

Abstract
Pyroptosis is a form of programmed cell death associated with activation of inflammasomes and inflammatory caspases, proteolytic cleavage of gasdermin proteins (forming pores in the plasma membrane), and selective release of proinflammatory mediators. Induction of pyroptosis results in amplification of inflammation, contributing to the pathogenesis of chronic cardiovascular diseases such as atherosclerosis and diabetic cardiomyopathy, and acute cardiovascular events, such as thrombosis and myocardial infarction. While engagement of pyroptosis during sepsis-induced cardiomyopathy and septic shock is expected and well documented, we are just beginning to understand pyroptosis involvement in the pathogenesis of cardiovascular diseases with less defined inflammatory components, such as atrial fibrillation. Due to the danger that pyroptosis represents to cells within the cardiovascular system and the whole organism, multiple levels of pyroptosis regulation have evolved. Those include regulation of inflammasome priming, post-translational modifications of gasdermins, and cellular mechanisms for pore removal. While pyroptosis in macrophages is well characterized as a dramatic proinflammatory process, pyroptosis in other cell types within the cardiovascular system displays variable pathways and consequences. Furthermore, different cells and organs engage in local and distant crosstalk and exchange of pyroptosis triggers (oxidized mitochondrial DNA), mediators (IL-1β, S100A8/A9) and antagonists (IL-9). Development of genetic tools, such as Gasdermin D knockout animals, and small molecule inhibitors of pyroptosis will not only help us fully understand the role of pyroptosis in cardiovascular diseases but may result in novel therapeutic approaches inhibiting inflammation and progression of chronic cardiovascular diseases to reduce morbidity and mortality from acute cardiovascular events.
Keywords: pyroptosis, atherosclerosis, myocardial infarction, cardiomyopathy, cardiac macrophages, cardiomyocytes
1. Introduction
Cardiovascular diseases (CVD) are the leading cause of morbidity and mortality in the developed and developing world [1]. Excessive and poorly controlled inflammation drives the pathogenesis and poor outcome of chronic cardiovascular diseases (e.g. atherosclerosis, diabetic cardiomyopathy, hypertension) and contributes to acute cardiovascular events (stroke, myocardial infarction, thrombosis). Pyroptosis, a pore-forming mechanism of programmed or regulated cell death associated with release of proinflammatory mediators, is an important amplifier of inflammation in cardiovascular diseases. In this work, we review the evidence for pyroptosis as a defining point in the pathogenesis and outcome of cardiovascular diseases and characterize involvement of multiple cell types in initiation, progression, and control of pyroptosis in relation to CVD. We highlight recently described regulators of pyroptosis and identify ongoing research on targeting pyroptosis as a therapeutic intervention in cardiovascular diseases.
2. A place for pyroptosis in the landscape of programmed cell death within cardiovascular system.
2.1. Definition and specific features of pyroptosis.
The term pyroptosis was introduced by Brad Cookson and Molly Brennan in 2001 to describe a form of a proinflammatory programmed cell death [2]. The original definition of pyroptosis described caspase-1-dependent cell death following invasion of macrophages by Salmonella typhimurium [3,4], hypoxic injury in the central nervous system [5], and plaque rupture in cardiovascular system [6]. Our understanding of pyroptosis activation progressively expanded as the varieties of inflammasomes were found to activate inflammatory caspases (caspase-1, −4, −5, and −11) [7]. Execution of pyroptosis is determined by caspase-mediated cleavage of the 52-kDa protein Gasdermin D (GSDMD) resulting in formation of the 31-kDa N-GSDMD fragment that oligomerizes and forms pores in the cell membrane [8,9]. Binding of the N-GSDMD to the membrane lipid, formation of the 33-mer prepore and transition into the functional pore has been described through structural analyses, which also revealed the electrostatic selectivity of the GSDMD pores and preferential release of IL-1β [10]. Furthermore, ROS-dependent S-palmitoylation of Cys191 residue of human GSDMD, augmented by LPS priming, was recently identified as an obligate requirement for GSDMD pore formation [11,12]. Besides GSDMD, proteolytic cleavage of other members of Gasdermin family (GSDMA, GSDMB, GSDMC, GSDME (also known as DFNA5) and PJVK (also known as DFNB59) leads to release of proinflammatory mediators, such as IL-1β, and subsequent cell lysis, sharing the characteristic features of executors of pyroptosis [13]. Recent studies revealed further mechanisms leading to the same cell fate and expanded the definition of pyroptosis to include non-canonical inflammasome (caspase-1-independent cleavage of GSDMD) pathway as well as cleavage of GSDMD by caspase-8 and cleavage of GSDME by caspase-3 in addition to the canonical inflammasome caspase-1-dependent pathway [14]. Thus, pyroptosis can be described as a mechanism of programmed cell death that “critically depends on the formation of plasma membrane pores by members of the gasdermin protein family, often (but not always) as a consequence of inflammatory caspase activation.” [15].
2.2. The role of inflammasome priming for pyroptosis outcome.
Under homeostatic conditions, most cells within the cardiovascular system, especially cardiomyocytes, express relatively low levels of pattern recognition receptors and inflammasome components [16]. Pyroptosis-inducing inflammasome activation is positively regulated by inflammasome priming, which is induced by engagement of TLRs or IL-1 receptors [16,17]. The transcriptional phase of inflammasome priming primarily depends on NFкB and increases expression levels for IL1B, IL18, NLRP3, ASC, CASP1 gene products [18,19]. Non-transcriptional or rapid priming involves post-translational modifications of NLRP3 [16,17]. Thus, an ongoing acute or chronic inflammation is likely to shift the balance of cell responses to external or internal triggers towards pyroptosis. This implicates pyroptosis as a feedforward step in fatal inflammatory conditions.
2.3. Mitochondrial DNA (mtDNA) as a trigger and/or as a mediator of pyroptosis.
Contracting hearts have a high energy demand, which is primarily met by oxidative phosphorylation in mitochondria [20]. Chronic CVD and acute cardiovascular events create ischemic conditions that result in mitochondrial impairment due to hypoxia and excessive ROS accumulation, mtDNA oxidation and breach into cytosol or extracellular space [20]. Oxidized cytosolic mtDNA activates the cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) signaling pathway and NLRP3 inflammasome leading to pyroptosis [21,22]. Extracellular mtDNA may be released from cardiomyocytes as part of homeostatic turnover or following myocardial infarction, from neutrophils during NETosis and from other dying cells [23–25]. Depending on the cellular source and process, mtDNA may be present in circulation and extracellular space in naked form, in association with mitochondrial proteins, or enveloped into phospholipid vesicles [25,26]. Although extracellular mtDNA does not always activate pyroptosis, recognition of its CpG DNA repeats following partial degradation by endosomal TLR9 after phagocytosis or clathrin-mediated endocytosis may lead to inflammasome priming and indirectly contribute to pyroptosis-amplified inflammation [20,27–29]. Thus the source, localization, and form of the mtDNA, may determine the extent of pyroptosis activation and outcome in cardiovascular pathologies.
2.4. Crosstalk between pyroptosis and other forms of programmed cell death (PCD).
Pyroptosis shares several molecular mediators and features with other forms of cell death. Notably, the first observation of pyroptosis was initially labeled as apoptosis [30]. The key distinction of pyroptosis from apoptosis is the membrane pore formation and pro-inflammatory outcome [31] and (Table 1). However, the pathways upstream of the pore formation engage in intricate crosstalk switching the forms of programmed cell death based on the context of the initial trigger and cell type. For example, caspase-8, associated with extrinsic apoptosis may cleave GSDMD and induce pyroptosis in macrophages [32,33]. Activation of caspase-3, either directly by caspase-8 downstream of death receptors TNFR1 and Fas or as a result of mitochondrial permeabilization and activation of caspase-9, is usually associated with apoptosis. Yet, recent studies demonstrated that caspase-3 can also induce pyroptosis or secondary necrosis by cleaving GSDME [34,35].
Table 1.
Common and distinct features of pyroptosis, apoptosis, and necroptosis.
| Forms of cell death | Pyroptosis | Apoptosis | Necroptosis | PANoptosis | Refs. |
|---|---|---|---|---|---|
| Relation to inflammation | Pro-inflammatory | Anti-inflammatory | Pro-inflammatory | Pro-inflammatory | [2,31,36–38] |
| Triggers | Bacterial toxins, intracellular LPS, Trimethylamine N-oxide (TMAO), ox-mtDNA | Death ligands (FasL), mitochondrial stress | Death ligands, intracellular viral RNA | Viral proteins, TNF, IFNγ, nuclear export inhibitors Selinexor (KPT-330) and Eltanexor (KPT-8602) | [3,21,22, 50–52] |
| Sensors | PAMPs, DAMPs, NLRP1, NLRP3, NLRC4 | Death receptors (TNFR1, Fas), | Death receptors (TNFR1, Fas), TLRs, ZBP1 | TAK1, ZBP1 | [36], [53], [54], [55] |
| Mediators | Inflammasome (NLRP1, NLRP3, NLRC4, AIM2) | Apoptosome (APAF1) | Necrosome (RIPK1, RIPK3, MLKL) | ZBP1 and AIM2 PANoptosome | [31] |
| Key effectors and executioners | Caspase-1, caspase-4, caspase-5, caspase-8, caspase-11 | Caspase-3, caspase-7, caspase-8 | MLKL | Caspase-1, caspase-3, caspase-8, caspase-6, MLKL | [7,36–38,43,56] |
| Pores and pore executioners | Plasma membrane (~10–20 nm), Gasdermin D and other Gasdermins | Mitochondrial outer membrane permeabilization, BAX/BAK | Plasma membrane (~4 nm), MLKL | Plasma membrane pores and rupture | [8–10,53] |
| Specific features | Membrane pores, intact nuclei | Membrane blebbing, nuclear condensation | Cell swelling, membrane rupture | Cell swelling, membrane pores and rupture | [31] |
| Inhibitors | NIX, CLMP, , IL-9, disulfiram, dimethyl fumarate, necrosulfonamide | cFLIP, XIAP, zVAD | cFLIP, necrostatin-1, necrosulfonami de (in human cells) | Dickkopf-1, FUNDC1 | [47,48,55,57–61] |
To describe cell death and pathways shared by (P)yroptosis, (A)poptosis, and (N)ecroptosis, the concept of PANoptosis was recently introduced [36–38]. The biological role of PANoptosis cannot be individually accounted for by the three PCD pathways alone [39,40]. PANoptosis is regulated by PANoptosomes [41,42], multifaceted macromolecular complexes that integrate GSDMD, caspases−1, −3 and −8, and RIP kinases 1 (RIPK1) and 3 (RIPK3) and activate all three pathways to execute cell death. To date, two prototypical PANoptosomes have been biochemically identified: the Z-DNA binding domain protein 1 (ZBP1)-PANoptosome [42,43] and the AIM2-PANoptosome [39]. PANoptosomes have different sensors but share many key components associated with PCD. An important differentiating characteristic of PANoptosis from other forms of cell death is that deletion of the key component of PANoptosome ZBP1 blocks cell death, whereas deletion of individual components of other PCD pathways does not rescue cells [38]. While the roles of PANoptosis in cancer and infectious diseases, including infections with SARS-CoV-2 and cytokine storm have already been described in detail [43–46] we are just beginning to understand how PANoptosis may be involved and regulated in cardiovascular diseases. In diabetic retinopathy, Dickkopf-1 exerts protective effects by inhibiting PANoptosis and retinal neovascularization [47]. Similarly, FUNDC1, a mitochondrial membrane protein participating in the regulation of mitochondrial integrity, protects murine cardiomyocytes from PANoptosis in a model of doxorubicininduced heart injury [48]. Conversely, targeting the PANoptosome with 3,4-Methylenedioxy-β-Nitrostyrene reduces PANoptosis and protects the kidney against renal ischemia reperfusion injury [49]. Further investigation is warranted to characterize the roles of PANoptosis in human cardiovascular system and whether it would be possible to change the course of CVD by targeting PANoptosis.
3. Involvement of pyroptosis in the pathogenesis of cardiovascular diseases.
The most common underlying condition of chronic vascular diseases (coronary heart disease, peripheral artery disease, cerebrovascular diseases) is atherosclerosis, which develops due to a combination of metabolic factors and immune cell responses to deposition of low density lipoprotein cholesterol within arterial walls [62]. Acute cardiovascular events (myocardial infarction and stroke), caused by spontaneous atherosclerotic plaque rupture and thrombotic occlusion of major blood vessels, lead to massive cell death and critical organ injury due to ischemia and ischemia/reperfusion injury, mitochondrial damage, and accumulation of ROS. There is increasing evidence that pyroptosis, as a form of programmed cell death in response to external triggers, mediates amplification of inflammatory cascades and contributes to the pathogenesis of chronic and acute CVD. In Table 2 and the text below, we list specific examples of pyroptosis involvement in CVD with specific attention to cell types, triggers, mediators and inhibitors of pyroptosis.
Table 2.
