

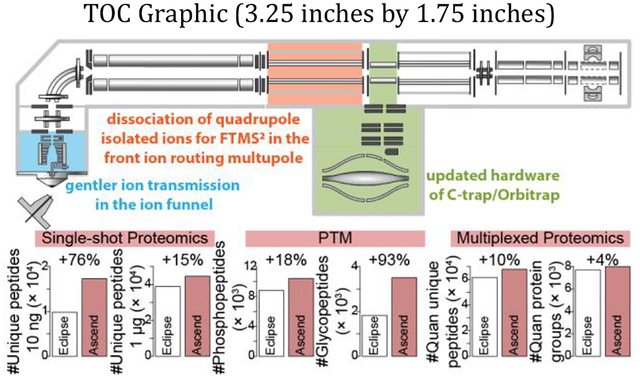
Abstract
Mass spectrometry (MS)-based proteomics is a powerful technology to globally profile protein abundances, activities, interactions, and modifications. The extreme complexity of proteomics samples, which often contain hundreds of thousands of analytes, necessitates continuous development of MS techniques and instrumentation to improve speed, sensitivity, precision, accuracy, among other analytical characteristics. Here, we systematically evaluated an Orbitrap Ascend Tribrid mass spectrometer in the context of shotgun proteomics, and we compared its performance to that of the previous generation of Tribrid instruments - Orbitrap Eclipse. The updated architecture of Orbitrap Ascend includes a second ion routing multipole (IRM) in front of the redesigned C-trap/Orbitrap and a new ion funnel that allows gentler ion introduction, among other changes. These modifications in Ascend hardware configuration enabled an increase in parallelizable ion injection time during higher-energy collisional dissociation (HCD) Orbitrap tandem MS (FTMS2) analysis of ~ 5 ms. This enhancement was particularly valuable in the analyses of limited sample amounts, where improvements in sensitivity resulted in up to 140% increase in the number identified tryptic peptides. Further, analysis of phosphorylated peptides enriched from the K562 human cell line yielded up to ~ 50% increase in the number of unique phosphopeptides and localized phosphosites. Strikingly, we also observed a ~ 2-fold boost in the number of detected N-glycopeptides, likely owing to the improvements in ion transmission and sensitivity. In addition, we performed the multiplexed quantitative proteomics analyses of TMT11-plex labeled HEK293T tryptic peptides and observed 9–14% increase in the number of quantified peptides. In conclusion, Orbitrap Ascend consistently outperformed its predecessor Orbitrap Eclipse in various bottom-up proteomic analysis, and we anticipate it will generate reproducible and in-depth datasets for numerous proteomic applications.
Graphical Abstract
High complexity of human and other proteomes necessitates the continuous advancement of mass spectrometry (MS)-based proteomic technology for more in-depth and high-throughput analyses of protein abundances, activities, interactions, and modifications1,2. With the human genome containing ~ 20,000 entries (~ 70,000 considering splice variants)3, achieving deep and comprehensive proteome coverage has been an analytical challenge for mass spectrometry in terms of speed, sensitivity, precision, and accuracy4. The ability to rapidly acquire high-quality tandem mass spectra is one of the most critical characteristics that deals with this challenge, and hybrid mass spectrometers featuring Orbitraps and other mass analyzers are uniquely suited for the task5–9.
Among Orbitrap-equipped mass spectrometers, the Tribrid architecture, combining Orbitrap, quadrupole and ion trap mass analyzers, provides flexible and diverse modes of operation. This instrument efficiently pipelines ion with its ability to perform ion isolation, dissociation, and mass analysis in parallel5. The basic layout of the Orbitrap Tribrid mass spectrometer series includes an ion inlet proceeded by a quadrupole mass filter, then the C-trap/Orbitrap, followed by the ion routing multipole (IRM) for initial ion accumulation and higher-energy collisional dissociation (HCD), with the quadrupole linear ion trap in the back of the instrument.
The design of the latest addition in the Orbitrap Tribrid series, the Orbitrap Ascend, improves upon this architecture by adding a second IRM in front of the redesigned C-trap/Orbitrap and the new ion funnel that allows gentler ion introduction, among other changes. Collectively, these updates increase scan speed and sensitivity of the instrument, resulting in generation of a greater number of higher quality tandem mass spectra. Here, we systematically evaluated the new Orbitrap Ascend in the context of various shotgun proteomics experiments and compared its performance to that of the previous generation Tribrid instrument, the Orbitrap Eclipse. We acquired data using multiple Orbitrap MS2 (FTMS2) resolution settings and evaluated varying peptide loading amounts and liquid chromatography (LC) elution lengths for single-shot proteomics. A substantial improvement in the number of detected peptides was observed in analyses of limited peptide amounts (≥ 10 ng) and at low FTMS2 resolution. Analyses of post-translationally modified (PTM) peptides, including phosphorylation and N-glycosylation, demonstrated 14–127% increases in the number of modified peptides and PTM sites. We also explored the multiplexed quantitative proteomics by analyzing TMT11-plex labeled HEK293T tryptic peptides and observed 9–14% improvement in the number of quantified peptides. Together, enhanced sensitivity and scan parallelization of Orbitrap Ascend translated into promising improvements in the results of various bottom-up proteomics analyses.
EXPERIMENTAL SECTION
Solvents and Chemicals.
Water (Optima, W6–4), acetonitrile (ACN, Optima, A9554), methanol (Optima, A454SK-4), formic acid (Pierce, PI28905), TMT10plex label reagents (90406) and TMT11–131C label reagents (A34807) were purchased from Fisher Scientific. Trifluoroacetic acid (TFA, 302031–100ML), tris(2-carboxyethyl)phosphine hydrochloride (TCEP HCl, C4706), 2-chloroacetamide (C0267), glycolic acid (124737), and ammonium hydroxide solution (338818) were purchased from Sigma Aldrich. Lysyl EndopeptidaseR (LysC, 129–02541) was purchased from Wako. Trypsin (V5113) and intact MS-compatible human protein extracts (V6941) was purchased from Promega.
Lysis, Digestion, and Desalting.
Proteins from cell lines were solubilized in 8 M Urea and 100 mM tris buffer. 10 mM TCEP and 40 mM 2-chloroacetamide were added for reduction and alkylation. LysC was added at a protein-enzyme ratio of 50:1. After 4 h of incubation at room temperature, trypsin was added at a protein-enzyme ratio of 50:1 for overnight digestion at room temperature. The digestion was quenched by adding 10% TFA to pH of ~ 1. Peptides were desalted using Strata-X 33 μm polymeric reversed phase SPE Cartridge (Phenomenex, 8B-S100-AAK). Desalted peptides were dried by SpeedVac (Thermo Scientific).
Mouse brain tissue was frozen in liquid nitrogen immediately upon collection, pulverized with mortar and pestle, and lysed as described above. Proteins were precipitated through the addition of 90% methanol (volume/volume), sequentially digested with LysC and trypsin, and desalted, as described above.
PTM Enrichment.
Phosphopeptides were enriched using ReSyn Biosciences Ti-IMAC HP beads (MR-THP005). The Ti-IMAC beads were washed three times by 1 mL 80% ACN with 6% TFA. Peptide samples were mixed with beads at a bead-peptide ratio of 2:1 in 1 mL 80% ACN with 6% TFA, followed by a vortex for 20 min at room temperature. The beads were washed by 1 mL 80% ACN with 6% TFA for three times, 1 mL 80% ACN for one time, 1 mL 80% ACN with 0.5 M glycolic acid for one time, and 1 mL 80% ACN for one time. Phosphopeptides were eluted from beads by 300 μL 50% ACN with 1% ammonium hydroxide for two times. Eluted phosphopeptides were dried by SpeedVac, resuspended in 0.2% formic acid, desalted using Strata-X 33 μm polymeric reversed phase SPE Cartridge, and dried by SpeedVac again.
Glycopeptides were enriched using SAX-ERLIC solid-phase extraction columns (Thermo Scientific), according to the published protocol10.
TMT Labeling.
