissolved in 1% Tween-80 and 0.5% hydroxypropyl methylcellulose (Methocel, Modernist Pantry, Eliot, ME, USA) via intraperitoneal injection in 100 ul daily doses for 5 days prior to infection with MuAstV1.
MuAstV1 inoculum preparation and quantification
Inoculum was prepared using homogenized feces (100 mg/ml in PBS) using a Next Advance Bullet Blender (Next Advance, Inc. Troy, NY, USA) (4 min, level 4) before the samples were centrifuged at 14,000 rpm for 2 minutes and then 0.22 μm-filtered. To quantitate virus levels, RNA was extracted using the MagMAX-96 AI/ND Viral RNA Isolation Kit (Thermo Fisher, Waltham, MA, USA) for fecal filtrates (100 mg/ml in PBS, 50 μl) or the MagMAX Pathogen RNA/DNA Kit (Thermo Fisher) for tissue homogenate (1 mg/ml in PBS, 300 μl) using the KingFisher Flex Purification System (Thermo Fisher Scientific, Waltham, MA, USA). Copies of the MuAstV genome were quantified using a g-block standard (Integrated DNA Technologies) in a one-step quantitative RT-PCR using TaqMan Fast Advanced Master Mix Virus (Applied Biosystems, Waltham, MA, USA) with primers (F: TACATCGAGCGGGTGGTCGC, R: GTGTCACTAACGCGCACCTTTTCA) and probe ((6-FAM)-TTTGGCATGTGGGTTAA-(MB GNFQ)2 under the following conditions: 50 °C for 5 minutes, 95 °C for 20 seconds followed by 40 cycles of 95 °C for 3 seconds and 60 °C for 30 seconds on a BioRad CFX96 Real-Time System (BioRad, Hercules, CA, USA)5.
Fecal Lipocalin-2
Levels of fecal lipocalin-2 were determined by the Mouse Lipocalin-2/NGAL DuoSet enzyme-linked immunosorbent assay (R&D Systems, Minneapolis, MN, USA), using 100 μl of clarified fecal homogenates (100 mg/ml in PBS) plated in duplicate and in accordance with the manufacturer’s instructions. Optical density of each well was obtained on a Thermo Multiskan Ascent Microplate Reader (Thermo Fisher) set to 450 nm.
GSEA on scRNA-seq
Duodenums were collected from infected and uninfected C57BL6 mice at 6 days after MuAstV infection (n = 4/group). Intestines were harvested and live, clusters of differentiation 45−, EPCAM + cells were sorted and ~ 25,000 cells were loaded onto the Chromium controller (10x Genomics, Pleasanton, CA, USA) to partition single cells into gel beads. Single-cell transcriptomic libraries were generated using the 5′ Gene Expression Kit (V2, 10x Genomics) and libraries were sequenced on the Illumina NovaSeq (San Diego, CA), generating ~ 500 M reads per sample (NCBI BioProject PRJNA573959). 10x gene expression data were first processed as previously described21. For pairwise cluster comparisons of interest, genes were then ranked as a function of the product of their average log fold change, the absolute value of the difference in percent expression, and the inverse of the scaled, FDR-adjusted q value. These gene rank lists were then analyzed using preranked GSEA.
Tissue analyses
Levels of 13 proinflammatory chemokines and cytokines were quantified by the LEGENDplex Mouse Inflammation Panel (Bio-Legend, San Diego, CA, USA), using 25 ul of clarified duodenal homogenate (1 mg/ml in PBS) plated in duplicate and in accordance with the manufacturer’s instructions. LEGENDplex data were acquired using the BD LSR Fortessa (BD Biosciences, Franklin, NJ, USA) and analyzed using LEGENDplex Data Analysis Software (Version 8.0). Mouse organs were formalin-fixed and then embedded in paraffin, sectioned at 4 μm, and stained with a customized ISH probe specific for the MuAstV1 genome (Advanced Cell Diagnostics, Neward, CA, USA), Mm-Ido1-C2 ISH probe (315971-C2, Advanced Cell Diagnostics), alcian blue, anti-RFP (ab124754, Abcam), or anti-Muc2 rabbit polyclonal antibodies (GTX100664, GeneTex, Irvine, CA, USA) by the St. Jude Veterinary Pathology Core. Muc2 expression was determined from whole duodenal homogenates (1 mg/ml in PBS) collected from WT and Ido−/− animals by first extracting RNA by TRIzol LS Reagent (Thermo Fisher). Superscript IV VILO (Invitrogen, Waltham, MA, USA) was then used to generate complementary DNA before QuantiTect primer assays were used with QuantiFast SYBR Green PCR kit (Qiagen, Hilden, Germany) to quantify host transcripts including mouse Muc2 (Mm_Muc2_2_SG; QT01060773) and 18s (Mm_Rn18s_3_SG; QT02448075). Delta-delta Ct method was used to resolve the differential expression between WT and Ido−/− animals relative to the housekeeping gene, 18s.
