

SUMMARY
Broadly neutralizing antibodies (bNAbs) against HIV can reduce viral transmission in humans, but an effective therapeutic will require unusually high breadth and potency of neutralization. We employ the OSPREY computational protein design software to engineer variants of two apex-directed bNAbs, PGT145 and PG9RSH, resulting in increases in potency of over 100-fold against some viruses. The top designed variants improve neutralization breadth from 39% to 54% at clinically relevant concentrations (IC80 < 1 μg/mL) and improve median potency (IC80) by up to 4-fold over a cross-clade panel of 208 strains. To investigate the mechanisms of improvement, we determine cryoelectron microscopy structures of each variant in complex with the HIV envelope trimer. Surprisingly, we find the largest increases in breadth to be a result of optimizing side-chain interactions with highly variable epitope residues. These results provide insight into mechanisms of neutralization breadth and inform strategies for antibody design and improvement.
Graphical Abstract
In brief
Broadly neutralizing antibodies against HIV are promising therapeutics and targets for vaccine elicitation. Using the OSPREY design software, Holt et al. design antibody variants with improved breadth and potency of virus neutralization. They solve bound structures for these variants and provide insight into mechanisms of breadth and potency.
INTRODUCTION
Broad and potent antibodies against HIV-1 show therapeutic promise for preventing viral transmission or infection1–4 and have been shown to suppress viremia in humans.5–8 The HIV-1 envelope (Env) apex, comprised of variable loops V1 and V2, is a common target site for anti-HIV-1 broadly neutralizing antibodies (bNAbs)9–11 despite the high antigen sequence variation at the V1V2 region12 and the presence of a protective glycan shield.13,14 These bNAbs form an important category15,16 that contains the PG917–21 and PGT14522–26 antibody classes, members of which (e.g., PGDM140023) are among the broadest and most potent HIV-1 NAbs thus far identified. Their utility as therapeutics or for prevention, however, would be improved if their potency and breadth were increased. Recent clinical data suggest an in vitro IC80 of <1 μg/mL for viral strains (measured by TZM-bl assay) to be associated with prevention of transmission in humans.8
Extensive structural characterization of bNAb lineages has suggested that breadth of neutralization is conferred by favorable interactions with conserved epitope features. Both the eponymous PG917,18 and PGT14522,24,25 monoclonal antibodies achieve neutralization breadth by targeting conserved structural features on the Env apex.17,27 PG9 uses its long, axe-like CDRH3 loop to form hydrogen bonds with the C strand of the Env V2 region in a parallel beta-strand conformation and also to interact productively with several apex glycans, including those at Env residues N160, N156, and, in some cases, N173.17 The beta-strand interaction allows PG9 to maintain favorable contacts with the V2 region despite variation in Env side-chain identities. We previously28 improved the potency of PG9 by incorporating mutations from the related PG16 antibody to improve interactions with hybrid-type glycans at N173, yielding the antibody PG9–16-RSH (herein, PG9RSH). PGT145 uses its long, needlelike CDRH3 loop to insert sulfated tyrosines into the Env apex hole24,25 to contact sites of conserved positive charge, both on the C strand and deeper beneath the surface of the Envtrimer.20,24,25 These strategies contrast with those used by members of the VRC38 class of antibodies (a member of the same V1V2 bNAb category), which rely on specific V2 C-strand side-chain interactions to neutralize HIV and, perhaps as a result, exhibit much narrower breadth of neutralization.29
The relationship between breadth and potency of neutralization is of considerable interest for antibody design. Although some studies have indicated that improving neutralization against a single antigen can lead to improved neutralization breadth,1,30–32 other evidence suggests the existence of a tradeoff between breadth and potency.33,34 Exploration of this relationship from a structural perspective is made more challenging by the relative scarcity of high-resolution structure information compared with the extreme antigenic diversity of targets like HIV. One reasonable hypothesis is that concomitant increases in potency and breadth require designing improved interactions with conserved antigen residues.
Methods for improving breadth and potency of antibodies against various targets including HIV-1, dengue, influenza, and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) have included affinity maturation,30–35 structure-based in silico design,32,36,37 sequence-based in silico design,38 and hybrid approaches.1,28,39,40 Previously, we have proposed a knockin humanized mouse model,41 surface matrix screening,42 and a structure-based matrix method43 to yield improved antibodies. In silico methods are attractive due to their ability to find improved variants without huge experimental effort and have found success in improving both breadth and potency of neutralization. Researchers have improved the breath and potency of cross-reactive anti-dengue,38 cross-reactive anti-influenza,37 and anti-HIV-132 antibodies using computational strategies. Hybrid methods that include in silico design have also achieved remarkable improvements.1
In this work, we design the PG9RSH and PGT145 anti-HIV-1 bNAbs for improved potency and breadth using our computational protein design software OSPREY.44 We use predicted potency for the BG505 strain as a proxy for predicted neutralization breadth, and we computationally optimize interactions with both conserved and non-conserved epitope residues. We present and characterize three bNAb single-mutation variants, compare them with both wild-type (PG9RSH and PGT145) and best-in-class (PGDM1400) antibodies, and observe measured improvements in breadth or potency relative to wild type. One of these three point mutations has been previously characterized in PG9 but not in PG9RSH.32 We determine cryoelectron microscopy (cryo-EM) structures for these three designed variants to provide atomic-level insight into increases in breadth and potency. The largest improvements in median potency (≈ 3-fold IC50, ≈ 4-fold IC80) occurred for PG9RSH variant DU025, which achieves neutralization breadth and potency rivaling that of the best-in-class antibody PGDM1400. Surprisingly, we find that the largest improvements in breadth occurred for a variant that optimizes interactions with variable epitope residues. This variant, PGT145 DU303, lost subtype potency for clade B but nonetheless improved overall breadth of neutralization from 39% (wild type) to 54% at clinically relevant concentrations (IC80 < 1 μg/mL). For this designed antibody, we observed increases in potency of >100-fold for six pseudoviruses across five clades. Moreover, the median improvement in IC80 (across 208 strains) was over 3-fold.
RESULTS
OSPREY predicts mutations with improved affinity for PGT145 and PG9RSH
We used OSPREY to design variants of the PGT145 and PG9 antibodies using structures of each antibody bound to the BG505 SOSIP Env trimer (PDB: 5U1F, 5VJ6). We predicted the effect of antibody mutations on antigen-binding affinity by computing K*scores45–48 for both wild-type and mutant antibodies at 9 positions (Table S1), pruning or computing scores for approximately 14,000 sequences. An increase in score relative to wild type predicts an increase in Ka , i.e., improved binding. In general, antibody binding affinity for the Env spike correlates well with neutralization potency.49 These designs predicted that PGT145 N(100l)D (variant DU303), PG9RSH N(100f)Y, and PG9RSH Y(100k)D (variant DU025), among other substitutions, would improve the neutralization potency of PGT145 and PG9RSH.
Double-mutation designs of PGT145 at residues F(100d) and N(100l) predicted that negatively charged substitutions at position 100l would improve binding affinity (Figure S1; Table S2). To avoid destabilization of the antibody CDRH3 conformation, sequences for which the lower bound on the partition function for the antibody state (ZAb ) was less than 1017:7 were excluded from consideration. Mutations N(100l)D, F(100d)H/N(100l)D, and N(100l)E were all predicted to increase the K*score. Notably, these substitutions place a negatively charged side chain at residue 100l, perhaps leveraging proximity to the (generally) positively charged Env residues 166 and 169 (see Figure 3C). Additional designs of PGT145 are discussed in Document S2.
Figure 1. Large-panel neutralization breadth and potency for PGT145 and PG9RSH variants.
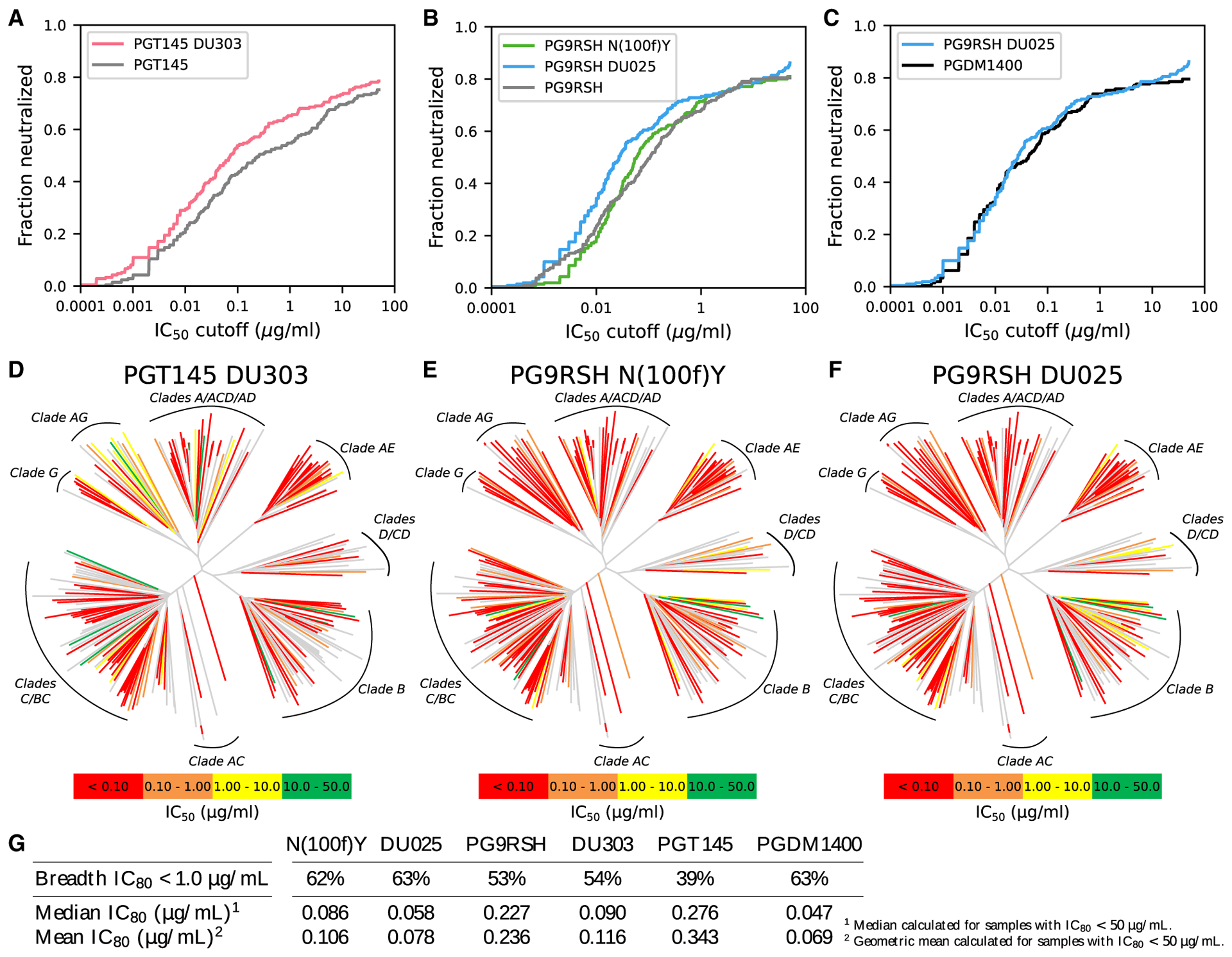
Neutralization breadth and potency of PGT145 and PG9RSH variants assayed on a panel of 208 pseudoviruses.