Examples of cardiovascular diseases associated with pyroptosis in specific cell types.
| Cardiovascular Disease | Cell type(s) involved | Pyroptosis agonists and mediators | Pyroptosis Antagonists | Refs. |
|---|---|---|---|---|
| Atherosclerosis | Macrophages | Ox-LDL, AIM2, caspase-1, caspase-11, −4/5, GSDMD, GSDME | NIX | [6,65–67,70,116] |
| VSMC | GSDMD, caspase-1, AIM2 | Belnacasan/VX-765 (caspase-1 inhibitor) | [71,73] | |
| Endothelial cells | TMAO, mitochondrial SDHB, ROS, NLPR3, caspase-1 | [51] | ||
| Myocardial infarction | Cardiac fibroblasts | NLRP3 | CLMP | [57,76] |
| Cardiomyocyte | NLRP3, ROS, doxorubicin, GSDMD | GDF11, HOXA3, | [79,117,118] | |
| Neutrophils | GSDMD | [77,78] | ||
| Diabetic cardiomyopathy | Cardiomyocytes | ROS, TXNIP, chemerin and its receptor CMKLR1, NLRP3, caspase-1 | [83,84] | |
| Sepsis and septic shock, sepsis-induced cardiomyopathy | Platelets | S100A8/A9, TLR4, NLRP3, caspase-1, GSDMD | Paquinimod (inhibits S100A8/A9 binding to TLR4) MitoTempo, a specific scavenger of mitochondrial superoxide |
[54] |
| Cardiomyocytes | LPS+nigericin, NLRP3, caspase-1, GSDMD, ROS | VX-765, MCC950 | [50] | |
| Macrophages | GSDMD, Tissue Factor | [98] | ||
| Neutrophils | GSDMD undergoes secondary cleavage by elastase. | [95,97,119] | ||
| Endothelial cells | GSDMD | ILC2-derived IL-9 protects EC from pyroptosis | [58] | |
| Atrial fibrillation | Cardiomyocytes | TMAO, NLRP3, caspase-1, GSDMD | [102–104,106] | |
| Macrophages | TMAO, caspase-1, GSDMD | Akkermansia muciniphila | [106] | |
| Cardiac fibroblast | TMAO in co-culture with macrophages | Akkermansia muciniphila | [106] | |
| Hypertension | Cardiomyocytes | GSDMD, IL-18 | [108] | |
| Pulmonary artery hypertension | Pulmonary artery VSMC | Hypoxia, GSDMD | Disulfiram | [110] |
| Endothelial cells | Caspase-4, −11, GSDMD | [111] | ||
| Preeclampsia | Trophoblasts | NLRP3, GSDMD, IL-1β, IL-18 | [109] | |
| Abdominal aortic aneurism | VSMC | NLRP3, caspase-1, GSDMD | Disulfiram, α7 nicotinic acetylcholine receptor | [112,113] |
3.1. Atherosclerosis
Atherosclerosis is characterized by abnormal deposition of lipids and lipoproteins within arterial walls and associated with accumulation of foam cells derived from infiltrated monocytes and transdifferentiated vascular smooth muscle cells (VSMC) [63]. Inflammation drives atherosclerosis through continuing recruitment of monocytes into atherosclerotic plaques. Formation of mature plaques (atheromas), characterized by necrotic core and fibrous caps, is a chronic process that eventually disrupts arterial blood flow. The major risks of atherosclerosis are recurrent cardiovascular events (myocardial infarction and stroke), triggered by plaque rupture, as a result of thinning of the fibrous caps or superficial erosions [63]. A recent large-scale (>10,000 patients) clinical study CANTOS, using canakinumab, a therapeutic monoclonal antibody targeting interleukin-1β, reduced the rate of recurrent cardiovascular events [64].
Macrophage cell death in ruptured plaques was initially, and mistakenly, described as caspase-1-mediated apoptosis [6]. As subsequent studies showed, transition of atherosclerotic plaques to inflammatory phenotype in ApoE−/− mice requires GSDMD [65,66] or GSDME [67]. Importantly, deletion of GSDMD reduced the burden of atherosclerotic plaques, reduced leukocyte infiltration, and switched cell death from pyroptosis to apoptosis [66]. Exposure of macrophages to ox-LDL or cholesterol crystals activates NLRP3 inflammasome and caspase-1 [55]. Inhibition of the mitochondrial outer membrane protein NIX, known to be involved in mitophagy, leads to pyroptosis of macrophages exposed to ox-LDL [55]. Besides caspase-1, other caspases, such as caspase-11 in mice and caspase-4/5 in humans may activate GSDMD and trigger macrophage pyroptosis via non-canonical (caspase-1-independent) pathway and contribute to the pathogenesis of atherosclerosis [56].
Macrophage proliferation and inflammation contributes to accelerated atherosclerosis progression and increased risk of atherosclerotic CVD in clonal hematopoiesis associated with the somatic V617F mutation in Janus kinase 2 (JAK2) non-receptor tyrosine kinase [68,69]. Increased AIM2 inflammasome activation, IL-1β expression and markers of pyroptosis in the atherosclerotic plaques was documented in atherosclerosis prone Ldlr−/− mice with bone marrow cells expressing the mutant JAK2 [70]. Deletion of AIM2, Caspase-1/11, or GSDMD in the JAK2 mutant bone marrow or treatment with the IL-1 receptor antagonist anakinra improved features of plaque stability, including macrophage accumulation, cap thickness, and necrotic core area [70]. However, deletion of NLRP3 inflammasome in the JAK2 mutant bone marrow cells had no significant effect on atherosclerotic plaques [70], suggesting a dominant role of AIM2 bot not NLRP3 inflammasome in macrophage pyroptosis and progression of atherosclerosis in the context of clonal hematopoiesis.
Detection of cleaved GSDMD and caspase-1 colocalization with macrophage (CD68) and VSMC (α-smooth muscle actin) markers in human carotid lesions validates the hypothesis that pyroptosis is involved in atherosclerosis pathogenesis [6,71]. Since the roles of VSMC, especially transdifferentiated VSMC, in progression of atherosclerosis become more apparent [72], the findings that ox-LDL induces expression of AIM2 and GSDMD in VSMC and triggers caspase-1-mediated VSMC death suggest that pyroptosis in VSMC may be an important factor in promoting inflammation [71,73].
The most dramatic forms of pyroptosis are observed in response to infections, especially in the presence of LPS or intracellular bacteria [3,4,32,33,50]. Although there is no infectious agent that can be defined as a cause of atherosclerosis according to Koch’s postulates, chronic and acute infections with various viruses and bacteria may modify progression of atherosclerosis [74]. The finding that trimethylamine N-oxide (TMAO), a phosphatidylcholine metabolite released by intestinal microbiota, promotes plaque growth and triggers death of endothelial cells via mitochondrial damage and activation of NLRP3 and caspase-1, illustrates that diet-induced dysbiosis and/or intestinal microbiota metabolic factors may advance atherosclerosis via pyroptosis [51].
3.2. Myocardial infarction.
Severe occlusion of the coronary arteries supplying blood oxygen to heart due to thrombosis and/or underlying atherosclerosis results in ischemia of heart muscle and may lead to acute myocardial infarction characterized by ischemic death of cardiomyocytes and surrounding cardiac fibroblasts. Reperfusion-associated accumulation of ROS and leukocyte infiltration may result in increased heart tissue damage, loss of heart function, and death. Clinical studies and animal models of AMI established the negative impact of NLRP3 inflammasome activation, severe inflammation associated with neutrophil accumulation, increased IL-1β and IL-18 and other mediators on severity of myocardial infarction [75,76]. Our group determined that activation of GSDMD occurs in the early phase of AMI and is essential for recruitment of neutrophils and monocytes to ischemic hearts in AMI whereas genetic deletion of GSDMD reduces cell death and IL-1β secretion and attenuates myocardial injury [77]. An elegant study combining the AMI models with parabiosis and bone marrow transfer from caspase-1 and GSDMD-deleted mice demonstrated that neutrophils respond to heart injury by accumulating in the bone marrow and secreting IL-1β to promote granulopoiesis [78]. Hematopoietic deletion of caspase-1 or GSDMD improved heart function, likely by blocking sustained influx of neutrophils into the inflamed heart.
There is increasing evidence for involvement of pyroptosis of stromal cells (cardiomyocytes and fibroblasts) in AMI pathogenesis. Pyroptosis in cardiomyocytes during AMI was shown to occur after hypoxic conditions disrupted an inhibitory pathway mediated by growth differentiation factor 11 (GDF11), a member of the superfamily of the transforming growth factor β [79]. Under homeostatic conditions, GDF11 inhibits pyroptosis by TGFβ-SMAD2/3 induction of HOXA3, which in turn represses transcription of NLRP3. Following experimental AMI, hypoxic conditions lead to decreased GDF11 expression thus enabling NLRP3 expression and cardiomyocyte pyroptosis [79]. Furthermore, increased expression of IL1-β, IL-18 mRNA and activation of NLRP3 inflammasome in cardiac fibroblasts was associated with myocardial infarction [76]. Deletion of CXADR-like membrane protein (CLMP) resulted in pyroptosis in cardiac fibroblasts in mice post AMI [57].
3.3. Diabetes and diabetic cardiomyopathy
Diabetic cardiomyopathy is one of the leading causes of heart failure and death in diabetes mellitus (DM) [80]. High levels of glucose have been previously shown to induce oxidative stress via aldose reductase and other glucose toxicity pathways [81,82]. ROS-dependent activation of NLRP3 inflammasome appears to contribute to diabetic cardiomyopathy as miRNA-mediated NLRP3 gene silencing improved heart function and reversed cardiac remodeling in a rat model of diabetic cardiomyopathy [83]. The role of ROS-induced thioredoxin interacting protein TXNIP and its interaction with NLRP3 was confirmed by siRNA-mediated silencing of TXNIP resulting in decreased caspase-1 activation and IL-1β production [83]. A recent study suggested that activation of NLRP3 inflammasome, dependent on chemerin and its G-protein coupled receptor CMKLR1, triggered cardiomyocyte pyroptosis and played a critical role in a model of diabetic cardiomyopathy in rats [84]. However, both studies relied primarily on analyses of NLRP3 inflammasome and caspase-1 cleavage and did not provide a definitive proof of cardiomyocyte pyroptosis since they lacked analyses of GSDMD involvement [83,84]. Thus, more detailed analyses, involving GSDMD deletion and characterization of membrane pore formation is necessary to prove that pyroptosis is responsible for diabetic cardiomyopathy.
In addition to diabetic cardiomyopathy, platelet abnormalities are another hallmark of DM, which contribute to the increased thrombotic events and development of atherosclerosis [81]. Our previous study has suggested platelet apoptosis was induced by hyperglycemia-mediated mitochondrial ROS via p53-caspase-3 pathway in DM [82]. In addition to myocardial pyroptosis, it would be intriguing to investigate whether platelet pyroptosis also ensue in DM, and how it may contribute to diabetic cardiovascular disease.
3.4. Sepsis and sepsis-induced cardiomyopathy
Sepsis is a systemic and dysregulated host response to infection, often leading to multiorgan failure and death [85]. During the systemic inflammatory responses, excessive dilation and increased permeability of blood vessels place a greater demand on the heart in septic patients and contribute to myocardial dysfunction characterized by decreased systolic contractility and cardiomyopathy [86]. Myocardial dysfunction is a key component of septic shock, a failure of the cardiovascular system to maintain adequate tissue perfusion and oxygenation, leading to mortality of often more than 70% [86,87]. The pathophysiology of sepsis-induced cardiomyopathy involves inflammation, immune and endothelial cell dysfunction, and mitochondrial impairment [87,88].
Recent studies suggested that pyroptosis is an important pathogenetic mechanism of sepsis-induced cardiomyopathy. NLRP3 inflammasome-mediated excessive inflammation has been shown to contribute to myocardial dysfunction during experimental sepsis [89], [90]. Furthermore, Nlrp3 deletion protected mice from experimental sepsis while IL-1β has been shown to reduce cardiomyocyte contractility and induce cardiomyocyte atrophy [91]. A study using an LPS-induced cardiomyopathy model showed that accumulation of cleaved GSDMD in the heart tissues coincides with release of cardiac troponin, creatin kinase isoenzymes MB (CK-MB) and LDH into circulation, indicative of decreased cardiac function and cardiac damage [50]. Global deletion of GSDMD attenuated LPS-induced myocardial dysfunction and inflammation while also reducing mitochondrial dysfunction, ROS accumulation and NLRP3 inflammasome activation [50], thereby suggesting a mechanism for amplification of sepsis-induced cardiomyopathy.
A number of cell types, include macrophages [92], endothelial cells [93,94], neutrophils [95,96], and platelets [54] undergo pyroptosis during sepsis. The importance of careful evaluation of pyroptosis pathways and consequences in specific cell types is urgently needed to understand whether pyroptosis could be a tangible target for intervention. For example, while global deletion of Gsdmd or inhibition with disulfiram protected mice from multiple organ injury in experimental sepsis, likely by reducing release of neutrophil extracellular traps (NET) [95], neutrophil-specific deletion of Gsdmd using MRP8-Cre mice did not protect mice from sepsis, but, rather unexpectedly, increased multiorgan dysfunction [97].