Caco-2 cells and IDO1−/−characterization
The human intestinal adenocarcinoma cell line Caco-2 was obtained from ATCC (HTB-37). Cells were propagated in minimum essential medium (MEM; Corning, Corning, NY, USA) supplemented with 20% fetal bovine serum (HyClone), GlutaMax-I (Gibco), and 1 mM sodium pyruvate (Gibco, Waltham, MA, USA). IDO1−/− Caco-2 cells were created using CRISPR-Cas9 technology. Briefly, 400,000 Caco-2 cells were transiently transfected with precomplexed ribonuclear proteins (RNPs) consisting of 100 pmol of chemically modified sgRNA (5′ – UCUGCAUAAACCAAAAUAGG- 3′, Synthego) targeted the 4th exon of IDO1, 35 pmol of Cas9 protein (St. Jude Protein Production Core), and 500 ng of pMaxGFP (Lonza, Bend, OR, USA) via nucleofection (Lonza 4D-Nucleofector X-unit) according to the manufacturer’s recommended protocol using solution P3 and program CM-138 in a small (20 ul) cuvette. Cells were single-cell sorted by FACS (St. Jude Flow Cytometry and Cell Sorting Shared Resource) to enrich for GFP positive (transfected) cells into 96-well tissue culture treated plates 5 days post nucleofection. Cells were clonally expanded and screened for the desired targeted modification (out-of-frame indels) via targeted deep sequencing using gene-specific primers with partial Illumina adapter overhangs (CAGE435.IDO1.F– 5′ TGCAGTGAGCCGAGATCCTGCCACT-3′ and CAGE435.IDO1.R– 5′ CTGAGAGGAAAGCTGCGCTGCCCAA-3′, overhangs not shown) as previously described52. Next-generation sequencing (NGS) analysis of clones was performed using CRIS.py53. Knockout clones were identified as clones containing only out-of-frame indels. Final clones were authenticated using the PowerPlex® Fusion System (Promega, Madison, WI, USA) performed at the Hartwell Center for Biotechnology at St. Jude Children’s Research Hospital. Final clones tested negative for mycoplasma with the MycoAlert Plus Mycoplasma Detection Kit (Lonza).
To measure transepithelial resistance, cells were plated in transwells in a 24-well plate and were read using an EVOM Ohmeter (World Precision, Sarasota, FL. USA).
IDO1 and sucrase-isomaltase (SI) expression were determined from lysates collected from WT and IDO1−/− cells by first extracting RNA with an RNeasy Mini RNA extraction kit (Qiagen). Superscript IV VILO (Invitrogen) was then used to generate complementary DNA before QuantiTect primer assays were used with QuantiFast SYBR Green PCR kit (Qiagen) to quantify host transcripts including mouse IDO1 (Hs_IDO1_1_SG; QT00000504), SI (Hs_SI_1_SG; QT00061474), and GAPDH (Hs_GAPDH_1_SG; QT00079247). Delta-delta Ct method was used to resolve the differential expression between WT and IDO1−/− cells relative to the housekeeping gene, GAPDH.