(A and B) Breadth/potency curves for PGT145 and PG9RSH variants and controls, respectively. Curves represent the fraction of pseudoviruses that were neutralized with IC50 smaller than the given cutoff. An increase in breadth and potency is indicated by a shift upward and left. PGT145 variant DU303 and PG9RSH variants N(100f)Y and DU025 improve breadth and potency relative to wild type. For IC80 curves, see Figure S3.
(C) Breadth/potency curves for PG9RSH DU025 and PGDM1400. Despite slightly weaker median and mean neutralization potency, the overall breadth and potency of DU025 rival that of the best-in-class PGDM1400 antibody.
(D–F) Neutralization dendrograms for variants DU303, PG9RSH N(100f)Y, and DU025, and respectively. Pseudoviruses are grouped into clades by sequence similarity, forming a tree graph. Internal branches in the tree, which denote groups of viruses, are colored gray. Terminal branches, corresponding to a single pseudovirus, are colored by IC50, where high neutralization potency is indicated by warm colors, low potency is indicated by cool colors, and lack of neutralization is indicated by gray.
(G) Summary of large-panel neutralization breadth and potency for variants and controls, measured by IC80.
A single-residue design of PG9RSH at residue N(100f) predicted that substitutions to Trp, Met, Tyr, His, or Phe would improve binding affinity (Figure S2A; Table S3). The mutation N(100f)Y was discovered previously and was shown to improve breadth and potency of neutralization in PG9.50 An additional single-residue design of PG9RSH at residue Y(100k) predicted that large or negatively charged substitutions (Trp, Asp, or Glu) would improve binding affinity (Figure S2B; Table S4). Additional designs of PG9RSH are discussed in Document S2.
Neutralization assessment reveals improvements in breadth and potency
Based on these designs, we selected 10 and 34 variants of antibodies PGT145 and PG9RSH, respectively, for small-panel neutralization assays (Tables S5 and S6; Data S1). These variants were selected by prioritizing variants with high K*scores and (to a lesser extent) high unbound-state partition functions,51 and in some cases, promising mutations from different designs were combined. We then selected variants DU303 (PGT145 N(100l)D), PG9RSH N(100f)Y, and DU025 (PG9RSH Y(100k)D) for assay against a large panel of 208 pseudoviruses to further characterize their potency and breadth of neutralization (Data S2). These variants were selected based on the number of pseudoviruses neutralized with an IC80 ≤50 μg/mL, the median IC80 value, the number of pseudoviruses neutralized with an IC50 ≤ 50 μg/mL, and the median IC50 value (listed in order of importance). For example, variants N(100f)Y and DU017 were selected over DU014 because DU014 performs comparatively poorly as measured by IC80. Although its IC50 neutralization breadth appears to be greater, the additional neutralized virus is neutralized relatively poorly (IC50 = 36.1 μg/mL) and disappears when measured using IC80. Furthermore, the median IC50 and IC80 values of DU014 are greater than for N(100f)Y (less potent). Additionally, the sequence and neutralization diversity of the set of variants to be characterized was considered. DU303, DU025, and PG9RSH N(100f)Y improved over wild-type activity in both breadth and potency of neutralization, although PG9RSH N(100f)Y achieved only small increases in breadth.
DU303 neutralized more pseudovirus strains with higher potency (Figure 1A). DU303 increased neutralization potency against the BG505 strain by 3- and 25-fold as measured by IC50 and IC80, respectively: IC50 decreased from 0.010 to 0.003 μg/mL, and IC80 decreased from 0.253 to 0.010 μg/mL (Data S2). Median neutralization potency across the large panel increased by 2- and 3-fold (IC50 and IC80): median IC50 decreased from 0.053 to 0.024 μg/mL, and median IC80 decreased from 0.276 to 0.090 μg/mL (Tables S7 and S8). DU303 improved neutralization breadth relative to PGT145: the percentage of tested pseudoviruses with measurable neutralization (IC50 < 50μg/mL) increased from 75% to 79% (Table S7). Interestingly, the improvement in breadth relative to PGT145 was more pronounced when evaluated at a cutoff with clinical relevance8: the percentage of viruses neutralized with IC80 < 1 μg/mL increased from 39% to 54% (Table S8).
PG9RSH N(100f)Y increased median potency of neutralization but only slightly increased breadth (Figure 1B). PG9RSH N(100f) Y increased neutralization potency against the BG505 strain by 2.8-fold as measured by IC80 but showed no appreciable change in IC50: IC80 decreased from 0.065 to 0.023 μg/mL, and IC50 remained at 0.009 μg/mL (Data S2). Median neutralization potency across the large panel increased by 1.9- and 2.6-fold (IC50 and IC80): median IC50 decreased from 0.047 to 0.025 μg/mL, and median IC80 decreased from 0.227 to 0.086 μg/mL (Tables S7 and S8). PG9RSH N(100f)Y slightly improved neutralization breadth relative to PG9RSH: the percentage of tested pseudoviruses with measurable neutralization (IC50 < 50 μg/mL) increased from 81% to 83% (Table S7). However, the improvement in breadth relative to PG9RSH was larger when evaluated for pseudoviruses neutralized with an IC80 < 1 μg/mL: breadth increased from 53% to 62% (Table S8).
DU025 increased potency and breadth of neutralization (Figure 1B), and the resulting breadth-potency plot is qualitatively similar to that for the best-in-class PGDM1400 antibody (Figure 1C). DU025 increased neutralization potency against the BG505 strain by 2.2- and 6.5-fold as measured by IC50 and IC80, respectively: IC50 decreased from 0.009 to 0.004 μg/mL, and IC80 decreased from 0.065 to 0.010 μg/mL. Median neutralization potency across the large panel increased by 2.7- and 3.9-fold (IC50 and IC80): median IC50 decreased from 0.047 to 0.017 μg/mL, and median IC80 decreased from 0.227 to 0.058 μg/mL (Tables S7 and S8). DU025 also improved neutralization breadth relative to PG9RSH: the percentage of tested pseudoviruses with measurable neutralization (IC50 < 50 μg/mL) increased from 81% to 87% (Table S7). Again, the improvement in breadth relative to PG9RSH was larger when evaluated at the clinically relevant cutoff of IC80 < 1 μg/mL: breadth increased from 53% to 63% (Table S8). While the mean and median potencies for DU025 remained slightly weaker than for PGDM1400, overall, the breadth and potency of DU025 rivaled that of best-in-class antibodies.
Strains with positively charged side chains at Env residue 169 are neutralized more potently by DU303
To further characterize the changes in neutralization activity for variant antibodies relative to PGT145 and PG9RSH, we computed the fold decrease in IC50 for each tested pseudovirus (Figure S4). We observed increases in potency of >100-fold for seven virus-antibody pairs across five clades, including 988-, 643-, and 228-fold improvements in IC50 for strains CAP256.206.C9, 16936–2.21, and CH038.12, respectively (Table S9). Analysis revealed a wide distribution of improvements in neutralization for DU303, with populations of strains with either slightly reduced or greatly improved neutralization. Conversely, both PG9RSH N(100f)Y and DU025 were characterized by a narrow distribution of fold decrease in IC50 over tested pseudoviruses. The largest fold decrease in IC50 for DU303 was 988-fold, occurring for the clade C pseudovirus CAP256.206.C9. The largest improvements in neutralization for PG9RSH N(100f)Y and DU025 were 63- and 110-fold, respectively, both occurring for the clade C pseudovirus ZM233.6. Examination of the fold change in neutralization by clade (Figures S5A, S6, and S7C) revealed the improvements in breadth and potency for DU303 to be non-uniform: marked improvements are evident for clades C/BC, A/AD, and D/CD, but decreases in neutralization occurred for clade B. On the other hand, the same analysis for both PG9RSH N(100f)Y (Figures S5B, S6, and S7A) and DU025 (Figures S5C, S6, and S7B) revealed most clades to show slight increases in neutralization with no clear pattern of decreased neutralization.
A gradient-boosted trees classifier predicted the changes in neutralization of pseudoviruses between wild-type PGT145 and DU303 and indicated these changes to be associated with the amino acid identity at Env residue 169. To investigate Env sequence features that may explain changes in neutralization for DU303, PG9RSH N(100f)Y, and DU025 relative to their ancestors, we trained gradient-boosting tree models (similar to the approach in Rawi et al.52) to predict the sign of the change in neutralization for each variant based on pseudovirus Env sequences. Models were evaluated using repeated 10-fold nested cross-validation (Table S10; Figure S8). The model for DU303 performed well, with a mean area under the curve (AUC) of 0:807 ± 0:128, but models for PG9RSH N(100f)Y and DU025 performed poorly, with mean AUCs of 0:521 ± 0:134 and 0:571 ± 0:134, respectively. To identify residues on Env that were important for the improved neutralization of DU303, we evaluated the permutation importance (PI)53 of Env residues. K169 and R169 were the important (PI > 0.05) features that were associated with an increase in neutralization for DU303 relative to PGT145 (Figure S9). Separating the large-panel data by residue identity at Env residue 169 revealed two distinct populations of viruses (Figure S10): Viruses with a positively charged residue at Env residue 169 were more potently neutralized by DU303 than by PGT145, while viruses without a positively charged residue at this position were less potently neutralized by DU303. We propose a structural mechanism for this observation in Document S2 supported by homology modeling.
Cryo-EM structures of BG505 DS-SOSIP.664 bound by DU303, PG9RSH N(100f)Y, and DU025 reveal improved side-chain interactions
We solved cryo-EM structures of PGT145 variant DU303, PG9RSH N(100f)Y, and PG9RSH variant DU025 in complex with the BG505 DS-SOSIP.664 Env trimer (Figure 2). Three-dimensional reconstructions yielded resolutions of 3.58, 3.40, and 3.75 Å , respectively (Table S11), and local resolutions ranged between 3 and 9.3, 2.7 and 6.3, and 3.18 and 7.5 Å , respectively (Figures S11–S13). The trimer apex and antibody CDRH3 loop were well resolved in all cases and indicated binding modes consistent with previous structures of PGT145 and PG9RSH (Figure 2). These structures revealed details for key interactions between the HIV Env apex and the DU303, PG9RSH N(100f)Y, and DU025 variant antibodies. An extended structural analysis can be found in Document S2.
Figure 2. Cryo-EM structures of PG9RSH and PGT145 variants in complex with BG505 DS-SOSIP Env trimer.