Engagement of pyroptosis in one cell type with subsequent release of proinflammatory mediators instigates multiple cell types, often in distant organs or system-wide [54,98]. Sepsis induces inflammasome activation and pyroptosis in macrophages, resulting in systemic release of tissue factor, which acts on platelets to trigger disseminated intravascular coagulation and host death [92]. Therefore, one could speculate that a change in hemodynamics due to platelet hyperactivity and clotting may be an additional factor contributing to myocardial dysfunction in sepsis. On the other hand, cross-talk between cell types dampen the inflammation and lead to recovery, as sepsis-induced IL-33 expansion of innate lymphoid cells ILC2 illustrates a negative feedback mechanism involving IL-9 mediated prevention of pyroptosis in lung endothelial cells [58].
3.5. Where there’s smoke, there’s fire: CVD with emerging pyroptosis involvement.
As our attention to pyroptosis is growing, we are likely to find characteristic features of pyroptosis in CVD beyond those listed above. There is plenty of evidence that stroke, an acute cardiovascular event caused by arterial blockage or vascular rupture [99], results in massive neuronal and microglial cell death, including by pyroptosis, in response to hypoxia and/or hemorrhage. Since the pyroptotic death of brain cells is beyond the scope of this work, the readers are directed to other sources [100].
There is a growing evidence of intricate relationships between inflammation and atrial fibrillation, the most common cardiac arrhythmia that is associated with the risk of thrombosis, stroke, and heart failure [101]. Infections, obesity, hypertension, coronary artery diseases, and prior surgeries are among the factors that initiate inflammatory responses in the atrial tissue leading to dysfunctional electrical and structural remodeling prior to atrial fibrillation. Atrial tissue and cardiomyocytes from patients with atrial fibrillation were found to have increased NLRP3-mediated caspase-1 activation, whereas genetic cardiomyocyte-specific modification of NLRP3 inflammasome in animals correlated with dysfunctional electrical and structural remodeling and sensitivity to atrial fibrillation [102]. Follow up studies documented GSDMD cleavage in atrial tissues and allowed to establish the role of NLRP3 in linking atrial fibrillation with prior cardiac surgeries or obesity [103,104]. Paradoxically, homozygous deletion of GSDMD gene in human patients with non-familial atrial fibrillation increased the risk of thromboembolic stroke [105]. The hypothesis put forward by the authors of the study stating that deletion of GSDMD impairs clearance of injured atrial myocytes and promotes thromboembolic stroke requires further testing.
An intriguing study using rats suggested that cold exposure, a known trigger of atrial fibrillation, elevates plasma levels of gut metabolites, such as TMAO, which increases M1 macrophage infiltration of atrial tissues and triggers caspase-1-dependent cleavage of GSDMD not only in macrophages, but also in cardiomyocytes and cardiac fibroblasts when they are co-cultured with macrophages [106]. Remarkably, cold exposure reduced gut colonization by anaerobic intestinal symbiont Akkermansia muciniphila known to degrade mucin and control plasma levels of TMAO, whereas oral supplementation with these bacteria reversed cold-induced sensitivity to atrial fibrillation [106].
Pyroptosis activation is relatively poorly described in CVD characterized by increased blood pressure despite the evident role of macrophages in the pathogenesis of pulmonary artery hypertension and T cell role in systemic hypertension induced by high salt diet and angiotensin II [107]. So far, relatively few studies suggesting involvement of pyroptosis in the pathogenesis of systemic hypertension, pulmonary artery hypertension, and preeclampsia rely on more than a single readout of pyroptosis [108–111]. Hence, more investigations are likely to follow to determine how pyroptosis may contribute to the pathogenesis of hypertension.
Aortic aneurysm is a life-threatening condition characterized by a balloon-like building in the aorta due to remodeling of the extracellular matrix and aortic wall cell composition, developing often as a complication of hypertension and atherosclerosis. Inflammation-associated pyroptosis of VSMC, evidenced by induction of NPRL3, caspase-1 and GSDMD has been recently reported in a mouse model of angiotensin II-induced abdominal aortic aneurysm [112,113]. Interestingly, aneurysm development, VSMC pyroptosis and inflammation could be inhibited not only by disulfiram [112], but also by PNU-282987, a specific agonist of α7 nicotinic acetylcholine receptor [113]. These findings are so far limited to a single mouse model of aortic aneurysm and need further exploration to determine whether pyroptosis could be a valid target to reduce aortic aneurysm rupture.
In light of apparent involvement of apoptosis in the onset and potential inflammatory state of congenital heart diseases [114,115], it is rather surprising that we have yet to see publications on potential involvement of pyroptosis in those conditions. The hemodynamic conditions of severe congenital heart diseases can lead to myocardial injury, heart failure and hypoxia. These events are likely to be associated with inflammatory conditions predisposing to inflammasome activation and possible downstream activation of pyroptosis. Panoptosis as described above, may ultimately play an important role in all these cardiovascular processes.
4. Cell-type specific features of pyroptosis in CVD.
Differential expression of sensors (PAMP, DAMP), mediators (NRLP, caspases) and executioners (gasdermins) as well as pro-inflammatory mediators determine sensitivity to pyroptosis and its consequences in a cell-type specific mode [16,120]. While some cells display characteristic features of canonical and non-canonical pyroptosis pathways, there is a considerable difference in the resistance to pro-pyroptotic signals and pyroptosis sequela between stromal and immune cell types (Table 2 and Figure 1).
Figure 1:
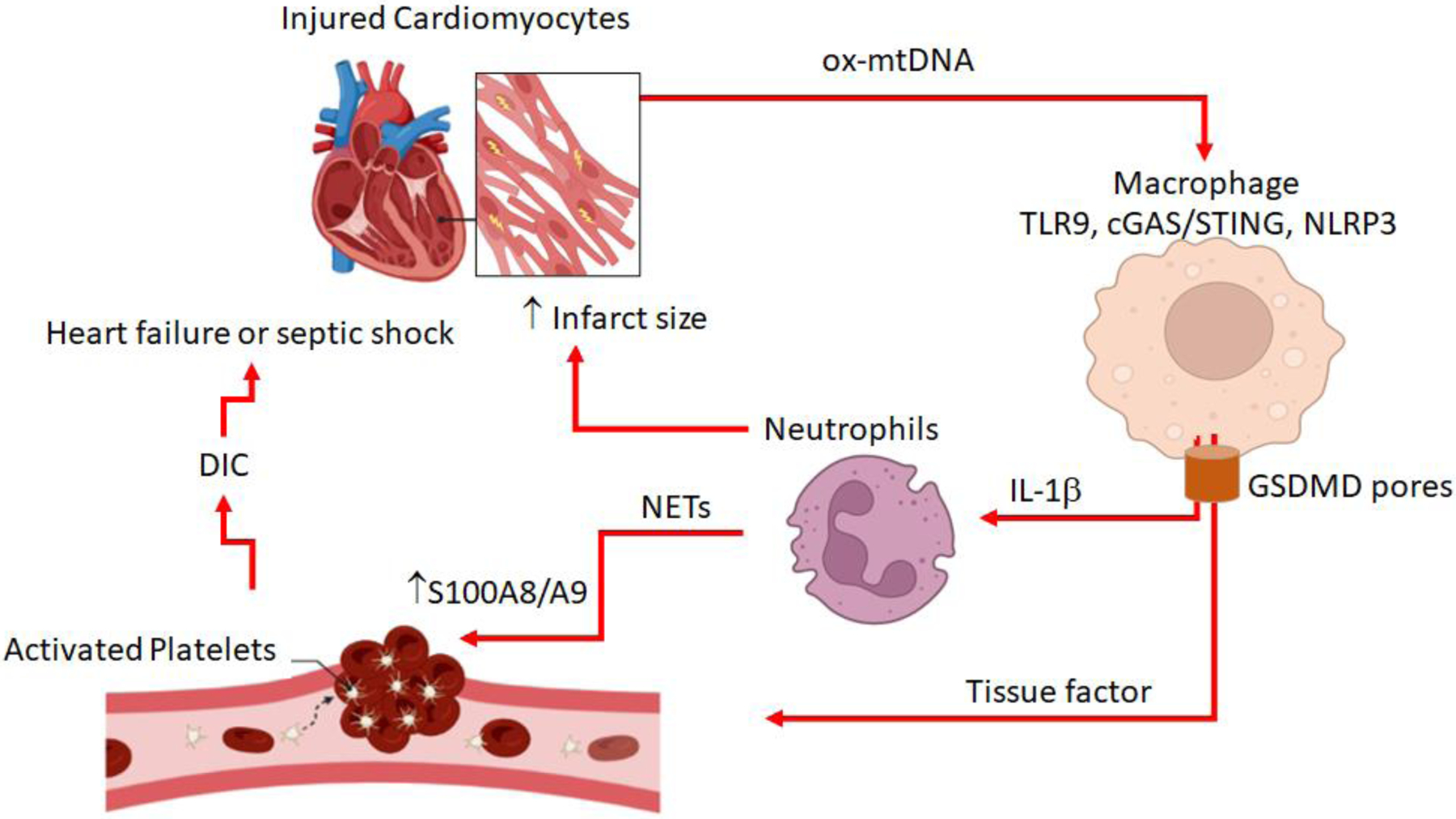
A simplified cartoon highlighting the role of cardiac mtDNA as a trigger and mediator of pyroptosis and complex interactions of various cell types with regards to pyroptosis after myocardial infarction or sepsis. The illustration is based on [54,77,92,149] and drawn with BioRender templates.
4.1. Macrophages and monocytes as targets, drivers, and regulators of pyroptosis in CVD.
Molecular mechanisms and consequences of pyroptosis in macrophages have been studied in the greatest detail. With the cardiovascular system, it is very important to consider macrophage origin, i.e. whether they are yolk sac or fetal liver derived tissue resident macrophages or adult bone marrow derived monocytes or recruited macrophages. Under homeostatic conditions, the majority of cardiac and intravascular macrophages can be traced to embryonic, primarily fetal liver, origin. The balance shifts toward bone marrow derived monocytes and macrophages in myocardial infarction or chronic inflammatory conditions, including atherosclerosis.
Compared to other cells, monocytes and bone marrow derived macrophage express the highest levels of caspase-1, precursors of IL-1β and IL-18, and a spectrum of inflammasome related proteins. Their arsenal of the TLRs, and potential for rapid transcriptional regulation of pyroptosis-associated genes, poise them as drivers of pyroptosis during infections and cardiovascular diseases (myocardial infarction, stroke, and atherosclerosis). Furthermore, macrophage polarization (oversimplified by the M1 vs. M2 paradigm), plasticity, migration and ability to produce massive amounts of IL-1β suggest that they can drive pyroptosis in other cell types through inflammasome priming. Besides being sensitive targets of pyroptosis, macrophages play a central role in propagation of pyroptosis to other cells of cardiovascular system through release of inflammatory mediators via pyroptotic and non-pyroptotic mechanisms when challenged by microbial products [92,106]. However, resident cardiac macrophages are likely to prevent spontaneous pyroptosis in cardiomyocytes and other cells under homeostatic conditions in the heart by taking up and degrading damaged mitochondria from cardiomyocytes [23].
4.2. Distinct cleavage of GSDMD by elastase changes plasma membrane pores in pyroptotic neutrophils.
Neutrophils are the most abundant leukocytes in human blood and are rapidly recruited to the sites of inflammation [121]. By responding to microbial stimuli, chemokines and cytokines and other cues from immune and stromal cells, these terminally differentiated short-lived phagocytes have important proinflammatory and repair functions in CVD [122]. Activation of NLRP3, NLRP1b, NLRC4, AIM2 or Pyrin inflammasomes in neutrophils has been reported to result in caspase-1-mediated cleavage of GSDMD and canonical pyroptosis associated with release of mature IL-1β through membrane pores [53]. However, several studies noted that instead of pyroptotic cell death observed in macrophages, activation of inflammasomes, caspase-1 or caspase−4/−11 with subsequent cleavage of GSDMD in neutrophils results in release of NETs rather than cell death [123,124]. The distinctive feature of neutrophil pyroptosis pathways is that cleaved N-GSDMD does not localize to the plasma membrane, but rather accumulates in the LC3+ autophagosomes and azurophilic granules [125]. There, N-GSDMD contributes to cytoplasmic release of elastase and undergoes secondary cleavage resulting in a smaller 28-kDa fragment [125]. Caspase-independent cleavage of GSDMD by neutrophil elastase following lysosomal membrane permeabilization has also been reported [119]. The detailed analysis of pyroptosis in neutrophils will be presented in the accompanying review in this series by Mohamed Lamkanfi and Etienne Meunier. In this section, we will briefly outline aspects of neutrophil pyroptosis and associated pathways that are most relevant to CVD.
Rapid and prominent infiltration of heart tissue by neutrophils augments severity and expansion of injury during AMI [122]. Deletion of Casp1 or Gsdmd improved heart functions, reduced mortality and release of IL-1β and LDH from unfractionated CD11b+ leukocytes in a mouse model of AMI [77,78]. However, no significant difference in LDH or IL-1β release was found between the neutrophils isolated from the ischemic hearts of wild-type vs. Gsdmd−/− mice [77]. On the other hand, AMI resulted in accumulation of reverse-migrating inflammasome-primed neutrophils in the bone marrow [78]. GSDMD-dependent release of IL1-β in neutrophils in the bone marrow after AMI stimulated granulopoiesis and contributed to sustained influx of neutrophils into post-myocardial infarction heart [78].