TMT labeling was performed as described previously11. Briefly, 6 mg HEK293T tryptic peptides were resuspended in 30% acetonitrile, split into 11 fractions, and labeled each with 10 mg of TMT 11-plex. To check mixing ratios, 1 μg of each sample was pooled, desalted with Strata-X 33 μm polymeric reversed phase SPE Cartridge, and analyzed by mass spectrometry. After accounting for normalization factors, samples were mixed 1:1 across channels and desalted.
High-pH Fractionation.
TMT labeled peptides were fractionated using XBridge Peptide ethylene bridged hybrid (BEH) C18 Column, 130Å, 3.5 μm, 4.6 mm X 150 mm column (Waters) and Agilent 1260 Infinity Binary LC. Mobile phase A consisted of 10 mM ammonium formate in water (pH 10). Mobile phase B consisted of 10 mM ammonium formate in 80% methanol (pH 10). The LC was set to 0.8 mL/min. Mobile phase B was set to 35% for 2 min, increased to 75% for 6 min, and further increased to 100% for 5 min. The LC was re-equilibrated at 0% mobile phase B for 5 min. 24 fractions were collected and then combined into 12 fractions.
Capillary LC-MS.
LC separation was performed on a Vanquish Neo System (Thermo Scientific) with an in-house packed reversed-phase BEH C18 column (50 cm length × 75 μm inner diameter × 1.7 μm particle size) at 55 °C and 300 nL/min flow rate12. Mobile phase A consisted of 0.2% formic acid in water. Mobile phase B consisted of 0.2% formic acid in 80% ACN. Eluting peptides were ionized by electrospray ionization and then analyzed by an Orbitrap Ascend Tribrid mass spectrometer (tune version 4.0.4084.16, Thermo Scientific) and an Orbitrap Eclipse Tribrid mass spectrometer (tune version 4.0.12491, Thermo Scientific). Spray voltage was set to 2 kV. Ion transfer tube temperature was set to 275 °C. Source RF was set to 30.
For single-shot proteomics with data dependent acquisition (DDA), the MS1 scan resolution was set to 60,000 (at m/z 200) for all methods that use the Orbitrap for tandem MS (FTMS2) and 240,000 for all methods that use the ion trap for tandem MS (ITMS2). MS1 scan range was 300−1,350 m/z, AGC target was 250%, maximum injection time was 50 ms. Precursor ions with charge states of 2–5 were isolated with the quadrupole mass filter and fragmented by higher-energy collisional dissociation (HCD) at a normalized collision energy (NCE) of 25%. On the Orbitrap Eclipse trapping and HCD occur in the one (rear) IRM, on the Orbitrap Ascend trapping and HCD for MS2 scans occurs in the new IRM in front of the C-Trap. Quadrupole isolation was 1.1 m/z for FTMS2 and 0.5 m/z for ITMS2. For FTMS2 scans on the Orbitrap Ascend, first mass was set to 150, AGC target was 300%, maximum injection time was 11 ms for resolution of 7,500, 27 ms for resolution of 15,000, and 59 ms for resolution of 30,000. For FTMS2 scans on Orbitrap Eclipse, first mass was set to 150, AGC target was 300%, maximum injection time was 11 ms for resolution of 7,500, 22 ms for resolution of 15,000, and 54 ms for resolution of 30,000. For ITMS2 scans on both instruments, scan rate was set to turbo, scan range was 150–1,350 m/z, AGC target was 250%, and maximum injection time was 14 ms.
For single-shot proteomics with data independent acquisition (DIA), the MS1 scan resolution was set to 60,000 (at m/z 200). MS1 scan range was 300–1350 m/z, AGC target was 250%, maximum injection time was 50 ms. Precursors were isolated with an isolation width of 10 m/z covering 400–1040 m/z. Precursors were fragmented by HCD at an NCE of 25%±5%. MS2 scans were acquired by Orbitrap with a resolution of 15,000. Maximum injection time was 22 ms for the Eclipse and 27 ms for the Ascend.
For phosphoproteomics, MS1 scan range was 300−1,350 m/z, AGC target was 250%, maximum injection time was 50 ms. Cycle time was set to 1 s. Dynamic exclusion was set to 10 s for method with 10 min elution length and 20 s for all other methods. Precursor ions with charge states of 2–6 were isolated with the quadrupole mass filter using 0.7 m/z width and fragmented by HCD at an NCE of 30%. For FTMS2 scans on Orbitrap Ascend, first mass was set to 150, AGC target was 300%, maximum injection time was 11 ms for resolution of 7,500, 27 ms for resolution of 15,000, and 59 ms for resolution of 30,000. For FTMS2 scans on Orbitrap Eclipse, first mass was set to 150, AGC target was 300%, maximum injection time was 11 ms for resolution of 7,500, 22 ms for resolution of 15,000, and 54 ms for resolution of 30,000.
For glycoproteomics, MS1 scan range was 300−2,000 m/z, AGC target was 250%, maximum injection time was 123 ms. Cycle time was set to 2 s in TopSpeed mode, and dynamic exclusion was set to 20 s. Precursor ions with charge states of 2–6 were isolated by quadrupole mass filter at 1.3 m/z width and fragmented by HCD at fixed NCE of 36%. The fragments were scanned in the Orbitrap at resolutions of 15,000, 30,000, or 60,000 with normalized AGC of 200% and defined first mass of 150. Injection times for resolutions of 15,000, 30,000, and 60,000 were set to 27 vs 22, 59 vs 54 and 123 vs 118 ms for Ascend and Eclipse, respectively. If fragments of m/z of 204.0867, 138.0545, 366.1396, 274.0921, 292.1027, 126.055, 144.0655, 168.0654 or 186.076 (± 10 ppm) were detected, an additional MS2 scan was triggered. The resolutions and injection times for the triggered scans were set to be the same as described above for the survey scans, except precursors were fragmented using stepped HCD of 35±15 % and scan range was set to 150–4,000.
For TMT experiments, MS1 scan range was 400−1,600 m/z, AGC target was 250%, maximum injection time was 50 ms. Cycle time was set to 2.5 s. Precursor ions with charge states of 2–5 were isolated by quadrupole mass filter at 0.5 m/z width and fragmented by collision-induced dissociation (CID) at an NCE of 34%, activation time of 10 ms, and activation Q of 0.25. MS2 analysis was performed in ion trap at the turbo scan rate. MS2 scan range was 150−1,600 m/z, AGC target was 300%, maximum injection time was 23 ms. Top10 most abundant ions from MS2 were isolated at 2 m/z width and fragmented at an HCD NCE of 55% after synchronous precursor selection (SPS) if the MS2 spectrum passes the real time search (RTS) filter9,13. Static modifications were set to Carbamidomethyl on cysteine (C) and TMT6plex on lysine and N-terminus (Kn). Variable modifications were set to Oxidation on methionine (M). Maximum missed cleavages were set to 1. Maximum variable mods/peptide were set to 2. Maximum search time was set to 35 ms. The scoring threshold was set to 1.4 XCorr, 0.1 dCn, and 20 ppm precursor tolerance. MS3 analysis was performed in the Orbitrap at resolutions of 15,000 and 30,000 when turboTMT was enabled for super resolution of the very narrow m/z band containing reporter ions (phase-constrained spectrum deconvolution method, ΦSDM). A higher resolution of 45,000 (50,000 res on the Eclipse) was used when turboTMT was disabled.
Data Processing.
For single-shot proteomics, raw files were searched through a custom release of the software suite COMPASS (v1.4.3.31)14. This in-house software suite was modified to enable high-throughput processing using resources provided by University of Wisconsin-Madison Center for High Throughput Computing. For DIA, data were process by FragPipe (v19.0) using the default workflow with DIA-NN15–17. For phosphoproteomics, raw files were searched through FragPipe (v19.0)15,16. Digest mass range was 500–6,500. Isotope error was 0/1/2. PTMProphet was used for modification localization18. All other parameters were set to default. For glycoproteomics, raw files were searched through FragPipe (v19.0) with default settings for HCD N-linked glycosylation with the exception of peptide length, which was set to 6–65 and max peptide mass which was set to 6,500 Da. For TMT data, raw files were searched through MaxQuant (v2.2.0.0)19,20. Default parameters were used.