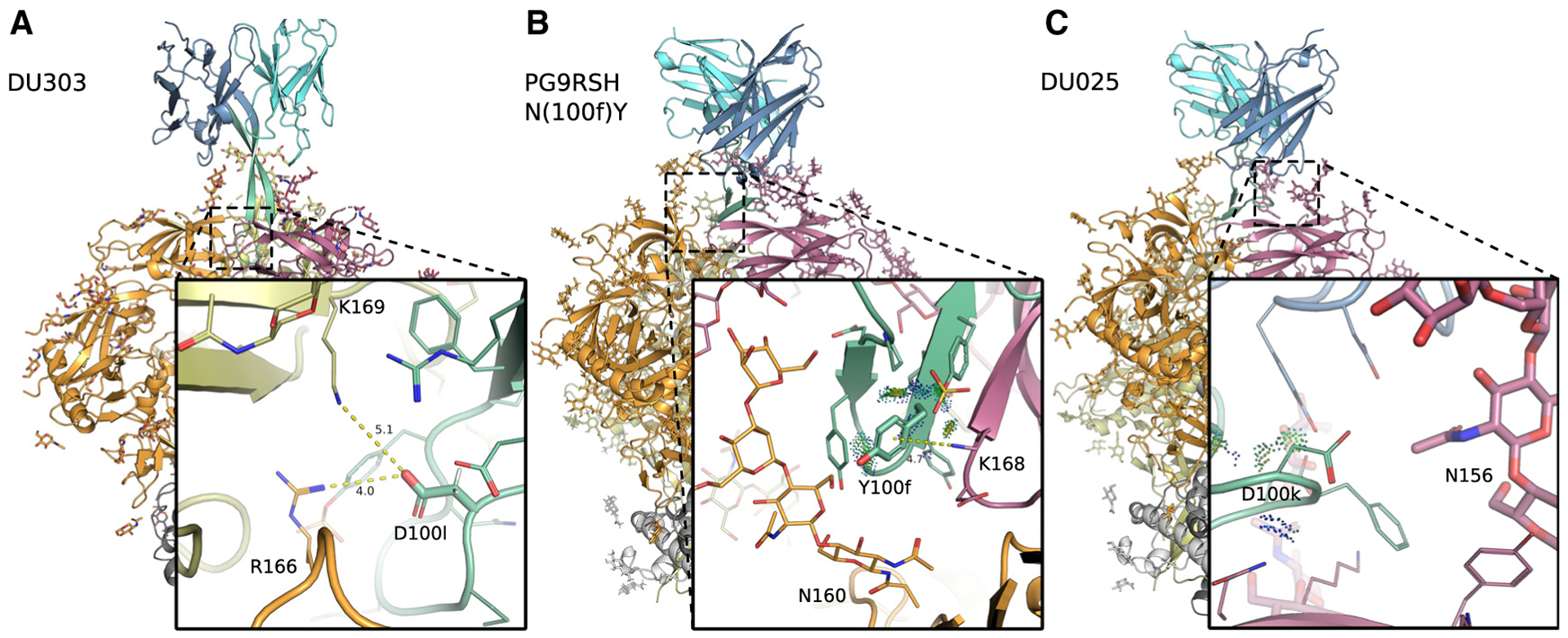
Backbone shown in ribbon representation with glycans, and amino acids shown as sticks or lines. Env subunits colored with warm colors and grays, and antibodies shown in cool colors. CDRH3 (residues 95–102) is shown in green. Distances (Å) are shown with dotted yellow lines, and energetic interactions are shown with Probe dots. Members of the PG9RSH (A and B) and PGT145 lineages (C) interact with the trimer apex and are characterized by a negatively charged CDRH3 that is hammer-like or extended, respectively.
(A) PGT145 mutation N(100l)D forms more favorable electrostatic interactions with gp120.
(B) The PG9RSH N(100f)Y mutation creates interactions with a side-chain nitrogen from gp120 residue K168, forming geometry consistent with a π-cation interaction.
(C) The PG9RSH Y(100k)D mutation forms long-range interactions with polar and positively charged residues Q170 and K305.
DU303 improves side-chain interactions with HIV Env residues 166 and 169 by introducing the N(100l)D mutation. Cryo-EM maps show well-resolved electron density for gp120 residues R166 and K169 but reveal ambiguity in the precise side-chain placements of residues D(100l) and F(100d) (Figure S14). The atomic model of DU303 indicates that D(100l) could form electrostatic interactions with gp120 residues R166 and K169: the side-chain nitrogen of K169 lies 5.1 Å from a side-chain carboxyl oxygen of D(100l) (Figure 3D). Similarly, one of the side-chain nitrogens of R166 lies 4 Å from a side-chain carboxyl oxygen of D(100l). The position of these side chains suggests that the negatively charged D(100l) forms favorable interactions with positively charged residues on gp120 to improve breadth and potency of neutralization.
Figure 3. OSPREY design ensembles correctly predicted structural features for PG9RSH and PGT145 variants.
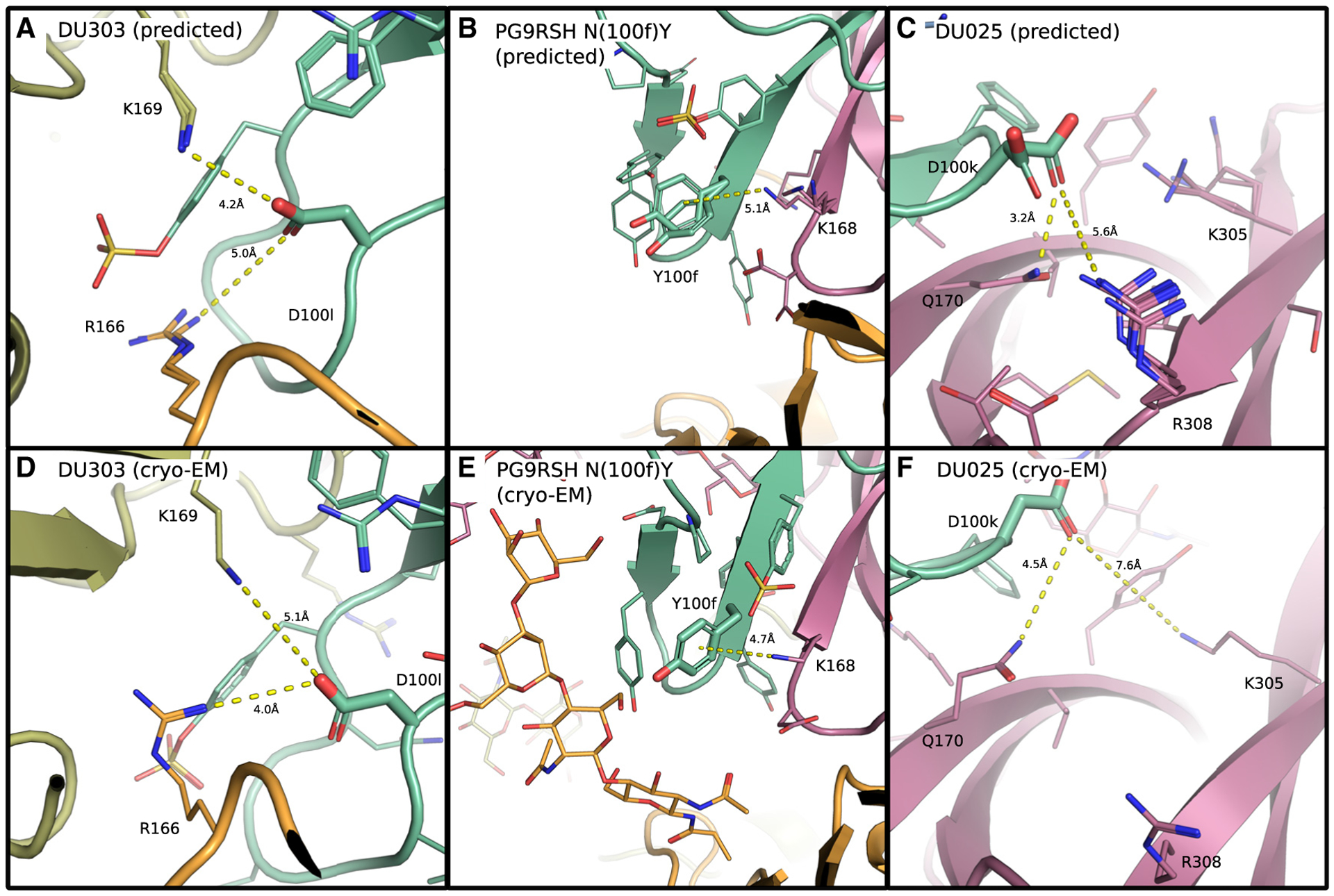
Ten members of the low-energy ensemble (LEE) predicted by OSPREY are shown for variants of PGT145 (A) and PG9RSH (B and C) above corresponding cryo-EM structures (D–F). Backbones are shown as ribbons with amino acids shown as lines or as sticks with Env subunits colored with warm colors, and antibody CDRH3 loops (residues 95–102) are shown in green. Distances (Å) are shown with dotted yellow lines.
(A) PGT145 mutation N(100l)D is predicted to form electrostatic interactions with gp120 residues R166 and K169. A carboxyl oxygen of D(100l) lies 5 and 4.2 Å from side-chain nitrogens of gp120 residues R166 and K169, respectively. Despite a lateral translation of the CDRH3 loop relative to the trimer apex, the LEE correctly predicts features of the experimental structure (F).
(B) The PG9RSH N(100f)Y mutation creates interactions with gp120 residue K168. The side-chain amino nitrogen of K168 lies 5.1 Å from the ring plane of Y(100f), forming geometry consistent with a π-cation interaction. The LEE correctly predicts interactions found in the experimentally determined structure (D).
(C) The PG9RSH Y(100k)D mutation is predicted to form electrostatic interactions with polar and charged residues on gp120. A carboxyl oxygen of D(100k) lies 3.2 Å from the side-chain nitrogen of Q170 and 5.6 Å from R308. The LEE correctly predicts interactions with Q170, but a translation and rotation of the CDRH3 loop places R308 further away (E).
PG9RSH N(100f)Y improves side-chain interactions with Env residue 168. Electron density maps show well-resolved density for gp120 residues D167, K168, and K169, along with the first two N-acetylglucosamine (GlcNAc) sugars of gp120 glycan N160 (Figure S15A). Density corresponding to bNAb residues is more ambiguous: peaks between the modeled side-chain locations of residues Y(100f) and Y(100a) suggest the presence of alternate rotamer configurations. Examination of low-density peaks (0.5 σ) reveals a small peak in density of the second GlcNAc of glycan N160 near the modeled location of Y(100f), suggesting interactions between Y(100f) and the glycan shield (Figure S15B). The atomic model of PG9RSH N(100f)Y fit to the density map indicates that the primary interaction between Y(100f) and gp120 is a π-cation interaction with residue K168 (Figure 3E). The ammonium nitrogen of K168 lies 4.7 Å from the center of the Y(100f) π system, and the angle between the distance vector and the ring normal vector is approximately 20°, which is representative of typical π-cation geometry.54 Y(100f) also forms van der Waals interactions with antibody residues P99, Y(100a), and TYS(100h). The side-chain geometry suggests that the aromatic Y(100f) side chain participates in a π-cation interaction with the positively charged K168 to improve potency of neutralization.