Neutrophil pyroptosis has also been linked to occlusion of blood vessels and acute lung injury during sickle cell disease. Caspase-4 activation of GSDMD and GSDMD-dependent NET release in liver followed by intravascular NET travel to the lungs, resulting in neutrophil-platelet aggregation [126]. However, it has also been reported that NET formation by viable neutrophils after inflammasome activation is independent of gasdermin D and pyroptotic cell death [127].
We anticipate that the use of neutrophil-specific conditional knockouts of GSDMD and upstream mediators will uncover additional mechanisms of pyroptosis regulation in neutrophils that are distinct from macrophages [97,128], especially in the context of CVD.
4.3. Multilevel regulation of pyroptosis in cardiomyocytes.
There are many reports claiming induction of pyroptosis in cardiomyocytes based on limited characterization the morphology and criteria of pyroptosis and lack of exclusion of other forms of cell death. Furthermore, limited availability and viability of primary cardiomyocytes in culture resulted on reliance on the cardiomyoblast cell line H9c2 in those studies. In our analyses of the literature on cardiomyocyte pyroptosis, we relied on the adopted definition of pyroptosis and guidance for evaluation of cardiomyocyte cell death [15,129].
Compared to macrophages, cardiomyocytes express relatively low levels of pro-caspase-1, IL-1β, and IL-18. Thus, activation of inflammasomes restricted to cardiomyocytes has been considered less inflammatory (reviewed in [16]). A study demonstrating effects of TMAO on primary rat macrophages, fibroblasts and cardiomyocytes, pyroptosis in cardiomyocytes and fibroblasts was detectable only in the presence of macrophages in the Transwell culture system, with the lowest extent of caspase-1 and GSDMD cleavage in cardiomyocytes in comparison with fibroblasts and macrophages [106]. In addition, transcriptional repression of NLRP3 inflammasome by GDF11-TGFβ-SMAD2/3-HOXA3 pathway renders cardiomyocytes relatively resistant to pyroptosis [79]. Cardiomyocytes may also reduce the risk of going into pyroptosis or other forms of programmed cell death by eliminating compromised mitochondria through exophers [23]. Those autophagy-derived specialized vesicles containing dysfunctional mitochondria are phagocytosed and silently eliminated by a network of cardiac macrophages under homeostatic conditions [23]. Yet, activation of NLRP3 inflammasome, GSDMD cleavage and other features of pyroptosis have been described in cardiomyocytes after ischemia/reperfusion in vivo and hypoxia/reoxygenation in vitro [117] as well as in human cardiomyocytes during dilated cardiomyopathy [118]..
Careful analyses of cell death morphology, along with multiparameter imaging to co-localize active caspase-1 and cleaved GSDMD to cardiomyocytes will help in verifying cardiomyocyte pyroptosis in AMI and other heart diseases. The use of induced pluripotent stem cell-derived cardiomyocytes or application of cardiomyocyte-specific conditional Gsdmd-knockouts by crossing the mice with cardiac-specific α-myosin heavy chain Cre drivers with Gsdmdfl/fl mice will help with mechanistic studies [130].
4.4. Pyroptosis in platelets.
Platelets, small anucleate cell fragments released by megakaryocytes, play central roles in safeguarding vascular integrity [131]. Traditional view of platelets as initiators of clotting and hemostasis by aggregation has been greatly expanded in recent years. It has become clear that platelets are intricately involved in regulation of innate and adaptive immune responses and the pathogenesis of acute and chronic inflammatory disorders [132]. Activation of caspase-1 by NLRP3 inflammasome in platelets resulting in increased release of IL-1β and IL-18 was described in humans infected with dengue virus [133] and in rats after experimental sepsis induced by cecal ligation and puncture (CLP) [134]. In both studies, excessive activation of platelet inflammasome resulted in increased vascular permeability and inflammation, although it is not clear whether the process of pyroptosis was indeed engaged. Pyroptosis in platelets was also described in patients with primary immune thrombocytopenia (ITP), a common acquired autoimmune disease characterized by low platelet counts and increased risk of bleeding. Reduced platelet antioxidant activity was associated with expression and activation of NLRP3 inflammasome, active caspase-1 and increased platelet IL-1β release [135]. Unfortunately, the study identified caspase−1+/annexin V− platelets as pyroptotic cells but did not assess GSDMD cleavage and formation of membrane pores, essential features of pyroptosis.
Notably, our group recently confirmed expression of NLRP3, ASC, IL-1β and GSDMD in septic platelets using a series of complementary techniques and demonstrated the deleterious effects of platelet GSDMD-dependent pyroptosis in severe sepsis [54]. In human patients with sepsis and in a mouse model of CLP-induced sepsis, platelets displayed characteristic features of pyroptosis (cell swelling, plasma membrane rupture, and release of cytosolic content) associated with cleavage of GSDMD. Platelet pyroptosis was apparently triggered by recognition of S100A8/A9 alarmin protein by TLR4 and resulted in release of ox-mtDNA from platelets, subsequently contributing to NET formation and amplifying the vicious feedforward cycle as additional S100A8/A9 was released from NETs [54]. Importantly, mice with platelet-specific condition deletion of GSDMD (PF4creGsdmdfl/fl) had significantly reduced numbers of swollen platelets, attenuated NET formation, reduced plasma levels of IL-1β, and improved survival after CLP [54]. Platelet pyroptosis was also effectively inhibited by paquinimod, which prevents binding of S100A9 to TLR4, as evidenced by reduced activation of NLRP3 and caspase-1 and diminished cleavage of GSDMD [54]. Furthermore, a mitochondrial-specific ROS scavenger, MitoTempo, inhibited sepsis-induced caspase-1 activity, release of ox-mtDNA, and NET formation, suggesting that mitochondrial ROS activate caspase-1 and induce platelet pyroptosis.
Pyroptosis occurring in cells other than platelets, i.e., macrophages, may result in release of tissue factor, which hyperactivates platelets and causes disseminated intravascular coagulation and death [92]. Conversely, platelets may regulate pyroptosis in other cells as yet to be fully characterized combination of constitutive heat-sensitive soluble platelet factors enhances macrophage and neutrophil NLPR3 transcription, caspase-1 activity and IL-1β secretion [136]. Furthermore, platelet-derived microparticles loaded with NLRP3 enhance pyroptosis of endothelial cells in antiphospholipid syndrome [137].
As we are still learning the mechanisms and sequela of canonical pyroptosis in platelets (Figure 2), further investigation is called for to determine whether the non-canonical pathway of pyroptosis (i.e., requiring caspase−4/5 in humans and caspase−11 in mice) has a place in platelets. It remains to be seen whether platelet pyroptosis occurs and is important in CVD with ROS burden but without direct infectious etiology.
Figure 2.
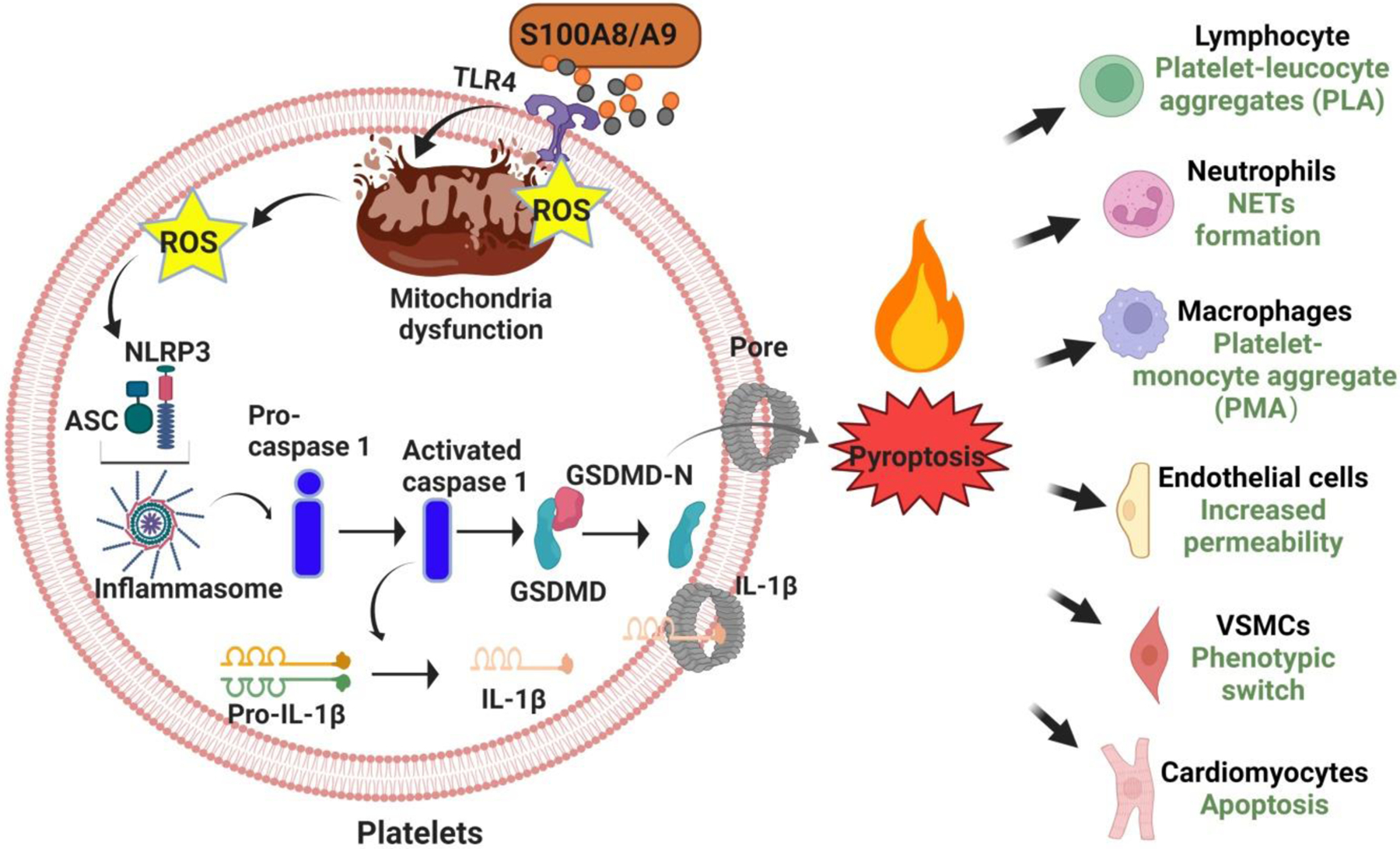
Mechanisms and possible roles of platelet pyroptosis in CVD.
4.5. Stromal cells (fibroblasts and endothelial) cells are relatively refractory to pyroptosis triggers.
Cardiac fibroblasts play essential roles for maintenance of heart homeostasis and regulation of adaptive or pathogenic remodeling. Although pyroptosis was demonstrated in cardiac fibroblast after myocardial infarction [57,76] and atrial fibrillation [106], they seem to be relatively refractory to triggers of pyroptosis and require inflammasome priming by inflammatory mediators or other signals, likely coming from macrophages.
Endothelial cells undergo pyroptosis in atherosclerosis, apparently in response to the TMAO exposure [51] and after experimental endotoxemia [93]. However, endothelial cells are resistant to intracellular LPS as the crosstalk of STING and NLRP3 pathways in response to mtDNA suppresses EC proliferation with little evidence of pyroptosis [138].
4.6. Pyroptosis of mural cells (VSMC and pericytes) may affect vascular integrity and barrier function.
Collectively called mural cells, VSMC and pericytes are involved in regulation of vascular development, remodeling, and integrity [139,140]. Caspase-1-dependnet pyroptosis of pericytes after thrombosis has been shown to contribute to increased vascular permeability and disruption of blood-brain barrier [141,142]. Similar pathways of pyroptosis have been described in VSMC during atherosclerosis, pulmonary artery hypertension, and abdominal aortic aneurysm [71,110,112,113]. However, many questions remain unresolved given VSMC heterogeneity and plasticity. For example, it is not known whether pyroptosis sensitivity, pathways and consequences differ between contractile vs. synthetic VSMC after vascular injury and remodeling. It is also not clear whether VSMC susceptibility to pyroptosis is increased after their trans-differentiation and acquisition of macrophage phenotype in atherosclerotic plaques. Single cell functional and multiomics analyses of vascular cells may help address those questions in the near future.
4.7. Lymphocytes and innate lymphoid cells.
Low levels of caspase-1 and IL-1β expression in lymphocytes suggest limited role of pyroptosis in this cell type in relation to CVD. However, the finding that group 2 innate lymphoid cells protect endothelial cells from pyroptosis via IL-9 in sepsis [58] may generate an interest in using IL-9 for minimizing the impact of pyroptosis in other conditions.
5. Tools for pyroptosis research and intervention in cardiovascular diseases.
Development of small molecule inhibitors, fluorescent reporters, and genetic tools, along with communal guidelines, particularly benefited apoptosis research and spurred interest in other forms of programmed cell death. The explosion of publications on cell death prompted discussion and adoption of recommendations for nomenclature of the forms of regulated and guidelines for evaluating myocardial cell death [15,129]. At the same time, identification of gasdermins as key mediators of pyroptosis led to expansion and refinement of available tools and approaches to characterizing pyroptotic pathways and development of genetic and small molecule inhibitors of pyroptosis [8,9,59]. In addition to the global Gsdmd- and Gsdme-knockout mice [8,9,67], cell type-specific conditional or inducible deletion of GSDMD is now feasible using Gsdmdfl/fl mice [54,108,128].