RESULTS AND DISCUSSION
Instrument Architecture.
The main components of Orbitrap Ascend Tribrid mass spectrometer consist of an electrodynamic ion funnel, an advanced ion beam guide, a quadrupole mass filter, two ion-routing multipoles (IRMs), a C-trap, a dual-pressure linear ion trap, and an high field Orbitrap mass analyzer (Figure 1a). While the basic layout of the analyzers remains the same as the legacy Tribrid instruments, the Ascend was substantially overhauled. The C-trap/Orbitrap components and electronics were updated to those used in the Orbitrap Exploris 480 instruments. Previous Tribrid generations utilized the Q Exactive version of these components. While dimensions of the Orbitrap analyzer remained the same, with the Exploris-based components the central electrode is now held at 4 kV. Further, the Ascend also benefits from faster polarity switching and improved vacuum systems with these updated C-trap/Orbitrap assemblies.
Figure 1. Overview of Orbitrap Ascend Tribrid mass spectrometer.
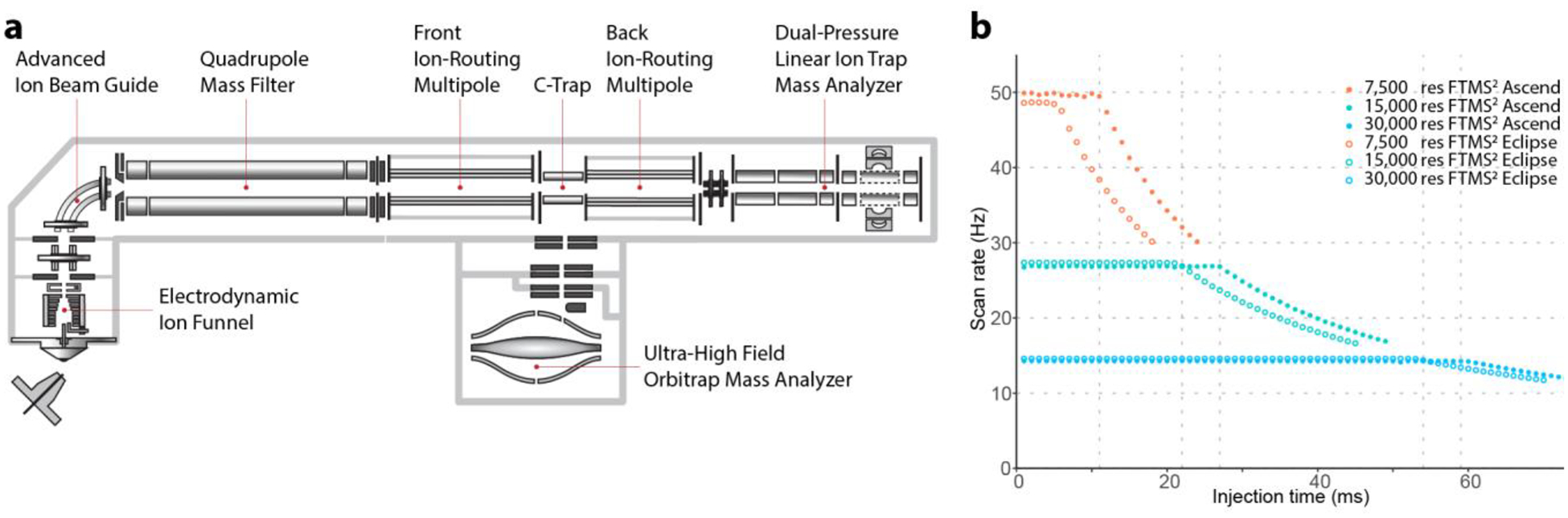
(a) Schematic of Orbitrap Ascend Tribrid mass spectrometer. (b) Scan rates of FTMS2 for different resolutions of Orbitrap Ascend and Orbitrap Eclipse by measuring peptide MRFA (524.4 m/z) from direct infusion of Thermo FlexMix calibration solution.
The largest change to the instrument is a new ion routing multipole (the front IRM) installed between the quadrupole mass filter and C-trap. This additional IRM allows parallel ion injection and accumulation during FTMSn ion manipulation and Orbitrap injection. Note earlier hybrid and Tribrid mass spectrometers included a single IRM located downstream of the C-trap. This old hardware configuration necessitated quadrupole-isolated ions to pass through the C-trap to enter the IRM for ion accumulation and possible HCD fragmentation. Ions from this rear IRM then need to be moved back to the C-trap for injection into the Orbitrap. Only after the C-trap was reset from ejection, could the instrument start injecting the next packet of ions into the rear IRM. With the new IRM in front of the C-trap, the next packet of ions can start accumulating as soon as the ions are passed from this IRM into the C-trap, thereby gaining ~ 5 ms of parallelizable injection time during higher-energy collisional dissociation (HCD) FTMS2 analysis. This time gain is equal to the time spent transferring and injecting ions into the Orbitrap (Figure 1b). The data in Figure 1b were measured by directly infusing Thermo FlexMix calibration solution, isolating the peptide MRFA (524.4 m/z) using quadrupole with an isolation width of 2 m/z, and scanning in the Orbitrap after HCD fragmentation with an NCE of 30%. Thanks to the presence of the front IRM, the Ascend can now record FTMS2 spectra at up to 50 Hz when utilizing up to 11 ms of ion injection time, while the FTMS2 scan rate of Orbitrap Eclipse only reaches ~ 38 Hz (Table 1). The hardware/software updates further reduce ion transfer time and improve ion transmission, resulting in faster ITMS2 spectral acquisition rates (Supplemental Figure 1). These data were collected by the same way as described above except the linear ion trap was used for MS2 scanning. These gains grow substantially from ~ 5 ms, as the complexity of the FTMSn scan increases. On the Ascend instrument, ions can be accumulated in the front IRM during all MSn ion manipulation steps (e.g., MS3 isolation and activation, ion/ion reactions). The back IRM is reserved for dissociating ions isolated in the linear ion trap and for MSn scans, where n is ≥ 3.
Table 1.
Details of FTMS2 settings.
| instrument | FTMS2 resolution | ion injection time (ms) | max scan rate (Hz) |
|---|---|---|---|
| Orbitrap Ascend | 7,500 | 11 | ~ 50 |
| Orbitrap Ascend | 15,000 | 27 | ~ 27 |
| Orbitrap Ascend | 30,000 | 59 | ~ 14 |
| Orbitrap Ascend | 60,000 | 123 | ~ 7 |
| Orbitrap Eclipse | 7,500 | 11 (5) | ~ 38 (49) |
| Orbitrap Eclipse | 15,000 | 22 | ~ 27 |
| Orbitrap Eclipse | 30,000 | 54 | ~ 14 |
| Orbitrap Eclipse | 60,000 | 118 | ~ 7 |
Note, this table specifies the ion injection time used for each FTMS2 resolution (at m/z 200). For 7,500 resolution FTMS2 on Eclipse, although the injection time of 5 ms in parentheses is the maximum parallelizable injection time and the scan rate of ~ 49 Hz can be achieved, the injection time of 11 ms was used to ensure sufficient ion intensity, which results in a reduced scan rate of ~ 38 Hz.
The electrode lengths and profiles are similar between the two IRMs, with the front IRM being slightly longer than the back (~ 17 cm vs. 12 cm). This combination of lengths is optimal for the instrument footprint and permits running the front IRM at slightly reduced pressure, depending on the application. Both IRMs are capable of operating at pressures ranging from 1–20 mTorr (same as the Orbitrap Eclipse). The Ascend IRM RF frequencies are capable of “switching” from a high frequency of ~ 3 MHz down to a low frequency of ~ 1 MHz, a feature that allows scan range to extend to 16,000 m/z (not examined or tested in this publication). For all the experiments performed in this study, the ion guides operated at the higher frequency, which affords a better mass range and transmission efficiency in the “standard” mass range of < 2,000 m/z.