DU025 may improve long-range side-chain interactions or glycan interactions by introducing the Y(100k)D mutation. The electron density around the side chains of D(100k), Q170, K305, and Y173 are well resolved, along with the core of glycan N156 (Figure S16A). Interestingly, three unassigned density peaks arise in the groove between the V2 and V3 loops at both 1.2 and 3 σ, which could indicate the presence of solvent at this interface (Figure S16B). Furthermore, a bridge of density at 1 σ arises between the modeled locations of Env residues Q170 and R308, hinting at long-range or solvent-mediated interactions. These data suggest that residue D(100k) may form long-range or solvent-mediated interactions with residues Q170 and K305, which lie at distances of 4.1 and 6.8 Å , respectively, in the atomic model.
OSPREY predictions are validated by cryo-EM structures
OSPREY designs of antibody variants correctly predicted side-chain interactions. For DU303, predicted interaction distances between D(100l) and K169 and R169 in the OSPREY low-energy ensemble (LEE) differed by at most one angstrom from distances in the experimental model (Figures 3A and 3D). The side-chain orientations of this system were qualitatively similar between the LEE and the experimental model, indicating that OSPREY correctly predicted the structural consequences of the N(100l) D substitution. For PG9RSH N(100f)Y, side-chain locations in the LEE were qualitatively similar to those in the experimental model (Figures 3B and 3E). Interestingly, multiple rotamers of Y(100f) and Y(100a) appeared in the LEE, resulting in a conformation in which these side chains have rotated and stacked. The overall correspondence between the LEE and the experimental model indicated that OSPREY correctly predicted the structural consequences of the N(100f)Y substitution. Designs of DU025 predicted interactions with Q170 and R308, but differences in loop backbone conformation resulted in a change of environment near residue 100k. As a result, although the design ensemble correctly predicted that D(100k) creates long-range electrostatic interactions and correctly predicted a favorable interaction with Q170 (Figures 3C and 3F), the predicted interaction between D(100k) and R308 is not supported by the cryo-EM structure. Instead, shifts in the backbone create interactions between D(100k) and K305. However, the overall quality and type of interactions formed by D(100k) in the design ensemble were in fact consistent with the experimental structure. For an extended analysis of structural correspondence between the LEE and cryo-EM structures, see Document S2.
DISCUSSION
In this work, we tested our ability to redesign apex-directed anti-HIV bNAbs for improved neutralization breadth using the OSPREY protein design software. Ideally, to model structural variation, we would incorporate high-resolution co-crystal structures of bNAb:Env complexes from all tested strains into the design process, but such structures are scarce and difficult to obtain. In fact, at the time of design, the BG505 strain was the only HIV genotype represented in experimental structures of PGT145 and PG9 complexed with trimeric Env. Additionally, complex structures for poorly neutralized strains may be difficult to solve due to affinity issues. Previous studies indicated that improving neutralization potency against a single virus strain, either by affinity maturation30 or by design,1,31,32 can result in improved neutralization breadth. Therefore, we used the predicted affinity for the BG505 DS-SOSIP.664 trimer as a proxy for neutralization breadth during the design process. Assessment on a panel of 208 Env pseudoviruses indicated that three designed variants exhibited improved neutralization breadth and potency. We solved structures of these three variants bound to the BG505 DS-SOSIP.664 trimer to investigate the mechanisms of improved neutralization potency. We additionally investigated relationships between Env epitope residue characteristics and neutralization potency to draw conclusions about mechanisms of improved breadth. Surprisingly, mutations that optimized interactions with variable epitope residues resulted in the largest improvements in breadth.
Single mutations improve antibody neutralization of BG505
Experimental characterization of bNAb variants showed that DU303, PG9RSH N(100f)Y, and DU025 improved or maintained neutralization potency for the BG505 pseudovirus, with the most notable improvements observed by IC80 measurements. Cryo-EM structures of each variant bound to the BG505 trimer indicate that OSPREY designs improved side-chain interactions. The PGT145 N(100l)D mutation (DU303) improved electrostatic interactions with the Env apex residues R166 and K169, improving charge complementarity. The PG9RSH N(100f)Y substitution created a p-cation interaction with Env residue K168 and may also interact with glycan N160. Improved stability of the CDRH3 loop has also been suggested for this mutation.32 Finally, the PG9RSH Y(100k)D mutation (DU025) improved side-chain interactions with the polar Env residue Q170 and glycan N156. The interface around residue D(100k) is difficult to resolve, which may be indicative of a mobile or solvent-accessible environment. Unassigned electron-density peaks suggest that D(100k) may also form solvent-mediated interactions with K305 and perhaps even R308. All three designs were successful in improving neutralization potency against BG505 by optimizing side-chain interactions. The general correspondence between OSPREY-generated design ensembles and cryo-EM structures also indicated that our algorithms accurately modeled both environment and side-chain interactions at the PG9RSH and PGT145 epitopes. This is interesting given the low resolution of the design input structures and may be due to the fact that our algorithms are more sensitive to the input backbone conformation than the side chains, which are more difficult to resolve experimentally.
bNAb variants show different patterns of improvement in neutralization breadth
The three best antibody variants improved neutralization breadth across a panel of 208 HIV pseudoviruses but differed in the pattern and extent of change in breadth. DU303 improved neutralization for most clades but sacrificed some subtype potency for clade B. PG9RSH N(100f)Y and DU025, on the other hand, increased neutralization in a relatively uniform manner across all clades. Interestingly, DU303 and DU025 improved overall breadth to a greater extent despite relatively low conservation of their Env epitope residues. Conversely, PG9RSH N(100f)Y resulted in smaller improvements despite the high conservation of Env residues that interact with the mutated antibody residue 100f (Figure S17).
PGT145 DU303 improved breadth by improving potency against “sensitive” strains containing a lysine or arginine at residue 169 while slightly decreasing potency against “resistant” strains with different substitutions at this epitope residue. Sensitive strains were more potently neutralized by ≈ 5-fold (geometric mean), while resistant strains were less potently neutralized by ≈ 2-fold (Figure S10). Because the effect of improving neutralization against sensitive strains was larger than the effect of decreasing neutralization against resistant strains, and because there were more sensitive strains than resistant strains, the aggregate effect of the N(100l)D substitution was a gain of breadth and potency. These observations explain the loss of subtype potency for clade B, in which Env residue 169 is predominantly hydrophobic (valine, methionine) and unlikely to interact favorably with D(100l).
PG9RSH DU025 improved breadth of neutralization by improving interactions with variable residues on the Env V2 and V3 loops and with the conserved glycan N156. Analysis across a large panel of pseudoviruses revealed no major decreases in subtype potency, despite the relative variability of the epitope residues in proximity to residue D(100k) (Figure S17). This could indicate that the Y(100k)D mutation improves breadth by improving interactions with the conserved glycan N156 or by improving interactions with the variable Env residues 170 and 305 in a manner that is tolerant of variation. Most panel strains have polar or charged residues at Env positions 170 (Q, K, or R) and 305 (K, R, or T), and it is possible that D(100k) interacts favorably with any of these amino acids, especially if interactions were to be solvent mediated.
PG9RSH N(100f)Y slightly improved breadth of neutralization by improving interactions with Env residue 168 and glycan N160. Overall, the slight improvement in breadth did not appear to sacrifice subtype potency, likely because the N(100f)Y substitution interacts with highly conserved Env features. The improvement in breadth was small relative to DU303 or DU025, suggesting that these interactions may already be highly optimized in parent PG9RSH.
Improvements in breadth did not require residue conservation
These examples confirm previous observations1,30–32 that antibody neutralization breadth can be increased by improving potency for a single “design antigen.” One intuitive explanation for this phenomenon is that the design antigen contains residues that are conserved across the entire antigen population. However, for our designs, epitope residue conservation did not appear to be critical for improving breadth. At least one variant (DU303) optimized interactions with epitope residues that were among the least conserved across the 208-strain test panel. Another (DU025) interacts with an epitope containing a conserved glycan, but structures suggest interactions with multiple non-conserved residues.
There are several possible explanations for increased breadth without epitope-residue conservation. First, the energetic benefit for strains with residues that match the BG505 sequence could be larger than the energetic cost for strains that do not match. For example, for DU303, strains containing Env K169 (like BG505) have improved electrostatic interactions with antibody residue D(100k). Although strains with Env V169 would not realize the same energetic benefit, they would be unlikely to suffer from large steric clashes because of the smaller size of valine relative to lysine. As a result, optimizing for design antigen residues would improve overall breadth, even though they are non-conserved. If this is correct, we would expect improvements in breadth to be less likely to occur when the design antigen contains small epitope residues at a design site.
Second, the presence of long-range, possibly solvent-mediated interactions in these designs could allow for favorable energetic interactions with multiple amino acids at epitope sites. As a result, the local chemical environment around design residues could be more conserved than the epitope amino acid identity. In this case, we would expect improvements in breadth to be less likely to occur when optimizing close-range interactions with the design antigen.
Finally, it is possible that natural selection has already optimized interactions with highly conserved residues. The difference between Env residue identity distributions for donor virus populations and test panels depends in part on residue conservation. If natural selection is effectively optimizing antibody residues for donor virus populations, we would expect improvements in test panel neutralization breadth to be larger or more likely when optimizing interactions with non-conserved epitope residues. Further research will be necessary to evaluate these hypotheses.
Comparison to previous work
Various other methods for computational antibody design have been proposed and implemented. Hybrid knowledge-based and computational-structure-based design methods like RAbD,55 OptCDR56 and OptMAVEn,57,58 and abDesign59 graft experimentally determined CDR loop backbone structures, then use computational structure-based design to optimize loop sequences. However, these approaches are likely most useful for de novo antibody design, and the variability of antibody CDRH3 loops makes these approaches suboptimal for antibodies that rely heavily on CDRH3 contacts for neutralization55 such as PG9RSH or PGT145. Deep learning approaches60 have been shown to model loops more effectively than some grafting methods. Straightforward applications of general protein design frameworks or algorithms like Rosetta32 or variants of dead-end elimination or A*36 have also been used to design antibodies in a more conservative manner. In some cases, modifications are necessary: Tidor and co-workers found that relying on electrostatic interactions improved the performance of their algorithms.36 Our approach—a straightforward application of OSPREY—is most similar to these latter works, although unlike the designs using Rosetta,32 our algorithms provably return optimal sequences and structures with respect to the input model. Finally, some approaches explicitly use multistate design to optimize for neutralization across a wide panel of antigens.37,61 However, this requires either a varied set of experimentally determined antibody:antigen structures or extensive homology modeling, which can degrade performance by inaccurately modeling important structural interactions. Although this is a promising future direction, our results and others30,32 indicate that improved neutralization breadth can also be achieved through design for potency against one representative antigen.