The use of genetically modified mice with targeted deletions and mutations in genes encoding inflammasome components, specific caspases and gasdermins, as well as factors driving clonal hematopoiesis, enabled mechanistic studies on the roles of pyroptosis and upstream pathways that are extremely difficult or impossible to carry out in larger animals or clinical settings. Yet, it is important to be vigilant about the limitations of mouse and other animal models, when translating the findings to design of therapeutic interventions of CVD in humans. The differences in lipoprotein profiles and characteristics of the plaque progression between humans and the most frequently used mouse models of atherosclerosis (ApoE−/− and Ldlr−/− mice on high fat diet) are well characterized and acknowledged as limitations. The advent of advanced genetic engineering tools, such as CRISPR-Cas9 gene editing, spurred interest and effort to develop and adopt alternative models such as genetically-modified rodents (hamsters and guinea pigs) [143], rabbits [144] and pigs [145]. Similar concerns due were raised for other models of CVD due to physiological, morphological, and genetic differences between mice and humans, prompting studies of inflammasome and pyroptosis involvement in atrial fibrillation in dogs [102] and myocardial infarction in pigs [146]. Those complex animal models are important for advancement to clinical trials, but they are not likely to completely replace mouse models for basic research and early preclinical studies due to time, cost, and ethical considerations, when advantages and limitations of the mouse models are carefully assessed [147,148].
Given the complexity of the pyroptosis pathways and extensive crosstalk between different forms of the programmed cell death, factors that target pyroptosis triggers, prevent activation of inflammasomes and inhibit inflammatory caspases may provide broader protection from pyroptosis and necroptosisand related sequelae. Paquinimod, an inhibitor of S100A8/A9 binding to TLR4, breaks the vicious cycle of platelet pyroptosis in severe sepsis [54] and shows protective effects in a model of myocardial infarction by reducing IL-1β-dependent granulopoiesis [149]. Necrosulfonamide, an inhibitor of necroptosis in human cells, which binds to human but not mouse MLKL, was shown to bind to Cys191 of human GSDMD and to inhibit pyroptosis in human and mouse macrophages as well as improve survival in experimental sepsis [60]. The cell permeable peptide Ac-FLTD-CMK that inhibits inflammatory caspases−1, −4, −5, and −11 is widely used to prevent GSDMD cleavage. In addition, a caspase-1-specific prodrug inhibitor belnacasan, also known as VX-765 (Vertex Pharmaceuticals) is widely used in preclinical research showing protective effects against myocardial infarction in combination with platelet inhibitors in rats [71,150]. It progressed to Phase 2 clinical trials for various indications but showed poor efficacy. Another elegant approach is worth mentioning as it uses the pyroptosis pores to deliver nanobodies that can dismantle ASC specs of inflammasomes and reduce inflammation [151].
6. Reduction of recurrent cardiovascular events in patients using IL-1β-specific monoclonal antibody canakinumab indicates that limiting inflammation is an important goal in management of chronic CVD [64]. It remains to be determined whether inhibition of pyroptosis reducing cell death and release of IL-1β and IL-18 could be more effective than inhibition of IL-1β alone and achieve better results in chronic CVD and acute cardiovascular events. Several small molecule inhibitors of NLRP3 inflammasome activation reached clinical trials in hope of blocking inflammation upstream of IL-1β release in CVD and COVID-19 [152]. One of the candidates, dapansutrile, demonstrated safety and improvement of left ventricular ejection fraction in patients with heart failure in a Phase 1b study [153]. Targeting of the Cys191 residue of human GSDMD by necrosulfonamide [60] or its succination by dimethyl fumarate [61] could be effective approaches to inhibit GSDMD-dependent pore formation and reduce severe inflammation. Disulfiram, an FDA approved drug for treatment of alcohol addiction, was identified in a high-throughput screening as a potent inhibitor of GSDMD pore formation through prevention of S-palmitoylation of Cys191 of GSDMD [12,59] and showed efficacy in several preclinical models of sepsis[95], abdominal aortic aneurism [112] and pulmonary hypertension [110] Therefore, repurposing disulfiram for inhibition of pyroptosis and reduction of inflammation seems an easy way to enter the clinical trials with CVD in sight. Several important questions would need to be addressed before GSDMD-targeted therapeutics can be approved: What is the fate and impact of the “zombie” Gsdmd−/− macrophages loaded with IL-1β that were reported to accumulate in a model of atherosclerosis exacerbated by clonal hematopoiesis [70]? Does inhibition of GSDMD raise the risks of acute cardiovascular events similar to the increased risks of thromboembolic stroke in atrial fibrillation patients with GSDMD gene [105]? Does inhibition of GSDMD pores impair host immunity to any infectious agents?
Conclusions
Pyroptosis contributes to the pathogenesis of cardiovascular diseases by amplifying inflammatory responses to bacterial products, hypoxia and ischemia/reperfusion, metabolic stress, and mitochondrial DNA. Inflammasome-dependent activation of inflammatory caspases that cleave gasdermin D and other gasdermins resulting in formation of selective pores at the plasma membrane has been described in tissue resident and circulating immune cells, cardiomyocytes, fibroblasts, endothelial cells, pericytes, and vascular smooth muscle cells. Cell-specific gene expression signatures and complex networks of local and distal cell interactions determine sensitivity to pyroptosis and its consequences. In some circumstances, concomitant activation of other forms of PCD besides pyroptosis is involved in pathophysiology of CVD. Detailed characterization of pyroptosis cascades, gasdermin-specific small molecule inhibitors of pore formation and upstream inhibitors opens a new perspective for designing novel therapeutic strategies to dampen severe inflammation during chronic cardiovascular diseases and reduce the risks and mortality due to acute cardiovascular events.
Supplementary Material
Highlights.
Pyroptosis, a form of regulated/programmed cell death, contributes to the pathogenesis of chronic and acute cardiovascular diseases by intensifying inflammation.
Multiple cell types within the cardiovascular system respond variably and coordinately, to triggers of pyroptosis.
Cell-specific mechanisms exist to regulate pyroptosis during homeostasis or cardiovascular disease.
Pyroptosis is an attractive target for pharmacological intervention to reduce excessive inflammation during cardiovascular disease.
Acknowledgements
This work was supported by the National Institutes of Health National Heart, Lung, and Blood Institute R01 grants HL122815, HL115247, and HL150515 (JH), the National Key R&D Programmes of China Grants 2021YFC2701205 (WHT) and 2017YFC1700402 (YX), National Natural Science Foundation of China Grant 82270350 (YX), 82100141 (MS), 82101655 (CC), 82022003, 81970437 (WHT), Guangzhou Science and Technology Project 202201011007 (CC), Fundamental Research Funds for the Central Universities Grant 22120220162 (YX), the Frontier Science Research Center for Stem Cells, Ministry of Education (YX), and China Postdoctoral Science Foundation Grant 2021M700934 (CC).
Abbreviations:
- AMI
acute myocardial infarction
- CVD
cardiovascular diseases
- CLP
cecal ligation and puncture
- DM
diabetes mellitus
- GSDMD
Gasdermin D
- GSDME
Gasdermin E
- ITP
immune thrombocytopenia
- LDH
lactate dehydrogenase
- NET
neutrophil extracellular trap
- Ox-LDL
oxidized low density lipoprotein
- PCD
programmed cell death
- ROS
reactive oxygen species
- SDHB
succinate dehydrogenase complex subunit B
- TMAO
Trimethylamine N-oxide
- VSMC
vascular smooth muscle cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest
T.O. Yarovinsky reported consulting fees from CaroGen Corporation outside the submitted work. Wai Ho Tang, Meiling Su, and Chaofei Chen are named inventors on a technical patent related to the therapeutic evaluation of the inhibitors of platelets pyroptosis in sepsis (202210225079.2).
References
- [1].Mensah GA, Roth GA, Fuster V, The Global Burden of Cardiovascular Diseases and Risk Factors: 2020 and Beyond, J. Am. Coll. Cardiol 74 (2019) 2529–2532. [DOI] [PubMed] [Google Scholar]
- [2].Cookson BT, Brennan MA, Pro-inflammatory programmed cell death, Trends Microbiol. 9 (2001) 113–114. [DOI] [PubMed] [Google Scholar]
- [3].Hersh D, Monack DM, Smith MR, Ghori N, Falkow S, Zychlinsky A, The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1, Proc. Natl. Acad. Sci. U. S. A 96 (1999) 2396–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Brennan MA, Cookson BT, Salmonella induces macrophage death by caspase-1-dependent necrosis, Mol. Microbiol 38 (2000) 31–40. [DOI] [PubMed] [Google Scholar]
- [5].Hara H, Fink K, Endres M, Friedlander RM, Gagliardini V, Yuan J, Moskowitz MA, Attenuation of transient focal cerebral ischemic injury in transgenic mice expressing a mutant ICE inhibitory protein, J. Cereb. Blood Flow Metab 17 (1997) 370–375. [DOI] [PubMed] [Google Scholar]
- [6].Kolodgie FD, Narula J, Burke AP, Haider N, Farb A, Hui-Liang Y, Smialek J, Virmani R, Localization of apoptotic macrophages at the site of plaque rupture in sudden coronary death, Am. J. Pathol 157 (2000) 1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Man SM, Karki R, Kanneganti TD, Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases, Immunol. Rev 277 (2017) 61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F, Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death, Nature. 526 (2015) 660–665. [DOI] [PubMed] [Google Scholar]
- [9].Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, Dixit VM, Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling, Nature. 526 (2015) 666–671. [DOI] [PubMed] [Google Scholar]
- [10].Xia S, Zhang Z, Magupalli VG, Pablo JL, Dong Y, Vora SM, Wang L, Fu TM, Jacobson MP, Greka A, Lieberman J, Ruan J, Wu H, Gasdermin D pore structure reveals preferential release of mature interleukin-1, Nature. 593 (2021) 607–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Balasubramanian A, Ghimire L, Hsu AY, Kambara H, Liu X, Hasegawa T, Xu R, Tahir M, Yu H, Lieberman J, Luo HR, Palmitoylation of gasdermin D directs its membrane translocation and pore formation in pyroptosis, BioRxiv. (2023) 2023.02.21.529402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Du G, Healy LB, David L, Walker C, Fontana P, Dong Y, Devant P, Puthenveetil R, Ficarro SB, Banerjee A, Kagan JC, Lieberman J, Wu H, ROS-dependent palmitoylation is an obligate licensing modification for GSDMD pore formation, BioRxiv. (2023) 2023.03.07.531538. [Google Scholar]
- [13].Broz P, Pelegrín P, Shao F, The gasdermins, a protein family executing cell death and inflammation, Nat. Rev. Immunol 20 (2020) 143–157. [DOI] [PubMed] [Google Scholar]