For confocal imaging, Caco-2 cells were seeded onto glass coverslips, and once confluent, cells were fixed with 100% ice-cold methanol, blocked with normal goat serum (NGS) in PBS at room temperature for 1 hour, and stained for sodium–potassium ATPase (ab167390; Abcam) and ezrin (MA5–13862; Invitrogen) for 1 hour followed by anti-mouse immunoglobulin (Ig) G-Alexa Fluor 488 or anti-rabbit IgG-Alexa Fluor 555 (Invitrogen) secondary antibodies and DAPI (4′,6′-diamidino-2-phenylindole; Sigma, St. Louis, MO, USA) in 1% NGS for 30 minutes at room temperature. Following staining, coverslips were mounted onto slides with Prolong Gold Antifade Mountant (Invitrogen) and sealed. Z-stack images were acquired with a Nikon TE2000 inverted microscope with a 60x objective lens and resolved using Nikon NIS Elements software (Nikon, Brighton, MI, USA). Antibodies against IDO1 (Clone D8W5E, 51851S, Cell Signaling, Danvers, MA, USA) and MUC2 (GTX100664, GeneTex) were also used to stain Caco-2 cells following the same procedures and then visualized on the ECHO Revolve Microscope.
For western blot quantification of cellular proteins, Caco-2 monolayers were lysed in 100 ul of RIPA buffer (Abcam) and 1x protease inhibitor cocktail (Pierce, Waltham, MA, USA) for 15 minutes at room temperature and centrifuged at 14,000 × g for 5 minutes at 4 °C. Protein concentrations were determined using BCA Protein Assay Kit (Pierce). Equal protein concentrations of the soluble fraction were separated by SDS-PAGE (4–20% gel) under reducing conditions. Following transfer to nitrocellulose and probing for ezrin (MA5–13862; Invitrogen), occludin (71–1500, Invitrogen) and ZO-1 (33–9100, Invitrogen), and β-actin (A5441, Sigma). The blot was imaged on a Licor Odyssey Fc and band densitometry was measured using Image Studio version 5.2 software (LI-COR Biotechnology, Lincoln, NE, USA).
HAstV1 and Reovirus infections
HAstV1 viral stocks were propagated in Caco-2 cells and infectious titers were determined using a fluorescent-focus assay using Caco-2 cells as previously described54. WT and IDO1−/− Caco-2 cells were plated and once confluent, the cells were inoculated with HAstV1, Reovirus type 1 Lang (T1L), or mock (PBS). At 24 hours after infection, cells were fixed with 100% ice-cold methanol and then blocked with 5% NGS in PBS at room temperature for 1 hour. Detection of HAstV1 and Reovirus was performed by capsid staining using the mouse monoclonal 8E7 (Thermo Fisher) and IgG rabbit antisera, respectively, followed by anti-mouse IgG-Alexa Fluor 488 or anti-rabbit IgG-Alexa Fluor 488 (Invitrogen) and DAPI (Sigma) in 1% NGS for 30 minutes at room temperature. Reovirus T1L and IgG rabbit antisera were kind gifts from Dr. Terence Dermody, University of Pittsburgh. For each well, two images at 20X magnification were collected using an EVOS FL microscope (Thermo Fisher Scientific) and quantified in Image J (Rockville, MD, USA).
HAstV1 capsid spike binding
Recombinant GFP-Spike was expressed and purified as described previously55. 24-well plates containing fibronectintreated glass coverslips were seeded with 100,000 Caco-2 cells per well and allowed to adhere for 3 days. 250-μl samples containing 400 nM GFP-Spike were incubated for 1 hour at room temperature in PBS. Samples of PBS alone and 400 nM GFP were used as controls for autofluorescence and nonspecific binding, respectively. Media was aspirated from the cell monolayer, and then GFP-Spike was added to Caco-2 cells and incubated at 4 °C for 1 hour. Cells were washed with PBS and fixed with 2% paraformaldehyde in PBS for 15 minutes. Cells were washed with PBS and then stained with DAPI (VWR, Radnor, PA, USA) in PBS for 1 hour. Coverslips were washed with PBS, dried, and mounted in SlowFade Diamond Antifade Mountant (Thermo-Fisher) on glass microscope slides.