Future directions
Until more structures of bNAbs in complex with HIV Env trimers from diverse strains become available, methods for structure-based design for breadth will likely continue to rely on information gleaned from one or a few representative antigens. Can the method presented herein be applied to any antigen genotype or site to successfully improve breadth? If not, how can we differentiate between genotypes or sites that are more “canonical” (i.e., for which design for potency can result in increased breadth) versus those that are less? We have suggested several hypotheses based on our new data, but additional work will be required to evaluate these hypotheses. Orthogonally, our method could potentially be used not only for therapeutic antibody improvement but also to assess the projected importance of rare mutations in antibodies targeted for elicitation through vaccination or, alternatively, to identify important immunogen variants.
Limitations of the study
As mentioned above, due to the scarcity of trimeric structures, these designs relied exclusively on structures of antibodies bound to the BG505 SOSIP trimer. Additionally, like most other structures of bNAbs in complex with trimeric Env, our solved structures of improved antibody variants also include the BG505 SOSIP trimer. Although we attempt to mitigate these limitations by characterizing neutralization over a large set of strains and by homology modeling approaches, our understanding of the mechanisms of neutralization breadth for these antibodies and others would be improved by complex structures that span a more diverse set of stable Env trimers.62 Finally, neutralization potency and breadth are not themselves sufficient for therapeutic efficacy—other properties, including thermostability and lack of autoreactivity, are critical.1 Further characterization of the antibodies developed herein will be needed to assess their therapeutic potential.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Bruce R. Donald (brd+cell22@cs.duke.edu).
Materials availability
TZM-bl cells (https://www.aidsreagent.org, cat# 8129) are available through the NIH HIV Reagent Program, Division of AIDS, NIAID, NIH, contributed by Dr. John C. Kappes, Dr. Xiaoyun Wu and Tranzyme Inc.
Data and code availability
Neutralization data are included in this manuscript in the supplemental information. Cryo-EM maps and structures have been deposited in the EMDB (EMDB: EMD-299248, EMD-29264, EMD-29288) and the PDB (PDB: 8FK5, 8FL1, 8FLW), respectively, and are publicly available as of the date of publication.
All code used and discussed in this manuscript is available from the Harvard Dataverse repository (Harvard Dataverse: https://doi.org/10.7910/DVN/NXD2JR) as of the date of publication. For new empirical designs we recommend using the latest version of OSPREY available for free at http://www.cs.duke.edu/donaldlab/opsrey.php. All computer code for the OSPREY system is also available on GitHub at https://github.com/donaldlab/OSPREY3, and is open/source and free. The version of record is archived with Zenodo (Zenodo: https://doi.org/10.5281/zenodo.7633931).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
These cells are a HeLa cell line generated from JC.53 cells that expresses CD4, CCR5, and CXCR4, with galactosidase and luciferase reporter genes under the HIV-1 promoter. For long-term storage store at or below −100° C, preferably in FBS supplemented with 40% DMEM and 10% DMSO. Propagate in DMEM supplemented with 10% FBS, 100 U per mL penicillin and 0.1 mg per mL streptomycin, incubate at 37°C. For more information see the NIH AIDS Reagent Program (https://www.aidsreagent.org, cat# 8129).
METHOD DETAILS
Redesign of PG9RSH and PGT145
Designs to obtain improved variants of PG9RSH and PGT145 were performed by defining sets of accessible conformations (conformation spaces) for unliganded antibody, unliganded Env trimer, and complexed antibody:Env states, followed by approximation of binding affinity using the K* algorithm45,46,48 or an early version of the EWA* algorithm72 in OSPREY.
Conformation spaces were defined for PG9RSH or PGT145 based on an EM structure of the PG9 and 8ANC195 bNAbs in complex with the BG505 SOSIP.664 Env trimer (PDB: 5VJ6)73 or a Cryo-EM structure of PGT145 and sCD4 in complex with the DS-SOSIP.664 (Based on PDB: 5U1F),25 respectively. The structure of PGT145 contained modeled locations of amino acid side chains and glycans, which were not present in the deposited structure due to resolution limitations.
Structures were inspected to determine whether they were suitable for design or needed refinement. MolProbity71 analysis of the CDRH3 region of PG9 (5VJ6) revealed a few major clashes, one of which involved antibody backbone atoms, indicating that the atomic model may represent an inaccurate backbone conformation. As a result, the PG9 input structure was all-atom minimized using Yasara74 to relax steric clashes, and the 8ANC195 antibody was removed along with some distal regions of gp160. This step was performed due to the very low resolution of the structure, coupled with the evident backbone clashes. Similar analysis of the PGT145 structure based on 5U1F (with added side chains) showed only a few clashes, each of which involved only side chain conformations. We chose not to perform all-atom minimization of this structure, because we did not consider the relaxation of side chain conformations to be worth changing the backbone conformation without experimental evidence. The PGT145 input structure was modified by removing sCD4 along with parts of the PGT145 antibody and gp160 distal from peptide contacts of the PGT145:Env interface.
Hydrogens were added to both input structures using Reduce.75 Backbone coordinates for the complex were defined by the resulting modified PG9:Env and PGT145:Env structures, and coordinates for the unliganded antibody and unliganded Env states were obtained by removing atoms corresponding to the Env and antibody, respectively. Design residues (See Table S1) were modeled as continuously flexible48 in OSPREY, for which rotamers from the Penultimate Rotamer Library76 were allowed to adopt any side-chain conformation such that all χ;-angles are within ± 9° of their modal c-angles. All other side-chain coordinates were obtained from the input structures. Rotamers and energetic interactions for sulfated tyrosines were modeled using the methods reported in Wang.77 Briefly, a rotamer library was constructed, partial charges and force-field parameters were computed with Antechamber in AMBER,78 and solvation parameters were computed using an extended version of the EEF1 solvation model. For each model we computed ε-approximate bounds on the K score to a guaranteed accuracy51 of ε<0:683 using the K*45,46,48 or EWAK*72 algorithms.
Antibody variant expression and purification
DNA sequences of heavy and light chain variable regions of antibodies PG9RSH and PGT145 and variants were synthesized and subcloned into the pVRC8400 vector. For antibody expression, equal amounts of antibody heavy and light chain plasmid DNA were transfected into Expi293 cells using Turbo293 transfection reagent (Speed BioSystems). The transfected cells were incubated in shaker incubator at 120 rpm, 37° C, 9% CO2. The culture supernatants were harvested, filtered, and loaded on a protein A (GE Healthcare) column at 5 days post transfection. After washing the column with PBS, each antibody was eluted with an IgG elution buffer (Pierce) and immediately neutralized with one-tenth volume of 1M Tris-HCl pH 8.0. Eluted antibodies were dialyzed against PBS overnight and were confirmed by SDS-PAGE before use.
Pseudovirus neutralization assays
Antibody neutralization was evaluated with the single-round infection assay of TZM-bl cells.79 Antibodies were serially diluted into wells of a 384-well plate, a constant amount of pseudovirus was added, plates were incubated for 60 min, and TZM-bl cells, which cells express luciferase upon viral infection, were added. Plates were incubated for 48 h, lysed, and measured for luciferase activity. The antibody concentration required to achieve 50% neutralization of infection (IC50) was calculated using a dose-response curve fit with a 5-parameter nonlinear function. For small-panel neutralization assays we used a panel of 10 HIV-1 Env pseudoviruses from clades A, B, and C. For large-panel neutralization assays we used a previously described panel9,29,80 of 208 geographically and genetically diverse HIV-1 Env pseudoviruses representing the major subtypes and circulating recombinant forms. All IC50 values reported here are from small (10 viruses) or large (complete set of 208 viruses) neutralization panels run at the VRC. In some cases, multiple runs were averaged. We report both the potency (measured as the median or geometric mean IC50 or IC80 for strains with measurable neutralization) and the breadth of neutralization (the number or percentage of strains with measurable neutralization). These summary statistics were computed in this way to conform to the literature standard and to enable straightforward comparison. Sources of error include the fact that neutralization IC50 values are known to vary up to 3-fold between repeat assays.80
Cryo-EM data collection, structure determination, and refinement
The BG505 DS-SOSIP.664 Env trimer was incubated with molar excess of antigen-binding fragment (Fab) for each of the improved V2-apex directed antibodies. Grids were prepared by depositing 2 μL of each complex at 2 mg/mL final concentration on C-flat 1.2/1.3 grids (emsdiasum.com) and vitrified with an FEI Vitrobot Mark IV with a wait time of 30 s, blot time of 3 s, and blot force of 1. Data collections were performed on a Titan Krios electron microscope with Leginon using a Gatan K3 direct detection device. Exposures 2 were collected in movie mode for 2 s with the total dose of 63.75 e°−/Å2 fractionated over 40 raw frames. cryoSPARC v3.1 was used for frame alignment, CTF estimation, 2D classifications, ab initio 3D reconstruction, homogeneous refinement, and nonuniform 3D refinement. 3D reconstruction and final refinements were performed using C1 symmetry.
Coordinates from PDB: 5V8L and PDB: 3U4E were used for initial fits to the reconstructed maps. This was followed by simulated annealing and real space refinement in Phenix v1.19 with the sharpened map from cryoSPARC v3.1 and with a density modified map from Phenix Resolve and manually fit with Coot v0.9.8 and then improved through iterative rounds. Geometry and map fitting parameters were evaluated using Molprobity v4.5.1 and EMRinger. Maps and structures were deposited to the EMDB (EMDB: EMD-29248, EMD-29264, EMD-29288) and PDB (PDB: 8FK5, 8FL1, 8FLW).
QUANTIFICATION AND STATISTICAL ANALYSIS
Predicting change in neutralization from Env sequence
We modified a previously reported method for predicting bNAb neutralization from Env sequence52 to predict the consequences of bNAb single mutations (increased or decreased neutralization). We constructed models using gradient-boosted decision trees in scikit-learn,81 which uses a boosting approach to construct ensemble models of CART decision trees.
To define labels corresponding to the change in neutralization relative to wild-type for each antibody we computed the log-ratio of neutralization for each antibody (DU303, PG9RSH N(100f)Y, DU025) and its corresponding ancestor (PGT145, PG9RSH): We defined labels y for binary classification where y = 1 if z>0, y = 0 otherwise. We processed Env using BioPython.82 To generate Env sequence features X we first augmented the Env protein sequences by identifying potential N-glycosylation sites, defined as sites containing the amino acid motif N-X-S/T, where X represents any amino acid. This resulted in 957 categorical features with an alphabet size of 21. Final features were obtained by one-hot encoding, resulting in a total of 4939 binary features.