- [14].Wu Y, Zhang J, Yu S, Li Y, Zhu J, Zhang K, Zhang R, Cell pyroptosis in health and inflammatory diseases, Cell Death Discov. 8 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, Annicchiarico-Petruzzelli M, Antonov AV, Arama E, Baehrecke EH, Barlev NA, Bazan NG, Bernassola F, Bertrand MJM, Bianchi K, Blagosklonny MV, Blomgren K, Borner C, Boya P, Brenner C, Campanella M, Candi E, Carmona-Gutierrez D, Cecconi F, Chan FKM, Chandel NS, Cheng EH, Chipuk JE, Cidlowski JA, Ciechanover A, Cohen GM, Conrad M, Cubillos-Ruiz JR, Czabotar PE, D’Angiolella V, Dawson TM, Dawson VL, De Laurenzi V, De Maria R, Debatin KM, Deberardinis RJ, Deshmukh M, Di Daniele N, Di Virgilio F, Dixit VM, Dixon SJ, Duckett CS, Dynlacht BD, El-Deiry WS, Elrod JW, Fimia GM, Fulda S, García-Sáez AJ, Garg AD, Garrido C, Gavathiotis E, Golstein P, Gottlieb E, Green DR, Greene LA, Gronemeyer H, Gross A, Hajnoczky G, Hardwick JM, Harris IS, Hengartner MO, Hetz C, Ichijo H, Jäättelä M, Joseph B, Jost PJ, Juin PP, Kaiser WJ, Karin M, Kaufmann T, Kepp O, Kimchi A, Kitsis RN, Klionsky DJ, Knight RA, Kumar S, Lee SW, Lemasters JJ, Levine B, Linkermann A, Lipton SA, Lockshin RA, López-Otín C, Lowe SW, Luedde T, Lugli E, MacFarlane M, Madeo F, Malewicz M, Malorni W, Manic G, Marine JC, Martin SJ, Martinou JC, Medema JP, Mehlen P, Meier P, Melino S, Miao EA, Molkentin JD, Moll UM, Muñoz-Pinedo C, Nagata S, Nuñez G, Oberst A, Oren M, Overholtzer M, Pagano M, Panaretakis T, Pasparakis M, Penninger JM, Pereira DM, Pervaiz S, Peter ME, Piacentini M, Pinton P, Prehn JHM, Puthalakath H, Rabinovich GA, Rehm M, Rizzuto R, Rodrigues CMP, Rubinsztein DC, Rudel T, Ryan KM, Sayan E, Scorrano L, Shao F, Shi Y, Silke J, Simon HU, Sistigu A, Stockwell BR, Strasser A, Szabadkai G, Tait SWG, Tang D, Tavernarakis N, Thorburn A, Tsujimoto Y, Turk B, Vanden Berghe T, Vandenabeele P, Vander Heiden MG, Villunger A, Virgin HW, Vousden KH, Vucic D, Wagner EF, Walczak H, Wallach D, Wang Y, Wells JA, Wood W, Yuan J, Zakeri Z, Zhivotovsky B, Zitvogel L, Melino G, Kroemer G, Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018, Cell Death Differ. 25 (2018) 486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN, Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease, Physiol. Rev 99 (2019) 1765–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].McKee CM, Coll RC, NLRP3 inflammasome priming: A riddle wrapped in a mystery inside an enigma, J. Leukoc. Biol 108 (2020) 937–952. [DOI] [PubMed] [Google Scholar]
- [18].Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, Hornung V, Latz E, Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression, J. Immunol 183 (2009) 787–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhu Q, Kanneganti T-D, Cutting Edge: Distinct Regulatory Mechanisms Control Proinflammatory Cytokines IL-18 and IL-1β, J. Immunol 198 (2017) 4210–4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zhou B, Tian R, Mitochondrial dysfunction in pathophysiology of heart failure, J. Clin. Invest 128 (2018) 3716–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, jia Lin X, Wong J, Ding S, Seki E, Schnabl B, Hevener AL, Greenberg HB, Kisseleva T, Karin M, New mitochondrial DNA synthesis enables NLRP3 inflammasome activation, Nature. 560 (2018) 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Xian H, Watari K, Sanchez-Lopez E, Offenberger J, Onyuru J, Sampath H, Ying W, Hoffman HM, Shadel GS, Karin M, Oxidized DNA fragments exit mitochondria via mPTP- and VDACdependent channels to activate NLRP3 inflammasome and interferon signaling, Immunity. 55 (2022) 1370–1385.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Nicolás-Ávila JA, Lechuga-Vieco AV, Esteban-Martínez L, Sánchez-Díaz M, Díaz-García E, Santiago DJ, Rubio-Ponce A, Li JLY, Balachander A, Quintana JA, Martínez-de-Mena R, Castejón-Vega B, Pun-García A, Través PG, Bonzón-Kulichenko E, García-Marqués F, Cussó L, A-González N, González-Guerra A, Roche-Molina M, Martin-Salamanca S, Crainiciuc G, Guzmán G, Larrazabal J, Herrero-Galán E, Alegre-Cebollada J, Lemke G, Rothlin CV, JimenezBorreguero LJ, Reyes G, Castrillo A, Desco M, Muñoz-Cánoves P, Ibáñez B, Torres M, Ng LG, Priori SG, Bueno H, Vázquez J, Cordero MD, Bernal JA, Enríquez JA, Hidalgo A, A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart, Cell. 183 (2020) 94–109.e23. [DOI] [PubMed] [Google Scholar]
- [24].Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, Malech HL, Ledbetter JA, Elkon KB, Kaplan MJ, Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease, Nat. Med 22 (2016) 146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Riley JS, Tait SW, Mitochondrial DNA in inflammation and immunity, EMBO Rep. 21 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].De Gaetano A, Solodka K, Zanini G, Selleri V, Mattioli AV, Nasi M, Pinti M, Molecular Mechanisms of mtDNA-Mediated Inflammation, Cells. 10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, Lien E, Nilsen NJ, Espevik T, Golenbock DT, TLR9 signals after translocating from the ER to CpG DNA in the lysosome, Nat. Immunol 5 (2004) 190–198. [DOI] [PubMed] [Google Scholar]
- [28].Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ, Circulating mitochondrial DAMPs cause inflammatory responses to injury, Nature. 464 (2010) 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Liu J, Jia Z, Gong W, Circulating Mitochondrial DNA Stimulates Innate Immune Signaling Pathways to Mediate Acute Kidney Injury, Front. Immunol 12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zychlinsky A, Prevost MC, Sansonetti PJ, Shigella flexneri induces apoptosis in infected macrophages, Nature. 358 (1992) 167–169. [DOI] [PubMed] [Google Scholar]
- [31].Ketelut-Carneiro N, Fitzgerald KA, Apoptosis, Pyroptosis, and Necroptosis-Oh My! The Many Ways a Cell Can Die, J. Mol. Biol 434 (2022). [DOI] [PubMed] [Google Scholar]
- [32].Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, Brooks A, Xia S, Wu H, Kelliher MA, Berger SB, Gough PJ, Bertin J, Proulx MM, Goguen JD, Kayagaki N, Fitzgerald KA, Lien E, Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death, Science. 362 (2018) 1064–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, Rongvaux A, Bunnell SC, Shao F, Green DR, Poltorak A, Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection, Proc. Natl. Acad. Sci. U. S. A 115 (2018) E10888–E10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K, Shao F, Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin, Nature. 547 (2017) 99–103. [DOI] [PubMed] [Google Scholar]
- [35].Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES, Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death, Nat. Commun 8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Malireddi RKS, Kesavardhana S, Kanneganti TD, ZBP1 and TAK1: Master Regulators of NLRP3 Inflammasome/Pyroptosis, Apoptosis, and Necroptosis (PAN-optosis), Front. Cell. Infect. Microbiol 9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Frank D, Vince JE, Pyroptosis versus necroptosis: similarities, differences, and crosstalk, Cell Death Differ. 26 (2019) 99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wang Y, Kanneganti TD, From pyroptosis, apoptosis and necroptosis to PANoptosis: A mechanistic compendium of programmed cell death pathways, Comput. Struct. Biotechnol. J 19 (2021) 4641–4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lee SJ, Karki R, Wang Y, Nguyen LN, Kalathur RC, Kanneganti TD, AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence, Nature. 597 (2021) 415–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gullett JM, Tweedell RE, Kanneganti TD, It’s All in the PAN: Crosstalk, Plasticity, Redundancies, Switches, and Interconnectedness Encompassed by PANoptosis Underlying the Totality of Cell Death-Associated Biological Effects, Cells. 11 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Christgen S, Zheng M, Kesavardhana S, Karki R, Malireddi RKS, Banoth B, Place DE, Briard B, Sharma BR, Tuladhar S, Samir P, Burton A, Kanneganti TD, Identification of the PANoptosome: A Molecular Platform Triggering Pyroptosis, Apoptosis, and Necroptosis (PANoptosis), Front. Cell. Infect. Microbiol 10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Samir P, Malireddi RKS, Kanneganti TD, The PANoptosome: A Deadly Protein Complex Driving Pyroptosis, Apoptosis, and Necroptosis (PANoptosis), Front. Cell. Infect. Microbiol 10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zheng M, Karki R, Vogel P, Kanneganti TD, Caspase-6 Is a Key Regulator of Innate Immunity, Inflammasome Activation, and Host Defense, Cell. 181 (2020) 674–687.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Karki R, Sharma BR, Lee E, Banoth B, Malireddi RKS, Samir P, Tuladhar S, Mummareddy H, Burton AR, Vogel P, Kanneganti TD, Interferon regulatory factor 1 regulates PANoptosis to prevent colorectal cancer, JCI Insight. 5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, Zheng M, Sundaram B, Banoth B, Malireddi RKS, Schreiner P, Neale G, Vogel P, Webby R, Jonsson CB, Kanneganti TD, Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes, Cell. 184 (2021) 149–168.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Camilli S, Lockey R, Kolliputi N, Nuclear Export Inhibitors Selinexor (KPT-330) and Eltanexor (KPT-8602) Provide a Novel Therapy to Reduce Tumor Growth by Induction of PANoptosis, Cell Biochem. Biophys (2023). [DOI] [PubMed] [Google Scholar]
- [47].Xu X, Lan X, Fu S, Zhang Q, Gui F, Jin Q, Xie L, Xiong Y, Dickkopf-1 exerts protective effects by inhibiting PANoptosis and retinal neovascularization in diabetic retinopathy, Biochem. Biophys. Res. Commun 617 (2022) 69–76. [DOI] [PubMed] [Google Scholar]
- [48].Bi Y, Xu H, Wang X, Zhu H, Ge J, Ren J, Zhang Y, FUNDC1 protects against doxorubicininduced cardiomyocyte PANoptosis through stabilizing mtDNA via interaction with TUFM, Cell Death Dis. 13 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Uysal E, Dokur M, Kucukdurmaz F, Altınay S, Polat S, Batcıoglu K, Sezgın E, Sapmaz Erçakallı T, Yaylalı A, Yılmaztekin Y, Cetın Z, Saygılı İ, Barut O, Kazımoglu H, Maralcan G, Koc S, Guney T, Eser N, Sökücü M, Dokur SN, Targeting the PANoptosome with 3,4-Methylenedioxyβ-Nitrostyrene, Reduces PANoptosis and Protects the Kidney against Renal İschemia-Reperfusion Injury, J. Invest. Surg 35 (2022) 1824–1835. [DOI] [PubMed] [Google Scholar]
- [50].Dai S, Ye B, Zhong L, Chen Y, Hong G, Zhao G, Su L, Lu Z, GSDMD Mediates LPS-Induced Septic Myocardial Dysfunction by Regulating ROS-dependent NLRP3 Inflammasome Activation, Front. Cell Dev. Biol 9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wu P, Chen JN, Chen JJ, Tao J, Wu SY, Xu GS, Wang Z, Wei DH, Yin WD, Trimethylamine N-oxide promotes apoE−/− mice atherosclerosis by inducing vascular endothelial cell pyroptosis via the SDHB/ROS pathway, J. Cell. Physiol 235 (2020) 6582–6591. [DOI] [PubMed] [Google Scholar]
- [52].Kuriakose T, Man SM, Subbarao Malireddi RK, Karki R, Kesavardhana S, Place DE, Neale G, Vogel P, Kanneganti TD, ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways, Sci. Immunol 1 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chauhan D, Demon D, Vande Walle L, Paerewijck O, Zecchin A, Bosseler L, Santoni K, Planès R, Ribo S, Fossoul A, Gonçalves A, Van Gorp H, Van Opdenbosch N, Van Hauwermeiren F, Meunier E, Wullaert A, Lamkanfi M, GSDMD drives canonical inflammasome-induced neutrophil pyroptosis and is dispensable for NETosis, EMBO Rep. 23 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Su M, Chen C, Li S, Li M, Zeng Z, Zhang Y, Xia L, Li X, Zheng D, Lin Q, Fan X, Wen Y, Liu Y, Chen F, Luo W, Bu Y, Qin J, Guo M, Qiu M, Sun L, Liu R, Wang P, Hwa J, Tang WH, Gasdermin D-dependent platelet pyroptosis exacerbates NET formation and inflammation in severe sepsis, Nat. Cardiovasc. Res 1 (2022) 732–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nũez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E, NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals, Nature. 464 (2010) 1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Jiang M, Sun X, Liu S, Tang Y, Shi Y, Bai Y, Wang Y, Yang Q, Yang Q, Jiang W, Yuan H, Jiang Q, Cai J, Caspase-11-Gasdermin D-Mediated Pyroptosis Is Involved in the Pathogenesis of Atherosclerosis, Front. Pharmacol 12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Han X, Zhao ZA, Yan S, Lei W, Wu H, Lu XA, Chen Y, Li J, Wang Y, Yu M, Wang Y, Zheng Y, Wang H, Shen Z, Hu S, CXADR-like membrane protein protects against heart injury by preventing excessive pyroptosis after myocardial infarction, J. Cell. Mol. Med 24 (2020) 13775–13788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Lai D, Tang J, Chen L, Fan EK, Scott MJ, Li Y, Billiar TR, Wilson MA, Fang X, Shu Q, Fan J, Group 2 innate lymphoid cells protect lung endothelial cells from pyroptosis in sepsis, Cell Death Dis. 9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, Ruan J, Luo X, Lou X, Bai Y, Wang J, Hollingsworth LR, Magupalli VG, Zhao L, Luo HR, Kim J, Lieberman J, Wu H, FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation, Nat. Immunol 21 (2020) 736–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, Kondolf HC, Benson BL, Chirieleison SM, Huang AY, Dubyak GR, Xiao TS, Li X, Abbott DW, Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis, Sci. Immunol 3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Humphries F, Shmuel-Galia L, Ketelut-Carneiro N, Li S, Wang B, Nemmara VV, Wilson R, Jiang Z, Khalighinejad F, Muneeruddin K, Shaffer SA, Dutta R, Ionete C, Pesiridis S, Yang S, Thompson PR, Fitzgerald KA, Succination inactivates gasdermin D and blocks pyroptosis, Science. 369 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Wolf D, Ley K, Immunity and Inflammation in Atherosclerosis, Circ. Res 124 (2019) 315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Libby P, The changing landscape of atherosclerosis, Nature. 592 (2021) 524–533. [DOI] [PubMed] [Google Scholar]