To characterize performance of the new front IRM, we assessed a range of HCD NCE values by analyzing a complex mixture of unmodified tryptic peptides separated over a 70 min LC gradient (Supplemental Figure 2a). The range of front HCD NCE values between 21–27% yielded a comparable number of identified unique peptides. The NCE values are typical in analysis of tryptic peptides on other Tribrid instruments21–23, indicating the added front IRM works the same as the previous ones.
Other modifications on the Ascend include new ion funnel optics, similar to those found in the Orbitrap IQ-X Tribrid, which allow for gentler ion introduction. Briefly, the ion funnel lenses taper down to a radius of 3 mm at the exit of the ion funnel, while the ion funnel design on older Tribrid instruments tapers down to 2 mm. The larger radius produces a weaker axial field, which in turn results in lower ion energies as the beam exits the ion funnel. The gentler funnel is paired with a higher capacity ion transfer tube, and together they enable the same bright ion flux with less ion-source dissociation, relative to the Eclipse. The ion funnel exit aperture was also increased from 2 mm to 2.5 mm. This alteration slightly improves the ion beam flux and helps load up the downstream chamber with more gas to improve desolvation of large ions (m/z values approaching 16,000).
We assessed the effect of the new ion funnel on the shotgun proteomic analysis. We observed no significant differences in the total number of identified unique peptides for source RF values between 28 and 52, when analyzing 1 μg tryptic peptides separated over a 70 min gradient (Supplemental Figure 2b). However, when we directly infused the purified standard peptide VILEVAEEFYK, the Ascend generated fewer in-source fragments than the Eclipse, especially at higher RF values (Supplemental Figure 2c–e). We speculate that while marginally important to the analysis of unmodified tryptic peptides, the redesigned ion funnel with reduced in-source fragmentation will be more valuable for analyses of labile post-translationally modified peptides and small molecules.
Single-shot proteomics.
The hardware changes allow Ascend to either extend ion injection time while maintaining the same scan rate or collect spectra faster under certain fixed maximum injection times. To evaluate improvements in the number of detected peptides afforded by the new instrument, we performed a series of experiments comparing the Ascend to its predecessor, Orbitrap Eclipse, using complex mixtures of human tryptic peptides prepared from the HAP1 human cell line. Note in all experiments we used the same chromatographic setup on the Ascend and the Eclipse (the capillary column and the LC system). Four methods were assessed on both instruments: 1) 7,500 res FTMS2; 2) 15,000 res FTMS2; 3) 30,000 res FTMS2; 4) turbo scan ITMS2. For 7,500 res FTMS2 method, we set injection times to 11 ms on both instruments. The value was chosen because although 5 ms is the maximum parallelizable ion injection time for 7,500 res FTMS2 on Eclipse, practically it is too short to accumulate enough ions. For the other three methods, the instruments could achieve optimal parallelization with the corresponding optimal injection times (Table 1). We applied the four methods with an elution length of 40 min to the analyses of a range of peptide loading amounts – 10 ng, 100 ng, and 1,000 ng (Figure 2a–d). Comparing the results obtained on the Ascend to those of the Eclipse, we observed a 138.7% increase in the number of unique peptides identified from the 10 ng injections when utilizing the FTMS2 method with a resolution of 7,500. 19.3% and 14.6% more unique peptides were identified with the 7,500 resolution FTMS2 method when loading 100 ng and 1,000 ng, respectively (Figure 2a). Similarly, we identified more unique peptides in analysis of 10 ng (54.8% increase), 100 ng (6.8% increase), and 1,000 ng (2.9% increase) peptide loading amounts using the ITMS2 method (Figure 2d). For the 15,000 and 30,000 resolution FTMS2 methods, we observed 76.1% and 25.1% more unique peptides detected in the analyses of 10 ng loading amount, respectively. However, we found no significant increases in the number of unique peptides detected for larger loading amounts when using these two higher resolution Orbitrap methods (Figure 2b–c). Based on these results, we conclude that when the Ascend operates in a fully parallelized manner, the gain of extra 5 ms in ion injection time noticeably benefits sensitivity, which is especially evident when sample amount and parallelizable injection time is limited.
Figure 2. Single-shot proteomics comparing Orbitrap Ascend and Orbitrap Eclipse.
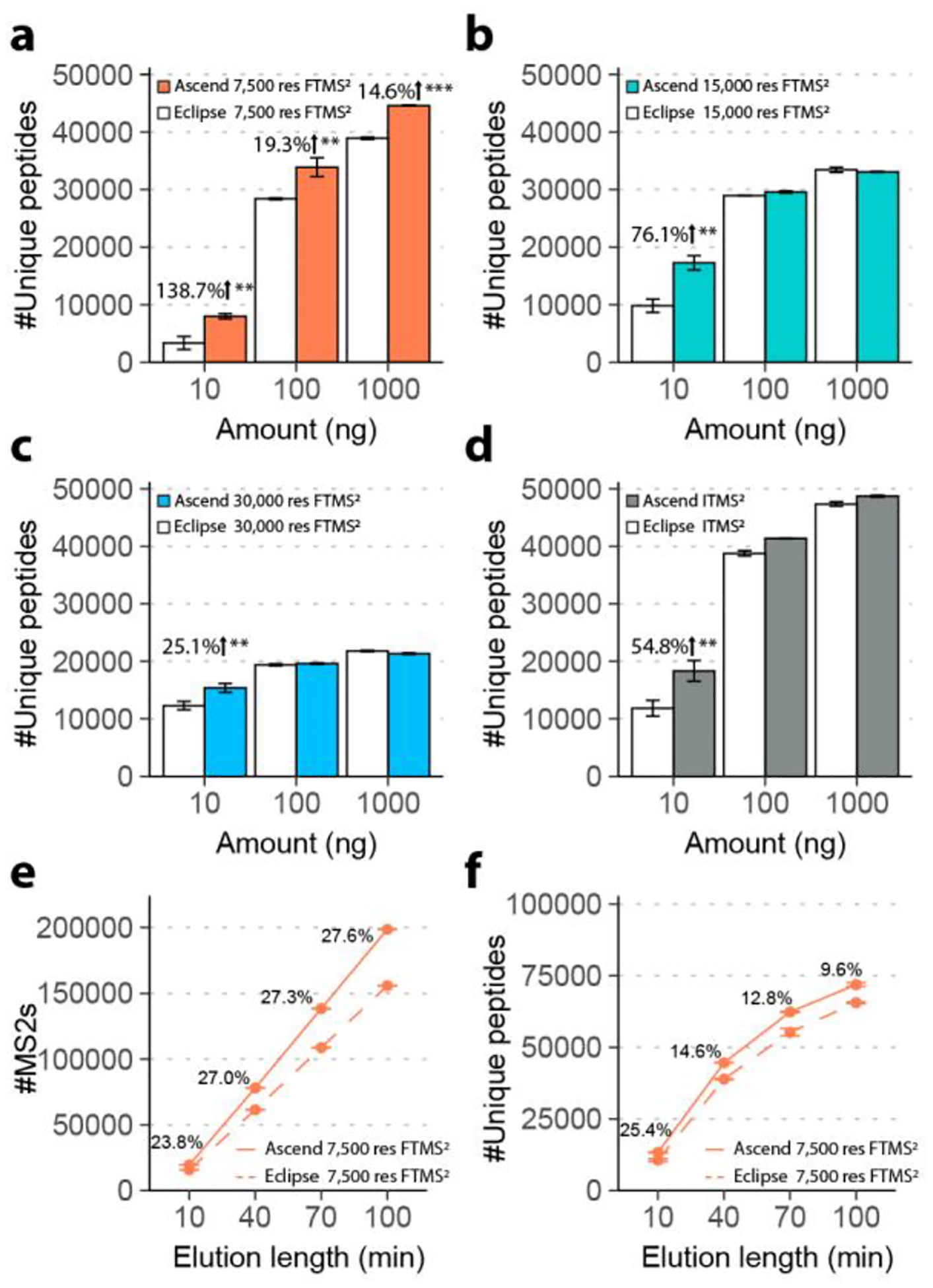
The number of identified unique peptides from serial dilution analysis, injecting 10–1,000 ng of tryptic peptides and eluting with a length of 40 min, performed using (a) 7,500 res FTMS2, (b) 15,000 res FTMS2, (c) 30,000 res FTMS2, and (d) ITMS2. (e) The number of 7,500 res FTMS2 scans acquired over a range of elution lengths. (f) The number of unique peptides identified over a range of elution length using 7,500 res FTMS2. Student’s t-tests were performed to calculate p values for comparison between the two instruments. *, p < 0.05. **, p < 0.01. ***, p < 0.001. Error bars represented one standard deviation of uncertainty. n=3.