Z-stack images were acquired by using identical acquisition parameters with a Zeiss Axio Imager equipped with an AxioCam 506 monochrome camera using an oil-immersion 100/1.4NA Plan Apo objective lens (Zeiss, Oberkochen, Germany). Z-stack images contained nine slices at 1-μm intervals, with GFP and DAPI channels exposed for 240 ms and 17 ms, respectively. GFP signal was collected with a Zeiss Fset38 filter cube, and DAPI signal was collected with a Zeiss Fset49 filter cube. After acquisition, images were deconvolved with AutoQuant X 3D deconvolution software (Media Cybernetics Version X3.1.3, Rockville, MD, USA) for five iterations, using the Z-montage option. GFP spot objects and nuclei were quantified separately using Imaris Software version 9.9.1 (Bitplane Inc., Belfast, U.K.). GFP and nuclei spot objects creation algorithms were generated using the Spots Creation Wizard. The same creation parameters were applied across all quantified images for each type of object, GFP, or nuclei. Creation algorithms are available upon request. Z-stack slices three through nine from the resulting 32-bit float images were converted to a single RGB image after linear histogram adjustments were made to the GFP channel such that minimum values were 300 and maximum values were 9,000 to help reduce background fluorescence, and also to the DAPI channel such that minimum values were 0 and maximum values were 20,000 to improve the visibility of cell nuclei. Images are representative of data from two independent experiments.
Statistical analyses
To compare across experimental groups, Kruskal-Wallis Test with Dunn’s multiple comparisons, Mann-Whitney U for non-normally distributed datasets, and unpaired t-test with Welch’s correction for normally distributed data with unequal variances were performed using GraphPad Prism version 9.5.0. Two-tailed and one-tailed p values are indicated, depending on the hypothesis.
Supplementary Material
ACKNOWLEDGMENTS
The authors also thank Lee-Ann Van de Velde for helping to obtain and genotype animals, Dr. Ben Abrams of the University of California, Santa Cruz Life Sciences Microscopy Center Core Facility (RRID: SCR_021135) and the St. Jude Veterinary Pathology Core and Flow Cytometry and Cell Sorting Shared Resources.
FUNDING
Funding for this research included National Institutes of Health Allergy and Infectious Diseases grants R21 AI135254 and R01 AI166434 to S.S-C., T32 AI106700 and K22 AI156116 and K22 AI156116-S1 to V.C., U01 AI50747, R01 AI154470, and U01 AI44616 to P.G.T. and J.C.C., R01 AI144090 to R.M.D., as well as General Medical Sciences grant T32 GM133391 to S.L. and N.P. Funding was also provided from American Lebanese Syrian Associated Charities (ALSAC) to S.S.C.
Footnotes
DECLARATIONS OF COMPETING INTEREST
The authors have no competing interests to declare.
APPENDIX A. SUPPLEMENTARY DATA
Supplementary data to this article can be found online at https://doi.org/10.1016/j.mucimm.2023.05.011.