- [64].Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ, Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease, N. Engl. J. Med 377 (2017) 1119–1131. [DOI] [PubMed] [Google Scholar]
- [65].Opoku E, Traughber CA, Zhang D, Iacano AJ, Khan M, Han J, Smith JD, Gulshan K, Gasdermin D Mediates Inflammation-Induced Defects in Reverse Cholesterol Transport and Promotes Atherosclerosis, Front. Cell Dev. Biol 9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Puylaert P, Van Praet M, Vaes F, Neutel CHG, Roth L, Guns PJ, De Meyer GRY, Martinet W, Gasdermin D Deficiency Limits the Transition of Atherosclerotic Plaques to an Inflammatory Phenotype in ApoE Knock-Out Mice, Biomedicines. 10 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Wei Y, Lan B, Zheng T, Yang L, Zhang X, Cheng L, Tuerhongjiang G, Yuan Z, Wu Y, GSDMEmediated pyroptosis promotes the progression and associated inflammation of atherosclerosis, Nat. Commun 14 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, Baber U, Mehran R, Fuster V, Danesh J, Frossard P, Saleheen D, Melander O, Sukhova GK, Neuberg D, Libby P, Kathiresan S, Ebert BL, Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease, N. Engl. J. Med 377 (2017) 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Wang W, Liu W, Fidler T, Wang Y, Tang Y, Woods B, Welch C, Cai B, Silvestre-Roig C, Ai D, Yang YG, Hidalgo A, Soehnlein O, Tabas I, Levine RL, Tall AR, Wang N, Macrophage Inflammation, Erythrophagocytosis, and Accelerated Atherosclerosis in Jak2 V617F Mice, Circ. Res 123 (2018) E35–E47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Fidler TP, Xue C, Yalcinkaya M, Hardaway B, Abramowicz S, Xiao T, Liu W, Thomas DG, Hajebrahimi MA, Pircher J, Silvestre-Roig C, Kotini AG, Luchsinger LL, Wei Y, Westerterp M, Snoeck HW, Papapetrou EP, Schulz C, Massberg S, Soehnlein O, Ebert B, Levine RL, Reilly MP, Libby P, Wang N, Tall AR, The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis, Nature. 592 (2021) 296–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Li Y, Niu X, Xu H, Li Q, Meng L, He M, Zhang J, Zhang Z, Zhang Z, VX-765 attenuates atherosclerosis in ApoE deficient mice by modulating VSMCs pyroptosis, Exp. Cell Res 389 (2020). [DOI] [PubMed] [Google Scholar]
- [72].Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AAC, Greene ES, Straub AC, Isakson B, Randolph GJ, Owens GK, KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis, Nat. Med 21 (2015) 628–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Pan J, Han L, Guo J, Wang X, Liu D, Tian J, Zhang M, An F, AIM2 accelerates the atherosclerotic plaque progressions in ApoE−/− mice, Biochem. Biophys. Res. Commun 498 (2018) 487–494. [DOI] [PubMed] [Google Scholar]
- [74].Pothineni NVK, Subramany S, Kuriakose K, Shirazi LF, Romeo F, Shah PK, Mehta JL, Infections, atherosclerosis, and coronary heart disease, Eur. Heart J 38 (2017) 3195–3201. [DOI] [PubMed] [Google Scholar]
- [75].Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J, Hongo M, Noda T, Nakayama J, Sagara J, Taniguchi S, Ikeda U, Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury, Circulation. 123 (2011) 594–604. [DOI] [PubMed] [Google Scholar]
- [76].Sandanger Ø, Ranheim T, Vinge LE, Bliksøen M, Alfsnes K, Finsen AV, Dahl CP, Askevold ET, Florholmen G, Christensen G, Fitzgerald KA, Lien E, Valen G, Espevik T, Aukrust P, Yndestad A, The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury, Cardiovasc. Res 99 (2013) 164–174. [DOI] [PubMed] [Google Scholar]
- [77].Jiang K, Tu Z, Chen K, Xu Y, Chen F, Xu S, Shi T, Qian J, Shen L, Hwa J, Wang D, Xiang Y, Gasdermin D inhibition confers antineutrophil-mediated cardioprotection in acute myocardial infarction, J. Clin. Invest 132 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Sreejit G, Nooti SK, Jaggers RM, Athmanathan B, Ho Park K, Al-Sharea A, Johnson J, Dahdah A, Lee MKS, Ma J, Murphy AJ, Nagareddy PR, Retention of the NLRP3 Inflammasome-Primed Neutrophils in the Bone Marrow Is Essential for Myocardial Infarction-Induced Granulopoiesis, Circulation. 145 (2022) 31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Li Z, Xu H, Liu X, Hong Y, Lou H, Liu H, Bai X, Wang L, Li X, Monayo SM, Mokembo JN, Jha NK, Yang B, Zhang Y, GDF11 inhibits cardiomyocyte pyroptosis and exerts cardioprotection in acute myocardial infarction mice by upregulation of transcription factor HOXA3, Cell Death Dis. 11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Jia G, Hill MA, Sowers JR, Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity, Circ. Res 122 (2018) 624–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Domingueti CP, Dusse LMSA, Carvalho MDG, De Sousa LP, Gomes KB, Fernandes AP, Diabetes mellitus: The linkage between oxidative stress, inflammation, hypercoagulability and vascular complications, J. Diabetes Complications 30 (2016) 738–745. [DOI] [PubMed] [Google Scholar]
- [82].Tang WH, Stitham J, Jin Y, Liu R, Lee SH, Du J, Atteya G, Gleim S, Spollett G, Martin K, Hwa J, Aldose reductase-mediated phosphorylation of p53 leads to mitochondrial dysfunction and damage in diabetic platelets, Circulation. 129 (2014) 1598–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Luo B, Li B, Wang W, Liu X, Xia Y, Zhang C, Zhang M, Zhang Y, An F, NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model, PLoS One. 9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Xie Y, Huang Y, Ling X, Qin H, Wang M, Luo B, Chemerin/CMKLR1 Axis Promotes Inflammation and Pyroptosis by Activating NLRP3 Inflammasome in Diabetic Cardiomyopathy Rat, Front. Physiol 11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Singer M, Deutschman CS, Seymour C, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, Der Poll T, Vincent JL, Angus DC, The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3), JAMA. 315 (2016) 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Walley KR, Sepsis-induced myocardial dysfunction, Curr. Opin. Crit. Care 24 (2018) 292–299. [DOI] [PubMed] [Google Scholar]
- [87].Romero-Bermejo FJ, Ruiz-Bailen M, Gil-Cebrian J, Huertos-Ranchal MJ, Sepsis-induced cardiomyopathy, Curr. Cardiol. Rev 7 (2011) 163–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Hollenberg SM, Singer M, Pathophysiology of sepsis-induced cardiomyopathy, Nat. Rev. Cardiol 18 (2021) 424–434. [DOI] [PubMed] [Google Scholar]
- [89].Kalbitz M, Fattahi F, Grailer JJ, Jajou L, Malan EA, Zetoune FS, Huber-Lang M, Russell MW, Ward PA, Complement-induced activation of the cardiac NLRP3 inflammasome in sepsis, FASEB J. 30 (2016) 3997–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Li N, Zhou H, Wu H, Wu Q, Duan M, Deng W, Tang Q, STING-IRF3 contributes to lipopolysaccharide-induced cardiac dysfunction, inflammation, apoptosis and pyroptosis by activating NLRP3, Redox Biol. 24 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Busch K, Kny M, Huang N, Klassert TE, Stock M, Hahn A, Graeger S, Todiras M, Schmidt S, Chamling B, Willenbrock M, Groß S, Biedenweg D, Heuser A, Scheidereit C, Butter C, Felix SB, Otto O, Luft FC, Slevogt H, Fielitz J, Inhibition of the NLRP3/IL-1β axis protects against sepsis-induced cardiomyopathy, J. Cachexia. Sarcopenia Muscle 12 (2021) 1653–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Wu C, Lu W, Zhang Y, Zhang G, Shi X, Hisada Y, Grover SP, Zhang X, Li L, Xiang B, Shi J, Li XA, Daugherty A, Smyth SS, Kirchhofer D, Shiroishi T, Shao F, Mackman N, Wei Y, Li Z, Inflammasome Activation Triggers Blood Clotting and Host Death through Pyroptosis, Immunity. 50 (2019) 1401–1411.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Liu H, Tang D, Zhou X, Yang X, Chen AF, PhospholipaseCγ1/calcium-dependent membranous localization of Gsdmd-N drives endothelial pyroptosis, contributing to lipopolysaccharide-induced fatal outcome, Am. J. Physiol. Heart Circ. Physiol 319 (2020) 1482–1495. [DOI] [PubMed] [Google Scholar]
- [94].Ju J, Liu Y, Liang H, Yang B, The role of pyroptosis in endothelial dysfunction induced by diseases, Front. Immunol 13 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Silva CMS, Wanderley CWS, Veras FP, Sonego F, Nascimento DC, Gonçalves AV, Martins TV, Cólon DF, Borges VF, Brauer VS, Damasceno LEA, Silva KP, Toller-Kawahisa JE, Batah SS, Souza ALJ, Monteiro VS, Oliveira AER, Donate PB, Zoppi D, Borges MC, Almeida F, Nakaya HI, Fabro AT, Cunha TM, Alves-Filho JC, Zamboni DS, Cunha FQ, Gasdermin D inhibition prevents multiple organ dysfunction during sepsis by blocking NET formation, Blood. 138 (2021) 2702–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Zhang H, Chen Z, Zhou J, Gu J, Wu H, Jiang Y, Gao S, Liao Y, Shen R, Miao C, Chen W, NAT10 regulates neutrophil pyroptosis in sepsis via acetylating ULK1 RNA and activating STING pathway, Commun. Biol 5 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Liu F, Ghimire L, Balasubramanian A, Hsu AY, Zhang Z, Yu H, Ma F, Luo HR, Neutrophilspecific depletion of gasdermin D does not protect against murine sepsis, Blood. 141 (2023) 550–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Burzynski LC, Clarke MCH, Death Is Coming and the Clot Thickens, as Pyroptosis Feeds the Fire, Immunity. 50 (2019) 1339–1341. [DOI] [PubMed] [Google Scholar]
- [99].Peisker T, Koznar B, Stetkarova I, Widimsky P, Acute stroke therapy: A review, Trends Cardiovasc. Med 27 (2017) 59–66. [DOI] [PubMed] [Google Scholar]
- [100].Gou X, Xu D, Li F, Hou K, Fang W, Li Y, Pyroptosis in stroke-new insights into disease mechanisms and therapeutic strategies, J. Physiol. Biochem 77 (2021) 511–529. [DOI] [PubMed] [Google Scholar]
- [101].Hu YF, Chen YJ, Lin YJ, Chen SA, Inflammation and the pathogenesis of atrial fibrillation, Nat. Rev. Cardiol 12 (2015) 230–243. [DOI] [PubMed] [Google Scholar]
- [102].Yao C, Veleva T, Scott L, Cao S, Li L, Chen G, Jeyabal P, Pan X, Alsina KM, Abu-Taha I, Ghezelbash S, Reynolds CL, Shen YH, Lemaire SA, Schmitz W, Müller FU, El-Armouche A, Tony Eissa N, Beeton C, Nattel S, Wehrens XHT, Dobrev D, Li N, Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation, Circulation. 138 (2018) 2227–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Heijman J, Muna AP, Veleva T, Molina CE, Sutanto H, Tekook M, Wang Q, Abu-Taha IH, Gorka M, Künzel S, El-Armouche A, Reichenspurner H, Kamler M, Nikolaev V, Ravens U, Li N, Nattel S, Wehrens XHT, Dobrev D, Atrial Myocyte NLRP3/CaMKII Nexus Forms a Substrate for Postoperative Atrial Fibrillation, Circ. Res 127 (2020) 1036–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Scott L, Fender AC, Saljic A, Li L, Chen X, Wang X, Linz D, Lang J, Hohl M, Twomey D, Pham TT, Diaz-Lankenau R, Chelu MG, Kamler M, Entman ML, Taffet GE, Sanders P, Dobrev D, Li N, NLRP3 inflammasome is a key driver of obesity-induced atrial arrhythmias, Cardiovasc. Res 117 (2021) 1746–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].PS H, JF C, GW L, EY C, JJ C, FC C, CK W, YC W, JJ H, CT T, Copy number variation of gasdermin D gene is associated with atrial fibrillation-related thromboembolic stroke, Europace. 25 (2023) 5–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Luo Y, Zhang Y, Han X, Yuan Y, Zhou Y, Gao Y, Yu H, Zhang J, Shi Y, Duan Y, Zhao X, Yan S, Hao H, Dai C, Zhao S, Shi J, Li W, Zhang S, Xu W, Fang N, Gong Y, Li Y, Akkermansia muciniphila prevents cold-related atrial fibrillation in rats by modulation of TMAO induced cardiac pyroptosis, EBioMedicine. 82 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].De Miguel C, Pelegrín P, Baroja-Mazo A, Cuevas S, Emerging Role of the Inflammasome and Pyroptosis in Hypertension, Int. J. Mol. Sci 22 (2021) 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].You J, Li X, Dai F, Liu J, Zhang Q, Guo W, GSDMD-mediated pyroptosis promotes cardiac remodeling in pressure overload, Clin. Exp. Hypertens 45 (2023). [DOI] [PubMed] [Google Scholar]