To investigate how throughput is improved with the higher scan rate of the Ascend, we tested a range of LC elution lengths – 10, 40, 70, and 100 min – while loading 1 μg of tryptic peptides. For the FTMS2 method with a resolution of 7,500, the Ascend acquired > 20% more MS2 spectra than the Eclipse, which translated into detection of additional 9.6% (100 min analysis) to 25.4% (10 min analysis) unique peptides (Figure 2e–f). Nearly equal number of unique peptides (> 62,000) were identified in 70 min method on the Ascend as were in 100 min method on the Eclipse. For the ITMS2 method, we observed only a slight increase in the number of MS2 scans and unique peptides detected by the Ascend over the Eclipse, likely owing to the reduced ion transfer time and improved ion transmission of the former. We obtained equal detection depth on both instruments when analyzing 1 μg injections using the higher FTMS2 resolution method (15,000 and 30,000, Supplemental Figure 3). We conclude that for the analyses where the sample amount is not limited, the gains in sensitivity and the throughput of the Ascend are more consequential for shorter analysis time when utilizing lower FTMS2 resolution.
Additionally, we conducted a set of experiments with 1 μg HAP1 peptides analyzed over the elution lengths of 40- and 70-min using data independent acquisition (DIA). The same sample was fractionated into 8 fractions by offline high-pH fractionation and then analyzed using the same 15,000 res DDA method mentioned above. The Ascend performed slightly better than the Eclipse: 8,664 vs 8,451 protein groups (2.5% more) for the 40-min-elution method; 9,177 vs 8,907 protein groups (3.0% more) for the 70-min-elution method (Supplemental Figure 4). Although moderate, these differences are consistent with the increased scan rate of the Orbitrap Ascend.
PTM analyses.
Post-translational modifications (PTMs), such as phosphorylation and glycosylation, are important regulators of protein functions and play vital roles in various molecular and cellular processes. Detection of PTM-containing peptides, as well as localization of the modified amino-acid residues remains challenging. To evaluate if PTM analysis benefits from the sensitivity gain and higher scan rate of Orbitrap Ascend, we performed phosphoproteomic and N-glycoproteomic analyses on both the Ascend and the Eclipse.
For phosphoproteomics, we loaded 400 ng of enriched phosphopeptides from K562 cell line on the column, eluted over varying gradient lengths between 10 and 100 min, and analyzed on both instruments with multiple Orbitrap MS2 resolutions. For 15,000 res FTMS2, a 40 min elution length yielded the optimal number of localized phosphosites on both the Ascend (6,630) and the Eclipse (5,056), with the Ascend’s results corresponding to 31.1% boost in the number of unique localized phosphosites (Figure 3a). Extending the run time did not improve the results due to the concurrent reduction in analyte intensity and the limited maximum MS2 injection time, kept fixed in all experiments at 22 and 27 ms for the Ascend and the Eclipse, respectively. Across methods tested, the 100 min 30,000 res FTMS2 method yielded the highest number of localized phosphosites on both the Ascend (8,222) and the Eclipse (7,154), representing a 14.9% increase afforded by the Ascend (Figure 3b). The 30,000 res FTMS2 method benefited from longer elution lengths, as they were better complemented by its longer maximum ion injection times. In all experiments, we observed higher spectra quality with data collected on the Ascend, as compared to the Eclipse (Figure 3c–d, Supplemental Figure 5&6). Note, although the Ascend and the Eclipse generated comparable number of phosphopeptides using 30,000 res method, the Ascend was more successful at localizing phosphosites because of improved signal-to-noise in MS2 spectra (Supplemental Figure 7). Additionally, the 15,000 and 30,000 res FTMS2 methods outperformed 7,500 res FTMS2 method due to the shorter injection times and the resultant low sensitivity of the 7,500 res FTMS2 method (Supplemental Figure 8).
Figure 3. Phosphoproteomics and glycoproteomics comparing Orbitrap Ascend and Orbitrap Eclipse.
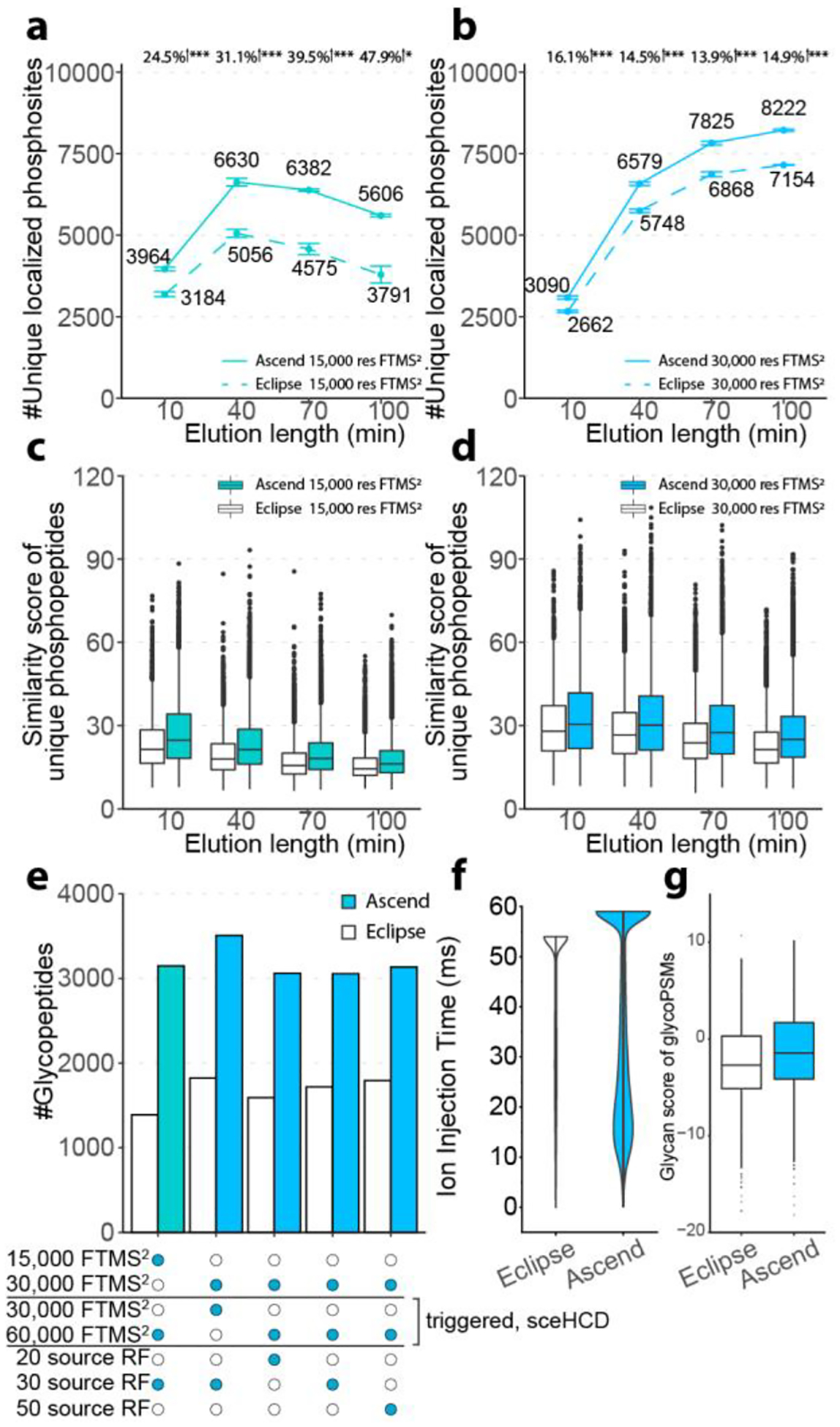
The number of unique localized phosphosites detected over a range of elution lengths by (a) 15,000 res FTMS2 and (b) 30,000 res FTMS2. Student’s t-tests were performed to calculate p values for comparison between the two instruments. *, p < 0.05. **, p < 0.01. ***, p < 0.001. Error bars represented one standard deviation of uncertainty. n=3 for elution length of 10 to 70 and n=2 for elution length of 100. Hyper score of unique phosphopeptides detected over a range of elution lengths by (c) 15,000 res FTMS2 and (d) 30,000 res FTMS2. (e) The number of glycopeptides identified by various 70 min data acquisition methods. (f) MS2 ion injection times of 30,000 res FTMS2/sceHCD 30,000 res FTMS2 method. (g) Glycan scores, as reported by MSFragger - Glyco, of 30,000 res FTMS2/sceHCD 30,000 res FTMS2 method.