For training we optimized three hyperparameters, leaving the rest at default values. We used an early stopping criterion implemented in sci-kit learn for training: 10% of the training data was held out as an additional validation set, and training was halted if the score on the validation set did not improve for a user-specified number of iterations. We optimized the maximum depth of the CART decision trees in the ensemble, the “learning rate” - a scaling of the contribution of each decision tree to the overall decision function, and the number of iterations of no improvement required for the early stopping criterion. Hyperparameters were optimized by 10-fold cross-validation (repeated 5 times) and parameters were selected by computing the average accuracy, AUC, or F1 score on the validation set.
Variable importance, measured by mean decrease in impurity (MDI) and permutation importance (PI), was evaluated for DU303 on a model trained using the entire available dataset. The MDI variable importance measure is analogous to the Gini importance - for each feature its MDI is defined as the average decrease in impurity over all nodes that correspond to the feature.83 In this case our splitting criterion is the Friedman Mean-Squared Error, Equation 35 in Friedman.83 The MDI importance was computed using the scikit-learn implementation. PI was computed by randomly permuting each feature and then computing the difference in loss between using scrambled and original features using the scikit-learn implementation (sklearn.inspection.permutation_importance).
Visualization and figure generation
Structure and density was visualized using PyMOL, USCF Chimera,65 and King,64 and images were generated with PyMOL. Analysis of neutralization data was performed using Python, and accompanying figures were generated using the Matplotlib84 and Seaborn85 libraries.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |
|---|---|---|---|
| Antibodies | |||
| PG9RSH-(DU001 – DU010, N(100fY), DU012 – DU034) | NIH/VRC, this paper | N/A | |
| PGT145-(DU301 – DU310) | NIH/VRC, this paper | N/A | |
| Monoclonal anti-HIV-1 Env PGT145 | NIH AIDS Reagent Program22 | CAT# 12703, RRID: AB_2491054 | |
| Monoclonal anti-HIV-1 Env PG9–16-RSH | NIH/VRC28 | N/A | |
| Monoclonal anti-HIV-1 Env PGDM1400 | Dennis R. Burton, Scripps23 | N/A | |
| Bacterial and virus strains | |||
| VRC 208 virus panel | NIH/VRC63 | N/A | |
| VRC 10 virus panel (PGT145) | NIH/VRC | N/A | |
| VRC 10 virus panel (PG9RSH) | NIH/VRC | N/A | |
| Chemicals, peptides, and recombinant proteins | |||
| BG505 DS-SOSIP.664 | NIH/VRC | N/A | |
| Critical commercial assays | |||
| Turbo293 Ab Transfection Kit | Speed Biosystems | Cat# PXX1005 | |
| Human Antibody Capture Kit | GE Healthcare Life Sciences | Cat# BR-1008–39 | |
| Deposited data | |||
| Cryo-EM map: PG9RSH N(100f)Y in complex with BG505 Env Trimer | EMDB | 29248 | |
| Cryo-EM structure: PG9RSH N(100f)Y in complex with BG505 Env Trimer | PDB | 8FK5 | |
| Cryo-EM map: PG9RSH DU025 in complex with BG505 Env Trimer | EMDB | 29264 | |
| Cryo-EM structure: PG9RSH DU025 in complex with BG505 Env Trimer | PDB | 8FL1 | |
| Cryo-EM map: PGT145 DU303 in complex with BG505 Env Trimer | EMDB | 29288 | |
| Cryo-EM structure: PGT145 DU303 in complex with BG505 Env Trimer | PDB | 8FLW | |
| Code required for replication of designs and analysis | Harvard Dataverse | https://doi.org/10.7910/DVN/NXD2JR | |
| Experimental models: Cell lines | |||
| TZM-bl cells | NIH AIDS Reagent Program | Cat# 8129 | |
| Expi293F cells | ThermoFisher Scientific Inc | Cat# A14527 | |
| Recombinant DNA | |||
| pVRC8400 | https://www.addgene.org/ | Cat# 63160 | |
| pVRC8400-PG9RSH-(DU001 – DU034) Heavy | NIH/VRC, this paper | N/A | |
| pVRC8400-PG9RSH-(DU001 – DU034) Light | NIH/VRC, this paper | N/A | |
| pVRC8400-PGT145-(DU301 – DU310) Heavy | NIH/VRC, this paper | N/A | |
| pVRC8400-PGT145-(DU301 – DU310) Light | NIH/VRC, this paper | N/A | |
| Software and algorithms | |||
| OSPREY | Hallen et al.44 | https://github.com/donaldlab/OSPREY3 | |
| The PyMol Molecular Graphics System | Schrodinger, LLC | https://pymol.org/2/ | |
| KING | Chen et al.64 | https://github.com/rlabduke/javadev | |
| USCF Chimera | Pettersen et al.65 | https://www.cgl.ucsf.edu/chimera/ | |
| Leginon | Suloway et al.66 | https://sbgrid.org/software/titles/leginon | |
| cryoSPARC | Punjani et al.67 | https://cryosparc.com/ | |
| EMRinger | Barad et al.68 | https://github.com/fraser-lab/EMRinger | |
| Phenix | Adams et al.69 | https://sbgrid.org/software/ | |
| Coot | Emsley and Cowtan.70 | https://sbgrid.org/software/ | |
| MolProbity | Williams et al.71 | http://molprobity.biochem.duke.edu/ | |
Highlights.
Antibody variants designed using OSPREY improve neutralization breadth and potency
Structural and statistical analyses highlight improved side-chain interactions
Improvements in breadth result from interaction with variable epitope residues
ACKNOWLEDGMENTS
We thank J. Baalwa, D. Ellenberger, F. Gao, B. Hahn, K. Hong, J. Kim, F. McCutchan, D. Montefiori, L. Morris, E. Sanders-Buell, G. Shaw, R. Swan-strom, M. Thomson, S. Tovanabutra, C. Williamson, and L. Zhang for contributing the HIV-1 Env plasmids used in our neutralization panel and N. Jean-Baptiste, R. Carroll, B. Flach, K. McKee, C. Moore, G. Padilla, J. Rathmann, S. O’Connell, S. O’Dell, S.D. Schmidt, C. Whittaker, and A.B. McDermott for assistance with neutralization assessments on the 208-strain panel. We thank J.D. Jou, A. Ojewole, and other members of the Donald lab for helpful discussions of antibody design strategies, and we thank A. Bartesaghi and J. Richardson for advice and guidance on the interpretation of cryo-EM densities. We thank Catherine Ehrhart for her invaluable help in producing models of antibody:Env complexes using AlphaFold. We gratefully acknowledge grant support from the NIH (R01 GM078031, R01 GM118543, and R35 GM144042 to B.R.D.). VRC research was funded by the Intramural Research Program of the Vaccine Research Center, NIAID, NIH. Some of this work was performed at the Simons Electron Microscopy Center and National Resource for Automated Molecular Microscopy located at the New York Structural Biology Center, supported by grants from the Simons Foundation (SF349247), NYSTAR, and the NIH National Institute of General Medical Sciences (GM103310).
Footnotes
DECLARATION OF INTERESTS
B.R.D. and M.S.F. are founders of Ten63 Therapeutics. B.R.D., S.W., A.U.L., G.T.H., M.S.F., P.D.K., J.G., and N.A.D. are inventors on a patent application filed by Duke University.
SUPPLEMENTAL INFORMATION
supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112711.
REFERENCES
- 1.Rudicell RS, Kwon YD, Ko SY, Pegu A, Louder MK, Georgiev IS,Wu X, Zhu J, Boyington JC, Chen X, et al. (2014). Enhanced Potency of a Broadly Neutralizing HIV-1 Antibody In Vitro Improves Protection against Lentiviral Infection In Vivo. J. Virol 88, 12669–12682. 10.1128/JVI.02213-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Julg B, Tartaglia LJ, Keele BF, Wagh K, Pegu A, Sok D, Abbink P, Schmidt SD, Wang K, Chen X, et al. (2017). Broadly neutralizing antibodies targeting the HIV-1 envelope V2 apex confer protection against a clade C SHIV challenge. Sci. Transl. Med 9, eaal1321. 10.1126/scitranslmed.aal1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pegu A, Borate B, Huang Y, Pauthner MG, Hessell AJ, Julg B, Do-ria-Rose NA, Schmidt SD, Carpp LN, Cully MD, et al. (2019). A Meta-analysis of Passive Immunization Studies Shows that Serum-Neutralizing Antibody Titer Associates with Protection against SHIV Challenge. Cell Host Microbe 26, 336–346.e3. 10.1016/j.chom.2019.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mahomed S, Garrett N, Baxter C, Abdool Karim Q, and Abdool Karim SS (2021). Clinical Trials of Broadly Neutralizing Monoclonal Antibodies for Human Immunodeficiency Virus Prevention: A Review. J. Infect. Dis 223, 370–380. 10.1093/infdis/jiaa377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trkola A, Kuster H, Rusert P, Joos B, Fischer M, Leemann C, Man-rique A, Huber M, Rehr M, Oxenius A, et al. (2005). Delay of HIV-1 rebound after cessation of antiretroviral therapy through passive transfer of human neutralizing antibodies. Nat. Med 11, 615–622. 10.1038/nm1244. [DOI] [PubMed] [Google Scholar]
- 6.Ng CT, Jaworski JP, Jayaraman P, Sutton WF, Delio P, Kuller L,Anderson D, Landucci G, Richardson BA, Burton DR, et al. (2010). Passive neutralizing antibody controls SHIV viremia and enhances B cell responses in infant macaques. Nat. Med 16, 1117–1119. 10.1038/nm.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caskey M, Klein F, Lorenzi JCC, Seaman MS, West AP, Buckley N, Kremer G, Nogueira L, Braunschweig M, Scheid JF, et al. (2015). Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature 522, 487–491. 10.1038/nature14411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corey L, Gilbert PB, Juraska M, Montefiori DC, Morris L, Karuna ST, Edupuganti S, Mgodi NM, deCamp AC, Rudnicki E, et al. (2021). Two Randomized Trials of Neutralizing Antibodies to Prevent HIV-1 Acquisition. N. Engl. J. Med 384, 1003–1014. 10.1056/NEJMoa2031738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Georgiev IS, Doria-Rose NA, Zhou T, Kwon YD, Staupe RP, Mo-quin S, Chuang GY, Louder MK, Schmidt SD, Altae-Tran HR, et al. (2013). Delineating antibody recognition in polyclonal sera from patterns of HIV-1 isolate neutralization. Science 340, 751–756. 10.1126/science.1233989. [DOI] [PubMed] [Google Scholar]
- 10.Landais E, Huang X, Havenar-Daughton C, Murrell B, Price MA,Wickramasinghe L, Ramos A, Bian CB, Simek M, Allen S, et al. (2016). Broadly Neutralizing Antibody Responses in a Large Longitudinal Sub-Saharan HIV Primary Infection Cohort. PLoS Pathog. 12, e1005369. 10.1371/journal.ppat.1005369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walker LM, Simek MD, Priddy F, Gach JS, Wagner D, Zwick MB, Phogat SK, Poignard P, and Burton DR (2010). A Limited Number of Antibody Specificities Mediate Broad and Potent Serum Neutralization in Selected HIV-1 Infected Individuals. PLoS Pathog. 6, e1001028. 10.1371/journal.ppat.1001028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foley B, Leitner T, Apetrei C, Hahn B, Mirachi I, Mullins J, Ram-baut A, Wolinsky S, and Korber B (2018). HIV Sequence Compendium 2018 Contracting Officer’s Representative Los Alamos HIV Sequence Database and Analysis Staff. Technical Report. https://www.hiv.lanl.gov/. [Google Scholar]