- [109].Bin Cheng S, Nakashima A, Huber WJ, Davis S, Banerjee S, Huang Z, Saito S, Sadovsky Y, Sharma S, Pyroptosis is a critical inflammatory pathway in the placenta from early onset preeclampsia and in human trophoblasts exposed to hypoxia and endoplasmic reticulum stressors, Cell Death Dis. 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Hu S, Wang L, Xu Y, Li F, Wang T, Disulfiram attenuates hypoxia-induced pulmonary hypertension by inhibiting GSDMD cleavage and pyroptosis in HPASMCs, Respir. Res 23 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Wu Y, Pan B, Zhang Z, Li X, Leng Y, Ji Y, Sun K, Chen AF, Caspase-4/11-Mediated Pulmonary Artery Endothelial Cell Pyroptosis Contributes to Pulmonary Arterial Hypertension, Hypertens. (Dallas, Tex. 1979) 79 (2022) 536–548. [DOI] [PubMed] [Google Scholar]
- [112].Liao F, Wang L, Wu Z, Luo G, Qian Y, He X, Ding S, Pu J, Disulfiram protects against abdominal aortic aneurysm by ameliorating vascular smooth muscle cells pyroptosis, Cardiovasc. Drugs Ther (2022). [DOI] [PubMed] [Google Scholar]
- [113].Fu H, rui Shen Q, Zhao Y, Ni M, can Zhou C, kuai Chen J, Chi C, jie Li D, Liang G, ming Shen F, Activating α7nAChR ameliorates abdominal aortic aneurysm through inhibiting pyroptosis mediated by NLRP3 inflammasome, Acta Pharmacol. Sin 43 (2022) 2585–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Jiang HK, Qiu GR, Li-Ling J, Xin N, Sun KL, Reduced ACTC1 expression might play a role in the onset of congenital heart disease by inducing cardiomyocyte apoptosis, Circ. J 74 (2010) 2410–2418. [DOI] [PubMed] [Google Scholar]
- [115].Wienecke LM, Cohen S, Bauersachs J, Mebazaa A, Chousterman BG, Immunity and inflammation: the neglected key players in congenital heart disease?, Heart Fail. Rev 27 (2022) 1957–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Peng X, Chen H, Li Y, Huang D, Huang B, Sun D, Effects of NIX-mediated mitophagy on ox-LDLinduced macrophage pyroptosis in atherosclerosis, Cell Biol. Int 44 (2020) 1481–1490. [DOI] [PubMed] [Google Scholar]
- [117].Shi H, Gao Y, Dong Z, Yang J, Gao R, Li X, Zhang S, Ma L, Sun X, Wang Z, Zhang F, Hu K, Sun A, Ge J, GSDMD-Mediated Cardiomyocyte Pyroptosis Promotes Myocardial I/R Injury, Circ. Res 129 (2021) 383–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Zeng C, Duan F, Hu J, Luo B, Huang B, Lou X, Sun X, Li H, Zhang X, Yin S, Tan H, NLRP3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy, Redox Biol. 34 (2020) 101523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Kambara H, Liu F, Zhang X, Liu P, Bajrami B, Teng Y, Zhao L, Zhou S, Yu H, Zhou W, Silberstein LE, Cheng T, Han M, Xu Y, Luo HR, Gasdermin D Exerts Anti-inflammatory Effects by Promoting Neutrophil Death, Cell Rep. 22 (2018) 2924–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Cao A, Kagan JC, Gasdermin Pore Forming Activities that Promote Inflammation from Living and Dead Cells, J. Mol. Biol 434 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Kolaczkowska E, Kubes P, Neutrophil recruitment and function in health and inflammation, Nat. Rev. Immunol 13 (2013) 159–175. [DOI] [PubMed] [Google Scholar]
- [122].Silvestre-Roig C, Braster Q, Ortega-Gomez A, Soehnlein O, Neutrophils as regulators of cardiovascular inflammation, Nat. Rev. Cardiol 17 (2020) 327–340. [DOI] [PubMed] [Google Scholar]
- [123].Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, von Pein JB, Broz P, Sweet MJ, Schroder K, Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps, Sci. Immunol 3 (2018). [DOI] [PubMed] [Google Scholar]
- [124].Son S, Yoon SH, Chae BJ, Hwang I, Shim DW, Choe YH, Hyun YM, Yu JW, Neutrophils Facilitate Prolonged Inflammasome Response in the DAMP-Rich Inflammatory Milieu, Front. Immunol 12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Karmakar M, Minns M, Greenberg EN, Diaz-Aponte J, Pestonjamasp K, Johnson JL, Rathkey JK, Abbott DW, Wang K, Shao F, Catz SD, Dubyak GR, Pearlman E, N-GSDMD trafficking to neutrophil organelles facilitates IL-1β release independently of plasma membrane pores and pyroptosis, Nat. Commun 11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Vats R, Kaminski TW, Brzoska T, Leech JA, Tutuncuoglu E, Katoch O, Jonassaint J, Tejero J, Novelli EM, Pradhan-Sundd T, Gladwin MT, Sundd P, Liver-to-lung microembolic NETs promote gasdermin D-dependent inflammatory lung injury in sickle cell disease, Blood. 140 (2022) 1020–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Stojkov D, Claus MJ, Kozlowski E, Oberson K, Schären OP, Benarafa C, Yousefi S, Simon H-U, NET formation is independent of gasdermin D and pyroptotic cell death, Sci. Signal 16 (2023). [DOI] [PubMed] [Google Scholar]
- [128].Li S, Wu Y, Yang D, Wu C, Ma C, Liu X, Moynagh PN, Wang B, Hu G, Yang S, Gasdermin D in peripheral myeloid cells drives neuroinflammation in experimental autoimmune encephalomyelitis, J. Exp. Med 216 (2019) 2562–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Mishra PK, Adameova A, Hill JA, Baines CP, Kang PM, Downey JM, Narula J, Takahashi M, Abbate A, Piristine HC, Kar S, Su S, Higa JK, Kawasaki NK, Matsui T, Guidelines in Cardiovascular Research: Guidelines for evaluating myocardial cell death, Am. J. Physiol. - Hear. Circ. Physiol 317 (2019) H891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Doetschman T, Azhar M, Cardiac-specific inducible and conditional gene targeting in mice, Circ. Res 110 (2012) 1498–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Ho-Tin-Noé B, Boulaftali Y, Camerer E, Platelets and vascular integrity: how platelets prevent bleeding in inflammation, Blood. 131 (2018) 277–288. [DOI] [PubMed] [Google Scholar]
- [132].Nicolai L, Gaertner F, Massberg S, Platelets in Host Defense: Experimental and Clinical Insights, Trends Immunol. 40 (2019) 922–938. [DOI] [PubMed] [Google Scholar]
- [133].Hottz ED, Lopes JF, Freitas C, Valls-De-Souza R, Oliveira MF, Bozza MT, Da Poian AT, Weyrich AS, Zimmerman GA, Bozza FA, Bozza PT, Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation, Blood. 122 (2013) 3405–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Cornelius DC, Baik CH, Travis OK, White DL, Young CM, Austin Pierce W, Shields CA, Poudel B, Williams JM, NLRP3 inflammasome activation in platelets in response to sepsis, Physiol. Rep 7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Wang S, Liu Y, Li G, Feng Q, Hou M, Peng J, Reduced intracellular antioxidant capacity in platelets contributes to primary immune thrombocytopenia via ROS-NLRP3-caspase-1 pathway, Thromb. Res 199 (2021) 1–9. [DOI] [PubMed] [Google Scholar]
- [136].Rolfes V, Ribeiro LS, Hawwari I, Böttcher L, Rosero N, Maasewerd S, Santos MLS, Próchnicki T, de S. Silva CM, de S. Wanderley CW, Rothe M, Schmidt SV, Stunden HJ, Bertheloot D, Rivas MN, Fontes CJ, Carvalho LH, Cunha FQ, Latz E, Arditi M, Franklin BS, Platelets Fuel the Inflammasome Activation of Innate Immune Cells, Cell Rep. 31 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Di L, Zha C, Liu Y, Platelet-derived microparticles stimulated by anti-β2GPI/β2GPI complexes induce pyroptosis of endothelial cells in antiphospholipid syndrome, Platelets. 34 (2023) 2156492. [DOI] [PubMed] [Google Scholar]
- [138].Huang LS, Hong Z, Wu W, Xiong S, Zhong M, Gao X, Rehman J, Malik AB, mtDNA Activates cGAS Signaling and Suppresses the YAP-Mediated Endothelial Cell Proliferation Program to Promote Inflammatory Injury, Immunity. 52 (2020) 475–486.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Bennett MR, Sinha S, Owens GK, Vascular Smooth Muscle Cells in Atherosclerosis, Circ. Res 118 (2016) 692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Armulik A, Genové G, Betsholtz C, Pericytes: developmental, physiological, and pathological perspectives, problems, and promises, Dev. Cell 21 (2011) 193–215. [DOI] [PubMed] [Google Scholar]
- [141].Rashad S, Niizuma K, Sato-Maeda M, Fujimura M, Mansour A, Endo H, Ikawa S, Tominaga T, Early BBB breakdown and subacute inflammasome activation and pyroptosis as a result of cerebral venous thrombosis, Brain Res. 1699 (2018) 54–68. [DOI] [PubMed] [Google Scholar]
- [142].Liang Y, Song P, Chen W, Xie X, Luo R, Su J, Zhu Y, Xu J, Liu R, Zhu P, Zhang Y, Huang M, Inhibition of Caspase-1 Ameliorates Ischemia-Associated Blood-Brain Barrier Dysfunction and Integrity by Suppressing Pyroptosis Activation., Front. Cell. Neurosci 14 (2020) 540669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Zhao Y, Qu H, Wang Y, Xiao W, Zhang Y, Shi D, Small rodent models of atherosclerosis, Biomed. Pharmacother 129 (2020). [DOI] [PubMed] [Google Scholar]
- [144].Fan J, Wang Y, Chen YE, Genetically Modified Rabbits for Cardiovascular Research., Front. Genet 12 (2021) 614379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Getz GS, Reardon CA, Pig and Mouse Models of Hyperlipidemia and Atherosclerosis, Methods Mol. Biol 2419 (2022) 379–411. [DOI] [PubMed] [Google Scholar]
- [146].Van Hout GPJ, Bosch L, Ellenbroek GHJM, De Haan JJ, Van Solinge WW, Cooper MA, Arslan F, De Jager SCA, Robertson AAB, Pasterkamp G, Hoefer IE, The selective NLRP3-inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction, Eur. Heart J 38 (2017) 828–836. [DOI] [PubMed] [Google Scholar]
- [147].Azushima K, Gurley SB, Coffman TM, Modelling diabetic nephropathy in mice, Nat. Rev. Nephrol 14 (2018) 48–56. [DOI] [PubMed] [Google Scholar]
- [148].Bacmeister L, Schwarzl M, Warnke S, Stoffers B, Blankenberg S, Westermann D, Lindner D, Inflammation and fibrosis in murine models of heart failure, Basic Res. Cardiol 114 (2019). [DOI] [PubMed] [Google Scholar]
- [149].Sreejit G, Abdel-Latif A, Athmanathan B, Annabathula R, Dhyani A, Noothi SK, QuaifeRyan GA, Al-Sharea A, Pernes G, Dragoljevic D, Lal H, Schroder K, Hanaoka BY, Raman C, Grant MB, Hudson JE, Smyth SS, Porrello ER, Murphy AJ, Nagareddy PR, Neutrophil-Derived S100A8/A9 Amplify Granulopoiesis After Myocardial Infarction, Circulation. 141 (2020) 1080–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Yang XM, Downey JM, Cohen MV, Housley NA, Alvarez DF, Audia JP, The Highly Selective Caspase-1 Inhibitor VX-765 Provides Additive Protection Against Myocardial Infarction in Rat Hearts When Combined With a Platelet Inhibitor, J. Cardiovasc. Pharmacol. Ther 22 (2017) 574–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Bertheloot D, Wanderley CW, Schneider AH, Schiffelers LD, Wuerth JD, Tödtmann JM, Maasewerd S, Hawwari I, Duthie F, Rohland C, Ribeiro LS, Jenster L, Rosero N, Tesfamariam YM, Cunha FQ, Schmidt FI, Franklin BS, Nanobodies dismantle post-pyroptotic ASC specks and counteract inflammation in vivo, EMBO Mol. Med 14 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Toldo S, Mezzaroma E, Buckley LF, Potere N, Di Nisio M, Biondi-Zoccai G, Van Tassell BW, Abbate A, Targeting the NLRP3 inflammasome in cardiovascular diseases, Pharmacol. Ther 236 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Wohlford GF, Van Tassell BW, Billingsley HE, Kadariya D, Canada JM, Carbone S, Mihalick VL, Bonaventura A, Vecchié A, Chiabrando JG, Bressi E, Thomas G, Ho AC, Marawan AA, Dell M, Trankle CR, Turlington J, Markley R, Abbate A, Phase 1B, Randomized, Double-Blinded, Dose Escalation, Single-Center, Repeat Dose Safety and Pharmacodynamics Study of the Oral NLRP3 Inhibitor Dapansutrile in Subjects With NYHA II-III Systolic Heart Failure, J. Cardiovasc. Pharmacol 77 (2020) 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.