For N-glycoproteomics, we loaded ~ 1 μg of enriched N-glycopeptides from mouse brain on the column, separated over an elution length of 70 min, and analyzed with five different data acquisition methods (see Experimental Section for additional details): 1) 15,000 res fixed HCD FTMS2 scan followed by triggered 60,000 res FTMS2 using stepped HCD (15k/60k sceHCD) with source RF of 30; 2) 30,000 res fixed HCD FTMS2 followed by triggered 30,000 res FTMS2 using stepped HCD (30k/30k sceHCD) with source RF of 30; 3) 30,000 res fixed HCD FTMS2 followed by triggered 60,000 res FTMS2 using stepped HCD (30k/60k sceHCD) with source RF of 20; 4) 30,000 res fixed HCD FTMS2 followed by triggered 60,000 res FTMS2 using stepped HCD with source RF of 30; 5) 30,000 res fixed HCD FTMS2 followed by triggered 60,000 res FTMS2 using stepped HCD with source RF of 50. The resolution for MS1 was set to 60,000 for all methods. Stepped collision energy HCD (sceHCD) was used for triggered FTMS2 scans if signature oxonium ions were detected. We observed a pronounced increase of 1.7- to 2.3-fold in the number of detected N-glycopeptides for all tested methods (Figure 3e). The 30k/30ksceHCD method generated the highest number of N-glycopeptides on both the Ascend and the Eclipse (3,508 vs 1,822). This method generated 2.3-folds more MS2 spectra on Ascend over Eclipse (56,878 vs. 24,461). While the MS2 AGC targets were identical, the percentage of MS2 scans that reached the AGC target before the allowed maximum injection time was 65.8% on the Ascend, while only 41.1% on the Eclipse (Figure 3f). These results indicate that the Ascend not only could accumulate ions longer for MS2 scans, while maintaining maximum parallelization, but also generated more MS2 scans with enough ions to reach the AGC target. These differences are likely attributed to the improved ion transmission and the reduced in-source fragmentation of the Ascend. The hypothesis is further supported by the observation that the glycan scores reported by MSFragger were considerably improved in the spectra obtained by the Ascend, over those from the Eclipse, indicating higher spectral quality for the scans generated by the former instrument (Figure 3g). Overall, we can conclude that due to the boost in sensitivity and corresponding spectral quality, the hardware and other advancements present on the Ascend uniquely benefit analyses of PTM-containing peptides.
Multiplexed quantitative proteomic analyses.
Multiplexed quantification of stable isotope labeled peptides (e.g., tandem mass tags TMT), remains a workhorse of quantitative bottom-up proteomics and is especially popular for large-scale proteomic studies24. To assess whether the Orbitrap Ascend could outperform the Orbitrap Eclipse in multiplexed quantitative proteomic analyses, we prepared TMT11-plex labeled HEK293T tryptic peptides and analyzed the sample using an RTS FTMS3 method. In this method, peptides are identified in ITMS2 scans and then quantified in higher resolution FTMS3 scans. This sequence is expected to be sped up on the Ascend, thanks to the presence of the second IRM that improves parallelizable injection time during the FTMS3 scans. Additionally, we employed the turboTMT (ΦSDM) feature for scans at resolutions of 15,000 and 30,000, which enables super resolution to differentiate the 6.3 mDa mass difference of the tags in the narrow m/z band containing reporter ions25,26. The resolutions of 45,000 and 50,000 were used on the Ascend and the Eclipse, respectively, when ΦSDM was disabled.
First, we evaluated the instrument performance in the 70 min analyses of 1 μg unfractionated labeled peptides. For the 15,000 res FTMS3 method, we observed an improved signal-to-noise across all channels (Supplemental Figure 9a). On the Ascend, more MS3 scan events were triggered (Ascend 24,150 vs Eclipse 22,249). Further, more and higher percent of MS3 spectra contained non zero-intensity reporter in all channels (Ascend 22,659 and 93.8% vs Eclipse 18,501 and 83.2%). The Ascend yielded a greater number and a higher percent of quantifiable 126/127C (Ascend 23,078 and 95.6% vs Eclipse 19,291 and 86.7%) and 126/131N (Ascend 23,071 and 95.5% vs Eclipse 19,249 and 86.5%) pairs (Supplemental Figure 9b). Similar improvements were observed for the 30,000 res FTMS3 method (Supplemental Figure 9c–d). The median cumulative signal-to-noise levels were higher on the Ascend for the 15,000 and 30,000 res (Supplemental Figure 9e). For the 15,000 res FTMS3 method, 8.6% more unique peptides were quantified by the Ascend (Ascend 12,064 vs. Eclipse 11,110). For the 30,000 res FTMS3 method, the percentage increase was 13.2% (Ascend 11,362 vs. Eclipse 10,041). For the 45,000 res FTMS3 method (50,000 res on Eclipse) there was a 12.9% improvement (Ascend 10,501 vs. Eclipse 9,299) (Figure 4a). The higher number of quantified unique peptides resulted in the higher number of quantified protein groups. We observed increases of 6.5% (Ascend 2,466 vs. Eclipse 2,316), 8.4% (Ascend 2,359 vs. Eclipse 2,176), and 7.5% (Ascend 2,217 vs. Eclipse 2,062) quantified protein groups for the 15,000 res, 30,000 res, and 45,000 (50,000) res FTMS3 methods, respectively (Figure 4b).
Figure 4. Multiplexed quantitative proteomic analyses comparing Orbitrap Ascend and Orbitrap Eclipse using TMT11-plex labeling.