REFERENCES
- 1.Farkas T, Fey B, Keller G, Martella V & Egyed L Molecular detection of novel astroviruses in wild and laboratory mice. Virus Genes 45, 518–525 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Yokoyama CC et al. Adaptive immunity restricts replication of novel murine astroviruses. J Virol 86, 12262–12270 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ng TFF et al. Identification of an astrovirus commonly infecting laboratory mice in the US and Japan. PLoS One 8, e66937 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marvin SA et al. Type I interferon response limits astrovirus replication and protects against increased barrier permeability in vitro and in vivo. J Virol 90, 1988–1996 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cortez V et al. Characterizing a murine model for astrovirus using viral isolates from persistently infected immunocompromised mice. J Virol 93. e00223–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Compton SR, Booth CJ & Macy JD Murine Astrovirus Infection and transmission in neonatal CD1 mice. J Am Assoc Lab Anim Sci 56, 402–411 (2017). [PMC free article] [PubMed] [Google Scholar]
- 7.Ingle H et al. Murine astrovirus tropism for goblet cells and enterocytes facilitates an IFN-λ response in vivo and in enteroid cultures. Mucosal Immunol 14, 751–761 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sebire NJ et al. Pathology of astrovirus associated diarrhoea in a paediatric bone marrow transplant recipient. J Clin Pathol 57, 1001–1003 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koci MD et al. Astrovirus induces diarrhea in the absence of inflammation and cell death. J Virol 77, 11798–11808 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baldridge MT et al. Expression of Ifnlr1 on intestinal epithelial cells is critical to the antiviral effects of interferon lambda against Norovirus and reovirus. J Virol 91. e02079–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin JD et al. Distinct roles of type I and Type III interferons in intestinal immunity to homologous and heterologous rotavirus infections. PLoS Pathog 12, e1005600 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nice TJ et al. Interferon-λ cures persistent murine Norovirus infection in the absence of adaptive immunity. Science 347, 269–273 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johansson C et al. Type I interferons produced by hematopoietic cells protect mice against lethal infection by mammalian reovirus. J Exp Med 204, 1349–1358 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sen A, Sharma A & Greenberg HB Rotavirus degrades multiple interferon (IFN) type receptors to inhibit IFN signaling and protects against mortality from endotoxin in suckling mice. J Virol 92. e01394–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barro M & Patton JT Rotavirus NSP1 inhibits expression of type I interferon by antagonizing the function of interferon regulatory factors IRF3, IRF5, and IRF7. J Virol 81, 4473–4481 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stanifer ML, Kischnick C, Rippert A, Albrecht D & Boulant S Reovirus inhibits interferon production by sequestering IRF3 into viral factories. Sci Rep 7, 10873 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morelli M, Dennis AF & Patton JT Putative E3 ubiquitin ligase of human rotavirus inhibits NF-κB activation by using molecular mimicry to target β-TrCP. MBio 6. e02490–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kolawole AO et al. Astrovirus replication in human intestinal enteroids reveals multi-cellular tropism and an intricate host innate immune landscape. PLoS Pathog 15, e1008057 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Triana S et al. Single-cell transcriptomics reveals immune response of intestinal cell types to viral infection. Mol Syst Biol 17, e9833 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guix S et al. Type I interferon response is delayed in human astrovirus infections. PLoS One 10, e0123087 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cortez V et al. Astrovirus infects actively secreting goblet cells and alters the gut mucus barrier. Nat Commun 11, 2097 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kulkarni DH et al. Goblet cell associated antigen passages are inhibited during Salmonella typhimurium infection to prevent pathogen dissemination and limit responses to dietary antigens. Mucosal Immunol 11, 1103–1113 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shan M et al. Mucus enhances gut homeostasis and oral tolerance by delivering immunoregulatory signals. Science 342, 447–453 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hassanain HH, Chon SY & Gupta SL Differential regulation of human indoleamine 2,3-dioxygenase gene expression by interferons-gamma and - alpha. Analysis of the regulatory region of the gene and identification of an interferon-gamma-inducible DNA-binding factor. J Biol Chem 268, 5077–5084 (1993). [PubMed] [Google Scholar]