- 13.Lee JH, Ozorowski G, and Ward AB (2016). Cryo-EM structure of anative, fully glycosylated, cleaved HIV-1 envelope trimer. Science 351, 1043–1048. 10.1126/science.aad2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stewart-Jones GBE, Soto C, Lemmin T, Chuang GY, Druz A,Kong R, Thomas PV, Wagh K, Zhou T, Behrens AJ, et al. (2016). Trimeric HIV-1-Env Structures Define Glycan Shields from Clades A, B, and G. Cell 165, 813–826. 10.1016/j.cell.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwong PD, and Mascola JR (2012). Human Antibodies that NeutralizeHIV-1: Identification, Structures, and B Cell Ontogenies. Immunity 37, 412–425. 10.1016/j.immuni.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwong PD, and Mascola JR (2018). HIV-1 Vaccines Based on Antibody Identification, B Cell Ontogeny, and Epitope Structure. Immunity 48, 855–871. 10.1016/j.immuni2012.04.029. [DOI] [PubMed] [Google Scholar]
- 17.McLellan JS, Pancera M, Carrico C, Gorman J, Julien JP, Khayat R, Louder R, Pejchal R, Sastry M, Dai K, et al. (2011). Structure of HIV-1 gp120 V1/V2 domain with broadly neutralizing antibody PG9. Nature 480, 336–343. 10.1038/nature10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walker LM, Phogat SK, Chan-Hui PY, Wagner D, Phung P, Goss JL, Wrin T, Simek MD, Fling S, Mitcham JL, et al. (2009). Broad and potent neutralizing antibodies from an african donor reveal a new HIV-1 vaccine target. Science 326, 285–289. 10.1126/science.1178746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonsignori M, Hwang KK, Chen X, Tsao CY, Morris L, Gray E,Marshall DJ, Crump JA, Kapiga SH, Sam NE, et al. (2011). Analysis of a Clonal Lineage of HIV-1 Envelope V2/V3 Conformational Epitope-Specific Broadly Neutralizing Antibodies and Their Inferred Unmutated Common Ancestors. J. Virol 85, 9998–10009. 10.1128/jvi.05045-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andrabi R, Voss JE, Liang CH, Briney B, McCoy LE, Wu CY, Wong CH, Poignard P, and Burton DR (2015). Identification of Common Features in Prototype Broadly Neutralizing Antibodies to HIV Envelope V2 Apex to Facilitate Vaccine Design. Immunity 43, 959–973. 10.1016/j.immuni.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gorman J, Soto C, Yang MM, Davenport TM, Guttman M, Bailer RT, Chambers M, Chuang GY, DeKosky BJ, Doria-Rose NA, et al. (2016). Structures of HIV-1 Env V1V2 with broadly neutralizing antibodies reveal commonalities that enable vaccine design. Nat. Struct. Mol. Biol 23, 81–90. 10.1038/nsmb.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walker LM, Huber M, Doores KJ, Falkowska E, Pejchal R, Julien JP, Wang SK, Ramos A, Chan-Hui PY, Moyle M, et al. (2011). Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 477, 466–470. 10.1038/nature10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sok D, van Gils MJ, Pauthner M, Julien JP, Saye-Francisco KL,Hsueh J, Briney B, Lee JH, Le KM, Lee PS, et al. (2014). Recombinant HIV envelope trimer selects for quaternary-dependent antibodies targeting the trimer apex. Proc. Natl. Acad. Sci. USA 111, 17624–17629. 10.1073/pnas.1415789111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee JH, Andrabi R, Su CY, Yasmeen A, Julien JP, Kong L, Wu NC, McBride R, Sok D, Pauthner M, et al. (2017). A Broadly Neutralizing Antibody Targets the Dynamic HIV Envelope Trimer Apex via a Long, Rigidified, and Anionic b-Hairpin Structure. Immunity 46, 690–702. 10.1016/j.immuni.2017.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Q, Acharya P, Dolan MA, Zhang P, Guzzo C, Lu J, Kwon A,Gururani D, Miao H, Bylund T, et al. (2017). Quaternary contact in the initial interaction of CD4 with the HIV-1 envelope trimer. Nat. Struct. Mol. Biol 24, 370–378. 10.1038/nsmb.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rantalainen K, Berndsen ZT, Murrell S, Cao L, Omorodion O,Torres JL, Wu M, Umotoy J, Copps J, Poignard P, et al. (2018). Co-evolution of HIV Envelope and Apex-Targeting Neutralizing Antibody Lineage Provides Benchmarks for Vaccine Design. Cell Rep. 23, 3249–3261. 10.1016/j.celrep.2018.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gorman J, Chuang GY, Lai YT, Shen CH, Boyington JC, Druz A,Geng H, Louder MK, McKee K, Rawi R, et al. (2020). Structure of Super-Potent Antibody CAP256-VRC26.25 in Complex with HIV-1 Envelope Reveals a Combined Mode of Trimer-Apex Recognition. Cell Rep. 31, 107488. 10.1016/j.celrep.2020.03.052. [DOI] [PubMed] [Google Scholar]
- 28.Pancera M, Shahzad-Ul-Hussan S, Doria-Rose NA, McLellan JS,Bailer RT, Dai K, Loesgen S, Louder MK, Staupe RP, Yang Y, et al. (2013). Structural basis for diverse N-glycan recognition by HIV-1neutralizing V1-V2-directed antibody PG16. Nat. Struct. Mol. Biol 20, 804–813. 10.1038/nsmb.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cale EM, Gorman J, Radakovich NA, Crooks ET, Osawa K, Tong T, Li J, Nagarajan R, Ozorowski G, Ambrozak DR, et al. (2017). Viruslike Particles Identify an HIV V1V2 Apex-Binding Neutralizing Antibody that Lacks a Protruding Loop. Immunity 46, 777–791.e10. 10.1016/j.immuni.2017.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barbas CF, Hu D, Dunlop N, Sawyer L, Cababa D, Hendry RM,Nara PL, and Burton DR (1994). In vitro evolution of a neutralizing human antibody to human immunodeficiency virus type 1 to enhance affinity and broaden strain cross-reactivity. Proc. Natl. Acad. Sci. USA 91, 3809–3813. 10.1073/pnas.91.9.3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Diskin R, Scheid JF, Marcovecchio PM, West AP Jr., Klein F, Gao H, Gnanapragasam PNP, Abadir A, Seaman MS, Nussenzweig MC, and Bjorkman PJ (2011). Increasing the potency and breadth of an HIV antibody by using structure-based rational design. Science 334, 1289–1293. 10.1126/science.1213782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Willis JR, Sapparapu G, Murrell S, Julien JP, Singh V, King HG,Xia Y, Pickens JA, LaBranche CC, Slaughter JC, et al. (2015). Redesigned HIV antibodies exhibit enhanced neutralizing potency and breadth. J. Clin. Invest 125, 2523–2531. 10.1172/JCI80693. Cell Reports 42, 112711, July 25, 2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu NC, Grande G, Turner HL, Ward AB, Xie J, Lerner RA, andWilson IA (2017). In vitro evolution of an influenza broadly neutralizing antibody is modulated by hemagglutinin receptor specificity. Nat. Commun 8, 15371. 10.1038/ncomms15371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Starr TN, Czudnochowski N, Liu Z, Zatta F, Park YJ, Addetia A,Pinto D, Beltramello M, Hernandez P, Greaney AJ, et al. (2021). SARS-CoV-2 RBD antibodies that maximize breadth and resistance to escape. Nature 597, 7874. 10.1038/s41586-021-03807-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rappazzo CG, Tse LV, Kaku CI, Wrapp D, Sakharkar M, Huang D, Deveau LM, Yockachonis TJ, Herbert AS, Battles MB, et al. (2021). Broad and potent activity against SARS-like viruses by an engineered human monoclonal antibody. Science 371, 823–829. 10.1126/science.abf4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lippow SM, Wittrup KD, and Tidor B (2007). Computational design ofantibody-affinity improvement beyond in vivo maturation. Nat. Biotechnol 25, 1171–1176. 10.1038/nbt1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sevy AM, Wu NC, Gilchuk IM, Parrish EH, Burger S, Yousif D,Nagel MB, Schey KL, Wilson IA, Crowe JE Jr., et al. (2019). Multistate design of influenza antibodies improves affinity and breadth against seasonal viruses. Proc. Natl. Acad. Sci. USA 116, 1597–1602. 10.1073/pnas.1806004116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tharakaraman K, Robinson LN, Hatas A, Chen YL, Siyue L, Ra-guram S, Sasisekharan V, Wogan GN, and Sasisekharan R (2013). Redesign of a cross-reactive antibody to dengue virus with broad-spectrum activity and increased in vivo potency. Proc. Natl. Acad. Sci. USA 110, E1555–E1564. 10.1073/pnas.1303645110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barderas R, Desmet J, Timmerman P, Meloen R, and Casal JI(2008). Affinity maturation of antibodies assisted by in silico modeling. Proc. Natl. Acad. Sci. USA 105, 9029–9034. 10.1073/pnas.0801221105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simonelli L, Pedotti M, Beltramello M, Livoti E, Calzolai L, Sallusto F, Lanzavecchia A, and Varani L (2013). Rational Engineering of a Human Anti-Dengue Antibody through Experimentally Validated Computational Docking. PLoS One 8, e55561. 10.1371/journal.pone.0055561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kratochvil S, Shen CH, Lin YC, Xu K, Nair U, Da Silva Pereira L,Tripathi P, Arnold J, Chuang GY, Melzi E, et al. (2021). Vaccination in a humanized mouse model elicits highly protective PfCSP-targeting anti-malarial antibodies. Immunity 54, 2859–2876.e7. 10.1016/j.immuni.2021.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kwon YD, Chuang GY, Zhang B, Bailer RT, Doria-Rose NA, Gin-din TS, Lin B, Louder MK, McKee K, O’Dell S, et al. (2018). Surface-Matrix Screening Identifies Semi-specific Interactions that Improve Potency of a Near Pan-reactive HIV-1-Neutralizing Antibody. Cell Rep. 22, 1798–1809. 10.1016/j.celrep.2018.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kwon YD, Asokan M, Gorman J, Zhang B, Liu Q, Louder MK, Lin BC, McKee K, Pegu A, Verardi R, et al. (2021). A matrix of structure-based designs yields improved VRC01- class antibodies for HIV-1 therapy and prevention.mAbs13. 10.1080/19420862.2021.1946918. [DOI] [PMC free article] [PubMed]
- 44.Hallen MA, Martin JW, Ojewole A, Jou JD, Lowegard AU, Frenkel MS, Gainza P, Nisonoff HM, Mukund A, Wang S, et al. (2018). OSPREY 3.0: Open-Source Protein Redesign for You, with Powerful New Features”. J. Comput. Chem 39, 2494–2507. 10.1002/jcc.25522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lilien RH, Stevens BW, Anderson AC, and Donald BR (2005). Anovel ensemble-based scoring and search algorithm for protein redesign and its application to modify the substrate specificity of the gramicidin synthetase a phenylalanine adenylation enzyme. J. Comput. Biol 12, 740–761. 10.1089/cmb.2005.12.740. [DOI] [PubMed] [Google Scholar]