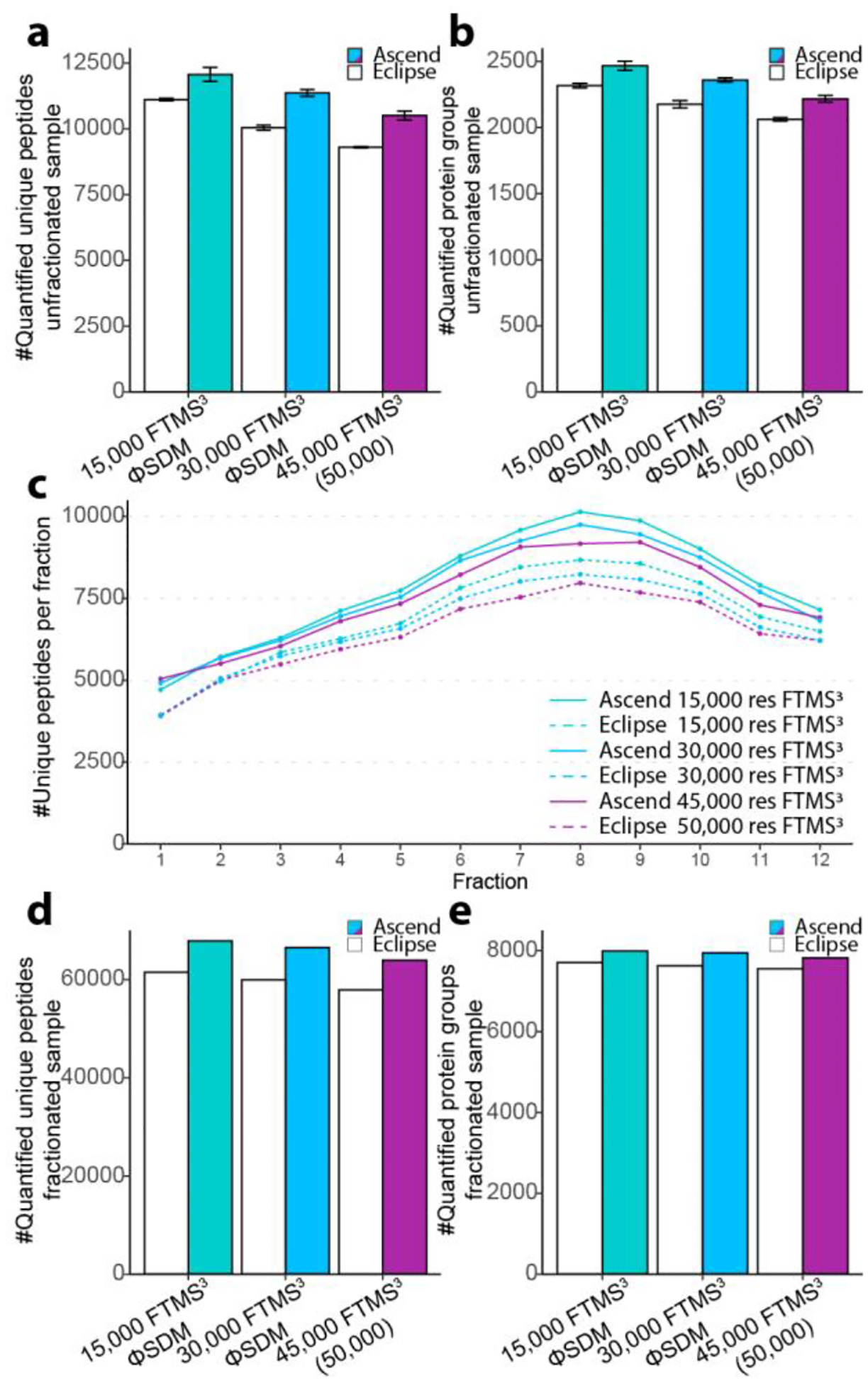
(a) The number of quantified unique peptides detected in unfractionated HEK293T tryptic peptide samples (n=3). (b) The number of quantified protein groups detected in analysis of unfractionated HEK293T tryptic peptide samples (n=3). (c) The number of identified peptides detected in each fraction of the HEK293T tryptic peptide samples. (d) The number of quantified unique peptides in analysis of fractionated HEK293T tryptic peptide samples. (e) The number of quantified protein groups in analysis of fractionated HEK293T tryptic peptide samples.
Next, we fractionated the labeled sample offline by reversed-phase high-pH liquid chromatography into 24 fractions and concatenated them into 12 fractions. We injected 500 ng of the fractionated peptides and analyzed them in the same manner as described above for the single-shot TMT experiments. Consistently, the Ascend identified and quantified a greater number of unique peptides across all fractions (Figure 4c). In total, we quantified an additional 10.4% unique peptides at 15,000 res (Ascend 67,872 vs. Eclipse 61,502), 10.9% at 30,000 res (Ascend 66,498 vs. Eclipse 59,959), and 10.5% at 45,000 (50,000) res (Ascend 63,940 vs. Eclipse 57,885) (Figure 4d). These improvements translated into 3.8% (Ascend 7,994 vs. Eclipse 7,705), 4.2% (Ascend 7,948 vs. Eclipse 7,629), and 3.5% (Ascend 7,820 vs. Eclipse 7,552) increases in the number of quantified protein groups for the 15,000 res, 30,000 res, and 45,000 (50,000) res FTMS3 methods, respectively (Figure 4e). Together these results illustrate that the addition of the second IRM and the resulting increase in spectral acquisition rates and scan parallelization produce consistent, albeit moderate, improvements in the number of quantified TMT-labeled peptides and proteins.
CONCLUSIONS
Here, we evaluated an Orbitrap Ascend Tribrid mass spectrometer and systematically compared this new instrument to its predecessor the Orbitrap Eclipse in the context of shotgun proteomics. We observed substantial improvements in various single-shot proteomics analyses, PTM analyses, and multiplexed quantitative proteomics experiments. The additional 5 ms of parallelizable ion injection time for FTMS2 with HCD benefited analyses of limited peptide loading amount, which on the Ascend yielded > 17,000 unique peptide identifications, a 76.1% increase over the results produced by the Eclipse. The faster ion transfer times and improved ion transmission of the Ascend also benefit ITMS2. Further, the extra parallelizable injection time combined with gentler ion transmission benefited a phosphopeptide analysis workflow, generating up to ~ 50% more localized phosphosites and > 2-fold increase in the number of identified N-glycopeptides. We also observed a boost in the number of quantified unique peptides and protein groups in multiplexed quantitative proteomics analyses, which could potentially increase throughput - a particularly valuable advantage for fractionation methods. Future work will focus on PTM analyses, especially glycoproteomics, in addition to investigating multiple activation strategies and MSn methodologies. We envision the stark improvements can be afforded by the new Ascend instrument in other cases where samples are limited, e.g., single cell proteomics. Lastly, further investigations should consider biomolecules other than proteins and peptides that can take full advantage of the gentler ion transmission and improved MSn acquisition, such as hydrophilic small molecules and lipids27.
Supplementary Material
ACKNOWLEDGMENT
We are grateful for support from NIH P41 GM108538 (J.J.C.).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Supplemental results. (PDF).
Notes
J.J.C. is a consultant for Thermo Fisher Scientific, 908 Devices, and Seer. D.B., J.H., R.H., M.W.S., V.Z., and G.C.M are employees of Thermo Fisher Scientific.
Data Availability
Raw data are available in MassIVE repository (MSV000091467).