- 25.Mehraj V & Routy JP Tryptophan catabolism in chronic viral infections: handling uninvited guests. Int J Tryptophan Res 8, 41–48 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mellor AL & Munn DH Tryptophan catabolism and regulation of adaptive immunity. J Immunol 170, 5809–5813 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Ciorba MA Indoleamine 2,3 dioxygenase in intestinal disease. Curr Opin Gastroenterol 29, 146–152 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manco R et al. Clump sequencing exposes the spatial expression programs of intestinal secretory cells. Nat Commun 12, 3074 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nyström EEL et al. An intercrypt subpopulation of goblet cells is essential for colonic mucus barrier function. Science 372, eabb1590 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haber AL et al. A single-cell survey of the small intestinal epithelium. Nature 551, 333–339 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Al-Sadi R et al. Mechanism of interleukin-1β induced-increase in mouse intestinal permeability in vivo. J Interferon Cytokine Res 32, 474–484 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chassaing B et al. Fecal lipocalin 2, a sensitive and broadly dynamic non-invasive biomarker for intestinal inflammation. PLoS One 7, e44328 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ball HJ et al. Characterization of an indoleamine 2,3-dioxygenase-like protein found in humans and mice. Gene 396, 203–213 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Van de Velde LA, Gingras S, Pelletier S & Murray PJ Issues with the specificity of immunological reagents for murine IDO1. Cell Metab 23, 389–390 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Sen GC Viruses and interferons. Annu Rev Microbiol 55, 255–281 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Hoshi M et al. The absence of IDO upregulates type I IFN production, resulting in suppression of viral replication in the retrovirus-infected mouse. J Immunol 1950, 3305–3312 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Kim SB et al. Blockage of indoleamine 2,3-dioxygenase regulates Japanese encephalitis via enhancement of type I/II IFN innate and adaptive T-cell responses. J Neuroinflammation 13, 79 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Milano J et al. Modulation of Notch processing by γ-secretase inhibitors causes intestinal goblet cell Metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicol Sci 82, 341–358 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Alvarado DM et al. Epithelial indoleamine 2,3-dioxygenase 1 modulates aryl hydrocarbon receptor and Notch signaling to increase differentiation of secretory cells and alter mucus-associated microbiota. Gastroenterology 157, 1093–1108.e11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rhee SJ, Walker WA & Cherayil BJ Developmentally regulated intestinal expression of IFN-γ and its target genes and the age-specific response to enteric salmonella infection. J Immunol 175, 1127–1136 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Moser LA, Carter M & Schultz-Cherry S Astrovirus increases epithelial barrier permeability independently of viral replication. J Virol 81, 11937–11945 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Astroviridae - positive sense RNA viruses - positive sense RNA viruses, ICTV. Available at: https://talk.ictvonline.org/ictv-reports/ictv_9th_report/positive-sense-rna-viruses-2011/w/posrna_viruses/247/astroviridae (2011) [Date accessed: 01 June 2022].
- 43.Janowski AB Beyond the gastrointestinal tract: the emerging and diverse tissue tropisms of astroviruses. Viruses 13, 732 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barceló-Batllori S et al. Proteomic analysis of cytokine induced proteins in human intestinal epithelial cells: implications for inflammatory bowel diseases. Proteomics 2, 551–560 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Lucas TM, Richner JM & Diamond MS The interferon-stimulated gene Ifi27l2a restricts West Nile virus infection and pathogenesis in a cell-type- and region-specific manner. J Virol 90, 2600–2615 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strine MS & Wilen CB Tuft cells are key mediators of interkingdom interactions at mucosal barrier surfaces. PLoS Pathog 18, e1010318 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bomidi C, Robertson M, Coarfa C, Estes MK & Blutt SE Single-cell sequencing of rotavirus-infected intestinal epithelium reveals cell-type specific epithelial repair and tuft cell infection. Proc Natl Acad Sci U S A 118. e2112814118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roach SN et al. Respiratory influenza virus infection causes dynamic tuft cell and innate lymphoid cell changes in the small intestine. J Virol 96, e0035222 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tschurtschenthaler M et al. Type I interferon signalling in the intestinal epithelium affects Paneth cells, microbial ecology and epithelial regeneration. Gut 63, 1921–1931 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Katlinskaya YV et al. Type I interferons control proliferation and function of the intestinal epithelium. Mol Cell Biol 36, 1124–1135 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Van Winkle JA et al. Homeostatic interferon-lambda response to bacterial microbiota stimulates preemptive antiviral defense within discrete pockets of intestinal epithelium. Elife 11, e74072 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sentmanat MF, Peters ST, Florian CP, Connelly JP & Pruett-Miller SM A survey of validation strategies for CRISPR-Cas9 editing. Sci Rep 8, 888 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Connelly JP & Pruett-Miller SM CRIS.py: a versatile and high-throughput analysis program for CRISPR-based genome editing. Sci Rep 9, 4194 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marvin S, Meliopoulos V & Schultz-Cherry S Human astrovirus propagation, purification and quantification. Bio-Protocol 4, e1078 (2014). [Google Scholar]
- 55.Bogdanoff WA et al. Structure of a human astrovirus capsid-antibody complex and mechanistic insights into virus neutralization. J Virol 91. e01859–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.