- 46.Georgiev I, Lilien RH, and Donald BR (2008). The minimized dead-end elimination criterion and its application to p rotein redesign in a hybrid scoring and search algorithm for computing partition funct ions over molecular ensembles. J. Comput. Chem 29, 1527–1542. 10.1002/jcc.20909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Donald Bruce R. (2011). Algorithms in Structural Molecular Biology (The MIT Press; ). [Google Scholar]
- 48.Gainza P, Roberts KE, and Donald BR (2012). Protein design usingcontinuous rotamers. PLoS Comput. Biol 8, e1002335. 10.1371/journal.pcbi.1002335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dennis RB, Williamson RA, and Parren PWHI (2000). MINIREVIEWAntibody and Virus: Binding and Neutralization. Virology 270, 1–3. 10.1006/viro.2000.0239. [DOI] [PubMed] [Google Scholar]
- 50.Willis JR, Sapparapu G, Murrell S, Julien JP, Singh V, King HG, Xia Y, Pickens JA, LaBranche CC, Slaughter JC, et al. (2015). Redesigned HIV antibodies exhibit enhanced neutralizing potency and b readth. J. Clin. Invest 125, 2523–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ojewole AA, Jou JD, Fowler VG, and Donald BR (2018). BBK*(Branch and Bound Over K*): A Provable and Efficient Ensemble-Based Protein Design Algorithm to Optimize Stability and Binding Affinity Over Large Sequence Spaces. J. Comput. Biol 25, 1557–8666. 10.1089/cmb.2017.0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rawi R, Mall R, Shen CH, Farney SK, Shiakolas A, Zhou J, Bens-mail H, Chun TW, Doria-Rose NA, Lynch RM, et al. (2019). Accurate Prediction for Antibody Resistance of Clinical HIV-1 Isolates. Sci. Rep 9, 14696. 10.1038/s41598-019-50635-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Breiman L (2001). Random Forests. Mach. Learn 45, 5–32. [Google Scholar]
- 54.Minoux H, and Chipot C (1999). Cation-p interactions in proteins: Can simple models provide an accurate description? J. Am. Chem. Soc 121, 10366–10372. 10.1021/ja990914p. [DOI] [Google Scholar]
- 55.Adolf-Bryfogle J, Kalyuzhniy O, Kubitz M, Weitzner BD, Hu X, Adachi Y, Schief WR, and Dunbrack RL Jr. (2018). RosettaAntibodyDesign (RAbD): A general framework for computational antibody design. PLoS Comput. Biol 14, e1006112. 10.1371/journal.pcbi.1006112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pantazes RJ, and Maranas CD (2010). OptCDR: a general computational method for the design of antibody complementarity determining regions for targeted epitope binding. Protein Eng. Des. Sel 23, 849–858. 10.1093/protein/gzq061. [DOI] [PubMed] [Google Scholar]
- 57.Tong L, Pantazes RJ, and Maranas CD (2014). OptMAVEn – A NewFramework for the de novo Design of Antibody Variable Region Models Targeting Specific Antigen Epitopes. PLoS One 9, e105954. 10.1371/journal.pone.0105954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chowdhury R, Allan MF, and Maranas CD (2018). OptMAVEn-2.0: Denovo Design of Variable Antibody Regions Against Targeted Antigen Epitopes. Antibodies 7, 23. 10.3390/antib7030023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lapidoth GD, Baran D, Pszolla GM, Norn C, Alon A, Tyka MD, and Fleishman SJ (2015). AbDesign: An algorithm for combinatorial backbone design guided by natural conformations and sequences. Proteins 83, 1385–1406. 10.1002/prot.24779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ruffolo JA, Sulam J, and Gray JJ (2022). Antibody structure prediction using interpretable deep learning. Patterns 3, 100406. 10.1016/j.patter.2021.100406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Alexander M (2015). Sevy et al. “Design of Protein Multi-specificity Usingan Independent Sequence Search Reduces the Barrier to Low Energy Sequences”. PLoS Comput. Biol 11, e1004300. 10.1371/journal.pcbi.1004300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rawi R, Rutten L, Lai YT, Olia AS, Blokland S, Juraszek J, Shen CH, Tsybovsky Y, Verardi R, Yang Y, et al. (2020). Automated Design by Structure-Based Stabilization and Consensus Repair to Achieve Prefusion-Closed Envelope Trimers in a Wide Variety of HIV Strains. Cell Rep. 33, 108432. 10.1016/j.celrep.2020.108432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chuang GY, Zhou J, Acharya P, Rawi R, Shen CH, Sheng Z,Zhang B, Zhou T, Bailer RT, Dandey VP, et al. (2019). Structural Survey of Broadly Neutralizing Antibodies Targeting the HIV-1 Env Trimer Delineates Epitope Categories and Characteristics of Recognition. Structure 27, 196–206.e6. 10.1016/j.str.2018.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen VB, Davis IW, and Richardson DC (2009). KiNG (Kinemage,Next Generation): A versatile interactive molecular and scientific visualization program. Protein Sci. 18, 2403–2409. 10.1002/pro.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera - A visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 66.Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J,Stagg S, Potter CS, and Carragher B (2005). Automated molecular microscopy: The new Leginon system. J. Struct. Biol 151, 41–60. 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 67.Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). Cryo-SPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296. 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- 68.Barad BA, Echols N, Wang RYR, Cheng Y, DiMaio F, Adams PD, and Fraser JS (2015). EMRinger: side chain–directed model and map validation for 3D cryo-electron microscopy. Nat. Methods 12, 943–946. 10.1038/nmeth.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Adams PD, Afonine PV, Bunkó czi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Cryst 66, 213–221. 10.1107/s0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Cryst 60, 2126–2132. 10.1107/s0907444904019158. [DOI] [PubMed] [Google Scholar]
- 71.Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL,Deis LN, Verma V, Keedy DA, Hintze BJ, Chen VB, et al. (2018). MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315. 10.1002/pro.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lowegard AU, Frenkel MS, Holt GT, Jou JD, Ojewole AA, andDonald BR (2020). Novel, provable algorithms for efficient ensemble-based computational protein design and their application to the redesign of the c-Raf- RBD:KRas protein-protein interface. PLoS Comput. Biol 16, e1007447. 10.1371/journal.pcbi.1007447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Julien JP, Lee JH, Cupo A, Murin CD, Derking R, Hoffenberg S,Caulfield MJ, King CR, Marozsan AJ, Klasse PJ, et al. (2013). Asymmetric recognition of the HIV-1 trimer by broadly neutralizing antibody PG9. Proc. Natl. Acad. Sci. USA 110, 4351–4356. 10.1073/pnas.1217537110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Krieger E, Joo K, Lee J, Lee J, Raman S, Thompson J, Tyka M, Baker D, and Karplus K (2009). Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: four approaches that performed well in CASP8. Proteins: Struct., Funct., Bioinf 77, 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Word JM, Lovell SC, Richardson JS, and Richardson DC (1999). Asparagine and glutamine: Using hydrogen atom contacts in the choice of side-chain amide orientation. J. Mol. Biol 285, 1735–1747. 10.1006/jmbi.1998.2401. [DOI] [PubMed] [Google Scholar]
- 76.Lovell SC, Word JM, Richardson JS, and Richardson DC (2000). The penultimate rotamer library. Proteins 40, 389–408. [PubMed] [Google Scholar]
- 77.Wang S (2021). Computational Protein Design with Non-proteinogenic Amino Acids and Small Molecule Ligands, with Applications to ProteinProtein Interaction Inhibitors, Anti-microbial Enzyme Inhibitors, and Antibody Design (Duke University). PhD thesis. [Google Scholar]
- 78.Case DA, Darden TA, Cheatham TE III, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Pearlman DA, Crowley M, et al. (2006). AMBER 9 (University of California; ), p. 45. [Google Scholar]
- 79.Sarzotti-Kelsoe M, Bailer RT, Turk E, Lin CL, Bilska M, Greene KM, Gao H, Todd CA, Ozaki DA, Seaman MS, et al. (2014). Optimization and validation of the TZM-bl assay for standardized assessments Cell Reports 42, 112711, July 25, 2023 of neutralizing antibodies against HIV-1. J. Immunol. Methods 409, 131–146. 10.1016/j.jim.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu X, Yang ZY, Li Y, Hogerkorp CM, Schief WR, Seaman MS,Zhou T, Schmidt SD, Wu L, Xu L, et al. (2010). Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329, 856–861. 10.1126/science.1187659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pedregosa F, Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, et al. (2011). Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res 12, 2825–2830. [Google Scholar]
- 82.Cock PJ, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A,Friedberg I, Hamelryck T, Kauff F, Wilczynski B, et al. (2009). Bio-Cell Reports 42, 112711, July 25, 2023 python: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423. 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Friedman JH (2001). Greedy function approximation: A gradient boosting machine. Ann. Statist 29, 1189–1232. 10.1214/aos/1013203451. [DOI] [Google Scholar]
- 84.Hunter JD (2007). Matplotlib: A 2D graphics environment. Comput. Sci. Eng 9, 90–95. 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
- 85.Waskom Michael L. (2021). “seaborn: statistical data visualization”.J. Open Source Softw 6, 3021. 10.21105/joss.03021. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Neutralization data are included in this manuscript in the supplemental information. Cryo-EM maps and structures have been deposited in the EMDB (EMDB: EMD-299248, EMD-29264, EMD-29288) and the PDB (PDB: 8FK5, 8FL1, 8FLW), respectively, and are publicly available as of the date of publication.
All code used and discussed in this manuscript is available from the Harvard Dataverse repository (Harvard Dataverse: https://doi.org/10.7910/DVN/NXD2JR) as of the date of publication. For new empirical designs we recommend using the latest version of OSPREY available for free at http://www.cs.duke.edu/donaldlab/opsrey.php. All computer code for the OSPREY system is also available on GitHub at https://github.com/donaldlab/OSPREY3, and is open/source and free. The version of record is archived with Zenodo (Zenodo: https://doi.org/10.5281/zenodo.7633931).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.