REFERENCES
- (1).Meissner F; Geddes-McAlister J; Mann M; Bantscheff M The Emerging Role of Mass Spectrometry-Based Proteomics in Drug Discovery. Nat Rev Drug Discov 2022, 21 (9), 637–654. 10.1038/s41573-022-00409-3. [DOI] [PubMed] [Google Scholar]
- (2).Bekker-Jensen DB; Kelstrup CD; Batth TS; Larsen SC; Haldrup C; Bramsen JB; Sørensen KD; Høyer S; Ørntoft TF; Andersen CL; Nielsen ML; Olsen JV An Optimized Shotgun Strategy for the Rapid Generation of Comprehensive Human Proteomes. Cell Systems 2017, 4 (6), 587–599.e4. 10.1016/j.cels.2017.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Aebersold R; Agar JN; Amster IJ; Baker MS; Bertozzi CR; Boja ES; Costello CE; Cravatt BF; Fenselau C; Garcia BA; Ge Y; Gunawardena J; Hendrickson RC; Hergenrother PJ; Huber CG; Ivanov AR; Jensen ON; Jewett MC; Kelleher NL; Kiessling LL; Krogan NJ; Larsen MR; Loo JA; Ogorzalek Loo RR; Lundberg E; MacCoss MJ; Mallick P; Mootha VK; Mrksich M; Muir TW; Patrie SM; Pesavento JJ; Pitteri SJ; Rodriguez H; Saghatelian A; Sandoval W; Schlüter H; Sechi S; Slavoff SA; Smith LM; Snyder MP; Thomas PM; Uhlén M; Van Eyk JE; Vidal M; Walt DR; White FM; Williams ER; Wohlschlager T; Wysocki VH; Yates NA; Young NL; Zhang B How Many Human Proteoforms Are There? Nat Chem Biol 2018, 14 (3), 206–214. 10.1038/nchembio.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Eliuk S; Makarov A Evolution of Orbitrap Mass Spectrometry Instrumentation. Annual Rev. Anal. Chem 2015, 8 (1), 61–80. 10.1146/annurev-anchem-071114-040325. [DOI] [PubMed] [Google Scholar]
- (5).Senko MW; Remes PM; Canterbury JD; Mathur R; Song Q; Eliuk SM; Mullen C; Earley L; Hardman M; Blethrow JD; Bui H; Specht A; Lange O; Denisov E; Makarov A; Horning S; Zabrouskov V Novel Parallelized Quadrupole/Linear Ion Trap/Orbitrap Tribrid Mass Spectrometer Improving Proteome Coverage and Peptide Identification Rates. Anal. Chem 2013, 85 (24), 11710–11714. 10.1021/ac403115c. [DOI] [PubMed] [Google Scholar]
- (6).Kelstrup CD; Jersie-Christensen RR; Batth TS; Arrey TN; Kuehn A; Kellmann M; Olsen JV Rapid and Deep Proteomes by Faster Sequencing on a Benchtop Quadrupole Ultra-High-Field Orbitrap Mass Spectrometer. J. Proteome Res 2014, 13 (12), 6187–6195. 10.1021/pr500985w. [DOI] [PubMed] [Google Scholar]
- (7).Scheltema RA; Hauschild J-P; Lange O; Hornburg D; Denisov E; Damoc E; Kuehn A; Makarov A; Mann M The Q Exactive HF, a Benchtop Mass Spectrometer with a Pre-Filter, High-Performance Quadrupole and an Ultra-High-Field Orbitrap Analyzer. Molecular & Cellular Proteomics 2014, 13 (12), 3698–3708. 10.1074/mcp.M114.043489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kelstrup CD; Bekker-Jensen DB; Arrey TN; Hogrebe A; Harder A; Olsen JV Performance Evaluation of the Q Exactive HF-X for Shotgun Proteomics. J. Proteome Res 2018, 17 (1), 727–738. 10.1021/acs.jproteome.7b00602. [DOI] [PubMed] [Google Scholar]
- (9).Yu Q; Paulo JA; Naverrete-Perea J; McAlister GC; Canterbury JD; Bailey DJ; Robitaille AM; Huguet R; Zabrouskov V; Gygi SP; Schweppe DK Benchmarking the Orbitrap Tribrid Eclipse for Next Generation Multiplexed Proteomics. Anal. Chem 2020, 92 (9), 6478–6485. 10.1021/acs.analchem.9b05685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Bermudez A; Pitteri SJ Enrichment of Intact Glycopeptides Using Strong Anion Exchange and Electrostatic Repulsion Hydrophilic Interaction Chromatography. In Mass Spectrometry of Glycoproteins; Delobel A, Ed.; Methods in Molecular Biology; Springer US: New York, NY, 2021; Vol. 2271, pp 107–120. 10.1007/978-1-0716-1241-5_8. [DOI] [PubMed] [Google Scholar]
- (11).Navarrete-Perea J; Yu Q; Gygi SP; Paulo JA Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)Proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3. J. Proteome Res 2018, 17 (6), 2226–2236. 10.1021/acs.jproteome.8b00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Shishkova E; Hebert AS; Westphall MS; Coon JJ Ultra-High Pressure (>30,000 Psi) Packing of Capillary Columns Enhancing Depth of Shotgun Proteomic Analyses. Anal. Chem 2018, 90 (19), 11503–11508. 10.1021/acs.analchem.8b02766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Schweppe DK; Eng JK; Yu Q; Bailey D; Rad R; Navarrete-Perea J; Huttlin EL; Erickson BK; Paulo JA; Gygi SP Full-Featured, Real-Time Database Searching Platform Enables Fast and Accurate Multiplexed Quantitative Proteomics. J. Proteome Res 2020, 19 (5), 2026–2034. 10.1021/acs.jproteome.9b00860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wenger CD; Phanstiel DH; Lee MV; Bailey DJ; Coon JJ COMPASS: A Suite of Pre- and Post-Search Proteomics Software Tools for OMSSA. Proteomics 2011, 11 (6), 1064–1074. 10.1002/pmic.201000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Kong AT; Leprevost FV; Avtonomov DM; Mellacheruvu D; Nesvizhskii AI MSFragger: Ultrafast and Comprehensive Peptide Identification in Mass Spectrometry–Based Proteomics. Nat Methods 2017, 14 (5), 513–520. 10.1038/nmeth.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Teo GC; Polasky DA; Yu F; Nesvizhskii AI Fast Deisotoping Algorithm and Its Implementation in the MSFragger Search Engine. J. Proteome Res 2021, 20 (1), 498–505. 10.1021/acs.jproteome.0c00544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Demichev V; Szyrwiel L; Yu F; Teo GC; Rosenberger G; Niewienda A; Ludwig D; Decker J; Kaspar-Schoenefeld S; Lilley KS; Mülleder M; Nesvizhskii AI; Ralser M Dia-PASEF Data Analysis Using FragPipe and DIA-NN for Deep Proteomics of Low Sample Amounts. Nat Commun 2022, 13 (1), 3944. 10.1038/s41467-022-31492-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Shteynberg DD; Deutsch EW; Campbell DS; Hoopmann MR; Kusebauch U; Lee D; Mendoza L; Midha MK; Sun Z; Whetton AD; Moritz RL PTMProphet: Fast and Accurate Mass Modification Localization for the Trans-Proteomic Pipeline. J. Proteome Res 2019, 18 (12), 4262–4272. 10.1021/acs.jproteome.9b00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Cox J; Mann M MaxQuant Enables High Peptide Identification Rates, Individualized p.p.b.-Range Mass Accuracies and Proteome-Wide Protein Quantification. Nat Biotechnol 2008, 26 (12), 1367–1372. 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- (20).Tyanova S; Temu T; Cox J The MaxQuant Computational Platform for Mass Spectrometry-Based Shotgun Proteomics. Nat Protoc 2016, 11 (12), 2301–2319. 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- (21).Hebert AS; Richards AL; Bailey DJ; Ulbrich A; Coughlin EE; Westphall MS; Coon JJ The One Hour Yeast Proteome. Molecular & Cellular Proteomics 2014, 13 (1), 339–347. 10.1074/mcp.M113.034769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Trujillo EA; Hebert AS; Brademan DR; Coon JJ Maximizing Tandem Mass Spectrometry Acquisition Rates for Shotgun Proteomics. Anal. Chem 2019, 91 (20), 12625–12629. 10.1021/acs.analchem.9b02979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Muehlbauer LK; Jen A; Zhu Y; He Y; Shishkova E; Overmyer KA; Coon JJ Rapid Multi-Omics Sample Preparation for Mass Spectrometry. Anal. Chem 2023, acs.analchem.2c02042. 10.1021/acs.analchem.2c02042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Li J; Cai Z; Bomgarden RD; Pike I; Kuhn K; Rogers JC; Roberts TM; Gygi SP; Paulo JA TMTpro-18plex: The Expanded and Complete Set of TMTpro Reagents for Sample Multiplexing. J. Proteome Res 2021, 20 (5), 2964–2972. 10.1021/acs.jproteome.1c00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Grinfeld D; Aizikov K; Kreutzmann A; Damoc E; Makarov A Phase-Constrained Spectrum Deconvolution for Fourier Transform Mass Spectrometry. Anal. Chem 2017, 89 (2), 1202–1211. 10.1021/acs.analchem.6b03636. [DOI] [PubMed] [Google Scholar]
- (26).Kelstrup CD; Aizikov K; Batth TS; Kreutzman A; Grinfeld D; Lange O; Mourad D; Makarov AA; Olsen JV Limits for Resolving Isobaric Tandem Mass Tag Reporter Ions Using Phase-Constrained Spectrum Deconvolution. J. Proteome Res 2018, 17 (11), 4008–4016. 10.1021/acs.jproteome.8b00381. [DOI] [PubMed] [Google Scholar]
- (27).He Y; Brademan DR; Hutchins PD; Overmyer KA; Coon JJ Maximizing MS/MS Acquisition for Lipidomics Using Capillary Separation and Orbitrap Tribrid Mass Spectrometer. Anal. Chem 2022, acs.analchem.1c05552. 10.1021/acs.analchem.1c05552. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data are available in MassIVE repository (MSV000091467).